UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
OR
For the fiscal year ended
OR
OR
Date of event requiring this shell company report . . . . . . . . . . . . . . .
For the transition period from to
Commission file number:
Medicenna Therapeutics Corp.
(Exact name of Registrant as specified in its charter)
N/A
(Translation of Registrant’s name into English)
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
Telephone:
E-mail: ewilliams@medicenna.com
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| | | The |
Securities registered or to be registered pursuant to Section 12(g) of the Act
None
(Title of Class)
Securities for which there is a reporting obligation pursuant to section 15(d) of the Act
None
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
☐ Yes ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
☐ Yes ☒
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
☒
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files)
☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ Accelerated filer ☐
Emerging growth company
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act.
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP | ☐ | | ☒ | Other | ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
(APPLICABLE ONLY TO ISSUERS INVOLVED IN BANKRUPTCY PROCEEDINGS DURING THE PAST FIVE YEARS)
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Sections 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court.
☒ Yes ☐ No
Unless otherwise noted or the context indicates otherwise “we”, “us”, “our”, the “Company” or “Medicenna” refer to Medicenna Therapeutics Corp. and its subsidiaries.
Unless otherwise indicated, financial information in this Annual Report on Form 20-F (this “Annual Report”) has been prepared in accordance with International Financial Reporting Standards (“IFRS”), as issued by the International Accounting Standards Board (“IASB”). Unless otherwise noted herein, all references to “$,” “C$,” “Canadian dollars,” or “dollars” are to the currency of Canada and “US$,” “United States dollars,” or “U.S. dollars” are to the currency of the United States.
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”) and as such, we have elected to comply with certain reduced U.S. public company reporting requirements.
Unless otherwise indicated, the Company has obtained the market and industry data contained in this Annual Report from its internal research, management’s estimates and third-party public information and other industry publications. While the Company believes such internal research, management’s estimates and third-party public information is reliable, such internal research and management’s estimates have not been verified by any independent sources and the Company has not verified any third party public information. While the Company is not aware of any misstatements regarding the market and industry data contained in this Annual Report, such data involves risks and uncertainties and are subject to change based on various factors, including those described under “Cautionary Statement Regarding Forward-Looking Information and Statements” and “Item 3.D. Risk Factors”.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report contains forward-looking statements that are subject to risks and uncertainties. These forward-looking statements include information about possible or assumed future results of our business, financial condition, results of operations, liquidity, plans and objectives. In some cases, you can identify forward-looking statements by terminology such as “will, ” “seek,” “believe,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “expect,” “predict,” “potential,” or the negative of these terms or other similar expressions. The statements we make regarding the following matters are forward-looking by their nature and are based on certain of the assumptions noted below:
| ● |
the intentions, plans and future actions of the Company; |
| ● |
statements relating to the business and future activities of the Company; |
| ● |
anticipated developments in operations of the Company; |
| ● |
market position, ability to compete and future financial or operating performance of the Company; |
| ● |
the timing and amount of funding required to execute the Company’s business plans; |
| ● |
capital expenditures; |
| ● |
the effect on the Company of any changes to existing or new legislation or policy or government regulation; |
| ● |
the availability of labor; |
| ● |
requirements for additional capital; |
| ● |
goals, strategies and future growth; |
| ● |
the adequacy of financial resources; |
| ● |
expectations regarding revenues, expenses and anticipated cash needs; and |
| ● |
general market conditions and macroeconomic trends driven by the COVID-19 pandemic and/or geopolitical conflicts, including supply chain disruptions, market volatility, inflation, and labor challenges, among other factors. |
The preceding list is not intended to be an exhaustive list of all of our forward-looking statements. The forward-looking statements are based on our beliefs, assumptions and expectations of future performance, taking into account the information currently available to us. These statements are only predictions based upon our current expectations and projections about future events. There can be no assurance that such statements will prove to be accurate and actual results and future events could differ materially from those anticipated in such statements. There are important factors that could cause our actual results, levels of activity, performance or achievements to differ materially from the results, levels of activity, performance or achievements expressed or implied by the forward-looking statements, including, but not limited to, those factors identified under the Risk Factors listed below in Item 3.D. of this Annual Report. Furthermore, unless otherwise stated, the forward-looking statements contained in this Annual Report are made as of the date hereof, and we have no intention and undertake no obligation to update or revise any forward-looking statements, whether as a result of new information, future events, changes or otherwise, except as required by law.
GLOSSARY
“AACR” means the America Association for Cancer Research;
“Articles” means the articles of continuance dated November 13, 2017 which govern the Company;
“ASCO” means the American Society of Clinical Oncology;
“ATM” means at-the-market;
“Board” means the Board of Directors of the Company;
“By-law” means the Company’s by-law no. 2 dated July 31, 2020;
“CBCA” means the Canada Business Corporations Act;
“CED” means convection-enhanced delivery;
“CEO” means the Chief Executive Officer of the Company;
“CFO” means the Chief Financial Officer of the Company;
“cGMP” means Good Manufacturing Practices;
“Common Shares” means the Common Shares of the Company;
“CPRIT” means Cancer Prevention and Research Institute of Texas;
“Director” means a member of the Board of Directors of the Company;
“ECA” means External Control Arm;
“Eligible Person” means a director, officer, employee or service provider of the Company or any related entity (being a person that controls or is controlled by the Company or that is controlled by the same person that controls the Company) that options may be granted to under the Company’s Stock Option Plan;
“EMA” means European Medicines Agency;
“ENA” means EORTC (European Organisation for Research and Treatment of Cancer) – NCI (National Cancer Institute) AACR;
“Exchange Act” means the Securities Exchange Act of 1934;
“FDA” means U.S. Food and Drug Administration;
“GBM” means glioblastoma;
“IFRS” means International Accounting Standards Board;
“IL-2” means interleukin-2;
“IL-4” means interleukin-4;
“IL4R” means interleukin-4 receptor;
“IL-13” means interleukin-13;
“IMPD” means Investigational Medical Product Dossier;
“KOL” means key opinion leader;
“MBI” means Medicenna Biopharm Inc., a Delaware corporation;
“mOS” means median overall survival;
“MTD” maximum tolerated dose;
“MTI” means Medicenna Therapeutics Inc., a British Columbia corporation;
“MTI Reverse Takeover” means the reverse takeover of A2 Acquisition Corp. by the shareholders of MTI;
“Nasdaq” means the Nasdaq Capital Market;
“Nasdaq Rules” means the rules of the Nasdaq Capital Market;
“NHP” means non-human primate;
“NIH” means the National Institutes of Health;
“NK” means natural killer;
“Officer” means an executive officer of the Company;
“Options” means the stock options of the Company;
“OS-24” means overall survival at 24 months;
“Phase 1/2 ABILITY Study” means a Beta-only IL-2 Immuno Therapy Study;
“Preferred Shares” means the Preferred Shares of the Company;
“rGBM” means recurrent glioblastoma;
“RRIF” means Registered Retirement Income Fund;
“RRSP” means Registered Retirement Savings Plan;
“Shareholders” means holders of Common Shares of the Company;
“Stanford” means the Leland Stanford Junior University;
“Stock Option Plan” means the Company’s Stock Option Plan, which was approved for adoption by shareholders on September 21, 2017, which amended, restated and superseded the previous stock option plan adopted by the Company in 2015;
“TFSA” means Tax-Free Savings Account;
“TME” means tumor microenvironment;
“TMZ” means temozolomide;
“Tregs” means regulatory T cells;
“TSX” means the Toronto Stock Exchange; and
“USPTO” means the United States Patent and Trademark Office.
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISORS
Not required.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not required.
3.A.
[Reserved]
3.B. Capitalization and Indebtedness
Not required.
3.C. Reasons for the Offer and Use Of Proceeds
Not required.
3.D. Risk Factors
Following is a list of risks that the Company faces in its normal course of business. The risks and uncertainties set out below are not the only ones the Company is facing. There are additional risks and uncertainties that the Company does not currently know about or that the Company currently considers immaterial which may also impair the Company’s business operations and cause the price of the Common Shares of the Company to decline. If any of the following risks actually occur, the Company’s business may be harmed and the Company’s financial condition and results of operations may suffer significantly. Investors should carefully consider the risk factors set out below and consider all other information contained herein and in the Company's other public filings before making an investment decision. The risks set out below are not an exhaustive list and should not be taken as a complete summary or description of all the risks associated with the Company's business and the biotechnology business generally.
Risks Related to the Company’s Business, Industry, and Financial Position
The Company has no sources of product revenue and there is uncertainty regarding its ability to maintain operations and research and development without sufficient funding.
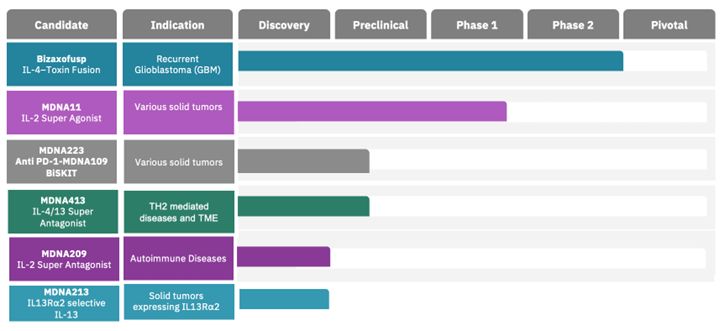
The Company has no sources of product revenue and cannot predict when or if it will generate product revenue. The Company’s ability to generate product revenue and ultimately become profitable depends upon its ability, alone or with partners, to successfully develop the product candidates, obtain regulatory approval, and commercialize products, including any of the current product candidates, or other product candidates that may be developed, in-licensed or acquired in the future. The Company does not anticipate generating revenue from the sale of products for the foreseeable future. The Company expects research and development expenses to increase in connection with ongoing activities, particularly as MDNA11 is advanced from the dose escalation portion of the Phase 1/2 ABILITY Study into the Phase 2 dose expansion cohorts as well as advancing a lead BiSKIT candidate into IND enabling studies.
The Company will require significant additional capital resources to expand its business, in particular the further development of its proposed products. Advancing its product candidates or acquisition and development of any new products or product candidates will require considerable resources and additional access to capital markets. In addition, the Company’s future cash requirements may vary materially from those now expected.
The Company can potentially seek additional funding through corporate collaborations and licensing arrangements, through public or private equity or debt financing, or through other transactions. However, if clinical trial results are neutral or unfavorable, or if capital market conditions in general, or with respect to life sciences companies such as Medicenna, are unfavorable, the Company’s ability to obtain significant additional funding on acceptable terms, if at all, will be negatively affected. Additional financing that it may pursue may involve the sale of the Common Shares or financial instruments that are exchangeable for, or convertible into, the Common Shares, which could result in significant dilution to its shareholders. If sufficient capital is not available, the Company may be required to delay the implementation of its business strategy, which could have a material adverse effect on its business, financial condition, prospects or results of operations.
The Company will need substantial additional funding which may not be available on terms acceptable to the Company or at all. If the Company is unable to raise capital when needed, the Company would be forced to delay, reduce, terminate or eliminate product development programs.
The Company expects research and development expenses to increase in connection with ongoing activities, particularly as MDNA11 is advanced into the Phase 2 portion of the Phase 1/2 ABILITY Study as well as advancing a lead BiSKIT candidate into IND enabling studies. In addition, if the Company obtains regulatory approval for any of its product candidates, the Company expects to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. Furthermore, the Company will need to obtain additional funding in connection with continuing operations. If the Company is unable to raise capital when needed or on attractive terms, the Company could be forced to delay, reduce, terminate or eliminate its product development programs, potentially including the ongoing Phase 1/2 ABILITY Study.
As of March 31, 2023, the Company had cash and cash equivalents of $33.6 million.
Developing pharmaceutical products, which includes manufacturing, quality control, conducting preclinical studies and clinical trials, is expensive. Our operations have consumed significant amounts of cash since inception. As we continue to advance MDNA11 or future product candidates into clinical trials and launch and commercialize any product candidates for which we receive regulatory approval, we expect research and clinical development expenses, as well as selling, general and administrative expenses to increase substantially. In connection with our ongoing activities, we believe that our existing cash and cash equivalents will be sufficient to fund our operating requirements through calendar Q3 2024. However, circumstances may cause us to consume capital more rapidly than we anticipate. We will require additional capital for the further development and potential commercialization of future product candidates.
We have incurred significant losses in every quarter since our inception and anticipate that we will continue to incur significant losses in the future.
Investment in a biotechnology company is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect or an acceptable safety profile, gain regulatory approval or become commercially viable. We have not generated any revenue from product sales to date, and all of our product candidates are in early clinical or preclinical development. We continue to incur significant expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in every reporting period since our inception. We expect to continue to incur significant expenses and operating losses for the foreseeable future as we seek to identify, acquire and conduct research and development of future product candidates, and potentially begin to commercialize any future products that may achieve regulatory approval. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our financial condition. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. Our prior losses and expected future losses have had, and will continue to have, an adverse effect on our financial condition. If any of our future product candidates fail in clinical trials or do not gain regulatory approval, or if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
Risks Related to the Discovery, Development and Commercialization of our Product Candidates
Our product candidates are in early stages of development and may fail in development or suffer delays that materially and adversely affect their commercial viability. If we are unable to complete development of, or commercialize our product candidates, if approved, or experience significant delays in doing so, our business will be materially harmed.
We are in the early stages of development efforts for MDNA11 and our BiSKIT platform. We have no products on the market and all of our product candidates, with the exception of bizaxofusp which is not in active development by the Company, are still in early clinical, preclinical or drug discovery stages, and we may not ever obtain regulatory approval for any of our product candidates. We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA. Before obtaining regulatory approval for the commercial distribution of our product candidates, we or an existing or future collaborator must conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. Additionally, our BiSKIT platform is in earlier stages of discovery and preclinical development and may never advance to clinical-stage development. If we do not receive marketing approvals and successfully commercialize our product candidates, if approved, we may not be able to continue our operations.
The success of the Company’s product candidates will depend on several factors, including the following:
| ● |
securing additional funding to continue development; |
| ● |
successful completion of preclinical studies and clinical trials; |
| ● |
demonstrating a superior product profile compared with competitors; |
| ● |
receipt of marketing approvals from the FDA, Health Canada and similar regulatory authorities outside the United States and Canada; |
| ● |
establishing commercial manufacturing capabilities by identifying and securing arrangements with third party manufacturers for the product candidates; |
| ● |
maintaining patent and trade secret protection and regulatory exclusivity for the product candidates; |
| ● |
launching commercial sales of the product candidates, if and when approved, whether alone or in collaboration with others; |
| ● |
acceptance of the products, if and when approved, by patients, the medical community and third party payers; |
| ● |
effectively competing with other therapies; and |
| ● |
a continued acceptable safety profile of the products following approval. |
If the Company does not achieve one or more of these factors in a timely manner or at all, the Company could experience significant delays or an inability to successfully commercialize its product candidates, if approved, which would materially harm its business.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, results of earlier studies and trials may not be predictive of future trial results, and the Company’s product candidates may not have favorable results in later trials or in the commercial setting.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials may not be predictive of the results of later-stage clinical trials. In the case of bizaxofusp, the promising results seen in the Phase 2b clinical study may not be replicated in a randomized, controlled Phase 3 clinical study. Success in preclinical or animal studies and early clinical trials does not ensure that later large-scale efficacy trials will be successful nor does it predict final results. This is applicable to MDNA11 as the promising preclinical data may not be replicated in the Phase 1/2 ABILITY Study. Favorable results in early trials may not be repeated in later trials. There is no assurance the FDA, the EMA or other similar government bodies will view the results as the Company does or that any future trials of its product candidates for other indications will achieve positive results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials.
The Company will be required to demonstrate through larger-scale clinical trials that any product candidate is safe and effective before it can seek regulatory approvals for commercial sale of its product. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical and post-approval trials. If bizaxofusp, MDNA11 and other product candidates fail to demonstrate sufficient safety and efficacy in future clinical trials, the Company’s operations and financial condition will be adversely impacted.
The Company may not achieve its publicly announced milestones according to schedule, or at all.
From time to time, the Company may announce the timing of certain events expected to occur, such as the anticipated timing of results from clinical trials. These statements are forward-looking and are based on the best estimates of management at the time relating to the occurrence of such events. However, the actual timing of such events may differ from what has been publicly disclosed. The timing of events such as initiation or completion of a clinical trial, filing of an application to obtain regulatory approval or announcement of additional clinical trials for a product candidate may ultimately vary from what is publicly disclosed. These variations in timing may occur as a result of different events, including the ability to recruit patients in a clinical trial in a timely manner, the nature of results obtained during a clinical trial or during a research phase, problems with a contract development and manufacturing organizations (“CDMO”) or a contract research organization (“CRO”), or any other event having the effect of delaying the publicly announced timeline. The Company undertakes no obligation to update or revise any forward-looking information, whether as a result of new information, future events or otherwise, except as otherwise required by law. Any variation in the timing of previously announced milestones could have a material adverse effect on the business plan, financial condition or operating results and the trading price of the Common Shares.
If the Company’s competitors develop and market products that are more effective than the Company’s existing product candidates or any future product candidates it may develop, or if they obtain marketing approval before it does, the Company’s products may be rendered obsolete or uncompetitive.
