
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM _____________________ TO _____________________ |
Commission File Number:
(Exact name of Registrant as specified in its Charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
|||
|
|
|
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the Registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
☒ |
|
Smaller reporting company |
|
||
|
|
|
|
Emerging growth company |
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
The aggregate market value of voting common stock held by non-affiliates of the Registrant (assuming for purposes of this calculation, without conceding, that all executive officers and directors are "affiliates") was approximately $
The number of shares of the Registrant’s common stock outstanding as of June 29, 2023 was
Table of Contents
|
|
Page |
|
|
|
Item 1. |
1 |
|
Item 1A. |
47 |
|
Item 1B. |
101 |
|
Item 2. |
101 |
|
Item 3. |
101 |
|
Item 4. |
101 |
|
|
|
|
|
102 |
|
Item 5. |
102 |
|
Item 6. |
102 |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
103 |
Item 7A. |
119 |
|
Item 8. |
119 |
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
119 |
Item 9A. |
119 |
|
Item 9B. |
121 |
|
Item 9C. |
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
121 |
|
|
|
|
122 |
|
Item 10. |
122 |
|
Item 11. |
126 |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
129 |
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
130 |
Item 14. |
131 |
|
|
|
|
|
132 |
|
Item 15. |
132 |
|
Item 16. |
134 |
i
EXPLANATORY NOTE
As used in this Annual Report on Form 10-K, unless otherwise stated in this report or the context otherwise requires, references to the “Company,” “Peak Bio,” “we,” “us” and “our” refer to Peak Bio, Inc., a Delaware corporation formerly known as Ignyte Acquisition Corp. (“Ignyte”) and its subsidiaries.
On November 1, 2022, we consummated the previously announced Business Combination (pursuant to that certain Business Combination Agreement, dated as of April 28, 2022, by and among the Company, Peak Bio Co., Ltd., a corporation organized the laws of the Republic of Korea and Ignyte Korea Co., Ltd. a corporation organized under the laws of the Republic of Korea, the “Business Combination Agreement”). Pursuant to the terms of the Business Combination Agreement, a business combination (herein referred to as the “Business Combination”) between the Company and Peak Bio Co., Ltd. was effected through the Share Swap (as such term is defined in the Business Combination Agreement) resulting in Peak Bio Co., Ltd. as a wholly-owned subsidiary of the Company. In connection with the Business Combination, the Company changed its name from Ignyte Acquisition Corp. to Peak Bio, Inc.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K may contain forward- looking statements as defined by the Private Securities Litigation Reform Act of 1995. These statements are based on the beliefs and assumptions of management. Although we believe that its plans, intentions and expectations reflected in or suggested by these forward-looking statements are reasonable, we cannot assure you that it will achieve or realize these plans, intentions or expectations. Forward-looking statements are inherently subject to risks, uncertainties and assumptions. Generally, statements that are not historical facts, including statements concerning our possible or assumed future actions, business strategies, events or results of operations, are forward-looking statements. These statements may be preceded by, followed by or include the words “believes,” “estimates,” “expects,” “projects,” “forecasts,” “may,” “will,” “should,” “seeks,” “plans,” “scheduled,” “anticipates” or “intends” or similar expressions.
Forward-looking statements are not guarantees of performance. You should not put undue reliance on these statements which speak only as of the date hereof. You should understand that the following important factors, among others, could affect our future results and could cause those results or other outcomes to differ materially from those expressed or implied in our forward-looking statements:
These and other factors that could cause actual results to differ from those implied by the forward-looking statements in this Annual Report on Form 10-K are more fully described in the “Risk Factors” section. The risks described in “Risk Factors” are not
ii
exhaustive. New risk factors emerge from time to time and it is not possible for us to predict all such risk factors, nor can we assess the impact of all such risk factors on its business or the extent to which any factor or combination of factors may cause actual results to differ materially from those contained in any forward-looking statements. All forward- looking statements attributable to us or persons acting on its behalf are expressly qualified in their entirety by the foregoing cautionary statements. We undertake no obligations to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
iii
PART I
Item 1. Business.
SUMMARY
Peak Bio is a clinical-stage biopharmaceutical company focused on commercializing innovative therapeutics that aim to improve and address significant unmet medical need for patients with inflammatory, rare and specialty diseases and cancer. We will continue to explore and partner with researchers, clinicians, patient advocacy groups, academic institutions, governmental agencies, and our investors to continue to expand treatment options and partnerships to meet those expectations.
We will continue to grow our clinical pipeline by executing on our clinical plans for our existing program, and ideally add new clinical assets through acquisition, and through our internal oncology platform engine. To achieve this, we believe Peak Bio’s management team, with more than a combined 50 plus years of industry experience in small molecule, antibodies, and antibody-drug-conjugates (ADC) drug development and having successfully led companies that created therapeutics in above categories during their tenures, are well suited to drive this strategic initiative. During his career, Dr. Huh has founded or co-founded companies such as pH Pharma and BridgeBio (NASDAQ: BBIO) and been a partner of McKinsey & Co (Healthcare/ Technology sector). He has held various leadership positions including Chairman at companies such as Pliant Therapeutics (NASDAQ: PLRX), CytomX Therapeutics (NASDAQ: CTMX), Geron Corporation (NASDAQ: GERN), Epizyme (NASDAQ: EPZM), Chief Executive Officer of BiPar Sciences (acquired by Sanofi) and has served on the Board of Directors for Facet Biotech (acquired by Abbott) and Nektar Therapeutics (NASDAQ: NEKTAR).
Our lead product candidate, PHP-303 is a small molecule, 5th generation, phase 2 clinical-ready neutrophil elastase (NE) inhibitor (NEI). PHP- 303 is a potentially novel, oral, once daily, 0.65 nanomolar (nM; in vitro IC50 value for inhibition of human NE), selective, small molecule reversible inhibitor of NE designed to inhibit its bioactive form (von Nussbaum et al., 2015, Chem Med Chem 10:1163) that Peak Bio is developing for the treatment of alpha-1 antitrypsin (AAT) deficiency (AATD), a genetic disorder that may result in lung disease or liver disease and, potentially, acute respiratory distress syndrome (ARDS). Peak Bio has received a non-dilutive pre-clinical grant from the Department of Defense (DoD) to explore animal models of COVID-19-related ARDS. Currently, we are focusing our clinical efforts on developing PHP-303 for the treatment of the genetic disorder, AATD, a potentially life-threatening rare, genetic condition that results in severe debilitating diseases, including early-onset pulmonary emphysema. Scientific data indicate that the increased risk of lung tissue injury in AATD patients may be due to inadequately controlled NE caused by the insufficient amounts of AAT, the major antiprotease that inhibits NE, that these patients produce. We believe that by inhibiting NE, PHP-303 has the potential to reduce the destruction of lung tissue and stabilize clinical deterioration in AATD patients.
We are progressing, likely, to an end of Q2 2023 start for our phase 2 clinical study in AATD for which NE is hypothesized to be an important determinant of the disease progression (lung damage) in AATD. NE is a proteolytic enzyme that is required during the inflammatory response, but if inadequately opposed by endogenously produced antiproteases, such as AAT, can lead to tissue injury in the lungs, liver, and other major organs. Moreover, if chronically left unchecked this protracted inflammatory cascade, in which NE plays a major role, can lead to increased acute and/or chronic exacerbations and cause further tissue and organ damage that manifests immediately or in the long-term. This can ultimately result in increased morbidity and mortality in a genetic disorder such as AATD. The impact of deregulated NE inhibition is likely important in other diseases where inflammatory responses are left unchecked.
We have been able to deliver higher doses, with no serious adverse events (SAEs) reported, (than in earlier phase 1 trials) of PHP-303 in both single-ascending dose (SAD) and multiple-ascending dose (MAD) phase 1 human clinical trials. We have demonstrated dose-dependent pharmacokinetics and have achieved the recommended phase 2 dose for PHP-303. A maximal tolerated dose for PHP-303 in the phase1 trials was not achieved which suggests we could have still obtained higher NE inhibition with higher doses of PHP-303 and may explain the lack of adverse effects at the tested dose levels. The drug has been tested in nearly 200 subjects including the most recent SAD and MAD studies. PHP-303 is a once daily, orally-dosed and reversible (von Nussbaum et al., 2015) NE inhibitor that achieves greater than 90% inhibition of the bioactive form of NE at doses (10 or 20 mg) deemed to be below the maximal tolerated dose, which has yet to be determined (See clinical characterization of PHP-303 section below; von Nussbaum & Lee, 2015, Bioorg & Med Chem Let 25: 4370–438;4381). At this time, the pharmacokinetic profile and phase 1 clinical study results of PHP-303 support the clinical evaluation of PHP-303 as an investigational therapy for the treatment of AATD in the chronic setting. That PHP-303 has already been dosed in nearly 200 subjects with no reported SAEs further supports phase 2 evaluation in AATD.
In addition, we have leveraged two decades of industry learnings in expanding an important area of the antibody-drug-conjugates (ADC) field allowing for highly targeted treatments in cancer. Despite the continued scientific advancements in the cancer field that has led to the many incremental improvements in patient cancer survival there continues to be a need for ADCs
1
that not only deliver antibody-directed payloads selectively to their tumors, but to also release them via improved linker technology avoiding the potential for significant off-target toxicities. Secondly, we believe that adding an immunomodulatory effect to our toxin(s) that engages our immune systems to assist in the cancer killing would contribute to a long-term tumor regression.
These incremental improvements in cancer treatments for patients and specifically ADCs have also led to the growing commercial success of ADCs currently on the market and likely for those currently in development. A quick scan of the deal flow associated with ADCs over the past 5 years is encouraging both from their continued clinical and commercial success. We believe Peak Bio is well-positioned to take advantage of this field with novel payload platform driven ADC based therapeutics. We are poised to launch off a platform of proprietary in-house technologies that differentiate our ADCs from existing on-market and in-development antibody or ADC programs. Why do we postulate that our approach could be a very important next step in the ADC field? Our programs have taken the traditional approach of an ADC and added an important component which we believe is attributed to the immunomodulatory effects of our novel toxin targeting alternative splicing. In essence, we hypothesize that our combination of Antibody + Linker + Peak Bio Toxin with Immune Modulation is potentially a better ‘Mouse Trap’ based on the clinical successes of checkpoint inhibitors that activate immune cell mediated killing of tumors.
Our most advanced platform in oncology utilizes our toxin, PH-1 or Thailanstatin (a spliceosome modulator) to generate a pipeline of proprietary ADC product candidates that are differentiated from traditional ADC-based therapies so that we may address unmet need in cancer patients. Differentiation is the first, and necessary step, towards the development of therapies serving an unmet need in patients. For e.g., the tumor may already be resistant to an approved ADC with payload A but may still respond to an investigational ADC with payload B, as the mechanism of action (MoA) is different. In that regard, PH-1 is a novel ADC payload and targets the proper splicing of introns. These mis-spliced RNAs are subjected to mRNA decay depriving cancer cells of thousands of essential proteins vital to survival and proliferation. In addition, PH-1 creates mis-spliced proteins or neoepitopes which the immune cells can target well after the initial “chemotherapy” is delivered, in essence creating a second mechanism for cancer killing.
Our first product candidate is an ADC targeting Trop2, which is an antigen broadly expressed in solid tumors of epithelial origin. Our Trop2 ADC and other undisclosed discovery-stage product candidates are based on our proprietary PH-1 platform of toxin payloads targeting RNA splicing. We will continue to identify cancer targets that are well suited to our technology. The goal over time and with the appropriate investment, Peak Bio desires to create a series of differentiated next generation cancer therapies targeting difficult to treat cancers and contribute to increase cancer survival to the benefit of patients, care givers, our potential future partners with the added benefit to our investors.
Even though Peak Bio’s PH1 platform approach has been initiated, and our first nominated ADC targeting Trop2 has been nominated, however we are still working on two additional toxins that are in early R&D to add to our armamentarium of novel toxins.
We believe that Peak Bio and the management team are well positioned to continue to work with our researchers, clinicians, patient advocacy groups, academic institutions, governmental agencies, and our investors to continue to address the significant unmet medical need for patients with AATD, ARDS and cancer with our approaches.
PHP-303
Overview
Peak Bio is a clinical-stage biopharmaceutical company focused on commercializing innovative therapeutics that aim to improve and address significant unmet medical need for patients with inflammatory, rare and specialty diseases and cancer. We will continue to explore and partner with researchers, clinicians, patient advocacy groups, academic institutions, governmental agencies, and our investors to continue to expand treatment options and partnerships to meet those expectations. Peak Bio will continue to grow our clinical and preclinical pipeline by executing on our clinical plans for our existing program, ideally add new clinical assets through acquisition and through our internal oncology platform engine.
We are developing PHP-303 for the treatment of alpha-1 antitrypsin deficiency (AATD), a genetic disorder that can result in lung disease or liver disease and exploring opportunities with PHP-303 for the treatment of acute respiratory distress syndrome (ARDS). We believe our portfolio is well diversified because our product candidates employ different mechanisms of action and target separate indications. We intend to develop and potentially commercialize our rare disease product candidates and potentially future acquired opportunities to maximize potential future sales and marketing synergies. We will also consider potentially seeking strategic partnerships and relationships for further potential clinical development and/or commercialization of these assets.
2
As part of our strategic business plan, we sought and acquired a clinical stage asset that is a small molecule, a neutrophil elastase (NE) inhibitor (NEI). The Peak Bio senior management team, in addition to their business acumen and drug development and commercialization experiences across a multitude of therapeutic areas and technologies, has long-standing relationships with senior executives of large pharmaceutical, smaller biotech companies, key academic institutions and investment banks, which we believe enhances our ability to identify and acquire additional product candidates.
We acquired PHP-303 from Bayer in 2017 through our existing executives’ professional longstanding relationships with Bayer. PHP-303 products’ data package included substantial pre-clinical, clinical, and manufacturing data sets from Bayer, a well-known, well-regarded, multinational healthcare company. We have since completed two additional clinical studies (see clinical studies a Summary of PHP-303 Clinical Development Program table below), including a single ascending dose (SAD) and a multiple ascending dose (MAD) studies, that verify tolerability, and NE inhibition by PHP-303.
Our Pipeline
The following table summarizes our pipeline. We have global commercial rights to all of our product candidates.
Our portfolio consists of the following product candidates:
PHP-303: PHP-303 is a potentially novel, oral, once daily, small molecule inhibitor of NE that Peak Bio is initially developing for two indications, AATD and ARDS. PHP-303 was licensed from Bayer, who previously conducted multiple non-clinical and clinical trials summarized below. These data support a potential clinical benefit of PHP-303 for treatment of patients with AATD, a genetic disorder that may result in lung or liver disease, or ARDS. We recently received a non-dilutive preclinical grant from the Department of Defense to study PHP-303 treatment for ARDS related to COVID-19 to support advancing PHP-303 potentially in this second clinical indication.
AATD is a potentially life-threatening rare, genetic condition caused by a lack of alpha-1 antitrypsin (AAT) a protein (antiprotease) that protects the lungs from enzymatic degradation by endogenous proteases (such as NE). The disease manifests as early-onset pulmonary emphysema, caused by irreversible destruction of lung tissue supporting gas exchange (https://www.lung.org/lung-health-diseases/lung-disease-lookup/alpha-1-antitrypsin-deficiency/learn-about-alpha-1-antitrypsin-defiency). There are an estimated 70,000-100,000 patients in the United States and 120,000 patients in Europe with severe AATD (https://www.rarediseaseadvisor.com/disease-info-pages/alpha-1-antitrypsin-deficiency-epidemiology-aatd; Torres-Duran et al. 2018, Orpha J of Rare Dis 13:114). PHP-303 is designed to selectively inhibit NE, a neutrophil enzyme, which is the major protease responsible for the destruction of lung tissue in AATD.
3
The graphic below highlights potential disease targets for NEIs such as PHP-303. The unchecked imbalance of NE that occurs in many disease states is depicted, and in addition to AATD, it highlights ARDS as another disease indication for PHP-303 for which we have initiated some early preclinical work.
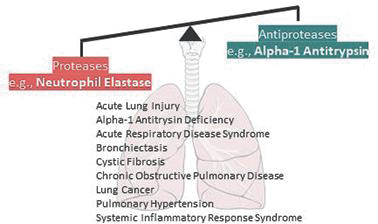
ARDS is a serious lung condition characterized by acute, diffuse, inflammatory lung injury resulting from a range of predisposing etiologies (https://www.uptodate.com/contents/image?imageKey=PULM%2F58759 Gonzales et al., 2015 June 4, Austin J Vasc Med 2:1). ARDS generally progresses from a stage of damage to one where “air” (gas) exchange unit becomes compromised, which is followed by a proliferative repair response by the body’s fibroblasts, ultimately leading to lung fibrosis and scarring likely resulting in an increase in morbidity, mortality and healthcare costs. The estimated incidence of ARDS in the US ranges from 64.2- 78.9 cases/100,000 people with 75% of these patients presenting with moderate to severe disease (reviewed in Diamond et al., 2021, Acute Respiratory Distress Syndrome – StatPearls—NCBI). The incidence of ARDS ranges from 1.5-79 cases/100,000 people in European countries (reviewed in Confalonieri et al., 2017, Eur Res Rev 26:160). The emergence of COVID-19 has led to an increase in the incidence of ARDS in a significant number of hospitalized COVID-19 patients, who may also have other significant complications in the renal, cardiovascular, gastrointestinal, and/or central nervous systems (Gibson et al., 2020, Med J Aust 213:54). At this juncture in the pandemic, we are now seeing incidences of not only acute ARDS associated from COVID-19 but also increased reports of “long- COVID”, which we are just beginning to understand (https://www.uptodate.com/contents/covid-19-epidemiology-clinical-features-and-prognosis-of-the-critically-ill-adult?topicRef=127926&source=related_link). In the future, we will be exploring the potential benefits of PHP-303 treatment across the spectrum of ARDS-related syndromes focusing not only on acute ARDS but also on the more protracted or chronic ARDS-Covid-19 situation.
Neutrophils (a type of immune cell) play an important role in the body’s defense against foreign invaders (Brinkmann et al., 2004, Science 303:1532; Potey et al., 2019, J Pathol, 247: 67; Rosales, 2018 Front Physiol, 9:article 113). They are one of the first immune cells to be recruited to a site of infection. One of the key enzymes involved in neutrophil movement and function is NE which is contained within and associated with the surface of neutrophils. During a normal immune response, NE is released from neutrophils to help degrade tissue allowing neutrophils to reach sites of infection. NE also influences cytokine signaling pathways to increase the immune response. Most importantly, NE directly destroys pathogens by 3 mechanisms: by release into the extracellular matrix, within neutrophils after pathogen engulfment, and by association with neutrophil extracellular traps (NETs). NETs, released from neutrophils, are large web-like structures comprised of sticky circulating free (cf) DNA to which histones and other antimicrobial proteins (including NE and other proteases), are attached. NETs can have both defensive and pro-inflammatory functions: they trap and kill extracellular pathogens, thereby limiting their spread, but can also cause direct or indirect tissue injury.
Under highly pro-inflammatory states, as occurs in patients with COVID-19- associated ARDS, neutrophils become dysregulated and vast numbers invade the area directly releasing excessive amounts of NE and large numbers of NETs (Janoff 1985, Am Rev Respir Dis, 132:417-433; Barnes et al., 2020; Pechous, 2017, Front in Cell and Infect Microbio|, 7:Article 16). The body’s endogenous antiprotease production/release that typically acts to limit damage of NE are overwhelmed and the inadequately opposed high concentrations of NE in the microenvironment cause tissue damage leading to fibrosis. There is also evidence that this large increase in NET production contributes to thrombosis (NETs, and particularly cfDNA, serve as scaffolds that activate platelets and coagulation), which results in further tissue damage.
We believe the inhibition of NE has the potential to protect AATD and ARDS patients from further lung damage by decreasing the impact of high NE tissue concentrations that are insufficiently opposed by endogenous antiproteases. In AATD,
4
the body is unable to produce adequate levels of AAT for sufficient inhibition of NE. In ARDS, NE production and release overwhelms endogenous antiprotease activity leading to tissue damage. Thus, both diseases are, at least in part, due to an overabundance of NE in lung that causes damage. A selective NE inhibitor such as PHP-303 may inhibit this excess NE and provide a potential therapy for AATD and ARDS patients. Furthermore, because NE is necessary for the production of NETs, an NE inhibitor would also be expected to decrease the process of NET formation (NETosis) in patients with COVID-19-associated ARDS, potentially reducing lung injury, thrombosis and cytokine storm.
Peak Bio plans to conduct a Phase 2 proof-of-concept clinical trial in patients with severe AATD with trial initiation, potentially, in 2023. This study has approved Clinical Trial Applications in the UK and Ireland with all approvals necessary to initiate the study shortly after funding is received. Additionally, we have a relationship with and have a research agreement with the Alpha-1 Project. We believe that this relationship will assist in accessing patients, clinicians, thought leaders for our future planned clinical trials.
Preclinical studies characterizing PHP-303 in models of acute lung injury and COVID-19 infection are currently on-going and are being funded by the US Department of Defense (DoD). These studies will inform the role of NE and NETosis in COVID-19 and non-COVID-19 associated ARDS.
Our Strategy
Potentially develop and directly commercialize PHP-303 for AATD treatment and build upon skills sets and relationships with pharmaceutical, biotech, academia, and advocacy groups to build our pipeline/portfolio.
Efficiently advance our specialty disease product candidate(s) and explore strategic relationships with third parties for further possible clinical development and/or commercialization.
5
Leverage our expertise in business development to expand our pipeline of product candidates.
6
Summary of PHP-303 clinical development program
Clinical Stage and Test No. (Report)
|
Nation (Number of organization)
|
Target
|
Test purpose
|
Number of
|
Administration
|
Test Design
|
Primary and
|
Whether
|
Persons/
|
Number of
|
Phase 1 BAY 85-8501 /14431 |
Germany (1) |
Healthy Volunteers |
Evaluation of |
N = 37 |
Single-dose |
Single- |
Primary: safety |
Yes |
Bayer |
7/27 treated subjects — 11 AEs; 5/10 subjects – 6 AEs |
|
|
|
|
|
|
|
|
|
|
|
Phase 1 BAY85-8501/ 14433 |
Germany (1) |
Healthy Volunteers |
Evaluation of |
N = 12 |
Single-dose |
Single- |
Primary: safety, |
Yes |
Bayer |
7/12 over 48 treatment doses – 12 AEs |
|
|
|
|
|
|
|
|
|
|
|
Phase 1 BAY85-8501 /16332 |
Germany (1) |
Healthy Volunteers |
Evaluation of |
N = 34 |
Single |
Single- |
Primary: safety, |
Yes |
Bayer |
7/26 treated subjects – 7 AEs; 3/8 placebo subjects - 4 AEs |
|
|
|
|
|
|
|
|
|
|
|
Phase 1 PHP-303-N101 (Sponsor: pH Pharma) |
|
|
Safety, |
N = 48 |
Ascending |
Single |
Primary: safety, |
Yes |
pH |
10/36 subjects – 8 AEs; 5/12 subjects – 12 AEs |
7
Clinical Stage and Test No. (Report)
|
Nation (Number of organization)
|
Target
|
Test purpose
|
Number of
|
Administration
|
Test Design
|
Primary and
|
Whether
|
Persons/
|
Number of
|
|
|
|
|
|
|
|
|
|
|
|
Phase 1 PHP-303-N102 (Sponsor: pH Pharma) |
|
Overweight or obese but otherwise healthy male and female subjects |
Safety, |
N = 50 |
PHP-303 Oral |
Phase 1, |
Primary: safety, |
Yes |
pH |
14/40 subjects - 32 AEs; 1/10 subjects - 1 AE |
|
|
|
|
|
|
|
|
|
|
|
Phase 2a/ BAY85-8501/ 16359 |
Germany, U.K., Italy, Spain (28) |
Non-cystic fibrosis bronchiectasis patients |
Safety, efficacy |
N=94 |
28 days One dose per |
Multi- |
Primary: safety |
Partially, |
Bayer |
92 subjects analyzed for safety 11/45 treated subjects - 14 AEs; 12/47 subjects - 7 AEs |
Clinical characterization of PHP-303.
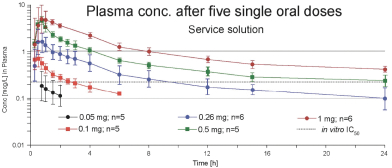
Nearly 200 subjects have been exposed to one or more doses of PHP-303, and the data shows that PHP-303 was tolerated with no SAEs reported. PHP-303 is rapidly absorbed in the fasted state, where median peak concentrations of drug were achieved in < 1 hour and half-life was in the range of 110 to 175 hours. Exposure pharmacokinetics appeared to increase proportionally with increasing dose to 40 mg. With multiple dosing, steady-state concentrations are reached by Day 21. Food delayed the rate of absorption of PHP-303 and therefore moderately decreased maximum serum concentration, but there was no effect on overall exposure to PHP-303. PHP-303 administered in an oral, daily schedule causes inhibition of NE, suggesting potential benefit in several NE and/or NET mediated diseases including AATD and ARDS.
PHP-303 tolerability and adverse effects
The phase 1 clinical studies were not designed to evaluate statistical significance on clinically approvable endpoints. The phase 2 clinical study results and analysis are described in (Watz et al., 2019, Pulm Pharmacol Ther, 56:86). Some 186 subjects have received one or more doses of PHP-303 in clinical studies conducted by Bayer and Peak Bio. These include 141 healthy volunteers and 45 subjects with NCFB, a chronic condition characterized by lung inflammation and airway damage. Doses studied range from 0.05 mg to 20 mg daily, with maximum study duration of 28 days. No serious adverse events (SAEs) have been attributed to drug administration in this program. Across all studies, 84 drug-related adverse events were reported. The most commonly reported adverse events (AEs) observed across clinical studies of PHP-303 include headache, nasopharyngitis, and cough. There was no apparent dose relationship to the reported adverse events and subjects who received placebo had similar frequencies and types of adverse events. Mild, sporadic, and transient elevations in LFTs, lipase, and CPK were uncommonly observed, but these events did not appear to be drug related.
8
Confirmation of NE inhibitory effect of PHP-303 in vivo
Study 16332, (an earlier Bayer study), evaluated whole blood NE activity using an ex vivo zymosan challenge assay. Samples were collected at baseline and several time points after dosing. Samples were incubated with zymosan, a yeast cell wall component that activates neutrophils and release of NE.
PHP-303 resulted in dose and time-dependent inhibition of human NE (hNE) activity. At steady-state repeat dosing, inhibition of hNE exceeded 50% (relative to placebo) at lowest or trough concentration (Day 14 pre-dose) at all dose levels, and approximated 90 to 100% maximal inhibition after the last dose at the mid and high dose levels. Importantly, daily dosing of PHP-303 at doses of 0.5 mg or 1.0 mg yielded long lasting ( > 24 hours) inhibition of NE at levels exceeding 50%. Similar effects on systemic inhibition of hNE activity were observed in the Phase 2a Study 16359, (conducted by Bayer), in patients with NCFB; a chronic condition characterized by lung inflammation and airway damage. [NOTE: NCFB is a type of bronchiectasis (permanent dilation of the airways of the lungs) that arises as a result of chronic inflammation and is not due to the genetic condition cystic fibrosis.]
Changes in neutrophil elastase activity in accordance with plasma concentration and time of PHP-303 tablets (ex-vivo zymosane challenge) and the envisaged inhibitory range for neutrophil elastase
(IC50, IC90 values based on in vitro assay)
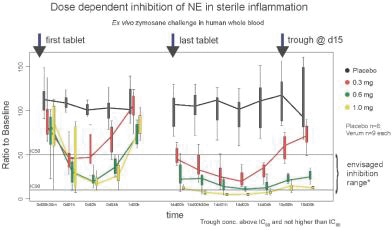
(Source: Clinical Trial Investigator Data Collection, PHP-303_16332 Clinical Test Results Report
Drug Concentration in Blood by Dose After a Single Oral Dose PHP-303 in Liquid Form (Source: PHP-303-14431 Clinical Test Result Report: PHP-303 Data Package _Page 45)
Phase 2a study in non-Cystic Fibrosis bronchiectasis:
Bayer performed Study 16359 to explore the potential benefit of PHP-303 at a daily dose of 1 mg, administered once daily for 28 days in patients with NCFB. The drug was tolerated with no SAEs reported and showed inhibition of systemic NE, but it had no clear benefit on any pulmonary function endpoint. Interestingly, post-bronchodilator lung function test FEV1 increased slightly over the 28-day treatment interval (+26 ml) in drug treated, but not placebo treated subjects (-51ml, p = Not Significant).
9
The lack of clinical benefit may reflect the relatively limited overall drug exposure time or limited local exposure in the relevant lung compartment. The sponsor discontinued further development of the compound in this indication.
Peak Bio (pH Pharma) Conducted Phase 1 Clinical Trials with PHP-303
Peak Bio, (pH Pharma) after acquiring PHP-303 from Bayer, and to better characterize and improve the chances for future clinical success, repeated both the Phase 1 dose trials of single ascending dose (SAD) and multiple ascending dose (MAD) studies. The studies reconfirmed that PHP-303 was tolerated with no SAEs reported in either study. In both studies dose proportional PK exposure was observed. Dose-dependent NE inhibition was greatest in the 10 and 20 mg cohorts and steady state was achieved between 11 and 18 days in the MAD study. The studies were conducted at Alta Sciences in Kansas City, MO.
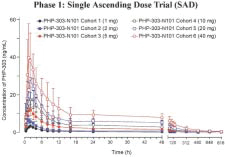
Phase 1: Multiple Ascending Dose Trial in overweight Obese Subjects (MAD)
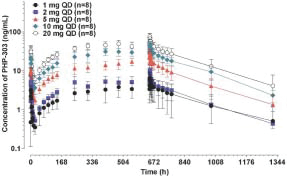

Neutrophil Elastase Inhibition Activity
As part of the MAD study, NE activity was measured to determine what doses of PHP-303 had the highest suppression of NE. The study demonstrated an Inverse Correlation between Drug Concentration and Neutrophil Elastase Activity. Sustained, dose-dependent suppression of NE activity was observed and appeared to be more complete at doses of 10mg and 20mg of
10
PHP-303. Those two dose cohorts demonstrated greater than 90% NE inhibition over a 24-hour period suggesting that a once-daily dose of 10-20mg can progress to our Phase 2 trials. Again, the PHP-303 doses of 10mg and 20mg demonstrated rapid onset < 2-4 hr and the drug was tolerated with few adverse events and no SAEs over a 28-day dosing period. The results suggest that PHP-303 has acceptable PK and tolerability profiles making it amenable for long-term chronic therapy in a disease such as AATD.
Phase 1: Multiple Ascending Dose Trial in overweight Obese Subjects (MAD) — Neutrophil Elastase Activity
Peak Bio Clinical Trial Strategy
Based on its preclinical and clinical data package, Peak Bio licensed PHP-303 (formerly Bay 85-8501) from Bayer and performed two additional phase 1 studies to extend the dose range in single ascending and multiple ascending dose trials. The drug demonstrates PK and tolerability profiles that make it suitable for an oral, once daily dosing schedule. Based on these data the company is preparing to initiate a phase 2a proof of concept study in patients with AATD and performing collaborative proof of principle studies in preclinical models of acute lung injury.
The clinical development and commercialization of Peak Bio’s products will be regulated by the Food and Drug Administration (USA), the Medicines and Healthcare Products Regulatory Agency (MHRA) in the UK and European Medicines Agency (for countries in the European Union). Other countries have national authorities such as Canada (Health Canada) that also regulate clinical development and commercialization of therapeutic products.
Peak Bio has approved Clinical Trial Applications in both UK and Ireland for a planned Phase 2 trial in patients with a genetic disease – alpha-1 antitrypsin deficiency disease. The specific gene responsible for the disease is found in individuals of Scandinavian origin, thus our target geographies for commercialization are in Western Europe and North America. We have not yet filed applications for clinical trials in the USA or Canada, nor anticipate the need to file in these jurisdictions in order to complete the approved clinical trial. Peak Bio does maintain an active IND with the FDA to support the follow-up to the completed clinical trials in healthy volunteers.
Peak Bio has filed a request for Orphan Drug Designation with the US FDA. We are in the process of responding to FDA questions. Under the Orphan Drug Act of 1983 (the “Orphan Drug Act”), the FDA may designate a product as an orphan drug if it is intended to treat a rare disease or condition, defined as one occurring in a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. If Peak Bio is able to obtain approval from the FDA, PHP-303 will be eligible the following benefits:
11
Ongoing healthcare legislative and regulatory reform measures may have a material adverse effect on our business and results of operations. Changes in regulations, statutes or the interpretation of existing regulations governing the regulatory clearance or approval, manufacture, and marketing of regulated products or the pricing, coverage and reimbursement thereof could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. We cannot predict the likelihood, nature, or extent of health reform initiatives that may arise from future legislation or administrative action. However, we expect these initiatives to increase pressure on drug pricing. Further, certain broader legislation that is not targeted to the health care industry may nonetheless adversely affect our profitability. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
Even if we obtain FDA approval of any of our product candidates, we may never obtain approval or commercialize such products outside of the United States, which would limit our ability to realize their full market potential. In order to market any products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties, and costs for us and may require additional preclinical studies or clinical trials which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. Satisfying these and other regulatory requirements is costly, time consuming, uncertain, and subject to unanticipated delays. In addition, our failure to obtain regulatory approval in any country may delay or have negative effects on the process for regulatory approval in other countries. We do not have any product candidates approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, our ability to realize the full market potential of our products will be harmed.
Drug marketing and reimbursement regulations may materially affect our ability to market and secure reimbursement for our products. We intend to seek approval to market our product candidates in both the United States and in selected foreign jurisdictions. If we obtain approval in one or more foreign jurisdictions for our product candidates, we will be subject to rules and regulations in those jurisdictions. In some foreign countries, particularly those in the EU, the pricing of drugs is subject to governmental control and other market regulations which could put pressure on the pricing and usage of our product candidates. In these countries, pricing negotiations with governmental authorities can take considerable time after obtaining marketing approval of a product candidate. In addition, market acceptance and sales of our product candidates will depend significantly on the availability of adequate coverage and reimbursement from third-party payors for our product candidates and may be affected by existing and future health care reform measures.
Even if we receive regulatory approval of any product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense, and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our product candidates. If any of our product candidates are approved, they will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies and submission of safety, efficacy, and other post-market information, including both federal and state requirements in the United States and requirements of comparable foreign regulatory authorities. In addition, we will be subject to continued compliance with current Good Manufacturing Practice, or cGMP, and Good Clinical Practice, or GCP, requirements for any clinical trials that we conduct post-approval.
Manufacturers and their facilities are required to comply with extensive FDA and comparable foreign regulatory authority requirements, including ensuring that quality control and manufacturing procedures conform to cGMP regulations. As such, we
12
and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any marketing application, and previous responses to inspection observations. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control.
Any regulatory approvals that we receive for our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a risk evaluation and mitigation strategies, or REMS, program as a condition of approval of our product candidates, which could entail requirements for long-term patient follow-up, a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, we will have to comply with requirements including submissions of safety and other post-marketing information and reports and registration.
The FDA may impose consent decrees or withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Products may be promoted only for the approved indications and in accordance with the provisions of the approved label. However, companies may share truthful and not misleading information that is not inconsistent with the labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses and a company that is found to have improperly promoted off-label uses may be subject to significant liability. The policies of the FDA and of other regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government
If we receive U.S. orphan drug designation for PHP-303 for the AATD indication and apply for and receive MHRA and/or EMA orphan drug designation, we may not be able to realize the benefits of such designation, including potential marketing exclusivity of our product candidates, if approved.
Regulatory authorities in some jurisdictions, including the United States and other major markets, may designate drugs intended to treat conditions or diseases affecting relatively small patient populations as orphan drugs. Under the Orphan Drug Act of 1983, the FDA may designate a product candidate as an orphan drug if it is intended to treat a rare disease or condition, which is generally defined as having a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In order to obtain orphan designation in the European Economic Area, or EEA, the product must fulfill certain challenging criteria. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as an orphan medicinal product if it meets the following criteria: (1) such product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; (2) either the prevalence of such condition must not be more than five in 10,000 persons in the EU when the application is made, or without the benefits derived from orphan status, it must be unlikely that the marketing of the medicine would generate sufficient return in the EU to justify the investment needed for its development; and (3) there exists no satisfactory method of diagnosis, prevention or treatment of such condition
13
authorized for marketing in the EU or if such a method exists, the product will be of significant benefit to those affected by the condition, as defined in Regulation (EC) 847/2000. A potential/ future designation of any of our product candidates as an orphan drug does not mean that any regulatory agency will accelerate regulatory review of, or ultimately approve, that product candidate, nor does it limit the ability of any regulatory agency to grant orphan drug designation to product candidates of other companies that treat the same indications as our product candidates.
Generally, if a product candidate with an orphan drug designation receives the first marketing approval for the indication for which it has such designation, the product is entitled to a period of marketing exclusivity, which precludes the FDA or foreign regulatory authorities from approving another marketing application for a product that constitutes a similar medicinal product treating the same indication for that marketing exclusivity period, except in limited circumstances. The applicable period is seven years in the United States and ten years in the EEA. The ten year period of market exclusivity in the EEA can be extended by a further two years if the product qualifies for a pediatric extension, but can be reduced to a period of six years if the orphan designation criteria are no-longer met after the fifth year Orphan drug exclusivity may be revoked if any regulatory agency determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the product to meet the needs of patients with the rare disease or condition.
Even if we obtain orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product candidate from competition because different drugs can be approved for the same condition in the United States or EEA. Even after an orphan drug is approved, the FDA or EMA may subsequently approve another drug for the same condition if the FDA or EMA, as applicable, concludes that the latter drug is not a similar medicinal product or is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care.
Our business is subject to risks associated with conducting business internationally. We source research and development, manufacturing, consulting, and other services from companies based throughout the United States, the EU, and select Asian countries and we will be planning and conducting our clinical trials in the United States, Canada, certain European countries, in the near-term and in the future.
Accordingly, our future results could be harmed by a variety of factors, including: economic weakness, including inflation, or political instability in varying economies and markets; differing regulatory requirements for drug approvals in non-European Union (EU) countries; differing jurisdictions could present different issues for securing, maintaining, or obtaining freedom to operate for our intellectual property in such jurisdictions; such jurisdictions; potentially reduced protection for intellectual property rights; difficulties in compliance with non-US laws and regulations; changes in non-U.S. regulations and customs, tariffs, and trade barriers; changes in non-U.S. currency exchange rates of the USD and currency controls; changes in a specific country’s or region’s political or economic environment, trade protection measures, import or export licensing requirements or other restrictive actions by the USA or non-U.S. governments; differing reimbursement regimes and price controls in certain non-U.S. markets; negative consequences from changes in tax laws; compliance with tax, employment, immigration, and labor laws for employees living or traveling outside of the USA; business interruptions resulting from geo-political actions, including war and terrorism, health epidemics and other widespread outbreaks of contagious disease, or natural disasters, including earthquakes, typhoons, hurricanes, floods, and fires; and business interruptions resulting from the COVID-19 pandemic or any other similar pandemic.
The United Kingdom’s (UK) withdrawal from the European Union (commonly referred to as Brexit) on January 3, 2020, may adversely impact our ability to obtain regulatory approvals of our product candidates and in particular PHP-303 in AATD in the European Union and may require us to incur additional expenses in order to develop, manufacture and commercialize our product candidates in the European Union. There is considerable uncertainty resulting from a lack of precedent and the complexity of the UK and EU’s intertwined legal regimes as to how Brexit (following the Transition Period) will impact the life sciences industry in Europe, including our Company, including with respect to ongoing or future clinical trials, among other aspects. Since a significant proportion of the clinical and regulatory framework for PHP-303 utilizes Irish investigators and the fact that the UK (Brexit) would be applicable to our business and our product candidate for AATD is derived from EU directives and regulations, the withdrawal could materially impact the regulations with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the UK or the EU.
