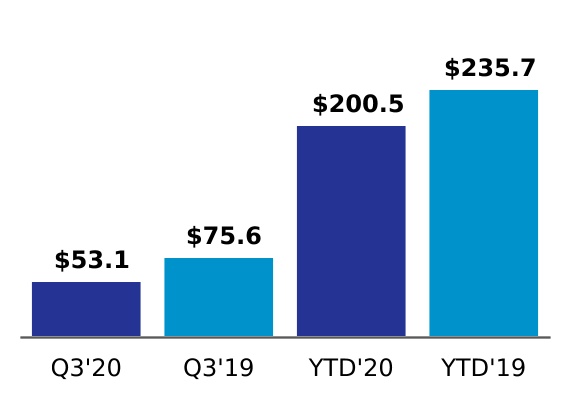000089986612/312020Q3FALSE00008998662020-01-012020-09-30xbrli:shares00008998662020-10-27iso4217:USD00008998662020-09-3000008998662019-12-31iso4217:USDxbrli:shares0000899866alxn:NetProductSalesMember2020-07-012020-09-300000899866alxn:NetProductSalesMember2019-07-012019-09-300000899866alxn:NetProductSalesMember2020-01-012020-09-300000899866alxn:NetProductSalesMember2019-01-012019-09-300000899866alxn:OtherRevenueMember2020-07-012020-09-300000899866alxn:OtherRevenueMember2019-07-012019-09-300000899866alxn:OtherRevenueMember2020-01-012020-09-300000899866alxn:OtherRevenueMember2019-01-012019-09-3000008998662020-07-012020-09-3000008998662019-07-012019-09-3000008998662019-01-012019-09-300000899866us-gaap:CommonStockMember2020-06-300000899866us-gaap:AdditionalPaidInCapitalMember2020-06-300000899866us-gaap:TreasuryStockMember2020-06-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2020-06-300000899866us-gaap:RetainedEarningsMember2020-06-3000008998662020-06-300000899866us-gaap:TreasuryStockMember2020-07-012020-09-300000899866us-gaap:CommonStockMember2020-07-012020-09-300000899866us-gaap:AdditionalPaidInCapitalMember2020-07-012020-09-300000899866us-gaap:RetainedEarningsMember2020-07-012020-09-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2020-07-012020-09-300000899866us-gaap:CommonStockMember2020-09-300000899866us-gaap:AdditionalPaidInCapitalMember2020-09-300000899866us-gaap:TreasuryStockMember2020-09-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2020-09-300000899866us-gaap:RetainedEarningsMember2020-09-300000899866us-gaap:CommonStockMember2019-06-300000899866us-gaap:AdditionalPaidInCapitalMember2019-06-300000899866us-gaap:TreasuryStockMember2019-06-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2019-06-300000899866us-gaap:RetainedEarningsMember2019-06-3000008998662019-06-300000899866us-gaap:TreasuryStockMember2019-07-012019-09-300000899866us-gaap:CommonStockMember2019-07-012019-09-300000899866us-gaap:AdditionalPaidInCapitalMember2019-07-012019-09-300000899866us-gaap:RetainedEarningsMember2019-07-012019-09-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2019-07-012019-09-300000899866us-gaap:CommonStockMember2019-09-300000899866us-gaap:AdditionalPaidInCapitalMember2019-09-300000899866us-gaap:TreasuryStockMember2019-09-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2019-09-300000899866us-gaap:RetainedEarningsMember2019-09-3000008998662019-09-300000899866us-gaap:CommonStockMember2019-12-310000899866us-gaap:AdditionalPaidInCapitalMember2019-12-310000899866us-gaap:TreasuryStockMember2019-12-310000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2019-12-310000899866us-gaap:RetainedEarningsMember2019-12-310000899866us-gaap:TreasuryStockMember2020-01-012020-09-300000899866us-gaap:CommonStockMember2020-01-012020-09-300000899866us-gaap:AdditionalPaidInCapitalMember2020-01-012020-09-300000899866us-gaap:RetainedEarningsMember2020-01-012020-09-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2020-01-012020-09-300000899866us-gaap:CommonStockMember2018-12-310000899866us-gaap:AdditionalPaidInCapitalMember2018-12-310000899866us-gaap:TreasuryStockMember2018-12-310000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2018-12-310000899866us-gaap:RetainedEarningsMember2018-12-3100008998662018-12-310000899866us-gaap:TreasuryStockMember2019-01-012019-09-300000899866us-gaap:CommonStockMember2019-01-012019-09-300000899866us-gaap:AdditionalPaidInCapitalMember2019-01-012019-09-300000899866us-gaap:RetainedEarningsMember2019-01-012019-09-300000899866us-gaap:AccumulatedOtherComprehensiveIncomeMember2019-01-012019-09-300000899866alxn:CumulativeEffectPeriodOfAdoptionAdjustmentMemberus-gaap:RetainedEarningsMember2018-12-310000899866alxn:CumulativeEffectPeriodOfAdoptionAdjustmentMember2018-12-310000899866alxn:AchillionMember2020-01-282020-01-280000899866alxn:DanicopanMemberalxn:AchillionMember2020-01-282020-01-280000899866alxn:ACH5528Phase3Memberalxn:AchillionMember2020-01-282020-01-280000899866alxn:AchillionMember2020-01-280000899866alxn:AcquiredIprdMemberalxn:AchillionMember2020-01-280000899866alxn:AchillionMember2020-04-012020-06-30xbrli:pure0000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMembersrt:MinimumMember2020-01-282020-01-280000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMembersrt:MaximumMember2020-01-282020-01-280000899866alxn:AchillionMembersrt:MinimumMember2020-01-280000899866srt:MaximumMemberalxn:AchillionMember2020-01-280000899866alxn:AchillionMember2020-09-300000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMembersrt:MinimumMember2020-01-012020-09-300000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMembersrt:MaximumMember2020-01-012020-09-300000899866alxn:AchillionMember2020-07-012020-09-300000899866alxn:AchillionMember2020-01-012020-09-300000899866alxn:AcquiredIprdMember2020-04-012020-06-300000899866alxn:PortolaPharmaceuticalsIncMember2020-07-022020-07-020000899866alxn:PortolaPharmaceuticalsMember2020-03-012020-03-310000899866alxn:PortolaPharmaceuticalsMember2020-04-012020-04-300000899866alxn:PortolaPharmaceuticalsMember2020-07-020000899866alxn:PortolaPharmaceuticalsIncMember2020-07-020000899866alxn:PortolaPharmaceuticalsIncMemberalxn:AndexxaProductLineMember2020-07-022020-07-020000899866alxn:PurchasedTechnologyMemberalxn:PortolaPharmaceuticalsIncMember2020-07-020000899866alxn:PortolaPharmaceuticalsIncMemberalxn:AcquiredIprdMember2020-09-300000899866alxn:PurchasedTechnologyMemberalxn:PortolaPharmaceuticalsIncMember2020-07-022020-07-020000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2017-02-280000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2020-07-022020-07-020000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2020-07-020000899866alxn:PortolaPharmaceuticalsIncMembersrt:MinimumMember2020-07-022020-07-020000899866alxn:PortolaPharmaceuticalsIncMembersrt:MaximumMember2020-07-022020-07-020000899866alxn:PortolaPharmaceuticalsIncMember2020-01-012020-09-300000899866alxn:PortolaPharmaceuticalsIncMember2020-07-012020-09-300000899866alxn:PortolaPharmaceuticalsIncMember2019-07-012019-09-300000899866alxn:PortolaPharmaceuticalsIncMember2019-01-012019-09-300000899866us-gaap:InventoriesMember2020-09-300000899866us-gaap:InventoriesMember2019-12-310000899866us-gaap:OtherAssetsMember2020-09-300000899866us-gaap:OtherAssetsMember2019-12-310000899866alxn:PreApprovalInventoryMember2020-09-300000899866alxn:PreApprovalInventoryMember2019-12-310000899866srt:MinimumMemberus-gaap:LicensingAgreementsMember2020-01-012020-09-300000899866srt:MaximumMemberus-gaap:LicensingAgreementsMember2020-01-012020-09-300000899866us-gaap:LicensingAgreementsMember2020-09-300000899866us-gaap:LicensingAgreementsMember2019-12-310000899866us-gaap:PatentsMember2020-01-012020-09-300000899866us-gaap:PatentsMember2020-09-300000899866us-gaap:PatentsMember2019-12-310000899866alxn:PurchasedTechnologyMembersrt:MinimumMember2020-01-012020-09-300000899866alxn:PurchasedTechnologyMembersrt:MaximumMember2020-01-012020-09-300000899866alxn:PurchasedTechnologyMember2020-09-300000899866alxn:PurchasedTechnologyMember2019-12-310000899866us-gaap:OtherIntangibleAssetsMember2020-01-012020-09-300000899866us-gaap:OtherIntangibleAssetsMember2020-09-300000899866us-gaap:OtherIntangibleAssetsMember2019-12-310000899866alxn:AcquiredIprdMember2020-09-300000899866alxn:AcquiredIprdMember2019-12-310000899866alxn:PurchasedTechnologyMember2020-04-012020-06-300000899866alxn:KanumaProductLineMember2020-06-300000899866alxn:CreditAgreementMemberus-gaap:RevolvingCreditFacilityMember2018-06-070000899866alxn:SeniorSecuredTermLoanMemberus-gaap:LineOfCreditMemberalxn:CreditAgreementMember2018-06-070000899866alxn:CreditAgreementMember2019-01-012019-03-310000899866alxn:SeniorSecuredTermLoanMemberus-gaap:LineOfCreditMemberalxn:CreditAgreementMember2020-07-012020-09-300000899866alxn:SeniorSecuredTermLoanMemberus-gaap:LineOfCreditMemberalxn:CreditAgreementMember2020-01-012020-09-300000899866alxn:SeniorSecuredTermLoanMemberus-gaap:LineOfCreditMemberalxn:CreditAgreementMember2020-09-300000899866alxn:CreditAgreementMemberus-gaap:RevolvingCreditFacilityMember2020-09-300000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2017-02-012017-02-280000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2018-05-310000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMembersrt:MinimumMember2017-02-280000899866alxn:PortolaPharmaceuticalsIncMemberalxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMembersrt:MaximumMember2017-02-280000899866alxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2020-09-300000899866alxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2020-07-012020-09-300000899866alxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2020-01-012020-09-300000899866alxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMembersrt:ScenarioForecastMember2020-10-012020-12-3100008998662020-07-032020-09-300000899866alxn:HealthCareRoyaltyPartnersMemberalxn:RoyaltyBasedFinancingAgreementMember2020-07-032020-09-300000899866us-gaap:CommercialPaperMember2019-12-310000899866us-gaap:CorporateBondSecuritiesMember2019-12-310000899866us-gaap:USGovernmentAgenciesDebtSecuritiesMember2019-12-310000899866us-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:DebtSecuritiesMember2019-12-310000899866us-gaap:CashAndCashEquivalentsMember2020-09-300000899866us-gaap:CashAndCashEquivalentsMember2019-12-310000899866alxn:MarketableSecuritiesMember2020-09-300000899866alxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:DesignatedAsHedgingInstrumentMember2020-01-012020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberalxn:ForeignExchangeForwardOpenExpenseMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberalxn:InterestRateSwapTwoMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberalxn:InterestRateSwapTwoMembersrt:MinimumMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberalxn:InterestRateSwapTwoMembersrt:MaximumMember2020-09-300000899866alxn:InterestRateSwapFourMemberus-gaap:DesignatedAsHedgingInstrumentMember2020-09-300000899866alxn:InterestRateSwapFourMemberus-gaap:DesignatedAsHedgingInstrumentMembersrt:MinimumMember2020-09-300000899866alxn:InterestRateSwapFourMemberus-gaap:DesignatedAsHedgingInstrumentMembersrt:MaximumMember2020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ForeignExchangeForwardMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2020-07-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ForeignExchangeForwardMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2019-07-012019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:InterestRateContractMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2020-07-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:InterestRateContractMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2019-07-012019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ForeignExchangeForwardMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2020-01-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ForeignExchangeForwardMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2019-01-012019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:InterestRateContractMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2020-01-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:InterestRateContractMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2019-01-012019-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2020-07-012020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2019-07-012019-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2020-01-012020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2019-01-012019-09-300000899866us-gaap:SalesMemberus-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2020-07-012020-09-300000899866us-gaap:SalesMemberus-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2019-07-012019-09-300000899866us-gaap:SalesMemberus-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2020-01-012020-09-300000899866us-gaap:SalesMemberus-gaap:ForeignExchangeForwardMemberus-gaap:CashFlowHedgingMember2019-01-012019-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMember2020-07-012020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMember2019-07-012019-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMember2020-01-012020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMember2019-01-012019-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMemberus-gaap:InterestExpenseMember2020-07-012020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMemberus-gaap:InterestExpenseMember2019-07-012019-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMemberus-gaap:InterestExpenseMember2020-01-012020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:CashFlowHedgingMemberus-gaap:InterestExpenseMember2019-01-012019-09-300000899866us-gaap:SalesMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2020-07-012020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMember2020-07-012020-09-300000899866us-gaap:NondesignatedMember2020-07-012020-09-300000899866us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:NondesignatedMember2019-07-012019-09-300000899866us-gaap:NondesignatedMember2020-01-012020-09-300000899866us-gaap:NondesignatedMember2019-01-012019-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentAssetsMember2020-09-300000899866us-gaap:OtherNoncurrentLiabilitiesMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:InterestRateContractMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMember2020-09-300000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:InterestRateContractMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMemberus-gaap:OtherNoncurrentAssetsMember2020-09-300000899866us-gaap:OtherNoncurrentLiabilitiesMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMember2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentAssetsMember2019-12-310000899866us-gaap:OtherNoncurrentLiabilitiesMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:InterestRateContractMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMember2019-12-310000899866us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:InterestRateContractMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMemberus-gaap:OtherNoncurrentAssetsMember2019-12-310000899866us-gaap:OtherNoncurrentLiabilitiesMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:InterestRateContractMember2019-12-310000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMemberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:ForeignExchangeForwardMember2019-12-310000899866alxn:ModernaLLCMember2014-01-012014-12-310000899866alxn:ModernaLLCMember2019-07-012019-09-300000899866alxn:ModernaLLCMember2019-01-012019-09-300000899866alxn:ModernaLLCMember2019-12-092019-12-090000899866alxn:DicernaMember2018-10-012018-10-310000899866alxn:DicernaMember2020-07-012020-09-300000899866alxn:DicernaMember2020-01-012020-09-300000899866alxn:DicernaMember2019-07-012019-09-300000899866alxn:DicernaMember2019-01-012019-09-300000899866alxn:DicernaMember2020-09-300000899866alxn:DicernaMember2019-12-310000899866alxn:CaelumBiosciencesMember2019-01-012019-01-310000899866alxn:CaelumBiosciencesMember2019-10-012019-12-310000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-10-012019-12-310000899866alxn:CollaborativeAgreementAmendmentMemberalxn:CaelumBiosciencesMember2019-10-012019-12-310000899866alxn:CaelumBiosciencesMember2019-01-012019-12-310000899866alxn:CaelumBiosciencesMember2020-01-012020-09-300000899866alxn:CaelumBiosciencesOptionMember2020-01-012020-09-300000899866alxn:CaelumBiosciencesOptionMember2019-01-012019-12-310000899866alxn:ZealandMember2019-03-012019-03-310000899866alxn:ZealandMember2020-07-012020-09-300000899866alxn:ZealandMember2020-01-012020-09-300000899866alxn:ZealandMember2019-07-012019-09-300000899866alxn:ZealandMember2019-01-012019-09-300000899866alxn:ZealandMember2020-09-300000899866alxn:ZealandMember2019-12-310000899866alxn:EidosMemberMember2019-09-012019-09-300000899866alxn:EidosMemberMember2020-07-012020-09-300000899866alxn:EidosMemberMember2020-01-012020-09-300000899866alxn:EidosMemberMember2019-07-012019-09-300000899866alxn:EidosMemberMember2019-01-012019-09-300000899866alxn:EidosMemberMember2020-09-300000899866alxn:EidosMemberMember2019-12-310000899866alxn:StealthMember2019-10-012019-10-310000899866alxn:StealthMember2020-07-012020-09-300000899866alxn:StealthMember2020-01-012020-09-300000899866alxn:StealthMember2020-09-300000899866alxn:StealthMember2019-12-310000899866alxn:PortolaPharmaceuticalsMember2020-07-012020-09-300000899866alxn:PortolaPharmaceuticalsMember2020-01-012020-09-300000899866alxn:PortolaPharmaceuticalsMember2020-07-022020-07-020000899866alxn:InozymeMember2020-07-172020-07-170000899866alxn:InozymeMember2020-01-012020-09-300000899866alxn:InozymeMember2020-07-012020-09-300000899866alxn:InozymeMember2020-09-300000899866us-gaap:CommonStockMember2017-02-280000899866us-gaap:CommonStockMember2019-10-220000899866us-gaap:CommonStockMember2020-07-280000899866us-gaap:SubsequentEventMemberus-gaap:CommonStockMember2020-10-012020-10-290000899866us-gaap:SubsequentEventMemberus-gaap:CommonStockMember2020-10-270000899866us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2019-12-310000899866us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2019-12-310000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMember2019-12-310000899866us-gaap:AccumulatedTranslationAdjustmentMember2019-12-310000899866us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2020-01-012020-09-300000899866us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2020-01-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMember2020-01-012020-09-300000899866us-gaap:AccumulatedTranslationAdjustmentMember2020-01-012020-09-300000899866us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2020-09-300000899866us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMember2020-09-300000899866us-gaap:AccumulatedTranslationAdjustmentMember2020-09-300000899866us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2018-12-310000899866us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2018-12-310000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMember2018-12-310000899866us-gaap:AccumulatedTranslationAdjustmentMember2018-12-310000899866us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2019-01-012019-09-300000899866us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2019-01-012019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMember2019-01-012019-09-300000899866us-gaap:AccumulatedTranslationAdjustmentMember2019-01-012019-09-300000899866us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2019-09-300000899866us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMember2019-09-300000899866us-gaap:AccumulatedTranslationAdjustmentMember2019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2020-07-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2019-07-012019-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2020-01-012020-09-300000899866us-gaap:AccumulatedGainLossCashFlowHedgeIncludingNoncontrollingInterestMemberus-gaap:ReclassificationOutOfAccumulatedOtherComprehensiveIncomeMember2019-01-012019-09-300000899866us-gaap:CashEquivalentsMemberus-gaap:MoneyMarketFundsMember2020-09-300000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel1Memberus-gaap:MoneyMarketFundsMember2020-09-300000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel2Memberus-gaap:MoneyMarketFundsMember2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CashEquivalentsMemberus-gaap:MoneyMarketFundsMember2020-09-300000899866alxn:MutualFundsMemberalxn:MarketableSecuritiesMember2020-09-300000899866us-gaap:FairValueInputsLevel1Memberalxn:MutualFundsMemberalxn:MarketableSecuritiesMember2020-09-300000899866us-gaap:FairValueInputsLevel2Memberalxn:MutualFundsMemberalxn:MarketableSecuritiesMember2020-09-300000899866us-gaap:FairValueInputsLevel3Memberalxn:MutualFundsMemberalxn:MarketableSecuritiesMember2020-09-300000899866us-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMember2020-09-300000899866us-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMemberus-gaap:FairValueInputsLevel1Member2020-09-300000899866us-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMemberus-gaap:FairValueInputsLevel2Member2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMember2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel1Member2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel2Member2020-09-300000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentAssetsMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel1Memberus-gaap:OtherNoncurrentAssetsMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel2Memberus-gaap:OtherNoncurrentAssetsMember2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentAssetsMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMember2020-09-300000899866us-gaap:FairValueInputsLevel1Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Member2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMember2020-09-300000899866us-gaap:FairValueInputsLevel1Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMember2020-09-300000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Member2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:OtherCurrentLiabilitiesMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:FairValueInputsLevel1Memberus-gaap:OtherCurrentLiabilitiesMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Member2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:InterestRateContractMemberus-gaap:OtherCurrentLiabilitiesMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:OtherLiabilitiesMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:FairValueInputsLevel1Memberus-gaap:OtherLiabilitiesMember2020-09-300000899866us-gaap:InterestRateContractMemberus-gaap:FairValueInputsLevel2Memberus-gaap:OtherLiabilitiesMember2020-09-300000899866us-gaap:FairValueInputsLevel3Memberus-gaap:InterestRateContractMemberus-gaap:OtherLiabilitiesMember2020-09-300000899866alxn:ContingentConsiderationMemberalxn:AcquisitionRelatedContingentConsiderationMember2020-09-300000899866alxn:ContingentConsiderationMemberus-gaap:FairValueInputsLevel1Memberalxn:AcquisitionRelatedContingentConsiderationMember2020-09-300000899866alxn:ContingentConsiderationMemberalxn:AcquisitionRelatedContingentConsiderationMemberus-gaap:FairValueInputsLevel2Member2020-09-300000899866alxn:ContingentConsiderationMemberus-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMember2020-09-300000899866us-gaap:CashEquivalentsMemberus-gaap:MoneyMarketFundsMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel1Memberus-gaap:MoneyMarketFundsMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel2Memberus-gaap:MoneyMarketFundsMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CashEquivalentsMemberus-gaap:MoneyMarketFundsMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:CommercialPaperMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel1Memberus-gaap:CommercialPaperMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel2Memberus-gaap:CommercialPaperMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CashEquivalentsMemberus-gaap:CommercialPaperMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:CorporateBondSecuritiesMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel1Memberus-gaap:CorporateBondSecuritiesMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:CorporateBondSecuritiesMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CashEquivalentsMemberus-gaap:CorporateBondSecuritiesMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel1Memberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel2Memberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CashEquivalentsMemberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:CashEquivalentsMemberalxn:OtherGovernmentObligationsMember2019-12-310000899866us-gaap:CashEquivalentsMemberus-gaap:FairValueInputsLevel1Memberalxn:OtherGovernmentObligationsMember2019-12-310000899866us-gaap:CashEquivalentsMemberalxn:OtherGovernmentObligationsMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CashEquivalentsMemberalxn:OtherGovernmentObligationsMember2019-12-310000899866alxn:MutualFundsMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberalxn:MutualFundsMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel2Memberalxn:MutualFundsMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberalxn:MutualFundsMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:CommercialPaperMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberus-gaap:CommercialPaperMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel2Memberus-gaap:CommercialPaperMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CommercialPaperMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:CorporateBondSecuritiesMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberus-gaap:CorporateBondSecuritiesMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:CorporateBondSecuritiesMemberus-gaap:FairValueInputsLevel2Memberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:CorporateBondSecuritiesMemberalxn:MarketableSecuritiesMember2019-12-310000899866alxn:OtherGovernmentObligationsMemberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberalxn:OtherGovernmentObligationsMemberalxn:MarketableSecuritiesMember2019-12-310000899866alxn:OtherGovernmentObligationsMemberus-gaap:FairValueInputsLevel2Memberalxn:MarketableSecuritiesMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberalxn:OtherGovernmentObligationsMemberalxn:MarketableSecuritiesMember2019-12-310000899866alxn:MarketableSecuritiesMemberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberalxn:MarketableSecuritiesMemberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:FairValueInputsLevel2Memberalxn:MarketableSecuritiesMemberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberalxn:MarketableSecuritiesMemberus-gaap:CertificatesOfDepositMember2019-12-310000899866us-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMember2019-12-310000899866us-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMemberus-gaap:FairValueInputsLevel1Member2019-12-310000899866us-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:OtherAssetsMemberus-gaap:EquitySecuritiesMember2019-12-310000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel1Member2019-12-310000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentAssetsMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel1Memberus-gaap:OtherNoncurrentAssetsMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:FairValueInputsLevel2Memberus-gaap:OtherNoncurrentAssetsMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentAssetsMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherCurrentLiabilitiesMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMember2019-12-310000899866us-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherNoncurrentLiabilitiesMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:OtherCurrentLiabilitiesMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:FairValueInputsLevel1Memberus-gaap:OtherCurrentLiabilitiesMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:OtherCurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:InterestRateContractMemberus-gaap:OtherCurrentLiabilitiesMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:OtherLiabilitiesMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:FairValueInputsLevel1Memberus-gaap:OtherLiabilitiesMember2019-12-310000899866us-gaap:InterestRateContractMemberus-gaap:FairValueInputsLevel2Memberus-gaap:OtherLiabilitiesMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:InterestRateContractMemberus-gaap:OtherLiabilitiesMember2019-12-310000899866alxn:ContingentConsiderationMemberalxn:AcquisitionRelatedContingentConsiderationMember2019-12-310000899866alxn:ContingentConsiderationMemberus-gaap:FairValueInputsLevel1Memberalxn:AcquisitionRelatedContingentConsiderationMember2019-12-310000899866alxn:ContingentConsiderationMemberalxn:AcquisitionRelatedContingentConsiderationMemberus-gaap:FairValueInputsLevel2Member2019-12-310000899866alxn:ContingentConsiderationMemberus-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMember2019-12-310000899866us-gaap:OtherCurrentLiabilitiesMemberalxn:OtherContingentPaymentsMember2019-12-310000899866us-gaap:FairValueInputsLevel1Memberus-gaap:OtherCurrentLiabilitiesMemberalxn:OtherContingentPaymentsMember2019-12-310000899866us-gaap:OtherCurrentLiabilitiesMemberus-gaap:FairValueInputsLevel2Memberalxn:OtherContingentPaymentsMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberus-gaap:OtherCurrentLiabilitiesMemberalxn:OtherContingentPaymentsMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMember2019-12-310000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMember2020-01-012020-09-300000899866us-gaap:FairValueInputsLevel3Memberalxn:AcquisitionRelatedContingentConsiderationMember2020-09-300000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-01-012019-01-310000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-12-310000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMembersrt:MinimumMember2019-01-012019-01-310000899866srt:MaximumMemberus-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-01-012019-01-310000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-12-012019-12-310000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2020-04-012020-06-300000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2020-07-012020-09-300000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2020-04-012020-09-300000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2020-01-012020-09-300000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-07-012019-09-300000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-01-012019-09-300000899866alxn:SolirisMembercountry:US2020-07-012020-09-300000899866alxn:SolirisMembercountry:US2019-07-012019-09-300000899866alxn:SolirisMembercountry:US2020-01-012020-09-300000899866alxn:SolirisMembercountry:US2019-01-012019-09-300000899866alxn:SolirisMembersrt:EuropeMember2020-07-012020-09-300000899866alxn:SolirisMembersrt:EuropeMember2019-07-012019-09-300000899866alxn:SolirisMembersrt:EuropeMember2020-01-012020-09-300000899866alxn:SolirisMembersrt:EuropeMember2019-01-012019-09-300000899866srt:AsiaPacificMemberalxn:SolirisMember2020-07-012020-09-300000899866srt:AsiaPacificMemberalxn:SolirisMember2019-07-012019-09-300000899866srt:AsiaPacificMemberalxn:SolirisMember2020-01-012020-09-300000899866srt:AsiaPacificMemberalxn:SolirisMember2019-01-012019-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:SolirisMember2020-07-012020-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:SolirisMember2019-07-012019-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:SolirisMember2020-01-012020-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:SolirisMember2019-01-012019-09-300000899866alxn:SolirisMember2020-07-012020-09-300000899866alxn:SolirisMember2019-07-012019-09-300000899866alxn:SolirisMember2020-01-012020-09-300000899866alxn:SolirisMember2019-01-012019-09-300000899866alxn:UltomirisMembercountry:US2020-07-012020-09-300000899866alxn:UltomirisMembercountry:US2019-07-012019-09-300000899866alxn:UltomirisMembercountry:US2020-01-012020-09-300000899866alxn:UltomirisMembercountry:US2019-01-012019-09-300000899866alxn:UltomirisMembersrt:EuropeMember2020-07-012020-09-300000899866alxn:UltomirisMembersrt:EuropeMember2019-07-012019-09-300000899866alxn:UltomirisMembersrt:EuropeMember2020-01-012020-09-300000899866alxn:UltomirisMembersrt:EuropeMember2019-01-012019-09-300000899866alxn:UltomirisMembersrt:AsiaPacificMember2020-07-012020-09-300000899866alxn:UltomirisMembersrt:AsiaPacificMember2019-07-012019-09-300000899866alxn:UltomirisMembersrt:AsiaPacificMember2020-01-012020-09-300000899866alxn:UltomirisMembersrt:AsiaPacificMember2019-01-012019-09-300000899866alxn:UltomirisMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2020-07-012020-09-300000899866alxn:UltomirisMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2019-07-012019-09-300000899866alxn:UltomirisMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2020-01-012020-09-300000899866alxn:UltomirisMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2019-01-012019-09-300000899866alxn:UltomirisMember2020-07-012020-09-300000899866alxn:UltomirisMember2019-07-012019-09-300000899866alxn:UltomirisMember2020-01-012020-09-300000899866alxn:UltomirisMember2019-01-012019-09-300000899866alxn:StrensiqProductLineMembercountry:US2020-07-012020-09-300000899866alxn:StrensiqProductLineMembercountry:US2019-07-012019-09-300000899866alxn:StrensiqProductLineMembercountry:US2020-01-012020-09-300000899866alxn:StrensiqProductLineMembercountry:US2019-01-012019-09-300000899866alxn:StrensiqProductLineMembersrt:EuropeMember2020-07-012020-09-300000899866alxn:StrensiqProductLineMembersrt:EuropeMember2019-07-012019-09-300000899866alxn:StrensiqProductLineMembersrt:EuropeMember2020-01-012020-09-300000899866alxn:StrensiqProductLineMembersrt:EuropeMember2019-01-012019-09-300000899866alxn:StrensiqProductLineMembersrt:AsiaPacificMember2020-07-012020-09-300000899866alxn:StrensiqProductLineMembersrt:AsiaPacificMember2019-07-012019-09-300000899866alxn:StrensiqProductLineMembersrt:AsiaPacificMember2020-01-012020-09-300000899866alxn:StrensiqProductLineMembersrt:AsiaPacificMember2019-01-012019-09-300000899866alxn:StrensiqProductLineMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2020-07-012020-09-300000899866alxn:StrensiqProductLineMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2019-07-012019-09-300000899866alxn:StrensiqProductLineMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2020-01-012020-09-300000899866alxn:StrensiqProductLineMemberalxn:OtherNonUsExcludingEuropeAndAsiaPacificMember2019-01-012019-09-300000899866alxn:StrensiqProductLineMember2020-07-012020-09-300000899866alxn:StrensiqProductLineMember2019-07-012019-09-300000899866alxn:StrensiqProductLineMember2020-01-012020-09-300000899866alxn:StrensiqProductLineMember2019-01-012019-09-300000899866alxn:AndexxaProductLineMembercountry:US2020-07-012020-09-300000899866alxn:AndexxaProductLineMembercountry:US2019-07-012019-09-300000899866alxn:AndexxaProductLineMembercountry:US2020-01-012020-09-300000899866alxn:AndexxaProductLineMembercountry:US2019-01-012019-09-300000899866alxn:AndexxaProductLineMembersrt:EuropeMember2020-07-012020-09-300000899866alxn:AndexxaProductLineMembersrt:EuropeMember2019-07-012019-09-300000899866alxn:AndexxaProductLineMembersrt:EuropeMember2020-01-012020-09-300000899866alxn:AndexxaProductLineMembersrt:EuropeMember2019-01-012019-09-300000899866alxn:AndexxaProductLineMembersrt:AsiaPacificMember2020-07-012020-09-300000899866alxn:AndexxaProductLineMembersrt:AsiaPacificMember2019-07-012019-09-300000899866alxn:AndexxaProductLineMembersrt:AsiaPacificMember2020-01-012020-09-300000899866alxn:AndexxaProductLineMembersrt:AsiaPacificMember2019-01-012019-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:AndexxaProductLineMember2020-07-012020-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:AndexxaProductLineMember2019-07-012019-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:AndexxaProductLineMember2020-01-012020-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:AndexxaProductLineMember2019-01-012019-09-300000899866alxn:AndexxaProductLineMember2020-07-012020-09-300000899866alxn:AndexxaProductLineMember2019-07-012019-09-300000899866alxn:AndexxaProductLineMember2020-01-012020-09-300000899866alxn:AndexxaProductLineMember2019-01-012019-09-300000899866alxn:KanumaProductLineMembercountry:US2020-07-012020-09-300000899866alxn:KanumaProductLineMembercountry:US2019-07-012019-09-300000899866alxn:KanumaProductLineMembercountry:US2020-01-012020-09-300000899866alxn:KanumaProductLineMembercountry:US2019-01-012019-09-300000899866alxn:KanumaProductLineMembersrt:EuropeMember2020-07-012020-09-300000899866alxn:KanumaProductLineMembersrt:EuropeMember2019-07-012019-09-300000899866alxn:KanumaProductLineMembersrt:EuropeMember2020-01-012020-09-300000899866alxn:KanumaProductLineMembersrt:EuropeMember2019-01-012019-09-300000899866alxn:KanumaProductLineMembersrt:AsiaPacificMember2020-07-012020-09-300000899866alxn:KanumaProductLineMembersrt:AsiaPacificMember2019-07-012019-09-300000899866alxn:KanumaProductLineMembersrt:AsiaPacificMember2020-01-012020-09-300000899866alxn:KanumaProductLineMembersrt:AsiaPacificMember2019-01-012019-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:KanumaProductLineMember2020-07-012020-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:KanumaProductLineMember2019-07-012019-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:KanumaProductLineMember2020-01-012020-09-300000899866alxn:OtherNonUsExcludingEuropeAndAsiaPacificMemberalxn:KanumaProductLineMember2019-01-012019-09-300000899866alxn:KanumaProductLineMember2020-07-012020-09-300000899866alxn:KanumaProductLineMember2019-07-012019-09-300000899866alxn:KanumaProductLineMember2020-01-012020-09-300000899866alxn:KanumaProductLineMember2019-01-012019-09-300000899866us-gaap:ProductMember2020-07-012020-09-300000899866us-gaap:ProductMember2019-07-012019-09-300000899866us-gaap:ProductMember2020-01-012020-09-300000899866us-gaap:ProductMember2019-01-012019-09-300000899866alxn:IntangibleAssetImpairmentMember2020-04-012020-06-300000899866alxn:IntellectualPropertyElectionMember2019-01-012019-09-300000899866alxn:ValuationAllowanceReleaseMember2019-01-012019-09-3000008998662018-01-012018-12-310000899866us-gaap:CollaborativeArrangementMemberalxn:CaelumBiosciencesMember2019-01-310000899866alxn:ZealandPharmaMemberalxn:ZealandMember2019-03-012019-03-310000899866alxn:ZealandPharmaMemberalxn:ZealandMember2019-03-310000899866alxn:ZealandPharmaMemberalxn:ZealandMember2019-01-012019-03-310000899866alxn:ZealandPharmaMemberalxn:ZealandMember2020-01-012020-09-300000899866alxn:AffibodyABDomainalxn:AffibodyMember2019-04-012019-04-300000899866alxn:AffibodyABDomainalxn:EidosMemberMember2019-09-012019-09-300000899866alxn:AffibodyABDomainalxn:EidosMemberMember2019-07-012019-09-300000899866alxn:AffibodyABDomainalxn:EidosMemberMember2020-01-012020-09-300000899866alxn:DicernaPharmaceuticalCollaborationAgreementMemberalxn:DicernaMember2018-10-012018-10-310000899866alxn:DicernaPharmaceuticalCollaborationAgreementMemberalxn:DicernaMember2018-10-012018-12-310000899866alxn:DicernaPharmaceuticalCollaborationAgreementMemberalxn:DicernaMember2019-10-012019-12-310000899866alxn:DicernaPharmaceuticalCollaborationAgreementMemberalxn:DicernaMember2020-09-300000899866alxn:LicenseAgreement1Memberalxn:HalozymeTherapeuticsIncMember2017-10-012017-12-310000899866alxn:LicenseAgreement1Memberalxn:HalozymeTherapeuticsIncMember2020-09-300000899866alxn:SyntimmuneIncMember2018-11-012018-11-300000899866alxn:SyntimmuneIncMemberalxn:SubcutaneousFormulationMember2018-11-012018-11-300000899866alxn:CollaborationandLicenseAgreementMember2020-01-012020-09-300000899866alxn:AstellasPharamMember2020-07-012020-09-300000899866alxn:LonzaGroupAGMember2020-09-300000899866alxn:OtherThirdPartyManufacturersMember2020-09-300000899866alxn:SECMember2020-07-022020-07-020000899866alxn:DOJAndOIGMember2019-04-012019-04-300000899866us-gaap:SubsequentEventMemberalxn:CanadianPatentedMedicinePricesReviewBoardMember2020-10-270000899866alxn:CanadianPatentedMedicinePricesReviewBoardMember2020-09-300000899866us-gaap:EmployeeSeveranceMember2020-07-012020-09-300000899866us-gaap:OtherRestructuringMember2020-07-012020-09-300000899866us-gaap:EmployeeSeveranceMember2019-07-012019-09-300000899866us-gaap:OtherRestructuringMember2019-07-012019-09-300000899866us-gaap:EmployeeSeveranceMember2020-01-012020-09-300000899866us-gaap:OtherRestructuringMember2020-01-012020-09-300000899866us-gaap:EmployeeSeveranceMember2019-01-012019-09-300000899866us-gaap:OtherRestructuringMember2019-01-012019-09-300000899866us-gaap:EmployeeSeveranceMember2020-06-300000899866us-gaap:OtherRestructuringMember2020-06-300000899866us-gaap:EmployeeSeveranceMember2019-12-310000899866us-gaap:OtherRestructuringMember2019-12-310000899866us-gaap:EmployeeSeveranceMember2020-09-300000899866us-gaap:OtherRestructuringMember2020-09-30
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
| | | | | |
| ☒ | Quarterly report pursuant to Section 13 or 15 (d) of the Securities Exchange Act of 1934 |
For the quarterly period ended September 30, 2020
or
| | | | | |
| ☐ | Transition report pursuant to Section 13 or 15 (d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission file number: 0-27756
ALEXION PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
| | | | | |
| Delaware | 13-3648318 |
| (State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) |
121 Seaport Boulevard, Boston Massachusetts 02210
(Address of Principal Executive Offices) (Zip Code)
475-230-2596
(Registrant’s telephone number, including area code)
N/A
(Former name, former address, and former fiscal year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Common Stock $0.0001 par value | ALXN | NASDAQ Stock Market LLC |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act.
Large accelerated filer x Accelerated filer ☐ Non-accelerated filer ☐
Smaller reporting company ☐ Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No x
| | | | | | | | |
| Common Stock | $0.0001 par value | 218,845,432 |
| Class | Outstanding as of October 27, 2020 |
Alexion Pharmaceuticals, Inc.
Table of Contents
| | | | | | | | | | | |
| | | Page |
| PART I. | | |
| Item 1. | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| Item 2. | | |
| Item 3. | | |
| Item 4. | | |
| PART II. | | |
| Item 1. | | |
| Item 1A. | | |
| Item 2. | | |
| Item 5. | | |
| Item 6. | | |
| SIGNATURES | | |
Alexion Pharmaceuticals, Inc.
