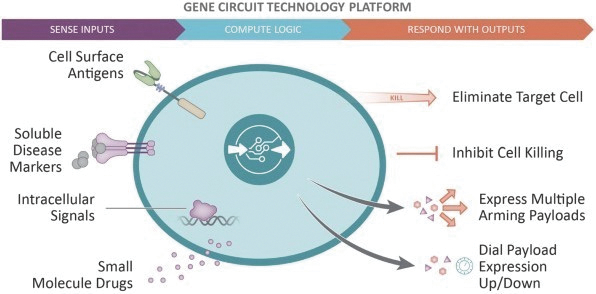
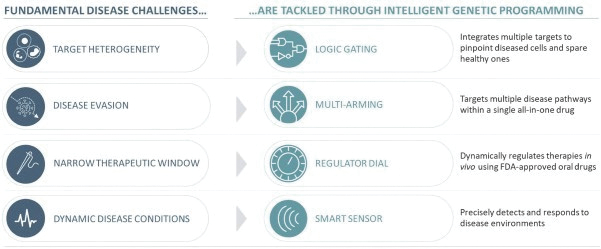
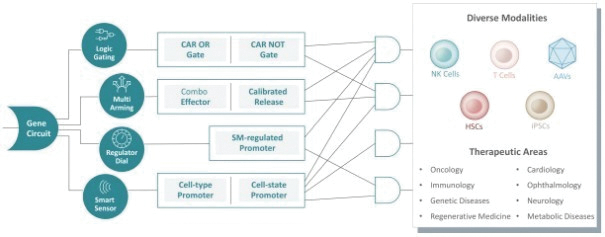
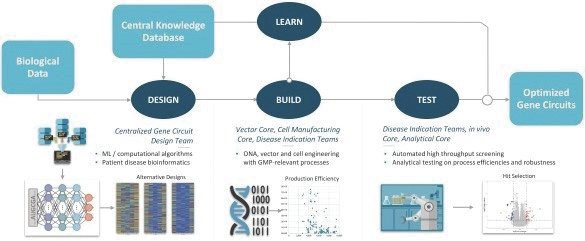
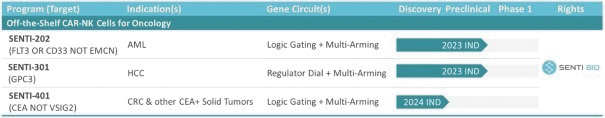
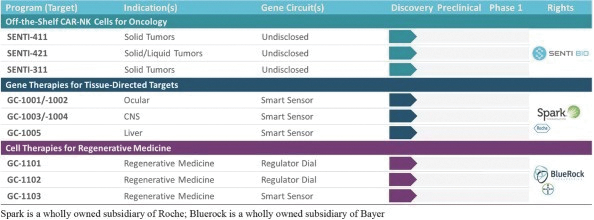
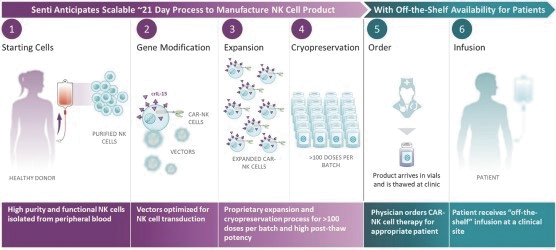
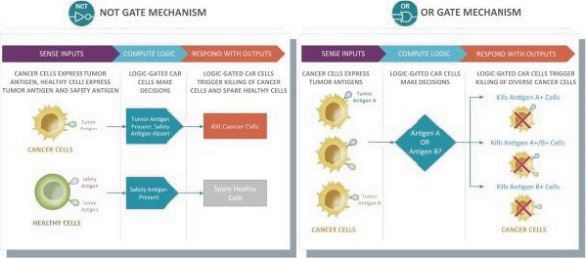
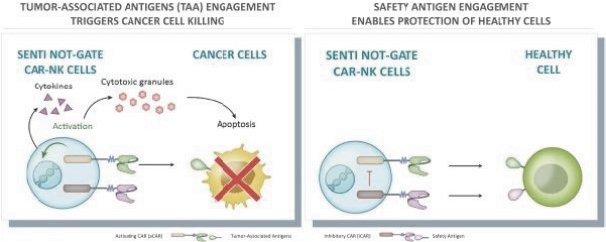
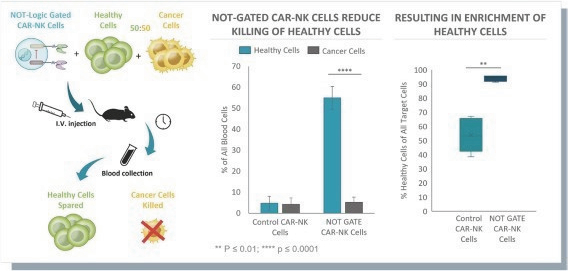
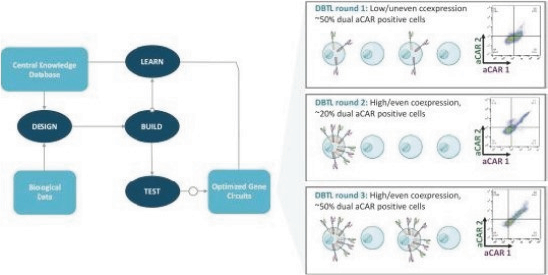
