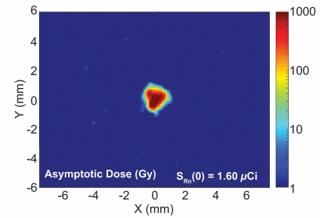
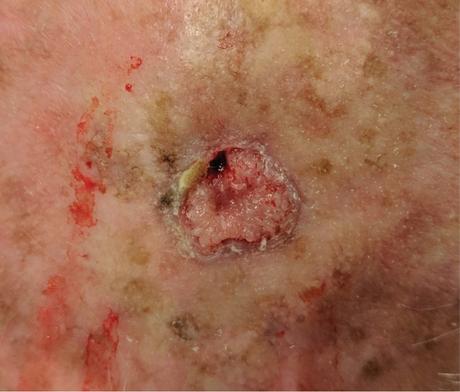
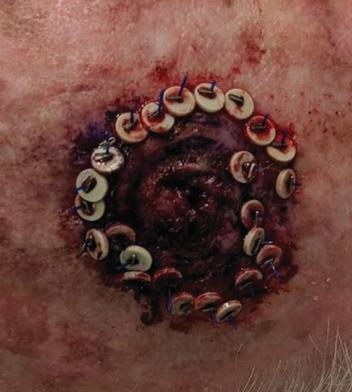
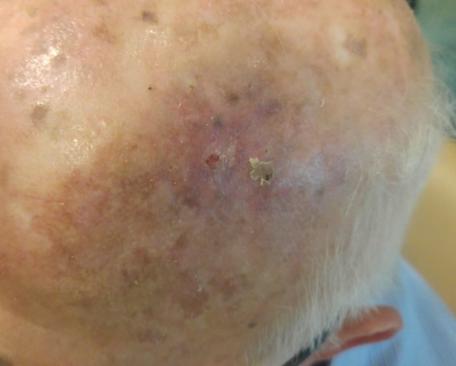
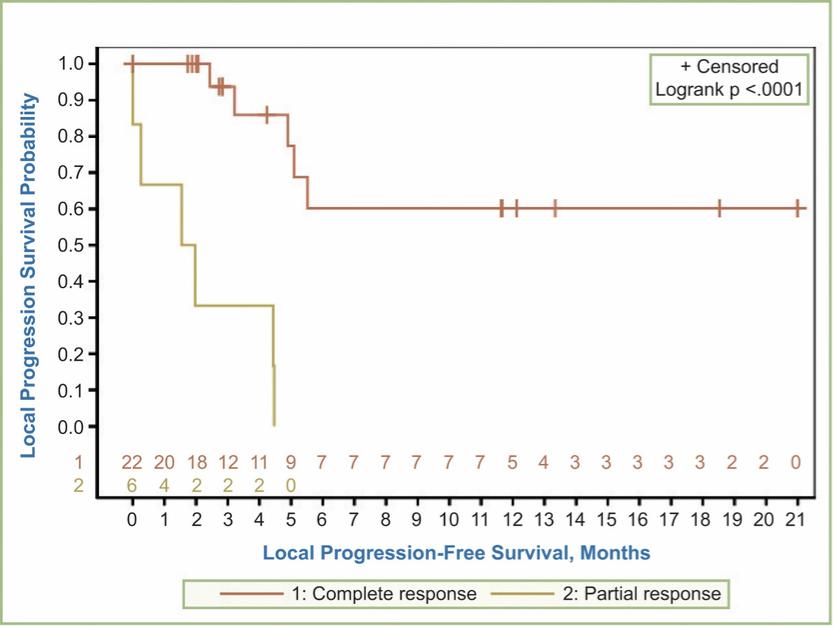
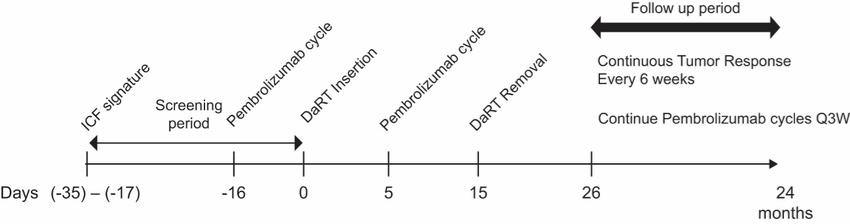
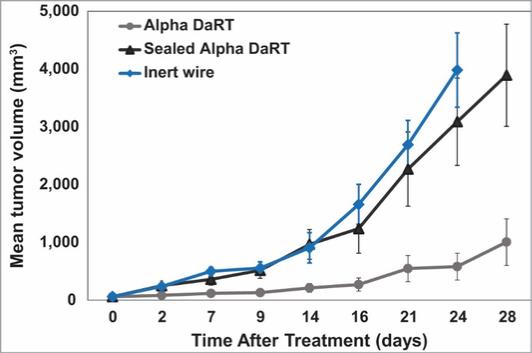
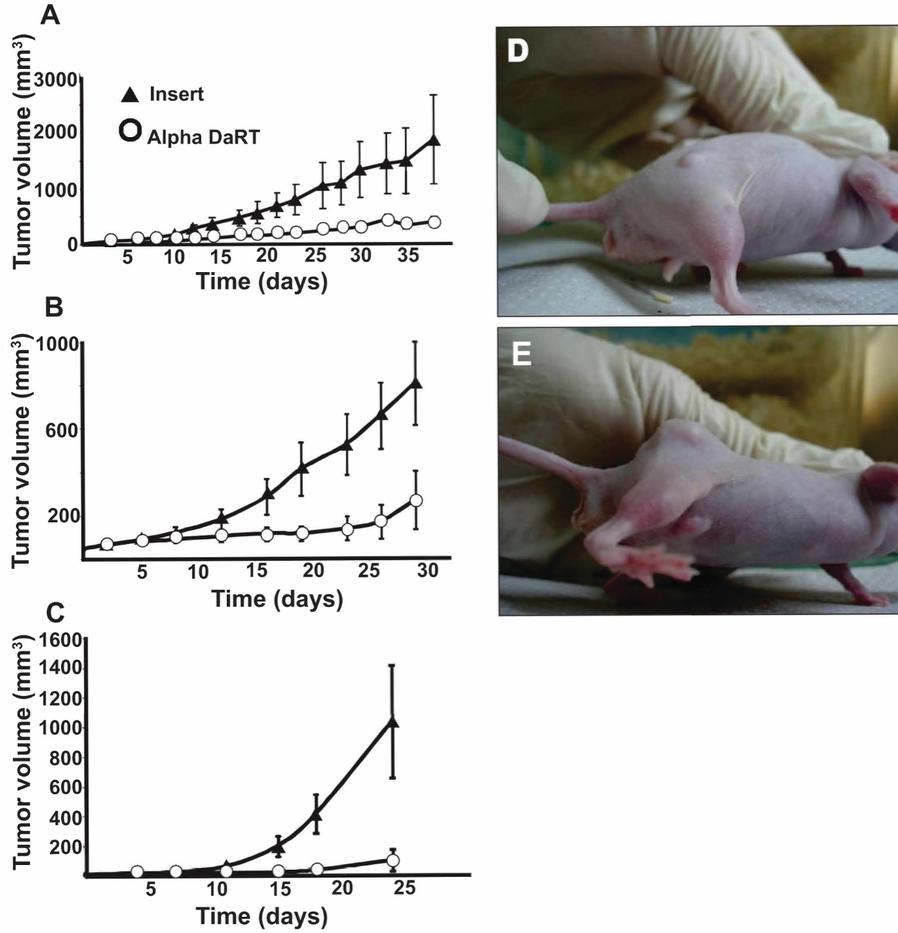
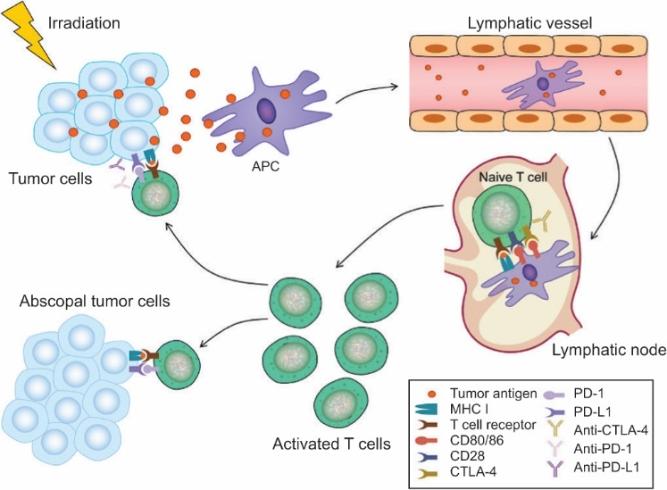
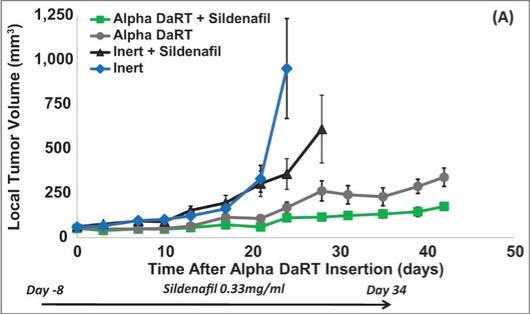
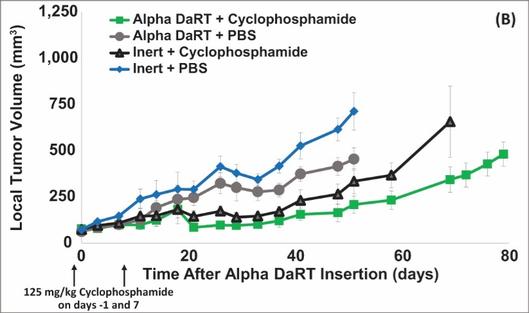
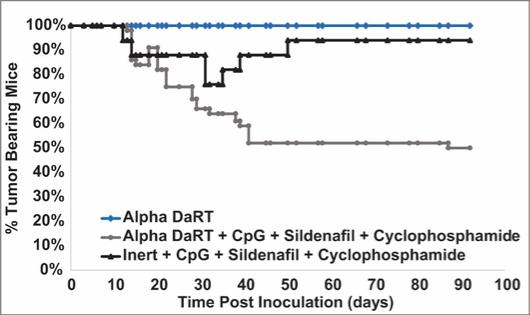