Condensed Consolidated Balance Sheets
(unaudited)
(amounts in millions, except per share amounts)
| | | | | | | | | | | |
| | September 30, | | December 31, |
| | 2020 | | 2019 |
| Assets | | | |
| Current Assets: | | | |
| Cash and cash equivalents | $ | 2,268.0 | | | $ | 2,685.5 | |
| Marketable securities | 28.9 | | | 64.0 | |
| Trade accounts receivable, net | 1,437.1 | | | 1,243.2 | |
| Inventories | 729.0 | | | 627.6 | |
| Prepaid expenses and other current assets | 604.8 | | | 456.1 | |
| Total current assets | 5,067.8 | | | 5,076.4 | |
| Property, plant and equipment, net | 1,214.5 | | | 1,163.3 | |
| Intangible assets, net | 3,056.6 | | | 3,344.3 | |
| Goodwill | 5,100.7 | | | 5,037.4 | |
| Right of use operating assets | 220.1 | | | 204.0 | |
| Deferred tax assets | 2,270.5 | | | 2,290.2 | |
| Other assets | 618.2 | | | 429.0 | |
| Total assets | $ | 17,548.4 | | | $ | 17,544.6 | |
| Liabilities and Stockholders' Equity | | | |
| Current Liabilities: | | | |
| Accounts payable | $ | 89.2 | | | $ | 74.0 | |
| Accrued expenses | 978.4 | | | 892.7 | |
| Current portion of long-term debt | 138.6 | | | 126.7 | |
| Other current liabilities | 124.8 | | | 100.9 | |
| Total current liabilities | 1,331.0 | | | 1,194.3 | |
| Long-term debt, less current portion | 2,453.3 | | | 2,375.0 | |
| Contingent consideration | 398.1 | | | 192.4 | |
| Deferred tax liabilities | 1,818.2 | | | 2,081.4 | |
| Noncurrent operating lease liabilities | 175.8 | | | 164.1 | |
| Other liabilities | 297.1 | | | 265.6 | |
| Total liabilities | 6,473.5 | | | 6,272.8 | |
| Commitments and contingencies (Note 16) | | | |
| Stockholders' Equity: | | | |
Common stock, $0.0001 par value; 290.0 shares authorized; 239.7 and 237.8 shares issued at September 30, 2020 and December 31, 2019, respectively | — | | | — | |
| Additional paid-in capital | 9,030.4 | | | 8,804.7 | |
Treasury stock, at cost, 20.7 and 16.5 shares at September 30, 2020 and December 31, 2019, respectively | (2,543.7) | | | (2,105.9) | |
| Accumulated other comprehensive loss | (119.2) | | | (66.8) | |
| Retained earnings | 4,707.4 | | | 4,639.8 | |
| Total stockholders' equity | 11,074.9 | | | 11,271.8 | |
| Total liabilities and stockholders' equity | $ | 17,548.4 | | | $ | 17,544.6 | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Condensed Consolidated Statements of Operations
(unaudited)
(amounts in millions, except per share amounts)
| | | | | | | | | | | | | | | | | | | | | | | |
| | Three months ended September 30, | | Nine months ended September 30, |
| | 2020 | | 2019 | | 2020 | | 2019 |
| Net product sales | $ | 1,588.3 | | | $ | 1,263.1 | | | $ | 4,477.4 | | | $ | 3,605.8 | |
| Other revenue | 0.4 | | | — | | | 0.7 | | | 1.0 | |
| Total revenues | 1,588.7 | | | 1,263.1 | | | 4,478.1 | | | 3,606.8 | |
| Costs and expenses: | | | | | | | |
| Cost of sales (exclusive of amortization of purchased intangible assets) | 144.7 | | | 95.2 | | | 401.3 | | | 280.2 | |
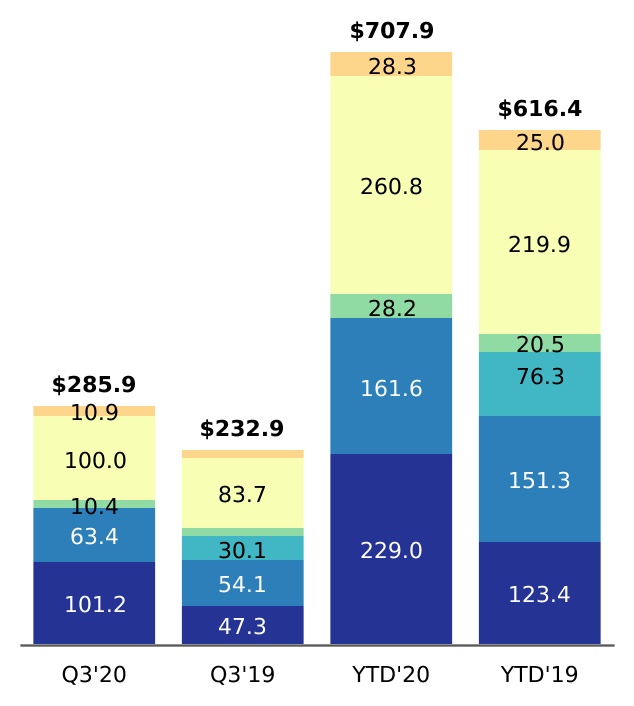
| Research and development | 285.9 | | | 232.9 | | | 707.9 | | | 616.4 | |
| Selling, general and administrative | 334.2 | | | 299.3 | | | 955.5 | | | 880.1 | |
| Acquired in-process research and development | — | | | — | | | — | | | (4.1) | |
| Amortization of purchased intangible assets | 53.1 | | | 75.6 | | | 200.5 | | | 235.7 | |
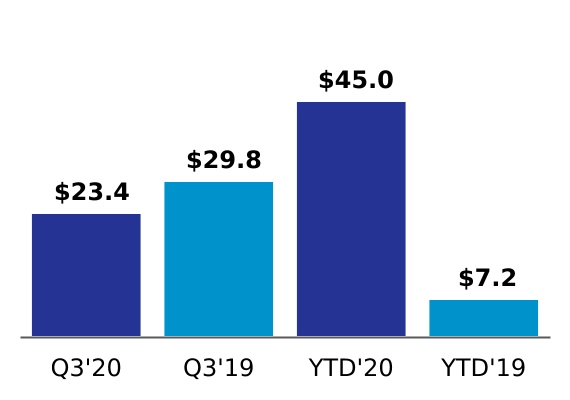
| Change in fair value of contingent consideration | 23.4 | | | 29.8 | | | 45.0 | | | 7.2 | |
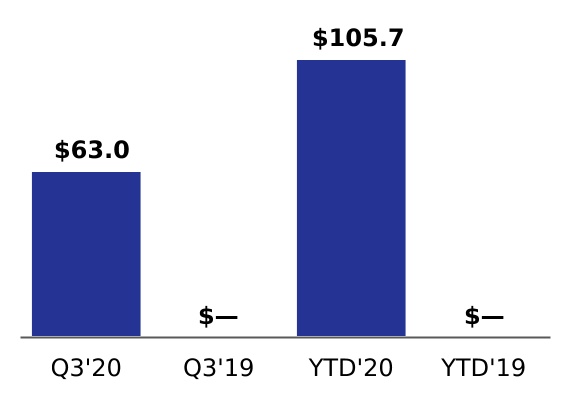
| Acquisition-related costs | 63.0 | | | — | | | 105.7 | | | — | |
| Restructuring expenses | 14.3 | | | 0.3 | | | 13.5 | | | 11.9 | |
| Impairment of intangible assets | — | | | — | | | 2,053.3 | | | — | |
| Gain on sale of asset | (14.8) | | | — | | | (14.8) | | | — | |
| Total costs and expenses | 903.8 | | | 733.1 | | | 4,467.9 | | | 2,027.4 | |
| Operating income | 684.9 | | | 530.0 | | | 10.2 | | | 1,579.4 | |
| Other income and expense: | | | | | | | |
| Investment income, net | 11.5 | | | 23.0 | | | 47.8 | | | 50.6 | |
| Interest expense | (27.6) | | | (17.9) | | | (77.0) | | | (56.1) | |
| Other income and (expense) | (1.9) | | | 0.4 | | | (2.6) | | | 2.9 | |
| Income (loss) before income taxes | 666.9 | | | 535.5 | | | (21.6) | | | 1,576.8 | |
| Income tax expense (benefit) | 88.8 | | | 67.9 | | | (89.2) | | | 61.5 | |
| Net income | $ | 578.1 | | | $ | 467.6 | | | $ | 67.6 | | | $ | 1,515.3 | |
| Earnings per common share | | | | | | | |
| Basic | $ | 2.64 | | | $ | 2.09 | | | $ | 0.31 | | | $ | 6.77 | |
| Diluted | $ | 2.62 | | | $ | 2.08 | | | $ | 0.30 | | | $ | 6.72 | |
| Shares used in computing earnings per common share | | | | | | | |
| Basic | 219.1 | | | 223.3 | | | 220.4 | | | 223.8 | |
| Diluted | 220.6 | | | 224.5 | | | 221.9 | | | 225.4 | |
| | | | | | | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Condensed Consolidated Statements of Comprehensive Income
(unaudited)
(amounts in millions)
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended September 30, | | Nine months ended September 30, |
| 2020 | | 2019 | | 2020 | | 2019 |
| Net income | $ | 578.1 | | | $ | 467.6 | | | $ | 67.6 | | | $ | 1,515.3 | |
| Other comprehensive (loss) income, net of tax: | | | | | | | |
| Foreign currency translation | 1.2 | | | (2.5) | | | (5.3) | | | (3.0) | |
| Unrealized gains on debt securities | — | | | — | | | 0.1 | | | 0.2 | |
Unrealized (gains) losses on hedging activities, net of tax of $(2.8), $3.5, $(14.3) and $(10.8), respectively | (9.5) | | | 12.2 | | | (47.2) | | | (35.7) | |
| Other comprehensive (loss) income, net of tax | (8.3) | | | 9.7 | | | (52.4) | | | (38.5) | |
| Comprehensive income | $ | 569.8 | | | $ | 477.3 | | | $ | 15.2 | | | $ | 1,476.8 | |
| | | | | | | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Condensed Consolidated Statements of Changes in Stockholders’ Equity
(unaudited)
(amounts in millions)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended September 30, 2020 | Common Stock | | Additional
Paid-In
Capital | | Treasury Stock
at Cost | | Accumulated Other
Comprehensive
Income (Loss) | | Retained Earnings | | Total
Stockholders’
Equity |
| | Shares Issued | | Amount | | Shares | | Amount | |
| Balances, June 30, 2020 | 239.2 | | | $ | — | | | $ | 8,942.4 | | | 20.1 | | | $ | (2,470.0) | | | $ | (110.9) | | | $ | 4,129.3 | | | $ | 10,490.8 | |
| Repurchase of common stock | — | | | — | | | — | | | 0.6 | | | (73.5) | | | — | | | — | | | (73.5) | |
| Issuance of common stock under stock option and stock purchase plans | 0.2 | | | — | | | 10.5 | | | — | | | — | | | — | | | — | | | 10.5 | |
| Issuance of restricted common stock | 0.3 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Share-based compensation expense | — | | | — | | | 70.3 | | | — | | | (0.2) | | | — | | | — | | | 70.1 | |
| Portola replacement equity awards attributable to the pre-combination period | — | | | — | | | 7.2 | | | — | | | — | | | — | | | — | | | 7.2 | |
| Net income | — | | | — | | | — | | | — | | | — | | | — | | | 578.1 | | | 578.1 | |
| Other comprehensive loss | — | | | — | | | — | | | — | | | — | | | (8.3) | | | — | | | (8.3) | |
| Balances, September 30, 2020 | 239.7 | | | $ | — | | | $ | 9,030.4 | | | 20.7 | | | $ | (2,543.7) | | | $ | (119.2) | | | $ | 4,707.4 | | | $ | 11,074.9 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended September 30, 2019 | Common Stock | | Additional
Paid-In
Capital | | Treasury Stock
at Cost | | Accumulated Other
Comprehensive
Income (Loss) | | Retained Earnings | | Total
Stockholders’
Equity |
| | Shares Issued | | Amount | | Shares | | Amount | |
| Balances, June 30, 2019 | 237.3 | | | $ | — | | | $ | 8,677.0 | | | 13.1 | | | $ | (1,738.8) | | | $ | (57.9) | | | $ | 3,283.2 | | | $ | 10,163.5 | |
| Repurchase of common stock | — | | | — | | | — | | | 3.1 | | | (334.6) | | | — | | | — | | | (334.6) | |
| Issuance of common stock under stock option and stock purchase plans | 0.1 | | | — | | | 1.9 | | | — | | | — | | | — | | | — | | | 1.9 | |
| Issuance of restricted common stock | 0.1 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Share-based compensation expense | — | | | — | | | 59.1 | | | — | | | — | | | — | | | — | | | 59.1 | |
| Net income | — | | | — | | | — | | | — | | | — | | | — | | | 467.6 | | | 467.6 | |
| Other comprehensive income | — | | | — | | | — | | | — | | | — | | | 9.7 | | | — | | | 9.7 | |
| Balances, September 30, 2019 | 237.5 | | | $ | — | | | $ | 8,738.0 | | | 16.2 | | | $ | (2,073.4) | | | $ | (48.2) | | | $ | 3,750.8 | | | $ | 10,367.2 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Nine months ended September 30, 2020 | Common Stock | | Additional Paid-In Capital | | Treasury Stock at Cost | | Accumulated Other Comprehensive Income (Loss) | | Retained Earnings | | Total Stockholders’ Equity |
| Shares Issued | | Amount | | Shares | | Amount | |
| Balances, December 31, 2019 | 237.8 | | | $ | — | | | $ | 8,804.7 | | | 16.5 | | | $ | (2,105.9) | | | $ | (66.8) | | | $ | 4,639.8 | | | $ | 11,271.8 | |
| Repurchase of common stock | — | | | — | | | — | | | 4.2 | | | (434.3) | | | — | | | — | | | (434.3) | |
| Issuance of common stock under stock option and stock purchase plans | 0.4 | | | — | | | 23.4 | | | — | | | — | | | — | | | — | | | 23.4 | |
| Issuance of restricted common stock | 1.5 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Share-based compensation expense | — | | | — | | | 195.1 | | | — | | | (3.5) | | | — | | | — | | | 191.6 | |
| Portola replacement equity awards attributable to the pre-combination period | — | | | — | | | 7.2 | | | — | | | — | | | — | | | — | | | 7.2 | |
| Net income | — | | | — | | | — | | | — | | | — | | | — | | | 67.6 | | | 67.6 | |
| Other comprehensive loss | — | | | — | | | — | | | — | | | — | | | (52.4) | | | — | | | (52.4) | |
| Balances, September 30, 2020 | 239.7 | | | $ | — | | | $ | 9,030.4 | | | 20.7 | | | $ | (2,543.7) | | | $ | (119.2) | | | $ | 4,707.4 | | | $ | 11,074.9 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Nine months ended September 30, 2019 | Common Stock | | Additional Paid-In Capital | | Treasury Stock at Cost | | Accumulated Other Comprehensive Income (Loss) | | Retained Earnings | | Total Stockholders’ Equity |
| Shares Issued | | Amount | | Shares | | Amount | |
| Balances, December 31, 2018 | 236.2 | | | $ | — | | | $ | 8,539.1 | | | 12.7 | | | $ | (1,689.9) | | | $ | (9.7) | | | $ | 2,325.8 | | | $ | 9,165.3 | |
| Repurchase of common stock | — | | | — | | | — | | | 3.5 | | | (383.5) | | | — | | | — | | | (383.5) | |
| Issuance of common stock under stock option and stock purchase plans | 0.3 | | | — | | | 23.2 | | | — | | | — | | | — | | | — | | | 23.2 | |
| Issuance of restricted common stock | 1.0 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Share-based compensation expense | — | | | — | | | 175.7 | | | — | | | — | | | — | | | — | | | 175.7 | |
| Net income | — | | | — | | | — | | | — | | | — | | | — | | | 1,515.3 | | | 1,515.3 | |
| Other comprehensive loss | — | | | — | | | — | | | — | | | — | | | (38.5) | | | — | | | (38.5) | |
| Adoption of new accounting standards | — | | | — | | | — | | | — | | | — | | | — | | | (90.3) | | | (90.3) | |
| Balances, September 30, 2019 | 237.5 | | | $ | — | | | $ | 8,738.0 | | | 16.2 | | | $ | (2,073.4) | | | $ | (48.2) | | | $ | 3,750.8 | | | $ | 10,367.2 | |
The accompanying notes are an integral part of these condensed consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Condensed Consolidated Statements of Cash Flows
(unaudited)
(amounts in millions)
| | | | | | | | | | | |
| | Nine months ended September 30, |
| | 2020 | | 2019 |
| Cash flows from operating activities: | | | |
| Net income | $ | 67.6 | | | $ | 1,515.3 | |
| Adjustments to reconcile net income to net cash flows from operating activities: | | | |
| Depreciation and amortization | 254.3 | | | 286.2 | |
| Change in fair value of contingent consideration | 45.0 | | | 7.2 | |
| Payments of contingent consideration | — | | | (100.0) | |
| Share-based compensation expense | 195.6 | | | 176.8 | |
| Deferred taxes (benefit) | (174.2) | | | (136.1) | |
| Unrealized foreign currency loss (gain) | 0.9 | | | (3.3) | |
| Unrealized gain on forward contracts | (3.9) | | | (15.3) | |
| Unrealized gain on strategic equity investments | (4.6) | | | (20.6) | |
| Gain on sale of asset | (14.8) | | | — | |
| Gain on derecognition of Portola strategic equity investment | (29.7) | | | — | |
| Inventory obsolescence charge | 24.6 | | | — | |
| Impairment of intangible assets | 2,053.3 | | | — | |
| Other | 10.7 | | | (2.3) | |
| Changes in operating assets and liabilities, excluding the effect of acquisitions: | | | |
| Accounts receivable | (183.5) | | | (199.1) | |
| Inventories | (10.3) | | | (105.9) | |
| Prepaid expenses, right of use operating assets and other assets | (92.3) | | | (42.2) | |
| Accounts payable, accrued expenses, lease liabilities and other liabilities | 23.7 | | | 207.1 | |
| Net cash provided by operating activities | 2,162.4 | | | 1,567.8 | |
| Cash flows from investing activities: | | | |
| Purchases of available-for-sale debt securities | (19.4) | | | (51.2) | |
| Proceeds from maturity or sale of available-for-sale debt securities | 184.2 | | | 211.0 | |
| Purchases of mutual funds related to nonqualified deferred compensation plan | (14.1) | | | (13.4) | |
| Proceeds from sale of mutual funds related to nonqualified deferred compensation plan | 9.6 | | | 11.4 | |
| Purchases of property, plant and equipment | (29.2) | | | (124.7) | |
| Payment for acquisition of businesses, net of cash and restricted cash acquired | (2,111.9) | | | — | |
| Purchases of strategic equity investments and options | (38.1) | | | (63.7) | |
| Purchase of intangible assets | — | | | (16.0) | |
| Net cash used in investing activities | (2,018.9) | | | (46.6) | |
| Cash flows from financing activities: | | | |
| Payments on term loan | (97.9) | | | (65.4) | |
| Payments on revolving credit facility | — | | | (250.0) | |
| Repurchases of common stock | (434.3) | | | (383.5) | |
| Net proceeds from issuance of common stock under share-based compensation arrangements | 22.1 | | | 23.2 | |
| Other | (28.0) | | | (3.7) | |
| Net cash used in financing activities | (538.1) | | | (679.4) | |
| Effect of exchange rate changes on cash and cash equivalents and restricted cash | (0.5) | | | (6.1) | |
| Net change in cash and cash equivalents and restricted cash | (395.1) | | | 835.7 | |
| Cash and cash equivalents and restricted cash at beginning of period | 2,723.6 | | | 1,367.4 | |
| Cash and cash equivalents and restricted cash at end of period | $ | 2,328.5 | | | $ | 2,203.1 | |
| | | |
| | | |
| | | |
| | | | | | | | | | | |
| | Nine months ended September 30, |
| | 2020 | | 2019 |
| | | |
| Supplemental cash flow disclosures from investing and financing activities: | | | |
| Contingent consideration issued in acquisitions | $ | 155.0 | | | $ | — | |
| Fair value of equity shares in Portola settled at closing of the acquisition | $ | 47.8 | | | $ | — | |
| Fair value of replacement equity awards issued to Portola employees attributable to the pre-combination period | $ | 7.2 | | | $ | — | |
| Exchange of intellectual property rights for equity shares in Inozyme | $ | 14.8 | | | $ | — | |
Fair value of strategic investment and purchase option acquired, less upfront cash paid | $ | — | | | $ | 27.6 | |
| Operating ROU lease assets obtained in exchange for operating lease liabilities | $ | 21.2 | | | $ | 23.9 | |
| Accounts payable and accrued expenses for purchases of property, plant and equipment and intangible assets | $ | 14.3 | | | $ | 3.1 | |
The following provides a reconciliation of cash and cash equivalents and restricted cash reported within the condensed consolidated balance sheets to the total of such amounts shown in the condensed consolidated statement of cash flows:
| | | | | | | | | | | |
| | Nine months ended September 30, |
| | 2020 | | 2019 |
| | | |
| Cash and cash equivalents | $ | 2,268.0 | | | $ | 2,171.3 | |
| Restricted cash included in other current assets | 60.4 | | | 31.5 | |
| Restricted cash included in other noncurrent assets | 0.1 | | | 0.3 | |
| Total cash and cash equivalents and restricted cash reported in the condensed consolidated statement of cash flows | $ | 2,328.5 | | | $ | 2,203.1 | |
Amounts included in restricted cash primarily represent funds placed in escrow as a result of the judicial order issued by the Federal Court of Canada related to SOLIRIS pricing (Note 16, Commitments and Contingencies).
The accompanying notes are an integral part of these condensed consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
1.Business
Alexion Pharmaceuticals, Inc. (Alexion, the Company, we, our or us) is a global biopharmaceutical company focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialization of life-changing medicines.
As a leader in rare diseases for more than 25 years, Alexion has developed and commercializes two approved complement inhibitors to treat patients with paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS), as well as the first and only approved complement inhibitor to treat anti-acetylcholine receptor (AChR) antibody-positive generalized myasthenia gravis (gMG) and neuromyelitis optica spectrum disorder (NMOSD) in patients who are anti-aquaporin-4 (AQP4) antibody positive. Alexion also has two highly innovative enzyme replacement therapies and the first and only approved therapies for patients with life-threatening and ultra-rare metabolic disorders, hypophosphatasia (HPP) and lysosomal acid lipase deficiency (LAL-D). With the acquisition of Portola Pharmaceuticals, Inc. (Portola) in July 2020, we added the first and only approved Factor Xa inhibitor reversal agent for patients treated with rivaroxaban or apixaban when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding.
In addition to our marketed therapies, we have a diverse pipeline resulting from internal innovation and business development. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on the core therapeutic areas of hematology, nephrology, neurology, metabolic disorders, cardiology, ophthalmology and acute care. We were incorporated in 1992 under the laws of the State of Delaware.
2.Basis of Presentation and Principles of Consolidation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States for interim financial information and with the instructions to Form 10-Q and Article 10 of Regulation S-X. Accordingly, they do not include all of the information and footnotes required by accounting principles generally accepted in the United States for complete financial statements. These accounting principles were applied on a basis consistent with those of the consolidated financial statements contained in the Company's Annual Report on Form 10-K for the year ended December 31, 2019. In our opinion, the accompanying unaudited condensed consolidated financial statements include all adjustments, consisting of only normal recurring adjustments, necessary for a fair statement of our financial statements for interim periods presented in accordance with accounting principles generally accepted in the United States. The condensed consolidated balance sheet as of December 31, 2019 was derived from audited annual financial statements but does not include all disclosures required by accounting principles generally accepted in the United States. These interim financial statements should be read in conjunction with the audited financial statements for the year ended December 31, 2019 included in our Annual Report on Form 10-K for the year ended December 31, 2019. The results of operations for the three and nine months ended September 30, 2020 are not necessarily indicative of the results to be expected for the full year or any other future periods.
The financial statements of our subsidiaries with functional currencies other than the U.S. dollar are translated into U.S. dollars using period-end exchange rates for assets and liabilities, historical exchange rates for stockholders' equity and weighted average exchange rates for operating results. Translation gains and losses are included in accumulated other comprehensive income (loss), net of tax, in stockholders' equity. Foreign currency transaction gains and losses are included in the results of operations in other income and expense.
The accompanying unaudited condensed consolidated financial statements include the accounts of Alexion Pharmaceuticals, Inc. and its subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Our significant accounting policies are described in Note 1 of the notes to the consolidated financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2019. Updates to our accounting policies, including impacts from the adoption of new accounting standards, are discussed below in this Note 2.
Reclassifications
Certain items in the prior period’s condensed consolidated financial statements have been reclassified to conform to the current presentation.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Use of Estimates
Preparation of the condensed consolidated financial statements in conformity with U.S. GAAP requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, revenues, expenses and disclosure of contingent liabilities in our condensed consolidated financial statements.
Due to the COVID-19 pandemic, there has been uncertainty and disruption in the global economy and financial markets. The full extent to which the COVID-19 pandemic will directly or indirectly impact our business, results of operations and financial condition, including sales, expenses, reserves and allowances, manufacturing, clinical trials, research and development costs and employee-related amounts, will depend on future developments that are highly uncertain. We are not aware of any specific event or circumstance that would require an update to our estimates, judgments and assumptions or a revision of the carrying value of our assets or liabilities as of October 29, 2020, the date of issuance of this Quarterly Report on Form 10-Q. These estimates may change, as new events occur and additional information is obtained. Actual results may differ from these estimates under different assumptions or conditions and such differences may be material.
New Accounting Pronouncements
Accounting Standards Update (ASU) 2019-12, “Income Taxes: Simplifying the Accounting for Income Taxes”: In December 2019, the Financial Accounting Standards Board (FASB) issued a new standard intended to simplify the accounting for income taxes by eliminating certain exceptions related to the approach for intraperiod tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. The new standard also simplifies aspects of the accounting for franchise taxes and enacted changes in tax laws or rates and clarifies the accounting for transactions that result in a step-up in the tax basis of goodwill. The standard is effective for annual periods beginning after December 15, 2020 and interim periods within, with early adoption permitted. Adoption of the standard requires certain changes to be made prospectively, with some changes to be made retrospectively. We do not expect the adoption of this standard to have a material impact on our financial condition and results of operations.
ASU 2020-01, “Investments - Equity Securities, Investments - Equity Method and Joint Ventures, and Derivatives and Hedging - Clarifying the Interactions Between Topic 321, Topic 323, and Topic 815”: In January 2020, the FASB issued a new standard intended to clarify the interactions between Accounting Standards Codification (ASC) 321, ASC 323 and ASC 815. The new standard addresses accounting for the transition into and out of the equity method and measurement of certain purchased options and forward contracts to acquire investments. The standard is effective for annual and interim periods beginning after December 15, 2020, with early adoption permitted. Adoption of the standard requires changes to be made prospectively. We do not expect the adoption of this standard to have a material impact on our financial condition and results of operations.
ASU 2020-04, “Reference Rate Reform, Facilitation of the Effects of Reference Rate Reform on Financial Reporting": In response to concerns about structural risks of interbank offered rates, and, particularly, the risk of cessation of the London Interbank Offered Rate (LIBOR), regulators around the world have undertaken reference rate reform initiatives to identify alternative reference rates that are more observable or transaction-based and less susceptible to manipulation. In March 2020, the FASB issued a new standard that provides optional guidance for a limited time to ease the potential burden in accounting for the effects of reference rate reform, including optional expedients and exceptions for the accounting implications of contracts, hedging relationships, and other transactions affected by reference rate reform if certain criteria are met.
The amendments in this new standard only apply to contracts and hedging relationships that reference LIBOR or another reference rate expected to be discontinued due to reference rate reform. The expedients and exceptions provided by the standard do not apply to contract modifications made and hedging relationships entered into or evaluated after December 31, 2022. We are currently reviewing our contracts impacted by reference rate reform and are assessing the impact of this standard on our financial condition and results of operations.
Recently Adopted Accounting Pronouncements
ASU 2018-15, "Customer's Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract": In August 2018, the FASB issued a new standard on a customer's accounting for implementation, set-up, and other upfront costs incurred in a cloud computing arrangement (CCA) that aligns the requirements for capitalizing implementation costs in a CCA service contract with existing internal-use software guidance. The standard also provides classification guidance on these implementation costs as well as additional
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
quantitative and qualitative disclosures. The standard is effective for interim and annual periods beginning after December 15, 2019, with early adoption permitted, and can be adopted prospectively or retrospectively.
We adopted the new standard on January 1, 2020 on a prospective basis. The adoption of this standard had no impact on our financial statements at the date of adoption; however, we anticipate the adoption of this standard will result in an increase in capitalized assets related to qualifying CCA implementation costs in future periods.
Qualifying CCA implementation, set-up and other upfront costs incurred after January 1, 2020 are capitalized as other assets in our condensed consolidated balance sheets. These assets will be expensed over the term of the hosting arrangement and such expense will be presented within the same line item in our condensed consolidated statements of operations as the expense for fees for the associated hosting arrangement. These capitalized costs will be evaluated for impairment when events or changes in circumstances indicate that the carrying value of the capitalized implementation costs is not recoverable. For the nine months ended September 30, 2020, capitalized CCA implementation costs were not material.
ASU 2016-13, "Measurement of Credit Losses on Financial Instruments": In June 2016, the FASB issued a new standard intended to improve reporting requirements specific to loans, receivables and other financial instruments. The new standard requires that credit losses on financial assets measured at amortized cost be determined using an expected loss model, instead of the current incurred loss model, and requires that credit losses related to available-for-sale debt securities be recorded through an allowance for credit losses and limited to the amount by which carrying value exceeds fair value. The new standard also requires enhanced disclosure of credit risk associated with financial assets. The standard is effective for interim and annual periods beginning after December 15, 2019 with early adoption permitted.
We adopted the new standard on January 1, 2020 and have completed our assessment of the standard based on the composition of our portfolio of financial instruments and current and forecasted economic conditions at that date. Our significant financial assets that are within the scope of the new standard consist of trade accounts receivable and available for sale debt securities. We have not historically experienced any material credit losses associated with our trade accounts receivable or available for debt securities.
We monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. We disaggregate our trade accounts receivable population into pools of similar risk characteristics based on underlying customer type and geographical location. Current expected credit loss allowances are estimated for each risk pool based on available information, including i) historical credit loss experience, ii) current economic conditions and, iii) reasonable and supportable forecasts of future economic conditions that may affect the collectibility of the recorded amounts. Based on the relevant facts and economic conditions as of the date of adoption, we concluded that the expected credit losses on our trade accounts receivable were immaterial. Additionally, unrealized losses on our available for sale investment portfolio were immaterial.
As of September 30, 2020, we reassessed our estimated credit losses on our trade accounts receivable, including consideration of the potential impacts of the COVID-19 global pandemic. Based on the relevant facts and economic conditions as of September 30, 2020, we concluded that the expected credit losses on our trade accounts receivable continued to be immaterial.
3.Acquisitions
Business Combinations
Achillion Pharmaceuticals, Inc.
In October 2019, Alexion entered into a definitive agreement to acquire Achillion Pharmaceuticals, Inc. (Achillion), a clinical-stage biopharmaceutical company focused on the development of oral Factor D inhibitors. Achillion was developing oral small molecule Factor D inhibitors to treat people with complement alternative pathway-mediated rare diseases, such as PNH and C3 glomerulopathy (C3G). Achillion had two clinical stage medicines in development, including danicopan (ACH-4471/ALXN2040) and ACH-5228 (ALXN2050).
The acquisition of Achillion closed on January 28, 2020. Under the terms of the agreement, we acquired all outstanding common stock of Achillion for $6.30 per share, or an aggregate of $926.2, inclusive of the settlement of Achillion's outstanding equity awards. The acquisition was funded with cash on hand. The transaction includes the potential for additional consideration in the form of non-tradeable contingent value rights (CVRs), which will be paid to Achillion shareholders if certain clinical and regulatory milestones are achieved within specified periods. These
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
include $1.00 per share for the U.S. Food and Drug Administration (FDA) approval of danicopan and $1.00 per share for the initiation of a Phase III clinical trial in ACH-5228.
The transaction was accounted for as a business combination. The following table summarizes the total consideration transferred to acquire Achillion and the estimated fair value of the identified assets acquired and liabilities assumed at the acquisition date:
| | | | | |
| Consideration | |
| Upfront payment to shareholders and option holders | $ | 926.2 | |
| Upfront payment, fair value of equity compensation attributable to the post-combination service period | (20.0) | |
| Upfront cash paid, net | 906.2 | |
| |
| Contingent consideration | 160.7 | |
| Contingent consideration, fair value of equity compensation attributable to the post-combination service period | (5.7) | |
| Total consideration | $ | 1,061.2 | |
| |
| Assets Acquired and Liabilities Assumed | |
| Cash and cash equivalents | $ | 68.5 | |
| Marketable securities | 106.1 | |
| In-process research & development assets (IPR&D) | 918.0 | |
| Goodwill | 37.8 | |
| Deferred tax liabilities, net | (62.9) | |
| Other assets and liabilities, net | (6.3) | |
| Total net assets acquired | $ | 1,061.2 | |
Our accounting for this acquisition was finalized during the second quarter of 2020. Measurement period adjustments increased goodwill by $3.1 during the second quarter of 2020 due to purchase price allocation increases to deferred tax liabilities, net. Measurement period adjustments were recorded as a result of studies completed during the second quarter of 2020 to determine the tax deductibility of certain acquisition-related costs and the valuation of historical net operating loss and income tax credit carryforwards.
The initial fair value estimate of the contingent consideration in the form of non-tradeable CVRs was $160.7, which was recorded as a noncurrent liability in our condensed consolidated balance sheet, including $5.7 related to compensation attributable to the post-combination service period. We determined the fair value of these milestone-related payment obligations using various estimates, including probabilities of success prior to expiration of the specified period, discount rates and the amount of time until the conditions of the milestone payments are expected to be met. This fair value measurement was based on significant inputs not observable in the market, representing Level 3 measurements within the fair value hierarchy. The resulting probability-weighted cash flows were discounted using a cost of debt rate ranging from 2.1% to 2.3%. The range of estimated milestone payments upon closing of the acquisition is from zero, if no milestones are achieved for any product, to $306.3 if certain development and regulatory milestones are achieved.
Subsequent to the acquisition date, we have adjusted the contingent consideration to fair value with changes in fair value recognized in operating earnings. Changes in fair values reflect new information about the probability and timing of meeting the conditions of the milestone payments. In the absence of new information, changes in fair value will only reflect the interest component of contingent consideration related to the passage of time as development work progresses towards the potential achievement of the milestones. At September 30, 2020, the fair value of the contingent consideration for the Achillion acquisition was $209.4 based on the probability-weighted cash flows, discounted using a cost of debt ranging from 3.9% to 4.3%. Changes in fair value of the contingent consideration associated with the Achillion acquisition for the three and nine months ended September 30, 2020 was $19.3 and $48.6, respectively.
The aggregate fair value of equity compensation attributable to the post-combination service period was $25.7. This amount was excluded from the total consideration transferred and was recognized as a charge to acquisition-related costs in our condensed consolidated statements of operations. These amounts were associated with the
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
accelerated vesting of stock options previously granted to Achillion employees. Excluding the $5.7 of contingent consideration related to equity compensation attributable to the post-combination service period, such amounts were paid during the first quarter 2020.
Intangible assets associated with IPR&D relate to two development-stage programs, ACH-4471 (ALXN2040) and ACH-5228 (ALXN2050). The estimated fair value of $918.0 was determined using the excess earnings valuation method, a variation of the income valuation approach. The excess earnings valuation method estimates the value of an intangible asset equal to the present value of the incremental after-tax cash flows attributable to that intangible asset. Some of the more significant assumptions utilized in our asset valuations included the estimated net cash flows for each asset, including net revenues, cost of sales, research and development and other operating expenses, the potential regulatory and commercial success rates, competitive trends impacting the assets, and tax rates. The fair value using the excess earnings valuation method was determined using an estimated weighted average cost of capital for Achillion of 11.5%, which represents a rate of return that a market participant would expect for these assets. These fair value measurements were based on significant inputs not observable in the market and thus represent Level 3 fair value measurements. In the second quarter 2020, we recognized an impairment charge of $11.0 to write off our ACHN-4471 (ALXN2040) IPR&D asset due to clinical results received during the quarter.
The excess of purchase price over the fair value of the assets acquired and liabilities assumed represents the goodwill resulting from the acquisition. The goodwill, which is not tax-deductible, has been recorded as a noncurrent asset and is not amortized, but is subject to an annual review for impairment. The factors that contributed to the recognition of goodwill include the value of the acquired workforce, synergies that are specific to our business and not available to market participants, and early research in preclinical Factor D inhibitors, as well as the effects of the establishment of a deferred tax liability for the acquired IPR&D intangible assets, which has no tax basis.
We recorded a net deferred tax liability of $62.9, inclusive of measurement period adjustments recorded during the second quarter 2020. This amount was primarily comprised of $205.3 of deferred tax liabilities relating to the IPR&D acquired, offset by $142.4 of deferred tax assets related to net operating loss carryforwards (NOLs), income tax credits, and other temporary differences.
Achillion's results of operations are included in the condensed consolidated financial statements from the date of acquisition. For the three and nine months ended September 30, 2020 we recorded $24.6 and $51.4 of pre-tax operating losses exclusive of transaction costs, $19.3 and $48.6, respectively, of changes in contingent consideration and zero and $11.0, respectively, of impairment charges, associated with the operations of Achillion in our condensed consolidated statements of operations. We also recorded acquisition-related costs in connection with the acquisition during the three and nine months ended September 30, 2020 as presented below. No revenues were recorded in the results of operations during the three and nine months ended September 30, 2020 as neither ALXN2040 nor ALXN2050 has been approved for commercial sale by any regulatory agency.
Portola Pharmaceuticals, Inc.
In May 2020, Alexion entered into a definitive merger agreement to acquire Portola Pharmaceuticals, Inc. (Portola), a commercial-stage biopharmaceutical company focused on life-threatening blood-related disorders. Portola’s commercialized medicine, ANDEXXA®, marketed as ONDEXXYA® in Europe, is the first and only approved Factor Xa inhibitor reversal agent, and has demonstrated transformative clinical value by rapidly reversing the anticoagulant effects of Factor Xa inhibitors rivaroxaban and apixaban in severe and uncontrolled bleeding. The acquisition provides the opportunity to grow Alexion's commercial portfolio and is a strategic fit with our existing expertise in acute care, hematology and neurology.
Alexion completed the acquisition through a tender offer and subsequent merger of Portola which closed on July 2, 2020. Under the terms of the tender offer and merger agreement, Alexion purchased all outstanding common stock of Portola for $18.00 per share, or an aggregate of approximately $1,380.8, including the settlement of certain of Portola's outstanding equity awards but excluding shares of Portola stock held by Alexion at closing. The acquisition was funded by cash on hand.
Prior to the acquisition of Portola, in March 2020 and April 2020, we purchased $14.5 and $3.6, respectively, of common stock of Portola, which we recorded at fair value. Upon the closing of the acquisition of Portola, the fair value of the equity investment of $47.8 was derecognized and included in the fair value of consideration transferred. For additional information on our Portola equity investment, please see Note 10, Other Investments.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The aggregate fair value of equity compensation attributable to the post-combination service period was $11.1. This amount was excluded from the total consideration transferred and was recognized as a charge to acquisition-related costs in our condensed consolidated statements of operations. These amounts were primarily associated with the accelerated vesting of stock options previously granted to Portola employees and were paid during the third quarter 2020.
We issued $41.5 of equity compensation replacement awards, of which the portion attributable to services performed prior to the acquisition date, or $7.2, was allocated to purchase consideration. The remaining fair value is attributable to future services and will be expensed as share-based compensation over the remaining service periods. Expense associated with the accelerated-vesting of the replacement awards in connection with employee terminations will be recognized as acquisition-related employee separation costs.
In connection with the acquisition, Alexion also paid $196.9 to settle certain debt held by Portola that was subject to preexisting change of control provisions.
The transaction was accounted for as a business combination. The following table summarizes the total consideration transferred to acquire Portola and the estimated fair value of the identified assets acquired and liabilities assumed at the acquisition date:
| | | | | |
| Consideration | |
| Upfront payment to shareholders and equity holders | $ | 1,380.8 | |
| Upfront payment, fair value of equity compensation attributable to the post-combination service period | (11.1) | |
| Upfront cash paid, net | 1,369.7 | |
| |
| Fair value of equity shares held by Alexion at closing | 47.8 | |
| Fair value of replacement equity awards attributable to the pre-combination period | 7.2 | |
| Total consideration to acquire outstanding equity, net | 1,424.7 | |
| |
| Total consideration to settle preexisting debt | 196.9 | |
| Total consideration | $ | 1,621.6 | |
| |
| Assets Acquired and Liabilities Assumed | |
| Cash and cash equivalents | $ | 288.5 | |
| Marketable securities | 17.8 | |
Inventories, including noncurrent portion of $169.1 and validation batches of $60.9 | 362.5 | |
| Intangible assets | 1,051.0 | |
| Goodwill | 25.5 | |
| Deferred tax assets, net | 116.0 | |
| Other assets | 41.9 | |
| Accounts payable and accrued expenses | (75.6) | |
Long-term debt, including current portion of $7.7 | (182.0) | |
| Other liabilities | (24.0) | |
| Total net assets acquired | $ | 1,621.6 | |
Our accounting for this acquisition is preliminary. The fair value estimates for the assets acquired and liabilities assumed were based on preliminary calculations, and our estimates and assumptions are subject to change as we obtain additional information for our estimates during the measurement period. The areas that rely on preliminary estimates that are not yet finalized relate to the valuation of acquired intangible assets, an ongoing assessment of the condition of certain inventory and deferred taxes, net, as a result of ongoing studies to determine the tax deductibility of certain acquisition-related costs and the valuation of historical net operating loss and income tax credit carryforwards.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
We acquired $362.5 of ANDEXXA inventory, inclusive of $60.9 of validation batches manufactured under processes which are subject to regulatory approval and expected to be commercially saleable following approval. The estimated fair value of raw material inventory was valued at replacement cost, which is equal to the value a market participant would pay to acquire the inventory. The estimated fair value of work-in-process and finished goods inventory was based on the expected selling price of the inventory, adjusted for incremental costs to complete the manufacturing process, for direct selling efforts, and for a normal profit on the remaining manufacturing and selling costs. Additionally, as the inventory acquired, inclusive of validation batches, is expected to be realized over a period of approximately 3.0 years, the fair value of the inventory was determined using a discount rate of 17.5%, representing the rate of return that a market participant would expect for the inventory, which shares risk that is similar to the underlying intellectual property. These fair value measurements were based on significant inputs not observable in the market and thus represent Level 3 fair value measurements. The acquired inventory, inclusive of the acquisition-date fair value step-up, will be expensed within cost of sales as the inventory is sold to customers. We classified the ANDEXXA inventory that is expected to be utilized beyond our normal operating cycle as a long-term asset. The fair value of the non-current portion of inventory, in addition to the validation batches, are classified within other assets in our condensed consolidated balance sheet.
Intangible assets consist of purchased technology of $1,036.0 and IPR&D of $15.0. The purchased technology intangible asset relates to Portola's lead product ANDEXXA. The estimated fair value was determined using the excess earnings valuation method, a variation of the income valuation approach. The excess earnings valuation method estimates the value of an intangible asset equal to the present value of the incremental after-tax cash flows attributable to that intangible asset. Some of the more significant assumptions utilized in our asset valuation included the estimated net cash flows for ANDEXXA, including net revenues, cost of sales, research and development and other operating expenses, the potential regulatory and commercial success rates associated with ANDEXXA's current conditional approval status and planned extension into the urgent surgery setting, competitive trends impacting the assets, and tax rates. The fair value using the excess earnings valuation method was determined using a discount rate commensurate with the risks of ANDEXXA of 17.5%, which represents a rate of return that a market participant would expect for the asset. The acquired purchased technology intangible asset is being amortized over an estimated useful life of approximately 10 years. IPR&D relates to the cerdulatinib development-stage asset. The estimated fair value of the IPR&D asset was determined using a relief from royalty (RFR) method, a variation of the income approach that is based on the cost savings that accrue to the owner of an intangible asset who would otherwise have to pay royalties on revenues earned through the use of the asset. The RFR method was modified to reflect the cash flow forecast of Portola's pre-existing in-license of cerdulatinib from Astellas Pharma, Inc. The acquired fair value of $15.0 represents an increase in the value of the asset relative to when it was initially in-licensed by Portola. Some of the more significant assumptions utilized in the IPR&D asset valuation included the estimated net revenue, royalty rate, and tax rates. The fair value using the RFR method was determined using an estimated discount rate commensurate with the risks of cerdulatinib of 17.5%, which represents a rate of return that a market participant would expect for the asset. These fair value measurements were based on significant inputs not observable in the market and thus represent Level 3 fair value measurements.
In connection with the acquisition, we assumed royalty-based debt which requires repayment through tiered royalties on future net worldwide sales of ANDEXXA. Total potential royalty payments are capped at $290.6, of which $13.7 were paid by Portola prior to the acquisition. The fair value of the remaining $276.9 in royalty-based payments as of the date of acquisition was $182.0. The estimated fair value was measured using Level 3 inputs and was calculated using a real options method, which runs simulations using various estimates, including probability-weighted net sales of ANDEXXA and volatility. Using the simulation results, the fair value was calculated based on the expected probability-weighted risk-neutral royalties, discounted at our estimated cost of debt, ranging from 3.3% to 7.1%, commensurate with the cost of debt at each period in which the royalty-based payments are estimated to be made.
We recorded net deferred tax assets of $116.0. This amount was primarily comprised of $301.0, $42.0, $42.4 and $39.1 of deferred tax assets relating to net operating loss carryforwards (NOLs), income tax credits, royalty-based debt, and other temporary differences, respectively, offset by $245.1 and $63.4 of deferred tax liabilities relating to intangible assets acquired and inventory fair value adjustments, respectively.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The excess of purchase price over the fair value of the assets acquired and liabilities assumed represents the goodwill resulting from the acquisition. The goodwill, which is not tax-deductible, has been recorded as a noncurrent asset and is not amortized, but is subject to an annual review for impairment. The factors that contributed to the recognition of goodwill primarily include the value of the acquired workforce and the effects of the establishment of a deferred tax liability for the fair value step-up of acquired inventory and intangible assets which exceed the incremental book value of acquired deferred tax assets over their fair value.
Portola's results of operations are included in the condensed consolidated financial statements from the date of acquisition. For the three and nine months ended September 30, 2020, we recorded $38.9 of revenue associated with ANDEXXA in our condensed consolidated statements of operations. For the three and nine months ended September 30, 2020, we recorded $35.4 of pre-tax operating losses excluding transaction costs and $25.9 of intangible asset amortization, associated with the operations of Portola in our condensed consolidated statements of operations. We also recorded acquisition-related costs in connection with the acquisition during the three and nine months ended September 30, 2020 as presented below.
Pro forma financial information (unaudited)
The following unaudited pro forma information presents the combined results of Alexion, Achillion, and Portola as if the acquisitions of Achillion and Portola had been completed on January 1, 2019, with adjustments to give effect to pro forma events that are directly attributable to the acquisitions. The unaudited pro forma results do not reflect operating efficiencies or potential cost savings that may have resulted from the consolidation of operations. Accordingly, the unaudited pro forma financial information is not necessarily indicative of the results of operations had we completed the transaction on January 1, 2019.
| | | | | | | | | | | | | | | | | | | | | | | |
| | Three months ended September 30 | | Nine months ended September 30 |
| | 2020 | | 2019 | | 2020 | | 2019 |
| Pro forma revenue | $ | 1,589.5 | | | $ | 1,299.9 | | | $ | 4,526.5 | | | $ | 3,694.2 | |
| Pro forma net income (loss) | $ | 625.4 | | | $ | 372.0 | | | $ | (23.2) | | | $ | 1,063.7 | |
The unaudited pro forma consolidated results for the three and nine months ended September 30, 2020 and 2019 primarily include the following pro forma adjustments related to non-recurring activity, net of tax:
•Reclassification of Alexion, Achillion and Portola acquisition-related costs. Acquisition-related costs of $72.2 and $143.5, respectively, were excluded from net income for the three and nine months ended September 30, 2020. Expenses of $129.3 were included in net income for the nine months ended September 30, 2020.
•Incremental amortization expense related to Portola purchased technology intangible assets for the three and nine months ended September 30, 2019 was $19.9 and $59.6, respectively, and for the nine months ended September 30, 2020 was $39.7.
•Incremental cost of goods sold related to Portola inventory fair value step-up adjustments calculated based on the fair value of finished goods inventory for the three and nine months ended September 30, 2019 was $10.1 and $16.7, respectively, and for the nine months ended September 30, 2020 was $10.9.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Acquisition-Related Costs
Acquisition-related costs recorded within the condensed consolidated statement of operations associated with our acquisitions of Achillion and Portola for the three and nine months ended September 30, 2020 and 2019 include the following:
| | | | | | | | | | | | | | | | | | | | | | | |
| | Three months ended September 30, | | Nine months ended September 30, |
| | 2020 | | 2019 | | 2020 | | 2019 |
Transaction costs (1) | $ | 1.8 | | | $ | — | | | $ | 7.4 | | | $ | — | |
| Integration costs | 4.6 | | | — | | | 5.6 | | | — | |
| Fair value of equity compensation attributable to the post-combination service period | 11.1 | | | — | | | 36.8 | | | — | |
Employee separation costs (2) | 45.5 | | | — | | | 55.9 | | | — | |
| $ | 63.0 | | | $ | — | | | $ | 105.7 | | | $ | — | |
(1) Transaction costs primarily include legal fees related to the acquisition of Portola as well as costs incurred to effectuate the settlement of the Achillion outstanding options
(2) Employee separation costs include liabilities recognized, and subsequent changes in estimates recorded for, severance payments, one-time short-term retention awards agreed to in connection with the acquisition of Achillion and share-based compensation expense relating to awards accelerated in connection with terminations of Portola employees.
Acquisition-related costs attributable to the Achillion acquisition for the nine months ended September 30, 2020 were $38.0. There were immaterial acquisition-related costs attributable to the Achillion acquisition for the three months ended September 30, 2020. Acquisition-related costs attributable to the Portola acquisition for three and nine months ended September 30, 2020 were $63.0 and $67.7, respectively.
4.Inventories
The components of inventory are as follows:
| | | | | | | | | | | |
| | September 30, | | December 31, |
| | 2020 | | 2019 |
| Raw materials | $ | 94.2 | | | $ | 41.2 | |
| Work-in-process | 256.6 | | | 180.8 | |
| Finished goods | 510.5 | | | 405.6 | |
| Total inventories | $ | 861.3 | | | $ | 627.6 | |
| | | |
| Balance Sheet Classification: | | | |
| Inventories | $ | 729.0 | | | $ | 627.6 | |
| Other assets | $ | 132.3 | | | $ | — | |
Total inventories include ANDEXXA inventory acquired in connection with the July 2, 2020 Portola acquisition, but exclude acquired ANDEXXA validation batches of $60.9 that were manufactured under processes which are subject to regulatory approval. The acquired ANDEXXA inventory includes the acquisition-date fair value step-up, which is expensed within cost of sales as the inventory is sold to customers. For additional information on our acquisition of Portola, please see Note 3, Acquisitions.
We classify our inventory costs as long-term when we expect to utilize the inventory beyond our normal operating cycle and include these costs in other assets in our condensed consolidated balance sheets. Inventories classified as long-term relate to ANDEXXA inventory acquired in connection with the Portola acquisition.
As of September 30, 2020 and December 31, 2019, the carrying value of capitalized inventory manufactured at production facilities that have not yet received regulatory approval was $117.2 and $60.5, respectively.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
5.Intangible Assets and Goodwill
The following table summarizes the carrying amount of our intangible assets and goodwill, net of accumulated amortization and impairment charges:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | September 30, 2020 | | December 31, 2019 |
| Estimated
Life (years) | | Cost | | Accumulated
Amortization | | Net | | Cost | | Accumulated
Amortization | | Net |
| Licensing rights | 3-8 | | $ | 57.0 | | | $ | (37.5) | | | $ | 19.5 | | | $ | 57.0 | | | $ | (34.7) | | | $ | 22.3 | |
| Patents | 7 | | 10.5 | | | (10.5) | | | — | | | 10.5 | | | (10.5) | | | — | |
| Purchased technology | 6-16 | | 5,746.5 | | | (3,631.5) | | (a) | 2,115.0 | | | 4,710.5 | | | (1,388.7) | | | 3,321.8 | |
| Other intangibles | 5 | | 0.4 | | | (0.3) | | | 0.1 | | | 0.4 | | | (0.2) | | | 0.2 | |
| Acquired IPR&D | Indefinite | | 922.0 | | | — | | | 922.0 | | | — | | | — | | | — | |
| Total | | | $ | 6,736.4 | | | $ | (3,679.8) | | | $ | 3,056.6 | | | $ | 4,778.4 | | | $ | (1,434.1) | | | $ | 3,344.3 | |
| Goodwill | Indefinite | | $ | 5,103.6 | | | $ | (2.9) | | | $ | 5,100.7 | | | $ | 5,040.3 | | | $ | (2.9) | | | $ | 5,037.4 | |
(a) Includes an impairment charge of $2,042.3 recognized during the second quarter 2020 related to the KANUMA intangible asset |
In connection with our acquisition of Achillion during the first quarter 2020, we acquired IPR&D programs with a fair value of $918.0 and recorded goodwill of $37.8. For additional information on our acquisition of Achillion, please see Note 3, Acquisitions. In the second quarter 2020, we recognized an impairment charge of $11.0 to write off the cost basis of our ACHN-4471 (ALXN2040) acquired in-process research and development asset due to clinical results received during the quarter.
In connection with our acquisition of Portola during the third quarter 2020, we acquired purchased technology and IPR&D programs with a fair value of $1,036.0 and $15.0, respectively and recorded goodwill of $25.5. For additional information on our acquisition of Portola, please see Note 3, Acquisitions.
Amortization expense for the three months ended September 30, 2020 and 2019 was $54.1 and $77.5, respectively. Amortization expense for the nine months ended September 30, 2020 and 2019 was $203.4 and $239.2, respectively. As of September 30, 2020, assuming no changes in the gross cost basis of intangible assets, the total estimated amortization expense for finite-lived intangible assets is $54.1 for the three months ending December 31, 2020, and approximately $216.0 for each of the years ending December 31, 2021 through December 31, 2025.
During the quarter ended June 30, 2020, based on continued challenges expanding patient growth and new alternative commercial opportunities, the Company revised its strategic view of KANUMA and determined that we have exhausted commercially viable initiatives related to KANUMA and will have difficulty expanding patient growth over the long term as we focus on promoting other commercial programs and growing our pipeline. As a result, we no longer expect to increase the number of KANUMA patients in the long term at the rate previously assumed. This determination resulted in reduced cash flow projections for KANUMA, which indicated that the related intangible asset value was not fully recoverable on an undiscounted cash flows basis. On June 30, 2020, the Company utilized market participant assumptions to determine its best estimate of the fair value of the intangible asset related to KANUMA that, when compared with its related carrying value, resulted in an impairment charge of $2,042.3.
The estimated fair value of the KANUMA asset as of June 30, 2020 was determined using the excess earnings method, a variation of the income approach. The excess earnings method estimates the value of an intangible asset equal to the present value of the incremental after-tax cash flows attributable to that intangible asset over its remaining economic life. Long term cash flow projections for the asset require the use of significant estimates and judgements, including discount rates and revenue growth rates, and were based on the Company’s most recent strategic plan. The fair value of the asset was determined using an estimated weighted average cost of capital of 10.0%, which reflects the risks inherent in future cash flow projections and represents a rate of return that a market participant would expect for this asset. The estimated revenue growth rates fluctuate over the life of the asset, with a weighted average growth rate in the low single digits. The Company believes its assumptions are consistent with the plans and estimates that a market participant would use to manage the business. The estimated fair value of the KANUMA intangible asset as of June 30, 2020 was $820.0 and will continue to be amortized over its remaining estimated useful life. This fair value measurement was based on significant inputs not observable in the market and thus represents a Level 3 fair value measurement.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The following summarizes the changes in the carrying amount of goodwill:
| | | | | |
| September 30, 2020 |
| Balance at December 31, 2019 | $ | 5,037.4 | |
| Goodwill resulting from the acquisitions of Achillion and Portola | 63.3 | |
| Balance at September 30, 2020 | $ | 5,100.7 | |
6.Debt
Credit Agreement
On June 7, 2018, we entered into an Amended and Restated Credit Agreement (the Credit Agreement), with Bank of America, N.A. as Administrative Agent. The Credit Agreement amended and restated our credit agreement dated as of June 22, 2015 (the Prior Credit Agreement).
The Credit Agreement provides for a $1,000.0 revolving credit facility and a $2,612.5 term loan facility. The revolving credit facility and the term loan facility mature on June 7, 2023. Beginning with the quarter ending June 30, 2019, we are required to make payments of 5.00% of the original principal amount of the term loan facility annually, payable in equal quarterly installments.
In connection with entering into the Credit Agreement and the Prior Credit Agreement, we paid an aggregate of $53.1 in financing costs in 2018. Financing costs are amortized as interest expense over the life of the debt. Amortization expense associated with deferred financing costs for the three months ended September 30, 2020 and 2019 was $1.2 and $1.3, respectively. Amortization expense associated with deferred financing costs for the nine months ended September 30, 2020 and 2019 was $3.6 and $3.8, respectively. Remaining unamortized deferred financing costs as of September 30, 2020 and December 31, 2019 were $12.2 and $15.8, respectively.
We made principal payments of $32.6 and $97.9 on the term loan during the three and nine months ended September 30, 2020, respectively, and as of September 30, 2020, we had $2,416.6 outstanding on the term loan. We had no outstanding borrowings under the revolving credit facility as of September 30, 2020. As of September 30, 2020, we had open letters of credit of $1.0 that offset our availability in the revolving credit facility.
