UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
| ý | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended January 31, 2013
OR
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______________ to _______________
Commission file number: 0-17085
PEREGRINE PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 95-3698422 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) |
| 14282 Franklin Avenue, Tustin, California | 92780-7017 | ||
| (Address of principal executive offices) | (Zip Code) |
(714) 508-6000
(Registrant’s telephone number, including area code)
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ý No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one)
| Large Accelerated Filer | o | Accelerated Filer | ý | |
| Non- Accelerated Filer | o | Smaller reporting company | o |
(Do not check if a smaller reporting company)
Indicate by checkmark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes o No ý
As of March 11, 2013, there were 137,110,758 shares of common stock, $0.001 par value, outstanding.
PEREGRINE PHARMACEUTICALS, INC.
TABLE OF CONTENTS
| Page | ||
| No. | ||
| PART I - FINANCIAL INFORMATION | 1 | |
| Item 1. | Consolidated Financial Statements. | 1 |
| Item 2. | Management’s Discussion and Analysis of Financial Condition And Results of Operations. | 15 |
| Item 3. | Quantitative and Qualitative Disclosures About Market Risk. | 24 |
| Item 4. | Controls and Procedures. | 24 |
| PART II - OTHER INFORMATION | 25 | |
| Item 1. | Legal Proceedings. | 25 |
| Item 1A. | Risk Factors. | 25 |
| Item 2. | Unregistered Sales of Equity Securities and Use of Proceeds. | 39 |
| Item 3. | Defaults Upon Senior Securities. | 39 |
| Item 4. | Mine Safety Disclosures. | 39 |
| Item 5. | Other Information. | 39 |
| Item 6. | Exhibits. | 40 |
| SIGNATURES | 41 | |
The terms “we,” “us,” “our,” “the Company,” and “Peregrine,” as used in this Report on Form 10-Q refers to Peregrine Pharmaceuticals, Inc. and its wholly-owned subsidiary, Avid Bioservices, Inc.
| i |
PART I - FINANCIAL INFORMATION
Item 1. Consolidated Financial Statements.
PEREGRINE PHARMACEUTICALS, INC.
CONDENSED CONSOLIDATED BALANCE SHEETS
| JANUARY 31, 2013 | APRIL 30, 2012 | |||||||
| Unaudited | ||||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | 26,255,000 | $ | 18,033,000 | ||||
| Trade and other receivables, net | 1,983,000 | 2,353,000 | ||||||
| Inventories, net | 4,635,000 | 3,611,000 | ||||||
| Prepaid expenses and other current assets, net | 878,000 | 795,000 | ||||||
| Total current assets | 33,751,000 | 24,792,000 | ||||||
| Property, net | 2,783,000 | 2,900,000 | ||||||
| Other assets | 623,000 | 570,000 | ||||||
| TOTAL ASSETS | $ | 37,157,000 | $ | 28,262,000 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable | $ | 1,620,000 | $ | 3,492,000 | ||||
| Accrued clinical trial and related fees | 1,478,000 | 2,111,000 | ||||||
| Accrued payroll and related costs | 2,949,000 | 2,468,000 | ||||||
| Deferred revenue | 5,061,000 | 3,651,000 | ||||||
| Customer deposits | 6,729,000 | 4,865,000 | ||||||
| Other current liabilities | 930,000 | 1,052,000 | ||||||
| Total current liabilities | 18,767,000 | 17,639,000 | ||||||
| Deferred revenue | 292,000 | 361,000 | ||||||
| Other long-term liabilities | 699,000 | 779,000 | ||||||
| Commitments and contingencies | ||||||||
| STOCKHOLDERS' EQUITY: | ||||||||
| Preferred stock-$0.001 par value; authorized 5,000,000 shares; non-voting; nil shares outstanding | – | – | ||||||
| Common stock-$0.001 par value; authorized 325,000,000 shares; outstanding – 133,770,614 and 101,421,365, respectively | 134,000 | 101,000 | ||||||
| Additional paid-in capital | 376,720,000 | 347,506,000 | ||||||
| Accumulated deficit | (359,455,000 | ) | (338,124,000 | ) | ||||
| Total stockholders’ equity | 17,399,000 | 9,483,000 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY | $ | 37,157,000 | $ | 28,262,000 | ||||
See accompanying notes to condensed consolidated financial statements.
| 1 |
PEREGRINE PHARMACEUTICALS, INC.
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| THREE MONTHS ENDED JANUARY 31, | NINE MONTHS ENDED JANUARY 31, | |||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| Unaudited | Unaudited | Unaudited | Unaudited | |||||||||||||
| REVENUES: | ||||||||||||||||
| Contract manufacturing revenue | $ | 6,961,000 | $ | 3,203,000 | $ | 17,157,000 | $ | 12,796,000 | ||||||||
| License revenue | 78,000 | 78,000 | 272,000 | 372,000 | ||||||||||||
| Total revenues | 7,039,000 | 3,281,000 | 17,429,000 | 13,168,000 | ||||||||||||
| COSTS AND EXPENSES: | ||||||||||||||||
| Cost of contract manufacturing | 3,651,000 | 2,484,000 | 9,378,000 | 9,219,000 | ||||||||||||
| Research and development | 5,437,000 | 9,180,000 | 18,471,000 | 26,758,000 | ||||||||||||
| Selling, general and administrative | 3,112,000 | 2,710,000 | 9,469,000 | 8,371,000 | ||||||||||||
| Total costs and expenses | 12,200,000 | 14,374,000 | 37,318,000 | 44,348,000 | ||||||||||||
| LOSS FROM OPERATIONS | (5,161,000 | ) | (11,093,000 | ) | (19,889,000 | ) | (31,180,000 | ) | ||||||||
| OTHER INCOME (EXPENSE): | ||||||||||||||||
| Interest and other income | 255,000 | 9,000 | 307,000 | 31,000 | ||||||||||||
| Interest and other expense | (8,000 | ) | (6,000 | ) | (53,000 | ) | (88,000 | ) | ||||||||
| Loss on early extinguishment of debt | – | – | (1,696,000 | ) | – | |||||||||||
| NET LOSS | $ | (4,914,000 | ) | $ | (11,090,000 | ) | $ | (21,331,000 | ) | $ | (31,237,000 | ) | ||||
| WEIGHTED AVERAGE COMMON SHARES OUTSTANDING: | ||||||||||||||||
| Basic and Diluted | 131,489,994 | 87,149,770 | 114,726,569 | 78,443,114 | ||||||||||||
| BASIC AND DILUTED LOSS PER COMMON SHARE | $ | (0.04 | ) | $ | (0.13 | ) | $ | (0.19 | ) | $ | (0.40 | ) | ||||
| COMPREHENSIVE LOSS | $ | (4,914,000 | ) | $ | (11,090,000 | ) | $ | (21,331,000 | ) | $ | (31,237,000 | ) | ||||
See accompanying notes to condensed consolidated financial statements.
| 2 |
PEREGRINE PHARMACEUTICALS, INC.
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
| NINE MONTHS ENDED JANUARY 31, | ||||||||
| 2013 | 2012 | |||||||
| Unaudited | Unaudited | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (21,331,000 | ) | $ | (31,237,000 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Share-based compensation | 2,320,000 | 2,438,000 | ||||||
| Depreciation and amortization | 806,000 | 664,000 | ||||||
| Amortization of discount on notes payable and debt issuance costs | – | 33,000 | ||||||
| Loss on early extinguishment of debt | 1,696,000 | – | ||||||
| Loss on disposal of property | 8,000 | 2,000 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Trade and other receivables, net | 370,000 | (596,000 | ) | |||||
| Inventories, net | (1,024,000 | ) | 2,540,000 | |||||
| Prepaid expenses and other current assets, net | (83,000 | ) | (285,000 | ) | ||||
| Other non-current assets | 2,000 | 748,000 | ||||||
| Accounts payable | (1,883,000 | ) | (119,000 | ) | ||||
| Accrued clinical trial and related fees | (633,000 | ) | 750,000 | |||||
| Accrued payroll and related expenses | 481,000 | 643,000 | ||||||
| Deferred revenue | 1,341,000 | (3,174,000 | ) | |||||
| Customer deposits | 1,864,000 | 704,000 | ||||||
| Other current liabilities | (64,000 | ) | (29,000 | ) | ||||
| Other long-term liabilities | (80,000 | ) | (224,000 | ) | ||||
| Net cash used in operating activities | (16,210,000 | ) | (27,142,000 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Property acquisitions | (686,000 | ) | (1,103,000 | ) | ||||
| (Increase) decrease in other assets | (55,000 | ) | 34,000 | |||||
| Net cash used in investing activities | (741,000 | ) | (1,069,000 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from issuance of common stock, net of issuance costs of $885,000 and $1,007,000, respectively | 26,166,000 | 26,190,000 | ||||||
| Proceeds from issuance of notes payable, net of issuance costs of $251,000 | 14,749,000 | – | ||||||
| Proceeds from exercise of stock options and under Employee Stock Purchase Plan | 291,000 | 96,000 | ||||||
| Principal payments on notes payable | (15,000,000 | ) | (1,333,000 | ) | ||||
| Payment of final fee on notes payable | (975,000 | ) | – | |||||
| Principal payments on capital leases | (58,000 | ) | (56,000 | ) | ||||
| Net cash provided by financing activities | 25,173,000 | 24,897,000 | ||||||
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | 8,222,000 | (3,314,000 | ) | |||||
| CASH AND CASH EQUIVALENTS, beginning of period | 18,033,000 | 23,075,000 | ||||||
| CASH AND CASH EQUIVALENTS, end of period | $ | 26,255,000 | $ | 19,761,000 | ||||
| Schedule of non-cash investing and financing activities: | ||||||||
| Accounts payable for purchase of property | $ | 11,000 | $ | 13,000 | ||||
| Fair market value of warrants issued in connection with notes payable | $ | 470,000 | $ | – | ||||
See accompanying notes to condensed consolidated financial statements.
| 3 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited)
| 1. | ORGANIZATION AND BUSINESS |
Peregrine Pharmaceuticals, Inc. (“Peregrine” or “Company”) is a biopharmaceutical company developing first-in-class monoclonal antibodies focused on the treatment and diagnosis of cancer. We are advancing two oncology programs with our lead product candidates, bavituximab and Cotara, for the treatment of various cancers. In addition, we are advancing our lead molecular imaging agent, 124I-PGN650, in an exploratory clinical trial for the imaging of multiple solid tumor types. Peregrine also has in-house manufacturing capabilities through its wholly-owned subsidiary Avid Bioservices, Inc. (“Avid”), a contract manufacturing organization that provides development and biomanufacturing services for Peregrine and its third-party clients.
| 2. | BASIS OF PRESENTATION |
The accompanying interim unaudited condensed consolidated financial statements have been prepared in accordance with United States generally accepted accounting principles (“U.S. GAAP”) and with the rules and regulations of the U.S. Securities and Exchange Commission (“SEC”) related to quarterly reports on Form 10-Q. Accordingly, they do not include all of the information and disclosures required by U.S. GAAP for a complete set of financial statements. These interim unaudited condensed consolidated financial statements and notes thereto should be read in conjunction with the audited consolidated financial statements and notes thereto included in the Company’s Annual Report on Form 10-K for the year ended April 30, 2012. The condensed consolidated balance sheet at April 30, 2012 has been derived from audited financial statements at that date. The unaudited financial information for the interim periods presented herein reflects all adjustments which, in the opinion of management, are necessary for a fair presentation of the financial condition and results of operations for the periods presented, with such adjustments consisting only of normal recurring adjustments. Results of operations for interim periods covered by this quarterly report on Form 10-Q may not necessarily be indicative of results of operations for the full fiscal year.
The interim unaudited condensed consolidated financial statements include the accounts of Peregrine Pharmaceuticals, Inc., and its wholly-owned subsidiary, Avid Bioservices, Inc. All intercompany accounts and transactions have been eliminated in the interim unaudited condensed consolidated financial statements.
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts, as well as disclosures of commitments and contingencies in the financial statements and accompanying notes. Actual results could differ from those estimates.
Going Concern
Our interim unaudited condensed consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The financial statements do not include any adjustments relating to the recoverability of the recorded assets or the classification of liabilities that may be necessary should it be determined that we are unable to continue as a going concern.
At January 31, 2013, we had $26,255,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced negative cash flows from operations since our inception and we expect the negative cash flows from operations to continue for the foreseeable future. Our net loss incurred during the nine-month period ended January 31, 2013 amounted to $21,331,000 and our net losses incurred during the past three fiscal years ended April 30, 2012, 2011 and 2010, amounted to $42,119,000, $34,151,000, and $14,494,000, respectively. Therefore, unless and until we are able to generate sufficient revenues from Avid’s contract manufacturing services and/or from the sale and/or licensing of our products under development, we expect such losses to continue for the foreseeable future.
Therefore, our ability to continue to fund our clinical trials and development efforts is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, issuing additional equity or debt.
| 4 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
Historically, we have funded a significant portion of our operations through the issuance of equity. During the nine months ended January 31, 2013, we raised $27,051,000 in gross proceeds under an At Market Sales Issuance Agreement (Note 8), of which $25,555,000 was raised subsequent to September 25, 2012, primarily to replace the $15,000,000 of initial funding we repaid on September 25, 2012 under an earlier loan facility we entered into on August 30, 2012 (Note 7). Subsequent to January 31, 2013 and through March 12, 2013, we raised an additional $4,805,000 in aggregate gross proceeds under two separate At Market Sales Issuance Agreements (Note 8).
With respect to our ability to raise additional capital from the issuance of equity, as of March 12, 2013, we have an effective shelf registration statement on Form S-3, under which we may issue, from time to time, in one or more offerings, shares of our common stock for gross proceeds of up to $145,525,000. However, our ability to raise additional capital in the equity markets is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse clinical trials results, and significant delays in one or more clinical trials. If our ability to access the capital markets becomes severely restricted, it could have a negative impact on our business plans, including our clinical trial programs and other research and development activities. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
In addition to financing our operations through the issuance of equity, we may also secure additional funding through the issuance of debt, licensing or partnering our products in development, or increasing revenue from our wholly-owned subsidiary, Avid. While we will continue to explore these potential opportunities, there can be no assurances that we will be successful in securing debt financing, licensing or partnering our products in development, or generating additional revenue from Avid to complete the research, development, and clinical testing of our product candidates.
| 3. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Adoption of Recent Accounting Pronouncements
Effective May 1, 2012, we adopted Financial Accounting Standards Board’s (“FASB”) Accounting Standards Update (“ASU”) No. 2011-05, Comprehensive Income (Topic 220): Presentation of Comprehensive Income and ASU No. 2011-12, Comprehensive Income (Topic 220): Deferral of the Effective Date for Amendments to the Presentation of Reclassifications of Items Out of Accumulated Other Comprehensive Income in ASU No. 2011-5. In these updates, an entity has the option to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. In both choices, an entity is required to present each component of net income along with total net income, each component of other comprehensive income along with a total for other comprehensive income, and a total amount for comprehensive income. ASU No. 2011-05 eliminates the option to present the components of other comprehensive income as part of the statement of changes in stockholders’ equity. The amendments in ASU No. 2011-05 do not change the items that must be reported in other comprehensive income or when an item of other comprehensive income must be reclassified to net income. The adoption of ASU Nos. 2011-05 and 2011-12 did not have a material impact on our consolidated financial statements. We have presented comprehensive loss in the accompanying interim unaudited condensed consolidated statements of operations and comprehensive loss.
Pending Adoption of Recent Accounting Pronouncements
In February 2013, the FASB issued ASU No. 2013-02, Other Comprehensive Income (Topic 220): Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income. ASU No. 2013-02 does not change the current requirements for reporting net income or other comprehensive income in financial statements, however, it does require an entity to report the effect of significant reclassifications out of accumulated other comprehensive income on the respective line items in net income if the amounts are required to be reclassified in their entirety to net income. For other amounts that are not required to be reclassified in their entirety to net income in the same reporting period, an entity is required to cross-reference to other disclosures that provide additional detail about those amounts. This guidance will be effective for reporting periods beginning after December 15, 2012, which will be our fiscal year 2014 (or May 1, 2013). We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
| 5 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
Revenue Recognition
We currently derive revenue from two sources: (i) contract manufacturing services provided by Avid, and (ii) licensing revenue related to agreements associated with Peregrine’s technologies under development.