Competition from pharmaceutical companies, biotechnology companies and universities is intense and is expected to increase. Many of the Company’s competitors and potential competitors have substantially greater product development capabilities and financial, scientific, marketing and human resources than the Company does. Our future success depends in part on our ability to maintain a competitive position, including our ability or the ability of our partners to further progress bizaxofusp, MDNA11 and our BiSKIT platform through the necessary preclinical and clinical trials towards regulatory approval for sale and commercialization. MDNA11 is in a particularly competitive space and the need to differentiate the program clinically is important to its future success. Other companies may succeed in commercializing their products earlier than we are able to commercialize our product candidates, if approved, or they may succeed in developing products that are more effective than our product candidates, if approved. While the Company will seek to expand its technological capabilities in order to remain competitive, there can be no assurance that developments by others will not render its product candidates, if approved, non-competitive or that the Company or its licensors will be able to keep pace with technological developments. Competitors have developed technologies that could be the basis for competitive products. Some of those products may have an entirely different approach or means of accomplishing the desired therapeutic effect than the Company’s product candidates and may be more effective or less costly than its product candidates. In addition, other forms of medical treatment may offer competition to the product candidates, if approved. The success of the Company’s competitors and their products and technologies relative to its technological capabilities and competitiveness could have a material adverse effect on the future preclinical and clinical trials of its product candidates, including its ability to obtain the necessary regulatory approvals for the conduct of such trials.
The Company may not be able to secure a partnership for bizaxofusp which would halt future development.
The Company is seeking a partner to continue the clinical development and commercialization of bizaxofusp. The Company does not have the financial resources to complete the necessary development work internally and should it not be able to secure a partnership, further development of bizaxofusp may not continue.
The Company is subject to extensive government regulation that will increase the cost and uncertainty associated with gaining regulatory approval of its product candidates.
Securing regulatory approval for the manufacture and sale of human therapeutic products in the United States, Canada and other markets is a long and costly process that is controlled by that particular country’s national regulatory agency. Approval in the United States, Canada or Europe does not assure approval by other national regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country. Other national regulatory agencies may have similar regulatory approval processes, but each is different.
Prior to obtaining regulatory approval to market a drug product, every national regulatory agency has a variety of statutes and regulations which govern the principal development activities. These laws require controlled research and testing of product candidates, government review and approval of a submission containing preclinical and clinical data establishing the safety and efficacy of the product candidate for each use sought, approval of manufacturing facilities including adherence to cGMP during production and storage and control of marketing activities, including advertising and labelling. There can be no assurance that MDNA11 or bizaxofusp will be approved or successfully commercialized, if approved, in any given country. There can be no assurance that the Company’s product candidates will prove to be safe and effective in clinical trials under the standards of the regulations in the various jurisdictions or receive applicable regulatory approvals from applicable regulatory bodies.
Negative results from clinical trials or studies of third parties and adverse safety events involving the targets of the Company’s product candidates may have an adverse impact on future commercialization efforts.
From time to time, studies or clinical trials on various aspects of biopharmaceutical products are conducted by academic researchers, competitors or others. The results of these studies or trials, when published, may have a significant effect on the market for the biopharmaceutical product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to the Company’s product candidates, or the therapeutic areas in which the Company’s product candidates compete, could adversely affect the share price and ability to finance future development of the Company’s product candidates, and the business and financial results could be materially and adversely affected.
The Company faces the risk of product liability claims, which could exceed its insurance coverage and cause product recalls, each of which could deplete cash resources.
The Company is exposed to the risk of product liability claims alleging that use of its product candidates MDNA11, bizaxofusp, and in the future, the BiSKIT platform, caused an injury or harm. These claims may arise at any point in the development, testing, manufacture, marketing or sale of product candidates and may be made directly by patients involved in clinical trials of product candidates, by consumers or healthcare providers or by individuals, organizations or companies selling the product candidates, if approved. Product liability claims can be expensive to defend, even if the product or product candidate did not actually cause the alleged injury or harm.
Insurance covering product liability claims becomes increasingly expensive as a product candidate moves through the development pipeline to commercialization. Currently the Company maintains clinical trial liability insurance coverage of US$5 million. However, there can be no assurance that such insurance coverage is or will continue to be adequate or available at a cost acceptable to the Company or at all. The Company may choose or find it necessary under its collaborative agreements to increase the insurance coverage in the future, but may not be able to secure greater or broader product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any liability for damages resulting from a product liability claim could exceed the amount of the coverage, require payment of a substantial monetary award from the Company’s cash resources and have a material adverse effect on the business, financial condition and results of operations. Moreover, a product recall, if required, could generate substantial negative publicity about the products and business, inhibit or prevent commercialization of other products and product candidates, if approved, or negatively impact existing or future collaborations.
If the Company is unable to enroll subjects in clinical trials, it will be unable to complete its clinical trials on a timely basis.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, the ability to obtain and maintain patient consents, the risk that enrolled subjects will drop out before completion, competing clinical trials, and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications the Company is investigating. Furthermore, the Company relies on CROs and clinical trial sites to ensure the proper and timely conduct of its clinical trials, and while it has agreements governing their committed activities, the Company has limited influence over their actual performance.
If the Company experiences delays in the completion or termination of any clinical trial of its product candidates or any future product candidates, the commercial prospects of its product candidates will be harmed and its ability to generate product revenues from any of these product candidates, if approved, will be delayed. In addition, any delays in completing clinical trials will increase costs, slow down product candidate development and approval processes and may shorten any periods during which the Company may have the exclusive right to commercialize its product candidates, if approved, all of which may allow the Company’s competitors to bring products to market before it does. Delays may further jeopardize the Company’s ability to commence product sales, which will impair its ability to generate revenues and may harm the business, results of operations, financial condition and cash flows and future prospects. In addition, many of the factors that may cause a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of its product candidates or its future product candidates.
Even if any product candidates we develop receive marketing approval, they may fail to achieve the degree of market acceptance by physicians, patients, healthcare payers, and others in the medical community necessary for commercial success.
The commercial success of any of our product candidates, if approved, will depend upon its degree of market acceptance by physicians, patients, third party payers, and others in the medical community. Even if any product candidates we may develop receive marketing approval, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, healthcare payers, and others in the medical community. The degree of market acceptance of any product candidates we may develop, if approved for commercial sale, will depend on a number of factors, including:
| ● |
the efficacy and safety of such product candidates as demonstrated in pivotal clinical trials and published in peer-reviewed journals; |
| ● |
the potential and perceived advantages compared to alternative treatments; |
| ● |
the ability to offer our products for sale at competitive prices; |
| ● |
the ability to offer appropriate patient access programs, such as co-pay assistance; |
| ● |
the extent to which physicians recommend our products to their patients; |
| ● |
convenience and ease of dosing and administration compared to alternative treatments; |
| ● |
the clinical indications for which the product candidate is approved by the FDA, EMA or other comparable foreign regulatory agencies; |
| ● |
product labeling or product insert requirements of the FDA, EMA or other comparable foreign regulatory authorities, including any limitations, contraindications or warnings contained in a product’s approved labeling; |
| ● |
restrictions on how the product is distributed; |
| ● |
the timing of market introduction of competitive products; |
| ● |
publicity concerning our products or competing products and treatments; |
| ● |
the effectiveness of marketing and distribution efforts by us and other licenses and distributors; |
| ● |
sufficient governmental and third-party coverage or reimbursement; and |
| ● |
the prevalence and severity of any side effects. |
If any product candidates we develop and for which we receive marketing approval do not achieve an adequate level of acceptance by physicians, healthcare payers, patients and the medical community, we will not be able to generate significant revenue, and we may not become or remain profitable. The failure of any of our product candidates, if approved, to find market acceptance would harm our business prospects.
The Company’s discovery, testing, development and manufacturing processes involve the use of hazardous and radioactive materials which may result in potential environmental exposure.
Although our current laboratory and manufacturing activities are handled by third parties, the Company’s discovery, testing and development processes may, in the future, involve the direct controlled use of hazardous and radioactive materials. Accordingly, the Company may become subject to federal, provincial, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, the Company could be held liable for any damages that result and any such liability could exceed the Company’s resources. The Company is not specifically insured with respect to this liability. There can be no assurance that the Company will not be required to incur significant costs to comply with environmental laws and regulations in the future, or that the operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
Significant disruption in availability of key components for ongoing clinical studies could considerably delay completion of potential clinical trials, product testing and regulatory approval of potential product candidates.
The Company relies on third parties to supply ingredients and excipients for the manufacture and formulation of its product candidates, compatible syringes or infusion systems for drug administration, catheters required to deliver the product candidate to the brain as well as imaging software to accurately place catheters in the tumor (“Components”). Each of the suppliers of these Components in turn need to comply with applicable regulatory requirements. Any significant disruption in supplier relationships could harm the Company’s business. Any significant delay in the supply of a Component for an ongoing or future clinical study could considerably delay initiation or completion of a clinical trial, drug manufacturing, drug testing and regulatory approval of a product candidate or future product candidate. If the Company or its suppliers are unable to purchase these Components after regulatory approval has been obtained for the product candidates, or the suppliers decide not to manufacture these Components or provide support for any of the Components, clinical trials or the commercial launch of that product candidate, if approved, would be delayed or there would be a shortage in supply, which would impair the ability to generate revenues from the sale of the product candidates, if approved. It may take several years to establish an alternative source of supply for such Components and to have any such new source approved by the FDA and other regulatory agencies.
Preliminary and interim data from our clinical trials that we may announce or publish from time to time may change as patient data are further examined, audited or verified and more patient data become available.
From time to time, we may announce or publish preliminary or interim data from our clinical trials. Preliminary and interim data remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary or interim data. Preliminary and interim results of a clinical trial are not necessarily predictive of final results. There can be no assurance that favorable interim or preliminary data will result in favorable final data. Preliminary and interim data are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues, patient data are further examined and reviewed, more patient data become available, and we prepare and issue our final clinical study report. As a result, preliminary and interim data should be viewed with caution until the final, complete data are available. Material adverse changes in the final data compared to the preliminary or interim data could significantly harm our business, prospects, financial condition and results of operations.
Biologics carry unique risks and uncertainties, which could have a negative impact on future results of operations.
The successful discovery, development, manufacturing and sale of biologics is a long, expensive and uncertain process. There are unique risks and uncertainties with biologics. For example, access to and supply of necessary biological materials, such as cell lines, may be limited and governmental regulations restrict access to and regulate the transport and use of such materials. In addition, the development, manufacturing and sale of biologics is subject to regulations that are often more complex and extensive than the regulations applicable to other pharmaceutical products. Manufacturing biologics, especially in large quantities, is often complex and may require the use of innovative technologies. Such manufacturing also requires facilities specifically designed and validated for this purpose and sophisticated quality assurance and quality control procedures. Biologics are also frequently costly to manufacture because production inputs are derived from living animal or plant material, and some biologics cannot be made synthetically. Failure to successfully discover, develop, manufacture and sell our biological candidates would adversely impact our business and future results of operations.
Bizaxofusp has been granted Fast Track designation by the FDA and we may seek Fast Track designation for one or more of our other drug or biologic candidates in the future. Even if we apply for Fast Track designation in the future, we might not receive such designation, and even if we do, such designation may not actually lead to a faster development or regulatory review or approval process, and further, such designation could be withdrawn by the FDA.
If a drug or biologic candidate is intended for the treatment of a serious condition and nonclinical or clinical data demonstrate the potential to address an unmet medical need for this condition, a product sponsor may request an FDA Fast Track designation from the FDA. If we seek Fast Track designation for a drug or biologic candidate, we may not receive it from the FDA. However, even if we receive Fast Track designation, Fast Track designation does not ensure that we will receive marketing approval or that approval will be granted within any particular time frame. We may not experience a faster development or regulatory review or approval process with Fast Track designation compared to conventional FDA procedures. In addition, the FDA may withdraw Fast Track designation if the designation is no longer supported by data from our clinical development program. Fast Track designation alone does not guarantee qualification for the FDA’s priority review procedures.
Even if we obtain regulatory approval for a product, we will remain subject to ongoing regulatory requirements. Maintaining compliance with ongoing regulatory requirements may result in significant additional expense to us, and any failure to maintain such compliance could subject us to penalties and cause our business to suffer.
If any of our drug or biologic candidates are approved, we will be subject to ongoing regulatory requirements with respect to manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing clinical trials and submission of safety, efficacy and other post-approval information, including both federal and state requirements in the United States and requirements of comparable foreign regulatory authorities.
Manufacturers and manufacturers’ facilities are required to continuously comply with FDA and comparable foreign regulatory authority requirements, including ensuring that quality control and manufacturing procedures conform to cGMP regulations and corresponding foreign regulatory manufacturing requirements. As such, we and our CDMOs will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any BLA, NDA or other marketing authorization application.
Any regulatory approvals that we receive for our drug or biologic candidates may be subject to limitations on the approved indicated uses for which the drug or biologic candidate may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase IV clinical trials, and surveillance to monitor the safety and efficacy of the drug or biologic candidate. In addition, if the FDA or a comparable foreign regulatory authority approves any of our drug or biologic candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and record keeping for the products will be subject to extensive and ongoing regulatory requirements. Any new legislation addressing drug safety issues could result in delays in product development or commercialization, or increased costs to assure compliance.
We must also comply with requirements concerning advertising and promotion for any of our drug or biologic candidates for which we hope to obtain marketing approval. Promotional communications with respect to prescription drugs and biologics are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved labeling. If we are not able to comply with post-approval regulatory requirements, we could have marketing approval for any of our products withdrawn by regulatory authorities and our ability to market any future products could be limited, which could adversely affect our ability to achieve or sustain profitability. Thus, the cost of compliance with post-approval regulations may have a negative effect on our operating results and financial condition.
In addition, later discovery of previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or failure to comply with applicable regulatory requirements may result in a variety of risks. For example, a regulatory agency or enforcement authority may, among other things:
| ● |
impose restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● |
impose requirements to conduct post-marketing studies or clinical trials; |
| ● |
issue warning or untitled letters if the regulator is the FDA, or comparable notice of violations from foreign regulatory authorities; |
| ● |
issue consent decrees, injunctions or impose civil or criminal penalties; |
| ● |
require the payment of fines, restitution or disgorgement of profits or revenues; |
| ● |
suspend or withdraw regulatory approval; |
| ● |
suspend any of our ongoing clinical trials; |
| ● |
refuse to approve pending applications or supplements to approved applications submitted by us; |
| ● |
impose restrictions on our operations, including closing our or our CDMOs’ manufacturing or analytical testing facilities; or |
| ● |
require product seizure or detention, recalls or refuse to permit the import or export of products. |
Any government investigation of alleged violations of law would require us to expend significant time and resources in response and could generate adverse publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to develop and commercialize our products and our value and our operating results would be adversely affected. In addition, regulatory authorities’ policies (such as those of the FDA or EMA) may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug or biologic candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are otherwise not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
We currently have limited marketing and sales experience. If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our drug or biologic candidates, we may be unable to generate any revenue.
Although some of our employees may have marketed, launched and sold other pharmaceutical products in the past while employed at other companies, we have no experience selling and marketing our drug or biologic candidates, and we currently have no marketing or sales organization. To successfully commercialize any products that may result from our development programs, we will need to find one or more collaboration partners to commercialize our products or invest in and develop these capabilities, either on our own or with others, which would be expensive, difficult and time consuming. Any failure or delay in the timely development of our internal commercialization capabilities could adversely impact the potential for success of our products.
If commercialization collaboration partners do not commit sufficient resources to commercialize our future drugs or biologics, and if we are unable to develop the necessary marketing and sales capabilities on our own, we will be unable to generate sufficient product revenue to sustain or grow our business. We may be competing with companies that currently have extensive and well-funded marketing and sales operations, particularly in the markets our drug or biologic candidates are intended to address. Without appropriate capabilities, whether directly or through third-party collaboration partners, we may be unable to compete successfully against these more established companies.
Failure to obtain or maintain adequate reimbursement or insurance coverage for approved products, if any, could limit our ability to market those products and decrease our ability to generate revenue.
The pricing, coverage, and reimbursement of our approved drugs, if any, must be sufficient to support our commercial efforts and other development programs, and the availability and adequacy of coverage and reimbursement by third-party payors, including governmental and private insurers, are essential for most patients to be able to afford medical treatments. Sales of our approved drugs, if any, will depend substantially, both domestically and abroad, on the extent to which the costs of our approved drugs, if any, will be paid for or reimbursed by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or government payors and private payors. If coverage and reimbursement are not available, or are available only in limited amounts, we may have to subsidize or provide drugs for free or we may not be able to successfully commercialize our drugs.
In addition, there is significant uncertainty related to the insurance coverage and reimbursement for newly approved drugs. In the United States, the principal decisions about coverage and reimbursement for new drugs are typically made by the Centers for Medicare and Medicaid Services (“CMS”) an agency within the United States Department of Health and Human Services, as CMS decides whether and to what extent a new drug will be covered and reimbursed under Medicare. Private payors tend to follow the coverage reimbursement policies established by CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for novel drug or biologic candidates such as ours and what reimbursement codes our drug or biologic candidates may receive if approved. Moreover, Congress recently enacted and President Biden signed into law new authorities for CMS to negotiate drug prices annually for certain prescription drugs and biological products, subject to statutory criteria and a future selection process that is in the process of being developed by CMS. It is unclear how these forthcoming changes in the way that CMS does business with certain members of the biopharmaceutical industry may impact coverage or reimbursement decisions across the industry as a whole.
Outside the United States, international operations are generally subject to extensive governmental price controls and other price-restrictive regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada and other countries has and will continue to put pressure on the pricing and usage of drugs. In many countries, the prices of drugs are subject to varying price control mechanisms as part of national health systems. Price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our drugs, if any. Accordingly, in markets outside the United States, the potential revenue may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and private payors in the United States and abroad to limit or reduce healthcare costs may result in restrictions on coverage and the level of reimbursement for new drugs and, as a result, they may not cover or provide adequate payment for our drugs, if any. We expect to experience pricing pressures in connection with drugs due to the increasing trend toward managed healthcare, including the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, and prescription drugs or biologics in particular, has and is expected to continue to increase in the future. As a result, profitability of our drugs, if any, may be more difficult to achieve even if any of them receive regulatory approval.