The amount outstanding under the term loan of $2,416.6 as of September 30, 2020 is subject to variable interest rates, which are based on current market rates, and as such, the Company believes the carrying amount of the obligation approximates fair value.
We were in compliance with all applicable covenants under the Credit Agreement as of September 30, 2020.
Royalty-based Financing
In connection with our acquisition of Portola during the third quarter 2020, we assumed royalty-based debt relating to a royalty sales agreement Portola had entered into with HealthCare Royalty Partners (HCR) whereby HCR acquired a tiered royalty interest in future worldwide net sales of ANDEXXA. Portola received $50.0 upon closing of the agreement in February 2017 and an additional $100.0 following the U.S. regulatory approval of ANDEXXA in May 2018. Tiered royalties ranging from 4.2% to 8.5% are required to be paid to HCR based on net worldwide sales of ANDEXXA. The applicable rate decreases as worldwide net annual sales levels increase above defined thresholds. Total potential royalty payments are capped at 195% of the funding received less certain transaction expenses, or $290.6. As of the date of acquisition, the remaining due to HCR was $276.9 in royalty-based payments.
We recorded the HCR debt at its fair value of $182.0 upon closing of the acquisition, representing an initial debt discount of $94.9. We have also recognized a deferred tax asset of $42.4 related to the royalty-based debt as of the acquisition date. For additional information on our acquisition of Portola, please see Note 3, Acquisitions. Interest expense is recognized using the effective interest rate method over the estimated period the related debt will be paid. This requires estimation of the timing and amount of future royalty payments to be generated from future sales of ANDEXXA. We reassess the expected royalty payments each reporting period and account for any changes through an adjustment to the effective interest rate on a prospective basis. The assumptions used in determining the expected repayment term of the debt require that we make estimates that could impact the short and long term classification of the debt carrying values.
Each period, we amortize the initial debt discount using the effective interest rate implied from the projected timing of royalty payments to HCR. The effective interest rate for the HCR royalty-based debt as of September 30, 2020 was 11.4%. During the three and nine months ended September 30, 2020, we recognized interest expense
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
associated with the amortization of the debt discount of $4.9. We made royalty-based debt payments of $1.7 during the three and nine months ended September 30, 2020. As of September 30, 2020, the carrying value of the royalty-based debt includes approximately $3.0 of royalty payments on third quarter sales of ANDEXXA which will be paid during the fourth quarter 2020.
As of September 30, 2020, the carrying value of the HCR royalty-based debt was $185.2, of which $11.7 was recorded within current portion of long-term debt and $173.5 was recorded within long-term debt, less current portion on our condensed consolidated balance sheet. Our payment obligations for HCR royalty-based debt are as follows:
| | | | | |
| Nine Months Ended September 30, 2020 |
| Total repayment obligation as of the acquisition date | $ | 276.9 | |
| Less: interest to be accreted in future periods | (90.0) | |
| Less: payments made | (1.7) | |
| Carrying value as of September 30, 2020 | $ | 185.2 | |
The carrying value of the royalty based debt as of September 30, 2020 approximates fair value due to the proximity to the acquisition date.
7.Earnings Per Common Share
Basic earnings per common share (EPS) is computed by dividing net income by the weighted-average number of shares of common stock outstanding. For purposes of calculating diluted EPS, the denominator reflects the potential dilution that could occur if stock options, unvested restricted stock units or other contracts to issue common stock were exercised or converted into common stock, using the treasury stock method.
The following table summarizes the calculation of basic and diluted EPS for the three and nine months ended September 30, 2020 and 2019:
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended | | Nine months ended |
| | September 30, | | September 30, |
| | 2020 | | 2019 | | 2020 | | 2019 |
| Net income used for basic and diluted calculation | $ | 578.1 | | | $ | 467.6 | | | $ | 67.6 | | | $ | 1,515.3 | |
| Shares used in computing earnings per common share—basic | 219.1 | | | 223.3 | | | 220.4 | | | 223.8 | |
| Weighted-average effect of dilutive securities: | | | | | | | |
| Stock awards | 1.5 | | | 1.2 | | | 1.5 | | | 1.6 | |
| Shares used in computing earnings per common share—diluted | 220.6 | | | 224.5 | | | 221.9 | | | 225.4 | |
| Earnings per common share: | | | | | | | |
| Basic | $ | 2.64 | | | $ | 2.09 | | | $ | 0.31 | | | $ | 6.77 | |
| Diluted | $ | 2.62 | | | $ | 2.08 | | | $ | 0.30 | | | $ | 6.72 | |
We exclude from diluted EPS the weighted-average number of securities whose effect is anti-dilutive. Excluded from the calculation of EPS for the three and nine months ended September 30, 2020 were 2.5 and 1.8 shares of common stock, respectively, because their effect is anti-dilutive. Excluded from the calculation of EPS for the three and nine months ended September 30, 2019 were 3.3 and 3.1 shares of common stock, respectively, because their effect is anti-dilutive.
8.Marketable Securities
The proceeds from maturities and sales of available-for-sale debt securities and resulting realized gains and losses are summarized below. In the second quarter of 2020, we liquidated all of our available-for-sale debt securities to fund the acquisition of Portola. Additionally, we liquidated all available-for-sale debt securities acquired in connection with the Portola acquisition.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended | | Nine months ended |
| September 30, | | September 30, |
| 2020 | | 2019 | | 2020 | | 2019 |
Proceeds from maturities and sales (1) | $ | 25.5 | | | $ | 645.4 | | | $ | 1,042.4 | | | $ | 2,271.7 | |
| Realized gains | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Realized losses | $ | — | | | $ | — | | | $ | — | | | $ | — | |
(1) Proceeds from maturities and sales of available-for-sale debt securities include securities previously classified as cash and cash equivalents and marketable securities in the condensed consolidated balance sheet. |
We utilize the specific identification method in computing realized gains and losses.
As a result of our liquidation of all available-for-sale debt securities during the second and third quarters of 2020, we have no remaining available-for-sale debt securities as of September 30, 2020. The amortized cost, gross unrealized holding gains, gross unrealized holding losses and fair value of available-for-sale debt securities by type of security as of December 31, 2019 were as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2019 |
| | Amortized Cost | | Gross Unrealized Holding Gains | | Gross Unrealized Holding Losses | | Fair Value |
| Commercial paper | | $ | 246.9 | | | $ | — | | | $ | — | | | $ | 246.9 | |
| Corporate bonds | | 24.3 | | | — | | | — | | | 24.3 | |
| Other government-related obligations: | | | | | | | | |
| U.S. | | 70.4 | | | — | | | — | | | 70.4 | |
| Bank certificates of deposit | | 27.4 | | | — | | | — | | | 27.4 | |
| Total available-for-sale debt securities | | $ | 369.0 | | | $ | — | | | $ | — | | | $ | 369.0 | |
The aggregate fair value of available-for-sale debt securities in an unrealized loss position as of December 31, 2019 was $21.5. We did not have any investments in a continuous unrealized loss position for more than twelve months as of December 31, 2019.
The fair values of available-for-sale debt securities by classification in the condensed consolidated balance sheets were as follows:
| | | | | | | | | | | |
| September 30, 2020 | | December 31, 2019 |
| Cash and cash equivalents | $ | — | | | $ | 328.1 | |
| Marketable securities | — | | | 40.9 | |
| $ | — | | | $ | 369.0 | |
We sponsor a nonqualified deferred compensation plan which allows certain highly-compensated employees to elect to defer income to future periods. Participants in the plan earn a return on their deferrals based on several investment options, which mirror returns on underlying mutual fund investments. We choose to invest in the underlying mutual fund investments to offset the liability associated with our nonqualified deferred compensation plan. These mutual fund investments are valued at net asset value per share and are carried at fair value with gains and losses included in investment income. The changes in the underlying liability to the employee are recorded in operating expenses. As of September 30, 2020 and December 31, 2019, the fair value of these investments was $28.9 and $23.1, respectively.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
9.Derivative Instruments and Hedging Activities
We operate internationally and, in the normal course of business, are exposed to fluctuations in foreign currency exchange rates. The exposures result from portions of our revenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, primarily the Euro and Japanese Yen. We are also exposed to fluctuations in interest rates on outstanding borrowings under our revolving credit facility, if any, and term loan facility. We manage these exposures within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes.
We enter into foreign exchange forward contracts, with durations of up to 60 months, to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, and certain forecasted expenses that are denominated in currencies other than the U.S. dollar. The purpose of these hedges is to reduce the volatility of exchange rate fluctuations on our operating results. These hedges are designated as cash flow hedges upon contract inception. As of September 30, 2020, we had open revenue related foreign exchange forward contracts with notional amounts totaling $1,029.0 that qualified for hedge accounting with current contract maturities through June 2022. As of September 30, 2020, we had open expense related foreign exchange forward contracts with notional amounts totaling $10.0 that qualified for hedge accounting with contract maturities through September 2022.
To achieve a desired mix of floating and fixed interest rates on our term loan, we enter into interest rate swap agreements that qualify for and are designated as cash flow hedges. These contracts convert the floating interest rate on a portion of our debt to a fixed rate, plus a borrowing spread.
The following table summarizes the total interest rate swap contracts executed as of September 30, 2020:
| | | | | | | | | | | | | | |
| Type of Interest Rate Swap Contract | Notional Amount | Effective Date | Termination Date | Fixed Interest Rate or Rate Range |
| Floating to Fixed | $450.0 | December 2018 | December 2022 | 2.60% - 2.79% |
| Floating to Fixed | $1,300.0 | December 2019 | December 2022 | 2.37% - 2.83% |
The amount of gains and (losses) recognized in the condensed consolidated statements of operations for the three and nine months ended September 30, 2020 and 2019 from foreign exchange and interest rate swap contracts that qualified as cash flow hedges were as follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended | | Three months ended |
| | September 30, 2020 | | September 30, 2019 |
| Financial Statement Line Item in which the Effects of Cash Flow Hedges are Recorded | Net Product Sales | | Interest Expense | | Net Product Sales | | Interest Expense |
| Total amount presented in the Condensed Consolidated Statements of Operations | $ | 1,588.3 | | | $ | (27.6) | | | $ | 1,263.1 | | | $ | (17.9) | |
| Impact of cash flow hedging relationships: | | | | | | | |
| Foreign exchange forward contracts | $ | (5.0) | | | $ | — | | | $ | 11.0 | | | $ | — | |
| Interest rate swap contracts | $ | — | | | $ | (11.3) | | | $ | — | | | $ | 3.4 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Nine months ended | | Nine months ended |
| | September 30, 2020 | | September 30, 2019 |
| Financial Statement Line Item in which the Effects of Cash Flow Hedges are Recorded | Net Product Sales | | Interest Expense | | Net Product Sales | | Interest Expense |
| Total amount presented in the Condensed Consolidated Statements of Operations | $ | 4,477.4 | | | $ | (77.0) | | | $ | 3,605.8 | | | $ | (56.1) | |
| Impact of cash flow hedging relationships: | | | | | | | |
| Foreign exchange forward contracts | $ | 15.9 | | | $ | — | | | $ | 27.1 | | | $ | — | |
| Interest rate swap contracts | $ | — | | | $ | (25.9) | | | $ | — | | | $ | 12.5 | |
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The impact on accumulated other comprehensive income (AOCI) and earnings from foreign exchange and interest rate swap contracts that qualified as cash flow hedges, for the three and nine months ended September 30, 2020 and 2019 were as follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended | | Nine months ended |
| | September 30, | | September 30, |
| | 2020 | | 2019 | | 2020 | | 2019 |
| Foreign Exchange Forward Contracts: | | | | | | | |
| (Loss) gain recognized in AOCI, net of tax | $ | (22.9) | | | $ | 30.5 | | | $ | (3.0) | | | $ | 40.8 | |
| (Loss) gain reclassified from AOCI to net product sales, net of tax | $ | (4.0) | | | $ | 8.6 | | | $ | 12.2 | | | $ | 21.0 | |
| Interest Rate Swap Contracts: | | | | | | | |
| Gain (loss) recognized in AOCI, net of tax | $ | 0.1 | | | $ | (7.1) | | | $ | (52.6) | | | $ | (45.9) | |
| (Loss) gain reclassified from AOCI to interest expense, net of tax | $ | (8.8) | | | $ | 2.6 | | | $ | (20.1) | | | $ | 9.6 | |
Assuming no change in foreign exchange rates from market rates at September 30, 2020, $13.6 of losses recognized in AOCI will be reclassified to revenue over the next 12 months. Assuming no change in LIBOR-based interest rates from market rates at September 30, 2020, $45.6 of losses recognized in AOCI will be reclassified to interest expense over the next 12 months. Amounts recognized in AOCI for expense related foreign exchange forward contracts were immaterial as of September 30, 2020.
We enter into foreign exchange forward contracts, with durations of up to 6 months, designed to limit the balance sheet exposure of monetary assets and liabilities. We enter into these hedges to reduce the impact of fluctuating exchange rates on our operating results. Hedge accounting is not applied to these derivative instruments as gains and losses on these hedge transactions are designed to offset gains and losses on underlying balance sheet exposures. As of September 30, 2020, the notional amount of foreign exchange contracts where hedge accounting is not applied was $1,253.0.
We recognized a (loss) gain of $(6.2) and $1.4, in other income and (expense) for the three months ended September 30, 2020 and 2019, respectively, associated with the foreign exchange contracts not designated as hedging instruments. We recognized a gain (loss) of $6.8 and $(1.2), in other income and (expense) for the nine months ended September 30, 2020 and 2019, respectively, associated with the foreign exchange contracts not designated as hedging instruments. These amounts were partially offset by gains or losses on monetary assets and liabilities.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The following tables summarize the fair value of outstanding derivatives as of September 30, 2020 and December 31, 2019:
| | | | | | | | | | | | | | | | | | | | | | | |
| September 30, 2020 |
| | Derivative Assets | | Derivative Liabilities |
| Balance Sheet
Location | | Fair
Value | | Balance Sheet
Location | | Fair
Value |
| Derivatives designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | $ | 3.0 | | | Other current liabilities | | $ | 16.8 | |
| Foreign exchange forward contracts | Other assets | | 0.4 | | | Other liabilities | | 0.3 | |
| Interest rate swap contracts | Prepaid expenses and other current assets | | — | | | Other current liabilities | | 45.6 | |
| Interest rate swap contracts | Other assets | | — | | | Other liabilities | | 57.6 | |
| Derivatives not designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | 8.7 | | | Other current liabilities | | 8.0 | |
| Total fair value of derivative instruments | | | $ | 12.1 | | | | | $ | 128.3 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2019 |
| | Derivative Assets | | Derivative Liabilities |
| Balance Sheet
Location | | Fair
Value | | Balance Sheet
Location | | Fair
Value |
| Derivatives designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | $ | 12.7 | | | Other current liabilities | | $ | 6.2 | |
| Foreign exchange forward contracts | Other assets | | 0.6 | | | Other liabilities | | 1.1 | |
| Interest rate swap contracts | Prepaid expenses and other current assets | | — | | | Other current liabilities | | 19.5 | |
| Interest rate swap contracts | Other assets | | — | | | Other liabilities | | 41.9 | |
| Derivatives not designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | 17.2 | | | Other current liabilities | | 20.4 | |
| Total fair value of derivative instruments | | | $ | 30.5 | | | | | $ | 89.1 | |
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Although we do not offset derivative assets and liabilities within our condensed consolidated balance sheets, our International Swap and Derivatives Association agreements provide for net settlement of transactions that are due to or from the same counterparty upon early termination of the agreement due to an event of default or other termination event. The following tables summarize the potential effect on our condensed consolidated balance sheets of offsetting our foreign exchange forward contracts and interest rate contracts subject to such provisions:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | September 30, 2020 |
| | | | | | | | Gross Amounts Not Offset in the Condensed Consolidated Balance Sheet | | |
| Description | | Gross Amounts of Recognized Assets/Liabilities | | Gross Amounts Offset in the Condensed Consolidated Balance Sheet | | Net Amounts of Assets/Liabilities Presented in the Condensed Consolidated Balance Sheet | | Derivative Financial Instruments | | Cash Collateral Received (Pledged) | | Net Amount |
| Derivative assets | | $ | 12.1 | | | $ | — | | | $ | 12.1 | | | $ | (11.4) | | | $ | — | | | $ | 0.7 | |
| Derivative liabilities | | $ | (128.3) | | | $ | — | | | $ | (128.3) | | | $ | 11.4 | | | $ | — | | | $ | (116.9) | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2019 |
| | | | | | | | Gross Amounts Not Offset in the Condensed Consolidated Balance Sheet | | |
| Description | | Gross Amounts of Recognized Assets/Liabilities | | Gross Amounts Offset in the Condensed Consolidated Balance Sheet | | Net Amounts of Assets/Liabilities Presented in the Condensed Consolidated Balance Sheet | | Derivative Financial Instruments | | Cash Collateral Received (Pledged) | | Net Amount |
| Derivative assets | | $ | 30.5 | | | $ | — | | | $ | 30.5 | | | $ | (21.4) | | | $ | — | | | $ | 9.1 | |
| Derivative liabilities | | $ | (89.1) | | | $ | — | | | $ | (89.1) | | | $ | 21.4 | | | $ | — | | | $ | (67.7) | |
10.Other Investments
Other investments include strategic investments in equity securities of certain biotechnology companies which we acquired in connection with strategic business development transactions, including license and option agreements. These investments are included in other assets in our condensed consolidated balance sheets.
Moderna
During 2014, we purchased $37.5 of preferred stock of Moderna Therapeutics, Inc. (Moderna), a privately held biotechnology company, which was initially recorded at cost. We began recording the investment at fair value, with the effects of a holding period restriction estimated using an option pricing valuation model, upon Moderna's completion of its initial public offering (IPO) in 2018. During the three and nine months ended September 30, 2019, we recognized an unrealized gain of $12.3 and $13.1, respectively, in investment income to adjust our investment in Moderna to fair value. On December 9, 2019, we sold our investment in Moderna for $114.7 in net proceeds, resulting in a realized gain of $77.2 on our initial investment.
Dicerna
In October 2018, we purchased $10.3 of Dicerna Pharmaceuticals Inc. (Dicerna) common stock in connection with an agreement that we entered into with Dicerna, a publicly-traded biopharmaceutical company. As our equity investment in Dicerna common stock has a readily determinable fair value, we are recording the investment at fair value. During the three and nine months ended September 30, 2020, we recognized an unrealized loss of $6.2 and $3.4, respectively, in investment income to adjust our equity investment in Dicerna to fair value. During the three and nine months ended September 30, 2019, we recognized an unrealized loss of $1.1 and unrealized gain of $3.1, respectively, in investment income to adjust our equity investment in Dicerna to fair value.
The fair value of this investment was $15.0 and $18.4 as of September 30, 2020 and December 31, 2019, respectively.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Caelum
In January 2019, we purchased $41.0 of preferred stock of Caelum Biosciences (Caelum), a privately-held biotechnology company, and a $16.1 option to acquire the remaining equity in Caelum, based on Phase II data, in connection with an agreement that we entered into with Caelum. Following discussions with the FDA, Caelum changed its clinical development plan for CAEL-101 in the fourth quarter 2019. A Phase II trial for CAEL-101 subsequently commenced during the first quarter of 2020 and met its primary objectives, supporting the safety and tolerability of CAEL-101 and confirmed the dose and regimen to be adopted for the Phase III studies. In December 2019, we amended the terms of the agreement with respect to the option to acquire the remaining equity in Caelum based on data from the modified Phase II/III trials. We accounted for the amendment as an exchange transaction as the terms of the modified option were determined to be substantially different than the terms of the original option. In conjunction with this amendment, we recognized a gain of $32.0 during the fourth quarter 2019 in other income and (expense), which reflects an increase in the fair value of the option, less $20.0 in incremental upfront funding which we accrued as of December 31, 2019 and paid during the first quarter 2020, and $4.1 associated with the change in the fair value of contingent payments which we also modified as part of the amendment. See Note 16, Commitments and Contingencies, for additional information on the agreement. As our equity investment in Caelum and the option to acquire the remaining equity in Caelum do not have a readily determinable fair value, we only adjust the carrying value of the assets for impairment and any subsequent changes resulting from an observable price change in an orderly transaction for identical or similar equity securities of the same issuer.
There were no observable price changes associated with these assets during the three and nine months ended September 30, 2020 and 2019. As of September 30, 2020, the carrying value of the investment and option, respectively, was $41.0 and $64.0. As of December 31, 2019, the carrying value of the investment and option, respectively, was $41.0 and $64.0. The investment and option were not impaired as of September 30, 2020.
Zealand
In March 2019, we purchased $13.8 of Zealand Pharma A/S (Zealand) common stock in connection with an agreement that we entered into with Zealand, a publicly-traded biopharmaceutical company based in Copenhagen, Denmark. See Note 16, Commitments and Contingencies, for additional information on the agreement. As our equity investment in Zealand common stock has a readily determinable fair value, we are recording the investment at fair value. During the three and nine months ended September 30, 2020, we recognized an unrealized gain of $1.8 and $0.8, respectively, in investment income to adjust our equity investment in Zealand to fair value. During the three and nine months ended September 30, 2019, we recognized an unrealized gain of $3.7 and $7.3, respectively, in investment income to adjust our equity investment in Zealand to fair value.
The fair value of this investment was $30.5 and $28.5 as of September 30, 2020 and December 31, 2019, respectively.
Eidos
In September 2019, we purchased $19.9 of Eidos Therapeutics, Inc. (Eidos) common stock, in connection with an agreement that we entered into with Eidos, a publicly-traded biopharmaceutical company and subsidiary of BridgeBio Pharma, Inc. See Note 16, Commitments and Contingencies, for additional information on the agreement. As our equity investment in Eidos common stock has a readily determinable fair value, we are recording the investment at fair value. During the three and nine months ended September 30, 2020, we recognized an unrealized gain of $3.4 and $0.3, respectively, in investment income to adjust our equity investment in Eidos to fair value. During the three and nine months ended September 30, 2019, we recognized an unrealized loss of $2.9, in investment income to adjust our equity investment in Eidos to fair value.
The fair value of this investment was $28.1 and $27.8 as of September 30, 2020 and December 31, 2019, respectively.
Stealth
In October 2019, we purchased $9.6 of Stealth BioTherapeutics Corp. (Stealth) common stock, in connection with an agreement that we entered into with Stealth, a publicly traded clinical-stage biotechnology company. As our equity investment in Stealth common stock has a readily determinable fair value, we are recording the investment at fair value. During the three and nine months ended September 30, 2020, we recognized an unrealized loss of $0.7 and $2.6, respectively, in investment income to adjust our equity investment in Stealth to fair value.
The fair value of this investment was $1.8 and $4.4 as of September 30, 2020 and December 31, 2019, respectively.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Portola
In March 2020 and April 2020, we purchased $14.5 and $3.6, respectively, of common stock of Portola Pharmaceuticals, Inc., a publicly traded commercial-stage biological company which we acquired on July 2, 2020. As our equity investment in Portola common stock had a readily determinable fair value, we recorded the investment at fair value. During the three and nine months ended September 30, 2020, we recognized a net gain of $0.1 and $29.7 in investment income, respectively, relating to our Portola investment. Upon the closing of the acquisition of Portola on July 2, 2020, the fair value of the equity investment of $47.8 was derecognized and included in the fair value of consideration transferred, resulting in a realized gain of $29.7 on our initial investment.
Inozyme
On July 17, 2020, we sold certain intellectual property rights and assets focusing on ENPP1 gene deficiencies to Inozyme Pharma (Inozyme), a publicly traded biopharmaceutical company, in exchange for $14.8 of Inozyme Pharma common stock, which was initially recorded at its IPO offering price, net of the effects of a nine month holding period restriction. We recognized the $14.8 of consideration received as a gain within gain on sale of asset in our condensed consolidated statement of operations. As our equity investment in Inozyme common stock has a readily determinable fair value, we are recording the investment at fair value, with the effects of a nine month holding period restriction estimated using an option pricing valuation model. During the three and nine months ended September 30, 2020, we recognized an unrealized gain of $10.2, in investment income to adjust our investment to fair value.
The fair value of this investment was $25.0 as of September 30, 2020.
11.Stockholders' Equity
Share Repurchases
In November 2012, our Board of Directors authorized a share repurchase program. In February 2017, our Board of Directors increased the amount that we are authorized to expend on future repurchases to $1,000.0 under our repurchase program, which superseded all prior repurchase programs. The entire amount authorized pursuant to this February 2017 Board approval has been utilized. On October 22, 2019, the Board of Directors approved a share repurchase authorization of up to $1,000.0. On July 28, 2020, the Board of Directors approved a new share repurchase authorization of up to an additional $1,500.0. The repurchase program does not have an expiration date and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at our discretion. Under the program, we repurchased 0.6 and 3.1 shares of our common stock at a cost of $73.5 and $334.6 during the three months ended September 30, 2020 and 2019, respectively. During the nine months ended September 30, 2020 and 2019, we repurchased 4.2 and 3.5 shares of our common stock at a cost of $434.3 and $383.5, respectively.
Subsequent to September 30, 2020, we repurchased 0.4 shares of common stock under our repurchase program at a cost of $45.2. As of October 27, 2020, there is a total of $2,055.9 remaining for repurchases under the repurchase program.
12.Other Comprehensive Income and Accumulated Other Comprehensive Income
The following tables summarize the changes in AOCI, by component, for the nine months ended September 30, 2020 and 2019:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Defined Benefit Pension Plans | | Unrealized Gains (Losses) from Debt Securities | | Unrealized Gains (Losses) from Hedging Activities | | Foreign Currency Translation Adjustment | | Total Accumulated Other Comprehensive Income (Loss) |
| Balances, December 31, 2019 | $ | (9.2) | | | $ | (0.1) | | | $ | (40.1) | | | $ | (17.4) | | | $ | (66.8) | |
| Other comprehensive income (loss) before reclassifications | — | | | 0.1 | | | (55.1) | | | (5.3) | | | (60.3) | |
| Amounts reclassified from other comprehensive income | — | | | — | | | 7.9 | | | — | | | 7.9 | |
| Net other comprehensive income (loss) | — | | | 0.1 | | | (47.2) | | | (5.3) | | | (52.4) | |
| Balances, September 30, 2020 | $ | (9.2) | | | $ | — | | | $ | (87.3) | | | $ | (22.7) | | | $ | (119.2) | |
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Defined Benefit Pension Plans | | Unrealized Gains (Losses) from Debt Securities | | Unrealized Gains (Losses) from Hedging Activities | | Foreign Currency Translation Adjustment | | Total Accumulated Other Comprehensive Income (Loss) |
| Balances, December 31, 2018 | $ | (2.6) | | | $ | (0.3) | | | $ | 9.6 | | | $ | (16.4) | | | $ | (9.7) | |
| Other comprehensive income (loss) before reclassifications | — | | | 0.2 | | | (5.1) | | | (3.0) | | | (7.9) | |
| Amounts reclassified from other comprehensive income | — | | | — | | | (30.6) | | | — | | | (30.6) | |
| Net other comprehensive income (loss) | — | | | 0.2 | | | (35.7) | | | (3.0) | | | (38.5) | |
| Balances, September 30, 2019 | $ | (2.6) | | | $ | (0.1) | | | $ | (26.1) | | | $ | (19.4) | | | $ | (48.2) | |
The table below provides details regarding significant reclassifications from AOCI during the three and nine months ended September 30, 2020 and 2019:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Details about Accumulated Other Comprehensive Income Components | Amount Reclassified From Accumulated Other Comprehensive Income during the three months ended September 30, | | Amount Reclassified From Accumulated Other Comprehensive Income during the nine months ended September 30, | Affected Line Item in the Condensed Consolidated Statements of Operations |
| 2020 | | 2019 | | 2020 | | 2019 |
| Unrealized Gains (Losses) on Hedging Activity | | | | | | | | |
| Foreign exchange forward contracts | $ | (5.0) | | | $ | 11.0 | | | $ | 15.9 | | | $ | 27.1 | | Net product sales |
| Interest rate swap contracts | (11.3) | | | 3.4 | | | (25.9) | | | 12.5 | | Interest expense |
| (16.3) | | | 14.4 | | | (10.0) | | | 39.6 | | |
| 3.5 | | | (3.2) | | | 2.1 | | | (9.0) | | Income tax (benefit) expense |
| $ | (12.8) | | | $ | 11.2 | | | $ | (7.9) | | | $ | 30.6 | | |
13.Fair Value Measurement
Authoritative guidance establishes a valuation hierarchy for disclosure of the inputs to the valuation used to measure fair value. This hierarchy prioritizes the inputs into three broad levels as follows. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on our own assumptions used to measure assets and liabilities at fair value.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The following tables present information about our assets and liabilities that are measured at fair value on a recurring basis as of September 30, 2020 and December 31, 2019, and indicate the fair value hierarchy of the valuation techniques we utilized to determine such fair value.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Fair Value Measurement at
September 30, 2020 |
Balance Sheet
Classification | Type of Instrument | Total | | Level 1 | | Level 2 | | Level 3 |
| Cash equivalents | Money market funds | $ | 424.9 | | | $ | — | | | $ | 424.9 | | | $ | — | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| Marketable securities | Mutual funds | $ | 28.9 | | | $ | 28.9 | | | $ | — | | | $ | — | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| Other assets | Equity securities | $ | 100.4 | | | $ | 75.4 | | | $ | 25.0 | | | $ | — | |
| Prepaid expenses and other current assets | Foreign exchange forward contracts | $ | 11.7 | | | $ | — | | | $ | 11.7 | | | $ | — | |
| Other assets | Foreign exchange forward contracts | $ | 0.4 | | | $ | — | | | $ | 0.4 | | | $ | — | |
| Other current liabilities | Foreign exchange forward contracts | $ | 24.8 | | | $ | — | | | $ | 24.8 | | | $ | — | |
| Other liabilities | Foreign exchange forward contracts | $ | 0.3 | | | $ | — | | | $ | 0.3 | | | $ | — | |
| | | | | | | | |
| | | | | | | | |
| Other current liabilities | Interest rate contracts | $ | 45.6 | | | $ | — | | | $ | 45.6 | | | $ | — | |
| Other liabilities | Interest rate contracts | $ | 57.6 | | | $ | — | | | $ | 57.6 | | | $ | — | |
| | | | | | | | |
| Contingent consideration | Acquisition-related contingent consideration | $ | 398.1 | | | $ | — | | | $ | — | | | $ | 398.1 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Fair Value Measurement at
December 31, 2019 |
Balance Sheet
Classification | Type of Instrument | Total | | Level 1 | | Level 2 | | Level 3 |
| Cash equivalents | Money market funds | $ | 635.9 | | | $ | — | | | $ | 635.9 | | | $ | — | |
| Cash equivalents | Commercial paper | $ | 227.9 | | | $ | — | | | $ | 227.9 | | | $ | — | |
| Cash equivalents | Corporate bonds | $ | 20.6 | | | $ | — | | | $ | 20.6 | | | $ | — | |
| Cash equivalents | Bank certificates of deposit | $ | 19.2 | | | $ | — | | | $ | 19.2 | | | $ | — | |
| Cash equivalents | Other government-related obligations | $ | 60.4 | | | $ | — | | | $ | 60.4 | | | $ | — | |
| Marketable securities | Mutual funds | $ | 23.1 | | | $ | 23.1 | | | $ | — | | | $ | — | |
| Marketable securities | Commercial paper | $ | 19.0 | | | $ | — | | | $ | 19.0 | | | $ | — | |
| Marketable securities | Corporate bonds | $ | 3.7 | | | $ | — | | | $ | 3.7 | | | $ | — | |
| Marketable securities | Other government-related obligations | $ | 10.0 | | | $ | — | | | $ | 10.0 | | | $ | — | |
| Marketable securities | Bank certificates of deposit | $ | 8.2 | | | $ | — | | | $ | 8.2 | | | $ | — | |
| Other assets | Equity securities | $ | 79.0 | | | $ | 51.2 | | | $ | 27.8 | | | $ | — | |
| Prepaid expenses and other current assets | Foreign exchange forward contracts | $ | 29.9 | | | $ | — | | | $ | 29.9 | | | $ | — | |
| Other assets | Foreign exchange forward contracts | $ | 0.6 | | | $ | — | | | $ | 0.6 | | | $ | — | |
| Other current liabilities | Foreign exchange forward contracts | $ | 26.6 | | | $ | — | | | $ | 26.6 | | | $ | — | |
| Other liabilities | Foreign exchange forward contracts | $ | 1.1 | | | $ | — | | | $ | 1.1 | | | $ | — | |
| Other current liabilities | Interest rate contracts | $ | 19.5 | | | $ | — | | | $ | 19.5 | | | $ | — | |
| Other liabilities | Interest rate contracts | $ | 41.9 | | | $ | — | | | $ | 41.9 | | | $ | — | |
| Contingent consideration | Acquisition-related contingent consideration | $ | 192.4 | | | $ | — | | | $ | — | | | $ | 192.4 | |
| Other current liabilities | Other contingent payments | $ | 24.0 | | | $ | — | | | $ | — | | | $ | 24.0 | |
There were no securities transferred between Level 1, 2 and 3 during the nine months ended September 30, 2020.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Valuation Techniques
We classify mutual fund investments and equity securities, which are valued based on quoted market prices in active markets with no valuation adjustment, as Level 1 assets within the fair value hierarchy.
Cash equivalents and marketable securities classified as Level 2 within the valuation hierarchy include money market funds, commercial paper, U.S. and foreign government-related debt, corporate debt securities and certificates of deposit. We estimate the fair values of these marketable securities by taking into consideration valuations obtained from third-party pricing sources. These pricing sources utilize industry standard valuation models, including both income and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. These inputs include market pricing based on real-time trade data for similar securities, issuer credit spreads, benchmark yields, and other observable inputs. We validate the prices provided by our third-party pricing sources by understanding the models used, obtaining market values from other pricing sources and analyzing pricing data in certain instances.
Other investments in equity securities of publicly traded companies which are subject to holding period restrictions are carried at fair value using an option pricing valuation model and classified as Level 2 equity securities within the fair value hierarchy. The most significant assumptions within the option pricing valuation model are the term of the restrictions and the stock price volatility, which is based upon the historical volatility of the applicable company or similar companies. We also use a constant maturity risk-free interest rate to match the remaining term of the restrictions on such investments.
Our derivative assets and liabilities include foreign exchange and interest rate derivatives that are measured at fair value using observable market inputs such as forward rates, interest rates, our own credit risk as well as an evaluation of our counterparties’ credit risks. Based on these inputs, the derivative assets and liabilities are classified within Level 2 of the valuation hierarchy.
Contingent consideration liabilities related to business acquisitions and derivative liabilities associated with other contingent payments are classified as Level 3 within the valuation hierarchy and are valued based on various estimates, including probability of success, discount rates and amount of time until the conditions of the milestone payments are anticipated to be met.
As of September 30, 2020, there has not been any impact to the fair value of our derivative liabilities due to our own credit risk. Similarly, there has not been any significant adverse impact to our derivative assets based on our evaluation of our counterparties’ credit risks.
Acquisition-Related Contingent Consideration
In connection with our prior business combinations, we may be required to pay future consideration that is contingent upon the achievement of specified development, regulatory approvals or sales-based milestone events. We determine the fair value of these obligations using various estimates that are not observable in the market and represent a Level 3 measurement within the fair value hierarchy. As of September 30, 2020, the resulting probability-weighted cash flows were discounted using a cost of debt ranging from 3.9% to 4.3% for developmental and regulatory milestones and a weighted average cost of capital of 9.0% for sales-based milestones. As of December 31, 2019, the resulting probability-weighted cash flows were discounted using a weighted average cost of capital of 9.0% for sales-based milestones.
Each reporting period, we adjust the contingent consideration to fair value with changes in fair value recognized in operating earnings. Changes in fair values reflect new information about the probability and anticipated timing of meeting the conditions of the milestone payments. In the absence of new information, changes in fair value will only reflect the interest component of contingent consideration related to the passage of time.
As of September 30, 2020, estimated future contingent milestone payments related to our business combinations range from zero if no milestone events are achieved, to a maximum of $905.6 if all development, regulatory and sales-based milestones are reached.
As of September 30, 2020, the fair value of acquisition-related contingent consideration was $398.1. Amounts issued during the nine months ended September 30, 2020, represent the fair value of the non-tradeable CVRs recorded in connection with the acquisition of Achillion. See Note 3, Acquisitions. The following table represents a roll-forward of our acquisition-related contingent consideration:
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
| | | | | |
| Nine Months Ended September 30, 2020 |
| Balance as of December 31, 2019 | $ | 192.4 | |
| Amounts issued | 160.7 | |
| Changes in fair value | 45.0 | |
| Balance as of September 30, 2020 | $ | 398.1 | |
Other Contingent Payments
In January 2019, we entered into an agreement with Caelum, a biotechnology company that is developing CAEL-101 for light chain (AL) amyloidosis. Under the terms of the agreement, we acquired a minority equity interest in preferred stock of Caelum and an exclusive option to acquire the remaining equity in Caelum based on Phase II data, for pre-negotiated economics. We paid $30.0 during the first quarter 2019 and agreed to pay up to an additional $30.0 in contingent development milestones prior to our exercise of the option to acquire the remaining equity in Caelum. These contingent payments met the definition of a derivative liability and were initially recorded at fair value of $27.1, based on the probability-weighted cash flows, discounted using a cost of debt ranging from 3.3% to 3.5%.
In December 2019, following FDA feedback which resulted in the redesign and expansion of Caelum's planned clinical development program for CAEL-101, we amended the terms of our existing option agreement with Caelum. The amendment modified the terms of the option to acquire the remaining equity in Caelum based on data from the expanded Phase II/III trials. The amendment also modified the development-related milestone events associated with the initial $30.0 in contingent payments, provided for an additional $20.0 in upfront funding, which we accrued as of December 31, 2019 and paid during the first quarter 2020, as well as funding of $60.0 in exchange for an additional equity interest at fair value upon achievement of a specific development-related milestone event. During the second quarter 2020, we paid an aggregate of $15.0 of contingent payments to Caelum related to two development-based milestones and during the third quarter 2020, we paid a $15.0 contingent payment to Caelum related to another development-based milestone.
Each reporting period, we adjust the derivative liability associated with the contingent payments to fair value with changes in fair value recognized in other income and (expense). Changes in fair values reflect new information about the probability and anticipated timing of meeting the conditions of the milestone payments. In the absence of new information, changes in fair value will only reflect the interest component of the liability related to the passage of time. The aggregate $30.0 milestone payments made during the second and third quarter 2020 settled the derivative liability and reduced the derivative liability balance to zero. We recorded $3.6 and $6.0 of expense in other income and (expense) during the three and nine months ended September 30, 2020, respectively, related to the change in the fair value of the liability. We recorded $0.2 and $0.5 of expense in other income and (expense) during the three and nine months ended September 30, 2019, respectively, related to the change in the fair value of the liability.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
14.Revenue Recognition
Disaggregation of Revenue
The Company disaggregates revenue from contracts with customers into product and geographical regions as summarized below.
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended September 30, | | Nine months ended September 30, |
| 2020 | | 2019 | | 2020 | | 2019 |
| SOLIRIS | | | | | | | |
| United States | $ | 562.8 | | | $ | 496.8 | | | $ | 1,672.3 | | | $ | 1,456.8 | |
| Europe | 254.3 | | | 255.5 | | | 765.7 | | | 800.2 | |
| Asia Pacific | 86.0 | | | 118.0 | | | 255.5 | | | 329.2 | |
| Rest of World | 139.2 | | | 120.2 | | | 347.2 | | | 347.1 | |
| Total | $ | 1,042.3 | | | $ | 990.5 | | | $ | 3,040.7 | | | $ | 2,933.3 | |
| | | | | | | |
| ULTOMIRIS | | | | | | | |
| United States | $ | 170.7 | | | $ | 65.1 | | | $ | 460.3 | | | $ | 143.9 | |
| Europe | 46.6 | | | 21.1 | | | 112.4 | | | 21.1 | |
| Asia Pacific | 69.6 | | | 3.7 | | | 186.3 | | | 3.7 | |
| Rest of World | 2.4 | | | — | | | 4.2 | | | — | |
| Total | $ | 289.3 | | | $ | 89.9 | | | $ | 763.2 | | | $ | 168.7 | |
| | | | | | | |
| STRENSIQ | | | | | | | |
| United States | $ | 149.3 | | | $ | 118.0 | | | $ | 418.1 | | | $ | 323.7 | |
| Europe | 19.3 | | | 19.0 | | | 61.6 | | | 56.0 | |
| Asia Pacific | 16.1 | | | 14.0 | | | 44.7 | | | 36.0 | |
| Rest of World | 4.7 | | | 3.3 | | | 21.5 | | | 10.0 | |
| Total | $ | 189.4 | | | $ | 154.3 | | | $ | 545.9 | | | $ | 425.7 | |
| | | | | | | |
| ANDEXXA | | | | | | | |
| United States | $ | 36.2 | | | $ | — | | | $ | 36.2 | | | $ | — | |
| Europe | 2.7 | | | — | | | 2.7 | | | — | |
| Asia Pacific | — | | | — | | | — | | | — | |
| Rest of World | — | | | — | | | — | | | — | |
| Total | $ | 38.9 | | | $ | — | | | $ | 38.9 | | | $ | — | |
| | | | | | | |
| KANUMA | | | | | | | |
| United States | $ | 15.8 | | | $ | 16.0 | | | $ | 47.6 | | | $ | 45.1 | |
| Europe | 9.5 | | | 6.3 | | | 25.4 | | | 19.4 | |
| Asia Pacific | 1.1 | | | 1.3 | | | 2.9 | | | 3.4 | |
| Rest of World | 2.0 | | | 4.8 | | | 12.8 | | | 10.2 | |
| Total | $ | 28.4 | | | $ | 28.4 | | | $ | 88.7 | | | $ | 78.1 | |
| | | | | | | |
| Total Net Product Sales | $ | 1,588.3 | | | $ | 1,263.1 | | | $ | 4,477.4 | | | $ | 3,605.8 | |
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Contract Balances and Receivables
Contract liabilities primarily relate to consideration received and/or billed for goods that have not been delivered to the customer and for which the performance obligation has not yet been completed. These amounts are included within other current liabilities in the condensed consolidated balance sheets.
The following table provides information about receivables and contract liabilities from our contracts with customers.
| | | | | | | | | | | |
| September 30, 2020 | | December 31, 2019 |
| Receivables, which are included in "Trade accounts receivable, net" | $ | 1,437.1 | | | $ | 1,243.2 | |
| Contract liabilities, which are included in "Other current liabilities" | $ | 22.7 | | | $ | 6.8 | |
Contract balances and receivables associated with collaboration agreements assumed through the acquisition of Portola in the third quarter 2020, which were included in the table above, were not material as of September 30, 2020.
15.Income Taxes
Coronavirus Aid, Relief and Economic Security Act
In response to the market volatility and instability resulting from the COVID-19 pandemic, the Coronavirus Aid, Relief, and Economic Security Act (CARES Act) was signed into law on March 27, 2020. The CARES Act lifts certain deduction limitations originally imposed by the Tax Cuts and Jobs Act of 2017 (2017 Tax Act). Under the 2017 Tax Act, federal net operating losses (NOLs) generated after 2017 could not be carried back and utilization was limited to 80% of taxable income. The CARES Act allows for a five-year carryback of federal NOLs generated in 2018 through 2020 and eliminates the 80% taxable income limitation by allowing corporate entities to fully utilize NOL carryforwards to offset taxable income in 2018 through 2020. In addition, the CARES Act generally allows taxpayers to deduct interest up to 50% of adjusted taxable income (30% limit under the 2017 Tax Act) for tax years 2019 and 2020. The CARES Act also allows taxpayers with prior year alternative minimum tax (repealed by the 2017 Tax Act) (AMT) credits to accelerate refund claims to tax years beginning in 2018 and 2019 instead of recovering the credits over a period of years, as originally enacted by the 2017 Tax Act.
Additionally, the CARES Act raises the corporate charitable deduction limit to 25% of taxable income and provides a technical correction to the 2017 Tax Act to generally provide qualified improvement property a 15-year cost-recovery period and allow 100% bonus depreciation. The enactment of the CARES Act did not result in any material adjustments to our income tax provision for the three and nine months ended September 30, 2020, or to our U.S. federal and state net deferred tax liabilities as of September 30, 2020.
Tax Rate
The following table provides a comparative summary of our income tax expense and effective income tax rate for the three and nine months ended September 30, 2020:
| | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended | | Nine months ended |
| September 30, | | September 30, |
| 2020 | | 2019 | | 2020 | | 2019 |
| Income tax expense (benefit) | $ | 88.8 | | $ | 67.9 | | | $ | (89.2) | | | $ | 61.5 | |
| Effective income tax rate | 13.3 | % | | 12.7 | % | | 413.0 | % | | 3.9 | % |
Income tax expense (benefit) is attributable to the U.S. federal, state and foreign income taxes on our profitable operations.
During the second quarter 2020, we recognized an impairment charge of $2,042.3 related to the KANUMA intangible asset, resulting in a deferred tax benefit of $377.3. See Note 5, Intangible Assets and Goodwill, for additional information on the impairment charge. These deferred tax benefits increased the effective tax rate for the nine months ended September 30, 2020 by approximately 399.6%.
The income tax expense for the nine months ended September 30, 2019 includes one-time tax benefits recorded during the first quarter 2019, in connection with the future integration of intellectual property of Wilson
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Therapeutics into the Alexion corporate structure. The deferred tax benefits included $95.7 and $30.3 associated with a tax election made with respect to intellectual property of Wilson Therapeutics and a valuation allowance release and corresponding recognition of net operating losses, respectively. These deferred tax benefits decreased the effective tax rate for the nine months ended September 30, 2019 by approximately 8.0%. Additionally, during the third quarter 2019, we completed a comprehensive analysis of our current and prior year estimates related to our foreign-derived intangible income based on additional guidance provided in the proposed regulations issued by the U.S. Treasury Department in 2019. The analysis resulted in income tax benefits of approximately $29.9, including $17.0 related to prior year estimated tax benefits, which were recorded as a change in estimate in income tax expense in our consolidated statements of operations during the three and nine months ended September 30, 2019. These tax benefits decreased the effective tax rate for the three and nine months ended September 30, 2019 by approximately 5.6% and 1.9%, respectively.
In April 2020 we became aware of a European withholding tax regulation that could be interpreted to apply to certain of our previous intra-group transactions. We continue to evaluate whether the interpretation of this regulation applies to our facts and circumstances, and, based on our preliminary analysis, we recorded an immaterial reserve related to this matter during the second quarter of 2020.
In August 2020, we received a notice of examination from the Dutch Tax Authorities (“DTA”) regarding certain matters relating to our 2014 through 2017 tax years. Discussions with the DTA are currently ongoing and, at Alexion’s request, we have expanded these discussions to include the 2018 and 2019 tax years as well. We have not received any final findings on any of these tax years, nor have we received any proposed adjustments or any tax assessments related to any of the tax years under discussion with the DTA. Accordingly, we have not recorded any related reserves and an estimate of the possible loss or range of loss, if any, cannot be made at this time.
During the third quarter of 2020, the U.S. Treasury Department released certain proposed and final regulations which were originally enacted under the 2017 Tax Act. The impact of these regulations did not have a material impact on our financial condition and results of operations.
In 2017, the Internal Revenue Service (IRS) commenced an examination of our U.S. income tax returns for 2015. During the second quarter of 2020 we received a Revenue Agent Report (RAR) and held discussions with the IRS regarding a proposed adjustment related to the valuation of certain intellectual property that was contributed into our captive partnership during 2015. The Company agreed with the adjustment outlined in the RAR and recognized a previously unrecognized tax benefit in the second quarter of 2020 that did not result in a significant impact to the financial statements. The IRS concluded its examination during the third quarter 2020 without additional adjustments.
We have recorded tax on the undistributed earnings of our controlled foreign corporation (CFC) subsidiaries. To the extent CFC earnings may not be repatriated to the U.S. as a dividend distribution due to limitations imposed by law, we have not recorded the related potential withholding, foreign, local, and U.S. state income taxes.
We continue to maintain a valuation allowance against certain deferred tax assets where realization is not certain.
16.Commitments and Contingencies
Asset Acquisition and In-License Agreements
We have entered into asset purchase agreements, license agreements, and option arrangements in order to advance and obtain technologies and services related to our business. These agreements generally require us to pay an initial fee and certain agreements call for future payments upon the attainment of agreed upon development, regulatory and/or commercial milestones. These agreements may also require minimum royalty payments based on sales of products developed from the applicable technologies, if any.
