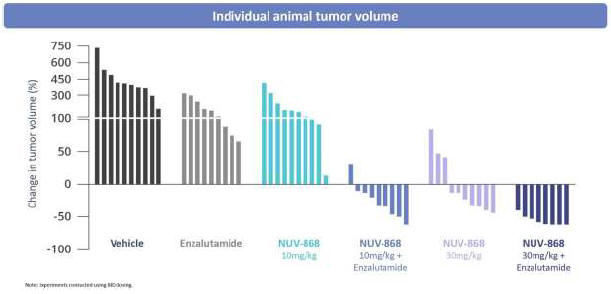
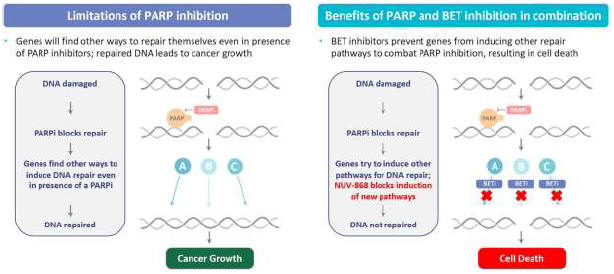
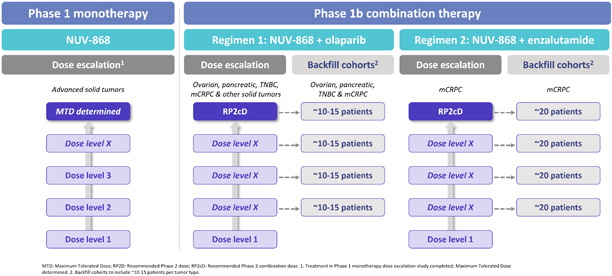
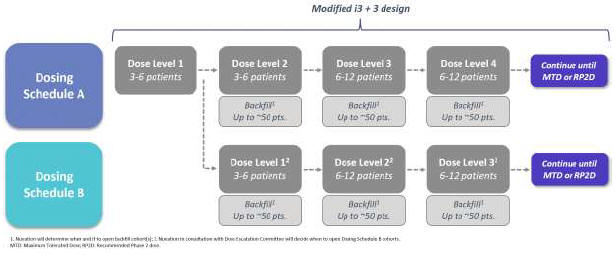
Introductory Note
This Amendment No. 1 on Form
8-K/A
(this “Amendment No. 1”) includes an additional Item 8.01 and amends Item 9.01 of the Current Report on Form 8-K
filed by Nuvation Bio Inc. (the “Company”) on April 10, 2024 (the “Original Report”), in which the Company reported, among other events, the completion of the acquisition of AnHeart. This Amendment No. 1 (i) includes an updated description of the Company’s business reflecting this acquisition and (ii) amends the financial statements provided under Items 9.01(a) and 9.01(b) in the Original Report to include (a) the audited consolidated financial statements of AnHeart for the years ended December 31, 2023 and 2022, (b) the unaudited condensed consolidated financial statements of AnHeart for the quarterly periods ended March 31, 2024 and 2023 and (c) the unaudited pro forma condensed combined financial information of the Company as of and for the year ended December 31, 2023 and the quarterly period ended March 31, 2024 . This Amendment No. 1 does not amend any other item of the Original Report or purport to provide an update or a discussion of any developments at the Company subsequent to the filing date of the Original Report. Capitalized terms used but not defined herein have the meanings given in the O
rig
inal Report. Item 8.01. |
Other Events. |
The description of the Company’s business set forth in Item 1 of its Annual Report on Form
10-K
filed on Februar
y 29, 2024 (the “Form 10-K”)
does not reflect the completion of the acquisition of AnHeart on April 9, 2024. The following description of the Company’s business has been updated to reflect the completion of this acquisition and supersedes in its entirety Item 1 set forth in the Form 10-K.
