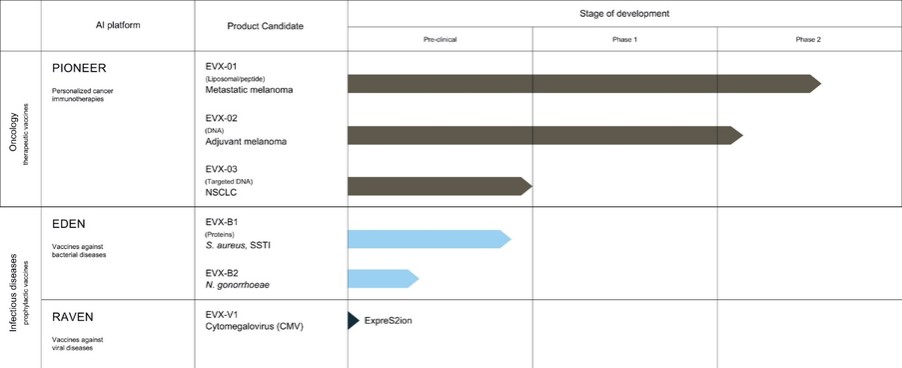
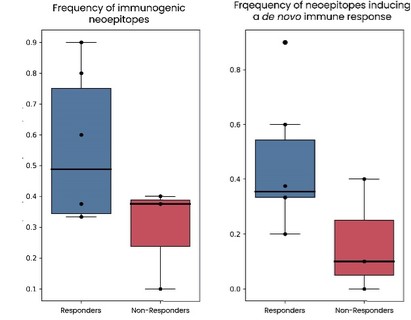
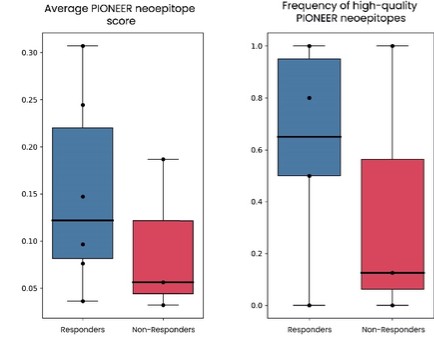
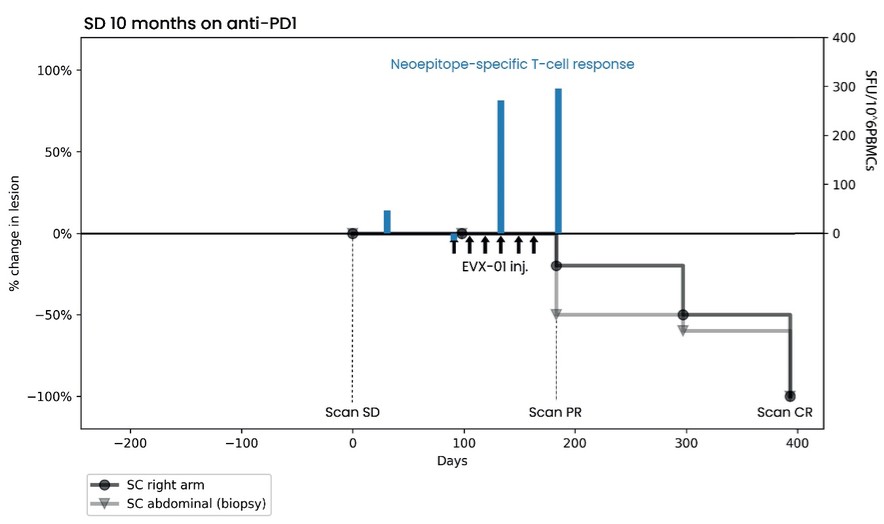
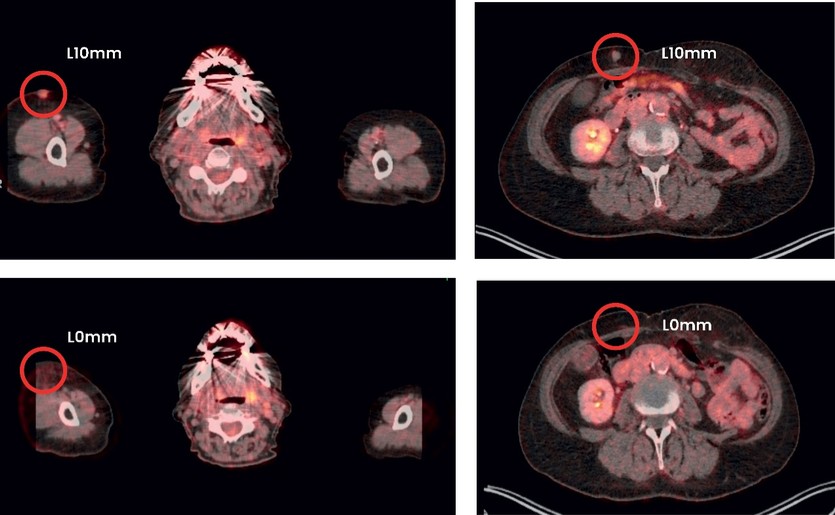
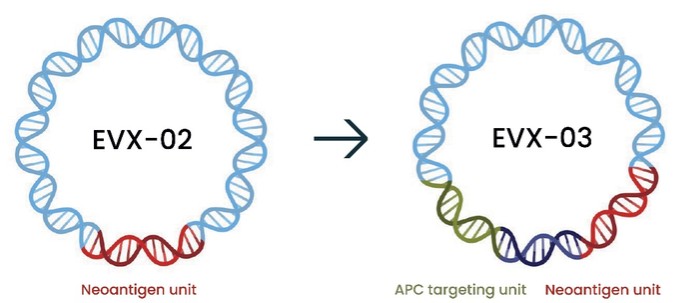
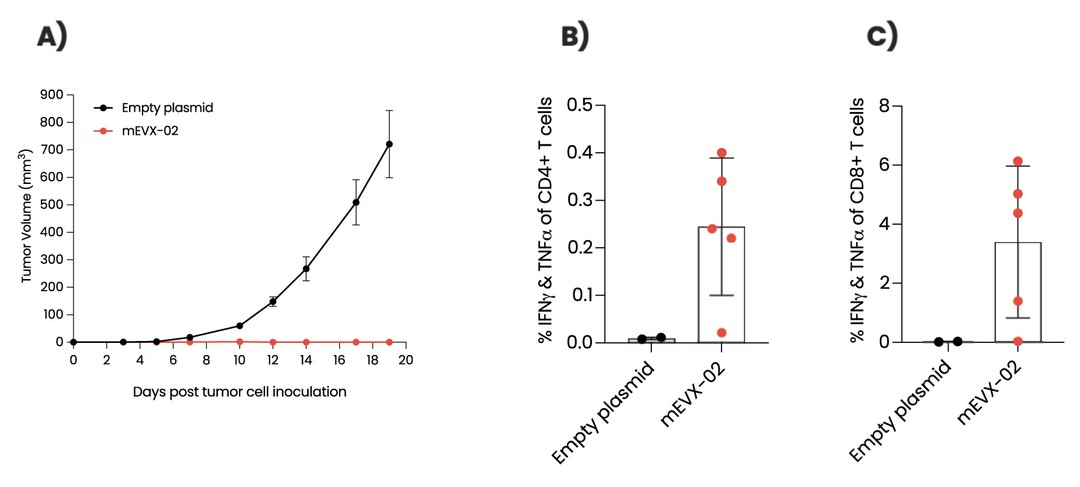
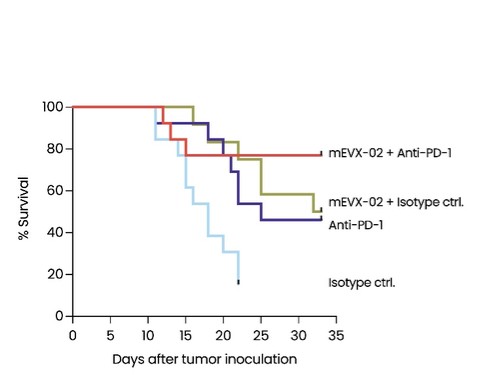
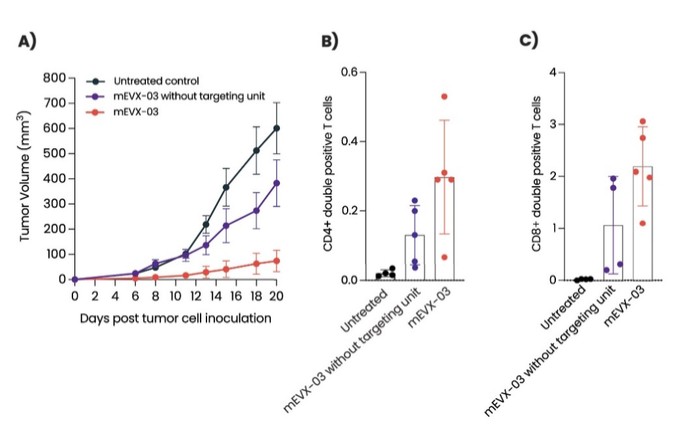
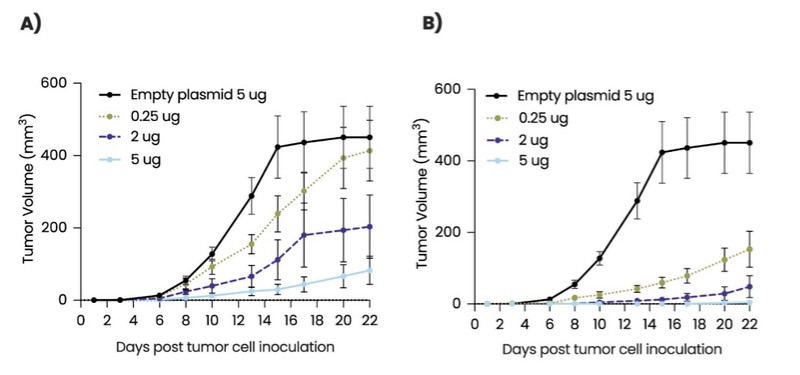