We recognize revenue in accordance with the authoritative guidance for revenue recognition. We recognize revenue when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller’s price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple deliverables.
Contract Manufacturing Revenue
Revenue associated with contract manufacturing services provided by Avid is recognized once the service has been rendered and/or upon shipment (or passage of title) of the product to the customer. On occasion, we recognize revenue on a “bill-and-hold” basis in accordance with the authoritative guidance. Under “bill-and-hold” arrangements, revenue is recognized once the product is complete and ready for shipment, title and risk of loss has passed to the customer, management receives a written request from the customer for “bill-and-hold” treatment, the product is segregated from other inventory, and no further performance obligations exist.
In addition, we also follow the authoritative guidance when reporting revenue as gross when we act as a principal versus reporting revenue as net when we act as an agent. For transactions in which we act as a principal, have discretion to choose suppliers, bear credit risk and perform a substantive part of the services, revenue is recorded at the gross amount billed to a customer and costs associated with these reimbursements are reflected as a component of cost of sales for contract manufacturing services.
Any amounts received prior to satisfying our revenue recognition criteria are recorded as deferred revenue in the accompanying interim unaudited condensed consolidated financial statements. We also record a provision for estimated contract losses, if any, in the period in which they are determined.
License Revenue
Revenue associated with licensing agreements primarily consists of non-refundable upfront license fees, non-refundable annual license fees and milestone payments. Non-refundable upfront license fees received under license agreements, whereby continued performance or future obligations are considered inconsequential to the relevant license technology, are recognized as revenue upon delivery of the technology. If a licensing agreement has multiple elements, we analyze each element of our licensing agreements and consider a variety of factors in determining the appropriate method of revenue recognition of each element.
Multiple Element Arrangements. Prior to the adoption of ASU No. 2009-13 on May 1, 2011, if a license agreement has multiple element arrangements, we analyze and determine whether the deliverables, which often include performance obligations, can be separated or whether they must be accounted for as a single unit of accounting in accordance with the authoritative guidance. Under multiple element arrangements, we recognize revenue for delivered elements only when the delivered element has stand-alone value and we have objective and reliable evidence of fair value for each undelivered element. If the fair value of any undelivered element included in a multiple element arrangement cannot be objectively determined, the arrangement would be accounted for as a single unit of accounting, and revenue is recognized over the estimated period of when the performance obligation(s) are performed.
In addition, under certain circumstances, when there is objective and reliable evidence of the fair value of the undelivered items in an arrangement, but no such evidence for the delivered items, we utilize the residual method to allocate the consideration received under the arrangement. Under the residual method, the amount of consideration allocated to delivered items equals the total arrangement consideration less the aggregate fair value of the undelivered items, and revenue is recognized upon delivery of the undelivered items based on the relative fair value of the undelivered items.
For new licensing agreements or material modifications of existing licensing agreements entered into after May 1, 2011, we follow the provisions of ASU No. 2009-13. If a licensing agreement includes multiple elements, we identify which deliverables represent separate units of accounting, and then determine how the arrangement consideration should be allocated among the separate units of accounting, which may require the use of significant judgment.
| 6 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
If a licensing agreement includes multiple elements, a delivered item is considered a separate unit of accounting if both of the following criteria are met:
| 1. | The delivered item has value to the licensing partner on a standalone basis based on the consideration of the relevant facts and circumstances for each agreement; |
| 2. | If the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the Company’s control. |
Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence (“VSOE”), of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, we use our best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement.
Milestone Payments. Prior to the adoption of ASU No. 2010-17 on May 1, 2011, milestone payments were recognized as revenue upon the achievement of the specified milestone, provided that (i) the milestone event was substantive in nature and the achievement of the milestone was not reasonably assured at the inception of the agreement, (ii) the fees were non-refundable, and (iii) there was no continuing performance obligations associated with the milestone payment.
Effective May 1, 2011, we adopted on a prospective basis the Milestone Method under ASU No. 2010-17 for new licensing agreements or material modifications of existing licensing agreements entered into after May 1, 2011. Under the Milestone Method, we recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following criteria:
| 1. | The consideration is commensurate with either the entity’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone; |
| 2. | The consideration relates solely to past performance; and |
| 3. | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to the Company.
The provisions of ASU No. 2010-17 do not apply to contingent consideration for which payment is either contingent solely upon the passage of time or the result of a counterparty’s performance. We will assess the nature of, and appropriate accounting for, these payments on a case-by-case basis in accordance with the applicable authoritative guidance for revenue recognition.
Any amounts received prior to satisfying our revenue recognition criteria are recorded as deferred revenue in the accompanying interim unaudited condensed consolidated financial statements.
Fair Value Measurements
We determine fair value measurements in accordance with the authoritative guidance for fair value measurements and disclosures for all assets and liabilities within the scope of this guidance. This guidance, which among other things, defines fair value, establishes a consistent framework for measuring fair value and expands disclosure for each major asset and liability category measured at fair value on either a recurring or nonrecurring basis. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The guidance prioritizes the inputs used in measuring fair value into the following hierarchy:
| 7 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
| · | Level 1 – Quoted prices in active markets for identical assets or liabilities. |
| · | Level 2 – Observable inputs other than quoted prices included in Level 1, such as assets or liabilities whose values are based on quoted market prices in markets where trading occurs infrequently or whose values are based on quoted prices of instruments with similar attributes in active markets. |
| · | Level 3 – Unobservable inputs that are supported by little or no market activity and significant to the overall fair value measurement. |
As of January 31, 2013 and April 30, 2012, we do not have any Level 2 or Level 3 financial assets or liabilities and our cash and cash equivalents are carried at fair value based on quoted market prices for identical securities (Level 1 input).
Customer Deposits
Customer deposits primarily represent advance billings and/or payments received from Avid’s third-party customers prior to the initiation of contract manufacturing services.
Research and Development Expenses
Research and development costs are charged to expense when incurred in accordance with the authoritative guidance for research and development costs. Research and development expenses primarily include (i) payroll and related costs associated with research and development personnel, (ii) costs related to clinical and preclinical testing of our technologies under development, (iii) costs to develop and manufacture the product candidates, including raw materials and supplies, product testing, depreciation, and facility related expenses, (iv) expenses for research services provided by universities and contract laboratories, including sponsored research funding, and (v) other research and development expenses.
Accrued Clinical Trial and Related Fees
We accrue clinical trial and related fees based on work performed in connection with advancing our clinical trials, which relies on estimates and/or representations from clinical research organizations (“CRO”), hospitals, consultants, and other clinical trial related vendors. We maintain regular communication with our vendors, including our CRO vendors, and gauge the reasonableness of estimates provided. However, actual clinical trial costs may differ from estimated clinical trial costs and are adjusted for in the period in which they become known. There were no material adjustments for a change in estimate to research and development expenses in the accompanying interim unaudited condensed consolidated financial statements for the three and nine months ended January 31, 2013 and 2012.
Share-based Compensation
We account for stock options and other share-based awards granted under our equity compensation plans in accordance with the authoritative guidance for share-based compensation. The estimated fair value of share-based payments to employees in exchange for services is measured at the grant date, using a fair value based method, and is recognized as expense on a straight-line basis over the requisite service periods. Share-based compensation expense recognized during the period is based on the value of the portion of the share-based payment that is ultimately expected to vest during the period.
In addition, we periodically grant stock options and other share-based awards to non-employee consultants, which we account for in accordance with the authoritative guidance for share-based compensation. The cost of non-employee services received in exchange for share-based awards are measured based on either the fair value of the consideration received or the fair value of the share-based award issued, whichever is more reliably measurable. In addition, guidance requires share-based compensation related to unvested options and awards issued to non-employees to be recalculated at the end of each reporting period based upon the fair market value on that date until the share-based award has vested, and any adjustment to share-based compensation resulting from the remeasurement is recognized in the current period. See Note 9 for further discussion regarding share-based compensation.
Basic and Dilutive Net Loss Per Common Share
Basic net loss per common share is computed by dividing our net loss by the weighted average number of common shares outstanding during the period excluding the dilutive effects of stock options, common shares expected to be issued under our employee stock purchase plan, and warrants in accordance with the authoritative guidance. Diluted net loss per common share is computed by dividing the net loss by the sum of the weighted average number of common shares outstanding during the period plus the potential dilutive effects of stock options, common shares expected to be issued under our employee stock purchase plan, and warrants outstanding during the period calculated in accordance with the treasury stock method, but are excluded if their effect is anti-dilutive. Because the impact of options, awards and warrants are anti-dilutive during periods of net loss, there was no difference between basic and diluted loss per share amounts for the three and nine months ended January 31, 2013 and 2012.
| 8 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
The calculation of weighted average diluted shares outstanding excludes the dilutive effect of outstanding stock options, common shares expected to be issued under our employee stock purchase plan, and warrants, to purchase up to an aggregate of 3,387,483 and 3,500,609 shares of common stock for the three months and nine months ended January 31, 2013, respectively, and 50,577 and 87,083 shares of common stock for the three and nine months ended January 31, 2012, respectively, since their impact are anti-dilutive during periods of net loss.
The calculation of weighted average diluted shares outstanding also excludes weighted average outstanding stock options and warrants to purchase up to an aggregate of 6,053,421 and 6,044,158 shares of common stock for the three and nine months ended January 31, 2013, respectively, and 5,971,867 and 6,046,536 shares of common stock for the three and nine months ended January 31, 2012, respectively, as their exercise prices were greater than the average market price of our common stock during the respective periods, resulting in an anti-dilutive effect.
Subsequent to January 31, 2013 and through March 12, 2013, we issued an aggregate of 3,333,556 shares of our common stock (Note 8), which are not included in the calculation of basic and dilutive net loss per common share for the three and nine months ended January 31, 2013.
| 4. | TRADE AND OTHER RECEIVABLES |
Trade and other receivables are recorded at the invoiced amount net of an allowance for doubtful accounts, if necessary. Trade and other receivables, net, consist of the following at January 31, 2013 and April 30, 2012:
| January 31, 2013 | April 30, 2012 | |||||||
| Trade receivables(1) | $ | 1,964,000 | $ | 2,264,000 | ||||
| Other receivables, net | 19,000 | 89,000 | ||||||
| Trade and other receivables, net | $ | 1,983,000 | $ | 2,353,000 | ||||
______________
| (1) | Represents amounts billed for contract manufacturing services provided by Avid. |
We continually monitor our allowance for doubtful accounts for all receivables. We apply judgment in assessing the ultimate realization of our receivables and we estimate an allowance for doubtful accounts based on various factors, such as, the aging of accounts receivable balances, historical experience, and the financial condition of our customers. Based on our analysis, an allowance for doubtful accounts amounted to $19,000 as of January 31, 2013 and April 30, 2012.
| 9 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
| 5. | PROPERTY |
Property, net consists of the following at January 31, 2013 and April 30, 2012:
| January 31, 2013 | April 30, 2012 | |||||||
| Leasehold improvements | $ | 1,383,000 | $ | 1,383,000 | ||||
| Laboratory equipment | 5,339,000 | 4,967,000 | ||||||
| Furniture, fixtures, office equipment and software | 2,553,000 | 2,287,000 | ||||||
| 9,275,000 | 8,637,000 | |||||||
| Less accumulated depreciation and amortization | (6,492,000 | ) | (5,737,000 | ) | ||||
| Property, net | $ | 2,783,000 | $ | 2,900,000 | ||||
Depreciation and amortization expense for three and nine months ended January 31, 2013 was $280,000 and $806,000, respectively, and $238,000 and $664,000 for the three and nine months ended January 31, 2012, respectively.
| 6. | INVENTORIES |
Inventories are stated at the lower of cost or market and primarily include raw materials, direct labor and overhead costs (work-in-process) associated with our wholly-owned subsidiary, Avid. Cost is determined by the first-in, first-out method. Inventories consist of the following at January 31, 2013 and April 30, 2012:
| January 31, 2013 | April 30, 2012 | |||||||
| Raw materials, net | $ | 2,125,000 | $ | 1,966,000 | ||||
| Work-in-process | 2,510,000 | 1,645,000 | ||||||
| Total inventories, net | $ | 4,635,000 | $ | 3,611,000 | ||||
| 7. | NOTE PAYABLE |
On August 30, 2012, we entered into a loan and security agreement (the “Loan Agreement”) with Oxford Finance LLC, MidCap Financial SBIC LP, and Silicon Valley Bank (collectively, the “Lenders”) for up to $30,000,000 in total funding available in two $15,000,000 tranches. The Loan Agreement was secured by a first-priority security interest in substantially all of our assets, excluding our intellectual property and our rights under license agreements granting us rights to intellectual property. On August 30, 2012, we received initial funding of $15,000,000 under the Loan Agreement, excluding debt issuance costs of $251,000.
Subsequently, on September 24, 2012, we received a written notice of default (“Notice of Default”) from the Lenders, with respect to the Loan Agreement. The Notice of Default was triggered by a material adverse change under the Loan Agreement due to our discovery of major discrepancies in treatment group coding by an independent third-party vendor responsible for distribution of blinded investigational product used in our bavituximab Phase II second-line non-small cell lung cancer clinical trial. Pursuant to the terms of the Notice of Default, all amounts due under the Loan Agreement were declared immediately due and payable by the Lenders. On September 25, 2012, we paid the Lenders all obligations declared due and payable under the Loan Agreement, including outstanding principal of $15,000,000, accrued interest thereon at the Loan Agreement’s applicable fixed rate of 7.95% per annum, plus a final payment fee equal to 6.5% of the principal amount funded (or $975,000), upon which, the Loan Agreement was terminated.
In addition, under the Loan Agreement, we issued to the Lenders six-year warrants to purchase shares of our common stock upon the funding of each tranche in an amount equal to 4.50% of the amount of such tranche divided by the exercise price, which is the lower of the average closing price of our common stock for the 10 business days immediately prior to the funding date for such tranche or the closing price on the day prior to such funding date. Therefore, upon the initial funding under the Loan Agreement, we issued the Lenders warrants to purchase an aggregate of 273,280 shares of our common stock at a per share price of $2.47, which are exercisable on a cash or cashless basis, and will expire on August 30, 2018. The fair value of the warrants issued was $470,000 and was calculated using a Black-Scholes valuation model with the following assumptions: risk-free interest rate of 0.87%; expected volatility of 80.20%; expected term of six years; and a dividend yield of 0%. The fair value of the warrants issued was initially recorded as a debt discount with a corresponding increase to additional paid-in capital. As of January 31, 2013, the warrants issued under the Loan Agreement were outstanding and exercisable (Note 10).
| 10 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
Upon the termination of the Loan Agreement, we recorded a loss on the early extinguishment of debt of $1,696,000, which consisted of the final payment fee of $975,000, the unamortized debt discount associated with the fair value of the warrants issued to the Lenders of $470,000, and the unamortized aggregate debt issuance costs of $251,000. The loss on the early extinguishment of debt is included in the accompanying interim unaudited condensed consolidated statements of operations and comprehensive loss for the nine months ended January 31, 2013.
| 8. | STOCKHOLDERS’ EQUITY |
December 2010 AMI Agreement.
On December 29, 2010, we entered into an At Market Sales Issuance Agreement (the “December 2010 AMI Agreement”) with McNicoll, Lewis & Vlak LLC (now known as MLV & Co. LLC, “MLV”), pursuant to which we may sell shares of our common stock at market prices through MLV, as agent, in registered transactions from the Company’s shelf registration statement on Form S-3 (File No. 333-171252) filed with the SEC on December 29, 2010, for aggregate gross proceeds of up to $75,000,000.