Risks Related to the Company’s Reliance on Third Parties
The Company relies and will continue to rely on third parties to plan, conduct and monitor preclinical studies and clinical trials, and their failure to perform as required could cause substantial harm to the Company’s business.
The Company relies and will continue to rely on third parties to conduct a significant portion of clinical development and planned preclinical activities. Preclinical activities include in vivo studies providing access to specific disease models in different species, pharmacology and toxicology studies, and assay development. Clinical development activities include trial design, regulatory submissions, clinical patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management. If there is any dispute or disruption in the Company’s relationship with third parties, or if the third party is unable to provide quality services in a timely manner and at a reasonable cost, or is unable to secure access to specific disease models or is unable to acquire and maintain inventory of different species required for pre-clinical testing, any active development programs could face delays. Further, if any of these third parties fails to perform as expected or if their work fails to meet regulatory requirements, testing could be delayed, cancelled or rendered ineffective.
The Company relies on contract manufacturers over whom the Company has limited control. If the Company is subject to quality, cost or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, business operations could suffer significant harm.
The Company has limited manufacturing experience and relies on CDMOs to manufacture MDNA11 and bizaxofusp for clinical trials and the BiSKIT Platform for preclinical development as well as for manufacturing, testing, filling, packaging, storing and shipping of its product candidates in compliance with cGMP, regulations applicable to its products. The FDA ensures the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packing of a drug product.
There can be no assurances that the CDMOs selected will be able to meet future timetables and requirements. If the Company is unable to arrange for alternative third-party manufacturing sources on commercially reasonable terms or in a timely manner, it may delay the development of the product candidates. Further, contract manufacturers must operate in compliance with cGMP and failure to do so could result in, among other things, the disruption of product supplies. The Company’s dependence upon third parties for the manufacture of its product candidates may adversely affect profit margins and ability to develop and deliver product candidates, if approved, on a timely and competitive basis.
Our reliance on third-party manufacturers also exposes us to the following additional risks:
| ● |
we may be unable to identify manufacturers to manufacture our drug or biologic candidates on acceptable terms or at all, because the number of qualified potential manufacturers is limited. Following NDA or BLA approval, a change in the manufacturing site could require additional approval from the FDA. This approval would require new testing and compliance inspections; |
| ● |
our third-party manufacturers might be unable to timely formulate and manufacture our product or produce the quantity and quality required to meet our clinical and commercial needs, if any; |
| ● |
our future third-party manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our drug or biologic candidates; |
| ● |
if any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own or be able to license, or we may have to share, the intellectual property rights to any improvements made by our third-party manufacturers in the manufacturing process for our drug or biologic candidates; |
| ● |
while we currently carry insurance in an amount and on terms and conditions that are customary for similarly situated companies and that are satisfactory to our board of directors, we and/or our third-party manufacturers may not have sufficient insurance coverage in the event of any inadvertent destruction of or loss of any drug substance by them, which could result in delays in production and/or our clinical trials and/or result in additional costs to us; and |
| ● |
our third-party manufacturers could breach or terminate their agreements with us. |
Our employees, independent contractors, principal investigators, CROs, consultants or vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, CROs, consultants or vendors may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates: FDA regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA; manufacturing standards; federal and state health care fraud and abuse laws and regulations; or laws that require the true, complete and accurate reporting of financial information or data. In addition, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials or creating fraudulent data in our preclinical studies or clinical trials, which could result in regulatory sanctions and serious harm to our reputation.
It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished potential profits and future earnings, and curtailment of our operations, any of which could adversely affect our business, financial condition, results of operations or prospects.
If the Company breaches any of the agreements under which it licenses rights to product candidates or technology from third parties, it can lose license rights that are important to its business. The Company’s current license agreements may not provide an adequate remedy for breach by the licensor.
The Company is seeking a partnership for bizaxofusp, developing MDNA11 and other earlier stage preclinical and discovery drug candidates pursuant to license agreements with NIH and Stanford (collectively, the “Licensors”). The Company is subject to a number of risks associated with its collaboration with the Licensors, including the risk that the Licensors may terminate a license agreement upon the occurrence of certain specified events. Each license agreement requires, among other things, that the Company makes certain payments and use reasonable commercial efforts to meet certain clinical and regulatory milestones. If the Company fails to comply with any of these obligations or otherwise breaches this or similar agreements, the Licensors or any future licensors may have the right to terminate the license in whole. The Company may also suffer the consequences of non-compliance or breaches by Licensors in connection with the license agreements. Such non-compliance or breaches by such third parties may in turn result in breaches or defaults under the Company’s agreements with other collaboration partners, and the Company may be found liable for damages or lose certain rights, including rights to develop and/or commercialize a product or product candidate, if approved. Loss of the Company’s rights to the licensed intellectual property or any similar license granted to it in the future, or the exclusivity rights provided therein, may harm the Company’s financial condition and operating results.
The Company is subject to the restrictions and conditions of the CPRIT agreement. Failure to comply with the CPRIT agreement may adversely affect the Company’s financial condition and results of operations.
The Company obtained a grant from CPRIT to fund a portion of its historical operations. If the Company is found to have used any grant proceeds for purposes other than intended, is in violation of the terms of the grant, or relocates its bizaxofusp related operations outside of the State of Texas, then the Company may be required to repay the grant proceeds received. A failure to maintain compliance with the grant, including maintaining a presence in the state of Texas for three years after the grant is complete, may require the Company to reimburse all or a portion of the CPRIT grant which may cause a halt or delay in ongoing operations, which may adversely affect the Company’s financial condition and operating results.
Risks Related to Intellectual Property and Litigation
The Company’s success depends upon its ability to protect its intellectual property and its proprietary technology.
The Company’s success depends, in part, on its ability and its licensors’ ability to obtain patents, maintain trade secrets protection and operate without infringing on the proprietary rights of third parties or having third parties circumvent its rights. Certain licensors and the institutions that they represent have filed and are actively pursuing certain applications for certain Canadian and foreign patents. The patent position of pharmaceutical and biotechnology firms is uncertain and involves complex legal and financial questions for which, in some cases, certain important legal principles remain unresolved. There can be no assurance that the patent applications made in respect of the owned or licensed products will result in the issuance of patents, that the term of a patent will be extendable after it expires in due course, that the licensors or the institutions that they represent will develop additional proprietary products that are patentable, that any patent issued to the licensors or the Company will provide it with any competitive advantages, that patents of others will not impede its ability to do business or that third parties will not be able to circumvent or successfully challenge the patents obtained in respect of the licensed products. The cost of obtaining and maintaining patents is high and may affect the Company’s financial condition. Furthermore, there can be no assurance that others will not independently develop competitor products which duplicate any of the owned/licensed products under pending patent protection or, if patents are issued to such owned/licensed products, will not design around such patents. There can be no assurance that the Company’s processes or products or those of its licensors do not or will not infringe upon the patents of third parties or that the scope of its patents or those of its licensors will successfully prevent third parties from developing similar and competitive products.
Much of the Company’s know-how and technology may not be patentable, though it may constitute trade secrets. There can be no assurance, however, that the Company will be able to meaningfully protect its trade secrets. To help protect its intellectual property rights and proprietary technology, the Company requires employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance that these agreements will provide meaningful protection for its intellectual property rights or other proprietary information in the event of any unauthorized use or disclosure.
The Company’s potential involvement in intellectual property litigation could negatively affect its business.
The Company’s future success and competitive position depends in part upon its ability to maintain its intellectual property portfolio. There can be no assurance that any patents will be issued on any existing or future patent applications. Even if such patents are issued, there can be no assurance that any patents issued or licensed to the Company will not be successfully challenged. The Company’s ability to establish and maintain a competitive position may require that it successfully prosecute claims against others who it believes are infringing its rights and successfully defend claims brought by others who believe that the Company is infringing their rights. In addition, enforcement of its patents in foreign jurisdictions will depend on the legal procedures in those jurisdictions. Even if the Company is successful in intellectual property litigation, the Company’s involvement in such litigation could have a material adverse effect on its ability to out-license any products that are the subject of such litigation and could result in significant expense, which could materially adversely affect the use or licensing of related intellectual property and divert the efforts of its valuable technical and management personnel from their principal responsibilities, whether or not such litigation is resolved in its favor.
The Company’s reliance on third parties requires it to share its trade secrets, which increases the possibility that a competitor will discover them.
Because the Company relies on third parties to develop its products, it must share trade secrets with them. The Company seeks to protect its proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with its collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically restrict the ability of the Company’s collaborators, advisors, employees and consultants to publish data potentially relating to the Company’s trade secrets. The Company’s academic collaborators typically have rights to publish data, provided that the Company is notified in advance and may delay publication for a specified time in order to secure its intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by the Company, although in some cases it may share these rights with other parties. The Company also conducts joint research and development programs which may require it to share trade secrets under the terms of research and development collaboration or similar agreements. Despite the Company’s efforts to protect its trade secrets, its competitors may discover its trade secrets, either through breach of these agreements, independent development or publication of information including its trade secrets in cases where the Company does not have proprietary or otherwise protected rights at the time of publication. A competitor’s discovery of the Company’s trade secrets may impair its competitive position and could have a material adverse effect on its business and financial condition.
Product liability claims are an inherent risk of the Company’s business, and if the Company’s clinical trial and product liability insurance prove inadequate, product liability claims may harm its business.
Human therapeutic products involve an inherent risk of product liability claims and associated adverse publicity. There can be no assurance that the Company will be able to obtain or maintain product liability insurance on acceptable terms or with adequate coverage against potential liabilities. Such insurance is expensive, difficult to obtain and may not be available in the future on acceptable terms, or at all. An inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims could have a material adverse effect on the Company’s business by preventing or inhibiting the commercialization of its products, licensed and owned, if a product is withdrawn or a product liability claim is brought against the Company.
Intellectual property rights do not necessarily address all potential threats to our business.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business. The following examples are illustrative:
| ● |
others may be able to make compounds or formulations that are similar to our product candidates but that are not covered by the claims of any patents, should they issue, that we own or control; |
| ● |
we might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own or control; |
| ● |
we might not have been the first to file patent applications covering certain of our inventions; |
| ● |
others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| ● |
it is possible that our pending patent applications will not lead to issued patents; |
| ● |
issued patents that we own or control may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges; |
| ● |
our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights and then use the information learned from such activities to develop competitive drugs for sale in our major commercial markets; |
| ● |
we may not develop additional proprietary technologies that are patentable; and |
| ● |
the patents of others may have an adverse effect on our business. |
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and prospects.
Risks Related to our Common Shares
Our Common Share price has been volatile in recent years and may continue to be volatile.
The market prices for securities of biotechnology companies, including ours, have historically been volatile. In the year ended March 31, 2023, our Common Shares traded on the TSX at a high of $2.38 and a low of $0.54 per share and on the Nasdaq at a high of US$1.88 and a low of US$0.40 per share. A number of factors could influence the volatility in the trading price of our Common Shares, including changes in the economy or in the financial markets, industry related developments, the results of product development and commercialization, changes in government regulations, and developments concerning proprietary rights, litigation and cash flow. Our quarterly losses may vary because of the timing of costs for clinical trials, manufacturing and preclinical studies. Also, the reporting of clinical data or the lack thereof, adverse safety events involving our products and public rumors about such events could cause our share price to decline or experience periods of volatility. Each of these factors could lead to increased volatility in the market price of our Common Shares. In addition, changes in the market prices of the securities of our competitors may also lead to fluctuations in the trading price of our Common Shares.
Future sales or issuances of equity securities or the conversion of securities into Common Shares could decrease the value of the Common Shares, dilute investors’ voting power, and reduce earnings per share.
The Company may sell additional equity securities in future offerings, including through the sale of securities convertible into equity securities, to finance operations, acquisitions or projects, and issue additional Common Shares if outstanding securities are converted into Common Shares, which may result in dilution.
The Company’s board of directors will have the authority to authorize certain offers and sales of additional securities without the vote of, or prior notice to, shareholders. Based on the need for additional capital to fund expected expenditures and growth, it is likely that the Company will issue additional securities to provide such capital.
Sales of substantial amounts of securities, or the availability of such securities for sale, as well as the issuance of substantial amounts of Common Shares upon conversion or exchange of outstanding convertible or exchangeable securities, could adversely affect the prevailing market prices for securities and dilute investors’ earnings per share. A decline in the future market prices of the Company’s securities could impair its ability to raise additional capital through the sale of securities should it desire to do so.
In the past, following periods of volatility in the market price of a company’s securities, shareholders have instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm the Company’s profitability and reputation.
The market price for the Common Shares may also be affected by the Company’s ability to meet or exceed expectations of analysts or investors. Any failure to meet these expectations, even if minor, may have a material adverse effect on the market price of the Common Shares.
Failure to meet Nasdaq’s continued listing requirements could result in the delisting of our Common Shares, negatively impact the price of our Common Shares and negatively impact our ability to raise additional capital.
If we fail to satisfy the continued listing requirements of the Nasdaq Capital Market, such as corporate governance requirements or the minimum closing bid price requirement, the exchange may take steps to delist our Common Shares. Such a delisting would likely have a negative effect on the price of our Common Shares and would impair shareholders’ ability to sell or purchase our Common Shares when they wish to do so. In the event of a delisting notification, we anticipate that we would take actions seeking to restore our compliance with applicable exchange requirements, such as stabilize our market price, improve the liquidity of our Common Shares, prevent our Common Shares from dropping below such exchange’s minimum bid price requirement, or prevent future non-compliance with such exchange’s listing requirements.
On October 25, 2022, we received a letter from the Nasdaq Stock Market, LLC indicating that, for the last 30 consecutive business days, the bid price for our Common Shares had closed below the minimum $1.00 per share required for continued inclusion on the Nasdaq Capital Market under the Nasdaq Listing Rules. The notice had no effect on the listing or trading of our Common Shares.
Under Nasdaq Listing Rule 5810(c)(3)(A), if during the 180 calendar day period following the date of the notice, the closing bid price of our Common Shares is at or above $1.00 for a minimum of 10 consecutive business days, we would regain compliance with the minimum bid price requirement and our Common Shares would continue to be eligible for listing on the Nasdaq Capital Market, absent noncompliance with any other requirement for continued listing.
On April 25, 2023, we were granted an additional 180-day extension, to regain compliance with the minimum bid price requirement, which expires on October 23, 2023. If we are unable to meet the minimum closing bid price requirement under Nasdaq Listing Rule 5810(c)(3)(A) by then, Nasdaq will provide notice that our securities will be subject to delisting. At such time, we may appeal the delisting determination to a Nasdaq Hearings Panel (the “Panel”). The Company would remain listed pending the Panel’s decision.
We intend to monitor the closing bid price of our Common Shares and consider our available options if the closing bid price of our Common Shares remains below $1.00 per share, including effecting a reverse stock split. There can be no assurance that we will be able to regain compliance with the minimum bid price requirement during the additional 180-day compliance period with respect to the minimum bid price requirement, maintain compliance with the other listing requirements, or maintain the listing of our Common Shares on Nasdaq.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our Common Shares will depend, in part, on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover our company downgrade our Common Shares or publish inaccurate or unfavorable research about our business, our share price would likely decline. In addition, if our operating results fail to meet the forecast of analysts, our share price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our Common Shares could decrease, which might cause our share price and trading volume to decline.
Any future profits will likely be used for the continued growth of the business and products and will not be used to pay dividends on the issued and outstanding shares.
The Company will not pay dividends on the issued and outstanding Common Shares in the foreseeable future. If the Company generates any future earnings, such cash resources will be retained to finance further growth and current operations. The board of directors will determine if and when dividends should be declared and paid in the future based on the Company’s financial position and other factors relevant at the particular time. Until the Company pays dividends, which it may never do, a shareholder will not be able to receive a return on his or her investment in the Common Shares unless such Common Shares are sold. In such event, a shareholder may only be able to sell Common Shares at a price less than the price such shareholder originally paid for them, which could result in a significant loss of such shareholder’s investment.
If the Company is treated as a passive foreign investment company, United States shareholders may be subject to adverse U.S. federal income tax consequences.
Under the U.S. Internal Revenue Code of 1986, as amended (the “Code”), the Company will be classified as a passive foreign investment company (“PFIC”) in respect of any taxable year in which either (i) 75% or more of its gross income consists of certain types of “passive income” or (ii) 50% or more of the average quarterly value of its assets is attributable to “passive assets” (assets that produce or are held for the production of passive income). For purposes of these tests, passive income includes dividends, interest, gains from the sale or exchange of investment property and certain rents and royalties. In addition, for purposes of the above calculations, if the Company directly or indirectly owns at least 25% by value of the shares of another corporation, the Company will be treated as if it held its proportionate share of the assets and received directly its proportionate share of the income of such other corporation. PFIC status is a factual determination that needs to be made annually after the close of each taxable year, on the basis of the composition of the Company’s income, the relative value of its active and passive assets, and its market capitalization. For this purpose, the Company’s PFIC status depends in part on the application of complex rules, which may be subject to differing interpretations, relating to the classification of the Company’s income and assets. Based on the Company’s interpretation of the law, the Company’s recent financial statements, and taking into account expectations about the Company’s income, assets and activities, the Company believes that it may have been a PFIC for the taxable year ended March 31, 2023 and expects that it will be a PFIC for the current taxable year. The determination of whether the Company is a PFIC for the taxable year ended March 31, 2023 and the current taxable year will depend, in part, on whether the Company receives government grants (including certain grants similar to those previously awarded by CPRIT) during the taxable year ended March 31, 2024, and the Company’s determination of whether such grants (if received) constitute passive income for PFIC testing purposes. A separate determination must be made after the close of each taxable year as to whether the Company is a PFIC for that year, and as a result, its PFIC status may change from year to year.
