PART I
ABOUT THIS ANNUAL REPORT
Unless the context otherwise requires, the terms “Benitec,” the “Company,” “we,” “us,” “our” and similar terms used in this Annual Report on Form 10-K refer (i), prior to the Re-domiciliation (as defined herein) to Benitec Biopharma Limited (BBL), an Australian corporation, and its subsidiaries, and (ii), following the Re-domiciliation, to Benitec Biopharma Inc., a Delaware corporation, and its subsidiaries (including Benitec Limited). Any references to “Benitec Limited” or “BBL” refer to Benitec Biopharma Limited, an Australian corporation. On August 14, 2020, BBL reorganized as a Proprietary Limited company and changed its name to Benitec Biopharma Proprietary Limited.
All references to “$” in this Annual Report refer to U.S. dollars. All references to “A$” in this Annual Report mean Australian dollars. As of June 30, 2022, the rate of exchange of U.S. dollars to Australian dollars was 1.4512 AUD.
Our fiscal year-end is June 30. References to a particular “fiscal year” are to our fiscal year ended June 30 of that calendar year.
INDUSTRY AND MARKET DATA
This Annual Report includes information with respect to market and industry conditions and market share from third-party sources or based upon estimates using such sources when available. We believe that such information and estimates are reasonable and reliable. We also believe the information extracted from publications of third- party sources has been accurately reproduced. However, we have not independently verified any of the data from third-party sources. Similarly, our internal research is based upon our understanding of industry conditions, and such information has not been verified by any independent sources.
TRADEMARKS AND TRADENAMES
We have proprietary and licensed rights to trademarks used in this Annual Report which are important to our business, many of which are registered under applicable intellectual property laws. Our trademarks include:
| • | BENITEC BIOPHARMA ® |
| • | BENITEC ® |
| • | GIVING DISEASE THE SILENT TREATMENT ® |
| • | SILENCING GENES FOR LIFE ® |
Solely for convenience, trademarks and trade names referred to in this Annual Report appear without the “
®
” or “™” symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent possible under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, any other companies. Each trademark, trade name or service mark of any other company appearing in this Annual Report is the property of its respective holder. 1
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report contains forward-looking statements that are subject to a number of risks and uncertainties, many of which are beyond our control. All statements, other than statements of historical fact included in this Annual Report, regarding our strategy, future operations, financial position, projected costs, prospects, plans and objectives of management are forward-looking statements. When used in this Annual Report, the words “could,” “believe,” “anticipate,” “intend,” “estimate,” “expect,” “may,” “continue,” “predict,” “potential,” “project,” or the negative of these terms, and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain such identifying words. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. These risks, uncertainties and factors include:
| • | the success of our plans to develop and potentially commercialize our product candidates; |
| • | the timing of the initiation and completion of preclinical studies and clinical trials; |
| • | the timing and sufficiency of patient enrollment and dosing in any future clinical trials; |
| • | the timing of the availability of data from clinical trials; |
| • | the timing and outcome of regulatory filings and approvals; |
| • | unanticipated delays; |
| • | sales, marketing, manufacturing and distribution requirements; |
| • | market competition and the acceptance of our products in the marketplace; |
| • | regulatory developments in the United States, France and Canada; |
| • | the development of novel AAV vectors; |
| • | the plans of licensees of our technology; |
| • | the clinical utility and potential attributes and benefits of ddRNAi and our product candidates, including the potential duration of treatment effects and the potential for a “one shot” cure; |
| • | our dependence on our relationships with collaborators and other third parties; |
| • | expenses, ongoing losses, future revenue, capital needs and needs for additional financing, and our ability to access additional financing given market conditions and other factors, including our capital structure; |
| • | our ability to continue as a going concern; |
| • | the length of time over which we expect our cash and cash equivalents to be sufficient to execute on our business plan; |
| • | our intellectual property position and the duration of our patent portfolio; |
| • | the impact of local, regional, and national and international economic conditions and events; and |
| • | the impact of the current COVID-19 pandemic, the disease caused by the SARS-CoV-2 virus, which may adversely impact our business and preclinical and future clinical trials; |
as well as other risks detailed under the caption “Risk Factors” in this Annual Report and in other reports filed with the SEC. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Annual Report, we caution you that these statements are based on a combination of facts and important factors currently known by us and our expectations of the future, about which we cannot be certain.