In January 2019, we entered into an agreement with Caelum, a biotechnology company that is developing CAEL101 for light chain (AL) amyloidosis. Under the terms of the agreement, we acquired a minority equity interest in preferred stock of Caelum and an exclusive option to acquire the remaining equity in Caelum based on Phase II data, for pre-negotiated economics. We paid $30.0 in the first quarter 2019 and agreed to pay up to an additional $30.0 in contingent development milestones prior to the exercise the option to acquire the remaining equity in Caelum. These contingent payments meet the definition of a derivative liability and were initially recorded at fair value of $27.1. We allocated the total consideration of $57.1, inclusive of the fair value of the contingent payments, to the equity investment in Caelum and the option to acquire the remaining equity in Caelum based on the relative fair values of the assets. Following discussions with the FDA, Caelum changed its clinical development plan for
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
CAEL-101 in the fourth quarter 2019. A Phase II trial for CAEL-101 subsequently commenced during the first quarter of 2020 and met its primary objectives, supporting the safety and tolerability of CAEL-101 and confirmed the dose and regimen to be adopted for the Phase III studies. In September 2020, Alexion and Caelum announced the initiation of the Cardiac Amyloid Reaching for Extended Survival (CARES) program. This includes two parallel Phase III trials to evaluate the survival benefits of CAEL-101. In December 2019, we amended the terms of the agreement with Caelum to modify the option to acquire the remaining equity in Caelum based on data from the modified Phase II/III trials. The amendment also modified the development-related milestone events associated with the initial $30.0 in contingent payments, provided for an additional $20.0 in upfront funding, which we accrued as of December 31, 2019 and paid during the first quarter 2020, as well as funding of $60.0 in exchange for an additional equity interest at fair value upon achievement of a specific development-related milestone event. The agreement with Caelum also provides for additional payments, in the event Alexion exercises the purchase option, for up to $500.0, which includes an upfront option exercise payment and potential regulatory and commercial milestone payments. During the second quarter 2020, we paid an aggregate of $15.0 of contingent payments to Caelum related to two development-based milestones and during the third quarter 2020, we paid a $15.0 contingent payment to Caelum related to another development-based milestone.
In March 2019, we entered into an agreement with Zealand which provides us with exclusive worldwide licenses, as well as development and commercial rights, for subcutaneously delivered preclinical peptide therapies directed at up to four complement pathway targets. Pursuant to the agreement, Zealand will lead joint discovery and research efforts through the preclinical stage, and Alexion will lead development efforts beginning with the investigational new drug filing and Phase I studies. In addition to the agreement, we made an equity investment in Zealand (see Note 10, Other Investments). Under the terms of the agreement, we made an upfront payment of $40.0 for an exclusive license to the lead target and the equity investment, as well as for preclinical research services to be performed by Zealand in relation to the lead target. The market value of the equity investment was $13.8 as of the date of acquisition, which we recorded in other assets in our condensed consolidated balance sheets. We also recognized prepaid research and development expense of $5.0 within the condensed consolidated balance sheets associated with the research activities to be performed by Zealand. Due to the early stage of the asset we are licensing, we recorded the upfront license payment of $21.2 as research and development expense during the first quarter 2019. As of September 30, 2020, we could be required to pay up to $610.0, for the lead target, upon the achievement of specified development, regulatory and commercial milestones, as well as royalties on commercial sales. In addition, we could be required to pay up to an additional $115.0 in development and regulatory milestones if both a long-acting and short-acting product are developed with respect to the lead target. Each of the three subsequent targets can be selected for an option fee of $15.0 and has the potential for additional development, regulatory and commercial milestones, as well as royalty payments, at a reduced price to the lead target.
In April 2019, we entered into an agreement with Affibody AB (Affibody), through which Alexion obtained an exclusive worldwide license, as well as development and commercial rights, to ABY-039, a bivalent antibody-mimetic that targets the neonatal Fc receptor (FcRn). Under the terms of the agreement, we made an upfront payment of $25.0 for the exclusive license to ABY-039. Due to the early stage of the asset we licensed, we recorded the upfront license payment as research and development expense during the second quarter 2019. In February 2020, based on data from our Phase I study, we terminated the agreement to co-develop ABY-039 with Affibody.
In September 2019, we entered into an agreement with Eidos through which Alexion obtained an exclusive license to develop and commercialize AG10 in Japan. AG10 is a small molecule designed to treat the root cause of transthyretin amyloidosis (ATTR) and is currently in a Phase III study in the U.S. and Europe for ATTR cardiomyopathy (ATTR-CM). In addition, we made an equity investment in Eidos (see Note 10, Other Investments). Under the terms of the agreement, we made an upfront payment of $50.0 for the exclusive license to AG10 in Japan and the equity investment. The market value of the equity investment was $19.9 as of the date of acquisition, which we recorded in other assets in our condensed consolidated balance sheets. Due to the early stage of the asset we are licensing, we recorded the upfront license payment of $30.1 as research and development expense during the third quarter 2019. As of September 30, 2020, we could also be required to pay $30.0 upon achievement of a Japanese-based regulatory milestone as well as royalties on commercial sales.
In October 2018, we entered into a collaboration agreement with Dicerna that provides us with exclusive worldwide licenses and development and commercial rights for two preclinical RNA interference (RNAi) subcutaneously delivered molecules for complement-mediated diseases, as well as an exclusive option for other preclinical RNAi molecules for two additional targets within the complement pathway. In addition to the collaboration agreement, we made an equity investment in Dicerna. Under the terms of the agreements, we made an upfront payment of $37.0 for the exclusive licenses and the equity investment. The market value of the equity investment
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
was $10.3 as of the date of acquisition, which we recorded in other assets in our condensed consolidated balance sheets. Due to the early stage of the assets we are licensing, we recorded the upfront license payment of $26.7 as research and development expense during the fourth quarter 2018. In December 2019, we exercised our option for exclusive rights to two additional targets within the complement pathway under an existing agreement with Dicerna, which expands our existing research collaboration and license agreement with Dicerna to include a total of four targets within the complement pathway. In connection with the option exercise, we paid Dicerna $20.0, which we recorded as research and development expense in the fourth quarter 2019. As of September 30, 2020, excluding accrued milestones, we could be required to pay up to $604.1 for amounts due upon the achievement of specified research, development, regulatory and commercial milestones on the four licensed targets, as well as royalties on commercial sales.
In December 2017, we entered into a collaboration and license agreement with Halozyme Therapeutics, Inc. that allows us to use drug-delivery technology in the development of subcutaneous formulations for our portfolio of products for up to four targets. Under the terms of the agreement, we made an upfront payment of $40.0 for an exclusive license to two of the four potential targets and due to the early stage of the assets we are licensing, we recorded an expense for the upfront payment during the fourth quarter 2017. During the second quarter 2020, we forfeited our rights to one of the two targets we initially licensed. As of September 30, 2020, we could be required to pay up to $155.0 for the remaining licensed target upon achievement of specified development, regulatory and sales-based milestones, as well as royalties on commercial sales. Each of the two subsequent targets can be licensed for an option fee of $8.0, with contingent payments of up to $160.0 per target, subject to development, regulatory and commercial milestones, as well as royalties on commercial sales.
In connection with our prior acquisition of Syntimmune, Inc., a clinical-stage biotechnology company developing an antibody therapy targeting the FcRn, we could be required to pay up to $800.0 upon the achievement of specified development, regulatory and commercial milestones, of which $130.0 is specific to the subcutaneous formulation.
In addition, excluding accrued milestones, as of September 30, 2020, we have other license agreements under which we may be required to pay up to an additional $114.0 for currently licensed targets, if certain development, regulatory and commercial milestones are met, including up to $71.5 for the development of cerdulatinib in multiple indications pursuant to an in-licensing agreement with Astellas Pharma, Inc. which was assumed through the acquisition of Portola in the third quarter 2020. Additional amounts may be payable if we elect to acquire licenses to additional targets, as applicable, under the terms of these agreements.
Asset Sale and Out-License Arrangements
In connection with prior asset sale and out-license arrangements, including those assumed by Alexion through the acquisition of Portola in the third quarter 2020, Alexion is entitled to receive contingent payments upon the achievement of various regulatory and commercial milestones and other events, as well as royalties on commercial sales. The amount of contingent consideration related to these agreements is fully constrained and therefore has not been recognized as of September 30, 2020.
Manufacturing Agreements
We have various manufacturing development and license agreements to support our clinical and commercial product needs.
We rely on Lonza, a third party manufacturer, to produce a portion of commercial and clinical quantities of our commercial products and product candidates. We have various manufacturing and license agreements with Lonza, with remaining total non-cancellable future commitments of approximately $1,432.3 through 2031. This amount includes $105.5 of undiscounted, fixed payments applicable to our Contract Manufacturing Organization (CMO) embedded lease arrangement with Lonza. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of SOLIRIS that was manufactured at the Alexion Rhode Island Manufacturing Facility (ARIMF facility) prior to the sale of the facility and a payment with respect to sales of SOLIRIS manufactured at Lonza facilities. We also pay Lonza a royalty on the sales of ULTOMIRIS.
In addition to our commitments with Lonza, as of September 30, 2020 we have non-cancellable commitments of approximately $78.6 through 2021 with other third party manufacturers.
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
Contingent Liabilities
We are currently involved in various claims, disputes, lawsuits, investigations, administrative proceedings and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. In accordance with generally accepted accounting principles, if the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims, proceedings and litigation, accruals are based on our best estimates based on information available at the time of the assessment. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation, court decisions or settlement of claims (and offers of settlement), we may reassess the potential liability related to these matters and may revise these estimates, which could result in a material adverse adjustment to our operating results. Costs associated with our involvement in legal proceedings are expensed as incurred. The outcome of any such proceedings, regardless of the merits, is inherently uncertain. If we were unable to prevail in any such proceedings, our consolidated financial position, results of operations, and future cash flows may be materially impacted.
We have received, and may in the future receive, notices from third parties claiming that their patents may be infringed by the use, development, manufacture, importation or sale of our products or product candidates. Under the guidance of ASC 450, Contingencies, we record a royalty accrual based on our best estimate of the fair value percent of net sales of our products that we could be required to pay the owners of patents for technology used in the manufacture and sale of our products. A costly license, or inability to obtain a necessary license, could have a material adverse effect on our financial results.
In May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the Securities and Exchange Commission (SEC) requesting information related to our grant-making activities and compliance with the Foreign Corrupt Practices Act (FCPA) in various countries. In addition, in October 2015, we received a request from the Department of Justice (DOJ) for the voluntary production of documents and other information pertaining to Alexion’s compliance with the FCPA. The SEC and DOJ also sought information related to Alexion’s recalls of specific lots of SOLIRIS and related securities disclosures.
The investigations focused on operations in various countries, including Brazil, Colombia, Japan, Russia and Turkey, and Alexion's compliance with the FCPA and other applicable laws.
In May 2020, DOJ informed us that it has closed its inquiry into these matters.
On July 2, 2020, we reached a civil settlement with the SEC fully resolving the SEC’s investigation into possible violations of the FCPA. Alexion neither admitted nor denied any wrongdoing in connection with the settlement but agreed to pay $21.5 to the SEC, consisting of amounts attributable to disgorgement, civil penalties, and pre-judgment interest. In connection with this settlement, in July 2020, we paid $21.5 to the SEC.
Following the settlement with the SEC, the Ministry of Health in Turkey initiated an investigation regarding the matters referenced in the SEC Order as they relate to the Company’s operations in Turkey between 2010 and 2015. We are cooperating with this investigation.
Alexion is committed to continually focusing on its compliance program and continues to enhance its comprehensive company-wide program that is designed to enhance our business processes, structures, controls, training, talent, and systems across Alexion’s global operations.
As previously reported, on December 29, 2016, a shareholder filed a putative class action against the Company and certain former employees in the U.S. District Court for the District of Connecticut, alleging that defendants made misrepresentations and omissions about SOLIRIS. On April 12, 2017, the court appointed a lead plaintiff. On July 14, 2017, the lead plaintiff filed an amended putative class action complaint against the Company and seven current or former employees. Defendants moved to dismiss the amended complaint on September 12, 2017. Plaintiffs filed an opposition to defendants’ motion to dismiss on November 13, 2017, and defendants filed a reply brief in further support of their motion on December 28, 2017. On March 26, 2019, the court held a telephonic status conference. During that conference, the court informed counsel that it was preparing a ruling granting the defendants’ pending motion to dismiss. The court inquired of plaintiffs’ counsel whether they intended to seek leave to amend their complaint, and indicated that if they wished to file a second amended complaint, they would be allowed to do so. On April 2, 2019, the court granted plaintiffs until May 31, 2019 to file a second amended complaint, thereby rendering moot defendants’ pending motion to dismiss. On June 2, 2019, plaintiffs filed a second amended complaint against the same defendants. The complaint alleges that defendants engaged in securities
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
fraud, including by making misrepresentations and omissions in its public disclosures concerning the Company’s SOLIRIS sales practices, management changes, and related investigations, between January 30, 2014 and May 26, 2017, and that the Company's stock price dropped upon the purported disclosure of the alleged fraud. The plaintiffs seek to recover unspecified monetary relief, unspecified equitable and injunctive relief, interest, and attorneys’ fees and costs. Defendants’ filed a motion to dismiss the amended complaint on August 2, 2019; plaintiffs’ filed their opposition to that motion on October 2, 2019; and defendants’ filed their reply in further support of their motion on November 15, 2019. Given the early stage of these proceedings, we cannot presently predict the likelihood of obtaining dismissal of the case (or the ultimate outcome of the case if the motion to dismiss is denied by the court), nor can we estimate the possible loss or range of loss at this time.
In December 2016, we received a subpoena from the U.S. Attorney's Office for the District of Massachusetts requesting documents relating generally to our support of Patient Services, Inc. (PSI) and National Organization for Rare Disorders (NORD), 501(c)(3) organizations that provide financial assistance to Medicare patients taking drugs sold by Alexion; Alexion’s provision of free drug to Medicare patients; and Alexion compliance policies and training materials concerning the anti-kickback statute and information on donations to PSI and NORD from 2010 through 2016. In April 2019, we entered into a civil settlement agreement with the DOJ and the Office of Inspector General (OIG) of the U.S. Department of Health and Human Services to resolve this matter. As part of the settlement agreement, Alexion paid $13.1 to the DOJ and OIG. OIG did not require a Corporate Integrity Agreement with Alexion because it made fundamental organizational changes, including hiring a new executive leadership team, replacing half of the members of its Board of Directors, and effecting a significant change in the workforce.
In May 2017, Brazilian authorities seized records and data from our São Paulo, Brazil offices as part of an investigation being conducted into Alexion’s Brazilian operations. We are cooperating with this inquiry.
In June 2017, we received a demand to inspect certain of our books and records pursuant to Section 220 of the General Corporation Law of the State of Delaware on behalf of a purported stockholder. Among other things, the demand sought to determine whether to institute a derivative lawsuit against certain of the Company’s directors and officers in relation to the investigation by our Audit and Finance Committee announced in November 2016 and the investigations instituted by the SEC, DOJ, U.S. Attorney’s Office for the District of Massachusetts, and Brazilian law enforcement officials that are described above. We have responded to the demand. Given the early stages of this matter, an estimate of the possible loss or range of loss cannot be made at this time.
On September 27, 2017, a hearing panel of the Canadian Patented Medicine Prices Review Board (PMPRB) issued a decision in a previously pending administrative pricing matter that we had excessively priced SOLIRIS in a manner inconsistent with the Canadian pricing rules and guidelines. In its decision, the PMPRB ordered Alexion to decrease the price of SOLIRIS to an upper limit based upon pricing in certain other countries, and to forfeit excess revenues for the period between 2009 and 2017. The amount of excess revenues for the period between 2009 and 2017 was not determined to be a material amount and was paid in 2018. In October 2017, Alexion filed an application for judicial review of the PMPRB’s decision in the Federal Court of Canada. On May 23, 2019, the Federal Court of Canada dismissed Alexion's application for judicial review and, as a consequence, affirmed the decision of the PMPRB that we had excessively priced SOLIRIS. On June 21, 2019, Alexion filed a notice of appeal of the Federal Court of Canada's ruling, and, on October 17, 2019, Alexion filed a memorandum of fact and law in support of the appeal. On December 3, 2019, the Attorney General of Canada filed its memorandum of fact and law in support of the Federal Court of Canada's dismissal of Alexion's appeal of the PMPRB's decision. On December 19, 2019, the intervenor, the Minister of Health for the Province of British Columbia, filed a separate memorandum of fact and law in support of the Federal Court of Canada's decision. The Canadian Federal Court of Appeal heard the appeal on October 21 and 22, 2020, but has not issued a decision as of the date of this filing. Pursuant to an order made by the Federal Court of Canada, as of October 27, 2020, we have placed approximately $61.5 in escrow to secure our obligations pending the final resolution of all appeals in this matter. This amount reflects the difference between the list price for SOLIRIS and the price determined by the PMPRB to be non-excessive for sales of SOLIRIS in Canada for the period beginning September 2017 through September 30, 2020. In addition, on a quarterly basis, until the appeals process has concluded, Alexion will be required to place amounts into escrow for each vial of SOLIRIS sold in the applicable quarter equal to the list price for SOLIRIS and the price determined by the PMPRB to be non-excessive. Our revenues in Canada have been reduced by $42.2 cumulatively to date, which is our current best estimate of our liability through September 30, 2020 if we lose the appeal of this matter (the amount of our ultimate liability, however, may be greater than this estimate when the appeal process for this matter is concluded).
Chugai Pharmaceutical Co., Ltd. has filed three lawsuits against Alexion. The first was filed in November 2018 in the United States District Court for the District of Delaware against Alexion Pharmaceuticals, Inc. alleging that
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
ULTOMIRIS infringes one U.S. patent held by Chugai Pharmaceutical Co., Ltd. Upon issuance of a new U.S. patent on November 12, 2019, Chugai filed a second lawsuit in the United States alleging that ULTOMIRIS infringes the new patent. The parties have agreed to consolidate the November 2018 and November 2019 lawsuits. Chugai filed a third lawsuit in December 2018 in the Tokyo District Court against Alexion Pharma GK (a wholly-owned subsidiary of Alexion) in Japan, and alleges that ULTOMIRIS infringes two Japanese patents held by Chugai Pharmaceutical Co., Ltd. Chugai’s complaints seek unspecified damages and certain injunctive relief. On March 5, 2020, the Supreme Court of Japan dismissed Chugai's appeal against an earlier IP High Court of Japan decision which held that one of the Chugai patents-in-suit is invalid. Subsequently Chugai filed a correction to the claims of this patents-in-suit and Alexion has countered that the corrected claims are still invalid and not infringed. In all cases, Alexion has denied the charges and countered that the patents are neither valid nor infringed. A trial date for the U.S. case which was initially set for July 2021 has been re-scheduled for October 2021. The case is still at the briefing stage in Japan. Given the early stages of these litigations, an estimate of the possible loss or range of loss cannot be made at this time.
On February 28, 2019, Amgen Inc. (Amgen) petitioned the U.S. Patent and Trademark Office (PTO) to institute Inter Partes Review (IPR) of three patents owned by Alexion that relate to SOLIRIS: U.S. Patent Nos. 9,725,504; 9,718,880; and 9,732,149. In each case, Amgen alleged the patented subject matter was anticipated and/or obvious in view of prior art, and that the patent claims are therefore invalid. On August 30, 2019, the PTO instituted IPRs of each of the three patents. On May 28, 2020, we entered into a Confidential Settlement and License Agreement (the “Settlement Agreement”) with Amgen to settle the three IPRs at the Patent Trial and Appeal Board (“PTAB”) of the PTO. Pursuant to the Settlement Agreement, Alexion and Amgen have terminated each of the pending IPRs. In addition, effective March 1, 2025 (or an earlier date in certain circumstances), the Company grants to Amgen (and its affiliates and certain partners) a non-exclusive, royalty-free, license under U.S. patents and patent applications related to eculizumab and various aspects of the eculizumab product that Alexion currently markets and sells under the tradename SOLIRIS. This license will allow Amgen (and its affiliates and certain partners), effective March 1, 2025, the right to make, have made, use, import, have imported, sell, have sold, offer for sale, have offered for sale, distribute, and have distributed in, or for, the U.S., an eculizumab product.
In connection with an ongoing matter, in August 2019, the Brazilian Federal Revenue Service provided a Notice of Tax and Description of the Facts (the “Tax Assessment”) to two Alexion subsidiaries (the "Brazil Subsidiaries"), as well as to two additional entities, a logistics provider utilized by Alexion and a distributor. The Tax Assessment focuses on the importation of SOLIRIS vials pursuant to Alexion’s free drug supply to patients program (referred to as Global Access to Medicines, or GATM) in Brazil. In September 2019, the Brazil Subsidiaries filed defenses to the Tax Assessment disputing the basis for liability under the Tax Assessment, based on, among others, the following: in connection with the operation of GATM, during the period from September 2014 to June 2019: (i) the importers responsible for the importation of the GATM SOLIRIS vials into Brazil were correctly identified and (ii) the correct customs value was utilized for the purpose of importing the GATM SOLIRIS vials provided to the patients free of charge. The defenses filed by Alexion are pending judgment at the first level of administrative appeals within the Brazilian federal administrative proceeding system. There are three separate levels of administrative appeals within the Brazilian federal administrative proceeding system and, if the outcome of these administrative appeals is unfavorable, the final decision of the federal administrative proceeding system can be disputed to the federal court systems in Brazil (at this time, Alexion intends to appeal the Tax Assessment if it is not overturned in the course of administrative appeals). Given the early stage of these proceedings, Alexion is unable to predict the duration, scope or outcome of this matter, but we expect that a final resolution will take three years or more. While it is possible that a loss related to the Tax Assessment may be incurred, given its ongoing nature, we cannot reasonably estimate the potential magnitude of any such possible loss or range of loss, or the cost of the ongoing administrative appeals (and potential appeals to the federal court system) of the Tax Assessment. Any determination that any aspects of the importation of free of charge medications into Brazil as set forth in the Tax Assessment are not, or were not, in compliance with existing laws or regulations could result in the imposition of fines, civil penalties and, potentially criminal penalties, and/or other sanctions against us, and could have an adverse impact on our Brazilian operations.
In connection with Alexion’s acquisition of Portola, we have assumed litigation to which Portola was a party. Among the litigation assumed is a securities fraud class action filed against Portola and certain of its officers, directors and underwriters (“Defendants”) under the Securities Act of 1933 and the Securities Exchange Act of 1934. Specifically, on January 16, 2020, February 7, 2020, and February 28, 2020, stockholders filed three putative class actions in the U.S. District Court for the Northern District of California, captioned Hayden v. Portola Pharmaceuticals, Inc., et al., No. 3:20-cv-00367-VC (N.D. Cal.); McCutcheon v. Portola Pharmaceuticals, Inc., et al., No. 3:20-cv-00949 (N.D. Cal.); and Southeastern Pennsylvania Transportation Authority v. Portola Pharmaceuticals, Inc., et
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
al., No. 3:20-cv-01501 (N.D. Cal.). These cases have since been consolidated, and on April 22, 2020, the Court issued an Order appointing the Alameda County Employees’ Retirement Association (“ACERA”) as Lead Plaintiff in the litigation. ACERA filed its amended consolidated complaint on May 20, 2020, asserting that Defendants made misrepresentations and omissions in public disclosures (including in materials issued in connection with the August 7, 2019 securities offering) concerning Portola’s sales of andexanet alfa, marketed as ANDEXXA in the United States and ONDEXXYA in Europe, between January 8, 2019 and February 26, 2020. Specifically, plaintiffs allege that Defendants made materially false and/or misleading statements about the demand for ANDEXXA, usage of ANDEXXA by hospitals and healthcare organizations, and about Portola’s accounting for its return reserves. Plaintiffs contend that the alleged fraud was revealed on January 9, 2020, when Portola announced its preliminary unaudited financial results for the fourth quarter of 2019, and again on February 26, 2020, when Portola issued its fourth quarter 2019 financial results. In July 2020, Portola and the Portola Defendants filed a motion to dismiss with the Court. The court heard oral argument on September 24, 2020 and granted defendants’ pending motion to dismiss, but with leave for plaintiffs to amend further their complaint. Plaintiffs have until November 5, 2020 to file a second amended complaint. Plaintiffs seek to recover unspecified monetary relief, interest, and attorneys’ fees and costs. Given the early stage of these proceedings, we cannot presently predict the likelihood of obtaining dismissal of the case which the Company intends to submit following the filing of the second amended complaint (or the ultimate outcome of the case if that motion to dismiss is denied by the court), nor can we estimate the possible loss or range of loss at this time.
17. Restructuring and Related Expenses
During the third quarter 2020, we initiated restructuring activities primarily within our commercial organization as part of an initiative intended to redefine our operating model. The actions are intended to reallocate resources necessary to align our organization with our diversifying portfolio of new products and strategic objectives, and will include investments in digital capabilities, technologies and solutions to support a more virtual and digital customer experience and tailored to the markets in which we operate.
The actions are expected to be substantially completed by the end of 2021, with the cumulative pretax costs to be incurred by the Company to implement the program estimated to be approximately $25.0. We expect that the pretax costs will primarily result in cash outlays, as the costs primarily relate to employee separation expenses.
In the first quarter 2019, we initiated corporate restructuring activities to re-align our international commercial organization through re-prioritization of certain geographical markets and to implement operational excellence through strategic reallocation of resources. Actions under the previous restructuring programs have been completed.
The following table summarizes the total expenses recorded related to the restructuring activities by type of activity and the locations recognized within the consolidated statements of operations:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended September 30, | | Three months ended September 30, |
| 2020 | | 2019 |
| Employee Separation Costs | | | | Other | | Total | | Employee Separation Costs | | | | Other | | Total |
| Restructuring expenses | $ | 14.3 | | | | | $ | — | | | $ | 14.3 | | | $ | (2.8) | | | | | $ | 3.1 | | | $ | 0.3 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Nine months ended September 30, | | Nine months ended September 30, |
| 2020 | | 2019 |
| Employee Separation Costs | | | | Other | | Total | | Employee Separation Costs | | | | Other | | Total |
| Restructuring expenses | $ | 14.0 | | | | | $ | (0.5) | | | $ | 13.5 | | | $ | 8.7 | | | | | $ | 3.2 | | | $ | 11.9 | |
Alexion Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(amounts in millions, except per share amounts)
The following table presents a reconciliation of the restructuring reserve recorded within accounts payable and accrued expenses on the Company's consolidated balance sheets as of September 30, 2020
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended September 30, | | Nine months ended September 30, |
| 2020 | | 2020 |
| Employee Separation Costs | | Other | | Total | | Employee Separation Costs | | Other | | Total |
| Liability, beginning of period | $ | 1.6 | | | $ | — | | | $ | 1.6 | | | $ | 3.3 | | | $ | 3.5 | | | $ | 6.8 | |
| Charges | 14.3 | | | — | | | 14.3 | | | 14.3 | | | — | | | 14.3 | |
| Settlements | — | | | — | | | — | | | (1.4) | | | (3.0) | | | (4.4) | |
| Adjustments to previous estimates | — | | | — | | | — | | | (0.3) | | | (0.5) | | | (0.8) | |
| Liability, end of period | $ | 15.9 | | | $ | — | | | $ | 15.9 | | | $ | 15.9 | | | $ | — | | | $ | 15.9 | |
The restructuring reserve of $15.9 and $6.8 is recorded in accounts payable and accrued expenses on the Company's condensed consolidated balance sheet as of September 30, 2020 and December 31, 2019, respectively. The accrued amounts are expected to be paid in the next twelve months. We currently estimate incurring up to an additional $11.0 in restructuring expenses related to the third quarter 2020 action.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Item 2.MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
Note Regarding Forward-Looking Statements
This quarterly report on Form 10-Q contains forward-looking statements. Words such as “anticipates,” "may," "forecasts," “expects,” “intends,” “plans,” "potentially," “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. Forward-looking statements are not guarantees of future performance and are subject to certain risks, uncertainties, and assumptions that are difficult to predict; therefore, actual results may differ materially from those expressed or forecasted in any such statements. Such forward-looking statements are based on current expectations, estimates and projections about our industry and business, management's beliefs, and certain assumptions made by our management, and may include, but are not limited to, statements regarding:
•the potential benefits and commercial potential of ULTOMIRIS®, SOLIRIS®, STRENSIQ®, KANUMA® and ANDEXXA® for approved indications and any expanded uses;
•sales of our products in various markets worldwide, pricing for our products, level of insurance coverage and reimbursement for our products, timing regarding development and regulatory approvals for our products or for additional indications or in additional territories;
•plans for clinical trials (and proof of concept trials and exploratory clinical studies), status of our ongoing clinical trials for our product candidates, commencement dates for new clinical trials, clinical trial results and evaluation of our clinical trial results by regulatory agencies;
•potential benefits offered by product candidates, including improved dosing intervals and potential to improve treatment in a number of IgG-mediated and neurological diseases;
•the medical and commercial potential of additional indications for our products;
•the expected timing for the completion and/or regulatory approval of our facilities and facilities of our third-party manufacturers;
•future expansion of our commercial organization and transition to third parties in certain jurisdictions to perform sales, marketing and distribution functions;
•future governmental and regulatory decisions that directly or indirectly impact drug pricing (and discounts) and the adoption, implementation and interpretation of healthcare laws and regulations (and the impact on our business);
•plans, prospects and expected timing for future regulatory approval of products and product candidates;
•competitors, potential competitors and future competitive products (including biosimilars);
•plans to grow our product pipeline (and diversify our business, including through acquisitions) and anticipated benefits to the Company;
•future objective to expand business and sales;
•future plans to retain earnings and not pay dividends;
•expected decisions to appeal certain litigation and intellectual property decisions;
•expectations to realize the carrying value of product inventory;
•impact of accounting standards;
•future costs, operating expenses (including research and development, sales, general and administrative and restructuring expenses) and capital requirements, capital investment, sufficiency of cash to fund operations for at least the next 12 months, ability to make payment on our credit facility and make contingent payment obligations, the sufficiency of our existing capital resources and projected cash needs, price approval and funding processes in various countries;
•the sources of expected increases in cash flow from operations, if any;
•anticipated impact of interest rate changes on financial statements;
•anticipated future milestone, contingent and royalty payments and lease payments (and, in each case, expected impact on liquidity);
•anticipated impact of the COVID-19 pandemic on our business;
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
•timing and anticipated amounts of future tax payments and benefits (including the potential recognition of unrecognized tax benefits), as well as timing of conclusion of tax audits;
•collection of accounts receivable and impact of any delay in the future in collecting accounts receivable on financial condition and operations, as well as the ability of counterparties to our derivatives to perform their obligations;
•the safety and efficacy of our products and our product candidates;
•the adequacy of our pharmacovigilance and drug safety reporting processes;
•the uncertainties involved in the drug development process and manufacturing;
•performance and reliance on third party service providers;
•our future research and development activities, plans for acquired programs, our ability to develop and commercialize products with our collaborators, anticipated regulatory approval of acquisitions and anticipated closing of acquisitions;
•periods of patent, regulatory and market exclusivity for our products;
•the scope of our intellectual property and the outcome of any challenges or opposition to our intellectual property; and
•estimates of the capacity of manufacturing and other service facilities to support our business operations, products and product candidates.
Such risks and uncertainties include, but are not limited to, the impact of the COVID-19 pandemic on our business (including our financial results and clinical trials), increased competition, actions by regulatory agencies, product candidates not receiving regulatory approvals, the possibility that expected tax benefits will not be realized, changes in healthcare and tax laws and regulations following the U.S. 2020 presidential election, assessment of impact of recent accounting pronouncements, potential declines in sovereign credit ratings or sovereign defaults in countries where we sell our products, delay of collection or reduction in reimbursement due to adverse economic conditions or changes in government and private insurer regulations and approaches to reimbursement, uncertainties surrounding legal proceedings, company investigations and government investigations and assessments, pending securities class action litigations, the investigation of our Brazilian operations by Brazilian authorities, the tax assessment by the Brazilian Federal Revenue Service and potential future tax assessments or liabilities by other revenue or tax regulators, risks related to the short and long-term effects of other government healthcare measures, intellectual property lawsuits, and the effect of shifting foreign exchange rates, as well as those risks and uncertainties discussed later in this report under the section entitled “Risk Factors.” Unless required by law, we undertake no obligation to update publicly any forward-looking statements, whether because of new information, future events or otherwise. However, readers should carefully review the risk factors set forth in this and other reports or documents we file from time to time with the SEC.
Overview
Alexion is a global biopharmaceutical company focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialization of life-changing medicines.
As a leader in rare diseases for more than 25 years, Alexion has developed and commercializes two approved complement inhibitors to treat patients with paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS), as well as the first and only approved complement inhibitor to treat anti-acetylcholine receptor (AChR) antibody-positive generalized myasthenia gravis (gMG) and neuromyelitis optica spectrum disorder (NMOSD) in patients who are anti-aquaporin-4 (AQP4) antibody positive. Alexion also has two highly innovative enzyme replacement therapies and the first and only approved therapies for patients with life-threatening and ultra-rare metabolic disorders, hypophosphatasia (HPP) and lysosomal acid lipase deficiency (LAL-D). With the acquisition of Portola Pharmaceuticals, Inc. (Portola) in July 2020, we added the first and only approved Factor Xa inhibitor reversal agent for patients treated with rivaroxaban or apixaban when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding.
In addition to our marketed therapies, we have a diverse pipeline resulting from internal innovation and business development. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and development efforts on the core therapeutic areas of hematology, nephrology, neurology, metabolic disorders, cardiology, ophthalmology and acute care.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Recent Developments
On October 12, 2020, we announced that the U.S. Food and Drug Administration (FDA) approved the ULTOMIRIS (ravulizumab-cwvz) 100 mg/mL intravenous (IV) formulation for the treatment of adults with paroxysmal nocturnal hemoglobinuria (PNH) and for atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy for adult and pediatric (one month age and older) patients. ULTOMIRIS 100 mg/mL is an advancement in the treatment experience for patients with aHUS and PNH, as it reduces average annual infusion times by approximately 60 percent compared to ULTOMIRIS 10 mg/mL while delivering comparable safety and efficacy.
On September 25, 2020, we announced that Japan's Ministry of Health, Labour, and Welfare (MHLW) approved ULTOMIRIS for the treatment of adults and children living with aHUS.
On September 21, 2020, we announced that the Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion, recommending marketing authorization in the European Union for a new 100 mg/mL IV advanced formulation of ULTOMIRIS.
On September 14, 2020, Alexion and Caelum Biosciences (Caelum) announced the initiation of the Cardiac Amyloid Reaching for Extended Survival (CARES) Phase III clinical program to evaluate CAEL-101, a first-in-class amyloid fibril targeted therapy, in combination with standard-of-care therapy in light chain (AL) amyloidosis. The CARES clinical program includes two parallel Phase III studies - one in patients with Mayo stage IIIa disease and one in patients with Mayo stage IIIb disease and will collectively enroll approximately 370 patients globally. Enrollment is underway in both studies. The primary objective of the clinical program is to assess overall survival.
COVID-19 Pandemic
During the first quarter of 2020, the World Health Organization (WHO) declared the COVID-19 public health crisis a pandemic and recommended containment and mitigation measures worldwide. On March 13, 2020, U.S. President Trump announced a National Emergency relating to the pandemic. Government authorities worldwide have recommended or imposed various social distancing, quarantine and isolation measures on large portions of the population. While the impact of the COVID-19 pandemic to date on our business has been less than we had initially forecast, it is evolving rapidly and its future effects are difficult to predict with meaningful precision as the impact will depend on many factors beyond the Company’s control and knowledge. As the pandemic continues, we are taking steps that are designed to respond proactively to evolving events and planning for COVID-19 uncertainties. We remain focused on continuing to serve patients, protecting the health and safety of our employees and the communities in which we live and work, and supporting our patients in clinical trials.
In early March 2020, we activated a task force designed to assess, mitigate and manage the risks related to COVID-19 to avoid or minimize business disruption, including safeguarding of our facilities, and to ensure the safety and sense of security for our staff. In early March 2020, Alexion closed all sites to non-essential employees and the Company has suspended all travel indefinitely. In early June 2020, Alexion gradually allowed re-entry to certain sites in some geographies through a pilot program, including Switzerland, Germany, Australia, and Japan in accordance with local government laws, regulations and restrictions and our own safety procedures and checklists. In September 2020, we extended our global guidance to employees to strongly encourage working remotely until July 2021, while offering limited access to physical sites through pilot programs. Office sites are being reconfigured to maintain physical distancing and we expect to adopt and implement additional precautions commensurate with any expansion of employees returning to worksites. To date, our remote working arrangements have not significantly affected our ability to maintain critical business operations.
We are focused on protecting patient and customer safety as well as providing an uninterrupted supply of medicines for patients around the world. We have taken proactive measures that are designed to mitigate the risk of potential supply interruptions, and we strive to maintain sufficient inventory levels to continue serving current and new patients receiving our medicines for approved indications, as well as those participating in ongoing clinical trials. We and our third-party contract manufacturing partners continue to operate manufacturing facilities at near normal levels.
We are monitoring the demand for our products as there may continue to be fewer patient/doctor in-person interactions, fewer visits between our representatives and health care providers and the potential inability of patients to access hospitals or infusion centers (voluntarily or involuntarily), all of which may result in a reduction in future sales. We have been proactively engaging with healthcare professionals virtually and through enhanced digital channels in an effort to mitigate this risk, however we have noted that the new patient productivity and initiation queue has decreased since the COVID-19 outbreak, particularly in neurological indications. Additionally, we continue to actively monitor potential further impacts on our business such as growth in unemployment and loss of commercial insurance coverage and/or growth in Medicaid with higher discounts.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
We have preclinical studies and clinical testing ongoing across the globe. We have a business continuity plan for our preclinical and clinical trials, including a pandemic response plan. A number of clinical trial sites are restricting site visits and imposing restrictions on the initiation of new trials and patient visits to protect both site staff and patients from possible COVID-19 exposure. Given the safety concerns around COVID-19 and the associated risk to maintaining normal clinical trial operations, we are making decisions study-by-study and country-by-country to minimize the risk to the patients and facilities, and there has been and may continue to be an impact on the timing of trials that are under active enrollment. The majority of clinical trials that were paused at the onset of the pandemic have resumed, however we are continuing to experience impact to enrollment for some studies. We are actively implementing remote and local procedures per recent guidance of the FDA.
In May 2020, Alexion initiated a global Phase III study to investigate ULTOMIRIS® (ravulizumab-cwvz) in a subset of adult patients with COVID-19 who are hospitalized with severe pneumonia or acute respiratory distress syndrome (ARDS). The study is actively enrolling patients and is expected to enroll approximately 270 patients across the U.S., E.U. and Japan and is evaluating the impact of ULTOMIRIS on survival, duration of mechanical ventilation, and duration of hospital stay compared to best supportive care. This follows the FDA's rapid review and acceptance of Alexion’s investigational new drug (IND) application for ULTOMIRIS for severe COVID-19. In recognition of the urgent needs of some patients and in order to streamline the emergency access process, Alexion has opened emergency Expanded Access Programs (EAP) in the U.S. and France for SOLIRIS in severe COVID-19 pneumonia.
The extent to which the COVID-19 pandemic impacts our business, including our commercial results and clinical trials, will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the virus, the duration of the outbreak, governmental regulations and restrictions, travel restrictions and actions to contain the outbreak or treat its impact. We continue to be responsive to the ever-changing situation while remaining true to our core values.
Products and Development Programs
We focus our product development programs on life-transforming therapeutics for rare diseases and devastating conditions for which current treatments are either non-existent or inadequate. We have developed or are developing innovative products for, among others, the following indications:
| | | | | |
Paroxysmal Nocturnal Hemoglobinuria (PNH) | PNH is a chronic, progressive, debilitating and life-threatening ultra-rare blood disorder characterized by intravascular hemolysis (destruction of red blood cells) that is mediated by an uncontrolled activation of the complement system, a part of the immune system. PNH red blood cells are exquisitely vulnerable to activated complement, resulting in chronic intravascular hemolysis. Chronic intravascular hemolysis in patients with PNH may be associated with life-threatening thromboses, recurrent pain, kidney disease, disabling fatigue, impaired quality of life, severe anemia, pulmonary hypertension, shortness of breath and intermittent episodes of dark-colored urine (hemoglobinuria). A small sub-set of PNH patients on C5-inhibitor treatment may experience clinically evident extravascular hemolysis (PNH-EVH). |
| | | | | |
Atypical Hemolytic Uremic Syndrome (aHUS) | aHUS is a severe and life-threatening, ultra-rare genetic disease characterized by chronic uncontrolled complement activation and thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body, causing a reduction in platelet count (thrombocytopenia) and life-threatening damage to the kidney, brain, heart and other vital organs. |
| | | | | |
Generalized Myasthenia Gravis (gMG) | Myasthenia Gravis (MG) is a debilitating, complement-mediated neuromuscular disease in which patients suffer profound muscle weakness throughout the body, resulting in slurred speech, impaired swallowing and choking, double vision, upper and lower extremity weakness, disabling fatigue, shortness of breath due to respiratory muscle weakness and episodes of respiratory failure. |
| | | | | |
Hypophosphatasia (HPP) | HPP is an ultra-rare genetic and progressive metabolic disease in which patients experience devastating effects on multiple systems of the body, leading to debilitating or life-threatening complications. HPP is characterized by defective bone mineralization that can lead to deformity of bones and other skeletal abnormalities, as well as systemic complications such as profound muscle weakness, seizures, pain, and respiratory failure leading to premature death in infants. |
| | | | | |
Lysosomal Acid Lipase Deficiency (LAL Deficiency or LAL-D) | LAL-D is a serious, life-threatening ultra-rare disease associated with premature mortality and significant morbidity. LAL-D is a chronic disease in which genetic mutations result in decreased activity of the LAL enzyme that leads to marked accumulation of lipids in vital organs, blood vessels, and other tissues, resulting in progressive and systemic organ damage including hepatic fibrosis, cirrhosis, liver failure, accelerated atherosclerosis, cardiovascular disease, and other devastating consequences. |
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
| | | | | |
Neuromyelitis Optica Spectrum Disorder (NMOSD) | NMOSD is a severe and ultra-rare autoimmune disease of the central nervous system (CNS) that primarily affects the optic nerves and the spinal cord. Each relapse of the disorder results in a stepwise accumulation of disability, including blindness and paralysis, and sometimes premature death. Complement activation due to anti-AQP4 antibodies is one of the primary underlying causes of the destruction of vital cells in the central nervous system in patients with NMOSD.
|
| | | | | |
Anticoagulant Effects of Factor Xa Inhibitors | Factor Xa Inhibitors (i.e. apixaban, rivaroxaban and edoxaban), may rarely cause patients to be hospitalized with life-threatening or uncontrolled bleeding. Potential events include intracranial hemorrhage; intraocular, pericardial, intraspinal, intraarticular bleeding at critical sites; major gastrointestinal, retroperitoneal, or genitourinary bleeding; and bleeding associated with major blunt or penetrating injury. |
| | | | | |
Wilson Disease | Wilson disease is a rare disorder, characterized by excess copper stored in various body tissues, that can lead to severe liver disease, including cirrhosis and acute liver failure, as well as debilitating neurological morbidities such as impaired movement, gait, speech, swallowing, and psychiatric disorders. |
| | | | | |
Warm Autoimmune Hemolytic Anemia (WAIHA) | WAIHA is a rare autoimmune disorder caused by pathogenic Immunoglobulin G (IgG) antibodies that react with and cause the premature destruction of red blood cells at normal body temperature. The disease is often characterized by profound, and potentially life-threatening anemia and other acute complications, including severe and life-threatening hemolysis, severe weakness, enlarged spleen and/or liver, rapid heart rate (tachycardia), chest pain, heart failure and fainting (syncope). |
| | | | | |
Amyotrophic Lateral Sclerosis (ALS) | ALS is a progressive neurodegenerative disease of the CNS characterized by the loss of upper (brain) and lower (spinal cord) motor neurons. Ongoing loss of motor neurons and muscle strength leads to loss of independence, paralysis and death, typically due to respiratory insufficiency. |
| | | | | |
C3 Glomerulopathy (C3G) | C3G is a rare, chronic disease affecting the kidneys in which the alternative pathway of the complement system is dysregulated due to genetic mutations or autoantibodies affecting the regulation of the alternative pathway. This lack of regulation results in the alternative pathway overactivation and the excessive deposition of C3 protein fragments in the glomeruli, a key filtration component of the kidney, often leading to serious kidney damage. |
| | | | | |
Relapsed/refractory B-and T-cell malignancies | The B cell non-Hodgkin lymphomas (NHLs) are a diverse group of disorders of proliferating malignant B cells. Collectively, NHL is the eighth leading type of cancer in the U.S., with B-cell lymphomas diagnosed in approximately 85% to 90% of patients. In 2019, an estimated 74,200 new cases of NHL will be diagnosed along with nearly 20,000 deaths. Patients present with fever, night sweats, and unintentional weight loss. With progressive disease, the malignant cells metastasize from lymph nodes and bone marrow to other organs, ultimately resulting in organ failure. Front-line therapy consists of combination chemotherapy with rituximab. Experimental agents are in development to address patients who relapse or are refractory to front-line therapy. There are no curative therapies in the relapsed/refractory setting.
The T cell lymphomas are represented by diverse histologies depending on the malignant cell of origin. T-cell NHL comprises approximately 10% to 15% of the total cases of NHL, resulting in approximately 7,000 to 10,000 cases a year in the U.S. Symptoms are similar to those of the B cell malignancies, however, the 5 year overall survival rates are typically lower for T cell NHL vs B cell NHL with rates of approximately 35% being found in several studies. These are highly aggressive lymphomas for which there is no optimal standard of care. |
| | | | | |
COVID-19 | Coronaviruses are a family of viruses that can cause illnesses such as the common cold, severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS). In 2019, a new coronavirus was identified as the cause of a disease outbreak that likely originated in China. The virus is now known as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the disease it causes is called coronavirus disease 2019 (COVID-19). In March 2020, the World Health Organization (WHO) declared the COVID-19 outbreak a pandemic. Signs and symptoms of COVID-19 may appear two to 14 days after exposure and can include fever, cough, shortness of breath or difficulty breathing. The severity of COVID-19 symptoms can range from very mild to very severe. Some people may have no symptoms at all and spread the infection inadvertently. People who are older or who have pre-existing diagnosed or undiagnosed medical conditions, such as heart disease, lung disease and/or diabetes, or who have a compromised or overreacting immune system may be at higher risk of serious illness or complications and may require assisted ventilation as well as urgent critical care. |
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Marketed Products
Our marketed products consist of the following:
| | | | | | | | |
| Product | Therapeutic Area | Approved Indication |
| Hematology | Paroxysmal Nocturnal Hemoglobinuria (PNH) |
|
| Hematology/Nephrology | Atypical Hemolytic Uremic Syndrome (aHUS)
|
|
| Hematology | Paroxysmal Nocturnal Hemoglobinuria (PNH) |
| Hematology/Nephrology | Atypical Hemolytic Uremic Syndrome (aHUS) |
| Neurology | Generalized Myasthenia Gravis (gMG) |
| Neurology | Neuromyelitis Optica Spectrum Disorder (NMOSD) |
| Metabolic Disorders | Hypophosphatasia (HPP) |
| Metabolic Disorders | Lysosomal Acid Lipase Deficiency (LAL-D) |
| Acute Care | Reversal of anticoagulation in patients treated with Factor Xa inhibitors when experiencing life-theatening or uncontrolled bleeding. |
ULTOMIRIS (ALXN1210/ravulizumab-cwvz)
ULTOMIRIS is an innovative, long-acting C5 inhibitor discovered and developed by Alexion that works by inhibiting the C5 protein in the terminal complement cascade. In clinical studies, ULTOMIRIS demonstrated rapid, complete, and sustained reduction of free C5 levels.
In December 2018, ULTOMIRIS was approved by the FDA as a new treatment option for adult patients with PNH in the U.S.
ULTOMIRIS was approved as a new treatment option for adult patients with PNH by Japan's MHLW in June 2019. ULTOMIRIS was approved by the European Commission (EC) in July 2019 as a treatment for adult patients with PNH with hemolysis with clinical symptoms indicative of high disease activity, and also for adult patients who are clinically stable after having been treated with SOLIRIS for at least the past six months.
In October 2019, the FDA approved the use of ULTOMIRIS as a treatment for adult and pediatric (one month of age or older) patients with aHUS to inhibit complement-mediated TMA.
