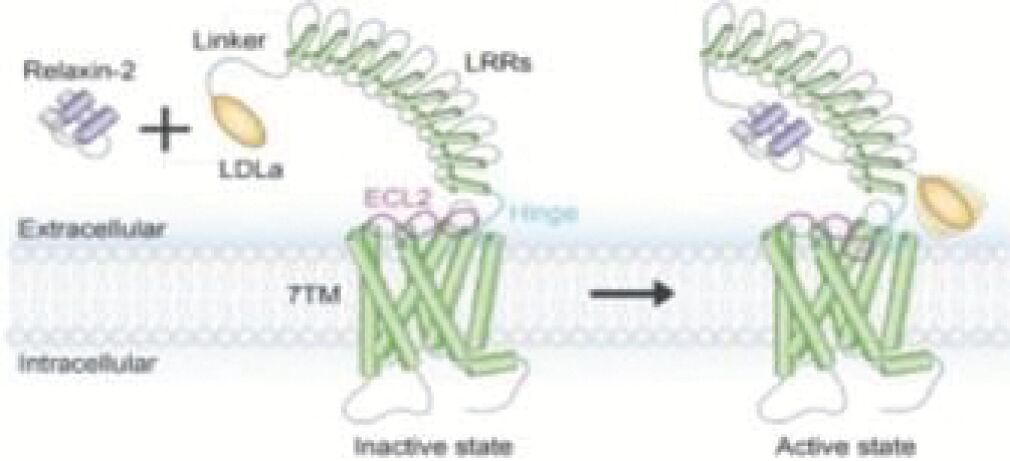
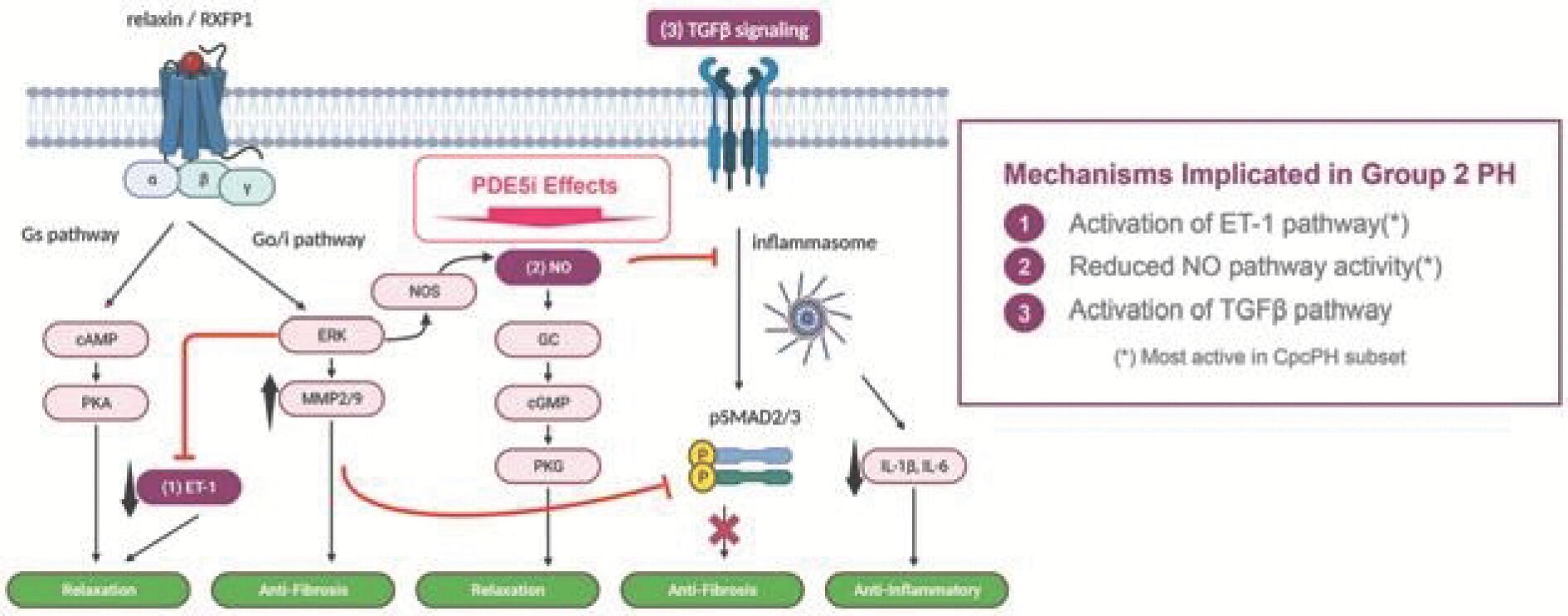
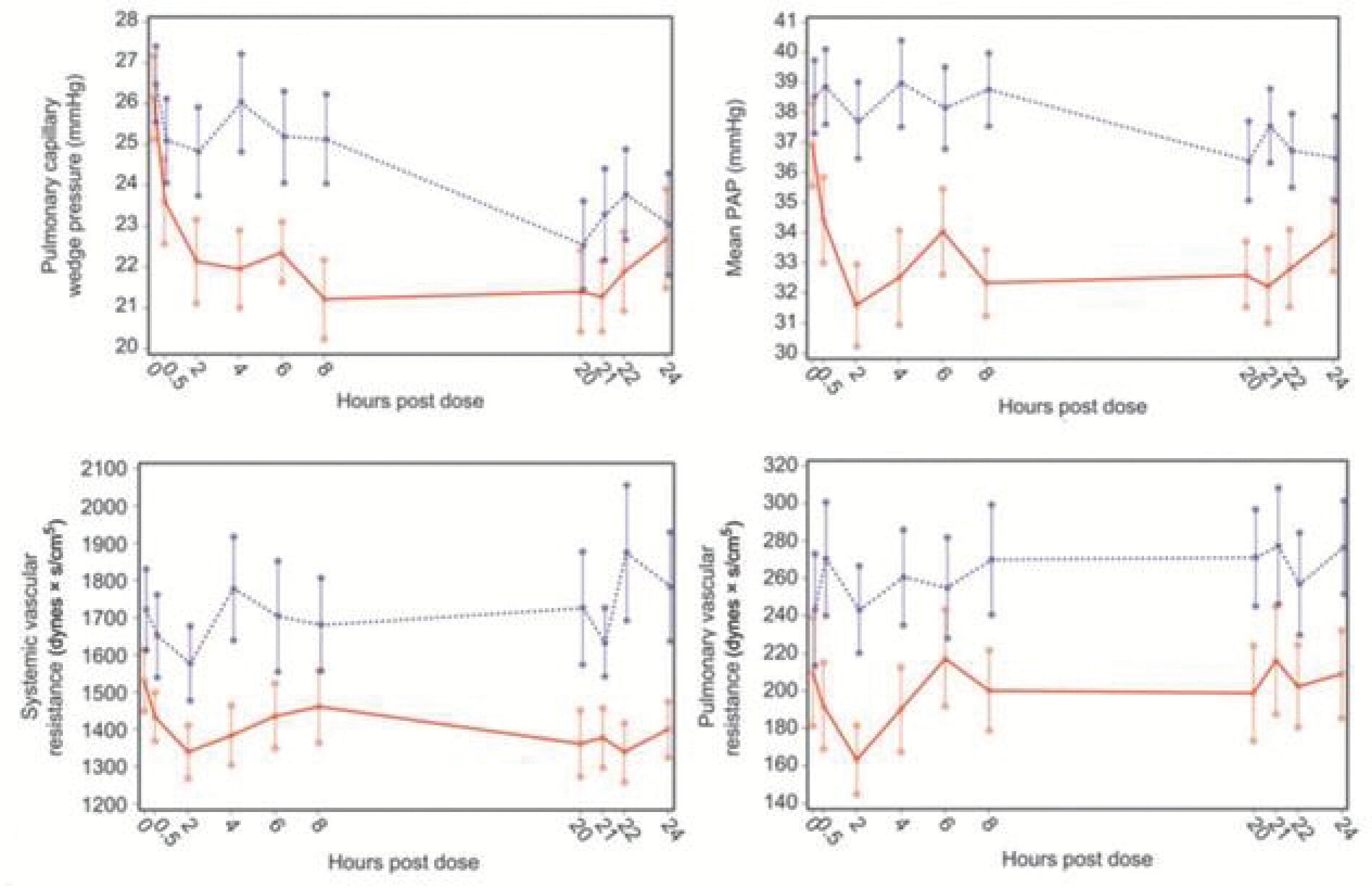
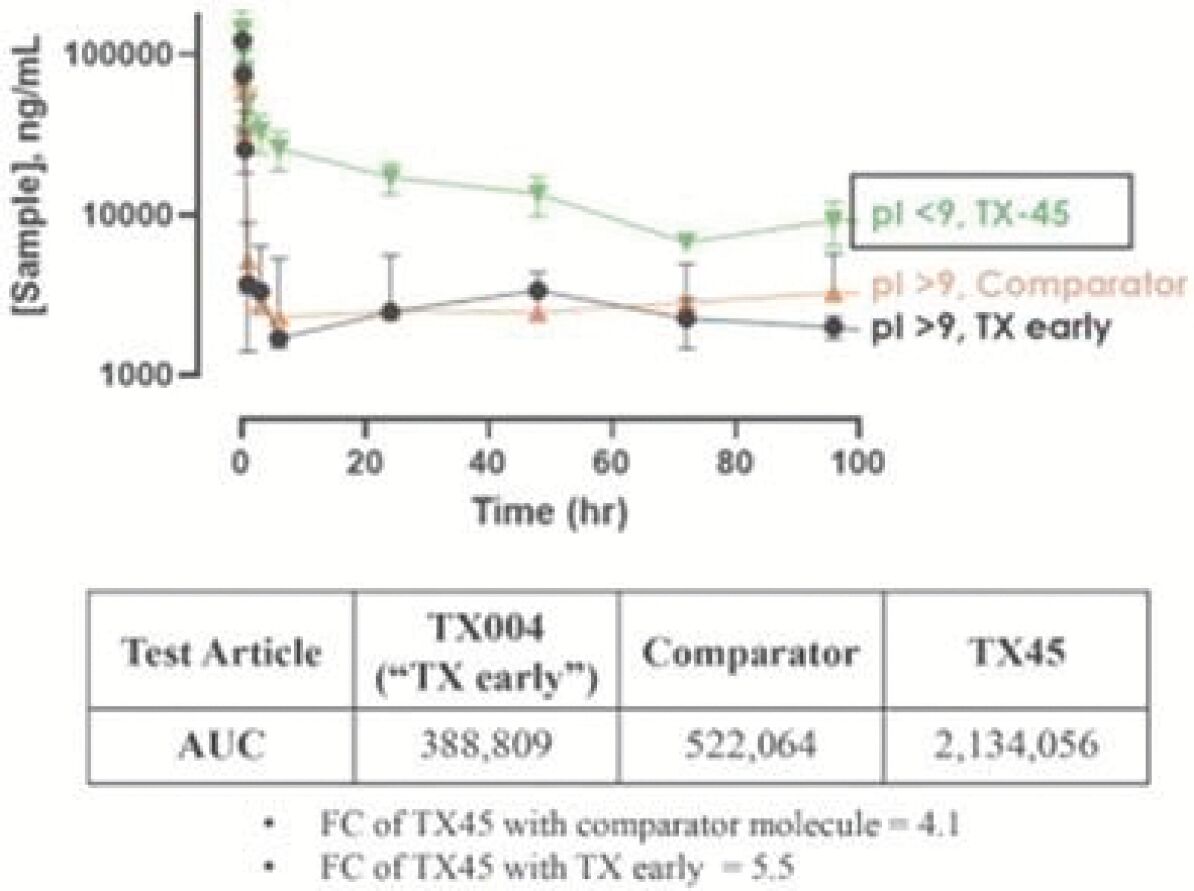
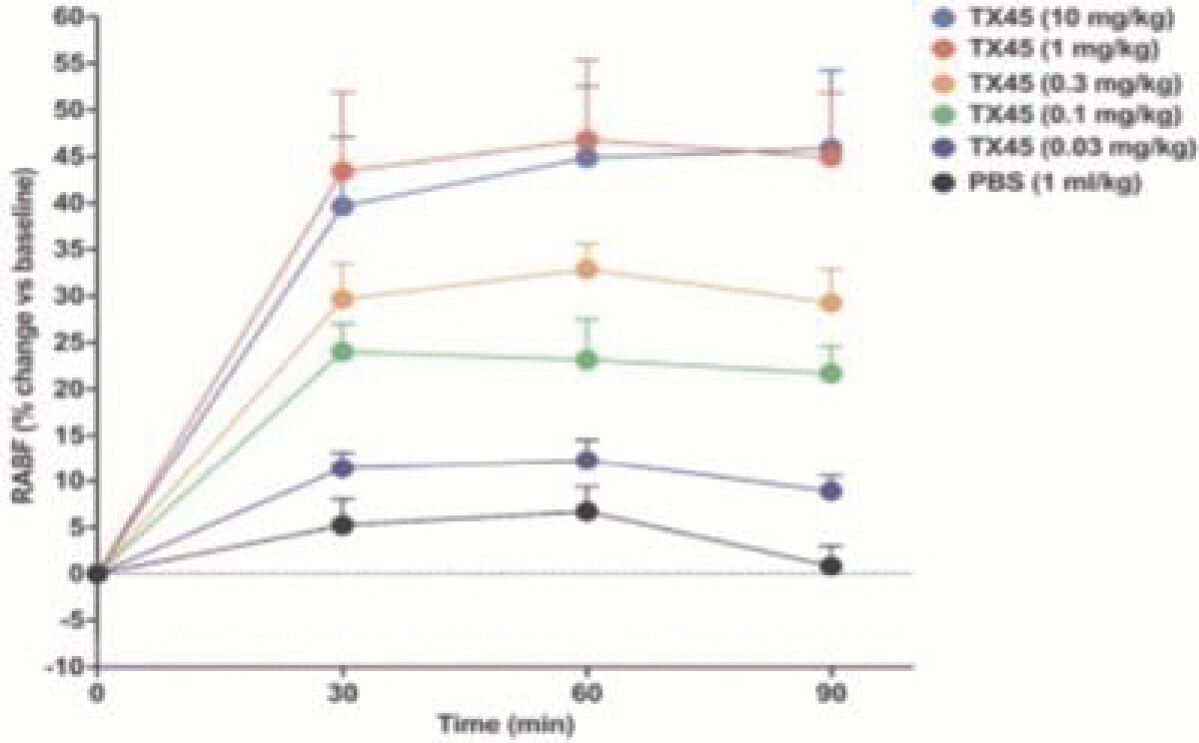
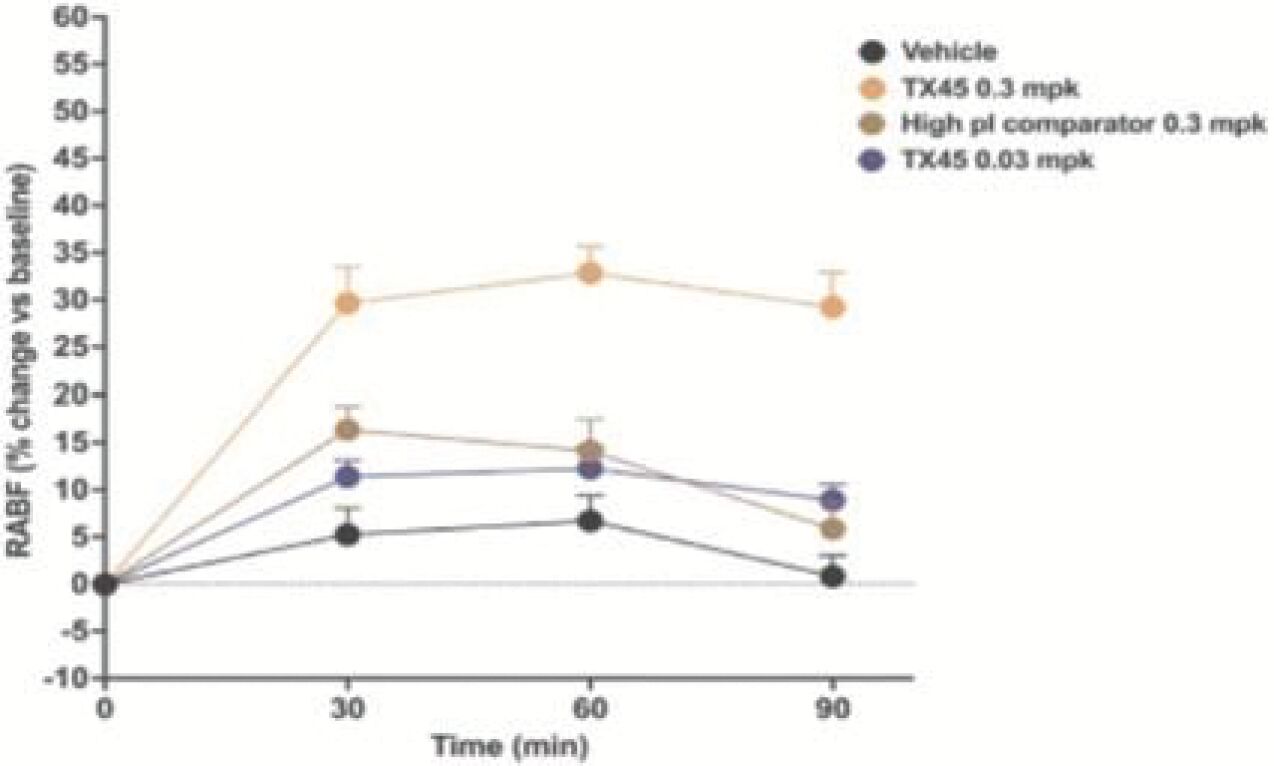
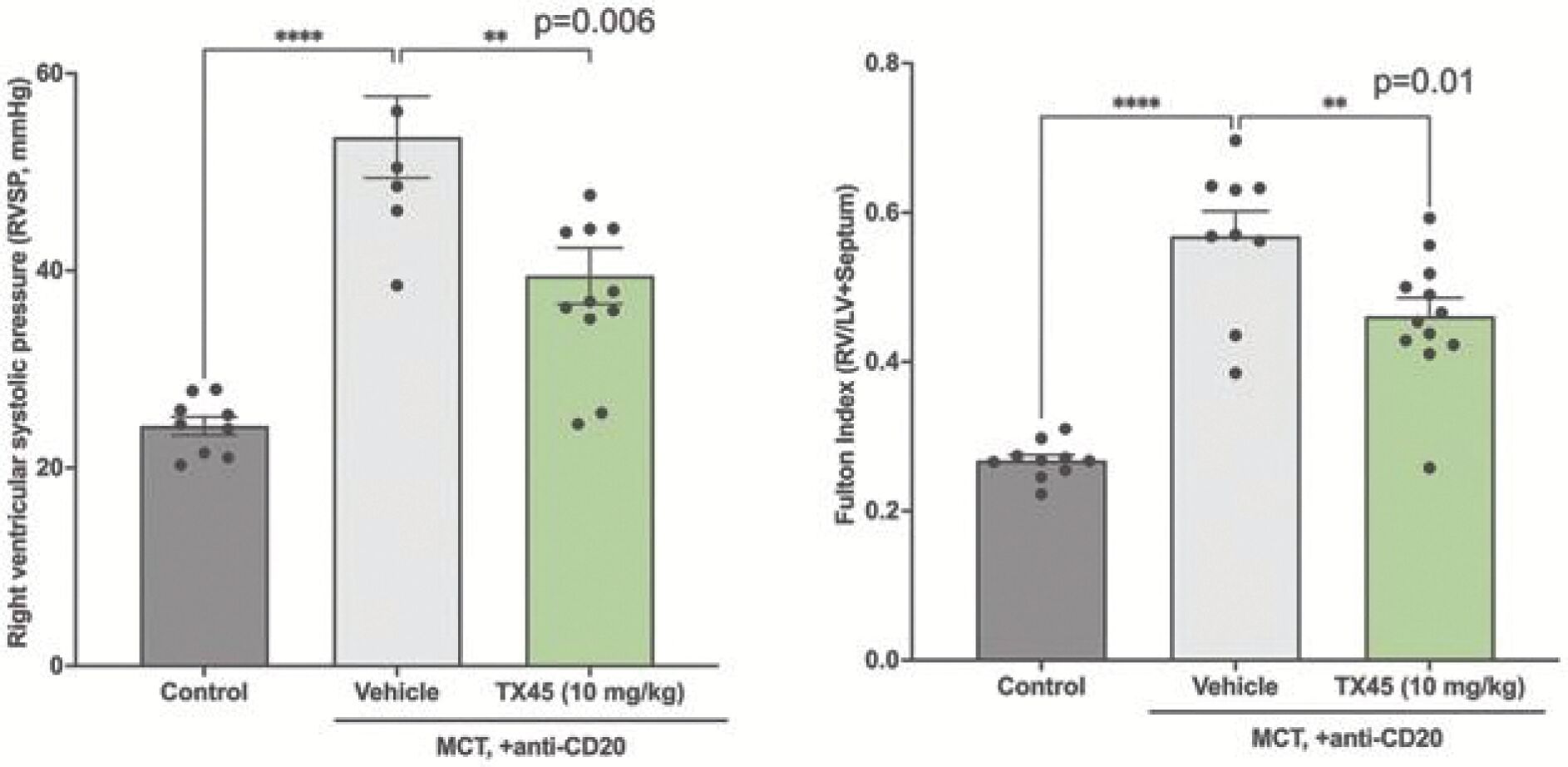
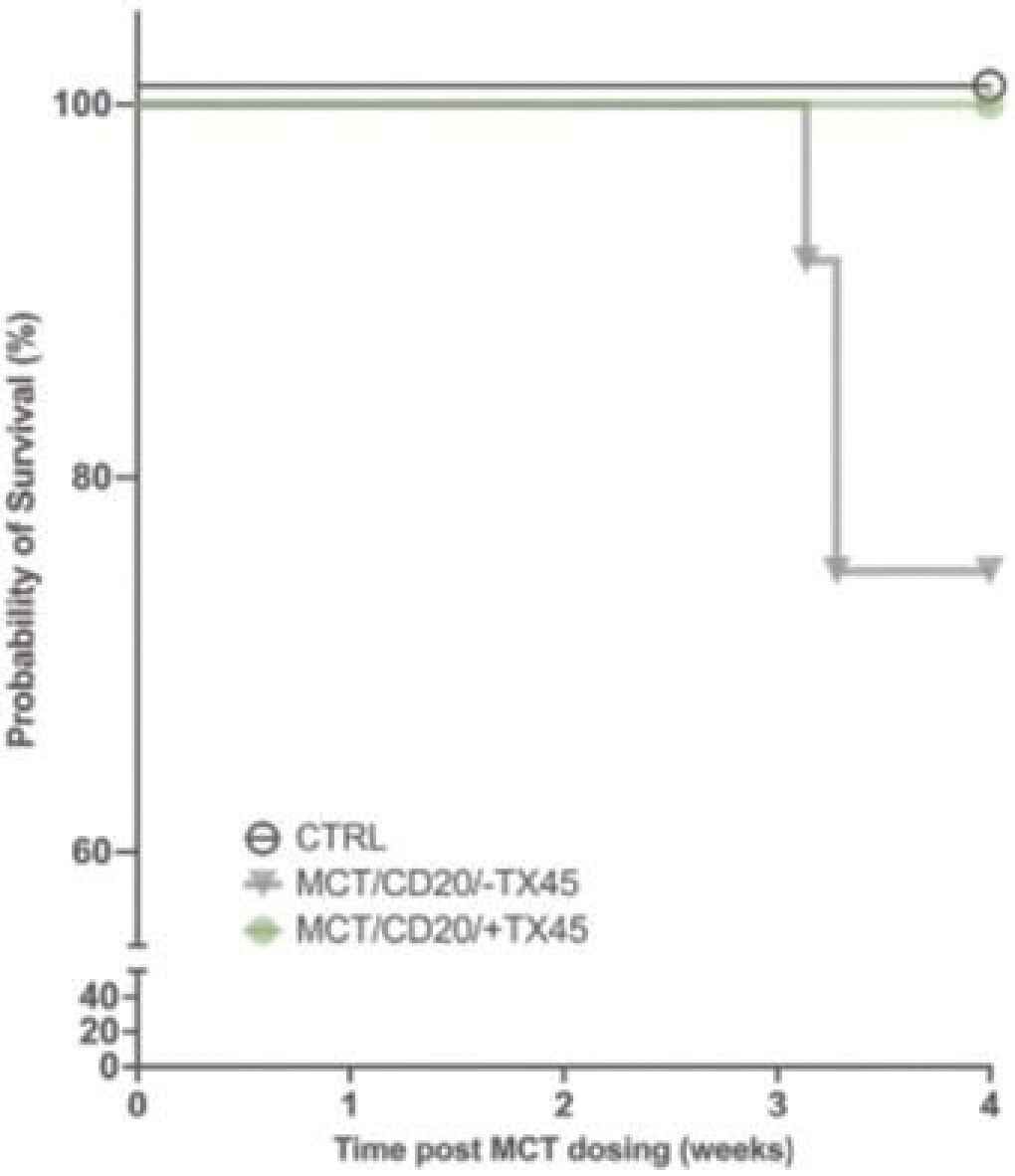