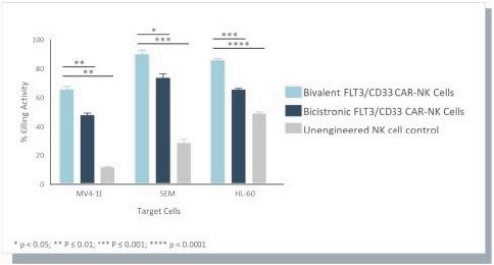
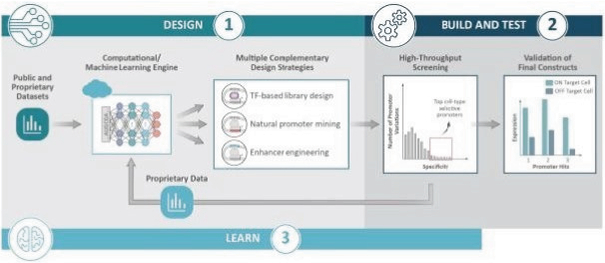
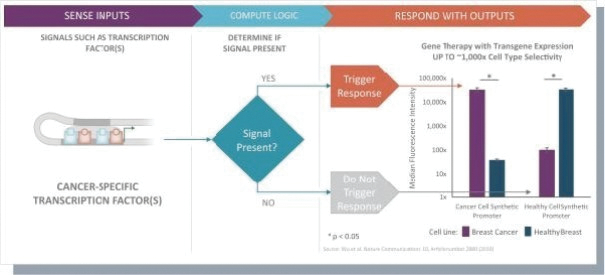
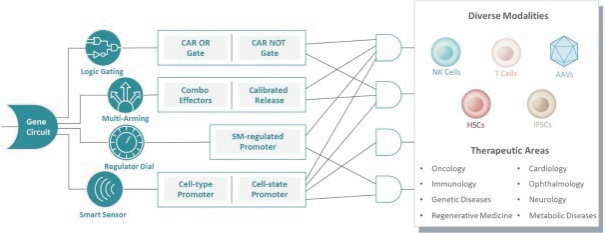
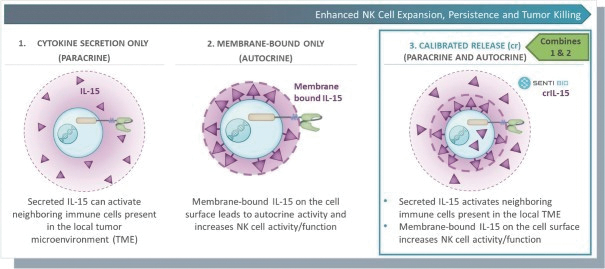
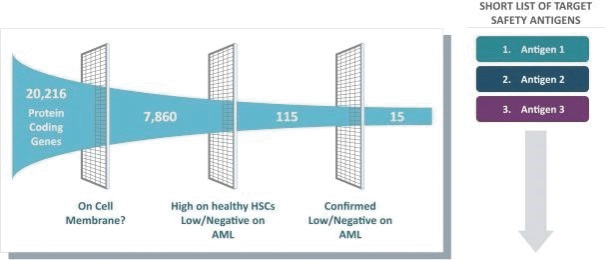
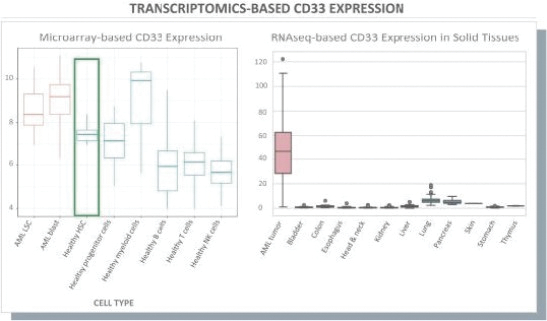
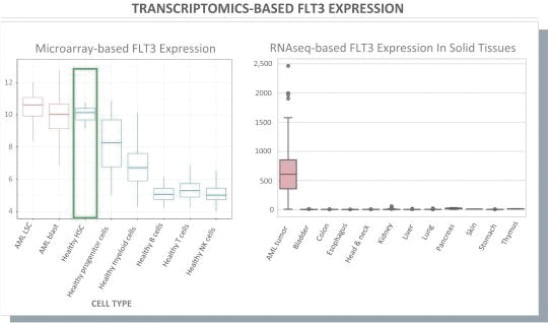
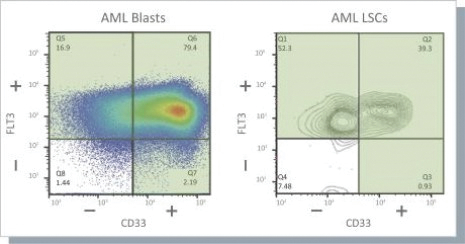
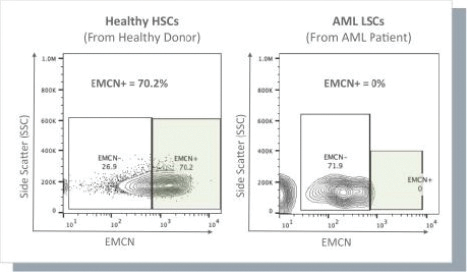
