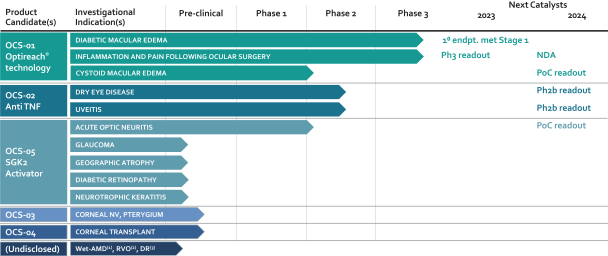
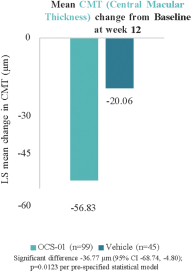
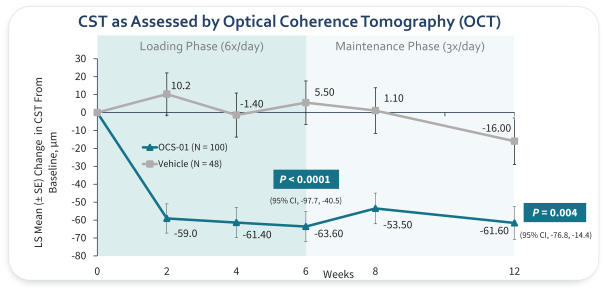
The information in this preliminary prospectus is not complete and may be changed. The selling securityholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we are not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion, dated May 30, 2023
PRELIMINARY PROSPECTUS

5,000,000 Ordinary Shares
We are offering 5,000,000 of our ordinary shares, CHF 0.01 nominal value.
Our ordinary shares are listed on the Nasdaq Global Market under the symbol “OCS.” The last reported sale price of our ordinary shares on the Nasdaq Global Market on May 26, 2023 was $12.00 per share. The final offering price per ordinary share will be determined through negotiations between us and the representatives of the underwriters for the offering, and by reference to the prevailing market prices of our ordinary shares on the Nasdaq Global Market after taking into account market conditions and other factors.
We are a “foreign private issuer” under applicable Securities and Exchange Commission (the “SEC”) rules and an “emerging growth company” as that term is defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”) and are eligible for reduced public company disclosure requirements.
Investing in our ordinary shares involves risks that are described in the “Risk Factors” section beginning on page 14 of this prospectus.
| Per Share | Total | |||||||
| Public offering price |
$ | $ | ||||||
| Underwriting discounts and commissions (1) |
$ | $ | ||||||
| Proceeds, before expenses, to us |
$ | $ | ||||||
| (1) | See the section titled “Underwriting” for additional information regarding compensation payable to the underwriters. |
We have granted the underwriters an option to purchase up to an additional 750,000 ordinary shares from us at the public offering price, less the underwriting discounts and commissions.
Neither the SEC nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the ordinary shares on or about , 2023.
| BofA Securities | SVB Securities | |||||
| Wedbush PacGrow | Baird | H.C. Wainwright & Co. |
Pareto Securities | |||
PROSPECTUS DATED , 2023