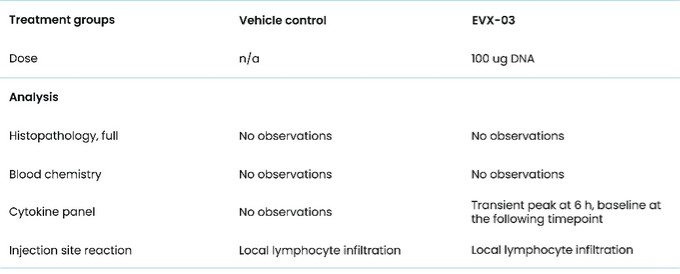
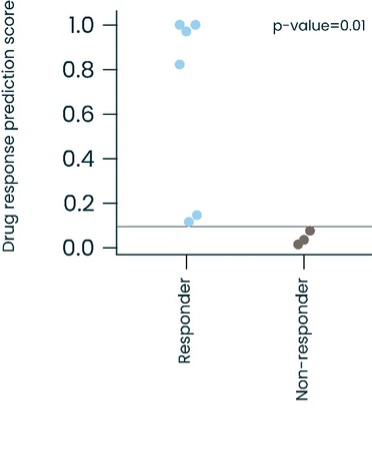
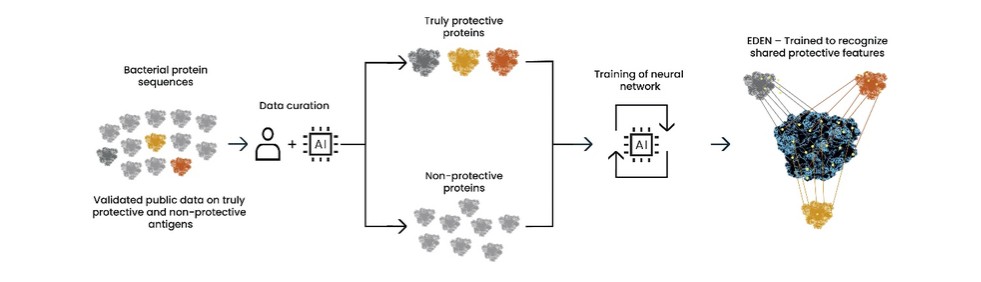
As of December 31 2022, there were 2,743,093 warrants outstanding. If these warrants are exercised then an additional 2, 743,093 ordinary shares will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act. If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of the ADSs could decline. Any sales of securities by these security holders could have a negative effect on the trading price of our ordinary shares and ADSs. In addition, as of December 31, 2022, there were 351,036 warrants issued to EIB under the loan agreement with EIB, which are expected to be cash settled. For a more detailed description of the EIB warrants see the section herein entitled “Our EIB warrants.”