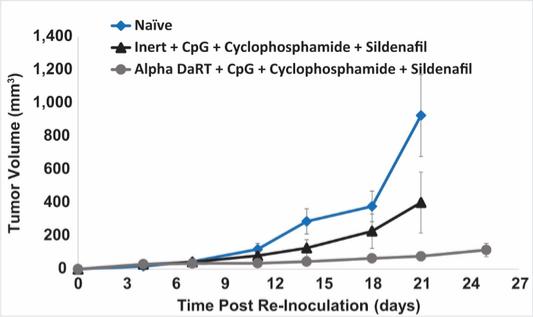
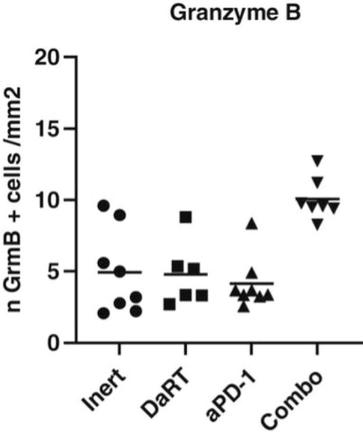
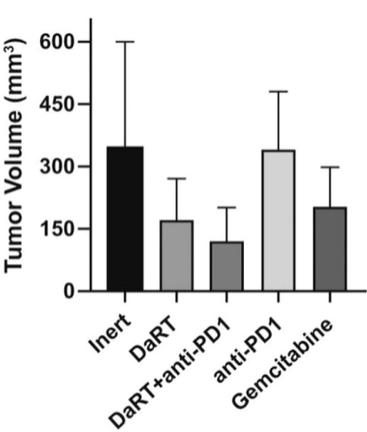
There are several companies developing targeted alpha-based radiopharmaceuticals for the treatment of cancer, including Bayer AG, or Bayer, Novartis AG, Fusion Pharmaceuticals Inc., RayzeBio, Inc., Actinium Pharmaceuticals, Inc., RadioMedix, Inc, Orano and Telix Pharmaceuticals Limited. These companies are targeting a wide range of solid and hematologic malignancies using various alpha emitting isotopes, including Radium-223, Actinium-225 and Thorium-227. The first and only approved alpha particle-based therapy is Bayer’s Xofigo, a salt of Radium-223 that cannot easily and robustly be attached to a targeting molecule, but naturally localizes to regions where cancer cells are infiltrating bone. Xofigo was approved in 2013 for the treatment of bone metastases associated with prostate cancer.