In June 2020, the EC approved ULTOMIRIS for the treatment of adults and children with a body
weight of 10kg or above with aHUS who are complement inhibitor treatment-naïve or have received SOLIRIS (eculizumab) for at least three months and have evidence of response to eculizumab.
In September 2020, ULTOMIRIS was approved by Japan's MHLW as a new treatment option for adult and pediatric patients with aHUS.
In October 2020, ULTOMIRIS 100 mg/mL formulation was approved by the FDA for the treatment of adult patients with PNH and for adults and pediatric (one month of age or older) patients with aHUS to inhibit complement-mediated TMA.
SOLIRIS (eculizumab)
SOLIRIS is an innovative C5 inhibitor discovered and developed by Alexion that works by inhibiting the C5 protein in the terminal complement cascade. SOLIRIS is a humanized monoclonal antibody that effectively blocks terminal complement activity at the doses currently prescribed.
SOLIRIS is approved for the treatment of PNH and aHUS in pediatric and adult patients in the U.S., Europe, Japan and in several other countries. Alexion is sponsoring multinational registries to gather information regarding the natural history of patients with PNH and aHUS and the longer-term outcomes during SOLIRIS treatment.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
In 2017, the FDA and EC regulatory authorities approved SOLIRIS for the treatment of gMG in adults who are anti-acetylcholine receptor (AChR) antibody-positive. Additionally, in 2017, the MHLW in Japan approved SOLIRIS as a treatment for patients with gMG who are AChR antibody-positive and whose symptoms are difficult to control with high-dose intravenous immunoglobulin therapy or plasmapheresis (PLEX).
In June 2019, SOLIRIS became the first FDA-approved treatment option for adult patients with NMOSD who are AQP4 auto antibody positive. In August 2019, the EC approved SOLIRIS as the first treatment in Europe for NMOSD in adults who are AQP4 antibody-positive with a relapsing course of the disease. In November 2019, the Japanese MHLW approved SOLIRIS as a treatment for the prevention of relapse in patients with AQP4 antibody-positive NMOSD, including Neuromyelitis Optica.
STRENSIQ (asfotase alfa)
STRENSIQ, a targeted enzyme replacement therapy, is the first and only approved therapy for patients with HPP and is designed to directly address underlying causes of HPP by aiming to restore the genetically defective metabolic process, thereby preventing or reversing the severe and potentially life-threatening complications in patients with HPP. STRENSIQ is approved in the U.S. for patients with perinatal-, infantile- and juvenile-onset HPP, in Europe for the treatment of patients with pediatric-onset HPP, and in Japan for the treatment of patients with HPP. Alexion is sponsoring a multinational registry to gather information regarding the natural history of patients with HPP and the longer-term outcomes during STRENSIQ treatment.
KANUMA (sebelipase alfa)
KANUMA, a recombinant form of the human LAL enzyme, is the only enzyme-replacement therapy that is approved for the treatment for patients with LAL-D. KANUMA is approved in the U.S. for the treatment of patients with LAL-D, in Europe for long-term enzyme replacement therapy in patients with LAL-D, and in Japan for the treatment of patients with LAL-D. Alexion is sponsoring a multinational registry to gather information regarding the natural history of patients with LAL-D and the longer-term outcomes during KANUMA treatment.
ANDEXXA (coagulation factor Xa - [recombinant] inactivated-zhzo)
ANDEXXA is approved by the FDA as a reversal agent for patients treated with rivaroxaban or apixaban, when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding. ANDEXXA was approved under the FDA’s Accelerated Approval Pathway, and received conditional marketing authorization in the EU based on the change from baseline in anti-Factor Xa activity in healthy volunteers and in patients through the ANDEXXA-4 trial demonstrating hemostatic efficacy. Continued approval for this indication is contingent upon post-marketing study results that verify that clinical benefit is conferred to patients.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Clinical Development Programs
Our ongoing clinical development programs include the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Product | Mechanism of Action | Development Area | Indication | Phase I | Phase II | Phase III | Phase IV | Filed |
| ULTOMIRIS (ALXN1210/ravulizumab-cwwz) (Intravenous) | Anti-C5 | Neurology
Pulmonology | gMG/NMOSD/ALS
COVID-19 | | | l | | |
| | | | | | | | |
ULTOMIRIS (ALXN1210/ravulizumab-cwvz)
(Subcutaneous) | Anti-C5 | Hematology/Nephrology | PNH/aHUS | | | l | | |
| | | | | | | | |
| ALXN1720 (Subcutaneous) | Anti-C5 | Next Generation Subcutaneous Complement Inhibitor | | l | | | | |
| | | | | | | | |
ALXN1830
(SYNT001)
(Subcutaneous) | Anti-FcRN | FcRN | | l | | | | |
| | | | | | | | |
ALXN1840
(WTX101) | High-affinity, specific Cu binder | Metabolic Disorders | Wilson disease | | | l | | |
| | | | | | | | |
ALXN2040
(ACH-4771) | Factor D Inhibitor | Hematology/Nephrology | PNH-EVH / C3G | | l | | | |
| | | | | | | | |
ALXN2050
(ACH-5228) | Factor D Inhibitor | Hematology/Nephrology | PNH | | l | | | |
| | | | | | | | |
ALXN2070
(ANDEXXA) | Anti Factor Xa Reversal | Acute Care | Reversal of anticoagulation in patients treated with Factor Xa inhibitors when experiencing life-theatening or uncontrolled bleeding. | | | | l | |
| | | | | | | | |
ALXN2075
(Cerdulatinib) | Dual spleen tyrosine kinase and janus kinase (SYK/JAK) inhibitor | Acute Care/ Oncology | Relapsed/refractory chronic lymphocytic leukemia or B-cell or T-cell NHL | | l | | | |
In addition to our ongoing development programs, Alexion holds a minority interest in and an option to acquire Caelum Biosciences (Caelum), a biotechnology company that is developing CAEL-101 for light chain (AL) amyloidosis. CAEL-101 is a first-in-class chimeric monoclonal antibody (mAb) designed to improve organ function by reducing or eliminating amyloid deposits in the tissues and organs of patients with AL amyloidosis, a rare systemic disorder caused by an abnormality of plasma cells in the bone marrow. A Phase Ia/Ib study for CAEL-101 has been completed. Following discussions with the FDA, a Phase II trial for CAEL-101 commenced during the first quarter of 2020 and met its primary objectives, supporting the safety and tolerability of CAEL-101 and confirmed the dose and regimen to be adopted for the Phase III studies. In September 2020, Alexion and Caelum announced the initiation of the Cardiac Amyloid Reaching for Extended Survival (CARES) program. This includes two parallel Phase III trials to evaluate the survival benefits of CAEL-101. Enrollment is underway in both studies.
ULTOMIRIS (ALXN1210/ravulizumab-cwvz)
ULTOMIRIS is an innovative, long-acting C5 inhibitor discovered and developed by Alexion that works by inhibiting the C5 protein in the terminal complement cascade. In clinical studies, ALXN1210 demonstrated rapid, complete, and sustained reduction of free C5 levels.
Intravenous (IV)
In January 2019, Alexion announced that the Phase III, global, single arm, multicenter study evaluating the safety and efficacy of ALXN1210 administered by IV infusion every 8 weeks to adult patients with aHUS who had never been treated with a
complement inhibitor (inhibitor-naïve patients) met its primary objective. In the study's initial 26-week treatment period, 53.6 percent of patients demonstrated complete TMA response. A second Phase III, single arm, multicenter study to evaluate the safety, efficacy, pharmacokinetics (PK), and pharmaco-dynamics (PD) of ALXN1210 administered by IV infusion every 8 weeks in inhibitor-naïve pediatric patients (including adolescents) with aHUS is ongoing.
In November 2019, the Extension Application to register the ULTOMIRIS 100 mg/ml formulation (which is a higher concentration formulation of ULTOMIRIS than the formulation currently commercialized) was submitted to the EMA. In August 2020, Alexion
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
submitted an application to the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) to register the ULTOMIRIS 100mg/ml formulation. In September 2020, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending marketing authorization in the European Union for the 100 mg/ml formulation.
In March 2019, Alexion initiated a Phase III double-blind, placebo-controlled, multicenter study to evaluate the safety and efficacy of ALXN1210 in adult patients for the treatment of gMG. Additionally, in December 2019, Alexion initiated a Phase III, single arm, open-label, multicenter study to evaluate the safety and efficacy of ALXN1210 in adult patients with NMOSD.
In March 2020, Alexion initiated a Phase III, randomized, double-blind, placebo-controlled, multicenter study to evaluate the efficacy and safety of ALXN1210 in patients with ALS.
In May 2020, following the FDA's acceptance of Alexion's IND application, Alexion initiated a Phase III open-label, randomized, controlled clinical trial of ALXN1210 in adult patients with COVID-19 who are hospitalized with severe pneumonia or Acute Respiratory Distress Syndrome (ARDS). The trial is investigating the role of terminal complement inhibition in managing patients with severe COVID-19.
In addition to aHUS, NMOSD, gMG, ALS and COVID-19, Alexion plans to initiate: (i) Phase III studies of ALXN1210 in adult and pediatric hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA) in the fourth quarter 2020; (ii) a Phase III study of ALXN1210 in complement mediated thrombotic microanglopathy (CM-TMA) and (iii) a proof of concept basket study in renal indications, including Lupus Nephritis (LN) and Immunoglobulin A Nephropathy (IgAN).
Subcutaneous (SC) Delivery
In March 2019, Alexion initiated a single, PK-based Phase III study of ALXN1210 delivered subcutaneously once per week to PNH patients to support regulatory approval submissions in both PNH and aHUS. In June 2020, Alexion announced that the ongoing study met its primary objective of pharmacokinetic-based non-inferiority of ULTOMIRIS SC versus intravenous (IV) ULTOMIRIS at Day 71. Pending completion of the study and collection of required 12-month safety data, Alexion expects to file for approval in the U.S. and E.U. for the SC formulation and device combination in PNH and aHUS in the third quarter of 2021.
ALXN1720 Subcutaneous (SC) Delivery
ALXN1720 is a novel humanized bi-specific minibody antibody that binds selectively and with high affinity to C5. ALXN1720 is designed for subcutaneous administration as a concentrated formulation for the treatment of disease states involving dysregulated terminal complement activity. In September 2019, Alexion initiated a Phase I healthy volunteer study of ALXN1720 to assess its safety and tolerability. This trial was paused due to the COVID-19 pandemic, but was re-initiated in August 2020. Additionally, we plan to initiate trials in gMG and dermatomyositis (DM), a rare autoimmune inflammatory myopathy characterized by chronic inflammation and degeneration of muscle and skin.
ALXN1810 Subcutaneous (SC) Delivery
ALXN1810 combines ravulizumab-cwvz with recombinant human hyaluronidase enzyme (rHuPH20), licensed from Halozyme Therapeutics, Inc., to potentially further extend the dosing interval for ALXN1210 SC from once per week to once every two weeks or more. Alexion completed a SC healthy volunteer study with ALXN1810 in December 2018. We are no longer pursuing this combination therapy.
ALXN1830 (SYNT001)
ALXN1830 (SYNT001) is a humanized monoclonal antibody that is designed to inhibit the interaction of the neonatal Fc receptor (FcRn) with IgG and IgG immune complexes and has the potential to improve treatment in a number of rare IgG-mediated diseases. Alexion re-initiated a Phase II trial of the IV formulation in WAIHA in early 2020. In addition, Alexion initiated a Phase I study of a SC formulation of ALXN1830 in healthy volunteers in December 2019. Due to the COVID-19 pandemic, Alexion discontinued the Phase II trial in WAIHA and paused the Phase I healthy volunteer study. Phase I healthy volunteer study and Phase II studies in WAIHA and gMG exclusively with the SC formulation are planned to start in 2021.
ALXN1840 (WTX101)
ALXN1840 (WTX101), an innovative product candidate that addresses the underlying cause of Wilson disease, is a first-in-class oral copper-binding agent with a unique mechanism of action and the ability to access and bind copper from serum and remove it from the liver.
In February 2020, Alexion completed enrollment in a Phase III study of ALXN 1840 for the treatment of Wilson disease. ALXN1840 has received Fast Track designation in the U.S.
Alexion planned to initiate a Phase II study for ALXN1840 in Primary Biliary Cholangitis, a chronic liver disease resulting from progressive destruction of
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
the bile ducts in the liver, however, we are no longer pursuing this study.
ALXN2040 (danicopan/ACH-4771)
ALXN2040 is an oral Factor D inhibitor designed to treat diseases associated with dysregulation of the complement alternative pathway. ALXN2040 as an add on therapy for PNH patients with clinically evident extravascular hemolysis (EVH) has received orphan drug and breakthrough therapy designation by the FDA and both orphan and PRIME designation by EMA. Two Phase II studies of ALXN2040 as an oral add-on therapy for PNH-EVH are ongoing, and a Phase III trial is planned to commence by the end of 2020. Two Phase II studies evaluating the safety, efficacy, pharmacokinetics, and pharmaco-dynamics of ALXN2040 in patients with C3G are in the process of being wound down. We will no longer continue to pursue ALXN2040 as a treatment for C3G. Alexion plans to develop ALXN2050 in C3G, which has improved PK/PD and inhibition of the alternative pathway of the complement system as compared to ALXN2040.
We plan to initiate a Phase II study of ALXN2040 in Geographic Atrophy, a chronic and progressive degeneration of the portion of the retina responsible for central and color vision, in 2021.
ALXN2040 was added to the pipeline portfolio as a result of the acquisition of Achillion.
ALXN2050 (ACH-5228)
ALXN2050 is an oral Factor D inhibitor designed to treat rare diseases associated with dysregulation of the complement alternative pathway. ALXN2050 is in Phase II trials as a potential monotherapy treatment for PNH and is being evaluated for development in other alternative pathway-mediated rare diseases. Additionally, Alexion plans to initiate proof-of-concept trials of ALXN2050 in patients with various renal diseases in 2021.
ALXN2050 was added to the pipeline portfolio as a result of the acquisition of Achillion.
ALXN2060 (AG10)
In September 2019, Alexion entered into an agreement with Eidos Therapeutics, Inc. (Eidos), through which Alexion obtained an exclusive license to develop and commercialize AG10 in Japan for transthyretin amyloidosis (ATTR). AG10 is an orally administered small molecule in development designed to target the root cause of ATTR by stabilizing transthyretin (TTR) in the blood. Eidos is currently evaluating AG10 in a Phase III study in the United States and Europe for ATTR cardiomyopathy (ATTR-CM) and plans to initiate a Phase III study in ATTR polyneuropathy (ATTR-PN) by the end of 2020. Alexion plans to expand the AG10 program into Japan through the initiation of a clinical trial for which data would
serve as the basis for seeking regulatory approval to commercialize AG10 in Japan.
ALXN2070 (ANDEXXA)
The acquisition of Portola added Portola’s commercialized medicine, ANDEXXA, for which additional clinical trials are currently being conducted to obtain full regulatory approvals and expand approved indications. It has obtained accelerated approval in the US and conditional marketing authorization in the EU based on the change from baseline in anti-Factor Xa activity in healthy volunteers, and in patients through the ANNEXA-4 trial. Full approval in the US and EU for current indications requires completion of ANNEXA-I, a Phase IV randomized controlled clinical trial currently underway evaluating the safety and efficacy of ANDEXXA versus standard of care in patients presenting with acute intracranial hemorrhage while taking an oral Factor Xa inhibitor. A Japan J-NDA filing is under evaluation. A US sBLA submission for a label expansion for the reversal of edoxaban and enoxaparin associated bleeds is anticipated for late 2020 or early 2021.
Additionally, we plan to initiate a Phase II study in 2021 for the reversal of anticoagulation in patients taking apixaban, rivaroxaban, edoxaban, or enoxaparin who require urgent surgery.
ALXN2075 (Cerdulatinib)
ALXN2075 (Cerdulatinib) is a small molecule SYK/JAK kinase inhibitor in development for treatment of hematological malignancies. We are reviewing preliminary clinical data and considering Follicular Lymphoma as a next step in the program. A Phase II study is ongoing. ALXN2075 was added to the pipeline portfolio as a result of the acquisition of Portola.
SOLIRIS (eculizumab)
SOLIRIS is an innovative C5 inhibitor discovered and developed by Alexion that works by inhibiting the C5 protein in the terminal complement cascade. SOLIRIS is a humanized monoclonal antibody that effectively blocks terminal complement activity at the doses currently prescribed.
In June 2020, Japan’s MHLW granted SAKIGAKE designation for SOLIRIS in Guillain-Barré syndrome (GBS). Results from the Japanese Eculizumab Trial for GBS (JET-GBS study) suggested the potential efficacy and safety of SOLIRIS as a treatment for GBS. Complement activation may play a role in the pathophysiology of GBS. Alexion plans to initiate a Phase III study of SOLIRIS in GBS in Japan in 2021.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Manufacturing
We utilize both internal manufacturing facilities and third-party contract manufacturers to supply clinical and commercial quantities of our products and product candidates. Our internal manufacturing capability includes our Ireland facilities, a fill/finish facility in Athlone, and a packaging facility in Dublin, as well as a production facility in Georgia. Third party contract manufacturers, including Lonza Group AG and its affiliates (Lonza), provide bulk drug substance as well as other manufacturing services like purification, product filling, finishing, packaging, and labeling.
We have various agreements with Lonza through 2031, with remaining total non-cancellable commitments of approximately $1,432.3. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangements. Under an existing arrangement, we pay Lonza a royalty on sales of SOLIRIS that was manufactured at the Alexion Rhode Island Manufacturing Facility (ARIMF) prior to the sale of the facility in 2018. We also pay Lonza a royalty on the sales of ULTOMIRIS and a payment with respect to sales of SOLIRIS manufactured at Lonza facilities. Lonza also manufactures ANDEXXA for Alexion. Lonza is in the process of qualifying a new manufacturing facility in New Hampshire that would, if and when such facility is approved by regulatory authorities, manufacture STRENSIQ for commercial use (and commitments entered into under this arrangement are included in the non-cancellable commitments amount noted in the first sentence of this paragraph).
In addition, we have non-cancellable commitments of approximately $78.6 through 2021 with other third-party manufacturers.
In April 2014, we purchased a fill/finish facility in Athlone, Ireland, which has been refurbished to become our first company-owned fill/finish facility. We have also completed construction of a new biologics manufacturing facility at this site and we are currently pursuing regulatory approval.
In May 2015, we announced plans to construct a new biologics manufacturing facility on our existing property in Dublin, Ireland. Construction of this facility has been completed and we are currently pursuing regulatory approval.
While we continue to actively engage with regulators, the timing of regulatory approvals for each of these facilities may be delayed as a result of the COVID-19 pandemic.
Critical Accounting Policies and Estimates
The significant accounting policies and basis of preparation of our consolidated financial statements are described in Note 1, Business Overview and Summary of Significant Accounting Policies of the Consolidated Financial Statements included in our Form 10-K for the year ended December 31, 2019. Under accounting principles generally accepted in the U.S., we are required to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and disclosure of contingent assets and liabilities in our financial statements. We believe the most complex judgments result primarily from the need to make estimates about the effects of matters that are inherently uncertain and are significant to our consolidated financial statements. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. We evaluate our estimates, judgments and assumptions on an ongoing basis. Actual results could differ materially from those estimates.
We believe the judgments, estimates and assumptions associated with the following critical accounting policies have the greatest potential impact on our consolidated financial statements:
•Revenue recognition;
•Contingent liabilities;
•Share-based compensation;
•Valuation of acquired assets, including goodwill, intangible assets and inventory;
•Valuation of contingent consideration; and
•Income taxes.
For a complete discussion of these critical accounting policies, refer to “Critical Accounting Policies and Use of Estimates” within “Item 7 - Management's Discussion and Analysis of Financial Condition and Results of Operations” included within our Annual Report on Form 10-K for the year ended December 31, 2019. There have been no significant changes to the critical accounting policies.
Valuation of Acquired Assets, Including Goodwill, Intangible Assets and Inventory
We have recorded goodwill, acquired intangible assets and inventory related to our business combinations. When identifiable intangible assets, including IPR&D, are acquired, we determine the fair values of the assets as of the acquisition date. An income approach, which generally relies upon forecasted cash flow models, is typically used in these valuations if quoted market prices are not available. These valuations require the use of
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
significant estimates and assumptions including but not limited to:
•timing and costs to complete the in-process projects;
•timing and probability of success of clinical events or regulatory approvals;
•estimated future cash flows from product sales resulting from completed products and in-process projects, which consider competitive trends impacting the assets;
•tax rates; and
•discount rates
We may also utilize a cost approach, which estimates the costs that would be incurred to replace the assets being purchased. Significant inputs into the cost approach include estimated rates of return on historical costs that a market participant would expect to pay for these assets.
Intangible assets with indefinite lives are not amortized, but are tested for impairment at least annually or when a triggering event occurs that could indicate a potential impairment. Intangible assets with definite useful lives are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if triggering events occur. When performing our impairment assessment for definite-lived intangible assets, we rely upon cash flow projections attributable to the asset to determine if the carrying value of the asset is recoverable, on an undiscounted cash flow basis. If the carrying value of a definite lived intangible asset is not recoverable, or if there is an indicator of impairment on an indefinite-lived intangible asset, we will recognize an impairment in the amount by which the carrying value of the asset exceeds its fair value. We calculate the fair value of these assets using discounted cash flow models which require the use of significant estimates and judgements which include, but are not limited to, probability of success of clinical events or regulatory approvals, discount rates, and estimated future cash flows from product sales. Changes to assumptions used in our cash flow projections could result in an impairment. Impairments are recorded within impairment of intangible assets in our consolidated statements of income.
During the quarter ended June 30, 2020, based on continued challenges expanding patient growth and new alternative commercial opportunities, the Company revised its strategic view of KANUMA and determined that we have exhausted commercially viable initiatives related to KANUMA and will have difficulty expanding patient growth over the long term as we focus on promoting other commercial programs and growing our pipeline. As a result, we no longer expect to increase the number of KANUMA patients in
the long term at the rate previously assumed. This determination resulted in reduced cash flow projections for KANUMA, which indicated that the related intangible asset value was not fully recoverable on an undiscounted cash flows basis. On June 30, 2020, the Company utilized market participant assumptions to determine its best estimate of the fair value of the intangible asset related to KANUMA that, when compared with its related carrying value, resulted in an impairment charge of $2,042.3.
The estimated fair value of the KANUMA asset as of June 30, 2020 was determined using the excess earnings method, a variation of the income approach. The excess earnings method estimates the value of an intangible asset equal to the present value of the incremental after-tax cash flows attributable to that intangible asset over its remaining economic life. Long term cash flow projections for the asset require the use of significant estimates and judgements, including discount rates and revenue growth rates, and were based on the Company’s most recent strategic plan. The fair value of the asset was determined using an estimated weighted average cost of capital of 10.0%, which reflects the risks inherent in future cash flow projections and represents a rate of return that a market participant would expect for this asset. The estimated revenue growth rates fluctuate over the life of the asset, with a weighted average growth rate in the low single digits. The Company believes its assumptions are consistent with the plans and estimates that a market participant would use to manage the business. The estimated fair value of the KANUMA intangible asset as of June 30, 2020 was $820.0 and will continue to be amortized over its remaining estimated useful life. This fair value measurement was based on significant inputs not observable in the market and thus represents a Level 3 fair value measurement.
Inventory acquired in connection with the acquisition of a business is measured at fair value as of the acquisition date and requires the use of significant estimates, which include the expected selling price of the inventory, estimated costs to complete the manufacturing process and dispose of the inventory, a reasonable profit allowance for the remaining manufacturing and selling effort, estimated holding periods based on internal forecasts of demand and discount rates. We exclude from the value of acquired inventory any excess quantities that we do not expect to be salable based on future projected inventory requirements. If future demand or market conditions are lower than our projections as of the acquisition date, we may be required to write down the value of inventory through a charge to cost of sales in the period the revision is made.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination and is not amortized. Goodwill is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We are organized and operate as a single reporting unit and therefore the goodwill impairment test is performed using our overall market value, as determined by our traded share price, compared to our book value of net assets.
New Accounting Pronouncements
For information on new accounting pronouncements adopted in the current period and recently issued standards, see Note 2, Basis of Presentation and Principles, to our condensed consolidated financial statements included elsewhere in this Quarterly Report on Form 10-Q.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Results of Operations
Net Product Sales
Net product sales by significant geographic region for the three and nine months ended September 30, 2020 and 2019 are as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three months ended | | | | Nine months ended | | |
| September 30, | | % | | September 30, | | % |
| 2020 | | 2019 | | Change | | 2020 | | 2019 | | Change |
| SOLIRIS | | | | | | | | | | | |
| United States | $ | 562.8 | | | $ | 496.8 | | | 13.3 | % | | $ | 1,672.3 | | | $ | 1,456.8 | | | 14.8 | % |
| Europe | 254.3 | | | 255.5 | | | (0.5) | % | | 765.7 | | | 800.2 | | | (4.3) | % |
| Asia Pacific | 86.0 | | | 118.0 | | | (27.1) | % | | 255.5 | | | 329.2 | | | (22.4) | % |
| Rest of World | 139.2 | | | 120.2 | | | 15.8 | % | | 347.2 | | | 347.1 | | | — | % |
| Total | $ | 1,042.3 | | | $ | 990.5 | | | 5.2 | % | | $ | 3,040.7 | | | $ | 2,933.3 | | | 3.7 | % |
| | | | | | | | | | | |
| ULTOMIRIS | | | | | | | | | | | |
| United States | $ | 170.7 | | | $ | 65.1 | | | 162.2 | % | | $ | 460.3 | | | $ | 143.9 | | | 219.9 | % |
| Europe | 46.6 | | | 21.1 | | | 120.9 | % | | 112.4 | | | 21.1 | | | 432.7 | % |
| Asia Pacific | 69.6 | | | 3.7 | | | ** | | 186.3 | | | 3.7 | | | ** |
| Rest of World | 2.4 | | | — | | | ** | | 4.2 | | | — | | | ** |
| Total | $ | 289.3 | | | $ | 89.9 | | | 221.8 | % | | $ | 763.2 | | | $ | 168.7 | | | 352.4 | % |
| | | | | | | | | | | |
| STRENSIQ | | | | | | | | | | | |
| United States | $ | 149.3 | | | $ | 118.0 | | | 26.5 | % | | $ | 418.1 | | | $ | 323.7 | | | 29.2 | % |
| Europe | 19.3 | | | 19.0 | | | 1.6 | % | | 61.6 | | | 56.0 | | | 10.0 | % |
| Asia Pacific | 16.1 | | | 14.0 | | | 15.0 | % | | 44.7 | | | 36.0 | | | 24.2 | % |
| Rest of World | 4.7 | | | 3.3 | | | 42.4 | % | | 21.5 | | | 10.0 | | | 115.0 | % |
| Total | $ | 189.4 | | | $ | 154.3 | | | 22.7 | % | | $ | 545.9 | | | $ | 425.7 | | | 28.2 | % |
| | | | | | | | | | | |
| ANDEXXA | | | | | | | | | | | |
| United States | $ | 36.2 | | | $ | — | | | ** | | $ | 36.2 | | | $ | — | | | ** |
| Europe | 2.7 | | | — | | | ** | | 2.7 | | | — | | | ** |
| Asia Pacific | — | | | — | | | ** | | — | | | — | | | ** |
| Rest of World | — | | | — | | | ** | | — | | | — | | | ** |
| Total | $ | 38.9 | | | $ | — | | | ** | | $ | 38.9 | | | $ | — | | | ** |
| | | | | | | | | | | |
| KANUMA | | | | | | | | | | | |
| United States | $ | 15.8 | | | $ | 16.0 | | | (1.3) | % | | $ | 47.6 | | | $ | 45.1 | | | 5.5 | % |
| Europe | 9.5 | | | 6.3 | | | 50.8 | % | | 25.4 | | | 19.4 | | | 30.9 | % |
| Asia Pacific | 1.1 | | | 1.3 | | | (15.4) | % | | 2.9 | | | 3.4 | | | (14.7) | % |
| Rest of World | 2.0 | | | 4.8 | | | (58.3) | % | | 12.8 | | | 10.2 | | | 25.5 | % |
| Total | $ | 28.4 | | | $ | 28.4 | | | — | % | | $ | 88.7 | | | $ | 78.1 | | | 13.6 | % |
| | | | | | | | | | | |
| Total Net Product Sales | $ | 1,588.3 | | | $ | 1,263.1 | | | 25.7 | % | | $ | 4,477.4 | | | $ | 3,605.8 | | | 24.2 | % |
** Percentages not meaningful
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Net Product Sales
| | | | | | | | | | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
SOLIRIS net product sales
| | | | | | | | | | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
ULTOMIRIS net product sales
| | | | | | | | | | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
STRENSIQ net product sales
| | | | | | | | | | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
ANDEXXA net product sales
| | | | | | | | | | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
KANUMA net product sales
| | | | | | | | | | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
The increase in net product sales for the three and nine months ended September 30, 2020, as compared to the same periods in 2019, was primarily due to an increase in unit volumes. The increase in unit volumes was primarily due to increased global demand for SOLIRIS therapy, with sales to patients with gMG and NMOSD being the largest drivers. As a result of continued patient conversion, ULTOMIRIS unit volumes for PNH and aHUS also increased due to an increase in PNH and aHUS patients on ULTOMIRIS therapy, inclusive of the loading dose impact required in a patient's first year on therapy. Partially offsetting this increase was the continued conversion of PNH and aHUS patients from SOLIRIS to ULTOMIRIS, resulting in a decrease in SOLIRIS PNH and aHUS revenues for the three and nine months ended September 30, 2020, as compared to the same periods in 2019. Additional unit volume increases were due primarily to increased demand for STRENSIQ during 2020 and contributions of $38.9 from ANDEXXA as a result of the Portola acquisition that closed on July 2, 2020.
As a result of patient conversion from SOLIRIS to ULTOMIRIS, we expect variability in our revenues in future quarters due to the extended ULTOMIRIS dosing interval and infusion timing which may result in either one or two infusions in a quarter. ULTOMIRIS loading doses for PNH patients will result in increased revenues during a patient’s first year on therapy. The ULTOMIRIS annual maintenance dose for PNH and aHUS requires fewer vials as compared to the annual dose for SOLIRIS. Due to the decision to price ULTOMIRIS lower than SOLIRIS on an annual basis, we anticipate U.S. revenues will be unfavorably impacted by the lower annual cost per patient in maintenance years, with the impact more pronounced for aHUS due to the greater decrease in vials for aHUS ULTOMIRIS patients.
Foreign exchange had a negative impact of 2.0% and 1.5% for the three and nine months ended September 30, 2020, respectively, as compared to the same periods in 2019, with the largest impact relating to the weakening of the Brazilian Real versus the U.S. Dollar.
In response to the COVID-19 pandemic, we have taken proactive measures that are designed to mitigate the risk of potential supply interruptions, and we strive to maintain sufficient inventory levels to continue serving current and new patients receiving our medicines for approved indications. Additionally, we expect fewer patient/doctor interactions, fewer visits between our representatives and health care providers and the potential inability of patients to access hospitals or infusion centers (voluntarily or involuntarily) which may result in a reduction in future sales. However, we are proactively engaging with healthcare professionals virtually and through
enhanced digital channels in an effort to mitigate this risk. We have observed that the new patient productivity and initiation queue has decreased since the COVID-19 outbreak, particularly in neurological indications.
Cost of Sales (exclusive of amortization of purchased intangible assets)
Cost of sales includes manufacturing costs, actual and estimated royalty expenses associated with sales of our products, and amortization of licensing rights.
The following table summarizes cost of sales for the three and nine months ended September 30, 2020 and 2019:
| | | | | |
| 2020 Cost of Sales |
| 2019 Cost of Sales |
| ● | Cost of sales as a percentage of net product sales |
Cost of sales as a percentage of net product sales was 9.1% and 7.5% for the three months ended September 30, 2020 and 2019, respectively, and 9.0% and 7.8% for the nine months ended September 30, 2020 and 2019.
The increase in cost of sales as a percentage of net product sales for the three months ended September 30, 2020 as compared to the same period in 2019, was primarily associated with the amortization of the ANDEXXA inventory fair value step-up recognized in purchase accounting for Portola.
The increase of cost of sales as a percentage of net product sales for the nine months ended September 30, 2020 as compared to the same period in 2019, was primarily due to the amortization of the ANDEXXA inventory fair value step-up recognized in purchase accounting for Portola, inventory obsolescence reserves recorded, and higher period costs related to COVID-19 manufacturing capacity levels.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
As a result of the Portola acquisition completed on July 2, 2020, we expect cost of goods sold to increase in future periods as compared to prior periods due to the amortization of ANDEXXA inventory fair value step-up adjustments.
Research and Development Expense
| | | | | |
| 2020 Research and Development Expense (R&D) |
| 2019 Research and Development Expense (R&D) |
| ● | R&D as a % of net product sales |
Our research and development expense includes personnel, facility and direct costs associated with the research and development (R&D) of our product candidates, as well as product development costs.
R&D expenses are comprised of costs paid for clinical development, product development and discovery research, as well as costs associated with certain strategic licensing agreements and R&D-related asset purchase agreements which we have entered into with third parties. Clinical development costs are comprised of costs to conduct and manage clinical trials related to product candidates. Product development costs are those incurred in performing duties related to manufacturing development and regulatory functions, including manufacturing of material for clinical and research activities and other administrative costs incurred during product development. Discovery research costs are incurred in conducting laboratory studies and performing preclinical research for other uses of our current products and other new product candidates. Upfront payments include upfront payments related to strategic licensing agreements and R&D-related asset purchase agreements. Subsequent milestone payments incurred under such agreements which relate to R&D activities are classified as clinical, discovery or product development costs based on the nature of the underlying milestone event. Clinical development costs have been accumulated and allocated to each of our programs, while product development and discovery research costs have not been allocated.
Other R&D expenses consist of costs to compensate personnel, to maintain our facilities and equipment, and other occupancy costs associated with our research and development efforts. These costs relate to efforts for our clinical and preclinical candidates, our product development and our discovery research efforts. These costs have not been allocated directly to each program.
The following graph provides information regarding R&D expenses for the three and nine months ended September 30, 2020 and 2019:
| | | | | | | | | | | |
| Clinical Development | | Discovery |
| Product Development | | Payroll and Benefits |
| Upfront Payments | | Facilities and Other |
For the three months ended September 30, 2020, the increase in research and development expense, as compared to the same period in the prior year, was primarily related to the following:
•Increase of $53.9 in clinical development primarily driven by increased clinical expenses related to ALXN1210 for multiple ongoing studies; shared expenses related to investments in enhanced systems and processes to support our clinical trial expansion; and clinical expenses related to assets acquired from Achillion and Portola in 2020. See chart below for additional details by program.
•Increase of $16.3 in payroll and benefits primarily related to headcount increases.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
•Decrease of $30.1 in upfront payments relating to the license payment made during the third quarter 2019 in connection with the arrangement we entered into with Eidos. No upfront payments related to licensing arrangements were made during the three months ended September 30, 2020.
For the nine months ended September 30, 2020, the increase in research and development expense, as compared to the same period in the prior year, was primarily related to the following
•Increase of $105.6 in clinical development primarily driven by increased clinical expenses related to ALXN1210 for multiple ongoing studies; shared expenses related to investments in enhanced systems and processes to support our clinical trial expansion; and clinical expenses related to assets acquired from Achillion and Portola in 2020. See chart below for additional details by program.
•Increase of $40.9 in payroll and benefits primarily related to headcount increases.
•Decrease of $76.3 in upfront payments relating to the license payments made during the nine months ended September 30, 2019 in connection with the arrangements we entered into with Zealand Pharma A/S, Affibody AB, and Eidos. No upfront payments related to licensing arrangements were made during the nine months ended September 30, 2020.
The following graph summarizes R&D expenses related to our clinical development programs. Please refer to "Clinical Development Programs" above for a description of certain of these programs:
The successful development of our drug candidates is uncertain and subject to a number of risks. We cannot guarantee that results of clinical
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
trials will be favorable or sufficient to support regulatory approvals for any of our product development programs. We could decide to abandon development or be required to spend considerable resources not otherwise contemplated. For additional discussion regarding the risks and uncertainties regarding our research and development programs, please refer to Item 1A “Risk Factors” in this Quarterly Report on Form 10-Q.
Selling, General and Administrative Expense
| | | | | |
| 2020 Selling General and Administrative Expense (SG&A) |
| 2019 Selling General and Administrative Expense (SG&A) |
| ● | SG&A as a % of net product sales |
Our selling, general and administrative expense includes commercial and administrative personnel, corporate facility and external costs required to support the marketing and sales of our commercialized products. These selling, general and administrative costs include: corporate facility operating expenses and depreciation; marketing and sales operations in support of our products; human resources; finance, legal, information technology and support personnel expenses; and other corporate costs such as telecommunications, insurance, audit, government affairs and our global corporate compliance program.
The graph below provides information regarding selling, general and administrative expense:
| | | | | |
| Salary, benefits and other labor expense |
| External selling, general and administrative expense |
For the three months ended September 30, 2020 as compared to the three months ended September 30, 2019, the increase of $34.9 in selling, general and administrative expense, was primarily attributable to an increase in salary, benefits and other labor expenses related to increases in headcount offset by decreases in travel and entertainment (T&E) expenses as a result of the COVID-19 pandemic.
For the nine months ended September 30, 2020 as compared to the nine months ended September 30, 2019, the increase of $75.4 in selling, general and administrative expense, was primarily related to the following:
•Increase in salary, benefits and other labor expenses of $53.7, primarily related to increases in headcount offset by decreases in T&E expenses as a result of the COVID-19 pandemic.
•Increase in external selling, general and administrative expense of $21.7, primarily related to increased costs relating to businesses acquired. In addition, there was an increase of $21.5 in litigation charges recorded during the first quarter 2020 in connection with legal proceedings, offset by a decrease of $22.0 in professional services.
Amortization of Purchased Intangible Assets
For the three months ended September 30, 2020 and 2019, we recorded $53.1 and $75.6, respectively, and $200.5 and $235.7 for the nine months ended September 30, 2020 and 2019, respectively, in amortization expense related to
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
purchased intangible assets. Amortization expense is primarily associated with intangible assets related to STRENSIQ, KANUMA and ANDEXXA.
The decrease in amortization of purchased intangible assets for the three and nine months ended September 30, 2020, as compared to the same periods in 2019, was primarily due to a decrease in amortization associated with the KANUMA and STRENSIQ intangible assets, both of which are explained below. This decrease was partially offset by an increase in amortization associated with the ANDEXXA intangible asset acquired in connection with the Portola acquisition, which closed during the third quarter 2020.
During the second quarter 2020, we recorded an impairment charge of $2,042.3 to write-down the KANUMA intangible asset to fair value. The lower asset value is contributing to lower amortization expense for the three and nine months ended September 30, 2020 as compared to the three and nine months ended September 30, 2019.
During the third quarter 2019, the U.S. patent term extension to a composition of matter patent for STRENSIQ was granted, which resulted in an increase in the estimated useful life of the STRENSIQ intangible asset and is contributing to lower amortization for the three and nine months ended September 30, 2020 as compared to the three and nine months ended September 30, 2019.
We expect amortization for finite-lived intangible assets to decrease in future periods as compared to prior periods as a result of the KANUMA impairment charge recognized during the second quarter of 2020, offset by amortization expense associated with the ANDEXXA intangible asset.
Change in Fair Value of Contingent Consideration
For the three and nine months ended September 30, 2020, the expense resulting from the change in fair value of contingent consideration associated with our business combinations was $23.4 and $45.0, respectively, as compared to $29.8 and $7.2 for the three and nine months ended September 30, 2019. The change in the fair value of
contingent consideration will fluctuate based on the timing of recognition of changes in the probability of achieving and the expected timing of milestone payments in connection with our business combinations.
For the three and nine months ended September 30, 2020 changes in the fair value of contingent consideration expense reflected changes in the expected timing and probability of achieving contingent milestone payments, and the interest component of contingent consideration related to the passage of time. For the three and nine months ended September 30, 2019, changes in the fair value of contingent consideration expense reflected changes in the expected timing of achieving contingent milestone payments and the interest component of contingent consideration related to the passage of time.
Acquisition-related Costs
For the three and nine months ended September 30, 2020, we recorded $63.0 and $105.7, respectively, of acquisition-related costs in connection with the Achillion and Portola acquisitions. Acquisition-related costs consist primarily of transaction costs, costs associated with the accelerated vesting of equity awards previously granted to Achillion and Portola employees, and Achillion and Portola employee separation costs. No
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
acquisition-related costs were incurred during the three and nine months ended September 30, 2019.
Impairment of Intangible Assets
For the nine months ended September 30, 2020, we recorded impairment charges of $2,053.3, relating to ACHN-4471 (ALXN2040) and primarily KANUMA. No impairment charges were incurred during the three months ended September 30, 2020 and 2019 and nine months ended September 30, 2019.
During the quarter ended June 30, 2020, based on continued challenges expanding patient growth and new alternative commercial opportunities, the Company revised its strategic view of KANUMA and determined that we have exhausted commercially viable initiatives related to KANUMA and will have difficulty expanding patient growth over the long term as we focus on promoting other commercial programs and growing our pipeline. As a result, we no longer expect to increase the number of KANUMA patients in the long term at the rate previously assumed. This determination resulted in reduced cash flow projections for KANUMA, which indicated that the related intangible asset value was not fully recoverable on an undiscounted cash flows basis. On June 30, 2020, the Company utilized market participant assumptions to determine its best estimate of the fair value of the intangible asset related to KANUMA that, when compared with its related carrying value, resulted in an impairment charge of $2,042.3.
During the quarter ended June 30, 2020, we recognized an impairment charge of $11.0 to write off the cost basis of our ACHN-4471 (ALXN2040) acquired in-process research and development asset due to clinical results received during the quarter.
Restructuring Expenses
During the third quarter 2020, we initiated restructuring activities primarily within our commercial organization as part of an initiative intended to redefine our operating model. The actions are intended to reallocate resources necessary to align our organization with our diversifying portfolio of new products and strategic objectives, and will include
investments in digital capabilities, technologies and solutions to support a more virtual and digital customer experience and tailored to the markets in which we operate.
The actions are expected to be substantially completed by the end of 2021, with the cumulative pretax costs to be incurred by the Company to implement the program estimated to be approximately $25.0. We expect that the pretax costs will primarily result in cash outlays, as the costs primarily relate to employee separation expenses.
In the first quarter 2019, we initiated corporate restructuring activities to re-align our international commercial organization through re-prioritization of certain geographical markets and to implement operational excellence through strategic reallocation of resources. Actions under the previous restructuring programs have been completed.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Other Income and (Expense)
The following table provides information regarding other income and expense:
| | | | | |
| Investment (expense) Income |
| Interest Expense |
| Other Income and (expense) |
For the three and nine months ended September 30, 2020, we recognized investment income of $11.5 and $47.8, respectively, primarily related to the recognition of gains of $8.6 and $35.0, respectively, on our strategic equity investments recorded at fair value. Gains recorded during the three months ended September 30, 2020 on our strategic equity investments were primarily driven by an unrealized gain from our equity investment in Inozyme Pharma (Inozyme). Gains recorded during the nine months ended September 30, 2020 on our strategic equity investments were primarily driven by an unrealized gain from our Inozyme equity investment and a realized gain from our Portola equity investment which was derecognized and included in the fair value of consideration transferred in connection with the Portola acquisition. For the three and nine months ended September 30, 2019, we recognized investment income of $23.0 and $50.6, respectively, primarily related to the recognition of unrealized gains of $12.0 and $20.6, respectively, on our strategic equity investments recorded at fair value, with the largest component related to our Moderna Therapeutics Inc. equity investment, which was sold in the fourth quarter of 2019.
For the three and nine months ended September 30, 2020, interest expense was $27.6
and $77.0, respectively as compared to $17.9 and $56.1 for the three and nine months ended September 30, 2019. The increase in interest expense for the three and nine months ended September 30, 2020 as compared to the same periods in the prior year is primarily due to an increase in the average interest rates associated with our interest rate swaps, partially offset by lower term loan borrowings and lower interest rates, which are based on market rates, on the unhedged portion of our outstanding term loan.
Income Taxes
| | | | | |
| 2020 Income tax benefit (expense) |
| 2019 Income tax benefit (expense) |
| ● | Effective tax rate |
During the three and nine months ended September 30, 2020, we recorded an income tax expense (benefit) of $88.8 and $(89.2) and an effective tax rate of 13.3% and 413.0%, respectively, compared to an income tax expense of $67.9 and $61.5 and an effective tax rate of 12.7% and 3.9%, respectively, for the three and nine months ended September 30, 2019.
Income tax (benefit) expense is attributable to the U.S. federal, state and foreign income taxes on our profitable operations.
During the second quarter 2020, we recognized an impairment charge of $2,042.3 related to the KANUMA intangible asset, resulting in a deferred tax benefit of $377.3. See Note 5, Intangible Assets and Goodwill, for additional information on the impairment charge. These deferred tax benefits increased the effective tax rate for the nine months
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
ended September 30, 2020 by approximately 399.6%.
The income tax expense for the nine months ended September 30, 2019 includes one-time tax benefits recorded during the first quarter 2019, in connection with the future integration of intellectual property of Wilson Therapeutics into the Alexion corporate structure. The deferred tax benefits included $95.7 and $30.3 associated with a tax election made with respect to intellectual property of Wilson Therapeutics and a valuation allowance release and corresponding recognition of net operating losses, respectively. These deferred tax benefits decreased the effective tax rate for the nine months ended September 30, 2019 by approximately 8.0%. Additionally, during the third quarter 2019, we completed a comprehensive analysis of our current and prior year estimates related to our foreign-derived intangible income based on additional guidance provided in the proposed regulations issued by the U.S. Treasury Department in 2019. The analysis resulted in income tax benefits of approximately $29.9, including $17.0 related to prior year estimated tax benefits, which were recorded as a change in estimate in income tax expense in our consolidated statements of operations during the three and nine months ended September 30, 2019. These tax benefits decreased the effective tax rate for the three and nine months ended September 30, 2019 by approximately 5.6% and 1.9%, respectively.
In April 2020 we became aware of a European withholding tax regulation that could be interpreted to apply to certain of our previous intra-group transactions. We continue to evaluate whether the interpretation of this regulation applies to our facts and circumstances, and, based on our preliminary analysis, we recorded an immaterial reserve related to this matter during the second quarter of 2020.
In August 2020, we received a notice of examination from the Dutch Tax Authorities (“DTA”) regarding certain matters relating to our 2014 through 2017 tax years. Discussions with the DTA are currently ongoing and, at Alexion’s request, we have expanded these discussions to include the 2018 and 2019 tax years as well. We have not received any final findings on any of these tax years, nor have we received any proposed adjustments or any tax assessments related to any of the tax years under discussion with the DTA. Accordingly, we have not recorded any related reserves and an estimate of the possible loss or range of loss, if any, cannot be made at this time.
During the third quarter of 2020, the U.S. Treasury Department released certain proposed and final regulations which were originally enacted under the 2017 Tax Act. The impact of these regulations did
not have a material impact on our financial condition and results of operations.
In 2017, the Internal Revenue Service (IRS) commenced an examination of our U.S. income tax returns for 2015. During the second quarter of 2020 we received a Revenue Agent Report (RAR) and held discussions with the IRS regarding a proposed adjustment related to the valuation of certain intellectual property that was contributed into our captive partnership during 2015. The Company agreed with the adjustment outlined in the RAR and recognized a previously unrecognized tax benefit in the second quarter of 2020 that did not result in a significant impact to the financial statements. The IRS concluded its examination during the third quarter 2020 without additional adjustments.
We continue to benefit from a reduced tax rate as a result of our centralized global supply chain and technical operations in Ireland.