Additionally, on November 28, 2021, we entered into a Share Sale and Restriction Agreement with, Dr. Lars Staal Wegner, our former Chief Executive Officer, Dr. Niels Iversen Møller our Co-Founder, and current member of our Board of Directors, and Andreas Mattsson, our Co-Founder and Chief AI and Culture Officer, pursuant to which Dr. Wegner agreed to exercise 211,849 warrants in each of the two week exercise windows established under our Articles of Association that are expected to open two trading days following publication of our annual report and interim quarterly financial reports in March 2022, May 2022, August 2022 and November 2022, respectively.
Under the terms of this agreement, Dr. Wegner, Dr. Møller and Mr. Mattsson further agreed with us that in the corresponding open trading window related to each such exercise consisting of the four-week period commencing on the third full trading day after the date of publication of our annual report or interim financial reports in March 2022, May 2022, August 2022 and November 2022, each a Trading Window, Dr. Wegner would sell such Ordinary Shares and Dr. Møller and Mr. Mattsson will purchase such ordinary shares, with each of Dr. Møller and Mr. Mattsson purchasing fifty per cent (50%) of such shares, at a purchase price per share equal to the Volume Weighted Average Price, or VWAP, of our ADSs at the close of the market on the date of exercise as reported on Nasdaq.
Under the terms of the agreement, Dr. Møller and Mr. Mattsson agreed that during each Trading Window each of them will sell 328,731 ADSs representing ordinary shares at a price equal to the prevailing market price thereof on the date of such sale as reported on Nasdaq. Furthermore, pursuant the terms of the agreement, Dr. Møller and Mr. Mattsson are required to sell such shares and are prohibited from exercising any subsequent influence over how, when, or whether to affect the trade(s). As of December 31, 2022, due to market conditions, Dr. Møller and Mr. Mattsson had only sold 43,196 of such ADSs representing ordinary shares, thereby, leaving a total of 285,535 ADSs subject to future sale under this arrangement.
In addition, on June 7, 2022, we entered into the LPC Purchase Agreement pursuant to which we issued 428,572 commitment shares represented by ADS’ (“Commitment Shares”) to Lincoln Park as consideration for a commitment fee of $1,200,000 for Lincoln Park’s agreement to purchase ordinary shares represented by ADSs under the LPC Purchase Agreement. As of the date of this Form 20-F we have not issued any additional ordinary shares represented by ADSs to Lincoln Park. In accordance with the terms of the LPC Purchase Agreement, we filed a selling shareholder Form F-1 Registration Statement with the SEC on July 7, 2002, which was declared effective by the SEC on August 26, 2022 registering the potential future sale by Lincoln Park of up to 4,649,250 ADSs represented ordinary shares inclusive of the 428,572 Commitment Shares. As of the date of this Form 20-F, Lincoln Park has only sold an aggregate of 2,000 of such Commitment Shares thereby leaving 426,572 of such Commitment Shares available for sale.
Additionally, on October 3, 2022, we entered into a Capital on DemandTM Sales Agreement, or the Sales Agreement, with JonesTrading Institutional Services LLC,or JonesTrading, pursuant to which we may sell from time to time, at our option, ADSs representing ordinary shares through or to JonesTrading, as sales agent or principal. The ADSs are offered pursuant to a prospectus supplement, dated October 3, 2022, or the Prospectus Supplement, which was filed with the SEC on such date and our Form F-3 (Registration No. 333-265132) shelf registration statement filed with the SEC on May 20, 2022 and declared effective by the SEC on June 3, 2022. Pursuant to the Prospectus Supplement, we may offer and sell up to an aggregate of $14,439,000 of ADSs. Sales of the ADSs made pursuant to the Sales Agreement, are made by any method deemed to be an “at the market offering”, or ATM, as defined in Rule 415(a)(4) promulgated under the Securities Act. JonesTrading is not required to sell any specific number or dollar amount of ADSs, but has agreed to use its commercially reasonable efforts to sell the ADSs from time to time, based upon our instructions, including any price, time or size limits or other customary parameters or conditions we may impose. As of the date of this Form 20-F, we have sold a total of 2,417,446 ADSs under this ATM program for an aggregate purchase price of $4,713,910, thereby leaving up to an aggregate $3 million of ADSs available for future sale under this ATM program, depending in the development in the share price.
Sales of ADSs or our ordinary shares as restrictions end or pursuant to the above described agreements or pursuant to registration rights may make it more difficult for us to finance our operations through the sale of equity securities in the future at a time and at a price that we deem appropriate. These sales also could cause the trading price of the ADSs to fall and make it more difficult for holders of ADSs to sell the ADSs.