14
PHP-303 for the Treatment of AATD
Overview
We are developing PHP-303 for the treatment of AATD, a potentially life-threatening rare, genetic condition that results in severe debilitating symptoms, including early-onset pulmonary emphysema. PHP-303 is a novel, selective, oral, once-daily, small molecule that is designed to inhibit the bioactive form of NE. Scientific data indicate that the increased risk of lung tissue injury in AATD patients may be due to inadequately controlled NE caused by insufficient AAT. We believe that by inhibiting NE, PHP-303 has the potential to reduce the destruction of lung tissue and stabilize clinical (lung) deterioration in AATD patients.
Background of Alpha-1-Antitrypsin Deficiency: AATD is a rare genetic disease that results in quantitative and/or qualitative defects in the AAT protein (https://www.lung.org/lung-health-diseases/lung-disease-lookup/alpha-1-antitrypsin-deficiency). Individuals can be characterized by the genotype of the SERPINA1 gene. In general, single nucleotide polymorphisms give rise to gene variants resulting in AAT proteins with single amino acid alterations. Although the AATD is an autosomal recessive disease, protein levels are regulated in an autosomal codominant manner, such that each allele contributes 50 percent to the serum AAT level. Most severely affected AATD patients include individuals who are homozygous for the Z allele (PI*ZZ), the null allele or the F (PI*FF) allele. These individuals experience emphysema at young age of onset with risk dramatically increased by exposure to cigarette smoke or occupational exposures. Patients with PI*ZZ genotype are also at high risk of liver cirrhosis, due to abnormal intracellular protein folding of mutant ATT resulting in damage to liver cells. The F allele results in a functionally abnormal protein without anti-protease activity, although AAT levels are normal. Non-smoker heterozygotes (PI*MZ or PI*SZ genotypes) experience a lower risk of lung disease, though risk increases in smokers.
There are estimated to be 70,000 to 100,000 individuals with AATD in the US. Worldwide, more than 3 million people are at risk of severe deficiency of AAT (https://www.rarediseaseadvisor.com/disease-info-pages/alpha-1-antitrypsin-deficiency-epidemiology-aatd). Similar to smoking related chronic obstructive pulmonary disease (COPD), AATD patients present clinically with dyspnea, cough, sputum production and wheezing. Lung function testing reveals fixed airflow obstruction and reduced diffusing capacity. Two distinct features of AATD are younger age of onset and a particular pattern of emphysema on lung imaging. The presentation of emphysema in a non-smoker or an individual with a family history of liver disease are also
15
suggestive. Laboratory diagnosis of AATD has also advanced: current approaches favor simultaneous testing of the serum AAT level and targeted genotyping for the most common variants. The natural history of AATD is variable, with liver dysfunction accounting for most mortality in patients less than 40 years old. Longitudinal studies demonstrate progressive loss of lung function in older individuals, with annual rates of FEV1 decline of 44 -110 ml/year in non-smokers and much higher rates in smokers with AATD. Mortality rates increase dramatically as FEV1 falls below 35% predicted levels.
In addition to smoking abstinence or cessation, supportive treatment for COPD includes nutritional support, pulmonary rehabilitation, prophylactic vaccines and supplemental oxygen. Guidelines support administration of bronchodilators and corticosteroids. Replacement therapy for AAT is used for individuals with low serum levels of AAT and airflow obstruction. Pooled human AAT is administered by weekly infusion and is associated with adverse events and vein collapse necessitating a central line with long-term weekly intravenous infusions and include such products as Prolastin, Aralast, Zemaira, Trypsone, Alfalastin, Glassia, and Respreeza. These agents have been approved in the United States and Europe based on biochemical efficacy or demonstration of increased plasma levels of AAT. The 2015 RAPID trial demonstrated that replacement therapy reduced the rate of decline of lung density assessed by High- Resolution CT imaging, suggesting likely clinical benefit. This effect was sustained for a four-year treatment period (Chapman et al., 2015, Lancet 386:360). Lung transplantation is an option for selected subjects with advanced emphysema. Experimental therapies in development for AATD include inhibitors of NE, (such as PHP-303), RNA interference agents, AAT correctors and gene therapy. None of these experimental approaches has yet demonstrated compelling clinical benefit nor gained regulatory approval.
Our Approach
Our product candidate for treating AATD is PHP-303, a 0.65 nM (in vitro IC50 value for inhibition of human NE), selective, oral, once-daily, small molecule that is designed to inhibit the bioactive form of NE (von Nussbaum et al., 2015). We believe that by inhibiting NE, PHP-303 has the potential to reduce the enzymatic destruction of lung tissue in these patients. pH Pharma (now Peak Bio Co., Ltd.) has established a research agreement with the Alpha-1 Project, a for-profit organization that pursues treatments for AATD to enhance the lives of these patients. We believe that this relationship with the Alpha-1 Project will help garner access to patients, potential investigators, clinicians, and important information on advances in the treatment of AATD. The convenient once-daily, oral dosing of PHP-303 could provide a significant advantage compared to the current treatments for AATD which are surgery or weekly intravenous AAT augmentation therapy.
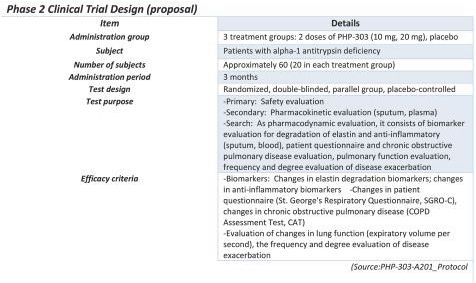
Planned Phase 2 Clinical Trial in AATD
We plan to evaluate the safety, tolerability, pharmacokinetic properties, and pharmacodynamic properties (elastin degradation and anti-inflammatory biomarkers, patient questionnaire, lung function) for two doses (10 mg and 20 mg) of PHP-303 in AATD patients and determine the optimal dosage for patients based on these parameters. pH Pharma, now Peak Bio Co., Ltd., has contracted with a principal investigator at the Royal College of Surgeons in Ireland and with a principal investigator at the University of Birmingham to conduct the Phase 2 AATD trial in Ireland and UK, each of whom have numerous publications and experience in the area of AATD clinical trials (For e.g., European Respiratory Journal 2019 53: 1900138; DOI: 10.1183/13993003.00138-2019). Data from this trial will inform the design of a pivotal trial with registrational intent.
16
PHP-303 for the Treatment of ARDS
Acute respiratory distress syndrome, ARDS, is acute, severe, and results in lung injury. It is defined as respiratory failure with bilateral lung opacities, not due to cardiac failure or fluid overload, occurring within one week of a known clinical insult or the onset of new pulmonary symptoms. Severity correlates with blood oxygen levels with mortality reaching 50% for individuals with severe disease. Although numerous conditions can predispose to ARDS; sepsis, aspiration pneumonitis, bacterial or viral pneumonia, trauma and inhalational injuries are commonly recognized complications that follow. The recent emergence of COVID-19. a pathogen with the ability to directly damage lung epithelial cells. has led to a large number of ARDS cases.
The National Center for Biotechnology Information (NCBI; Nov. 2019) estimated, that in the US, the incidence of ARDS ranged from 64.2 to 78.9 cases/100,000 people with 75% of those cases being moderate to severe (reviewed in Diamond et al., 2021, Acute Respiratory Distress Syndrome – StatPearls—NCBI). The incidence of ARDS in European countries has been reported as a range of 1.5 to 79 cases per 100,000 population and that ARDS rates were higher in North America, Oceania, and Europe compared to South America, Africa, and Asia (reviewed in Confalonieri et al., 2017, Eur Res Rev 26:160). We will be able to estimate the potential of PHP-303 therapy in ARDS, only when we know how COVID-19 has impacted the numbers (and severity) of ARDS worldwide.
The pathophysiology of ARDS begins with an exudative or fluid leakage in the lungs, a phase also termed diffuse alveolar damage. Prominent features included damage to the alveolar-capillary membrane and alveolar filling with fluid, protein debris and cellular infiltrates (Gonzales et al., 2015; Mathay & Zemans, 2011, Annu Rev Pathol, 6:147). Alveolar type II cell hyperplasia and formation of hyaline membranes are also recognized. A fibroproliferative phase follows characterized by resolution of alveolar and interstitial edema, continued proliferation of type II alveolar cells, squamous metaplasia, myofibroblast infiltration and collagen deposition. Some patients progress to a fibrotic stage with destruction of lung units and replacement by fibrous tissue (scarring) and cyst formation leading to greater morbidity and potentially mortality.
Accumulating evidence suggests that release of inflammatory cytokines and proteolytic enzymes from neutrophils, platelets, monocytes and macrophages may contribute to lung inflammation and epithelial damage characteristic of ARDS. This suggestive evidence is a central theme and hypothesis for the utility of PHP-303, in ARDS. NET formation has been correlated with COVID-19-associated ARDS severity and mortality in both preclinical models and human data. (Barnes et al., 2020, J Exp Med, 217; Adrover et al, 2022, JCI Insight,7:e15734). NET formation has also been implicated in small and large vessel thrombotic events recognized in the context of acute lung injury.
We are presently performing preclinical studies to examine the role of NE, NET formation and treatment with PHP-303 in several lung injury models. These studies are designed to determine if PHP-303 can inhibit NETosis in activated freshly isolated neutrophils; reach effective concentrations in the lung; and demonstrate efficacy in preclinical animal models of lung damage/COVID-19 infection with evidence of a decrease in NETosis.
The results of these studies will guide further preclinical and potentially clinical development of PHP-303 for the treatment of ARDS. We postulate that by inhibiting NE, PHP-303 has the potential to reduce the destruction of lung tissue and alleviate clinical deterioration in ARDS patients, particularly in those patients in which the development of their diseases is associated with high levels of NETosis.
Material Agreements
The Bayer Agreement
In March 2017, the Company entered into an Assignment, License, Development and Commercialization Agreement with Bayer (the “Bayer Agreement”) in regard to the assignment by Bayer to the Company of Bayer’s patents covering a neutrophil elastase inhibitor compound (which we refer to as PHP-303) and a license by Bayer to the Company of Bayer’s know-how for the development, manufacture and commercialization of the compound.
The Bayer Agreement is terminable by either party for material breach by the other party or in the event of bankruptcy or insolvency of the other party, in each case, subject to an opportunity to cure of 90 and 60 days respectively. The Bayer Agreement is also terminable by the Company at any time for convenience or in the event of Company safety concerns.
Alpha-1 Project Research Agreement
On June 28, 2019, the Alpha-1 Project, Inc. (TAP) entered into a sponsored Research Agreement with pH Pharma Co, Ltd, now referred to as Peak Bio Co., Ltd. TAP is a for-profit entity focused on identifying, funding, providing expertise and accelerating diagnostic and therapeutic interventions for patients with the rare disease AATD. Peak Bio proposed use of its
17
molecule PHP-303 for research activities for developing novel therapeutics based on PHP-303, a neutrophil elastase inhibitor targeting AAT and AATD. Peak Bio Co., Ltd. has entered into a funding agreement with TAP to support funding for activities related to Phase 2 clinical trials for PHP-303. $100,000 USD was provided by TAP for the research effort and for that consideration and upon execution of the agreement Peak Bio Co., Ltd. issued TAP 4,800 shares of its common stock at the most recent share price at that time.
The funding is for the sole purpose of the clinical trial activities of the PHP-303 in the treatment of AATD, however, Peak Bio Co., Ltd. shall be solely responsible for the management, conduct, oversight and generation of a final report of the Research Plan and results. TAP has the right to participate in any future external grant funding activities of Peak Bio Co., Ltd. and TAP may elect to participate in such funding on a “most favored nations” basis. Peak Bio and TAP formed a “steering committee” to oversee the funded activities during the term of the agreement and to act as a forum for TAP to provide reasonable comments and input on the scientific progress of the research.
TAP and Peak Bio Co., Ltd. have rights to publications of completed data within scope of not compromising of Peak Bio’s confidential information or proprietary know-how or trade secrets or that could compromise securing patent protection of any inventions arising from the research plan. In addition, Peak Bio Co., Ltd. is required to acknowledge the support of TAP in any future publications from the research. Each of TAP and Peak Bio retained ownership and control of their respective works of authorship, know-how, information, and data, and intellectual property therein, that were in existence as of the date of the Research Agreement or are later generated outside of scope of the research plan. Peak Bio Co., Ltd. will own all arising data and intellectual property arising from the research plan, and accordingly, TAP hereby assigns to Peak Bio Co., Ltd. (and shall cooperate with Peak Bio Co., Ltd. to execute assignment documents as necessary to perfect the assignment to Peak Bio Co., Ltd. of any and all such intellectual property rights arising out of the research plan). TAP will acquire no ownership interest or other rights or licenses of any kind whatsoever in any intellectual property, data or results or any patents or patent applications or know-how arising out of the research plan.
TAP will be entitled to receive milestone payments as a percentage of total funding, with such payments due if, as and when there occur the following events in the development and commercialization of any product derived from the research plan. Milestone payments in aggregate will not exceed 350% of any money funded by TAP to Peak Bio for regulatory approval, achievement of first commercial sale and after cumulative net sales considerations. To date, the amount of the funded research proceeds provided to Peak Bio Co., Ltd. by TAP is $100,000 USD that would be subject to this payment calculation.
In January 2021, we entered into an agreement with the U.S. DoD to perform “Preclinical Studies of PHP-303, a Neutrophil Elastase Inhibitor to Treat Severe COVID-19 Associated Acute Respiratory Distress Syndrome and Lung Injury”. We have been awarded up to $3,954,626 in expense reimbursement for preclinical studies to obtain pharmacokinetic data with PHP-303, a neutrophil elastase (NE) inhibitor, and to determine if PHP-303 can inhibit NETosis and/or oppose the damaging effects of the large amounts of NE released into tissue during this biological process. And, if PHP-303 does inhibit NETosis, does this improve outcomes in animal models of acute lung injury including a COVID-19 model. Through this effort with the DoD, we intend to identify whether, based on preclinical data, PHP-303 looks like a potential treatment for ARDS that occurs in a subset of some of the most ill COVID-19 patients. The work being done under this grant could also establish a preclinically-based foundation of data for the potential utility of PHP-303 treatment of patients with ARDS due to other underlying bacterial/viral infections and/or in other disease states in which high levels of neutrophil elastase and/or NETosis are part of the disease etiology. COVID-19 is most easily spread among individuals in close contact and has a disproportional impact on persons of advanced age, so PHP-303 may offer significant benefit to active-duty warfighters, veterans, and their families aligning our capabilities with the DoD’s mission to respond to the COVID-19 pandemic and to provide medical countermeasures for the warfighter and the nation against present and future biological and chemical agents of concern. If the studies performed under this grant indicate that PHP-303 could be a promising treatment for COVID-19-related ARDS, we are positioned such that we could take PHP-303 into a phase 2 clinical trial. PHP-303 is already in clinical development. We believe our partnership with the DoD could enable us to leverage our compound, PHP-303, in additional therapeutic applications, such as in a future viral pandemic if it involves lung damage induced by inadequate endogenous opposition of NE and/or NETosis. We own all study data generated under the DoD Agreement, whether generated by us or the DoD, and the DoD will have no ownership interest in any inventions resulting from the agreement. Accordingly, any therapeutic or prototype developed under the agreement will be owned by us. Under the DoD agreement, we are required to use commercially reasonable efforts to complete specified research activities for the prototype project based on the estimated cost for such prototype. In connection with the DoD Agreement, we are eligible to receive up to $3,954,626 in the aggregate from DoD, subject to continued compliance with the terms of the DoD agreement and future pricing strategy. We are not obligated to pay any royalties or other future consideration under this agreement. The DoD agreement was extended to expire March 31, 2023, subject to completion of the prototype project as determined by a DoD official in accordance with key technical goals established for the project or results that justify completion. The DoD may terminate the agreement in its entirety for convenience or in whole or in part for our material breach of the agreement.
18
The following statement are required to accompany any public release of information pertaining to the agreement:
Manufacturing
We have been using raw materials and finished products supplied directly from Bayer in Germany, which has the highest level of CMC (manufacturing/quality) technology among global pharmaceutical companies and have used them in our preclinical and initial clinical trials. For our finished products, we recently transferred technology to Catalent, the largest contract producer in the United States, for manufacture, allowing for the establishment of a very stable and efficient partnership for the supply of clinical investigational drugs and the development of commercial products.
|
Although PHP-303 is currently in the early clinical development stage, we took over the technology after reaching a high process development level in Bayer to the extent that high-purity raw materials of several kilograms (Kg) or more can be produced, so late clinical supply is possible. The finished product has also been developed with a stable formulation, so our CMC development process has been completed from adding of high-capacity tablets to large-scale commercial production. |
We do not own or operate facilities for the manufacturing of our product candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We have entered into manufacturing agreements with a number of drug substance, drug product, and other manufacturers and suppliers for PHP-303, and we intend to enter into additional manufacturing agreements as necessary. Following our license of PHP-303, we acquired certain clinical trial materials and we plan to outsource production of further clinical supplies to our own manufacturing partners. We also intend to outsource certain product formulation trials. We expect that drug product pre-validation and validation batches will be manufactured to satisfy regulatory requirements when we progress products to late-stage trials.
We do not yet have any contractual relationships for the manufacture of commercial supplies of PHP-303, and we intend to enter into contractual relationships for commercial supplies following approvals of our investigational therapies. Any batches of product candidates for commercialization will need to be manufactured in facilities, and by processes, that comply with the requirements of the FDA, the EMA, and the regulatory agencies of other jurisdictions in which we are seeking approval. We utilize our internal resources and experienced consultants to manage our manufacturing contractors and ensure they are compliant with current good manufacturing practices.
Commercialization, Sales, and Marketing
We do not have our own marketing, sales, or distribution capabilities. In order to commercialize our product candidates, if approved for commercial sale, we must either develop a sales and marketing infrastructure or collaborate with third parties that have sales and marketing experience. We plan to establish our own sales and marketing organization in the United States and Europe for PHP-303 and may seek to directly commercialize our future product candidates. In markets for which commercialization may be less capital efficient for us, we may selectively pursue arrangements with third parties in order to maximize the commercial potential of PHP-303, and our future product candidates. We intend to seek to and may enter into one or more strategic relationships with third parties for PHP-303 to undertake the next phase of clinical development and, if approved, for commercialization, enter into strategic relationships with third parties for further clinical development and/or commercialization of PHP-303.
19
Competition AATD
We compete directly with other biopharmaceutical and pharmaceutical companies that focus on the treatment of AATD or ARDS. We may also face competition from academic research institutions, governmental agencies, and other various public and private research institutions. We expect to face increasingly intense competition as new technologies become available. Any product candidates, including PHP-303 that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Current approved treatments for alpha-1 antitrypsin deficiency

We consider PHP-303’s current closest potential competitors for the treatment of severe AATD to be alpha1-proteinase inhibitors that are administered intravenously in AAT augmentation therapy. Currently, there are four inhibitors on the market in the United States: Grifols’s Prolastin-C, Shire’s Aralast, CSL’s Zemaira and Kamada’s Glassia. Kamada is also investigating an inhaled version of augmentation therapy and Apic Bio and Adverum are in the early stages of developing gene-therapy approaches for AATD. Santhera has in licensed an inhaled NEI and is planning a multiple ascending dose study, with the initial indication targeted being cystic fibrosis.
In Phase 2 development is the Mereo BioPharma compound, alvelestat, which was licensed from AstraZeneca. On May 9, 2022, Mereo announced positive top-line efficacy and safety results from its “ASTRAEUS” a Phase 2 study of the investigational oral NEI, alvelestat (MPH-966), in patients with severe AATD-associated emphysema. The double-blind, placebo-controlled study evaluated two different doses of alvelestat (high or low [120mg] dose) administered twice daily versus placebo over a 12-week period (with evaluation at weeks 4, 8 and 12), and its effect on three primary biomarker endpoints associated with AATD-related lung disease, blood NE activity, a NE-driven target breakdown product of fibrogen, A@-val360, and the elastin breakdown product, desmosine. At the high dose, alvelestat demonstrated statistically significant changes versus placebo in all three primary biomarker endpoints that included ~90% inhibition of NE at the high undisclosed dose.
It is our belief these Mereo data support target and pathway engagement in AATD patients by a NEI at clinically available doses. To the extent these are class effects, these data potentially de-risk the Peak Bio PHP-303 AATD program. Based on PHP-303’s attributes, Peak Bio plans to move it forward into phase 2 trials with oral administration at doses of 10 mg and 20 mg once daily. We have been and continue to follow closely any biomarkers assessed in AATD clinical trials, and data around them, but do believe that the regulatory approach still needs better assessment at this juncture. As we learn more, we intend to capture as much intelligence around a good clinical and regulatory strategy moving forward. Overall, the Mereo NEI positive results contribute to our belief that PHP-303 is well positioned for possible clinical development.
20
As discussed previously, PHP-303 has demonstrated greater than 90% NE inhibition at doses of 5mg, 10mg and 20mg as a once daily oral administration. Additionally, PHP-303 appears to inhibit the bioactive form of NE. This will be explored more in-depth as we progress the program forward.

Competition ARDS (Acute/Subacute, Covid-19)
The competitive nature of ARDS treatments (Acute, Sub-acute, and Chronic) as they relate specifically to traditional definitions of ARDS or most recently the global pandemic associated with COVID-19-associated ARDS, has many facets. There is an evolving science around COVID-19 and ARDS in which it is potentially important to consider the roles of Acute, Chronic, and now Sub-Acute COVID-19 and the various treatment options for these conditions that are both on the market and still experimental. In many instances there is cross-over in the treatments associated with ARDS and COVID-19 and very dependent on severity of disease. Since we are focused on pharmaceutical treatments and specifically immunomodulation and the impact of the inflammatory cascade associated with ARDS in COVID-19 and ARDS we will forgo discussion of other therapies involving mechanical ventilation strategies, the evolving utility of ECMO and antiviral therapies.
Immuno-modulators: As discussed previously, a subgroup of patients with severe COVID-19 and/or ARDS demonstrate a serious immune response known as cytokine release syndrome. Current suggestions are to screen all patients with severe COVID-19 for this syndrome, even though it’s usually rare.
Corticosteroids and monoclonal antibodies: To treat hyper-inflammation, immune-modulator drugs such as corticosteroids or monoclonal antibodies are emerging as therapies but with equivocal results. For example, the monoclonal antibody therapies being utilized either experimentally or approved were originally developed to treat conditions such as rheumatoid arthritis. They may limit lung inflammation but also inhibit immune responses and pathogen clearance, according to observational studies on SARS/MERS patients.
Due to the lack of reliable evidence from large-scale randomized clinical trials, there has been uncertainty with regards to the effectiveness of corticosteroids in COVID-19 patients. Preliminary findings from a recent study report shows that a low dosage of dexamethasone reduces 28-day mortality in patients with COVID-19 who are receiving respiratory support. The results have many caveats but generally suggest corticosteroid therapy may be beneficial. However, it is unclear which ARDS patients are most likely to benefit from this treatment, because ARDS patients consist of heterogeneous populations with likely different inflammatory pathways leading to ARDS.
With regards to monoclonal antibody approaches there are two monoclonal antibodies, tocilizumab and sarilumab [antibodies that block interlukin-6 receptor (IL-6R)], that have been re-purposed for the treatment of ARDS in COVID-19 patients. Both agents were approved for the treatment of rheumatoid arthritis. Tocilizumab has been tested in several COVID-19 clinical studies with sometimes conflicting results, but overall the data suggests a mortality benefit (reviewed in https://www.uptodate.com/contents/covid-19-management-in-hospitalized-adults?search=covid-19-management-inhospitalized%20adults&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1). Sarilumab treatment, in COVID-19 trails, on the other hand, has had less robust effects. COVID-19 has now moved into its endemic phase and some new stains are better at evading our immune system responses, therefore the search for new treatments continues.
21
We believe that PHP-303 could potentially serve as one of several novel approaches to the treatment of ARDS and may also have potential utility in treating “long-COVID”. We now see a growing cohort of individuals suffering from this condition. Long-COVID, or similar conditions, are likely to greatly impact patients’ long-term quality of life including hampering their ability to even do the simplest of tasks associated with daily living or work. Resources-permitting, we may investigate the role of PHP-303 in inflammation, and inflammation involving long COVID-19.
Antibody-drug-conjugates (ADC):
Peak Bio has leveraged two decades of industry learnings in expanding an important area of the antibody-drug-conjugate (ADC) field allowing for highly targeted treatments in cancer. Despite the continued scientific advancements in the cancer field that has led to the many incremental improvements in patient cancer survival, there continues to be a need for ADCs that not only deliver antibody-directed payloads selectively to their tumors, but to also release them via improved linker technology avoiding the potential for significant off-target toxicities. Secondly, based on the success of immune checkpoint inhibitors, we believe that adding an immunomodulatory effect to our toxin(s) that engages our immune systems to assist in the cancer killing would contribute to increased tumor regression.
These incremental improvements in cancer treatments for patients and specifically ADCs has also led to the growing commercial success of ADCs currently on the market and likely for those currently in development. A quick scan of the deal flow associated with ADCs over the past 5 years is encouraging both from their continued clinical and commercial success. We believe, Peak Bio is well-positioned to take advantage of this field with our proprietary ADC based therapeutics. We are poised to launch our platform of proprietary in-house technologies that enable us to design ADCs that we believe potentially offers improved ADC characteristics such as a dual mechanism of action (MoA), immune stimulation, and being refractory to multi-drug resistance (MDR)- related forms of resistance.
Antibody drug conjugates are an established therapeutic approach in oncology where an antibody is used to selectively deliver a potent toxin directly to tumor cells. The goal is to focus and maximize the ADC’s activity at the tumor site, sparing normal tissues and organs, resulting in a wide therapeutic index. There are four important aspects of an ADC approach/ program- 1) an antigen, a carbohydrate or protein moiety that is expressed preferentially on tumor cells, or cells in the tumor microenvironment contributing to its survival, 2) an antibody, a protein from the immunoglobulin family that is highly selective for seeking out the tumor antigen wherever tumor cells reside, 3) a toxin that is often a small molecule or a protein (also called payload or warhead) that is 10-10,000 times more potent than conventional chemotherapy, or sometimes a chemotherapy itself and 4) a linker that serves to attach the small molecule to the antibody.
Cell-surface receptor internalization and recycling is a process that is physiological to normal and cancer cells and most ADCs that complex with target antigen receptors are internalized within the cancer cells, delivering, and releasing the payload, triggering cell death. Additionally, some ADCs also may have a feature engineered into their linkers that allow the payload to be released in the tumor environment by exploiting some feature specific to a tumor, for e.g., low pH conditions, or high tumor expression of certain enzymes such as beta-glucuronidase.
ADCs have demonstrated therapeutic efficacy in clinical trials and an increasing number of ADCs are standards of care in various hematologic and solid cancers (see below). Most of ADC research has primarily focused on antigen and target discovery as opposed to payload discovery where Peak Bio is making progress.
22
FDA-approved ADCs through 2022
ADC
|
Trade name
|
Target
|
Company
|
Indication
|
Approval Year
|
Microtubule inhibitor payload class |
|||||
|
|
|
|
|
|
Brentuximab vedotin |
Adcetris |
CD30 |
Seattle Genetics, Takeda |
relapsed HL and relapsed sALCL |
2011 |
|
|
|
|
|
|
Trastuzumab emtansine |
Kadcyla |
HER2 |
Genentech, |
HER2-positive metastatic breast cancer (mBC) following treatment with trastuzumab and a maytansinoid |
2013 |
|
|
|
|
|
|
Polatuzumab vedotin-piiq |
Polivy |
CD79 |
Genentech, |
relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) |
2019 |
|
|
|
|
|
|
Enfortumab vedotin |
Padcev |
Nectin-4 |
Astellas/ Seattle |
adult patients with locally advanced or metastatic urothelial cancer who have received a PD-1 or PD-L1 inhibitor, and a Pt-containing therapy |
2019 |
|
|
|
|
|
|
Belantamab mafodotin-blmf |
Blenrep |
BCMA |
GlaxoSmithKline |
adult patients with relapsed or refractory multiple myeloma |
2020 |
|
|
|
|
|
|
Tisotumab vedotin-tftv |
Tivdak |
Tissue factor |
Seagen Inc |
Recurrent or metastatic cervical cancer |
2021 |
|
|
|
|
|
|
Disitamab vedotin |
Aidixi |
Her2 |
Remegen |
HER2 expressing urothelial cancer |
2021 |
|
|
|
|
|
|
Mirvetuximab soravtansine |
Elahere |
FR alpha |
Immunogen |
Platinum-resistant ovarian cancer |
2022 |
|
|||||
DNA-acting payload class |
|||||
|
|
|
|
|
|
Gemtuzumab ozogamicin |
Mylotarg |
CD33 |
Pfizer/ Wyeth |
relapsed acute myelogenous leukemia (AML) |
2017
2000 |
|
|
|
|
|
|
Inotuzumab ozogamicin |
Besponsa |
CD22 |
Pfizer/ Wyeth |
relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia |
2017 |
|
|
|
|
|
|
Loncastuximab tesirine-lpyl |
Zynlonta |
CD19 |
ADC |
Large B-cell lymphoma |
2021 |
|
|||||
Topoisomerase I inhibitor payload class |
|||||
|
|
|
|
|
|
Trastuzumab deruxtecan |
Enhertu |
HER2 |
AstraZeneca/ Daiichi Sankyo |
adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2 based regimens. unresectable or metastatic breast cancer patients with HER2-low lesions and NSCLC patients with HER2-mutations |
2019 2022 |
|
|
|
|
|
|
Sacituzumab govitecan |
Trodelvy |
Trop-2 |
Immunomedics |
adult patients with metastatic triple-negative breast cancer (mTNBC) who have received at least two prior therapies for patients with relapsed or refractory metastatic disease |
2020 |
|
|||||
Peptide toxin class |
|||||
|
|
|
|
|
|
Moxetumomab pasudotox |
Lumoxiti |
CD22 |
AstraZeneca |
adults with relapsed or refractory hairy cell leukemia (HCL) |
2018 |
23
Peak Bio antibody catalog:
We leverage our team’s deep experience and proficiency in oncology research for selecting our target antigens. If there is scientific validation from academia or industry in peer-reviewed journals or clinical validation by any form of oncology therapeutic, then these targets are given weighted preference. Here the primary focus is directed towards engineering in desired features in combination with our novel payload(s). Also, we utilize our expertise in data mining of publicly available clinical data sets to seek out under-represented targets that may be relevant to hematologic and solid cancers.
Once short-listed, we perform literature searches for published monoclonal antibodies that have been described to target those candidates. Using such information, we generate a catalog of proof-of-concept (POC) antibodies to be used in combination with our proprietary toxins to create differentiated ADCs. Where needed, we may also generate our own monoclonal antibodies in normal or humanized mice. To date, we have generated over twenty POC antibodies and expressed them in monomeric IgG format in Chinese hamster ovary cells for exploratory evaluation at laboratory scale. Using processes described above and platforms such as Oncomine and Megasampler, we have identified over 50 cancer-associated targets that we intend to evaluate with antibody-based therapeutics.
Need for new ADC Toxin strategies:
The ADC field started with the most potent toxins- for e.g., calicheamicin (Wyeth/ Pfizer). After observing the pre-clinical and clinical toxicities of these toxin warheads, the field moved down the potency scale towards the maytansines and the auristatins- monomethyl auristatin E/ F abbreviated MMAE/ MMAF (Seattle Genetics/ SeaGen), Auristatin Au101 (Pfizer)- and towards the camptothecins (Immunogen). This is where the moderate potency payload containing ADCs achieved clinical successes with multiple different targets. Those ADC programs working with the more potent toxin warheads focused on linker stability and identifying targets with low to normal tissue expression to achieve acceptable therapeutic indices.
Of the 14 ADCs that are currently in the clinic, eight feature microtubule inhibitors- vedotin/ MMAE (5), mafodotin/ MMAF (1), soravtansine/ DM4 (1) and emtansine/ DM1 (1); two feature topoisomerase inhibitors- govitecan (1) and deruxtecan/ DXd (1); three feature DNA-acting payloads- ozogamicin/ calicheamicin (2) and tesirine (1); and lastly, one featuring a peptide toxin from the bacterium Pseudomonas aeruginosa- pasudotox (1).
Based on published results, even after opting for lower potency, these payloads have been linked to the following toxicities in multiple approved ADCs- MMAE (neutropenia, peripheral neuropathy and gastrointestinal), MMAF (thrombocytopenia and ocular), DM1 (thrombocytopenia, neutropenia and gastrointestinal), calicheamicin (thrombocytopenia, gastrointestinal and hepatic veno-occlusive disease) and DXd (stomatitis and interstitial lung disease).
Research and investment into novel payloads are also necessary from the viewpoint of durable efficacy and reducing the potential for resistance. Like several chemotherapies, ADC payloads such as MMAE are substrates of MDR pumps (also called ABC transporters or P-glycoprotein) and MDR-mediated resistance to ADC therapy are being highlighted in scientific publications. Finally, topoisomerase I mutations associated with resistance to camptothecin/ irinotecan family of payloads have also been identified.
The above factors limit the ability of current ADC payloads to maintain durable tumor regression and reemphasize the need for new ADC payloads in drug development. We, therefore, focused on payload research, to provide optionality for our patients.
24
Our strategy was to select for a payload with nanomolar potency with sufficient cytotoxic ability and select for MoA that would include a second complementary punch to provide additional potency.
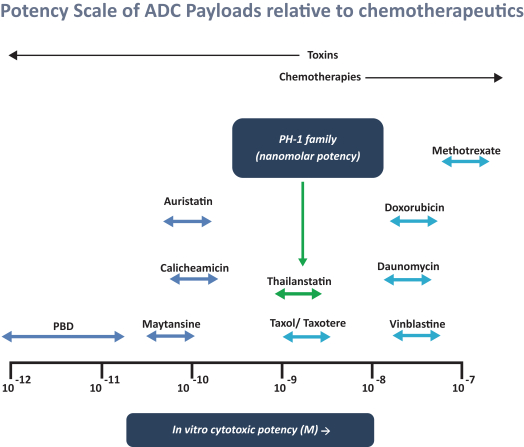
Given the clinical success of various checkpoint inhibitors, it was reasonable that the modulation of the innate or adaptive immune system by an ADC payload could perform this complementary, yet orthogonal function. Immune cells are nature’s defense against foreign invaders- bacteria and viruses- but fail to identify and destroy cancer cells as a) the latter are derived from self, and b) immune cells are prone to active suppression by the tumor. We leveraged our understanding of oncology, immunology, and immuno-oncology to prioritize biologies that would have this dual activity. It is this concept that we believe allows for a potentially more robust cancer therapeutic approach.
25
In theory, these dual acting payloads would have:
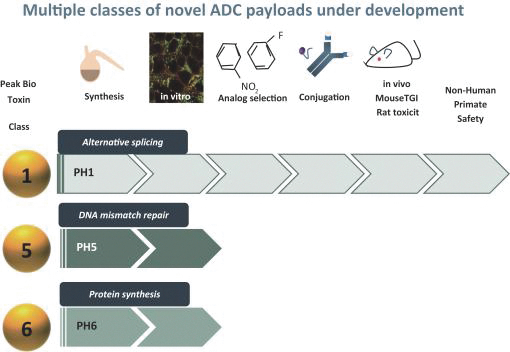
PH-1 family of payloads targeting splicing:
a) Biology of splicing:
In higher organisms, eukaryotes, coding regions of the genome called exons are interrupted by noncoding sequences known as introns or “junk DNA”. Genes are expressed by a two-step process. A first step called transcription that expresses deoxyribonucleic acid (DNA) as an intermediate called ribonucleic acid (RNA). It is at this intermediate step that introns are removed to generate a mature and functional mRNA molecule. The splicing machinery, known as spliceosomes, comprises five small nuclear ribonucleoprotein particles (snRNPs) that interact with more than 200 different auxiliary and regulatory factors that work in concert to precisely remove introns and connect the coding exons end-to-end and generate the final “mature” RNA. The removal of introns from mRNA is referred to as alternative splicing (AS) or simply splicing. In step two, the mature RNA is translated into various functional proteins.
Over the past 15 years, the role of alternative splicing in human disease has become apparent. When the human genome project was completed, in silico analysis predicted that at least 75% of human genes underwent splicing and that 15-50% of genetic diseases were related to aberrant splicing events. With growing knowledge in the areas of algorithms that accurately predict splicing, and advances in areas of high-throughput validation of spliced protein isoforms (proteomics and immunopeptidomics), we now know that this percentage is even higher.
26
We now know splicing has been implicated in malignant progression of hematologic and solid tumors, enhancing development of features such as increased cell proliferation, invasion, and recruitment of tumor blood vessels. This happens in different ways:
Therefore, we hypothesized that ADC payloads targeting splicing may have the following effects:
Thus, having identified a biology for ADC payload that may simultaneously a) induce cytotoxicity by a mechanism different from conventional ADCs, and b) stimulate and activate immune cells, we turned our focus towards spliceosome modulators.
b) Thailanstatin payloads:
In nature, bacteria and fungi are the source of many toxins.
One such bacterium Pseudomonas sp. 2663 produced a small molecule toxin termed FR901464 or Spliceostatin A. FR901464 biosynthesis by Pseudomonas sp. 2663 was performed by a cluster of genes called fr9. Screening of fr9-like gene clusters in other bacteria identified a bacterium by the name of Burkholderia thailandensis MSMB43, that produced the toxin Thailanstatin. Research groups then proceeded to purify Thailanstatins A, B and C from fermentation broth and demonstrated its cytotoxic effect on cell lines and confirmed its anti-splicing MoA.
We focused on Thailanstatin as an ideal ADC payload with the potential to induce cytotoxicity and immune activation creating two distinct ways to enhance the killing of targeted cancer cells.
27
Over time, we generated and evaluated a series of 13 non-natural Thailanstatin (Th) analogs through structure activity relationship (SAR) studies and optimized for potency and metabolic stability. These naked analogs were evaluated for potency and permeability against a panel of a dozen cell lines. Those analogs that were amenable to linker addition and suited for ADC development were given preference. Test conjugations of Th linker-toxin analogs were performed with clinical-grade Trastuzumab, purified to remove free toxin, and laboratory-grade ADC preparations were evaluated against a panel of Her2-high, Her2-low, and Her2-negative cell lines to determine baseline levels of ADC potency and specificity. Using this approach, we made SAR-based changes in three generations, making modifications, and optimizing for potency, stability, specificity, and conjugation ability as we went along. The first Generation yielded analog 3 (ThA3), second Generation yielded analog 9 (ThA9), and the third Generation gave us analog 13 (ThA13). Based on our results, a derivative of ThA13 was selected as our final analog.
Unlike conventional ADC toxins where linkers and toxins are separate and modular, and one linker is applied to multiple toxins for e.g., alanine-alanine, valine-valine, valine-alanine, or valine-citrulline formats; Th—compatible linkers had to be designed and then built into the synthesis route of the toxin analog. Subsequently, the synthetic route for each toxin analog and its derivative linker toxin was determined, then optimized for better yield at each step. Unlike other ADCs, where the linker and the toxin are coupled in the last steps, Th-linker toxins were assembled during the chemosynthetic process. Furthermore, where possible, we made both non-cleavable and cleavable versions of linkers (L) for conjugation to either lysine or cysteine amino acids.