We continue to maintain a valuation allowance against certain other deferred tax assets where realization is not certain. We periodically evaluate the likelihood of realizing deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Financial Condition, Liquidity and Capital Resources
The following table summarizes the components of our financial condition as of September 30, 2020 and December 31, 2019:
| | | | | | | | | | | |
| September 30, 2020 | December 31, 2019 | $
Change |
| Cash and cash equivalents | $ | 2,268.0 | | $ | 2,685.5 | | $ | (417.5) | |
Marketable securities | $ | 28.9 | | $ | 64.0 | | $ | (35.1) | |
Long-term debt (includes current portion & revolving credit facility) | $ | 2,601.8 | | $ | 2,514.5 | | $ | 87.3 | |
| | | |
| Current assets | $ | 5,067.8 | | $ | 5,076.4 | | $ | (8.6) | |
| Current liabilities | $ | 1,331.0 | | $ | 1,194.3 | | $ | 136.7 | |
| Working capital | $ | 3,736.8 | | $ | 3,882.1 | | $ | (145.3) | |
The aggregate decrease in cash and cash equivalents and marketable securities of $452.6 at September 30, 2020 as compared to December 31, 2019 was primarily attributable to $2,023.0 of cash utilized to fund the Achillion and Portola acquisitions, net of $480.9 of cash and cash equivalents and marketable securities acquired in the transactions and net of $196.9 of cash paid to settle preexisting debt held by Portola. Additionally, the decrease is due to cash utilized to repurchase shares of common stock, payments on our term loan facility, purchases of property, plant, and equipment and strategic equity investments, partially offset by cash generated from operations and proceeds from the sale of available-for-sale debt securities.
Excluding the impact of any significant future asset acquisitions, licenses or collaboration agreements, we expect our annual operating expenses to increase as a percentage of sales in 2020 as compared to 2019, primarily due to the impairments of intangible assets of $2,053.3 recorded during the second quarter 2020. We also expect reduced capital investment in 2020 as compared to 2019. We anticipate that cash generated from operations and our existing available cash, cash equivalents and marketable securities should provide us adequate resources to fund our operations as currently planned for at least the next twelve months.
We have financed our operations and capital expenditures primarily through positive cash flows from operations. We expect to continue to be able to fund our operations, including principal and interest payments on our Amended and Restated Credit Agreement, royalty-based debt and contingent payments associated with our in-licenses and acquisitions principally through our cash flows from
operations. We may, from time to time, also seek additional funding through a combination of equity or debt financings or from other sources, if necessary for future acquisitions or other strategic purposes. New sources of financing through equity and/or debt financing(s), especially in light of increased volatility within the global financial markets as a result of the COVID-19 pandemic, may not always be available on acceptable terms, or at all, and we may be required to obtain certain consents in connection with completing such financings.
Financial Instruments
Until required for use in the business, we may invest our cash reserves in money market funds, bank deposits, and high quality marketable debt securities in accordance with our investment policy. The stated objectives of our investment policy are to preserve capital, provide liquidity consistent with forecasted cash flow requirements, maintain appropriate diversification and generate returns relative to these investment objectives and prevailing market conditions.
Financial instruments that potentially expose us to concentrations of credit risk are cash equivalents, marketable securities, accounts receivable and our derivative contracts. At September 30, 2020, four customers accounted for 65.6% of the accounts receivable balance, with these individual customers accounting for 11.2% to 21.5% of the accounts receivable balance. At December 31, 2019, four customers accounted for 66.9% of the accounts receivable balance, with these individual customers accounting for 11.6% to 20.3% of the accounts receivable balance.
For the three months ended September 30, 2020, three customers accounted for 56.1% of our net product sales with these individual customers accounting for 14.3% to 16.2% of our net product sales. For the nine months ended September 30, 2020, four customers accounted for 58.2% of our net product sales with these individual customers accounting for 10.1% to 17.0% of our net product sales. For the three months ended September 30, 2019, three customers accounted for 46.6% of our net product sales with these individual customers accounting for 14.2% to 16.7% of our net product sales. For the nine months ended September 30, 2019, three customers accounted for 45.7% of our net product sales with these individual customers accounting for 13.8% to 16.9% of our net product sales.
We continue to monitor economic conditions, including volatility associated with international economies and the COVID-19 pandemic, and the associated impacts on the financial markets and our business. Substantially all of our accounts receivable are due from wholesale distributors, public hospitals
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
and other government entities. We monitor the financial performance of our customers so that we can appropriately respond to changes in their credit worthiness. We operate in certain jurisdictions where weakness in economic conditions can result in extended collection periods. We continue to monitor these conditions and assess their possible impact on our business. As a result of the COVID-19 pandemic, we have experienced an increase in requests for extended payment terms with certain customers. To date, we have not experienced any significant losses with respect to collection of our accounts receivable and do not currently anticipate any material credit losses on our accounts receivable as a result of the pandemic.
We manage our foreign currency transaction risk and interest rate risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. As of September 30, 2020, we had foreign exchange forward contracts with notional amounts totaling $2,292.0. These outstanding foreign exchange forward contracts had a net fair value liability of $13.0, of which $12.1 is included in other current assets and other noncurrent assets and $25.1 is included in other current liabilities and other noncurrent liabilities. As of September 30, 2020, we had interest rate swap contracts with notional amounts totaling $1,750.0. These outstanding interest rate swap contracts had a net fair value liability of $103.2 included in other current liabilities and other noncurrent liabilities. The counterparties to these contracts are large domestic and multinational commercial banks, and we believe the risk of nonperformance is not material.
As of September 30, 2020, our financial assets and liabilities were recorded at fair value. We have classified our financial assets and liabilities as Level 1, 2 or 3 within the fair value hierarchy. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Our Level 1 assets consist of mutual fund investments and equity securities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Our Level 2 assets consist primarily of money market funds, equity securities subject to holding period restrictions and derivative contracts. Our Level 2 liabilities consist also of derivative contracts. Level 3 inputs are unobservable inputs based on our own assumptions used to measure assets and liabilities at fair value. Our Level 3 liabilities consist of contingent consideration related to business acquisitions and derivative liabilities associated with other contingent payments.
Business Combinations and Contingent Consideration Obligations
On July 2, 2020, we completed the acquisition of Portola. Under the terms of the agreement, we acquired all outstanding common stock of Portola for $18.00 per share, or an aggregate of approximately $1,380.8, including the settlement of certain of Portola's outstanding equity awards but excluding shares of Portola stock held by Alexion at closing. The acquisition was funded with cash on hand. In connection with the acquisition, we also paid $196.9 to settle certain debt held by Portola that was subject to preexisting change of control provisions. The repayment of Portola's debt was funded with cash acquired from Portola. Additionally, we assumed royalty-based debt which requires repayment through tiered royalties on future net worldwide sales of ANDEXXA.
On January 28, 2020, we completed the acquisition of Achillion. Under the terms of the agreement, we acquired all outstanding common stock of Achillion for $6.30 per share, or an aggregate of $926.2, inclusive of the settlement of Achillion's outstanding equity awards. The acquisition was funded with cash on hand. The transaction includes the potential for additional consideration in the form of non-tradeable contingent value rights (CVRs), which will be paid to Achillion shareholders if certain clinical and regulatory milestones are achieved within specified periods. These include $1.00 per share for the U.S. Food and Drug Administration (FDA) approval of danicopan and $1.00 per share for the initiation of a Phase III clinical trial in ACH-5228.
At September 30, 2020, the purchase agreements for our business combinations, including Achillion, include contingent payments totaling up to $905.6 that will become payable if and when certain development and commercial milestones are achieved. Of these milestone amounts, $670.6 of the contingent payments relate to development and regulatory milestones and $235.0 of the contingent payments relate to commercial milestones, respectively. We do not expect these amounts to have a significant impact on our liquidity in the near-term, and, during the next 12 months, we do not expect to make milestone payments associated with our prior business combinations.
As additional future payments become probable, we will evaluate methods of funding payments, which could be made from available cash and marketable securities, cash generated from operations, or proceeds from the sale of equity securities or debt.
Asset Acquisitions and In-License Agreements
In January 2019, we entered into an agreement with Caelum, a biotechnology company that is developing CAEL101 for AL amyloidosis. Under the
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
terms of the agreement, we acquired a minority equity interest in preferred stock of Caelum and an exclusive option to acquire the remaining equity in Caelum based on Phase II data, for pre-negotiated economics. We paid $30.0 in the first quarter 2019 and agreed to pay up to an additional $30.0 in contingent development milestones prior to the exercise the option to acquire the remaining equity in Caelum. These contingent payments meet the definition of a derivative liability and were initially recorded at fair value of $27.1. We allocated the total consideration of $57.1, inclusive of the fair value of the contingent payments, to the equity investment in Caelum and the option to acquire the remaining equity in Caelum based on the relative fair values of the assets. Following discussions with the FDA, Caelum changed its clinical development plan for CAEL-101 in the fourth quarter 2019. A Phase II trial for CAEL-101 subsequently commenced during the first quarter of 2020 and met its primary objectives, supporting the safety and tolerability of CAEL-101 and confirmed the dose and regimen to be adopted for the Phase III studies. In September 2020, Alexion and Caelum announced the initiation of the CARES program. This includes two parallel Phase III trials, the primary objective of these trials are to evaluate the survival benefits of CAEL-101. In December 2019, we amended the terms of the agreement with Caelum to modify the option to acquire the remaining equity in Caelum based on data from the modified Phase II/III trials. The amendment also modified the development-related milestone events associated with the initial $30.0 in contingent payments, provided for an additional $20.0 in upfront funding, which we accrued as of December 31, 2019 and paid during the first quarter 2020, as well as funding of $60.0 in exchange for an additional equity interest at fair value upon achievement of a specific development-related milestone event. The agreement with Caelum also provides for additional payments, in the event Alexion exercises the purchase option, for up to $500.0, which includes an upfront option exercise payment and potential regulatory and commercial milestone payments. During the second quarter 2020, we paid an aggregate of $15.0 of contingent payments to Caelum related to two development-based milestones and during the third quarter 2020, we paid a $15.0 contingent payment to Caelum related to another development-based milestone.
In March 2019, we entered into an agreement with Zealand which provides us with exclusive worldwide licenses, as well as development and commercial rights, for subcutaneously delivered preclinical peptide therapies directed at up to four complement pathway targets. Pursuant to the agreement, Zealand will lead joint discovery and research efforts through the preclinical stage, and Alexion will lead development efforts beginning with
the investigational new drug filing and Phase I studies. In addition to the agreement, we made an equity investment in Zealand (see Note 10, Other Investments). Under the terms of the agreement, we made an upfront payment of $40.0 for an exclusive license to the lead target and the equity investment, as well as for preclinical research services to be performed by Zealand in relation to the lead target. As of September 30, 2020, we could be required to pay up to $610.0, for the lead target, upon the achievement of specified development, regulatory and commercial milestones, as well as royalties on commercial sales. In addition, we could be required to pay up to an additional $115.0 in development and regulatory milestones if both a long-acting and short-acting product are developed with respect to the lead target. Each of the three subsequent targets can be selected for an option fee of $15.0 and has the potential for additional development, regulatory and commercial milestones, as well as royalty payments, at a reduced price to the lead target.
In September 2019, we entered into an agreement with Eidos through which Alexion obtained an exclusive license to develop and commercialize AG10 in Japan. AG10 is a small molecule designed to treat the root cause of ATTR and is currently in a Phase III study in the U.S. and Europe for ATTR-CM. In addition, we made an equity investment in Eidos (see Note 10, Other Investments). Under the terms of the agreement, we made an upfront payment of $50.0 for the exclusive license to AG10 in Japan and the equity investment. As of September 30, 2020, we could also be required to pay $30.0 upon achievement of a Japanese-based regulatory milestone as well as royalties on commercial sales.
In October 2018, we entered into a collaboration agreement with Dicerna that provides us with exclusive worldwide licenses and development and commercial rights for two preclinical RNA interference (RNAi) subcutaneously delivered molecules for complement-mediated diseases, as well as an exclusive option for other preclinical RNAi molecules for two additional targets within the complement pathway. In addition to the collaboration agreement, we made an equity investment in Dicerna. Under the terms of the agreements, we made an upfront payment of $37.0 for the exclusive licenses and the equity investment. In December 2019, we exercised our option for exclusive rights to two additional targets within the complement pathway under an existing agreement with Dicerna, which expands our existing research collaboration and license agreement with Dicerna to include a total of four targets within the complement pathway. In connection with the option exercise, we paid Dicerna $20.0, which we recorded as research and development expense in the fourth quarter 2019. As of September 30, 2020, excluding accrued milestones, we could be required to pay up
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
to $604.1 for amounts due upon the achievement of specified research, development, regulatory and commercial milestones on the four licensed targets, as well as royalties on commercial sales.
In December 2017, we entered into a collaboration and license agreement with Halozyme Therapeutics, Inc. that allows us to use drug-delivery technology in the development of subcutaneous formulations for our portfolio of products for up to four targets. Under the terms of the agreement, we made an upfront payment of $40.0 for an exclusive license to two of the four potential targets and due to the early stage of the assets we are licensing, we recorded an expense for the upfront payment during the fourth quarter 2017. During the second quarter 2020, we forfeited our rights to one of the two targets we initially licensed. As of September 30, 2020, we could be required to pay up to $155.0 for the remaining licensed target upon achievement of specified development, regulatory and sales-based milestones, as well as royalties on commercial sales. Each of the two subsequent targets can be licensed for an option fee of $8.0, with contingent payments of up to $160.0 per target, subject to development, regulatory and commercial milestones, as well as royalties on commercial sales.
In connection with our prior acquisition of Syntimmune, Inc., a clinical-stage biotechnology company developing an antibody therapy targeting the FcRn, we could be required to pay up to $800.0 upon the achievement of specified development, regulatory and commercial milestones, of which $130.0 is specific to the subcutaneous formulation.
In addition, excluding accrued milestones, as of September 30, 2020, we have other license agreements under which we may be required to pay up to an additional $114.0 for currently licensed targets, if certain development, regulatory and commercial milestones are met, including up to $71.5 for the development of cerdulatinib in multiple indications pursuant to an in-licensing agreement with Astellas Pharma, Inc. which was assumed through the acquisition of Portola in the third quarter 2020. Additional amounts may be payable if we elect to acquire licenses to additional targets, as applicable, under the terms of these agreements.
We do not expect the payments associated with milestones under our asset acquisitions and licensing agreements to have a significant impact on our liquidity in the near-term. During the next 12 months, we may make milestone payments related to these arrangements of approximately $78.3, excluding milestones which were accrued as of September 30, 2020.
As additional future payments become probable, we will evaluate methods of funding payments, which could be made from available cash and marketable
securities, cash generated from operations or proceeds from the sale of equity securities or debt.
Operating and Financing Lease Liabilities
Operating and financing lease liabilities are recorded at lease commencement based on the present value of fixed, or in substance fixed, lease payments over the expected lease term. Lease liabilities are amortized over the lease term.
As of September 30, 2020, we have $275.5 of total financing and operating lease liabilities recorded on our condensed consolidated balance sheets. The total undiscounted lease commitments as of September 30, 2020 were $328.3, of which, $10.0 is payable during the remainder of 2020. We do not expect the payments associated with the maturity of lease liabilities to have a significant impact on our liquidity in the near-term.
Long-term Debt
On June 7, 2018, we entered into an Amended and Restated Credit Agreement (the Credit Agreement), with Bank of America, N.A. as Administrative Agent. The Credit Agreement amended and restated our credit agreement dated as of June 22, 2015 (the Prior Credit Agreement).
The Credit Agreement provides for a $1,000.0 revolving credit facility and a $2,612.5 term loan facility. The revolving credit facility and the term loan facility mature on June 7, 2023. Beginning with the quarter ending June 30, 2019, we are required to make payments of 5.00% of the original principal amount of the term loan facility annually, payable in equal quarterly installments.
As of September 30, 2020, we had $2,416.6 outstanding on the term loan. We had no outstanding borrowings under the revolving credit facility as of September 30, 2020. As of September 30, 2020, we had open letters of credit of $1.0 that offset our availability in the revolving credit facility.
In connection with our acquisition of Portola during the third quarter 2020, we assumed royalty-based debt relating to a royalty sales agreement Portola had entered into with HealthCare Royalty Partners (HCR) whereby HCR acquired a tiered royalty interest in future worldwide net sales of ANDEXXA. Portola received $50.0 upon closing of the agreement in February 2017 and an additional $100.0 following the U.S. regulatory approval of ANDEXXA in May 2018. Tiered royalties ranging from 4.2% to 8.5% are required to be paid to HCR based on net worldwide sales of ANDEXXA. The applicable rate decreases as worldwide net annual sales levels increase above defined thresholds. Total potential royalty payments are capped at 195% of the funding received less certain transaction expenses, or $290.6.
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
As of September 30, 2020, the royalty-based debt carrying value of $185.2 includes accrued interest of $3.2, net of unamortized debt discount of $90.0. The maximum remaining royalty payments are capped at $275.2, as of September 30, 2020.
Manufacturing Obligations
We have supply agreements with Lonza relating to the manufacture of SOLIRIS, STRENSIQ, ULTOMIRIS and ANDEXXA which requires payments to Lonza at the inception of contract and upon the initiation and completion of product manufactured. On an ongoing basis, we evaluate our plans for future levels of manufacturing by Lonza, which depends upon our commercial requirements and the progress of our clinical development programs.
We have various agreements with Lonza, with remaining total non-cancellable commitments of approximately $1,432.3 through 2031. Certain commitments may be canceled only in limited circumstances. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of SOLIRIS that was manufactured at Alexion Rhode Island Manufacturing Facility (ARIMF) prior to its sale and a payment with respect to sales of SOLIRIS manufactured at Lonza facilities. We also pay Lonza a royalty on the sales of ULTOMIRIS.
In addition to Lonza, we have non-cancellable commitments of approximately $78.6 through 2021 with other third party manufacturers.
Taxes
We have recorded tax on the undistributed earnings of our controlled foreign corporation (CFC) subsidiaries. To the extent CFC earnings may not be repatriated to the U.S. as a dividend distribution due to limitations imposed by law, we have not recorded the related potential withholding, foreign local, and U.S. state income taxes.
Common Stock Repurchase Program
In November 2012, our Board of Directors authorized a share repurchase program. In February
2017, our Board of Directors increased the amount that we are authorized to expend on future repurchases to $1,000.0 under our repurchase program, which superseded all prior repurchase programs. The entire amount authorized pursuant to this February 2017 Board approval has been utilized. On October 22, 2019, the Board of Directors approved a share repurchase authorization of up to $1,000.0. On July 28, 2020, the Board of Directors approved a new share repurchase authorization of up to an additional $1,500.0. The repurchase program does not have an expiration date and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at our discretion. Under the program, we repurchased 0.6 and 3.1 shares of our common stock at a cost of $73.5 and $334.6 during the three months ended September 30, 2020 and 2019, respectively. During the nine months ended September 30, 2020 and 2019, we repurchased 4.2 and 3.5 shares of our common stock at a cost of $434.3 and $383.5, respectively.
Subsequent to September 30, 2020, we repurchased 0.4 shares of common stock under our repurchase program at a cost of $45.2. As of October 27, 2020, there is a total of $2,055.9 remaining for repurchases under the repurchase program.
Cash Flows
The following summarizes our net change in cash and cash equivalents:
| | | | | | | | | | | | | | | | | |
| | Nine months ended September 30, | | $ |
| | 2020 | | 2019 | | Change |
| Net cash provided by operating activities | $ | 2,162.4 | | | $ | 1,567.8 | | | $ | 594.6 | |
| Net cash used in investing activities | (2,018.9) | | | (46.6) | | | (1,972.3) | |
| Net cash used in financing activities | (538.1) | | | (679.4) | | | 141.3 | |
| Effect of exchange rate changes on cash and restricted cash | (0.5) | | | (6.1) | | | 5.6 | |
| Net change in cash and cash equivalents and restricted cash | $ | (395.1) | | | $ | 835.7 | | | $ | (1,230.8) | |
Alexion Pharmaceuticals, Inc.
(amounts in millions, except per share amounts)
Operating Activities
Cash flows provided by operations for the nine months ended September 30, 2020 was $2,162.4 compared to $1,567.8 for the nine months ended September 30, 2019. The increase in cash provided by operating activities was primarily due to cash generated from operations, including the timing of cash receipts, payments and other changes in working capital, during the nine months ended September 30, 2020 as compared to the same period in the prior year. Additionally, the same period in the prior year included upfront payments made in connection with agreements entered into with Zealand, Affibody and Eidos and a payment of a sales-based milestone to the former equity holders of Enobia Pharma Corp.
Investing Activities
Cash flows used in investing activities for the nine months ended September 30, 2020 was $2,018.9 compared to $46.6 for the nine months ended September 30, 2019. The increase in cash used in investing activities as compared to the prior year was primarily due to payments for the acquisition of Achillion and Portola, net of cash acquired, of $2,111.9. Partially offsetting these impacts were decreases in purchases of property, plant and equipment and purchases of strategic equity investments and options during the nine months ended September 30, 2020 as compared to the same period in the prior year.
Financing Activities
Cash flows used in financing activities for the nine months ended September 30, 2020 was $538.1 compared to $679.4 for the nine months ended September 30, 2019. The decrease in cash used for financing activities was primarily due to a decrease in payments on our revolving credit facility, partially offset by an increase of $50.8 in common stock repurchases and an increase of $32.5 in payments on term loan during the nine months ended September 30, 2020 as compared to the same period in the prior year.
Contractual Obligations
Other than potential contingent payments related to our acquisition of Achillion and obligations related to the acquisition of Portola, primarily consisting of $368.1 of manufacturing commitments with Lonza through 2028 and royalty-based debt assumed in connection with the acquisition, there have been no significant changes to the disclosure of payments we have committed to make under our contractual obligations as summarized in our Annual Report on Form 10-K for the twelve months ended December 31, 2019, in the section titled “Management's Discussion and Analysis of Financial Condition and Results of Operations” under the caption “Contractual Obligations.”
Item 3.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
(amounts in millions, except
percentages)
Interest Rate Risk
We have historically invested our cash in a variety of financial instruments, principally money market funds, bank deposits, corporate bonds, municipal bonds, commercial paper and government-related obligations which are subject to interest rate risk and could decline in value if interest rates fluctuate. Our investment portfolio has historically been comprised of marketable debt securities of highly rated financial institutions and investment-grade debt instruments, and we have guidelines to limit the term-to-maturity of our investments. During the second quarter of 2020, we liquidated all of our available-for-sale debt securities to fund the acquisition of Portola. As of September 30, 2020, our investment portfolio primarily consists of money market funds and mutual funds. Based on the type of securities we hold, we do not believe a change in interest rates would have a material impact on our financial statements. If interest rates were to increase or decrease by 1%, the fair value of our investment portfolio would increase (decrease) by an insignificant amount.
On June 7, 2018, we entered into an Amended and Restated Credit Agreement (the Credit Agreement), with Bank of America N.A. as administrative agent. The Credit Agreement amended and restated our credit agreement dated as of June 22, 2015 (the Prior Agreement). Loans under the Credit Agreement bear interest at our option, at either the base rate or a Eurodollar rate, in each case plus an applicable margin. Under the Credit Agreement, the applicable margins on base rate loans range from 0.25% to 1.00% and the applicable margins on Eurodollar loans range from 1.25% to 2.00%, in each case based on our consolidated net leverage ratio (as calculated in accordance with the Credit Agreement).
Changes in interest rates related to the Credit Agreement could have a material effect on our financial statements.
To achieve a desired mix of floating and fixed interest rates on our term loan, we entered into a number of interest rate swap agreements that qualified for and are designated as cash flow hedges. As of September 30, 2020 we had cash flow hedges with aggregate amounts of approximately 72.4% of our current outstanding term loan covering periods over the next twelve months. If interest rates were to increase or decrease by 1%, interest expense over the next year would increase or decrease by $6.1, based on the unhedged portion of our outstanding term loan as of September 30, 2020.
Foreign Exchange Market Risk
Our operations include activities in many countries outside the U.S. As a result, our financial results are impacted by factors such as changes in foreign currency exchange rates or weak economic conditions in the foreign markets where we operate. We have exposure to movements in foreign currency exchange rates, the most significant of which are the Euro, and Japanese Yen, against the U.S. dollar. We are a net receiver of many foreign currencies, and our consolidated financial results benefit from a weaker U.S. dollar and are adversely impacted by a stronger U.S. dollar relative to foreign currencies in which we sell our products.
Our monetary exposures on our balance sheet arise primarily from cash, accounts receivable, and payables denominated in foreign currencies. Approximately 39.6% and 39.2% of our net product sales were denominated in foreign currencies for the three and nine months ended September 30, 2020, and our revenues are also exposed to fluctuations in the foreign currency exchange rates over time. In certain foreign countries, we may sell in U.S. dollar, but our customers may be impacted adversely by fluctuations in foreign currency exchange rates which may also impact the timing and amount of our revenue.
Both positive and negative impacts to our international product sales from movements in foreign currency exchange rates are only partially mitigated by the natural, opposite impact that foreign currency exchange rates have on our international operating expenses. Additionally, we have operations based in Europe and accordingly, our expenses are impacted by fluctuations in the value of the Euro against the U.S. dollar.
We currently have a derivative program in place intended to achieve the following: (1) limit the foreign currency exposure of our monetary assets and liabilities on our balance sheet, using contracts with durations of up to 6 months and (2) hedge a portion of our forecasted product sales (in some currencies), including intercompany sales, and certain forecasted expenses using contracts with durations of up to 60 months. The objective of this program is to reduce the volatility of our operating results due to fluctuation of foreign exchange. This program utilizes foreign exchange forward contracts intended to reduce, not eliminate, the volatility of operating results due to fluctuations in foreign exchange rates.
As of September 30, 2020 and December 31, 2019, we held foreign exchange forward contracts with notional amounts totaling $2,292.0 and $3,078.5, respectively. As of September 30, 2020 and December 31, 2019, our outstanding foreign exchange forward contracts had a net fair value of $(13.0) and $2.8, respectively.
We do not use derivative financial instruments for speculative trading purposes. The counterparties to these foreign exchange forward contracts are large domestic and multinational commercial banks. We believe the risk of counterparty nonperformance is not material.
Based on our foreign currency exchange rate exposures as of September 30, 2020, a hypothetical 10% adverse fluctuation in exchange rates would decrease the fair value of our foreign exchange forward contracts that are designated as cash flow hedges by approximately $109.2. The resulting loss on these forward contracts would be offset by the gain on the underlying transactions and therefore would have minimal impact on future anticipated earnings and cash flows. Similarly, adverse fluctuations in exchange rates that would decrease the fair value of our foreign exchange forward contracts that are not designated as hedge instruments would be offset by a positive impact of the underlying monetary assets and liabilities.
Credit Risk
As a result of our foreign operations, we are exposed to changes in the general economic conditions in the countries in which we conduct business. The majority of our receivables are due from wholesale distributors, public hospitals and other government entities. We monitor the financial performance and creditworthiness of our large customers so that we can properly assess and respond to changes in their credit profile. We continue to monitor these conditions, including the volatility associated with international economies, the COVID-19 pandemic and the relevant financial markets, and assess their possible impact on our business. Although collection of our accounts receivables from certain countries may extend beyond our standard credit terms and while we have begun to see an increase in requests for extended payment terms with certain customers as a result of the COVID-19 pandemic, we have not experienced any significant losses with respect to collection of our accounts receivable and we do not expect any such delays to have a material impact on our financial condition or results of operations.
Item 4.CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (Exchange Act), as of September 30, 2020. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of September 30, 2020, our disclosure controls and procedures were effective to provide reasonable assurance that information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure, and ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms.
Changes in Internal Control over Financial Reporting
Other than the item noted below, there has been no change in our internal control over financial reporting that occurred during the quarter ended September 30, 2020 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
In July 2020, the Company acquired Portola. Management is continuing to integrate the Portola internal controls over financial reporting with those of the Company and intends to exclude the internal controls of Portola from its annual assessment of the effectiveness of the Company’s internal control over financial reporting for 2020.
PART II. OTHER INFORMATION
Item 1.LEGAL PROCEEDINGS.
For a discussion of legal matters as of September 30, 2020, see Note 16, “Commitments and Contingencies,” Contingent Liabilities, within our notes to the condensed consolidated financial statements included in this Quarterly Report on Form 10-Q, which is incorporated into this item by reference.
Item 1A.Risk Factors.
(amounts in millions, except percentages)
You should carefully consider the following risk factors before you decide to invest in Alexion securities and our business, because the risks described below may have a material impact on our business, operating results, financial condition, and cash flows. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations. If any of the following risks actually occurs, our business, financial condition and results of operations could be materially and adversely affected.
Risks Related to Revenue Concentration and Conversion
We depend on revenue from sales of our C5 complement inhibitors and, if we are unable to continue to increase revenues from sales of our C5 complement inhibitors, our business would be materially harmed and our future operating results may be adversely impacted.
Since 2007, our revenue has depended primarily on the sales of SOLIRIS, a C5 complement inhibitor with a 2-week dosing schedule. In December 2018, we obtained our first regulatory approval in the U.S. to sell ULTOMIRIS, a long-acting C5 complement inhibitor, with an 8-week dosing schedule (and we recently obtained approval in the US for a 100mg/mL formulation of ULTOMIRIS). Our C5 complement inhibitors accounted for 85.9% of our total revenues for the fiscal year ended December 31, 2019. Unless we are able to develop or acquire and commercialize new products beyond these C5 complement inhibitors, and/or materially increase sales of STRENSIQ and KANUMA (two additional currently approved products) and ANDEXXA®(another approved product that was obtained through the acquisition of Portola in July 2020), we will remain dependent on sales of SOLIRIS and ULTOMIRIS as a source of our revenue. We expect our revenues for 2020 will continue to depend on our ability to sell our C5 complement inhibitors.
The commercial success of our C5 complement inhibitors and our ability to generate revenue depends on several factors, including: the safety and efficacy of our C5 complement inhibitors; coverage or reimbursement by government or third-party payers for our C5 complement inhibitors; pricing for our complement inhibitors; the analysis by doctors, payers and patients of the cost of our C5 complement inhibitors relative to the perceived benefits; manufacturing and uninterrupted supply; the introduction and success of competing products (including novel products and biosimilars to SOLIRIS); the size of patient populations and the number of
patients diagnosed who may be treated with our C5 complement inhibitors; the impact of legal, administrative, regulatory or legislative developments that impact the price or use of C5 complement inhibitors; and our ability to develop, obtain regulatory approval for and commercialize our C5 complement inhibitors for new indications. Any of these or other factors may cause revenues from sales of our C5 complement inhibitors to decrease, which would harm our business results.
While SOLIRIS and ULTOMIRIS are studied for indications beyond those currently approved by regulatory authorities and ULTOMIRIS is being studied for subcutaneous administration, there is no guarantee that we can obtain regulatory approval or achieve any commercial sales of SOLIRIS or ULTOMIRIS for such other indications or for subcutaneous administration of ULTOMIRIS. Nor can we guarantee that, even if regulatory approval is obtained for such additional indications and routes of administration, physicians and patients will accept SOLIRIS or ULTOMIRIS as a treatment for such indications or means of administration, or that payers will pay for or reimburse the costs of these therapies.
If we are not able to maintain revenues from sales of SOLIRIS and ULTOMIRIS, or such revenues decrease, our operating results would be negatively impacted and our ability to fund research and development, commercialize or acquire new products would be harmed, which would limit our ability to diversify our revenue base and our stock price could be adversely affected. In addition, as a result of having our revenue concentrated in SOLIRIS and ULTOMIRIS, our future revenues and results of operations can be significantly harmed by, among other factors, the introduction of one or more biosimilar products or other competitive products that treat the same indications, adverse developments in the commercialization and sale of our C5 inhibitors or a change in reimbursement policies by payers for the C5 complement inhibitors. For example, a biosimilar has been introduced in Russia and a CD19-directed cytolytic antibody treatment and an IL-6R antibody, were both recently approved for NMOSD patients in the US and certain other jurisdictions.
We aim to facilitate the conversion of patients from SOLIRIS to ULTOMIRIS. If we are unable to achieve our conversion objectives, our business may be harmed. In addition, even if we are successful, due to the pricing of ULTOMIRIS, our revenues may decrease unless we are able to increase the number of patients using our C5 inhibitors.
ULTOMIRIS has been approved for patients with PNH and aHUS in certain jurisdictions, including in the U.S., Europe and Japan.
One of our principal business objectives is to facilitate the conversion of PNH and aHUS patients
from SOLIRIS to ULTOMIRIS. While clinical trials in PNH patients demonstrated that ULTOMIRIS is non-inferior to SOLIRIS at an 8 week dosing interval (compared to a 2 week dosing interval for SOLIRIS), existing patients taking SOLIRIS for PNH or aHUS and their physicians may decline to switch to ULTOMIRIS. If we are unable to facilitate conversion to ULTOMIRIS prior to the loss of intellectual property or regulatory exclusivities for SOLIRIS, our future revenues could be adversely impacted if we were to face biosimilar competition for SOLIRIS.
We have established what we believe is a globally sustainable and durable pricing strategy for ULTOMIRIS that is intended to facilitate such patient conversions (for example, in the U.S. the cost of current labeled maintenance therapy for ULTOMIRIS for adult PNH patients of average weight, represents on an annual basis an approximate 10% decrease relative to the cost of SOLIRIS). However, in the first year of PNH conversion to ULTOMIRIS, due to the loading doses required, there is an approximate 10% premium to the cost of SOLIRIS. We have also priced ULTOMIRIS for patients with aHUS in the U.S. at a cost relative to the cost of SOLIRIS for patients with aHUS in the U.S. that is approximately 30% less on an annual basis for an average adult patient on maintenance therapy (unlike PNH, the cost in the first year of aHUS conversion to ULTOMIRIS is approximately 20% less than the cost of SOLIRIS). If we achieve our goal of facilitating the conversion of patients from SOLIRIS (which accounted for approximately $3,946.4, or 79.1%, of our revenues in 2019) to ULTOMIRIS, due to these discounts we anticipate that U.S. revenue attributable to each patient that converts from SOLIRIS to ULTOMIRIS will decrease. In addition, as a result of the decreased cost for ULTOMIRIS relative to SOLIRIS on a per patient basis, in order to maintain or increase C5 complement inhibitor revenues in the future as we succeed in converting patients from SOLIRIS to ULTOMIRIS, we must increase the total number of patients utilizing SOLIRIS, including gMG and NMOSD patients, and ULTOMIRIS.
Finally, as a result of patient conversion from SOLIRIS to ULTOMIRIS, we expect variability in our revenues in future quarters due to the extended ULTOMIRIS dosing interval and infusion timing which may result in either one or two infusions in a quarter.
Due to the decision to price ULTOMIRIS lower than SOLIRIS on an annual basis, we anticipate U.S. revenues will be unfavorably impacted by the lower annual cost per patient in maintenance years, with the impact more pronounced for aHUS due to the greater decrease in vials for aHUS ULTOMIRIS patients.
Risks Related to the COVID-19 Pandemic
Our business may be adversely affected by the ongoing COVID-19 pandemic.
Our business could be adversely affected by health epidemics in regions where we have operations, sales or other business activities, including regions where we have offices, manufacturing facilities, clinical trial sites and where our third party manufacturers, vendors and suppliers operate and where patients and potential patients are located. The outbreak of a novel strain of virus, which causes the disease called COVID-19, has evolved into a global pandemic. The ultimate impact of the COVID-19 pandemic on our business operations and financial results is highly uncertain and will depend on future developments, which cannot be accurately predicted, including the duration of the pandemic, the ultimate geographic spread of the disease, additional or modified government actions, new information that will emerge concerning the severity and impact of the coronavirus and the actions taken to address its impact, among others.
We have initiated a global Phase III study to investigate ULTOMIRIS in a subset of adults with COVID-19, who are hospitalized with severe pneumonia or acute respiratory distress syndrome (ARDS). The trial will evaluate the impact of ULTOMIRIS on survival, duration of mechanical ventilation, and duration of hospital stay compared to best supportive care. We cannot guarantee that the clinical trial will provide sufficient evidence that ULTOMIRIS is safe and effective for use as a treatment for adult COVID-19 patients hospitalized with severe pneumonia or ARDS. In addition, there can be no guarantee that the FDA or any other regulatory authority will approve ULTOMIRIS as a treatment for COVID-19.
As a result of the COVID-19 pandemic, we expect that we may experience disruptions that could severely impact our business and results of operations, including:
•Government and healthcare policies and federal, state, local or foreign regulations to address the COVID-19 pandemic may adversely affect our sales and revenue. Due to quarantines, travel restrictions, hospital policies and patient concerns regarding exposure to COVID-19, we have observed fewer patient/doctor interactions, we have also noted that the new patient productivity and initiation queue has decreased since the COVID-19 outbreak (particularly in our neurological indications) and our representatives are having fewer in-person visits with health care providers, including for infusion of our products which may affect our sales in the future. A decrease in the demand of our products could cause our cost of goods sold to increase due to expiration of inventory on hand, an increase in manufacturing
overhead allocated to inventory sold and other factors. Our net product sales could also be adversely impacted by the negative effects the COVID-19 pandemic has had on the global economy, which could result in (i) an increased number of patients utilizing our patient access programs to receive free drug due to loss of employer-based health insurance, or other factors impacting their ability to afford our medicines; and (ii) patients increasingly seeking Medicaid coverage for our products, which would lead to higher gross-to-net revenue reductions compared to commercial insurance providers. We are monitoring the impacts on our business of the growth in unemployment and loss of commercial insurance coverage and/or growth in Medicaid with higher discounts.
•Due to financial demands in addressing the COVID-19 pandemic, payors have requested and may continue to request extended credit terms, may extend payment dates beyond those experienced in the past, or may not be able to timely reimburse us for our products or at all, and such actions could have a material adverse impact on our cashflow from operations. Additionally, federal and state governments and foreign jurisdictions where we operate could increase tax rates to offset the economic impact and cost of addressing the COVID-19 pandemic. Any increase in such tax rates could have an adverse impact on our business.
•We believe that the COVID-19 pandemic has had, and will likely continue to have, an impact on various aspects of our clinical programs and trials. For example, our Phase I and Phase II trials for ALXN1830 had been paused due to COVID-19. Patient dosing and study monitoring may be paused, delayed or temporarily halted (and clinical trial re-start schedules may be delayed beyond the dates we anticipated) due to changes in policies at various clinical sites and federal, state, local or foreign laws, rules and regulations, including quarantines or other travel restrictions, prioritization of healthcare resources toward pandemic efforts, including diminished attention of physicians serving as our clinical trial investigators and reduced availability of site staff supporting the conduct of our clinical trials, or other reasons related to the COVID-19 pandemic. If the COVID-19 pandemic continues for an extended period of time, other aspects of our clinical trials may be adversely affected, delayed or interrupted, including, for example, site initiation, patient recruitment and enrollment, availability of
clinical trial materials, and data analysis. Some patients and clinical investigators may not be able to comply with clinical trial protocols and patients may choose to withdraw from our studies, or we may choose to or be required to pause enrollment and/or patient dosing in our ongoing clinical trials in order to preserve health resources and protect trial participants. Any such disruption could negatively impact the results generated in the trial, the development of our pipeline programs and the timing and probability of paying milestones associated with prior acquisitions and active license agreements (which may lead to litigation over milestone payments).
•We currently utilize third parties to, among other things, manufacture our products and product candidates, supply raw materials and consumables, perform quality testing and provide supply chain services. We also manufacture certain of our products and product candidates and perform various services at our manufacturing facilities. If any of these processes or services are adversely impacted by the COVID-19 outbreak, our ability to manufacture and supply our products to patients or manufacture product candidates for our clinical trials and conduct our research and development operations may be materially affected.
•The potential economic and financial impacts of the pandemic, including a deterioration in economic conditions that may negatively impact revenue and our liquidity, increase expenses and result in market capitalization declines, and disruption to our business, may result in the impairment of our long-lived and other assets, including goodwill, intangible assets and equity investments without readily determinable fair values. The impairment of significant assets could have a material impact on our deferred tax assets and liabilities. In addition, any impairment charge would have a negative impact on our financial results in the quarter that the charge is taken, and such charge may be material in amount.
•In accordance with business continuity plans and for the safety of our employees, we have directed most of our personnel to work remotely and we have generally restricted on-site staff to only those personnel and contractors who perform essential activities that must be completed on-site. Our increased reliance on personnel working from home may negatively impact productivity, or disrupt, delay, or otherwise adversely impact
our business. In addition, this could increase our cyber-security risk, create data accessibility concerns, and make us more susceptible to communication disruptions, any of which could adversely impact our business operations.
•While our essential R&D employees have been able to access our laboratory space, if employees and contractors conducting such activities were exposed to or contracted COVID-19, we may be required to restrict access to our laboratory space for an extended period of time as a result. Governmental authorities may also impose restrictions limiting access to our lab space. As a result, this could delay timely completion of preclinical and other R&D activities.
•Health regulatory agencies globally may experience disruptions in their operations as a result of the COVID-19 pandemic. The FDA and other foreign regulatory agencies may have slower response times or be under-resourced to continue to authorize and monitor our clinical trials or review regulatory submissions (or authorize the use of facilities for manufacturing and related services) and, as a result, review, inspection, and other timelines may be materially delayed.
•In addition, a recession or other sustained adverse market event resulting from the spread of COVID-19 could materially and adversely affect our business, the value of our common shares and the availability of credit to operate our business and execute business development transactions. As a result, we may face difficulties raising capital through sales of our common shares, accessing credit to support our business development activities or other capital initiatives or such sales of common stock or credit may only be available on unfavorable terms.
•The trading prices for our common shares and other biopharmaceutical companies have been highly volatile as a result of the COVID-19 pandemic and we expect this volatility may continue.
COVID-19, and the volatile regional and global economic conditions stemming from the pandemic, could also precipitate or aggravate the other risk factors discussed in this Quarterly Report on Form 10-Q, which could materially adversely affect our business, financial condition, results of operations, liquidity, and stock price.
Risks Related to Pricing and Reimbursement
Sales of our products depend on reimbursement by payers and these payers are subject to pressures to contain costs.
Our commercial success depends on setting a price for our products that will enable us to obtain reimbursement at anticipated levels. Our products are significantly more expensive than traditional drug treatments and almost all patients require governmental payers and/or private third-party payers to pay all or a portion of the cost of our products. There is a significant trend in the health care industry by public and private payers to contain or reduce their costs, including by taking the following steps, among others: decreasing the portion of costs payers will cover, ceasing to provide adequate payment for certain products or not covering certain products at all or requiring use of therapies that are less expensive. If payers implement any of the foregoing (or undertake similar measures) with respect to our products, it would have an adverse impact on our revenue and results of operations.
Our ability to set the price for our products varies significantly from country to country, including in those countries where pricing, coverage, reimbursement or funding of prescription drugs are subject to governmental control. We may be unable to timely or successfully negotiate coverage, pricing and reimbursement on terms that are favorable to us (or at all), or such coverage, pricing and reimbursement may differ in separate regions in the same country. In some countries, the proposed pricing for a drug must be approved before it may be lawfully marketed, which could delay market entry (or, if pricing is not approved, we may be unable to sell at all in a country where we have received regulatory approval for a product). In addition, authorities in some countries impose additional obligations, such as health technology assessments (HTAs), which assess how well a prescription drug works in relation to its cost. U.S. payers are increasingly considering new metrics, including HTAs, as the basis for reimbursement rates. If our products do not meet or surpass these metrics, these payers may not reimburse the use of our products or may reduce the rate of reimbursement for our products and as a result, revenue from such products may decrease. We have, in certain cases, voluntarily elected to reduce prices or establish price caps with payers. For example, in the past we have implemented strategic pricing decisions for STRENSIQ in the U.S. that limit annual treatment costs given weight-based dosing. In addition, in the future, we may implement further price caps on STRENSIQ or on our other products. And while we believe such price caps can provide value in the long term, they result in decreased revenue per patient and we may be unable to increase sales volume for such products in an amount sufficient to maintain revenues
commensurate with those prior to implementing the price cap.
In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement governmental controls on pharmaceutical pricing. On July 24, 2020, the executive branch issued four executive orders intended to address the affordability and accessibility of medicines, including the “Executive Order on Lowering Drug Prices by Putting America First” that directs the US federal government to implement regulations that would require Medicare to benchmark its payments for Medicare Part B and Part D products to a set of "Most Favored Nations." The implementing regulations in connection with these executive orders are pending and, until issued, we are not able to determine the potential impact on our US business and results of operations in the future. Certain states have also proposed measures that are designed to control the costs of pharmaceuticals that they reimburse. If the U.S. (through the federal or state governments), which accounted for approximately 25% of our revenue in 2019, were to move to a pricing system based on a "Most Favored Nations" model, negotiated prices with Medicare (that may apply to private payers as well) or to an international price index (or similar model) that were to apply to our products, we expect that our revenues for sales in the U.S. would be lower, and potentially materially lower than if the current pricing program remained in place.
Other countries, including many European countries and Canada, have established pricing and reimbursement policies that contain costs by referencing the price of the same or similar products in other countries. In these instances, if coverage or the level of reimbursement is reduced, limited or eliminated in one or more countries, we may be unable to obtain or maintain anticipated pricing or reimbursement in other countries or in new markets. This may create the opportunity for third-party cross-border trade or influence our decision whether to sell a product in one or more countries, thus adversely affecting our geographic expansion plans and revenues. See Note 16, Commitments and Contingencies, to the condensed consolidated financial statements for information about our lawsuit against the Patented Medicine Prices Review Board (PMPRB) to establish that Alexion did not excessively price SOLIRIS in Canada, which uses reference pricing.
Due to the cost of our therapies, any potential increase in the number of patients receiving our products (for example, we expect there may be increases in sales of SOLIRIS for patients with gMG and NMOSD as we continue the launch for those indications), may cause third-party payers to modify,
limit or eliminate coverage or reimbursement for our products because they may require an allocation of a greater percentage of the potential financial resources of any public or private payer for our products.
Further, health insurance programs may utilize coverage incentives and obstacles to discourage or prevent beneficiaries from using higher priced products such as ours, including:
•establishing formularies under which only selected drugs are covered (which may exclude one or more of our products);
•utilizing variable co-payments that make drugs that are not preferred by the payer more expensive for patients; and
•utilizing management controls, such as requirements for prior authorization or failure first on another treatment.
In countries where patients have access to insurance, their insurance co-payment amounts or other benefit limits may represent a barrier to obtaining or continuing use of our products or adoption of new treatment options, such as ULTOMIRIS and ANDEXXA. The imposition or continuation of the use of these types of limits or barriers by insurers or the imposition of similar limitations or barriers in the future may have an adverse impact on our revenue and results of operations. In some cases, we have financially supported non-profit organizations that assist patients in accessing treatments. Such organizations assist patients whose insurance coverage imposes high co-payment amounts or other expensive financial obligations. Such organizations’ ability to provide assistance to patients is dependent on funding from external sources, and we cannot guarantee that such funding will be provided at adequate levels, if at all. We have also provided our products without charge to patients who have no or limited insurance coverage for drugs. We are not able to predict the financial impact of the support we may provide for these and other charitable purposes; however, substantial support could have a material adverse effect on our profitability in the future.
As third-party payers attempt to contain health care costs, they are demanding price discounts or rebates and limiting both the types and variety of drugs that they may cover and the amounts that they will pay for drugs. As a result, they may not cover or provide adequate payment to patients for our products or they may demand discounts or rebates from us, which may be material.
In 2019, four customers accounted for 56.4% of our total revenues. If any one or more of these customers (or other customer who accounts for a significant percentage of revenue or accounts receivable) were to require significant discounts or
rebates, or were to discontinue purchasing our products (due to cost or otherwise), our results of operations may be materially and adversely impacted.
Risks Related to Intellectual Property
If we cannot obtain new patents, maintain our existing patents and protect the confidentiality and proprietary nature of our trade secrets and other intellectual property, our business and competitive position may be harmed.
Our success depends in part on our ability to obtain and maintain patent and regulatory protections for our products and investigational compounds, to preserve our trade secrets and other proprietary rights and to prevent third parties from infringing on our rights.
We have procured patent rights, through both ownership and license, that cover our products and investigational compounds, and will likely apply for additional patent protections in the future. However, our patent applications may not result in the issuance of patents in the U.S. or other countries. In addition, a patent may be issued in one country, but a counterpart patent may not be issued in another country. For example, the European Patent Office in September 2019 rejected a patent application relating to the composition of matter for SOLIRIS; related patents were granted in the U.S. and Japan.
Even if a patent is issued, that is not conclusive as to its inventorship, scope, validity or enforceability and therefore that patent may not afford adequate (or any) protection for our products. On the basis of such inconclusiveness, third parties may challenge our patents, have done so in the past and, in some cases, have been successful in such challenges. For example, on January 21, 2019, the Opposition Division of the European Patent Office determined, following multi-party opposition proceedings, to revoke one of our European patents that relates to the formulation of SOLIRIS. Further, on August 30, 2019, the U.S. Patent and Trademark Office instituted inter partes review (IPR) of three of our patents that relate to SOLIRIS. In May 2020, we entered into a Confidential Settlement and License Agreement with Amgen to settle the three IPRs (Settlement Agreement). Pursuant to the Settlement Agreement, Alexion and Amgen have terminated each of the pending IPRs and, effective March 1, 2025 (or an earlier date in certain circumstances), Alexion grants to Amgen (and its affiliates and certain partners) a non-exclusive, royalty-free, license under U.S. patents and patent applications related to eculizumab and various aspects of the eculizumab product that Alexion currently markets and sells under the tradename SOLIRIS. We may enter into similar agreements in the future to grant or clarify certain rights of third-parties in connection with our intellectual property rights in SOLIRIS or other
products or product candidates. In addition, under the settlement agreement with Amgen, if certain circumstances are satisfied, Amgen may have the right to market and sell an eculizumab product in the US prior to March 2025.