We have based the forward-looking statements included in this Annual Report on information available to us on the date of this Annual Report or on the date thereof. Except as required by law we undertake no obligation to revise or update any forward-looking statements, whether as a result of new information, future events or
2
otherwise. You are advised to consult any additional disclosures that we may make directly to you or through reports that we, in the future, may file with the SEC, including annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K.
All forward-looking statements included herein or in documents incorporated herein by reference are expressly qualified in their entirety by the cautionary statements contained or referred to elsewhere in this Annual Report.
Item 1. |
Business. |
Company Overview
We endeavor to become the leader in discovery, development, and commercialization of therapeutic agents capable of addressing significant unmet medical need via the application of the silence and replace approach to the treatment of genetic disorders.
Benitec Biopharma Inc. (“Benitec” or the “Company” or in the third person, “we” or “our”) is a development- stage biotechnology company focused on the advancement of novel genetic medicines with headquarters in Hayward, California. The proprietary platform, called DNA-directed RNA interference, or ddRNAi, combines RNA interference, or RNAi, with gene therapy to create medicines that facilitate sustained silencing of disease- causing genes following a single administration. The Company is developing ddRNAi-based therapeutics for chronic and life-threatening clinical indications including Oculopharyngeal Muscular Dystrophy (OPMD), a chronic, life-threatening genetic disorder.
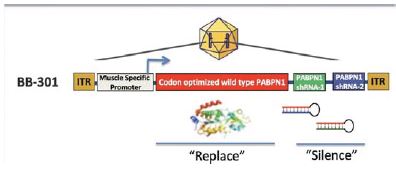
BB-301 is the most advanced ddRNAi-based genetic medicine currently under development by Benitec. BB-301 is an AAV-based gene therapy designed to both silence the expression of mutated, disease-causing genes (to slow, or halt, the underlying mechanism of disease progression) and simultaneously replace the mutant genes with normal, “wild type” genes (to drive restoration of function in diseased cells). This fundamental therapeutic approach to disease management is called “silence and replace.” The silence and replace mechanism offers the potential to restore the normative physiology of diseased cells and tissues and to improve treatment outcomes for patients suffering from the chronic, and potentially fatal, effects of OPMD. BB-301 has been granted Orphan Drug Designation in the United States and the European Union.
The targeted gene silencing effects of RNAi, in conjunction with the durable transgene expression achievable via the use of modified viral vectors, imbues the silence and replace approach with the potential to produce long-term silencing of disease-causing genes along with simultaneous replacement of wild type gene function following a single administration of the proprietary genetic medicine. We believe that this novel mechanistic profile of the current and future investigational agents developed by Benitec could facilitate the achievement of robust and durable clinical activity while greatly reducing the frequency of drug administration traditionally expected for medicines employed for the management of chronic diseases. Additionally, the achievement of long-term gene silencing and gene replacement may significantly reduce the risk of patient non-compliance during the course of medical management of potentially fatal clinical disorders.
We will require additional financing to continue the development of our product candidates through key inflection points and to continue to execute on our business plan. See “Risk Factors—Our auditors’ report expresses doubt about our ability to continue as a going concern.”
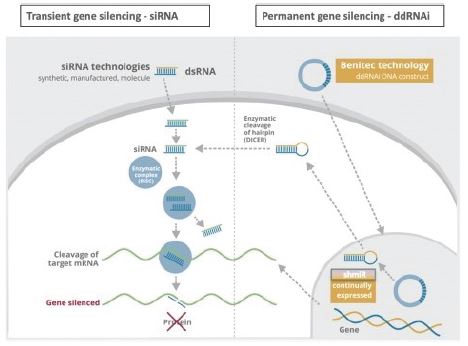
Our proprietary technology platforms are designated as DNA-directed RNA interference, or “ddRNAi”, and “silence and replace”. ddRNAi is designed to produce long-term silencing of disease-causing genes, by combining RNA interference, or RNAi, with viral delivery agents typically associated with the field of gene therapy (i.e., viral vectors). Modified AAV vectors are employed to deliver genetic constructs which encode short hairpin RNAs that are, then, serially expressed and processed to produce siRNA molecules within the transduced cell for the duration of the life of the target cell. These newly introduced siRNA molecules drive
3
long-term, and potentially permanent, silencing of the expression of the disease-causing gene. The silence and replace approach further bolsters the biological benefits of long-term silencing of disease-causing genes by incorporating multifunctional genetic constructs within the modified AAV vectors to create an AAV-based gene therapy agent that is designed to both silence the expression of mutated, disease-causing genes (to slow, or halt, the underlying mechanism of disease progression) and, simultaneously, replace the mutant genes with normal, “wild type” genes (to drive restoration of function in diseased cells). This fundamentally distinct therapeutic approach to disease management offers the potential to restore the underlying physiology of the treated tissues and, in the process, improve treatment outcomes for patients suffering from the chronic and, potentially, fatal effects of diseases like Oculopharyngeal Muscular Dystrophy (OPMD).