During the nine months ended January 31, 2013, we sold 31,662,214 shares of common stock at varying market prices under the December 2010 AMI Agreement for aggregate gross proceeds of $27,051,000 before deducting commissions and other issuance costs of $885,000. As of January 31, 2013, aggregate gross proceeds of up to $330,000 remained available under the December 2010 AMI Agreement.
Subsequent to January 31, 2013 and through March 12, 2013, we sold 201,154 shares of common stock at market prices under the December 2010 AMI Agreement for aggregate gross proceeds of $330,000. As of March 12, 2013, we had raised the full amount of gross proceeds available under the December 2010 AMI Agreement.
December 2012 AMI Agreement
On December 27, 2012, we entered into an At Market Sales Issuance Agreement (the “December 2012 AMI Agreement”) with MLV, pursuant to which we may sell shares of our common stock at market prices through MLV, as agent, for aggregate gross proceeds of up to $75,000,000, in registered transactions from the Company’s shelf registration statement on Form S-3 (File No. 333-180028), filed with the SEC on March 9, 2012. As of January 31, 2013, we had not sold any shares of common stock under the December 2012 AMI Agreement.
Subsequent to January 31, 2013 and through March 12, 2013, we sold 3,132,402 shares of common stock at market prices under the December 2012 AMI Agreement for aggregate gross proceeds of $4,475,000. As of March 12, 2013, aggregate gross proceeds of up to $70,525,000 remained available under the December 2012 AMI Agreement.
Shares of Common Stock Authorized And Reserved For Future Issuance
As of January 31, 2013, we had reserved 24,414,094 additional shares of our common stock, which may be issued under our equity compensation plans and outstanding warrant agreements, excluding shares of common stock that could potentially be issued under our current effective shelf registration statements, as further described in the following table:
| Number of Shares Reserved | ||||
| Common shares reserved for issuance under outstanding option grants and available for issuance under our stock incentive plans | 20,150,287 | |||
| Common shares reserved for and available for issuance under our Employee Stock Purchase Plan | 3,889,004 | |||
| Common shares issuable upon exercise of outstanding warrants | 374,803 | |||
| Total shares of common stock reserved for issuance | 24,414,094 | |||
| 11 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
| 9. | EQUITY COMPENSATION PLANS |
Stock Incentive Plans
We currently maintain seven stock incentive plans referred to as the 2011 Plan, the 2010 Plan, the 2009 Plan, the 2005 Plan, the 2003 Plan, the 2002 Plan, and the 1996 Plan (collectively referred to as the “Stock Plans”). On October 18, 2012, our stockholders approved an amendment to our 2011 Plan to increase the number of shares of our common stock reserved for issuance from 3,500,000 to 11,500,000 shares.
As of January 31, 2013, we had an aggregate of 20,150,287 shares of common stock reserved for issuance under the Stock Plans, of which, 15,487,701 shares were subject to outstanding options and 4,662,586 shares were available for future grants of share-based awards.
The following summarizes our stock option transaction activity for the nine months ended January 31, 2013:
| Stock Options | Shares | Weighted Average Exercisable Price | ||||||
| Outstanding, May 1, 2012 | 7,531,651 | $ | 2.90 | |||||
| Granted | 8,556,862 | $ | 0.87 | |||||
| Exercised | (92,497 | ) | $ | 0.86 | ||||
| Canceled or expired | (508,315 | ) | $ | 1.70 | ||||
| Outstanding, January 31, 2013 | 15,487,701 | $ | 1.83 | |||||
Employee Stock Purchase Plan
We have reserved a total of 5,000,000 shares of common stock to be purchased under our 2010 Employee Stock Purchase Plan (the “2010 ESPP”), of which 3,889,004 shares of common stock remain available for purchase as of January 31, 2013. Under the 2010 ESPP, we will sell shares to participants at a price equal to the lesser of 85% of the fair market value of stock at the (i) beginning of a six-month offering period or (ii) at the end of the six-month offering period. The 2010 ESPP provides for two six-month offering periods each year; the first offering period will begin on the first trading day on or after each November 1; the second offering period will begin on the first trading day on or after each May 1. During the nine months ended January 31, 2013, 548,111 shares of common stock were purchased under the 2010 ESPP at a purchase price per share of $0.39.
Share-Based Compensation
Total share-based compensation expense for the three and nine-month periods ended January 31, 2013 and 2012 are included in the accompanying interim unaudited condensed consolidated statements of operations and comprehensive loss as follows:
| Three Months Ended January 31, | Nine Months Ended January 31, | |||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| Cost of contract manufacturing | $ | 37,000 | $ | 5,000 | $ | 55,000 | $ | 10,000 | ||||||||
| Research and development | 539,000 | 299,000 | 1,151,000 | 920,000 | ||||||||||||
| Selling, general and administrative | 453,000 | 455,000 | 1,114,000 | 1,508,000 | ||||||||||||
| Total | $ | 1,029,000 | $ | 759,000 | $ | 2,320,000 | $ | 2,438,000 | ||||||||
| Share-based compensation from: | ||||||||||||||||
| Stock options | $ | 870,000 | $ | 720,000 | $ | 2,069,000 | $ | 2,356,000 | ||||||||
| Employee stock purchase plan | 159,000 | 39,000 | 251,000 | 82,000 | ||||||||||||
| $ | 1,029,000 | $ | 759,000 | $ | 2,320,000 | $ | 2,438,000 | |||||||||
As of January 31, 2013, the total estimated unrecognized compensation cost related to non-vested stock options was $6,045,000. This cost is expected to be recognized over a weighted average vesting period of 1.64 years based on current assumptions.
| 12 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
| 10. | WARRANTS |
During the quarter ended January 31, 2013, 118,444 warrants were exercised on a cashless basis in exchange for 46,427 shares of our common stock. The warrants were issued in December 2008 in connection with a three-year term loan we entered into during fiscal year 2009.
As of January 31, 2013, the following warrants to purchase an aggregate of 374,803 shares of our common stock were outstanding:
| Date Issued | Warrants Outstanding | Exercise Price Per Share | Expiration Date | |||
| December 19, 2008 | 101,523 | $1.4775 | December 19, 2013 | |||
| August 30, 2012 (Note 7) | 273,280 | $2.4700 | August 30, 2018 | |||
| Total Warrants Outstanding | 374,803 |
| 11. | SEGMENT REPORTING |
Our business is organized into two reportable operating segments and both operate in the U.S. Peregrine is engaged in the research and development of monoclonal antibodies for the treatment and diagnosis of cancer. Avid is engaged in providing contract manufacturing services for Peregrine, and third-party customers on a fee-for-service basis.
The accounting policies of the operating segments are the same as those described in Note 3. We evaluate the performance of our contract manufacturing services segment based on gross profit or loss from third-party customers. However, our products in the research and development segment are not evaluated based on gross profit or loss, but rather based on scientific progress of the technologies. As such, gross profit or loss is only provided for our contract manufacturing services segment in the below table. All revenues shown below are derived from transactions with third-party customers.
Segment information for the three and nine-month periods is summarized as follows:
| Three Months Ended January 31, | Nine Months Ended January 31, | |||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| Contract manufacturing services revenue | $ | 6,961,000 | $ | 3,203,000 | $ | 17,157,000 | $ | 12,796,000 | ||||||||
| Cost of contract manufacturing services | 3,651,000 | 2,484,000 | 9,378,000 | 9,219,000 | ||||||||||||
| Gross profit | 3,310,000 | 719,000 | 7,779,000 | 3,577,000 | ||||||||||||
| Revenue from products in research and development | 78,000 | 78,000 | 272,000 | 372,000 | ||||||||||||
| Research and development expense | (5,437,000 | ) | (9,180,000 | ) | (18,471,000 | ) | (26,758,000 | ) | ||||||||
| Selling, general and administrative expense | (3,112,000 | ) | (2,710,000 | ) | (9,469,000 | ) | (8,371,000 | ) | ||||||||
| Other income (expense), net | 247,000 | 3,000 | 254,000 | (57,000 | ) | |||||||||||
| Loss on early extinguishment of debt | – | – | (1,696,000 | ) | – | |||||||||||
| Net loss | $ | (4,914,000 | ) | $ | (11,090,000 | ) | $ | (21,331,000 | ) | $ | (31,237,000 | ) | ||||
| 13 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES
TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
FOR THE NINE MONTHS ENDED JANUARY 31, 2013 (unaudited) (continued)
Revenues generated from our contract manufacturing services segment were derived from a limited number of customers. The percentages below represent revenue derived from each customer as a percentage of total contract manufacturing services revenue:
| Three Months Ended January 31, | Nine Months Ended January 31, | |||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| United States (customer A) | 60% | 58% | 78% | 36% | ||||||||||||
| United States (customer B) | 37 | – | 20 | – | ||||||||||||
| United States (customer C) | – | 31 | – | 8 | ||||||||||||
| Germany (one customer) | – | 5 | – | 20 | ||||||||||||
| Denmark (one customer) | – | 4 | – | 28 | ||||||||||||
| Other customers | 3 | 2 | 2 | 8 | ||||||||||||
| Total | 100% | 100% | 100% | 100% | ||||||||||||
Revenue generated from our products in our research and development segment during the three and nine months ended January 31, 2013 and 2012, is directly related to license revenue recognized under licensing agreements with unrelated entities.
| 12. | COMMITMENTS AND CONTINGENCIES |
In the ordinary course of business, we are at times subject to various legal proceedings and disputes. Except as set forth below, we currently are not aware of any material litigation or other dispute nor, to management’s knowledge, is any litigation or other proceeding threatened against us that collectively is expected to have a material adverse effect on our consolidated cash flows, financial condition or results of operations.
Securities Related Class Action Lawsuits
On September 28, 2012, three complaints were filed in the U.S. District Court for the Central District of California (the “Court”) against us and certain of our executive officers and one consultant (collectively, the “Individual Defendants”) on behalf of certain purchasers of our common stock. The complaints have been brought as purported stockholder class actions, and, in general, include allegations that we and the Individual Defendants violated (i) Section 10(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Rule 10b-5 promulgated thereunder and (ii) Section 20(a) of the Exchange Act, by making materially false and misleading statements regarding the interim median overall survival results of our bavituximab Phase II second-line non-small cell lung cancer trial, thereby artificially inflating the price of our common stock. The plaintiffs are seeking unspecified monetary damages and other relief. On November 27, 2012, four prospective lead plaintiffs filed motions to consolidate, appoint a lead plaintiff, and appoint lead counsel. On February 5, 2013, the court appointed James T. Fahey as lead plaintiff in the action. The lead plaintiff has until April 8, 2013 to file an amended consolidated complaint against us. We believe that the various shareholder lawsuits are without merit, and we intend to vigorously defend the various actions and to seek dismissal of these complaints. Due to the early stage of these proceedings, we believe that the probability of an unfavorable outcome or loss related to these proceedings and an estimate of the amount or range of loss related to these claims, if any, from an unfavorable outcome are not determinable at this time.
Other Legal Matters
On September 24, 2012, we filed a lawsuit against Clinical Supplies Management, Inc. (“CSM”), in the U.S. District Court for the Central District of California (the “Court”). We had contracted with CSM in 2010 as our third-party vendor responsible for distribution of the blinded investigational product used in our bavituximab Phase II second-line non-small cell lung cancer trial. As part of the routine collection of data in advance of an end-of-Phase II meeting with regulatory authorities, we discovered major discrepancies between some patient sample test results and patient treatment code assignments. Consequently, we filed this lawsuit against CSM alleging breach of contract, negligence and negligence per se arising from CSM’s performance of its contracted services. We are seeking monetary damages. On March 7, 2013, we and CSM submitted to the Court a proposed stipulation pursuant to which the lawsuit would be stayed for up to 120 days during which time we and CSM would participate in an alternative dispute resolution process, pursuant to our contract with CSM. The proposed stipulation was approved by the Court on March 8, 2013.
| 14 |
| Item 2. | Management’s Discussion and Analysis of Financial Condition And Results of Operations. |
This Quarterly Report on Form 10-Q contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which represent our projections, estimates, expectations or beliefs concerning among other things, financial items that relate to management’s future plans or objectives or to our future economic and financial performance. In some cases, you can identify these statements by terminology such as “may”, “should”, “plans”, “believe”, “will”, “anticipate”, “estimate”, “expect” “project”, or “intend”, including their opposites or similar phrases or expressions. You should be aware that these statements are projections or estimates as to future events and are subject to a number of factors that may tend to influence the accuracy of the statements. These forward-looking statements should not be regarded as a representation by the Company or any other person that the events or plans of the Company will be achieved. You should not unduly rely on these forward-looking statements, which speak only as of the date of this Quarterly Report. We undertake no obligation to publicly revise any forward-looking statement to reflect circumstances or events after the date of this Quarterly Report or to reflect the occurrence of unanticipated events. You should, however, review the factors and risks we describe in the reports we file from time to time with the Securities and Exchange Commission (“SEC”) after the date of this Quarterly Report. Actual results may differ materially from any forward-looking statement.
Overview
We are a biopharmaceutical company developing first-in-class monoclonal antibodies focused on the treatment and diagnosis of cancer. We are advancing two oncology programs with our lead product candidates, bavituximab and Cotara, for the treatment of various cancers. In addition, we are advancing our lead imaging agent, 124I-PGN650, in an exploratory clinical trial for the imaging of multiple solid tumor types.
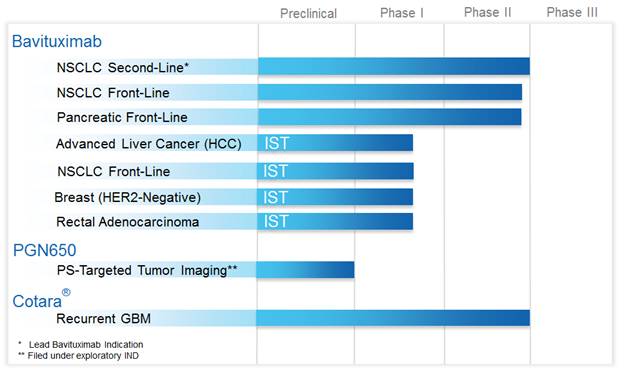
The following product pipeline reflects our current clinical trials focused on oncology, as further discussed below:
| 15 |
Bavituximab for the Treatment of Solid Tumors
Bavituximab is our lead phosphatidylserine (“PS”)-targeting antibody that has demonstrated broad therapeutic potential in combination with chemotherapy across multiple oncology indications and represents a new approach to treating cancer. PS is a highly immunosuppressive molecule usually located inside the membrane surface of healthy cells, but “flips” and becomes exposed on the outside of cells that line tumor blood vessels, creating a specific target for anti-cancer treatments. PS-targeting antibodies target and bind to PS and block this immunosuppressive signal, thereby enabling the immune system to recognize and fight the tumor.
As reflected in the above product pipeline, bavituximab’s therapeutic potential is currently being evaluated in three company-sponsored Phase II randomized trials in second-line non-small cell lung cancer (“NSCLC”), front-line NSCLC, and front-line pancreatic cancer, as well as in several investigator-sponsored trials (“IST”) in additional oncology indications. The following represents the current status of each of these clinical trials:
Phase IIb Trial – Bavituximab Plus Docetaxel in Second-Line NSCLC
We conducted a randomized, double-blind, placebo-controlled Phase IIb second-line NSCLC trial evaluating two dose levels of bavituximab plus docetaxel (“bavituximab-containing arms”) versus docetaxel plus placebo (“control arm”) as second-line treatment in 121 patients with Stage IIIb or Stage IV NSCLC. The trial was designed to evaluate overall response rate (“ORR”) measured in accordance with Response Evaluation Criteria In Solid Tumors (“RECIST”) criteria, progression-free survival (“PFS”), duration of response, overall survival (“OS”), and safety.
On September 24, 2012, we announced that during the course of preparing for an end-of-Phase II meeting with regulatory authorities and following the data announcement on September 7, 2012 from this Phase IIb trial, we discovered major discrepancies between some patient sample test results and patient treatment code assignments. As a result of these discrepancies, the data that we disclosed on or before September 7, 2012 should not be relied upon.
