Exhibit 10.32
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
AMENDED AND RESTATED AGREEMENT FOR THE SALE AND ASSIGNMENT OF RIGHTS
DATED AS OF DECEMBER 20, 2013
BY AND BETWEEN
NPS PHARMACEUTICALS, INC.
AND
DRUG ROYALTY L.P. 3
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
TABLE OF CONTENTS
1 |
DEFINITIONS | 2 |
| 1.1 | Defined Terms | 2 |
| 1.2 | Interpretation | 6 |
| 1.3 | Currency | 7 |
| 1.4 | Additional Definitions | 7 |
2 |
ROYALTY GRANT; CONSENT; AMENDMENT AND RESTATEMENT OF ORIGINAL PURCHASE AGREEMENT | 8 |
| 2.1 | Royalty Grant | 8 |
| 2.2 | Consent to the Revenue Interest Arrangement | 8 |
| 2.3 | Conditional Amendment and Restatement of the Original Purchase Agreement | 8 |
| 2.4 | Mutual Release | 9 |
| 2.5 | Tolling | 9 |
| 2.6 | Accrued Claims | 10 |
3 |
THE CLOSING | 10 |
| 3.1 | The Closing | 10 |
| 3.2 | NPS Closing Deliveries | 10 |
| 3.3 | DR3 Closing Deliveries | 11 |
4 |
REPRESENTATIONS, WARRANTIES OF NPS | 11 |
| 4.1 | Organization, Standing and Power | 11 |
| 4.2 | Authority, Execution and Delivery; Enforceability | 11 |
| 4.3 | No Conflicts | 11 |
| 4.4 | No Consent | 12 |
| 4.5 | Rights in DR3 Royalty Right | 12 |
| 4.6 | Agreements | 12 |
| 4.7 | Patents and Other Intellectual Property | 12 |
| 4.8 | Litigation | 13 |
| 4.9 | Regulatory Submissions | 14 |
| 4.1 | Prior Royalties | 14 |
| 4.11 | Reports | 14 |
| 4.12 | Disclosure | 14 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
5 |
REPRESENTATIONS, WARRANTIES OF DR3 | 14 |
| 5.1 | Organization | 14 |
| 5.2 | Authorization | 14 |
| 5.3 | No Conflicts | 14 |
| 5.4 | No Consent | 15 |
| 5.5 | No Litigation | 15 |
6 |
COVENANTS | 15 |
| 6.1 | Patent Obligations | 15 |
| 6.2 | Confidentiality | 16 |
| 6.3 | Infringement of NPS Technology | 17 |
| 6.4 | DR3 May Perform | 17 |
| 6.5 | Grant of Security Interest | 18 |
| 6.6 | Acknowledgment of Existing Patent Filings | 18 |
| 6.7 | Further Sale or Financing | 18 |
7 |
COMMERCIALIZATION AND UPDATES | 19 |
| 7.1 | Commercialization of Product | 19 |
| 7.2 | Updates | 19 |
| 7.3 | Notifications | 19 |
| 7.4 | No Assumption of Obligations | 20 |
| 7.5 | No Contravention of NPS's Residual Rights; End of Term | 20 |
| 7.6 | Actions upon Revocation | 20 |
8 |
PAYMENTS | 20 |
| 8.1 | Royalty Term | 20 |
| 8.2 | Royalty Payments and Royalty Statements | 21 |
| 8.3 | Late Payments | 21 |
| 8.4 | Royalty Withholding Taxes | 21 |
| 8.5 | Currency Conversion | 21 |
| 8.6 | Records, Audits | 22 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
9 |
TERM AND TERMINATION | 22 |
| 9.1 | Term of Agreement | 22 |
| 9.2 | Termination | 23 |
| 9.3 | Accrued Rights | 23 |
| 9.4 | Survival | 23 |
10 |
INDEMNIFICATION | 23 |
| 10.1 | Indemnification by NPS | 23 |
| 10.2 | Indemnification by DR3 | 23 |
| 10.3 | Procedure for Claims | 23 |
11 |
MISCELLANEOUS | 25 |
| 11.1 | Costs and Expenses | 25 |
| 11.2 | Further Assurances | 25 |
| 11.3 | Notices | 25 |
| 11.4 | Successors and Assigns | 26 |
| 11.5 | No Partnership | 26 |
| 11.6 | Entire Agreement | 26 |
| 11.7 | Amendments, Supplements, Waivers | 27 |
| 11.8 | Severability | 27 |
| 11.9 | Governing Law | 27 |
| 11.1 | Waiver of Jury Trial | 27 |
| 11.11 | Counterparts | 27 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
SCHEDULES
1.1(j) DR3 Account
4.6 Copies of Certain Agreements and Notifications
4.7 List of NPS Patents
4.7(a) Opinions of Counsel
4.8 Litigation
6.1(a) Jurisdictions Requiring Consent to License
6.2(d) Form 8-K
EXHIBITS
A Form of Restated Security Agreement
B Patent Filings
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
AMENDED AND RESTATED AGREEMENT FOR THE SALE AND ASSIGNMENT OF RIGHTS
THIS AMENDED AND RESTATED AGREEMENT FOR THE SALE AND ASSIGNMENT OF RIGHTS (this "Agreement") is made and entered into as of December 20, 2013, by and between NPS Pharmaceuticals, Inc., a Delaware corporation ("NPS"), and Drug Royalty L.P.3, a Cayman Islands limited partnership ("DR3"). NPS and DR3 are sometimes referred to herein individually as a "Party" and together as the "Parties".
RECITALS
WHEREAS, NPS, Takeda Pharma A/S, a Danish company ("Takeda Pharma") (formerly known as Nycomed Danmark ApS) and Takeda GmbH entered into that certain Termination and Transition Agreement, dated as of March 18, 2013 (the "Termination and Transition Agreement"), pursuant to which, among other things, Takeda terminated that certain License Agreement, dated as of July 2, 2007, by and between NPS (as assignee of NPS Allelix Corp., a Canadian corporation ("NPS Allelix")) and Takeda Pharma (the "License Agreement"), pursuant to Section 16.2 of the License Agreement and NPS acquired certain additional rights and assets related to the Product;
WHEREAS, NPS (both as an original party and as assignee of NPS Allelix) and DR3 are parties to that certain Agreement for the Sale and Assignment of Rights dated as of July 16, 2007 (the "Original Purchase Agreement"), pursuant to which NPS assigned to DR3 certain of its rights under the License Agreement, including the right to receive certain payments from Takeda Pharma under the License Agreement;
WHEREAS, NPS intends to develop and market the Product on its own in the territories previously licensed to Takeda Pharma utilizing in part the rights and assets acquired from Takeda Pharma, and grant to DR3 the right to receive royalties on Net Sales of the Product (the "Revenue Interest Arrangement");
WHEREAS, NPS has agreed to grant to DR3 the DR3 Royalty Right (as defined below) in consideration of the Consent (as defined below) and subject to the conditions specified herein, and DR3 and NPS wish to enter into this Agreement to effect this transaction; and
WHEREAS, NPS and DR3 further desire to conditionally amend and restate the Original Purchase Agreement to reflect the Revenue Interest Arrangement, subject to the terms and conditions set forth herein.
NOW, THEREFORE, in consideration of the foregoing premises and the mutual covenants contained in this Agreement, and other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the Parties, intending to be legally bound, hereby agree as follows:
-1-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- DEFINITIONS
- "Affiliate", in respect of any Person, means any other Person which, directly or indirectly through one or more intermediaries, controls, is controlled by or is under common control with the subject Person, and a Person that has an entity that is an Affiliate under the foregoing shall also be deemed to be an Affiliate of such Person.
- "Asahi" means Asahi Kasei Pharma Corporation.
- "Asahi Agreement" means the non-exclusive patent license agreement dated as of April 1, 2005 between NPS and Asahi.
- "Biologics License Application" or "BLA" means a Biologics License Application (including any supplements or amendments thereto) as defined in the United States Food, Drug, and Cosmetic Act, as amended from time to time, and the regulations promulgated thereunder.
- "Business Day" means any day that is not a Saturday, Sunday or other day on which banks are required or authorized by law to be closed in New York, New York.
- "Closing Document" means any document, instrument, undertaking or agreement required under this Agreement to be delivered by NPS at the Closing.
- "Control" means, when used in reference to intellectual property, other intangible property, or materials, that a Party owns or has a license or sublicense to such intellectual property, other intangible property or materials, and has the ability to grant a license or sublicense or other right to use to such intellectual property, other intangible property or materials, as applicable, as provided for herein, without (i) requiring the consent of a Third Party or (ii) violating the terms of any existing license agreement in respect of such intellectual property, other intangible property or materials with any Third Party.
- "Damages" means any damage, loss, claim, cost, liability, demand or expense (including reasonable out-of-pocket expenses of investigation and reasonable legal fees and expenses in connection with any action, suit or proceeding), but excluding punitive and consequential damages.
- "Dollar" means a U.S. dollar, and "$" shall be interpreted accordingly.
1.1 Defined Terms . For the purposes of this Agreement, the following terms shall have the respective meanings set out below, and grammatical variations of such terms shall have corresponding meanings:
-2-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- "DR3 Account" means the bank account specified in Schedule 1.1(j).
- "DR3 Royalty Right" means the right to receive the Royalty Payments.
- "Encumbrance" means any lien, charge, security interest, mortgage, option, privilege, pledge, trust or deemed trust (whether contractual, statutory or otherwise arising) or any other encumbrance, right or claim of any other Person of any kind whatsoever whether choate or inchoate, or any agreement (whether written or oral) to create any of the foregoing.
- "FDA" means the U.S. Food and Drug Administration or its successor.
- "First Commercial Sale" means, with respect to the Product, the first sale of the Product in a given country or other regulatory jurisdiction in the Territory by or on behalf of NPS, or any of its Affiliates, licensees or sublicensees, to a Third Party, after receipt of Regulatory Approval for the Product in such country or regulatory jurisdiction.
- "Gautvik Agreement" means the purchase and sale agreement dated April 1, 1996 between Allelix Biopharmaceuticals, Inc., Kaare M. Gautvik and Peter Alestrom.
- "Gautvik Patents" means those patents purchased by NPS pursuant to the Gautvik Agreement.
- "Governmental Authority" means any government, regulatory or administrative agency or commission, governmental department, ministry, bureau, commission or agency, court, tribunal, governmental arbitrator or arbitration board or other similar body, or other governmental authority or instrumentality, whether federal, provincial, state or municipal (domestic or foreign).
- "Knowledge of NPS" means the actual knowledge of NPS after due inquiry.
- "Know-How" means information, results and data of any type whatsoever, in any tangible or intangible form whatsoever, including without limitation, technical information, databases, practices, methods, techniques, specifications, formulations, formulae, knowledge, know-how, skill, experience, test data including pharmacological, medicinal chemistry, biological, chemical, biochemical, toxicological and clinical test data, analytical and quality control data, stability data, studies and procedures, and manufacturing process and development information, results and data relating to the Product or the device used to deliver the Product.
- "Laws" means all applicable laws, statutes, rules, regulations, ordinances, guidelines and other pronouncements having the effect of law of any Governmental Authority.
-3-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- "Material Adverse Effect" means a material adverse effect on (a) the ability of NPS to perform its obligations under this Agreement, (b) the validity or enforceability of this Agreement or the rights or remedies of DR3 hereunder, (c) the timing, amount or duration of the Royalty Payments, (d) the DR3 Royalty Right, or (e) the Products.
- "Net Sales" means, with respect to any period, the gross invoiced commercial sales or other commercial dispositions of the Product during such period by NPS, its Affiliates, licensees and sublicensees, to Third Parties, less the following deductions to the extent included in the gross invoiced sales price for the Product or otherwise directly paid, allowed, accrued, or incurred by NPS, its Affiliates, licensees or sublicensees, with respect to the Product during such period:
- quantity or cash discounts, credits, retroactive price reductions, rebates, allowances and adjustments granted, to the extent usual and customary in the pharmaceutical industry and consistent with NPS's usual course of dealing for its products other than the Product (including government mandated and managed healthcare negotiated rebates);
- amounts repaid, credited or written off by reason of rejections, recalls, billing errors and returns;
- sales, excise, turnover, inventory, value-added, and similar taxes assessed on the sale of the Product (other than income taxes of NPS, its Affiliates, licensees, sublicensees), and import and customs duties; and
- transportation, importation, shipping, insurance and other handling expenses.
- the Net Sales amount for Product sold in such transaction determined as provided above, with any non-cash consideration attributable to such transaction valued at fair market value;
- if there has been any arm's length sale of Product separate from any sale or disposition of other products or of services to a non-licensee or non-sublicensee Third Party, the Net Sales amount, determined as provided above, for the most contemporaneous such sale; or
- if there has been no such arm's length sale, the fraction of the overall value of such transaction reasonably attributed to Product sold in such transaction, with any non-cash consideration attributable to such transaction valued at fair market value.
Notwithstanding the foregoing, in any case where Product is sold or otherwise disposed of in a transaction that is not an arm's length sale exclusively for cash that is separate from any sale or disposition of other products or of services, Net Sales shall mean the greatest of:
-4-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- "NPS Know-How" means (a) any non-public or confidential sections of any marketing authorization application relating to any Product, and (b) any other non-public or confidential Know-How Controlled by NPS that is necessary or useful for the performance of pre-clinical or clinical development, for the filing of marketing authorizations in the Territory, or the commercialization, marketing or manufacture of a Product.
- "NPS Patents" means the Patents Controlled by NPS that are necessary or useful for the development, commercialization or manufacture of the Product in the Territory.
- "NPS Technology" means the NPS Patents and the NPS Know-How.
- "NPS Trademarks" means NATPARA™, PREOTACT™ and any other trademarks, trade dress, logos, slogans, and designs, whether or not registered in the Territory, used to identify or promote the Product in the Territory.
- "Product" means recombinant human parathyroid hormone (rhPTH 1-84) as set forth in the BLA No. 125511 (and regardless of the trademark under which it may be sold or distributed) as well as any invention, discovery or derivative or Know-How, whether or not patentable related thereto, including other formulations or indications. For clarity, "Product" includes rhPTH 1-84 as described in BLA No. 125511 but sold, distributed or otherwise commercialized outside of the United States.
For sake of clarity, (a) sales by NPS, its Affiliates, licensees or sublicensees to distributors and wholesalers shall be considered sales to Third Parties, and (b) amounts recovered by NPS or its Affiliates from any assertion of NPS Technology after deduction of the expenses specified in Section 6.3(b)(iii) shall be deemed Net Sales without further deductions.
-5-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- When a reference is made in this Agreement to an "Article", "Section", "Schedule" or "Exhibit", such reference shall be to an Article, Section, Schedule or Exhibit to this Agreement unless otherwise indicated.
- The words "include," "includes" and "including" when used herein shall be deemed in each case to be followed by the words "without limitation" and shall not be construed to limit any general statement which it follows to the specific or similar items or matters immediately following it.
1.2 Interpretation . The following initially capitalized terms shall the meaning set forth below. Other initially capitalized terms shall have the meaning ascribed to such terms elsewhere in this Agreement.
-6-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- None of the Parties hereto shall be or be deemed to be the drafter of this Agreement for the purposes of construing this Agreement against another party.
- Unless otherwise specified in this Agreement, words importing one gender include all genders.
- All uses of the words "hereto", "herein", "hereof', "hereby" and "hereunder" and similar expressions refer to this Agreement.
- The headings and captions in this Agreement are for convenience and reference purposes only and shall not be considered a part of, or affect the construction or interpretation of, any provision of this Agreement.
1.3 Currency . Unless specified otherwise, all references to monetary amounts in this Agreement are to references to the lawful currency of the United States of America.
1.4 Additional Definitions . The following terms have the meanings set forth in the corresponding Sections of this Agreement:
|
Term |
Section |
|
Agreement |
Preamble |
|
Audit |
8.6 |
|
Closing |
3.1 |
|
Consent |
2.2 |
|
DR3 |
Preamble |
|
DR3 Released Parties |
2.4(b) |
|
indemnified party |
10.3(a) |
|
Launch |
7.2 |
|
License Agreement |
Recitals |
|
NPS |
Preamble |
|
NPS Allelix |
Recitals |
|
NPS Released Parties |
2.4(a) |
|
Original Purchase Agreement |
Recitals |
|
Party or Parties |
Preamble |
|
Patent Filings |
6.6 |
|
Regulatory Milestone |
2.2 |
|
Revocation |
2.2 |
|
Revenue Interest Arrangement |
Recitals |
|
Royalty Payments |
2.1 |
|
Royalty Statements |
8.2 |
|
Royalty Term |
8.1 |
|
Takeda Pharma |
Recitals |
|
Term |
9.1 |
|
Termination and Transition Agreement |
Recitals |
|
Third Party Claim |
10.3(a) |
-7-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- ROYALTY GRANT; CONSENT; AMENDMENT AND RESTATEMENT OF ORIGINAL PURCHASE AGREEMENT
- Until such time as either DR3 delivers a Revocation or the Consent becomes irrevocable pursuant to Section 2.2, the Original Purchase Agreement shall be tolled and the Parties' rights and obligations with respect to the NPS Technology and the Product, and DR3's right to receive any payments with respect to the Product, shall be governed by this Agreement.
- In the event that DR3 delivers a Revocation in accordance with Section 2.2, this Agreement shall terminate and the Parties' rights and obligations with respect to the NPS Technology and the Product, and DR3's right to receive any payments with respect to the Product, shall be governed by the Original Purchase Agreement (for clarity, prior to its amendment and restatement by this Agreement) and this Agreement shall terminate pursuant to Section 9.2. In such event, any Royalty Payments made to DR3 prior to the effective date of such termination of this Agreement shall, for purposes of the Original Purchase Agreement, be credited against the Purchaser Royalty Interest (as such term is defined in Section 8.1 of the Original Purchase Agreement).
2.1 Royalty Grant . NPS shall pay to DR3 a royalty of [* * *] of Net Sales of Product in the Territory for all or any portion of the calendar year falling within the Royalty Term ("Royalty Payments"), on the terms specified in Article 8.
2.2 Consent to the Revenue Interest Arrangement . Subject to the terms and conditions hereof, DR3 hereby grants its consent to the Revenue Interest Arrangement pursuant to Section 7.5 of the Original Purchase Agreement and the Revenue Interest Arrangement shall be deemed to be a "New Arrangement" as such term is defined in Section 7.5(a) of the Original Purchase Agreement (the "Consent"), which may only be revoked pursuant to the next sentence. In the event that NPS or any of its Affiliates fails to submit a Marketing Authorization Application (that NPS reasonably expects to be validated and accepted for review) to the European Medicines Agency for the Product in the European Union by [* * *] (the "Regulatory Milestone"), DR3 may revoke the Consent by providing written notice to NPS of such revocation on or before [* * *] (a "Revocation"). In connection with the Regulatory Milestone, NPS currently intends to pursue approval of the Product for hypo-parathyroidism and may amend, modify or withdraw any pre-existing regulatory filings or marketing approvals for the Product. In the event of a Revocation, this Agreement shall be of no further force or effect and the Original Purchase Agreement shall become active. In such event, as of the date DR3 delivers a Revocation to NPS, at DR3's election either (i) the twelve (12) month period referenced in Section 7.5(a) of the Original Purchase Agreement shall commence, or (ii) the provisions of Section 7.5(c) of the Original Purchase Agreement shall commence as though such twelve (12) month period had already expired. In the event (i) that NPS achieves the Regulatory Milestone or (ii) that NPS doesn't achieve the Regulatory Milestone and DR3 fails to exercise the Revocation by [* * *], the Consent shall be irrevocable.
2.3 Conditional Amendment and Restatement of the Original Purchase Agreement.
-8-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- If the Consent becomes irrevocable pursuant to Section 2.2, NPS and its Affiliates, and its and their respective officers, directors, shareholders, employees, agents, predecessors, successors and assigns (collectively, the "NPS Released Parties") shall automatically be fully, finally and irrevocably relinquished, released and discharged by DR3, on behalf of itself and its Affiliates, and its and their respective officers, directors, limited partners, employees, agents, predecessors, successors and assigns, from any and all claims, damages, liabilities, obligations, and causes of action, including indemnification claims, known or unknown, suspected or unsuspected, in law or equity, that were asserted, or that could have been asserted by DR3 and its Affiliates, and its and their respective officers, directors, shareholders, employees, agents, predecessors, successors and assigns, against the NPS Released Parties arising out of the Original Purchase Agreement and the Security Agreement, whether accrued or not; provided, however, that nothing in this Section 2.4(a) shall be deemed to affect the enforceability of DR3's rights or the obligations of the Parties under this Agreement or the Restated Security Agreement.
- If the Consent becomes irrevocable pursuant to Section 2.2, DR3 and its Affiliates, and its and their respective officers, directors, shareholders, employees, agents, predecessors, successors and assigns (collectively, the "DR3 Released Parties") shall automatically be fully, finally and irrevocably relinquished, released and discharged by NPS, on behalf of itself and its Affiliates, and its and their respective officers, directors, limited partners, employees, agents, predecessors, successors and assigns, from any and all claims, damages, liabilities, obligations, and causes of action, including indemnification claims, known or unknown, suspected or unsuspected, in law or equity, that were asserted, or that could have been asserted by NPS and its Affiliates, and its and their respective officers, directors, shareholders, employees, agents, predecessors, successors and assigns, against the DR3 Released Parties arising out of the Original Purchase Agreement and the Security Agreement, whether accrued or not; provided, however, that nothing in this Section 2.4(b) shall be deemed to affect the enforceability of NPS's rights or the obligations of the Parties under this Agreement or the Restated Security Agreement.
- Until a Revocation, DR3 and its Affiliates, and its and their respective officers, directors, shareholders, employees, agents, predecessors, successors and assigns, shall not assert any claims against the NPS Released Parties arising out of the Original Purchase Agreement and the Security Agreement whether accrued or not.
2.4 Mutual Release.
2.5 Tolling.
-9-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- Until a Revocation, NPS and its Affiliates, and its and their respective officers, directors, shareholders, employees, agents, predecessors, successors and assigns shall not assert any claims against the DR3 Released Parties arising out of the Original Purchase Agreement and the Security Agreement whether accrued or not.
- THE CLOSING
3.1 The Closing . The closing of the transactions contemplated by this Agreement, including the grant by NPS to DR3 of the DR3 Royalty Right, the grant by DR3 to NPS of the Consent, the conditional amendment and restatement of the Original Purchase Agreement described in Section 2.3, and the conditional amendment and restatement of the Security Agreement shall take place on the date hereof (the "Closing").
3.2 NPS Closing Deliveries . DR3 acknowledges that, at the Closing, NPS has delivered or has caused to be delivered to DR3:
- a current Certificate of Good Standing for NPS from the Delaware Department of State;
- the Restated Security Agreement, duly executed and delivered by NPS;
- a patent security agreement in a form sufficient for the recordation thereof by the United States Patent and Trademark Office, together with an appropriately completed recordation form, each duly executed and delivered by NPS;
- a trademark security agreement in a form sufficient for the recordation thereof by the United States Patent and Trademark Office, together with an appropriately completed recordation form, each duly executed and delivered by NPS;
- standard corporate existence and authority opinions and enforceability opinions with respect to this Agreement, the Restated Security Agreement, the patent security agreement referred to in Section 3.2(c), the trademark security agreement referred to in Section 3.2(d), a perfection and registration opinion with respect to the Restated Security Agreement and a no-conflict-with-laws- opinion with respect to this Agreement; and
2.6 Accrued Claims. Without an admission of liability by either Party, each Party acknowledges and agrees that, with respect to any claim a Party has against the other under the Original Purchase Agreement or the Security Agreement, the period of time between the date hereof and the effective date of a Revocation shall be excluded in any calculation of damages with respect to such claim.
-10-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- REPRESENTATIONS, WARRANTIES OF NPS
NPS hereby represents and warrants to DR3 as of the date hereof as follows and acknowledges that DR3 is relying on such representations and warranties in entering into this Agreement.
4.1 Organization, Standing and Power.
- NPS is a corporation duly incorporated, validly existing and in good standing under the laws of the State of Delaware and has full corporate power and authority and possesses all governmental franchises, licenses, permits, authorizations and approvals necessary to enable it to own, lease or otherwise hold its properties and assets and to carry on its business as presently conducted.
- NPS is not insolvent and no proceedings have been taken or authorized by NPS, or to the Knowledge of NPS been taken or threatened by any other Person, with respect to the bankruptcy, insolvency, liquidation, dissolution or winding up of NPS. NPS will not become insolvent or be put in insolvent circumstances or become unable to meet its obligations as they become due, in each case within the meaning of applicable bankruptcy, insolvency and similar Laws to which NPS is subject, by or as a result of entering into this Agreement. NPS is not entering into this Agreement for the purpose of injuring, obstructing, impeding, defeating, hindering, delaying, defrauding or oppressing the rights and claims of creditors or others against NPS.
4.2 Authority, Execution and Delivery; Enforceability . NPS has full power and authority to execute and deliver this Agreement and perform all of the obligations to be performed by NPS hereunder. The execution and delivery of this Agreement and the consummation of the transactions contemplated hereby have been duly authorized by all necessary corporate action of NPS. This Agreement has been duly executed and delivered by NPS and constitutes NPS's legal, valid and binding obligations, enforceable against NPS in accordance with its respective terms, subject to creditors' rights and general principles of equity.
4.3 No Conflicts . The execution and delivery of this Agreement by NPS do not and will not, and the consummation of the transactions contemplated hereby and the compliance by NPS with the terms hereof will not:
3.3 DR3 Closing Deliveries . NPS acknowledges that, at the Closing, DR3 has delivered or has caused to be delivered to NPS the Restated Security Agreement, duly executed and delivered by DR3.
-11-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- conflict with, result in a breach or violation of, constitute a default (with or without notice or lapse of time, or both) under, or give rise to a right of termination, cancellation or acceleration of any obligation or to a loss of a benefit under, any provision of (i) any applicable statute, law, ordinance, rule or regulation of any Governmental Authority, or any judgment, order, writ, decree, permit or license of any court or any Governmental Authority to which NPS or its properties or assets may be subject, (ii) any agreement (whether written or oral), commitment or instrument to which NPS is a party or by which NPS or any of its assets is bound (including any agreement to which NPS is a party relating to the NPS Technology, the NPS Trademarks or the DR3 Royalty Right), or (iii) the bylaws of NPS; or
- result in the creation or imposition of any Encumbrance on the NPS Technology, the NPS Trademarks or the DR3 Royalty Right except as contemplated in the Restated Security Agreement in favor of DR3.
- Other than the Milestone Payment (as such term is defined in the Termination and Transition Agreement), there are no other contracts, arrangements or understandings (whether written or oral) to which NPS or its Affiliates is a party relating to granting any Third Party any Royalty Right in the Territory.
- Other than DR3 under this Agreement and Takeda Pharma under the Termination and Transition Agreement (in respect of the Milestone Payment, as defined therein), no Person has any entitlement to receive, in whole or in part, any Royalty Right in the Territory.
- Schedule 4.7 sets forth a complete list, including status, of all Patents which comprise the NPS Patents. NPS is the exclusive owner of the entire right, title and interest in and to the NPS Patents free and clear of any Encumbrances other than the encumbrances in respect of the NPS Technology that are specifically created under or by the Asahi Agreement. NPS has not received written notice that any Person has
4.4 No Consent . No consent, approval, license, permit, order or authorization of, or registration, declaration or filing with, any Person is required to be obtained or made by NPS in connection with the execution and delivery by NPS of this Agreement, the performance by NPS of its obligations under this Agreement or the consummation of any of the transactions contemplated hereby, including the right of DR3 to receive the Royalty Payments.
4.5 Rights in DR3 Royalty Right.
4.6 Agreements . The Asahi Agreement expired according to its express terms as of October 8, 2013, and the Gautvik Agreement expired as of September 11, 2009. On October 4, 2010, Ypsomed AG delivered written notice to NPS under the Patent 151 License that it was allowing Patent No. EP 1519766A (filing date June 25, 2003) to lapse, and the Patent 151 License has now expired. A true, correct and complete certified copy of the Asahi Agreement, the Gautvik Agreement, the Patent 151 License and lapse notifications referred to in this Section 4.6 are attached hereto in Schedule 4.6.
4.7 Patents and Other Intellectual Property.
-12-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- To the Knowledge of NPS, no Third Party has a claim or has claimed any ownership rights or received any demand or claim by any Person that any of the NPS Technology or NPS Trademarks is infringing or may infringe upon any patent, copyright, trademark, trade secret or any other intellectual property rights of any Third Party.
- Except as set forth in Schedule 4.7(c), NPS has not entered into any contract, agreement, commitment or undertaking granting to any Person the right within the Territory (i) under the NPS Patents or (ii) to use the NPS Technology, in either case, to market or sell Products or any other product.
- NPS has not granted to any Governmental Authority a license relating to the NPS Technology and, to the Knowledge of NPS, there is no reason to believe that NPS is or will be required to grant any such license to any Governmental Authority.
- The Gautvik Patents have expired and Pat. No. EP 1519766 has been abandoned. NPS owns sufficient rights in and to Pat. No. EP 0515228 such that NPS's manufacture, use and sale of the Product in Denmark and Norway would not infringe it.
challenged the validity or enforceability of the NPS Patents. To the Knowledge of NPS, there is, and since July 16, 2007 there has been, no infringement of the NPS Patents by any Person. NPS has not received any demand or claim by any Person that such Person has any ownership interest in any of the NPS Patents, or that any of the NPS Patents are, or may be, invalid or unenforceable or that any Product infringes upon or may infringe upon any patent, copyright, trademark, trade secret or other intellectual property right of any Third Party. All appropriate patent fees required to be paid with respect to the applications listed on Schedule 4.7 have been paid. To the Knowledge of NPS, the sale of the Product in the Territory as previously sold by Takeda Pharma does not infringe any issued patent of any Third Party or infringes any other trademarks or trade secrets of any Third Party. Except as set forth in Schedule 4.7(a), NPS has not requested any written opinions of counsel relating to any Third Party patent or published patent application which may be considered to relate to any Product or device used to administer the Product.
4.8 Litigation . Except as set out on Schedule 4.8, there is no: (a) action, suit, claim or proceeding pending or, to the Knowledge of NPS, threatened against NPS, at law or in equity, (b) arbitration proceeding to which NPS is a party, or (c) any inquiry by any Governmental Authority pending or, to the Knowledge of NPS, threatened against NPS, which, if adversely determined, would question the validity or enforceability of the NPS Technology, the NPS Trademarks or the ability of NPS to grant the right for DR3 to receive Royalty Payments, or prevent the consummation of the transactions contemplated by this Agreement or otherwise adversely affect the right of DR3 to receive Royalty Payments. There is no action or suit by NPS pending or threatened in writing against others relating to the NPS Technology, the NPS Trademarks, the Products or any device used to administer the Product. None of the NPS Patents is subject to any outstanding
-13-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- REPRESENTATIONS, WARRANTIES OF DR3
DR3 hereby represents and warrants to NPS as of the date hereof as follows and acknowledges that NPS is relying on such representations and warranties in entering into this Agreement.
5.1 Organization . DR3 is a limited partnership duly organized, validly existing and in good standing under the laws of the Cayman Islands and has full organizational power and authority and possesses all governmental franchises, licenses, permits, authorizations and approvals necessary to enable it to own, lease or otherwise hold its properties and assets and to carry on its business as presently conducted.
5.2 Authorization . DR3 has full power and authority to execute and deliver this Agreement and to perform all of the obligations to be performed by DR3 hereunder. The execution and delivery of this Agreement and the consummation of the transactions contemplated hereby have been duly authorized by all necessary partnership action of DR3. This Agreement has been duly executed and delivered by DR3 and constitutes DR3's legal, valid and binding obligation, enforceable against DR3 in accordance with its terms.
decree, order, judgment, or stipulation restricting in any manner the use or licensing thereof by NPS. Schedule 4.8 sets forth a list of all patent office proceedings, including oppositions, interferences or re-examinations, relating to the NPS Patents.
4.9 Regulatory Submissions . NPS has submitted an orphan drug designation application for the Product to the European Medicines Agency, and has received orphan drug designation for the Product by the FDA. NPS has submitted BLA 125511 to the FDA for the Product and expects to receive twelve (12) years of data exclusivity in the United States.
4.10 Prior Royalties . To the Knowledge of NPS, all royalties required to be paid by Takeda Pharma pursuant to the License Agreement for any period ending on or prior to March 18, 2013 have been paid in full as and when due, and no portion of the Purchaser Royalty Interest (as defined in the Original Purchase Agreement) has accrued but not yet been paid to DR3. No royalties have been received by NPS from Takeda Pharma for the period from March 18, 2013 to the date hereof.
4.11 Reports . NPS has provided to DR3 true, correct and complete copies of all royalty reports received by NPS from Takeda Pharma as of the date hereof.
4.12 Disclosure . No representation or warranty made by NPS in this Agreement, when considered together with other representations and warranties made by NPS in this Agreement, contains any untrue statement of a material fact or omits to state any material fact necessary to make any such representation or warranty, in the light of the circumstances under which it is made, not misleading to a prospective buyer of the DR3 Royalty Right.
-14-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- COVENANTS
- NPS may transfer or license the NPS Technology or the NPS Trademarks to its Affiliates without DR3's prior written consent. Without the prior written consent of DR3 (such consent to be at DR3's sole discretion), NPS may not sell or otherwise transfer any NPS Technology or any NPS Trademark to any Third Party. NPS may not license the NPS Technology or the NPS Trademarks to any Third Party in the jurisdictions set forth on Schedule 6.1(a) without DR3's prior written consent (not to be unreasonably withheld).
- NPS shall (i) to the full extent allowed by Law, prosecute and maintain in full force and effect each pending patent application included in the NPS Patents in the Territory, and (ii) maintain and keep in full force and effect any issued Patents in the Territory. In addition, prior to issuance of a patent from a pending patent application included in the NPS Patents or abandonment of a patent application included in the NPS Patents, NPS shall file, to the extent permitted by Law, a continuation or divisional application of the patent application unless: (i) NPS has prior written consent from DR3 not to file such a continuation or divisional application, or (ii) as of the date of such issuance or abandonment, the term of all issued NPS Patents has expired. Notwithstanding the foregoing, NPS shall have the right to abandon, or allow to lapse,
5.3 No Conflicts . The execution and delivery of this Agreement by DR3 do not, and the consummation of the transactions contemplated hereby and the compliance by DR3 with the terms hereof will not conflict with, result in a breach or violation of, constitute a default (with or without notice or lapse of time, or both) under, or give rise to a right of termination, cancellation or acceleration of any obligation or to a loss of a material benefit under, any provision of: (a) any applicable statute, law, ordinance, rule or regulation of any Governmental Authority, or any judgment, order, writ, decree, permit or license of any court or any Governmental Authority to which DR3 or its properties or assets may be subject, (b) any material contract, commitment or instrument to which DR3 is a party or by which DR3 or any of its assets is bound, or (c) the governing documents of DR3.
5.4 No Consent . No consent, approval, license, permit, order or authorization of, or registration, declaration or filing with, any Person is required to be obtained or made by DR3 in connection with the execution and delivery by DR3 of this Agreement, the performance by DR3 of its obligations under this Agreement or the consummation of any of the transactions contemplated hereby.
5.5 No Litigation . There is no: (a) action, suit, claim or proceeding pending or, to the knowledge of DR3, threatened against DR3, at law or in equity, (b) arbitration proceeding to which DR3 is a party, or (c) any inquiry by any Governmental Authority pending or, to the knowledge of DR3, threatened against DR3, which, if adversely determined, would prevent the consummation of the transactions contemplated by this Agreement.
-15-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- All information furnished by DR3 to NPS or by NPS to DR3 in connection with this Agreement and the transactions contemplated hereby, as well as the terms, conditions and provisions of this Agreement and any other agreement delivered pursuant hereto, shall be kept confidential by NPS and DR3 and shall be used by NPS and DR3 only in connection with this Agreement and the transactions contemplated hereby, except in connection with the enforcement of rights under this Agreement and except to the extent that such information: (i) is already known (and not subject to confidentiality) by the Party to whom the information is disclosed or in the public domain at the time the information is disclosed; (ii) thereafter becomes lawfully obtainable from other sources other than as a result of a breach of an obligation of confidentiality; (iii) is required to be disclosed in any document to be filed with any Governmental Authority; or (iv) is required to be disclosed under securities Laws applicable to NPS, DR3 or their respective Affiliates, or by court or administrative order. Notwithstanding the foregoing, DR3 may disclose such information to its manager and partners and each of its and their respective Affiliates, directors, officers, investors, bankers, financing sources, ratings agencies, advisors, trustees, representatives and potential acquirers of all or part of (x) DR3, (y) an Affiliate of DR3 or (z) the DR3 Royalty Right; and NPS may disclose such information to its Affiliates, directors, officers, bankers, advisors, investors, financing sources, strategic partners and representatives, provided in each case that such Persons shall be informed of the confidential nature of such information and shall be obligated to keep such information confidential pursuant to the terms of this Section 6.2.
- The Parties shall use reasonable efforts, acting in good faith, to cooperate with each other with respect to the scope and substance of all disclosures regarding this Agreement to or as required by any Governmental Authority, including the United States Securities and Exchange Commission. In addition, the Parties will coordinate in advance with each other in connection with the redaction of certain provisions of this Agreement with respect to any United States Securities and Exchange Commission filings of this Agreement, and each Party shall use commercially reasonable efforts to seek confidential treatment for such terms if so requested by the other Party. Notwithstanding the foregoing, each Party shall ultimately retain control over the scope of information to be disclosed to any Governmental Authority for purposes of complying with any applicable Law.
any NPS Patent in the Territory, provided that (i) such abandonment or lapse would not reasonably be expected to cause, singly or in aggregate with other abandonments or lapses, a Material Adverse Effect, and (ii) in the event NPS intends to allow any NPS Patent in the Territory to lapse or become abandoned, NPS shall so notify DR3, and DR3 shall have the right (but not the obligation) to assume further responsibility for the prosecution, maintenance and defense of such NPS Patent at DR3's expense. For the avoidance of doubt, any Patent for which DR3 assumes responsibility pursuant to this Section 6.1(b) shall remain an NPS Patent.
6.2 Confidentiality.
-16-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- The Parties shall be free to publicly disclose information contained in any materials that have previously been approved for public disclosure by the other Party, without further approvals from the other Party hereunder, to the extent there have been no material additions or changes thereto.
- No announcement or other disclosure, public or otherwise, concerning the financial or other terms of this Agreement shall be made, either directly or indirectly, by any Party hereto without first obtaining the written approval of the other Party and agreement upon the nature and text of such announcement or disclosure, such approval and agreement not to be unreasonably withheld or delayed. Notwithstanding the foregoing, NPS may disclose the transactions contemplated by this Agreement in a form 8-K substantially in the form attached hereto as Schedule 6.2(d).
- In the event either Party becomes aware of any actual or threatened infringement or misappropriation of any NPS Technology, the Party first having knowledge of such infringement shall promptly notify the other, and the Parties shall thereupon consult together as to the action to be taken.
- Enforcement.
- NPS shall have the first right to initiate and prosecute any legal actions to enforce its right in the NPS Technology and prosecute apparent infringers.
- If, after three (3) months have passed from the date notice is given pursuant to Section 6.3(a), NPS has not initiated legal action or succeeded in stopping the infringement, and is not then in substantial and active settlement or licensing discussions with the infringer, then DR3 shall have the right, in the event such infringement has or is reasonably likely, in DR3's reasonable opinion, to have a negative effect on Net Sales of the Product, upon request to NPS, to compel NPS to initiate and prosecute such a legal action against any Person infringing or misappropriating the NPS Technology directly or contributorily, using counsel selected by DR3 and at DR3's expense, and DR3 shall have the right to control such legal action. Notwithstanding the foregoing, if NPS's ongoing evaluation of patent scope, validity, enforceability, and/or possible infringement defenses requires more than three (3) months from the date of such a notice, then NPS shall so inform DR3, and shall provide DR3 with weekly updates on the status of its evaluation. If NPS receives advice from outside counsel reasonably acceptable to DR3 that the assertion of a claim against the Third Party designated in a notice provided pursuant to this Section 6.3(b)(ii) would pose an unreasonable risk to the validity or enforceability of the NPS Patent portfolio and confirms the receipt of such advice in writing to DR3, then NPS shall not be obligated to commence an action as described in this Section 6.3(b)(ii) with respect to such notice.
