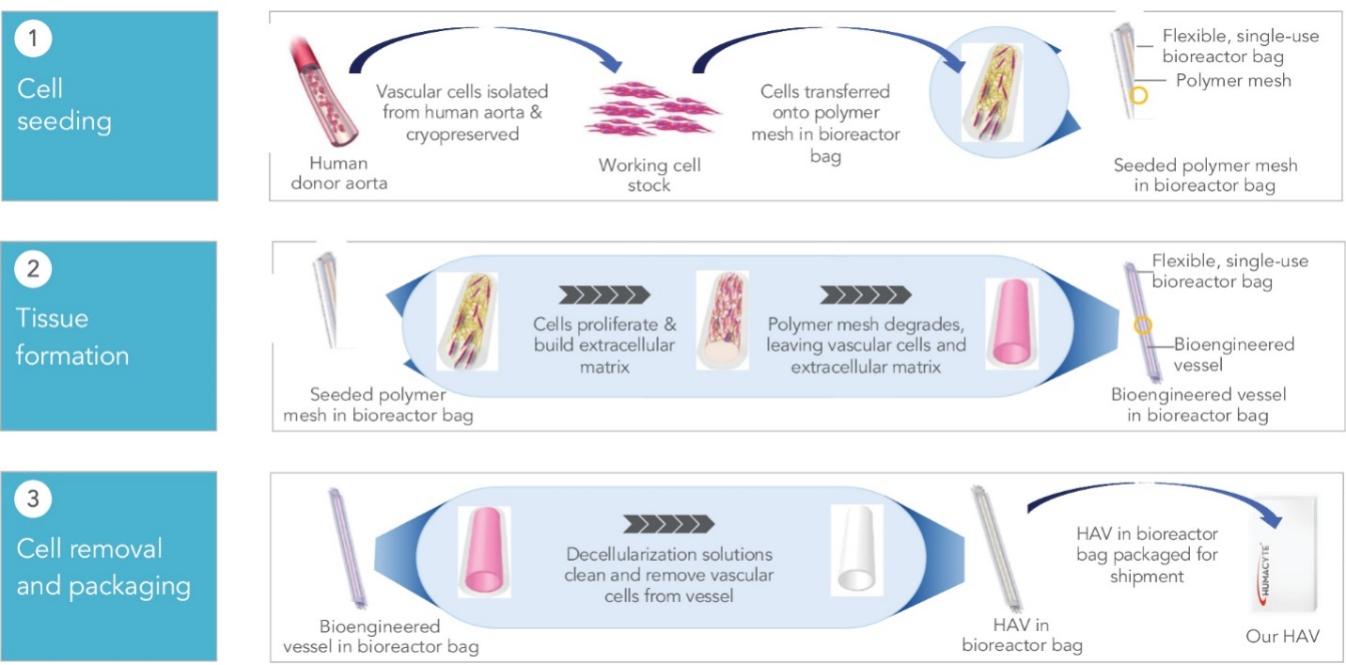
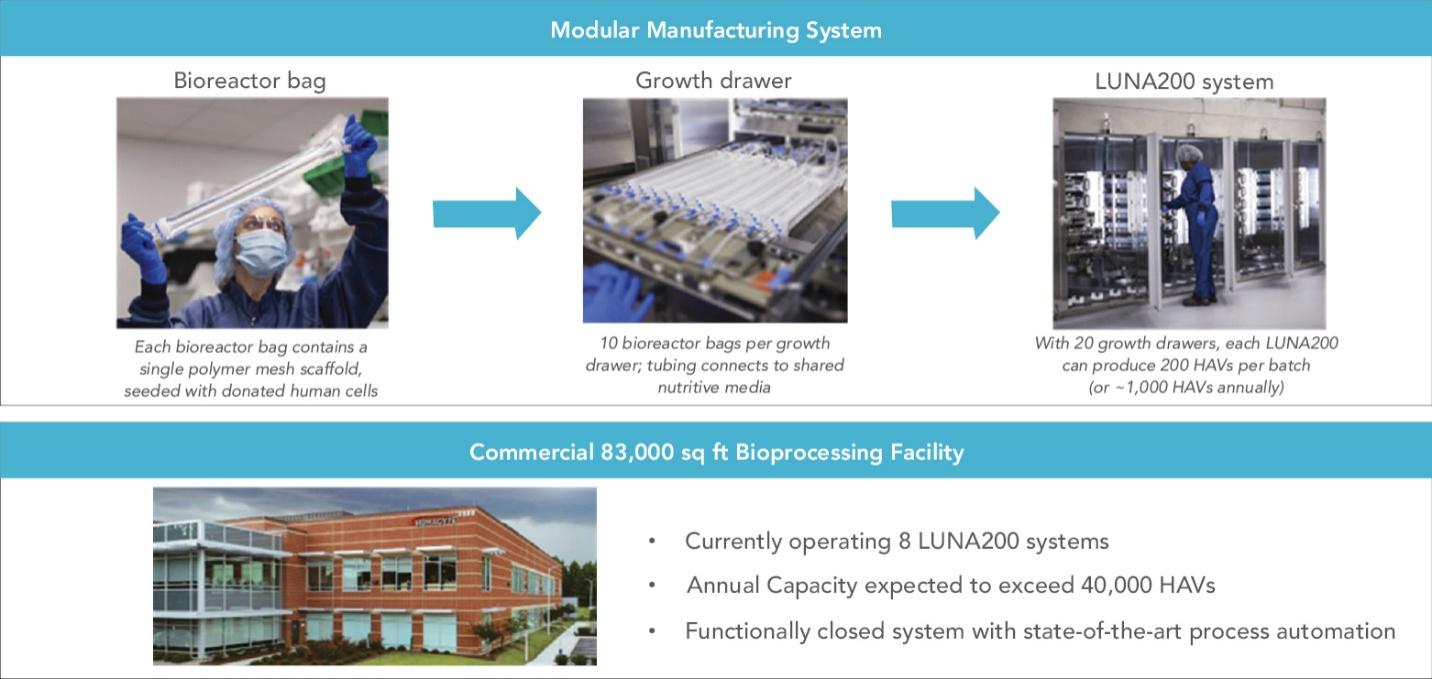
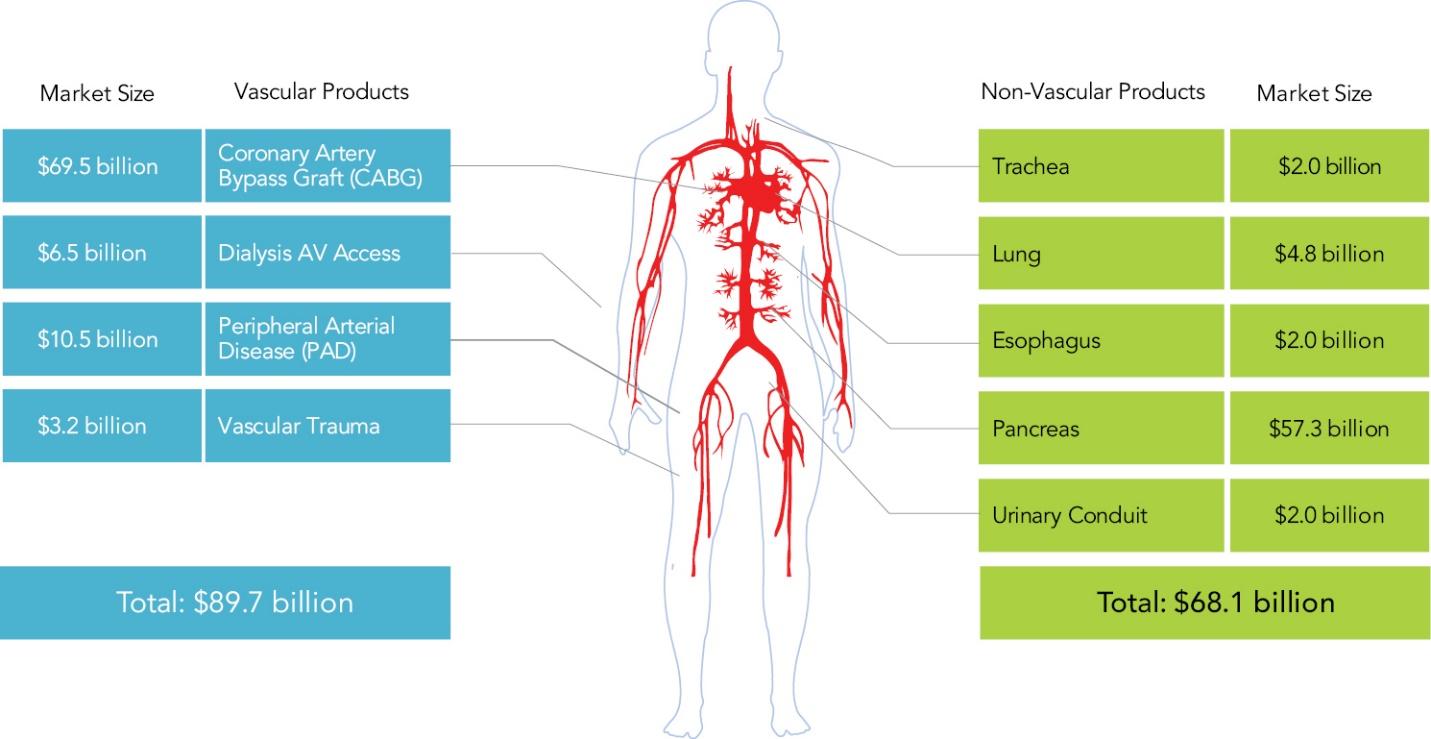
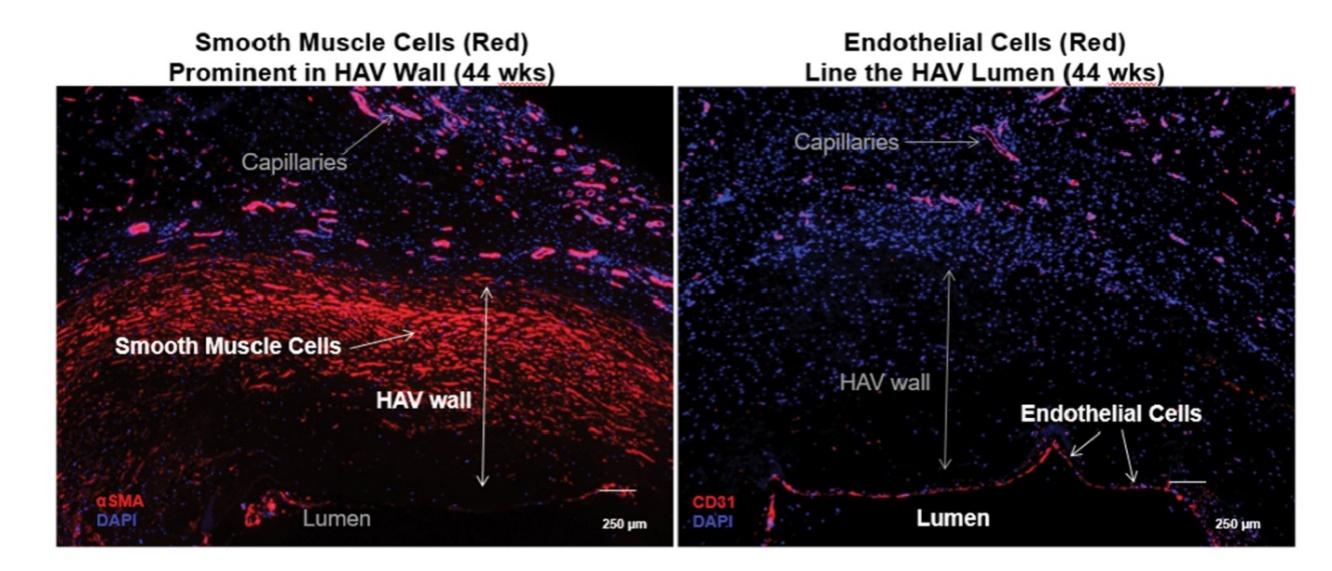
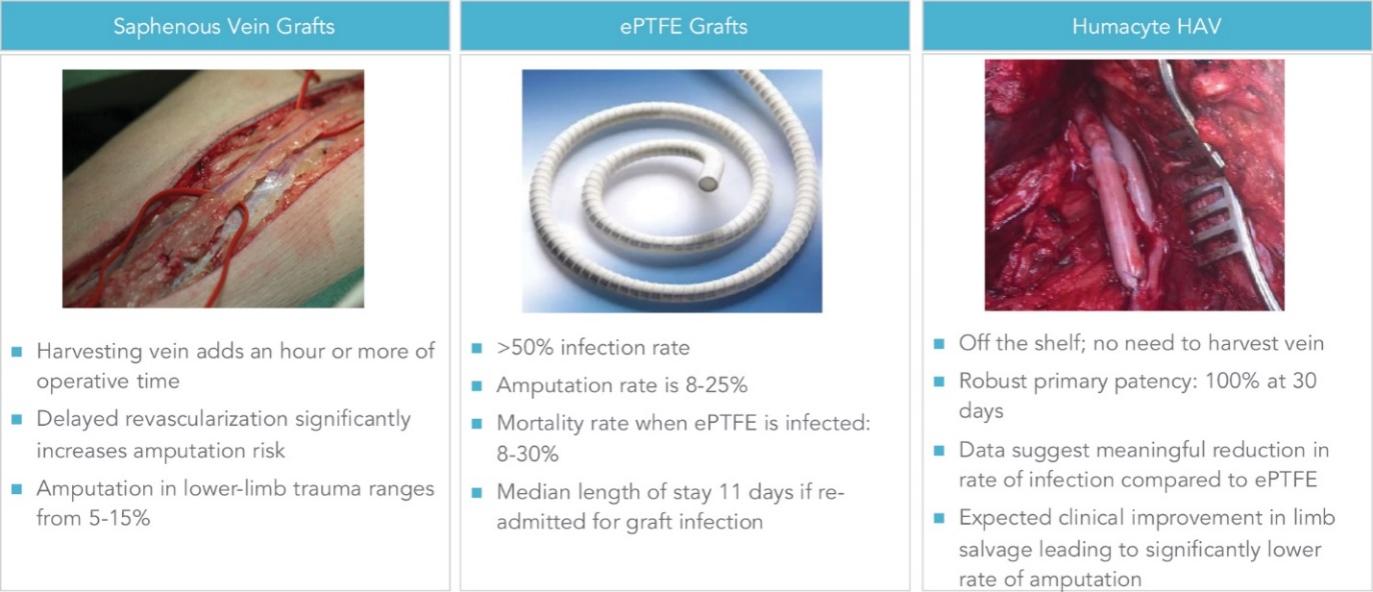
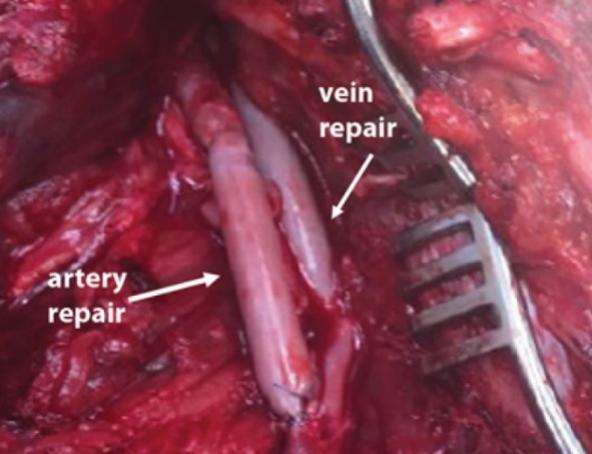
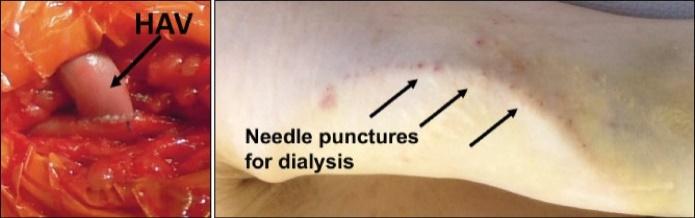
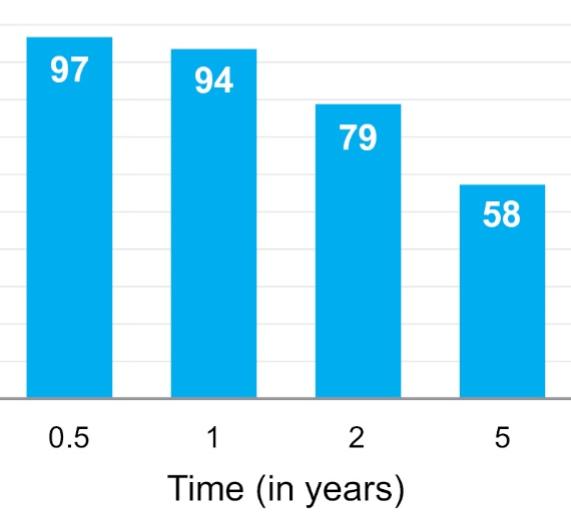
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is declared effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED SEPTEMBER 17, 2021
PRELIMINARY PROSPECTUS

Up to 90,069,340 Shares of Common Stock
Up to 5,177,500 Shares of Common Stock Issuable Upon Exercise of Warrants
This prospectus relates to the issuance by us of an aggregate of up to 5,177,500 shares of our common stock, $0.0001 par value per share (the “common stock”), which