Upon discovery of the discrepancies, we initiated an internal review of this Phase IIb trial, which included the testing of investigational product, patient samples, reviewing the operations of multiple vendors, among other activities. The initial results of this internal review were announced on January 7, 2013 and indicated that discrepancies were isolated to the control arm and 1mg/kg bavituximab-containing treatment arms of the trial and that there was no evidence of discrepancies in the 3mg/kg bavituximab-containing treatment arm of the trial. Based on the results of our internal review, we took a conservative approach toward analyzing the results from the trial, which included combining the control arm and 1mg/kg bavituximab-containing arm into one treatment arm (“combined control arm”), and comparing those results to the 3mg/kg bavituximab-containing treatment arm.
On February 19, 2013, we reported updated top-line data from this trial based upon the completion of the aforementioned internal review of discrepancies in the trial and updated patient survival data from the trial. Updated top-line data from this Phase IIb trial indicate a meaningful improvement in median OS of 11.7 months in the 3mg/kg bavituximab-containing arm compared to 7.3 months in the combined control arm. In addition, these results also demonstrated that bavituximab was well-tolerated with no significant differences in adverse events between the trial arms. We believe these updated top-line data support advancing the 3mg/kg bavituximab dose into Phase III development in second-line NSCLC, and therefore, we are actively preparing for an end-of-Phase II meeting with the United States Food and Drug Administration (“FDA”) which we planning to have by the end of the first half of calendar year 2013.
Phase IIb Trial – Bavituximab Plus Paclitaxel/Carboplatin in Front-Line NSCLC
Our randomized Phase II trial is designed to evaluate bavituximab plus carboplatin and paclitaxel versus carboplatin and paclitaxel alone as front-line therapy in 86 patients with Stage IIIb or Stage IV NSCLC. In March 2012, we announced top-line ORR, a primary endpoint, and current median PFS, a secondary endpoint, from this trial from 83 evaluable patients. Initial ORR and median PFS data from this study were deemed inconclusive and therefore, we believe median OS, another secondary endpoint, will be an important data point from this study and instrumental in determining our next steps in advancing bavituximab in front-line NSCLC in combination with carboplatin and paclitaxel. We anticipate announcing median OS from this trial in the first half of calendar year 2013.
| 16 |
Phase II Trial – Bavituximab Plus Gemcitabine in Pancreatic Cancer
In June 2012, we announced the completion of patient enrollment in our Phase II randomized trial evaluating bavituximab in combination with gemcitabine versus gemcitabine alone in 70 patients with previously untreated pancreatic cancer patients. The primary endpoint from this trial is median OS and the secondary endpoints are ORR and median PFS. On February 13, 2013, we announced top-line results from this trial showing that the combination of bavituximab and gemcitabine resulted in more than a doubling of ORR and an improvement in OS when compared to gemcitabine alone (“control arm”). In the trial, patients treated with a combination of bavituximab and gemcitabine had a 28% tumor response rate as compared to 13% in the control arm. In addition, we saw a modest improvement in median OS, the primary endpoint of the trial, which was 5.6 months for the bavituximab plus gemcitabine arm and 5.2 months for the control arm.
Based on these data, as well as other recent developments in the treatment of pancreatic cancer, we are actively evaluating the next steps for advancing our bavituximab pancreatic program.
Investigator-Sponsored Trials (“IST”)
With respect to our ISTs, our clinical collaborators are evaluating bavituximab with additional drug combinations and additional oncology indications. Enrollment is ongoing in each of the following ISTs:
| (i) | a Phase I/II trial evaluating bavituximab combined with sorafenib in patients with advanced hepatocellular carcinoma (“HCC”), or liver cancer; |
| (ii) | a Phase Ib trial evaluating bavituximab combined with pemetrexed and carboplatin in patients with front-line NSCLC; |
| (iii) | a Phase I trial evaluating bavituximab combined with paclitaxel in patients with HER2-negative metastatic breast cancer; and |
| (iv) | a Phase I trial evaluating bavituximab combined with capecitabine and radiation in patients with stage II or III rectal adenocarcinoma. |
In addition, we periodically evaluate our IST program based on a number of factors, including enrollment and changes in the standard of care of patients for each of our ongoing IST’s. As a result of our recent evaluation, during March 2013, we discontinued a Phase I/II IST evaluating bavituximab combined with cabazitaxel in patients with second-line castration resistant prostate cancer due to slow enrollment in the trial which we believe will continue due to two new oral drugs that had been approved for the same indication following the inception of this IST. We will continue to monitor our IST program as we look to evaluate new indications and combinations based on the broad therapeutic potential of bavituximab.
PS-Targeting Molecular Imaging Program (PGN650)
In addition to bavituximab’s therapeutic potential to treat multiple solid tumors, we believe these PS-targeting antibodies may have broad potential for the imaging and diagnosis of multiple diseases, including cancer. In April 2012, we filed an exploratory Investigational New Drug Application with the FDA to advance our lead molecular imaging agent, 124I-PGN650 (“PGN650”), into clinical development for the imaging of multiple solid tumor types. Our initial goal for the PGN650 program is to further validate the broad nature of the PS-targeting platform. The current trial will enroll up to 12 patients and results from this study may provide new insight into new indications and potential applications, including development of antibody drug conjugates, the ability of PGN650 to monitor the effectiveness of current standard cancer treatments, and the ability to potentially select patients that may benefit from bavituximab-based treatment.
Cotara for the Treatment of Brain Cancer
Cotara is a monoclonal antibody linked to a radioisotope that is administered as a single one-time infusion, directly into the tumor, destroying the tumor from the inside out, with minimal exposure to healthy tissue. In calendar year 2011, we reported what we believe to be promising median OS of 9.3 months in patients with glioblastoma multiforme (“GBM”) at first relapse following a single dose of Cotara in a Phase II clinical trial. Based on these data and data from earlier clinical studies, we have reached an agreement with the FDA on the design of a single pivotal trial to potentially support product registration for Cotara in the treatment of recurrent GBM. We are actively seeking potential partners as we look to further advance Cotara into Phase III development. Cotara has been granted orphan drug status and fast track designation for the treatment of GBM and anaplastic astrocytoma by the FDA.
Integrated Biomanufacturing Subsidiary
In addition to our clinical research and development efforts, we operate a wholly-owned cGMP (current Good Manufacturing Practices) contract manufacturing subsidiary, Avid Bioservices, Inc. (“Avid”). Avid is a Contract Manufacturing Organization that provides fully integrated services from cell line development to commercial cGMP biomanufacturing for Peregrine and Avid’s third-party clients. In addition to generating revenue from providing a broad range of biomanufacturing services to third-party clients, Avid is strategically integrated with Peregrine to manufacture all clinical products to support our clinical trials while also preparing for Phase III and potential commercial launch.
| 17 |
Going Concern
Our interim unaudited condensed consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The financial statements do not include any adjustments relating to the recoverability of the recorded assets or the classification of liabilities that may be necessary should it be determined that we are unable to continue as a going concern.
At January 31, 2013, we had $26,255,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced negative cash flows from operations since our inception and we expect the negative cash flows from operations to continue for the foreseeable future. Our net loss incurred during the nine-month period ended January 31, 2013 amounted to $21,331,000 and our net losses incurred during the past three fiscal years ended April 30, 2012, 2011 and 2010, amounted to $42,119,000, $34,151,000, and $14,494,000, respectively. Therefore, unless and until we are able to generate sufficient revenues from Avid’s contract manufacturing services and/or from the sale and/or licensing of our products under development, we expect such losses to continue for the foreseeable future.
Therefore, our ability to continue to fund our clinical trials and development efforts is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, issuing additional equity or debt.
Historically, we have funded a significant portion of our operations through the issuance of equity. During the nine months ended January 31, 2013, we raised $27,051,000 in gross proceeds under an At Market Sales Issuance Agreement, of which $25,555,000 was raised subsequent to September 25, 2012, primarily to replace the $15,000,000 of initial funding we repaid on September 25, 2012 under an earlier loan facility we entered into on August 30, 2012. Subsequent to January 31, 2013 and through March 12, 2013, we raised an additional $4,805,000 in aggregate gross proceeds under two separate At Market Sales Issuance Agreements.
With respect to our ability to raise additional capital from the issuance of equity, as of March 12, 2013, we have an effective shelf registration statement on Form S-3, under which we may issue, from time to time, in one or more offerings, shares of our common stock for gross proceeds of up to $145,525,000. However, our ability to raise additional capital in the equity markets is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse clinical trials results, and significant delays in one or more clinical trials. If our ability to access the capital markets becomes severely restricted, it could have a negative impact on our business plans, including our clinical trial programs and other research and development activities. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
In addition to financing our operations through the issuance of equity, we may also secure additional funding through the issuance of debt, licensing or partnering our products in development, or increasing revenue from our wholly-owned subsidiary, Avid. While we will continue to explore these potential opportunities, there can be no assurances that we will be successful in securing debt financing, licensing or partnering our products in development, or generating additional revenue from Avid to complete the research, development, and clinical testing of our product candidates.
Based on our current projections, which include projected cash inflows under signed contracts with existing customers of Avid, assuming we raise no additional capital from the capital markets or other potential sources, and excluding any third-party Phase III development costs, we believe we will have sufficient cash on hand to meet our obligations as they become due through at least the third quarter of our fiscal year 2014 ending January 31, 2014. There are a number of uncertainties associated with our financial projections, including but not limited to, termination of third party contracts, technical challenges, and regulatory decisions affecting bavituximab, any of which could reduce, delay or accelerate our future projected cash inflows and outflows. In addition, in the event our projected cash-inflows are reduced or delayed we might not have sufficient capital to operate our business beyond the third quarter of our fiscal year 2014. The uncertainties surrounding our future cash inflows have raised substantial doubt regarding our ability to continue as a going concern.
| 18 |
Results of Operations
The following table compares the interim unaudited condensed consolidated statements of operations for the three and nine-month periods ended January 31, 2013 and 2012. This table provides you with an overview of the changes in the condensed consolidated statements of operations for the comparative periods, which are further discussed below.
| Three Months Ended January 31, | Nine Months Ended January 31, | |||||||||||||||||||||||
| 2013 | 2012 | $ Change | 2013 | 2012 | $ Change | |||||||||||||||||||
| REVENUES: | ||||||||||||||||||||||||
| Contract manufacturing revenue | $ | 6,961,000 | $ | 3,203,000 | $ | 3,758,000 | $ | 17,157,000 | $ | 12,796,000 | $ | 4,361,000 | ||||||||||||
| License revenue | 78,000 | 78,000 | – | 272,000 | 372,000 | (100,000 | ) | |||||||||||||||||
| Total revenues | 7,039,000 | 3,281,000 | 3,758,000 | 17,429,000 | 13,168,000 | 4,261,000 | ||||||||||||||||||
| COSTS AND EXPENSES: | ||||||||||||||||||||||||
| Cost of contract manufacturing | 3,651,000 | 2,484,000 | 1,167,000 | 9,378,000 | 9,219,000 | 159,000 | ||||||||||||||||||
| Research and development | 5,437,000 | 9,180,000 | (3,743,000 | ) | 18,471,000 | 26,758,000 | (8,287,000 | ) | ||||||||||||||||
| Selling, general & administrative | 3,112,000 | 2,710,000 | 402,000 | 9,469,000 | 8,371,000 | 1,098,000 | ||||||||||||||||||
| Total costs and expenses | 12,200,000 | 14,374,000 | (2,174,000 | ) | 37,318,000 | 44,348,000 | (7,030,000 | ) | ||||||||||||||||
| LOSS FROM OPERATIONS | (5,161,000 | ) | (11,093,000 | ) | 5,932,000 | (19,889,000 | ) | (31,180,000 | ) | 11,291,000 | ||||||||||||||
| OTHER INCOME (EXPENSE): | ||||||||||||||||||||||||
| Interest and other income | 255,000 | 9,000 | 246,000 | 307,000 | 31,000 | 276,000 | ||||||||||||||||||
| Interest and other expense | (8,000 | ) | (6,000 | ) | (2,000 | ) | (53,000 | ) | (88,000 | ) | 35,000 | |||||||||||||
| Loss on the early extinguishment of debt | – | – | – | (1,696,000 | ) | – | (1,696,000 | ) | ||||||||||||||||
| NET LOSS | $ | (4,914,000 | ) | $ | (11,090,000 | ) | $ | 6,176,000 | $ | (21,331,000 | ) | $ | (31,237,000 | ) | $ | 9,906,000 | ||||||||
Results of operations for interim periods covered by this quarterly report on Form 10-Q may not necessarily be indicative of results of operations for the full fiscal year.
Total Revenues
Three and Nine Months: The increases in total revenues of $3,758,000 (115%) and $4,261,000 (32%) during the three and nine months ended January 31, 2013, respectively, compared to the same periods in the prior year were due to current year three and nine-month period increases in contract manufacturing revenue of $3,758,000 and $4,361,000, respectively, offset by a current year nine-month period decrease in license revenue of $100,000. The increases in contract manufacturing revenue were primarily due to a greater number of completed manufacturing runs released and shipped in the current year periods compared to the same prior year periods, which can primarily be attributed to an increase in the demand for Avid’s manufacturing services.
Based on the current commitments for manufacturing services from Avid’s third-party customers and the anticipated completion of in-process third-party customer manufacturing runs, we expect contract manufacturing revenue to exceed $20 million for fiscal year 2013. In addition, although we expect to continue to recognize license revenue during the remainder of fiscal year 2013, we do not expect license revenue to be significant based on current agreements.
| 19 |
Cost of Contract Manufacturing
Three and Nine Months: The increases in cost of contract manufacturing of $1,167,000 (47%) and $159,000 (2%) during the three and nine months ended January 31, 2013, respectively, compared to the same periods in the prior year were primarily due to current year three and nine-month period increases in contract manufacturing revenue, which were offset by current year improvements in the cost of contract manufacturing as a percentage of contract manufacturing revenue. During the current year three and nine-month periods, the cost of contract manufacturing as a percentage of contract manufacturing revenue improved to 52% and 55%, respectively, compared to 78% and 72%, for the prior year three and nine-month periods, respectively. The current year three and nine-month period improvements in cost of contract manufacturing as a percentage of revenue were primarily the result of the mix of services provided and the gross margins associated with these services provided during the current year periods.
Research and Development Expenses
Research and development expenses primarily include (i) payroll and related costs associated with research and development personnel, (ii) costs related to clinical and preclinical testing of our technologies under development, (iii) costs to develop and manufacture the product candidates, including raw materials and supplies, product testing, depreciation, and facility related expenses, (iv) expenses for research services provided by universities and contract laboratories, including sponsored research funding, and (v) other research and development expenses.