If any of our patents are narrowed, invalidated, revoked or become unenforceable, competitors may develop and market products similar to ours that do not conflict with or infringe our patents rights, which could have a material adverse effect on our financial condition. Even if we obtain and maintain patents, our business may be significantly harmed if the patents are not broad enough to protect our products from copycat products. In addition, we may in the future enter into agreements similar to the agreement with Amgen that provides certain intellectual property rights to our marketed products or products in our pipeline.
We may finance or collaborate in research and development projects conducted by third parties, including government organizations, hospitals, universities or other educational or research institutions, or other for-profit companies. Such third parties may be unwilling to grant us certain rights to technology or products developed through such projects. Disputes may also arise as to the rights to technology or products developed in collaboration with such third parties.
Significant legal questions exist concerning the extent and scope of patent protection for biopharmaceutical products and processes in the U.S. and elsewhere. Accordingly, there is no certainty that patent applications owned or licensed by us will issue as patents, or that our issued patents will afford meaningful protection against competitors. Once issued, patents are subject to challenge through both administrative and judicial proceedings in the U.S. and other countries. Such proceedings include re-examinations, inter partes reviews, post-grant reviews and interference proceedings before the U.S. Patent and Trademark Office, as well as opposition proceedings before the European Patent Office and other non-U.S. patent offices. Certain countries have laws that provide stronger bases for challenging third party patent rights than are available to challenge patents in other countries. Therefore, we may be able to defend our patents against a third-party claim in one country but counterpart patents may be invalidated in other countries and we may be able to invalidate a third-party patent in one country but not invalidate its counterpart patents in other countries. Litigation may be required to enforce, defend or obtain our patent and other intellectual property rights. Any administrative proceeding or litigation could require a significant commitment of our resources and, depending on outcome, could adversely affect the
scope, validity or enforceability of certain of our patent or other proprietary rights.
Some of the sensitive technology, techniques and proprietary compounds used in our business are protected as trade secrets. However, we may also rely on collaboration with, or discuss the potential for collaboration with, suppliers, outside scientists and other biopharmaceutical companies. Collaboration and discussion of potential collaboration or inadvertent disclosure of a trade secret present a strong risk of exposing our trade secrets. If our trade secrets were exposed, we may lose the protection and potential exclusive rights afforded by trade secret law, and such exposure may likely help our competitors and allow them to access technology without restriction and adversely affect our business prospects.
If we are found to be infringing third party patents, we may be forced to pay damages to the patent owner and/or obtain a license to continue the manufacture, sale or development of our products. If we cannot obtain a license, we may be prevented from the manufacture, sale or development of our products or product candidates, which may adversely affect our business.
Parts of our technology, techniques, proprietary compounds and potential product candidates, including those which are or may be in-licensed or developed in collaboration with third parties, may be found to infringe patents owned by or granted to others. We have, and may in the future, receive notices claiming our products infringe third party patents and third parties have and may in the future file civil lawsuits against us claiming infringement of their intellectual property rights. Chugai Pharmaceutical Co., Ltd. filed suits in the U.S. and Japan alleging that ULTOMIRIS infringes patents held by Chugai. See Note 16, Commitments and Contingencies to the footnotes to the condensed consolidated financial statements. Additional third parties may claim that the manufacture, use or sale of our products or product candidates infringes patents owned or granted to such third parties. We are aware of patents owned by third parties that might be claimed by such third parties to be infringed by the development and commercialization of our products or investigational compounds. In respect to some of these patents, we have invalidated patents or obtained licenses, or expect to obtain licenses. However, with regard to other patents, we have determined in our judgment that:
•our products and investigational compounds do not infringe the patents;
•the patents are not valid or enforceable; and/or
•we have identified and are testing various alternatives that should not infringe the patents and which should permit continued development and commercialization of our products and investigational compounds.
Any holder of these patents or other patents covering similar technology could sue us for damages, which may be material in amount, and seek to prevent us from manufacturing, selling or developing our products (and we may be, in certain cases, prevented from initiating product launches in certain jurisdictions or required to withdraw the product from the market after it has been launched). Intellectual property disputes, such as those initiated by Chugai, can be costly and time consuming to defend and there is no guarantee that we would prevail in such lawsuit. If we cannot successfully defend against any infringement claims, we may seek to invalidate the patent or seek a license to the technology prior to or during legal actions in order to reduce the risks in connection with the product launches (or at a later time after product introduction) and to reduce further costs and the risk of a court determination that our technology, techniques, proprietary compounds or potential product candidates infringe the third party’s patents. A required license may be costly or may not be available on acceptable terms, if at all. A costly license, or inability to obtain a necessary license, could have a material adverse effect on our business.
In some instances, we believe we may prevail in a patent infringement action. There can, however, be no assurance that the court will agree with our position or that it will decide any infringement case in our favor. Nor can we be certain, if we do not prevail in litigation, that we may be able to obtain a license to any third-party patent on commercially reasonable terms (or at all); successfully develop non-infringing alternatives on a timely basis (or at all); or license alternative non-infringing technology, if any exists, on commercially reasonable terms (or at all). Any impediment to our ability to manufacture, use or sell approved forms of our products or our product candidates could have a material adverse effect on our business and prospects.
It is possible that we could lose market exclusivity for a product earlier than expected, which may harm our competitive position.
In our industry, much of an innovative product’s commercial value is realized while it has market exclusivity.
Market exclusivity for our products depends in large part on patent rights and certain regulatory forms of protection. As noted above, patent protection can be uncertain as to the validity, scope and enforceability of many issued patents. Absent relevant patent protection for a product, once regulatory exclusivity periods expire, biosimilar or generic
versions of the product can be approved and marketed. For example, in 2019, a SOLIRIS biosimilar was approved in Russia for the treatment of patients with PNH and aHUS. We also believe that the manufacturer of a SOLIRIS biosimilar has commenced the process to obtain regulatory approval to market and sell a SOLIRIS biosimilar in Brazil and Turkey and, if approved, this biosimilar may compete with Soliris in Brazil and Turkey.
The market exclusivity of our products may be impacted by competitive products that are either innovative, biosimilar or generic copies. In our industry, the risk of biosimilar or generic challenges has been increasing. U.S. law includes an approval pathway for biosimilar versions of innovative biological products. Under the pathway, the FDA may approve products that are similar to (but not generic copies of) innovative biologics (SOLIRIS, ULTOMIRIS and ANDEXXA are each innovative biologics) on the basis of less extensive data than is required for a full biologic license application (and there are similar pathways for generic copies of small molecule therapies (the Factor D therapies acquired in connection with the Achillion transaction are, for example, small molecules)). The law provides a mechanism to challenge the patents that protect an innovator’s products. Such litigation may begin as early as four years after the innovative biological product is first approved by the FDA. Pathways for biosimilar products also exist in many other markets, including Europe, Japan and Russia. Other companies are developing and advancing SOLIRIS biosimilar programs, including conducting clinical trials. Competition, including from biosimilars approved for marketing, would likely result in a decrease in volume of sales of our products, as well as a decrease in prices and lower margins for our products. In addition, approval of a biosimilar that is a substitute for one of our products may increase the risk of accelerated market penetration by that biosimilar. Further, if patients or healthcare providers do not believe that ULTOMIRIS provides a compelling profile for patient conversion from SOLIRIS, a SOLIRIS biosimilar may not only be expected to have a material and negative impact on our SOLIRIS revenues and margins (which accounted for a significant percentage of our revenue in 2019), it may also have a material impact on ULTOMIRIS revenue and margins and the ability of ULTOMIRIS to gain market acceptance.
Our other products and product candidates in development and trials are also at risk from biosimilars and generic drugs. Other than SOLIRIS for the treatment of gMG and NMOSD and SOLIRIS and ULTOMIRIS as a treatment for PNH and aHUS, each of our products is currently the only approved drug for the disease(s) the product treats. If a competitive product is approved for sale, including a biosimilar or generic product or other therapy (such as the two non-
C5 therapies approved by certain regulatory authorities, including the FDA, as a treatment for NMOSD), our market share and our revenues could decline, particularly if the competitive product is perceived to be more effective or is less expensive than our product.
Risks Related to Our Products and Product Candidates
Our future commercial success depends on gaining regulatory approval for new products and obtaining approvals for existing products for new indications.
We invest significant amounts in acquiring new products and technologies and advancing our existing product candidates and technologies. Our success and revenue growth and diversification will depend in part on our identification, acquisition (including licenses from or collaborations with third parties), development and commercialization of new products and technologies, and approval of additional indications for our existing products and products under development. Product development is very expensive, takes significant time and involves a high degree of risk. Only a small number of research and development programs result in the commercialization of a product. For example, we have recently terminated our agreement to co-develop ABY-039 with Affibody and determined not to exercise the co-development option agreement with Stealth BioTherapeutics Corp., in each case due to results of clinical trials. In addition, our recent business development activities have focused on new technologies with which we have very limited experience, including a Factor Xa reversal agent and antibody therapeutics targeting the neonatal Fc receptor, which may make the development, approval and commercialization of such potential products challenging for us.
Our ability to maintain or grow and diversify revenues may be adversely affected if we are delayed or unable to successfully develop the products in our pipeline, if we are unable to gain approval for SOLIRIS and ULTOMIRIS for additional indications, for new routes of administration (subcutaneous delivery) and in new jurisdictions, obtain marketing approval for STRENSIQ, KANUMA and ANDEXXA in additional territories, obtain approval for different dosing regimens (or expansion of indications included in the product label) or acquire or license products and technologies from third parties.
Even if we are successful in developing new products or addressing new indications, we cannot market any of those products unless and until we obtain all required regulatory approvals in each jurisdiction where we plan to sell these therapies. We must also maintain all such regulatory approvals for the period of time that we sell the product in each such jurisdiction. Our failure to obtain, or we have a delay in obtaining, approval or we fail to maintain
approvals once obtained, will prevent us from selling products and generating revenues for those products in such jurisdiction where we do not hold such approvals.
Our products and product candidates target diseases and conditions with small patient populations and we may not be effective at identifying patients.
The therapies that we have developed, acquired and that are in our product pipeline and in preclinical development target diseases and conditions that have small patient populations that have not been definitively determined. Further, in many cases there are either no or limited diagnostic tools for the indications we treat or may treat in the future. The lack of diagnostic tools, coupled with the fact that there is frequently limited awareness among certain health care providers concerning the rare diseases we treat, often means that a proper diagnosis can, and frequently does, take years to identify (or an appropriate diagnosis may never be made for certain patients). As a result, we may not be able to grow our revenues (even as we introduce new products or as existing products are approved for additional indications). There can be no guarantee that any of our programs will be effective at identifying patients that will benefit from our therapies, and even if we can identify patients that our therapies can help, the number of patients that our therapies treat may turn out to be lower than we expect, they may not be otherwise amenable to treatment with our products, or new patients may become increasingly difficult to identify, all of which may adversely affect our ability to grow and diversify revenue and adversely affect our results of operations and our business. In addition, even in instances where we do add patients, the number may be less than the number of patients that discontinue use of the applicable product in a given period resulting in a net loss of patients and potentially decreased revenue.
We may not be able to gain or maintain market acceptance of our products among the medical community, patients or payers, which could prevent us from maintaining profitability or growth.
Our products may not gain or maintain market acceptance among physicians, patients, payers and others. Although we have received regulatory approval for certain of our products in certain territories (and may receive approvals for additional products or in additional jurisdictions), such approvals do not guarantee future revenue. Physicians’ willingness to prescribe, and patients’ willingness to accept, our products, depends on many factors, including:
•prevalence and severity of adverse side effects in both clinical trials and commercial use;
•the timing of the market introduction of competitive drugs, biosimilars and generics;
•perceived safety of our products
•demonstrated clinical safety and efficacy compared to other drugs;
•perceived benefits relative to cost and/ or evaluations in HTAs (or similar assessments);
•pricing and availability of reimbursement from third-party payers, including governmental entities;
•convenience and ease of administration;
•effectiveness of our marketing strategy;
•publicity concerning our products and our other product candidates (and those of competitive products); and
•availability of alternative treatments.
The likelihood of physicians to prescribe SOLIRIS and ULTOMIRIS for patients with aHUS may also depend on how quickly SOLIRIS or ULTOMIRIS can be delivered to the hospital or clinic and our distribution methods may not be sufficient to satisfy this need. In addition, while SOLIRIS as a treatment for aHUS is recommended by some regulatory authorities to be used for the duration of a patient’s lifetime, we are aware that some healthcare providers prescribe SOLIRIS for aHUS for a shorter time period and, in some cases, may prescribe SOLIRIS for aHUS in emergency or acute situations only (and the same may occur in connection with the use of ULTOMIRIS for aHUS). Decisions such as this by aHUS patients and healthcare providers to use our products for a period that is less than the remaining lifetime of the patient or in only acute circumstances may cause our SOLIRIS or ULTOMIRIS revenues, and revenues for our other products, to fluctuate and past sales of our products may not be indicative of future sales for such products.
If our products fail to achieve or maintain market acceptance among the medical community or patients in a particular country, we may not be able to market and sell our products successfully in such country, which may limit our ability to generate revenue and could harm our overall business.
If our products harm patients, or are perceived to harm patients even when such harm is unrelated to our products, our regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The testing, manufacturing, marketing and sale of biologics and small molecule therapies for use in humans may cause harm to patients, which exposes us to product liability risks and regulatory penalties.
Our products and our product candidates generally treat patients with rare diseases and, as a result, we generally are able to test our products in only a small number of patients. As more patients use our products, including more children and adolescents, new risks and side effects may be discovered, the rate of known risks or side effects may increase, and risks previously viewed as less significant could be determined to be significant.
Under pharmacovigilance guidelines, we are required to timely report any adverse events that any patient using our products experiences, as well as any clinical evaluations of outcomes in the post-marketing setting. This information is required to be reported to appropriate regulatory agencies in accordance with relevant regulations and, as a result, any potential adverse events will be promptly brought to the attention of regulators that may likely require prompt remedial action (and any failure to report these adverse events or report such events in a timely manner may result in penalties being imposed on Alexion by regulators). In the event any new risks or adverse effects are discovered as patients are treated for approved indications, or as our products are studied in or used by patients for other indications, regulatory authorities may delay or revoke their approvals or require changes to labeling or reformulation of the products (or take other actions that may adversely impact sales of such products).
If previously unknown side effects are discovered or if there is an increase in negative publicity regarding known side effects of any of our products, it could significantly reduce demand for the product, harm our reputation, result in product withdrawals, recalls, delays or revocations of regulatory approvals or require us to take actions that could negatively affect sales and operating results, including conducting additional clinical trials and safety studies, making changes in labeling, reformulating our products or making changes and obtaining new approvals for our and our suppliers’ manufacturing facilities. Further, any investigation into the circumstances surrounding an adverse event may be costly and time consuming (even if it is ultimately determined that the adverse event is not the result of the use of our product).
There are also risks associated with our products; for example, use of C5 Inhibitors, such as SOLIRIS and ULTOMIRIS, is associated with an increased risk for certain types of infection, including meningococcal infection. In certain cases, a physician may not have the opportunity to timely vaccinate a patient in the event of an acute emergency episode, such as in a patient presenting with aHUS, which could result in the patient using SOLIRIS or ULTOMIRIS experiencing a life-threatening meningococcal infection (and even in certain cases in
which a vaccination can be delivered to the patient, it may not, eliminate all risk of meningococcal infection). In addition, ANDEXXA has been associated with thrombolic risks, ischemic risks, cardiac arrest and sudden death. Patients using our products and product candidates have died or suffered potentially life-threatening conditions either during or after ending their treatments, and these include patients who have died while participating in a clinical trial. In addition, many patients who use our products are already very ill and may suffer adverse events, including death, for reasons that may or may not be related to our products. We may be sued by patients who are harmed during the course of using our products, whether as a prescribed therapy, during a clinical trial, during an investigator-initiated study, or otherwise. Any such product liability lawsuit or injury claim, which could include class actions, could harm our reputation among patients, physicians, payers and others and require us to pay substantial amounts of money to injured patients, and even if successfully defended, could have a material adverse effect on our business, financial condition or results of operations due to the expense of defending any such claim. While we do have product liability insurance, it may not cover all potential types of liabilities or may not cover certain liabilities completely. Moreover, we may not be able to maintain our insurance on acceptable terms, or at all.
We anticipate that we will face increased competition from companies that will enter into the markets we currently serve and as our product pipeline expands into markets that are currently served by other companies.
We expect that the business environment in which we operate will become increasingly competitive. Currently, certain of our products are the only approved therapies for certain indications they treat. For example, SOLIRIS and ULTOMIRIS are the only approved treatments for PNH and aHUS in the U.S., Europe and Japan. In the future, we expect that SOLIRIS and ULTOMIRIS may compete with new drugs and biosimilars currently in development. Several companies are developing other therapies to treat PNH, aHUS, gMG and NMOSD, and other pharmaceutical companies have publicly stated that they are developing and intend to commercialize a SOLIRIS biosimilar. For example, the FDA and certain other foreign regulatory authorities recently approved a CD19-directed cytolytic antibody and an IL-6R antibody therapy indicated for the treatment of NMOSD in adult patients who are anti-aquaporin-4 antibody positive. The introduction of these and other competitive products may negatively impact our business, including our revenue and profitability. In addition, following the introduction of a SOLIRIS biosimilar in 2019 in Russia for the treatment of PNH and aHUS, we experienced a decrease in revenue from sales of SOLIRIS and expect that Russia will
account for a minor portion, if any, of future SOLIRIS revenue as a result of this competitive product. We also believe that the manufacturer of a SOLIRIS biosimilar has commenced the process to obtain regulatory approval to market and sell a SOLIRIS biosimilar in Brazil and Turkey and, if approved, this biosimilar may compete with Soliris in Brazil and Turkey and, like Russia, have an adverse impact on SOLIRIS revenues in Brazil and Turkey. STRENSIQ, KANUMA and ANDEXXA may also experience competition in the future. We are also aware of companies that have initiated or are planning to initiate studies for diseases and conditions that we are also targeting with our product pipeline. Our revenues could be negatively affected if patients or potential patients enroll in our clinical trials or clinical trials of other companies with respect to diseases and conditions that we also target with approved therapies.
Some of our competitors and future competitors may have significantly greater financial, technical and marketing resources than us and may commercialize competitive products that are cheaper, more effective, safer, have less frequent dosing schedules, or are easier and quicker to administer than our products. Our current and future competitors may develop products that are more broadly accepted or may receive patent protection that dominates, blocks or adversely affects our product development or business. These competitive products, including any biosimilars approved under alternative regulatory pathways (or generics that may be approved that compete with our small molecule therapies), may significantly reduce both the price that we receive for our marketed products and the volume of products that we sell, which may negatively impact our revenues and profitability. Given that a significant portion of our 2019 revenue was attributable to SOLIRIS, one or more competitive novel products or biosimilars could have a significant impact on our entire business.
In addition, we experience competition in drug development from universities and other research institutions, and pharmaceutical companies compete with us to attract universities and academic research institutions as drug development partners, including for licensing their proprietary technology. If our competitors successfully enter into such arrangements with academic institutions, we may be precluded from pursuing those unique opportunities and may not be able to find equivalent opportunities elsewhere.
If a company announces successful clinical trial results for a product that may be competitive with one of our products or product candidates, receives marketing approval of a competitive product, or gets to the market before we do with a competitive
product, our business may be harmed or our stock price may decline.
Risks Related to Business Operations
We rely on a limited number of facilities to produce our products and manufacturing issues at our facilities or the facilities of our third party service providers could cause product shortages, stop or delay commercialization of our products, disrupt or delay our clinical trials or regulatory approvals, and adversely affect our business.
The majority of our products and product candidates are biologics and the production of such biologic therapeutics that meet all product specification and regulatory requirements is particularly complex. Even slight deviations at any point in the production process may lead to production failures, product recalls and regulatory actions. For example, in 2013 and 2014 we undertook a voluntary recall of SOLIRIS due to the presence of visible particles in a limited number of vials. In addition, because the production process involves the use of materials that are derived from biological sources, the process can be affected by contaminants that could impact those biological micro-organisms. These manufacturing challenges are coupled with the fact that we have limited experience manufacturing commercial quantities of certain of our products (so we or our third party manufacturers may have limited previous experience resolving any issues in connection with the manufacture of certain of our products and any issues may take significant time to remediate or we may be unable to solve any manufacturing problems). In addition, with our acquisition of Achillion, we also have small molecules in clinical trials and we are planning future clinical trials in new indications and we expect that manufacture of these therapies and compliance with cGMP will pose similar challenges and we have limited experience manufacturing small molecules for clinical trials and for commercial sales.
If we and/or our third party manufacturers and suppliers fail to meet the highly technical requirements/specifications of manufacturing our biologic and small molecule products and our strict quality and control specifications, we (or they) may be unable to manufacture or supply our products. We depend on our third party manufacturers to perform effectively on a timely basis and to comply with regulatory requirements and meet our product specifications. For example, we rely on Lonza owned and operated facilities for the production of a significant portion of our products, including ANDEXXA which we recently acquired, and Lonza has undertaken the construction and operation of new facilities to meet demand for certain of our products (including a new facility that is being qualified for manufacturing in New Hampshire) and these facilities
must meet our production requirements and new facilities must be qualified by regulatory authorities before product can be sold. Our failure or the failure of our third-party manufacturers (including the Lonza facility in New Hampshire) to produce sufficient quantities of our products and product candidates or to meet our specifications and quality standards or those standards imposed by regulatory authorities could result in lost revenue, diminish our profitability, delay the development of our product candidates, delay regulatory approval, result in the rejection of our product candidates or result in supply shortages for our patients, which may lead to lawsuits, harm to our reputation or could accelerate introduction of competing products to the market. For example, we experienced unexpected chemistry, manufacturing and control (or CMC) issues with our ALXN 1830 program that resulted in a delay in the clinical trial timeline for that program. We may experience similar CMC issues in the future that may impact marketed products or other clinical trials.
If we underestimate demand for ULTOMIRIS, SOLIRIS, ANDEXXA or any of our products, or experience product interruptions at Alexion’s internal manufacturing facilities or a facility of a third party provider, including as a result of risks and uncertainties described in this Quarterly Report on Form 10-Q, we may not be able to increase our revenues and alternative therapies may gain greater market acceptance.
We also face external factors, many of which are beyond our control, that could cause production interruptions at our facilities or at the facilities of our third party providers, including natural disasters, public health crises (such as COVID-19), labor disputes, acts of terrorism or war.
The risks to our business of any manufacturing stops or interruptions (whether the result of internal or external factors of the nature identified above) are amplified because we rely on a limited number of facilities to produce our products and product candidates. Further, we expect that we will continue to rely on a very limited number of manufacturing facilities in the future for all of our products, including our complement inhibitors. Although we have business continuity plans, including with respect to inventory, to reduce the potential for manufacturing disruptions or delays and reduce the severity of a disruptive event, there is no guarantee that these plans will be adequate, which could adversely affect our business and operations.
We and our third party providers are required to maintain compliance with cGMP and other stringent operation and manufacturing requirements and are subject to inspections by the FDA and comparable agencies in other jurisdictions to confirm such compliance. Governmental authorities will generally
not permit products manufactured at a facility that is not registered by the applicable government agency to enter into the country and such products may be returned for failure to comply with such regulation, which may decrease or delay sales and result in the loss of inventory. Any delay, interruption or other issues that arise in the manufacture, fill-finish, packaging or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection or comply with ongoing operating regulations could significantly impair our ability to supply our products and product candidates. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions and damage our reputation.
Our efforts to bring more of our manufacturing operations under our control present additional risks. We have made significant investments in biologics manufacturing facilities, warehousing, fill-finish and other facilities at our sites in Athlone and Dublin, Ireland and at dedicated sites owned by third parties. We have commenced manufacturing operations at certain of these sites prior to receiving regulatory approval and we have $117.2 of product produced at such sites in inventory as of September 30, 2020. Despite the significant investment we have made in these facilities and operations, we cannot guarantee that we will be able to successfully and timely complete the appropriate validation processes or obtain the necessary regulatory approvals for these and other facilities, that we will be able to perform the intended manufacturing and supply chain services at these facilities for commercial or clinical use or that we will be able to use the product manufactured at these sites. Prior to such time, we may continue to rely on third parties for these services.
If our products are subject to any manufacturing issues, we may be unable to timely identify alternative manufacturers, and if we are able to timely identify alternative manufacturers, such alternative manufactures may not be able to satisfy our requirements. No guarantee can be made that regulators will approve additional third party providers in a timely manner or at all, or that any third party providers will be able to perform manufacturing or related services for sufficient product volumes for any country or territory. Further, due to the nature of the current market for third-party commercial manufacturing, many arrangements require substantial penalty payments by the customer for failure to use the manufacturing capacity for which it contracted. The payment of a substantial penalty could harm our financial condition and may restrict our ability to transition to internal manufacturing or manufacturing by other third parties. In addition, the terms and conditions to engage an additional third-party manufacturer may not be as favorable to us as
our current arrangements and may likely reduce the profit on the sales of any products to which they relate.
Any adverse developments affecting our manufacturing operations or the operations of our third-party providers could result in a product shortage of clinical or commercial requirements, withdrawal of our product candidates or any approved products, shipment delays, lot failures or recalls. We may also have to write-off inventory and incur other charges and expenses for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Each of these could have an adverse material impact on our business individually or in the aggregate.
We rely on a limited number of providers for our raw materials and supply chain services, which could result in our being unable to continue to successfully commercialize our products and our product candidates (if approved) and to advance our clinical pipeline.
Certain of the raw materials required in the manufacture and the formulation of our products are derived from biological sources. Such raw materials are difficult to procure and may be subject to contamination or recall. Access to and supply of sufficient quantities of raw materials which meet the technical specifications for the production process is challenging, and often limited to single-source suppliers. If a raw material manufacturer were unable to supply such materials, our business may be impacted because we rely on one or a limited number of such manufacturers for certain materials for our products. Finding an alternative supplier could take a significant amount of time and involve significant expense due to the nature of the products and the need to obtain regulatory approvals. The failure of these single-source suppliers to supply adequate quantities of raw materials for the production process in a timely manner, or at all, may impact our ability to produce sufficient quantities of our products for clinical or commercial requirements. A material shortage, delay, contamination, recall, or restriction on the use of certain biologically derived substances or any raw material used in the manufacture of our products could adversely impact or disrupt manufacturing and materially limit our ability to generate revenues.
In addition, KANUMA is a transgenic product and the facilities on which we rely to produce raw material for KANUMA are the only animal facilities in the world that produce the necessary egg whites from transgenic chickens. Natural disasters, disease, such as exotic Newcastle disease or avian influenza, or other catastrophic events could have a significant impact on the supply of unpurified KANUMA, or destroy our animal operations altogether. If our animal
operations are disrupted, it may be extremely difficult to set up another animal facility to supply the unpurified KANUMA.
We also depend on a very limited number of third party providers for supply chain services with respect to our clinical and commercial product requirements, including product filling, finishing, packaging and labeling.
Our third-party raw material providers and supply chain service providers operate as independent entities and we do not exercise control over any such third-party provider’s operations or their compliance with our internal or external specifications or the rules and regulations of regulatory agencies. Any contractual remedies we may have under agreements with these parties may not protect us from the harm suffered by our business or our patients if they fail to provide material or perform services that meet our specifications. Due to the highly specialized nature of the services performed by these third parties, particularly the supply of raw materials and other drug product, as well as the delivery and supply chain operations regarding our products, we do not believe that we could quickly find replacement suppliers or service providers and, even if we were able to identify additional third parties, the terms of any such arrangement may not be favorable to us. In either of these cases, our revenue, results of operations, business and reputation may be harmed and we may not be able to provide the therapies that our patients require.
The success of our business may also depend on the security of our products while in the supply chain for delivery to patients, which, as noted above, is dependent on third-party providers. For example, if our products are not fully and adequately secured from unauthorized access by third parties, any of our products may be tampered with or contaminated. If our products were exposed to any tampering or contamination, or if they are not transported in accordance with the required specifications, our patients may be harmed through use of our products, and such harm may be severe. In addition, if the supply chain is not secure (or our distributors do not exercise control over our products while in their possession), we are also at risk for our products being diverted to patients other than those who are the intended recipient or to patients who do not have a prescription to receive our therapies (or it may be used for treatment by physicians who have not completed the necessary REMs protocols in order to treat patients) or it may be sold by distributors, channels or other entities that are not authorized by Alexion to sell our products. In addition, an unauthorized distributor may not properly store or ship our products, thereby exposing patients to potential harm from use of the product that was not handled in
accordance with our standards. If any of the foregoing were to happen, we could be subject to costly litigation, significant monetary penalties, harm to our reputation and investigation by regulatory authorities (and potentially subject to regulatory action, including recall, product withdrawals, suspensions and monetary penalties).
The sale and use of counterfeit versions of our products could result in significant harm to patients, reduced sales of our products and harm to our reputation.
We are aware that counterfeit versions of our products have been sold by entities that are not affiliated with Alexion using product packaging suggesting that the product was manufactured by Alexion. If unauthorized third parties illegally distribute and sell counterfeit versions of our products, those products may not meet our very stringent product specifications (or the manufacturing, handling and distribution requirements for our products) and any patient that takes any counterfeit product may suffer serious adverse health consequences, including death. Our reputation and business could suffer harm as a result of counterfeit drugs sold under our brand name and could result in lost sales for us and decreased revenues.
If we are unable to establish and maintain effective sales, marketing and distribution capabilities or to enter into agreements with third parties to do so, we may be unable to successfully commercialize our products.
We currently market and sell our products in the U.S., the EU, Japan and several other territories through a direct sales force. In addition, in order to gain greater efficiencies in our operations, we are implementing a plan pursuant to which certain portions of our international commercial operations have already or will transition to a new operating model in which sales, distribution, marketing and related efforts in designated countries will be conducted by third-parties, and our direct sales and marketing presence will decrease (or be eliminated) in these regions.
Due to the fact that some of our products are new to the market, we do not have lengthy experience in marketing and selling these products to patients, healthcare providers and payers (for example, we are relatively new to certain therapeutic areas, such as neurology (gMG and NMOSD), and our sales force has had limited exposure in educating and targeting sales to patients and physicians in neurology practices). In addition, ANDEXXA is also new to the market, having been authorized by the FDA to be marketed in the US in mid-2018. This challenge is coupled with the fact that many members of our sales and marketing team are new to working with Alexion products (and ANDEXXA) and we are transitioning to third parties to
market, distribute and sell our product in certain countries. If we are unable to successfully market and sell our new products (and expand our sales and commercial operations) and to successfully sell our products in new therapeutic areas, as well as successfully implement the transition to third parties to sell, distribute and market our products in certain countries, our business and sales may be harmed. We cannot guarantee that we will be able to establish, maintain and expand our own capabilities or enter into and maintain any sales, marketing or distribution agreements with third-party providers on acceptable terms, if at all, or that we will be able to manage the transition to third-party sales, marketing and distribution in the relevant jurisdictions that will not cause any interruption or disruption in our business and sales of our products. We will not exercise the same degree of control over such third parties that we do over our direct sales force and the ability to direct the third party and provide incentives for such third party to market and sell our products may not be as strong as in the case of a direct sales force. This transition and greater reliance on third party sales force, marketers and distributors may also increase the risk of litigation with or liability to third parties that we had previously engaged to perform services for us in jurisdictions where we are implementing these operational changes.
Even if we hire qualified sales and marketing personnel necessary to support our objectives and enter into distribution agreements with third parties on acceptable terms, we may not hire such employees or enter into such agreements in an efficient manner or on a timely basis. We may not be able to forecast accurately the size and experience of the sales and marketing force and the scale of distribution capabilities necessary to successfully market and sell our products, which could result in decreased revenues or margins. In addition, as we launch new products, such as ULTOMIRIS, and we move into other therapeutic areas (such as neurology and reversal of Factor Xa inhibitors), and, if and when, the products we acquire in connection with acquisitions and development agreements with third parties move closer to regulatory approval, we may have a larger product portfolio and address more therapeutic areas and the foregoing risks may continue to apply and may increase. Our expenses associated with building up and maintaining the sales force and distribution capabilities around the world, and in transitioning from direct sales force to third party sales, marketing and distribution, may be disproportionate compared to the revenues we may be able to generate on sales or any savings or efficiencies we gain through use of such third-parties. We cannot guarantee that we will be successful in commercializing any of our products for the above referenced or other reasons.
Our efforts to expand our business and product offerings through acquisitions of businesses and technologies may not be successful.
Building our product pipeline is a key strategic objective to address revenue concentration risk in C5 complement inhibitors and we expect to regularly evaluate and, when appropriate, purchase businesses and acquire, co-develop or license technologies and products from third parties in an effort to expand and diversify our pipeline, product offerings, and our technologies. For example, we recently completed the acquisitions of Portola and Achillion. Acquisitions of new businesses or products and in-licensing of new technologies and products may involve numerous risks, including:
•substantial cash expenditures;
•potentially dilutive issuance of equity securities and incurrence of debt;
•assumption of material liabilities in connection with the target or purchased technology, some of which may be difficult or impossible to identify at the time of acquisition;
•difficulties in integrating the operations of the acquired companies;
•failure of any acquired businesses or products or in-licensed products or technologies to achieve the scientific, medical, commercial or other results we anticipate;
•diverting our management's attention away from other business opportunities and on-going operations;
•the potential loss of our key employees or key employees of the acquired companies;
•risks of entering disease areas and indications or modalities in which we have limited or no direct experience; and
•significant investments in resources and personnel to evaluate, integrate and develop acquisition and in-license programs.
A substantial portion of our strategic efforts are focused on opportunities for rare disorders, but the availability of such opportunities may be limited. We may not be able to identify opportunities that satisfy our strategic criteria or are acceptable to us or our stockholders. Several companies have publicly announced intentions to establish or develop rare disease programs and we may compete with these companies (some of which may be larger and may be able to provide more consideration than we can) for the same opportunities. For these and other reasons, we may not be able to acquire the rights to additional product candidates or approved products on
acceptable terms, or at all. In such event, we may not be able to further rebuild our pipeline and any future revenue may remain largely dependent on our existing products, which are subject to the risks noted above.
In addition, through our business development initiatives we have acquired or obtained rights to new technologies, including a therapy to reverse Factor Xa inhibitors, Factor D small molecules and two FcRN platforms (in February 2020, based on data from our Phase I study, we terminated the agreement to co-develop ABY-039 with Affibody, which was the developer of one such FcRN platform), among others. These technologies are intended to diversify our pipeline and revenue base (if products based on these technologies are, where applicable, approved by regulatory authorities), but we have limited experience with these technologies, including developing these therapies, operating clinical trials with these therapies, obtaining regulatory approval and commercializing these assets. If we are unable to successfully bring these products to market and to increase sales of approved medicines in the case of ANDEXXA, we may not be able to diversify our revenue or generate a return on our investments.
Even if we are able to successfully identify and complete acquisitions and other strategic transactions, we may not be able to integrate or take full advantage of them. An acquisition or other strategic transaction may or may not result in short-term or long-term benefits to us (such as our transactions with Affibody and Stealth). A commercial stage product or near-commercial stage product acquired may not result in the revenues that we anticipate or we may be unable to realize expected profits on sales due to expenses related to the manufacturing and/or sales of the commercial product. We may also incorrectly judge the value or worth of an acquired company or business or an acquired or in-licensed product, particularly if the acquired technology is in preclinical trials or early-stage clinical trials. Any therapies we acquire that are pre-clinical or in clinical trials may not result in a commercialized product and any revenues, if the product is commercialized, may not meet our projections or result in generating an adequate return on our investment.
The acquired business of Portola may underperform relative to our expectations, and we may not be able to successfully integrate our existing business with the business of Portola and achieve anticipated synergies.
We completed the acquisition of Portola on July 2, 2020. Through the acquisition, we acquired ANDEXXA, a commercial-stage product that is intended, in part, to diversify our revenue from reliance on C5 inhibitors. The acquired business of Portola may underperform relative to our expectations, which may cause our financial results to differ from
our own or the investment community’s expectations, and it may not result in the revenue or generate the operating income in the future that we anticipate. The ultimate success of the acquisition will depend, in part, on Alexion’s ability to successfully combine and integrate the Portola business and realize the anticipated benefits, including synergies, innovation opportunities and operational manufacturing and sales efficiencies, from the acquisition. If we are unable to achieve our objectives within the anticipated time frame, or at all, the anticipated benefits may not be realized fully or at all, or may take longer to realize than expected, and the value of Alexion’s common stock may decline. In addition, if the results of the Phase IV post-marketing trial required by the FDA does not meet the safety and efficacy requirements of the FDA, ANDEXXA (the product that generated almost all of Portola’s revenues) may be withdrawn from the market or otherwise subject to regulatory restrictions which may limit the ability to realize expected value from the transaction. Andexanet alfa also received conditional approval in the EU.
The integration of the two companies may result in material challenges, including, without limitation:
•the diversion of management’s attention from ongoing business concerns;
•managing a larger combined business that includes a new therapeutic area for Alexion;
•maintaining employee morale and retaining key management and other employees;
•retaining existing business and operational relationships, including customers, suppliers and employees and other counterparties, and attracting new business and operational relationships; and
•coordinating geographically separate organizations.
The financial results of ANDEXXA, and our ability to generate returns on our investment in Portola, will require that we successfully commercialize this product (which received conditional approval from the FDA in early 2019). ANDEXXA utilization will depend, in large part, on access for the therapy at both the institutional level (where adoption will be driven by hospitals and emergency care facilities including ANDEXXA in the approved protocols for care and by ANDEXXA being qualified for adequate reimbursement by payers for use) and at the individual prescriber level (where adoption will be driven by immediate physical access to the product as it will be used in the acute care setting and by acceptance and endorsement of use by individual physicians who will need to be satisfied with, among other things, ANDEXXA's safety and efficacy). In addition, ANDEXXA may not gain expected market acceptance due to, among other reasons: pricing and
reimbursement decisions and lower than anticipated usage at facilities and dosing. While we do have experience with obtaining institutional approval for our therapies and promoting acute care products (SOLIRIS and ULTOMIRIS for aHUS) we will need to effectively address the needs of institutions and prescribers, as noted above, in order to increase sales of ANDEXXA.
In order to support potential growth of the business, we will be required to make significant investments in our business operations.
To effectively manage our current and future potential growth, we must continue to effectively enhance and develop our global employee base and our operational and financial processes. Supporting our growth strategy may require significant capital expenditures and management resources, including investments in research, development, sales and marketing, clinical trial capabilities, manufacturing and other areas of our operations. Efforts to advance our product pipeline, including the increased number of clinical trials that are under way or will commence in the future, will require significant expense in the remainder of 2020 and in 2021. The development or expansion of our business, any acquired business or any acquired or in-licensed products may require a substantial capital investment by us and we may likely incur substantial expenses in advancing acquired products through development, trials, regulatory approval and to commercialization. We may not have the necessary funds for these capital expenditures and expenses or these funds might not be available to us on acceptable terms, or at all. We may also seek to raise funds by incurring additional indebtedness and selling shares of our capital stock, which could dilute current stockholders’ ownership interest in our company, or securities convertible into our capital stock, which could dilute current stockholders’ ownership interest in us upon conversion.
Completion of proof of concept trials, biomarker studies, preclinical studies or clinical trials does not guarantee advancement to the next phase of development or regulatory approval or successful commercialization.
Conducting clinical trials is a complex, time-consuming and expensive process and there are no guarantees that any trial will meet its endpoints or objectives. Completion of preclinical studies or clinical trials does not guarantee that we will initiate additional studies or trials for our product candidates, if further studies or trials are initiated, what the scope and phase of the trial will be or that they will be completed, or if these further studies or trials are completed, that the design or results may provide a sufficient basis to apply for or receive regulatory approvals or to commercialize products. Results of clinical trials could be inconclusive, requiring
additional or repeat trials. Data obtained from preclinical studies and clinical trials are subject to varying interpretations that could delay, limit or prevent regulatory approval. Many companies have believed their product candidates performed satisfactorily in clinical trials but nonetheless failed to obtain marketing approval of their drug candidate. If the design or results achieved in our clinical trials are insufficient to proceed to further trials or to sustain regulatory approval of our product candidates, our business could be materially adversely affected. Failure of a clinical trial to achieve its pre-specified primary endpoint or objective generally increases the possibility that additional studies or trials may be required if we even determine to continue development of the product candidate, reduces the likelihood of timely development of and regulatory approval to market the product candidate, and may decrease the chances for successfully achieving the primary endpoint(s) or objective(s) in scientifically similar indications.
We are currently planning and conducting several clinical trials of products and product candidates that we anticipate may be important to our goal of expanding our business and diversifying our product portfolio. These trials may not yield the anticipated results for a number of reasons and may not result in a product that obtains regulatory approval.
ULTOMIRIS may not be approved as a treatment for additional indications or in other jurisdictions and any clinical trials may not achieve the designated endpoints and prove to be effective for use in patients with these additional indications. For example, we have initiated Phase III clinical trials for ULTOMIRIS as a treatment for: (i) Amyotrophic Lateral Sclerosis (ALS) and (ii) patients with COVID-19 and severe pneumonia or acute respiratory distress syndrome (and we plan to initiate studies of ULTOMIRIS for patients with HSCT-TMA, CM-TMA and a proof of concept basket study in renal indications, Lupus Nephritis (LN) and Immunoglobulin A Nephropathy (IgAN)). There is no guarantee that the Phase III clinical trial for ALS and COVID-19 (or the additional studies that we are planning for ULTOMIRIS) will provide sufficient evidence to advance our research beyond these stages. Drug development is very uncertain. We had, for example, conducted an exploratory clinical study in Primary Progressive Multiple Sclerosis (PPMS) that we have decided to no longer pursue based on biomarker analysis.
In addition, we are also conducting clinical trials in therapeutic areas with which we have limited experience (for example, ALXN1840 (WTX101), a therapy for Wilson’s disease), Factor D small molecules, and with technology platforms with which we also have limited experience (for example, humanized monoclonal antibody that inhibits the
interaction of FcRn with Immunoglobulin G (IgG) and IgG immune complexes). Further, we plan to initiate a Phase II clinical trial for ALXN2040 in Geographic Atrophy (GA), which is our first clinical trial in opthalmology, an indication with which we have limited experience at Alexion. In addition, we intend to also initiate a proof of concept trial using a Factor D molecule, ALXN2050, in patients with various renal diseases. And we are pursuing trials to expand the label for ANDEXXA to treat additional acute care indications. Each of these clinical trials, and any other trial we commence, require significant financial expenses and operational resources, is subject to the risks highlighted above and the investments we have made in these technologies may not generate the expected returns.
Our clinical studies may be costly and lengthy, and there are many reasons why drug testing could be delayed or terminated.
For human trials, patients must be recruited and each product candidate must generally be tested at various doses and formulations for each clinical indication. Many of our programs focus on diseases and conditions with small patient populations making patient enrollment difficult and requiring a relatively large number of trial sites to meet enrollment requirements to power our clinical trials to our desired levels for efficacy and, in certain cases, superiority. Additionally, we can have multiple clinical trials running for the same indication, further challenging clinical trial enrollment. Insufficient patient enrollment in our clinical trials could delay or cause us to abandon a product development program. We may decide to abandon development of a product candidate or a study at any time due to unfavorable results or other reasons, including if there are concerns about patient safety (as patients have, and may in the future, suffer injuries during clinical trials). If initial trials do not produce adequate results, we may have to spend considerable resources repeating clinical trials or conducting additional trials, either of which may increase costs and delay revenue from those product candidates, if any. We may open clinical sites and enroll patients in countries where or for indications in which we have little experience.
Even if we were to complete clinical trials for one or more of our therapies, we or regulatory authorities may determine that the results are not be sufficient for filing a BLA or NDA or granting approval to market the therapy.
We rely on a small number of clinical research organizations to carry out our clinical trial related activities, and one contract research organization (CRO) is responsible for many of our studies. We rely on such parties to enroll clinical sites and patients, operate trials and accurately report their results. Our reliance on CROs may impact our ability to control the
timing, conduct, expense and quality of our clinical trials. In addition, we may be responsible for any errors in clinical trials by a CRO as a result of the performance of services in connection with a clinical trial on our behalf. And regulatory agencies, in connection with a potential product approval or as part of ongoing monitoring, will review a CRO's compliance with regulatory requirements relating to clinical trials and we may be subject to findings and regulatory action (including denial or delay of product approval) if a CRO fails to comply with regulations.
Additional factors that can cause delay, impairment or termination of our clinical trials or our product development efforts include:
•delay or failure in obtaining institutional review board (IRB) approval or the approval of other reviewing entities to conduct a clinical trial at each site;
•delay or failure in reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
•withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials;
•clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial;
•delay or failure in having patients complete a trial or return for post-treatment follow-up;
•lack of sufficient supplies of the product candidate;
•disruption of operations at the clinical trial sites;
•adverse medical events or side effects in treated patients;
•failure of patients taking the placebo to continue to participate in our clinical trials;
•insufficient clinical trial data to support safety and effectiveness of the product candidates;
•lack of effectiveness or safety of the product candidate being tested;
•decisions by regulatory authorities, the IRB, ethics committee, or us, or recommendation by a data safety monitoring board, to suspend or terminate clinical trials at any time for safety issues or for any other reason;
•failure to obtain the necessary regulatory approvals for the product candidate or the
approvals for the facilities in which such product candidate is manufactured; and
•decisions by competent authorities, IRBs or ethics committees to demand variations in protocols or conduct of clinical trials
We expect our operating results to fluctuate.
Our quarterly revenues, expenses and net income (loss) may fluctuate, even significantly, due to certain risks, including those described in these “Risk Factors” as well as the timing of charges and expenses that we may take, acquisitions and business development transactions (such as the Portola Pharmaceuticals, Achillion Pharmaceuticals, Wilson Therapeutics and Syntimmune acquisitions) and the impact of converting patients from SOLIRIS to ULTOMIRIS (as noted above). We may not be able to sustain or increase profitability on a quarterly or annual basis. Since we have a limited sales and operating history with certain of our products and for new indications of existing products (such as SOLIRIS as a treatment for NMOSD, the 100mg/mL formulation of ULTOMIRIS and ANDEXXA), we may not be able to accurately forecast demand for our products or for new indications and formulations. Product demand and, in the case of conversion to ULTOMIRIS, product preference and conversion, is dependent on a number of factors, many of which are beyond our control. For these reasons, we may not be able to accurately forecast demand for our products. You should not consider our financial performance, including our revenue growth, in recent periods as indicative of our future performance.
We cannot guarantee that we will achieve our financial goals, including our ability to maintain profitability on a quarterly or annual basis in the future.
In the future, we may not generate sufficient revenues or control expenses to achieve our financial goals. Our investors and investment analysts may have widely varying expectations that may be materially higher or lower than actual revenues and profits and if our revenues and profits are different from these expectations, our stock price may experience significant volatility. Our revenues and profits are also subject to foreign exchange rate fluctuations due to the global nature of our operations and our results of operations could be adversely affected due to unfavorable foreign exchange rates. Although we use derivative instruments to manage foreign currency risk, our efforts to reduce currency exchange losses may not be successful.
In addition, we have in the past provided, and expect to continue to provide, financial guidance and anticipated customers for certain indications for future periods and if our actual operating results fail to meet or exceed the guidance that we have previously provided to our investors, our stock price could drop
suddenly and significantly. Financial guidance and anticipated customer levels are based on certain assumptions about future performance and such guidance is not a guarantee that the targets set forth will be achieved. In addition, due to the potential impact of COVID-19 on our business, operations and results of operations (including our revenues), the estimates, judgments and inputs required to generate guidance and customer levels are increasingly uncertain and therefore accurately forecasting performance is even more challenging in light of the current health crisis.
As we attempt to grow and expand our business, we may have substantial expenses as we continue our research and development efforts and our efforts to develop the assets we have acquired through acquisitions, collaborations and in-licenses, continue to undertake additional business development activities, continue to conduct clinical trials and continue to develop and expand manufacturing, sales, marketing and distribution capabilities worldwide, some of which could be delayed, scaled-back or eliminated to control expenses and/or achieve our financial objectives. Additionally, business development activities may include milestone and royalty obligations and may require substantial investment in research and development to achieve product approval. These expenses may increase and such increases may exceed analyst and investor expectations.