Many of our current or potential competitors, either alone or with their collaboration partners, have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, medical device and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient enrollment in clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
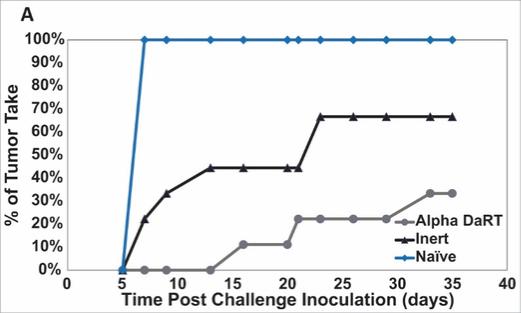
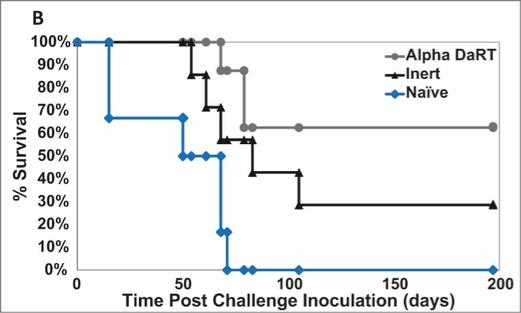
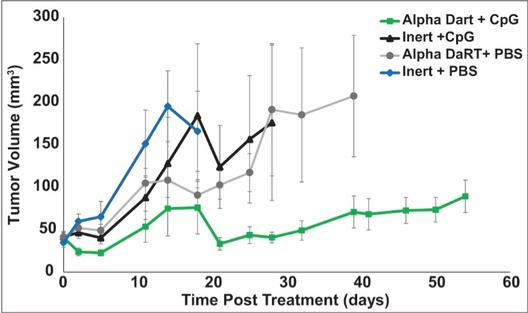
We could see a reduction or elimination in our commercial opportunity if our competitors develop and commercialize treatments that are safer, more effective, have fewer or less severe side effects, are more convenient to administer, are less expensive or have a more favorable label than our Alpha DaRT technology. Our competitors also may obtain FDA or other regulatory approval for their treatments more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. The key competitive factors affecting the success of all of our product candidates, if approved, are likely to be their efficacy, safety, convenience and ease of use, price, the effectiveness of imaging diagnostics, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Our Intellectual Property
As of December 31, 2021, our patent portfolio included 83 issued patents, and 76 pending patent applications including two allowed patent applications.
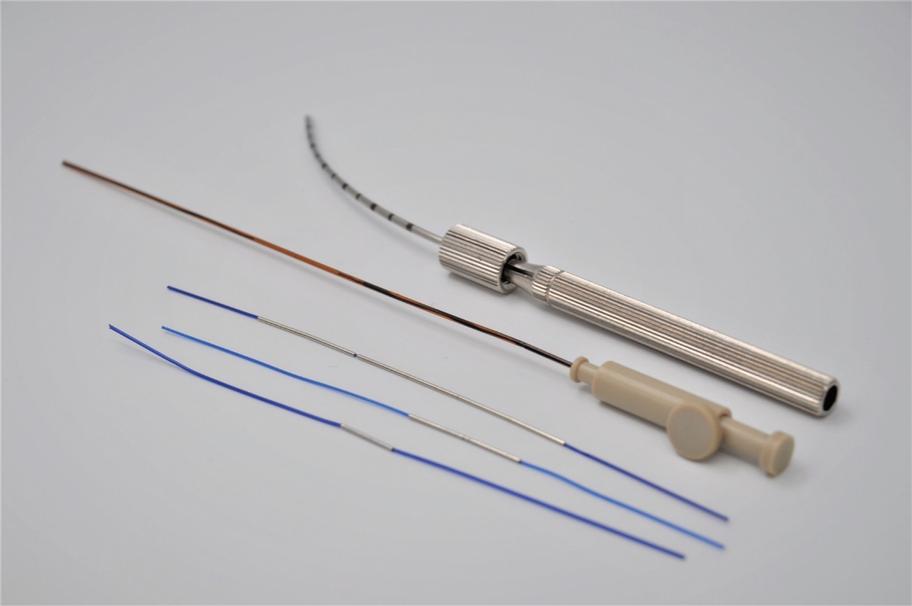
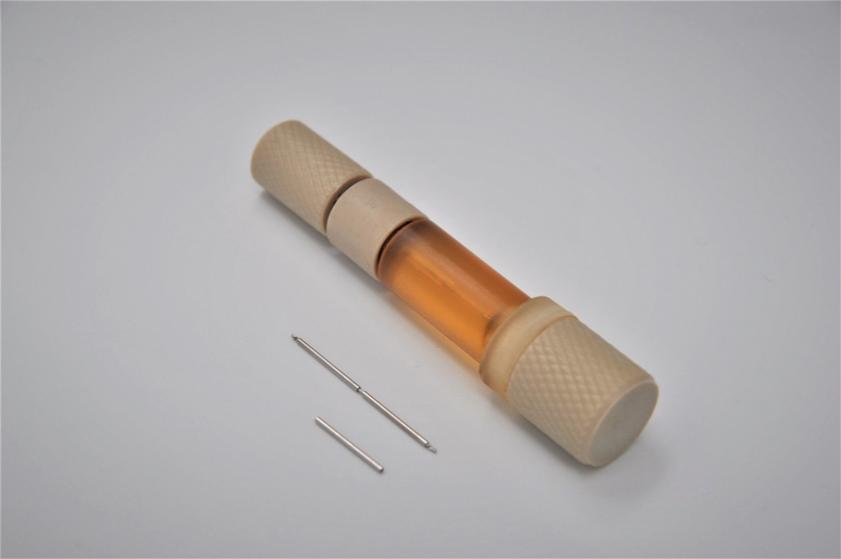
Pursuant to an Intellectual Property Purchase Agreement dated February 2, 2016, we acquired from Althera Medical Ltd. a patent portfolio which now totals 80 patents, including some patent applications whose prosecution was completed following our acquisition. These patents were all assigned to us and are recorded in our name. The patents relate primarily to a device, method of treatment, or method of production of the Alpha DaRT product itself, specifically, to intratumoral diffusing alpha-emitter radiation therapy wherein a probe is loaded with radioisotopes which undergo