The Thailanstatin ThA13 suite comprising the PH-1 family of validated linker-toxins (L-Ts) comes with a set of seven related molecules with distinct ADC features that have been extensively characterized in vitro and in vivo as Her2 ADCs:
All above ThA13 L-Ts and ADCs derived from them are collectively referred to as the PH-1 ADC platform. Stability and performance of these L-Ts has been characterized on at least two different antibodies targeting different antigens, Her2 and Trop2, yielding similar results. After proving selectivity on target-positive (vs target-negative cells), a measure of off-target activity, we tested their ability to shrink pre-implanted target-positive 200 mm3 sized-tumors in therapeutic mode. Of these, the lead L-T that yielded the maximum anti-tumor growth inhibition (TGI) in in vivo xenograft studies as a Her2 or Trop2 ADC conjugate was the non-cleavable L-T ThA13L22, later renamed PH1.
After reviewing the adverse effects associated with various non-cleavable vs cleavable ADCs for e.g., T-DM1 vs T-DXd, we concluded that PH1 ADCs in non-cleavable format are likely to be associated with fewer serious toxicities due reduced systemic exposure of the free payload. To corroborate this viewpoint, we refer to the recent meta-analysis performed by Wynn et al (DOI: 10.1200/JCO.2022.40.16_suppl.3032 Journal of Clinical Oncology 40, no. 16_suppl (June 01, 2022) 3032-3032) of commercially available ADCs that showed that ADCs with non-cleavable linkers were associated with significantly less toxicity than those with cleavable linkers. ADCs with cleavable linkers tended to have greater instances of >Grade 3 adverse events (AEs). 47% of patients (total 1082) treated with 7 cleavable L-Ts developed AEs > grade 3 compared to 34% of patients (total 1335) treated with 2 non-cleavable L-Ts. This was significantly different (weighted risk difference -12.9%; 95% Confidence Interval ranging from -17.1% to -8.8%). There was also a significant difference favoring non-cleavable ADCs for > grade 3 neutropenia (-9.1%; 95% CI -12% to -6.2%) and > grade 3 anemia.
We therefore decided to proceed with the non-cleavable L-T PH1 as a) our non-cleavable format was associated with better TGI conjugated to Her2 and Trop2 antibodies, b) 57% and 14% of TNBC patients treated with cleavable Trop2 ADC (Trodelvy®) presented with >Grade 3 hematologic and gastrointestinal AEs, respectively (Bardia et al 2019)., and c) non-cleavable ADCs were likely to be associated with less systemic exposure and toxicity.
Microtubule inhibitor payloads are known to induce immunogenic cell death and/ or induce anti-tumor immunity in combination with checkpoint inhibitors for e.g., MMAE in Adcetris® and Tivdak®, and DM1 in Kadcyla®. Therefore, we compared PH1 with DM1 in their abilities to induce immunogenicity over and above that of vehicle control treatment.
We performed an unbiased comparison of PH1-, DM1- and DMSO- treated human gastric cancer cells by RNA sequencing of all genes and looked for sequences that would give rise to aberrant proteins (neoepitopes). After identifying the normal and novel RNA species, we highlighted the neoepitope-containing species that increased in response to DM1 vs DMSO and PH1 vs DMSO treatments (red dots in figure below). As expected, DM1-treated cells contained 89 more neoepitope-containing RNA species than control, proving that microtubule inhibitor payloads are indeed immunogenic. However, PH1-treated cells contained
28
765 neoepitope-containing species, suggesting that PH1 payload may be highly proficient at recruiting immune cells to the tumor and impacting immune-cell mediated cancer cell death. We believe that this ability to recruit immune cells may evolve into an important future differentiator for our PH1 program and current and ensuing ADC constructs.
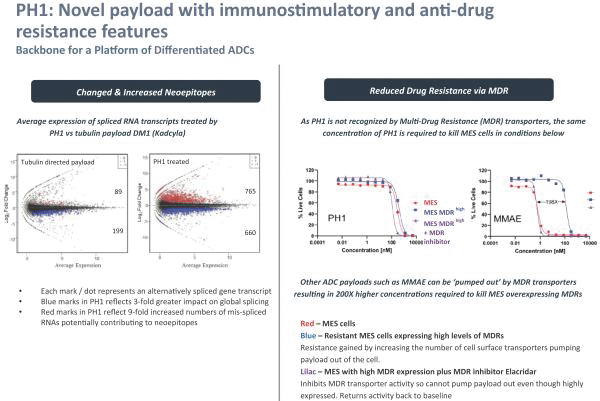
When we looked for genes that were negatively impacted and reduced in quantity (blue dots in figure above), we found 660 different RNA species were depleted in PH1-treated cells. Likely due to the combined effects of our payload targeting splicing with NMD-mediated degradation, these RNAs encompassed genes fueling proliferation, growth, and malignancy, and therefore, vital to the survival of the cancer cell. This was due to PH1’s global impact on splicing and largely reflected this payload’s MoA as opposed to DM1, where the payload functions by targeting microtubules.
We then evaluated PH1’s performance vs auristatins such as MMAE that are substrates of multi-drug transporter (MDR) pumps. MMAE is actively pumped out of the cancer cells, giving rise to ADC resistance. Even within the normal course of ADC administration there are concerns about increased resistance to these payloads over time and why potentially this attribute could serve as an important market differentiator.
We evaluated PH1 and MMAE’s ability to kill MES cells with normal vs high levels of MDR. We found that MMAE, not PH1, was recognized by these pumps, and the presence of high levels of these pumps reduced the in vitro cytotoxicity (IC50) of MMAE 198-fold. The presence of high levels of these pumps had no significant effect on the cytotoxic potency of PH1, as the latter were not substrates and therefore not recognized by MDRs nor pumped out of the cell. The MDR-specific inhibitor Elacridar prevented MDR pumps in MDR-high MES cells from pumping MMAE payload out of the cell, allowing its accumulation, and returning MMAE’s cell killing potency back to baseline. This finding confirmed that the loss of MMAE’s potency was specific to increase in the elevated number of MDR pumps and did not occur even in the presence of increased numbers, when we blocked MDR’s ability to pump out the payload using Elacridar. This is important because MDR transporters are known to be implicated in the emergence of resistance against many chemotherapies, including some ADC payloads. Furthermore, if MDRs recognized PH1, it would have reduced its potency, and restricted its cytotoxicity to only targets that were highly expressed in cancer cells.
c) Properties of PH1 ADCs:
We used the Her2-targeted antibody Trastuzumab to tease out the differentiating properties of PH1 ADCs. As trastuzumab is an FDA-approved therapeutic, both as a naked antibody and as an ADC (trastuzumab emtansine, also known as T-DM1, or
29
Kadcyla®), with well-published pre-clinical TGI and toxicology profiles in animal models, we decided to use clinical grade Trastuzumab for conjugation with PH1. The resulting ADC, Tras PH1, was benchmarked against Kadcyla® to determine how our payload would fare relative to microtubule targeting payload DM1 on the same antibody backbone.
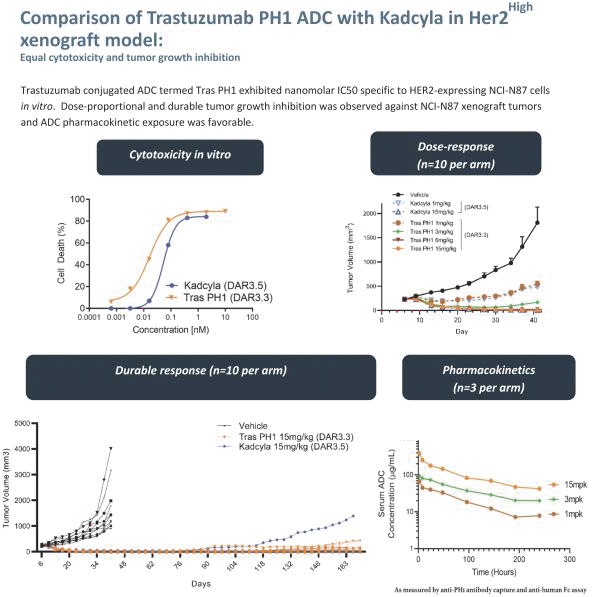
When conjugated at drug-to-antibody ratio (DAR) of 3.3, Tras PH1 ADC demonstrated cytotoxic potency in the sub-nanomolar range. The ADCs were then evaluated against pre-established Her2-high expressing tumors in athymic mice; mice that lack an intact immune system to prevent rejection of human tumors. We then paid attention to the ADC dose that a) showed statistically significant TGI and b) shrank established 200 mm3 tumors, and we evaluated both the short- (30-day) and the long-term or durable (5 months or more) responses of the two ADCs.
The short-term TGI of both ADCs was indistinguishable in doses ranging from 1- 15 mg/kg. Both ADCs showed statistically significant tumor growth inhibition at 1 mg/kg and both ADCs shrank established 200 mm3 tumors equally at 3 mg/kg or higher doses. The results suggested that a DAR-matched ADC containing PH1 was at least as effective as DM1 in vitro and in vivo.
When we followed the mice for extended observations, we noted that in the high-dose 15 mg/kg- treated animals, Kadcyla®-treated tumors occasionally rebounded within 3-months and Tras PH1-treated tumors rebounded in around 5 months. Tras PH1 ADC showed dose-dependent pharmacokinetics and the linker was stable in mouse circulation.
30
Previously, we showed that PH1 had an increased propensity to stimulate neoepitopes due to its anti-splicing MoA. In order to evaluate the immunogenic potential of our payload, we evaluated tumor growth inhibition in syngeneic mice with an intact immune system. We used murine MC38 colorectal cancer cells that were genetically engineered to swap out the mouse Her2 gene with its human counterpart, so that our ADCs targeting human Her2 could be evaluated in this tumor model.
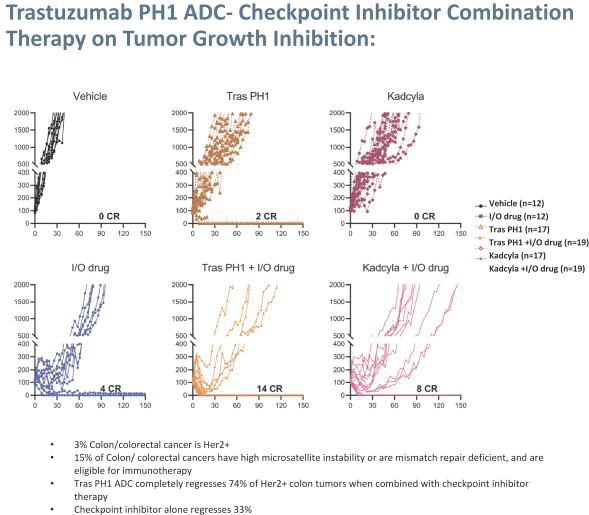
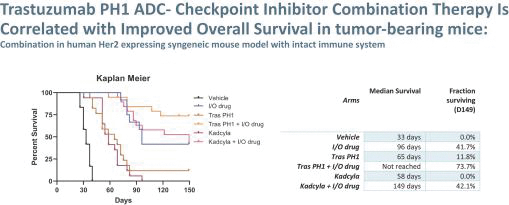
Also, 15% of colorectal cancer patients are eligible to receive checkpoint inhibitor therapy and we selected this particular murine cell line as it is responsive to different immuno-oncology (I/O) therapies.
31
We then evaluated a DAR-matched Tras PH1 ADC with Kadcyla®, separately and in combination with checkpoint inhibitor therapy (termed I/O drug) and compared short- and long-term responses. TGI of Tras PH1 and Kadcyla® were largely similar, except for a small proportion of complete regressions observed only in Tras PH1-treated mice. As anticipated, the tumor model responded to standard-of-care I/O drug administered as a single agent.
When administered as a combination with checkpoint inhibitor therapy, the Tras PH1 ADC induced complete regressions (CRs) in 14 mice whereas 5 tumors rebounded after initial shrinkage (n=19 mice per arm). As a result, 73% of Tras PH1 + I/O treated mice showed complete regressions and were still on study at 5 months and the median survival was not reached. In Kadcyla® combination arm, there were 8 CRs, and 11 tumor rebounds, and 42% of Kadcyla® + I /O treated mice were tumor-free at 5 months. The median survival of Kadcyla® combination was 149 days.
The above results support our theory that immunostimulatory ADC payloads will induce longer and deeper responses due to greater immune cell engagement with tumor cells. In checkpoint blocked tumor cells, this deep response may require checkpoint alleviation. Also, the Tras PH1 combo-treated CR mice rejected a rechallenge with a fresh round of tumor cells, suggesting the presence of anti-tumor immunity. This immunity rejected MC38 cells with or without human Her2, suggesting that the immune response had spread beyond the original protein that the Her2 ADC targeted. This phenomenon of epitope spreading is characteristic of immune B and T cells that surveil many surrounding epitopes of the cancer cell and are not restricted to the target protein.
32
This is an advantage of immunostimulatory payloads such as PH1 that attract immune cells to the tumor. As payload delivery, and therefore cytotoxicity is directly proportional to the amount of target antigen receptors, target heterogeneity (for e.g., high-Her2 and low-Her2 expressing cells) within the same tumor is often a problem. It is likely that ADCs may not be able to deliver sufficient cytotoxic payload to kill the tumor cells with lower expression.
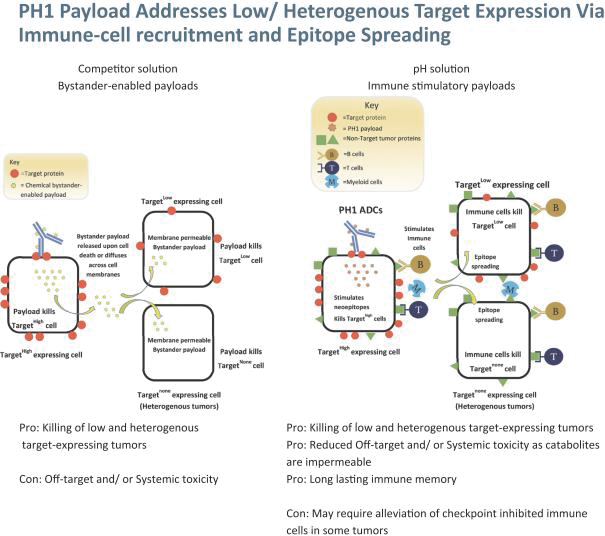
To solve this problem, different ADC programs have taken various approaches:
All above approaches have consequences relating to off-target cell killing.
We have focused our efforts and prefer that our payloads have inherent immunostimulatory properties that attract immune cells. Having derived from self, immune T and B cells do not have the toxicity concerns of a payload gaining access to the systemic circulation.
Our DAR-matched Trastuzumab ADC was then evaluated in non-human primates (NHP) to assess the toxicology and toxicokinetic (TK) properties of Tras PH1 ADC.
33
The Maximally Tolerated single IV Dose (MTD) of Tras PH1 ADC was 20 mg/kg. Below 15 mg/kg dose, there were no Tras PH1 ADC-related clinical signs, changes in body weight, food consumption, or clinical pathology parameters (hematology, serum chemistry). Below 15 mg/kg dose, there were no test article-related organ weight changes, nor macroscopic or microscopic findings. At MTD, moderate elevations in liver enzymes and moderate decreases in platelets were noted; and yet both changes were completely reversed to baseline after 10 days. These changes were also noted and published for Kadcyla® at the highest non-severe toxic dose (HNSTD) by Poon, et al.
Remarkably at MTD with Tras PH1 ADC, histology of bone marrow smears was within normal limits and there was no evidence of neutropenia by hematology. Also, no gross lesions were observed in eyes and optic nerves (ocular toxicity) and sciatic nerves (peripheral neuropathy) of animals treated at MTD with Tras PH1 ADC.
The toxicology data suggested PH1 ADCs would also be differentiated from conventional payload ADCs by toxicology parameters, in addition to pharmacology (TGI).
These findings support differentiated features, creating a pipeline of PH1 ADCs against multiple targets using our catalog of POC antibodies.
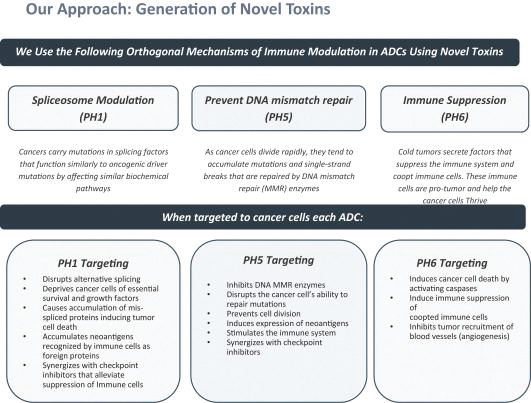
PH5 payloads targeting DNA mismatch repair (MMR) and/ or DNA damage response (DDR):
Biology of MMR:
Cancer cells are associated with uncontrolled cell division. Before cells divide, they replicate their DNA to forward one chromosome copy to each daughter cell. Largely, DNA replication is a robust process controlled by enzymes with precise fidelities, low error rates, and the presence of correction mechanisms termed DNA mismatch repair (MMR). Due to rapid and frequent cell division, cancer cells tend to accumulate errors such as mutations, single-, and double-stranded DNA breaks, that are corrected in real time by MMR enzymes. Errors left uncorrected trigger a set of cellular responses collectively termed the DNA damage response (DDR). The DDR engages signaling pathways that regulate the recognition of DNA damage, the recruitment of DNA repair factors, the initiation and coordination of DNA repair pathways, and transition through the cell division cycle. If the cells are at a significant survival disadvantage, DDR processes activate apoptosis and trigger cell death.
34
When cancer cells are treated with DNA-damaging chemotherapeutic agents for e.g., such as the DNA alkylating agent platinum, cancer cells activate DDR and MMR processes, and when the errors are significant in terms of cellular liability and cannot be repaired, they are committed to programmed cell death.
In adult cancer patients, cancer cells are likely to be actively involved in cell division compared to normal differentiated cells. Therefore, ADC payloads that target DNA DDR and/or MMR is likely to preferentially target proliferating cancer cells. If we prevent the repair mechanisms, cancer cells are likely to be committed to cell death because of the errors they incorporate. We may even choose to accelerate the process by combining with certain chemotherapies.
Conversely, mutations in MMR and DDR genes may provide a selective advantage to the cancer cell by not correcting the mutation that would offer a significant growth or survival advantage. MMR-deficiency (dMMR) is common in many colorectal, gastrointestinal, and endometrial cancers and found in lower frequency in other solid cancers of breast, prostate, bladder, and thyroid. Here, dMMR patients can have increasing numbers of microsatellite repeats, also called high microsatellite instability (MSI-H). Both dMMR and MSI-H are considered biomarkers and predict response to checkpoint therapy and may go hand in hand with the neoepitopes that are formed when errors in DNA go uncorrected.
It is therefore likely that an ADC payload targeting MMR/ DDR biology may have a dual punch, inducing apoptosis in targeted cells on the one hand and activating the immune system by the other. This biology is compatible with Peak Bio philosophy of generating ADC payloads with multiple, orthogonal MoAs.
We are currently evaluating the first generation of PH5 linker-toxins against an undisclosed MMR/ DDR target. The toxin is bystander-enabled for killing the neighboring cell and may be adapted for low and heterogenous target expression. This is in addition to potential killing by immune activation via neoepitopes.
PH6 payloads targeting immune suppression:
Protein synthesis is integral to most biological functions. Even slow-growing, stem cell-like progenitors of tumor cells that divide less frequently synthesize proteins to support vital functions. DNA is transcribed into RNA and RNA is translated into protein. Theoretically, both inhibitors of transcription and translation may function as ADC payloads if one can partition them selectively to cancer cells using target-specific antibodies that can differentiate them from a normal cell. PH6 is an undisclosed payload that prevents protein synthesis at the stage of transcription.
Tumors containing an active population of immune cells capable of responding to immunogenic stimuli and killing cancer cells are referred to as immune “hot” tumors. Conversely, those tumors that have a low population of immune cells or have immune cells that are actively suppressed or co-opted into working for the tumor are referred to as immune “cold”. An extreme form of immune cold tumors called immune desert reflects tumors where immune cells are confined to the tumor periphery.
Immune cold tumors are hard to target and are typically unresponsive to immunotherapy. Checkpoint inhibitor therapy and immune stimulation approaches have largely been unsuccessful due the immune cells being suppressed or co-opted. These tumors have regulatory T cells (T-regs) that suppress T cell activation or express soluble factors that induce immune deserts. In this case, Peak Bio is testing payloads that a) induce cytotoxicity of tumor cells, and b) suppress immunosuppressive immune cells. This dual action protein synthesis inhibitor payload may potentially have a second function where tumor immunogenicity is increased by killing co-opted immune cells or suppressing function(s) of immunosuppressive cells.
We are currently evaluating the first generation of PH6 linker-toxins against an undisclosed target and validating its second MoA. Due to the varied effects of new protein synthesis inhibition, this toxin may also prevent the formation or recruitment of new blood vessels to the tumor.
Antibody-based Platforms:
Our objective is to use our expertise in antibodies and our novel technologies to develop our product pipeline and discover new product candidates for the treatment of cancer and related diseases. Our strategy includes initiatives to:
35
36
Summarized below is an average R&D workflow for a typical ADC program:
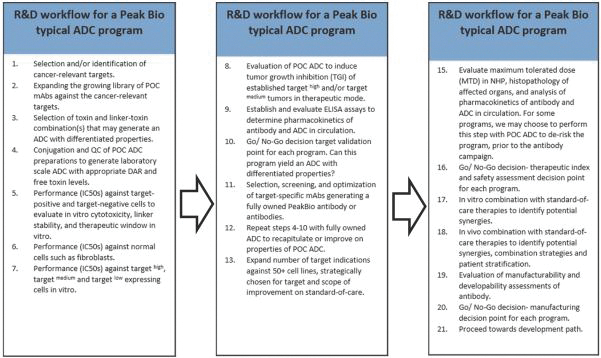
Peak Bio has additional discovery research programs directed towards identifying and developing new mAb-based products and technologies to treat cancer. Our discovery programs are currently focused on identifying and screening cancer-relevant targets, mAbs, ADCs, antibody-PROTACs and bispecific antibody therapies.
Our preclinical candidates:
Trop2 PH1 ADC is a Clinically validated target: Trophoblast antigen 2 (Trop2) or Tumor- Associated Calcium Signal Transducer 2 (TACSTD2) is a transmembrane glycoprotein that is highly expressed in many cancers over and above that of levels observed in normal healthy tissue, making this protein a prime ADC target. Trop2 levels are elevated in several solid tumor cancers (see table below). Trop2 overexpression in metastatic tissues makes it an attractive and potential therapeutic target for late-stage diseases.
37
Table 2: Expression of Trop2 target in various cancers
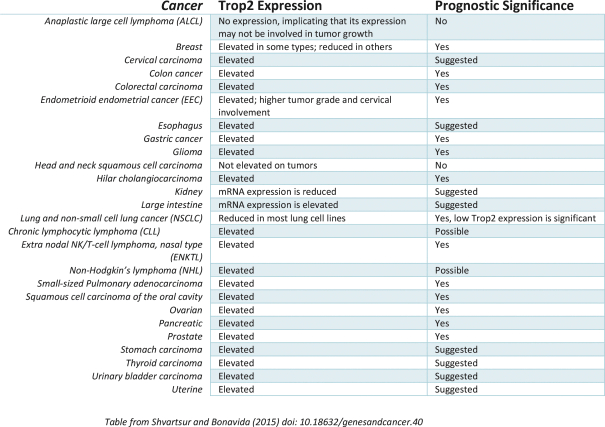
The Trop2 ADC approach has been clinically validated and has outperformed standard-of-care in at least two cancer settings- metastatic triple negative breast cancer (TNBC) and in advanced urinary bladder (urothelial) cancer. The Trop2 ADC Trodelvy®, also known as Sacituzumab govitecan or IMMU-132, has obtained approvals in the above indications after demonstrating significant improvement in clinical efficacy. Due to the potential of targeting Trop2 in multiple cancer settings (see table above), different companies have tried to carve out their niche using the advantages/ properties of their payloads (see table below). While Datopotamab DXd is currently being tested in Phase 1b clinical trial on NSCLC patients and have expanded to Phase 2 and Phase 3 clinical trials, others such as BAT8003 and PF-06664178 have discontinued their Trop2 programs for different reasons. The status of the other Trop2 ADC programs is unknown.
38
Table 3: ADC-based Trop2 therapeutics in clinical trials
Product (alias) |
Company |
Description |
Clinical stage |
Trodelvy® (Sacituzumab govitecan/ IMMU-132) |
Gilead (formerly Immunomedics) |
Humanized IgG1 mAb conjugated to irinotecan metabolite (SN-38) warhead via a maleimide–PEG–acid-sensitive cleavable carbonate linker |
Approved for metastatic TNBC, Accelerated approval for advanced urothelial cancer Several combination trials ongoing (Phase 2 and 3) |
Datopotamab deruxtecan (DS-1062) |
Daiichi Sankyo, AstraZeneca |
Humanized IgG1 mAb conjugated via a thioether bond to DNA topoisomerase I inhibitor exatecan derivative (DXd) warhead using an enzymatically cleavable tetrapeptide linker |
Phase 1 ongoing (TNBC) Phase 2 ongoing (NSCLC) Phase 3 ongoing (NSCLC) |
|
|
|
|
SKB264 |
Klus Pharma, Merck |
Humanized IgG1 mAb conjugated to topoisomerase I inhibitor belotecan via a cleavable linker |
Phase 1 ongoing |
|
|
|
|
JS-108/ DAC-002 |
Shanghai Junshi Bioscience Co., Ltd. DAC Biotech |
Humanized IgG1 mAb conjugated to tubulysin B analog Tub196 warhead via a 2,3-disubstituted long side chain hydrolysis-resistant linker |
Phase 1 (SCLC) Phase 1 (solid tumors) |
|
|
|
|
BIO-106 |
BioOneCure Therapeutics |
mAb targeting Trop2 conjugated to unknown tubulin inhibitor payload |
Phase 1 (solid tumors) |
|
|
|
|
LCB84 |
Legochem (South Korea) |
mAb targeting cleaved Trop2 conjugated to MMAE |
Unknown |
|
|
|
|
BAT8003 |
Bio-Thera Solutions (Guangzhou, China) |
Humanized IgG1 mAb with afucosylated Fc conjugated to microtubule-binding maytansine derivative batansine via a non-cleavable linker |
Phase 1. Batansine technology discontinued after Her2 ADC Phase 3 failure |
|
|
|
|
PF-06664178 |
Pfizer |
Humanized IgG1 mAb conjugated to microtubule inhibitor auristatin (Aur0101) warhead using a cleavable linker and site-specific transglutaminase |
Discontinued in phase 1 for business reasons |
The Trop2 ADCs under development have topoisomerase I- targeting payloads such as the irinotecan active metabolite SN38 (Trodelvy®), deruxtecan (DS-1062), and belotecan (SKB264).
Members of the camptothecin family of topoisomerase inhibitors such as irinotecan/ SN38 and topotecan are substrates of the MDR family of transporters and may be pumped out of the cancer cell, giving rise to resistance. Non-transport mechanisms of resistance have also been described wherein patients under Trodelvy® therapy for 6 months had progressed due to resistance mutations in the topoisomerase I (Top1) gene. In a study performed at Massachusetts General Hospital, Top1 mutations such as E418K and -p.-122 frameshift rendered cancer cells refractory to topoisomerase inhibition, and Trop2 T256R mutations reducing cell surface translocation of Trop2, resulted in resistance to Trodelvy® therapy and metastasis to liver and peri-aortic lymph nodes. Since our payload has a different MoA, it will not be subject to these topoisomerase-specific forms of resistance.
Furthermore, the immunostimulatory properties of the PH1 payload may induce:
39
Properties of Peak Bio Trop2 PH1 ADC:
After evaluating the Trop2 ADCs that are currently FDA-approved or heading towards approval in clinical trials, we investigated the potential of a differentiated Trop2 PH1 ADC with favorable resistance and immunogenicity characteristics from PH1 payload.
We optimized a fully- humanized antibody that was selective for the human and non-human primate versions of Trop2 but did not recognize rodent forms of Trop2. While compatible with evaluation of cytotoxic potency in vitro and evaluation of anti-xenograft tumor growth inhibition in athymic mice, this meant we had to engineer mouse cell lines with human Trop2 to test the immunostimulatory MoA in syngeneic mice models.
Since our antibody did not recognize rodent Trop2, standard evaluations of body weight loss in rodent models such as mice and rats would not provide meaningful toxicology data other than to reflect uncoupling of the payload from the ADC. NHP model would provide the relevant toxicology data.
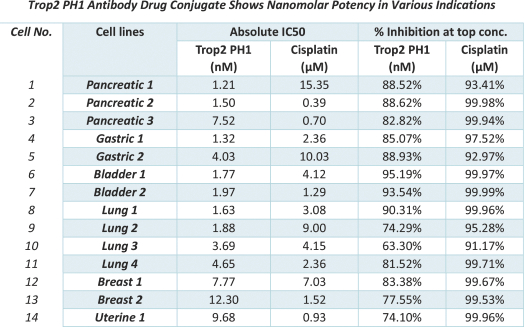
Even without the immunostimulatory mechanism, our investigational Trop2 PH1 ADC demonstrated nanomolar cytotoxic potency against cancer cells in vitro. In a parallel arm of the same study, cisplatin, a conventional chemotherapy exhibited micromolar cytotoxic potency. Also, these studies demonstrated that the potency of our Trop2 ADC was specific to the target and did not kill lung cancer cell lines that lacked Trop2. This is important to prevent off-target effects of our ADC against normal cells that lack Trop2.
As previously mentioned in our Thailanstatins section, PH1 belongs to the lysine non-cleavable class of payloads and was specially selected to reduce off-target effects of our Trop2 ADCs. While Trop2 is elevated significantly in solid tumors, there is small yet significant Trop2 expression in normal lung epithelium, prostate, skin, tongue, and salivary glands. This may be relevant as stomatitis (inflammation of the tongue and mouth) was observed as the dose-limiting toxicity in the TROPION PanTumor01 clinical trial for DS-1062. This meant that in addition to preventing off-target effects in our Trop2 PH1 ADCs, we may have to mitigate potential on-target effects.
In PH1, we selected a payload that potentially prevents reduced on and off-target effects by generating metabolites that are impermeable to neighboring cells. The low expression of Trop2 in normal tissue, potency range of PH1, and impermeability of generated active payload species are designed to limit the side effects of incidental Trop2 targeting with our ADC. Trodelvy® and DS-1062 are both bystander-enabled to extract maximal tumor cell killing. However, the payload’s function in Trop2 PH1 ADC is to stimulate initial tumor debulking, induce neoepitopes and stimulate the immune system whereupon the immune-mediated cell-killing MoA would kick in. In addition to opting for a non-bystander payload, we also opted for lower drug-to-antibody (DAR) ratio of 4 to reduce on-target toxicity to normal cells. By not opting for chemical bystander activity and by opting for lower DAR, we introduced control elements to differentiate our ADC program from a toxicology standpoint. Heterogenous Trop2 expression in cancer tissue would be addressed by immunostimulatory and epitope-spreading features of PH1 described previously.
40
Further supporting our hypothesis, not only did our cysteine and lysine cleavable Trop2 ADC versions kill cancer cell lines non-specifically i.e., they had higher baseline activity against non-target cells, but also had lower TGI in vivo in animal models. Therefore, our best strategy was to allow toxin accumulation within the target cell and have an inactive or impermeable payload species when released by lysed target cells. The active payload species of PH1 ADCs such as the Trop2 PH1 ADC would only be “cytotoxic” when internalized as an ADC by the target cancer cells, and impermeable as the active payload species to neighboring cells or other organs when in blood circulation, further reducing the potential for off-target effects. We therefore decided to proceed with PH1 for the Trop2 ADC program and tested low DAR ADCs for TGI against human tumors in animal xenograft models.
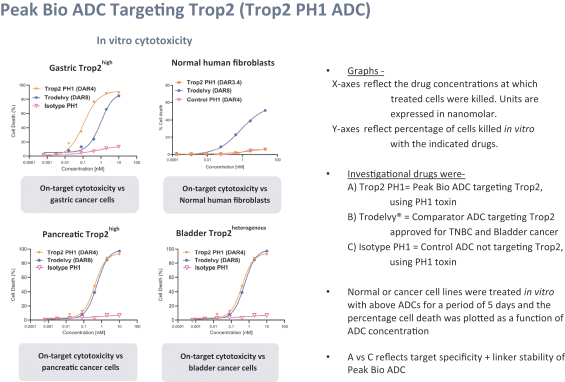
To demonstrate target-specific killing of cancer cells, we compared the cytotoxic potency of our Trop2 PH1 ADC with an ADC made from an isotype control mAb (not targeting Trop2) conjugated to the same PH1 L-T at a similar DAR, against gastric, pancreatic and bladder cancer cell lines. As the isotype antibody targets viral proteins and is characteristically absent on cancer cells, the wide margin between on-target and off-target killing against all above cell lines can be attributed to the stability of our linker. In this context, if the linker fell apart on Trop2 PH1 and Isotype control ADCs, it would release the toxin and kill the cells whether Trop2 was present on the cells or not, and this would be observable as activity for the Isotype PH1 ADC.
As Trop2 expression is mainly observed in cells of epithelial origin, we evaluated the cytotoxic potency of our Trop2 PH1 vs Isotype PH1 ADC and found no significant killing against normal human fibroblasts. Some cell death was observed upon confluence in all cell lines and occurred even on untreated cells.
Trodelvy® is the first-in-class Trop2 ADC with an acid-labile carbonate linker. It was included as an experimental arm in the above cytotoxicity assays and demonstrated potent in vitro activity against gastric, pancreatic and bladder cancer cell lines. Trodelvy® showed some off-target killing against normal human fibroblast cells in this setting.
To further corroborate our in vitro observations, we evaluated Trop2 PH1 ADC and Trodelvy® against the same Trop2high gastric carcinoma cell-line derived xenograft (CDx) grown as tumors in mice. For the studies to translate to a clinical setting, Trodelvy® (DAR 7.6) was administered on Day 1 and Day 8 as 10 mg/kg doses (QWx2). Trop2 PH1 ADCs at lower DARs (2 and 4) were tested only at 3 mg/kg. This was purely to evaluate the TGI from the Trop2 PHI ADC’s cytotoxic MoA alone in the absence of PH1’s immunostimulatory MoA, in xenograft tumor-bearing athymic mice lacking an immune system. The purpose of the experiment was to evaluate whether Trop2 PH1 ADC’s first MoA alone was sufficient for TGI in Trop2high expressing tumors.
41
When administered in therapeutic mode, against pre-established tumors of 200 mm3 size, all three ADCs induced tumor regression between 3-6 weeks. The TGIs for 10mg/kg Trodelvy®, 3mg/kg Trop2 PH1 ADC (DAR 2) and 3mg/kg Trop2 PH1 ADC (DAR 4) were 79 + 2.1%, 80.5 + 1.8%, and 87.7 + 1.0% at 21 days and 83.3 + 2.4%, 78.5 + 3.2%, and 90.3 + 1.8% at 41 days, respectively. At these times, the TGI associated with the Trop2 PH1 ADC (DAR 4) arm was significantly different from the Trop2 PH1 ADC (DAR2) and Trodelvy® arms (p<0.05) and is indicated in the table.
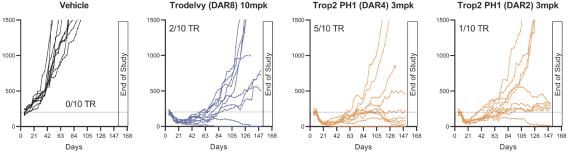
|
|
|
|
At DAR4- tumor |
|
At DAR2- Stable |
||||
Model: Nude mice bearing human gastric tumors |
|
Group |
|
TGI ± Std |
|
p Value |
|
TGI ± Std |
|
p Value |
Tumor shrinkage below this line was considered |
|
Trop2 PH1 |
|
87.7 ± 1.0 |
|
|
|
90.3 ± 1.8 |
|
|
Dosing regimen: Two doses in the first week |
|
Trop2 PH1 |
|
80.5 ± 1.8 |
|
1.40e-04 |
|
78.5 ± 3.2 |
|
1.42e-03 |
TR= tumor regression |
|
Trodelvy |
|
79.0 ± 2.1 |
|
2.79e-05 |
|
83.3 ± 2.4 |
|
3.54e-02 |
Upon extended observation, some tumors from each arm rebounded across all treatment groups, Trodelvy® and Trop2 PH1 ADCs. Around 2 months after treatment, 80% of Trodelvy®-treated tumors rebounded, until finally, only 20% of the mice showed significant tumor regression at 5+ months. 50% of Trop2 PH1 ADC (DAR4) showed tumor regression at 5+ months and 50% of Trop2 PH1 ADC (DAR2) showed stable disease in the same time frame with their tumors failing to grow past 400 mm3.
We conclude that Trop2 PH1 ADC has effective TGI at low DAR and dose even without PH1’s immunostimulatory MoA. Tumor cells that escape treatment tend to rebound. This is why we had envisioned the second immunostimulatory MoA when we conceptualized PH1.
To determine whether Trop2 PH1 ADC had retained the immunostimulatory activity characterized previously on PH1 payload alone or demonstrated by our POC Tras PH1 ADC, we evaluated the combination of the DAR4 Trop2 PH1 ADC with checkpoint inhibitor therapy against a syngeneic bladder cancer (urothelial) mouse model.
42
First-line checkpoint inhibitor therapy is standard-of-care (SOC) for platinum-ineligible patients that have recurrent, resistant, or metastatic urothelial cancer. Also, Trodelvy® is approved for treatment of metastatic urothelial cancers. Therefore, we used a urothelial model that was sensitive to checkpoint blockade and evaluated whether Trop2 PH1 ADC combination would result in an improvement upon SOC in this syngeneic mouse model.
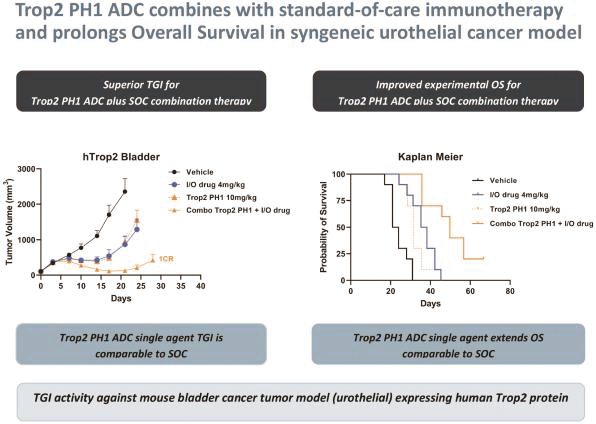
In these studies, Trop2 PH1 ADC showed single agent tumor growth inhibition that was equivalent to SOC for bladder cancer. At day 14, the combination was significantly superior in terms of TGI (p=0.01) and prolonged overall survival (OS) (p=0.013). Therefore, like the POC Tras PH1 ADC, our Trop2 PH1 ADC retained the ability to combine with checkpoint inhibitors and prolong OS.
To further de-risk our program, we performed toxicology studies in non-human primate model and determined the tolerability of our Trop2 PH1 ADC in NHP. We evaluated our ADCs at DARs of 2 and 4 and performed a repeat-dose study wherein three ADC doses were intravenously administered every 3 weeks followed by a 3-week recovery period. For an idea of maximal cumulative effects, animals were evaluated 2 days after receiving all 3 doses. Reversibility was addressed in another set of animals that received all 3 doses but were allowed a 3-week recovery period. As Trop2 PH1 ADC was a likely candidate for pipeline nomination, histopathology was performed unilaterally for all tissues in both sets of animals.