- Any and all amounts recovered by NPS or its Affiliates or DR3 with respect to any infringement or misappropriation action in respect of the NPS Technology (whether initiated or controlled by NPS or DR3) shall be applied first to reimburse the Parties for their out-of-pocket expenses (including reasonable attorneys' fees) in prosecuting such action. The remainder shall be deemed Net Sales under this Agreement and be treated accordingly.
6.3 Infringement of NPS Technology.
-17-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- NPS shall enter into and perform its obligations under the Restated Security Agreement, including, as general and continuing security for the due payment and performance of all of NPS' obligations under this Agreement, the grant to DR3 of a legal, valid and enforceable first priority security interest in and to the NPS Technology and NPS Trademarks, on the terms of and subject to the conditions contained in the Restated Security Agreement.
- As soon as possible following the Closing and, in any case, within sixty (60) days from the date hereof, NPS shall take such actions as are required to perfect the registers (for certainty, including, as required, ensuring that NPS' name and address are correctly referenced therein) in the patent and other offices in which the NPS Patents and NPS Trademarks are registered in the United States in order to, or in order to enable DR3 to, within such sixty (60)-day period, record on such registers the documents referred to in Section 3.2(c) and Section 3.2(d); provided that this Section 6.5(b) shall not require NPS to perfect such registers with respect to the PREOTACT trademarks unless a Revocation occurs.
6.4 DR3 May Perform . If NPS fails to observe or perform any covenant, condition or agreement contained in this Agreement, and such failure shall continue unremedied for a period of thirty (30) days after notice thereof from DR3 to NPS, DR3 may (but shall not be obliged to) perform, or cause performance of, such covenant, condition or agreement, provided that DR3 shall in any event first have given NPS written notice of its intent to do the same.
6.5 Grant of Security Interest.
6.6 Acknowledgment of Existing Patent Filings . Each Party hereto hereby acknowledges that in connection with the Original Purchase Agreement, the Parties have previously entered into certain agreements and made certain registrations and filings to perfect the security interest granted to DR3 and record the conditional assignment agreements executed in connection therewith (copies of which are attached hereto as Exhibit B; collectively, the "Patent Filings"). As of the date hereof, each Party hereby acknowledges and agrees that (i) the Patent Filings shall be and remain in full force and effect and shall constitute the legal, valid, binding and enforceable obligations of their parties thereto in accordance with their terms, (ii) such Patent Filings shall only be enforced in accordance with the circumstances specified in this Agreement and the Restated Security Agreement, and (iii) no further filing, registration or amendment is necessary to perfect the interest and rights granted under the Patent Filings. The execution, delivery and effectiveness of this Agreement shall not operate as a waiver of any right, power or remedy of the Parties under the Patent Filings.
-18-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- COMMERCIALIZATION AND UPDATES
7.1 Commercialization of Product . NPS is and will be solely responsible for the commercialization of the Product in the Territory at NPS's expense. NPS shall have the right to commercialize the Product through Affiliates, sublicensees or distributors. NPS shall launch, market and sell the Product in the countries of the Territory in which NPS at its sole discretion finds the marketing financially feasible as soon as reasonably possible after receipt of marketing authorization, pricing and reimbursement approval for the Product in such country (but only if such foregoing approvals are obtained). Subject to the immediately preceding sentence, NPS will promote, market, sell and distribute the Product in the Territory by applying efforts and resources as reasonably required to capture the commercial potential of the Product throughout the Territory and at least equal to the efforts and resources normally used by a similarly situated pharmaceutical company for a product owned by it which has a similar market potential and is at a similar stage in its product life cycle as the Product. All efforts of NPS's Affiliates, sublicensees or distributors will be considered efforts of NPS for purposes of determining NPS's compliance with such obligations.
7.2 Updates . In addition to the notifications provided pursuant to Section 7.3, NPS shall keep DR3 reasonably informed of material regulatory, clinical and commercial developments with respect to the Product in the Territory by providing a written summary report to DR3 on a semiannual basis until First Commercial Sale has occurred in both the United States and the European Union (the "Launch"), and thereafter on an annual basis. Additionally, until the Launch has occurred, at DR3's request the Parties shall hold quarterly update calls.
7.3 Notifications.
Right other than the Royalty Payments, provided that such sale or financing (i) has no adverse impact on Royalty Payments, (ii) does not, and would not reasonably be expected to, result, in a Material Adverse Effect and (iii) is junior and subordinate in all respects to the Royalty Payments. In the event that NPS or its Affiliates sells or finances such right in the future in accordance with the previous sentence, (A) DR3 shall negotiate an intercreditor agreement in good faith with any such prospective purchaser or financing source, and (B) the prospective purchaser or financing source shall (as a condition to such financing or sale) enter into an intercreditor agreement on terms and conditions reasonably satisfactory to DR3 in order to ensure that the security interests of DR3 and such prospective purchaser or financing source are properly segregated and that DR3's right to receive the Royalty Payments remain senior secured obligations of NPS.
-19-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- NPS shall promptly (but in no event later than five (5) Business Days after receipt) notify DR3 and provide DR3 with a copy of any correspondence or other reports or complaints submitted to or received by NPS or any of its Affiliates in respect of any actions, suits, claims, investigations or proceedings commenced or threatened in writing by a Governmental Authority or other Third Party against, relating to, involving or otherwise affecting the Product, Net Sales, the Royalty Payments or any other matters reasonably related thereto, in each case to the extent the foregoing would reasonably be expected to result, either individually or in the aggregate, in a Material Adverse Effect.
- Upon a Revocation, DR3 shall take such actions, and execute such documents, certificates and instruments, as reasonably requested by NPS and at NPS' expense to terminate the additional security interests granted by NPS with respect to (i) the NPS Patents in the United States and Japan, (ii) any NPS Know-How that is applicable only to the United States or Japan, and (iii) the NPS Trademarks in the United States and Japan.
- Upon a Revocation, NPS shall take any and all actions necessary, at NPS's expense, to effect and record the assignment of the PREOTACT trademarks, together with all applications and registrations therefor and all goodwill symbolized thereby, to NPS from Takeda Pharma in the following countries: all the countries of the European Union, European countries outside of the European Union, the Commonwealth of Independent States (formerly the USSR) and Turkey.
- Upon a Revocation, NPS shall immediately and automatically grant to DR3 an exclusive, royalty-free, perpetual, irrevocable license to use the PREOTACT trademark in connection with the promotion, sale or other
a copy of any written communication or a written account of any oral communication), but in no event later than five (5) Business Days after receipt of such communication.
7.4 No Assumption of Obligations . DR3 expressly does not assume or agree to become responsible for any obligation or liability of NPS of any kind whatsoever with respect to the development and commercialization of the Product, whether presently in existence or arising or asserted hereafter. All such obligations and liabilities (for certainty, including any and all existing and/or potential obligations under any license agreement that NPS enters into with respect to the Product and any product liability or intellectual property infringement in respect of the commercialization of Products or devices used to administer the Product) shall be retained by and remain obligations and liabilities of NPS.
7.5 No Contravention of NPS's Residual Rights; End of Term . At the end of the Term, DR3 shall take such actions, and execute such documents, certificates and instruments, as reasonably requested by NPS to terminate the security interests granted by NPS hereunder and pursuant to the Restated Security Agreement.
-20-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- PAYMENTS
8.1 Royalty Term . NPS's obligation to make Royalty Payments to DR3 shall commence on a country-by-country basis upon the First Commercial Sale of the Product in a country in the Territory and shall expire on a country-by-country basis upon the later of: (a) the last to expire of any NPS Patent in such country or (b) the expiration of any period of regulatory exclusivity applicable to the Product in such country (the "Royalty Term").
8.2 Royalty Payments and Royalty Statements . NPS shall calculate all amounts payable as Royalty Payments with respect to Net Sales at the end of each calendar quarter, which amounts shall be converted to Dollars at such time in accordance with Section 8.5. NPS shall pay to DR3 the royalty amount due for Net Sales during a given calendar quarter by deposit to the DR3 Account or such other account designated by DR3 from time to time in writing within forty-five (45) days after the end of such calendar quarter. Each payment of royalties due to DR3 shall be accompanied by a statement of the number of units of Product sold on a country-by-country basis, the amount of gross sales of the Product on a country-by-country basis in the Territory during the applicable calendar quarter (including such amounts expressed in local currency and as converted to Dollars), an itemized calculation of Net Sales in the Territory showing deductions provided for in the definition of "Net Sales" on a country-by-country basis during such calendar quarter and a calculation of the amount of royalty payment due on such Net Sales for such calendar quarter for the Territory and on a country-by-country basis (the "Royalty Statements"). NPS shall deliver the Royalty Statements to DR3 in accordance with Section 11.3 by e-mail addressed to DRINotices@dricapital.com. Without limiting the generality of the foregoing, NPS shall require its Affiliates, licensees and sublicensees to account for its Net Sales.
8.3 Late Payments . If NPS fails to pay any portion of any amount due under this Agreement by the date such amount is due, NPS shall be obligated to pay DR3, in addition to the amount due, interest at an interest rate of 1.0% per month over the prime rate reported in the eastern edition of the Wall Street Journal (or an alternative source reasonably selected by DR3, if the Wall Street Journal should cease to report the prime rate or cease to exist as a periodical) on such date (or the maximum rate permitted by applicable Law, if lesser), compounded monthly, accruing from the date the payment was due through the date of actual payment.
8.4 Royalty Withholding Taxes . Any income or other taxes which NPS is required by Law to pay or withhold on behalf of DR3 with respect to any Royalty Payments payable to DR3 shall be deducted from the amount of such payments due, and paid or withheld, as appropriate, by NPS on behalf of DR3. Any such tax required by applicable Law to be paid or withheld shall be an expense of, and borne solely by, DR3.
commercialization of Products in the Original Territory (as defined in the Original Purchase Agreement). For so long as NPS owns the PREOTACT trademark, all goodwill arising from any use of the PREOTACT trademark made pursuant to this Section 7.6(c) shall inure to the benefit of NPS.
-21-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
NPS shall furnish DR3 with reasonable evidence of such payment or amount withheld, in electronic or written form, as soon as practicable after such payment is made or such amount is withheld. The Parties will reasonably cooperate in completing and filing documents required under the provisions of any applicable tax laws or under any other applicable Law in connection with the making of any required tax payment or withholding payment, or in connection with any claim to a refund of or credit for any such payment.
8.5 Currency Conversion . For the purpose of calculating any sums due under, or otherwise reimbursable pursuant to, this Agreement (including the calculation of Net Sales expressed in currencies other than Dollars), NPS shall convert any amount expressed in a foreign currency into Dollar equivalents, calculated using the average of the exchange rate certified by the Federal Reserve Bank of New York for customs purposes for such currency (found at http://www.federalreserve.gov/releases/h10/Hist or any other publication as agreed to by the Parties) over the entire calendar quarter in which the Net Sales were made.
8.6 Records, Audits . NPS and its Affiliates shall keep, and NPS shall require its licensees and sublicensees to keep, full, true and accurate records and books of account containing all particulars that may be necessary for the purpose of confirming the accuracy of, and calculating, as applicable, all royalties (including records of Net Sales) and NPS shall maintain complete and accurate records in sufficient detail to permit DR3 to confirm the accuracy of all amounts payable hereunder, in each case for a minimum period of six (6) years or such longer period as required by applicable Law. DR3 shall have a right to request an audit of NPS in order to confirm the accuracy of any of the foregoing (an "Audit"). Upon the written request by DR3 to Audit NPS, DR3 shall have the right to engage an independent, internationally recognized, accounting firm to perform a review as is reasonably necessary to enable such accounting firm to calculate or otherwise confirm the accuracy of any of the foregoing for the calendar year(s) requested by DR3; provided that (i) such accountants shall be given access to, and shall be permitted to examine and copy such books and records of NPS upon fifteen (15) days prior written notice to NPS, and at all reasonable times on such Business Days, (ii) prior to any such examination taking place, such accountants shall enter into a confidentiality agreement with NPS reasonably acceptable to NPS in order to keep all information and data contained in such books and records strictly confidential and shall not disclose such information or copies of such books and records to any third person including DR3, but shall only use the same for the purpose of the reviews and/or calculations which they need to perform in order to determine any amounts being reviewed, and (iii) such accountants shall use reasonable efforts to minimize any disruption to NPS's business. NPS shall make personnel available during regular business hours to answer queries on all such books and records required for the purpose of the Audit. The accountants shall deliver a copy of its findings to each of the Parties within ten (10) Business Days of the completion of the review, and, in the absence of fraud or manifest error, the findings of such accountant shall be final and binding on each of the Parties. Any underpayments by NPS shall be paid to DR3 within ten
-22-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- TERM AND TERMINATION
9.1 Term of Agreement . The term of this Agreement shall commence on the date of the Closing and shall expire on the earlier of: (a) the expiration of the Royalty Term in all countries of the Territory or (b) such time as the aggregate amount of the payments received by DR3 in respect of the (i) Purchaser Royalty Interest (as defined in the Original Purchase Agreement) after July 16, 2007 and (ii) the Royalty Payments made pursuant to this Agreement is equal to $125,000,000 (the "Term").
9.2 Termination . This Agreement shall terminate if DR3 exercises the Revocation in accordance with Section 2.1.
9.3 Accrued Rights . Termination or expiration of this Agreement for any reason will be without prejudice to any rights that will have accrued to the benefit of a Party prior to the effective date of such termination. Such termination will not relieve a Party from obligations that are expressly indicated to survive the termination or expiration of this Agreement.
9.4 Survival . Notwithstanding anything to the contrary contained herein, the following provisions shall survive any expiration or termination of this Agreement: Sections 2.6, 6.2, 6.5, 7.4, 7.5, 7.6, 8.2 (to the extent arising prior to expiration or termination), 8.3, 8.4, 8.5, 8.6 (but only for a period of one (1) year after the expiration or termination of this Agreement), 9.3, 9.4 and Article 10 and Article 11. Except as set forth in this Section 9.4, or otherwise expressly set forth herein, upon termination or expiration of this Agreement all other rights and obligations of the Parties shall cease.
- INDEMNIFICATION
10.1 Indemnification by NPS . NPS shall indemnify DR3, and its officers, directors, managers, partners, trust beneficiaries, agents and representatives, against, and hold each of them harmless from, any Damages suffered or incurred by any such Person arising from, relating to or otherwise in respect of:
- any breach of any representation or warranty of NPS contained in this Agreement; and
- any breach of any covenant of NPS contained in this Agreement.
10.2 Indemnification by DR3 . DR3 shall indemnify NPS, its directors, officers, shareholders and representatives, against, and hold them harmless from, any Damages suffered or incurred by any such Person arising from, relating to or otherwise in respect of:
(10) Business Days of notification of the results of such inspection. Any overpayments made by NPS shall be refunded by DR3 within ten (10) Business Days of notification of the results of such inspection. The cost of the accountant shall be the responsibility of DR3 unless the accountants calculation shows that the actual royalties payable of Net Sales are different, by more than five percent (5%) of the amounts as previously calculated by NPS.
-23-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- any breach of any representation or warranty of DR3 contained in this Agreement; and
- any breach of any covenant of DR3 contained in this Agreement.
- Third Party Claims. In order for a Person (the "indemnified party") to be entitled to any indemnification provided for under Section 10.1 or 10.2 in respect of, arising out of or involving a claim made by any Person against the indemnified party (a "Third Party Claim"), such indemnified party must notify the indemnifying party in writing (and in reasonable detail) of the Third Party Claim within ten (10) Business Days after receipt by such indemnified party of notice of the Third Party Claim; provided, however, that failure to give such notification shall not affect the indemnification provided hereunder except to the extent the indemnifying party shall have been actually and materially prejudiced as a result of such failure. Thereafter, the indemnified party shall deliver to the indemnifying party, within five (5) Business Days after the indemnified party's receipt thereof, copies of all notices and documents (including court papers) received by the indemnified party relating to the Third Party Claim.
- Assumption. If a Third Party Claim is made against an indemnified party, the indemnifying party shall be entitled to participate in the defense thereof and, if it so chooses, to assume the defense thereof with counsel selected by the indemnifying party; provided, however, that such counsel is not reasonably objected to by the indemnified party. Should the indemnifying party so elect to assume the defense of a Third Party Claim, the indemnifying party shall not be liable to the indemnified party for any legal expenses subsequently incurred by the indemnified party in connection with the defense thereof. If the indemnifying party assumes such defense, the indemnified party shall have the right to participate in the defense thereof and to employ counsel, at its own expense, separate from the counsel employed by the indemnifying party, it being understood that the indemnifying party shall control such defense. The indemnifying party shall be liable for the fees and expenses of counsel employed by the indemnified party for any period during which the indemnifying party has not assumed the defense thereof. If the indemnifying party chooses to defend or prosecute a Third Party Claim, all the indemnified parties shall cooperate in the defense or prosecution thereof. Such cooperation shall include the retention and (upon the indemnifying party's request) the provision to the indemnifying party of records and information that are reasonably relevant to such Third Party Claim, and making employees available on a mutually convenient basis to provide additional information and explanation of any material provided hereunder. Whether or not the indemnifying party assumes the defense of a Third Party Claim, the indemnified party shall not admit any liability with respect to, or settle, compromise or discharge, such Third Party Claim without the indemnifying party's prior written consent (which consent shall not be unreasonably withheld). If the indemnifying party assumes the defense of a Third Party Claim, the indemnified party shall agree to any settlement, compromise or discharge of a Third Party Claim that the indemnifying party may recommend and that by its terms obligates the indemnifying
-24-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- Other Claims. In the event any indemnified party should have a claim against any indemnifying party under Section 10.1 or 10.2 that does not involve a Third Party Claim being asserted against or sought to be collected from such indemnified party, the indemnified party shall deliver notice of such claim with reasonable promptness to the indemnifying party. The failure by any indemnified party so to notify the indemnifying party shall not relieve the indemnifying party from any liability that it may have to such indemnified party under Section 10.1 or 10.2, except to the extent that the indemnifying party demonstrates that it has been materially prejudiced by such failure.
- MISCELLANEOUS
11.1 Costs and Expenses . Each Party shall be responsible for and bear all of its own costs and expenses (including attorney fees and any brokers, finders or investment banking fees or prior commitment in respect thereof) incurred in connection with this Agreement.
11.2 Further Assurances . After the Closing, from time to time, as and when requested by any Party, each Party shall execute and deliver, or cause to be executed and delivered, all such documents, certificates and instruments, and shall take, or cause to be taken, all such further or other actions, as such other Party may deem reasonably necessary, desirable or appropriate to carry out all of the provisions of this Agreement and to consummate all of the transactions contemplated by this Agreement.
11.3 Notices . All notices or other communications required or permitted to be given hereunder shall be in writing and shall be delivered by hand or sent electronically, by facsimile or e-mail (with proof of electronic transmission), or sent, postage prepaid, by registered, certified or express mail or overnight courier service and shall be deemed given when so delivered by hand, facsimile or e-mail, or if mailed, three Business Days after mailing (one Business Day in the case of express mail or overnight service), as follows:
- if to DR3:
Drug Royalty L.P. 3.
c/o DRI Capital Inc.
22 St. Clair Avenue East
Suite 200
Toronto ON M4T 2S5
Attention of: Behzad Khosrowshahi
Fax No.: (416) 863-5161
E-mail: DRINotices@dricapital.com
- if to DR3:
party to pay the full amount of the liability in connection with such Third Party Claim, which releases the indemnified party completely in connection with such Third Party Claim and that would not otherwise adversely affect the indemnified party.
-25-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- if to NPS:
NPS Pharmaceuticals Inc.
550 Hills Drive, 3rd Floor
Bedminster, New Jersey 07921
United States of America
Attention of: General Counsel
Fax No.: (908) 450-5344
E-mail: estratemeier@npsp.comwith a copy (which shall not constitute notice) to:
Morgan Lewis and Bockius LLP
502 Carnegie Center
Princeton, New Jersey 08540-6241
Attention of: Randall B. Sunberg
Fax No.: (609) 919-6701
E-Mail: rsunberg@morganlewis.com
with a copy to:
Attention of: Gary Margolis
Fax No.: (416) 863-5161
E-Mail: gm@dricapital.com
with a copy (which shall not constitute notice) to:
King & Spalding LLP
601 S. California Ave.
Palo Alto, CA 94304
Attention of: Emma Maconick
Fax No.: (650) 422 6800
E-mail: emaconick@kslaw.com
or to such other address or addresses as DR3 or NPS may from time to time designate by notice as provided herein.
11.4 Successors and Assigns . This Agreement shall inure to the benefit of and be binding upon the successors and permitted assigns of each of the Parties hereto. This Agreement may not be assigned in whole or in part by either Party without the prior written consent of the other Party; provided, however, that (i) DR3 may assign this Agreement in whole or in part without the prior written consent of NPS: (a) by way of security to a financial institution or other lender, (b) to any Person that directly, or indirectly through one or more intermediaries, controls or is controlled by, or is under common control with, DR3, (c) to a special purpose vehicle created to be bankruptcy remote or for financing purposes or (d) as part of a sale of a material part of DR3, an Affiliate of DR3 or of the DR3 Royalty Right, in any case whether by way of reorganization or otherwise, and DR3 shall give prompt notice of any such assignment to NPS within ten (10) Business Days after the occurrence thereof and (ii) NPS shall be permitted to assign this Agreement in whole or in part without the prior written consent
-26-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
- Except as otherwise provided herein, this Agreement shall be governed by and construed, interpreted and enforced in accordance with the laws of the State of New York, without giving effect to the principles of conflicts of law thereof except as set forth in Section 5-1401 of the New York General Obligations Law.
- Each Party hereby irrevocably and unconditionally submits to the non-exclusive jurisdiction of the courts of the State of New York located in New York County, New York and the U.S. federal district courts in the Southern District of the State of New York.
of DR3 to any Affiliate to which the NPS Patents are also assigned in accordance with Section 6.1, provided that such assignment could not reasonably be expected to result in or give rise to a material adverse effect on the validity or enforceability of this Agreement or the rights or remedies of DR3 hereunder.
11.5 No Partnership . Nothing in this Agreement shall be deemed in any way or for any purpose to constitute either Party as a partner of the other Party in the conduct of any business. For all purposes of this Agreement, the Parties are independent contractors. Except to the limited extent expressly provided in this Agreement, neither Party shall have the authority to bind, obligate or represent the other Party.
11.6 Entire Agreement . Subject to Section 2.3, this Agreement, the Restated Security Agreement and the Closing Documents, including the Schedules and Exhibits hereto and thereto, together constitute the entire agreement and understanding between the Parties hereto with respect to the subject matter hereof and supersede all prior agreements and understandings, whether written or oral, relating to such subject matter.
11.7 Amendments, Supplements, Waivers . This Agreement may be amended or supplemented only by a written agreement signed by DR3 and NPS. Any waiver of, or consent to depart from, the requirements of any provision of this Agreement shall be effective only if it is in writing and signed by the Party giving it, and only in the specific instance and for the specific purpose for which it has been given. No failure on the part of any Party to exercise, and no delay in exercising, any right under this Agreement shall operate as a waiver of such right. No single or partial exercise of any such right shall preclude any other or further exercise of such right or the exercise of any other right.
11.8 Severability . If any provision of this Agreement is held to be invalid or unenforceable, the remaining provisions shall nevertheless be given full force and effect.
11.9 Governing Law.
11.10 Waiver of Jury Trial . Each Party hereby waives to the fullest extent permitted by applicable Law, any right it may have to a trial by jury in respect of any claim, demand, action or cause of action directly or indirectly arising out of, under or in connection with this Agreement or any transaction contemplated hereby, and each Party hereby agrees and consents that any such claim, demand, action or cause of action shall be decided by court trial without a jury, and that either Party hereto may file an original
-27-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
counterpart or a copy of this Section 11.10 with any court as written evidence of the consent of the signatories hereto to the waiver of their right to trial by jury. Each Party: (a) certifies that no representative, agent or attorney of any other Party has represented, expressly or otherwise, that such other Party would not, in the event of litigation, seek to enforce the foregoing waiver; and (b) acknowledges that it and the other Party hereto have been induced to enter into this Agreement by, among other things, the mutual waivers and certifications in this Section 11.10.
11.11 Counterparts . This Agreement may be executed in one or more counterparts, all of which when taken together shall be considered one and the same agreement. This Agreement may be delivered by either Party by facsimile or by electronic delivery and, if so executed and delivered, shall be legally valid and binding on the Party executing in such manner.
[Signature Page Follows]
-28-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
IN WITNESS WHEREOF, the Parties, by their duly authorized officers, have executed this Agreement.
NPS PHARMACEUTICALS, INC.
By: /s/ Luke Beshar
Name: Luke Beshar
Title: EVP & CFO
DRUG ROYALTY L.P. 3, by its General Partner, DRC MANAGEMENT LLC 3
By: /s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
[Signature Page to Amended and Restated Agreement for the Sale and Assignment of Rights]
-29-
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 1.1(j)
DR3 Account
Bank Name: [* * *]
Account Name: [* * *]
Account #: [* * *]
ABA #: [* * *]
Swift Code: [* * *]
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 4.6
Copies of Certain Agreements and Notifications
[See attached.]
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
NPS PHARMACEUTICALS, INC.
CERTIFICATE OF SECRETARY
I, Edward H. Stratemeier, Secretary of NPS Pharmaceuticals, Inc., a Delaware
corporation (the "Company"), do hereby certify that:
1. I am the duly elected, qualified and acting Senior Vice President, General Counsel and Secretary of the Company on the date hereof.
2. Attached hereto are true, correct and complete copies of the following documents:
Exhibit A
Non-Exclusive Patent License Agreement between NPS Pharmaceuticals, Inc. and Asahi Kasei Pharma Corporation, effective April I,
2005 (Asahi Agreement)
Exhibit B
Purchase and Sale Agreement between Allelix Biopharmaceuticals, Inc. and Kaare M. Gautvik and Peter Alestrom, effective Apri11,
1996 (Gautvik Agreement)
Exhibit C
Development Agreement between NPS Allelix Corp. and Ypsomed AG, effective January 7, 2004 (the "Patent 151
License"') (except for the last page of Appendix C)
Exhibit D
Notification of lapse of Patent No. EP 1519766A (filing date June 25, 2003) under the Patent 151 License
IN WITNESS WHEREOF, I have signed this Certificate this 20th day of December 2013.
/s/ Edward H. Stratemeier
Edward H. Stratemeier
Senior Vice President, General Counsel
and Secretary
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
EXHIBIT A
Asahi Agreement
(See attached)
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
EXHIBIT B
Gautvik Agreement
(See attached)
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
EXHIBIT C
Patent 151 License
(See attached)
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
EXHIBIT D
Patent 151 Lapse Notification
(See attached)
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 4.7
List of NPS Patents
"ESSENTIALLY PURE HUMAN PARATHYROID HORMONE"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Australia |
6 May `92 |
16049/92 |
20 Sept `01 |
639856 |
6 May `12 |
|
Austria |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Belgium |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Canada |
12 May `92 |
2068438 |
12 Nov `96 |
2068438 |
12 May `12 |
|
Denmark |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Denmark (SPC) |
20 Oct `06 |
CA200600028 |
25 March `08 |
CR200600028 |
22 May `17 |
|
European Patent Convention |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Finland |
22 May `92 |
922,356 |
12 April `01 |
106802 |
22 May `12 |
|
France |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Germany |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Great Britain |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Greece |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Hong Kong |
22 May `92 |
92304692.4 |
25 March `98 |
1004340 |
22 May `12 |
|
Ireland |
1 July `92 |
921,672 |
28 May `04 |
83494 |
30 June `12 |
|
Italy |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Japan |
22 May `92 |
156100/92 |
19 Sep `96 |
2563726 |
22 May `12 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
|
Korea |
22 May `92 |
1019920008730 |
5 Nov `97 |
1001289520000 |
22 May `12 |
|
Liechtenstein |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Luxembourg |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Monaco |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Netherlands |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
New Zealand |
21 May `92 |
19920242855 |
11 Aug `94 |
242855 |
21 May `12 |
|
Norway |
21 May `92 |
92304692.4 |
9 November `98 |
304,190 |
21 May `12 |
|
Norway (SPC) |
24 Oct `06 |
2006013 |
26 Nov `09 |
2006013 |
21 May `17 |
|
Philippines |
21 May `92 |
44335 |
13 June `95 |
28922 |
13 June `12 |
|
Portugal |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
South Africa |
22 May `92 |
92/03735 |
27 Jan `93 |
92/03735 |
22 May `12 |
|
Spain |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Sweden |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
Switzerland |
22 May `92 |
92304692.4 |
25 March `98 |
0 515 228 |
22 May `12 |
|
United States |
23 May `91 |
07/707114 |
4 May `93 |
5208041 |
23 May `11 |
"PARATHYROID HORMONE FORMULATION"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Australia |
16 Dec `94 |
12698/95 |
8 January `98 |
681737 |
16 Dec `14 |
|
Canada |
16 Dec `94 |
2,179,207 |
14 November `00 |
2,179,207 |
16 Dec `14 |
|
China |
16 Dec `94 |
94194608.8 |
6 August `03 |
94194608.8 |
16 Dec `14 |
|
Europe |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Austria |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Belgium |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Denmark |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Finland |
16 Dec `94 |
962593 |
15 Sep `09 |
120291 |
16 Dec `14 |
|
France |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Germany |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Great Britain |
16 Dec `94 |
95903732.6 |
3Oct `07 |
0735896 |
16 Dec `14 |
|
Greece |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Hong Kong |
16 Dec `94 |
98111127.6 |
23 Nov `07 |
1010336 |
16 Dec `14 |
|
Hong Kong |
8 June `98 |
05103473.5 |
28 May `10 |
HK1070816B |
8 June `18 |
|
Ireland |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Italy |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Japan |
16 Dec `94 |
517064/95 |
12 May `00 |
3065662 |
16 Dec `14 |
|
Liechtenstein |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Lithuania |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Luxembourg |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Monaco |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Netherlands |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
New Zealand |
16 Dec `94 |
277463 |
23 August `97 |
277463 |
16 Dec `14 |
|
Norway |
16 Dec `94 |
P962634 |
27 Dec `07 |
324905 |
16 Dec `14 |
|
Portugal |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
South Korea |
16 Dec `94 |
1019960703288 |
02 September `03 |
1003984610000 |
16 Dec `14 |
|
Spain |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Sweden |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
Switzerland |
16 Dec `94 |
95903732.6 |
3 Oct `07 |
0735896 |
16 Dec `14 |
|
U.S. |
23 Dec `93 |
08/172,206 |
5 March `96 |
5,496,801 |
23 Dec `13 |
"PROTEIN FORMULATION (PTH) (MULTI-DOSE INJECTION)"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Albania |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Austria |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Australia |
26 April `99 |
35916/99 |
29 Jan `04 |
766514 |
26 April `19 |
|
Australia DIV |
16 Jan `04 |
2004200156 |
13 April `06 |
2004200156 |
26 April `19 |
|
Australia DIV |
26 April `99 |
2006201087 |
|||
|
Belgium |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Brazil |
26 April `99 |
BR19999909958 |
|||
|
Canada |
26 April `99 |
2,329,800 |
15 June `04 |
2,329,800 |
26 April `19 |
|
China |
26 April `99 |
99807362.8 |
17 Oct `04 |
ZL99807362.8 |
26 April `19 |
|
Cyprus |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Denmark |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Europe |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Finland |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
France |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Germany |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Great Britain |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Greece |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Ireland |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Italy |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Hong Kong |
26 April `99 |
02102980.6 |
18 March `05 |
HK1041218 |
26 April `19 |
|
Japan |
26 April `99 |
2000-545551 |
28 April `11 |
4733267 |
26 April `19 |
|
Japan |
26 April `99 |
2011-011811 |
|||
|
Liechtenstein |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Latvia |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Lithuania |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Luxembourg |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Macedonia |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Mexico |
26 April `99 |
010640 |
23 Jan `06 |
233893 |
26 April `19 |
|
Monaco |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Netherlands |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
New Zealand |
26 April `99 |
508269 |
29 Mar `04 |
508269 |
26 April `19 |
|
Portugal |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Romania |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Singapore |
26 April `99 |
200006124-2 |
31 May `05 |
76839 |
26 April `19 |
|
Slovenia |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
South Africa |
26 April `99 |
2000/6040 |
24 April `02 |
2000/6040 |
26 April `19 |
|
South Korea |
26 April `99 |
1020007012037 |
8 May `06 |
1005798720000 |
26 April `19 |
|
Spain |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Sweden |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
|
Switzerland |
26 April `99 |
99917715.7 |
18 Aug `04 |
1079803 |
26 April `19 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
"A COMBINED PHARMACEUTICAL PREPARATION COMPRISING
PARATHYROID HORMONE AND A
BONE RESORPTION INHIBITOR"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Australia |
8 June `98 |
79458/98 |
30 Jan `03 |
753477 |
8 June `18 |
|
Australia Div. |
5 Sep `02 |
2002300896 |
13 July `06 |
2002300896 |
8 June `18 |
|
Austria Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Austria |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Belgium |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Belgium Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Canada |
8 June `98 |
2,294,101 |
3 Aug `10 |
2,294,101 |
8 June `18 |
|
Canada Div. |
8 June `98 |
2,698,626 |
|||
|
Cyprus |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Cyprus Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
CY1110287 |
8 June `18 |
|
Denmark |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Denmark Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Europe |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Europe Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Finland |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Finland Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
France |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
France Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Germany |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Germany Div. |
8 June `98 |
69841279.6-08 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Great Britain |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Great Britain Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Greece Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Greece |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Hong Kong |
8 June `98 |
00107524.0 |
24 March `05 |
1029738 |
8 June `18 |
|
Ireland |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Ireland Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Italy |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Italy Div. |
8 June `98 |
19750BE/2010 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Japan |
8 June `98 |
HEI 11-504259 |
(2002-504140) |
||
|
Japan |
8 June `98 |
2010-000066 |
(2010-100644) |
||
|
Liechtenstein |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Liechtenstein Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Luxembourg |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Luxembourg Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Monaco |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Monaco Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Netherlands |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Netherlands Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Portugal |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Portugal Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
South Africa |
8 June `98 |
98/4947 |
31 March `99 |
98/4947 |
8 June `18 |
|
Spain |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Spain Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Sweden |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Sweden Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
Switzerland |
8 June `98 |
98929965.6 |
8 Sept `04 |
1001802 |
8 June `18 |
|
Switzerland Div. |
8 June `98 |
04017622.4 |
4 Nov `09 |
1473040 |
8 June `18 |
|
U.S. |
14 Aug `98 |
09/125,247 |
4 Sep `01 |
6,284,730 |
8 June `18 |
|
U.S. Div Con |
18 March `03 |
10/389,797 |
28 March `06 |
7,018,982 |
8 June `18 |
|
US Div Con 2 |
19 Dec `05 |
11/305,339 |
24 March `09 |
7,507,715 |
8 June `18 |
|
US Div |
9 Jan `09 |
12/351,558 |
6 July `10 |
7,749,543 |
8 June `18 |
|
US Div |
23 June `10 |
12/822,089 |
10 April `12 |
8,153,588 |
8 June `18 |
|
US DIV |
24 Feb `12 |
13/405,093 |
(2012/0148684) |
** Designated contracting states: Austria, Belgium, Cyprus, Denmark, Finland, France, Germany, Great Britain, Greece, Ireland, Italy, Liechtenstein, Luxembourg, Monaco, Netherlands, Portugal, Spain, Sweden, Switzerland.
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
"METHOD OF ADMINISTERING THERAPEUTIC INJECTIONS"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Canada 35296/125.2 |
18 October `04 |
2542715 |
|||
|
Europe* |
18 October 2004 |
04795465.6 |
(EP1687048A) |
||
|
US 50821/125.3 |
15 October `04 |
10/966364 |
4 May `10 |
7,708,732 |
11 May `27 |
* Designated contracting states: Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, Hungary, Ireland, Liechtenstein, Luxembourg, Monaco, Poland, Portugal, Romania, Slovenia, Slovakia, Sweden, Switzerland, Turkey, with Albania, Croatia, Lithuania, Latvia and Macedonia as extension states.
"PARATHYROID HORMONE FORMULATIONS AND USES THEREOF"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Albania |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Austria |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Belgium |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Bosnia-Herzegovina |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Bulgaria |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Croatia |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Cyprus |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Czech Republic |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Denmark |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Estonia |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Europe |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
26 Oct `27 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Finland |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
France |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Germany |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Great Britain |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Greece |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Hong Kong |
27 Oct `08 |
11100351.0 |
16 March `11 |
1146465 |
27 Oct `28 |
|