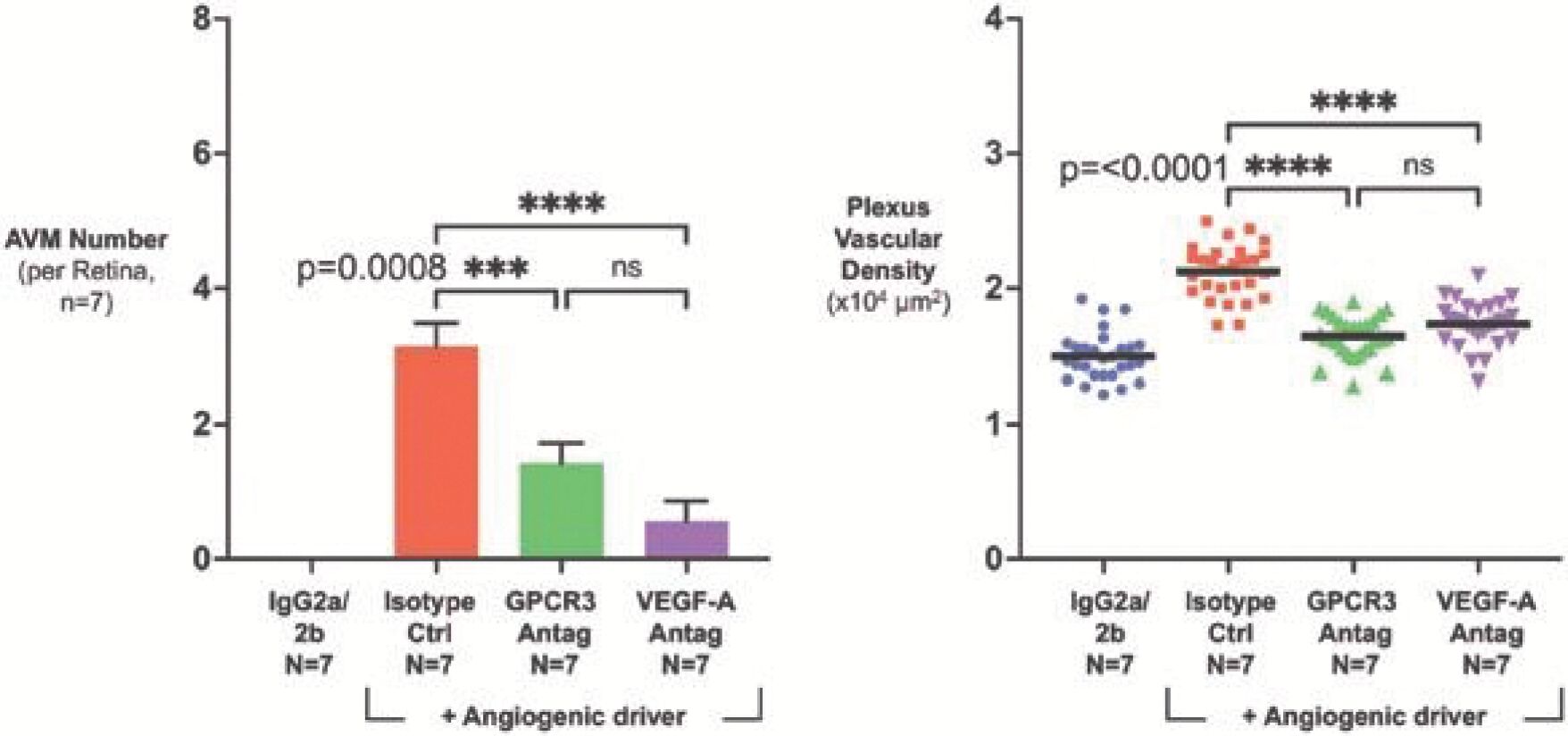
The information in this preliminary prospectus is not complete and may be changed. Neither we nor the selling securityholders may sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED July 19, 2024
PRELIMINARY PROSPECTUS

Up to 2,969,583 Shares of Common Stock
