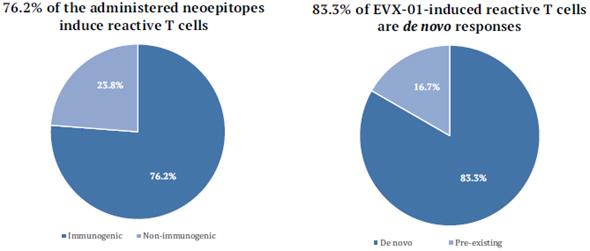
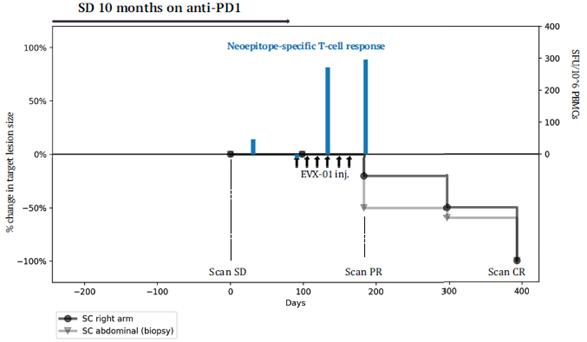
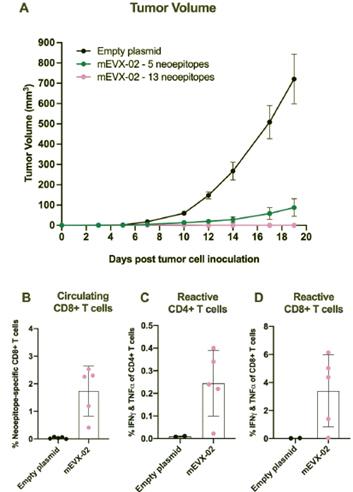
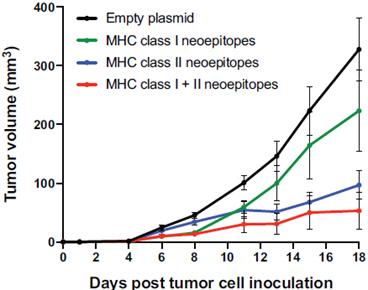
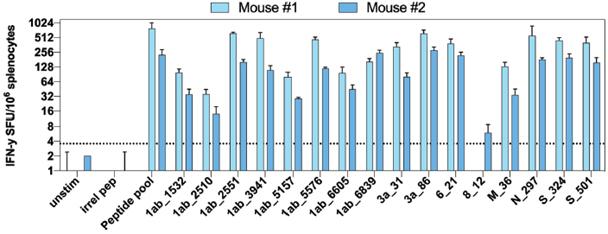
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the United States Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion October 26, 2021
5,167,958 American Depositary Shares

Representing 5,167,958 Ordinary Shares
We are offering 5,167,958 ADSs, with each ADS representing one ordinary share DKK 1 nominal value.
Our ADSs representing our ordinary shares are listed on The Nasdaq Capital Market, or Nasdaq, under the symbol “EVAX.” On October 22, 2021, the last reported closing trading price of the ADSs, on Nasdaq, was $7.74 per ADS. The actual offering price will be as determined between us and the underwriters at the time of pricing, and may be at a discount to the current market price. Accordingly, the recent market price used throughout this prospectus may not be indicative of the actual public offering price.
Investing in the ADSs involves a high degree of risk. See “Risk Factors” beginning on page 18 of this prospectus for a discussion of information that should be considered in connection with an investment in the ADSs.
| PER ADS |
| TOTAL |
| |||
Public offering price | $ | $ | |||||
Underwriting discounts and commissions(1) | $ | $ | |||||
Proceeds to us before expenses | $ | $ | |||||
| (1) | We have agreed to reimburse the underwriters for certain expenses incurred in the offering. See “Underwriting” for details. We refer you to “Underwriting” beginning on page for additional information regarding underwriting compensation. |
We have granted the underwriters an option to purchase up to an additional 775,193 ADSs from us at the public offering price, less underwriting discounts, within 30 days of the date of this prospectus.
We are an “emerging growth company” and a “foreign private issuer” as defined under the U.S. federal securities laws and, as such, will be eligible for reduced public company disclosure requirements. See “Prospectus Summary – Implications of Being an Emerging Growth Company and a Foreign Private Issuer” for additional information.
None of the Securities and Exchange Commission, any state securities commission, the Danish Financial Supervisory Authority, nor any other foreign securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Delivery of the ordinary shares and the ADSs is expected to be made on or about , 2021.
Joint Book-Running Managers | ||
Oppenheimer & Co. | Raymond James | |
Lead-Manager | ||
Ladenburg Thalmann | ||
Co-Manager | ||
Lake Street | ||
Prospectus dated , 2021 |