Three and Nine Months: The decreases in research and development (“R&D”) expenses of $3,743,000 (41%) and $8,287,000 (31%) during the three and nine months ended January 31, 2013, respectively, compared to the same periods in the prior year were due to the following changes associated with each of our following technologies under development:
| Technology Platform | R&D Expenses – Three Months Ended January 31, | R&D Expenses – Nine Months Ended January 31, | ||||||||||||||||||||||
| 2013 | 2012 | $ Change | 2013 | 2012 | $ Change | |||||||||||||||||||
| PS-Targeting (bavituximab and PGN650) | $ | 4,506,000 | $ | 7,904,000 | $ | (3,398,000 | ) | $ | 17,027,000 | $ | 23,749,000 | $ | (6,722,000 | ) | ||||||||||
| Cotara® | 931,000 | 1,276,000 | (345,000 | ) | 1,444,000 | 3,009,000 | (1,565,000 | ) | ||||||||||||||||
| Total R&D Expenses | $ | 5,437,000 | $ | 9,180,000 | $ | (3,743,000 | ) | $ | 18,471,000 | $ | 26,758,000 | $ | (8,287,000 | ) | ||||||||||
| o | PS-Targeting (bavituximab and PGN650) – The decreases in PS-targeting program expenses of $3,398,000 and $6,722,000 during the three and nine months ended January 31, 2013, respectively, compared to the same prior year periods were primarily due to decreases in third-party vendor costs regarding our three separate company-sponsored Phase II trials using bavituximab in combination with chemotherapy for the treatment of patients with (i) front-line non-small cell lung cancer (“NSCLC”), (ii) second-line NSCLC, and (iii) pancreatic cancer, as the majority of patients in these trials were enrolled prior to May 1, 2012. In addition, the current year three and nine-month period decreases were supplemented with decreases in costs associated with a prior completed Phase II trial using bavituximab for the treatment of patients with previously untreated genotype-1 hepatitis C virus (HCV) infection that completed enrollment in September 2011. These decreases in clinical trial expenses were further supplemented with a decrease in manufacturing costs incurred in the prior year associated with preparing bavituximab for potential later-stage clinical trials combined with a decrease in sponsored research fees associated with our preclinical anti-viral program. These decreases in PS-targeting program expenses were offset by increases in payroll and related expenses associated with our lead PS-targeting molecular imaging agent, PGN650. |
| o | Cotara – The decreases in Cotara related expenses of $345,000 and $1,565,000 during the three and nine months ended January 31, 2013, respectively, compared to the same prior year periods were primarily related to increased development and manufacturing costs incurred in the prior year periods associated with preparing Cotara for potential later-stage clinical trials for the treatment of recurrent glioblastoma multiforme (“GBM”). These current period decreases were further supplemented by decreases in expenses associated with our Phase II trial for GBM (or brain cancer), which trial completed patient enrollment in December 2010. |
| 20 |
During the remainder of the current fiscal year, we expect to continue to direct the majority of our research and development expenses towards our PS-targeting technology platform although it is extremely difficult for us to reasonably estimate all future research and development costs associated with each of our technologies due to the number of unknowns and uncertainties associated with preclinical and clinical trial development. These unknown variables and uncertainties include, but are not limited to:
| · | the uncertainty of future clinical trial results; |
| · | the uncertainty of the ultimate number of patients to be treated in any current or future clinical trial; |
| · | the uncertainty of the U.S. Food and Drug Administration allowing our studies to move forward from Phase II clinical studies to Phase III clinical studies; |
| · | the uncertainty of the rate at which patients are enrolled into any current or future study. Any delays in clinical trials could significantly increase the cost of the study and would extend the estimated completion dates; |
| · | the uncertainty of terms related to potential future partnering or licensing arrangements; |
| · | the uncertainty of protocol changes and modifications in the design of our clinical trial studies, which may increase or decrease our future costs; and |
| · | the uncertainty of our ability to raise additional capital to support our future research and development efforts. |
We or our potential partners will need to do additional development and clinical testing prior to seeking any regulatory approval for commercialization of our product candidates as all of our products are in discovery, preclinical or clinical development. Testing, manufacturing, commercialization, advertising, promotion, exporting, and marketing, among other things, of our proposed products are subject to extensive regulation by governmental authorities in the United States and other countries. The testing and approval process requires substantial time, effort, and financial resources, and we cannot guarantee that any approval will be granted on a timely basis, if at all. Companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in conducting advanced human clinical trials, even after obtaining promising results in earlier trials. Furthermore, the United States Food and Drug Administration may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Even if regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which it may be marketed. Accordingly, we or our potential partners may experience difficulties and delays in obtaining necessary governmental clearances and approvals to market our products.
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist primarily of payroll and related expenses, share-based compensation expense, director fees, legal fees, audit and accounting fees, patent fees, investor and public relation fees, insurance, and other expenses relating to the general management, administration, and business development activities of the Company.
Three Months: The increase in selling, general and administrative expenses of $402,000 (15%) during the three months ended January 31, 2013, compared to the same period in the prior year was primarily due to increases in payroll and related expenses and corporate legal fees of $376,000 and $124,000, respectively. The increase in payroll and related expenses is attributed to increases in compensation and other employee-related benefits and the increase in corporate legal fees is primarily attributable to the lawsuits described in this Quarterly Report on Form 10-Q under Part II, Item 1, “Legal Proceedings”. These increases in selling, general and administrative expenses were offset with incremental current year three-month period decreases in insurance and other corporate related expenses.
Nine Months: The increase in selling, general and administrative expenses of $1,098,000 (13%) during the nine months ended January 31, 2013, compared to the same period in the prior year was primarily due to increases in payroll and related expenses and corporate legal fees of $1,009,000 and $214,000, respectively. The increase in payroll and related expenses is attributed to increases in compensation and other employee-related benefits and the increase in corporate legal fees is primarily attributable to the lawsuits described in this Quarterly Report on Form 10-Q under Part II, Item 1, “Legal Proceedings”. These increases in selling, general and administrative expenses were further supplemented with incremental current year nine-month period increases in audit and accounting fees, market research fees, annual shareholder meeting expenses, and other corporate related expenses. These increases in selling, general and administrative expenses were offset with a current year nine-month period decrease in share-based compensation expense (non-cash) of $394,000 associated with the amortization of the fair value of options granted.
Interest and Other Income
Three and Nine Months: The increase in interest and other income of $246,000 and $276,000 during the three and nine months ended January 31, 2013, respectively, compared to the same periods in the prior year were due to current year three and nine-month period increases in interest income of $16,000 and $47,000, respectively, and a current year three and nine-month period increase in other income of $230,000.
| 21 |
Loss on Early Extinguishment of Debt
Nine Months: The increase in loss on early extinguishment of debt of $1,696,000 during the nine months ended January 31, 2013, compared to the same period in the prior year was directly related to the term loan we entered into during the quarter ended October 31, 2012 that was subsequently repaid in full and terminated in the same quarter under an event of default (as described in Note 7 to the accompanying interim unaudited condensed consolidated financial statements). Upon the termination of the term loan, we recorded a loss on the early extinguishment of debt of $1,696,000, which consisted of a final payment fee of $975,000, the unamortized debt discount associated with the fair value of the warrants issued to the lenders under the term loan of $470,000, and unamortized aggregate debt issuance costs of $251,000. The loss on the early extinguishment of debt is included in the accompanying interim unaudited condensed consolidated statements of operations and comprehensive loss for the nine months ended January 31, 2013.
Critical Accounting Policies and Estimates
Our discussion and analysis of our consolidated financial position and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of our consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses, and related disclosures. We review our estimates and assumptions on an ongoing basis. We base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for our judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what we anticipate and different assumptions or estimates about the future could change our reported results. During the nine months ended January 31, 2013, there were no significant changes in our critical accounting policies as previously disclosed by us in Part II, Item 7 of our Annual Report for the fiscal year ended April 30, 2012.
Liquidity and Capital Resources
At January 31, 2013, we had $26,255,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced negative cash flows from operations since our inception and we expect the negative cash flows from operations to continue for the foreseeable future. Our net loss incurred during the nine-month period ended January 31, 2013, amounted to $21,331,000 and our net losses incurred during the past three fiscal years ended April 30, 2012, 2011 and 2010, amounted to $42,119,000, $34,151,000, and $14,494,000, respectively. Therefore, unless and until we are able to generate sufficient revenues from Avid’s contract manufacturing services and/or from the sale and/or licensing of our products under development, we expect such losses to continue for the foreseeable future.
Therefore, our ability to continue to fund our clinical trials and development efforts is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, issuing additional equity or debt.
Historically, we have funded a significant portion of our operations through the issuance of equity. During the nine months ended January 31, 2013, we raised $27,051,000 in gross proceeds under an At Market Sales Issuance Agreement, of which $25,555,000 was raised subsequent to September 25, 2012, primarily to replace the $15,000,000 of initial funding we repaid on September 25, 2012 under an earlier loan facility we entered into on August 30, 2012. Subsequent to January 31, 2013 and through March 12, 2013, we raised an additional $4,805,000 in aggregate gross proceeds under two separate At Market Sales Issuance Agreements.
With respect to our ability to raise additional capital from the issuance of equity, as of March 12, 2013, we have an effective shelf registration statement on Form S-3, under which we may issue, from time to time, in one or more offerings, shares of our common stock for gross proceeds of up to $145,525,000. However, our ability to raise additional capital in the equity markets is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse clinical trials results, and significant delays in one or more clinical trials. If our ability to access the capital markets becomes severely restricted, it could have a negative impact on our business plans, including our clinical trial programs and other research and development activities. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
| 22 |
In addition to financing our operations through the issuance of equity, we may also secure additional funding through the issuance of debt, licensing or partnering our products in development, or increasing revenue from our wholly-owned subsidiary, Avid. While we will continue to explore these potential opportunities, there can be no assurances that we will be successful in securing debt financing, licensing or partnering our products in development, or generating additional revenue from Avid to complete the research, development, and clinical testing of our product candidates.
Based on our current projections, which include projected cash inflows under signed contracts with existing customers of Avid, assuming we raise no additional capital from the capital markets or other potential sources, and excluding any third-party Phase III development costs, we believe we will have sufficient cash on hand to meet our obligations as they become due through at least the third quarter of our fiscal year 2014 ending January 31, 2014. There are a number of uncertainties associated with our financial projections, including but not limited to, termination of third party contracts, technical challenges, and regulatory decisions affecting bavituximab, any of which could reduce, delay or accelerate our future projected cash inflows and outflows. In addition, in the event our projected cash-inflows are reduced or delayed we might not have sufficient capital to operate our business beyond the third quarter of our fiscal year 2014. The uncertainties surrounding our future cash inflows have raised substantial doubt regarding our ability to continue as a going concern.
Significant components of the changes in cash flows from operating, investing, and financing activities for the nine months ended January 31, 2013 compared to the same prior year period are as follows:
Cash Used In Operating Activities. Net cash used in operating activities represents our (i) net loss, as reported, (ii) less non-cash operating expenses, and (iii) net changes in the timing of cash flows as reflected by the changes in operating assets and liabilities, as described in the below table:
| NINE MONTHS ENDED | ||||||||
| January 31, 2013 | January 31, 2012 | |||||||
| Net loss, as reported | $ | (21,331,000 | ) | $ | (31,237,000 | ) | ||
| Less non-cash expenses and adjustments to net loss: | ||||||||
| Share-based compensation | 2,320,000 | 2,438,000 | ||||||
| Depreciation and amortization | 806,000 | 664,000 | ||||||
| Loss on early extinguishment of debt | 1,696,000 | – | ||||||
| Amortization of discount on notes payable and debt issuance costs | – | 33,000 | ||||||
| Loss on disposal of property | 8,000 | 2,000 | ||||||
| Net cash used in operating activities before changes in operating assets and liabilities | $ | (16,501,000 | ) | $ | (28,100,000 | ) | ||
| Net change in operating assets and liabilities | $ | 291,000 | $ | 958,000 | ||||
| Net cash used in operating activities | $ | (16,210,000 | ) | $ | (27,142,000 | ) | ||
Net cash used in operating activities decreased $10,932,000 to $16,210,000 for the nine months ended January 31, 2013, compared to net cash used in operating activities of $27,142,000 for the nine months ended January 31, 2012. This decrease in net cash used in operating activities was due to a decrease of $11,599,000 in net loss reported during the current nine-month period after taking into consideration non-cash operating expenses offset with a net change in operating assets and liabilities of $667,000. The net change in operating assets and liabilities was primarily due to decreases in accounts payable and accrued clinical trial and related fees combined with an increase in inventories, which were offset by increases in customer deposits and deferred revenue associated with payments received from Avid’s third-party customers. The decrease in our current nine-month net loss was primarily due to a current period increase in total revenues combined with a current period decrease in research and development expenses, offset by current period increases in cost of contract manufacturing, selling, general and administrative expenses and loss on early extinguishment of debt.
Cash Used In Investing Activities. Net cash used in investing activities decreased $328,000 to $741,000 for the nine months ended January 31, 2013, compared to net cash used in investing activities of $1,069,000 for the nine months ended January 31, 2012. This net decrease was due to a decrease in property acquisitions of $417,000 offset by an increase in other assets of $89,000.
| 23 |
Cash Provided By Financing Activities. Net cash provided by financing activities increased $276,000 to $25,173,000 for the nine months ended January 31, 2013, compared to net cash provided by financing activities of $24,897,000 for the nine months ended January 31, 2012. Net cash provided by financing activities for the nine months ended January 31, 2013, consisted of $26,166,000 in net proceeds from the sale of shares of our common stock under an At Market Issuance Sales Agreement combined with $291,000 in aggregate net proceeds received from stock option exercises and from the purchase of shares under our 2010 Employee Stock Purchase Plan, which were offset with principal payments on capital leases of $58,000. In addition, during the current year nine-month period, we received gross proceeds of $15,000,000 under a term loan, excluding debt issuance costs of $251,000, which principal amount was repaid in full during the current year nine-month period upon the termination of the term loan agreement on September 25, 2012 (as described in Note 7 to the accompanying interim unaudited condensed consolidated financial statements). In addition, we paid a final payment fee of $975,000 upon the termination of the term loan.
Net cash provided by financing activities for the nine months ended January 31, 2012, consisted of $26,190,000 in aggregate net proceeds from the sale of shares of our common stock under an At Market Issuance Sales Agreement and a registered direct public offering. In addition, we received net proceeds of $96,000 from the purchase of shares under our 2010 Employee Stock Purchase Plan. These prior year net proceeds were offset with aggregate principal payments on notes payable and capital leases of $1,389,000.
Commitments
At January 31, 2013, we had no material capital commitments.
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
Changes in United States interest rates would affect the interest earned on our cash and cash equivalents, however, they would not have an effect on our capital leases, which have fixed interest rates and terms.
Based on our overall cash and cash equivalents interest rate exposure at January 31, 2013, a near-term change in interest rates, based on historical movements, would not have a material adverse effect on our financial position or results of operations.
Item 4. Controls And Procedures.
The Company maintains disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e)) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that are designed to ensure that information required to be disclosed in its reports filed under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including its Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
The Company carried out an evaluation, under the supervision and with the participation of management, including its Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of its disclosure controls and procedures as of January 31, 2013, the end of the period covered by this Quarterly Report. Based on that evaluation, the Company’s Chief Executive Officer and Chief Financial Officer concluded that its disclosure controls and procedures were effective at the reasonable assurance level as of January 31, 2013.
There were no significant changes in the Company’s internal controls over financial reporting, during the quarter ended January 31, 2013, that have materially affected, or are reasonably likely to materially affect, the Company’s internal controls over financial reporting.
| 24 |
PART II - OTHER INFORMATION
Item 1. Legal Proceedings.
In the ordinary course of business, we are at times subject to various legal proceedings and disputes. Except as set forth below, we currently are not aware of any material litigation or other dispute nor, to management’s knowledge, is any litigation or other proceeding threatened against us that collectively is expected to have a material adverse effect on our consolidated cash flows, financial condition or results of operations.
Securities Related Class Action Lawsuits
On September 28, 2012, three complaints were filed in the U.S. District Court for the Central District of California (the “Court”) against us and certain of our executive officers and one consultant (collectively, the “Individual Defendants”) on behalf of certain purchasers of our common stock. The complaints have been brought as purported stockholder class actions, and, in general, include allegations that we and the Individual Defendants violated (i) Section 10(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Rule 10b-5 promulgated thereunder and (ii) Section 20(a) of the Exchange Act, by making materially false and misleading statements regarding the interim median overall survival results of our bavituximab Phase II second-line non-small cell lung cancer trial, thereby artificially inflating the price of our common stock. The plaintiffs are seeking unspecified monetary damages and other relief. On November 27, 2012, four prospective lead plaintiffs filed motions to consolidate, appoint a lead plaintiff, and appoint lead counsel. On February 5, 2013, the court appointed James T. Fahey as lead plaintiff in the action. The lead plaintiff has until April 8, 2013 to file an amended consolidated complaint against us. We believe that the various shareholder lawsuits are without merit, and we intend to vigorously defend the various actions and to seek dismissal of these complaints. Due to the early stage of these proceedings, we believe that the probability of an unfavorable outcome or loss related to these proceedings and an estimate of the amount or range of loss related to these claims, if any, from an unfavorable outcome are not determinable at this time.