If the Company is a PFIC for any taxable year during which a United States shareholder holds the Common Shares, the Company will continue to be treated as a PFIC with respect to such United States shareholder in all succeeding years during which the United States shareholder owns the Common Shares, regardless of whether the Company continues to meet the PFIC test described above, unless the United States shareholder makes a specified election once the Company ceases to be a PFIC. If the Company is classified as a PFIC for any taxable year during which a United States shareholder holds the Common Shares, the United States shareholder may be subject to adverse tax consequences regardless of whether the Company continues to qualify as a PFIC, including ineligibility for any preferred tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred, and additional reporting requirements. In certain circumstances, a United States shareholder may alleviate some of the adverse tax consequences attributable to PFIC status by making either a “qualified electing fund,” (“QEF”) election or a mark-to-market election (if the Common Shares constitute “marketable” securities under the Code). If the Company determines that it is a PFIC for this year or any future taxable year, the Company currently expects that it would provide the information necessary for United States shareholders to make a QEF election.
Each United States shareholder should consult its own tax advisors regarding the PFIC rules and the United States federal income tax consequences of the acquisition, ownership and disposition of the Common Shares.
It may be difficult for United States investors to obtain and enforce judgments against the Company because of the Company’s Canadian incorporation and presence.
The Company is a corporation existing under the federal laws of Canada. Most of the Company’s directors and officers, and several of the experts, are residents of Canada, and all or a substantial portion of their assets, and a substantial portion of the Company’s assets, are located outside the United States. Consequently, it may be difficult for holders of the Company’s securities who reside in the United States to effect service of process within the United States upon those directors, officers and experts who are not residents of the United States. It may also be difficult for holders of the Company’s securities who reside in the United States to entertain actions or enforce judgments of courts of the United States predicated upon the Company’s civil liability of the Company or its directors, officers and experts under the United States federal securities laws or the securities laws of any state or jurisdiction of the United States. Generally, original actions to enforce liabilities under U.S. federal securities laws may not be brought in a Canadian or other court. Such actions must be brought in a court in the United States with applicable jurisdiction. Persons obtaining judgments against the Company in United States courts, including judgments obtained under U.S. federal securities laws, will then be required to bring an application in a Canadian court to enforce such judgments in Canada. In addition, the protections afforded by Canadian securities laws may not be available to investors in the United States.
As a Foreign Private Issuer, the Company is subject to different U.S. securities laws and rules than a domestic U.S. issuer, which may limit the information publicly available to its U.S. shareholders.
The Company is a foreign private issuer under applicable U.S. federal securities laws and, therefore, is not required to comply with all of the periodic disclosure and current reporting requirements of the U.S. Securities Exchange Act of 1934, as amended (the “Exchange Act”), and related rules and regulations. As a result, the Company does not file the same reports that a U.S. domestic issuer would file with the United States Securities and Exchange Commission (the “SEC”), although it is required to file with or furnish to the SEC the continuous disclosure documents that the Company is required to file in Canada under Canadian securities laws. In addition, the Company’s officers, directors and principal shareholders are exempt from the reporting and “short swing” profit recovery provisions of Section 16 of the Exchange Act. Therefore, the Company’s shareholders may not know on as timely a basis when its officers, directors and principal shareholders purchase or sell securities of the Company as the reporting periods under the corresponding Canadian insider reporting requirements are longer. In addition, as a foreign private issuer, the Company is exempt from the proxy rules under the Exchange Act.
The Company may lose foreign private issuer status in the future, which could result in significant additional costs and expenses.
The Company may, in the future, lose foreign private issuer status if a majority of its Common Shares are held in the United States and the Company fails to meet the additional requirements necessary to avoid loss of foreign private issuer status, such as if: (i) a majority of the Company’s directors or executive officers are U.S. citizens or residents; (ii) a majority of the Company’s assets are located in the United States; or (iii) the Company’s business is administered principally in the United States. The regulatory and compliance costs to the Company under U.S. securities laws as a U.S. domestic issuer may be significantly more than the costs incurred as a foreign private issuer.
Regulatory Risks
Changes in government regulations, although beyond the Company’s control, could have an adverse effect on the Company’s business.
The Company depends upon the validity of its licenses and access to the data for the timely completion of clinical research. Any changes in the drug development regulatory environment or shifts in political attitudes of a government are beyond the Company’s control and may adversely affect its business. The Company’s business may also be affected in varying degrees by such factors as government regulations with respect to intellectual property, regulation or export controls. Such changes remain beyond the Company’s control and the effect of any such changes cannot be predicted. These factors could have a material adverse effect on the Company’s ability to further develop and commercialize its product candidates, if approved.
Failure to comply with the U.S. Foreign Corrupt Practices Act (“FCPA”), the Canadian Corruption of Foreign Public Officials Act (“CFPOA”), and other global anti-corruption and anti-bribery laws could subject the Company to penalties and other adverse consequences.
The FCPA and the CFPOA, as well as any other applicable domestic or foreign anti-corruption or anti-bribery laws to which the Company is or may become subject, generally prohibit corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity and requires companies to maintain accurate books and records and internal controls, including at foreign-controlled subsidiaries.
Compliance with these anti-corruption laws and anti-bribery laws may be expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, these laws present particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and physicians and other hospital employees are considered to be foreign officials. Certain payments by other companies to hospitals in connection with clinical trials and other work have been deemed to be improper payments to governmental officials and have led to FCPA enforcement actions.
The Company’s internal control policies and procedures may not protect it from reckless or negligent acts committed by the Company’s employees, future distributors, licensees or agents. The Company can make no assurance that they will not engage in prohibited conduct, and the Company may be held liable for their acts under applicable anti-corruption and anti-bribery laws. Noncompliance with these laws could subject the Company to investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension or debarment from contracting with certain persons, the loss of export privileges, whistleblower complaints, reputational harm, adverse media coverage, and other collateral consequences. Any investigations, actions or sanctions or other previously mentioned harm could have a material negative effect on the Company’s business, operating results and financial condition.
If we fail to comply with healthcare laws, we could face substantial penalties and our business, operations and financial conditions could be adversely affected.
Healthcare providers, physicians and payers play a primary role in the recommendation and prescription of any product candidates for which we may obtain marketing approval. Our future arrangements with payers and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute any product candidates for which we may obtain marketing approval. Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payers, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. Restrictions under applicable federal, state and foreign healthcare laws and regulations may affect our ability to operate and expose us to areas of risk, including:
| ● |
the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons, or entities from knowingly and willfully soliciting, offering, receiving, or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order, or arranging for or recommending the purchase, lease or order of, any good or service, for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● |
federal civil and criminal false claims laws and civil monetary penalty laws, including the U.S. civil False Claims Act (which can be enforced through “qui tam,” or whistleblower actions, by private citizens on behalf of the federal government), which prohibit any person from, among other things, knowingly presenting, or causing to be presented to the federal government claims for payment that are false or fraudulent or knowingly making, using, or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the U.S. federal government; |
| ● |
The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payer (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, which impose requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization; |
| ● |
the federal Physician Payments Sunshine Act and its implementing regulations, which require manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually to the U.S. Department of Health and Human Services under the Open Payments Program, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain advanced non-physician healthcare practitioners (such as physician assistants and nurse practitioners), and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members, as well as other state and foreign laws regulating marketing activities; |
| ● |
U.S. federal laws that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under federal health care programs; |
| ● |
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| ● |
analogous state and foreign laws and regulations, such as state and foreign anti-kickback, false claims, consumer protection and unfair competition laws which may apply to pharmaceutical business practices, including, but not limited to, research, distribution, sales and marketing arrangements as well as submitting claims involving healthcare items or services reimbursed by any third party payer, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restricts payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to file reports with states regarding pricing and marketing information, such as the tracking and reporting of gifts, compensations and other remuneration and items of value provided to healthcare professionals and entities; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could, despite our efforts to comply, be subject to challenge under one or more of such laws. Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. In addition, the approval and commercialization of any of our product candidates outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
Our actual or perceived failures to comply with applicable data protection laws and regulations, and the increasing use of social media, could lead to government enforcement actions, private litigation and/or adverse publicity and could negatively affect our operating results and business.
We are subject to data protection laws and regulations that address privacy and data security. The legislative and regulatory landscape for data protection continues to evolve, and in recent years there has been an increasing focus on privacy and data security issues. In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws and federal and state consumer protection laws govern the collection, use, disclosure and protection of health-related and other personal information.
Failure to comply with data protection laws and regulations could result in government enforcement actions, which could include civil or criminal penalties, private litigation and/or adverse publicity and could negatively affect our operating results and business. Complying with the enhanced obligations imposed by applicable international and U.S. privacy laws and regulations may result in significant costs to our business and require us to amend certain of our business practices. Further, enforcement actions and investigations by regulatory authorities related to data security incidents and privacy violations continue to increase. The future enactment of more restrictive laws, rules or regulations and/or future enforcement actions or investigations could have a materially adverse impact on us through increased costs or restrictions on our businesses, and non-compliance could result in regulatory penalties and significant legal liability.
Additionally, despite our efforts to monitor evolving social media communication guidelines and comply with applicable rules, there is risk that the use of social media by us or our employees to communicate about our drug or biologic candidates or business may cause us to be found in violation of applicable requirements, including but not limited to FDA prohibitions on the promotion of unapproved medical products. In addition, our employees may knowingly or inadvertently make use of social media in ways that may not comply with our internal policies or other legal or contractual requirements, which may give rise to liability, lead to the loss of trade secrets or other intellectual property, or result in public exposure of personal information of our employees, clinical trial patients, future customers and others. Our potential patient population may also be active on social media and use these platforms to comment on the perceived effectiveness of, or adverse experiences with, our drug or biologic candidates. Negative posts or comments about us or our drug or biologic candidates on social media could seriously damage our reputation, brand image and goodwill.
General Risk Factors
The Company’s significant shareholders may have material influence over its governance and operations.
Dr. Fahar Merchant and Ms. Rosemina Merchant (collectively, the “Merchants”), hold a significant interest in the Company’s outstanding Common Shares on a fully diluted basis. For as long as the Merchants maintain a significant interest in the Company, they may be in a position to affect the Company’s governance and operations. In addition, the Merchants may have significant influence over the passage of any resolution of the Company’s shareholders (such as those that would be required to amend the constating documents or take certain other corporate actions) and may, for all practical purposes, be able to ensure the passage of any such resolution by voting for it or prevent the passage of any such resolution by voting against it. The effect of this influence may be to limit the price that investors are willing to pay for the Common Shares. In addition, the potential that the Merchants may sell their Common Shares in the public market (commonly referred to as “market overhang”), as well as any actual sales of such Common Shares in the public market, could adversely affect the market price of the Common Shares.
The Company’s operations could be adversely affected by events outside of its control, such as health pandemics, natural disasters, geopolitical conflict and macroeconomic challenges.
Uncertainty remains regarding the severity, extent, and duration of the COVID-19 pandemic including the emergence of variant strains. While emergency measures and restrictions in response to the COVID-19 pandemic have been eased or, in certain cases, eliminated, resurgence in new COVID-19 cases, or the emergence and progression of new variants, may cause governmental authorities or companies to strengthen or re-introduce additional emergency measures and restrictions, which could materially adversely affect our business and continue to impact our operations and our employees, suppliers, partners, merchants and their customers. While we have implemented business continuity plans and taken additional steps and measures, the Company has been impacted by supply chain delays with respect to both the cGMP manufacturing and IND enabling studies and clinical trial enrollment timelines may be impacted by future waves of COVID-19. It is unknown whether and how the Company may further be affected if the pandemic persists for an extended period of time. Any other global health emergency or pandemic could raise similar issues and uncertainties. The Company’s operations could also be negatively affected by natural disasters including earthquakes, typhoons, floods and fires, the impact of which is unknown, but could have a material adverse effect on the Company’s operations.
Recent geopolitical conflicts, including the Russian invasion of Ukraine, have threatened peace and have helped to fuel global uncertainty. The outcome of the conflict is uncertain and is likely to have wide ranging consequences on the peace and stability of the region and the world economy. Certain countries including Canada and the United States, have imposed strict financial and trade sanctions against Russia and such sanctions may have far reaching effects on the global economy. The long-term impacts of the conflict and the sanctions imposed on Russia remain uncertain.
Combined with the lingering effects of the pandemic, the Russian war against Ukraine has put further pressure on the global economic order, further exacerbating inflation and global supply chain challenges and leading to an increase in market volatility. These supply chain issues could continue to negatively affect the Company’s ability to secure necessary products and supplies, while inflationary pressures could drastically increase the Company’s costs. Market volatility could also create material adverse effects for the Company as its ability to access public capital markets or private financing may be restricted owing to negative market conditions or the Company may be unable to access capital on acceptable terms, all of which could negatively impact the price of the Company’s Common Shares.
The Company’s success depends on its ability to effectively manage its growth.
The Company may be subject to growth-related risks including pressure on its internal systems and controls. The Company’s ability to manage its growth effectively will require the Company to continue to implement and improve its operational and financial systems and to expand, train and manage its employee base. Inability to deal with this growth could have a material adverse impact on its business, operations and prospects. The Company may experience growth in the number of its employees and the scope of its operating and financial systems, resulting in increased responsibilities for its personnel, the hiring of additional personnel and, in general, higher levels of operating expenses. In order to manage its current operations and any future growth effectively, the Company will also need to continue to implement and improve its operational, financial and management information systems and to hire, train, motivate, manage and retain its employees. There can be no assurance that the Company will be able to manage such growth effectively, that its management, personnel or systems will be adequate to support its operations or that the Company will be able to achieve the increased levels of revenue commensurate with the increased levels of operating expenses associated with this growth.
In the future, the Company may acquire businesses or products or form strategic alliances and the Company may not realize the benefits of such acquisitions.
The Company may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that the Company believes will complement or augment its existing business. If the Company acquires businesses with promising products or technologies, the Company may not be able to realize the benefit of acquiring such businesses if the Company is unable to successfully integrate them with its existing operations and company culture. The Company may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent it from realizing their expected benefits or enhancing the Company’s business. The Company cannot assure investors that, following any such acquisition, it will achieve the expected synergies to justify the transaction.
The Company is highly dependent upon certain key personnel and their loss could adversely affect its ability to achieve its business objective.
The loss of Dr. Fahar Merchant, the President and Chief Executive Officer, Rosemina Merchant, the Chief Development Officer, or other key members of the scientific and operating staff could harm the Company. Employment agreements exist with Dr. Merchant and Ms. Merchant, although such employment agreements do not guarantee their retention. The Company also depends on scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability. In addition, the Company believes that future success will depend in large part upon its ability to attract and retain highly skilled scientific, managerial, medical, clinical and regulatory personnel. Agreements have been entered into with scientific and clinical collaborators and advisors, key opinion leaders and academic partners in the ordinary course of business as well as with physicians and institutions who are recruiting patients into the MDNA11 clinical trial and will recruit patients into future clinical trials. Notwithstanding these arrangements, there is significant competition for these types of personnel from other companies, research and academic institutions, government entities and other organizations. The loss of the services of any of the executive officers or other key personnel could potentially harm the Company’s business, operating results or financial condition.
The Company is subject to foreign exchange risk relating to the relative value of the United States dollar.
A material portion of the Company’s expenses and cash and cash equivalents are denominated in United States dollars. As a result, the Company is subject to foreign exchange risks relating to the relative value of the Canadian dollar as compared to the United States dollar. A decline in the Canadian dollar would result in an increase in the actual amount of its expenses and adversely impact financial performance. An increase in the Canadian dollar would result in a decrease in the Canadian value of our US dollar denominated cash and cash equivalents which would adversely impact our cash balance and financial performance.
The Company’s disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
The Company’s disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by the Company in reports it files or submits under applicable securities laws is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified under applicable securities laws. The Company believes that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making may be faulty, and that breakdowns may occur because of simple error or mistake. Additionally, controls may be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in the Company’s control system, misstatements or insufficient disclosures due to error or fraud may occur undetected.
Any failure to maintain an effective system of internal controls may result in material misstatements of the Company’s consolidated financial statements or cause the Company to fail to meet the reporting obligations or fail to prevent fraud; and in that case, shareholders could lose confidence in the Company’s financial reporting, which would harm the business and could negatively impact the price of the Common Shares.
Effective internal controls are necessary to provide reliable financial reports and prevent fraud. If there is a failure to maintain an effective system of internal controls, the Company might not be able to report financial results accurately or prevent fraud; and in that case, shareholders could lose confidence in the Company’s financial reporting, which would harm the business and could negatively impact the price of the Common Shares. While the Company believes that it will have sufficient personnel and review procedures to maintain an effective system of internal controls, no assurance can be provided that potential material weaknesses in internal controls could arise. Even if it is concluded that the internal controls over financial reporting provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with IFRS, as issued by the International Accounting Standards Board (“IASB”), because of its inherent limitations, internal control over financial reporting may not prevent or detect fraud or misstatements. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm results of operations or cause a failure to meet future reporting obligations.
Our internal computer systems, or those used by our contractors or consultants or third parties on which we rely, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems, and those of the third parties on which we rely, are vulnerable to damage from cyber-attacks, computer viruses, malware, natural disasters, terrorism, war and telecommunication and electrical failures. The risk of a security breach or disruption, particularly through cyber-attacks, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions have increased. If such an event were to occur and cause interruptions in our operations or those of the third parties, it could result in a material disruption of our product development programs and our business operations. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. In some cases, data cannot be reproduced. Likewise, we rely on third parties for the manufacture of our product candidates and to conduct clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach results in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, we could incur significant liability and damage to our reputation and the further development and commercialization of our future product candidates could be delayed. Our insurance coverage may not be adequate to cover all the costs related to such breaches or attacks.