Hungary |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Iceland |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Ireland |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Italy |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Liechtenstein |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Latvia |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Lithuania |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Luxembourg |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Macedonia |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Malta |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Mexico |
27 Oct `08 |
MX/a/2010/004388 |
30 May `11 |
287021 |
27 Oct `28 |
|
Monaco |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Netherland |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Norway |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Poland |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Portugal |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Republic of Serbia |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Romania |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Russia |
27 Oct `08 |
2010 121165/15 |
16 March `11 |
2467762 |
27 Oct `28 |
|
Slovak Republic |
27 Oct `08 |
08842081.5 |
16 March `11 |
E9913 |
27 Oct `28 |
|
Slovenia |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
South Africa |
27 Oct `08 |
08842081.5 |
16 March `11 |
2010/02812 |
27 Oct `28 |
|
Spain |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Sweden |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Switzerland |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
|
Turkey |
27 Oct `08 |
08842081.5 |
16 March `11 |
2219665 |
27 Oct `28 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
SPC SUMMARY
|
Based on EP 0 515 228 |
||||
|
Country |
Application Number |
Filing Date |
Status |
Expiry |
|
Denmark |
CA 2006 00028 |
20 Oct. 2006 |
Granted 25 March 2008 |
22 May, 2017 |
|
Norway |
SPC/NO 2006 013 |
24 Oct. 2006 |
Granted 7 Dec. 2009 |
21 May 2017 |
|
Based on EP 1 079 803 |
||||
|
Country |
Application Number |
Filing Date |
Status |
Expiry Date |
|
Austria |
E273693-3 |
19 Oct. 2006 |
Grant Date 18 July 2007 |
April 24, 2021 |
|
Belgium |
2006C/033 |
19 Oct. 2006 |
Grant Date 5 June 2007 |
April 24, 2021 |
|
Cyprus |
CY06/006 |
23 Oct. 2006 |
Grant Date March 2007 |
April 23, 2021 |
|
Finland |
L20060013 |
13 Oct. 2006 |
Grant Date 29 Aug 2011 |
April 24, 2021 |
|
Germany |
12 2006 000 057.7 |
18 Oct. 2006 |
Grant Date Feb. 18 2009 |
April 24, 2021 |
|
Greece |
20060800026 |
20 Oct. 2006 |
Grant Date May 30 2007 |
April 24, 2021 |
|
France |
06C0032 |
20 Oct 2006 |
Grant Date 24 April 2007 |
April 24, 2021 |
|
Ireland |
2006/032 |
17 Oct. 2006 |
Grant Date 23 May 2007 |
April 23, 2021 |
|
Italy |
60605 |
23 Oct. 2006 |
Grant Date June 5 2007 |
April 22, 2021 |
|
Latvia |
C/LV2006/0009 |
17 Oct. 2006 |
Grant Date 20 March 2007 |
April 24, 2021 |
|
Lithuania |
PA 2006 007 |
23 Oct. 2006 |
Grant No. C1079803 Grant Date 04 June 2007 |
April 25, 2021 |
|
Luxembourg |
91281 |
19 Oct. 2006 |
Grant No. 91291 Grant Date 19 Dec. 2006 |
April 24, 2021 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
|
The Netherlands |
300243 |
17 Oct. 2006 |
Grant Date December 7 2006 |
April 23, 2021 |
|
Portugal |
240 |
20 Oct. 2006 |
Granted |
April 24, 2021 |
|
Romania |
C/022 |
10 April 2007 |
Grant Date Dec 23 2004 |
April 24, 2021 |
|
Slovenia |
200640015 |
18 Oct. 2006 |
Grant Date 16 July 2009 |
April 24, 2021 |
|
Sweden |
0690029-4 |
24 Oct. 2006 |
Grant Date 5 Feb 2008 |
23 April 2021 |
|
United Kingdom |
SPC/GB06/035 |
13 Oct. 2006 |
Grant Date 1 June 2007 |
23 April 2021 |
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 4.7(a)
Opinions of Counsel
- December 10, 1998---Vossius & Partner---PTH Search
- October 2, 2004-Plougmann & Vingtoft-Risk Assessment Report Concerning Lyophilised Parathyroid Hormone (PTH) Formulation
- July 30, 2007-Kraus & Weisert-Freedom to Operate Analysis in Europe for the Product "Preotact".
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 4.8
Litigation
Albanian patent 01282, the Albanian part of European patent number 1079803, serial number 99917715.7 is the subject of a suit for reinstatement against the Albanian Patent Office.
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 6.1(a)
Jurisdictions Requiring Consent to License
- Australia
- Belgium
- Canada
- Denmark
- Finland
- France
- Germany
- Iceland
- Italy
- Japan
- Luxembourg
- Netherlands
- Norway
- Spain
- Sweden
- Switzerland
- United Kingdom
- U.S.A.
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Schedule 6.2(d)
Form 8-K
[See attached.]
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
|
December 20, 2013 |
|
Date of Report (Date of earliest event reported) |
|
NPS PHARMACEUTICALS, INC. |
|
(Exact name of registrant as specified in its charter) |
|
Delaware |
0-23272 |
87-0439579 |
|
(State or other jurisdiction of incorporation) |
(Commission File Number)
|
(I.R.S. Employer Identification Number) |
|
550 Hills Drive, 3rd Floor |
|
Bedminster, NJ 07921 |
|
(Address of principal executive offices) |
|
|
(908) 450-5300 |
|
|
(Registrant's telephone number, including area code) |
||
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
⃞
Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)⃞
Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)⃞
Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))⃞
Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Item 1.01. Entry into a Material Definitive Contract.
On December 20, 2013, NPS Pharmaceuticals, Inc. (the "Company" or "NPS") entered into an amendment and restatement (the "Amendment and Restatement") to its Agreement for the Sale and Assignment of Rights (the "2007 Agreement"), dated as of July 16, 2007, between NPS Allelix Corp., Drug Royalty L.P. 3 ("DRLP3"), an investment fund managed by DRI Capital Inc. ("DRI"), and the Company.
Under the 2007 Agreement, the Company sold to DRLP3 its rights to receive future royalty payments arising from the sale of recombinant human parathyroid hormone 1-84 [rDNA origin] ("PTH") under its license agreement ("Takeda License Agreement") with Takeda Pharma A/S, formerly Nycomed Danmark ApS ("Takeda"). On March 18, 2013, pursuant to the previously disclosed Termination and Transition Agreement between NPS and Takeda, NPS' license agreement with Takeda was terminated and NPS re-acquired exclusive rights worldwide to develop and commercialize PTH. Preotact is the brand name that Takeda had used to market PTH for the treatment of osteoporosis in certain of its licensed territories. NPS is developing PTH in the U.S. under the trade name Natpara for the treatment of hypoparathyroidism. NPS filed a BLA for Natpara with the FDA in October 2013.
Pursuant to the Amendment and Restatement, (i) DRLP3 has consented to the commercialization of PTH by the Company, (ii) the terms of the 2007 Agreement are tolled, and (iii) the parties' rights and obligations regarding PTH and related technology are governed by the Amendment and Restatement.
The Company will be required to pay royalties in the mid single digits to DRLP3 based upon sales of PTH by the Company and its licensees (if any) worldwide, excluding Israel. The Company has agreed to undertake certain efforts to commercialize PTH. If the Company does not submit a Marketing Authorization Application to the European Medicines Agency for PTH in the European Union by an agreed upon date, DRLP3 will have the right to revoke the consent granted in the Amendment and Restatement, reinstate the 2007 Agreement, and either cause the Company to enter into a new license agreement with a third party with respect to PTH on terms that are substantially similar and no more extensive (when taken as a whole) than the terms contained in the terminated Takeda License Agreement, or negotiate such an agreement on NPS' behalf.
The Company's obligation to pay royalties to DRLP3 under the Amendment and Restatement shall expire on a country-by-country basis upon the later of (i) the last to expire patent controlled by the Company with claims covering PTH in such country or (ii) the expiration of any period of regulatory exclusivity applicable to PTH in such country. The Company's obligation to pay royalties to DRLP3 under the Amendment and Restatement shall terminate in its entirety once cumulative royalty payments made to DRLP3 by Takeda and the Company total $125 million. As of September 30, 2013, $45.5 million in royalties had been paid to DRLP3.
DRLP3 continues to maintain a security interest in NPS patents that contain claims covering PTH and certain other NPS intellectual property related to PTH. In the event of a default by NPS under the Amendment and Restatement, DRLP3 would be entitled to enforce its security interest against NPS and such intellectual property.
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
|
Date: |
December 20, 2013 |
NPS PHARMACEUTICALS, INC. |
|
|
|
|||
|
|
|
By: |
/s/ Edward Stratemeier |
|
Edward Stratemeier |
|||
|
Senior Vice President, General Counsel and |
|||
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
Exhibit A
Form of Restated Security Agreement
[See attached.]
Confidential Treatment Requested. Confidential portions of this document have
been redacted and have been separately filed with the Commission.
AMENDED AND RESTATED SECURITY AGREEMENT
THIS AMENDED AND RESTATED SECURITY AGREEMENT is made as of December 20, 2013.
BETWEEN:
NPS PHARMACEUTICALS, INC.,
a corporation existing under the laws of the State of Delaware
(collectively with its successors and permitted assigns, "Debtor"),
- and -
DRUG ROYALTY L.P. 3,
a Cayman Islands limited partnership
(collectively with its successors and permitted assigns, "Secured Party").
WHEREAS Debtor and Secured Party are parties to that certain Agreement for the Sale and Assignment of Rights dated as of July 16, 2007 (as amended, restated, supplemented or otherwise modified prior to the date hereof, the "Original Purchase Agreement");
AND WHEREAS in connection with the Original Purchase Agreement, Debtor and Secured Party entered into a security agreement dated as of October 2, 2009 (the "Original Security Agreement"), pursuant to which Debtor granted to Secured Party a first priority security interest in the Collateral (as defined therein);
AND WHEREAS Debtor and Secured Party have agreed to amend and restate the Original Purchase Agreement in accordance with the terms and conditions of that certain Amended and Restated Agreement for the Sale and Assignment of Rights (as such agreement may be amended, modified, supplemented or restated from time to time, the "A&R Purchase Agreement"); and
AND WHEREAS it is a condition to the effectiveness of the A&R Purchase Agreement that the Debtor and Secured Party amend and restate the Original Security Agreement on the terms and conditions set forth herein.
NOW THEREFORE in consideration of the respective covenants, promises and agreements of the parties herein contained and other good and valuable consideration (the receipt and sufficiency of which are hereby acknowledged by each of the parties), the parties hereby amend and restate the Original Security Agreement in its entirety as set forth herein and further agree as follows:
ARTICLE 1
INTERPRETATION
1.1 Defined Terms
Terms that are defined in the A&R Purchase Agreement and not otherwise defined herein have, unless the context otherwise requires, the respective meanings specified in the A&R Purchase Agreement and, in addition, the following terms have the following meanings (and grammatical variations of such terms shall have corresponding meanings):
"A&R Purchase Agreement" has the meaning specified in the recitals of this Agreement;
"Additional Collateral" means the NPS Trademarks, the NPS Patents in the United States and Japan and NPS Know-How specific to the United States and Japan.
"Agreement" means this Amended and Restated Security Agreement, the recitals, all attached exhibits and Schedules, and any agreement, exhibit or Schedule supplementing or amending this Agreement;
"Collateral" means all of Debtor's right, title and interest in, to and under the NPS Technology and the NPS Trademarks, whether now owned or hereinafter acquired, and includes Proceeds therefrom and, where the context permits, any reference to "Collateral" shall be deemed to be a reference to "Collateral or any part thereof";
"Event of Default" means a Major Default or a Minor Default;
"Financing Statements" has the meaning specified in Section 3.1(e);
"including", when used herein, means "including without limitation" and shall not be construed to limit any general statement which it follows to the specific or similar items or matters immediately following it;
"IP Office Filings" has the meaning specified in Section 3.1(g);
"Major Default" means the occurrence of any one or more of the following events:
(a) any breach attributable to Debtor of any representation or warranty made in Section 4.1 or 4.5 of the A&R Purchase Agreement; provided, however, that a breach of Section 4.1(a) of the A&R Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
(b) any breach by Debtor of any of its covenants under Section 6.1(a) of the A&R Purchase Agreement; or
(c) the cessation or threatened cessation by Debtor of its business generally or the admission by Debtor of its inability to, or its actual failure to, pay its debts generally, including circumstances where Debtor (i) is adjudged bankrupt or
2
insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee.
"Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a Major Default:
(a) the failure of Debtor to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
(b) the failure of Debtor to perform any non-monetary Obligation or covenant under this Agreement or the A&R Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
(c) any breach attributable to Debtor of any representation or warranty made in the A&R Purchase Agreement;
"Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of Debtor to Secured Party arising pursuant to (i) the A&R Purchase Agreement, (ii) in the case of a Revocation, the Original Purchase Agreement, and/or (iii) this Agreement.
"parties" means Debtor and Secured Party, and "party" means either of them;
"Proceeds" means property in any form derived, directly or indirectly, from any dealing with the Collateral or other proceeds (together with any reissue, continuation, continuation-in-part or extension of the Patents) thereof; and includes any accounts, payment intangibles or other general intangibles arising from any sale, transfer, license, lease or other dealing in any of the Collateral and any payment or value representing indemnity or compensation for loss or damage to the Collateral or other Proceeds, including insurance proceeds and proceeds (as such term is defined in the UCC);
"Security Interest" has the meaning specified in Section 2.1;
"Transaction Documents" means this Agreement, the A&R Purchase Agreement and each other Closing Document; and
"UCC" means (i) the Uniform Commercial Code as in effect from time to time in the State of New York, (ii) with respect to enforcement, the Uniform Commercial Code as in effect from time to time in any other state whose law is applicable with respect to Secured Party's enforcement of its rights hereunder and (iii) insofar as any references to the UCC is used in the context of perfection, the Uniform Commercial Code as in effect from time to time in the state that is the "location" of Debtor under the UCC, in each case including any legislation that may be substituted therefor (as any such substituted legislation may be amended from time to time).
3
1.2 Meanings under the UCC
All terms used herein and not otherwise defined pursuant to Section 1.1 (including any definitions incorporated herein by reference to the A&R Purchase Agreement) and which are defined in the UCC shall, unless the context otherwise requires, have the respective meanings assigned to such terms in the UCC.
1.3 Sections and Headings
(a) The division of this Agreement into Articles and Sections and the insertion of headings are for convenience of reference only and shall not affect the interpretation of this Agreement.
(b) All uses of the words "hereto", "herein," "hereof," "hereby" and "hereunder" and similar expressions refer to this Agreement and not to any particular Section or portion of it.
(c) References to an "Article", "Section" or "Schedule" are references to an Article or Section of or a Schedule to this Agreement unless otherwise indicated.
1.4 Applicable Law
(a) This Agreement shall be governed by, and interpreted and enforced in accordance with, the laws of the State of New York and the federal laws of the United States applicable therein, without giving effect to the principles of conflicts of law thereof except as set forth in Section 5-1401 of the New York General Obligations Law.
(b) EACH PARTY HEREBY WAIVES, TO THE FULLEST EXTENT PERMITTED BY APPLICABLE LAW, ANY RIGHT IT MAY HAVE TO A TRIAL BY JURY IN RESPECT OF ANY CLAIM, DEMAND, ACTION OR CAUSE OF ACTION DIRECTLY OR INDIRECTLY ARISING OUT OF, UNDER OR IN CONNECTION WITH THIS AGREEMENT OR ANY TRANSACTION CONTEMPLATED HEREBY; AND EACH PARTY HEREBY AGREES AND CONSENTS THAT ANY SUCH CLAIM, DEMAND, ACTION OR CAUSE OF ACTION SHALL BE DECIDED BY COURT TRIAL WITHOUT A JURY, AND THAT ANY PARTY HERETO MAY FILE AN ORIGINAL COUNTERPART OR A COPY OF THIS SECTION 1.4(b) WITH ANY COURT AS WRITTEN EVIDENCE OF THE CONSENT OF THE SIGNATORIES HERETO TO THE WAIVER OF THEIR RIGHT TO TRIAL BY JURY. Each party (i) certifies that no representative, agent or attorney of any other party has represented, expressly or otherwise, that such other party would not, in the event of litigation, seek to enforce the foregoing waiver and (ii) acknowledges that it and the other parties hereto have been induced to enter into this agreement by, among other things, the mutual waivers and certifications in this Section 1.4(b).
1.5 Consent to Jurisdiction
Each of Debtor and Secured Party (a) hereby irrevocably submits to the jurisdiction of the United States District Court for the Southern District of New York for the purposes of any suit, action or proceeding arising out of or relating to this Agreement and to the extent that such
4
United States district court lacks jurisdiction despite the consent herein, to the jurisdiction of the courts of the State of New York sitting in New York County and the appellate courts therefrom, and (b) hereby waives, and agrees not to assert in any such suit, action or proceeding, any claim that it is not personally subject to the jurisdiction of such court, that the suit, action or proceeding is brought in an inconvenient forum or that the venue of the suit, action or proceeding is improper. Each of Debtor and Secured Party consents to process being served in any such suit, action or proceeding by mailing a copy thereof to such party at the address in effect for notices to it under this Agreement and agrees that such service shall constitute good and sufficient service of process and notice thereof. Nothing in this Section 1.5 shall affect or limit any right to serve process in any other manner permitted by law.
1.6 Severability
If any provision of this Agreement is held to be illegal or invalid, the remaining provisions hereof shall nevertheless be given full force and effect, and all the provisions hereof are hereby declared to be separate, severable and distinct.
ARTICLE 2
SECURITY INTEREST
2.1 Grant of Security Interest
As general and continuing collateral security for the due payment and performance of the Obligations, Debtor hereby pledges, mortgages, charges and assigns (by way of security) to Secured Party, and grants to Secured Party, a security interest in the Collateral (collectively, the "Security Interest").
2.2 Attachment
Debtor and Secured Party acknowledge and agree that value has been given for the granting of the Security Interest and that they have not agreed to postpone the time for attachment. The security interest in the portion of the Collateral, as previously granted by Debtor pursuant to the Original Security Agreement, was attached and perfected, and this Agreement is being entered into not to reflect a new obligation with respect to such existing Collateral but to reflect the changes from the Original Security Agreement and the grant of Additional Collateral and removal of a portion of the Collateral (as such term was defined in the Original Security Agreement).
2.3 Conditional Assignment of Title of Certain Patents
(a) Debtor hereby grants, bargains, sells, assigns and transfers to Secured Party the Patents listed in Schedule 2.3 (collectively, the "Supplementary European Patents") such that title thereto and ownership therein shall, effective upon the occurrence of (i) a Major Default or (ii) subject to Section 2.3(b), a Minor Default, belong to and be vested in Secured Party.
(b) If a Minor Default occurs, the grant, bargain, sale, assignment and transfer contemplated in Section 2.3(a) shall not occur unless or until Secured Party has first sought recourse under Section 5.1(a) and any remedy obtained under such recourse has not satisfied the
5
Obligations of Debtor or any judgement obtained pursuant to such recourse remains unsatisfied by Debtor for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement) (such 15th Business Day, the "Assignment Date").
(c) Nothing contained in this Section 2.3 shall be construed to afford Secured Party any recourse to the Supplementary European Patents prior to (i) the occurrence of a Major Default or (ii) the Assignment Date, as applicable.
ARTICLE 3
REPRESENTATIONS AND WARRANTIES OF DEBTOR
3.1 Representations and Warranties
Debtor represents and warrants to Secured Party as follows and acknowledges that Secured Party is relying on such representations and warranties in connection with its entering into the A&R Purchase Agreement and the other transactions contemplated therein:
(a) "NPS Pharmaceuticals, Inc." is the only business name used by Debtor and has been the only business name used by Debtor for at least 12 months prior to the date hereof;
(b) the jurisdiction in which Debtor is located for purposes of Sections 9-301 and 9-307 of the UCC is the State of Delaware;
(c) except as set forth in Schedule 3.1(c), the Collateral has not been abandoned or adjudged invalid or unenforceable, in whole or in part;
(d) other than the Assignment and the "Security Interest" granted by Debtor under the Original Security Agreement, Debtor has not made a previous assignment, sale, transfer or agreement constituting a present or future assignment, sale or transfer of all or any portion of its right, title and interest in and to any Collateral for purposes of granting a security interest or as collateral that has not been terminated or released;
(e) copies of all UCC-1 financing statements filed by the Secured Party in the office of the Secretary of State of the State of Delaware to perfect the Security Interest (collectively, the "Financing Statements") are attached as Appendix 3.1(e). Debtor hereby approves, ratifies and confirms any such Financing Statement filed by Secured Party prior to the execution and delivery of this Agreement, and Debtor waives any right to claim that any such prior filing was not in compliance with the UCC or not pursuant to a prior authenticated record;
(f) copies of all notices of security interest incorporating by reference the terms of this Agreement, together with all appropriate cover sheets and other necessary documentation, required to be filed with any patent or trademark offices in the United States of America in order to record this Agreement are attached as Schedule 3.1(f).
(g) all necessary documentation required to be filed with any patent or trademark office in the United Kingdom, France, Germany, Spain, Italy, The Netherlands, Austria and Greece in order to record this Agreement has been filed (the filings referred to in Section 3.1(f) and this Section 3.1(g) collectively, the "IP Office Filings");
6
(h) upon the filing of the Financing Statements with the appropriate agencies under the UCC, the Security Interest will constitute a perfected first priority security interest in the Collateral described on such financing statements in favour of Secured Party to the extent that a security interest therein may be perfected by filing in Delaware pursuant to the relevant UCC, prior to all other Encumbrances;
(i) except as have been obtained or made and are in full force and effect, no authorization, approval or other action by, and no notice to or filing with, any Governmental Authority or any other third party is required either:
(i) for the grant by Debtor of the Security Interest; or
(ii) for the perfection or maintenance of the Security Interest hereunder, including the first priority nature of such security interest (except with respect to the Financing Statements and the IP Office Filings) or the exercise by Secured Party of its rights and remedies hereunder;
(j) Debtor is the exclusive owner of the entire right, title, and interest in and to the Collateral, free and clear of any Encumbrances except as contemplated herein and in the A&R Purchase Agreement in favour of Secured Party; and
(k) the representations and warranties of set forth in Sections 4.1, 4.2, 4.5, 4.6, 4.7(a), 4.7(b), 4.7(d), 5.1 and 5.2 of the A&R Purchase Agreement are hereby repeated in this Agreement. The representations and warranties set forth in this Section 3.1(k) shall survive for the Term and shall expire thereafter.
3.2 Survival
The representations and warranties of Debtor contained in this Agreement shall survive for so long as any of the Obligations shall remain unpaid or unperformed, as applicable, and, notwithstanding any investigation made by or on behalf of Secured Party, shall continue in full force and effect for the benefit of Secured Party during such period.
ARTICLE 4
COVENANTS OF DEBTOR
4.1 Change of Name or Jurisdiction
Debtor shall not change its name as set out in Section 3.1(a), add any new business name or change its jurisdiction as set out in Section 3.1(b), or move the location of Debtor's principal place of business or the location of the books and records related to the Collateral, in each case without providing at least thirty days' prior written notice to Secured Party of such change or addition.
7
4.2 Creating and Preserving the Security Interest
(a) As soon as possible following the execution and delivery of this Agreement and, in any case, within 60 days from the date hereof, Debtor will cause the IP Office Filings to be filed with the appropriate Governmental Authorities in the jurisdictions specified therein.
(b) Debtor shall, from time to time at the request of Secured Party acting reasonably, make and do all such acts and things and execute and deliver all such instruments, agreements, financing statements and documents as Secured Party reasonably requests by notice in writing given to Debtor in order to create, preserve, perfect, validate or otherwise protect (including with respect to priority) the Security Interest, to enable Secured Party to exercise and enforce its rights and remedies hereunder and generally to carry out the provisions and purposes of this Agreement.
4.3 Verification of Collateral
Subject to the intangible nature of the Collateral, Secured Party shall have the right one time per year to verify the existence and state of the Collateral in any reasonable manner that Secured Party may consider appropriate, and Debtor agrees to furnish all reasonable assistance and information and to perform all such reasonable acts as Secured Party may reasonably request in connection therewith and for such purpose to grant to Secured Party or its agents reasonable access to all places where Collateral may be located and to all premises occupied by Debtor; provided, however, that nothing in this Section 4.3 shall limit Secured Party's ability to access any public records relating to the Collateral maintained by any patent or trademark office or other Governmental Authority.
4.4 Preservation of Collateral
Except in connection with licenses granted in jurisdictions not included in Schedule 6.1(a) of the A&R Purchase Agreement or to the extent licenses or transfers are otherwise permitted pursuant to the A&R Purchase Agreement (including licenses or transfers to which Secured Party has consented), Debtor shall not transfer or convey any interest in the Collateral or suffer, permit or cause any Encumbrances thereon. Without limiting the foregoing, in the event Debtor makes such a transfer or conveyance, Debtor shall take any and all actions, at Debtor's expense, to ensure that Secured Party's Security Interest is and remains perfected. Subject to Section 7.6 of the A&R Purchase Agreement and 7.3(c) of this Agreement in the event of a Revocation and subject to Section 6.1(b) of the A&R Purchase Agreement, Debtor agrees to take all steps in any proceeding before the applicable patent or trademark Governmental Authority or any other Governmental Authority in each case in any jurisdiction in which Debtor intends to commercialize the Product to maintain each application and registration of the Patents and subject to the next two sentences, the NPS Trademarks, and to maintain the accuracy and effectiveness of the filings, registrations or recordings with respect to the Collateral in favour of Secured Party, in such capacity. NPS's obligation to maintain the PREOTACT trademarks shall be limited to the following countries: all the countries of the European Union, European countries outside of the European Union, the Commonwealth of Independent States (formerly the USSR) and Turkey. In the event that the Consent (as defined in the A&R Purchase Agreement) becomes irrevocable in accordance with the A&R Purchase Agreement, NPS shall
8
no longer have any obligation to maintain the PREOTACT trademarks in any of the countries set forth in the previous sentence. Secured Party may from time to time, at its option, perform the acts specified in this Section 4.4 which Debtor fails to perform after being requested by Secured Party in writing to so perform (it being understood that no such request need be given after the occurrence and during the continuance of an Event of Default). Secured Party may from time to time take any other action which it deems necessary for the maintenance, preservation or protection of any of the Collateral.
4.5 Registration of Security; Acknowledgement of Existing Filings.
(a) Debtor shall undertake all such acts, execute all such documents and do all such things as Secured Party may request in order for this Agreement and the rights granted to Secured Party by this Agreement to be recorded with the patent and trademark offices or appropriate Governmental Authority office of any relevant jurisdiction, including the European Patent Office and the United States Patent and Trademark Office.
(b) Each party hereto hereby acknowledges that in connection with the Original Purchase Agreement, the parties have previously entered into certain agreements and made certain registrations and filings to perfect the security interest granted to Secured Party and record the conditional assignment agreements executed in connection therewith (copies of which are attached hereto as Exhibit A; collectively, the "Patent Filings"). As of the date hereof, each party hereby acknowledges and agrees that (i) the Patent Filings shall be and remain in full force and effect, and shall, to the extent they expressly or implicitly so purport, except to the extent as would not either individually or in the aggregate have a Material Adverse Effect (as such term is defined in the A&R Purchase Agreement), constitute the legal, valid, binding and enforceable obligations of the parties thereto in accordance with their terms (ii) such Patent Filings shall only be enforced in accordance with the circumstances specified in this Agreement and the A&R Purchase Agreement, and (iii) no further filing, registration or amendment is necessary to perfect the interest and rights granted under the Patent Filings. The execution, delivery and effectiveness of this Agreement shall not operate as a waiver of any right, power or remedy of the Parties under the Patent Filings.
ARTICLE 5
DEFAULT AND SECURED PARTY'S REMEDIES
5.1 Remedies Upon Default
Subject to Sections 5.2, 5.3, 5.5 and 5.6, upon the occurrence of any Event of Default and at any time during the continuance thereof, without any further notice or any other action on the part of Secured Party except as provided in the UCC or expressly set forth in this Agreement, Secured Party may avail itself of all of its rights and remedies as a secured party under the UCC and, in addition to and without limiting such rights and remedies, shall be entitled to avail itself of the following remedies:
(a) commence legal action to enforce payment or performance of the Obligations;
(b) dispose of the Collateral by private or public sale, lease, licence or otherwise upon such terms and conditions as Secured Party may determine in accordance with applicable law
9
and whether or not Secured Party has taken possession of the Collateral; provided, however that Secured Party shall give Debtor at least 30 days prior written notice of such disposal event;
(c) file such proofs of claim or other documents as may be necessary or desirable to have its claim lodged in any bankruptcy, winding-up, liquidation, dissolution or other proceedings (voluntary or otherwise) relating to Debtor; and
(d) take any other action, suit, remedy or proceeding authorized or permitted by this Agreement, the A&R Purchase Agreement, the UCC or by law or equity, including pursuant to the authority granted in any power of attorney granted hereunder or otherwise pursuant to this Agreement or any other Transaction Document.
5.2 ;Delayed Realization for Minor Defaults
Notwithstanding Section 5.1, if the Event of Default is a Minor Default, Secured Party shall not be entitled to avail itself of its remedy under Section 5.1(b) or any other similar remedy under the UCC permitting Secured Party to take possession of or realize on Collateral, unless and until it has first sought recourse under Section 5.1(a) and any remedy obtained under such recourse has not satisfied the Obligations of Debtor or any judgement obtained pursuant to such recourse remains unsatisfied by Debtor for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement). Nothing in this Section 5.2 or in any judgement shall diminish or extinguish the rights of Secured Party as a secured creditor. For the avoidance of doubt, this Section 5.2 shall not apply in respect of an Event of Default that is a Major Default.
5.3 Standstill
Notwithstanding Sections 5.1 and 5.2, if an Event of Default is the result of a breach of a representation and warranty attributable to Debtor, then Secured Party shall not take any action under Section 5.1 or 5.2 for a period of 60 days commencing on the date that notice of such breach is provided to Debtor. During such 60-day period, Secured Party will enter into good-faith negotiations with Debtor in an effort to determine whether any remedy other than enforcement of the security interest granted to Secured Party hereunder can be agreed upon.
5.4 Limitations
Upon the occurrence of any Event of Default and during the continuance thereof, Section 5.1 shall be inapplicable with respect to the NPS Know-How. In lieu of such rights, solely to the extent that Secured Party or a third party obtains title to the Collateral by the exercising of Secured Party of its rights hereunder, Debtor hereby grants, effective upon the occurrence of any Event of Default, without any further action during the continuance of such Event of Default, or if such Collateral has been disposed of, indefinitely, the non-exclusive right and license under the NPS Know-How to manufacture, market, sell, offer to sell, use, import and distribute, with right to sublicense, the Product.
10
5.5 Rights and Remedies are Not Mutually Exclusive
To the fullest extent permitted by law, Secured Party's rights and remedies, whether provided for in this Agreement or otherwise, are not mutually exclusive, are cumulative and not alternative, and may be exercised independently or in any combination.
5.6 No Obligation to Enforce
Secured Party shall not be under any obligation to, or liable or accountable for any failure to, enforce payment or performance of the Obligations or to seize, realize, take possession of or dispose of the Collateral and shall not be under any obligation to institute proceedings for any such purpose.
5.7 Cost of Enforcement
Debtor shall, upon demand, reimburse Secured Party for all reasonable costs and expenses incurred by Secured Party in the enforcement of any rights hereunder (including reasonable fees and expenses of counsel where Secured Party prevails) and all such costs shall form part of the Obligations. Additionally, Debtor shall promptly pay directly or reimburse Secured Party for costs, expenses and fees (including attorneys' fees) payable for analysis and diligence relating to the transfers and restructuring described in the recitals to this Agreement as well as the preparation and/or review of all required consent and assignment and assumption agreements, other than the preparation of this Agreement and the preparation and filing of the Financing Statements.
5.8 Debtor Power of Attorney
Debtor hereby irrevocably constitutes and designates Secured Party as and for Debtor's attorney in fact:
(a) to exercise, upon the occurrence of an Event of Default and during the continuance thereof, any of the transactions and rights exercisable and powers referenced in this Article 5; and
(b) to execute, upon the occurrence of an Event of Default and during the continuance thereof, all and any such instruments, documents and papers as Secured Party determines to be appropriate in connection with the exercise of such rights and remedies and to cause the sale, license, assignment, transfer or other disposition of the Collateral, including all filings, recordings or registrations with the applicable government offices required or appropriate to effect such dispositions of Collateral.
The within grant of a power of attorney, being coupled with an interest, shall be irrevocable until this Agreement is terminated.
11
ARTICLE 6
ACKNOWLEDGEMENT BY DEBTOR
6.1 Acknowledgements
Debtor:
(a) acknowledges receipt of a true copy of this Agreement;
(b) acknowledges receipt of a copy of all of the Financing Statements;
(c) acknowledges and agrees that, upon notice to Debtor, this Agreement may be assigned by Secured Party to any Person as permitted by Section 7.4 and, in such event, such Person shall be entitled to all of the rights and remedies of Secured Party as set forth in this Agreement or otherwise and Secured Party shall be released and discharged from its further obligations hereunder upon the assumption of same by the assignee;
(d) acknowledges and agrees that in connection with the exercise of Secured Party's rights and remedies hereunder, any use by Secured Party of the Collateral shall be coextensive with Debtor's rights therein and with respect thereto and without any liability for royalties or other related charges from Secured Party to Debtor;
(e) authorizes and requests that each appropriate patent, trademark or other Governmental Authority requested by Secured Party record this Agreement, including any amendments hereto, or copies hereof; provided that this provision shall not preclude the filing by Debtor of the IP Office Filings; and
(f) without in any way limiting any other indemnity provisions contained herein or in the Transaction Documents, Debtor hereby indemnifies and holds Secured Party harmless from and against any claim, suit, loss, damage or expense (including reasonable legal fees) (each, a "Claim") arising out of any alleged defect in any Product, including any such Claim due to purported infringement of the Collateral or any portion thereof upon the intellectual property rights or other rights of any other Person or entity; provided that Debtor shall not be liable to Secured Party for the payment of any portion of such claim, suit, loss, damage or expense to the extent resulting solely from Secured Party's acts or omissions.
12
ARTICLE 7
MISCELLANEOUS
7.1 Notice
(a) Any notice, demand, direction or other instrument required or permitted to be
given hereunder or under the UCC shall be in writing and shall be given in the manner specified in Section 11.3 of the A&R Purchase Agreement and addressed as follows:
(i) if to Debtor:
NPS Pharmaceuticals Inc.
550 Hills Drive, 3rd Floor
Bedminster, New Jersey 07921
United States of America
Attention of: General Counsel
Fax No.: (908) 450-5344
E-mail: estratemeier@npsp.com
with a copy (which shall not constitute notice) to:
Morgan Lewis and Bockius LLP
502 Carnegie Center
Princeton, New Jersey 08540-6241
Attention of: Randall B. Sunberg
Fax No.: (609) 919-6701
E-Mail: rsunberg@morganlewis.com
(ii) if to Secured Party:
Drug Royalty L.P. 3.
c/o DRI Capital Inc.
22 St. Clair Avenue East
Suite 200
Toronto ON M4T 2S5
Attention of: Behzad Khosrowshahi
Fax No.: (416) 863-5161
E-mail: DRINotices@dricapital.com
with a copy to:
Attention of: Gary Margolis
Fax No.: (416) 863-5161
E-Mail: gm@dricapital.com
13
with a copy (which shall not constitute notice) to:
King & Spalding LLP
601 S. California Ave.
Palo Alto, CA 94304
Attention of: Emma Maconick
Fax No.: (650) 422 6800
E-mail: emaconick@kslaw.com
(b) Either party may change its address for service from time to time by notice given in accordance with the foregoing.
7.2 Waiver
(a) Secured Party may waive, in whole or in part, any breach by Debtor of any of the provisions of this Agreement, any default by Debtor in the payment or performance of any of the Obligations or any of its rights and remedies, whether provided for herein or otherwise, provided that no such waiver shall be effective unless given by Secured Party to Debtor in writing.
(b) No waiver given in accordance with Section 7.2(a) shall be a waiver of any other or subsequent breach by Debtor of any of the provisions of this Agreement, of any other or subsequent default by Debtor in the payment or performance of any of the Obligations or any of the rights and remedies of Secured Party, whether provided for herein or otherwise.
(c) Secured Party may, at any time, grant extensions of time or other indulgences to, or accept compositions from or grant releases and discharges to, Debtor in respect of the Collateral or otherwise deal with Debtor or with the Collateral and other security held by Secured Party, all as Secured Party may see fit, and Debtor agrees that any such act or any failure by Secured Party to exercise any of its rights or remedies, whether provided for herein or otherwise, shall in no way affect or impair the Security Interest or the rights and remedies of Secured Party, whether provided for in this Agreement or otherwise.
7.3 Termination
(a) This Agreement may be terminated by written agreement made between Secured Party and Debtor.
(b) Upon indefeasible payment and performance of the Obligations, this Agreement shall terminate and all rights in the Collateral shall revert to Debtor.
(c) Upon a Revocation, the Secured Party's security interest with respect to (i) the NPS Patents in the United States and Japan, (ii) any NPS Know-How that is applicable only to the United States or Japan, and (iii) the NPS Trademarks in the United States and Japan shall terminate (the Collateral described in clause (i), (ii) and (iii) is collectively referred to as the "New Collateral"). It is acknowledged and agreed by all parties hereto that upon a Revocation, the liens and security interests granted to the Secured Party in all Collateral (other than the New Collateral) shall remain in full force and effect and shall continue to secure the Obligations without interruption, and that the Revocation shall not be deemed a novation of the rights and obligations contained herein.
14
(d) Upon termination of this Agreement in accordance with this Section 7.3 or upon a Revocation with respect to the Additional Collateral, Secured Party shall, at the request and expense of Debtor, make and do all such acts and things and execute and deliver all such financing statements, instruments, agreements and documents as Debtor considers reasonably necessary or desirable to discharge the Security Interest, to release and discharge the Collateral (or upon a Revocation, with respect to the Additional Collateral) therefrom and to record such release and discharge in all appropriate offices of public record.
7.4 Assignment
This Agreement shall inure to the benefit of and be binding upon the successors and permitted assigns of each of Secured Party and Debtor. This Agreement may not be assigned in whole or in part by either party without the prior written consent of the other party; provided, however, that Secured Party may assign this Agreement in whole or in part without the prior written consent of Debtor: (i) by way of security to a financial institution or other lender, (ii) to any Person that directly, or indirectly through one or more intermediaries, controls or is controlled by, or is under common control with, Secured Party, (iii) to a special purpose vehicle created to be bankruptcy remote or for financing purposes or (iv) as part of a sale of a material part of Secured Party, in any case whether by way of reorganization or otherwise, and Secured Party shall give prompt notice of any such assignment to Debtor within 10 Business Days after the occurrence thereof; provided, however, that Debtor may assign this Agreement in whole or in part without the prior written consent of Secured Party in connection with an assignment of the NPS Patents to Affiliates of NPS in accordance with Section 6.1(a) of the A&R Purchase Agreement; provided, further, that Debtor shall give prompt notice of any such assignment to Secured Party within 10 Business Days after the occurrence thereof. For clarity, the obligations provided in this Section 7.4 are in addition to any obligations of Debtor pursuant to Section 4.4.
7.5 Further Assurances
From time to time, as and when requested by any party, each party shall execute and deliver, or cause to be executed and delivered, all such documents, certificates and instruments, and shall take, or cause to be taken, all such further or other actions, as such other party may deem reasonably necessary, desirable or appropriate to carry out, or otherwise give full effect to, the provisions and intent of this Agreement.
7.6 Execution in Counterparts and Facsimile Delivery
This Agreement may be executed in one or more counterparts, all of which when taken together shall be considered one and the same agreement. This Agreement may be delivered by either party by facsimile or by electronic delivery and, if so executed and delivered, shall be legally valid and binding on the party executing in such manner.
[signature page follows]
15