a process of alpha-emitting radioactive decay solely in proximity to and/or within a tumor. These patents include three U.S. patents, one Canadian patent, three Japanese patents, one Chinese patent, one Hong Kong patent, two European patents each validated in 30 European countries, two Korean patents, three U.K. patents, three French patents, and three German patents. The three issued patents in the United States are expected to expire between 2025 and 2029, without accounting for any potential patent term adjustments or extensions or other forms of exclusivity.
We have two issued patents and 12 pending patent applications, including one allowed patent application, relating to the use of a polymer allowing daughter atoms to escape the source and penetrate the tumor where they emit alpha particles by diffusion. This increases the percentage of daughter radionuclides that reach the tumor. Of the two issued patents, one is an Australian patent, and the other is a South African patent. The foregoing patent applications are pending in the U.S., Europe, Japan, Canada, China, Korea, Russia, African Regional Intellectual Property Organization (ARIPO), Mexico, India, Hong Kong, and Singapore. These patents or patents issuing from the pending applications will begin to expire in 2038, exclusive of possible patent term adjustments or extensions or other forms of exclusivity.
We have 14 pending patent applications, including one allowed patent application, relating to the potential controlled release of a certain amount of Radium-224 from the Alpha DaRT source into the tumor. One of these patent application has been allowed in Australia and 13 of these patent applications are pending in the U.S., Europe, Japan, Canada, China, Korea, Russia, ARIPO, Mexico, India, Hong Kong, Singapore, and South Africa. Patents issuing from these pending applications will begin to expire in 2039, exclusive of possible patent term adjustments or extensions or other forms of exclusivity.