In addition, the unauthorized dissemination of sensitive personal information could expose us or other third parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm our business.
The Company may pursue opportunities for further research and development or additional business opportunities in order to develop its business and/or products.
From time to time, the Company may pursue opportunities for further research and development of other products. Such activities may distract management time and attention from the Company’s principal product candidates and business and the Company’s success in these activities will depend on its ability to identify suitable technical experts, market needs, and effectively execute any such research and development opportunities. Any research and development would be accompanied by risks as a result of the use of business efforts and funds. In the event that the Company chooses to raise debt capital to finance any such research or development opportunities, its leverage will be increased. There can be no assurance that the Company would be successful in overcoming these risks or any other problems encountered in connection with any research or development opportunities.
ITEM 4. INFORMATION ON THE COMPANY
4.A. History and Development of the Company
Name, Address and Incorporation
We were incorporated under the laws of Canada on February 2, 2015, under the name A2 Acquisition Corp. pursuant to the Business Corporations Act (Alberta). Prior to the completion of the MTI Reverse Takeover, the Company amended its articles, changing its name to “Medicenna Therapeutics Corp.” On November 13, 2017, Medicenna continued under the CBCA.
Our registered office is located at 2 Bloor St. W., 7th Floor, Toronto, Ontario M4W 3E2 and our telephone number is (416) 648-5555. Our agent for service of process in the United States is CT Corporation System, located at 218 Liberty Street, New York, New York 10005. Our website address is https://www.medicenna.com/. The information contained on, or that can be accessed through, our website is not a part of this Annual Report. We have included our website address in this Annual Report solely as an inactive textual reference.
General Development of the Business of the Company
Medicenna Therapeutics Corp., formerly A2 Acquisition Corp. (“A2”), is the resulting issuer following a “three-cornered” amalgamation involving A2, 1102209 B.C. Ltd., a wholly owned subsidiary of A2 incorporated pursuant to the Business Corporations Act (British Columbia) (“BCBCA”), and Medicenna Therapeutics Inc. (“MTI”), completed on March 1, 2017.
A2 was formed by articles of incorporation under the Business Corporations Act (Alberta) on February 2, 2015, and following its initial public offering, was a capital pool company (“CPC”) listed on the TSX Venture Exchange (“TSXV”). As a CPC, A2 had no assets other than cash and did not carry on any operations other than identifying and evaluating opportunities for the acquisition of an interest in assets or businesses for the completion of a qualifying transaction.
On March 1, 2017, A2 completed its qualifying transaction in accordance with the policies of the TSXV by way of reverse takeover of A2 by the shareholders of MTI (the “Qualifying Transaction”). In addition, on March 1, 2017 and prior to the completion of the Qualifying Transaction, the Company amended its articles as a result of (a) implementing a consolidation (the “Consolidation”) of its pre-Qualifying Transaction common shares (the “A2 Shares”) on the basis of one new common share of the Company (each, a “Common Share”) for every fourteen A2 Shares (1:14) and (b) changing its name to Medicenna Therapeutics Corp.
On August 2, 2017 Medicenna graduated to the main board of the Toronto Stock Exchange (“TSX”). On November 13, 2017, Medicenna continued under the Canada Business Corporations Act (“CBCA”).
On August 24, 2020, Medicenna began trading on the Nasdaq under the symbol “MDNA”.
On March 30, 2021, the Company set up its wholly owned subsidiary Medicenna Australia PTY Ltd (Australia).
Property, Plant and Equipment
We currently utilize short-term (less than 1 year) office leases for our head office in Toronto, Ontario, Canada and our office in Houston, Texas. We do not have any leasehold improvements, office furniture or capitalized equipment. We do not own or lease any other office space, laboratory facilities, manufacturing facilities or equipment and do not have any current plans to construct or acquire any facilities.
Recent Developments
On April 17, 2023, we announced that new preclinical data characterizing the Interleukin 13 (IL-13) Superkines, MDNA132 and MDNA213 (an improved version of MDNA132) and a series of next generation IL-13 Superkine therapies, were presented at the AACR Annual Meeting which took place in Orlando, Florida. The AACR poster included data demonstrating that both MDNA132 and MDNA213 exhibit highly selective binding to the IL-13 decoy receptor (IL-13Rα2) and, in a murine model, selectively accumulate in the tumor microenvironment (TME) for several days.
On April 25, 2023, the Company received an extension notice (the “Extension Notice”) from Nasdaq granting the Company’s request for a 180-day extension to regain compliance with the minimum bid price requirement (“Minimum Bid Requirement”) of US$1.00 per share under the Nasdaq Listing Rule 5450(a)(1). The Company has until October 23, 2023 to meet the requirement. The Extension Notice had no immediate effect on the listing or trading of the Company’s Common Shares on Nasdaq, and the Company’s operations are not affected by the receipt of the Extension Notice.
Three Year History
Year ended March 31, 2021
On April 15, 2020, Medicenna announced the closing of the full over-allotment option to purchase an additional 1,693,548 Common Shares of Medicenna at a price of $3.10 per share, in connection with its public offering of Common Shares, which initially closed on March 17, 2020. The total gross proceeds arising from this financing were $40.25 million.
On May 29, 2020, Medicenna announced presentation of data from its Phase 2b trial of bizaxofusp at the virtual 2020 Annual Meeting of ASCO. The oral poster discussion focused on additional data supporting the clinical efficacy of bizaxofusp in patients with rGBM with a median survival and OS-12 in the high dose and IL4R high patient population (n = 32) was 15.8 months and 62% vs 7.0 months and 18%, respectively, when compared to the eligibility matched synthetic control arm (“SCA”), also known as the external control arm (“ECA”).
On May 29, 2020, Medicenna announced presentation of data on MDNA11, one of its candidates from the IL-2 Superkine program, at the virtual 2020 ASCO Annual Meeting. The poster presentation focused on encouraging data in NHP for MDNA11.
On August 24, 2020, the Company’s Common Shares began trading on the Nasdaq under the symbol “MDNA”.
On September 30, 2020, Dr. Jack Geltosky, an experienced pharmaceutical licensing executive with a strong research and development background, was elected to Medicenna’s board of directors.
On October 15, 2020, we announced positive outcomes following the End of Phase 2 (“EOP2”) meeting with the FDA. The FDA agreed that we could conduct an innovative open-label hybrid Phase 3 registration trial that allows use of a substantial number of patients (two-thirds) from a matched ECA to support regulatory approval of bizaxofusp for rGBM.
On October 26, 2020, we also announced a Late Breaking Abstract poster presentation at the 32nd ENA Symposium on Molecular Targets and Cancer Therapeutics. Amongst an all-comer population, a single treatment with bizaxofusp resulted in at least 100% increase in both 12-month progression free survival (“PFS-12”) (27% versus 2 to 10%) and 2-year survival (20% vs 5 to10%) when compared to what is achieved with approved therapies. In a subset of all-comer patients treated with transient low dose bevacizumab, to reduce steroid use, mOS was 21.8 months and OS-24 was 44%.
On November 4, 2020 Medicenna held a positive Scientific Advice Meeting for MDNA11 (similar to a pre-IND meeting) with the United Kingdom Medicines and Healthcare products Regulatory Agency (“MHRA”). MHRA confirmed that our plans for CMC, pre-clinical and Phase 1/2a clinical trial were appropriate for submission of an IMPD in calendar 2021 in order to commence first in human studies with MDNA11 in the UK.
On December 9, 2020, we presented at an oral session at the 2nd Annual Glioblastoma Drug Development Summit. The presentation included updated data from the MDNA55 Phase 2b clinical trial, as well as an overview of the planned MDNA55 Phase 3 registration trial.
On December 11, 2020, we hosted a KOL call on MDNA55 featuring presentations by KOLs who provided an overview on the current treatment landscape for rGBM, highlighted the results from the MDNA55 Phase 2b clinical trial and addressed the advantages of the hybrid Phase 3 design agreed by the FDA.
On December 30, 2020, we announced that we entered into a sales agreement with SVB Securities LLC (f/k/a SVB Leerink LLC) (“SVB”) acting as sales agent, pursuant to which the Company may, from time to time sell, through the ATM offering, such number of common shares as would have an aggregate offering price of up to US$25.0 million (the “ATM Facility”). We plan to use the net proceeds of the ATM offering for general corporate purposes including, but not limited to working capital expenditures, research and development expenditures, and clinical trial expenditures. During the fourth quarter of fiscal 2021, a total of 1,398,357 common shares were sold under the ATM Facility for total gross proceeds of US$5.8 million ($7.1 million). As at March 31, 2022, US$16 million ($20.5 million) remained available under the ATM Facility.
On March 25, 2021, Medicenna presented preclinical data from the Company’s Superkine platform programs at the virtual Cytokine-Based Cancer Immunotherapies Summit.
Year ended March 31, 2022
On April 12, 2021, Medicenna announced new preclinical data demonstrating the potentially potent immune modulatory effects of MDNA19-MDNA413, an IL-2/IL-13 dual specific cytokine derived from the Company’s BiSKITs™ platform.
On June 23, 2021, Medicenna announced submission of a clinical trial application to the Human Research Ethics Committee in Australia to initiate a Phase 1/2 ABILITY Study to assess the safety, PK, PD and anti-tumor activity of MDNA11 in patients with advanced solid tumors.
On September 14, 2021, Medicenna announced that the first patient was dosed in the MDNA11 Phase 1/2 ABILITY Study.
On September 20, 2021, Medicenna announced that the USPTO had issued U.S. Patent No. 11,117,943, titled “Superagonists and Antagonists of Interleukin-2.” The patent provides intellectual property (“IP”) protection for methods of treating a wide range of cancers specified in the claims with IL-2 variants such as MDNA11.
On September 23, 2021, Medicenna announced the election of John H. Sampson, MD, PhD, MBA, a world-renowned clinician-scientist, to its board of directors.
On October 27, 2021, Medicenna announced that the FDA had allowed the Company to expand the Phase 1/2 ABILITY Study at clinical trial sites in the United States, under an Investigational New Drug (“IND”) application.
On December 17, 2021, Medicenna announced that Health Canada had approved the expansion of the Phase 1/2 ABILITY Study to clinical trial sites in Canada.
On December 22, 2021, Medicenna announced preliminary data from the Phase 1/2 ABILITY Study, which were subsequently updated as described above.
On January 26, 2022, Medicenna announced the peer-reviewed publication of preclinical data on MDNA11 entitled “Fine-tuned Long-Acting Interleukin-2 Superkine Potentiates Durable Immune Responses in Mice and Non-Human Primate” published in the Journal for ImmunoTherapy of Cancer.
On January 31, 2022, Medicenna announced the formation of its Scientific Advisory Board (“SAB”). The SAB consists of four highly accomplished leaders in oncology, immunotherapy and drug development: Sergio Quezada, PhD (Chairman), Burkhard Becher, PhD, David Mooney, PhD, and William Redmond, PhD.
On March 3, 2022, Medicenna announced the formation of its Clinical Advisory Board comprised of Paolo Ascierto, M.D., Lillian Siu, M.D., FRCPC, and Hussein Tawbi, M.D., PhD, and the appointment of Dr. Kapil Dhingra as a Strategic Advisor.
Year ended March 31, 2023
On April 8, 2022, Medicenna announced new preclinical data highlighting the potent anti-tumor efficacy of the next-generation BiSKIT, anti-PD1-MDNA109FEAA (now known as MDNA223), an anti-PD1 antibody fused to an IL-2 Superkine as well as on its long-acting dual IL-4/IL-13 super-antagonist, Fc-MDNA413, during poster sessions at the AACR Annual Meeting.
On May 2, 2022, Medicenna announced new clinical data from the third cohort of the Phase 1/2 ABILITY Study of MDNA11 and on May 11, 2022, Medicenna presented clinical data from the Phase 1/2 ABILITY Study during a poster presentation at the 9th Annual Frontiers in Cancer Immunotherapy Meeting, organized by the New York Academy of Sciences. These data were subsequently updated as described below.
On June 9, 2022, we announced that the USPTO had issued U.S. Patent No. 11,352,402 titled, “Interleukin-4 Receptor-Binding Fusion Proteins And Uses Thereof.” The patent provides IP protection for composition and methods of treating degenerative diseases via administration of a fusion protein comprising an IL-4 or IL-13 Superkine and an anti-apoptotic Bcl-2 family polypeptide. The patent’s term extends into at least 2038 without accounting for any potential extensions.
On July 12, 2022, the USPTO issued U.S. Patent No. 11,117,943, titled “Superagonists and Antagonists of Interleukin-2.” The patent provides IP protection for methods of treating leukemia using IL-2 muteins, such as MDNA209, that have an increased binding capacity for IL-2Rb and a decreased binding capacity for IL-2Rgc.
On July 27, 2022, Medicenna announced new clinical data on safety, PK, PD and anti-tumor activity from the Phase 1/2 ABILITY Study of MDNA11, which were presented at the Cytokine Based Drug Development Summit, held in Boston. These data were subsequently updated and are described below.
On August 11, 2022, we raised gross proceeds of US$20.0 million ($25.6 million) pursuant to an underwritten public offering of units, with each unit consisting of one Common Share and one Common Share purchase warrant. Each Common Share purchase warrant entitles the holder to purchase one Common Share at an exercise price of US$1.85 until expiration of the warrants on August 9, 2027.
On September 13, 2022, we entered into a Clinical Trial Collaboration and Supply Agreement (“CTCSA”) with Merck (known as MSD outside the United States and Canada) to evaluate MDNA11 in combination with KEYTRUDA® (pembrolizumab), an anti-PD-1 (programmed death receptor-1) therapy, in the ongoing Phase 1/2 ABILITY Study.
On September 22, 2022, Medicenna announced presentation of data demonstrating the anti-tumor activity of the MDNA223 and MDNA413. The data were featured in two separate poster presentations at the 10th Annual Meeting of the International Cytokine & Interferon Society, held in Big Island, Hawaii.
On September 28, 2022, Medicenna announced new clinical data on anti-tumor activity from the Phase 1/2 ABILITY study of MDNA11. These data were subsequently updated and are described below.
On November 10, 2022, the Company announced new safety, PK, and PD data from the first four dose escalation cohorts of the Phase 1/2 ABILITY Study of MDNA11. The data were featured in two posters presented at the Society for Immunotherapy of Cancer (“SITC”) 37th Annual Meeting held in Boston.
In December 2022, previously reported data from the Phase 1/2 ABILITY Study of MDNA11 were featured in an oral presentation at the 2022 Immunotherapy Bridge Conference. The presentation, titled “Early Results of an IL-2 Superkine (MDNA11) from the Phase 1/2 ABILITY Study in Advanced Solid Tumors” was delivered by Arash Yavari, M.B.B.S., DPhil., M.R.C.P., Principal Investigator at the Radcliffe Department of Medicine, University of Oxford and Principal Clinical Advisor to Medicenna.
Additional updates on the anti-tumor efficacy of cohorts 1-4 were provided on March 30, 2023.
On January 5, 2023, Medicenna announced that the USPTO had issued U.S. Patent No. 11,542,312 titled “IL-2 Superagonists in Combination with Anti-PD-1.” The patent provides IP protection for methods of treating cancer with an IL-2 Superkine such as MDNA11 and a PD1 (for example, pembrolizumab), PDL1 or CTLA-4 checkpoint inhibitor in combination, as planned in the on-going ABILITY Study, or as a single agent (such as MDNA223) using our BiSKIT™ platform. The patent’s term extends into at least 2039, without accounting for any potential extensions.
In January 2023, the full results of a single-arm Phase 2b trial of bizaxofusp in patients with recurrent glioblastoma were published in the peer-reviewed journal Neuro-Oncology. Results showed the trial met its primary endpoint, with median overall survival (“mOS”) in the primary and supportive analysis populations exceeding the trial’s pre-defined success criteria and the mOS historically achieved with currently approved therapies.
On February 17, 2023, Medicenna announced that that it had entered into a sales agreement with Oppenheimer & Co. Inc,. acting as sales agent, pursuant to which the Company may, from time to time, sell through an ATM offering on the Nasdaq such number of Common Shares that would have an aggregate offering price of up to US$10 million under the ATM prospectus supplement. Medicenna will determine, in its sole discretion, the time, minimum price and maximum number of Common Shares to be sold under the ATM offering.
On March 15, 2023, we announced the publication of an abstract at the 2023 AACR Annual Meeting which described preclinical studies characterizing a long-acting version of MDNA132 and BiSKITs™, comprising MDNA132 fused to an IL-2 super-agonist or anti-PD1 antibody. MDNA132 is an IL-13 Superkine designed to enable targeted delivery of immunotherapies to the tumor microenvironment. MDNA132 exhibits high affinity and selectivity for the IL13Rα2, which is highly overexpressed in various tumors such as pancreatic, prostate, bladder, colorectal, breast and lung cancer but minimally expressed in healthy tissues.
On March 30, 3023, we announced updated data from the Phase 1/2 ABILITY Study. These data include the most recent antitumor activity data from the trial’s first four dose escalation cohorts and initial PK/PD data from the fifth dose escalation cohort.
Significant Acquisitions During Fiscal Year Ending March 31, 2023
Except as set forth herein, the Company has not completed any significant acquisitions for which disclosure would be required.