3 x 6 mg/kg Q3W doses of both DAR2 and DAR4 Trop2 PH1 ADCs were well tolerated without clinical signs or body weight loss. In these treatment groups, a mild increase in liver enzymes and mild decrease in platelets were noted that reset to baseline within 7-10 days of administration of each dose. Histological evaluation of the bone marrow revealed no evidence of reduced cellularity (no bone marrow toxicity), although an altered myeloid:erythroid ratio was noted. The latter finding was probably due to the MoA of PH1 that induces an anti-tumor myeloid response. There were no other histologic findings below MTD.
At the >MTD of 18 mg/kg for Trop2 PH1 ADC, we did not observe the pathologies associated with other Trop2 ADCs in the clinic- e.g., neutropenia, gastrointestinal-, oral (stomatitis)- or lung- lesions with fibrotic or cellular infiltrates indicative of ILD were characteristically absent from our findings. Our NHP data suggests our Trop2 ADC is likely to be differentiated from a toxicology standpoint, in addition to pharmacology.
43
Unmet need and epidemiology
There is significant unmet need in Trop2-expressing cancers as is illustrated in the table below. Currently, the Trop2 ADC Trodelvy® has been only approved in TNBC and bladder cancer DS-1062 is currently in phase 1b clinical trial in NSCLC patients. Restricting ourselves to current indications in which Trop2 ADCs have FDA approvals (Trodelvy®) or have advanced phase 1 data in (DS-1062), the number of annual deaths worldwide and in USA alone account for 1.8 million and 134,500 patients, respectively (assuming 15% of all breast cancers are TNBC and 84% of all lung cancers are NSCLC in the table below). Prior to Trodelvy’s® approval in 2020, these patients did not benefit from standard-of-care in these indications. Therefore, there is significant unmet need in Trop2-expressing cancers based on these three indications alone.
Since Trop2 expression is elevated in multiple solid tumors, there is untapped clinical and market potential of expanding the scope of Trop2 ADC therapies to reach a wide number of cancer indications. We cannot predict the total number of patients current and future Trop2 ADC therapies may expand to; however, at maximum, Trop2 ADCs may have the potential to impact the lives of 13 million cancer patients annually.
Table 4: New cases and deaths for Trop2-relevant cancers (worldwide and USA statistics)
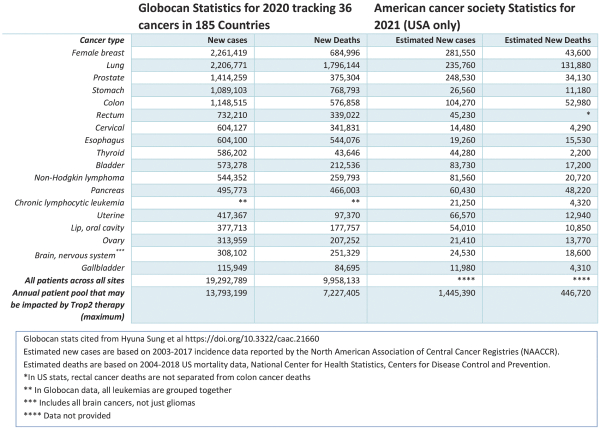
Our Preclinical Development Programs
We have evaluated multiple targets for our second candidate PH1 ADC. We are currently performing target validation for an ADC2 program against target M5. We may generate our own proprietary mAb against target M5 in humanized mice.
We are also in the process of evaluating the first Generation of two new research-stage toxins PH5 and PH6. These platforms may need optimization and need further structure-activity-relationship (SAR) studies requiring a second or third generation to be viable payloads for the Peak Bio pipeline.
Time and resource- permitting, we have identified additional opportunities utilizing our teams’ expertise to expand our portfolio by developing other modalities such as bispecifics and antibody-PROTACs. These are currently in early validation stages.
44
Intellectual Property:
IP Summary:
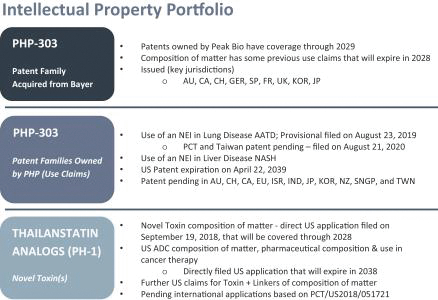
Oncology Platform PH-1 & PHP-303 Patent Status
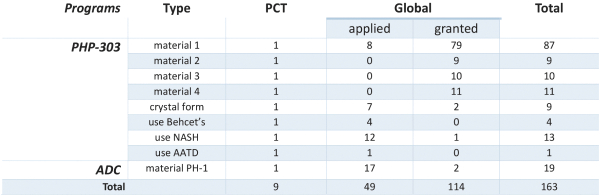
Peak Bio (Previously pH Pharma) secured patent protection for PH-1 & PHP-303 with over 114 patents granted in over 70 countries worldwide.
45
PH-1 & PHP-303 Patent Classes
Type
|
Content
|
Status
|
Material 1 |
Material patent of 4-(4-Cyano-2-Thioaryl) Dihydropyrimidinones including PHP-303 |
Granted in 79 countries including KR, US, EU, JP |
Material 2 |
Material patent of PHP-303 analogues (1,4-diaryl-pyrimidopyridazine-2,5-diones) |
Granted in 9 countries including US, EU, JP |
Material 3 |
Material patent of PHP-303 analogues (Triazolo and tetrazolo pyrimidine derivatives) |
Granted in 10 countries including US, EU, JP |
Material 4 |
Material patent of PHP-303 analogues (Sulfonic amide and sulfoximine-substituted diaryl-dihydropyrimidinones) |
Granted in 11 countries including US, EU, JP |
Crystal form |
Crystal form (A) of PHP-303 and method for producing PHP-303 |
Application in 7 countries including KR, EU, JP; granted in US |
Use |
PHP-303 and its analogues for treating Bechet’s disease |
Application in 7 countries including KR, US, EU, HK |
|
PHP-303 use in liver diseases as a NHE inhibitors |
Application in 7 countries including KR, EU, JP; granted in US |
|
PHP-303 use in lung disease including AATD as a NHE inhibitors |
PCT application including US, Taiwan |
PHP-303 Patent Status
As of March 1, 2022, our patent portfolio relating to our product candidate PHP-303 consisted of five issued U.S. patents and 114 issued foreign patents, and three pending patent applications. The patent of PHP-303 for its crystalline form (A) applicable to actual clinical trials has been registered in the United States and its screening is ongoing in other countries. The crystalline form patent will not expire earlier than 2036.
Peak Bio acquired full rights to patents from Bayer in 2017. We have global patent protection on 9 inventions on the compound that include the following:
We have acquired or exclusively licensed a comprehensive intellectual property portfolio from Bayer. We strive to protect and enhance the proprietary technologies, inventions, and improvements that we believe are important to our business, including seeking, maintaining, and defending patent rights, whether developed internally or acquired or licensed from third parties. Our policy is to seek to protect our proprietary position by, among other methods, pursuing and obtaining patent protection in the United States and in jurisdictions outside of the United States related to our proprietary technology, inventions, improvements, platforms, and our product candidates that are important to the development and implementation of our business.
PH-1 Patent Status
46
Item 1A. Risk Factors.
Investing in our securities involves risks. Before you make a decision to buy our securities, in addition to the risks and uncertainties discussed above under “Cautionary Note Regarding Forward-Looking Statements,” you should carefully consider the specific risks set forth herein. If any of these risks actually occur, it may materially harm our business, financial condition, liquidity and results of operations. As a result, the market price of our securities could decline, and you could lose all or part of your investment. Additionally, the risks and uncertainties described in this prospectus, any prospectus supplement or in any document incorporated by reference herein or therein are not the only risks and uncertainties that we face. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial may become material and adversely affect our business.
Summary Risk Factors
47
Risks Relating to Our Business and Programs
The potential ADC product candidates in our pipeline such as Trop2 PH1 ADC are in the preclinical and IND-enabling stages of development, with only PHP-303 having progressed to clinical stages of development. Trop2 PH1 ADC has never been tested in human subjects. We may be unable to advance any current or future potential product candidates through the completion of clinical development, obtain regulatory approval and ultimately commercialize any of our product candidates, or experience significant delays in doing so.
48
49
50
Our Peak Bio R&D Toxin and ADC Platform Engine is evolving and may not reach a state at which building a pipeline of product candidates is possible.
We may expend our limited resources and access to capital to pursue a particular product candidate; these decisions may prove to be wrong and may adversely impact our business. Because we have limited financial and managerial resources, we intend to focus our efforts on specific R&D programs, including our clinical development of product candidate(s) PHP-303, and Trop2 PH1 ADC, our lead oncology ADC candidate, and eventually advancing additional research programs progressing from our Peak Bio R&D Discovery Toxin and ADC Platform Engine.
51
The effects of health epidemics, geopolitical instability including the ongoing COVID-19 pandemic, the conflict in Eastern Europe and in regions where we, or the third parties on which we rely, have business operations could adversely impact our business, including our preclinical studies, anticipated clinical trials, and contract manufacturing capabilities. The COVID-19 pandemic and/or the geopolitical instability in Eastern Europe could materially affect our operations, which have been affected by supply chain issues subject to foreign policy changes and/or executive orders, and at our future clinical trial sites, as well as the business or operations of our CROs, CDMOs, or other third parties with whom we conduct business and could be adversely impacted and cause significant disruptions and delays which could greatly impact our business and may continue to impact planned preclinical and clinical plans.
52
Our planned preclinical research and clinical trials may be affected by the ongoing COVID-19 pandemic, should the pandemic continue, a new health crisis emerge, or until such time at which we intend to initiate such trials, including the following.
We depend on enrollment of patients in our clinical trials for our product candidates. If we are unable to enroll patients in our clinical trials, or enrollment is slower than anticipated, in particular for our product candidates with rare disease indications, our research and development efforts could be adversely affected.
53
Our business is subject to risks associated with conducting business internationally. We source research and development, manufacturing, consulting, and other services from companies based throughout the United States, the EU, and select Asian countries and we will be planning and conducting our clinical trials in the United States, Canada, certain European countries, in the near-term and in the future.
regulations, the withdrawal could materially impact the regulations with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the UK or the EU.
54
If PHP-303, our clinical stage asset, or other future product candidates acquired or those derived or developed from our Peak Bio R&D Toxin and ADC Platform, continue in clinical trials, or are eventually initiated in clinical trials in human subjects, they may not demonstrate the combination of safety and efficacy necessary to become approvable or commercially viable.
55
Our product candidates are at an early stage of development, and we may not be able to successfully develop and commercialize them.
Our ability to commercialize our product candidates depends on first receiving Food and Drug Administration (FDA) approval.
56
We are currently operating in a period of economic uncertainty and capital markets disruption, which has been significantly impacted by geopolitical instability, an ongoing military conflict between Russia and Ukraine, and record inflation. Our business, financial condition and results of operations could be materially adversely affected by any negative impact on the global economy and capital markets resulting from the conflict in Ukraine, geopolitical tensions or record inflation.
U.S. and global markets are experiencing volatility and disruption following the escalation of geopolitical tensions and the start of the military conflict between Russia and Ukraine. On February 24, 2022, a full-scale military invasion of Ukraine by Russian troops was reported. Although the length and impact of the ongoing military conflict is highly unpredictable, the conflict in Ukraine could lead to market disruptions, including significant volatility in commodity prices, credit and capital markets, as well as supply chain interruptions, which has led to record inflation globally. We are continuing to monitor inflation, the situation in Ukraine and global capital markets and assessing the potential impacts on our business.
The global economy has been, and may continue to be, negatively impacted by Russia’s invasion of Ukraine. As a result of Russia's invasion of Ukraine, the U.S., the European Union, the United Kingdom, and other G7 countries, among other countries, have imposed substantial financial and economic sanctions on certain industry sectors and parties in Russia. Broad restrictions on exports to Russia have also been imposed. These measures include: (i) comprehensive financial sanctions against major Russian banks; (ii) additional designations of Russian individuals with significant business interests and government connections; (iii) designations of individuals and entities involved in Russian military activities; and (iv) enhanced export controls and trade sanctions limiting Russia's ability to import various goods. Russian military actions and the resulting sanctions could continue to adversely affect the global economy and financial markets and lead to instability and lack of liquidity in capital markets, potentially making it more difficult for us to obtain additional funds. Further, there are current geopolitical tensions with China. Recently, the Biden administration has signed multiple executive orders regarding China. One particular executive order titled
57
Advancing Biotechnology and Biomanufacturing Innovation for a Sustainable, Safe, and Secure American Bioeconomy, signed on September 12, 2022, will likely impact the pharmaceutical industry to encourage U.S. domestic manufacturing of pharmaceutical products. Any additional executive orders or potential sanctions with China could materially impact our manufacturing partners.
Although our business has not been materially impacted by the ongoing military conflict between Russian and Ukraine, geopolitical tensions, or record inflation to date, it is impossible to predict the extent to which our operations, or those of our suppliers and manufacturers, will be impacted in the short and long term, or the ways in which the conflict may impact our business. The extent and duration of the conflict in Ukraine, geopolitical tensions, record inflation, sanctions and resulting market disruptions are impossible to predict, but could be substantial. Any such disruptions may also magnify the impact of other risks described herein.
Climate change or legal, regulatory or market measures to address climate change may negatively affect our business, results of operations, cash flows and prospects.
We believe that climate change has the potential to negatively affect our business and results of operations, cash flows and prospects. We are exposed to physical risks (such as extreme weather conditions or rising sea levels), risks in transitioning to a low-carbon economy (such as additional legal or regulatory requirements, changes in technology, market risk and reputational risk) and social and human effects (such as population dislocations and harm to health and well-being) associated with climate change. These risks can be either acute (short-term) or chronic (long-term).
The adverse impacts of climate change include increased frequency and severity of natural disasters and extreme weather events such as hurricanes, tornados, wildfires (exacerbated by drought), flooding, and extreme heat. Extreme weather and sea-level rise pose physical risks to our facilities as well as those of our suppliers. Such risks include losses incurred as a result of physical damage to facilities, loss or spoilage of inventory, and business interruption caused by such natural disasters and extreme weather events. Other potential physical impacts due to climate change include reduced access to high-quality water in certain regions and the loss of biodiversity, which could impact future product development. These risks could disrupt our operations and its supply chain, which may result in increased costs.
New legal or regulatory requirements may be enacted to prevent, mitigate, or adapt to the implications of a changing climate and its effects on the environment. These regulations, which may differ across jurisdictions, could result in us being subject to new or expanded carbon pricing or taxes, increased compliance costs, restrictions on greenhouse gas emissions, investment in new technologies, increased carbon disclosure and transparency, upgrade of facilities to meet new building codes, and the redesign of utility systems, which could increase our operating costs, including the cost of electricity and energy used by us. Our supply chain would likely be subject to these same transitional risks and would likely pass along any increased costs to us.
Environmental, social and governance matters may impact our business and reputation.
Governmental authorities, non-governmental organizations, customers, investors, external stakeholders and employees are increasingly sensitive to environmental, social and governance, or ESG, concerns, such as diversity and inclusion, climate change, water use, recyclability or recoverability of packaging, and plastic waste. This focus on ESG concerns may lead to new requirements that could result in increased costs associated with developing, manufacturing and distributing our products, when applicable. Our ability to compete could also be affected by changing customer preferences and requirements, such as growing demand for more environmentally friendly products, packaging or supplier practices, or by failure to meet such customer expectations or demand. While we strive to improve our ESG performance, we risk negative stockholder reaction, including from proxy advisory services, as well as damage to our brand and reputation, if we do not act responsibly, or if we are perceived to not be acting responsibly in key ESG areas, including equitable access to medicines and vaccines, product quality and safety, diversity and inclusion, environmental stewardship, support for local communities, corporate governance and transparency, and addressing human capital factors in our operations. If we do not meet the ESG expectations of our investors, customers and other stakeholders, we could experience reduced demand for our products, loss of customers, and other negative impacts on our business and results of operations.
Risks Relating to Development, Clinical Testing, Manufacturing and Regulatory Approval
Prior to our acquisition of PHP-303, we were not involved in its development and, as a result, we are dependent on Bayer having accurately reported the results and correctly collected and interpreted the data from all clinical trials conducted prior to our acquisition.
58
We face risks associated with the clinical development for PHP-303 and for our future ADC nominated candidates that are currently in preclinical development.
59
The success of our current product candidates will depend on many factors, including the following:
60
We may choose to, or may be required to, suspend, repeat, or terminate our clinical trials if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive or the trials are not well designed.
We are subject to environmental and other risks.
61
Risks Relating to Our Dependence on Third Parties
We rely, and expect to continue to rely, on third parties, including independent investigators and CROs, to conduct our clinical trials.
62
Manufacturing biotherapeutics is difficult and complex, and requires facilities specifically designed and validated for this purpose and we will use Contract Development Manufacturing Organizations (CDMOs) through various contract-manufacturing arrangements.
63
64
Because manufacturing processes and those of our contractors are highly complex and are subject to a lengthy FDA approval process, alternative qualified production capacity may not be available on a timely basis or at all. Difficulties or delays in our or our contractors’ manufacturing and supply of existing or new products could increase our costs, cause us to lose revenue or market share, damage our reputation and could result in a material adverse effect on our product sales, financial condition, and results of operations.
65
In some circumstances we may rely on current and future collaborators to assist in our R&D activities. If any of our partners do not satisfy their obligations under our agreements with them, or if they terminate our licenses, partnerships, or collaborations with them, we may not be able to develop or commercialize our licensed or partnered product candidates as planned.
66
Risks Relating to Intellectual Property
We rely on patents and other intellectual property rights to protect our product candidates, the obtainment, enforcement, defense, and maintenance of which may be challenging and costly. Failure to enforce or protect these rights adequately could harm our ability to compete and impair our business.
67
The issuance, scope, validity, enforceability, and commercial value of our and our current or future licensors’ patent rights are highly uncertain.
With respect to certain patents, we enjoy only limited geographical protection, and as a consequence we may not be able to protect our intellectual property rights throughout the world.
68
Our intellectual property rights may not adequately protect our technologies and product candidates and may not necessarily address all potential threats to our competitive advantage.
69
Our commercial success depends, in part, upon our ability to develop, manufacture, market, and sell our product candidates without alleged or actual infringement, misappropriation, or other violation of the patents and proprietary rights of third parties. Litigation relating to patents and other intellectual property rights in the biopharmaceutical and pharmaceutical industries is common, including patent infringement lawsuits and interferences, oppositions, and reexamination proceedings before the U.S. Patent and Trademark Office (the “USPTO”), and foreign patent offices.
70
71
We cannot guarantee that any of our, our licensors’, or the previous owners’ patent searches or analyses, including but not limited to the identification of relevant patents, the scope of patent claims, or the expiration of relevant patent applications or patents, are complete or thorough, nor can we be certain that we have identified each and every third-party patent and patent application in the United States, Europe and elsewhere that is relevant to or necessary for the commercialization of our product candidates in any jurisdiction.
72
Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our product candidates or the use of our product candidates.
If we or our licensors fail to maintain the patents and patent applications covering our product candidates or if we or our licensors otherwise allow our patents or patent applications to be abandoned or lapse, our competitors might be able to enter the market, which would hurt our competitive position and could impair our ability to successfully commercialize our product candidates in any indication for which they are approved.
73
We may be subject to claims challenging the inventorship of our patents and patent applications or ownership of our intellectual property.
74
Depending upon the timing, duration, and conditions of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the “Hatch-Waxman Amendments.”
75
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest and our competitive position may be adversely affected.
76
We may be subject to claims by third parties asserting that we or our employees have misappropriated third-party intellectual property, or claiming ownership of what we regard as our own intellectual property. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages and lose valuable intellectual property rights or personnel.
Risks Relating to Competitive Employment for Key Personnel and other Matters related to Managing Company Growth
77
We face intense competition and rapid technological change.
Our competitors may have superior products, manufacturing capability or marketing expertise.
78
We have no experience in commercializing products on our own.
Risks Relating to Commercialization
Market acceptance of our products is uncertain.
79
Our existing and future product candidates may not gain market acceptance, in which case our ability to generate product revenues will be compromised.
80
We expect to face competition.
81
82
We plan to seek orphan drug designation for PHP-303 and future rare disease product candidates.
83
We or any future collaboration partners may seek and fail to obtain breakthrough therapy designation by the FDA for PHP-303 or any future product candidates or access to the PRIME scheme by the EMA for PHP-303 or any future product candidates.
84
We likely will commercialize or co-commercialize our product candidates for rare diseases and potentially rare tumor types and seek strategic relationships with third parties for the development and/or commercialization of our other product candidates.
85
Risks Relating to Healthcare Laws and Other Legal Compliance Matters
86
87
We face product liability risks and may not be able to obtain adequate insurance.
88
Our business operations and current and future relationships with investigators, healthcare professionals, consultants, third-party payors, patient organizations, and customers will be subject to applicable healthcare regulatory laws, which could expose us to penalties.
89
Our employees and independent contractors, including principal investigators, CROs, CMOs, consultants, vendors, and any other third parties we may engage in connection with the development and commercialization of our product candidates may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could adversely affect our business.
90
We are subject to governmental regulation and other legal obligations related to privacy, data protection and data security. Our actual or perceived failure to comply with such obligations could harm our business.
91
We are also subject to evolving European privacy laws on electronic marketing and cookies.
Although the clinical trial process is designed to identify and assess potential side effects, it is always possible that a drug, even after regulatory approval, may exhibit unforeseen side effects. If our product candidates were to cause adverse side effects during clinical trials or after approval, we may be exposed to substantial liabilities.
92
We may be exposed to future liabilities and/or obligations with respect to sales or out-licensing arrangements or partnerships.
Additional Risks Relating to Ownership of Company Securities
Nasdaq has delisted our securities from trading on its exchange, which could limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions.
Currently, our Common Stock is publicly traded on the OTC Markets Pink tier. No assurance can be given that our securities will trade on a national securities exchange, such as the Nasdaq Stock Market LLC (the “Nasdaq”) or the New York Stock Exchange (the “NYSE”), or the OTC Markets’ OTCQB or OTCQX tiers in the future or that the trading prices of our Common Stock on the OTC Markets will be indicative of the prices of our Common Stock if our Common Stock were traded on a national securities exchange. As previously disclosed, on November 1, 2022, we received written notice from the Staff of the Listing Qualifications Department of Nasdaq (the “Staff”) stating that the Staff has determined that the Company has not complied with the requirements of IM-5101-2 because (i) the Company has not demonstrated that its common stock complies with the minimum 1,000,000 unrestricted publicly held shares requirement in Listing Rule 5505(a)(2) (the “Unrestricted Publicly Held Shares Requirement”) and (ii) the Company’s public warrants (the “Public Warrants”) do not qualify for initial listing since the security underlying the warrants, the Company’s Common Stock, does not qualify. The Company timely requested a hearing before the Nasdaq Hearings Panel (the “Panel”) and such hearing has been conducted, which ultimately stayed the suspension of the Company’s Common Stock and Public Warrants and the filing by Nasdaq of a Form 25-NSE (the “Form 25”), pending the Panel’s decision. On January 6, 2023, the Company received a determination letter (the “Determination Letter”) from the Panel to delist the Company’s Common Stock and Public Warrants from Nasdaq. The Determination Letter indicated Nasdaq will suspend trading in the Company’s Common Stock and Public Warrants effective at the open of business on January 10, 2023 and indicated that it intends to file a Form 25-NSE Notification of Delisting with the SEC once all applicable appeal and review periods have expired in order to effect the formal delisting of the Company’s securities from Nasdaq. The Determination Letter provided that the Company may request an appeal of the Panel’s decision to the Nasdaq Listing and Hearing Review Council within 15 days of the date of the Determination Letter. Since the Company did not request an appeal, the Form 25 was filed
93
thereafter by Nasdaq. Upon suspension from Nasdaq, the Company’s Common Stock began to trade on the OTC Markets “Pink” tier.
As a result of the delisting of our securities by Nasdaq, we may face significant material adverse consequences until we are able to list our securities on another national securities exchange, including:
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Since our Common Stock and warrants are no longer currently listed on the Nasdaq, they are not deemed “covered securities.” Therefore, we may be subject to regulation in each state in which we offer our securities, which could materially adversely affect our business, financial condition and results of operations.
We cannot assure you that an active public market for our Common Stock will develop or be sustained. The market price of our Common Stock could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including:
94
95
Our existing stockholders have significant control of our management and affairs, which they could exercise against your best interests.
Risks Relating to future indebtedness if debt financing is needed
To service any of our future indebtedness, we will require a significant amount of cash. Our ability to generate cash depends on many factors beyond our control.
96
Fluctuations in our operating results could affect the price of our common stock. Our operating results may vary from period to period for several reasons including:
We incur increased costs and obligations as a result of being a public company.
Because there are no current plans to pay cash dividends on our Common Stock for the foreseeable future, you may not receive any return on investment unless you sell your common stock for a price greater than that which you paid for it.
We intend to retain future earnings, if any, for future operations, expansion and debt repayment and there are no current plans to pay any cash dividends for the foreseeable future. The declaration, amount and payment of any future dividends on shares of our Common Stock will be at the sole discretion of our board of directors. Our board of directors may take into account general and economic conditions, our financial condition and results of operations, our available cash and current and anticipated cash needs, capital requirements, contractual, legal, tax, and regulatory restrictions, implications on the payment of dividends by us to our stockholders or by its subsidiaries to it and such other factors as our board of directors may deem relevant. In addition, our ability to pay dividends is limited by covenants of our existing and outstanding indebtedness and may be limited by covenants of any future indebtedness that we incur. As a result, you may not receive any return on an investment in our Common Stock unless you sell our Common Stock for a price greater than that which you paid for it.
97
If securities analysts do not publish research or reports about our business or if they downgrade our stock or our sector, our stock price and trading volume could decline.
The trading market for our Common Stock will rely in part on the research and reports that industry or financial analysts publish about us or our business. We will not control these analysts. In addition, some financial analysts may have limited expertise with our model and operations. Furthermore, if one or more of the analysts who do cover the downgrade of our stock or industry, or the stock of any of our competitors, or publish inaccurate or unfavorable research about our business, the price of our stock could decline. If one or more of these analysts ceases coverage of us or fails to publish reports on it regularly, we could lose visibility in the market, which in turn could cause its stock price or trading volume to decline.
Future sales, or the perception of future sales, by us or our stockholders in the public market could cause the market price for our Common Stock to decline.
The sale of shares of our Common Stock in the public market, or the perception that such sales could occur, could harm the prevailing market price of shares of our Common Stock. These sales, or the possibility that these sales may occur, also might make it more difficult for us to sell equity securities in the future at a time and at a price that it deems appropriate.
Certain holders of our Common Stock have entered into lock-up agreements (the “Lock-Up Agreements”) with us pursuant to which each such holder agreed, subject to certain exceptions, not to dispose of or hedge any of their common stock or securities convertible into or exchangeable for shares of our Common Stock during the period from the date of the closing of the Business Combination continuing through the date 180 days after the Closing Date.
Upon the expiration or waiver of the lock-ups described above, shares held by the stockholders party to the Lock-Up Agreements will be eligible for resale, subject to volume, manner of sale and other limitations under Rule 144.
As restrictions on resale end, the market price of shares of our Common Stock could drop significantly if the holders of these shares sell them or are perceived by the market as intending to sell them. These factors could also make it more difficult for us to raise additional funds through future offerings of our Common Stock or other securities.
In addition, Common Stock reserved for future issuance under our equity incentive plans will become eligible for sale in the public market once those shares are issued, subject to provisions relating to various vesting agreements, lock-up agreements and, in some cases, limitations on volume and manner of sale applicable to affiliates under Rule 144, as applicable. The aggregate number of shares of our Common Stock reserved for future issuance under our equity incentive plan is 3,756,816. We will file one or more registration statements on Form S-8 under the Securities Act of 1933, as amended (the “Securities Act”) to register shares of Common Stock or securities convertible into or exchangeable for shares of Common Stock issued pursuant to our equity incentive plans. Any such Form S-8 registration statements will automatically become effective upon filing. Accordingly, shares registered under such registration statements will be available for sale in the open market.
Depending upon market liquidity at the time, sales of shares of our Common Stock under the White Lion Purchase Agreement may cause the trading price of our Common Stock to decline. After White Lion has acquired shares under the White Lion Purchase Agreement, it may sell all, some or none of those shares. Sales to White Lion by us pursuant to the White Lion Purchase Agreement may result in substantial dilution to the interests of other holders of our Common Stock. The sale of a substantial number of shares of our Common Stock to White Lion, or anticipation of such sales, could make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales. However, we have the right to control the timing and amount of any sales of our shares to White Lion, and the White Lion Purchase Agreement may be terminated by us at any time at our discretion without penalty.
The sale of substantial amounts of shares of our Common Stock or warrants, or the perception that such sales could occur, could cause the prevailing market price of shares of our Common Stock to decline significantly. These sales, or the possibility that these sales may occur, also might make it more difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate. We believe the likelihood that warrant holders will exercise their Warrants is dependent upon the market price of our Common Stock.
In the future, we may also issue its securities in connection with investments or acquisitions. The amount of shares of Common Stock issued in connection with an investment or acquisition could constitute a material portion of our then-outstanding shares of Common Stock. Any issuance of additional securities in connection with investments or acquisitions may result in additional dilution to our stockholders.
98
Warrants will become exercisable for our Common Stock, which would increase the number of shares eligible for future resale in the public market and result in dilution to our existing stockholders.
Outstanding warrants to purchase an aggregate of 5,375,000 shares of our Common Stock will become exercisable on the date which is 30 days after the completion of the Business Combination. Each Warrant entitles the holder thereof to purchase one (1) share of our Common Stock at a price of $11.50 per whole share for the Public Warrants and private placement warrants initially issued to Ignyte Sponsor LLC and $0.01 per share for the PIPE Warrants (as defined below), subject to adjustment. Warrants may be exercised only for a whole number of shares of Common Stock. To the extent such warrants are exercised, additional shares of our Common Stock will be issued, which will result in dilution to the then existing holders of our Common Stock and increase the number of shares eligible for resale in the public market. Sales of substantial numbers of such shares in the public market could adversely affect the market price of our Common Stock.
Our management and auditors have expressed substantial doubt about our ability to continue as a going concern.
Our auditors’ report to our December 31, 2022 financial statements includes an explanatory paragraph that expressed substantial doubt about our ability to continue as a going concern. Our current cash level raises substantial doubt about our ability to continue as a going concern without immediate short-term financing required to get us through consummation of the Business Combination. Additionally, our management has independently determined that there is substantial doubt about Peak Bio’s ability to continue as a going concern because our cash flows generated from operations may not be sufficient to meet our current operating costs. In addition, our future financial statements may include similar qualifications about our ability to continue as a going concern. Peak Bio’s financial statements were prepared assuming that it will continue as a going concern and do not include any adjustments that may result from the outcome of this uncertainty. If Peak Bio is unable to meet its current operating costs, Peak Bio would need to seek or additional financing or modify or cease its operational plans. If Peak Bio seeks additional financing to fund its business activities in the future and there remains substantial doubt about its ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding to Peak Bio on commercially reasonable terms or at all.
The JOBS Act permits “emerging growth companies” like us to take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies.
We qualify as an “emerging growth company” as defined in Section 2(a)(19) of the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012, which we refer to as the “JOBS Act.” As such, we will take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies for as long as it continues to be an emerging growth company, including (i) the exemption from the auditor attestation requirements with respect to internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act, (ii) the exemptions from say-on-pay, say-on-frequency and say-on-golden parachute voting requirements and (iii) reduced disclosure obligations regarding executive compensation in its periodic reports and proxy statements. As a result, our stockholders may not have access to certain information they deem important. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year (a) following February 1, 2026, the fifth anniversary of the closing of Ignyte’s IPO, (b) in which we have total annual gross revenue of at least $1.235 billion or (c) in which we are deemed to be a large accelerated filer, which means the market value of our Common Stock that are held by non-affiliates exceeds $700 million as of the last business day of our prior second fiscal quarter, and (ii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the exemption from complying with new or revised accounting standards provided in Section 7(a)(2)(B) of the Securities Act as long as Porch is an emerging growth company. An emerging growth company can therefore delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies, but any such election to opt out is irrevocable. We have elected not to opt out of such extended transition period, which means that when a standard is issued or revised and it has different application dates for public or private companies, we, as an emerging growth company, can adopt the new or revised standard at the time private companies adopt the new or revised standard. This may make comparison of our financial statements with another public company which is neither an emerging growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible because of the potential differences in accounting standards used.
We cannot predict if investors will find our Common Stock less attractive because we will rely on these exemptions. If some investors find our Common Stock less attractive as a result, there may be a less active trading market for Common Stock and our stock price may be more volatile.
99
Anti-takeover provisions in our organizational documents could delay or prevent a change of control.
Certain provisions of our Amended and Restated Charter and Amended and Restated Bylaws have an anti-takeover effect and may delay, defer or prevent a merger, acquisition, tender offer, takeover attempt or other change of control transaction that a stockholder might consider in its best interest, including those attempts that might result in a premium over the market price for the shares held by our stockholders.
These provisions provide for, among other things:
These anti-takeover provisions could make it more difficult for a third party to acquire us, even if the third party’s offer may be considered beneficial by many of our stockholders. As a result, our stockholders may be limited in their ability to obtain a premium for their shares. These provisions could also discourage proxy contests and make it more difficult for you and other stockholders to elect directors of your choosing and to cause us to take other corporate actions you desire. See “Description of Securities.”
Our Amended and Restated Charter designates the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or stockholders.
Our Amended and Restated Charter provides that, subject to limited exceptions, any (1) derivative action or proceeding brought on behalf of us, (2) action asserting a claim of breach of a fiduciary duty owed by any director, officer, stockholder or employee to us or our stockholders, (3) action asserting a claim arising pursuant to any provision of the DGCL or our Amended and Restated Charter or our Amended and Restated Bylaws, or (4) action asserting a claim governed by the internal affairs doctrine shall, to the fullest extent permitted by law, be exclusively brought in the Court of Chancery of the State of Delaware or, if such court does not have subject matter jurisdiction thereof, another state or federal court located within the State of Delaware. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock shall be deemed to have notice of and to have consented to the provisions of our Amended and Restated Charter described above. This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or its directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and employees. Alternatively, if a court were to find these provisions of our Amended and Restated Charter inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business and financial condition.
100
Item 1B. Unresolved Staff Comments.
None
Item 2. Properties.
Our principal executive office is located in Pleasanton, California, which consists of general office space. We are in the process of seeking laboratory facilities to replace the previous facility in Palo Alto, California. We believe that our existing facilities are adequate to meet our needs and that existing needs and future growth can be accommodated by leasing alternative or additional space.
Item 3. Legal Proceedings.
The Company is not currently a party to any material legal proceedings. At each reporting date, the Company evaluates whether a potential loss amount or a potential range of loss is probable and reasonably estimable under the provisions of the authoritative guidance that addresses accounting for contingencies. The Company expenses the costs related to its legal proceedings as incurred.
Item 4. Mine Safety Disclosures.
Not applicable.
101
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information and Holders
Our Common Stock and Public Warrants were historically quoted on the Nasdaq under the symbols “IGNY” and “IGNYW,” respectively. On November 2, 2022, our Common Stock and Public Warrants were initially listed on the Nasdaq under the new trading symbols of “PKBO” and “PKBOW,” respectively. As of January 10, 2023, our Common Stock and Public Warrants have been suspended from trading on the Nasdaq and our Common Stock began trading on the OTC Markets “Pink” tier under the symbol “PKBO.”
As of March 31, 2023, there were approximately 131 record holders or our Common Stock and approximately 26 records holders of our warrants.
Dividends
We have not paid any cash dividends on our Common Stock to date. We may retain future earnings, if any, for future operations, expansion and debt repayment and have no current plans to pay cash dividends for the foreseeable future. Any decision to declare and pay dividends in the future will be made at the discretion of our board of directors and will depend on, among other things, our results of operations, financial condition, cash requirements, contractual restrictions and other factors that our board of directors may deem relevant. In addition, our ability to pay dividends may be limited by covenants of any existing and future outstanding indebtedness we or our subsidiaries incur. We do not anticipate declaring any cash dividends to holders of our Common Stock in the foreseeable future.
Stock Performance Graph
Not applicable.
Sale of Unregistered Securities
None.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Securities Authorized for Issuance Under Equity Incentive Plan
At the special meeting of Ignyte’s stockholders in lieu of our 2022 annual meeting held on October 25, 2022, our stockholders considered and approved the Peak Bio, Inc. 2022 Long-Term Incentive Plan (the “Incentive Plan”). The Incentive Plan was previously approved, subject to stockholder approval, by the Ignyte board of directors on April 27, 2022. The Incentive Plan became effective immediately upon the closing of the Business Combination. Pursuant to the Incentive Plan, 3,756,816 shares of Common Stock have been reserved for issuance under the Incentive Plan.
Item 6. Reserved
102
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of Peak Bio’s financial condition and results of operations together with Peak Bio’s audited consolidated financial statements and notes thereto included elsewhere in this Annual Report on Form 10-K. Certain of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report on Form 10-K, including information with respect to plans and strategy for Peak Bio’s business, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the section entitled “Risk Factors,” Peak Bio’s actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis. You should carefully read the section entitled “Risk Factors” to gain an understanding of the important factors that could cause actual results to differ materially from Peak Bio’s forward-looking statements. Please also see the section entitled “Cautionary Note Regarding Forward-Looking Statements.”
Unless otherwise indicated or the context otherwise requires, references in this Peak Bio’s Management’s Discussion and Analysis of Financial Condition and Results of Operations section to “Peak Bio,” “we,” “us,” “our” and other similar terms refer to Peak Bio Co., Ltd. (excluding the Non-Peak Bio Assets transferred in the Spin-Off) prior to the Business Combination and to Peak Bio, Inc. and its consolidated subsidiaries after giving effect to the Business Combination.
Overview
Peak Bio is a clinical-stage biopharmaceutical company focused on developing therapeutics addressing significant unmet need in the areas of oncology and inflammation. Our management team has a combined 50 years of industry experience in the areas of small molecules, antibodies, and antibody-drug-conjugates (ADC).
Our lead product candidate, PHP-303 is a small molecule, 5th generation Phase 2 clinical-ready neutrophil elastase (NE) inhibitor (NEI). We are planning a Phase 2 clinical study in Alpha-1 anti-trypsin deficiency (AATD) patients. We have completed two Phase 1 trials of PHP-303 in healthy volunteers testing higher doses of PHP-303 by single-ascending dose (SAD) and multiple-ascending dose (MAD). PHP-303 demonstrated dose- dependent pharmacokinetics and the recommended Phase 2 dose was achieved in these trials. A maximum tolerated dose for PHP-303 was not achieved in these Phase 1 trials.
In addition, we have leveraged two decades of industry learning in the antibody-drug-conjugate (ADC) field to develop a platform of proprietary technologies that enable us to design ADCs to have improved efficacy, safety, and tolerability relative to existing antibody or ADC therapies. Our most advanced platform, PH-1 or Thailanstatin is being used to generate a pipeline of proprietary ADC product candidates to address patient populations with improved efficacy relative to traditional ADC-based therapies. Our second product candidate is an ADC targeting Trop2, an antigen broadly expressed in solid tumors. We expect our Trop2 ADC to enter clinical development by late 2024. Our Trop2 ADC and other undisclosed discovery-stage product candidates are based on our proprietary PH-1 platform of toxin payloads targeting RNA splicing.
Despite commercial success of the ADCs currently on the market, there continues to be a need for ADCs that not only deliver antibody-directed payloads selectively to their tumors, but to also release them safely via improved linker technology and avoid off- target toxicities. Secondly, we believe that adding an immunomodulatory effect to our toxin(s) that engages our immune systems to assist in the cancer killing would contribute to improved tumor killing.