The Company’s risk factor disclosure set forth in its Quarterly Report on Form 10-Q
filed on May 14, 2024 was updated to reflect the acquisition of AnHeart and supersedes in its entirety the risk factor disclosure set forth in Item 1A of the Form 10-K.
Description of Our Business.
Business Combination of Panacea Acquisition Corp. and Nuvation Bio Inc.
On February 10, 2021, (the “Closing Date”), Nuvation Bio Inc., a Delaware corporation (“Legacy Nuvation Bio”), Panacea Acquisition Corp. (“Panacea”), and Panacea Merger Subsidiary Corp, a Delaware corporation and a direct, wholly owned subsidiary of Panacea (“
Merger
Sub”) consummated the transactions contemplated by an Agreement and Plan of Merger among them dated October 20, 2020 (“Merger Agreement”). Pursuant to the terms of the Merger Agreement, a business combination of Panacea and Legacy Nuvation Bio was effected through the merger of Merger Sub with and into Legacy Nuvation Bio, with Legacy Nuvation Bio surviving as a wholly owned subsidiary of Panacea (the “Merger”). On the Closing Date, Legacy Nuvation Bio changed its name to Nuvation Bio Operating Company Inc. and Panacea changed its name to Nuvation Bio Inc. (the “Company” or “Nuvation Bio”).
In connection with the closing of the Merger, our Class A common stock and warrants to purchase shares of our Class A common stock began trading on The New York Stock Exchange under the symbols “NUVB” and “NUVB.WS,” respectively, on February 11, 2021. The following disclosure gives effect to the Merger and includes the operations of Legacy Nuvation Bio prior to the Merger.
Business Overview
We are a late clinical-stage, global biopharmaceutical company tackling some of the greatest unmet needs in oncology by developing differentiated and novel product candidates. We were founded in 2018 by our chief executive officer, David Hung, M.D., who founded Medivation, Inc. and led its successful development of oncology drugs Xtandi® and talazoparib (now marketed as Talzenna®), leading to its $14.3 billion sale to Pfizer Inc. (“Pfizer”) in 2016.
