Executive Transition
On June 30, 2022, Steve R. Martin retired as our Chief Financial Officer and Senior Vice President (and Principal Financial and Accounting Officer). Mr. Martin agreed to continue his employment with us as an advisor to his successor in the Principal Financial and Accounting Officer role from July 1, 2022 through December 31, 2022.
On March 25, 2022, the Company’s board of directors (the “Board of Directors”) appointed Erin Butler to serve as (i) Vice President of Finance and Administration of the Company, effective April 1, 2022, and (ii) Principal Financial and Accounting Officer of the Company, effective July 1, 2022.
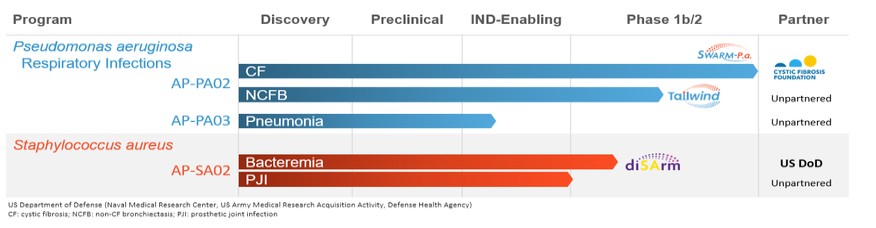
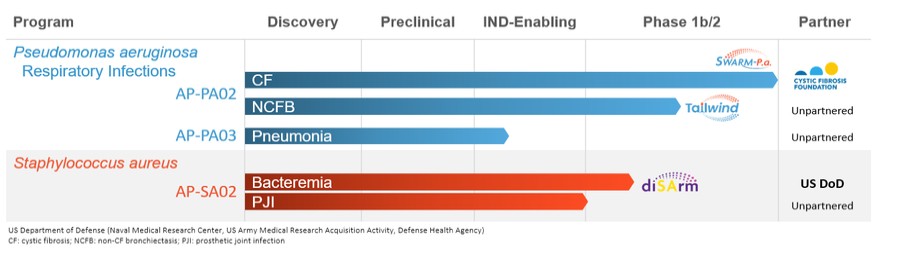
Pipeline
The following chart summarizes the status of our phage product candidate development programs and partners.
Strategy
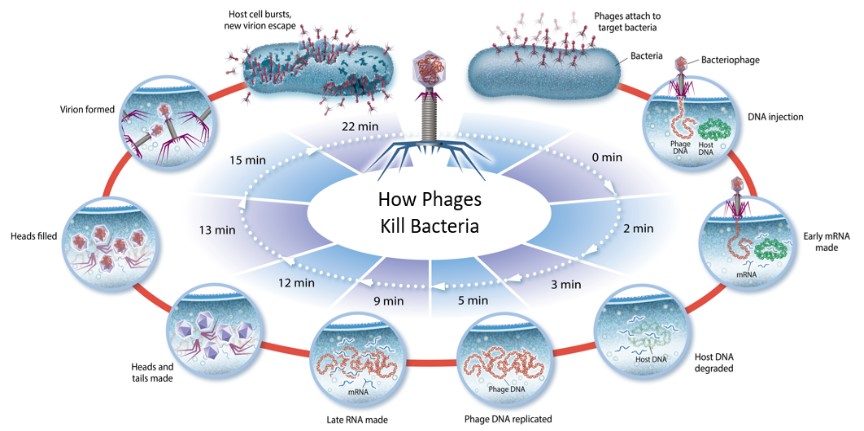
Our strategy is to demonstrate the safety, tolerability and efficacy of multiple phage products in randomized controlled clinical trials required for FDA approval and to support commercialization in multiple indications of high unmet medical need, including bacterial infections caused by multidrug-resistant and difficult-to-treat pathogens. Our fully integrated product development capabilities from bench to clinic enable the discovery of optimal product candidates, efficient process development and manufacturing, and rigorous clinical trials. Our microbiological surveillance and synthetic biology capabilities drive long-term product life cycle management. We intend to:
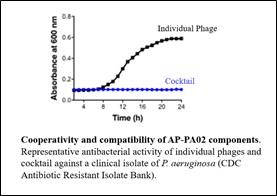
| ● | Advance clinical trials of AP-PA02 in patients with CF and chronic pulmonary P. aeruginosa infections. |
| ● | Develop bacteriophage therapeutics, including AP-PA02 and AP-PA03, for the treatment of other antibiotic-resistant and difficult-to-treat P. aeruginosa infections such as NCFB and hospitalized pneumonia. |
| ● | Initiate and advance clinical trials of AP-SA02 in patients with S. aureus bacteremia. |
| ● | Develop AP-SA02 for the treatment of other antibiotic-resistant and difficult-to-treat S. aureus infections such as PJI. |
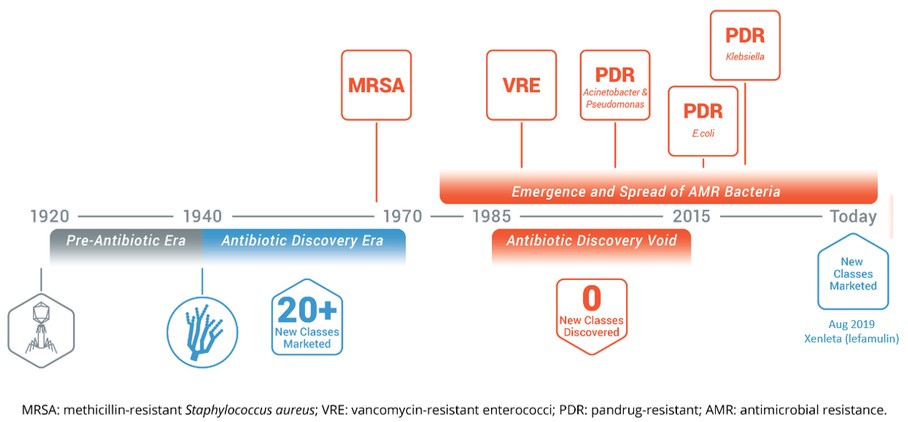
The Need for New Anti-Infective Therapies
The introduction of penicillin in the early 1940s marked the start of the antibiotic discovery era, during which more than 20 new classes of antibiotic were marketed over a period of three decades. The first case of the “superbug”, Methicillin-resistant S. aureus (“MRSA”), in the United States occurred in 1968. A void in the discovery of new classes of antibiotics lasting approximately 30 years drove the emergence and spread of antibiotic-resistant bacteria, including
8