We do not have any products available for commercial sale, and we have not generated any product revenue from our portfolio of product candidates or other sources. Our ability to generate revenue sufficient to achieve profitability, if ever, will depend on the successful development and eventual commercialization of our potential therapies, which we expect, if it ever occurs, will take a number of years. The research and development efforts require significant amounts of additional capital and adequate personnel infrastructure. There can be no assurance that our research and development activities will be successfully completed, or that our potential therapies will be commercially viable.
We have incurred significant losses since the commencement of our operations. Our net loss was $13.1 million and $8.3 million for the years ended December 31, 2022 and 2021, respectively. We expect to continue to incur significant expenses and operating losses for the foreseeable future as we continue our efforts to identify product candidates and seek regulatory approvals within our portfolio of product candidates. These losses have resulted primarily from costs incurred in connection with research and development activities and to a lesser extent from general and administrative costs associated with our operations. Our net losses may fluctuate significantly from period to period, depending on the timing of and expenditures on our research and development activities.
103
Recent Developments
Employment Agreements
In January 2022, we entered into an employment agreement with our founder and director. The effective date of the employment agreement was February 1, 2022 and was subject to the completion of the business combination with Ignyte (the “Business Combination”). As part of the agreement, we agreed to repay our founder and director $1.5 million in forwent salary over a period of four years. In addition, as part of the agreement, we agreed to repay $0.5 million of the $1.5 million loan outstanding upon closing of the Business Combination. The remaining $1.0 million plus accrued interest will be repaid pursuant to the discretion of our Board of Directors. We repaid $150,000 of the loan in December 2022. Further, the employment agreement provides for the payment of success fees in connection with future business or corporate development transactions (licensing, product development and acquisitions).
In March 2022, we entered into an employment agreement with our chief operating officer which was subject to the completion of the business combination with Ignyte. The agreement provides for confirmation of Peak Bio’s previously agreed upon success fee payment to Dr. LaMond upon consummation of the business combination with Ignyte in the amount of $250,000 and the payment of success fees in connection with future business or corporate development transactions (licensing, product development and acquisitions).
Spin-Off
Effective March 1, 2022, we spun off certain assets to a newly formed company in Korea (“SpinCo”) that we refer to in the Business Combination Agreement as “Non-Peak Bio Assets” (the “Spin-Off”). Those Non-Peak Bio Assets include PH-2 (Spliceostatin) toxin licensed from a third-party, the PH-3 (Callyspongiolide) toxin licensed from a third party, PH-4 (PBD Hybrid), and PHP-201 (Sovesudil).
As a result, we will require additional financing to fund our ongoing activities. We may raise this additional funding through the sale of equity, debt financings or other capital sources, including potential collaborations with other companies or other strategic transactions and funding under government contracts. We may be unable to raise additional funds or to enter into such agreements or arrangements on favorable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate certain of our research and development programs. There can be no assurances that other sources of financing would be available. The amount and timing of our future funding requirements will depend on many factors, including the pace and results of our research and development efforts.
VennDC, LLC (“Venn”)
In December 2019, a collaboration and license agreement (the “License Agreement”) was entered into with Venn to pursue research and development of certain payload and linker technologies that are useful for the development of antibody-drug conjugates. This collaboration was expected to allow Venn to further develop and commercialize such antibody-drug conjugates developed under the collaboration. Under the collaboration agreement with Venn, we received a $400,000 upfront payment and was expected to be eligible to receive reimbursement of costs and expenses incurred, certain development and regulatory milestone payments, royalties and commercial milestone payments with respect to licensed products for each product. Milestone payments were expected to be payable following the achievement of certain development, regulatory and commercial milestone events in each product, up to an aggregate of $107.1 million per product. Royalty payments were expected to be based on net sales of licensed products on a licensed product-by-licensed product basis. The initial term of the research collaboration was expected to be three years. During the years ended December 31, 2022 and 2021, we did not recognize any revenue related to the upfront payment as it was not probable that a significant reversal in the amount of cumulative revenue recognized would not occur. In addition, no reimbursement of costs and expenses incurred, and no other payments (for development and regulatory milestones, royalties, and commercial milestones with respect to licensed products for each product) were received by us during the years ended December 31, 2022 and 2021, as none of the performance obligations were satisfied by us. At December 31, 2021, we recorded a liability to accrued expense of $400,000 related to this payment. At December 31, 2022, we recorded a liability to related party loans of $400,000 related to this payment.
In April 2022, we entered into an agreement with our founder and director, Dr. Huh, in consideration of the repayment to be made by our founder and director to settle a contractual obligation for the upfront payment we received associated with the License Agreement with Venn. Per the agreement, we agreed to repay our founder and director $400,000, with interest to accrue on the unpaid principal balance at the rate of 1% per annum. The timing of the repayment will be determined and pursuant to the discretion of our Board of Directors.
104
In May 2022, our founder and director, Dr. Huh, repaid to Venn the $400,000 upfront payment and the License Agreement was terminated. At December 31, 2022, we recorded a liability to related party loans of $400,000 related to this payment.
Financing
In May 2022, we entered into an agreement with a certain investor in which the investor purchased an aggregate of 63,856 shares of Peak Bio Co., Ltd. common stock for aggregate gross proceeds of approximately $1.2 million.
From July through September 2022, we received proceeds from loans in the amount of $1.25 million from several lenders. The loans mature on the second anniversary and bear interest at a rate of 5.0% per annum. The loans were evidenced by promissory notes, which contain customary events of default relating to, among other things, payment defaults and breaches of representations and warranties. The loans may not be prepaid by us at any time prior to maturity without the consent of the lender. We will provide for the conversion of the principal and interest of the loans into shares of common stock at fair market value and 25% warrant coverage on common stock prior to the consummation of the Business Combination. Warrant coverage is conditioned on closing of the Business Combination and will be exercisable after the closing of the Business Combination with an exercise price of $0.01.
In September 2022, we received proceeds from a loan in the amount of $0.5 million from one of our director nominees. The loan matures on the second anniversary and bear interest at a rate of 5.0% per annum. The loan was evidenced by a promissory note, which contains customary events of default relating to, among other things, payment defaults and breaches of representations and warranties. The loan may be prepaid by us at any time prior to maturity without the consent of the lender.
PIPE Subscription Agreements
In November 2022, concurrently with the closing of the Business Combination, we entered into subscription agreements with certain investors in which the investors purchased, in a private placement, an aggregate of 302,500 shares of Common Stock and 281,325 warrants to purchase shares of Ignyte Common Stock, at an exercise price of $0.01 per share for aggregate gross proceeds of $3.025 million. The warrants are on terms substantially the same as the outstanding warrants that were included in the units issued in Ignyte's initial public offering, except that the warrants are non-redeemable, and the warrants are exercisable for one year.
Additional PIPE Subscription Agreement
In December 2022, we entered into a subscription agreement whereby we agreed to issue and sell to the investor party thereto, in a private placement, (i) 50,000 shares of our common stock at $10.00 per share and (ii) 46,500 warrants to purchase shares of our Common Stock, at an exercise price of $0.01 per share. The warrants are on terms substantially the same as the outstanding warrants that were included in the units issued in Ignyte’s initial public offering, except that the warrants are not redeemable, and the warrants are exercisable for one year.
Key Company Stockholder Agreements
In April 2022, we entered into a forward purchase agreement (the “Key Company Stockholder Forward Purchase Agreement”) with our founder and director, Hoyoung Huh (the “Key Company Stockholder”). Pursuant to the terms of the Key Company Stockholder Forward Purchase Agreement, the Key Company Stockholder would, subject to the receipt of margin financing within 180 days following the closing of the Business Combination, purchase shares of our common stock at a purchase price of $10.00 per share in a private placement (the “Key Company Stockholder Purchase”) for up to an aggregate amount of $10,000,000 (the “Subscription Amount”), subject to the conditions set forth in the Key Company Stockholder Forward Purchase Agreement.
In December 2022, we and the Key Company Stockholder entered into an amendment to the Key Company Stockholder Forward Purchase Agreement (the “Amendment to Key Company Stockholder Forward Purchase Agreement”), pursuant to which (i) the Key Company Stockholder Purchase is no longer subject to the receipt of margin financing as a condition precedent, (ii) the Key Company Stockholder agreed to fund the Subscription Amount on or prior to March 31, 2023 and (iii) the Key Company Stockholder Purchase would be consummated at a purchase price of $5.18 per share of our common stock. Accordingly, upon closing of such purchase, Dr. Huh will receive 1,930,501 shares of Common Stock in exchange for his $10 million investment in the Company.
In April 2023, we received notice from our founder and director informing us that he would not consummate the purchase of the Key Company Stockholder Forward Purchase Agreement as a result of our failure to satisfy the condition to be listed on
105
Nasdaq as required by the agreement. As a result we cancelled and forfeited the 1,930,501 shares of common stock being held in escrow.
In April 2023, we and our founder and director entered into a letter agreement to provide for the conversion of loans totaling $2,031,034 into future qualified financings on the same terms with other investors. Pursuant to the agreement, the amount converted would be based on the Key Company Stockholder's pro-rata portion of equity ownership in our outstanding common stock and would not exceed in the aggregate the amount of the debt outstanding owed.
Ignyte Acquisition Corp (Ignyte)
On November 1, 2022 (the “Closing Date”), we completed the transactions contemplated by that certain business combination agreement, dated as of April 28, 2022 (the “Business Combination Agreement”), by and among Ignyte, Ignyte Korea Co., Ltd., a corporation organized under the laws of the Republic of Korea (“Korean Sub”), and Peak Bio Co., Ltd. At the closing of the transactions, (i) the stockholders of Peak Bio Co., Ltd. transferred their respective shares of common stock to Korean Sub in exchange for shares of Ignyte common stock held by Korean Sub, and (ii) in the course of such share swap, Korean Sub distributed the shares of Peak Bio Co., Ltd. common stock to Ignyte in consideration of Ignyte common stock (which was in-turn delivered to the stockholders of Peak Bio Co., Ltd. as described in (i) above ((i) and (ii), collectively, the “Share Swap”). Upon consummation of the Share Swap, Peak Bio Co., Ltd. became a direct wholly-owned subsidiary of Ignyte. The transactions contemplated by the Business Combination Agreement are referred to herein as the “Business Combination.”
On the Closing Date, a purchaser (the “Original Subscriber”) purchased from us an aggregate of 50,000 shares of Ignyte Common Stock (the “Original PIPE Shares”), for a purchase price of $10.00 per share and an aggregate purchase price of $500,000, pursuant to a subscription agreement entered into effective as of April 28, 2020 (the “Original Subscription Agreement”).
On the Closing Date, a number of additional purchasers (each, a “New Subscriber”) purchased from us an aggregate of (i) 302,500 shares of Ignyte Common Stock (the “New PIPE Shares”) and (ii) 281,325 warrants (the “PIPE Financing Warrants”) to purchase shares of Ignyte Common Stock, at an exercise price of $0.01 per share, for a purchase price of $10.00 per share and an aggregate purchase price of $3,025,000, pursuant to separate subscription agreements entered into effective as of October 31, 2022 (each a “New Subscription Agreement”). The PIPE Financing Warrants are on terms substantially the same as the outstanding warrants that were included in the units issued in Ignyte’s initial public offering, except that the new warrants are not redeemable, and the warrants shall be exercisable for one year.
On the Closing Date, a number of Peak Bio’s lenders (each, a “Bridge Loan PIPE Subscriber” and together with the Original Subscriber and the New Subscribers, the “Subscribers”) purchased from us an aggregate of (i) 176,579 shares of Ignyte Common Stock (the “Bridge Loan PIPE Shares” and together with the Original PIPE Shares and the New PIPE Shares, the “PIPE Shares”) and (ii) 164,218 warrants (the “Bridge Loan PIPE Financing Warrants” and together with the PIPE Financing Warrants, the “PIPE Warrants”) to purchase shares of Ignyte Common Stock, at an exercise price of $0.01 per share, in consideration for their agreement to cancel an aggregate principal amount of $1,750,000 and the interest accrued thereon in promissory notes evidencing the loans such lenders had extended to Peak Bio Co., Ltd. between July and September 2022, pursuant to separate subscription agreements entered into effective as of October 31, 2022 (each a “Bridge Loan PIPE Subscription Agreement” and together with the Original Subscription Agreement and the New Subscription Agreements, the “Subscription Agreements”). The Bridge Loan PIPE Financing Warrants are on terms substantially the same as the outstanding warrants that were included in the units issued in Ignyte’s initial public offering, except that the new warrants are not redeemable, and the warrants shall be exercisable for one year.
Pursuant to the Subscription Agreements, we gave certain registration rights to the Subscribers with respect to the PIPE Shares and the PIPE Warrants. The sale of the PIPE Shares and PIPE Warrants was consummated concurrently with the Closing.
Upon the Closing, Ignyte as the registrant changed its name to “Peak Bio, Inc.”
Convertible Notes
On November 1, 2022, we issued a $1,512,500 convertible note to EarlyBirdCapital, Inc., the sole book running manager of Ignyte’s IPO, in lieu of the deferred underwriting fee that was payable at the Closing. The convertible note accrues interest at a rate of 8% per annum and is payable on October 31, 2023, provided however that we agree to make mandatory prepayments on this note (which shall first be applied to accrued interest and then to principal) from time to time in amounts equal to 15% of the gross proceeds received by us from any equity lines, forward purchase agreements or other equity financings consummated by us prior to the maturity date.
106
On the maturity date, the note holder may, in its sole and absolute discretion, convert all or part of the principal and/or accrued interest of this convertible note into shares of our common stock of at a per share conversion price equal to 90% of the volume weighted average price of a share of our common stock for the five trading days immediately prior to the maturity date.
In April 2023, we entered into separate subscription agreements for the issuance of convertible promissory notes (the "2023 Convertible Notes") in the aggregate principal amount of $2,195,034 and an aggregate amount of 3,658,390 warrants (the “Warrants”). The 2023 Convertible Notes will be convertible into shares of our common stock at $0.60 per share. For each share into which a 2023 Convertible Note is convertible, the investor received Warrants to purchase an equal amount of shares of our common stock at $0.60 per share. In connection with the issuance of the 2023 Convertible Notes and the Warrants, in consideration for its services in respect of the financing described above, the Company also issued the Placement Agent purchase warrants (the “Placement Agent Warrant”) to purchase 209,670 shares of the Company’s common stock at a price per share of $0.60. The Placement Agent Warrant has a 5-year term. In addition, the Company paid the Placement Agent a commission of approximately $125,000.
In April 2023, we entered into a subscription agreement with our founder and director to settle $1,130,775 in related party loans made to us. We issued a $1,130,775 related party unsecured convertible promissory note and warrants to purchase 1,884,625 shares of our common stock at $0.60 per share.
White Lion Common Stock Purchase and Registration Rights Agreements
On November 3, 2022, we entered into a Common Stock Purchase Agreement (the “White Lion Purchase Agreement”) and Registration Rights (the “White Lion RRA”) with White Lion Capital, LLC, a Delaware limited liability company (“White Lion”). Pursuant to the White Lion Purchase Agreement, we have the right, but not the obligation, to require White Lion to purchase, from time to time, up to $100,000,000 in aggregate gross purchase price of newly issued shares of our Common Stock, subject to certain limitations and conditions set forth in the White Lion Purchase Agreement. Capitalized terms used but not otherwise defined in this section shall have the meanings given to such terms by the White Lion Purchase Agreement and the White Lion RRA.
We are obligated under the White Lion Purchase Agreement and the White Lion RRA to file a registration statement with the SEC to register the Common Stock under the Securities Act, for the resale by White Lion of shares of Common Stock that we may issue to White Lion under the White Lion Purchase Agreement.
Subject to the satisfaction of certain customary conditions including, without limitation, the effectiveness of a registration statement registering the shares issuable pursuant to the White Lion Purchase Agreement, our right to sell shares to White Lion will commence on the effective date of the registration statement and extend until November 1, 2025. During such term, subject to the terms and conditions of the White Lion Purchase Agreement, we may notify White Lion when we exercise our right to sell shares (the effective date of such notice, a “Notice Date”).
The number of shares sold pursuant to any such notice may not exceed (i) the lower of (a) the Purchase Notice Fixed Limit (described below) and (b) the product of (1) the Average Daily Trading Volume (as defined in the White Lion Purchase Agreement), and (2) the applicable Percentage Limit (as defined in the White Lion Purchase Agreement). The Purchase Notice Fixed Limit is $500,000 upon payment of the Initial Commitment Shares (as defined in the White Lion Purchase Agreement) and can be increased in two tranches: (A) to $1,000,000 following an aggregate purchase of $5,000,000 shares and issuance by us to White Lion of an additional $250,000 in Commitment Shares, and (B) to $2,000,000 following an aggregate purchase of $10,000,000 shares and issuance by the for payment of an additional $250,000 in Commitment Shares (as defined in the White Lion Purchase Agreement).
The applicable Percentage Limit is 40% or 150% depending on the price we agree to sell shares to White Lion. At an applicable Percentage Limit of 40%, the Purchase Price to be paid by White Lion for any such shares will equal 97% of lowest daily volume-weighted average price of Common Stock during a period of two consecutive Trading Days following the applicable Purchase Notice Date (as defined in the White Lion Purchase Agreement) until an aggregate of $50,000,000 in Purchase Notice Shares (as defined in the White Lion Purchase Agreement) have been purchased under White Lion Purchase Agreement, at which point the Purchase Price (as defined in the White Lion Purchase Agreement) to be paid by White Lion will equal 98% of the lowest daily volume-weighted average price of Common Stock during a period of two consecutive Trading Days following the applicable Purchase Notice Date. At an applicable Percentage Limit of 150%, the Purchase Price to be paid by White Lion for any such shares will equal 94.5% of the lowest daily volume-weighted average price of Common Stock during a period of three consecutive Trading Days following the applicable Purchase Notice Date.
We will have the right to terminate the White Lion Purchase Agreement at any time after commencement, at no cost or penalty, upon three (3) Trading Days’ prior written notice. Additionally, White Lion will have the right to terminate the White
107
Lion Purchase Agreement upon three (3) days’ prior written notice to us if (i) there is a Fundamental Transaction (as defined in the White Lion Purchase Agreement), (ii) we are in breach or default in any material respect of the White Lion RRA, (iii) there is a lapse of the effectiveness, or unavailability of, the registration statement for a period of 45 consecutive Trading Days or for more than an aggregate of 90 Trading Days in any 365-day period, (iv) the suspension of trading of the Common Stock for a period of five (5) consecutive Trading Days, (v) the material breach of the White Lion Purchase Agreement by us, which breach is not cured within the applicable cure period or (vi) a Material Adverse Effect (as defined in the White Lion Purchase Agreement) has occurred and is continuing. No termination of the White Lion Purchase Agreement will affect the registration rights provisions contained in the White Lion RRA.
In consideration for the commitments of White Lion, as described above, we have agreed that it will issue to White Lion shares of Common Stock having a value of $250,000 based upon the Closing Sale Price (as defined in the White Lion Purchase Agreement) of Common Stock two Trading Days prior to the filing of the Initial Registration Statement as Initial Commitment Shares. We may increase the number of shares it may sell to White Lion by issuing additional Commitment Shares in two additional tranches of $250,000 each. The Company issued Initial Commitment Shares of 50,200 shares of Common Stock to White Lion, based upon the Closing Sale Price of our Common Stock of $4.98 per share on November 30, 2022.
Concurrently with the execution of the White Lion Purchase Agreement, we entered into the White Lion RRA with White Lion in which we have agreed to register the shares of Common Stock purchased by White Lion with the SEC for resale within 30 days of the consummation of a business combination. The White Lion RRA also contains usual and customary damages provisions for failure to file and failure to have the registration statement declared effective by the SEC within the time periods specified.
The White Lion Purchase Agreement and the White Lion RRA contain customary representations, warranties, conditions and indemnification obligations of the parties. The representations, warranties and covenants contained in such agreements were made only for purposes of such agreements and as of specific dates, were solely for the benefit of the parties to such agreements and may be subject to limitations agreed upon by the contracting parties.
In March 2023, we entered into an amendment to the White Lion Purchase Agreement to give the Company the right, but not the obligation to require White Lion to purchase shares of our common stock while trading on the OTC Market. Under the terms of the amendment, we will issue to White Lion within five (5) Trading Days following the effective date of the amendment fully paid, non-assessable shares of our Common Stock equal to the quotient obtained by dividing (i) $250,000 and (ii) the lowest traded sale price of the common stock of the 10 (ten) Trading Days prior to the effective date of the amendment, minus 50,200. In March 2023, we issued 412,763 shares of our common stock to White Lion.
In addition, in the event the Company does not issue Purchase Notices (as defined in the White Lion Purchase Agreement) to White Lion providing for the purchase of at least $1,000,000 of Purchase Shares (as defined in the White Lion Purchase Agreement) in the aggregate within 180 days following the effective date of the amendment, the Company will issue to White Lion an additional number of fully paid, non-assessable shares of common stock equal to the quotient obtained by dividing (i) $100,000 and (ii) the lowest Closing Sale Price (as defined in the White Lion Purchase Agreement) of Common Stock of the 10 (ten) Trading Days prior to the 180th day following the effective date of the amendment.
Forward Purchase Agreement
On December 29, 2022, we purchased 375,939 shares of our Common Stock at a price of $10.115 per share following the exercise of an investor’s right to sell up to 450,000 shares of Common Stock under a previously disclosed Forward Share Purchase Agreement entered into on October 25, 2022. The 375,939 shares of Common Stock have been retired. As a result of that exercise, funds in the amount of $4,551,750 being held in escrow in connection with the Forward Purchase Agreement were distributed as follows: $749,127 to us and $3,802,623 to the investor.
Release of Certain Lock-Up Restrictions
In order to comply with Nasdaq listing requirements, on January 4, 2023, we released 30% of the Target Consideration Shares (excluding shares held by Dr. Huh) from the six-month lock-up restrictions entered into in connection with the Business Combination (representing a total of 3,057,599 shares for release, of which, we estimate that approximately one-third will have been purchased below the closing price of our Common Stock of $4.21 on December 29, 2022). The release of those shares from lock-up restrictions may cause the market price of our securities to decline or increase the volatility in the market price of our securities. Additionally, since the Target Consideration Shares that may be offered for resale pursuant to this prospectus represent approximately 88% of the shares outstanding as of December 29, 2022 (after giving effect to redemptions and repurchases), the sale of all the securities being offered in this prospectus, or the perception that these sales could occur, could result in a significant decline in the public trading price of our securities.
108
Departure of Directors; Appointment of New Audit Committee Chair
On March 14, 2023, Brad Stevens notified us of his resignation from our Board of Directors (the “Board”), effective immediately. Mr. Stevens’ decision to resign from the Board is solely for personal reasons and is not the result of any disagreement with us with respect to any matter relating to our operations, policies or practices, or any disagreements in respect of accounting principles, financial statement disclosure, or any issue impacting the Audit Committee of the Board (the committee on which he served).
On June 21, 2023, Nevan Elam notified us of his resignation from our Board, effective immediately. Mr. Elam’s decision to resign from the Board is solely for personal reasons and is not the result of any disagreement with us with respect to any matter relating to our operations, policies or practices, or any disagreements in respect of accounting principles, financial statement disclosure, or any issue impacting the Audit Committee of the Board (the committee on which he served and chaired).
On June 22, 2023, the Board appointed Jim Neal as the chair of the Audit Committee and the Board appointed David Rosenberg as a member of the Audit Committee.
Issuance of Unregistered Securities
As previously disclosed, in April 2023 we issued to our founder and director warrants to purchase 1,884,625 shares of our common stock with an exercise price of $0.60 per share. On June 23, 2023, our founder and director exercised warrants to purchase 666,667 shares of our common stock at $0.60 per share for a total purchase price of $400,000.
Components of Results of Operations
Operating Expenses
Prior to April 1, 2022, the consolidated financial statements have been extracted from the accounting records of pH Pharma, Ltd. on a carve-out basis. The historical results of operations, financial position, and cash flows may not be indicative of what we would have been had we been a separate stand-alone entity, nor are they indicative of what the results of operations, financial position and cash flows may be in the future.
The majority of our operating expenses related to research and development (“R&D”). R&D expenses directly related to us were entirely attributed to us in the accompanying consolidated financial statements. R&D salaries, wages and benefits were allocated to us using methodologies based on the proportionate share of R&D expenses for the PHP-303 and PH-1 ADC Platform programs compared to the R&D expenses for pH Pharma Ltd as a whole. We also received services and support from other functions of pH Pharma Ltd. Our operations are dependent upon the ability of these other functions to provide these services and support. The costs associated with these services and support were allocated to us using methodologies based on the proportionate share of R&D expenses for the PHP-303 and PH-1 ADC Platform programs compared to the R&D expenses for pH Pharma Ltd as a whole. These allocated costs were primarily related to corporate administrative expenses, non-R&D employee related costs, including salaries and other benefits, for corporate and shared employees, and other expenses for shared assets for the following functional groups: information technology, legal, accounting and finance, human resources, facilities, and other corporate and infrastructural services. These allocated costs were primarily recorded as R&D expenses and general and administrative (“G&A”) expenses in the statements of operations and comprehensive loss.
The Spin-Off resulted in Peak Bio Co., Ltd. retaining the PHP-303 and PH-1 ADC Platform programs. Historically and throughout the periods presented, the PHP-303 and PH-1 ADC Platform programs have been owned by pH Pharma Co., Ltd and its subsidiaries (prior to the change of its name to Peak Bio Co., Ltd.). The PHP-303 and PH-1 ADC Platform programs have historically operated as a part of pH Pharma Co., Ltd and not as a separate stand-alone entity or group. The Spin-Off resulted in Peak Bio Co., Ltd. retaining approximately 90% of the equity outstanding in pH Pharma Co., Ltd., consisting of 8,283,613 shares of common stock and 693,000 stock options, before giving effect to the exchange ratio of 2.0719.
As of April 1, 2022, we concluded that all the assets and liabilities of the newly created Peak Bio Co., Ltd. legal entity were contributed by the parent company pH Pharma Ltd. No other assets or liabilities were considered to be attributable to Peak Bio Co., Ltd. or that would be transferred to Peak Bio Co., Ltd. upon the completion of the Business Combination, eliminating the necessity to allocate a portion of pH Pharma Ltd.’s assets and liabilities to Peak Bio Co., Ltd. on a carve-out basis. Therefore, there was no longer a need to allocate assets and liabilities, as well as expenses, from the parent for the consolidated financial statements.
Our consolidated financial statements for the year ended December 31, 2022 include the accounts of Peak Bio Co., Ltd. and its subsidiary, Peak Bio CA., Inc. All intercompany balances and transactions have been eliminated in consolidation.
109
Revenue
Our revenue has historically been generated through grants from government organizations. We currently have no commercially approved products. Grant revenue is recognized during the period that the research and development services occur, as qualifying expenses are incurred or conditions of the grants are met. We concluded that payments received under these grants represent conditional, nonreciprocal contributions, as described in ASC 958, Not-for-Profit Entities, and that the grants are not within the scope of ASC 606, Revenue from Contracts with Customers, as the organizations providing the grants do not meet the definition of a customer. Qualifying expenses are recognized when incurred as research and development expenses. Expenses for grants are tracked by using a project code specific to the grant, and the employees also track hours worked by using the project code.
Research and Development Expense
We expense research and development costs as incurred. Research and development expense consist primarily of costs related to personnel, including salaries and other personnel related expenses, contract manufacturing and supply, consulting fees, and the cost of facilities and support services used in drug development. Assets acquired that are used for research and development and have no future alternative use are expensed as in-process research and development.
General and Administrative Expenses
Our general and administrative expenses consist primarily of personnel costs, depreciation expense and other expenses for outside professional services, including legal fees relating to patent and corporate matters, human resources, audit and accounting services and facility-related fees not otherwise included in research and development expenses. Personnel costs consist of salaries, benefits and equity-based compensation expense, for our personnel in executive, finance and accounting, business operations and other administrative functions. We expect our general and administrative expenses to increase over the next several years to support our continued research and development activities, manufacturing activities, increased costs of expanding our operations and operating as a public company. These increases will likely include increases related to the hiring of additional personnel, fees to outside consultants, lawyers and accountants, and increased costs associated with being a public company such as expenses related to services associated with maintaining compliance with the applicable securities exchange listing rules and SEC requirements, director and officer insurance premiums and investor relations costs.
Change in fair value of derivative liabilities
Change in fair value of our derivative liabilities consists of changes in the fair value of certain conversion features associated with our convertible note that is required to be bifurcated and accounted for as free-standing derivative financial instruments and put options in our Forward Purchase Agreement, Key Company Stockholder Forward Purchase Agreement and White Lion Purchase Agreement.
Results of Operations for the Years Ended December 31, 2022 and 2021
The consolidated financial statements have been extracted from the accounting records of pH Pharma, Ltd. The historical results of operations, financial position, and cash flows may not be indicative of what we would have been had we been a separate stand-alone entity, nor are they indicative of what the results of operations, financial position and cash flows may be in the future.
The majority of our operating expenses related to research and development. Research and development expenses directly related to us were entirely attributed to us in the accompanying consolidated financial statements. research and development salaries, wages and benefits were allocated to us using methodologies based on the proportionate share of research and development expenses for the PHP-303 and PH-1 ADC Platform programs compared to the research and development expenses for pH Pharma Ltd as a whole. We also received services and support from other functions of pH Pharma Ltd. Our operations are dependent upon the ability of these other functions to provide these services and support. The costs associated with these services and support were allocated to us using methodologies based on the proportionate share of research and development expenses for the PHP-303 and PH-1 ADC Platform programs compared to the research and development expenses for pH Pharma Ltd as a whole. These allocated costs were primarily related to corporate administrative expenses, non-research and development employee related costs, including salaries and other benefits, for corporate and shared employees, and other expenses for shared assets for the following functional groups: information technology, legal, accounting and finance, human resources, facilities, and other corporate and infrastructural services. These allocated costs were primarily recorded as research and development expenses and general and administrative expenses in the statements of operations and comprehensive loss.
110
The following table provides selected financial information for the Company:
|
|
Year Ended December 31, |
|
|
Change |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
Amount |
|
|||
Revenues |
|
$ |
607,681 |
|
|
$ |
528,309 |
|
|
$ |
79,372 |
|
Operating expenses |
|
|
|
|
|
|
|
|
|
|||
Research and development |
|
|
3,924,253 |
|
|
|
7,124,077 |
|
|
|
(3,199,824 |
) |
General and administrative |
|
|
8,531,276 |
|
|
|
2,469,762 |
|
|
|
6,061,514 |
|
Total operating expenses |
|
|
12,455,529 |
|
|
|
9,593,839 |
|
|
|
2,861,690 |
|
Loss from operations |
|
|
(11,847,848 |
) |
|
|
(9,065,530 |
) |
|
|
(2,782,318 |
) |
Other income (expense), net |
|
|
(1,314,869 |
) |
|
|
855,021 |
|
|
|
(2,169,890 |
) |
Loss before income tax expense |
|
$ |
(13,162,717 |
) |
|
$ |
(8,210,509 |
) |
|
$ |
(4,952,208 |
) |
Revenue
Our revenue has historically been generated through grants from government organizations. The total revenue for government grants was $607,681 and $528,309, respectively, for the years ended December 31, 2022 and 2021.
Research and Development Expense
The following table summarizes our research and development expenses:
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Third-party direct project expenses |
|
|
|
|
|
|
||
PHP-303 |
|
$ |
362,221 |
|
|
$ |
1,063,702 |
|
PH-1 ADC Platform |
|
|
532,271 |
|
|
|
1,134,817 |
|
General program expenses and other pre-clinical programs |
|
|
222,365 |
|
|
|
891,493 |
|
Total third-party direct project expenses |
|
|
1,116,857 |
|
|
|
3,090,012 |
|
Other research and development costs |
|
|
|
|
|
|
||
Personnel costs |
|
|
1,363,572 |
|
|
|
2,555,631 |
|
Facilities and other costs |
|
|
1,443,824 |
|
|
|
1,478,434 |
|
Total other research and development costs |
|
|
2,807,396 |
|
|
|
4,034,065 |
|
Total research and development costs |
|
$ |
3,924,253 |
|
|
$ |
7,124,077 |
|
Research and development expense decreased by $3.2 million during the year ended December 31, 2022 compared to the prior year. The decrease was primarily due to decreases in direct project expenses related to the PHP-303 program of $701,481, the PH-1 ADC Platform of $602,546 and other general and pre-clinical programs of $669,128 as a result of delays in our ongoing and planned research activities due to the COVID-19 pandemic. In addition, there was a decrease in personnel costs of $1.2 million driven by a reduction of staff during 2021. We reduced our average headcount from 33 to 21 employees primarily as a result of scaling back our clinical activities as a result of the COVID-19 pandemic.
General and Administrative Expense
General and administrative expense increased by $6.1 million during the year ended December 31, 2022 compared to the prior year. The increase was primarily driven by a $2.5 million increase in professional fees related to expanding our operations and operating as a public company, $0.3 million related to increase in directors and officers insurance, $1.1 million in facilities expenses, and $2.1 million in deferred success fees, bonuses and unpaid backpay for executives.
Other Income (Expense), Net
Other income (expense), net decreased by $2.2 million during the year ended December 31, 2022 compared to the prior year. The decrease was primarily due to a $1.2 million fair value adjustment to convertible notes payable in 2022 and a $866,332 gain on extinguishment of debt related to our PPP loans in 2021.
111
Liquidity and Capital Resources
Sources of Liquidity
Since our inception, we have not generated any revenue from product sales and have incurred significant operating losses and negative cash flows from our operations. Our net loss was $13.1 million and $8.3 million for the years ended December 31, 2022 and 2021, respectively. At December 31, 2022 we had cash of $0.7 million. In March and April 2023, we received proceeds of $2.2 million from the issuance of convertible debt and $250,000 from a loan with our founder and director, Dr. Huh. Our primary uses of cash to date have been to fund our research and development activities, business planning, establishing and maintaining our intellectual property portfolio, capital investments and providing general and administrative support for our operations.
Funding Requirements
We expect to incur losses from operations for the foreseeable future primarily due to research and development expenses, including expenses related to conducting research activities, pre-clinical expenses and clinical trials. Our future capital requirements will depend on a number of factors, including:
We have not been capitalized with sufficient funding to conduct our operations. We entered into the White Lion Purchase Agreement on November 3, 2022, as amended, whereby we have the right, but not the obligation, to require White Lion to purchase, from time to time, up to $100 million in aggregate gross purchase price of newly issued shares of our Common Stock.
112
We expect to incur significant expenses and operating losses for the foreseeable future as we continue our efforts to identify product candidates and seek regulatory approvals within our gene therapy portfolio.
Additional financing will be needed to fund our ongoing activities. We may raise this additional funding through the sale of equity, debt financings or other capital sources, including potential collaborations with other companies or other strategic transactions and funding under government contracts. We may be unable to raise additional funds or enter into such other arrangements or arrangement when needed on favorable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate certain of our research and development programs. There can be no assurances that other sources of financing would be available. Due to these uncertainties, there is substantial doubt about our ability to continue as a going concern.
Our future operations are highly dependent on a combination of factors, including (i) the timely and successful completion of additional financing; (ii) the success of our research and development programs; (iii) the development of competitive therapies by other biotechnology and pharmaceutical companies, (iv) our ability to attract and retain key employees, (v) our ability to manage growth of the organization; (vi) our ability to protect our proprietary technology; and ultimately (vii) regulatory approval and market acceptance of our product candidates.
Cash Flows Discussion
The following table summarizes our cash flows for the periods indicated:
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Net cash used in operating activities |
|
$ |
(7,485,625 |
) |
|
$ |
(8,864,224 |
) |
Net cash used in investing activities |
|
|
(142,249 |
) |
|
|
(9,880 |
) |
Net cash provided by financing activities |
|
|
8,135,213 |
|
|
|
8,385,001 |
|
Net (decrease) increase in cash, cash equivalents and restricted cash |
|
$ |
507,339 |
|
|
$ |
(489,103 |
) |
Operating Activities
During the year ended December 31, 2022, net cash used in operating activities was $7.5 million, due to our operating loss of $13.1 million and an increase in working capital of $2.4 million, partially offset by non-cash items including amortization of right-of-use asset of $0.6 million, depreciation expense of $0.2 million, share-based compensation of $0.6 million, a non-cash change in fair value of convertible notes payable of $1.2 million, a non-cash loss on debt extinguishment of $0.5 million, and a non-cash financing fee of $0.25 million.
During the year ended December 31, 2021, net cash used in operating activities was $8.9 million, due to our operating loss of $8.3 million, a non-cash gain on extinguishment of debt of $0.9 million and an increase in working capital of $93,000, partially offset by depreciation expense of $0.2 million and share-based compensation of $4,890.
Investing Activities
During the year ended December 31, 2022, net cash used in investing activities was $142,249, primarily due to capital expenditures for furniture and fixtures related to the office in South San Francisco, California.
During the year ended December 31, 2021, net cash used in investing activities was $9,880, primarily due to purchases of research equipment and leasehold improvements related to the Palo Alto, California office and laboratory facility.
Financing Activities
During the year ended December 31, 2022 net cash provided by financing activities was driven by the net proceeds from pH Pharma Ltd of $1.3 million, proceeds from the issuance of long-term debt of $1.3 million, proceeds from a related party loan of $0.4 million, proceeds from the business combination of $3.9 million, and the issuance of common stock for $5.2 million partially offset by the $3.8 million settlement of the forward purchase agreement.
During the year ended December 31, 2021, net cash provided by financing activities was driven by the net proceeds from pH Pharma Ltd of $6.4 million, a related party loan of $1.5 million and a PPP loan of $0.5 million, which was forgiven during the year ended December 31, 2021.
113
Contractual Obligations and Commitments
In October 2021, we entered into a lease for laboratory and office facilities in Palo Alto, California that expires in March 2027 with a five-year renewal option and opened a secured letter of credit with a third-party financial institution in lieu of a security deposit for $177,000. Base rent for this sublease is approximately $89,000 monthly with annual escalations of 3%. In March 2023, the Company vacated the premises and returned possession of the premises to the landlord in April 2023. The Company is still responsible for the outstanding payments under the lease.
On March 1, 2022, we and pH Pharma Ltd entered into an administrative services and facilities agreement whereby pH Pharma Ltd will perform services, functions and responsibilities for us. Under the agreement, we will pay pH Pharma Ltd $100,000 per month through August 30, 2022 and $15,000 from September 1, 2022 through February 28, 2023 based on the estimated value of the level of service to be performed. Additionally, we will pay pH Pharma Ltd $3,000 per month in lease payments. At December 31, 2022 we recorded a liability to accounts payable of $426,673 related to this agreement.
Off-Balance Sheet Arrangements
We had no off-balance sheet arrangements as of December 31, 2022 and 2021.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements included within this Annual Report on Form 10-K, which we have prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the reported expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those described in greater detail below. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe that the following discussion represents our critical accounting policies.
Research and Development
We expect to continue to incur substantial research and development expenses as we continue to develop our product candidates. Research and development expense consists of:
We have multiple research and development projects ongoing at any one time. We utilize our internal resources, employees, and infrastructure across multiple projects. We record and maintain information regarding external, out-of-pocket research and development expenses on a project-specific basis.