Other Legal Matters
On September 24, 2012, we filed a lawsuit against Clinical Supplies Management, Inc. (“CSM”), in the U.S. District Court for the Central District of California (the “Court”). We had contracted with CSM in 2010 as our third-party vendor responsible for distribution of the blinded investigational product used in our bavituximab Phase II second-line non-small cell lung cancer trial. As part of the routine collection of data in advance of an end-of-Phase II meeting with regulatory authorities, we discovered major discrepancies between some patient sample test results and patient treatment code assignments. Consequently, we filed this lawsuit against CSM alleging breach of contract, negligence and negligence per se arising from CSM’s performance of its contracted services. We are seeking monetary damages. On March 7, 2013, we and CSM submitted to the Court a proposed stipulation pursuant to which the lawsuit would be stayed for up to 120 days during which time we and CSM would participate in an alternative dispute resolution process, pursuant to our contract with CSM. The proposed stipulation was approved by the Court on March 8, 2013.
Item 1A. Risk Factors.
The following risk factors below update, and should be considered in addition to, the risk factors previously disclosed by us in Part 1, Item 1A of our Annual Report for the fiscal year ended April 30, 2012.
If we cannot obtain additional funding, our product development and commercialization efforts may be reduced or discontinued and we may not be able to continue operations.
At January 31, 2013, we had $26,255,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced negative cash flows from operations since our inception and we expect the negative cash flows from operations to continue for the foreseeable future. Our net loss incurred during the nine-month period ended January 31, 2013 amounted to $21,331,000 and our net losses incurred during the past three fiscal years ended April 30, 2012, 2011 and 2010, amounted to $42,119,000, $34,151,000, and $14,494,000, respectively. Therefore, unless and until we are able to generate sufficient revenues from Avid’s contract manufacturing services and/or from the sale and/or licensing of our products under development, we expect such losses to continue for the foreseeable future.
Therefore, our ability to continue to fund our clinical trials and development efforts is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, issuing additional equity or debt.
| 25 |
Historically, we have funded a significant portion of our operations through the issuance of equity. During the nine months ended January 31, 2013, we raised $27,051,000 in gross proceeds under an At Market Sales Issuance Agreement, of which $25,555,000 was raised subsequent to September 25, 2012, primarily to replace the $15,000,000 of initial funding we repaid on September 25, 2012 under an earlier loan facility we entered into on August 30, 2012. Subsequent to January 31, 2013 and through March 12, 2013, we raised an additional $4,805,000 in aggregate gross proceeds under two separate At Market Sales Issuance Agreements.
With respect to our ability to raise additional capital from the issuance of equity, as of March 12, 2013, we have an effective shelf registration statement on Form S-3, under which we may issue, from time to time, in one or more offerings, shares of our common stock for gross proceeds of up to $145,525,000. However, our ability to raise additional capital in the equity markets is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse clinical trials results, and significant delays in one or more clinical trials. If our ability to access the capital markets becomes severely restricted, it could have a negative impact on our business plans, including our clinical trial programs and other research and development activities. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
In addition to financing our operations through the issuance of equity, we may also secure additional funding through the issuance of debt, licensing or partnering our products in development, or increasing revenue from our wholly-owned subsidiary, Avid. While we will continue to explore these potential opportunities, there can be no assurances that we will be successful in securing debt financing, licensing or partnering our products in development, or generating additional revenue from Avid to complete the research, development, and clinical testing of our product candidates.
Based on our current projections, which include projected cash inflows under signed contracts with existing customers of Avid, assuming we raise no additional capital from the capital markets or other potential sources, and excluding any third-party Phase III development costs, we believe we will have sufficient cash on hand to meet our obligations as they become due through at least the third quarter of our fiscal year 2014 ending January 31, 2014. There are a number of uncertainties associated with our financial projections, including but not limited to, termination of third party contracts, technical challenges, and regulatory decisions affecting bavituximab, any of which could reduce, delay or accelerate our future projected cash inflows and outflows. In addition, in the event our projected cash-inflows are reduced or delayed we might not have sufficient capital to operate our business beyond the end of the third quarter of our fiscal year 2014. The uncertainties surrounding our future cash inflows have raised substantial doubt regarding our ability to continue as a going concern.
We have had significant losses and we anticipate future losses.
We have incurred net losses in most fiscal years since we began operations in 1981. The following table represents net losses incurred for the nine months ended January 31, 2013, and for each of the past three fiscal years:
| Net Loss | ||||
| Nine months ended January 31, 2013 (unaudited) | $ | 21,331,000 | ||
| Fiscal Year 2012 | $ | 42,119,000 | ||
| Fiscal Year 2011 | $ | 34,151,000 | ||
| Fiscal Year 2010 | $ | 14,494,000 | ||
As of January 31, 2013, we had an accumulated deficit of $359,455,000. While we expect to continue to generate revenues from Avid’s contract manufacturing services, in order to achieve and sustain profitable operations, we must successfully develop and obtain regulatory approval for our products, either alone or with others, and must also manufacture, introduce, market and sell our products. The costs associated with clinical trials and product manufacturing is very expensive and the time frame necessary to achieve market success for our products is long and uncertain. Furthermore, as evidenced by the increase in our net loss over the past two fiscal years, the costs associated with advanced stage clinical trials can significantly increase due, in part, to expanded patient populations and the cost to prepare for potential commercialization. We do not expect to generate product or royalty revenues for at least the next two years, and we may never generate product and/or royalty revenues sufficient to become profitable or to sustain profitability.
| 26 |
The sale of substantial shares of our common stock may depress our stock price.
As of January 31, 2013, there were 133,770,614 shares of our common stock outstanding. Substantially all of these shares are eligible for trading in the public market, subject in some cases to volume and other limitations. The market price of our common stock may decline if our common stockholders sell a large number of shares of our common stock in the public market, or the market perceives that such sales may occur.
We could also issue up to 24,414,094 additional shares of our common stock that are reserved for future issuance under our stock incentive plans, employee stock purchase plan, and for outstanding warrants, as further described in the following table:
| Number of Shares Reserved | ||||
| Common shares reserved for issuance under outstanding option grants and available for issuance under our stock incentive plans | 20,150,287 | |||
| Common shares reserved for and available for issuance under our Employee Stock Purchase Plan | 3,889,004 | |||
| Common shares issuable upon exercise of outstanding warrants | 374,803 | |||
| Total shares of common stock reserved for issuance | 24,414,094 | |||
In addition, the above table does not include shares of common stock we could potentially issue from time to time, in one or more offerings, under our current effective shelf registration statements in exchange for remaining aggregate gross proceeds of up to $150,330,000 as of January 31, 2013.
Of the total options and warrants outstanding as of January 31, 2013, 10,386,617 would be considered dilutive to stockholders because we would receive an amount per share, which is less than the market price of our common stock at January 31, 2013.
In addition, we will need to raise substantial additional capital in the future to fund our operations. If we raise additional funds by issuing equity securities, the market price of our securities may decline and our existing stockholders may experience significant dilution.
Current economic conditions and capital markets are in a period of disruption and instability, which could adversely affect our ability to access the capital markets, and thus adversely affect our business and liquidity.
The current economic conditions and aftermath of the recent financial crisis have had, and will continue to have, a negative impact on our ability to access the capital markets, and thus have a negative impact on our business and liquidity. The shortage of liquidity and credit combined with the substantial losses in worldwide equity markets could lead to an extended worldwide recession. We may face significant challenges if conditions in the capital markets do not improve. Our ability to access the capital markets has been and continues to be severely restricted at a time when we need to access such markets, which could have a negative impact on our business plans, including our clinical trial programs and other research and development activities. Even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us. We cannot predict the occurrence of future disruptions or how long the current conditions may continue.
We and certain of our executive officers and one consultant have been named as defendants in litigation that could result in substantial costs and divert management’s attention.
On September 28, 2012, three complaints were filed in the U.S. District Court for the Central District of California against us and certain of our executive officers and one consultant (collectively, the “Individual Defendants”) on behalf of certain purchasers of our common stock. The complaints were brought as purported stockholder class actions, and, in general, include allegations that we and the Individual Defendants violated (i) Section 10(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and Rule 10b-5 promulgated thereunder and (ii) Section 20(a) of the Exchange Act, by making materially false and misleading statements regarding the interim median overall survival results of our bavituximab Phase II second-line non-small cell lung cancer trial, thereby artificially inflating the price of our common stock. The plaintiffs are seeking unspecified monetary damages and other relief. On February 5, 2013, the court appointed James T. Fahey as lead plaintiff in the action. The lead plaintiff has until April 8, 2013 to file an amended consolidated complaint against us.
| 27 |
There is no guarantee that we will be successful in defending consolidated lawsuit. Also, our insurance coverage may be insufficient, our assets may be insufficient to cover any amounts that exceed our insurance coverage, and we may have to pay damage awards or otherwise may enter into settlement arrangements in connection with such claims. A settlement of the lawsuit could involve the issuance of common stock or other equity, which may dilute your ownership interest. Any payments or settlement arrangements could have material adverse effects on our business, operating results, financial condition or your ownership interest. Even if the lead plaintiff’s claims are not successful, this litigation could result in substantial costs and significantly and adversely impact our reputation and divert management’s attention and resources, which could have a material adverse effect on our business, operating results, financial condition or partnering efforts. In addition, such consolidated lawsuit may make it more difficult to finance our operations, obtain certain types of insurance (including directors’ and officers’ liability insurance), and attract and retain qualified executive officers, other employees and directors.
Our highly volatile stock price and trading volume may adversely affect the liquidity of our common stock.
The market price of our common stock and the market prices of securities of companies in the biotechnology sector have generally been highly volatile and are likely to continue to be highly volatile.
The following table shows the high and low sales price and trading volume of our common stock for each of the last twelve (12) fiscal quarters ended January 31, 2013:
|
Common Stock Sales Price |
Common Stock Daily Trading Volume (000’s omitted) | |||||||
| High | Low | High | Low | |||||
| Quarter Ended January 31, 2013 | $2.78 | $0.69 | 62,489 | 739 | ||||
| Quarter Ended October 31, 2012 | $5.50 | $0.67 | 68,511 | 563 | ||||
| Quarter Ended July 31, 2012 | $1.89 | $0.42 | 11,875 | 276 | ||||
| Quarter Ended April 30, 2012 | $1.14 | $0.39 | 7,397 | 282 | ||||
| Quarter Ended January 31, 2012 | $1.53 | $0.85 | 7,162 | 138 | ||||
| Quarter Ended October 31, 2011 | $1.88 | $0.95 | 2,450 | 110 | ||||
| Quarter Ended July 31, 2011 | $2.48 | $1.56 | 1,012 | 144 | ||||
| Quarter Ended April 30, 2011 | $2.74 | $2.05 | 929 | 152 | ||||
| Quarter Ended January 31, 2011 | $3.10 | $1.46 | 3,434 | 105 | ||||
| Quarter Ended October 31, 2010 | $2.08 | $1.25 | 4,997 | 118 | ||||
| Quarter Ended July 31, 2010 | $4.14 | $1.51 | 9,520 | 140 | ||||
| Quarter Ended April 30, 2010 | $4.30 | $2.86 | 1,278 | 66 | ||||
The market price of our common stock may be significantly impacted by many factors, including, but not limited to:
| · | announcements of technological innovations or new commercial products by us or our competitors; |
| · | publicity regarding actual or potential company-sponsored clinical trial and investigator-sponsored clinical trial results relating to products under development by us or our competitors; |
| · | significant changes in our financial results or that of our competitors, including our abilities to continue as a going concern; |
| · | the offering and sale of shares of our common stock, either sold at market prices or at a discount under an equity transaction; |
| · | significant changes in our capital structure; |
| · | published reports by securities analysts; |
| · | announcements of licensing agreements, joint ventures, strategic alliances, and any other transaction that involves the development, sale or use of our technologies or competitive technologies; |
| 28 |
| · | developments and/or disputes concerning our patent or other proprietary rights; |
| · | regulatory developments, including possible delays, and product safety concerns; |
| · | results of litigation, disputes and other proceedings; |
| · | general stock trends in the biotechnology and pharmaceutical industry sectors; |
| · | public concerns as to the safety and effectiveness of our products; |
| · | economic trends and other external factors, including but not limited to, interest rate fluctuations, economic recession, inflation, foreign market trends, national crisis, and disasters; and |
| · | healthcare reimbursement reform and cost-containment measures implemented by government agencies. |
These and other external factors have caused and may continue to cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock, and may otherwise negatively affect the liquidity of our common stock.
The liquidity of our common stock will be adversely affected if our common stock is delisted from the NASDAQ Capital Market.
Our common stock is traded on The NASDAQ Capital Market. To maintain inclusion on The NASDAQ Capital Market, we must continue to meet the following six listing requirements:
| 1. | Stockholders’ equity of at least $2,500,000 or market value of at least $35,000,000 or net income of at least $500,000 in either our latest fiscal year or in two of our last three fiscal years; |
| 2. | Public float of at least 500,000 shares; |
| 3. | Market value of our public float of at least $1,000,000; |
| 4. | A minimum closing bid price of $1.00 per share of common stock, without falling below this minimum bid price for a period of thirty consecutive trading days; |
| 5. | At least two market makers; and |
| 6. | At least 300 stockholders, each holding at least 100 shares of common stock. |
On November 14, 2012, we received a deficiency notice from The NASDAQ Stock Market LLC indicating that the Company's minimum bid price had fallen below $1.00 for 30 consecutive business days, and therefore, we were not in compliance with NASDAQ Marketplace Rule 5550(a)(2). Pursuant to the NASDAQ notice, we were afforded an initial 180 calendar days, or until May 13, 2013, to regain compliance with this minimum bid price requirement. On December 11, 2012, we received a letter from The NASDAQ Stock Market LLC notifying us that we had regained compliance with NASDAQ Marketplace Rule 5550(a)(2), as the closing bid price of our common stock had been at or above $1.00 per share for at least 10 consecutive business days.
If our common stock were ever delisted, we would apply to have our common stock quoted on the OTCQX, the world’s largest interdealer quotation system, which is operated by OTC Market Groups, Inc. Upon any such delisting, our common stock would become subject to the regulations of the Securities and Exchange Commission relating to the market for penny stocks. A penny stock, as defined by the Penny Stock Reform Act, is any equity security not traded on a national securities exchange that has a market price of less than $5.00 per share. The penny stock regulations generally require that a disclosure schedule explaining the penny stock market and the risks associated therewith be delivered to purchasers of penny stocks and impose various sales practice requirements on broker-dealers who sell penny stocks to persons other than established customers and accredited investors. The broker-dealer must make a suitability determination for each purchaser and receive the purchaser’s written agreement prior to the sale. In addition, the broker-dealer must make certain mandated disclosures, including the actual sale or purchase price and actual bid offer quotations, as well as the compensation to be received by the broker-dealer and certain associated persons. The regulations applicable to penny stocks may severely affect the market liquidity for our common stock and could limit your ability to sell your securities in the secondary market.
Successful development of our products is uncertain. To date, no revenues have been generated from the commercial sale of our products and our products may not generate revenues in the future.
Our development of current and future product candidates is subject to the risks of failure inherent in the development of new pharmaceutical products and products based on new technologies. These risks include:
| 29 |
| · | delays in product development, clinical testing or manufacturing; |
| · | unplanned expenditures in product development, clinical testing or manufacturing; |
| · | failure in clinical trials or failure to receive regulatory approvals; |
| · | emergence of superior or equivalent products; |
| · | inability to manufacture on our own, or through others, product candidates on a commercial scale; |
| · | inability to market products due to third party proprietary rights; and |
| · | failure to achieve market acceptance. |
Because of these risks, our research and development efforts or those of our partners may not result in any commercially viable products. If significant portions of these development efforts are not successfully completed, required regulatory approvals are not obtained, or any approved products are not commercially successful, our business, financial condition and results of operations may be materially harmed.