consists of (i) up to 177,500 shares of common stock that are issuable upon the exercise of private placement warrants (the “Private Placement Warrants”) originally issued in a private placement to AHAC Sponsor LLC (the “Sponsor”), Oppenheimer & Co. Inc. (“Oppenheimer”) and Northland Securities, Inc. (“Northland”), in connection with the initial public offering of Alpha Healthcare Acquisition Corp. (“AHAC”), and (ii) up to 5,000,000 shares of common stock that are issuable upon the exercise of public warrants (the “Public Warrants” and, together with the Private Warrants, the “Warrants”). We will receive the proceeds from the exercise of any Warrants for cash.
This prospectus also relates to the offer and sale from time to time by the selling stockholders named in this prospectus or their permitted transferees (the “selling stockholders”) of up to 90,069,340 shares of common stock, which consists of (i) up to 17,500,000 shares of common stock issued on August 26, 2021 in a private placement pursuant to subscription agreements, dated February 17, 2021 (the “PIPE Shares”), (ii) up to 71,759,179 shares of common stock, required to be registered pursuant to that certain Investor Rights and Lock-up Agreement, dated August 26, 2021 (the “Investor Rights and Lock-up Agreement”), by and among us and certain of the selling stockholders and (iii) up to 810,161 shares of common stock purchased by a selling stockholder. We will not receive any proceeds from the sale of shares of common stock by the selling stockholders pursuant to this prospectus.
The selling stockholders may offer, sell or distribute all or a portion of the securities hereby registered publicly or through private transactions at prevailing market prices or at negotiated prices. We will not receive any of the proceeds from such sales of the shares of common stock, but we will receive the proceeds from the exercise of any Warrants for cash. We will bear all costs, expenses and fees in connection with the registration of these securities, including with regard to compliance with state securities or “blue sky” laws. The selling stockholders will bear all commissions and discounts, if any, attributable to their sale of shares of common stock. See the section titled “Plan of Distribution.”
Our common stock is listed on the Nasdaq Global Select Market (“Nasdaq”) under the symbol “HUMA.” On September 16, 2021, the last reported sales price of our common stock was $14.21 per share.
We are an “emerging growth company” and a “smaller reporting company” as defined under the U.S. federal securities laws. See “Prospectus Summary — Implications of Being an Emerging Growth Company and a Smaller Reporting Company.” This prospectus complies with the requirements that apply to an issuer that is an emerging growth company and a smaller reporting company.
Investing in our common stock involves risks. See “Risk Factors” beginning on page 4 of this prospectus to read about factors you should consider before investing in our common stock.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2021.