We expense research and development costs as incurred, including payments made to date under our license agreements. We believe that significant investment in product development is a competitive necessity and plan to continue these investments
114
in order to realize the potential of our product candidates. The successful development of our product candidates is highly uncertain. At this time, we cannot reasonably estimate or know the nature, timing, and costs of the efforts that will be necessary to complete the remainder of the development of our product candidates. As a result, we are not able to reasonably estimate the period, if any, in which material net cash inflows may commence from our product candidates. This uncertainty is due to the numerous risks and uncertainties associated with the conduct, duration, and cost of clinical trials, which vary significantly over the life of a project as a result of evolving events during clinical development, including:
Our expenditures are subject to additional uncertainties, including the terms and timing of regulatory approvals, and the expense of filing, prosecuting, defending, and enforcing any patent claims or other intellectual property rights. We may obtain unexpected results from our clinical trials. We may elect to discontinue, delay, or modify clinical trials of some product candidates or focus on others. A change in the outcome of any of the foregoing variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development, regulatory approval, and commercialization of that product candidate. For example, if the FDA or other regulatory authorities were to require us to conduct clinical trials beyond those which we currently anticipate, or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development. Drug development takes several years and millions of dollars in development costs.
Share-based Compensation
We recognize share-based compensation expense for grants under the pH Pharma Ltd stock option plan (the “Plan”), which provides for the granting of stock options to purchase common stock in pH Pharma Ltd to employees, directors, advisors, and consultants at a price to be determined by pH Pharma Ltd’ Board of Directors. The Plan is intended to encourage ownership of stock by employees and consultants of the Company and to provide additional incentives for them to promote the success of pH Pharma Ltd’ business. Under the provisions of the Plan, stock options will generally have a term of 7 years. The Board of Directors of pH Pharma Ltd, or its committee, is responsible for determining the individuals to be granted stock options, the number of stock options each individual will receive, the stock option price per share, and the exercise period of each stock option. Stock options granted pursuant to the Plan generally vest on the second-year anniversary date of grant and may be exercised in whole or in part for 100% of the shares vested at any time after the date of grant.
The Spin-Off was completed on March 1, 2022, prior to the execution of the Business Combination Agreement with Ignyte Acquisition Corp (“Ignyte”), with holders of stock options in the Plan retaining 693,000 stock options in us and 77,000 in the spun-out company pH Pharma Co., Ltd. Since this allocation of stock options was administrative in nature, it did not result in any incremental stock-based compensation expense under modification accounting.
The Black-Scholes option pricing model is used when estimating the grant date fair value for stock-based awards. Use of a valuation model requires management to make certain assumptions with respect to selected model inputs. Expected volatility was based on the historical volatility of a publicly traded set of peer companies of pH Pharma Ltd. The expected life was equal to the contractual life of the stock option. The risk-free interest rate is based on U.S. Treasury, zero-coupon issues with a remaining term equal to the expected life assumed at the date of grant.
Determination of the Fair Value of Convertible Notes
We have elected the fair value option for the accounting for the convertible promissory notes issued in 2022. Fair value adjustments to the convertible notes are included in our other income (expenses).
115
Derivative Instruments
ASC 815, Derivatives and Hedging Activities (“ASC 815”) requires companies to bifurcate certain conversion options and redemption features from their host instruments and account for them as free-standing derivative financial instruments should certain criteria be met. We do not use derivative instruments to hedge exposures to interest rate, market, or foreign currency risks. We evaluate all of our financial instruments, including notes payable and other derivative liabilities, to determine whether such instruments are derivatives or contain features that qualify as embedded derivatives. Embedded derivatives must be separately measured from the host contract if all the requirements for bifurcation are met. The assessment of the conditions surrounding the bifurcation of embedded derivatives depends on the nature of the host contract and the features of the derivatives. Bifurcated embedded derivatives are recognized at fair value, with changes in fair value recognized in the consolidated statement of operations each period. Bifurcated embedded derivatives are classified with the related host contract in our consolidated balance sheet.
Hybrid Instruments
We also follow ASC 480-10, Distinguishing Liabilities from Equity (“ASC 480-10”) when evaluating the accounting for our hybrid instruments. A financial instrument that embodies an unconditional obligation, or a financial instrument other than an outstanding share that embodies a conditional obligation, that the issuer must or may settle by issuing a variable number of its equity shares shall be classified as a liability (or an asset in some circumstances) if, at inception, the monetary value of the obligation is based solely or predominantly on any one of the following: (a) a fixed monetary amount known at inception; (b) variations in something other than the fair value of the issuer’s equity shares; or (c) variations inversely related to changes in the fair value of the issuer’s equity shares. Hybrid instruments meeting these criteria are not further evaluated for any embedded derivatives and are carried as a liability at fair value at each balance sheet date.
Warrants
We account for the Public Warrants and Private Warrants (as defined in Note 11) collectively (“Warrants”), as either equity or liability-classified instruments based on an assessment of the specific terms of the Warrants and the applicable authoritative guidance in FASB Accounting Standards Codification (“ASC”) 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the Warrants meet all of the requirements for equity classification under ASC 815, including whether the Warrants are indexed to our own common stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of our control, among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of issuance of the Warrants and as of each subsequent quarterly period end date while the Warrants are outstanding.
For issued or modified warrants that meet all of the criteria for equity classification, such warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all the criteria for equity classification, such warrants are required to be recorded at their initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the estimated fair value of liability-classified warrants are recognized as a non-cash gain or loss on the statements of operations.
We account for the Private Warrants in accordance with ASC 815-40 under which the Private Warrants do not meet the criteria for equity classification and must be recorded as liabilities. The fair value of the Private Warrants has been estimated using the Modified Black Scholes model. See Note 6 for further discussion of the pertinent terms of the Warrants used to determine the value of the Private Warrants and Representative’s Warrants.
We evaluated the Public Warrants in accordance with ASC 815-40, “Derivatives and Hedging — Contracts in Entity’s Own Equity” and concluded that they met the criteria for equity classification and are required to be recorded as part a component of additional paid-in capital at the time of issuance.
116
Income Taxes
We had $4.6 million, $4.3 million and $100 million of federal, state and South Korea net operating loss carryforwards, respectively, as of December 31, 2022. Our carryforwards are subject to review and possible adjustment by the appropriate taxing authorities. These carryforwards may generally be utilized in any future period but may be subject to limitations based upon changes in the ownership of our shares in a prior or future period. We have not quantified the amount of such limitations, if any. The Korean net operating losses are historical net operating losses generated in years prior to the Spin-Off.
Recently Issued Accounting Standards
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2, Summary of Significant Accounting Policies, to our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K.
Internal Control Over Financial Reporting
Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. GAAP. Under standards established by the Public Company Accounting Oversight Board, or PCAOB, a deficiency in internal control over financial reporting exists when the design or operation of a control does not allow management or personnel, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis. The PCAOB defines a material weakness as a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented, or detected and corrected, on a timely basis.
Impact of the COVID-19 Pandemic
In March 2020, the World Health Organization declared the outbreak of a novel coronavirus, or COVID-19, as a pandemic, which continues to spread throughout the U.S. and worldwide. As with many companies around the world, our day-to-day operations were disrupted with the imposition of work from home policies and requirements for physical distancing for any personnel present in our offices and laboratories. During the years ended December 31, 2022 and 2021, our operations were not significantly impacted by COVID-19, with the exception of research and development expense decreasing during the year ended December 31, 2021 in part due to delays in our ongoing and planned research activities due to COVID-19. Research and development expense during the year ended December 31, 2021 also decreased in part as we reduced our average headcount from 33 to 21 employees primarily as a result of scaling back our clinical activities as a result of COVID-19. The pandemic has also disrupted our sales and marketing activities as shelter-in-place orders, quarantines, travel restrictions and other public health safety measures have impacted our ability to interact with our existing and potential partners for our solutions. There is significant uncertainty as to the trajectory of the pandemic and its impact on our business in the future. We could be materially and adversely affected by the risks, or the public perception of the risks, related to the COVID-19 pandemic or similar public health crises. Such crises could adversely impact our ability to conduct on-site laboratory activities, expand our laboratory facilities, secure critical supplies such as reagents, laboratory tools or immunized animals required for discovery research activities, and hire and retain key personnel. The ultimate extent of the impact of any epidemic, pandemic, outbreak, or other public health crisis on our business, financial condition and results of operations will depend on future developments, which are highly uncertain and cannot be predicted, including new information that may emerge concerning the severity of such epidemic, pandemic, outbreak, or other public health crisis and actions taken to contain or prevent the further spread, among others. Accordingly, we cannot predict the extent to which our business, financial condition and results of operations will be affected. We remain focused on maintaining our operations, liquidity and financial flexibility and continue to monitor developments as we deal with the disruptions and uncertainties from the COVID-19 pandemic.
JOBS Act Accounting Election
We qualify as an “emerging growth company” as defined in the JOBS Act. An emerging growth company may take advantage of reduced reporting requirements that are not otherwise applicable to public companies. These provisions include, but are not limited to:
117
We may use these provisions until the last day of our fiscal year in which the fifth anniversary of the completion of this offering occurs. However, if certain events occur prior to the end of such five-year period, including if we become a “large accelerated filer,” our annual gross revenue exceeds $1.235 billion, or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to the end of such five-year period.
We have elected to take advantage of certain of the reduced disclosure obligations in this Annual Report on Form 10-K and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our shareholders may be different than the information you receive from other public companies in which you hold stock.
The JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards, until those standards apply to private companies. We have elected to take advantage of the benefits of this extended transition period and, therefore, we will not be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. Our financial statements may therefore not be comparable to those of companies that comply with such new or revised accounting standards. Until the date that we are no longer an emerging growth company or affirmatively and irrevocably opt out of the exemption provided by Section 7(a)(2)(B) of the Securities Act upon issuance of a new or revised accounting standard that applies to our financial statements and that has a different effective date for public and private companies, we will disclose the date on which we will adopt the recently issued accounting standard.
118
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Concentration of Credit Risk
We received 100% of our revenue through a grant from a government organization during the years ended December 31, 2022 and 2021. To date, no receivables have been written off.
Interest Rate Risk
As of December 31, 2022 and 2021, we had a cash balance of $0.7 million and $0.2 million, respectively, all of which were maintained in business checking accounts and money market accounts in the U.S. and South Korea. Our primary exposure to market risk is to interest income volatility, which is affected by changes in the general level of interest rates. As such rates are at a near record low, a 10% change in the market interest rates would not have a material effect on our business, financial condition or results of operations.
Foreign Currency Risk
We conduct our business in U.S. dollars and, thus, are not exposed to financial risks from exchange rate fluctuations between the U.S. dollar and other currencies.
Item 8. Financial Statements and Supplementary Data.
Our consolidated financial statements, together with the reports of our independent registered public accounting firms, appear beginning on page F-1 of this Annual Report on Form 10-K for the year ended December 31, 2022.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2022. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, mean controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms. Disclosure controls include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2022, our chief executive officer and chief financial officer concluded that, as of such date, our internal controls over financial reporting were not effective as of the end of the period covered by this Annual Report on Form 10-K due to material weaknesses as describe herein.
A material weakness is a control deficiency (within the meaning of the Public Company Accounting Oversight Board (United States) Auditing Standard No. 2) or combination of control deficiencies that result in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected. Management has identified the following material weaknesses:
119
Planned Remediation
Management continues to work to improve its controls related to our material weaknesses, specifically implementing improved processes and internal controls to ensure the proper application of accounting practices and guidance. We also intend to increase our accounting staff as soon as economically feasible and sustainable to remediate these material weaknesses. These material weaknesses will not be considered to be remediated until the applicable remediated controls are operating for a sufficient period of time and management has concluded that these controls are operating effectively.
Management’s Annual Report on Internal Controls Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act). Our internal control system was designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes, in accordance with generally accepted accounting principles in the United States. Due to inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness of the internal control over financial reporting to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies and procedures may deteriorate. Our management, under the supervision and with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our internal control over financial reporting as of the end of the period covered by this Annual Report on Form 10-K based on the framework in Internal Control---Integrated Framework (2013 framework) issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO. Based on such evaluation, our management concluded that our internal control over financial reporting was not effective as of the end of the period covered by this Annual Report on Form 10-K.
This Annual Report on Form 10-K does not include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. Our auditors will not be required to opine on the effectiveness of our internal control over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act of 2002 until we are no longer (i) an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012 or (ii) a non-accelerated filer, as defined under the Exchange Act.
Changes in Internal Control Over Financial Reporting
Except as noted above, there was no change in our internal control over financial reporting that occurred during the fiscal year ended December 31, 2022 covered by this Annual Report on Form 10-K that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitation on the Effectiveness of Internal Control
120
Our management, including our Interim Chief Executive Officer and Acting Chief Financial Officer, does not expect that our disclosure controls and procedures, or our internal controls, will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our Company have been detected.
Item 9B. Other Information.
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
None.
121
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
Management and Board of Directors
The following table sets forth certain information regarding our directors and executive officers as of March 31, 2023 who are responsible for overseeing the management of our business.
For biographical information concerning the executive officers and directors, see below.
|
|
|
|
|
Name |
|
Age |
|
Position |
Hoyoung Huh, MD, PhD |
|
53 |
|
Class II Director |
Stephen LaMond, PharmD, MBA |
|
61 |
|
Interim Chief Executive Officer, Chief Operating Officer and Secretary and Class II Director |
Timothy Cunningham, MBA, CPA |
|
61 |
|
Acting Chief Financial Officer |
Satyajit Mitra, PhD |
|
49 |
|
Executive Director, Head of Oncology |
Nevan Charles Elam, JD |
|
55 |
|
Class I Director (Lead Independent Director) (1) |
James Neal, MS, MBA |
|
68 |
|
Class I Director |
David Rosenberg |
|
49 |
|
Class III Director |
(1) Resigned effective June 21, 2023.
Executive Officers
Hoyoung Huh, MD, PhD, is the founder of Peak Bio Co., Ltd. (f/k/a pH Pharma) and has held positions of Chief Executive Officer and Board Chairman since founding pH Pharma in 2015. He currently serves as a director on the board of directors (the “Board”) of the Company. Dr. Huh is a Silicon Valley-based entrepreneur and investor in healthcare and technology-based businesses and has served as Lead Director of Pliant Therapeutics since December 2017. Dr. Huh was a Managing Director of Konus Advisory Group, Inc. from January 2012 to September 2014. Prior to founding Konus Advisory Group, Inc., Dr. Huh was Chief Executive Officer and Chairman of the board of directors of BiPar Sciences, Inc. from February 2008 until December 2010. In addition, Dr. Huh has been involved in the formation, management and board positions of multiple biotechnology and innovation-based companies. He previously served as the Chairman of the board of directors of Geron Corporation from September 2011 to December 2018, and CytomX Therapeutics, Inc. from February 2012 to December 2018, a member of the board of directors of Rezolute, Inc. (f/k/a AntriaBio, Inc.) from 2013 to January 2019, the Chairman of the board of directors of Epizyme, Inc. from October 2009 to February 2012, and as a member of the board of directors of Facet Biotech Corporation, Nektar Therapeutics, Inc., Addex Therapeutics Ltd. and EOS, S.p.A (Milano, Italy). Earlier in his career, Dr. Huh was a partner at McKinsey & Company. He holds A.B. in Biochemistry from Dartmouth College, an M.D. from Cornell University Medical College and a Ph.D. in Cell Biology and Genetics from Cornell University Sloan Kettering Institute. We believe Dr. Huh’s extensive management and operational experience as President and Chief Executive Officer of numerous biotechnology companies and his significant knowledge and expertise of biotechnology and pharmaceutical collaborations, qualifies Dr. Huh to serve as a director and Chairman of the Board of Peak Bio.
Stephen LaMond, PharmD, MBA, has been the Chief Operating Officer and Secretary of Peak Bio Co., Ltd. since March 1, 2022. He currently serves as Interim Chief Executive Officer and as a director of the Company. Prior to his current role, Dr. LaMond has been both an employee and independent consultant to Peak Bio (f/k/a pH Pharma). Peak Bio consists of the merged entity of Ignyte and the selected assets from pH Pharma. Dr. LaMond served as both a consultant and one of the original executives with pH Pharma and its affiliated companies serving in corporate and business development roles in addition to serving in a clinical program management capacity. Dr. LaMond has been directly involved with pH Pharma in both the U.S. and Korea and now at Peak Bio since 2016. Dr. LaMond has previously held management and executive roles in marketing, new product planning, corporate and business development at numerous companies including Tria Beauty, Corium International, Zoll Medical, GE Healthcare, Nektar Therapeutics and Pfizer Inc. across multiple therapeutic areas and has worked on some of the most innovative products over his tenure. Dr. LaMond received his PharmD and Executive MBA degrees from the University of Michigan, Ann Arbor and has executive finance training from Columbia and Stanford Universities. We believe Dr. LaMond’s
122
extensive experience in operations, business development, corporate development, marketing, regulatory and market access with biopharmaceutical and biotechnology companies qualifies him to serve on the Board.
Timothy Cunningham, MBA, CPA, serves as the Acting Chief Financial Officer of the Company following the closing of the Business Combination. He brings more than 30 years of finance and operations leadership experience in the life sciences and technology industries with a proven track record of driving growth. He is currently Chief Financial Officer at Danforth Advisors, a company that provides strategic and operational finance and accounting support for life science companies. Prior to joining Danforth, Mr. Cunningham served as Chief Financial Officer at Organogenesis, where he took the company public and raised over $250M in equity and debt financing to facilitate the company’s growth. Earlier, he held leadership positions with DialogTech, GFI Software SA, Metatomix, Mediabridge, IBM, PWC, and KPMG. Tim holds an MBA from Boston University, a BS in Accounting from Boston College and is a CPA in the state of Florida.
Satyajit Mitra, PhD, is the Executive Director and Head of Oncology for Peak Bio. As the Head of Oncology, he has been responsible for Peak Bio Co., Ltd.’s preclinical research activities at our CA research sites since January 2021. Dr. Mitra heads up a team of talented research associates, scientists, consultants, CROs and CDMOs and has been instrumental in advancing our novel toxin platform and ADC pipeline. From March 2019 to December 2020, Dr. Mitra headed up Cancer Biology at Peak Bio Co., Ltd. and was responsible for in vivo and pharmacology functions that led to the nomination of the current Peak Bio lead toxin (PH1), which then led to Peak Bio’s initial proof-of-concept (POC) efforts for an ADC. These POC efforts led to the nomination of Peak Bio’s first ADC pipeline candidate targeting Trop2. He previously served as a Senior Scientist, at VasGene Therapeutics, and was involved with IND-enabling studies for novel antibody targets. Dr. Mitra’s initial corporate scientific experience was at OncoMed Pharmaceuticals for 5 years where he worked on Target Validation. He was instrumental in identifying the first-in-class Wnt-pathway biologics, advancing these projects from early stage to an IND. In addition to Dr. Mitra’s oncology company experiences, he also previously worked as a research scientist at the University of Southern California in Los Angeles. He completed his postdoctoral fellowship at the Department of Immunology at Scripps Research Institute at La Jolla, California. He obtained his Ph.D. from the Centre for Cellular and Molecular Biology at Hyderabad, India an institute affiliated with the Jawaharlal Nehru University (JNU), New Delhi, India.
Non-Employee Directors
Nevan Charles Elam, JD, serves as a director on the Board of the Company. Mr. Elam currently serves as a director and as the Chief Executive Officer of Rezolute, Inc. Prior to Mr. Elam’s service with Rezolute, he has served various leadership roles throughout his career including as Chief Executive Officer of a European medical device company, co-founder and Chief Financial Officer of a software company, as well as a Senior Vice President at Nektar Therapeutics. Earlier in his career, Mr. Elam was a corporate partner in the law firm of Wilson Sonsini Goodrich & Rosati. He serves as a director of Savara, Inc. and Softhale in Belgium. Mr. Elam received his B.A. from Howard University and his J.D. from Harvard Law School. We believe that Mr. Elam’s experience advising pharmaceutical companies of their unique legal and regulatory obligations qualifies him to serve on our board of directors.
James Neal, MS, MBA serves as a director on the Board of the Company. He comes to Peak Bio’s board of directors as an experienced business professional serving as XOMA Corporation’s Chief Executive Officer and Chairman of the Board, joining that company in 2009. Mr. Neal brings more than 25 years’ experience in forming and maximizing business and technology collaborations globally and in bringing novel products and technologies to market. Prior to XOMA, Mr. Neal was Acting Chief Executive Officer of Entelos, Inc., a leading biosimulation company that acquired Iconix Biosciences, a privately held company where Mr. Neal was Chief Executive Officer. At Iconix, Mr. Neal established multi-year collaborations with Bristol-Myers Squibb, Abbott Labs, Eli Lilly and the U.S. Food and Drug Administration. From, 1999-2002, he was Executive Vice President of Incyte Genomics, leading the global commercial activities with pharmaceutical company collaborators and partners including Pfizer, Aventis and Schering-Plough, as well as sales, marketing and business development activities for the company. Earlier, he was associated with Monsanto Company in positions of increasing responsibility. Mr. Neal earned his B.S. in Biology and his M.S. in Genetics and Plant Breeding from the University of Manitoba, Canada, and holds an Executive MBA degree from Washington University in St. Louis, Missouri. We believe Mr. Neal’s significant experience with biopharmaceutical companies, including as a board member and CEO, qualifies him to serve on our board of directors.
David Rosenberg has been Ignyte’s Chairman of the Board and co-Chief Executive Officer since its formation. Mr. Rosenberg serves as a director on the Board of the Company. Mr. Rosenberg brings over 20 years of investment banking experience focused on growth companies. Since December 2011, Mr. Rosenberg has been Co-President and Co-Chief Executive Officer of Ladenburg Thalmann & Co. Inc., a leading underwriter of blank check companies or SPACs. Mr. Rosenberg is also a member of Board of Directors of Ladenburg Thalmann & Co. Inc. From 2006 to 2011, Mr. Rosenberg was a Managing Director and Co-Chief Operating Officer of Ladenburg Thalmann & Co. Inc. Since joining Ladenburg Thalmann in 2006, Mr. Rosenberg has managed more than 1,000 public offerings including but not limited to initial public offerings and follow on offerings raising in excess of $75 billion for small and mid-cap companies, as well as advising on numerous merger and acquisition transactions. Mr. Rosenberg also serves as member of the Board of Directors of Dianomi Therapeutics. Prior to joining Ladenburg Thalmann,
123
from 2004 to 2006, Mr. Rosenberg was co-founder and Chief Executive Officer of BroadWall Capital, LLC, an investment banking firm. Mr. Rosenberg received a B.A. from the University of Wisconsin-Madison. We believe Mr. Rosenberg is well qualified to serve on our board of directors because of his significant investment banking, equity capital markets and executive management experience.
Number and Terms of Office of Officers and Directors of Peak Bio
Our Board is divided into three classes with only one class of directors being elected in each year and each class serving a three-year term. The term of office of the first class of directors, consisting of Nevan Charles Elam and James Neal, will expire at our first annual meeting of stockholders. The term of office of the second class of directors, consisting of Hoyoung Huh and Stephen LaMond, will expire at the second annual meeting. The term of office of the third class of directors, consisting of David Rosenberg, will expire at the third annual meeting.
Our officers are appointed by the board of directors and serve at the discretion of the board of directors, rather than for specific terms of office. Our board of directors is authorized to appoint persons to the offices set forth in our bylaws as it deems appropriate. Our bylaws provide that our officers may consist of a Chairman of the Board, one or more Chief Executive Officers, Chief Financial Officer, President, Vice Presidents, Secretary, Treasurer, Assistant Secretaries and such other offices as may be determined by the board of directors.
Committees of the Board of Directors
The standing committees of our board of directors include an audit committee, a compensation committee and a nominating and corporate governance committee. Each of the committees reports to the board of directors as they deem appropriate and as the board of directors may request. The composition, duties and responsibilities of these committees are set forth below.
Audit Committee
The principal functions of the audit committee include, among other things:
Our audit committee consists of Nevan Elam and James Neal, with Nevan Elam serving as the chair of the audit committee. Each of Messrs. Elam and Neal will qualify as independent directors according to the rules and regulations of the SEC with respect to audit committee membership. Mr. Elam qualifies as our “audit committee financial expert,” as that term is defined in Item 401(h) of Regulation S-K. Our board of directors has adopted a written charter for the Audit Committee, which is available free of charge on our corporate website. The information on our website is not part of this Annual Report on Form 10-K.
124
Compensation Committee
The principal functions of the compensation committee include, among other things:
Our compensation committee consists of James Neal and Nevan Elam, with Mr. Neal serving as the chair of the compensation committee. Each of Messrs. Neal and Elam qualify as independent directors according to the rules and regulations of the SEC with respect to compensation committee membership. Our board of directors has adopted a written charter for the compensation committee, which is available free of charge on our corporate website. The information on our website is not part of this Annual Report on Form 10-K.
Nominating Committee
The principal functions of the nominating committee include, among other things:
The Nominating Committee will consider a number of qualifications relating to management and leadership experience, background and integrity and professionalism in evaluating a person’s candidacy for membership on the board of directors. The nominating committee may require certain skills or attributes, such as financial or accounting experience, to meet specific board needs that arise from time to time and will also consider the overall experience and makeup of its members to obtain a broad and diverse mix of board members. The nominating committee does not distinguish among nominees recommended by stockholders and other persons.
Our Nominating Committee consists of Nevan Elam and James Neal, with Mr. Elam serving as the chair of the Nominating Committee. We expect that our board of directors will adopt a written charter for the Nominating Committee, which is available free of charge on our corporate website. The information on our website is not part of this Annual Report on Form 10-K.
Code of Ethics
We have adopted a Code of Ethics applicable to our directors, executive officers and employees. Our code of business conduct and ethics is available free of charge on our corporate website. In addition, we intend to post on our website all disclosures that are required by law or the listing standards concerning any amendments to, or waivers from, any provision of the code. References to our website address do not constitute incorporation by reference of the information contained at or available through our website, and such information should not be considered to be a part of this Annual Report on Form 10-K. We also intend to disclose any amendments to or waivers of certain provisions of our Code of Ethics in a Current Report on Form 8-K. You may review these documents by accessing public filings at the SEC’s website at www.sec.gov.
Compensation Committee Interlocks and Insider Participation
125
None of our officers currently serves, or in the past year has served, as a member of the compensation committee of any entity that has one or more officers serving on our board of directors.
Compliance with Section 16(a) of the Exchange Act
Section 16(a) of the Exchange Act requires our executive officers, directors and persons who beneficially own more than 10% of a registered class of our equity securities to file with the Securities and Exchange Commission initial reports of ownership and reports of changes in ownership of our common stock and other equity securities. These executive officers, directors, and greater than 10% beneficial owners are required by SEC regulation to furnish us with copies of all Section 16(a) forms filed by such reporting persons. Based solely on our review of such forms furnished to us and written representations from certain reporting persons, we are not aware of any failure of any such person to comply with the requirements of Section 16(a) of the Exchange Act, except for the following which were not filed in a timely manner: one Form 3 for Brad Stevens prior to his resignation as a director and one Form 3 filed by SBI INVESTMENT KOREA CO., LTD. and its affiliated entities.
Item 11. Executive Compensation.
This section discusses the material components of the executive compensation program for our named executive officers who are identified in the 2022 Summary Compensation Table below. This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs.
Overview
This section discusses the material components of the executive compensation program for our executive officers who are named in the “2022 Summary Compensation Table” (the “named executive officers”). As an emerging growth company, we comply with the executive compensation disclosure rules applicable to “smaller reporting companies,” as such term is defined in the rules promulgated under the Securities Act. Our named executive officers for fiscal year 2022 were as follows:
1 |
Dr. LaMond will serve as Interim Chief Executive Officer of Peak Bio while Dr. Huh is taking a leave of absence during the pendency of a personal legal proceeding. Dr. Huh is the Chief Executive Officer of our principal subsidiary in Korea, Peak Bio Co., Ltd. |
2 |
Mr. Rosenberg was Co-Chief Executive Officer of Ignyte until closing of the Business Combination. |
3 |
Mr. Strupp was Co-Chief Executive Officer of Ignyte until closing of the Business Combination. |
2022 Compensation of Named Executive Officers
Summary Compensation Table
The following table sets forth certain summary information for the years indicated concerning total compensation earned by our named executive officers.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
126
Name and Principal Position |
|
Fiscal |
|
|
Salary |
|
|
Bonus |
|
|
Stock |
|
|
Option |
|
|
Non-Equity |
|
|
Change in |
|
|
All Other |
|
|
Total |
|
|||||||||
Hoyoung Huh, |
|
|
2022 |
|
|
|
1,669,838 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,669,838 |
|
Chief Executive Officer Peak Bio Co., Ltd. |
|
|
2021 |
|
|
|
216,113 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10,029 |
|
|
|
226,142 |
|
Stephen LaMond, |
|
|
2022 |
|
|
|
234,167 |
|
|
|
250,000 |
|
|
|
— |
|
|
|
193,508 |
|
|
|
— |
|
|
|
— |
|
|
|
15,000 |
|
|
|
692,675 |
|
Interim Chief Executive Officer and Chief Operating Officer |
|
|
2021 |
|
|
|
93,408 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
161,170 |
|
|
|
254,578 |
|
Satyajit Mitra, |
|
|
2022 |
|
|
|
206,232 |
|
|
|
110,991 |
|
|
|
— |
|
|
|
93,990 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
411,213 |
|
Executive Director, Head of Oncology |
|
|
2021 |
|
|
|
179,956 |
|
|
|
15,925 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
195,881 |
|
David Rosenberg, |
|
|
2022 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Former Co-Chief Executive Officer of Ignyte (4) |
|
|
2021 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
David Strupp, Jr., |
|
|
2022 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Former Co-Chief Executive Officer of Ignyte (5) |
|
|
2021 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
(1) |
Salary for Dr. Huh includes $1,524,852 of salary earned for past service but not yet paid and the Company entered into an employment agreement, updated as of January 10, 2022, to repay this amount. |
(2) |
Bonus amounts for 2022 represent success fees for the consummation of Peak Bio’s Business Combination. These fees were earned in 2022, but have not yet been paid. |
(3) |
All other compensation amounts consist of car allowances for Dr. Huh for 2021, severance of $81,650 for Dr. LaMond for 2021, consultant compensation of $15,000 and $79,520 for Dr. LaMond for 2022 and 2021, respectively, and a car allowance for Mr. Kim for 2021. |
(4) |
Mr. Rosenberg was Co-Chief Executive Officer of Ignyte until closing of the Business Combination. |
(5) |
Mr. Strupp was Co-Chief Executive Officer of Ignyte until closing of the Business Combination. |
2022 outstanding equity awards at fiscal year-end
The following table presents, for each of our named executive officers, information regarding outstanding stock options as of December 31, 2022.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
127
|
|
Option Awards1 |
|
|||||||||||||||||
Name |
|
Number of |
|
|
Number of |
|
|
Equity |
|
|
Option |
|
|
Option |
|
|||||
Hoyoung Huh, Chief Executive Officer of Peak Bio Co., Ltd. |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Stephen LaMond, Interim Chief Executive Officer and Chief Operating Officer January 25, 2022 |
|
|
14,618 |
|
|
|
16,882 |
|
|
|
— |
|
|
|
8.05 |
|
|
|
January 25, 2029 |
|
Satyajit Mitra, Executive Director, Head of Oncology June 11, 2019 |
|
|
9,000 |
|
|
|
— |
|
|
|
— |
|
|
|
6.10 |
|
|
|
June 11, 2026 |
|
January 25, 2022 |
|
|
7,100 |
|
|
|
8,200 |
|
|
|
|
|
|
|
8.05 |
|
|
|
January 25, 2029 |
|
David Rosenberg, Former Co-Chief Executive Officer of Ignyte |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
David Strupp, Jr., Former Co-Chief Executive Officer of Ignyte |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
(1) |
There were no outstanding stock awards as of the end of fiscal year 2021 |
(2) |
Stock options in Peak Bio Co., Ltd. were converted to stock options in Peak Bio, Inc. (f/k/a Ignyte Acquisition Corp.) at the Exchange Ratio set forth in the Business Combination Agreement |
Equity compensation
We will, from time-to-time, grant equity awards to our named executive officers, which are generally subject to vesting based on each named executive officer’s continued service. Each of our named executive officers currently holds outstanding options to purchase shares of common stock that were granted under Peak Bio Co., Ltd.’s form of stock option agreements issued in accordance with Korean law. Information on our named executive officer’s equity awards is set forth in the table above titled “2022 Outstanding Equity Awards at Fiscal Year-End.”
Director Compensation
For fiscal years 2022 and 2021, we did not provide director compensation to our non-employee directors. However, all of our non-employee directors are reimbursed for their reasonable out-of-pocket expenses related to their services as a member of our board of directors. In connection with the Business Combination, we intend to approve and implement a non-employee director compensation policy.
Potential payments upon termination or change of control
Employment Agreements
Dr. Huh’s employment agreement, updated as of January 10, 2022, provides for Dr. Huh to serve as Peak Bio’s Chief Executive Officer (Dr. LaMond will serve as Interim Chief Executive Officer of Peak Bio while Dr. Huh is taking a leave of absence during the pendency of a personal legal proceeding). The employment agreement terms, which were subject to completion of the Business Combination, provide for Dr. Huh to receive an annual base salary and to participate in a cash bonus plan with a target of up to 65% of base salary based on annual performance standards to be established by the board of directors. In addition, the employment agreement provides for repayment to Dr. Huh of backpay for years of forwent salary in the amount of $1,524,852 and repayment of an outstanding loan in the amount of $1,500,000 made by Dr. Huh to Peak Bio Co, Ltd. Further, the employment agreement provides for the payment of success fees in connection with future business or corporate development transactions (licensing, product development and acquisitions).
128
If Dr. Huh’s employment is terminated due to his death or disability, the employment agreement provides that Peak Bio will pay to Dr. Huh or Dr. Huh’s estate or designated beneficiary his accrued and unpaid salary plus his accrued and unused vacation pay.
If Dr. Huh’s employment is terminated by him for “good reason” or if Peak Bio terminates his employment without “cause,” then Peak Bio will pay to Dr. Huh his accrued and unpaid salary, his accrued and unused vacation pay, and continuation of his base salary for twelve months. For purposes hereof, “good reason” means, the occurrence of any of the following events: (i) the failure of Peak Bio or applicable subsidiary to pay any wages, or provide any benefits due to Dr. Huh within five (5) days after written notice thereof from Dr. Huh; (ii) a material change in Dr. Huh’s responsibilities, duties, reporting relationships or authorities as an employee of Peak Bio as they existed prior to such change; or (iii) a move of Dr. Huh’s principal place of work to a location more than fifty (50) miles distant therefrom. Termination with “cause” shall be deemed to exist if Dr. Huh engages in the following: (i) theft, dishonesty, misconduct or falsification of Peak Bio’s or its successor’s records or property; (ii) unauthorized use or disclosure of Peak Bio’s or its successor’s confidential or proprietary information or trade secrets; (iii) substantial negligence or misconduct; (iv) failure to perform such assigned duties and responsibilities as shall be consistent with the duties and responsibilities of an employee of Peak Bio in a similar job position after receipt of a written notice of specific deficiencies and failure to cure any such deficiencies within fifteen (15) days after the receipt of such notice; (v) a material breach by Dr. Huh of any agreement between Dr. Huh and Peak Bio, and such breach has not been cured by you within fifteen days (15) after written notice of breach by Peak Bio; (vi) commission of a felony or other crime involving moral turpitude; or (vii) Dr. Huh’s failure to cooperate in good faith with a governmental investigation of Peak Bio or its directors, officers or employees, if Peak Bio has requested his cooperation.
Dr. LaMond’s employment agreement, updated as of March 1, 2022, provides for Dr. LaMond to serve as Peak Bio ’s Chief Operating Officer. The employment agreement terms, which were subject to completion of the Business Combination, provide for Dr. LaMond to receive an annual base salary and to participate in a cash bonus plan with a target of up to 55% of base salary based on annual performance standards to be established by the board of directors. In addition, the employment agreement provides for confirmation of Peak Bio’s previously agreed upon success fee payment to Dr. LaMond upon consummation of the Business Combination in the amount of $250,000. Further, the employment agreement provides for the payment of success fees in connection with future business or corporate development transactions (licensing, product development and acquisitions).
Dr. LaMond is also eligible to participate in Peak Bio’s Long-Term Incentive Plan with a target recommended grant of 1.25% or greater of the outstanding shares of Peak Bio’s stock, subject to approval of Peak Bio’s board of directors.
The employment agreement permits Dr. LaMond to serve on up to three (3) outside boards of directors at the discretion of Peak Bio’s board of directors and to provide limited consulting services to non-affiliated third parties provided they are not in direct conflict with Peak Bio’s business activities.
If Dr. LaMond’s employment is terminated by Peak Bio without “cause,” then Peak Bio will pay Dr. LaMond his accrued and unpaid salary and continuation of his base salary for twelve months.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth information known to us regarding the beneficial ownership of our Common Stock as of December 31, 2022, after giving effect to the Closing, by:
Beneficial ownership for the purposes of the following table is determined in accordance with the rules and regulations of the SEC. A person is a “beneficial owner” of a security if that person has or shares “voting power,” which includes the power to vote or to direct the voting of the security, or “investment power”, which includes the power to dispose of or to direct the disposition of the security or has the right to acquire such powers within 60 days.
The beneficial ownership percentages set forth in the table below are based on 21,713,248 shares of Common Stock issued and outstanding as of December 31, 2022 and do not take into account the issuance of any shares of Common Stock upon the exercise of warrants or stock options.
Unless otherwise noted in the footnotes to the following table, and subject to applicable community property laws, the persons and entities named in the table have sole voting and investment power with respect to their beneficially owned common stock.
129
|
|
|
|
|
|
|
|
|
Name of Beneficial Owners(1) |
|
Number of |
|
|
Percentage of |
|
||
5% Stockholders: |
|
|
|
|
|
|
|
|
SBI Investment KOREA Co., Ltd. (5) |
|
|
3,621,489 |
|
|
|
18.3 |
% |
|
|
|
||||||
Executive Officers and Directors: |
|
|
|
|
|
|
|
|
Hoyoung Huh(2) |
|
|
6,800,349 |
|
|
|
34.4 |
% |
Stephen LaMond |
|
|
— |
|
|
|
— |
|
Timothy Cunningham |
|
|
— |
|
|
|
— |
|
Satyajit Mitra(3) |
|
|
18,647 |
|
|
|
|
* |
James Neal |
|
|
— |
|
|
|
— |
|
David I. Rosenberg(4) |
|
|
1,117,755 |
|
|
|
5.65 |
% |
Nevan Charles Elam |
|
|
— |
|
|
|
— |
|
All directors and executive officers as a group (7 individuals) |
|
|
7,936,751 |
|
|
|
36.55 |
% |
* |
Indicates less than 1 percent |
(1) |
Unless otherwise indicated, the business address of each of the individuals is c/o Peak Bio, Inc., 4900 Hopyard Road, Pleasanton, CA 94588. |
(2) |
Includes 1,930,501 shares of Common Stock held in escrow in the name of Hoyoung Huh for release upon payment under the Key Company Stockholder Forward Purchase Agreement, 6,427,409 shares of Common Stock held by Hoyoung Huh and 372,940 shares of Common Stock held by Hannol Ventures LLC of which Mr. Huh is the sole member and who has voting and dispositive power over such shares. |
(3) |
Includes 18,647 shares underlying options to purchase Common Stock that are fully vested and currently exercisable. |
(4) |
Includes 389,630 shares of Common Stock and 728,125 Private Warrants previously held by the Sponsor, of which David Rosenberg is a managing member.
|
(5) |
Includes 320,206 shares of Common Stock held by SBI Investment KOREA Co., Ltd. (“SBI”), 251,418 shares of Common Stock held by SBI Cross-border Advantage Fund, an affiliate of SBI, 599,202 shares of Common Stock held by SBI Healthcare Fund 1, an affiliate of SBI, 1,601,067 shares of Common Stock held by IBKC-SBI Bio Fund 1, an affiliate of SBI, 83,800 shares Common Stock held by SBI KIS 2016-1 Fund, an affiliate of SBI, 167,600 shares of Common Stock held 2014 KIF-SBI IT Investment Fund, an affiliate of SBI, 419,017 shares of Common Stock held by Global Gateway Fund 1, an affiliate of SBI and 179,179 shares of Common Stock held by 2019 SBI Job Creation Fund, an affiliate of SBI. The business address of SBI is 14th FL., NC Tower, 509, Teheran-ro, Gangnam-gu, Seoul, Korea. |
Item 13. Certain Relationships and Related Transactions, and Director Independence.