Because we have not begun the commercial sale of any of our products, our revenue and profit potential is unproven and our operating history makes it difficult for an investor to evaluate our business and prospects. Our technology may not result in any meaningful benefits to our current or potential partners. No revenues have been generated from the commercial sale of our products, and our products may not generate revenues in the future. Our business and prospects should be considered in light of the heightened risks and unexpected expenses and problems we may face as a company in an early stage of product development in an extremely competitive and rapidly evolving industry.
We are primarily focusing our activities and resources on the development of bavituximab and depend on its success.
We are focusing most of our near-term research and development activities and resources on bavituximab, and we believe a significant portion of the value of our Company relates to our ability to develop this drug candidate. The development of bavituximab is subject to many risks, including the risks discussed in other risk factors. If the results of clinical trials of bavituximab, the regulatory decisions affecting bavituximab, the anticipated or actual timing and plan for commercializing bavituximab, or, ultimately, the market acceptance of bavituximab do not meet our, your, analysts’ or others’ expectations, the market price of our common stock could be adversely affected.
Our product development efforts may not be successful.
Our product candidates have not received regulatory approval and are generally in research, preclinical and various clinical stages of development. If the results from any of the clinical trials are not positive, those results may adversely affect our ability to raise additional capital or obtain regulatory approval to conduct additional clinical trials, which will affect our ability to continue full-scale research and development for our antibody technologies. In addition, our product candidates may take longer than anticipated to progress through clinical trials, or patient enrollment in the clinical trials may be delayed or prolonged significantly, thus delaying the clinical trials. Patient enrollment is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to the clinical sites, competing studies of other investigational products, and the inclusion and exclusion eligibility criteria for the study. In addition, because our Cotara product candidate represents a departure from more commonly used methods for cancer treatment, potential patients and their doctors may be inclined to use conventional therapies, such as chemotherapy, rather than enroll patients in any future clinical study.
Clinical trials required for our product candidates are expensive and time consuming, and their outcome is uncertain.
In order to obtain FDA approval to market a new drug product, we or our potential partners must demonstrate proof of safety and efficacy in humans. To meet these requirements, we or our potential partners will have to conduct extensive preclinical testing and “adequate and well-controlled” clinical trials. Conducting clinical trials is a lengthy, time-consuming and expensive process. The length of time may vary substantially according to the type, complexity, novelty and intended use of the product candidate, and often can be several years or more per trial. Delays associated with products for which we are directly conducting preclinical or clinical trials may cause us to incur additional operating expenses. Moreover, we may continue to be affected by delays associated with the preclinical testing and clinical trials of certain product candidates conducted by our partners over which we have no control. The commencement and rate of completion of clinical trials may be delayed by many factors, including, for example:
| 30 |
| · | obtaining regulatory approval to commence a clinical trial; |
| · | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| · | slower than expected rates of patient recruitment due to narrow screening requirements; |
| · | the inability of patients to meet FDA or other regulatory authorities imposed protocol requirements; |
| · | the inability to retain patients who have initiated a clinical trial but may be prone to withdraw due to various clinical or personal reasons, or who are lost to further follow-up; |
| · | the inability to manufacture sufficient quantities of qualified materials under current good manufacturing practices, or cGMPs, for use in clinical trials; |
| · | shortages of chemotherapy or other drugs used in clinical trials in combination with bavituximab; |
| · | the need or desire to modify our manufacturing processes; |
| · | the inability to adequately observe patients after treatment; |
| · | changes in regulatory requirements for clinical trials; |
| · | the lack of effectiveness during the clinical trials; |
| · | unforeseen safety issues; |
| · | delays, suspension, or termination of the clinical trials due to the institutional review board responsible for overseeing the study at a particular study site; and |
| · | government or regulatory delays or “clinical holds” requiring suspension or termination of the trials. |
Even if we obtain positive results from preclinical or initial clinical trials, we may not achieve the same success in future trials. Clinical trials may not demonstrate statistically sufficient safety and effectiveness to obtain the requisite regulatory approvals for product candidates employing our technology.
Clinical trials that we conduct or that third-parties conduct on our behalf may not demonstrate sufficient safety and efficacy to obtain the requisite regulatory approvals for any of our product candidates. We expect to commence new clinical trials from time to time in the course of our business as our product development work continues. The failure of clinical trials to demonstrate safety and effectiveness for our desired indications could harm the development of that product candidate as well as other product candidates. Any change in, or termination of, our clinical trials could materially harm our business, financial condition and results of operations.
We rely on third parties to conduct our clinical trials and many of our preclinical studies. If those parties do not successfully carry out their contractual duties or meet expected deadlines, our drug candidates may not advance in a timely manner or at all.
In the course of our discovery, preclinical testing and clinical trials, we rely on third parties, including universities, investigators and clinical research organizations, to perform critical services for us. For example, we rely on third parties to conduct our clinical trials and many of our preclinical studies. Clinical research organizations and investigators are responsible for many aspects of the trials, including finding and enrolling patients for testing and administering the trials. Certain of our clinical trials are blind or double-blind. If the trial is blind, management does not have access to information regarding the trials’ administration and progress. We therefore must rely on third parties to conduct our clinical trials, but their failure to comply with all regulatory and contractual requirements, or to perform their services in a timely and acceptable manner, may compromise our clinical trials in particular or our business in general. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may not be available when we need them or, if they are available, may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner. Any failings by these third parties may compromise our clinical trials in particular or our business in general. Similarly, we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. These independent third parties may also have relationships with other commercial entities, some of which may compete with us. For example, if such third parties fail to perform their obligations in compliance with our clinical trial protocols, our clinical trials may not meet regulatory requirements or may need to be repeated. As a result of our dependence on third parties, we may face delays or failures outside of our direct control, as evidenced by the major discrepancies in treatment group coding by an independent third-party vendor responsible for distribution of blinded investigational product used in our bavituximab Phase II non-small cell lung cancer trial. These risks also apply to the development activities of our collaborators, and we do not control our collaborators’ research and development, clinical trials or regulatory activities. We do not expect any drugs resulting from our collaborators’ research and development efforts to be commercially available for many years, if ever.
| 31 |
In addition, we have prepaid research and development expenses to third parties that have been deferred and capitalized as pre-payments to secure the receipt of future preclinical and clinical research and development services. These pre-payments are recognized as an expense in the period that the services are performed. We assess our prepaid research and development expenses for impairment when events or changes in circumstances indicate that the carrying amount of the prepaid expense may not be recoverable or provide a future economic benefit, including the risk of third party nonperformance. If there are indicators that the third parties are unable to perform the research and development services, we may be required to take an impairment charge.
We do not have experience as a company conducting large-scale clinical trials, or in other areas required for the successful commercialization and marketing of our product candidates.
Results from early stage clinical trials of bavituximab and Cotara may not be indicative of successful outcomes in later stage trials. Negative or limited results from any current or future clinical trial could delay or prevent further development of our product candidates, which would adversely affect our business.
We have no experience as a Company in conducting large-scale, late-stage clinical trials, and our experience with early-stage clinical trials with small numbers of patients is limited. In part because of this limited experience, we cannot be certain that planned clinical trials will begin or be completed on time, if at all. Large-scale trials would require significant additional financial and management resources, and reliance on third-party clinical investigators, contract research organizations (“CROs”) or consultants. Relying on third-party clinical investigators or CROs may force us to encounter delays that are outside of our control. Any such delays could have a material adverse effect on our business.
We also do not currently have marketing, sales and distribution capabilities for our product candidates. Developing an internal sales and distribution capability would be an expensive and time-consuming process. We may enter into agreements with third parties that would be responsible for marketing and distribution. However, these third parties may not be capable of successfully selling any of our product candidates. The inability to commercialize and market our product candidates could materially affect our business.
Failure to recruit, enroll, and retain patients for clinical trials may cause the development of our product candidates to be delayed or development costs to increase substantially.
We have experienced, and expect to experience in the future, delays in patient enrollment in our clinical trials for a variety of reasons. The timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the study until its conclusion. The enrollment of subjects depends on many factors, including:
| · | the patient eligibility criteria defined in the protocol; |
| · | the size of the patient population required for analysis of the trial’s primary endpoints; |
| · | the proximity of patients to study sites; |
| · | the design of the trial; |
| · | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| · | our ability to obtain and maintain patient consents; |
| · | the risk that patients enrolled in clinical trials will drop out of the trials before completion; and |
| · | competition for patients by clinical trial programs for other treatments. |
Our clinical trials compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition reduces the number and types of subjects available to us, because some patients who might have opted to enroll in our trials opt to enroll in a trial being conducted by one of our competitors. Since the number of qualified clinical investigators is limited, we conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which reduces the number of subjects who are available for our clinical trials in such clinical trial site. Delays in patient enrollment in the future as a result of these and other factors may result in increased costs or may affect the timing or outcome of our clinical trials, which could prevent us from completing these trials and adversely affect our ability to advance the development of our product candidates.
| 32 |
Patient enrollment and patient care provided at international clinical sites may be delayed or otherwise adversely impacted by social, political and economic factors affecting the particular foreign country.
In the past we have conducted, and intend to conduct in the future, clinical trials globally including clinical sites in India and other countries. Our ability to successfully initiate, enroll and complete a clinical trial in any foreign country is subject to numerous risks unique to conducting business in foreign countries, including:
| · | difficulty in establishing or managing relationships with clinical research organizations and physicians; |
| · | different standards for the conduct of clinical trials and/or health care reimbursement; |
| · | our inability to locate qualified local consultants, physicians, and partners; |
| · | the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including the regulation of pharmaceutical products and treatment; and |
| · | general geopolitical risks, such as political and economic instability, and changes in diplomatic and trade relations. |
Because we intend in the future to conduct clinical trials in foreign counties, any disruption to our international clinical trial sites could significantly delay or jeopardize our product development efforts in those areas.
Success in early clinical trials may not be indicative of results obtained in later trials.
A number of new drugs and biologics have shown promising results in initial clinical trials, but subsequently failed to establish sufficient safety and effectiveness data to obtain necessary regulatory approvals. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval.
Data from our preclinical studies and Phase I and Phase II clinical trials should not be relied upon as evidence that later or larger-scale clinical trials will succeed. The Phase I studies we have completed to date have been designed to primarily assess safety in a small number of patients. In addition, the results we have obtained in the Phase II trials may not predict results for any future studies and may not predict future therapeutic benefit of our drug candidates. We will be required to demonstrate through larger-scale clinical trials that bavituximab and Cotara are safe and effective for use in a diverse population before we can seek regulatory approval for their commercial sale. There is typically an extremely high rate of attrition from the failure of drug candidates proceeding through clinical trials.
In addition, regulatory delays or rejections may be encountered as a result of many factors, including changes in regulatory policy during the period of product development.
If we successfully develop products but those products do not achieve and maintain market acceptance, our business will not be profitable.
Even if the FDA or other regulatory authorities approve bavituximab, Cotara, or any future product candidate for commercial sale, the degree of market acceptance of any approved product candidate by physicians, healthcare professionals and third-party payors and our profitability and growth will depend on a number of factors, including:
| · | our ability to provide acceptable evidence of safety and efficacy; |
| · | changes in the standard of care for the targeted indication; |
| · | relative convenience and ease of administration; |
| · | the prevalence and severity of any adverse side effects; |
| · | availability, cost and potential advantages of alternative treatments; |
| · | pricing and cost effectiveness, which may be subject to regulatory control; |
| · | effectiveness of our or our partners’ sales and marketing strategy; |
| · | the product labeling or product insert required by the FDA or regulatory authority in other countries; and |
| · | our ability to obtain sufficient third-party insurance coverage or reimbursement. |
| 33 |
In addition, if bavituximab, Cotara, or any future product candidate that we discover and develop does not provide a treatment regimen that is more beneficial than the current standard of care or otherwise provide patient benefit, that product likely will not be accepted favorably by the market. If any products we may develop do not achieve market acceptance, then we may not generate sufficient revenue to achieve or maintain profitability.
In addition, even if our products achieve market acceptance, we may not be able to maintain that market acceptance over time if new products or technologies are introduced that are more favorably received than our products, are more cost effective or render our products obsolete.
If we do not establish additional collaborations, we may have to alter our development plans.
Our drug development programs and potential commercialization of our drug candidates will require substantial additional cash to fund expenses. We either own or in-licensed all rights to our two lead drug candidates, bavituximab and Cotara, and are fully responsible for the associated development costs. Our strategy continues to include the potential of selectively collaborating with leading pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of some of our drug candidates and research programs. We may enter into one or more of such collaborations in the future, especially for target indications in which the potential collaborator has particular therapeutic expertise or that involve a large, primary care market that must be served by large sales and marketing organizations or for markets outside of North America. We face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on acceptable terms, or at all. Even if we successfully enter into a collaboration, we cannot provide assurance that our partner will perform its contractual obligations or will not terminate the agreement. If that were to occur, we may have to curtail the development of a particular drug candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of our sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all. If we do not have sufficient funds, we will not be able to bring our drug candidates to market and generate product revenue.
Healthcare reform measures and other statutory or regulatory changes could adversely affect our business.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could impact our business. For example, the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the “Affordable Care Act” or “ACA”), enacted in March 2010, substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. With regard to pharmaceutical products, among other things, ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare D program.
The pharmaceutical and biotechnology industries are subject to extensive regulation, and from time to time legislative bodies and governmental agencies consider changes to such regulations that could have significant impact on industry participants. For example, in light of certain highly-publicized safety issues regarding certain drugs that had received marketing approval, the U.S. Congress has considered various proposals regarding drug safety, including some which would require additional safety studies and monitoring and could make drug development more costly. We are unable to predict what additional legislation or regulation, if any, relating to safety or other aspects of drug development may be enacted in the future, or what effect such legislation or regulation would have on our business.
The coverage and reimbursement status of newly approved drugs is uncertain, and failure to obtain adequate coverage and reimbursement could limit our ability to market bavituximab and cotara and may decrease our ability to generate revenue.
There is significant uncertainty related to the third party coverage and reimbursement of newly approved drugs both nationally and internationally. The commercial success of bavituximab, Cotara, and any other of our future products, if any, in both domestic and international markets depends on whether third-party coverage and reimbursement is available for the ordering of our future products by the medical profession for use by their patients. Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to manage healthcare costs by limiting both coverage and the level of reimbursement of new drugs and, as a result, they may not cover or provide adequate payment for our future products. These payors may not view our future products as cost-effective, and reimbursement may not be available to consumers or may not be sufficient to allow our future products to be marketed on a competitive basis. Likewise, legislative or regulatory efforts to control or reduce healthcare costs or reform government healthcare programs could result in lower prices or rejection of our future products. Changes in coverage and reimbursement policies or healthcare cost containment initiatives that limit or restrict reimbursement for our future products may reduce any future product revenue.
| 34 |
Failure to obtain regulatory approval in foreign jurisdictions will prevent us from marketing bavituximab abroad.
We intend to market bavituximab in international markets either directly or through a potential future collaboration partner, if any. In order to market bavituximab in the European Union, Canada, Japan and many other foreign jurisdictions, we or a potential future collaboration partner must obtain separate regulatory approvals. We have, and potential future collaboration partners may have, had limited interactions with foreign regulatory authorities, and the approval procedures vary among countries and can involve additional testing at significant cost. The time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval processes may include all of the risks associated with obtaining FDA approval. We or a potential future collaboration partner may not obtain foreign regulatory approvals on a timely basis, if at all. We or a potential future collaboration partner may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize bavituximab or any other future products in any market.
Foreign governments often impose strict price controls, which may adversely affect our future profitability.