Traditional gene therapy is defined by the introduction of an engineered transgene to correct the pathophysiological derangements derived from mutated or malfunctioning genes. Mutated genes can facilitate the intracellular production of disease-causing proteins or hamper the production of critical, life-sustaining, proteins. The introduction of a new transgene can facilitate the restoration of production of normal proteins within the diseased cell, thus, restoring natural biological function. Critically, the implementation of this traditional method of gene therapy cannot eliminate the expression, or the potential deleterious effects of, the underlying mutant gene (as mutant proteins may be continually expressed and aggregate or drive the aggregation of other native proteins within the diseased cell). In this regard, the dual capabilities of the proprietary silence and replace approach to silence a disease-causing gene via ddRNAi and simultaneously replace the wildtype activity of a mutant gene via the delivery of an engineered transgene could facilitate the development of differentially efficacious treatments for a range of genetic disorders.
Overview of RNAi and the siRNA Approach
The mutation of a single gene can cause a chronic disease via the resulting intracellular production of a disease- causing protein (i.e., an abnormal form of the protein of interest), and many chronic and/or fatal disorders are known to result from the inappropriate expression of a single gene or multiple genes. In some cases, genetic disorders of this type can be treated exclusively by “silencing” the intracellular production of the disease-causing protein through well-validated biological approaches like RNA interference (“RNAi”). RNAi employs small nucleic acid molecules to activate an intracellular enzyme complex, and this biological pathway temporarily reduces the production of the disease-causing protein. In the absence of the disease-causing protein, normal cellular function is restored and the chronic disease that initially resulted from the presence of the mutant protein is partially or completely resolved. RNAi is potentially applicable to over 20,000 human genes and a large number of disease-causing microorganism-specific genes.
4
Figure 1
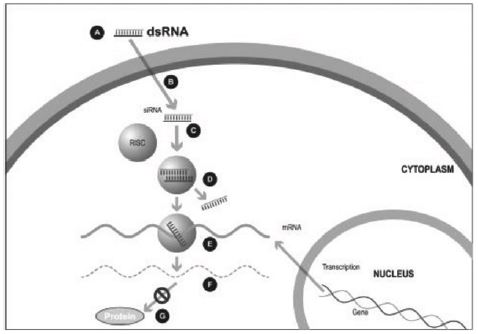
A small double stranded RNA, or dsRNA, molecule (A, Figure 1), comprising one strand known as the sense strand and another strand known as the antisense strand, which are complementary to each other, is synthesized in the laboratory. These small dsRNAs are called small interfering RNAs, or siRNAs. The sequence of the sense strand corresponds to a short region of the target gene mRNA. The siRNA is delivered to the target cell (B, Figure 1), where a group of enzymes, referred to as the RNA-Induced Silencing Complex, or RISC, process the siRNA (C, Figure 1), where one of the strands (usually the sense strand) is released (D, Figure 1). RISC uses the antisense strand to find the mRNA that has a complementary sequence (E, Figure 1) leading to the cleavage of the target mRNA (F, Figure 1). As a consequence, the output of the mRNA (protein production) does not occur (G, Figure 1). Several companies, including Alnylam Pharmaceuticals Inc. (“Alnylam”) and Arbutus Biopharma Corp. (“Arbutus”) utilize this approach in their RNAi product candidates.
Importantly, many genetic disorders are not amenable to the traditional gene silencing approach outlined in Figure 1, as the diseased cells may produce a mixture of the wild type protein of interest and the disease-causing mutant variant of the protein, and the underlying genetic mutation may be too small to allow for selective targeting of the disease-causing variant of the protein through the use of siRNA-based approaches exclusively. In these cases, it is extraordinarily difficult to selectively silence the disease-causing protein without simultaneously silencing the wild type intracellular protein of interest whose presence is vital to the conduct of normal cellular functions.
Our proprietary silence and replace technology utilizes the unique specificity and robust gene silencing capabilities of RNAi while overcoming many of the key limitations of siRNA-based approaches to disease management.