SCHEDULE 2.3
SUPPLEMENTARY EUROPEAN PATENTS
Note: For purposes of this Schedule 2.3, where the granted patents have been registered in Switzerland, the registration extends to Liechtenstein.
"EXCRETION OF HETEROLOGOUS PROTEINS FROM E. COLI"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Belgium |
: 30 August'89 |
89308753.6 |
31 July'96 |
0 357 391 |
30 August '09 |
|
Sweden |
: 30 August'89 |
89308753.6 |
31 July'96 |
0 357 391 |
30 August '09 |
|
Switzerland |
30 August '89 |
89308753.6 |
31 July'96 |
0 357 391 |
30 August '09 |
"ESSENTIALLY PURE HUMAN PARATHYROID HORMONE"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Belgium |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
|
Denmark |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
|
Denmark (SPC) |
20 Oct '06 |
CA200600028 |
25 March '08 |
CR200600028 |
22 May '17 |
|
Finland |
22 May '92 |
922,356 |
12 April '01 |
106802 |
22 May'12 |
|
Ireland |
1 July '92 |
921,672 |
28 May '04 |
83494 |
1 July'12 |
|
Luxembourg |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
|
Monaco |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
|
Norway |
21 May '92 |
92304692.4 |
9 November '98 |
304,190 |
21 May'12 |
|
Norway (SPC) |
24 Oct '06 |
2006013 |
7 Dec 2009 |
2006013 |
21 May'17 |
|
Portugal |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
|
Sweden |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
|
Switzerland |
22 May '92 |
92304692.4 |
25 March '98 |
0 515 228 |
22 May'12 |
"PARATHYROID HORMONE FORMULATION"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Publication No. |
Expiry Date. |
|
Belgium |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Denmark |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Ireland |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Liechtenstein |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Lithuania |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Luxembourg |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Monaco |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Portugal |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Sweden |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Switzerland |
16 December '94 |
95903732.6 |
03 October 2007 |
EP 735 896 |
16 December 2014 |
|
Finland |
16 December '94 |
962593 |
15 September 2009 |
120291 |
16 December 2014 |
|
Norway |
16 December '94 |
P962634 |
27 December 2007 |
324905 |
16 December 2014 |
"PROTEIN FORMULATION (PTH) (MULTI-DOSE INJECTION)"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Albania |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Belgium |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Cyprus |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Denmark |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Finland |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Ireland |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Latvia |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Lithuania |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Luxembourg |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Macedonia |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Monaco |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Portugal |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Romania |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Slovenia |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Sweden |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
|
Switzerland |
26 April '99 |
99917715.7 |
18 August '04 |
1079803 |
26 April '19 |
"A COMBINED PHARMACEUTICAL PREPARATION COMPRISING
PARATHYROID HORMONE AND A BONE RESORPTION INHIBITOR"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
Belgium |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Belgium Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Cyprus |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Cyprus Div |
8 June '98 |
4017622.4 |
8 Sept'04 |
CY1110287 |
8 June '18 |
|
Denmark |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Denmark Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1001802 |
8 June '18 |
|
Europe Div. ** |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Finland |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Finland Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Ireland |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Ireland Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Liechtenstein |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Liechtenstein Div |
8 June '98 |
04017622.4 |
4 Nov. '09 |
1473040 |
8 June '18 |
|
Luxembourg |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Luxembourg Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Monaco |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Monaco Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Portugal |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Portugal Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Sweden |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Sweden Div. |
8 June '98 |
04017622.4 |
4 Nov '09 |
1473040 |
8 June '18 |
|
Switzerland |
8 June '98 |
98929965.6 |
8 Sept'04 |
1001802 |
8 June '18 |
|
Switzerland Div. |
8 June '98 |
04017622.4 |
4 Nov. '09 |
1473040 |
8 June '18 |
** Designated contracting states: Belgium, Cyprus, Denmark, Finland, Ireland, Liechtenstein, Luxembourg, Monaco, Portugal, Sweden, Switzerland.
"METHOD OF ADMINISTERING THERAPEUTIC INJECTIONS"
|
Country |
Filing Date |
Serial No. |
Issue Date |
Publication No. |
Expiry Date |
|
Europe* |
18 October 2004 |
04795465.6 |
Pending |
EP1687048A |
* Designated contracting states: Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, Hungary, Ireland, Liechtenstein, Luxembourg, Monaco, Poland, Portugal, Romania, Slovenia, Slovakia, Sweden, Switzerland, Turkey, with Albania, Croatia, Lithuania, Latvia and Macedonia as extension states.
SPC SUMMARY
|
Based on EP 0 515 228 |
||||
|
Country |
Application Number |
Filing Date |
Status |
Expiry |
|
Denmark |
CA 2006 00028 |
20 Oct. 2006 |
Granted 25 March 2008 |
May 22, 2017 |
|
Norway |
SPC/NO 2006 013 |
24 Oct. 2006 |
Granted 7 Dec. 2009 |
May 21, 2017 |
|
Based on EP 1 079 803 |
||||
|
Country |
Application Number |
Filing Date |
Status |
Expiry Date |
|
Belgium |
2006C/033 |
19 Oct. 2006 |
Granted Grant Date 5 June 2007 |
April 24, 2021 |
|
Cyprus |
CY06/006 |
23 Oct. 2006 |
Granted Grant Date March 2007 |
April 23, 2021 |
|
Denmark |
CA 2006 00029 |
20 Oct. 2006 |
Abandoned |
|
|
Finland |
L20060013 |
13 Oct. 2006 |
Granted Grant Date 29 Aug 2011 |
April 24, 2021 |
|
Ireland |
2006/032 |
17 Oct. 2006 |
Granted Grant Date 23 May 2007 |
April 23, 2021 |
|
Latvia |
C/LV2006/0009 |
17 Oct. 2006 |
Granted Grant Date 20 March 07 |
April 24, 2021 |
|
Lithuania |
PA 2006 007 |
23 Oct. 2006 |
Granted Grant No. C1079803 Grant Date 04 June 07 |
April 25, 2021 |
|
Luxembourg |
91281 |
19 Oct. 2006 |
Granted Grant No. 91291 Grant Date 19 Dec. 06 |
April 24, 2021 |
|
Portugal |
240 |
20 Oct. 2006 |
Granted |
April 24, 2021 |
|
Romania |
C/022 |
10 April 2007 |
Granted Grant Date Dec 30, 2011 |
24 April 2021 |
|
Slovenia |
200640015 |
18 Oct. 2006 |
Granted Grant Date 16 July 2009 |
23 April 2021 |
|
Sweden |
0690029-4 |
24 Oct. 2006 |
Granted Grant Date 5 Feb 2008 |
23 April 2021 |
SCHEDULE 3.1(C)
COLLATERAL ABANDONED BY CONSENT
European patent application no. 07019169.7, filed September 28, 2007, became abandoned with consent of the Secured Party on April 9, 2009.
APPENDIX 3.1(E)
FINANCING STATEMENTS
See attached.
EXHIBIT A TO UCC-1 FINANCING STATEMENT DEBTOR:
NPS PHARMACEUTICALS, INC. All right, title and interest in, to and under the NPS Technology and the NPS Trademarks, whether now owned or
hereinafter acquired, and includes Proceeds therefrom (whether now owned by or owing to, or hereafter acquired by or arising in favor of the
Debtor collectively, the "Collateral"): The terms used herein shall have the respective meanings set out below, and grammatical variations of such terms
shall have corresponding meanings: "Know-How" means information, results and data of any type whatsoever, in any tangible or intangible form whatsoever, including
without limitation, technical information, databases, practices, methods, techniques, specifications, formulations, formulae, knowledge, know-how,
skill, experience, test data including pharmacological, medicinal chemistry, biological, chemical, biochemical, toxicological and clinical
test data, analytical and quality control data, stability data, studies and procedures, and manufacturing process and development information,
results and data relating to the Product or the device used to deliver the Product. "NPS Know-How" means (a) any non-public or confidential sections of any marketing authorization application relating to any
Product, and (b) any other non-public or confidential Know-How controlled by Debtor that is necessary or useful for the performance of pre-clinical or
clinical development, for the filing of marketing authorizations in the Territory, or the commercialization, marketing or manufacture of
a Product. "NPS Patents" means the Patents controlled by Debtor that are necessary or useful for the development, commercialization or
manufacture of the Product in the Territory and as listed on Schedule 2 attached hereto and made a part hereof. "NPS Technology" means the NPS Patents and the NPS Know-How. "NPS Trademarks" means NATPARA™, PREOTACT™ and any other trademarks, trade dress, logos, slogans, and
designs, whether or not registered in the Territory, used to identify or promote the Product in the Territory including without limitations the
trademarks listed on Schedule 1. "Proceeds" means the property in any form derived, directly or indirectly, from any dealing with the Collateral or other proceeds
(together with any reissue, continuation, continuation-in-part or extension of the patents) thereof; and includes any accounts, payment
intangibles or other general intangibles arising from any sale, transfer, license, lease or other dealing in any of the Collateral and any payment
or value representing indemnity or compensation for loss or damage to the Collateral or other proceeds, including insurance proceeds and
proceeds (as such term is defined in the UCC).
"Product" means recombinant human parathyroid hormone (rhPHT 1-84) as set forth in the BLA No. 125511 (and regardless of
the trademark under which it may be sold or distributed) as well as any invention, discovery or derivative or Know-How, whether or not
patentable related thereto, including other formulations or indications. "Territory" means all countries and territories of the world other than Israel. "UCC" means (i) the Uniform Commercial Code as in effect from time to time in the State of New York, (ii) with
respect to enforcement, the Uniform Commercial Code as in effect from time to time in any other state whose law is applicable with respect to
Secured Party's enforcement of its rights hereunder and (iii) insofar as any references to the UCC is used in the context of perfection, the
Uniform Commercial Code as in effect from time to time in the state that is the "location" of Debtor under the UCC, in each case including any
legislation that may be substituted therefor (as any such substituted legislation may be amended from time to time). All capitalized terms referenced herein and not otherwise defined herein shall have the meanings assigned to such terms in that certain
Amended and Restated Agreement for the Sale and Assignment of Rights dated as of December , 2013 by and among the Debtor in favor
of the Secured Party.
Schedule 1
TRADEMARKS Country Mark
Filing Date Serial No. Issue Date
Trademark No. U.S. NATPARA April 8,2011 85289949 September 18, 2012 4210947 U.S. NATPARA (RHPTH [1-84]) FOR INJECTION & DESIGN September 20, 2013 86071034 U.S. PREOTACT August 19, 2005 78696654 October 13, 2009 3697151 U.S. PREOTACT PARATHYROID HORMONE (RDNA ORIGIN) FOR INJECTION October 5, 2005 78726924 September 22, 2009 3687544 U.S. PREOTACT August 26, 2011 85408727
Schedule 2
PATENTS Country
Filing Date Serial No. Issue Date
Patent No.
Expiry Date U.S. 23 May '91 07/707114 4 May '93 5,208,041 23 May'11 U.S. 23 Dec '93 08/172,206 5 March '96 5,496,801 23 Dec'13 U.S. 14 Aug '98 09/125,247 4 Sep '01 6,284,730 8 June '18 U.S. Div Con 18 March'03 10/389,797 28 March '06 7,018,982 8 June '18 U.S.Div Con 2 19 Dec'05 11/305,339 24 March '09 7,507,715 8 June '18 U.S.Div 9 Jan '09 12/351,558 6 July'10 7,749,543 8 June'18 U.S.Div 23 June '10 12/822,089 10 April '12 8,153,588 8 June'18 U.S.Div 24 Feb'12 13/405,093 (2012/0148684) U.S. 15 Oct. '04 10/966,364 4 May '10 7,708,732 11 May'27
SCHEDULE 3.1(F) See attached.
PATENT SECURITY AGREEMENT THIS PATENT SECURITY AGREEMENT is made as of December 20, 2013, BETWEEN: NPS PHARMACEUTICALS, INC., (collectively with its successors and permitted assigns, "Debtor"), - and - DRUG ROYALTY L.P. 3, (collectively with its successors and permitted assigns, "Secured Party"). WHEREAS Secured Party and Debtor have entered into an Amended and Restated Security Agreement dated as
of December 20, 2013 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security
Agreement"); and WHEREAS pursuant to the Security Agreement, Debtor has granted to Secured Party a security interest in, among
other assets, certain intellectual property of Debtor, and Debtor has agreed to execute this Patent Security Agreement for recording with the
United States Patent and Trademark Office; NOW THEREFORE in consideration of the respective covenants, promises and agreements of the parties herein
contained and other good and valuable consideration (the receipt and sufficiency of which are hereby acknowledged by each of the parties),
the parties agree as follows: As general and continuing collateral security for the due payment and performance of the Obligations (as
such term is defined in the Security Agreement), Debtor hereby pledges, mortgages, charges and assigns (by way of security) to Secured
Party, and grants to Secured Party, a security interest in, the patents described in Schedule A hereto (as such Schedule may be
amended from time to time) and all reissues, divisions, continuations, continuations-in-part, extensions, renewals and re-examinations thereof
(collectively, the "Patents"). -2-
Debtor authorizes and requests that the United States Patent and Trademark Office record this Patent
Security Agreement. This Patent Security Agreement is being entered into in accordance with the terms of the Security
Agreement. Debtor hereby acknowledges and affirms that the rights and remedies of Secured Party with respect to the security interest in the
Patents made and granted hereby are more fully specified in the Security Agreement, the terms and provisions of which are incorporated by
reference herein. In the event of a conflict between any provision of this Patent Security Agreement and any provision of the Security
Agreement, the provision of the Security Agreement shall control. This Patent Security Agreement shall terminate and all rights in the Patents shall revert to Debtor upon the
termination of the Security Agreement, including termination of the Security Agreement in part, with respect to the Additional Collateral (as
such term is defined in the Security Agreement), in accordance with the terms thereof. This Patent Security Agreement shall inure to the benefit of and shall be binding on and enforceable by and
against the parties hereto and their respective successors and permitted assigns under the Security Agreement. This Patent Security Agreement shall be governed by, and interpreted and enforced in accordance with, the
laws of the State of New York and the federal laws of the United States applicable therein, without giving effect to the principles of conflicts of
law thereof except as set forth in Section 5-1401 of the New York General Obligations Law. This Patent Security Agreement may be executed in one or more counterparts, all of which when taken
together constitute one and the same agreement. [Signature Page Follows]
SCHEDULE A Country
Filing Date Serial No. Issue Date
Patent No.
Expiry Date U.S. 23 May '91 07/707114 4 May '93 5,208,041 23 May'11 U.S. 23 Dec '93 08/172,206 5 March '96 5,496,801 23 Dec'13 U.S. 14 Aug '98 09/125,247 4 Sep '01 6,284,730 8 June'18 U.S. Div Con 18 March'03 10/389,797 28 March '06 7,018,982 8 June'18 U.S.Div Con 2 19 Dec'05 11/305,339 24 March '09 7,507,715 8 June'18 U.S.Div 9 Jan '09 12/351,558 6 July'10 7,749,543 8 June '18 U.S.Div 23 June'10 12/822,089 10 April '12 8,153,588 8 June'18 U.S.Div 24 Feb'12 13/405,093 (2012/0148684) U.S. 15 Oct. '04 10/966,364 4 May '10 7,708,732 11 May'27
TRADEMARK SECURITY AGREEMENT THIS TRADEMARK SECURITY AGREEMENT is made as of December 20, BETWEEN: NPS PHARMACEUTICALS, INC., (collectively with its successors and permitted assigns, "Debtor"), - and - DRUG ROYALTY L.P. 3, (collectively with its successors and permitted assigns, "Secured Party"). WHEREAS Secured Party and Debtor have entered into an Amended and Restated Security Agreement dated as
of December 20, 2013 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security
Agreement"); and WHEREAS pursuant to the Security Agreement, Debtor has granted to Secured Party a security interest in, among
other assets, certain intellectual property of Debtor, and Debtor has agreed to execute this Trademark Security Agreement for recording with
the United States Patent and Trademark Office; NOW THEREFORE in consideration of the respective covenants, promises and agreements of the parties herein
contained and other good and valuable consideration (the receipt and sufficiency of which are hereby acknowledged by each of the parties),
the parties agree as follows: As general and continuing collateral security for the due payment and performance of the Obligations (as
such term is defined in the Security Agreement), Debtor hereby pledges, mortgages, charges and assigns (by way of security) to Secured
Party, and grants to Secured Party, a security interest in, the trademarks described in Schedule A hereto (as such Schedule may be
amended from time to time) (collectively, the "Trademarks"). -2-
Debtor authorizes and requests that the United States Patent and Trademark Office record this Trademark Security
Agreement. This Trademark Security Agreement is being entered into in accordance with the terms of the Security
Agreement. Debtor hereby acknowledges and affirms that the rights and remedies of Secured Party with respect to the security interest in the
Trademarks made and granted hereby are more fully specified in the Security Agreement, the terms and provisions of which are incorporated
by reference herein. In the event of a conflict between any provision of this Trademark Security Agreement and any provision of the Security
Agreement, the provision of the Security Agreement shall control. This Trademark Security Agreement shall terminate and all rights in the Trademarks shall revert to Debtor
upon the termination of the Security Agreement, including termination of the Security Agreement in part, with respect to the Additional
Collateral (as such term is defined in the Security Agreement), in accordance with the terms thereof. This Trademark Security Agreement shall inure to the benefit of and shall be binding on and enforceable by and
against the parties hereto and their respective successors and permitted assigns under the Security Agreement. This Trademark Security Agreement shall be governed by, and interpreted and enforced in accordance with,
the laws of the State of New York and the federal laws of the United States applicable therein, without giving effect to the principles of conflicts
of law thereof except as set forth in Section 5-1401 of the New York General Obligations Law. This Trademark Security Agreement may be executed in one or more counterparts, all of which when taken
together constitute one and the same agreement. [Signature Page Follows]
SCHEDULE A Country Mark Filing Date Serial No. Issue Date Trademark No. U.S. NATPARA April 8,2011 85289949 September 18, 2012 4210947 U.S. NATPARA (RHPTH [1-84]) FOR INJECTION & DESIGN September 20,2013 86071034 U.S. PREOTACT August 19, 2005 78696654 October 13, 2009 3697151 U.S. PREOTACT PARATHYROID HORMONE (RDNA ORIGIN) FOR INJECTION October 5, 2005 78726924 September 22, 2009 3687544 U.S. PREOTACT August 26, 2011 85408727
EXHIBIT A See attached.
UNDERTAKMG To: Drug Royalty L.P. 3 (the "Purchaser") Reference is made to the agreement for sale and assignment of rights made as of
the day of July, 2007 between the undersigned, NPS Pharmaceuticals, Inc. and the
Purchaser (the "Purchase Agreement"). Capitalized terms used and not otherwise defined herein have the meaning
specified in the Purchase Agreement. The undersigned herehy undertakes to, within 15 days of Closing, execute and deliver to the Purchaser or the
Purchaser's counsel: (a) a mortgage of the Patents to be registered in Spain; (b) a pledge of the Patents to be registered in Austria, in each
case on terms and conditions at least as good as the terms and conditions of the draft Spanish mortgage and draft Austrian pledge annexed to
this undertaking as Exhibits 1 and 2, and in each case in a form and language suitable for registration in the relevant country, and (c)
any documents or instruments as the Purchaser is advised by its counsel as are necessary to be executed by it in order to effect the
registration of the final forms of the Spanish mortgage and the Austrian pledge in the appropriate public registries in the relevant
country. DATED as of the 16th day of July, 2007. NPS ALLELIX CORP. by /s/ Val R. Antczak
CONDITIONAL ASSIGNMENT AGREEMENT BETWEEN
SECURED PARTY:
DRUG ROYALTY L.P. 3
List of Trademarks
List of NPS Patents
UNITED STATES IP OFFICE FILINGS