See (i) Note 7 – Related Party Transactions and Shared Service Costs, Note 13 – Debt and Note 14 – Subsequent Events of the notes accompanying our audited financial statements for the fiscal year ended December 31, 2022 and (ii) “Management’s Discussion and Analysis of Financial Condition and Results of Operations–Recent Developments” included elsewhere in this Annual Report on Form 10-K.
Procedures with Respect to Review and Approval of Related Person Transactions
Upon consummation of the Business Combination, our board of directors adopted a written related person transaction policy that sets forth the following policies and procedures for the review and approval or ratification of related person transactions.
A “Related Person Transaction” is a transaction, arrangement or relationship in which we or any of our subsidiaries was, is or will be a participant, the amount of which involved exceeds $120,000, and in which any related person had, has or will have a direct or indirect material interest. A “Related Person” means:
130
We also adopted policies and procedures designed to minimize potential conflicts of interest arising from any dealings it may have with its affiliates and to provide appropriate procedures for the disclosure of any real or potential conflicts of interest that may exist from time to time. Specifically, pursuant to our audit committee charter, the audit committee will have the responsibility to review related person transactions.
Item 14. Principal Accounting Fees and Services.
The following is a summary of fees paid or to be paid to Marcum LLP, or Marcum, the Company’s independent registered public accounting firm and Mayer Hoffman McCann P.C., or MHM, Peak Bio Co., Ltd.'s former independent registered public accounting firm, for services rendered. Substantially all of MHM’s personnel, who work under the control of MHM shareholders, are employees of wholly-owned subsidiaries of CBIZ, Inc., which provides personnel and various services to MHM in an alternative practice structure.
Audit Fees. Audit fees consist of fees billed for professional services rendered for the audit of our financial statements included in this Annual Report on Form 10-K, and review of financial statements included in our quarterly reports on Form 10-Q and services that are normally provided by our independent registered public accounting firm, in connection with regulatory filings and public offerings. The aggregate fees billed by Marcum for professional services rendered for the audit of our annual financial statements and other required filings with the SEC for each of the fiscal years ended December 31, 2022 and 2021 totaled $720,000. The aggregate fees billed by MHM for professional services rendered for the audit of our annual financial statements and other required filings with the SEC for the fiscal years ended December 31, 2022 and 2021, totaled $881,144 and $0, respectively. The above amounts include interim procedures and audit fees, as well as attendance at audit committee meetings.
Audit-Related Fees. Audit-related services consist of fees billed for assurance and related services that are reasonably related to performance of the audit or review of our financial statements and are not reported under “Audit Fees.” These services include attest services that are not required by statute or regulation and consultations concerning financial accounting and reporting standards. We did not pay Marcum or MHM for consultations concerning financial accounting and reporting standards during the years ended December 31, 2022 and 2021.
Tax Fees. We did not pay Marcum or MHM for tax planning and tax advice for the years ended December 31, 2022 and 2021.
All Other Fees. We did not pay Marcum or MHM for other services for the years ended December 31, 2022 and 2021.
Pre-Approval Policy
Our audit committee was formed upon the consummation of our Business Combination. As a result, the audit committee did not pre-approve all of the foregoing services, although any services rendered prior to the formation of our audit committee were approved by our board of directors. Since the formation of our audit committee, and on a going-forward basis, the audit committee has and will pre-approve all auditing services and permitted non-audit services to be performed for us by our auditors, including the fees and terms thereof (subject to the de minimis exceptions for non-audit services described in the Exchange Act which are approved by the audit committee prior to the completion of the audit).
131
PART IV
Item 15. Exhibits, Financial Statement Schedules.
132
Exhibit Number |
|
Description |
2.1 |
|
|
3.1 |
|
|
3.2 |
|
|
4.1 |
|
|
4.2 |
|
|
4.3* |
|
|
10.1 |
|
|
10.2 |
|
|
10.3 |
|
|
10.4 |
|
|
10.5 |
|
|
10.6 |
|
|
10.7 |
|
|
10.8 |
|
|
10.9 |
|
|
10.10 |
|
|
10.11 |
|
|
10.12 |
|
|
10.13 |
|
|
10.14 |
|
|
10.15 |
|
|
10.16 |
|
133
10.17 |
|
|
10.18 |
|
|
10.19 |
|
|
21.1 |
|
|
24.1* |
|
Power of Attorney (included on signature page to this Annual Report on Form 10-K). |
31.1* |
|
|
31.2* |
|
|
32.1** |
|
|
32.2** |
|
|
101.INS |
|
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
101.SCH |
|
Inline XBRL Taxonomy Extension Schema Document |
101.CAL |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
101.DEF |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
101.PRE |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
104 |
|
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
* Filed herewith.
** Furnished
Item 16. Form 10-K Summary
None.
134
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
PEAK BIO, INC. |
|
|
|
|
|
Date: June 29, 2023 |
|
By: |
/s/ Stephen LaMond |
|
|
|
Stephen LaMond |
|
|
|
Interim Chief Executive Officer (Principal Executive Officer) |
|
|
|
|
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Stephen LaMond and Timothy Cunningham and each or any one of them, their true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for them and in their name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the United States Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
|
|
|
|
|
Name |
|
Title |
|
Date |
|
|
|
||
/s/ Stephen LaMond |
|
Interim Chief Executive Officer and Director |
|
June 29, 2023 |
Stephen LaMond |
|
(principal executive officer) |
|
|
|
|
|
||
/s/ Timothy Cunningham |
|
Acting Chief Financial Officer |
|
June 29, 2023 |
Timothy Cunningham |
|
(principal financial and accounting officer) |
|
|
|
|
|
||
/s/ Hoyoung Huh |
|
Director |
|
June 29, 2023 |
Hoyoung Huh |
|
|
|
|
|
|
|
||
/s/ James Neal |
|
Director |
|
June 29, 2023 |
James Neal |
|
|
|
|
|
|
|
||
/s/ David Rosenberg |
|
Director |
|
June 29, 2023 |
David Rosenberg |
|
|
|
|
135
PEAK BIO
Consolidated Financial Statements
As of and for the Years Ended December 31, 2022 and 2021
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
Peak Bio, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Peak Bio, Inc. (the “Company”) as of December 31, 2022, the related consolidated statements of operations and comprehensive loss, equity (deficit) and cash flows for the year ended December 31, 2022, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2022, and the results of its operations and its cash flows for the year ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
We also have audited the adjustments to the December 31, 2021 financial statements and for the year ended December 31, 2021 to retrospectively apply the reclassification to shares and net loss per share prior to the reverse recapitalization, as described in Note 1, and the effects of the reverse recapitalization, as described in Note 1. In our opinion, such adjustments are appropriate and have been properly applied. We were not engaged to audit, review, or apply any procedures to the December 31, 2021 financial statements, or for the year ended December 31, 2021, of the Company other than with respect to the adjustments, and, accordingly, we do not express an opinion or any other form of assurance on the December 31, 2021 financial statements taken as a whole.
Explanatory Paragraph – Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 1, the Company has a significant working capital deficiency, has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Emphasis of Matter for Carve-out
As discussed in Note 2 to the consolidated financial statements, the Company’s business for the three months ended March 31, 2022 was a component of pH Pharma, Ltd. and was not a stand-alone entity. The consolidated financial statements of the Company reflect the assets, liabilities and expenses directly attributable to the Company for the three months ended March 31, 2022, as well as allocations deemed reasonable by management, to present the financial position, results of operations, and cash flows of the Company on a stand-alone basis and do not necessarily reflect the financial position, results of operations, and cash flows of the Company in the future or what they would have been had the Company been a separate stand-alone entity during the three months ended March 31, 2022.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
Adoption of New Accounting Standard
F-2
As discussed in Note 2 to the consolidated financial statements, the Company has changed its method of accounting for leases in 2022 due to the adoption of the guidance in ASC Topic 842, Leases (“Topic 842”), as amended, effective January 1, 2022, using the modified retrospective approach.
/s/ Marcum LLP
We have served as the Company’s auditor since 2022.
June 29, 2023
F-3
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
of Peak Bio Co., Ltd.
Opinion on the Financial Statements
We have audited, before the effects of the adjustments to retrospectively apply the reverse recapitalization described in Note 1, the accompanying carve-out consolidated balance sheet of Peak Bio Co., Ltd. (“the Company”) as of December 31, 2021, and the related carve-out consolidated statement of operations and comprehensive loss, carve-out consolidated statement of equity (deficit), and carve-out consolidated statement of cash flows for the year then ended, and the related carve-out notes (collectively referred to as the “carve-out consolidated financial statements”). The 2021 carve-out consolidated financial statements before the effects of the adjustments discussed in Note 1 are not presented herein. In our opinion, the carve-out consolidated financial statements, before the effects of the adjustments to retrospectively apply the reverse recapitalization described in Note 1, present fairly, in all material respects, the financial position of the Company as of December 31, 2021, and the results of their operations and their cash flows for the year then ended in conformity with accounting principles generally accepted in the United States of America.
We were not engaged to audit, review, or apply any procedures to the adjustments to retrospectively apply the reverse recapitalization described in Note 1 and, accordingly, we do not express an opinion or any other form of assurance about whether such adjustments are appropriate and have been properly applied. Those adjustments were audited by Marcum LLP.
Substantial Doubt About the Entity’s Ability to Continue as a Going Concern
The accompanying carve-out consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the carve-out consolidated financial statements, the Company had recurring losses from operations, negative cash flow from operations and is dependent on additional financing to fund operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are described in Note 1 to the carve-out consolidated financial statements. The carve-out consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
Emphasis of Matter for Carve-out consolidated financial statements
As discussed in Note 2 to the carve-out consolidated financial statements, the Company’s business is a component of pH Pharma, Ltd. and is not a stand-alone entity. The carve-out consolidated financial statements of the Company reflect the assets, liabilities and expenses directly attributable to the Company, as well as allocations deemed reasonable by management, to present the financial position, results of operations, and cash flows of the Company on a stand-alone basis and do not necessarily reflect the financial position, results of operations, and cash flows of the Company in the future or what they would have been had the Company been a separate, stand-alone entity during the periods presented.
Basis for Opinion
These carve-out consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s carve-out consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America (GAAS). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the carve-out consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audit included performing procedures to assess the risks of material misstatement of the carve-out consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the carve-out consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the carve-out consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
We conducted our audit in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether
F-4
the carve-out consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the carve-out consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the carve-out consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the carve-out consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
We served as the Company’s auditor in 2022.
/s/ Mayer Hoffman McCann P.C.
San Diego, California
June 17, 2022
F-5
PEAK BIO
CONSOLIDATED BALANCE SHEETS
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Assets |
|
|
|
|
|
|
||
Current assets |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Derivative asset |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Restricted cash |
|
|
|
|
|
|
||
Operating lease right-of-use asset |
|
|
|
|
|
|
||
Noncurrent assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
Liabilities and deficit |
|
|
|
|
|
|
||
Current liabilities |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued expenses |
|
|
|
|
|
|
||
Operating lease liability, current |
|
|
|
|
|
|
||
Insurance financing payable |
|
|
|
|
|
|
||
Derivative liability |
|
|
|
|
|
|
||
Convertible note payable, current |
|
|
|
|
|
|
||
Related party loan |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Operating lease liability, net of current portion |
|
|
|
|
|
|
||
Warrant liability |
|
|
|
|
|
|
||
Deferred tax liability |
|
|
|
|
|
|
||
Other noncurrent liabilities |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
Equity (deficit) |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, par value of $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Accumulated other comprehensive income |
|
|
|
|
|
|
||
Total deficit |
|
|
( |
) |
|
|
( |
) |
Total liabilities and deficit |
|
$ |
|
|
$ |
|
||
See accompanying notes to consolidated financial statements.
F-6
PEAK BIO
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Revenue |
|
|
|
|
|
|
||
Grant revenue |
|
$ |
|
|
$ |
|
||
Total revenue |
|
|
|
|
|
|
||
Operating expenses |
|
|
|
|
|
|
||
Research and development |
|
|
|
|
|
|
||
General and administrative |
|
|
|
|
|
|
||
Total operating expenses |
|
|
|
|
|
|
||
Loss from operations |
|
|
( |
) |
|
|
( |
) |
Other income (expense) |
|
|
|
|
|
|
||
Interest income |
|
|
|
|
|
|
||
Interest expense |
|
|
( |
) |
|
|
( |
) |
Fair value adjustment to convertible notes payable |
|
|
( |
) |
|
|
|
|
Fair value adjustment to warrant liability |
|
|
( |
) |
|
|
|
|
Fair value adjustment to derivative liability |
|
|
|
|
|
|
||
Other income |
|
|
|
|
|
|
||
Gain (loss) on extinguishment of debt |
|
|
( |
) |
|
|
|
|
Total other income (expense), net |
|
|
( |
) |
|
|
|
|
Loss before income tax expense |
|
|
( |
) |
|
|
( |
) |
Income tax benefit (expense) |
|
|
|
|
|
( |
) |
|
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Other comprehensive loss: |
|
|
|
|
|
|
||
Foreign currency translation |
|
|
( |
) |
|
|
|
|
Total comprehensive loss |
|
$ |
( |
) |
|
$ |
( |
) |
Basic and diluted weighted average shares outstanding |
|
|
|
|
|
|
||
Basic and diluted net loss per share |
|
$ |
( |
) |
|
$ |
( |
) |
See accompanying notes to consolidated financial statements.
F-7
PEAK BIO
CONSOLIDATED STATEMENTS OF EQUITY (DEFICIT)
|
|
Common Stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
Shares |
|
Amount |
|
|
Additional Paid-In Capital |
|
|
Accumulated |
|
|
Accumulated |
|
|
Accumulated Deficit |
|
|
Total Stockholders' |
|
|||||||
Balance, December 31, 2020, as previously reported |
|
|
— |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
( |
) |
Retroactive application of recapitalization |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
||||
Balance, December 31, 2020 |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|||
Capital contribution from parent |
|
|
— |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Foreign currency translation |
|
|
— |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance, December 31, 2021 |
|
|
|
|
|
|
|
|
|
$ |
— |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
||||
Capital contribution from parent |
|
|
— |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Issuance of common stock |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Shares acquired in reverse recapitalization net of transaction fees |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of shares to PIPE Subscribers |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of Shares for Debt Settlement |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of Shares for Financing Fee |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Settlement of Forward Purchase Agreement |
|
|
( |
) |
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Share-based compensation |
|
|
— |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Foreign currency translation |
|
|
— |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Net loss |
|
|
— |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance, December 31, 2022 |
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
||||
See accompanying notes to consolidated financial statements.
F-8
PEAK BIO
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
Year Ended |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Cash flows from operating activities |
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustment to reconcile net loss to net cash used in operating activities |
|
|
|
|
|
|
||
Share-based compensation |
|
|
|
|
|
|
||
Depreciation |
|
|
|
|
|
|
||
Loss (gain) on extinguishment of debt |
|
|
|
|
|
( |
) |
|
Change in fair value of convertible note payable |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
|
|
|
|
||
Change in fair value of derivative liability |
|
|
( |
) |
|
|
|
|
Amortization of right-of-use lease asset |
|
|
|
|
|
|
||
Issuance of shares for financing fee |
|
|
|
|
|
|
||
Loss on disposal of fixed assets |
|
|
|
|
|
|
||
Changes in operating assets and liabilities |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
( |
) |
|
|
|
|
Other noncurrent assets |
|
|
|
|
|
|
||
Accounts payable |
|
|
|
|
|
( |
) |
|
Accrued expenses and other current liabilities |
|
|
|
|
|
|
||
Operating lease liability |
|
|
( |
) |
|
|
|
|
Other noncurrent liabilities and deferred tax liability |
|
|
|
|
|
|
||
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
Cash flows from investing activities |
|
|
|
|
|
|
||
Purchase of property and equipment |
|
|
( |
) |
|
|
( |
) |
Net cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
Cash flows from financing activities |
|
|
|
|
|
|
||
Proceeds from issuance of common shares |
|
|
|
|
|
|
||
Proceeds from completion of business combination |
|
|
|
|
|
|
||
Settlement of forward purchase agreement |
|
|
( |
) |
|
|
|
|
Proceeds from net shareholder contributions |
|
|
|
|
|
|
||
Proceeds from long term convertible notes payable |
|
|
|
|
|
|
||
Proceeds from related party loan |
|
|
|
|
|
|
||
Repayment of related party loan |
|
|
( |
) |
|
|
|
|
Net cash provided by financing activities |
|
|
|
|
|
|
||
Net (decrease) increase in cash and cash equivalents |
|
|
|
|
|
( |
) |
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
( |
) |
|
|
|
|
Cash and cash equivalents, beginning of year |
|
|
|
|
|
|
||
Cash and cash equivalents, end of year |
|
$ |
|
|
$ |
|
||
Components of cash, cash equivalents and restricted cash |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
|
|
|
|
|
||
Restricted cash |
|
|
|
|
|
|
||
Total cash, cash equivalents and restricted cash |
|
|
|
|
|
|
||
Supplemental disclosures of non-cash financing activities: |
|
|
|
|
|
|
||
Cash paid for interest |
|
$ |
|
|
$ |
|
||
Cash paid for taxes |
|
$ |
|
|
$ |
|
||
Non-cash investing and financing activities: |
|
|
|
|
|
|
||
Purchase of property and equipment included in accounts payable |
|
$ |
|
|
$ |
|
||
Warrant liability assumed in Business Combination |
|
$ |
|
|
$ |
|
||
Related party loan assumed in Business Combination |
|
$ |
|
|
$ |
|
||
Convertible notes payable and derivative liability assumed in Business Combination |
|
$ |
|
|
$ |
|
||
Related party loan entered into for settlement of accrued expenses |
|
$ |
|
|
$ |
|
||
Shares issued for settlement of related party loan and accrued interest |
|
$ |
|
|
$ |
|
||
Shares issued for settlement of convertible notes payable and accrued interest |
|
$ |
|
|
$ |
|
||
Financing received for annual insurance policy |
|
$ |
|
|
$ |
|
||
Operating lease liabilities arising from obtaining right-of-use assets |
|
$ |
|
|
$ |
|
||
See accompanying notes to consolidated financial statements.
F-9
PEAK BIO
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The accompanying consolidated financial statements and notes have been prepared to include certain assets and liabilities of pH Pharma Co., Ltd (now Peak Bio Co., Ltd. or “Peak Bio”) (sometimes referred to as “pH Pharma Ltd” prior to the Spin-Off described below), on the basis described within Note 2, Summary of Significant Accounting Policies, with certain wholly-owned subsidiaries of Peak Bio, that were included following the Spin-Off as follows: Ph Pharma, Inc, as well as certain assets and liabilities allocated to Peak Bio, including the PHP- 303 and PH-1 ADC Platform programs.
The Spin-Off was completed on March 1, 2022, prior to the execution of the Business Combination Agreement with Ignyte Acquisition Corp. (“Ignyte”), with Peak Bio retaining the PHP-303 and PH-1 ADC Platform programs. Historically and throughout the periods presented, the PHP-303 and PH-1 ADC Platform programs have been owned by pH Pharma Co., Ltd and its subsidiaries (prior to the change of its name to Peak Bio Co., Ltd.).
Peak Bio is a clinical-stage biotechnology company focused on discovering, developing and delivering innovative therapies for multiple therapeutic areas. The Company has established a portfolio of potential therapies for the aging population. The Company’s pipeline includes the PHP-303 program for genetic disease, liver disease and inflammation, specifically for Alpha-1 antitrypsin deficiency (AATD) and acute respiratory distress syndrome (ARDS) including COVID-19. The Company’s pipeline also includes PH-1 ADC Platform for oncology.
Ignyte Acquisition Corp (Ignyte)
On November 1, 2022 (the “Closing Date”), the Company completed the transactions contemplated by the certain business combination agreement, dated as of April 28, 2022 (the “Business Combination Agreement”), by and among Ignyte, Ignyte Korea Co., Ltd., a corporation organized under the laws of the Republic of Korea (“Korean Sub”), and Peak Bio Co., Ltd. At the closing of the transactions, (i) the stockholders of Peak Bio transferred their respective shares of Peak Bio Common Stock to Korean Sub in exchange for shares of Ignyte Common Stock held by Korean Sub, and (ii) in the course of such share swap, Korean Sub distributed the shares of Peak Bio Common Stock to Ignyte in consideration of Ignyte Common Stock (which was in-turn delivered to the stockholders of Peak Bio as described in (i) above ((i) and (ii), collectively, the “Share Swap”). Upon consummation of the Share Swap, Peak Bio became a direct wholly-owned subsidiary of Ignyte. The transactions contemplated by the Business Combination Agreement are referred to herein as the “Business Combination.”
Pursuant to the Business Combination, the aggregate consideration payable to stockholders of Peak Bio at the closing date consisted of
Pursuant to the treatment of the Business Combination as a reverse recapitalization, Peak Bio assumed the liability position as it existed as of the closing date of the Business Combination. The net assets of the acquired entity were adjusted to include a derivative liability for the embedded put option (see Note 6). In connection with the Business Combination, an amount of $
The shares and net loss per share prior to the reverse recapitalization have been retroactively restated to reflect the exchange ratio of
The following table details the number of shares of common stock issued immediately following the consummation of the Business Combination:
F-10
|
|
Shares |
|
|
Common stock redeemable and outstanding prior to business combination |
|
|
|
|
Less: redemption of Ignyte shares |
|
|
( |
) |
Common stock of Ignyte |
|
|
|
|
Ignyte founder shares |
|
|
|
|
Shares issued for services and debt settlement |
|
|
|
|
Total Ignyte shares |
|
|
|
|
Peak Bio shareholders |
|
|
|
|
Total shares of common stock immediately after business combination |
|
|
|
|
The following table reconciles the elements of the Business Combination to the consolidated statement of cash flows for the year ended December 31, 2022:
|
|
Recapitalization |
|
|
Cash - Ignyte trust and cash, net of redemptions and PIPE proceeds |
|
$ |
|
|
Plus: restricted cash - Forward Share Purchase Agreement |
|
|
|
|
Less: cash transaction costs allocated to the Company's equity |
|
|
( |
) |
Total |
|
$ |
|
|
The following table reconciles the elements of the Business Combination to the consolidated statement of changes in stockholders' equity (deficit) for the year ended December 31, 2022:
|
|
Recapitalization |
|
|
Cash - Ignyte trust and cash, net of redemptions and PIPE proceeds |
|
$ |
|
|
Plus: restricted cash - Forward Share Purchase Agreement |
|
|
|
|
Less: fair value of private warrants |
|
|
( |
) |
Less: derivative liability on Forward Share Purchase Agreement |
|
|
( |
) |
Less: transaction costs allocated to the Company's equity |
|
|
( |
) |
Total |
|
$ |
|
|
The following table details the allocated assets acquired and liabilities assumed as follows:
Assets Acquired |
|
|
|
|
Cash - Ignyte trust and cash, net of redemptions |
|
$ |
|
|
Plus: restricted cash - Forward Share Purchase Agreement |
|
|
|
|
Other assets |
|
|
|
|
Assets acquired |
|
|
|
|
Liabilities Assumed |
|
|
|
|
Fair value of private warrants |
|
|
|
|
Derivative liability on Forward Share Purchase Agreement |
|
|
|
|
Other liabilities and accrued expenses |
|
|
|
|
Liabilities assumed |
|
|
|
|
Net assets acquired |
|
$ |
|
|
On the Closing Date, a purchaser (the “Original Subscriber”) purchased from the Company an aggregate of
On the Closing Date, a number of additional purchasers (each, a “New Subscriber”) purchased from the Company an aggregate of (i)
On the Closing Date, a number of Peak Bio’s lenders (each, a “Bridge Loan PIPE Subscriber” and together with the Original Subscriber and the New Subscribers, the “Subscribers”) purchased from the Company an aggregate of (i)
F-11
Common Stock (the “Bridge Loan PIPE Shares” and together with the Original PIPE Shares and the New PIPE Shares, the “PIPE Shares”) and (ii)
Pursuant to the Subscription Agreements, the Company gave certain registration rights to the Subscribers with respect to the PIPE Shares and the PIPE Financing Warrants. The sale of the PIPE Shares and PIPE Financing Warrants was consummated concurrently with the Closing.
Upon the Closing, Ignyte as the registrant changed its name to “Peak Bio, Inc.”
Risks and Uncertainties
The Company relies, and expects to continue to rely, on a small number of vendors to provide services, supplies and materials related to its research and development programs. These research and development programs could be adversely affected by a significant interruption in these services or the availability of materials.
Going Concern
Since inception, the Company has incurred significant net losses. The Company incurred net losses of $
The Company will need additional financing to fund its ongoing activities. The Company may raise this additional funding through the sale of equity, debt financings or other capital sources, including potential collaborations with other companies or other strategic transactions and funding under government contracts. The Company may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms, or at all. If the Company is unable to raise capital when needed or on attractive terms, the Company could be forced to delay, reduce or eliminate certain of the Company’s research and development programs. There can be no assurances that other sources of financing would be available. Due to these uncertainties, there is substantial doubt about the Company’s ability to continue as a going concern.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or classification of liabilities that might result from the outcome of the uncertainties discussed above.
The Company’s future operations are highly dependent on a combination of factors, including (i) the timely and successful completion of additional financing as discussed above; (ii) the success of its research and development programs; (iii) the development of competitive therapies by other biotechnology and pharmaceutical companies; (iv) the Company’s ability to manage growth of the organization; (v) the Company’s ability to protect its proprietary technology; and ultimately (vi) regulatory approval and market acceptance of the Company’s product candidates.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”) and are comprised of the Company’s activities distributed across multiple legal entities.
Basis of Presentation Prior to April 1, 2022
These consolidated financial statements were extracted from the accounting records of pH Pharma Ltd. on a carve-out basis prior to April 1, 2022. The historical results of operations, financial position, and cash flows may not be indicative of what such results
F-12
of operations, financial position, and cash flows would have been had the Company been a separate standalone entity, nor are they indicative of what the results of operations, financial position and cash flows may be in the future.
The accompanying carve-out consolidated financial statements reflect assets, liabilities, revenue, and expenses that are directly attributable to the Company, including the assets, liabilities, revenue and expenses of the PHP-303 and PH-1 ADC Platform programs. The assets and liabilities excluded from the accompanying carve-out consolidated financial statements consist of:
The majority of the Company’s operating expenses related to research and development (“R&D”). R&D expenses directly related to the Company were entirely attributed to the Company in the accompanying carve-out consolidated financial statements. R&D salaries, wages and benefits were allocated to the Company using methodologies based on the proportionate share of R&D expenses for the PHP-303 and PH-1 ADC Platform programs compared to the R&D expenses for pH Pharma Ltd as a whole. The Company also received services and support from other functions of pH Pharma Ltd. The Company’s operations are dependent upon the ability of these other functions to provide these services and support. The costs associated with these services and support were allocated to the Company using methodologies based on the proportionate share of R&D expenses for the PHP-303 and PH-1 ADC Platform programs compared to the total R&D expenses and certain administrative expenses for pH Pharma Ltd as a whole. These allocated costs were primarily related to corporate administrative expenses, non-R&D employee related costs, including salaries and other benefits, for corporate and shared employees, and other expenses for shared assets for the following functional groups: information technology, legal, accounting and finance, human resources, facilities, and other corporate and infrastructural services. These allocated costs were primarily recorded as R&D expenses and general and administrative (“G&A”) expenses in the statements of operations and comprehensive loss.
The Company believes the assumptions and allocations underlying the carve-out financial statements were reasonable and appropriate under the circumstances.
Basis of Presentation After April 1, 2022
The Spin-Off resulted in Peak Bio retaining the PHP-303 and PH-1 ADC Platform programs. Historically and throughout the periods presented, the PHP-303 and PH-1 ADC Platform programs have been owned by pH Pharma Co., Ltd and its subsidiaries (prior to the change of its name to Peak Bio Co., Ltd.). The PHP-303 and PH-1 ADC Platform programs have historically operated as a part of pH Pharma Co., Ltd and not as a separate stand-alone entity or group. The Spin-Off resulted in Peak Bio retaining approximately
As of April 1, 2022, as a result of the Spin-Off, the Company concluded that all the assets and liabilities of the newly created Peak Bio legal entity were contributed by the parent company pH Pharma Ltd. No other assets or liabilities were considered to be attributable to Peak Bio or that would be transferred to Peak Bio upon the completion of the Business Combination, eliminating the necessity to allocate a portion of pH Pharma Ltd.’s assets and liabilities to Peak Bio on a carve-out basis. Therefore, there was no longer a need to allocate assets and liabilities, as well as expenses, from the parent company for the consolidated financial statements.
The accompanying financial statements have been prepared in conformity with U.S. GAAP. Any reference in these notes to applicable guidance is meant to refer to U.S. GAAP, as found in the Accounting Standards Codification, or ASC, and Accounting Standards Update, or ASU, of the Financial Accounting Standards Board, or FASB. The Company’s consolidated financial statements for the year ended December 31, 2022 include the accounts of Peak Bio Co., Ltd. and its subsidiary, Peak Bio CA., Inc. All intercompany balances and transactions have been eliminated in consolidation.
F-13
Basis of Presentation After Consummation of Business Combination Agreement
The Business Combination was accounted for as a reverse recapitalization in accordance with U.S. GAAP (the “Reverse Recapitalization”). Under this method of accounting, Ignyte is treated as the “acquired” company and Peak Bio is treated as the acquirer for financial reporting purposes. Accordingly, for accounting purposes, the Reverse Recapitalization was treated as the equivalent of Peak Bio issuing stock for the net assets of Ignyte, accompanied by a recapitalization. The net assets of Ignyte are stated at historical cost, with
The consolidated assets, liabilities and results of operations prior to the Reverse Recapitalization are those of Peak Bio. At the closing date, and subject to the terms and conditions of the Business Combination Agreement, each share of Peak Bio's common stock, par value $
Segment Information
The Company currently operates in
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent liabilities at the date of the financial statements, and the reported amounts of expenses during the reporting periods. Actual results could differ from those estimates. Significant items subject to such estimates and assumptions, are used for, but not limited to, include stock-based compensation, derivative instruments, hybrid instruments, warrants, the valuation of pH Pharma Ltd common stock and the allocation of certain pH Pharma Ltd expenses in the consolidated financial statements.
Additionally, the Company assessed the impact the COVID-19 pandemic has had on its operations and financial results as of December 31, 2021. The Company’s analysis was informed by the facts and circumstances as they were known to the Company. This assessment considered the impact COVID-19 may have on financial estimates and assumptions that affect the reported amounts of assets and liabilities and revenue and expenses. Based on this assessment, the Company’s operations have not been significantly impacted, with the exception of research and development expense decreasing during the year ended December 31, 2021 in part due to delays in the Company’s ongoing and planned research activities due to COVID-19. Research and development expense during the year ended December 31, 2021 also decreased in part as the Company reduced its average headcount from
Fair Value Measurements
The Company records certain liability balances under the fair value measurements as defined by the Financial Accounting Standards Board (“FASB”) guidance. Current FASB fair value guidance emphasizes that fair value is a market-based measurement, not an entity-specific measurement. Therefore, a fair value measurement should be determined based on the assumptions that market participants would use in pricing the asset or liability. As a basis for considering market participant assumptions in fair value measurements, current FASB guidance establishes a fair value hierarchy that distinguishes between market participant assumptions based on market data obtained from sources independent of the reporting entity (observable inputs that are classified within Levels 1 and 2 of the hierarchy) and the reporting entity’s own assumptions that market participants assumptions would use in pricing assets or liabilities (unobservable inputs classified within Level 3 of the hierarchy).
F-14
Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities that the Company has the ability to access at measurement date.
Level 2 inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 inputs may include quoted prices for similar assets and liabilities in active markets, as well as inputs that are observable for the asset or liability (other than quoted prices), such as interest rates, foreign exchange rates, and yield curves that are observable at commonly quoted intervals.
Level 3 inputs are unobservable inputs for the asset or liability, which is typically based on an entity’s own assumptions, as there is little, if any, related market activity. In instances where the determination of the fair value measurement is based on inputs from different levels of the fair value hierarchy, the level in the fair value hierarchy within which the entire fair value measurement falls is based on the lowest level input that is significant to the fair value measurement in its entirety. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment and considers factors specific to the asset or liability.
Cash and cash equivalents
Cash equivalents include short-term, highly liquid instruments, consisting of money market accounts in the U.S.
Restricted Cash
The Company has a lease agreement for the premises it occupies in Palo Alto, California. A secured letter of credit in lieu of a lease deposit totaling $
Currency and currency translation
The consolidated financial statements are presented in U.S. dollars, the Company’s reporting currency. The functional currency of Peak Bio CA, Inc. is the U.S. dollar. The functional currency of Peak Bio Co., Ltd is the Korean Won. Adjustments that arise from exchange rate changes on transactions of each group entity denominated in a currency other than the functional currency are included in other income and expense in the consolidated statements of operations. Assets and liabilities of Peak Bio Co., Ltd are recorded in their Korean Won functional currency and translated into the U.S. dollar reporting currency of the Company at the exchange rate on the balance sheet date. Revenue, when recorded, and expenses of Peak Bio Co., Ltd are recorded in their Korean Won functional currency and translated into the U.S. dollar reporting currency of the Company at the average exchange rate prevailing during the reporting period. Resulting translation adjustments are recorded to other comprehensive income (loss).
Concentration of credit risk
The Company maintains its cash and cash equivalent balances in the form of business checking accounts and money market accounts in the U.S., the balances of which, at times, may exceed federally insured limits. The Federal Deposit Insurance Corporation (“FDIC”) insurance coverage limit is $
The Company received all of its total revenue through a grant from a government organization during the years ended December 31, 2022 and 2021.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is calculated over the estimated useful lives of the respective assets, which range from two to five years, or the lesser of the related initial term of the lease or useful life for leasehold improvements.
F-15
Impairment of Long-lived Assets
Long-lived assets consist primarily of property and equipment. The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate the carrying amount of an asset is not recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amount of the asset exceeds the fair value of the assets. Fair value would be assessed using discounted cash flows or other appropriate measures of fair value. The Company did not recognize any impairment losses for the years ended December 31, 2022 and 2021.
Derivative Instruments
ASC 815, Derivatives and Hedging Activities (“ASC 815”) requires companies to bifurcate certain conversion options and redemption features from their host instruments and account for them as free-standing derivative financial instruments should certain criteria be met. The Company does not use derivative instruments to hedge exposures to interest rate, market, or foreign currency risks. The Company evaluates all of its financial instruments, including notes payable and other derivative liabilities, to determine whether such instruments are derivatives or contain features that qualify as embedded derivatives. Embedded derivatives must be separately measured from the host contract if all the requirements for bifurcation are met. The assessment of the conditions surrounding the bifurcation of embedded derivatives depends on the nature of the host contract and the features of the derivatives. Bifurcated embedded derivatives are recognized at fair value, with changes in fair value recognized in the consolidated statement of operations each period. Bifurcated embedded derivatives are classified with the related host contract in the Company’s consolidated balance sheet.
Hybrid Instruments
The Company also follows ASC 480-10, Distinguishing Liabilities from Equity (“ASC 480-10”) when evaluating the accounting for its hybrid instruments. A financial instrument that embodies an unconditional obligation, or a financial instrument other than an outstanding share that embodies a conditional obligation, that the issuer must or may settle by issuing a variable number of its equity shares shall be classified as a liability (or an asset in some circumstances) if, at inception, the monetary value of the obligation is based solely or predominantly on any one of the following: (a) a fixed monetary amount known at inception; (b) variations in something other than the fair value of the issuer’s equity shares; or (c) variations inversely related to changes in the fair value of the issuer’s equity shares. Hybrid instruments meeting these criteria are not further evaluated for any embedded derivatives and are carried as a liability at fair value at each balance sheet date.
Warrants
The Company accounts for the Public Warrants and Private Warrants (as defined in Note 11) collectively (“Warrants”), as either equity or liability-classified instruments based on an assessment of the specific terms of the Warrants and the applicable authoritative guidance in FASB Accounting Standards Codification (“ASC”) 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the Warrants meet all of the requirements for equity classification under ASC 815, including whether the Warrants are indexed to the Company’s own common stocks and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of issuance of the Warrants and as of each subsequent quarterly period end date while the Warrants are outstanding.
For issued or modified warrants that meet all of the criteria for equity classification, such warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all the criteria for equity classification, such warrants are required to be recorded at their initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the estimated fair value of liability-classified warrants are recognized as a non-cash gain or loss on the statements of operations.
The Company accounts for the Private Warrants in accordance with ASC 815-40 under which the Private Warrants do not meet the criteria for equity classification and must be recorded as liabilities. The fair value of the Private Warrants has been estimated using the Modified Black Scholes model. See Note 6 for further discussion of the pertinent terms of the Warrants used to determine the value of the Private Warrants and Representative’s Warrants.
F-16
The Company evaluated the Public Warrants in accordance with ASC 815-40, “Derivatives and Hedging — Contracts in Entity’s Own Equity” and concluded that they met the criteria for equity classification and are required to be recorded as part a component of additional paid-in capital at the time of issuance.
Revenue recognition
The Company’s revenue is primarily generated through grants from government organizations.
The Company recognizes revenue from these contracts during the period that the research and development services occur, as qualifying expenses are incurred or conditions of the grant are met. The Company concluded that payments received under these grants represent conditional, nonreciprocal contributions, as described in ASC 958, Not-for-Profit Entities, and that the grants are not within the scope of ASC 606, Revenue from Contracts with Customers, as the organizations providing the grants do not meet the definition of a customer. Qualifying expenses are recognized when incurred as research and development expenses. Revenues and related expenses are presented gross in the statements of operations as the Company determined it is the principal in conducting the research and development services and the primary obligor relative to the research and development services it performed as lead technical expert. Expenses for grants are tracked by using a project code specific to the grant, and the employees also track hours worked by using the project code.
Research and Development Expenses
Research and development costs are expensed as incurred. Research and development expenses consist primarily of costs related to personnel, including salaries and other personnel related expenses, contract manufacturing and supply, consulting fees, and the cost of facilities and support services used in drug development. Assets acquired that are used for research and development and have no future alternative use are expensed as in-process research and development.
General and Administrative Costs
General and administrative expenses consist primarily of salaries and related benefits, including stock-based compensation, related to our executive, finance, business development, legal, human resources and support functions. Other general and administrative expenses include professional fees for auditing, tax, consulting and patent-related services, rent and utilities and insurance.
Share-based Compensation
The Company accounts for stock options granted in accordance with ASC 718, Compensation-Stock Compensation, or ASC 718. In accordance with ASC 718, compensation expense is measured at the estimated fair value of the stock options at grant date and is included as compensation expense over the vesting period during which an employee provides service in exchange for the award.
Stock-based compensation expense represents the cost of the grant date fair value of employee stock awards recognized over the requisite service period of the awards (usually the vesting period) on a straight-line basis. The Company estimates the fair value of each stock-based award on the date of grant using the Black-Scholes option pricing model. The Black-Scholes option pricing model incorporates various assumptions, such as the value of the underlying common stock, the risk-free interest rate, expected volatility, expected dividend yield, and expected life of the options. Forfeitures are recognized as a reduction of stock-based compensation expense as they occur.