In the standard RNAi approach, double-stranded siRNA is produced synthetically and, subsequently, introduced into the target cell via chemical modification of the RNA or alternative methods of delivery. While efficacy has been demonstrated in several clinical indications through the use of this approach, siRNA-based approaches maintain a number of limitations, including:
5
| • | Clinical management requires repeat administration of the siRNA-based therapeutic agent for multiple cycles to maintain efficacy; |
| • | Long-term patient compliance challenges due to dosing frequencies and treatment durations; |
| • | Therapeutic concentrations of siRNA are not stably maintained because the levels of synthetic siRNA in the target cells decrease over time; |
| • | Novel chemical modifications or novel delivery materials are typically required to introduce the siRNA into the target cells, making it complicated to develop a broad range of therapeutics agents; |
| • | Potential adverse immune responses, resulting in serious adverse effects; |
| • | Requirement for specialized delivery formulations for genetic disorders caused by mutations of multiple genes; and |
| • | siRNA acts only to silence genes and cannot be used to replace defective genes with normally functioning genes. |
Our Approach to the Treatment of Genetic Diseases—ddRNAi and Silence and Replace
Our proprietary silence and replace approach to the treatment of genetic diseases combines RNAi with wild type gene replacement to drive sustained silencing of disease-causing genes and concomitant restoration of functional wild type genes following a single administration of the therapeutic agent. Benitec employs ddRNAi in combination with classical gene therapy (i.e., transgene delivery via viral vectors) to overcome several of the fundamental limitations of RNAi.
The silence and replace approach to the treatment of genetic disorders employs adeno-associated viral vectors (“AAVs”) to deliver genetic constructs which may, after a single administration to the target tissues:
| • | Chronically express RNAi molecules inside of the target, diseased, cells (to serially silence the intracellular production of mutant, disease-causing, protein and the wild type protein of interest); |
| • | Simultaneously drive the expression of a wild type variant of the protein of interest (to restore native intracellular biological processes); and |
| • | AAV vectors can accommodate the multi-functional DNA expression cassettes containing the engineered wild type transgenes and the novel genes encoding short hairpinRNA/microRNA molecules (shRNA/miRNA) that are required to support the development of therapeutic agents capable of the achievement of the goals of the silence and replace approach to therapy. |
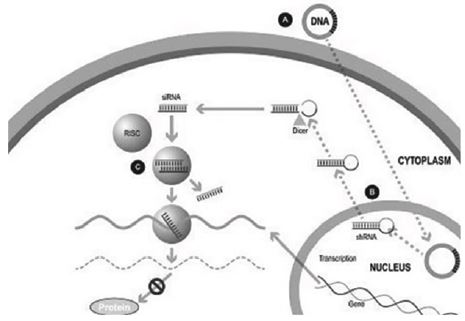
Our silence and replace technology utilizes proprietary DNA expression cassettes to foster continuous production of gene silencing shRNAs and wild type proteins (via expression of the wild type transgene). A range of viral and non-viral gene therapy vectors can be used to deliver the DNA construct into the nucleus of the target cell and, upon delivery, shRNA molecules are expressed and subsequently processed by intracellular enzymes into siRNA molecules that silence the expression of the mutant, disease-causing protein (Figure 2).
In the silence and replace approach (Figure 2):
| • | A DNA construct is delivered to the nucleus of the target cell by a gene therapy vector (A) such as an AAV vector; |
| • | Once inside of the nucleus, the DNA construct drives the continuous production of shRNA molecules (B) which are processed by an enzyme called Dicer into siRNAs (C); |
6
| • | The processed siRNA is incorporated into RISC and silences the target gene using the same mechanism shown in Figure 1; and |
| • | When the DNA expression cassette is additionally comprised of a wild type transgene, upon entry of the DNA construct into the nucleus of the target cell via the use of the AAV vector, the DNA construct also drives the continuous production of wild type protein (to restore native intracellular biological processes). |
Figure 2
Our strategy is to discover, develop and commercialize treatments that leverage the capabilities of ddRNAi and the silence and replace approach to disease management.
For selected product candidates, at the appropriate stage, we may collaborate with large biopharmaceutical companies to further co-develop and, if approved, commercialize our ddRNAi-based and silence and replace- based products to achieve broad clinical and commercial distribution. For specific clinical indications that we deem to be outside of our immediate areas of focus, we will continue to out-license, where appropriate, applications of our ddRNAi and silence and replace technology to facilitate the development of differentiated therapeutics, which could provide further validation of our proprietary technology and approach to disease management.