If we fail to attract and retain highly qualified personnel, we may not be able to successfully develop, manufacture or commercialize our products or products candidates.
The success of our business is dependent in large part on our continued ability to attract and retain our senior management, and other highly qualified personnel in our scientific, clinical, manufacturing, governmental regulations and commercial organizations and across the many geographies in which we operate. There is intense competition in the biopharmaceutical industry for these types of personnel.
Our business is specialized and global and we must attract and retain highly qualified individuals across many geographies and areas of expertise. We may not be able to continue to attract and retain the highly qualified personnel necessary to develop, manufacture and commercialize our products and product candidates. If we are unsuccessful in our recruitment and retention efforts, or if our recruitment efforts take longer than anticipated, our business may be harmed.
We may not achieve some or all of the expected benefits of our current and future restructuring plans and restructurings may adversely affect our business.
We initiated our most recent restructuring in the third quarter 2020, which is focused within our commercial organization as part of an initiative intended to redefine our operating model and reallocate resources necessary to align our organization with our diversifying portfolio of new products and strategic objectives. We may undertake additional restructurings in the future. Implementation of a restructuring plan may be costly and disruptive to our business, and we may not be able to obtain the estimated cost savings and benefits that were initially anticipated in connection with a restructuring in a timely manner, or at all. Additionally, as a result of any restructuring, we may experience a loss of continuity, loss of accumulated knowledge and/or inefficiency during transitional periods. Reorganization and restructuring can require a significant amount of management and other employees’ time and focus, which may divert attention from operating and growing our business. If we fail to achieve some or all of the expected benefits of restructuring, it could have an adverse effect on our business, financial condition, results of operations and cash flows.
If we fail to satisfy our debt service obligations or our contingent obligations, we may be unable to commercialize our products or continue or complete our product development.
We have significant debt service obligations. In addition to the obligations to make interest and principal payments under our credit facility throughout the term of the loans, any changes in interest rates related to this debt could significantly increase our annual interest expense and any hedging of this interest may not be effective to control expenses.
Our Amended and Restated Credit Agreement requires us to comply with certain financial covenants and negative covenants, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, and engage in certain investment, acquisition and disposition transactions, subject to limited exceptions. If an event of default occurs (due to, for example, the failure to comply with certain covenants in the Amended and Restated Credit Agreement), the interest rate may increase and the administrative agent may be entitled to take various actions, including the acceleration of amounts due under the Amended and Restated Credit Agreement. If the interest rate imposed under our Amended and Restated Credit Agreement were to increase as a result of a default, our expenses may increase and we may need to allocate additional funds to this interest expense (which may limit the use of these funds for other purposes, including growing our business or responding to changes in our business and industry). If some or all of the amounts outstanding under the Amended and Restated Credit Agreement were to be
accelerated by the lenders, we may not have sufficient cash on hand to pay the amounts due, we may not be able to refinance such debt on terms acceptable to us (or at all) and we may be required to sell certain assets on terms that are unfavorable to us.
In addition, we have substantial contingent liabilities, including milestone and royalty obligations associated with acquisitions and strategic transactions, and we have been, and in the future may again be, engaged in disputes with certain counterparties regarding potential milestone and royalty obligations. Our increased indebtedness, including increased interest expense, together with our significant contingent liabilities, could, among other things:
•make us more vulnerable to economic or industry downturns and competitive pressures;
•make it difficult for us to make payments on our credit facilities and require us to use cash flow from operations to satisfy our debt obligations, which may reduce the availability of our cash flow for other purposes, including business development efforts and research and development;
•limit our ability to incur additional debt or access the capital markets; and
•limit our flexibility in planning for, or reacting to changes in, our business.
Our ability to satisfy our obligations under the Amended and Restated Credit Agreement and meet our debt service obligations and our royalty and milestone obligations will depend upon our future performance, which will be subject to financial, business and other factors affecting our operations, many of which are beyond our control.
We may not be able to access the capital and credit markets on terms that are favorable to us or at all.
We may need to raise additional capital to supplement our existing funds and cash generated from operations for working capital, capital expenditure and debt service requirements, and other business activities (including business and technology acquisitions). The amount of capital we may need depends on many factors, including, the cost of any acquisition or any new collaborative, licensing or other commercial relationships that we may establish, the time and cost necessary to build new manufacturing facilities or enhance our manufacturing and related operations, amounts we may need to pay in connection with the resolution of any government investigation or litigation matter (including any securities class action matter or any product liability claim or any tax assessment), the cost of obtaining and maintaining the necessary regulatory approvals
for our manufacturing facilities, and the progress, timing and scope of our preclinical studies, clinical trials and product development and commercialization efforts. The capital and credit markets have experienced and may continue to experience extreme volatility and disruption. We may not receive additional funding when we need it or funding may only be available on unfavorable terms. If we cannot raise adequate funds to satisfy our working capital, capital requirements and debt repayment obligations (or royalty and milestone obligations) or business development activities, we may have to delay, scale-back or eliminate certain research, development, manufacturing, acquisition or commercial activities or sell certain assets and technologies.
We have incurred significant impairment charges, and may continue to incur such charges in the future for certain of our assets, including goodwill in connection with acquisitions, and such amounts may be material.
If the purchase price of a business acquisition exceeds the value of the assets (and liabilities) acquired, the acquirer must recognize goodwill in such amount. We may be required to recognize impairment charges for our goodwill and other intangible assets, and such charges may be material and have an adverse impact on our financial results in the period such charges are incurred and may also have an adverse impact on our reputation.
As of September 30, 2020, the net carrying value of our goodwill and other intangible assets, net totaled $8,157.3. As required by GAAP, we evaluate goodwill and intangible assets for impairment on an annual basis, or as facts and circumstances warrant. We have recorded charges that include inventory write-downs for failed quality specifications or recalls, impairments with respect to investments and acquisitions, fixed assets and long-lived assets, outcomes of litigation and other legal or administrative proceedings, regulatory matters and tax matters, and payments in connection with acquisitions and other business development activities, such as milestone payments. The impairment of tangible and intangible assets may be triggered by developments both within and outside our control. Deteriorating economic conditions, technological changes, disruptions to our business, inability to effectively integrate acquired businesses, unexpected significant changes or planned changes in the use of the assets, adverse clinical results, intensified competition, divestitures, market capitalization declines and other factors may impair our goodwill and other intangible assets.
As part of our standard quarterly procedures, we review facts and circumstances regarding our long-lived assets, including the KANUMA asset, to assess for potential indicators of impairment. During the quarter ended June 30, 2020, based on continued
challenges expanding patient growth and new alternative commercial opportunities, the Company revised its strategic view of KANUMA and determined that we have exhausted commercially viable initiatives related to KANUMA and will have difficulty expanding patient growth over the long term as we focus on promoting other commercial programs and growing our pipeline. While management is committed to continued access to KANUMA for existing patients and providing access to future patients diagnosed with LAL-D, as we grow our business and product offerings, including through the recent acquisition of Portola Pharmaceuticals, we will prioritize programs where the opportunity to find patients who can benefit from Alexion therapies is the greatest. Therefore, we no longer expect to increase the number of KANUMA patients at the rate we previously assumed in our cash flow projections for KANUMA. As a result of these developments during the second quarter 2020, management adjusted assumptions in our long term cash flow forecast model for KANUMA and recognized an impairment charge of $2,042.3 related to the associated intangible asset.
Cash flow models used in our assessments of intangible assets are based on the projected commercial sales of the underlying products which considers, where applicable, our commercial experience with the product to date. Cash flow models for products currently in development also include the likelihood of approval. These cash flow models require the use of significant estimates and judgements, which include, but are not limited to, probability of regulatory approval, market access assumptions, long-range pricing expectations and patient-related assumptions, including patient identification, conversion and retention rates. As we continue to develop and sell products that have a related intangible asset associated with it, new data may cause us to adjust the assumptions in our cash flow models. Changes to assumptions used in our net cash flow projections may result in material impairment charges in subsequent periods, similar to the impairment charge recognized in the second quarter 2020 related to KANUMA.
The efficiency of our corporate structure depends on the application of the tax laws and regulations in the countries where we operate and we may have exposure to additional tax liabilities or our effective tax rate could increase, which could have a material impact on our results of operations and financial position.
As a company with international operations, we are subject to income taxes, as well as non-income based taxes, in both the U.S. and various foreign jurisdictions. Significant judgment is required in determining our worldwide tax liabilities. Although we believe our estimates are reasonable at the time made, the final taxes we owe may differ from the
amounts recorded in our financial statements (and such differences may be material). If the IRS, or other taxing authority, disagrees with the positions we take (and such tax authorities have disagreed with certain positions we have taken for prior years, and may do so again the future), we could have additional tax liability, and this could have a material impact on our results of operations and financial position. Our effective tax rate could be adversely affected by changes in the mix of earnings in countries with different statutory tax rates, changes in the valuation of deferred tax assets and liabilities, changes in tax laws and regulations, changes in interpretations of tax laws, including pending tax law changes, changes in our manufacturing activities and changes in our future levels of research and development spending.
We have designed, and from time to time we modify, our corporate structure, the manner in which we develop and use our intellectual property, and our intercompany transactions between our affiliates in a way that is intended to enhance our operational and financial efficiency and increase our overall profitability. The application of the tax laws and regulations of various countries in which we operate and to our global operations is subject to interpretation. We also must operate our business in a manner consistent with our corporate structure to realize such efficiencies. The tax authorities of the countries in which we operate may challenge our methodologies for valuing developed technology or for transfer pricing or other operations. If tax authorities determine that the manner in which we operate results in our business not achieving the intended tax consequences, our effective tax rate could increase (and such increase may be material) and harm our financial position and results of operations. In addition, certain governments are considering and may adopt tax reform measures that significantly increase our worldwide tax liabilities. The Organization for Economic Co-operation and Development and other government bodies have focused on issues related to the taxation of multinational corporations, including, in the area of “base erosion and profit shifting,” where payments are made from affiliates in jurisdictions with high tax rates to affiliates in jurisdictions with lower tax rates. It is possible that these reform measures could increase our effective tax rate (and such increase may be material) and harm our financial position and results of operations over the next several years.
Our sales and operations are subject to a variety of risks relating to the conduct of our international business.
We have increased our international presence, including in emerging markets. Our operations in foreign countries subject us to a variety of risks, including:
•difficulties or the inability to obtain necessary foreign regulatory or reimbursement approvals of our products in a timely manner or at all;
•political or economic determinations or decisions that adversely impact pricing or reimbursement policies in foreign countries;
•economic problems or political instability;
•fluctuations in currency exchange rates;
•difficulties or inability to obtain financing in markets;
•unexpected changes in tariffs, trade barriers and regulatory requirements;
•customs and tax officials in foreign jurisdictions may disagree with the value we set when we or others import our products (including products that are donated for charitable purposes or used for clinical trials) and we may be required to pay additional duties or fines and such amounts may be substantial. For example, our offices in Brazil were visited by the Brazilian federal tax authorities and we received a written notice from such authorities requesting information with respect to the importation of SOLIRIS free of charge to patients in Brazil from 2014 to 2019. In connection with this matter, in August 2019, the Brazilian Federal Revenue Service provided a Notice of Tax and Description of the Facts to, among others, two Alexion subsidiaries. This notice focuses on: (i) the identity of the importer and (ii) the importation value of SOLIRIS vials in connection with Alexion’s free drug program in Brazil. See Note 16, Commitments and Contingencies to the condensed consolidated financial statements for more information on this matter);
•difficulties in establishing and enforcing contractual and intellectual property rights;
•compliance with complex import and export control laws;
•trade restrictions and restrictions on direct investments by foreign entities;
•compliance with local tax, employment and labor laws;
•costs and difficulties in recruiting and retaining qualified managers and employees to manage and operate the business in local jurisdictions;
•costs and difficulties in managing and monitoring international operations; and
•longer payment cycles
Additionally, our business and marketing methods are subject to the laws and regulations of the countries in which we operate, which may differ significantly from country to country and may conflict with U.S. laws and regulations. The FCPA and anti-bribery laws and regulations in the locations in which we operate our business are extensive and far-reaching, and we must maintain accurate records and control over the activities of our employees, distributors and third party service providers in countries where we operate. We have policies and procedures, and we are committed to continually focusing on our compliance program and we continue to enhance our comprehensive company-wide program and efforts, and these are designed to enhance our business processes, structures, controls, training, talent, and systems across Alexion’s global operations and to help us and our representatives, including our employees and our vendors and distributors, comply with such laws. We cannot, however, guarantee that these policies, programs and procedures will protect us against liability under the FCPA or other anti-bribery laws for actions taken by us, our employees or our representatives. Any determination that our operations or activities are not in compliance with existing laws or regulations, including the FCPA and the UK Anti-Bribery Act, could result in the imposition of fines, civil and criminal penalties, equitable remedies, including disgorgement, injunctive relief, and/or other sanctions against us, and remediation of such findings could have a material and adverse effect on our business operations. In addition, as our international operations expand, we are likely to become subject to new anti-corruption/anti-bribery laws or existing laws may govern our activities in new jurisdictions in which we commence operations. In addition, as we move from a direct sales force to third-party sales force, distributors and marketers in certain countries and regions, we may also have liability under the FCPA and anti-bribery laws and regulations for the actions of these third parties. Although we can impose contractual restrictions on what these third parties are authorized to do on our behalf, we will exercise only limited control over the actions of these third parties but may still face the same liabilities for their actions. Our failure, and the failure of others who we engage to act on our behalf, to comply, with the laws and regulations of the countries in which we operate, or will operate in the future, could materially harm our business.
Our business involves environmental risks and potential exposure to environmental liabilities.
As a biopharmaceutical company, our business involves the use of certain hazardous materials in our research, development, manufacturing and other activities. We and our third party providers are subject to various federal, state, local and foreign
environmental laws and regulations concerning the handling and disposal of non-hazardous and hazardous wastes, such as medical and biological wastes, and emissions and discharges into the environment (including air, soils and water sources). We also are subject to laws and regulations that impose liability and clean-up responsibility for releases of hazardous substances into the environment and a current or previous owner or operator of property may be liable for the costs of remediating its property or locations, without regard to whether the owner or operator knew of or caused the contamination. Although our safety procedures for handling and disposing of hazardous materials are designed to comply with the laws and regulations established by state, federal, local and foreign regulators, the risk of loss of, or accidental contamination or injury from, these materials cannot be eliminated. If an accident or environmental discharge occurs, or if we discover contamination caused by prior owners and operators of properties we acquire, we could be liable for remediation obligations, damages and fines that could exceed our insurance coverage and financial resources. Such obligations and liabilities, which to date have not been material, could have a material impact on our business and financial condition. Additionally, the cost of compliance with environmental and safety laws and regulations may increase in the future, and we may be required to dedicate more resources, including substantial financial resources, to comply with such laws and regulations or purchase supplemental insurance coverage, which may not be available on acceptable terms or at all.
Currency fluctuations and changes in exchange rates could adversely affect our revenue, increase our costs and negatively affect our profitability.
We conduct a substantial portion of our business in currencies other than the U.S. dollar. We are exposed to fluctuations in foreign currency exchange rates and such fluctuations affect our operating results. The exposures result from portions of our revenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, including the Euro, Japanese Yen, British Pound, Canadian dollar and Turkish Lira. We cannot predict fluctuations in currency exchange rates and such fluctuations in exchange rates (and inflation) could negatively affect our business, cash flow, results of operations, financial position and prospects. We manage a portion of our foreign currency transaction risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. While our hedging agreements may limit some of the exposure to exchange rate and interest rate fluctuations, such
attempts to mitigate these risks may be costly and not always successful and the results may have a material impact on our results of operations.
Risks Related to the Regulatory Environment
We operate in a highly regulated industry and if we or our third-party providers fail to comply with U.S. and foreign regulations, we or our third party providers could lose our approvals to market our products or our product candidates, and our business may be seriously harmed.
We and our current and future third-party vendors, including contract manufacturers, CROs, distributors and suppliers and logistic providers are subject to rigorous and extensive regulation by governmental authorities around the world, including the FDA, EMA, the competent authorities of the EU Member States and the MHLW. These regulations, many of which are complex, relate to almost all aspects of our business, including GCP, GLP, cGMP and pharmacovigilance rules (for additional information on the regulations relating to our business, see “Business - Government Regulation” in Item 1 in our Annual Report on Form 10-K for the year ended December 31, 2019). If we or a regulatory agency discover previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured (such as product contamination), or in the case of KANUMA, problems with animal operations, a regulatory agency may impose restrictions on that applicable product, the manufacturing facility or us. In 2013, we received a Warning Letter from the FDA relating to compliance with FDA’s cGMP requirements at one of our facilities, which was remediated. If we had failed to address the FDA’s concerns or if we (or one of our third-party contract manufacturers) were to receive another Warning Letter in the future relating to cGMP or other applicable regulations, the FDA or other regulatory authorities could take regulatory action, including fines, civil penalties, recalls, seizure of product, suspension of manufacturing operations, operating restrictions, injunctions, suspension of clinical trials, withdrawal of FDA (or other regulatory authority) approval and/or criminal prosecution.
If we or our third-party providers, including our product or raw material manufacturers, product fill-finish providers, packagers and labelers, fail to comply fully with applicable regulations, then we may be required to, among other things, initiate a recall or withdrawal of our products. In addition to our manufacturing operations and those of contract manufacturers’ manufacturing operations being subject to inspection and potential regulatory action for failure to comply with (among other regulations) cGMP, our animal operations may also be subject to FDA and U.S. Department of Agriculture, Animal and
Plant Health Inspection Service (USDA APHIS) inspection to evaluate whether our animal husbandry, containment, personnel, and record keeping practices are sufficient to ensure safety and security of our transgenic chickens and animal products (e.g., eggs, waste, etc.). Any failure to ensure safety and security of our transgenic chickens and/or animal products could result in regulatory action by the FDA or another regulatory body, including USDA APHIS.
Failure to comply with the laws and requirements that apply to our business, including statutes and regulations, administered by the FDA, the EC, the competent authorities of the EU Member States, the MHLW or other comparable agencies, could result in:
•a product recall;
•a product withdrawal;
•modification or revision to a product label;
•significant administrative and judicial sanctions, including, warning letters or untitled letters;
•significant fines and other civil penalties;
•suspension, variation or withdrawal of a previously granted approval for our products;
•interruption, suspension or termination of production;
•operating restrictions, such as a shutdown of production facilities or production lines, or new manufacturing requirements;
•suspension or termination of ongoing clinical trials;
•delays in approving or refusal to approve our products including pending BLAs, NDAs or BLA or NDA supplements for our products or a facility that manufactures our products;
•seizing or detaining product;
•requiring us or third-parties performing services for us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance;
•injunctions; and/or
•criminal prosecution.
In addition, we are subject to antitrust regulations with respect to our acquisitions, as well as our interactions with other participants in the markets we currently serve or may serve in the future. In addition, these antitrust laws are vigorously enforced in the U.S. and in other jurisdictions in which we operate. In connection with any business development transaction, acquisition, development agreement or collaboration that is reviewed by regulatory authorities, there can be no assurance that
these antitrust approvals will be obtained. In addition, the governmental entities from which these approvals are required may impose conditions on the completion of such business development transaction or require changes to the terms of the applicable transaction. Regulatory review process may cause a delay in the closing of a transaction beyond the time that we anticipate and communicate. Any conditions or unexpected delays in approval could have the effect of jeopardizing or delaying completion of the applicable transaction or reducing the anticipated benefits of the transaction (or, as noted above, may prohibit closing of the transaction).
Our product candidates require extensive clinical testing and regulatory approval and failure to satisfy regulatory requirements relating to safety and efficacy thresholds may prevent us from being able to market our products and limit our ability to grow our business and diversify our revenue.
We believe our future success may depend on our ability to develop and commercialize our product candidates and, to this end, we have recently acquired companies and technologies in an effort to expand our product pipeline. Our product candidates are in various stages of development and must satisfy the rigid safety and efficacy requirements of the FDA and other foreign regulatory agencies before they can be approved for sale to patients. To satisfy these standards, we must ensure, among other things, that we have appropriately established our protocol designs, obtained the necessary IRB approval, provide adequate patient enrollment rates, timely and appropriately report any adverse events and serious adverse events to the appropriate authorities and ensure compliance with cGCP. If we or our third-party clinical trial providers or third-party CROs do not successfully carry out these clinical activities, our clinical trials or the potential regulatory approval of a product candidate may be delayed or be unsuccessful.
If we discover safety or safety reporting issues with any of our approved products, or if we fail to comply with continuing U.S. and applicable foreign regulations as they relate to our products and operations, our revenue may decrease, an approved product could lose its marketing approval or sales could be suspended and our business could be materially harmed.
Following marketing approval of a pharmaceutical product, the safety profile of such product continues to be closely monitored by the FDA and other foreign regulatory authorities. Regulations continue to apply after product approval, and cover, among other things, testing, manufacturing, quality control, finishing, filling, labeling, advertising, promotion, risk mitigation, adverse event reporting requirements and export of biologics and small molecule compounds. Included in the post-approval marketing requirements are, for
example, the REMS program for both SOLIRIS and ULTOMIRIS in the U.S., and a REMS program can be updated from time to time by the FDA and such updates can be costly and burdensome to implement. In addition, continued approval for ANDEXXA for its currently approved indication in the US is contingent upon post-marketing study results that verify that clinical benefit is conferred to patients.
We are required to report any serious and unexpected adverse experiences and certain quality problems with our products to the FDA, the EMA, the MHLW and other health agencies. Adverse safety events involving our products may have a negative impact on our business. Discovery of safety issues with our products could result in product liability claims and could cause additional regulatory scrutiny and requirements for additional labeling or safety monitoring, withdrawal of products from the market and the imposition of fines or criminal penalties. In addition, governmental authorities are making greater amounts of safety information directly available to the public through periodic safety update reports, patient registries and other reporting requirements. The reporting of adverse safety events may also damage physician, patient and/or investor confidence in our products and our reputation. Any adverse events in connection with the use of our products could result in liabilities, loss of revenues, material write-offs of inventory, material impairments of intangible assets, goodwill and fixed assets, material restructuring charges, product liability claims and other adverse impacts on our results of operations.
Regulatory agencies periodically inspect our pharmacovigilance processes. If these regulatory agencies determine that we or other parties whom we do not control that perform pharmacovigilance-related services on our behalf, including clinical trial investigators, have not complied with the applicable reporting or other pharmacovigilance requirements, we may become subject to additional inspections, warning letters or other enforcement actions, including monetary fines, marketing authorization withdrawal and other penalties.
As a condition of approval for marketing our products, governmental authorities may require us to conduct additional studies. In connection with the approval of SOLIRIS we established a PNH Registry and an aHUS Registry to collect additional data on patients. Furthermore, in connection with the approval of STRENSIQ in the U.S., we agreed to conduct a prospective observational study in treated patients to assess the long-term safety of STRENSIQ therapy and to develop complementary assays. In the EU, in connection with the grant of authorization for STRENSIQ, we agreed to conduct a study of STRENSIQ in patients with HPP and to extend the studies ENB-008-10 and ENB-009-10 to provide
efficacy data in patients 13 to 18 years of age, and we completed this commitment to the EU. In the U.S., the FDA can also propose to withdraw approval for a product if it determines that such additional studies are inadequate or if new clinical data or information shows that a product is not safe for use in an approved indication.
In addition, similar or more stringent post-approval requirements and obligations may be imposed by the FDA and/or other regulatory agencies with respect to any of our future products that obtain regulatory approval. Compliance with these post-approval requirements could result in increased cost and expense and decrease our operating margins and, if we are unable to comply with these requirements, we may be subject to regulatory action by the applicable regulatory agency and the penalties may include fines and product withdrawals or restrictions in the use of a product.
If we fail to comply with applicable healthcare laws and regulations, including those related to healthcare fraud and abuse, we may be subject to investigations and civil or criminal penalties and our business could be adversely affected.
We are subject to healthcare “fraud and abuse” laws, such as the False Claims Act (FCA), the anti-kickback provisions of the federal Social Security Act, laws prohibiting off-label product promotion and other related federal and state laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, in cash or in kind to induce, or reward the purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid, or other federal healthcare programs. Liability may be established without a person or entity having actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. A conviction for violation of the Anti-kickback Statute requires mandatory exclusion from participation in federal healthcare programs. The majority of states also have statutes similar to the federal Anti-Kickback Statute and false claims laws that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer.
The FCA prohibits any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim. Pharmaceutical companies have been investigated and have reached substantial financial settlements with the Federal government under the FCA for a variety of alleged promotional and
marketing activities, such as allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees and other benefits to physicians to induce them to prescribe products; engaging in promotion of pharmaceuticals for uses that the FDA has not approved, or “off-label” uses; and submitting inflated best price information to the Medicaid Rebate Program.
We seek to comply with the Anti-Kickback Statute and FCA laws, including operating within any available safe harbors, but we cannot assure that our compliance program, policies and procedures will always protect us from acts committed by employees or third-party distributors or service providers.
There is also enhanced scrutiny of company-sponsored patient assistance programs, including insurance premium and co-pay assistance programs and donations to third-party charities that provide such assistance. In 2019, we settled an investigation by the Department of Justice relating to our support for 501(c)(3) entities. If we, or our vendors or donation recipients, are deemed to fail to comply with relevant laws, regulations or government guidance in the operation of these programs again in the future, we could be subject to significant fines or penalties.
Other related federal and state laws and regulations that may affect our ability to operate include, among others, the federal False Statements Statute, the federal Civil Monetary Penalties Law, HIPAA, the federal Open Payments program, state anti-kickback and false claims acts, and state and local disclosure requirements and marketing restrictions. Additional information about the scope of these requirements and potential penalties is provided under “Government Regulation - Fraud and Abuse” included in Part I, Item 1 in our Annual Report on Form 10-K for the year ended December 31, 2019.
In recent years, legislation has been adopted at the federal, state and local level requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports or make periodic public disclosures on sales, marketing, pricing, clinical trials, health care provider payments and other activities. For example, as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the PPACA), the federal government enacted the Open Payments (commonly known as the Sunshine Act) provisions. Open Payments requires pharmaceutical manufacturers to report annually to CMS payments or other transfers of value made by that entity to physicians and teaching hospitals. We also now have similar reporting obligations throughout the EU. Failure to comply with the reporting requirements may result in significant civil monetary penalties.
Violations of U.S. federal and state fraud and abuse laws (and comparable laws in foreign jurisdictions) may result in criminal, civil and administrative sanctions, including fines, damages, civil monetary penalties (which may be material in amount) and exclusion from federal healthcare programs (including Medicare and Medicaid). Any action initiated against us for violation of these laws, even if we successfully defend against it, could require the expenditure of significant resources and generate negative publicity, which could materially adversely affect our ability to operate our business and our financial results.
Finally, the FDA, the EU and EU Member States and the MHLW, among other regulatory agencies, impose restrictions on the promotion and marketing of drug products and prohibit pharmaceutical manufacturers from promoting products for indications other than those cleared or approved by regulatory authorities or for use in manner that is not consistent with the product label approved by regulatory agencies, or off-label promotion. In certain instances, physicians are, however, in their medical judgment permitted to use products for unapproved purposes and we are aware, for example, of such uses of SOLIRIS. Although we believe that our marketing materials and training programs for physicians do not constitute improper promotion, the FDA, the DOJ, other federal or state government agencies, the EU, EU Member States or the MHLW (or other foreign regulatory agencies) may disagree. If any governmental authority determines that our promotional materials, training or other activities constitute improper promotion of any of our products, it could request that we modify our training or promotional materials (which occurred in 2019 in Japan) or other activities or subject us to regulatory enforcement actions, including the issuance of a warning letter, product withdrawal or recall, injunction, seizure, civil fine and criminal penalties. It is also possible that other enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false or fraudulent claims for payment of government funds.
Our business and operations may be materially adversely affected by government investigations.
We are subject to the FCPA, the U.K. Bribery Act and other anti-corruption laws and regulations that generally prohibit companies and their intermediaries from making improper payments to government officials and/or other persons for the purpose of obtaining or retaining business and we operate in countries that are recognized as having a greater potential for governmental and commercial corruption.
While we have, and continue to, take steps that are intended to enhance our compliance and training programs, we cannot assure that our compliance program, policies and procedures will always protect us from acts committed by employees or third-parties acting on our behalf.
In May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the SEC requesting information related to our grant-making activities and compliance with the FCPA in various countries, including Brazil, Colombia, Japan, Russia and Turkey. In addition, in October 2015, we received a request from the DOJ for the voluntary production of documents and other information pertaining to our compliance with the FCPA. The SEC and DOJ also sought information related to our recalls of specific lots of SOLIRIS and related securities disclosures. DOJ informed us that it closed its inquiry into these matters. We settled the investigation with the SEC in July 2020, and made payment of approximately $21.5 in disgorgement, civil penalties, and pre-judgment interest in connection with the settlement. See Note 16, Commitments and Contingencies to our condensed consolidated financial statements included elsewhere in this Quarterly Report on Form 10-Q for additional information on this matter.
In May 2017, Brazilian authorities seized records and data from our Sao Paulo, Brazil offices as part of an investigation being conducted into our Brazilian operations. At this time, we are unable to predict the duration, scope or outcome of the open investigations. In addition, even though we have settled the DOJ investigation relating generally to our support of certain 501(c)(3) organizations that was initiated by the U.S. Attorney’s Office for the District of Massachusetts in December 2016 and the MHLW closed its 2018 investigation into our Japanese operations, we may be subject to similar investigations in the future by the same or other regulatory agencies and government authorities and the penalties imposed on us may be materially greater in amount or we may be subject to material limitations on our operations, activities and our business. In addition, any remedial actions that have been or will be taken with the intent to address the matters that were the subject of these or other governmental investigations may not prevent future investigations and potential liability as a result of such further investigations.
In addition, while we settled the SEC's investigation in July 2020 as described above (and the DOJ closed its inquiry into these matters), any future determination that our operations or activities are not, or were not, in compliance with existing U.S. or foreign laws or regulations could result in the imposition of a broad range of civil and criminal
sanctions against us and certain of our directors, officers and/or employees, including injunctive relief, disgorgement, substantial fines or penalties, imprisonment, and other legal or equitable sanctions, including exclusion from Medicare, Medicaid, and other governmental healthcare programs. In addition, settlement of investigations may lead to additional government actions. For example, following the settlement with the SEC in July 2020, the Ministry of Health in Turkey initiated an investigation regarding the matters referenced in the SEC Order as they relate to the Company’s operations in Turkey between 2010 and 2015. Any attempts to resolve some or all of any such matters in the future may not be successful. If we were to engage in settlement discussions with respect to any current or future investigation or litigation (and we may accrue amounts due to the nature of such discussions), but the matter is not settled, the ultimate resolution may result in monetary or other penalties materially greater or stricter than the amounts or terms that we proposed in discussions (or the amount that we accrued for such matter during negotiations). Additionally, remediation of any such findings resulting from any past or future investigations could have an adverse effect on our business operations, and we could experience interruptions of business, harm to our reputation, debarment from government contracts, loss of supplier, vendor or other third-party relationships, and necessary licenses and permits could be terminated. Other internal or government investigations or legal or regulatory proceedings, including lawsuits brought by private litigants, may also follow as a consequence. Cooperating with and responding to requests for information in connection with past and ongoing investigations, as well as responding to any future U.S., state or foreign governmental investigation or whistleblower lawsuit, has resulted and could continue to result in substantial expenses, and could divert management’s attention from other business concerns and could have a material adverse effect on our business and financial condition and growth prospects.
Our business could be adversely affected by litigation and regulatory enforcement actions.
We operate in many jurisdictions in a highly regulated industry and we could be subject to litigation, government investigations (as noted above) and enforcement and other legal actions on a variety of matters in the U.S. or foreign jurisdictions, including, without limitation, intellectual property, regulatory, product liability, tax and custom/import duties, environmental, whistleblower, Qui Tam, false claims, privacy, anti-kickback, anti-bribery, securities, commercial, product pricing, employment and other claims and legal proceedings which may arise from conducting our business. We are involved in certain legal proceedings. See Note 16, Commitments and
Contingencies to our condensed consolidated financial statements included elsewhere in this Quarterly Report on Form 10-Q for information on certain of these legal proceedings. In addition, in connection with any acquisitions, we may assume potential liability related to pending legal proceedings of the acquired company. For example, securities class action complaints were filed against Portola and certain officers of Portola alleging violation of the antifraud provisions of the Exchange Act of 1934 and the Securities Act of 1933 due to misrepresentations and omissions in public disclosures concerning sales of andexanet alfa between January 8, 2019 and February 26, 2020. Legal proceedings are inherently unpredictable, and the outcome can result in costly verdicts, fines and penalties, exclusion from federal healthcare programs and/or injunctive relief that affect how we operate our business. Defense of litigation claims can be expensive, time consuming and distracting, and it is possible that we could incur judgments or enter into settlements of claims for monetary damages or change the way we operate our business, which could have a material adverse effect on our product sales, business and results of operations. In addition, product liability is a major risk in testing, selling, using and marketing biotechnology and pharmaceutical products. We may face potential product liability exposure in human clinical trials and for products we sell after regulatory approval. Product liability claims, regardless of their merits, could be costly and divert management’s attention and could adversely affect our reputation and the demand for our products and result in significant monetary liability.
Changes in healthcare laws and implementing regulations, as well as changes in healthcare policy, may affect coverage and reimbursement of our products in ways that we cannot currently predict and these changes could adversely affect our business and financial condition.
In the U.S., there have been a number of legislative and regulatory initiatives focused on containing the cost of healthcare. The PPACA, for example, substantially changed the way healthcare is financed by both governmental and private insurers in the U.S., and significantly impacts the pharmaceutical industry. The PPACA contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, fraud and abuse enforcement and rules governing the approval of biosimilar and generic products (and allowing biosimilars access to the market in
accordance with the FDA’s Biosimilars Action Plan). These changes may impact existing government healthcare programs, industry competition, formulary composition, and may result in the development of new programs, including Medicare payment for performance initiatives, health technology assessments and improvements to the physician quality reporting system and feedback program. In 2016, CMS implemented changes to the Medicaid Drug Rebate Program under the PPACA. Moreover, in the future, Congress could enact legislation that further increases Medicaid drug rebates or other costs and charges associated with participating in the Medicaid Drug Rebate Program. The issuance of regulations and coverage expansion by various governmental agencies relating to the Medicaid Drug Rebate Program has and may continue to decrease revenues, increase our costs and the complexity of compliance, has been and may be time-consuming to comply with, and could have a material adverse effect on our results of operations.
Similar efforts to those in the United States, and in some cases even more aggressive efforts, are being taken by governments to control the costs of pharmaceutical drugs and regulate the industry in countries outside the U.S. In these markets outside the U.S., the pricing and reimbursement of pharmaceutical products is subject to direct or indirect governmental control and such government authorities are increasingly attempting to limit or regulate the price of drug products and due to their control over pricing are able to move quickly to implement pricing changes. In certain cases, governments may challenge the price we charge for our products already delivered to patients under applicable regulations in those countries (and if these governments prevail, we could be required to return amounts to the government or the government may take steps in an attempt to claw-back amounts that were previously paid to us and such amounts may be material).
We may face uncertainties as a result of federal and administrative efforts to repeal, substantially modify or invalidate some or all of the provisions of the PPACA. There is no assurance that the PPACA, as currently enacted or as amended in the future, will not adversely affect our business and financial results, and we cannot predict how future federal or state or foreign legislative or administrative changes relating to healthcare reform may affect our business. The recent COVID-19 pandemic may introduce temporary or permanent healthcare reform measures for which we cannot predict the financial implication on our business.
State governments have sought to put in place limits and caps on pharmaceutical prices and have also requested rebates for certain pharmaceuticals.
Attempts to decrease prices of pharmaceutical products may lead to increased use of managed care organizations by Medicaid programs which could lead to managed care organizations influencing prescription decisions for beneficiaries and a corresponding limitation on prices and reimbursement for our products.
Governments in countries where we operate have adopted or have also shown significant interest in pursuing legislative initiatives to reduce costs of healthcare. We expect that the implementation and enforcement of current laws and policies, the amendment of those laws and policies in the future, as well as the adoption of new laws and policies, could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales or revenues or successfully commercialize our product candidates, or could limit or eliminate our future spending on development projects and product candidates. The announcement or adoption of regulatory or legislative proposals could delay or prevent our entry into new markets, affect our reimbursement or sales in the markets where we are already selling our products and materially harm our business, financial condition and results of operations.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program, Medicare, or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We participate in and have certain price reporting obligations to the Medicaid Drug Rebate Program and we have obligations to report the average sales price under the Medicare program. Under the Medicaid Drug Rebate Program, we are required to pay a rebate to each state Medicaid program for quantities of our products that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our products under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS. Any failure to comply with these price reporting and rebate payment obligations could negatively impact our financial results.
Pricing and rebate calculations vary among products and programs. The calculations are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. We cannot assure you that our submissions will not be found by CMS or other applicable government authorities to be incomplete or incorrect. Governmental agencies may also make changes in
program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. Recalculations increase our costs for complying with the laws and regulations governing these programs, including the Medicaid Drug Rebate Program. Any corrections to our rebate calculations could result in an underage in our rebate liability for past quarters, and such amount may be material. Price recalculations also may affect the ceiling price at which we are required to offer our products to certain covered entities under the 340B pricing program.
We are liable for errors associated with our submission of pricing data. In addition to retroactive rebates and the potential for 340B program refunds, civil monetary penalties can be applied if we are found to have knowingly submitted any false pricing information to the government, if we are found to have made a misrepresentation in the reporting of our average sales price, or if we fail to submit the required pricing data on a timely basis. Such conduct also could be grounds for CMS to terminate our Medicaid drug rebate agreement, pursuant to which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement, federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs and any such actions could negatively impact our business and results of operations.
The Public Health Service’s 340B drug pricing program, and other comparable government and payer regulations, may have a negative impact on the price we can charge for our products and result in a decrease in revenues.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. The 340B pricing program is described in Pharmaceutical Pricing and Reimbursement in Item 1 Business in our Annual Report on Form 10-K for the year ended December 31, 2019. The 340B ceiling price is calculated using a statutory formula, which is based on, among other prices, the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. We are a participant in the 340B drug pricing program and are, for the applicable covered entities, subject to the price ceiling. Any changes to the 340B drug pricing program, including:
•the method of calculating the 340B ceiling price for our products;
•any expansion of the entities that qualify as covered entities; and
•any requirement that participating manufacturers agree to provide 340B discounted pricing on drugs used in an inpatient setting;
could have a material and negative impact our revenue and results of operations.
Pursuant to a final rule adopted on January 1, 2019, we could be subject to civil monetary penalties if the government finds that we knowingly and intentionally overcharged a 340B covered entity.
In addition, the agreement that manufacturers must sign to participate in the 340B pricing program obligates a manufacturer to offer the 340B price to covered entities if the manufacturer makes the drug available to any other purchaser at any price and to report to the government the ceiling prices for its drugs.
Beyond the Public Health Service’s 340B drug pricing program, federal law requires that a company must participate in the Department of Veterans Affairs Federal Supply Schedule (FFS) pricing program to be eligible to have its products paid for with federal funds. If we overcharge the government in connection with our FSS contract or Section 703 Agreement, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the FCA and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action with respect to our pricing or participation in government health programs, may be expensive, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which have affected and may affect our business. In the U.S., numerous federal and state laws and regulations, including state security breach notification laws, state health information privacy laws, and federal and state consumer protection laws, govern the collection, use, disclosure, and protection of personal information. Each of these laws is subject
to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations, we could be subject to penalties or sanctions, including criminal penalties if we, our affiliates, or our agents knowingly obtain or disclose individually identifiable health information from a HIPAA covered entity in a manner that is not authorized or permitted by HIPAA.
Numerous other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. Further, the EU's General Data Protection Regulation (GDPR) and implementing laws in the EU member states that govern the collection and processing of EU residents' personal data and among other requirements, imposes certain consent and data access rights. Such laws may impact, among other things, our ability to conduct clinical trials that involve EU personal data and engage in other activities that require the processing of EU personal data. These laws are complex, subject to interpretation by local authorities, and any determination that we breached such laws could lead to government enforcement actions, significant penalties, and these may adversely impact our operating results.
In May 2018, the GDPR, which applies in all EU Member States, went into effect. The regulation introduced comprehensive data protection requirements in the EU and substantial fines for breaches of the data protection rules. It increased our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new EU data protection rules.
Security breaches, cyber-attacks or other disruptions could expose us to liability and affect our business and reputation.
We are increasingly dependent on our information technology systems and infrastructure for our business. We collect, store and transmit sensitive information including intellectual property, proprietary business information and personal information in connection with business operations. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be an attractive target of criminal attack by third parties with a wide range of motives and expertise, including organized criminal groups, “hacktivists,” patient groups, disgruntled current or former employees and others. Cyber-attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be vulnerable to such attacks or may be breached, including due to employee error or malfeasance. We have implemented information security measures designed to protect patients’
personal information and other corporate information (including proprietary information) against the risk of inappropriate and unauthorized external use and disclosure. However, despite these measures, and due to the ever-changing information cyber-threat landscape, we may be subject to data breaches through cyber-attacks. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. If our information technology systems become compromised, we may not promptly discover the intrusion. Like other companies in our industry, we have experienced attacks to our data and systems, including malware and computer viruses. If our systems failed or were breached or disrupted, we could lose product sales, and suffer reputational damage and loss of customer confidence. Such incidents may result in notification obligations to affected individuals and government agencies, legal claims or proceedings, and liability under foreign, federal and state laws that protect the privacy and security of personal information. Our proprietary and confidential information may also be accessed. Any one of these events could cause our business to be materially harmed and our results of operations may be adversely impacted.
Additionally, in response to the ongoing COVID-19 pandemic, we have generally required all employees who are able to work from home to do so until further notice. As a result of these measures, and as our employees continue to work from home and access our systems remotely, we may be subject to heightened information security risks, including the risk of cyber attacks.
Negative public opinion and increased regulatory scrutiny of recombinant and transgenic products, genetically modified products and genetically modified animals generally may damage public perception of our KANUMA product.
KANUMA is a transgenic product produced in the egg whites of genetically modified chickens who receive copies of the human lysosomal acid lipase gene to produce recombinant human lysosomal acid lipase. The success of KANUMA may depend, in part, on public attitudes of the use of genetic engineering. Public attitudes may be influenced by claims and perceptions that these types of activities or products are unsafe, and our products may not gain sufficient acceptance by, or fall out of favor with, the public or the medical community. Negative public attitudes to genetic engineering activities in general could result in more restrictive legislation or regulations and could impede our ability to conduct our business, delay preclinical or clinical studies, or otherwise prevent us from commercializing our product.
Risks Related to Our Common Stock
Our stock price is volatile.
The trading price of our common stock has been volatile and may continue to be volatile in the future. Many factors could have an impact on our stock price, including fluctuations in our or our competitors’ operating results, clinical trial results or adverse events associated with our products or our competitors' products, product development by us or our competitors, changes in laws, including healthcare, tax or intellectual property laws, intellectual property developments, changes in reimbursement or drug pricing, the existence or outcome of litigation or government proceedings, including the Chugai lawsuits alleging patent infringement, acquisitions or other strategic transactions, and the perceptions of our investors that we are not performing or meeting expectations. In addition, the sales of our common stock by our officers, directors, or by any entities that an officer or director may be affiliated with, may have caused our stock price to drop in the past and any future sales by such officer, director or affiliate (or the perception that such sales could occur) may have a negative impact on our stock price. The trading price of the common stock of many biopharmaceutical companies, including ours, has experienced price and volume fluctuations, which have at times been unrelated to the operating performance of the companies whose stocks were affected.
Anti-takeover provisions in our charter and bylaws and under Delaware law could make a third-party acquisition of us difficult and may frustrate any attempt to remove or replace our current management.
Our corporate charter and by-law provisions may discourage certain types of transactions involving an actual or potential change of control that might be beneficial to us or our stockholders. Our bylaws provide that special meetings of our stockholders may be called only by the Chairman of the Board of Directors, the President, the Secretary, or a majority of the Board of Directors, or upon the written request of stockholders who together own of record 25.0% of the outstanding stock of all classes entitled to vote at such meeting. Our bylaws also specify that the authorized number of directors may be changed only by resolution of the Board of Directors. Our charter does not include a provision for cumulative voting for directors, which may have enabled a minority stockholder holding a sufficient percentage of a class of shares to elect one or more directors. Under our charter, our Board of Directors has the authority, without further action by stockholders, to designate up to five million shares of preferred stock in one or more series. The rights of the holders of common stock will be subject to, and may be adversely affected by, the rights of the holders of any class or series of preferred stock that may be issued in the future.
Because we are a Delaware corporation, the anti-takeover provisions of Delaware law could make it more difficult for a third party to acquire control of us, even if the change in control may be beneficial to stockholders. We are subject to the provisions of Section 203 of the Delaware General Laws, which prohibits a person who owns in excess of 15.0% of our outstanding voting stock from merging or
combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15.0% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Item 2.UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS.
ISSUER PURCHASE OF EQUITY SECURITIES (amounts in millions, except per share amounts)
The following table summarizes our common stock repurchase activity during the third quarter 2020:
| | | | | | | | | | | | | | | | | | | | | | | |
| Period | Total Number of Shares Purchased | | Average Price Paid per Share | | Total Number of Shares Purchased as Part of Publicly Announced Programs | | Maximum Dollar Value of Shares that May Yet Be Purchased Under the Program |
| July 1-31, 2020 | — | | | $ | — | | | — | | | $ | 2,174.7 | |
| August 1-31, 2020 | 0.2 | | | $ | 102.63 | | | 0.2 | | | $ | 2,148.0 | |
| September 1-30, 2020 | 0.4 | | | $ | 111.59 | | | 0.4 | | | $ | 2,101.1 | |
| Total | 0.6 | | | $ | 108.16 | | | 0.6 | | | |
In November 2012, our Board of Directors authorized a share repurchase program. In February 2017, our Board of Directors increased the amount that we are authorized to expend on future repurchases to $1,000.0 under our repurchase program, which superseded all prior repurchase programs. The entire amount authorized pursuant to this February 2017 Board approval has been utilized. On October 22, 2019, the Board of Directors approved a share repurchase authorization of up to $1,000.0. On July 28, 2020, the Board of Directors approved a new share repurchase authorization of up to an additional $1,500.0. The repurchase program does not have an expiration date and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at our discretion.
Item 5.OTHER INFORMATION.
None.
Item 6.EXHIBITS.
(a)Exhibits:
| | | | | |
| Confidential Release and Separation Agreement by and between Anne-Marie Law and Alexion Pharmaceuticals, Inc. effective September 26, 2020. |
| |
| Certificate of Chief Executive Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 Sarbanes Oxley Act of 2002. |
| |
| Certificate of Chief Financial Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 of Sarbanes Oxley Act of 2002. |
| |
| Certificate of Chief Executive Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act. |
| |
| Certificate of Chief Financial Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act. |
| |
| 101 | | The following materials from the Alexion Pharmaceuticals, Inc. Quarterly Report on Form 10-Q for the quarter ended September 30, 2020 formatted in Inline eXtensible Business Reporting Language (iXBRL): (i) the Condensed Consolidated Balance Sheets as of September 30, 2020 and December 31, 2019, (ii) the Condensed Consolidated Statements of Operations for the three and nine months ended September 30, 2020 and 2019, (iii) the Condensed Consolidated Statements of Comprehensive Income for the three and nine months ended September 30, 2020 and 2019, (iv) the Condensed Consolidated Statements of Cash Flows for the nine months ended September 30, 2020 and 2019, (v) the Condensed Consolidated Statements of Changes in Stockholders' Equity the three and nine months ended September 30, 2020 and 2019, and (vi) Notes to Condensed Consolidated Financial Statements. |
| 104 | The cover page from this Quarterly Report on Form 10-Q for the quarter ended September 30, 2020, formatted in Inline XBRL. |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | |
| ALEXION PHARMACEUTICALS, INC. |
| | |
| By: | /s/ Ludwig N. Hantson, Ph.D. |
| Date: October 29, 2020 | | Ludwig N. Hantson, Ph.D.
Chief Executive Officer
(principal executive officer) |
| | |
| By: | /s/ Aradhana Sarin, M.D. |
| Date: October 29, 2020 | | Aradhana Sarin, M.D.
Executive Vice President and Chief Financial Officer
(principal financial officer) |