Additional Information
Additional information about us may be found on SEDAR at www.sedar.com. The SEC maintains an Internet site that contains reports and other information regarding issuers, such as we, that file electronically, with the SEC at www.sec.gov. Additional information, including directors’ and officers’ remuneration and indebtedness, principal holders of our securities, options to purchase securities and securities authorized for issuance under equity compensation plans, is contained in our Management Information Circular for our most recent annual meeting of shareholders. Additional information may also be found in our audited financial statements and related management’s discussion and analysis for our most recently completed financial year.
4.B. Business Overview
Overview
Medicenna is an immunotherapy company developing novel, highly selective versions of interleukin-2 (“IL-2”), interleukin-4 (“IL-4”) and interleukin-13 (“IL-13”) tunable cytokines, called “Superkines”. These Superkines can be developed either on their own as short or long-acting therapeutics or fused with cell killing proteins to generate “Empowered Superkines” that precisely deliver potent payloads to cancer cells without harming adjacent healthy cells. Superkines can also be fused with a large variety of proteins, antibodies and even other Superkines to incorporate two synergistic therapeutic activities into one molecule, creating novel Bi-Functional SuperKine ImmunoTherapies referred to by Medicenna as “BiSKITs”™. Medicenna’s mission is to become the leader in the development and commercialization of Superkines, Empowered Superkines and BiSKITs™ for the treatment of a broad range of cancers and other diseases. The Company seeks to achieve its goals by drawing on its expertise, and that of world-class collaborators and advisors, to develop revolutionary medicines using evolutionary Superkines. Compared to naturally occurring cytokines – that bind to multiple receptors on many cell types – Superkines are engineered with unique selectivity toward specific receptor subtypes and defined target cell subsets to precisely activate or inhibit relevant signaling pathways or immune cells in order to improve therapeutic efficacy and safety.
Medicenna has built diverse platforms, each comprised of a pipeline of Superkine candidates in-licensed from Leland Stanford Junior University (“Stanford”). This includes the MDNA109 platform that consists of IL-2 agonists, IL-2 antagonists and partial agonists of IL-2. Additional assets from Stanford also include several super-agonists of IL-4 and IL-13 and dual IL-4/IL-13 antagonists. In addition, Medicenna has also independently developed therapeutic agents based on its Empowered Superkine and BiSKIT™ platforms.
The most advanced of the Superkine programs is the MDNA109 platform which is a genetically engineered IL-2 Superkine designed to specifically bind to CD122 (IL-2Rβ) with high affinity. To further enhance its selectivity, two additional mutations (FEAA) were incorporated in MDNA109 to abolish binding to CD25. To improve the PK properties of the highly selective version of MDNA109 (MDNA109FEAA), it was genetically fused to protein scaffolds such as the Fc domain of IgG1 (MDNA19) or recombinant human albumin (MDNA11) effectively increasing the size of the Superkine and improving its half-life to avoid frequent daily dosing which is required for an improved version of IL-2, Proleukin®.
We believe that, unlike Proleukin®, both MDNA11 and MDNA19, have superior PK properties, lack CD25 binding to improve safety and reduce immune suppression, potently stimulate effector T cells, reverse natural killer (“NK”) cell exhaustion and act with exceptional synergy when combined with checkpoint inhibitors and other therapeutic modalities.
Although MDNA19 was initially identified as the Company’s lead IL-2 candidate, a pilot NHP study comparing MDNA11 with MDNA19 demonstrated that the former had better PK and PD features. Medicenna is therefore advancing the clinical development of MDNA11 as it is a more promising molecule and has been selected as the lead IL-2 Superkine candidate. Medicenna has initiated the Phase 1/2 ABILITY Study (A Beta-only IL-2 ImmunoTherapY Study) with MDNA11 (the “ABILITY Study”). MDNA19 remains relevant for Medicenna as it provides unique design features in the development of our BiSKITs™ platform. Our BiSKITs™ platform allows us to develop designer Superkines by fusing them to other proteins, antibodies, cytokines or other Superkines resulting in two distinct but synergistic functions into one molecule: a BiSKIT™.
Complementing our MDNA109 platform is bizaxofusp (formerly MDNA55), Medicenna’s Empowered Superkine, for the treatment of recurrent glioblastoma (“rGBM”), the most common and uniformly fatal form of brain cancer. Bizaxofusp is a fusion of a circularly permuted version of IL-4, fused to a potent fragment of the bacterial toxin, Pseudomonas exotoxin (“PE”), and is designed to preferentially target tumor cells that over-express the interleukin 4 receptor (“IL-4R”). Bizaxofusp has been studied in five clinical trials in 132 patients, including 112 patients with rGBM, the results of which support our belief that it has superior efficacy when compared to the current standard of care (“SOC”). Bizaxofusp has secured Orphan Drug Status from the United States FDA and the EMA as well as Fast Track Designation from the FDA for the treatment of rGBM and other types of high-grade glioma. We continue to pursue a strategic partnership to facilitate bizaxofusp’s further development and commercialization.
OUR PRODUCT CANDIDATES
Superkines
Developed by scientists at Stanford, Medicenna has exclusively licensed highly selective versions of Superkines. These Superkines can be developed either on their own as short or long-acting therapeutics or fused with cell killing proteins in order to generate Empowered Superkines that precisely deliver potent toxins to cancer cells without harming adjacent healthy cells. Compared to naturally occurring cytokines – that bind to multiple receptor types on many cell types – Superkines are engineered with unique specificity toward defined target cell subsets to enable precise activation or inhibition of relevant immune cells in order to improve therapeutic efficacy and safety. Superkines can also be fused with a large variety of proteins, antibodies and even other Superkines to incorporate two synergistic mechanisms of action into one molecule: a BiSKIT™.

IL-2 Superkines
Recombinant IL-2 (also known as Proleukin®) was one of the first effective immunotherapies developed to treat cancer due to its ability to expand T cells, the central players in cell-mediated immunity. Originally discovered as a growth factor for T cells, IL-2 can also drive the generation of activated immune cells, immune memory cells and immune tolerance by virtue of its ability to bind to the IL-2 receptor.
The IL-2 receptor is composed of three different subunits, IL-2Rα (also known as CD25), IL-2Rβ (CD122) and IL-2Rγ (CD132). The arrangement of these different proteins determines the response to IL-2 signaling.
The IL-2β and IL-2γ components together make a receptor capable of binding IL-2, but only moderately so. When all three components are together, including IL-2Rα, the receptor binds IL-2 with a much higher affinity. This complete receptor is usually found on regulatory T cells (“Tregs”), as these T cells constantly express high levels of CD25, which dampens an ongoing immune response against the tumor. Furthermore, at high doses used for cancer patients, interaction of IL-2 with CD25 may cause life threatening adverse events, requiring administration of Proleukin® in an intensive care unit. The intermediate affinity receptor, composed of just the IL-2β and IL-2γ components, is more often found on Natural Killer (“NK”) cells and CD8 T cells responsible for attacking cancer cells.

Altering IL-2’s propensity for binding these receptors could encourage greater activation of cancer fighting immune cells and/or block the function of regulatory T cells. Medicenna’s MDNA109 (MDNA11) and MDNA209 platforms take advantage of this dynamic by binding to specific receptors and either activating (MDNA109) or blocking them (MDNA209). The majority of development has been focused on the MDNA109 platform candidates, in particular MDNA11 which is currently enrolling patients in the Phase 1/2 ABILITY study.
Like the MDNA109 platform, MDNA209 based therapeutics bind with exceptional affinity to IL-2Rβ, but have varying degrees of reduced affinity towards the common IL-2γ receptor which in turn blocks signaling and activation of NK cells and effector CD8 T cells. Therefore, we believe that the MDNA209 platform can offer a variety of candidates that are either partial agonists, partial antagonists or complete antagonists, enabling us to dampen the signaling properties of an over-active immune system to an amplitude that elicits desired therapeutic function without causing undesired toxicity. We believe MDNA209 variants can therefore be used to treat a host of autoimmune diseases such as multiple sclerosis and preliminary studies (Mitra et al., 2015) have shown that MDNA209 variants can also mitigate graft versus host disease (GvHD) following transplantation. Limited work on MDNA209 has been initiated but development timelines have not been established at this time.
MDNA11
MDNA109 (a precursor to MDNA19 and MDNA11) is an enhanced version of IL-2 that binds up to 200 times more effectively to IL-2Rβ, thus greatly increasing its ability to activate and proliferate the immune cells needed to fight cancer. Because it preferentially binds IL-2Rβ and not the receptor containing IL-2Rα, MDNA109 preferentially drives NK and effector CD8T cell responses over regulatory T cells.
Additionally, MDNA109 reverses NK cell exhaustion and acts with exceptional synergy when combined with checkpoint inhibitors.
One of the development challenges with MDNA109 was its short half-life, similar to native IL-2, which would require frequent dosing. In order to extend the half-life of MDNA109, Medicenna fused inactive protein scaffolds to MDNA109 to generate Fc-fusions (“Fc”) and Albumin fusions (“Alb”), whereby these fusions have better pharmacokinetic properties enabling less frequent dosing without sacrificing potential efficacy or safety.
Further modifications were made to MDNA109 in its extended half-life forms to enhance pharmacodynamics and further enhance selectivity in order to reduce binding to CD25 which is associated with the toxic side effect profile of Proleukin®. These modifications have provided us with two candidates in development, MDNA19 and MDNA11, of which MDNA11 has been selected as the lead candidate for clinical development while MDNA19 is being used in Medicenna’s BiSKIT program. MDNA11 is currently enrolling patients in the Phase 1/2 ABILITY study in Australia, Canada and the United States for the treatment of various solid tumors.
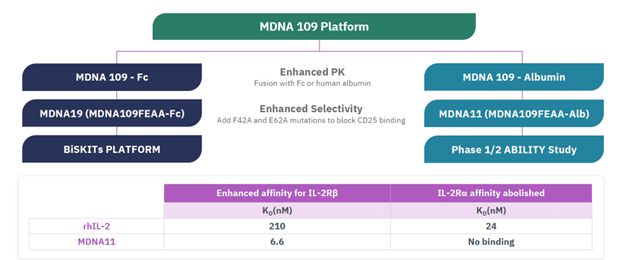
On May 29, 2020, Medicenna announced the virtual presentation of data on MDNA11 at the 2020 ASCO Annual Meeting. The poster presentation focused on encouraging data in NHP for MDNA11 and demonstrated that MDNA11 had better in-vitro and in-vivo characteristics than MDNA19 and was therefore selected as the lead candidate to move into clinical development.
On October 26, 2020, we announced a poster presentation at the 32nd ENA Symposium on Molecular Targets and Cancer Therapeutics. The presentation of preclinical results featured data on MDNA11 as well as data related to long acting bispecific IL-2/IL-13 Superkine that is designed to simultaneously activate cancer killing immune cells while reversing anti-inflammatory TME.
On November 4, 2020 Medicenna held a positive Scientific Advice Meeting for MDNA11 (similar to a pre-IND meeting) with the UK MHRA. MHRA confirmed that our plans for CMC, pre-clinical and Phase 1/2a clinical trial design would be appropriate for submission of an IMPD in calendar 2021 in order to commence first in human studies with MDNA11 in the UK.
On March 25, 2021, Medicenna presented preclinical data from the Company’s Superkine platform programs at the virtual Cytokine-Based Cancer Immunotherapies Summit. The presentation included data showing that treatment with MDNA11 alone or in combination with anti-PD-1 therapy led to tumor growth inhibition and complete responses in a murine MC38 tumor model.
On June 23, 2021, we announced that we had submitted a clinical trial application to a Human Research Ethics Committee in Australia to initiate a Phase 1/2 clinical study of MDNA11. Medicenna’s Phase 1/2 ABILITY Study is designed to assess the safety, PK, PD, and anti-tumor activity of various doses MDNA11 administered intravenously every 2 weeks, in patients with advanced solid tumors. The basket, dose finding study includes a dose escalation phase (Phase 1) followed by a dose expansion phase (Phase 2) with both an MDNA11 monotherapy arm as well as a combination arm designed to evaluate MDNA11 with a checkpoint inhibitor. The Phase 1 portion of the study includes patients with advanced and metastatic melanoma and renal cell carcinoma, where Proleukin® is known to have clinical activity, as well as cluster of other advanced solid tumors in order to explore safety, PK, PD, recommended dose for expansion (“RDE”) and the pan-tumor potential of MDNA11. The study also permits alternative dosing schedules, as well as options for intra-patient dose escalation.

On September 14, 2021, Medicenna announced that it had dosed the first patient in the Phase 1/2 ABILITY Study.
On September 20, 2021, Medicenna announced that the USPTO issued U.S. Patent No. 11,117,943, titled “Superagonists and Antagonists of Interleukin-2.” The patent provides intellectual property protection for methods of treating a wide range of cancers specified in the claims with IL-2 variants such as MDNA11, which is Medicenna’s selective, long-acting and novel IL-2 super-agonist. The patent’s term extends into at least 2032, without accounting for any potential extensions.
On October 7, 2021, Medicenna announced the presentation of new MDNA11 preclinical data at the AACR-NCI-EORTC Annual International Meeting. Data presented in the poster were from murine studies evaluating the anti-tumor activity of MDNA11 as monotherapy and in combination with anti-PD1 checkpoint inhibition in MC38 colon cancer model and NHP studies evaluating safety, PK, and PD of MDNA11.
On October 27, 2021, Medicenna announced that the FDA allowed it to proceed with the Phase 1/2 ABILITY Study and begin enrolling patients in the United States under its IND.
On December 17, 2021, Medicenna announced that Health Canada approved the expansion of the Phase 1/2 ABILITY Study to clinical trial sites in Canada.
On December 22, 2021, Medicenna announced preliminary data from the Phase 1/2 ABILITY (study of MDNA11, the Company’s selective, long-acting and novel IL-2 super-agonist). These data were subsequently updated in May 2022.
On January 26, 2022, Medicenna announced the peer-reviewed publication of preclinical data on MDNA11. The paper, which was published in the Journal for ImmunoTherapy of Cancer, is entitled, “Fine-tuned Long-Acting Interleukin-2 Superkine Potentiates Durable Immune Responses in Mice and Non-Human Primate.”
Key data and conclusions from the paper include:
In vitro studies:
| ● |
MDNA11 demonstrated a 30-fold increase in binding affinity for IL-2Rβ vs. rhIL-2. |
| ● |
MDNA11 showed no affinity for IL-2Rα at concentrations up to 2,000 nM MDNA11. |
| ● |
MDNA11 showed enhanced signaling in anti-cancer T and NK cells and reduced activation of pro-tumor Treg cells when compared to rhIL-2 as shown by 231-fold and 124-fold enhancements in CD8+/Treg and NK/Treg pSTAT EC50 ratios, respectively. |
Murine studies:
| ● |
The terminal half-life of MDNA11 in mice was 25 times greater than that of rhIL-2. |
| ● |
Cell depletion studies showed that both, CD8+ T cells and NK cells are important for MDNA11 mediated anti-tumor efficacy. |
| ● |
There was enhanced activation of CD8+ T cells within the tumors as demonstrated by significant increase in expression of intracellular interferon γ. |
| ● |
MDNA11 alone or in combination with checkpoint inhibitors generated durable complete responses and provided long-term protection against tumor re-challenge in murine cancer models. |
NHP studies:
| ● |
MDNA11 preferentially induced durable proliferation and expansion of anti-cancer immune effector cells (CD8+ T-cells, NK cells and non-Treg CD4+ T-cells), with limited stimulation of pro-tumor Treg cells. |
| ● |
Proliferation of anti-cancer immune effector cells remained elevated for at least 7 days following treatment with MDNA11. |
| ● |
MDNA11 was well tolerated. The main safety observations of reduced activity and diarrhea were primarily observed at the highest dose level following the first dose and were generally transient in nature. |
On May 2, 2022, Medicenna announced new clinical data from the third cohort of the Phase 1/2 ABILITY Study of MDNA11 and on May 11, 2022, Medicenna presented clinical data from the Phase 1/2 ABILITY Study during a poster presentation at the 9th Annual Frontiers in Cancer Immunotherapy Meeting, organized by the New York Academy of Sciences. These data were subsequently updated as described below.
On July 27, 2022, Medicenna announced new clinical data on safety, PK, PD and anti-tumor activity from the Phase 1/2 ABILITY Study of MDNA11 which were presented at the Cytokine Based Drug Development Summit, held in Boston. These data were subsequently updated as described below.
On September 13, 2022 we announced that we had entered into the CTCSA with Merck to evaluate MDNA11 in combination with KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 (programmed death receptor-1) therapy, in the ongoing Phase 1/2 ABILITY Study. Under the terms of the CTCSA, Medicenna will sponsor the study and Merck will supply KEYTRUDA®. The two companies will establish a Joint Development Committee to optimally advance the study’s combination arm.
On November 10, 2022, the Company announced new safety, PK, and PD data from the first four dose escalation cohorts of the Phase 1/2 ABILITY Study of MDNA11. The data were featured in two posters presented at the SITC 37th Annual Meeting. Additional updates on the anti-tumor efficacy of cohorts 1-4 were provided on March 30, 2023 and the cumulative data is summarized below.
In December 2022, previously reported data from the Phase 1/2 ABILITY Study of MDNA11 were featured in an oral presentation at the 2022 Immunotherapy Bridge Conference. The presentation, titled “Early Results of an IL-2 Superkine (MDNA11) from the Phase 1/2 ABILITY Study in Advanced Solid Tumors” was delivered by Arash Yavari, M.B.B.S., DPhil., M.R.C.P., Principal Investigator at the Radcliffe Department of Medicine, University of Oxford and Principal Clinical Advisor to Medicenna.
Additional updates on the anti-tumor efficacy of cohorts 1-4 were provided on March 30, 2023.