Equity-based compensation expense is classified in the statement of operations in the same manner in which the award recipients’ payroll costs are classified or in which the award recipients’ service payments are classified.
Prior to the Business Combination, there had been no public market of the Company’s common stock. Therefore, the grant date fair value of the Company's common stock was determined by the Company's board of directors with the assistance of management and a third-party valuation specialist (see "Common stock valuations"). Subsequent to the Business Combination, the Company now determines the fair value of common stock based on the closing market price on the date of grant.
Determination of the Fair Value of Convertible Notes
The Company has elected the fair value option in accordance with ASC 825, Financial Instruments, for the accounting for the convertible promissory notes issued in 2022. Fair value adjustments to the convertible notes are included in other income (expenses) in the consolidated statements of operations and comprehensive loss.
F-17
Net Loss Per Share
As a result of the Spin-Off, Peak Bio had
The Company has reported losses since inception and has computed basic net loss per share attributable to common stockholders by dividing net loss attributable to common stockholders by the weighted-average number of common shares outstanding for the period, without consideration for potentially dilutive securities. The Company computes diluted net loss per common share after giving consideration to all potentially dilutive common shares, including options to purchase common stock, outstanding during the period determined using the treasury-stock and if-converted methods, except where the effect of including such securities would be antidilutive. Because the Company has reported net losses since inception, these potential common shares have been anti-dilutive and basic and diluted loss per share have been the same.
The following table sets forth the potentially dilutive securities that have been excluded from the calculation of diluted net loss per share because to include them would be anti-dilutive (in common stock equivalent shares):
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Stock options to purchase common stock |
|
|
|
|
|
|
||
Income Taxes
Deferred income taxes reflect future tax effects of temporary differences between the tax and financial reporting basis of the Company’s assets and liabilities measured using enacted tax laws and statutory tax rates applicable to the periods when the temporary differences will affect taxable income. When necessary, deferred tax assets are reduced by a valuation allowance, to reflect realizable value, and all deferred tax balances are reported as long-term on the balance sheet. Accruals are maintained for uncertain tax positions, as necessary.
The Company uses a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken, or expected to be taken, in a tax return. The Corporation has elected to treat interest and penalties related to income taxes, to the extent they arise, as a component of income taxes.
ASC 740 prescribes the accounting for uncertainty in income taxes recognized in the financial statements. The Company regularly assesses the outcome of potential examinations in each of the taxing jurisdictions when determining the adequacy of the amount of unrecognized tax benefit recorded. The Company recognizes tax benefits from uncertain tax positions only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such positions are then measured based on the largest benefit which is more likely than not to be realized upon ultimate settlement. As of December 31, 2022 and 2021, the Company had no uncertain tax positions.
Common stock valuations
Prior to the Business Combination, the Company is required to periodically estimate the fair value of pH Pharma Ltd.’s common stock with the assistance of an independent third-party valuation firm when issuing stock options and computing the estimated stock-based compensation expense. The assumptions underlying these valuations represented management’s best estimates, which involved inherent uncertainties and the application of significant levels of management judgment.
In order to determine the fair value of pH Pharma Ltd.’s common stock, the Company considered, among other items, previous transactions involving the sale of pH Pharma Ltd.’s securities, the pH Pharma Ltd.’s business, financial condition and results of operations, economic and industry trends, the market performance of comparable publicly traded companies, and the lack of marketability of pH Pharma Ltd.’s common stock. Valuations are updated when facts and circumstances indicate that the most recent valuation is no longer valid, such as changes in the stage of development efforts, various exit strategies and their timing, and other scientific developments that could be related to the Company’s valuation, or, at a minimum, annually. Third-party valuations were performed in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation.
Recently Adopted Accounting Standards
F-18
Leases
In February 2016, the FASB issued Accounting Standards Update (“ASU”) No. 2016-02, “Leases” (“ASC 842”) to enhance the transparency and comparability of financial reporting related to leasing arrangements. Under this new lease standard, most leases are required to be recognized on the balance sheet as right-of-use assets and lease liabilities. Disclosure requirements have been enhanced with the objective of enabling financial statement users to assess the amount, timing, and uncertainty of cash flows arising from leases. Prior to January 1, 2019, U.S. GAAP did not require lessees to recognize assets and liabilities related to operating leases on the balance sheet. The new standard establishes a right-of-use model (“ROU”) that requires a lessee to recognize a ROU asset and corresponding lease liability on the balance sheet for all leases with a term longer than 12 months. Leases will be classified as finance or operating, with classification affecting the pattern and classification of expense recognition in the income statement as well as the reduction of the right of use asset.
The Company has adopted the standard effective January 1, 2022 and has chosen to use the effective date as our date of initial application. Prior to January 1, 2022, the Company accounted for leases under ASC 840, "Accounting for Leases". Consequently, financial information will not be updated and the disclosures required under the new standard will not be provided for dates and periods prior to January 1, 2022. The new standard provides a number of optional practical expedients in transition. The Company has elected to apply the ‘package of practical expedients’ which allow us to not reassess (i) whether existing or expired arrangements contain a lease, (ii) the lease classification of existing or expired leases, or (iii) whether previous initial direct costs would qualify for capitalization under the new lease standard. The Company has also elected to apply (i) the practical expedient which allows us to not separate lease and non-lease components, for new leases entered into after adoption and (ii) the short-term lease exemption for all leases with an original term of less than 12 months, for purposes of applying the recognition and measurements requirements in the new standard. For the impact to the Company’s consolidated financial statement upon adoption of the new leasing standard, see Note 8 to our consolidated financial statements.
At the inception of an arrangement, the Company determines whether the arrangement is or contains a lease based on specific facts and circumstances, the existence of an identified asset(s), if any, and the Company’s control over the use of the identified asset(s), if applicable. Operating lease liabilities and their corresponding right-of-use assets are recorded based on the present value of future lease payments over the expected lease term. The interest rate implicit in lease contracts is typically not readily determinable. As such, the Company will utilize the incremental borrowing rate, which is the rate incurred to borrow on a collateralized basis over a similar term an amount equal to the lease payments in a similar economic environment. As of the ASC 842 effective date, the Company’s incremental borrowing rate is approximately
The Company has elected to combine lease and non-lease components as a single component. Operating leases are recognized on the balance sheet as ROU lease assets, lease liabilities current and lease liabilities non-current. Fixed rents are included in the calculation of the lease balances while variable costs paid for certain operating and pass-through costs are excluded. Lease expense is recognized over the expected term on a straight-line basis.
Income Taxes
In December 2019, the FASB issued ASU 2019-12, "Simplifying the Accounting for Income Taxes" ("ASU 2019-12"). ASU 2019-12 eliminates certain exceptions related to the approach for intra-period tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. It also clarifies and simplifies other aspects of the accounting for income taxes. This guidance is effective for fiscal years beginning after December 15, 2021, and interim periods within fiscal years beginning after December 15, 2022. The adoption of ASU No. 2019-12 on January 1, 2022 did not have a material impact on the consolidated financial statements.
Grant Revenue
In November 2021, the FASB issued ASU 2021-10, Government Assistance (Topic 832), which requires business entities to disclose information about transactions with a government entity that are accounted for by applying a grant or contribution model by analogy. For transactions within scope, the new standard requires the disclosure of information about the nature of the transaction, including significant terms and conditions, as well as the amounts and specific financial statement line items affected by the transaction. The new guidance is effective for annual reporting periods beginning after December 15, 2021. The Company adopted ASU 2021-10 in the first quarter of 2022. The adoption of this update did not have a material impact on the Company’s financial statements and footnote disclosures.
F-19
Recently Issued Accounting Standards
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments-Credit Losses: Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”). ASU 2016-13 requires measurement and recognition of expected credit losses for financial assets. In April 2019, the FASB issued clarification to ASU 2016-13 within ASU 2019-04, Codification Improvements to Topic 326, Financial Instruments-Credit Losses, Topic 815, Derivatives and Hedging, and Topic 825, Financial Instruments, or ASU 2016-13. The guidance is effective for fiscal years beginning after December 15, 2022. The Company is currently assessing the potential impact of adopting ASU 2016- 02 on its financial statements and financial statement disclosures.
In August 2020, the FASB issued ASU 2020-06, “Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity” (“ASU 2020-06”), which simplifies the accounting for convertible instruments by removing major separation models required under current U.S. GAAP. ASU 2020-06 removes certain settlement conditions that are required for equity contracts to qualify for the derivative scope exception, which will permit more equity contracts to qualify for such exception and simplifies the diluted earnings per share calculation in certain areas. ASU 2020-06 is effective for public business entities that meet the definition of a SEC filer, excluding entities eligible to be smaller reporting companies as defined by the SEC, for fiscal years beginning after December 15, 2021, including interim periods within those fiscal years. For all other entities, the amendments are effective for fiscal years beginning after December 15, 2023, including interim periods within those fiscal years. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020, including interim periods within those fiscal years. The Company is currently assessing the potential impact of adopting ASU 2020-06 on its financial statements and financial statement disclosures.
Property and equipment consist of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Lab equipment |
|
$ |
|
|
$ |
|
||
Leasehold improvements |
|
|
|
|
|
|
||
Computer and office equipment |
|
|
|
|
|
|
||
Computer software |
|
|
|
|
|
|
||
Gross property and equipment |
|
$ |
|
|
$ |
|
||
Less: accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
Net property and equipment |
|
$ |
|
|
$ |
|
||
Depreciation expense, including an allocation of depreciation expense from pH Pharma Ltd, was $
Accrued expenses consist of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Contract research and development costs |
|
$ |
|
|
$ |
|
||
Professional Fees |
|
|
|
|
|
|
||
Employee compensation costs |
|
|
|
|
|
|
||
Income tax |
|
|
|
|
|
|
||
Other liabilities |
|
|
|
|
|
|
||
Total accrued expenses and other current liabilities |
|
$ |
|
|
$ |
|
||
Included in contract research and development costs as of December 31, 2021 was the liability related to the upfront payment received from VennDC, LLC (“Venn”). See Note 9, Collaborative and Licensing Agreements, for additional information. During the year ended December 31, 2022, the Company recorded a liability of $
F-20
The pH Pharma Ltd Stock Option Plan (the “Plan”) provides for the granting of stock options to purchase common stock in pH Pharma Ltd to employees, directors, advisors, and consultants at a price to be determined by pH Pharma Ltd’ Board of Directors. The Plan is intended to encourage ownership of stock by employees and consultants of the Company and to provide additional incentives for them to promote the success of pH Pharma Ltd’ business. Under the provisions of the Plan, stock options will generally have a term of
The Spin-Off was completed on March 1, 2022, prior to the execution of the Business Combination Agreement with Ignyte Acquisition Corp (“Ignyte”), with holders of stock options in the Plan retaining
The Black-Scholes option pricing model is used when estimating the grant date fair value for stock-based awards. Use of a valuation model requires management to make certain assumptions with respect to selected model inputs. Expected volatility was based on the historical volatility of a publicly traded set of peer companies of pH Pharma Ltd. The expected life was equal to the contractual life of the stock option. The risk-free interest rate is based on U.S. Treasury, zero-coupon issues with a remaining term equal to the expected life assumed at the date of grant. Forfeitures related to equity-based compensation awards are recognized as they occur, and the Company reverses any previously recognized compensation cost associated with forfeited awards in the period the forfeiture occurs.
The following table summarizes the stock option activity:
|
|
Number of Options |
|
|
Weighted-average exercise price per share |
|
|
Weighted average remaining contractual term (in years) |
|
|
Aggregate intrinsic value |
|
||||
Outstanding at December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
— |
|
|||
Granted |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Allocation of stock options in spin-off |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
Cancelled/Forfeited |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
Allocation of stock options in business combination |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Outstanding at December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Exercisable at December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
The fair value of the stock options granted is estimated on the date of grant using a Black-Scholes option pricing model with the following weighted average assumptions:
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Expected volatility |
|
|
% |
|
|
% |
||
Risk-free interest rate |
|
|
% |
|
|
% |
||
Expected term (in years) |
|
|
|
|
|
|
||
Expected dividend yield |
|
|
% |
|
|
% |
||
For the years ended December 31, 2022 and 2021, the share-based compensation expense allocated to the Company was $
The following table summarizes information related to share-based compensation expense recognized in the statements of operations and comprehensive loss related to the equity awards:
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Research and development |
|
$ |
|
|
$ |
— |
|
|
General and administrative |
|
|
|
|
|
|
||
Total equity-based compensation |
|
$ |
|
|
$ |
|
||
F-21
In accordance with ASC 820, Fair Value Measurements and Disclosures, fair value is defined as the exit price, or the amount that would be received for the sale of an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The guidance also establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs include those that market participants would use in valuing the asset or liability and are developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the factors that market participants would use in valuing the asset or liability.
The Company’s financial assets and liabilities are measured at fair value and classified within the fair value hierarchy which is defined as follows:
The categorization of a financial instrument within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
The Company believes the carrying amounts of its cash and cash equivalents, related party loan and debt approximate their fair values due to their near-term maturities. As of December 31, 2021, the Company did not have any assets or liabilities that were recorded at fair value on a recurring basis.
The following table presents a roll-forward of the fair value of the convertible notes payable, derivative liability, derivative asset and warrant liability that will continue to be measured at fair value on a recurring basis for which fair value is determined based on significant inputs not observable in the market, which represents a Level 3 measurement within the fair value hierarchy as of December 31, 2022. The valuation models used to determine the fair value at each reporting date require management judgment and pricing inputs from observable and unobservable markets, including projected share prices and probabilities of success. Significant deviations from these estimates and inputs could result in a material change in fair value.
|
|
Convertible Notes Payable |
|
|
Derivative Liability |
|
|
Derivative Asset |
|
|
Warrant Liability |
|
||||
Balance at December 31, 2021 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
Issuance of convertible note |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Assumption of Forward Purchase Agreement |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
Assumption of Key Company Stockholder Forward Purchase |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
Initial Measurement of White Lion Purchase Agreement |
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
||
Assumption of warrant liability in Business Combination |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Change in fair value |
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|||
Conversion to common stock |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
|
||
Balance at December 31, 2022 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
The derivative liability, accounted for under ASC 480, “Distinguishing Liabilities from Equity,” related to the $
The derivative liability, accounted for under ASC 480, “Distinguishing Liabilities from Equity,” for a put option related to the
F-22
Model based on the following assumptions: stock price of $
The derivative liability, accounted for under ASC 815, “Derivatives and Hedging,” for a put option related to the Key Company Stockholder Forward Purchase Agreement entered into on April 2022 was initially valued at $
The White Lion Purchase Agreement qualifies as a standby equity purchase agreement under ASC 815 “Derivatives and Hedging” and includes an embedded put option and an embedded forward option. The put option is recognized on inception and the forward option is recognized upon issuance of notice for the sale of the Company's Common Stock. As at December 31, 2022, there were no notices issued to White Lion for the sale of Common Stock of the Company. The derivative liability, accounted for under ASC 815, “Derivatives and Hedging,” for the put option related to the White Lion Purchase Agreement entered into on November 3, 2022 was initially valued at $
The Private Placement Warrants were accounted for under ASC 815, “Derivatives and Hedging,” pursuant to which the Private Placement Warrants do not meet the criteria for equity classification and must be recorded as liabilities. The Private Placement Warrants were valued using a Modified Black Scholes Option Pricing Model, which is considered to be a Level 3 fair value measurement, as the fair value was determined based on significant inputs not observable in the market. The fair value of the Private Placement Warrants was discounted to present value at December 31, 2022, utilizing the Company's stock price of $
The Company believes the carrying amounts of its cash and cash equivalents, related party loan and current note payable approximate their fair values due to their near-term maturities. There were
Transactions entered into between the Company and pH Pharma Ltd were included within the consolidated financial statements and are considered related party transactions and have been adjusted to Equity within the balance sheets and statements of cash flows as they represent an investment to the Company.
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Corporate allocations |
|
|
|
|
|
|
||
Research and development |
|
$ |
|
|
$ |
|
||
Selling, general and administrative |
|
|
|
|
|
|
||
Accounts payable and general financing activities |
|
|
|
|
|
|
||
Net increase in contributions from member |
|
$ |
|
|
$ |
|
||
F-23
On March 1, 2022, the Company and pH Pharma Ltd entered into an administrative services and facilities agreement whereby pH Pharma Ltd will perform services, functions and responsibilities for the Company. Under the agreement, the Company paid pH Pharma Ltd $
On January 1, 2022, the Company adopted ASC 842 using the modified retrospective transition approach allowed under ASU 2018-11 which releases companies from presenting comparative periods and related disclosures under ASC 842 (Note 2). The Company adopted the standard under the modified retrospective approach and the effective date is as of the initial application. Consequently, financial information was not updated, and the disclosures required under ASU 2016-02 are not provided for dates and periods prior to January 1, 2022. The Company is party to one operating lease for office and laboratory space. The Company’s does not have any finance leases. The Company has elected to apply the short-term lease exception to all leases of one year or less. As of December 31, 2022, this exception does not apply to any of the operating leases for office and laboratory space. Further, the Company has applied the guidance in ASC 842 to our corporate office and laboratory leases and has determined that these should be classified as operating leases. Consequently, as a result of the adoption of ASC 842, we recognized a ROU lease asset of approximately $
In October 2019, the Company entered into a 24-month sublease for laboratory and office facilities in San Francisco, California. Base rent for this sublease was approximately $
In October 2021, the Company entered into a lease for laboratory and office facilities in Palo Alto, California that expires in
Rent expense, including an allocation of costs from pH Pharma Ltd, for the years ended December 31, 2022 and 2021 was $
Quantitative information regarding the Company’s leases for the year ended December 31, 2022 is as follows:
|
|
Operating Lease |
|
|
Operating cash flows paid for amounts included in the measurement of lease liabilities |
|
$ |
|
|
Operating lease liabilities arising from obtaining right of use assets |
|
$ |
|
|
Weighted-average remaining lease terms (years) |
|
|
|
|
Weighted-average discount rate |
|
|
% |
|
Future lease payments under noncancelable leases are as follows at December 31, 2022:
|
|
Operating |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
Thereafter |
|
|
|
|
Total future minimum lease payments |
|
$ |
|
|
Less: imputed interest |
|
|
( |
) |
Total future minimum lease payments |
|
$ |
|
|
F-24
Bayer Acquisition Agreement
In March 2017, pH Pharma, Inc. entered into an assignment, license, development and commercialization agreement (the “Bayer Acquisition Agreement”) with Bayer, to acquire from Bayer all right, title and interest in and to PHP-303, including each and every invention and any priority rights relating to its patents.
Upon entering into the Bayer Acquisition Agreement, pH Pharma, Inc. made an upfront payment, and the Company has agreed to pay certain development and regulatory milestones and future royalties. Royalties will be payable on a licensed product-by-licensed product and country-by-country basis until the later of ten years after the first commercial sale of such licensed product in such country and expiration of the last patent covering such licensed product in such country that would be sufficient to prevent generic entry.
Either party may terminate the Bayer Acquisition Agreement upon prior written notice for the other party’s material breach that remains uncured for a specified period of time or insolvency. Bayer agreed not to assert any Bayer intellectual property rights that were included in the scope of the Bayer Acquisition Agreement against the Company.
Employment Agreements
In January 2022, the Company entered into an employment agreement with its founder and director. The effective date of the employment agreement was February 1, 2022, and is subject to the completion of the business combination with Ignyte. As part of the agreement, the Company agreed to repay its founder and director $
In March 2022, the Company entered into an employment agreement with its chief operating officer which is subject to the completion of the business combination with Ignyte. The agreement provides for confirmation of Peak Bio’s previously agreed upon success fee payment upon consummation of the business combination with Ignyte in the amount of $
Legal proceedings
The Company is not currently a party to any material legal proceedings. At each reporting date, the Company evaluates whether a potential loss amount or a potential range of loss is probable and reasonably estimable under the provisions of the authoritative guidance that addresses accounting for contingencies. The Company expenses the costs related to its legal proceedings as incurred.
Venn License Agreement
In December 2019, a collaboration and license agreement (the “License Agreement”) was entered into with Venn to pursue research and development of certain payload and linker technologies that are useful for the development of antibody-drug conjugates. This collaboration was expected to allow Venn to further develop and commercialize such antibody-drug conjugates developed under the collaboration. Under the collaboration agreement with Venn, the Company received a $
In April 2022, the Company entered into an agreement with its founder and director, Dr. Huh, in consideration of the repayment to be made by the Company’s founder and director to settle a contractual obligation for the upfront payment received by the Company associated with the License Agreement with Venn. Per the agreement, the Company agreed to repay its founder and director $
In May 2022, the Company’s founder and director repaid to Venn the $
F-25
During the years ended December 31, 2022 and 2021, the Company did
Preferred Stock
The Company is authorized to issue
Common Stock
The Company is authorized to issue
In May 2022, the Company entered into an agreement with a certain investor in which the investor purchased an aggregate of
PIPE Subscription Agreements
In November 2022, concurrently with the closing of the Business Combination, the Company entered into subscription agreements with certain investors in which the investors purchased, in a private placement, an aggregate of
Forward Purchase Agreement
On December 29, 2022, the Company purchased
Additional PIPE Subscription Agreement
In December 2022, the Company entered into a subscription agreement whereby the Company agreed to issue and sell to the investor party thereto, in a private placement, (i)
Key Company Stockholder Agreements
In April 2022, the Company entered into a forward purchase agreement (the “Key Company Stockholder Forward Purchase Agreement”) with its founder and director, Hoyoung Huh (the “Key Company Stockholder”). Pursuant to the terms of the Key Company Stockholder Forward Purchase Agreement, the Key Company Stockholder would, subject to the receipt of margin financing within
F-26
In December 2022, the Company and the Key Company Stockholder entered into an amendment to the Key Company Stockholder Forward Purchase Agreement (the “Amendment to Key Company Stockholder Forward Purchase Agreement”), pursuant to which (i) the Key Company Stockholder Purchase is no longer subject to the receipt of margin financing as a condition precedent, (ii) the Key Company Stockholder agreed to fund the Subscription Amount on or prior to March 31, 2023 and (iii) the Key Company Stockholder Purchase would be consummated at a purchase price of $
In April 2023, the Company received notice from its founder and director informing the Company that he would not consummate the purchase of the Key Company Stockholder Forward Purchase Agreement as a result of the Company’s failure to satisfy the condition to be listed on Nasdaq as required by the agreement. As a result the Company cancelled and forfeited the
In April 2023, the Company and its founder and director entered into a letter agreement to provide for the conversion of loans totaling $
White Lion Common Stock Purchase and Registration Rights Agreements
On November 3, 2022, the Company entered into a Common Stock Purchase Agreement (the “White Lion Purchase Agreement") and Registration Rights (the “White Lion RRA”) with White Lion Capital, LLC, a Delaware limited liability company (“White Lion”). Pursuant to the White Lion Purchase Agreement, the Company has the right, but not the obligation, to require White Lion to purchase, from time to time, up to $
The Company is obligated under the White Lion Purchase Agreement and the White Lion RRA to file a registration statement with the SEC to register the Common Stock under the Securities Act, for the resale by White Lion of shares of Common Stock that the Company may issue to White Lion under the White Lion Purchase Agreement.
Subject to the satisfaction of certain customary conditions including, without limitation, the effectiveness of a registration statement registering the shares issuable pursuant to the White Lion Purchase Agreement, the Company's right to sell shares to White Lion will commence on the effective date of the registration statement and extend until November 1, 2025. During such term, subject to the terms and conditions of the White Lion Purchase Agreement, the Company may notify White Lion when it exercises its right to sell shares (the effective date of such notice, a “Notice Date”).
The number of shares sold pursuant to any such notice may not exceed (i) the lower of (a) the Purchase Notice Fixed Limit (described below) and (b) the product of (1) the Average Daily Trading Volume (as defined in the White Lion Purchase Agreement), and (2) the applicable Percentage Limit (as defined in the White Lion Purchase Agreement). The Purchase Notice Fixed Limit is $
The applicable Percentage Limit is
The Company will have the right to terminate the White Lion Purchase Agreement at any time after commencement, at no cost or penalty, upon three (3) Trading Days’ prior written notice. Additionally, White Lion will have the right to terminate the White Lion Purchase Agreement upon three (3) days’ prior written notice to the Company if (i) there is a Fundamental Transaction (as defined in the White Lion Purchase Agreement), (ii) the Company is in breach or default in any material respect of the White Lion RRA, (iii) there is a lapse of the effectiveness, or unavailability of, the registration statement for a period of 45 consecutive Trading Days or for more than an aggregate of 90 Trading Days in any 365-day period, (iv) the suspension of trading of the Common Stock for a period of
F-27
five (5) consecutive Trading Days, (v) the material breach of the White Lion Purchase Agreement by the Company, which breach is not cured within the applicable cure period or (vi) a Material Adverse Effect (as defined in the White Lion Purchase Agreement) has occurred and is continuing. No termination of the White Lion Purchase Agreement will affect the registration rights provisions contained in the White Lion RRA.
In consideration for the commitments of White Lion, as described above, the Company has agreed that it will issue to White Lion shares of Common Stock having a value of $
Concurrently with the execution of the White Lion Purchase Agreement, the Company entered into the White Lion RRA with White Lion in which the Company agreed to register the shares of Common Stock purchased by White Lion with the SEC for resale within 30 days of the consummation of a business combination. The White Lion RRA also contains usual and customary damages provisions for failure to file and failure to have the registration statement declared effective by the SEC within the time periods specified.
The White Lion Purchase Agreement and the White Lion RRA contain customary representations, warranties, conditions and indemnification obligations of the parties. The representations, warranties and covenants contained in such agreements were made only for purposes of such agreements and as of specific dates, were solely for the benefit of the parties to such agreements and may be subject to limitations agreed upon by the contracting parties.
In March 2023, the Company entered into an amendment to the White Lion Purchase Agreement to give the Company the right, but not the obligation to require White Lion to purchase shares of the Company's common stock while trading on the OTC Market. Under the terms of the amendment, the Company will issue to White Lion within five (5) Trading Days following the effective date of the amendment fully paid, non-assessable shares of the Company's Common Stock equal to the quotient obtained by dividing (i) $
In addition, in the event the Company does not issue Purchase Notices (as defined in the White Lion Purchase Agreement) to White Lion providing for the purchase of at least $
F-28
Public Warrants
In November 2022, upon consummation of the Business Combination, the Company assumed
The Company may call the warrants for redemption:
If the Company calls the warrants for redemption as described above, the Company’s management will have the option to require all holders that wish to exercise warrants to do so on a “cashless basis.” In such event, each holder would pay the exercise price by surrendering the warrants for that number of shares of common stock equal to the quotient obtained by dividing (x) the product of the number of shares of common stock underlying the warrants, multiplied by the difference between the exercise price of the warrants and the “fair market value” (defined below) by (y) the fair market value. The “fair market value” for this purpose shall mean the average reported last sale price of the shares of common stock for the 5 trading days ending on the third trading day prior to the date on which the notice of redemption is sent to the holders of warrants.
Private Placement Warrants
In November 2022, upon consummation of the Business Combination, the Company assumed
The Private Placement Warrants are identical to the Public Warrants, except that the Private Placement Warrants are non-redeemable and may be exercised on a cashless basis, in each case so long as they continue to be held by the initial purchasers or their permitted transferees. Further, the Sponsor had agreed not to transfer, assign, or sell the Private Placement Warrants (including the shares of Common Stock issuable upon the exercise of the Private Placement Warrants), except to certain permitted transferees, until after the consummation of the Business Combination.
A summary of the Company's outstanding warrants at December 31, 2022 is as follows:
F-29
Description |
|
Number of Warrants |
|
|
Exercise price per share |
|
|
Expiration Date |
||
Private Placement Warrants |
|
|
|
|
$ |
|
|
11/1/2027 |
||
Public Warrants |
|
|
|
|
$ |
|
|
11/1/2027 |
||
Other Warrants |
|
|
|
|
$ |
|
|
11/1/2023 |
||
Outstanding Warrants |
|
|
|
|
|
|
|
|
||
Government grants
Department of Defense, US Army Medica Research Acquisition Activity – this grant is for work on a COVID-19 therapeutic with a potential of $
PPP loans pursuant to the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”)
In April 2020, the Company received proceeds from a loan in the amount of $
In April 2021, the Company received proceeds from another loan in the amount of $
The application for these funds required the Company to certify in good faith that the then-current economic uncertainty made the loan requests necessary to support the ongoing operations of the Company.
This certification further required the Company to take into account its current business activity and its ability to access other sources of liquidity sufficient to support ongoing operations in a manner that was not significantly detrimental to the business. The Company made this good faith assertion based upon various factors, including the degree of uncertainty introduced to the capital markets as a result of the COVID-19 pandemic and the Company’s dependency on its ability to raise capital to fund ongoing operations.
All or a portion of the loans may have been forgiven by the U.S. Small Business Administration (“SBA”) upon application by the Company upon documentation of expenditures in accordance with the SBA requirements. Under the CARES Act, loan forgiveness was available for the sum of eligible and documented payroll costs, covered rent payments, covered mortgage interest and covered utilities during the eight-week period beginning on the date of loan approval. If, despite the Company’s good-faith belief that given the circumstances the Company satisfied all eligibility requirements for the loans, the Company was later determined to have violated any applicable laws or regulations or it is otherwise determined that the Company was ineligible to receive the loans, the Company may have been required to repay the loans in their entirety and/or be subject to additional penalties. In the event the loans, or any portion thereof, were forgiven pursuant to the PPP, the amounts forgiven would be applied to outstanding principal.
The Company used all proceeds from the loans to retain employees, maintain payroll and make lease, rent and utility payments. Under the terms of the loans, the Company may have been eligible for full or partial loan forgiveness. The Company applied for forgiveness on the loan dated April 20, 2020 and the loan plus accrued interest was forgiven in full on April 30, 2021. The Company applied for forgiveness on the loan dated April 15, 2021 and the loan plus accrued interest was forgiven in full on October 5, 2021. The Company recorded a gain on extinguishment of debt in the amount of $
The Company has accounted for the loans as a debt instrument in accordance with ASC 470, Debt. At December 31, 2022 and 2021, there was
F-30
Related Party Loan
In May 2021, the Company received proceeds from a loan in the amount of approximately $
In August 2021, the Company received proceeds from a loan in the amount of approximately $
In January 2022, the Company entered into an employment agreement with its chairman and founding chief executive officer. As part of the agreement, the Company agreed to repay $
In April 2022, the Company entered into an agreement with its founder and director, in consideration of the repayment to be made by the Company’s founder and director to settle a contractual obligation for the upfront payment received by the Company associated with the License Agreement with Venn. Per the agreement, the Company agreed to repay its founder and director $
In May 2022, the Company’s founder and director repaid to Venn the $
In May 2022, the Company received proceeds from a loan in the amount of approximately $
In September 2022, the Company received proceeds from a loan in the amount of $
In November 2022, the related party loan entered into in September 2022 was amended resulting in the outstanding principal and accrued interest under the related party loan converting at a price of $
In November 2022, upon consummation of the Business Combination, the Company assumed a promissory note of $
Long-term Convertible Notes Payable
From July through September 2022, the Company received proceeds from loans in the amount of $
Upon issuance, the Company elected the fair value option to account for the promissory notes, including the component related to accrued interest. At December 31, 2022, the fair value of the promissory notes was $
F-31
In November 2022, the Company amended the terms to provide warrant coverage on the conversion of the loans from
On November 1, 2022, the Company issued a $
On the maturity date, the note holder may, in its sole and absolute discretion, convert all or part of the principal and/or accrued interest of this convertible note into shares of common stock of the Company at a per share conversion price equal to
Insurance Financing Payable
The Company obtained financing for certain Director & Officer liability insurance policy premiums. The agreement assigns First Insurance Funding (Lender) a first priority lien on and security interest in the financed policies and any additional premium required in the financed policies including (a) all returned or unearned premiums, (b) all additional cash contributions or collateral amounts assessed by the insurance companies in relation to the financed policies and financed by Lender, (c) any credits generated by the financed policies, (d) dividend payments, and (e) loss payments which reduce unearned premiums. If any circumstances exist in which premiums related to any Financed Policy could become fully earned in the event of loss, Lender shall be named a loss-payee with respect to such policy.
The total premiums, taxes and fees financed is approximately $
The components of (loss) income before income taxes are as follows:
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Domestic |
|
|
( |
) |
|
$ |
|
|
Foreign |
|
|
( |
) |
|
|
( |
) |
Total |
|
|
( |
) |
|
$ |
( |
) |
F-32
|
|
Year Ended December 31, |
|
|||||
Components of Tax Expense |
|
2022 |
|
|
2021 |
|
||
Current — Federal |
|
$ |
|
|
$ |
|
||
Current — State |
|
|
( |
) |
|
|
|
|
Total current |
|
|
( |
) |
|
|
|
|
Deferred — Federal |
|
$ |
( |
) |
|
$ |
( |
) |
Deferred — State |
|
|
( |
) |
|
|
— |
|
Deferred — Foreign |
|
|
( |
) |
|
|
— |
|
Change in Valuation Allowance |
|
|
|
|
|
— |
|
|
Total deferred |
|
|
( |
) |
|
|
( |
) |
(Benefit from) provision for income taxes |
|
$ |
( |
) |
|
$ |
|
|
Effective income tax rate |
|
|
% |
|
|
( |
)% |
|
The effective tax rate of the Company’s provision (benefit) for income taxes differs from the federal statutory rate for the years ended December 31, 2022 and 2021 as follows:
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Tax computed at federal statutory rate |
|
|
% |
|
|
% |
||
State Tax Provision/(Benefit) net of federal benefit |
|
|
% |
|
|
( |
)% |
|
Earnings in jurisdictions taxed at rates different from the statutory |
|
|
% |
|
|
% |
||
Permanent differences |
|
|
( |
)% |
|
|
% |
|
Return to Provision |
|
|
% |
|
|
— |
% |
|
Foreign tax credits |
|
|
— |
% |
|
|
% |
|
Change in valuation allowance |
|
|
( |
)% |
|
|
( |
)% |
Warrants issued on conversion |
|
|
( |
)% |
|
|
— |
% |
Income Tax Provision/(Benefit) |
|
|
% |
|
|
( |
)% |
|
The effective income tax rate is based upon the income for the year, the composition of the income in Korea, and adjustments, if any, for the potential tax consequences, benefits or resolutions of audits or other tax contingencies. The corporate income tax rate in Korea is more than our income tax rate in the United States. Our effective tax rate for the fiscal years 2022 and 2021 differed from the U.S. Federal statutory rate of
Deferred income taxes reflect the net tax effects of (a) temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes, and (b) operating losses and tax credit carryforwards. Significant components of deferred tax assets (liabilities) at December 31, 2022 and 2021 are as follows:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Deferred tax assets |
|
|
|
|
|
|
||
Federal Net Operating Loss |
|
|
|
|
$ |
|
||
State Net Operating Loss |
|
|
|
|
|
|
||
Foreign Net Operating Loss |
|
|
|
|
|
|
||
Foreign Tax Credits |
|
|
|
|
|
|
||
Foreign Accruals |
|
|
|
|
|
|
||
Accruals |
|
|
|
|
|
|
||
Capitalized Start up Costs |
|
|
|
|
|
|
||
Capitalized Section 174 R&D |
|
|
|
|
|
|
||
Right of Use Operating Lease ASC 842 |
|
|
|
|
|
|
||
Total deferred tax assets |
|
|
|
|
|
|
||
Deferred tax liabilities |
|
|
|
|
|
|
||
Right of Use Operating Lease ASC 842 |
|
|
( |
) |
|
|
|
|
Depreciation |
|
|
( |
) |
|
|
( |
) |
Total deferred tax liabilities |
|
|
( |
) |
|
|
( |
) |
Total net deferred tax assets |
|
|
|
|
|
|
||
Less: valuation allowance |
|
|
( |
) |
|
|
( |
) |
Net deferred tax assets |
|
|
|
|
$ |
( |
) |
|
F-33
Deferred income taxes reflect future tax effects of temporary differences between the tax and financial reporting basis of the Corporation’s assets and liabilities measured using enacted tax laws and statutory tax rates applicable to the periods when the temporary differences will affect taxable income. When necessary, deferred tax assets are reduced by a valuation allowance, if based on the weight of available positive and negative evidence, it is more likely than not that some portion or all the deferred tax assets will not be realized. As of December 31, 2022 and 2021, the Company has $
The Company decreased its valuation allowance by $
At December 31, 2022, the Company has U.S net operating losses (“NOL”) carryforwards of $
The Korean NOLs are historical NOLs generated in years prior to the acquisition that stay with the corporate entity. NOLs generated prior to 2020 have a
Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, if a corporation undergoes an "ownership change," the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income and taxes may be limited. In general, an "ownership change" generally occurs if there is a cumulative change in the Company’s ownership by "5-percent shareholders" that exceeds 50 percentage points over a rolling three-year period. The Company had an ownership change within the meaning of IRC Sec 382 in May of 2022. The Company has not performed an analysis to determine the annual limitation of the use of the U.S. NOLs going forward.
As of December 31, 2022, we have not provided taxes on undistributed earnings of our foreign subsidiaries, which may be subject to foreign withholding taxes upon repatriation, as we consider these earnings indefinitely reinvested. Our indefinite reinvestment determination is based on the future operational and capital requirements of our domestic and foreign operations. We expect our international cash and cash equivalents and marketable securities will continue to be used for our foreign operations and therefore do not anticipate repatriating these funds. As of December 31, 2022, it is not practical to calculate the unrecognized deferred tax liability on these earnings due to the complexities of the utilization of foreign tax credits and other tax assets.
Uncertain Tax Positions
ASC 740 prescribes the accounting for uncertainty in income taxes recognized in the financial statements. We regularly assess the outcome of potential examinations in each of the taxing jurisdictions when determining the adequacy of the amount of unrecognized tax benefit recorded. We recognize tax benefits from uncertain tax positions only if it more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such positions are then measured based on the largest benefit which is more likely than not to be realized upon ultimate settlement. As of December 31, 2022 the company has
The Company files income tax returns in the U.S., Korea and various state jurisdictions. The Company is not currently under audit for the open years through in the U.S. federal and state tax jurisdictions and in Korea. Carryforward attributes that were generated in earlier periods remain subject to examination to the extent the year in which they were used or will be used remains open for examination.
We have concluded that no subsequent events have occurred that require disclosure, except for those referenced below and as disclosed in Note 11 and Note 13 to the consolidated financial statements.
Leases
In October 2021, the Company entered into a lease for laboratory and office facilities in Palo Alto, California that expires in April 2027. In March 2023, the Company vacated the premises and returned possession of the premises to the landlord in April 2023. The Company is still responsible for the outstanding payments under the lease.
Debt
In March 2023, the Company received proceeds from a loan in the amount of $
In April 2023, the Company entered into separate subscription agreements (the “2023 Convertible Note and Warrant Subscription Agreements”) for the purchase of convertible promissory notes in the aggregate principal amount of $
F-34
“2023 Convertible Notes”) and an aggregate amount of
In April 2023, the Company entered into a subscription agreement with its founder and director to settle $
Departure of Directors; Appointment of New Audit Committee Chair
On March 14, 2023, Brad Stevens notified the Company of his resignation from the Company’s Board of Directors (the “Board”), effective immediately. On June 21, 2023, Nevan Elam notified the Company of his resignation from the Company’s Board, effective immediately. On June 22, 2023, the Board appointed Jim Neal as the chair of the Audit Committee and the Board appointed David Rosenberg as a member of the Audit Committee.
Issuance of Unregistered Securities
As previously disclosed, in April 2023 the Company issued to its founder and director warrants to purchase
F-35