Our cash and cash equivalents will be deployed to advance our product candidate BB-301 for OPMD, including the natural history lead-in study and Phase 1b/2a BB 301 treatment study, for the continued advancement of development activities for other existing and new product candidates, for general corporate purposes and for strategic growth opportunities.
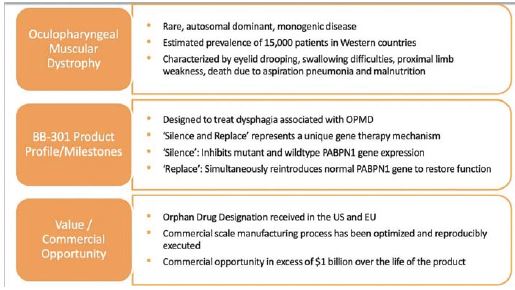
Oculopharyngeal Muscular Dystrophy—OPMD
OPMD is an insidious, autosomal-dominant, late-onset degenerative muscle disorder that typically presents in patients at 40-to-50 years of age. The disease is characterized by progressive swallowing difficulties (dysphagia)
7
and eyelid drooping (ptosis). OPMD is caused by a specific mutation in the poly(A)-binding protein nuclear 1, or PABPN1, gene. OPMD is a rare disease; however, patients have been diagnosed with OPMD in at least 33 countries. Patient populations suffering from OPMD are well-identified, and significant geographical clustering has been noted for patients with this disorder, which could simplify clinical development and global commercialization efforts.
BB-301 is an AAV-based gene therapy designed to both silence the expression of mutated, disease-causing genes (to slow, or halt, the underlying mechanism of disease progression) and simultaneously replace the mutant genes with normal, “wild type” genes (to drive restoration of function in diseased cells). This fundamental therapeutic approach to disease management is called “silence and replace” and this biological mechanism offers the potential to restore the underlying physiology of the treated tissues and, in the process, improve treatment outcomes for patients suffering from the chronic and, potentially, fatal effects of Oculopharyngeal Muscular Dystrophy (OPMD). BB-301 has been granted Orphan Drug Designation in the United States and the European Union.
On July 9, 2018, the Company entered into a License and Collaboration Agreement with Axovant. Pursuant to the Agreement, the Company granted Axovant an exclusive worldwide license to develop, manufacture, and commercialize products containing the Company’s product known as BB-301, which was designed for the potential treatment of Oculopharyngeal Muscular Dystrophy. As of September 3, 2019, the License and Collaboration Agreement with Axovant was terminated. As a result, all rights and licenses which Benitec had granted to Axovant to develop and commercialize BB-301 and related gene therapy product candidates terminated. We are now solely responsible for the costs in connection with the development and commercialization of the BB-301 product candidates.
Our Strengths
We believe that the combination of our proprietary ddRNAi technology and our deep expertise in the design and development of genetic medicines, and specifically ddRNAi-based therapeutics, will enable us to achieve and maintain a leading position in gene silencing and gene therapy for the treatment of human disease. Our key strengths include:
| • | A proprietary ddRNAi-based silence and replace technology platform that may potentially enable the serial development of single-administration therapeutics capable of facilitating sustained, long-term silencing of disease-causing genes and concomitant replacement of wild type gene function; |
| • | A proprietary AAV vector technology which improves the endosomal escape capability of virus produced in insect cells using a baculovirus system. This technology has broad application in AAV-based gene therapies; |
| • | The capabilities to drive the development of a pipeline of programs focused on chronic diseases with either large patient populations, or rare diseases, which may potentially support the receipt of Orphan Drug Designation, including OPMD; and |
8
| • | A growing portfolio of patents protecting improvements to our ddRNAi, and silence and replace, technology and product candidates through at least 2036, with additional patent life anticipated through at least 2040. |
Our Strategy
We endeavor to become the leader in discovery, development, and commercialization of therapeutic agents capable of addressing significant unmet medical need via the application of the silence and replace approach to the treatment of genetic disorders. We apply the following general strategy to drive the Company towards these goals:
| • | Selectively develop proprietary and partnered programs; and |
| • | Continue to explore and secure research and development partnerships with global biopharmaceutical companies supported by the differentiated nature of our scientific platform and intellectual property portfolio. |
Our senior leadership team will continue to explore partnership opportunities with global biopharmaceutical companies, as we expect that the unique attributes of the proprietary ddRNAi and silence and replace approaches, and the breadth of potential clinical indications amenable to our proprietary methods, to support the formation of collaborations over a broad range of diseases with significant unmet medical need.