In the dose escalation portion of the ABILITY Study, MDNA11 is administered intravenously, as a monotherapy, once every two weeks to patients with advanced solid tumors. The trial’s first two cohorts evaluated MDNA11 doses ≤ 10 µg/kg. The trial’s third cohort was administered a dose of 30 µg/kg. Patients in the fourth and fifth dose escalation cohorts receive two 30 µg/kg “priming” doses of MDNA11 before stepping up to receive fixed doses of 60 and 90 µg/kg, respectively.
Key data from patients enrolled in the trial’s four initial dose escalation cohorts include:
Demographics:
| - |
Patients enrolled in the study to date (N=14) have failed up to four lines of prior systemic therapy. |
| - |
11 of 14 patients have relapsed, were not tolerant to or did not respond to at least one prior immunotherapy with a checkpoint inhibitor. |
Safety:
| - |
MDNA11 treatment in Cohort 4 (comprised of two step-up doses of 30 µg/kg followed by fixed doses of 60 µg/kg every two weeks) was not associated with any dose-limiting toxicities. |
| - |
The Safety Review Committee approved dose escalation for Cohort 5 to the 90 µg/kg dose every two weeks following two priming doses at 30 µg/kg. |
| - |
Subsequent to the quarter end, the Safety Review Committee approved dose escalation for Cohort 6 to a target dose of 120 µg/kg dose every two weeks following three priming doses at 30, 60 and 90 µg/kg. |
| - |
Significant increases in eosinophil count from baseline have not been observed with MDNA11 treatment. Extremely high eosinophil count is associated with vascular leak syndrome which is a known side effect of high-dose recombinant human IL-2 (Proleukin®). |

Pharmacokinetics:
| - |
The PK data from the first three cohorts demonstrated similar trends following each of three repeat doses which suggests lack of immunogenicity or insignificant levels of anti-drug-antibodies. |
| - |
Dose dependent increase in the Cmax and Area Under the Curve were also observed. |
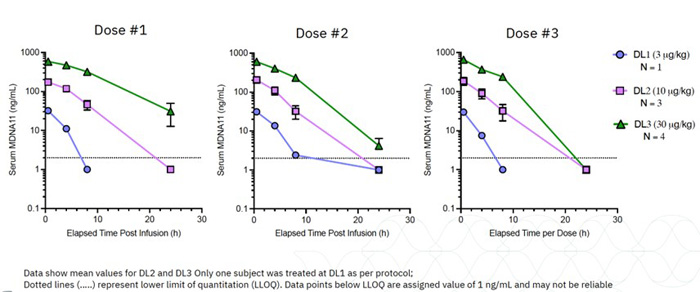
Pharmacodynamics:
| - |
In addition to dose-dependent increases in lymphocyte counts and lymphocyte kinetics, MDNA11 preferentially expanded anti-cancer NK and CD8+ T cells without stimulating proliferation of pro-tumor Treg cells. |


Anti-tumor Activity:
| - |
Of the 14 evaluable patients with at least one on-treatment imaging scan, five patients achieved tumor control (defined as stable disease, partial response, or complete response as per RECIST 1.1) during the monotherapy dose-escalation portion of the MDNA11 ABILITY Study as follows: |
| 1. |
Metastatic Leiomyosarcoma Stage IV (Dose Level 2 @ 10 µg/kg); stable disease. |
| 2. |
Metastatic Melanoma Grade 4C (initially enrolled at Dose Level 2 @ 10 µg/kg Q2W with subsequent intra-patient dose escalations to Dose Level 3 @30 µg/kg and Dose Level 4 @60 µg/kg), stable disease. |
| 3. |
Metastatic Sarcoma Stage IV (Dose Level 3 @ 30 µg/kg), stable disease |
| 4. |
Pancreatic Ductal Adenocarcinoma (PDAC) Stage IV (Dose Level 4 @ 60 µg/kg following 2 priming doses of 30 µg/kg), confirmed partial response. |
| 5. |
Non-clear cell 3L renal cell carcinoma patient (Dose Level 4 @ 60 µg/kg following 2 priming doses of 30 µg/kg), stable disease. |

Medicenna is currently enrolling patients in final (sixth) cohort of the dose escalation portion of the MDNA11 Phase 1/2 ABILITY Study and continues to follow-up patients in the lower dose escalation cohorts. Upon completion of the MDNA11 monotherapy dose-escalation phase (Phase 1), the study will commence enrolling patients in the dose-expansion phase (Phase 2). The dose expansion phase will evaluate both MDNA11 monotherapy as well as MDNA11 in combination with KEYTRUDA®.
It is expected that the dose escalation portion of the study will be completed in mid-calendar year 2023. An update on PK, PD, safety and efficacy data from all the six cohorts of the dose-escalation portion including initial anti-tumor activity data from the fifth and sixth dose escalation cohorts, is expected in calendar Q3 2023. The Phase 2 monotherapy dose expansion is expected to commence in calendar Q3 2023, with a clinical update from the Phase 2 monotherapy dose expansion expected in calendar Q4 2023, and the combination arm is expected to initiate in calendar Q4 2023. These timelines have been delayed from those originally disclosed due to additional dose escalation cohorts as well as implementation of step-up-dosing which requires extra time to reach the target dose, essentially extending the duration of the dose-limiting toxicity evaluation period from 4 weeks from first exposure to up to 10 weeks. If required, additional evaluation of MDNA11 dosing regimen (shorter duration and/or more rapid step-up to target dose) and schedule (Q3W instead of Q2W) for monotherapy and combination settings may also occur during the MDNA11 dose expansion portion of the study.
It is anticipated that following successful completion of the on-going Phase 1/2 ABILITY study, the Company will license the program to one or more potential partners who would continue further clinical development and commercialization of MDNA11. Alternatively, the Company may raise additional capital to fund Phase 2b and/or Phase 3 clinical trials. The Company expects the completion of clinical development of MDNA11, if undertaken by the Company, to last until at least 2027, with a projected aggregate cost of approximately US$150 million, incremental to the current funds available to the Company. Additional time and capital would also be required to obtain pre-market approval for MDNA11 and to complete business development, marketing and other pre-commercialization activities related to commercial launch.
IL-2 Superkine Competition
The development of next-generation IL-2 agonists for cancer immunotherapy is an area of intense interest within the biotechnology industry. The Company is aware of several IL-2 agonists in various stages of development. Due to the number of development programs only those with the therapeutic modality attributed solely to the IL-2 domain and in clinical stage of development are noted in the table below.
| Developer |
|
Name |
|
Stage |
| Alkermes |
|
ALKS 4230 |
|
Phase 3 |
| Sanofi |
|
Pegenzileukin |
|
Unknown |
| Cue Biopharma | CUE 101 & 102 | Phase 2 | ||
| Mabwell |
8MW2311 |
Phase 1/2 |
||
| Avenge Bio |
AVB-001 |
Phase 1/2 |
||
| Anaveon |
ANV419 |
Phase 1/2 |
||
| Selecxine |
SLC-3010 |
Phase 1/2 |
||
| BioNTech |
|
BNT151 |
|
Phase 1/2 |
| Xilio Therapeutics |
XTX202 |
Phase 1/2 |
||
| Ascendis Pharma |
|
Transcon IL-2 |
|
Phase 1/2 |
| Aulos |
AU-007 |
Phase 1/2 |
||
| Asher Bio |
AB821 |
Phase 1 |
||
| Synthekine |
|
STK012 |
|
Phase 1 |
| Jiangsu HengRui Medicine Co |
SHR-1916 |
Phase 1 |
||
| Werewolf |
WTX-124 |
Phase 1 |
Many of the programs in development that are ahead of Medicenna are engineered variants of IL-2 that attempt to reduce CD25 binding and/or extend the therapeutic window of native IL-2 and/or localize IL-2 activity at the tumor site. To our knowledge, MDNA11 is the only IL-2 product in clinical development where a significantly reduced CD25 binding and substantially enhanced CD122 binding have been achieved. Furthermore, MDNA11 relies on recombinant albumin to increase its half-life to allow for dosing every 2 or 3 weeks rather than PEGylation as many of its competitors. Albumin is also known to accumulate in tumors providing MDNA11 with enhanced localization at the tumor site.
IL-4 and IL-13 Superkines
Medicenna’s IL-4 and IL-13 Superkines, licensed from Stanford University, are engineered versions of wild type cytokines which possess enhanced affinity and selectivity for either the Type 1 or Type 2 IL4 receptors or dedicated IL13 receptors such as IL13Ra2. This selectivity is achieved through mutations of the IL-4 or IL-13 proteins to enhance affinity for binding to specific IL4R or IL13R subunits. Additional mutations have also been engineered to modulate their bioactivity, resulting in Superkines with enhanced signaling (super-agonists) or the ability to block signaling (super-antagonists).
MDNA413: An IL-4/IL-13 Dual Super-Antagonist
One promising IL-13 Superkine antagonist is MDNA413. Compared to wild type IL-13, MDNA413 has been engineered to have 2,000-fold higher selectivity for the Type 2 IL4R and which potently blocks IL-4 and IL-13 signaling (Moraga et al., 2015). Blocking of Type 2 IL4R by MDNA413 may be relevant not only for targeting solid tumors that overexpress this receptor, but also the Th2 biased tumour microenvironment, which shields the cancer from the immune system.
On April 8, 2022, Medicenna announced new preclinical data on its long-acting IL-13 super-antagonist, Fc-MDNA413, in an electronic poster at the AACR Annual Meeting. Fc-MDNA413 comprises of an IL-13 super-antagonist (MDNA413) fused to the Fc domain for half-life extension.
We believe that MDNA413’s ability to block IL-4/IL-13 signaling has the potential to address a significant unmet medical need for effective therapies against immunologically cold tumors which are often resistant to checkpoint inhibitors and other immunotherapeutic agents due to their immunosuppressive TME. MDNA413 has also been fused with MDNA19 (a long acting Fc-IL2 Superkine) as a novel BiSKIT™ candidate and was the basis of data presented at the 2021 AACR meeting as described below.
MDNA132 and MDNA213: High Affinity Cancer-Specific Targeting Ligands
Another promising IL-13 Superkine is MDNA132, and its variant, MDNA213. Unlike MDNA413, MDNA132 and MDNA213 are IL-13 ligands that have been engineered to increase affinity for IL13Ra2 overexpressed on certain solid tumors while exhibiting sharply decreased affinity for IL13Ra1. Medicenna believes MDNA132 and MDNA213 has superior targeting compared to other IL-13 variants in development, and is an attractively differentiated targeting domain for (a) cell-based immunotherapies (such as those using chimeric antigen receptors or CARs); (b) potent payloads used in antibody-drug conjugates (ADCs); (c) targeted fusion toxins or (d) radiopharmaceuticals. Development timelines for MDNA132 and MDNA213 have yet to be established. MDNA132 and/or MDNA213 are also being evaluated as a potential fusion protein in our BiSKITs™ platform.
On April 17, 2023, we announced that new preclinical data characterizing the Interleukin 13 (IL-13) Superkines, MDNA132 and MDNA213, and a series of next generation IL-13 Superkine therapies, were presented at the AACR Annual Meeting, which took place at from April 14, 2023 until April 19, 2023. The AACR poster included data demonstrating that both MDNA132 and MDNA213 exhibit highly selective binding to the IL-13 decoy receptor (IL-13Rα2) and, in a murine model, selectively accumulate in the tumor microenvironment (TME) for several days. MDNA132 and MDNA213 exhibit high affinity and selectivity for the IL13Rα2, which is overexpressed in various tumors such as pancreatic, prostate, bladder, colorectal, breast and lung cancer but minimally expressed in healthy tissues. High expression of IL13Rα2 in these tumors is generally associated with more aggressive cancer and poor survival outcomes.
Medicenna is currently screening and optimizing a variety of IL-2/IL-4/IL-13 superkines as part of our BiSKITs™ platform. Additional funding will be necessary to advance one or more of these product candidates into clinical trials.
BiSKITs™ (Bi-functional SuperKine ImmunoTherapies) Platform
Our BiSKITs™ platform allows us to develop designer Superkines by fusing them to other proteins, checkpoint inhibitors, antibodies or cytokines to our IL-2, IL-4 and/or IL-13 Superkines in order to combine two distinct and yet synergistic mechanisms of action into one molecule: a BiSKIT™.
On October 26, 2020, we announced a poster presentation at the 32nd ENA Symposium on Molecular Targets and Cancer Therapeutics. The presentation of preclinical results featured data on MDNA11 as well as data related to long acting bispecific IL-2/IL-13 Superkine that is designed to simultaneously activate cancer killing immune cells while reversing anti-inflammatory TME. Our bispecific IL-2/IL-13 Superkines are novel and demonstrate the potential of the BiSKITs™ platform to address a critical unmet need by effectively targeting immunologically “cold” tumors that are often resistant to immunotherapeutic agents. Data included in the poster and corresponding abstract showed that Medicenna’s IL-2/IL13 BiSKIT™ (MDNA19 fused to MDNA413) Superkine induced anti-tumor Th1 immune responses and inhibited pro-tumor IL-4/IL-13 signaling.
On April 12, 2021, we announced new preclinical data demonstrating the immune modulatory effects of MDNA19-413, an IL-2/IL-13 dual specific cytokine derived from the Company’s BiSKITs™ platform. Data presented in the poster suggest that this molecule simultaneously activates a pro-inflammatory anti-tumor response, due to its highly selective binding and signaling via the intermediate affinity IL-2 receptor (CD122/CD132), while inhibiting pro-tumoral immune pathways by blocking IL4/IL13 signaling via the Type 2 IL-4 receptor (IL-4Ra/IL-13Ra1).
On April 8, 2022, we announced new preclinical data highlighting the potent anti-tumor efficacy of the next-generation BiSKIT, anti-PD1-MDNA109FEAA, in an electronic poster at the AACR Annual Meeting. BiSKITs can target cancers where other immunotherapies have failed to be effective. One example of this is MDNA223, an IL-2 Superkine fused to a checkpoint inhibitor (anti-PD1). MDNA223 is a BiSKIT designed to activate cancer killing immune cells via the IL-2 receptor while simultaneously preventing their exhaustion through the validated method of blocking PD-1 signaling. Combining these two functions into a single molecule allows us to simultaneously engage both of these important targets on the same immune cells (also known as cis-binding).
On September 22, 2022, in vitro data presented at Cytokines 2022 demonstrated that MDNA223’s potency was similar to that of a control anti-PD1 antibody while displaying high-affinity for IL-2 receptor beta (IL-2Rβ) and no binding to IL-2 receptor alpha (IL-2Rα). This enhanced IL-2Rβ selectivity resulted in potent and preferential stimulation of anti-cancer CD8+ T cells over pro-tumor Treg cells. In vivo murine data showed MDNA223 exhibiting a prolonged PD response extending beyond the duration of PK exposure.

Data from murine tumor models of colon, skin and breast cancer using a mouse version of MDNA223 (i.e MDNA223m) showed dose-dependent and statistically significant improvements in efficacy compared to co-administration of the anti-PD-1 antibody and IL-2 super-agonist (MDNA19) at equivalent molar doses, demonstrating the advantage of exploiting the BiSKIT’s cis-binding potential. These data demonstrate the therapeutic synergy resulting from the BiSKIT’s ability to concurrently target PD1 and the IL-2 receptor on the same immune cells (cis-binding approach).

On January 5, 2023, Medicenna announced that the USPTO had issued U.S. Patent No. 11,542,312 titled “IL-2 Superagonists in Combination with Anti-PD-1.” The patent provides IP protection for methods of treating cancer with an IL-2 Superkine such as MDNA11 and a PD1 (for example, pembrolizumab), PDL1 or CTLA-4 checkpoint inhibitor in combination, as planned in the on-going ABILITY Study, or as a single agent using our BiSKIT™ platform. The patent’s term extends into at least 2039 without accounting for any potential extensions.
Medicenna is currently screening and optimizing a variety of IL-2/IL-4/IL-13 Superkines as part of our BiSKITs™ platform.
Bizaxofusp (formerly MDNA55)
Bizaxofusp is a novel, locally acting, anti-cancer therapeutic being developed by Medicenna for the treatment of tumors of the brain in adults, of which glioblastoma (“GBM”) is the most aggressive type. GBM is also the most common form of adult brain cancer, with 27,500 new cases diagnosed each year and the second most common cause of brain cancer deaths. Bizaxofusp has obtained Fast Track Designation from the FDA as well as Orphan Drug Designation from the FDA and the EMA.
Bizaxofusp: Structure and Mechanism of Action
Bizaxofusp is a targeted fusion protein for the treatment of tumors that over-express the IL4R. Bizaxofusp (see figure below) consists of a high-affinity circularly permuted variant of IL-4 (cpIL-4) fused with a truncated version of PE.

Bizaxofusp binds with high affinity to IL-4R overexpressed on the surface of tumor cells and is endocytosed. Following cleavage and activation by furin-like proteases found in the endosome of cancer cells, the catalytic domain of the truncated PE is released into the cytosol where it induces cell death via ADP-ribosylation of elongation factor-2 (see figure below).

Expression levels of IL4R are low on the surface of healthy and normal cells, but increase 10- to 100-fold on cancer cells. This differential expression of IL4R therefore provides bizaxofusp a wide therapeutic window.
The IL4R is an ideal target for the development of cancer therapeutics, as it is frequently and intensely expressed on a wide variety of human carcinomas. However, the IL4R target is currently under-exploited. Analysis of over 2,000 biopsies show IL4R over-expression in 20 different cancers affecting over a million cancer patients every year. Furthermore, the IL-4/IL4R bias is a marker for highly aggressive forms of cancer, plays a central role in the establishment of an immunosuppressive TME and is generally associated with poor survival outcomes. By disrupting this pro-tumoral IL-4/IL4R axis, bizaxofusp directly interferes with multiple networks that support cancer.