We intend to seek approval to market bavituximab in both the United States and foreign jurisdictions either directly or through a potential future collaboration partner. If we or a potential future collaboration partner obtain approval in one or more foreign jurisdictions, we or a potential future collaboration partner will be subject to rules and regulations in those jurisdictions relating to bavituximab. In some foreign countries, particularly in the European Union, prescription drug pricing is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a drug candidate. To obtain reimbursement or pricing approval in some countries, we or a potential future collaboration partner may be required to conduct a clinical trial that compares the cost-effectiveness of bavituximab to other available therapies. If reimbursement of bavituximab is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
If we cannot license or sell Cotara, it may be delayed or never be further developed in the U.S.
We have completed a single-arm Phase II study with Cotara for the treatment of brain cancer. In our most recent Phase II open-label, multicenter trial, 41 patients with glioblastoma multiforme (“GBM”) at first relapse were enrolled and received a single-treatment with Cotara. Median overall survival for patients treated with Cotara was 9.3 months. Based on these data and data from earlier clinical studies, we have reached an agreement with the FDA on the design of a single pivotal trial to potentially support product registration for Cotara. With this clear clinical path forward, we are actively pursuing a licensing or funding partner to further advance the program. In the event we are not able to secure a partnership for the program in the U.S., we may not be able to advance the project past its current stage of development. Because there are a limited number of companies, which have the financial resources, the internal infrastructure, the technical capability and the marketing infrastructure to develop and market a radiopharmaceutical-based oncology drug, we may not secure a suitable partner for Cotara. Furthermore, we cannot ensure that if we do secure a suitable licensing partner for the program, the financial terms that they propose will be acceptable to us.
| 35 |
Our manufacturing facilities may not continue to meet regulatory requirements and have limited capacity.
Before approving a new drug or biologic product, the FDA requires that the facilities at which the product will be manufactured comply with current Good Manufacturing Practices, or cGMP, requirements. To be successful, our therapeutic products must be manufactured for development and, following approval, in commercial quantities, in compliance with regulatory requirements and at acceptable costs. Currently, we manufacture all preclinical and clinical material through Avid Bioservices, Inc., our wholly-owned subsidiary. While we believe our current facilities are adequate for the manufacturing of product candidates for clinical trials, our facilities may not be adequate to produce sufficient quantities of any products for commercial sale.
If we are unable to establish and maintain a manufacturing facility or secure third-party manufacturing capacity within our planned time frame and cost parameters, the development and sales of our products, if approved, may be materially harmed.
We may also encounter problems with the following:
| · | production yields; |
| · | possible facility contamination; |
| · | quality control and quality assurance programs; |
| · | shortages of qualified personnel; |
| · | compliance with FDA or other regulatory authorities regulations, including the demonstration of purity and potency; |
| · | changes in FDA or other regulatory authorities requirements; |
| · | production costs; and/or |
| · | development of advanced manufacturing techniques and process controls. |
In addition, we or any third-party manufacturer will be required to register the manufacturing facilities with the FDA and other regulatory authorities, provided it had not already registered. The facilities will be subject to inspections confirming compliance with cGMP or other regulations. If any of our third-party manufacturers or we fail to maintain regulatory compliance, the FDA can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new drug product or biologic product, or revocation of a pre-existing approval. As a result, our business, financial condition and results of operations may be materially harmed.
We may have significant product liability exposure because we maintain only limited product liability insurance.
We face an inherent business risk of exposure to product liability claims in the event that the administration of one of our drugs during a clinical trial adversely affects or causes the death of a patient. Although we maintain product liability insurance for clinical studies in the amount of $5,000,000 per occurrence or $5,000,000 in the aggregate on a claims-made basis, this coverage may not be adequate. Product liability insurance is expensive, difficult to obtain and may not be available in the future on acceptable terms, if at all. Our inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims in excess of our insurance coverage, if any, or a product recall, could negatively impact our financial position and results of operations.
In addition, the contract manufacturing services that we offer through Avid expose us to an inherent risk of liability as the antibodies or other substances manufactured by Avid, at the request and to the specifications of our customers, could possibly cause adverse effects or have product defects. We obtain agreements from our customers indemnifying and defending us from any potential liability arising from such risk. There can be no assurance that such indemnification agreements will adequately protect us against potential claims relating to such contract manufacturing services or protect us from being named in a possible lawsuit. Although Avid has procured insurance coverage, there is no guarantee that we will be able to maintain our existing coverage or obtain additional coverage on commercially reasonable terms, or at all, or that such insurance will provide adequate coverage against all potential claims to which we might be exposed. A partially successful or completely uninsured claim against Avid would have a material adverse effect on our consolidated operations.
| 36 |
If we are unable to obtain, protect and enforce our patent rights, we may be unable to effectively protect or exploit our proprietary technology, inventions and improvements.
Our success depends in part on our ability to obtain, protect and enforce commercially valuable patents. We try to protect our proprietary positions by filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to developing our business. However, if we fail to obtain and maintain patent protection for our proprietary technology, inventions and improvements, our competitors could develop and commercialize products that would otherwise infringe upon our patents.
Our patent position is generally uncertain and involves complex legal and factual questions. Legal standards relating to the validity and scope of claims in the biotechnology and biopharmaceutical fields are still evolving. Accordingly, the degree of future protection for our patent rights is uncertain. The risks and uncertainties that we face with respect to our patents include the following:
| · | the pending patent applications we have filed or to which we have exclusive rights may not result in issued patents or may take longer than we expect to result in issued patents; |
| · | the claims of any patents that issue may not provide meaningful protection; |
| · | we may be unable to develop additional proprietary technologies that are patentable; |
| · | the patents licensed or issued to us may not provide a competitive advantage; |
| · | other parties may challenge patents licensed or issued to us; |
| · | disputes may arise regarding the invention and corresponding ownership rights in inventions and know-how resulting from the joint creation or use of intellectual property by us, our licensors, corporate partners and other scientific collaborators; and |
| · | other parties may design around our patented technologies. |
We may become involved in lawsuits to protect or enforce our patents that would be expensive and time consuming.
In order to protect or enforce our patent rights, we may initiate patent litigation against third parties. In addition, we may become subject to interference or opposition proceedings conducted in patent and trademark offices to determine the priority and patentability of inventions. The defense of intellectual property rights, including patent rights through lawsuits, interference or opposition proceedings, and other legal and administrative proceedings, would be costly and divert our technical and management personnel from their normal responsibilities. An adverse determination of any litigation or defense proceedings could put our pending patent applications at risk of not being issued.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. For example, during the course of this kind of litigation, confidential information may be inadvertently disclosed in the form of documents or testimony in connection with discovery requests, depositions or trial testimony. This disclosure could have a material adverse effect on our business and our financial results.
We may not be able to compete with our competitors in the Biotechnology Industry because many of them have greater resources than we do and they are further along in their development efforts.
The pharmaceutical and biotechnology industry is intensely competitive and subject to rapid and significant technological change. Many of the drugs that we are attempting to discover or develop will be competing with existing therapies. In addition, we are aware of several pharmaceutical and biotechnology companies actively engaged in research and development of antibody-based products that have commenced clinical trials with, or have successfully commercialized, antibody products. Some or all of these companies may have greater financial resources, larger technical staffs, and larger research budgets than we have, as well as greater experience in developing products and running clinical trials. We expect to continue to experience significant and increasing levels of competition in the future. In addition, there may be other companies which are currently developing competitive technologies and products or which may in the future develop technologies and products that are comparable or superior to our technologies and products.
| 37 |
Bavituximab is currently in clinical trials for the treatment of advanced solid tumors, including NSCLC and pancreatic cancer. Although we are not aware of any other monoclonal antibodies in clinical development targeting PS as a potential therapy for advanced solid tumors, there are a number of possible competitors with approved or developmental targeted agents used alone or in combination with standard chemotherapy for the treatment of cancer, including but not limited to, Avastin® (bevacizumab) and onartuzumab by Roche/Genentech, Gleevec® (imatinib) by Novartis, Tarceva® (erlotinib) by OSI Pharmaceuticals, Inc. and Roche/Genentech, Erbitux® (Cetuximab) by Eli Lilly and Company and Bristol-Myers Squibb Company, Rituxan® (rituximab) and Herceptin® (trastuzumab) by Roche/Genentech, Vectibix® (panitumumab) by Amgen, afatinib by Boehringer Ingelheim, Xalkori® (crizotinib) by Pfizer, iniparib by Sanofi-Aventis and Bipar Sciences, ganetespib by Synta Pharmaceuticals, and nivolumab and Yervoy® (ipilimumab) by Bristol-Myers Squibb Company. Additional possible competitors also exist with approved or developmental immunotherapies including but not limited to Provenge® (sipuleucel-T) and other Active Cellular Immunotherapy candidates by Dendreon, Emepepimut-S by Biomira and EMD Serono, and Astuprotimut-r by GlaxoSmithKline. There are a significant number of companies developing cancer therapeutics using a variety of targeted and non-targeted approaches. A direct comparison of these potential competitors will not be possible until bavituximab advances to later-stage clinical trials.
We are developing Cotara for the treatment of recurrent GBM, the most aggressive form of brain cancer. Since Cotara is a single-treatment approach that targets brain tumors from the inside out, it is a novel treatment dissimilar from other drugs approved or in development for this disease. Approved treatments for brain cancer include the Gliadel® Wafer (polifeprosan 20 with carmustine implant) from Eisai, Inc., Temodar® (temozolomide) from Merck, Avastin® (bevacizumab) from Roche/Genentech, and the NovoTTF-100A System by Novocure. Gliadel Wafers are inserted in the tumor cavity following surgical resection and releases a chemotherapeutic agent over time. Temodar is administered orally to patients with brain cancer. Avastin is a monoclonal antibody that targets vascular endothelial growth factor (“VEGF”) to prevent the formation of new tumor blood vessels. The NovoTTF-100A system is a portable, wearable device that delivers an anti-mitotic, anti-cancer therapy.
In addition, some products in development may compete with Cotara should they become approved for marketing. These products include, but are not limited to: Apocept a fully human fusion protein, being developed by Apogenix GmbH, rindopepimut, a peptide vaccine under development by Celldex, and DCVax® a dendritic cell-based vaccine being developed by Northwest Biotherapeutics. In addition, oncology products marketed for other indications such as Tarceva® (Genentech/OSI), Nexavar® (Bayer/Onyx), and afatinib by Boehringer Ingelheim are being tested in clinical trials for the treatment of brain cancer.
Avid Bioservices, Inc., our subsidiary, is exposed to risks resulting from its small customer base.
A significant portion of Avid Bioservices’ revenues has historically been derived from a small customer base. These customers typically do not enter into long-term contracts because their need for drug supply depends on a variety of factors, including the drug’s stage of development, their financial resources, and, with respect to commercial drugs, demand for the drug in the market. Our results of operations could be adversely affected if revenue from any one of our primary customers is significantly reduced or eliminated.
If we lose qualified management and scientific personnel or are unable to attract and retain such personnel, we may be unable to successfully develop our products or we may be significantly delayed in developing our products.
Our success is dependent, in part, upon a limited number of key executive officers, each of whom is an at-will employee, and upon our scientific researchers. For example, because of his extensive understanding of our technologies and product development programs, the loss of Mr. Steven W. King, our President & Chief Executive Officer and Director, would adversely affect our development efforts and clinical trial programs during the six to twelve month period that we estimate it would take to find and train a qualified replacement.
We also believe that our future success will depend largely upon our ability to attract and retain highly-skilled research and development and technical personnel. We face intense competition in our recruiting activities, including competition from larger companies with greater resources. We do not know if we will be successful in attracting or retaining skilled personnel. The loss of certain key employees or our inability to attract and retain other qualified employees could negatively affect our operations and financial performance.
| 38 |
Our Governance Documents and State Law provide certain anti-takeover measures which will discourage a third party from seeking to acquire us unless approved by the Board of Directors.
We adopted a shareholder rights plan, commonly referred to as a “poison pill,” on March 16, 2006. The purpose of the shareholder rights plan is to protect stockholders against unsolicited attempts to acquire control of us that do not offer a fair price to our stockholders as determined by our Board of Directors. Under the plan, the acquisition of 15% or more of our outstanding common stock by any person or group, unless approved by our board of directors, will trigger the right of our stockholders (other than the acquiror of 15% or more of our common stock) to acquire additional shares of our common stock, and, in certain cases, the stock of the potential acquiror, at a 50% discount to market price, thus significantly increasing the acquisition cost to a potential acquiror. In addition, our certificate of incorporation and by-laws contain certain additional anti-takeover protective devices. For example,
| · | no stockholder action may be taken without a meeting, without prior notice and without a vote; solicitations by consent are thus prohibited; |
| · | special meetings of stockholders may be called only by our Board of Directors; and |
| · | our Board of Directors has the authority, without further action by the stockholders, to fix the rights and preferences, and issue shares, of preferred stock. An issuance of preferred stock with dividend and liquidation rights senior to the common stock and convertible into a large number of shares of common stock could prevent a potential acquiror from gaining effective economic or voting control. |
Further, we are subject to Section 203 of the Delaware General Corporation Law, which, subject to certain exceptions, restricts certain transactions and business combinations between a corporation and a stockholder owning 15% or more of the corporation’s outstanding voting stock for a period of three years from the date the stockholder becomes a 15% stockholder.
Although we believe these provisions and our rights plan collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our Board of Directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board of Directors, which is responsible for appointing the members of our management.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
On January 8, 2013, MidCap Funding I, LLC, a holder of a warrant to purchase 118,444 shares of the Company’s common stock at an exercise price of $1.4775 per share (the “Warrant”) delivered a notice of election to exercise the Warrant in full on a cashless basis. In accordance with the terms of the Warrant, the fair market value of the Company’s common stock on the day immediately prior to the date of the notice of election to exercise was $2.43 per share, resulting in 46,427 shares of common stock being issued.
The shares of common stock above were offered and sold pursuant to an exemption from the registration requirements under Section 4(2) of the Securities Act of 1933, as amended (the "Securities Act") and Rule 506 of Regulation D promulgated thereunder since, among other things, the transaction did not involve a public offering and the shares of common stock were acquired by an accredited investor for investment purposes only and not with a view to or for sale in connection with any distribution thereof.
Item 3. Defaults Upon Senior Securities.
None
Item 4. Mine Safety Disclosures.
Not applicable
Item 5. Other Information.
None
| 39 |
Item 6. Exhibits.
| (a) | Exhibits: |
| 10.33 | Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Jeffrey L. Masten, dated December 27, 2012. (*)(**) |
| 10.34 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Steven W. King, effective December 27, 2012. (*)(**) |
| 10.35 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Paul J. Lytle, effective December 27, 2012. (*)(**) |
| 10.36 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Shelley P.M. Fussey, Ph.D., effective December 27, 2012. (*)(**) |
| 10.37 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Joseph Shan, effective December 27, 2012. (*)(**) |
| 10.38 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Mark R. Ziebell, effective December 27, 2012. (*)(**) |
| 31.1 | Certification of the Chief Executive Officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended.* |
| 31.2 | Certification of the Chief Financial Officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended. * |
| 32 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Rule 13a-14(b) and 15d-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. Section 1350. * |
| 101.INS | XBRL Instance Document. (*) (#) |
| 101.SCH | XBRL Schema Document. (*) (#) |
| 101.CAL | XBRL Calculation Linkbase Document. (*) (#) |
| 101.DEF | XBRL Definition Linkbase Document. (*) (#) |
| 101.LAB | XBRL Label Linkbase Document. (*) (#) |
| 101.PRE | XBRL Presentation Linkbase Document. (*) (#) |
| * | Filed herewith | |
| ** | This exhibit is a management contract or a compensation plan or arrangement. |
| # | Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files on Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
| 40 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| PEREGRINE PHARMACEUTICALS, INC. | ||||
| Date: March 12, 2013 | By: | /s/ STEVEN W. KING | ||
|
Steven W. King President, Chief Executive Officer, and Director |
||||
| Date: March 12, 2013 | By: | /s/ PAUL J. LYTLE | ||
|
Paul J. Lytle Chief Financial Officer (signed both as an officer duly authorized to sign on behalf of the Registrant and Principal Financial Officer and Chief Accounting Officer) |
||||
| 41 |