We leverage our team’s extensive expertise in medicinal chemistry, preclinical development, drug development, business development, manufacturing, and commercialization to pursue oncology targets validated by strong clinical or preclinical data and develop novel small molecules that improve the activity and overcome the liabilities of currently marketed drugs.
The foundations of our approach include:
| • | The pursuit of validated targets FDA-approved drugs, and we then attempt to design or acquire novel or potential best-in-class |
| • | Innovative medicinal chemistry expertise: |
| • | Human capital management |
The following table summarizes our product candidate pipeline:
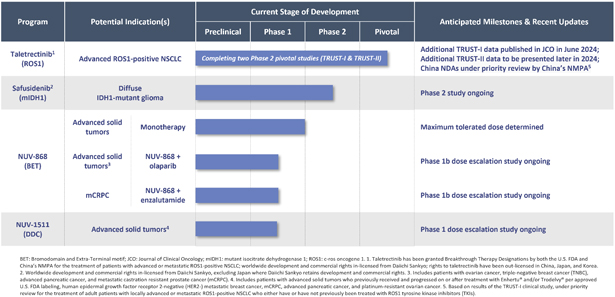
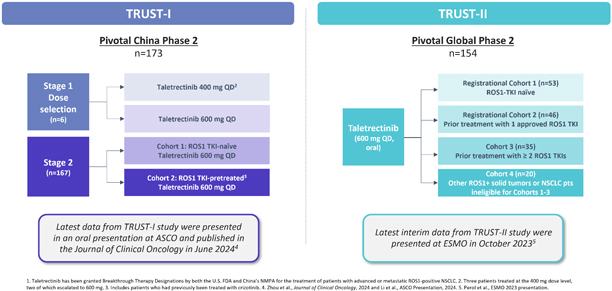
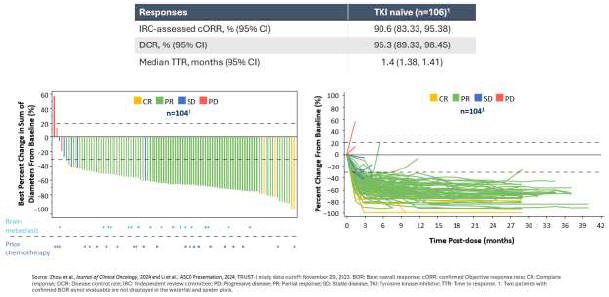
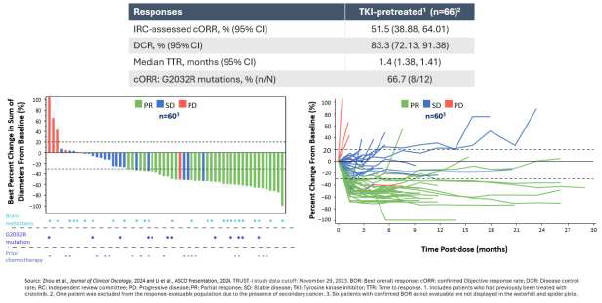
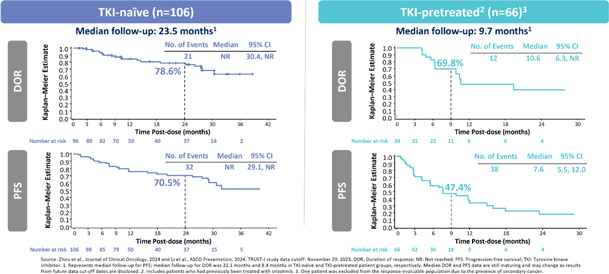
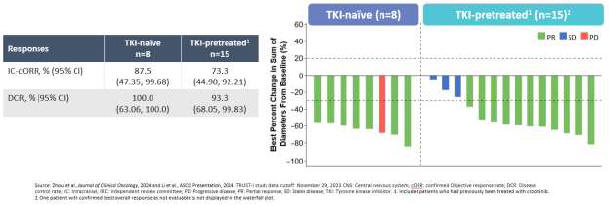
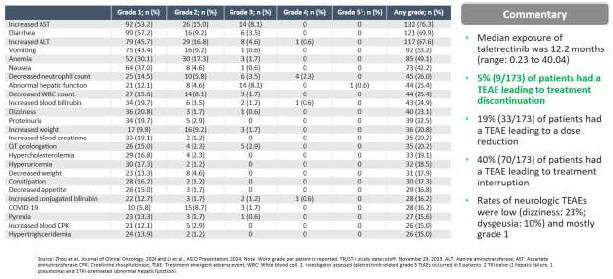
As a result of our acquisition of AnHeart Therapeutics Ltd. (“AnHeart”), which closed in April 2024, our most advanced clinical-stage product candidate is taletrectinib, an oral, potent, central nervous system-active, selective, next-generation
c-ros
oncogene 1 (“ROS1”) inhibitor. Taletrectinib is being evaluated for the treatment of patients with advanced ROS1-positive non-small
cell lung cancer (“NSCLC”) in two Phase 2 single-arm
pivotal studies: TRUST-I
in China, and TRUST-II,
a global study. Taletrectinib has been granted Breakthrough Therapy Designations by both the U.S. Food and Drug Administration (“FDA”) and China’s National Medical Products Administration (“NMPA”) for the treatment of patients with advanced or metastatic ROS1-positive NSCLC. Based on results of the TRUST-I
clinical study, China’s NMPA has accepted and granted Priority Review Designations to New Drug Applications for taletrectinib for the treatment of adult patients with locally advanced or metastatic ROS1-positive NSCLC who either have or have not previously been treated with ROS1 tyrosine kinase inhibitors (“TKIs”).