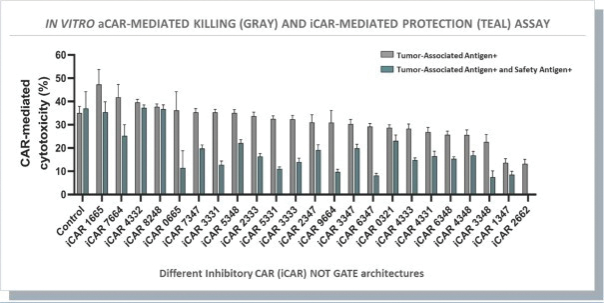
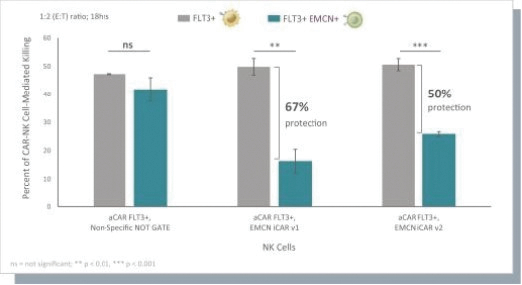
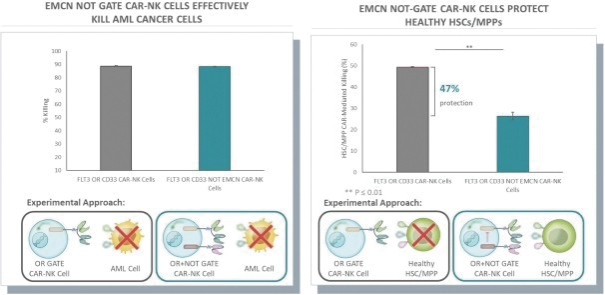
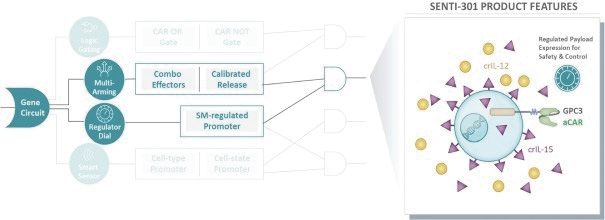
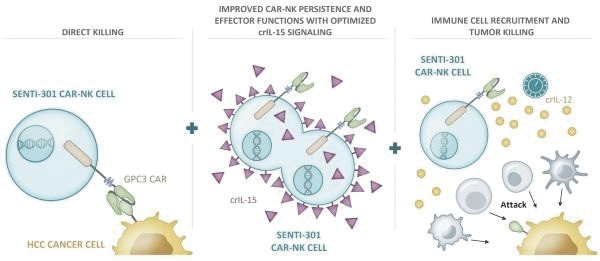
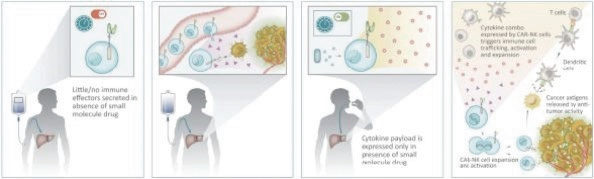
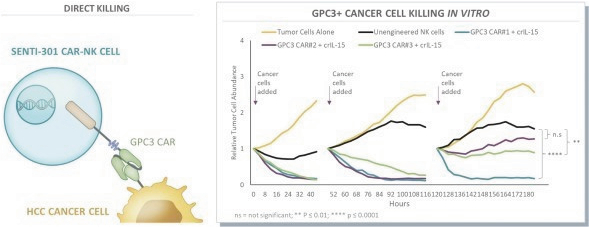
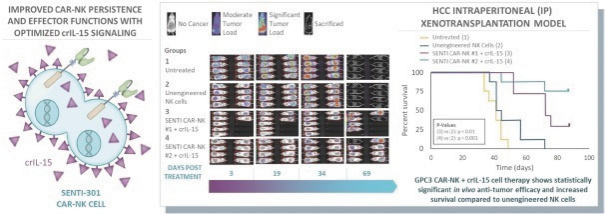
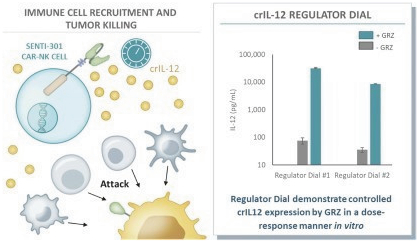
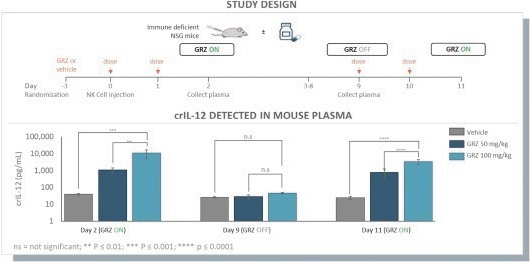
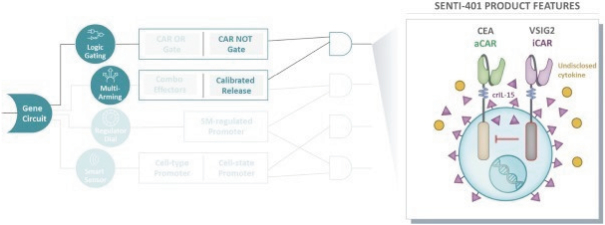