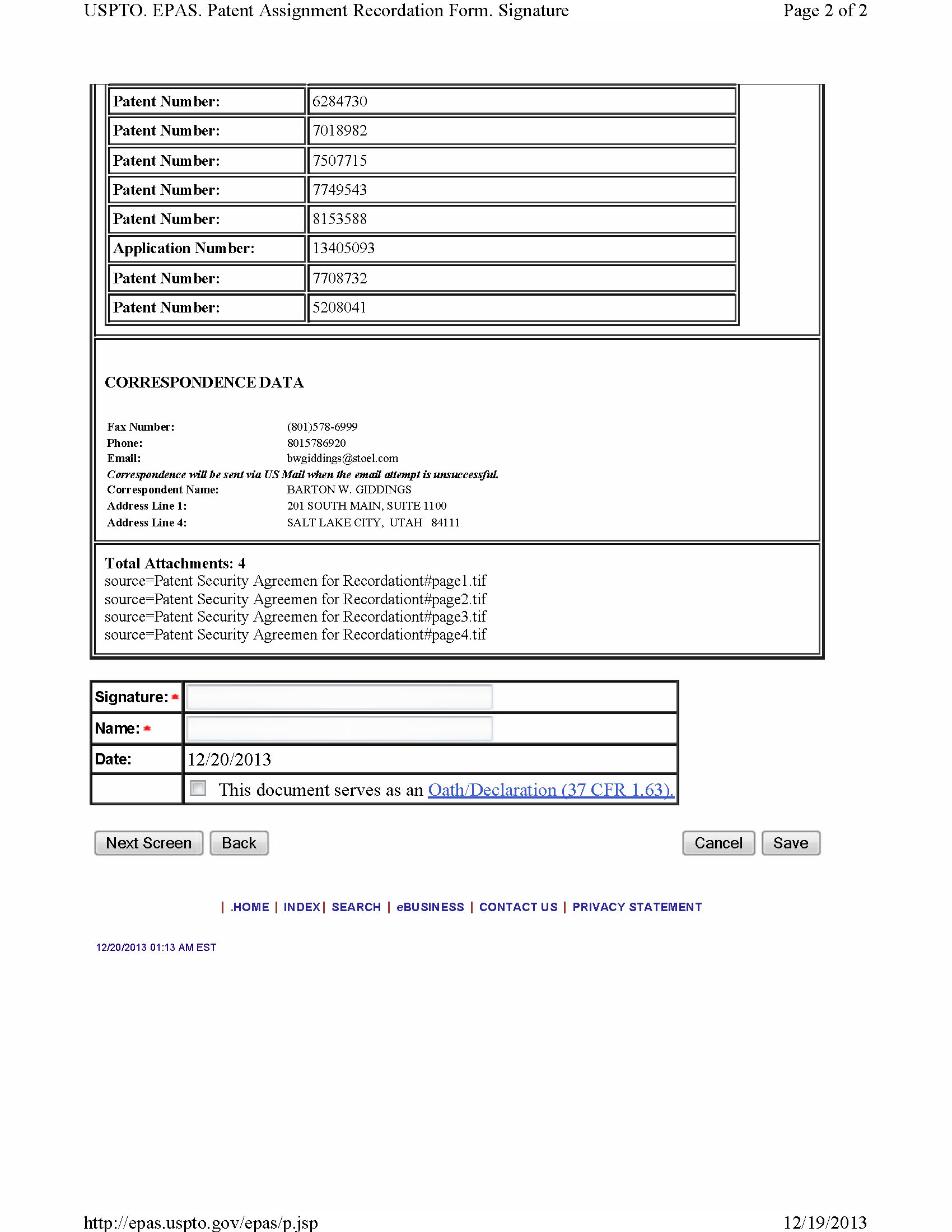
a corporation existing under the laws of the State of Delaware
a Cayman Islands limited partnership