We seek to actively protect our intellectual property and proprietary technology. These efforts are central to the growth of our business and include:
| • | Seeking and maintaining patents claiming our ddRNAi and silence and replace technologies and other inventions relating to our specific products in development or that are otherwise commercially and/or strategically important to the development of our business; |
| • | Protecting and enforcing our intellectual property rights; and |
| • | Strategically licensing intellectual property from third parties to advance development of our product candidates. |
Our Pipeline
The following table sets forth our current product candidate and the development status:
Table 1. Pipeline: Oculopharyngeal Muscular Dystrophy

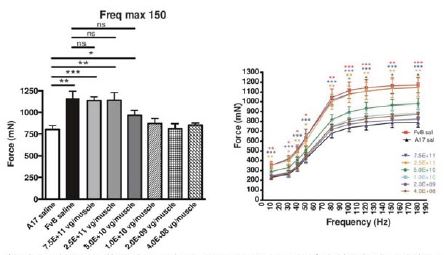
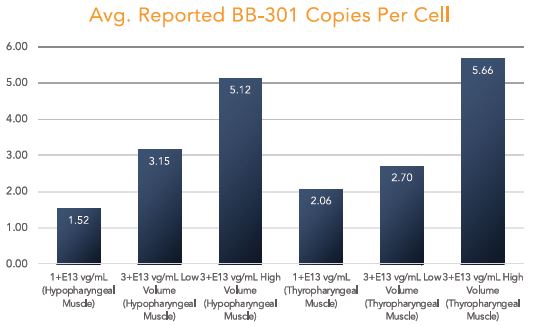
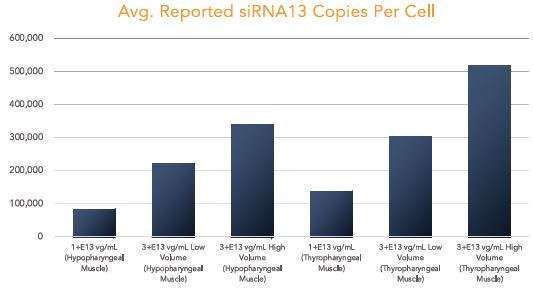
BB-301 is under development for the treatment of Oculopharyngeal Muscular Dystrophy and is currently undergoing evaluation in Clinical Trial Application (CTA)-enabling and Investigational New Drug (IND)-enabling studies. BB-301 is the lead pipeline program for Benitec, and the key attributes of BB-301 are outlined in in Figure 3.
9
Figure 3
In-House Development Programs
BB-301 for the Treatment of Oculopharyngeal Muscular Dystrophy
OPMD is an insidious, autosomal-dominant, late-onset, degenerative muscle disorder that typically presents in patients at 40-to-50 years of age. The disease is characterized by progressive swallowing difficulties (dysphagia) and eyelid drooping (ptosis). OPMD is caused by a specific mutation in the poly(A)-binding protein nuclear 1, or PABPN1, gene. OPMD is a rare disease, however, patients have been diagnosed with OPMD in at least 33 countries. Patient populations suffering from OPMD are well-identified, and significant geographical clustering has been noted for patients with this disorder, which could simplify clinical development and global commercialization efforts.
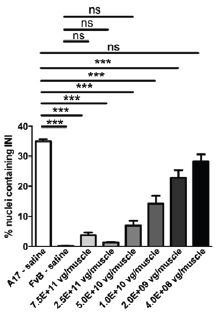
PABPN1 is a ubiquitous factor that promotes interaction between the poly(A) polymerase and CPSF (cleavage and polyadenylation specificity factor) and, thus, controls the length of mRNA poly(A) tails, mRNA export from the nucleus, and alternative poly(A) site usage. The characteristic genetic mutation underlying OPMD results in trinucleotide repeat expansion(s) within exon 1 of PABPN1 and results in an expanded poly-alanine tract at the N-terminal end of PABPN1. The mutation generates a protein with an N-terminal expanded poly-alanine tract of up to 18 contiguous alanine residues prone to the formation of aggregates called intranuclear inclusions (INIs). The INIs that sequester wild type PABPN1 could also contribute to loss of the function phenotype associated with OPMD.
Current OPMD Treatments and Investigational Therapeutic Agents in Development
No curative or disease-modifying therapies currently exist for OPMD patients. Surgical interventions can be undertaken for palliative purposes, including the use of cricopharyngeal myotomy.
10