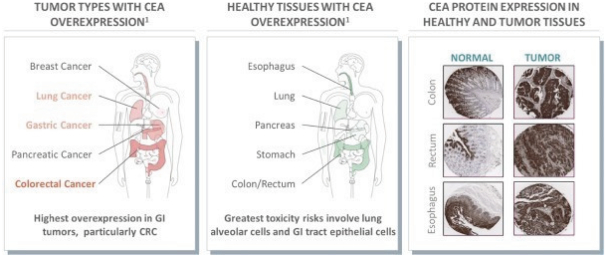
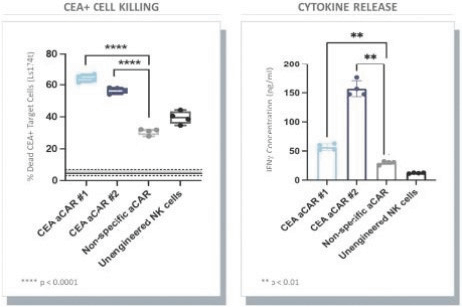
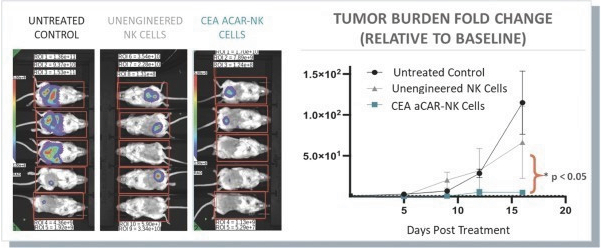
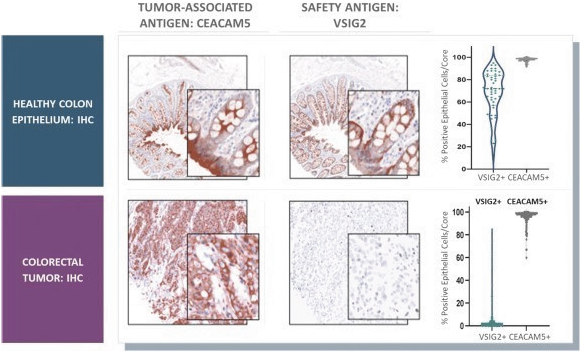
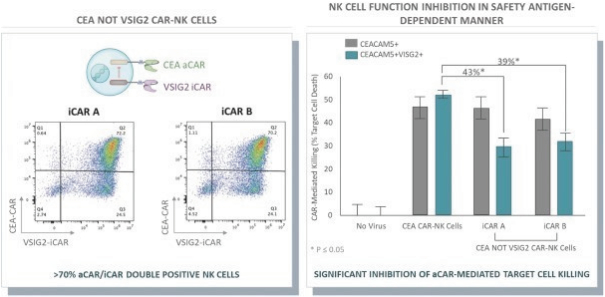
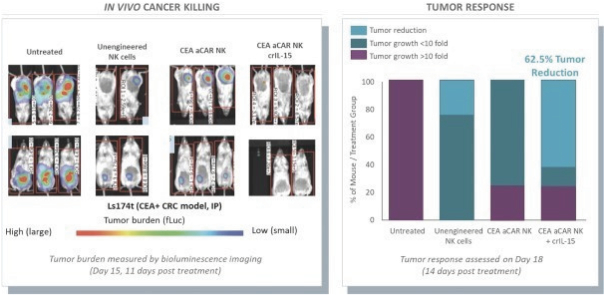
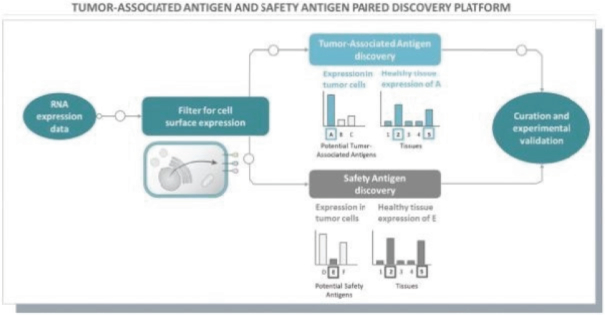
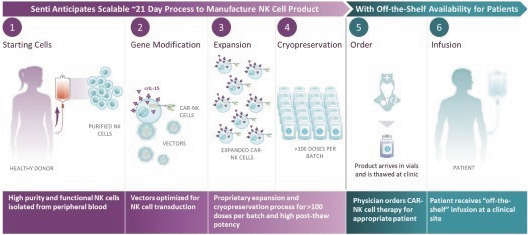
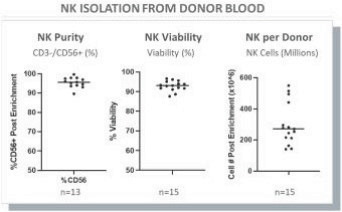
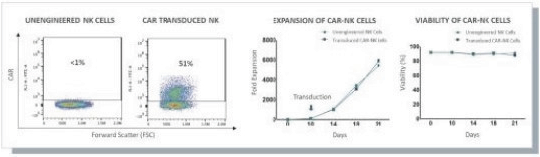
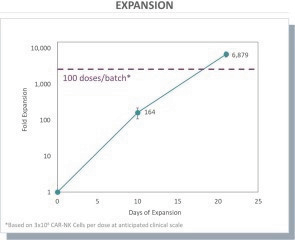
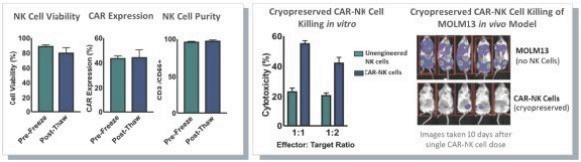
The information in this preliminary prospectus is not complete and may be changed. These securities may not be issued until the registration statement filed with the U.S. Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and does not constitute the solicitation of an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED September 12, 2022
PRELIMINARY PROSPECTUS

SENTI BIOSCIENCES, INC.