PATENTS

a corporation existing under the laws of the State of Delaware
a Cayman Islands limited partnership


TRADEMARKS
PATENT FILINGS
Name: Val R. Antczak
Title: Vice President and Secretary
Each a "Party" and together the "Parties".
WHEREAS
- NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
- NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) mcluding without limitation those patents, patent applications and supplementary protection certificates listed in the Schedule to this Agreement (the "Austrian Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such Austrian Patents (the "Related Rights");
- NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement");
- Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
- This Agreement sets out the terms of the assignment of the Austrian Patents, including their Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment by DRLP3 to NPS of US$1 (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:-
Defmitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee;
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a Major Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1(a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement.
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the Austrian Patents, including without limitation then-Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this Agreement is subject to the occurrence of an Event of Default. To ensure proper operation of this Agreement at such point in time NPS herewith agrees to duly execute the assignment deed Appendix .11 (the "Assignment Deed") with notarized and apostilled signatures concurrently with the execution of this Agreement and to hand over to DRLP3 the original of such
3
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the Austrian Patentsor any Related Rights, to a third party.
- Upon the occurrence of an Event of Default provided for under clause 2, DRLP3 shall be entitled to register the change of ownership in the Austria Patents as evidenced by the Assignment Deed with the Austrian Patent Office. NPS agrees to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the Austrian Patents, including their Related Rights, in DRLP3 and to record such assignment at the Austrian National Patent Register and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require. Any and all costs, expenses and taxes arising as a result of the registration of the change of ownership with the Austrian Patent Office, including, but not limited to, stamp duties, shall be borne by NPS.
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the Austrian Patents with the Austrian Patent Office in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
Assignment Deed. DRLP3 agrees to hold in strict confidence the Assignment Deed and to use the Assignment Deed solely in accordance with clause 6 below.
4
- To the extent that the parties fail to execute a pledge agreement granting DRLP3 a first ranking pledge over the Austrian Patents when this Agreement is executed, NPS agrees that it will execute such an agreement within 60 days of the execution of this Agreement. Further, NPS agrees that within the said 60 day period, it will execute any necessary powers of attorney or other documents that may be required by DRLP3 or its Austrian counsel to record the pledge granted over the Austrian Patents at the Austrian National Patent Office.
- This Agreement is governed by and construed in accordance with the laws of the province of Ontario and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Ontario Courts and Tribunals and all courts competent to hear appeals therefrom.
Date July 16, 2007
Having read the above, the Parties hereby sign:
5
NPS ALLELIX CORP.
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
6
SCHEDULE
The Austrian Patents
|
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
EP1001802B |
Dosage Regime |
Granted |
|
EP0515228B |
Pure PTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1687048A |
Administration Method |
Pending |
|
SZ 32/2006 (SPC) |
Filed 19 October 2006 |
7
Patent Pledge Agreement
between
NPS ALLELIX CORP.,
a corporation existing under the laws of the
Province of Ontario, Canada
("NPS" or the "Pledgor"),
- and -
DRUG ROYALTY L.P. 3,
a Cayman Island limited partnership
("DRLP" or the "Pledgee").
KEEP THE ORIGINAL OF THIS DOCUMENT AND ALL CERTIFIED COPIES THEREOF OUTSIDE OF THE REPUBLIC OF AUSTRIA. BRINGING THIS DOCUMENT OR ANY CERTIFIED COPY OF THIS DOCUMENT INTO THE REPUBLIC OF AUSTRIA AS WELL AS ANY WRITTEN CONFIRMATION OR WRITTEN REFERENCE TO THIS DOCUMENT MAY CAUSE THE IMPOSITION OF AUSTRIAN STAMP DUTY.
-2-
WHEREAS, NPS as vendor and DRLP as purchaser intend to enter info an agreement for the sale and assignment of rights;
WHEREAS, NPS as debtor and DRLP as secured party intend to enter into a security agreement for the creation of a security in certain collateral as defined therein;
WHEREAS, NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and supplementary protection certificates listed in the Schedule I to this Agreement (the "Austrian Patents").
NOW THEREFORE, IT IS AGREED as follows:
1. Definitions and Construction
1.1 Terms used but not otherwise defined herein shall have the meanings ascribed thereto in the Purchase Agreement, the Security Agreement and any other agreement referred to therein.
1.2 Additionally, terms used in this Agreement shall have the meaning as follows:
"Agreement" shall mean this patent pledge agreement;
"Austrian Patents" shall mean any and all patents registered with the official patent register as listed in Schedule I
"Business Day" shall mean a day (other than a Saturday or a Sunday) on which banks generally are open for business in New York and Vienna;
"Enforcement Event" shall mean the occurrence of an Event of Default , provided that if the Event of Default is:
(a)a Minor Default, the Enforcement Event shall only be deemed to have occurred if DRLP has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Pledgor's Obligations or any judgement obtained pursuant to such action remains unsatisfied by the Pledgor for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement); or
-3-
(b) the result of a breach of representation and warranty by the Pledgor, the Enforcement Event shall be deemed to have occurred after a period of 60 days commencing on the date on which notice of such breach is provided to the Pledgor, unless during that 60 day period the parties agree upon an alternative remedy in respect of that breach.;
and further provided that the Enforcement Event shall be deemed not to have occurred, if within 7 days of the date on which the Enforcement Event would otherwise have occurred (or have been deemed to have occurred) DRLP validly becomes the sole legal and beneficial owner of the Austrian Patents, free from all third party rights (other than Nycomed's rights under the License Agreement);
"Party" shall mean any of NPS and DRLP;
"Pledge" shall have the meaning as described in Clause 3.1;
"Pledgor" shall mean NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9, Canada;
"Pledgee" shall mean DRUG ROYALTY L.P. 3, a Cayman Island limited partnership incorporated under the laws of the Cayman Islands whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands;
"Purchase Agreement" shall mean the purchase agreement intended to be entered into by the Pledgor and the Pledgee after execution of this Agreement;
"Secured Claims" shall mean all present and future obligations and liabilities (whether actual or contingent and whether jointly or severally or in any other capacity whatsoever) of the Pledgor to the Pledgee under the Purchase Agreement, the Security Agreement as well as the present Agreement as of and from the date of execution of the Purchase Agreement and the Security Agreement;
and shall mean furthermore all present and future obligations and liabilities of the Pledgor to the Pledgee in connection with any assignment of the Austrian Patents;
-4-
and (for the avoidance of doubt) shall mean furthermore any obligations of the Pledgor to the Pledgee pursuant to Section 8 of this Agreement;
"Security Agreement" shall mean the security agreement intended to be entered into by the Pledgor and the Pledgee after execution of this Agreement;
"Secured Documents" shall mean the Purchase Agreement, the Security Agreement, the present Agreement and any and all ancillary agreements entered into by the Parties in connection therewith.
1.3 Construction
In this Agreement, unless the context otherwise requires:
(a) unless otherwise stated, a "Clause" is a reference to a Clause of this Agreement;
(b) unless otherwise stated, a "Schedule" is a reference to a Schedule of this Agreement and references to this Agreement include its Schedules;
(c) words importing the plural shall include the singular and vice versa;
(d) a reference to (or to any specified provision of) any agreement, deed or other instrument is to be construed as a reference to that agreement, deed or other instrument or that provision as from time to time amended, varied, supplemented, restated or novated but excluding for this purpose any amendment, variation, supplement or modification which is contrary to any provision of any of the Secured Documents;
(e) nothing in this Agreement shall prevent the Pledgor from carrying out a transaction not prohibited by the Secured Documents;
(f) to the extent not prohibited by mandatory provisions of Austrian law or any other applicable law nothing in this Agreement shall be construed in such a way that it would violate or contradict the provisions of the Secured Documents;
(g) References to any act or determination by a Party to be performed or made "reasonably" shall be deemed to be references to the Austrian law term "nach billigem Ermes-sen" and shall be construed according to this term, and any other reference relating to reasonability shall be construed to be a reference to the Austrian law concept of "Billigkeit".
2. Secured Claims
2.1 In case of an Enforcement Event, the Pledgor hereby unconditionally and irrevocably undertakes and agrees to pay to the Pledgee an amount of
-5-
€1,000,000 (one million euros) as contractual penalty, irrespective of fault and not being subject to judicial review, in accordance with the relevant provisions of the Austrian Civil Code (ABGB) and the Austrian Entrepreneur Code (UGB). Any other remedy (including but not limited to damage claims) the Pledgee may have against the Pledgor under the provisions of the Purchase Agreement and/or the Security Agreement shall remain unaffected hereby and subject to the applicable laws and contractual regulations as provided for therein.
2.2 This Pledge hereunder is constituted in order to secure the prompt and complete satisfaction of all Secured Claims.
2.3 The Parties agree that in case of a change, amendment, supplement or novation (Novation) of the Secured Claims, this Pledge shall not lapse but shall continue to. secure such changed, amended, supplemented or novated Secured Claims in accordance with Section 1378 of the Austrian General Civil Code (Allgemeines Burgerliches Gesetzbuch).
3. Pledge
3.1 The Pledgor hereby grants to the Pledgee a first ranking pledge over the Austrian Patents, together with all ancillary rights and claims associated with the Austrian Patents (the "Pledge") and the Pledgee accepts such Pledge.
3.2 The Pledge granted under this Agreement shall be in addition to and shall be independent of each other security and/or guarantee which the Pledgee may at any time hold for any of the Secured Claims. No prior security held by the Pledgee over the whole or any part of the Austrian Patents shall merge in the Pledge hereby constituted. In particular, nothing contained in this Agreement is intended to, or shall operate so as to, prejudice or affect any bill, note, guarantee, mortgage, pledge, charge or other security of any kind whatsoever which the Pledgee may have for the Secured Claims or any right, remedy or privilege of the Pledgee there under.
4. Operation of the Austrian Patents
The Pledgor shall not release, sell, transfer, assign, dispose of or otherwise deal in any way with any of the Austrian Patents other than within its ordinary course of business and in accordance with the provisions of the Secured Documents. Until the occurrence of an Enforcement Event, the Pledgor (acting in good faith) shall be free (without the further consent of the Pledgee) to apply any moneys it received in respect of the Austrian Patents subject to any applicable restrictions set out in the Secured Documents.
5. Valid Creation of the Pledge in accordance with Austrian law
5.1 Immediately upon execution of this Agreement, the Pledgor undertakes to execute as legally valid and binding agreement upon the Pledgor in notarized
-6-
and apostilled form a notice in form and substance as set out in Schedule II to the Austrian Patent Office and provide - once countersigned by the Pledgee -for submission of such notice with the Austrian Patent Office in order for the pledge over the Austrian Patents to become effective under Austrian law pursuant to section 452 of the Austrian Civil Code and section 43 of the Austrian Patent Act.
5.2 In addition and upon the request of the Pledgee, the Pledgor shall undertake any and all further action and shall deliver and issue all necessary documents and certificates with respect to the Austrian Patents which in the reasonable opinion of the Pledgee is necessary or desirable under all applicable laws to make the Pledge valid and enforceable in accordance with its terms.
5.3 Upon having actual knowledge thereof, the Pledgor shall immediately inform the Pledgee if the completion of the act of perfection as per Clause 5.1 by way of registration of the Pledge in the Patent Register turns out not to be sufficient for a valid and effective pledge of the Austrian Patents under Austrian law or any other laws applicable to the Austrian Patents and/or the Secured Documents and shall deliver and issue all necessary documents and certificates to the Pledgee as necessary to perfect the respective Pledge.
5.4 Upon the reasonable request of the Pledgee, the Pledgor shall provide to the Pledgee evidence reasonably requested showing that any perfection requirement as set out in Clauses 5.1 and 5.3 which shall be made by the Pledgor is complied with.
6. Enforcement of Pledge
6.1 Following the occurrence of an Enforcement Event, the Pledgee shall be entitled to avail itself of all rights and remedies that a pledgee has upon the default of a pledgor under Austrian law (including but not limited to enforcement pursuant to Section 461 of the Austrian Civil Code (Allgemeines Burgerliches Gesetzbuch)), In addition the Pledgee shall be entitled to realize the Pledge without writ, judgment or any other legal court action in a public auction (hereafter referred as "Public Auction") or by private sale (hereunder referred to as "Private Sale") by applying the provisions set forth in Section 466 lit a to e Austrian Civil Code and Section 368 of the Austrian Entrepreneurs Code (Unternehmensgesetzbuch), without the requirement of any further notice, calling, judicial warrant or decision, or the necessity to initiate any proceedings before the courts. The Pledgor herewith grants its express consent to such out-of-court enforcement.
6.2 Upon an Enforcement Event, the Pledgor shall be notified of any imminent realization of the Pledge by written notice prior to such realization. Such notice in order to be valid shall assert the time, place and the procedure of enforcement. Such notice, however, is not required if bankruptcy proceedings have been initiated against the Pledgor.
-7-
6.3 Any Private Sale or Public Auction shall only be made upon prior assessment of the value of the Austrian Patents by an independent certified public accountant from Deloitte & Touche after consultation with an Austrian patent lawyer. If the Pledgor and the Pledgee do not reach agreement on the identity of the Austrian patent lawyer within 10 Business Days after the lapse of the period as set out in section 6.2, such Austrian patent lawyer shall be appointed by the President of the Austrian chamber of patent Lawyers. The assessment of the Austrian Patent shall be made by the expert on reasonable grounds and assumptions and upon specification of the appropriate method of assessment. The costs of the expert's assessment shall be borne by the Pledgor or covered by the Pledge itself.
6.4 Once the expert has rendered its valuation or the Parties have mutually agreed upon the value of the Austrian Patents, the Pledgee shall notify the Pledgor (notwithstanding any other compulsory publication obligations provided by law) on the conditions of the realization of the Pledge, which may not take place within a period less than ten Business Days from the date when the valuation of the Austrian Patents has been completed.
6.5 Both in the case of a Public Auction or a Private Sale, the Austrian Patents must not be transferred at a price which is below the value assessed by the expert or agreed upon between the Parties. The Pledgor and any affiliates of the Pledgor are not entitled to participate in a Public Auction or in a Private Sale.
6.6 In case the Pledgee should seek to enforce the Pledge, the Pledgor shall render forthwith all reasonably necessary assistance in order to facilitate the prompt realisation of the Austrian Patents or any part thereof and/or the exercise by the Pledgee of any other right it may have as pledgee.
6.7 In case of enforcement of the Pledge, no rights of the Pledgee shall pass to the Pledgor by subrogation or otherwise unless and until all of the Secured Claims have been satisfied and discharged in full. Until then, the Pledgee shall apply all enforcement proceeds to the discharge of the Secured Claims.
6.8 The proceeds from the enforcement of the Pledge shall be paid to the Pledgee. Proceeds of the enforcement which exceeds the amount of the Secured Claims, including, in particular, any accrued interests since the Secured Claims have fallen due, are to be disbursed to the Pledgor.
6.9 The Pledgee may, in its sole reasonable discretion, determine which of several granted securities, if applicable, shall be used to satisfy the Secured Claims.
6.10 The Pledgor waives its rights of revocation (Anfechtbarkeit) and set-off (Aufrechenbarkeit) to the extent legally permissible.
6.11 In case of a Public Auction or a Private Sale, the Pledgor hereby irrevocably empowers and authorizes the Pledgee to take all judicial and non judicial
-8-
measures on behalf of the Pledgor necessary or appropriate for such realization, in particular, the Pledgor hereby irrevocably empowers and authorizes the Pledgee to sign and execute on its behalf an agreement on the sale and transfer of the Austrian Patents in the course of the Public Auction or the Private Sale, to sign all documents and make all legally binding declarations related thereto, to receive the transfer price and. to determine all other conditions of such agreement on the Pledgor's behalf. Furthermore, the Pledgor authorizes Pledgee to grant power of substitution to a person of the Pledgee's trust within the scope of this power of attorney.
7. Undertakings, Representations and Warranties
7.1 Unless otherwise permitted by any provision in any of the Secured Documents, the Pledgor hereby covenants with the Pledgee that without the prior consent of the Pledgee (not to be unreasonably withheld, delayed or conditioned where appropriate) the Pledgor shall:
7.1.1 not sell, transfer or in any other way dispose of any of the Austrian Patents or any interest therein, if such disposal is not permitted by the Secured Documents, and procure that none of its subsidiaries shall create or permit to subsist any encumbrance over all or any of the Austrian Patents,
7.1.2 effect promptly any payments to be made in respect of the Austrian Patents,
7.1.3 not, except as permitted or required under the Secured Documents, take any action or exercise any rights (or omit to take any action or exercise any rights) which could reasonably be expected materially and adversely to affect the rights of the Pledgee under and the preservation of the security interests intended to be created by this Agreement.
7.2 The Pledgor, represents and warrants to the Pledgee that
7.2.1 the Existing Austrian Patents are validly existing and legally enforceable;
7.2.2 it has good title to and is the sole owner of the Austrian Patents, free and clear of any third party rights (save for those rights granted to Nycomed Danmark APS, a corporation existing under the laws of the Kingdom of Denmark, pursuant to a licence agreement dated 2 July 2007 between the Pledgor and Nycomed Danmark APS), and it has not sold, assigned or otherwise disposed of or agreed to sell or otherwise dispose of the Austrian Patents or any of its rights or benefits in respect of the Austrian Patents;
7.2.3 the Austrian Patents are free of any encumbrances and are not subject to any prohibition to be pledged;
7.2.4 this Agreement provides for a valid first rank security over the Austrian Patents in accordance with all applicable laws.
-9-
8. No Liability and Indemnity
8.1 The Pledgee shall not be liable for any loss or damage suffered by the Pledgor save in respect of such loss or damage which is suffered as a result of the wilful misconduct (Vorsatz) or gross negligence (grobe Fahrlassigkeit) of the Pledgee.
8.2 The Pledgor shall defend and indemnify and hold harmless Pledgee against all claims, demands, proceedings, costs and expenses (including legal fees) referred to in clause 8.2 above which may be brought or asserted against Pledgee, its shareholders, directors, employees, agents or representatives or suffered or incurred by the Pledgee, its shareholders, directors, employees, agents or representatives. The obligations in this paragraph shall survive the termination or expiry of this Agreement.
9. Duration
This Agreement shall remain in full force and effect until release of the Austrian Patents in accordance with Clause 10.
10. Release of Pledge (Pfandfreigabe)
10.1 The Pledgee shall, at the request and cost of the Pledgor, release and cancel the Pledge and release the Austrian Patents from the Pledge upon the Secured Claims being discharged in full and the Pledgor not being under any further actual or contingent obligation to make advances or provide other financial accommodation to the Pledgee under the Secured Documents;
10.2 In connection with any release or cancellation described in ClauselO.l, the Pledgee shall (at the Pledgor's costs) do all such acts which are reasonably requested by the Pledgor in order to effect the release or cancellation the Pledge.
11. Transfer of Rights
The Pledgee, but not the Pledgor, is entitled to assign and pledge its rights under this Agreement and to assign and re- pledge the Pledge (Afterverpfandung) to third parties without the consent of the Pledgor. In case of such assignment or re-pledge the Pledgee shall inform the Pledgor to an address outside of Austria and the Pledgor shall take all action as may be instructed by the Pledgee from time to time.
12. Notices
12.1 Any notice or other communication under or in connection with this Agreement shall be in writing and shall be given in the form as set out in section 11.3 of the Purchase Agreement:
-10-
for the Pledgor:
NPS Allelix Corp.
c/o Blake, Cassels & Graydon LLP
199 Bay Street
Suite 2800, Commerce Court West
Toronto ON M5L 1A9 Canada
Attention of: Chris Hale
Fax No.: (416) 863-2653 '
with a copy (which shall not constitute notice) to:
NPS Pharmaceuticals Inc.
Morris Corporate Center 1.
4th Floor, Building B
300 Interpace Parkway
Parsippany, New Jersey 07054
United
States of America
Attention of: Vice-President, Corporate Development
Fax No.: (973)316-6463
for the Pledgee:
Drug Royalty L.P. 3
c/o Drug Royalty Corporation Inc.
Suite 3120, Royal Bank Plaza
Toronto, ON
Canada M5J 2J3
Attention of: Behzad Khosrowshahi
Fax No.:(416) 863-5161
E-mail: bk@drugroyalty.com
with a copy (which shall not constitute notice) to:
Davies Ward Phillips & Vineberg LLP
P.O. Box 63
Suite 4400, 1 First Canadian Place
Toronto, ON Canada M5X 1B1
Attention of: Gillian R. Stacey
Fax No.: (416) 863-0871
E-Mail: gstacey@dwpv.com
or to such other address as the recipient may notify or may have notified to the other Party in writing in accordance with the foregoing provided that such address is outside Austria.
-11-
12.2 Any notice or other communication under or in connection with this Agreement shall be in English or, if in any other language, accompanied by a translation into English. In the event of any conflict between the English text and the text in any other language, the English text shall prevail.
13. Austrian Stamp Duty
13.1 None of the Parties shall (i) bring an original or certified copy of this Agreement or any of the Secured Documents, or any other document that contains any written confirmation or written reference to this Agreement or any of the Secured Documents, apart from Schedule II which is intended to be filed with the Austrian Patent Office, (Ersatzbeurkundung oder rechtsbezeugende Urkunde) into the Republic of Austria or (ii) produce a document that contains any written confirmation or written reference to this Agreement or any of the Secured Documents within the Republic of Austria.
13.2 The Parties acknowledge that the bringing of this Agreement or any of the Secured Documents or any other document containing any written confirmation or any written reference to this Agreement or any of the Secured Documents, apart from Annex 2 which is intended to be filed with the Austrian Patent Office, (Ersatzbeurkundung oder rechtsbezeugende Urkunde) into the Republic of Austria or the production of any other document containing written confirmation or any written reference to this Agreement or any of the Secured Documents, apart from Annex 2 which is intended to be filed with the Austrian Patent Office, within the Republic of Austria may cause the imposition of Austrian stamp duty (Gebuhreri).
13.3 Any Party intentionally or negligently bringing or causing any other person (other than a Party) to bring this Agreement or any of the Secured Documents, either as original or certified copy, or any other written confirmation or written reference to this Agreement or any of the Secured Documents in another document, apart from Annex 2 which is intended to be filed with the Austrian Patent Office, into the Republic of Austria or producing any other document containing any written confirmation or written reference to this Agreement or any of the Secured Documents, apart from Annex 2 which is intended to be filed with the Austrian Patent Office, within the Republic of Austria shall be solely liable for any stamp duty resulting there from, provided, however, that nothing in this Clause shall prevent the Pledgee or the Security Agent from bringing or causing any other person (other than a Party) to bring this Agreement or any of the Secured Documents, either as original or certified copy, or any other written confirmation or written reference to this Agreement or any of the Secured Documents in another document, into the Republic of Austria or producing any other document containing any written confirmation or written reference to this Agreement or any of the Secured Documents in another document within the Republic of Austria if this is required and/or reasonably desirable for the enforcement or the preservation of any rights, powers and remedies under this Agreement or any of the Secured Documents, the perfection of the
-12-
Pledge created pursuant.to this Agreement, or if any proceedings are instituted by or against the Pledgee in connection with this Agreement or any of the Secured Documents. In any case, the Pledgee and the Pledgor agree not to contest the validity of an uncertified copy of this Agreement or any of the Secured Documents in any court and enforcement proceeding in Austria.
14. Miscellaneous
14.1 Changes and amendments of this Agreement including this subsection shall be made in writing, unless notarial form is required by the law.
14.2 This Agreement may be executed in any number of counterparts and by different parties hereto in separate counterparts, each of which when so executed shall be deemed to be an original and all of which taken together shall constitute one and the same Agreement.
14.3 If, at any time, any provision of this Agreement is or becomes illegal, invalid or unenforceable in any respect under the law of any jurisdiction, or if, at any time, any provision of this Agreement is or becomes incomplete neither the legality, validity or enforceability of the remaining provisions of this Agreement nor the legality, validity or enforceability of such provision under the law of any other jurisdiction shall in any way be affected or impaired thereby. Each insufficient (or incomplete) provision shall be replaced or completed by an effective, valid, practicable and enforceable provision in such a way that the new provision closely reflects the legal and economic effects, which the Parties have sought to achieve in the insufficient (or incomplete) provision.
14.4 No failure to exercise, nor any delay in exercising on the part of the Pledgee, any right or remedy hereunder shall operate as a waiver thereof, nor shall any single or partial exercise of any right or remedy prevent any further or other exercise thereof or the exercise of any other right or remedy. The rights and remedies provided hereunder are cumulative and not exclusive of any rights or remedies provided by law.
14.5 This Agreement shall be governed by and construed in accordance with the laws of Austria.
14.6 All disputes arising out of or in connection with this Agreement, including the question of its establishment and validity, shall be finally settled under the Rules of Arbitration and Conciliation of the International Arbitral Centre of the Austrian Federal Economic Chamber in Vienna, Austria, by three arbitrators appointed in accordance with said rules. The place of arbitration shall be Vienna, Austria. The language of the arbitration proceedings shall be English. All pleadings filed in the course of the proceedings and all attachments thereto shall be in English and if any original is in any other language shall include a certified translations of the relevant original document into English. The arbitration award shall be final and binding upon the Parties taking part in the arbitration proceedings.