This prospectus relates to the proposed offer and resale or other disposition from time to time by the selling stockholders identified in this prospectus of up to an aggregate of 2,969,583 shares of common stock, par value $0.0001 per share, of Tectonic Therapeutic, Inc.
We are registering the resale of the shares of common stock pursuant to the selling stockholders’ registration rights under the Subscription Agreement between us and the selling stockholders. Our registration of the resale of the shares of common stock covered by this prospectus does not mean that the selling stockholders will offer or sell all or any of the shares of common stock. The selling stockholders may offer, sell or distribute all or a portion of their shares of common stock from time to time directly or indirectly through one or more underwriters, broker-dealers or agents, and in one or more public or private transactions, which may involve crosses or block transactions. The shares of common stock may be sold in one or more transactions at fixed prices, at prevailing market prices at the time of the sale, at varying prices determined at the time of sale or at negotiated prices. See the section entitled “Plan of Distribution” for more information.
We will not receive any proceeds from any sale of common stock by the selling stockholders pursuant to this prospectus. We have agreed to bear the expenses in connection with the registration of the resale of the shares of common stock to be offered by this prospectus by the selling stockholders except for any underwriting discounts and commissions or transfer taxes relating to the sale of common stock, which will be borne by the selling stockholders.
Our common stock is listed on the Nasdaq Global Market (“Nasdaq”) under the symbol “TECX.” On July 12, 2024, the closing price for our common stock, as reported on Nasdaq, was $16.94 per share.
See the section entitled “Risk Factors” beginning on page 9 of this prospectus to read about factors you should consider before buying our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2024.