Up to 8,727,049 Shares of Common Stock
This prospectus relates to the potential offer and sale from time to time by Chardan Capital Markets LLC (“Chardan” or the “Selling Securityholder”) of up to 8,727,049 shares of our common stock, par value $0.0001 per share (“Senti Common Shares”) that have been or may be issued by us to Chardan pursuant to a ChEF Purchase Agreement, dated as of August 31, 2022, by and between us and Chardan (the “Purchase Agreement”) establishing a committed equity facility (the “Facility”). Such Senti Common Shares consist of (i) up to 8,627,049 Senti Common Shares that we may elect, in our sole discretion, to issue and sell to Chardan, from time to time under the Purchase Agreement and (ii) the 100,000 Senti Common Shares (such shares, the “Commitment Shares”) issued to Chardan as consideration for its execution and delivery of the Purchase Agreement on the Signing Date (as defined herein). The actual number of Senti Common Shares issuable will vary depending on the then current market price of Senti Common Shares sold to Chardan under the Facility. See “The Committed Equity Financing” for a description of the Purchase Agreement and the Facility and “Selling Securityholder” for additional information regarding Chardan and “Plan of Distribution (Conflicts of Interest)” for a description of compensation payable to Chardan.
We are not selling any securities under this prospectus and will not receive any of the proceeds from the sale of the Senti Common Shares by the Selling Securityholder. We may receive up to $50.0 million in aggregate gross proceeds from the Selling Securityholder under the Purchase Agreement in connection with sales of the Senti Common Shares to the Selling Securityholder pursuant to the Purchase Agreement after the date of this prospectus. However, the actual proceeds from the Selling Securityholder may be less than this amount depending on the number of Senti Common Shares sold and the price at which the Senti Common Shares are sold.
This prospectus provides you with a general description of such securities and the general manner in which Chardan may offer or sell the securities. More specific terms of any securities that Chardan may offer or sell may be provided in a prospectus supplement that describes, among other things, the specific amounts and prices of the securities being offered and the terms of the offering. The prospectus supplement may also add, update or change information contained in this prospectus.
Chardan may offer, sell or distribute all or a portion of the Senti Common Shares acquired under the Purchase Agreement and hereby registered publicly or through private transactions at prevailing market prices or at negotiated prices. We will bear all costs, expenses and fees in connection with the registration of the Senti Common Shares, including with regard to compliance with state securities or “blue sky” laws. The timing and amount of any sale are within the sole discretion of Chardan. Chardan is an underwriter under the Securities Act of 1933, as amended (the “Securities Act”), and any profit on sale of Senti Common Shares by them and any discounts, commissions or concessions received by them may be deemed to be underwriting discounts and commissions under the Securities Act. Although Chardan is obligated to purchase Senti Common Shares under the terms and subject to the conditions and limitations of the Purchase Agreement to the extent we choose to sell such Senti Common Shares to them (subject to certain conditions), there can be no assurances that we will choose to sell any Senti Common Shares to Chardan or that Chardan will sell any or all of the Senti Common Shares, if any, purchased under the Purchase Agreement pursuant to this prospectus. Chardan will bear all commissions and discounts, if any, attributable to its sale of Senti Common Shares. See “Plan of Distribution (Conflicts of Interest).”
You should read this prospectus and any prospectus supplement or amendment carefully before you invest in our securities.
Our common stock is listed on The Nasdaq Global Market (“Nasdaq”) under the symbol “SNTI”. On September 7, 2022, the last quoted sale price for the Senti Common Shares as reported on Nasdaq was $1.96 per share.
We are an “emerging growth company” under applicable federal securities laws and will be subject to reduced public company reporting requirements.
Investing in our securities involves a high degree of risk. Before buying any securities, you should carefully read the discussion of the risks of investing in our securities in “Risk Factors” beginning on page 8 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of the securities to be issued under this prospectus or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is .