-14-
Schedule I
List of Austrian Patents
|
Austrian Patent No. |
European Patent No. |
Subject |
Status |
|
E0273693 |
EP1079803B |
Formulation |
Granted |
|
E0275419 |
EP1001802B |
Dosage Regime |
Granted |
|
E0164394 |
EP0515228B |
Pure PTH |
Granted |
|
E0140973 |
EP0357391B |
DNA construct |
Granted |
|
Not yet known |
EP1473040A |
Dosage Regime |
Pending |
|
Not yet known |
EP0735896A |
Formulation |
Pending |
|
Not yet known |
EP1687048A |
Administration Method |
Pending |
|
SZ 32/2006 (SPC) |
SPC |
Filed 19 October 2006 |
-15-
Schedule II
Notice to the Austrian Patent Office
-16-








CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, with address c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada (hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. 3, a limited partnership incorporated under the laws of the Cayman Islands, whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents and patent applications listed in the Schedule to this Agreement (the "French Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such French Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing French Patents security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the French Patents, including their Related Rights, by NPS to DRLP3. French Patents
NOW THEREFORE in consideration of the respective covenants, promises and agreements of the Parties herein contained and other good and valuable consideration (the receipt and sufficiency of which are hereby acknowledged by each of the Parties), the Parties agree as follows:
Definitions
Terms that are defined in the Purchase Agreement and in the Security Agreement and not otherwise defined herein have, unless the context otherwise requires, the respective meanings specified in priority in the Purchase Agreement and in the Security Agreement and, in addition, the following terms have the following meanings (and grammatical variations of such terms shall have corresponding meanings)
- "Event of Defaulf' means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its
2
- "Minor Default" means the occurrence of any one or more of the following events to
the extent that it is not a Major Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1 (a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- "Parties" means NPS and DRLP3, and "Party" means either of them; (f) "Purchase Agreement" has the meaning specified in the recitals of this Agreement; (g)"Security Agreement" has the meaning specified in the recitals of this Agreement.
debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee;
3
Operative Clauses
- Subject to the condition provided for under Clause 3 below, NPS hereby agrees that all of its right, title and interest in the French Patents, including without limitation their Related Rights, be assigned to DRLP3.
- The operation of this Agreement does not constitute a waiver of any rights or remedies upon default provided between the parties in the Security Agreement or in other rights of DRLP3 as described in the Purchase Agreement.
- To the fullest extent permitted by law, rights and remedies available to DRLP3, whether provided for in this Agreement, in the Security Agreement, in the Purchase Agreement or otherwise, are not mutually exclusive, are cumulative and not alternative, and may be exercised independently or in any combination.
- Subject to the condition provided for under clause 5 below, provided that DRLP3 does not decide otherwise, NPS and DRLP3 hereby agree the French Patents, including without limitation their Related Rights, shall automatically be granted, assigned and transferred to DRLP3 such that title thereto and ownership therein shall belong to and be vested in DRLP3.
- Subject to clauses 6 and 7, the operation of this Agreement is subject to the occurrence of an Event of Default.
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and
4
- Such transfer, assignment and grant shall be effective upon the occurrence of an Event of Default, subject to clauses 4 to 7 above, regardless of any dispute or proceedings whatsoever that may arise between the Parties and regardless of the Expert proceeding set out in Clause to ascertain the value of the French Patents at the time of the transfer, assignment and grant (hereunder the "Value of the French Patents").
- Nothing contained in Clauses 4 to 7 shall be construed to afford DRLP3 any recourse to the French Patents prior to the occurrence of an Event of Default.
- Notwithstanding the effectiveness of the transfer, assignment and grant of the French Patents to DRLP3 upon the occurrence of an Event of Default as provided in Clause 4 to 7, NPS and DRLP3 hereby agree to appoint an Expert as listed in Schedule 2 with the mission to ascertain the Value of the French Patents at the time of the transfer to DRLP3,
- The Parties hereby agree that the decision of the Expert shall be final and binding upon the Parties and the Parties hereby waive any right to challenge the Value of the French Patents as ascertained by the Expert before any Governmental Authority Office, including any Court whatsoever.
- Any sums owed or to be owed to DRLP3 in accordance with Clause 7 of the Purchase Agreement until the Termination Date thereof as provided in Clause 8.1 of the Purchase Agreement and any Obligations shall be automatically deducted from the Value of the French Patents. In this respect, Clause 8.1 of the Purchase Agreement provides that the Termination date of the Purchase Agreement shall be: "(i) the date that the aggregate amount of the payments received by the Purchaser in respect of the Purchaser Royalty Interest is equal to the amount that is 2.5 times the amount of the Purchase Price actually paid by the Purchaser to the Vendor under Clause 2.3 of the Purchase Agreement; (ii) the expiration or termination of the License Agreement between NPS and Nycomed pursuant to Clause 16.1 or 16.2 thereof; or (iii) the termination of the License Agreement by Nycomed and the failure by the Vendor or the Purchaser to enter into a New License
furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
5
- Subject to the termination of this Agreement pursuant to Clause 11 herein, NPS covenants with DRLP3 that the French Patents is tree of hen, charge, pledge and encumbrance from any third party and that it shall not sell, assign, pledge or otherwise charge or transfer any of the French Patents to any third party.
- This Agreement and its assignment shall be recorded at the patent registry at the French patent office (Institut National de la Propriete Industrielle or INPI) or at any Governmental Authority office in France or in relevant jurisdictions, including the European Patent Office.
- NPS shall undertake at its own cost all such acts, execute all such documents and do all such things as DRLP3 may request in order for this Agreement and the rights granted to DRLP3 under this Agreement to be recorded with the French patent office (Institut National de la Propriete Industrielle or INPI) or appropriate Governmental Authority office in France or of any relevant jurisdiction, including the European Patent Office and, where required, to produce any additional documents that DRLP3 may require.
- NPS shall undertake at its own cost, upon the occurrence of an Event of Default,, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the French Patents at the French patent office (Institut National de la Propriete Industrielle or INPI) or at any Governmental Authority office in France or in relevant jurisdictions, including the European Patent Office and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may require.
- NPS hereby authorizes DRLP3 to take any action and make all such acts and things to create, preserve, perfect, validate or otherwise protect the French Patents and the rights acquired hereunder by DRLP 3 at the French patent office or at any Governmental Authority office in France or in relevant jurisdictions, including the European Patent Office, in the event that NPS fails to do so, at the expenses of NPS, including, without
Agreement within 12 months after such termination in accordance with Clause 7.5 of the Purchase Agreement."
6
- NPS hereby authorizes DRLP3 to take any action, including judicial proceedings, and make all such acts and things to create, preserve, perfect, validate or otherwise protect the French Patents and the rights acquired hereunder by DRLP3 at the French patent office or at any Governmental Authority office in France or in relevant jurisdictions, including the European Patent Office, in the event that NPS fails to do so, at the expenses of NPS, including, without limitation any infringement actions from any third party and opposition proceedings that may occur at any time during the execution of this Agreement.
- Subject to clauses 6 and 7, NPS shall, upon demand, reimburse DRLP3 for all reasonable costs and expenses incurred by DRLP3 in the enforcement of any rights hereunder (including without limitation fees and expenses of counsel at each step of the actions and proceedings where DRLP3 prevails) and all such costs shall form part of the Obligations.
- NPS hereby irrevocably constitutes and designates DRLP3 as and for NPS's attorney in fact:
- to execute, upon the occurrence of an Event of Default, subject to clauses 4 to 7 above, and during the continuance thereof, all and any such instraments, documents and papers as DRLP3.determines to be appropriate in connection with the exercise of such rights and remedies and to cause the sale, license, assignment, transfer or other disposition of the French Patents, including all filings, recordings or registrations with the applicable government offices required or appropriate to effect such dispositions of the French Patents.
- The within grant of a power of attorney, being coupled with an interest, shall be irrevocable until this Agreement is terminated.
limitation, the payment of taxes, renewals and annuity fees, and to enable DRLP3 to exercise and enforce its rights and remedies hereunder and generally to carry out the provisions and purposes of this Agreement;
7
- Any notice, demand, direction or other instrument required or permitted to be given hereunder shall be in writing and shall be given in the manner and addressed as set out in Clause 11.3 of the Purchase Agreement. Either party may change its address for service from time to time by notice given in accordance with the foregoing.
- This Agreement may be terminated by written agreement made between DRLP3 and NPS. Upon indefeasible fulfillment of the Obligations and full payment, in accordance with Clause 8.1 of the Purchase Agreement, this Agreement shall terminate and the French Patents shall belong to NPS.
- Upon termination of this Agreement, DRLP3 shall, at the request and expense of NPS, make and do all such acts and things and execute and deliver all such financing statements, instruments, agreements and documents as NPS considers reasonably necessary or desirable to discharge the French Patents, to release and discharge the French Patents therefrom and to record such release and discharge in all appropriate offices of public record.
- This Agreement shall inure to the benefit of and be binding upon the successors and permitted assigns of each of DRLP3 and NPS. This Agreement may not'be assigned in whole or in part by either party without the prior written consent of the other party; provided, however, that DRLP3 may assign this Agreement in whole or in part without the prior written consent of NPS: (i) by way of security to a financial institution or other lender, (ii) to any Person that directly, or indirectly through one or more intermediaries, controls or is controlled by, or is under common control with, DRLP3, (iii) to a special purpose vehicle or (iv) as part of a sale of a material part of DRLP3, in any case whether by way of reorganization or otherwise, and DRLP3 shall give prompt notice of any such assignment to NPS within 10 Business Days after the occurrence thereof.
- This Agreement is governed by and construed in accordance with the laws of the Province of Ontario and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Courts of the Province of Ontario and all courts competent to hear appeals therefrom.
8
Date July 16, 2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.,
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
9
SCHEDULE 1
The French Patents
|
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
SPC 06C0032 |
Formulation |
Granted |
|
EP1001802B |
Dosage Regime |
Granted |
|
EP0515228B |
Pure PTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1627048A |
Administration Method |
Pending |
10
SCHEDULE 2
NOMINATION OF THE EXPERT
Deloitte & Touche
11
CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9, Canada (hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. 3, a Cayman Island limited partnership incorporated under the laws of the Cayman Islands whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and supplementary protection certificates listed in the Schedule to this Agreement (the "German Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such German Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the German Patents, including their Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment by DRLP3 to NPS of US$1 (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy,
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a Major
Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1(a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NTS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement.
insolvency or similar legislation including for the appointment of a receiver or trustee;
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the German Patents, including without limitation their Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this Agreement is subject to the occurrence of an Event of Default.
3
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the German Patents, or any Related Rights, to a third party.
- This Agreement and its assignment shall be filed with the German National Patent Office. NPS undertakes, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to perfect and file this Agreement and its assignment with the German National Patent Office and, where required, to produce any additional documents that DRLP3 may reasonably require.
- NPS undertakes, upon the occurrence of an Event of Default, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the German Patents in DRLP3 and to record such assignment at the German National Patent Register and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require.
4
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the German Patents at the German Patent Office in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees,
- This Agreement is governed by and construed in accordance with the laws of the province of Ontario and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Ontario Courts and Tribunals and all courts competent to hear appeals therefrom.
Date July 16, 2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.,
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
5
SCHEDULE E
The German Patents
|
Patent No. |
German Patent No. |
Subject |
Status |
|
EP1079803B |
699 19 533.0 |
Formulation |
Granted |
|
EP1001802B |
698 26 132.1 |
Dosage Regime |
Granted |
|
EP0515228B |
692 24 858.7 |
Pure PTH |
Granted |
|
EP0357391B |
689 26 895.5 |
DNA construct |
Granted |
|
EP1473040A |
P 44 80 014.2 |
Dosage Regime |
Pending |
|
EP0735896A |
not assigned |
Formulation |
Pending |
|
EP1687048A |
Administration Method |
Pending |
|
|
12 2006 000 057.7 (SPC) |
Filed on 18 October 2006 |
6






CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite . 2800, Commerce Court West, Toronto ON M5L 1A9, Canada (hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. 3, a Cayman Island limited partnership incorporated under the laws of the Cayman Islands whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement');
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and supplementary protection certificates listed in the Schedule to this Agreement (the "Greek Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such Greek Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered, into an agreement, dated as of July. 16, 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, HPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the Greek Patents, including their Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment by DRLP3 to NPS of US$1 (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default/' means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase. Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or Otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy,
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a Major
Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice' thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1(a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement.
insolvency or similar legislation including for the appointment of a receiver or trustee;
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the. Greek Patents, including without limitation their Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this Agreement is subject to the occurrence of an Event of Default.
3
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take eff6ct unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains Unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event Of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the Greek Patents, or any Related Rights, to a third party.
- The consideration for the assignment under clause 1, as agreed by the Parties and provided for. in the Purchase Agreement and set out above, has already been fully provided and paid by DRLP3 to NPS, and NPS hereby acknowledges that it has received such price and agrees that DRLP3 is not required to make any further payment to NPS. The parties agree that the consideration is fair, reasonable and corresponds fully to the actual value of the assigned Greek Patents, including their Related Rights, and the conditions and prices of the relevant market.
- This Agreement and its assignment shall be recorded on the Greek National Patent Register (O.B.I.). NPS undertakes, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to record this Agreement and its assignment on the Greek National Patent Register (O.B.L) and, where required, to produce any additional documents that DRLP3 may reasonably require.
- NPS undertakes, upon the occurrence of an Event of Default, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents
4
- NPS hereby authorizes DRLP3, and grants a continuous and irrevocable power of attorney to DRLP3, to take any of the action referred to above in clauses 7 and 8 on behalf of NPS and in particular (but without limitation) to: (a) do all such acts and give any documents and signatures on behalf of NPS for the performance and the enforcement of all formalities required to record this Agreement and the assignment on the Greek National Patent Register (O.B.I.) and, where required, to produce any additional documents on behalf of NPS, (b) to do all such acts and give any documents and signatures on behalf of NPS ..for the performance and the enforcement of all formalities required to vest title in the Greek Patents, including their-Related-Rights, in itself (DRLP3) and to record such assignment at the Greek National Patent Register (O.B.I.) and, where required, to produce and sign on behalf of NPS any documents that be necessary or desirable to confirm that an Event of Default has taken place, (c) do all such acts and give any documents and signatures that might be needed in the future for the recording with the Greek National Patent Register (O.B.I.) of this Agreement with regard to any applications comprised in the Greek Patents upon their granting and validation in Greece, and (d) to carry out all or part of the present power by a substitute.
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the Greek Patents at the Greek Patent Office (O.B.I.) and for the maintenance of the Related Rights in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
and signatures for the performance and the enforcement of all formalities required to vest title in the Greek Patents, including their Related Rights, in DRLP3 and to record such assignment at the Greek National Patent Register (O.B.I.) and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require. NPS undertakes also to do all such acts and give any documents and signatures that might be needed in the future for the recording with the Greek National Patent Register (O.B.I.) of this Agreement with regard to EP 1473040 and EP 073589 upon their granting and validation in Greece.
5
- This Agreement is governed by and construed in accordance with the laws of the province of Ontario, and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Ontario Courts and Tribunals and all courts competent to hear appeals therefrom.
- NPS and DRLP2 both authorise Christina Panagoulea, Georgia Kotroni and Eleftheria Karabatou of "G.D. Kallimopoulos, K.Th. Loukopoulos, A.P. Chiotellis Law Firm", residents of Athens, 2 Ypsilantou Str., 106 75 Greece, jointly or each one separately, submit and record this Agreement at the Greek Patent Office (O.B.I.) and proceed to any action and perform all formalities necessary for the recording of this Agreement. NPS and DRLP3 hereby ratify and confirm everything whatsoever that said attorney(s) may lawfully do, namely any actions or omissions and authorize the above attorney(s) to carry out all or part of the present power by a substitute. NPS and DRLP3 appoint said attorney(s) to accept service of documents in Athens.
Date July 16, 2007
Having read the above, the Parties hereby sign:
6


SCHEDULE
The Greek Patents
|
European Patent |
Greek Patent No. |
Title |
Status |
|
No. |
|||
|
EP 1079803B |
GR 3050775 |
"Protein Formulations" |
Granted |
|
SPC 8000222 |
"PREOTACT' Active substance |
Granted |
|
|
EP 1001802B |
GR 3050819 |
"A COMBINED PHARMACEUTICS L PREPARATION COMPRISING PARATHYROID HORMONE AND A BONE RESORPTION INHIBITOR" |
Granted |
|
EP0515228B |
GR 3027113 |
"Essential pure human parathyroid hormone" . |
Granted |
|
EP1473040A |
"Use of humanparathyroid hormone" |
Pending |
|
|
EP0735896A |
"PARATHYROID HORMONE FORMULATION" |
Pending |
|
|
EP1687048A |
"Methods of Administering Therapeutic Injections" |
Pending |
|
|
SPC 8000222 (filed on 20 October 2006 with filing . number 20060800026) |
Filed 20 October 2006 |
8


CONDITIONAL ASSiGNMENT AGKEEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at with address c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada(hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. a Cayman Island limited partnership incorporated under the laws of the Cayman Islands whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may he amended", modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and SPCs listed in the Schedule to this Agreement (the "Italian Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such Italian Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing collateral security for the due payment and performance of
certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement');
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the Italian Patents, including their Related Rights, by NPS to DRLP3.
NOW IT IS HEREBY AGREED AS FOLLOWS:
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect; .
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) fdes a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee;
2
- "Minor Default" means the occurrence of any one or more of the
following events to the extent that it is not a Major Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1(a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the Italian Patents, and including without limitation their Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this Agreement is subject to the occurrence of an Event of Default.
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce
3
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the Italian Patents, or any Related Rights, to a third party.
- The purchase price for the assignment under clause 1, as agreed by the Parties, is EUR 10,000 less the damages
suffered by DRLP3 due to the Event(s) of Default; DRLP3 shall pay said amount within
- 30 days from the date on which the Parties agree in writing the amount of the damages due by NPS; or
- 30 days from the date on which an independent arbitrator, appointed according to the rules of the Italian Civil Procedure Code, settles the amount of the damages due by NPS.
- This Agreement and its assignment shall be recorded on the Italian National Patent Register before the payment of the purchase price according to clause 6 above. NPS undertakes to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to perfect and record this Agreement and. its assignment on the Italian Patent Office and, where required, to produce any additional documents that DRLP3 may reasonably require, including without limitation, any necessary powers of attorney that may be required by Italian counsel to record the Agreement. In particular, at the date of execution of this Agreement, NPS undertakes and agrees to execute the attached Simplified Conditional Assignment Form attached as Annex 1.
payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
4
- NPS undertakes, upon the occurrence of an Event of Default, subject to clauses 3 and 4, to Cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the Italian Patents, and their Related Rights, in DRLP3 and to perfect and record such assignment at the Italian Patent Office and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require, including without limitation, any necessary powers of attorney that may be required by Italian counsel to record the Agreement In particular, NPS undertakes and agrees that it will execute the attached Simplified Assignment Form attached as Annex 2.
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the Italian Patents, and their Related Rights, at the Italian Patent Office in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
- This Agreement is governed by and construed in accordance with the laws of the Province of Ontario and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, Validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Courts and Tribunals of the Province of Ontario and all courts competent to hear appeals therefrom.
Date July 16, 2007
Having read the above, the Parties hereby sign:
5



SCHEDULE
The Italian Patents
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
SPCCCPUB2006 934 . |
Granted |
|
|
EP100T802B |
Dosage Regime |
Granted |
|
EP0515228B |
PurePTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1687048A |
Administration Method |
Pending |
7
ANNEX 1
SIMPLIFIED CONDITIONAL ASSIGNMENT FORM
Between
NPS Allelix Corp., a company' existing under the laws of the Province of Ontario, "The Vendor"
Drug Royalty L.P. 3, a Cayman Island limited partnership "The Purchaser" (both "The Parties")
WHEREAS
- The Vendor is the registered proprietor of several Patents, as defined in the following Clause b);
- the Patents of which The Vendor grants full ownership, free and clear from any right of third parties, (hereinafter "The Collateral") are listed in Annex A
- the Purchaser wishes to acquire conditional ownership of The Collateral;
THE PARTIES AGREE AS FOLLOWS
- The Vendor agrees to sell, assign, transfer to The Purchaser all right, title and interest of The Vendor in and to The Collateral, such that title and ownership of The Collateral shall belong to The Purchaser;
- The Purchaser agrees to purchase from The Vendor all right, title and interest in and to The Collateral for the price determined according to the criteria set forth in Clause 4 of Ihe Conditional Assignment Agreement signed by The Parties on July 16, 2007 (BUR 10,000 less the sum due to the damages suffered by The Purchaser due to the Event(s) of Default);
- The Vendor shall undertake all such acts, execute all such documents and do all such things that The Purchaser may request in order to record this Form with the Italian Patent Office.
Date July 16, 2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.
________________________________
Name:
Title:
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC-3
________________________________
Name: Behzad Khosrowshahi
Title: Manager
8
ANNEX 2
SIMPLIFIED CONDITIONAL ASSIGNMENT FORM
Between
NPS Allelix Corp., a company' existing under the laws of the Province of Ontario, "The Vendor"
Drug Royalty L.P. 3, a Cayman Island limited partnership "The Purchaser" (both "The Parties")
WHEREAS
- The Vendor'is the registered proprietor of several Patents, as defined in the following Clause b);
- the Patents of which The Vendor grants full ownership, free and clear from any right of third parties, (hereinafter "The Collateral") are listed in Annex A
- the Purchaser wishes to acquire the full ownership of The Collateral;
THE PARTIES AGREE AS FOLLOWS
- The Vendor agrees to sell, assign, transfer to The Purchaser all right, title and interest of The Vendor in and to The Collateral, such that title and ownership of The Collateral shall belong to The Purchaser from the date of this Simplified Assignment Form;
- The Purchaser agrees to purchase from The Vendor all right, title and interest in and to The Collateral for the price determined according to the criteria set forth in Clause 4 of the Conditional Assignment Agreement signed by The Parties on July 16, 2007 (EUR 10,000 less the sum due to the damages suffered by The Purchaser due to the Event(s) of Default);
- The Vendor shall undertake all such acts, execute all such documents and do all such things that The Purchaser may request in order to record this Form with the Italian Patent Office.
Date July 16,2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.
________________________________
Name:
Title:
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC-3
________________________________
Name: Behzad Khosrowshahi
Title: Manager
9



Annex A
The Collateral
|
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
SPCCCPUB2006 934 |
Granted |
|
|
EP1001802B |
Dosage Regime |
Granted |
|
EP0515228B |
PurePTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1687048A |
Administration Method |
Pending |

CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, with address c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada (hereinafter "NPS"); and
(1) DRUG ROYALTY L.P. 3, a limited partnership incorporated under the laws of the Cayman Islands, whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and SPCs listed in the Schedule to this Agreement (the "Dutch Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16; 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) As further security for compliance with its Obligations under the Purchase Agreement NPS has agreed to grant DRLP3 a right of pledge on the Dutch Patents;
(F) This Agreement sets out the terms of the assignment of the Dutch Patents, including their Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment by DRLP3 to NPS of US$1 (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a
Major Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.
bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee;
3
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the Dutch Patents, including without limitation their Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this assignment under Clause 1 above is subject to the occurrence of an Event of Default.
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the event that, upon the occurrence of an Event of Default, subject to clauses 2, 3 and 4, for whatever reason the assignment provided for in Clause 1 above is not valid and/or effective and/or cannot be enforced, NPS shall forfeit a penalty of Euro 1,000,000 to DRLP3 which will become due and payable immediately upon the occurrence of such Event of Default.
- As security for the performance of its monetary Obligations and of the obligation under clause 1 of this deed (together the "Secured Obligations") NPS as pledgor agrees to grant to DRLP3 as pledgee, and hereby so grants to DRLP3, on the Dutch Patents a right of pledge ("een pandrecht"), which right of pledge DRLP3 hereby accepts.
4
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the Dutch Patents, or any Related Rights, to a third party.
- This Agreement and its assignment shall be recorded on the Dutch National Patent Register. NPS undertakes to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to perfect and record this Agreement and its assignment on the Dutch National Patent Register and, where required, to produce any additional documents that DRLP3 may reasonably require.
- In the event that NPS is in default ("verzuim"), as defined in Section 6:81 of the Dutch Civil Code, of performing one or more of the Secured Obligations, DRLP3 shall be authorised to sell the Dutch Patents or part thereof, in accordance with Section 3:248 of the Dutch Civil Code, without prejudice to DRLP3's rights under Section 3:251 (1) of the Dutch Civil Code, in order to recover the proceeds thereof. NPS shall not have the rights under Section 3:251 (1) of the Dutch Civil Code. DRLP3 does not bear the obligations referred to in Sections 3:249 and 3:252 of the Dutch Civil Code to give notice of an intended sale or to give notice following a sale,
- NPS undertakes, upon the occurrence of an Event of Default, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the Dutch Patents, including their Related Rights, in DRLP3 and to record such assignment at the Dutch National Patent Register and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require. NPS hereby authorizes DRLP3, and grants irrevocable power of attorney to DRLP3, to take any of the action referred to above on its behalf.
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the Dutch Patents at the Dutch Patent Office and for the maintenance of the Related Rights in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
5
- This Agreement is governed by and construed in accordance with the law of the Province of Ontario and the federal laws of Canada applicable therein, save that Clauses 5, 6 and 9 above are subject to the operation of Dutch law. All disputes arising out of or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent courts of the Province of Ontario and all courts competent to hear appeals therefrom, save that the Dutch Courts shall have exclusive jurisdiction in relation to any such disputes arising out of the operation of Clauses 5, 6 and 9 above.
Date July 16, 2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
6
SCHEDULE
The Dutch Patents
|
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
SPC 300243 |
Formulation |
Granted |
|
EP1001802B |
Dosage Regime |
Granted |
|
EP0515228B |
Pure PTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1627048A |
Administration Method |
Pending |
7
CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELLX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at with address c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada(hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. 3, a limited partnership incorporated under the laws of the Cayman Islands, whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and SPCs listed in the Schedule to this Agreement (the "Spanish Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such Spanish Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may
be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the Applications, including then Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment to be made by DRLP3 to NPS pursuant to clause 6 below (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a
Major Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1(a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement.
(iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation mcluding for the appointment of a receiver or trustee;
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the Spanish Patents, and including without limitation their Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this Agreement is subject to the occurrence of an Event of Default.
3
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the Spanish Patents, or any Related Rights, to a third party.
- The purchase price for the assignment under clause 1, as agreed by the Parties, is 3000 Euros, which DRLP3 pays in advance to NPS, and NPS hereby acknowledges that it has received such amount and agrees that DRLP3 is not required to make any further payment to NPS.
- This Agreement shall be raised to a public deed before a Spanish Notary Public and, subject to clauses 3 and 4, its assignment shall be recorded on the Spanish National Patent Register upon the occurrence of an Event of Default. NPS undertakes, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to perfect and record this Agreement and its assignment on the Spanish National Patent Register and, where required, to produce any additional documents that DRLP3 may reasonably require. Without prejudice to the generality of the foregoing, NPS agrees that it will within 60 days of the execution of this Agreement execute any necessary powers of attorney that may be required by Spanish counsel to record this Agreement at the Spanish National Patent Office. Further, to the extent that the parties fail to execute a mortgage granting DRLP3 a security interest over the Spanish Patents when
4
- NPS undertakes, upon the occurrence of an Event of Default, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the Spanish Patents in DRLP3 and to record such assignment at the Spanish National Patent Register and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require.
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the Spanish Patents, and their Related Rights, at the Spanish Patent Office in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
- This Agreement is governed by and construed in accordance with the laws of the Province of Ontario and the federal laws of Canada applicable therein.. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Ontario Courts and Tribunals Ontario and all courts competent to hear appeals therefrom.
this Agreement is executed, NPS agrees that it will execute such an agreement within the said 60 day period.
In relation to the above, NPS irrevocably empowers DRLP3 to appear before a Spanish notary public to execute any document and to comply with any formality necessary to have the assignment regulated hereby fully effective, subject to clauses 3 and 4, once an Event of Default has occurred.
Date July 16,2007
Having read the above, the Parties hereby sign:
5
NPS ALLELIX CORP.
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
6
SCHEDULE
The Spanish Patents
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
SPC 200600034 |
Formulation |
Granted |
|
EP1001802B |
Dosage Regime |
Granted |
|
EP0515228B |
Pure PTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1687048A |
Administration Regime |
Pending |
7
PATENT MORTGAGE - OUTSTANDING
POWER OF ATTORNEY
In the city of Salt Lake City, Utah, USA, on September, 2007.
BEFORE ME
Ms. Leslie Lancaster, Notary Public living and practicing in said city and duly entitled to formalise public deeds.
APPEARS
Mr. Barton W. Giddings, of legal age, married, of USA citizenship, with professional domicile at Salt Lake City, Utah, USA, with USA passport number 056408123, in force.
ACTS
In the name and on behalf of NPS ALLELIX CORP. a company duly incorporated and in existence in accordance with the laws of the Province of Ontario, with registered offices at c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada and registered with the Ontario Ministry of Government Services under number 00138941 (the "Grantor"), who is duly empowered to grant this power of attorney on behalf of the Grantor.
He is duly empowered for this act by virtue of his appointment as Assistant Corporate Secretary.
He has to my own judgement the necessary legal capacity for executing the present and for this purpose,
GRANTS
A power of attorney as broad and sufficient as required by law in favour of:
- Mr. Fernando de las Cuevas Castresana, of legal age, a Spanish citizen,.. married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and National Identity Card number 42.936.545-F, in force;
- Mr. Ralph Smith, of legal age, a British citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and holders of British passport number 704620075;
- Mr. Francisco Gil Duran, of legal age, a Spanish citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and National Identity Card number 5.405.248-H, in force;
- Ms. Valentina Rodriguez Hernandez, of legal age, a Venezuelan citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and Residence Permit number X-2868095-H, in force; and
- Ms. Alejandra Huidobro Andueza, of legal age, a Spanish citizen, single, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and National Identity Card number 2.912.275-S, in force.
so that any one of them, acting individually, in the name and on behalf of the Grantor and empowered hereby to totally or partially appoint their substitutes, may:
- Perform whatsoever acts and execute whatsoever public or private documents are necessary in order to constitute, create, modify, enforce and register in favor of DRUG ROYALTY L.P. 3, any patent mortgages, in any terms and conditions deemed appropriate, over the Spanish part of any of the following patents:
- European Patent number 99917715 (1079803).
- European Patent number 98929965.6 (1001802).
- European Patent number 92304692.4 (0515228).
- European Patent number 89308753.6 (0357391).
- European Patent number 4017622.4 (1473040).
- European Patent number 95903732.6 (0735896).
- European Patent number 04795465.6 (1687048).
- To appear before a Spanish notary public to execute a conditional assignment agreement (substantially in the form of the draft attached to this power of attorney) for the transfer, subject to condition precedent, of the patents identified in point 1 above, and to execute any kind of document and to comply any formality necessary to formalize satisfaction of the abovementioned condition precedent and the full effect of the transfer of the patents listed in point 1 above.
- Carry out, likewise, whatsoever related or complementary acts as may be necessary -including the execution of whatever document in order to authorize the registration of the documents referred to in points 1 and 2 above with the corresponding Movable Goods Registry, the Spanish Patents*, and Trademarks Office and/or any other relevant registry and filing of tax liquidations- for the fullest execution of the mandate received and to sign whatsoever public or private documents which may be necessary or appropriate to exercise the powers contained herein, including, if necessary, deeds of correction of possible errors.
IN WITNESS WHEREOF, the Grantor, having been informed of the content of this public deed of power of attorney, approves and signs it with me, the Notary Public, having complied with all the formalities required by applicable law where this power of attorney is granted.
2


POWER OF ATTORNEY
In the city of Toronto, Canada, on July 16, 2007.
BEFORE ME
Mr. Michael Uster, Notary Public living and practicing in said city and duly entitled to formalise public deeds.
APPEARS
Mr. Bezhad Khosrowshahi, of legal age, married, of Canadian citizenship, with professional domicile at Royal Bank Plaza, Suite 3120, South Tower, Box 122, 200 Bay St., Toronto, Ontario Canada M5J 2J3, with Canadian passport number BA002202, in force.
ACTS
In the name and on behalf of DRUG ROYALTY L.P. 3, a Cayman Island limited partnership existing under the laws of the Cayman Islands whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands, who is duly empowered to grant this power of attorney on behalf of the Grantor.
He is duly empowered for this act by virtue of him being the Manager of the Grantor's General Partner, DRC Management LLC3.
He has to my own judgement the necessary legal capacity for executing the present and for this purpose,
GRANTS
A power of attorney as broad and sufficient as required by law in favour of:
- Mr. Fernando de las Cuevas Castresana, of legal age, a Spanish citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and National Identity Card number 42.936.545-F, in force;
- Mr. Ralph Smith, of legal, age, a British citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and holders of British passport number 704620075;
- Mr. Francisco Gil Duran, of legal age, a Spanish citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and National Identity Card number 5.405.248-H, in force;
- Ms. Valentina Rodriguez Hernandez, of legal age, a Venezuelan citizen, married, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and Residence Permit number X-2868095-H, in force; and
- Ms. Alexandra Huidobro Andueza> of legal age, a Spanish citizen, single, lawyer, with professional domicile at Paseo de la Castellana 216, Madrid, Spain and National Identity Card number 2.912.275-S, in force.
so that any one of them, acting individually, in the name and on behalf of the Grantor and empowered hereby to totally or partially appoint their substitutes, may:
- Perform whatsoever acts and execute whatsoever public or private documents are necessary in order to accept, modify, enforce and register, in favor of the Grantor, any patent mortgages, in any terms and conditions deemed appropriate, over the Spanish part of any of the following patents:
- European Patent number 99917715 (1079803).
- European Patent number 98929965.6 (1001802).
- European Patent number 92304692.4 (0515228).
- European Patent number 89308753.6 (0357391).
- European Patent number 4017622.4 (1473040).
- European Patent number 95903732.6 (0735896).
- European Patent number 04795465.6 (1687048).
- To appear before a Spanish notary public to execute a conditional assignment agreement (substantially in the form of the draft attached to this power of attorney) for the acquisition, subject to condition precedent, of the patents identified in point 1 above, and to execute any kind of document and to comply any formality necessary to formalize satisfaction of the abovementioned condition precedent and the full effect of the transfer of the patents listed in point 1 above.
- Carry out, likewise, whatsoever related or complementary acts as may be necessary - including the execution of whatever document in order to register the documents referred to in points 1 and 2 with the corresponding Movable Goods Registry, the Spanish Patents and Trademarks Office and/or any other relevant registry and filing of tax liquidations - for the fullest execution of the mandate received and to sign whatsoever public or private documents which may be necessary or appropriate to exercise the powers contained herein, including, if necessary, deeds of correction of possible errors.
IN WITNESS WHEREOF, the Grantor, having been informed of the content of this public deed of power of attorney, approves and signs it with me, the Notary Public, having complied with all the formalities required by applicable law where this power of attorney is granted.
ATTESTATION OF THE NOTARY
I, the Notary Public, hereby certify and attest as follows:
- That the Grantor Is a Cayman Islands limited partnership existing under the laws of the Cayman Islands and complies with all legal conditions required by the laws of the Cayman Islands to continue its commercial activities and perform its corporate purpose; and
- That the appearer, whose personal particulars I know from his statements and from his personal documentation in force which he shows to me, and who signed in my presence this Power of Attorney, in the capacity in which he acts, is duly authorised and has the necessary legal capacity to execute this instrument in the name and on behalf of the Grantor according to the Laws of the Cayman Islands and pursuant to its by-laws and to his appointment as of the Grantor's General Partner, Manager of DRC Management, LLC3; and
- That in the execution of this Public Instrument, all the formalities and solemnities required by the Laws in force in the Cayman Islands, the place of the execution of the same, regarding this class of document, have been complied with.
SIGNATURE of
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
________________________________
[*]
SIGNATURE of the NOTORARY PUBLIC:
/s/ Michael Uster
Michael Uster
POWER OF ATTORNEY OF
NYCOMED DANMARK ApS - OUTSTANDING
CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at with address c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada(hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. 3, a Cayman Island limited partnership incorporated under the laws of the Cayman Islands whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patents, patent applications and SPCs listed in the Schedule to this Agreement (the "United Kingdom Patents"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such United Kingdom Patents (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may
be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver
certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the United Kingdom Patents,
including their Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment by DRLP3 to NPS of US$1 (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1 (a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a Major
Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1 (a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement.
(iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee;
Operative Clauses
3
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the United Kingdom Patents, or any Related Rights, to a third party.
- This Agreement and its assignment shall be recorded on the United Kingdom National Patent Register. NPS undertakes, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to perfect and record this Agreement and its assignment on the United Kingdom National Patent Register and, where required, to produce any additional documents that DRLP3 may reasonably require.
- NPS undertakes, upon the occurrence of an Event of Default, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the United Kingdom Patents, including their Related Rights, in DRLP3 and to record such assignment at the United Kingdom National Patent Register and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require.
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the United Kingdom Patents at the United Kingdom Patent Office and for the maintenance
4
- This Agreement is governed by and construed in accordance with the laws of the Province of Ontario and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Courts and Tribunals of the Province of Ontario and all courts competent to hear appeals therefrom.
of the Related Rights in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
Date July 16, 2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
5
SCHEDULE
The United Kingdom Patents
|
Patent No. |
Subject |
Status |
|
EP1079803B |
Formulation |
Granted |
|
SPC/GB06/035 |
Formulation |
Granted |
|
EP1001802B |
Dosage Regime |
Granted |
|
EP0515228B |
Pure PTH |
Granted |
|
EP0357391B |
DNA construct |
Granted |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1687048A |
Administration Method |
Pending |
6


CONDITIONAL ASSIGNMENT AGREEMENT
BETWEEN
(1) NPS ALLELIX CORP., a company incorporated under the laws of Ontario, Canada, whose registered office is at c/o Blake, Cassels & Graydon LLP, 199 Bay Street, Suite 2800, Commerce Court West, Toronto ON M5L 1A9 Canada (hereinafter "NPS"); and
(2) DRUG ROYALTY L.P. 3, a limited partnership incorporated under the laws of the Cayman Islands, whose registered office is at M&C Corporate Services Limited, PO Box 309GT, Ugland House, South Church Street, George Town, Grand Cayman, Cayman Islands (hereinafter "DRLP3").
Each a "Party" and together the "Parties".
WHEREAS
(A) NPS and DRLP3 have entered into an agreement for the sale and assignment of rights dated as of July 16, 2007 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Purchase Agreement");
(B) NPS is the registered proprietor of certain Patents (as defined in the Purchase Agreement) including without limitation those patent applications listed in the Schedule to this Agreement (the "Applications"). NPS is also the proprietor of any related Governmental Authority (as defined in the Purchase Agreement) approvals and agreements relating to any product that is the subject of any such Applications (the "Related Rights");
(C) NPS and DRLP3 have also entered into an agreement, dated as of July 16, 2007, as general and continuing collateral security for the due payment and performance of certain Obligations arising pursuant to the Purchase Agreement (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement");
(D) Pursuant to section 3.2(g) of the Purchase Agreement, NPS has agreed to deliver certain conditional assignment agreements to DRLP3, including this Agreement;
(E) This Agreement sets out the terms of the assignment of the Applications, including their Related Rights, by NPS to DRLP3.
IN CONSIDERATION OF the Parties entering into the Purchase Agreement AND IN FURTHER CONSIDERATION of the payment by DRLP3 to NPS of US$1 (the receipt and sufficiency of which is hereby acknowledged), IT IS HEREBY AGREED AS FOLLOWS:-
Definitions
- "Event of Default" means a Major Default or a Minor Default;
- "Major Default" means the occurrence of any one or more of the following events:
- any breach by NPS of any representation or warranty made by NPS in Section 4.1, 4.4, 4.5, 4.6(c), 4.6(d), 4.6(e) or 4.6(f) of the Purchase Agreement; provided, however, that a breach of Section 4.1(a) or 4.4 of the Purchase Agreement shall not be a Major Default unless such breach results in a Material Adverse Effect;
- any breach by NPS of any of its covenants under Section 7.2(b) of the Purchase Agreement and such breach shall continue unremedied for a period of 60 days after written notice thereof; or
- the cessation or threatened cessation by NPS of its business generally or the admission by NPS of its inability to, or its actual failure to, pay its debts generally, including circumstances where NPS (i) is adjudged bankrupt or insolvent, (ii) makes an assignment in bankruptcy or otherwise for the benefit of creditors, (iii) files a petition or proposal under bankruptcy, insolvency or similar legislation, or (iv) has instituted against it proceedings under bankruptcy, insolvency or similar legislation including for the appointment of a receiver or trustee;
2
- "Minor Default" means the occurrence of any one or more of the following events to the extent that it is not a Major
Default:
- the failure of NPS to pay any monetary Obligations within five Business Days of the date on which such Obligations are due or payable;
- the failure of NPS to perform any non-monetary Obligation or covenant under the Security Agreement or the Purchase Agreement and such failure shall continue unremedied for a period of 60 days after written notice thereof; or
- any breach by NPS of any representation or warranty made by NPS in the Purchase Agreement (for certainty, including without limitation a breach of Section 4.1 (a) or 4.4 of the Purchase Agreement if such breach does not result in a Material Adverse Effect;
- "Obligations" means all indebtedness, liabilities and obligations (whether direct, indirect, absolute, contingent or otherwise) of NPS to DRLP3 arising pursuant to the Purchase Agreement or the Security Agreement; and
- All other terms used in this Agreement shall have the same meanings as provided for in the Purchase Agreement and the Security Agreement.
Operative Clauses
- Subject to the condition provided for under clause 2 below, NPS hereby assigns all of its right, title and interest in the Applications, with respect to all confracting states designated in each of the Applications, and including without limitation their Related Rights, to DRLP3.
- Subject to clauses 3 and 4, the operation of this Agreement is subject to the occurrence of an Event of Default.
3
- If the Event of Default is a Minor Default, the operation of this Agreement shall not take effect unless or until DRLP3 has first sought to commence legal action to enforce payment or performance of the Obligations and any remedy obtained under such action has not satisfied the Obligations of NPS or any judgement obtained pursuant to such action remains unsatisfied by NPS for more than 15 Business Days after the rendering of such judgement (without the necessity of appeal from such judgement).
- If an Event of Default is the result of a breach of representation and warranty by NPS, then the operation of this Agreement shall not take effect for a period of 60 days commencing on the date on which notice of such breach is provided to NPS and furthermore shall not take effect if during that 60 day period the parties agree upon an alternative remedy in respect of that breach.
- In the meantime, NPS covenants with DRLP3 that it shall not sell, assign, pledge or otherwise charge or transfer any of the Applications, or any Related Rights, to a third party.
- This Agreement and its assignment shall be recorded with the European Patent Office (and, if DRLP3 so requests, the national patent offices of any relevant contracting state). NPS undertakes, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to perfect and record this Agreement and its assignment with the European Patent Office and/or national patent offices and, where required, to produce any additional documents that DRLP3 may reasonably require.
- NPS undertakes, upon the occurrence of an Event of Default,, subject to clauses 3 and 4, to cooperate in good faith with DRLP3, to do all such acts and give any documents and signatures for the performance and the enforcement of all formalities required to vest title in the Applications, including their Related Rights, in DRLP3 and to record such assignment with the European Patent Office (and, if DRLP3 so requests, the national patent offices of any relevant contracting state) and, where required, to produce any documents confirming that an Event of Default has taken place that DRLP3 may reasonably require.
4
- NPS hereby authorizes DRLP3 to do whatever is necessary for the maintenance of the Applications with the European Patent Office (and, if relevant, the national patent offices of any relevant contracting state) and for the maintenance of the Related Rights in the event that NPS fails to do so, including, without limitation, the payment of taxes, renewals and annuity fees.
- This Agreement is governed by and construed in accordance with the laws of the Province of Ontario and the federal laws of Canada applicable therein. All disputes arising out or in connection with this Agreement including those concerning its existence, validity, interpretation and performance are subject to the exclusive jurisdiction of the competent Courts and Tribunals of the Province of Ontario and all courts competent to hear appeals therefrom.
Date July 16, 2007
Having read the above, the Parties hereby sign:
NPS ALLELIX CORP.
/s/ Val R. Antczak
Name: Val R. Antczak
Title: Vice President and Secretary
DRUG ROYALTY L.P. 3,
by its General Partner
DRC MANAGEMENT LLC 3
/s/ Behzad Khosrowshahi
Name: Behzad Khosrowshahi
Title: Manager
5
SCHEDULE
The Applications
|
Application No |
Subject |
Status |
|
EP1473040A |
Dosage Regime |
Pending |
|
EP0735896A |
Formulation |
Pending |
|
EP1687048A |
Adminstration Method |
Pending |
Exhibit B
Patent Filings
[See attached.]

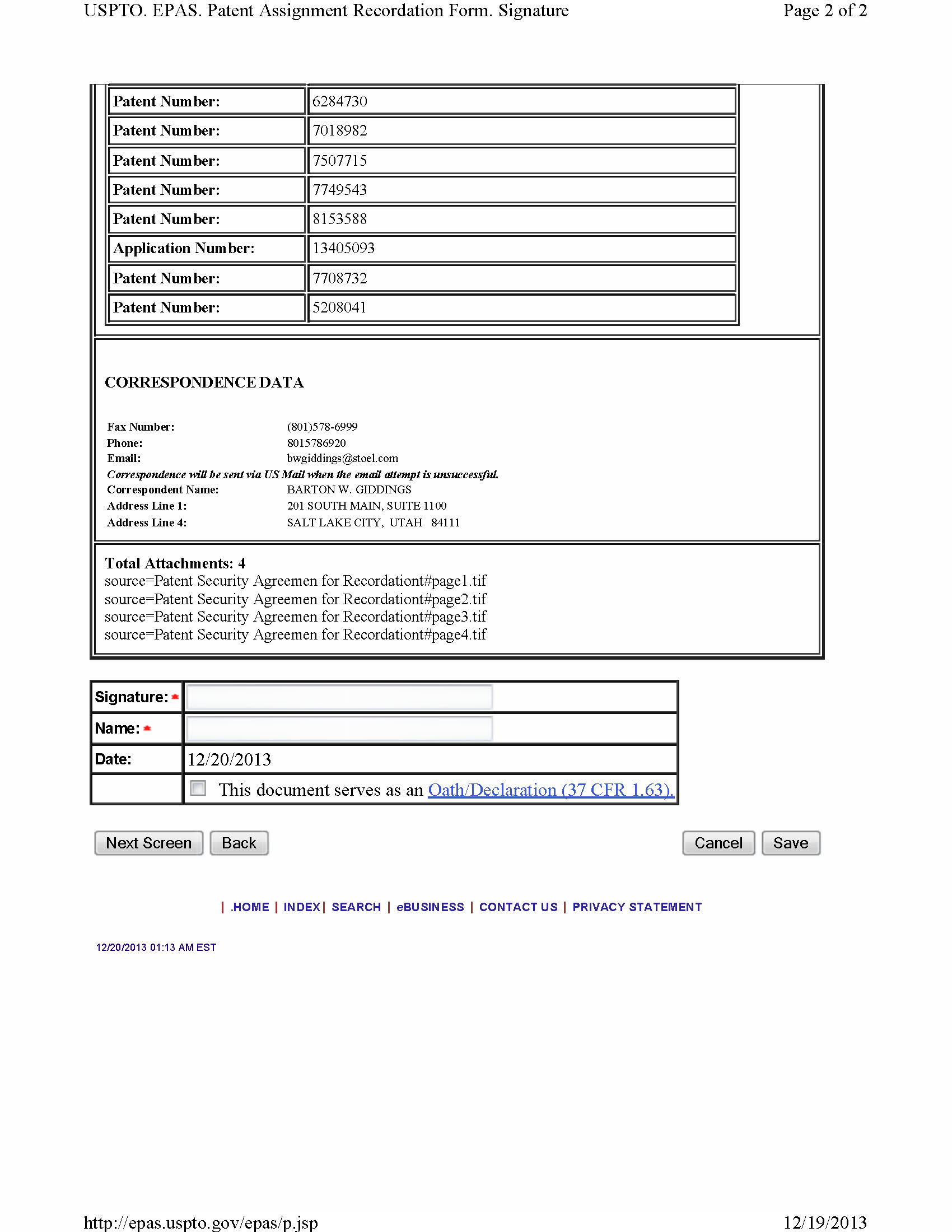
PATENT SECURITY AGREEMENT
THIS PATENT SECURITY AGREEMENT is made as of December 20, 2013,
BETWEEN:
NPS PHARMACEUTICALS, INC.,
a corporation existing under the laws of the State of Delaware
(collectively with its successors and permitted assigns, "Debtor"),
- and -
DRUG ROYALTY L.P. 3,
a Cayman Islands limited partnership
(collectively with its successors and permitted assigns, "Secured Party").
WHEREAS Secured Party and Debtor have entered into an Amended and Restated Security Agreement dated as of December 20, 2013 (as such agreement may be amended, modified, supplemented or restated from time to time, the "Security Agreement"); and
WHEREAS pursuant to the Security Agreement, Debtor has granted to Secured Party a security interest in, among other assets, certain intellectual property of Debtor, and Debtor has agreed to execute this Patent Security Agreement for recording with the United States Patent and Trademark Office;
NOW THEREFORE in consideration of the respective covenants, promises and agreements of the parties herein contained and other good and valuable consideration (the receipt and sufficiency of which are hereby acknowledged by each of the parties), the parties agree as follows:
- Grant of Security Interest
As general and continuing collateral security for the due payment and performance of the Obligations (as such term is defined in the Security Agreement), Debtor hereby pledges, mortgages, charges and assigns (by way of security) to Secured Party, and grants to Secured Party, a security interest in, the patents described in Schedule A hereto (as such Schedule may be amended from time to time) and all reissues, divisions, continuations, continuations-in-part, extensions, renewals and re-examinations thereof (collectively, the "Patents").
- Recordation
Debtor authorizes and requests that the United States Patent and Trademark Office record this Patent Security Agreement.
- Paramountry of Security Agreement
This Patent Security Agreement is being entered into in accordance with the terms of the Security Agreement. Debtor hereby acknowledges and affirms that the rights and remedies of Secured Party with respect to the security interest in the Patents made and granted hereby are more fully specified in the Security Agreement, the terms and provisions of which are incorporated by reference herein. In the event of a conflict between any provision of this Patent Security Agreement and any provision of the Security Agreement, the provision of the Security Agreement shall control.
- Termination
This Patent Security Agreement shall terminate and all rights in the Patents shall revert to Debtor upon the termination of the Security Agreement, including termination of the Security Agreement in part, with respect to the Additional Collateral (as such term is defined in the Security Agreement), in accordance with the terms thereof.
- Successors and Assigns
This Patent Security Agreement shall inure to the benefit of and shall be binding on and enforceable by and against the parties hereto and their respective successors and permitted assigns under the Security Agreement.
- Applicable Law
This Patent Security Agreement shall be governed by, and interpreted and enforced in accordance with, the laws of the State of New York and the federal laws of the United States applicable therein, without giving effect to the principles of conflicts of law thereof except as set forth in Section 5-1401 of the New York General Obligations Law.
- Execution in Counterparts and Facsimile Delivery
This Patent Security Agreement may be executed in one or more counterparts, all of which when taken together constitute one and the same agreement.
[Signature Page Follows]
IN WITNESS WHEREOF the parties have executed this Patent Security Agreement.
|
NPS Pharmaceuticals, Inc. Attention of: General Counsel |
NPS PHARMACEUTICALS, INC. |
|
Drug Royalty L.P. 3 Attention of: Behzad Khosrowshahi |
DRUG ROYALTY L.P. 3 |
Patent Security Agreement - Signature Page
SCHEDULE A
PATENTS
|
Country |
Filing Date |
Serial No. |
Issue Date |
Patent No. |
Expiry Date |
|
U.S. |
23 May '91 |
07/707114 |
4 May '93 |
5,208,041 |
23 May'11 |
|
U.S. |
23 Dec '93 |
08/172,206 |
5 March '96 |
5,496,801 |
23 Dec'13 |
|
U.S. |
14 Aug '98 |
09/125,247 |
4 Sep '01 |
6,284,730 |
8 June'18 |
|
U.S. Div Con |
18 March'03 |
10/389,797 |
28 March '06 |
7,018,982 |
8 June'18 |
|
U.S.Div Con 2 |
19 Dec'05 |
11/305,339 |
24 March '09 |
7,507,715 |
8 June'18 |
|
U.S.Div |
9 Jan '09 |
12/351,558 |
6 July'10 |
7,749,543 |
8 June '18 |
|
U.S.Div |
23 June'10 |
12/822,089 |
10 April '12 |
8,153,588 |
8 June'18 |
|
U.S.Div |
24 Feb'12 |
13/405,093 |
(2012/0148684) |
||
|
U.S. |
15 Oct. '04 |
10/966,364 |
4 May '10 |
7,708,732 |
11 May'27 |