Glioblastoma
GBM is an aggressive brain tumor characterized by rapid proliferation of undifferentiated cells, extensive infiltration, and a high propensity to recur. It is a rapidly progressing and universally fatal cancer. First-line treatment for primary GBM generally includes surgical resection of the bulk tumor to the maximal extent possible, followed by radiotherapy, often in combination with chemotherapy consisting of TMZ. The approval of TMZ represented a breakthrough in treatment; the drug offers improvements in overall survival (“OS”), although the actual benefits are modest. When used in combination with radiotherapy following surgery, TMZ provided a median survival of 58.4 weeks for newly diagnosed GBM patients compared to 48.4 weeks for radiotherapy alone. TMZ is less effective in GBM patients who harbor unmethylated O6-methylguanine-methyltransferase (“MGMT”) promoters in the tumor tissue; more than half of GBM patients have unmethylated MGMT promoters. In practice, even patients without MGMT promoter methylation are prescribed TMZ because of a lack of approved treatment alternatives.
Recurrent Glioblastoma (rGBM)
Unlike treatment of newly diagnosed GBM, no consensus exists regarding the optimal treatment of rGBM. Recurrence rates for newly diagnosed GBM patients treated with the current SOC is high, even in completely resected patients.
Drugs currently approved in the United States for treatment of rGBM are Gliadel® and bevacizumab (Avastin®). In a Phase 3 study, placing a Gliadel implant directly into the tumor cavity after surgical resection of the tumor, 56% of rGBM treated subjects survived 6 months and the median survival was 26 weeks. However, the majority of patients with rGBM are not candidates for additional surgery, resulting in a large unmet need for this patient population.
Avastin® is an anti-angiogenic antibody that targets the vascular endothelial growth factor receptors. It is indicated as a single agent for adult patients with rGBM but has not been shown to improve disease-related symptoms or survival. Avastin® was granted accelerated approval on the basis of an objective response rate of 28% following an open label Phase 2 study in 85 patients receiving Avastin® only. In 2013, Avastin® completed its confirmatory trial in newly diagnosed GBM patients and did not meet its primary endpoint of overall survival. Based on the results of this trial, Genentech, for Avastin®, did not receive approval in the European Union for newly diagnosed GBM; however, Avastin® remains indicated in the United States and Japan for rGBM.
Rationale for Development of Bizaxofusp for rGBM
Bizaxofusp has been initially developed for the treatment of rGBM. Using current treatment paradigms, most GBM patients experience tumor recurrence/progression after standard first line treatment. Treatment options for patients with rGBM are very limited and the outcome is generally unsatisfactory. Specifically, chemotherapy regimens for recurrent or progressive GBM have been unsuccessful, producing toxicity without benefit. As overall survival remains dismal, novel anti-cancer modalities, with greater tumor specificity, more robust cytotoxic mechanisms and novel delivery techniques are needed for the treatment of recurrent GBM.
Bizaxofusp is one such novel therapeutic that is intended to provide a targeted treatment approach whereby tumor cells are more sensitive to the toxic effects of the drug than normal cells. When combined with a novel precision delivery to the brain using CED, a single administration of bizaxofusp could be an ideal approach for the treatment of rGBM and other brain tumors that over-express the IL4R. Cells that do not express the IL4R target do not bind to bizaxofusp and are, therefore, not subject to the effects of the toxic payload.
Many features of bizaxofusp make it a potentially attractive choice for the treatment of recurrent GBM:
| 1. |
The majority of cancer biopsy and autopsy samples from adult and pediatric primary and metastatic brain cancers, including rGBM, have been shown to over-express the IL4R with little or no IL4R expression in normal adult and pediatric brain tissue. |
| 2. |
MGMT positive cancer cells (harboring unmethylated MGMT promoters) are common in GBM, making them resistant to TMZ. However, MGMT positive cancer tumors are extremely sensitive to bizaxofusp, suggesting that bizaxofusp could provide a treatment option for GBM patients who would not benefit from TMZ. |
| 3. |
GBM has a robust immunosuppressive TME and may comprise up to 40% of the tumor mass. It has been shown that malignant gliomas have a T-helper cell type-2 (“Th2”) bias and are heavily infiltrated by myeloid derived suppressor cells (“MDSCs”) and tumor associated macrophages (“TAMs”) and that the IL-4/IL4R bias mediates their immunosuppressive functions. Furthermore, IL4R is up-regulated on glioma-infiltrating myeloid cells but not in the periphery or in normal brain. Thus, purging Th2 cells, MDSCs, and TAMs using bizaxofusp may alleviate the immune block associated with cancer (in a manner similar to immunomodulators such as ipilumimab, pembrolizumab or nivolumab), thereby promoting anti-tumor immunity and aid in long-term disease control. |
The bizaxofusp program therefore offers a promising approach to address serious unmet needs for brain cancer patients. Furthermore, to our knowledge, bizaxofusp is the only treatment in development that has the potential to simultaneously target the bulk tumor and the immunosuppressive TME. Accordingly, we are of the view that bizaxofusp has the potential of altering the treatment paradigm for many brain cancer patients.
Convection Enhanced Delivery (“CED”) of Bizaxofusp
As with most protein therapeutics, bizaxofusp does not cross the blood-brain barrier, and therefore must be delivered directly to the tumor (also known as intra-tumoral therapy) via local one time infusion procedure called CED. Medicenna’s development platform includes rights to all oncology indications for bizaxofusp, a novel image guided CED of bizaxofusp and a novel formulation used to prepare an infusate for delivery of bizaxofusp in the brain. These technologies are protected by patents either owned or licensed by Medicenna.
Development History of Bizaxofusp
The targeting domain and payload for Medicenna’s lead candidate, bizaxofusp, were developed in the laboratories of Dr. Ira Pastan at the National Cancer Institute (“NCI”) and Dr. Raj Puri at Center for Biologics Evaluation and Research, at the FDA. The targeting domain (IL-4) was engineered to improve the binding affinity of IL-4 to the IL4R and thereby increase potency of bizaxofusp. The payload domain (pseudomonas toxin) of bizaxofusp was engineered in order to remove off-target binding components further improving safety. Preclinical and clinical development of bizaxofusp for the treatment of brain as well as other non-brain tumors is described in over 50 publications.
In March 2013, Medicenna acquired all clinical, regulatory and material assets for bizaxofusp from Sophiris Bio Inc. (formerly Protox Therapeutics, Inc.) (“Sophiris”). The acquisition was comprised of two IND applications with the FDA, Fast Track Designation from the FDA, Orphan Drug Designations from the FDA and the EMA, clinical data from 72 patients enrolled in three different brain cancer studies with recurrent high grade glioma (66 rGBM and 6 recurrent anaplastic astrocytoma (rAA) patients), clinical data from 14 patients enrolled in a Phase 1 solid tumor study and all cell banks and reference material required to manufacture bizaxofusp. In a majority of the 72 patients enrolled in three different brain cancer studies, bizaxofusp was delivered only once by intratumoral infusion using CED via ventricular catheters. Subsequent to the purchase agreement with Sophiris, Medicenna and the National Institutes of Health (“NIH”) entered into license agreements (the “NIH License Agreements”) covering composition, methods of use, combination therapy and delivery of bizaxofusp.
Phase 2b Study for Recurrent Glioblastoma
The Phase 2b trial with bizaxofusp using enhanced CED delivery was a multi-center, open-label, single-arm study in up to 52 patients (at least 46 intent-to-treat (“ITT”) patients evaluable for survival and 35 patients evaluable for response), with first or second recurrence or progression of GBM after surgery or radiotherapy ± adjuvant therapy or other experimental therapies.
The primary endpoint of the study was mOS comparing an expected null survival rate of 8.0 months (based on historical control) with an alternative pursue rate of 11.5 months (1-sided alpha = 0.10 and 80% power for approximately 46 ITT or per protocol subjects). IL4R expression levels in tumor biopsies and their potential impact on survival outcomes following treatment with bizaxofusp, were retrospectively evaluated.
In April 2017, Medicenna treated the first rGBM patient in the Phase 2b clinical trial of bizaxofusp and enrolled patients at eight clinical sites across the United States and 1 site in Europe with enrolment in the study (46 ITT patients) completed in April 2019 of which 44 patients met all the protocol eligibility requirements (per protocol population).
On September 28, 2017, it was announced that based on encouraging drug distribution and safety data observed Medicenna implemented an amended protocol allowing higher doses and volumes of bizaxofusp as well as an increase in study size to up to 52 subjects. This protocol amendment was based on a planned safety analysis following a unanimous recommendation from bizaxofusp’s Safety Review Committee.
It was reported on May 2, 2018 that half the patients in the study had been recruited and the data to date demonstrated solid safety results and early signals of efficacy based on the findings of the Safety Review and Clinical Advisory Committees. Following the Safety Review, Medicenna amended the protocol at the recommendation of clinical advisors to further improve the chances for demonstrating increased therapeutic benefit for patients. The amendment allowed the implementation of optimal methodologies including more personalized dosing based on the tumor load, incorporation of advanced imaging modalities to measure treatment responses more reliably, use of sub-therapeutic dose of Avastin® in patients that could not tolerate steroid use to control edema and inflammation and allowing investigators to administer a second dose of bizaxofusp where appropriate.
Following the amended protocol as announced on May 2, 2018 and after receiving the necessary regulatory and site approvals patient enrolment was resumed at higher doses provided that the pre-established MTD of 240 mg was not to be exceeded.
The protocol amendments announced September 28, 2017 and May 2, 2018 resulted in increased timelines for completion of the bizaxofusp Phase 2b clinical trial due to an increase in the original number of patients as well as a slowdown of patient recruitment while the necessary regulatory reviews and approvals were completed.
On April 30, 2019, Medicenna announced that enrolment in the study was complete with 46 evaluable patients (ITT population) of which 44 patients were subsequently identified as meeting protocol eligibility requirements without major deviations (per protocol population).
On May 29, 2020, Medicenna announced presentation of data from its Phase 2b trial of bizaxofusp in patients with rGBM, at the 2020 ASCO Annual Meeting. The oral poster discussion led by Dr. Ian F. Parney, MD, PhD (Mayo Clinic), and a presentation by Dr. John Sampson, MD, PhD (Robert H. and Gloria Wilkins Distinguished Professor of Surgery, Duke University School of Medicine), focused on additional data demonstrating clinical superiority of bizaxofusp in patients with rGBM.
Highlights from the ASCO presentation included:
| ● |
Comparison of bizaxofusp with an eligibility-matched External Control Arm (“ECA” or also known as Synthetic Control Arm, SCA) using propensity-score weighting (Li et al.), an unbiased approach to select patients that match the baseline characteristics of bizaxofusp treated patients based on 11 key baseline prognostic factors, demonstrated an improvement in mOS of 72%. When stratified by IL4R status, IL4R High subjects in the bizaxofusp arm demonstrated improved mOS by 116% (Table 1). |
Table 1.
| Propensity-Weighted Groups |
N |
mOS |
Improvement in mOS |
HR |
| Bizaxofusp All-comers |
43 |
12.4 |
72% |
0.63 |
| ECA All-comers |
40.8 |
7.2 |
||
| Bizaxofusp IL4R High |
17 |
13.2 |
116% |
0.52 |
| ECA IL4R High |
16.8 |
6.1 |
Irrespective of IL4R expression, subjects showed a tumor control rate (“TCR”) (tumor shrinkage or stabilization) of 76% based on modified RANO criteria; these subjects demonstrated mPFS of 4.6 months, PFS at six months (“PFS-6”) of 40%, PFS-12 of 33%, mOS of 15.0 months and OS-12 of 57%.
Additional updated results (not presented at ASCO) include the following:
Patients with Low IL4R expression (H-Score ≤ 60) had a similar TCR as patients with High IL4R expression (H-Score > 60); TCR of 75% vs. 76%, respectively. However, the majority of the IL4R Low patients (11 of 16) received high doses of bizaxofusp (180 – 240 mg; median 180 mg) whereas only 9 of 21 IL4R High patients received the high dose of bizaxofusp.
The IL4R Low group receiving high dose also showed improved survival (mOS Not Reached, OS-12 of 53%) when compared to the low dose group (mOS = 8 months, OS-12 = 13%).
The Proposed Population (n=32), comprised of all IL4R High (irrespective of dose) as well as IL4R Low patients receiving the high dose, were shown to benefit the most from a single treatment of bizaxofusp. Median survival and OS-12 in this population was 15.8 months and 62% vs 7.0 months and 18%, respectively, when compared to the eligibility matched ECA. (Table 2).
Table 2.
| Eligibility-Matched |
N |
mOS |
Improvement in mOS |
HR |
OS-12 |
| Proposed Population |
32 |
15.8 |
126% |
0.45 |
62% |
| ECA |
40 |
7.0 |
18% |
||
| Propensity-Weighted |
|||||
| Proposed Population |
32 |
15.7 |
118% |
0.52 |
NA |
| ECA |
33.9 |
7.2 |
NA |
||
TCR in the Proposed Population was 81% based on radiologic assessment by mRANO criteria.
These data indicate that bizaxofusp has the potential to benefit all rGBM patients treated at the high dose (180 – 240 mg; median 180 mg) irrespective of IL4R expression. The high dose was well tolerated in this and earlier clinical trials (MTD = 240 mg).
On September 29, 2020, Medicenna had an EOP2 meeting with the FDA to discuss future development and commercialization of bizaxofusp, if approved, for rGBM. On October 15, 2020, Medicenna announced that the FDA agreed that we could conduct an innovative open-label hybrid Phase 3 trial that allows use of a substantial number of patients (two-thirds) from a matched ECA to support marketing authorization of bizaxofusp for rGBM. The proposed Phase 3 clinical trial design includes a concurrent 3:1 randomized cohort (3 subjects receiving bizaxofusp for every 1 subject receiving SOC) and an additional matched ECA. The primary endpoint of overall survival (OS) will be determined by a 1:1 analysis of the bizaxofusp arm versus the pooled control arm, which will consist of ECA and subjects randomized to SOC. This hybrid trial design will also reduce the overall number of subjects needed to enroll in the study to achieve the primary endpoint, and notably reduce the number of subjects that would be randomized to SOC treatment under a conventional 1:1 randomization. By reducing the need to enroll control subjects, an ECA can increase efficiency, reduce delays, lower trial costs, and speed lifesaving therapies to market. The Company demonstrated promising results for bizaxofusp in a Phase 2b clinical trial when compared to a retrospective and a well-balanced ECA. Medicenna is pursuing strategic partnerships to assist with additional clinical development of bizaxofusp, as well as preparing the program for commercialization and its subsequent launch in various countries where marketing authorization has been granted.
On October 26, 2020, Dr. John Sampson, MD, PhD (Robert H. and Gloria Wilkins Distinguished Professor of Surgery, Duke University School of Medicine) updated clinical data from the Phase 2b trial of bizaxofusp in rGBM as a Late Breaking Abstract poster at the 32nd ENA Symposium on Molecular Targets and Cancer Therapeutics. Highlights from the poster included updated results following a longer follow-up duration and new data based on transient low-dose use of bevacizumab:
| ● |
Data from all trial participants showed a mOS of 11.9 months (expected 6-9 months) following treatment with bizaxofusp which is comparable to earlier reported mOS of 11.6 months, an OS-24 of 20% (expected 0-10%), and a PFS-12 of 27% (expected 2-10%). |
| ● |
In Medicenna’s proposed patient population, mOS was 14.0 months (comparable to mOS of 15 months reported earlier), OS-24 was 20%, and PFS-12 was 24%. The proposed patient population included all bizaxofusp -treated trial participants with high IL4R expression and participants with low IL4R expression that received a high dose of bizaxofusp treatment. |
| ● |
Unmethylated MGMT promoter affects more than 50% of GBM patients and is associated with treatment resistance and poorer survival outcomes. However, MGMT status did not negatively affect bizaxofusp treatment. In the proposed population (N=17), mOS was 14.9 months with an OS-24 of 22%. |
| ● |
Following bizaxofusp treatment, transient (median of 3 cycles) low dose (5 mg/Kg q2w or 7.5 mg/Kg q3w) administration of Avastin®, used for symptom control and steroid sparring in patients receiving high concentrations of bizaxofusp, further improved patient survival. Amongst all comers (N=9) and the proposed population (N=8), mOS was 21.8 months and 18.6 months and OS-24 was of 44% and 38%, respectively. |
On May 7, 2021, Medicenna announced the peer-reviewed publication of clinical data from the bizaxofusp Phase 2b rGBM trial in the journal, Clinical Cancer Research. The paper, entitled “Modified RANO, Immunotherapy RANO, and Standard RANO Response to Convection-enhanced Delivery of IL4R-targeted Immunotoxin bizaxofusp in Recurrent Glioblastoma,” was published in collaboration with researchers at several institutions including University of California Los Angeles and Duke University.
Results presented in the peer-reviewed paper show that the mOS of radiographically evaluable patients in the trial irrespective of dose or IL4R expression was 11.8 months, which is longer than what would be expected from currently approved drugs. Notably, the data also show a potential link between patients experiencing radiographic progression and those exhibiting insufficient bizaxofusp penetration into the tumor, suggesting that at least a portion of patients who did not respond well to bizaxofusp may have benefited from higher drug concentrations.
These analyses supplement previously presented findings observed in Medicenna’s proposed patient population showing an 81% tumor control rate (26/32) based on mRANO and a median OS of 15.7 months, which represents a >100% improvement compared to an ECA (median OS of 7.2 months). The proposed patient population included all bizaxofusp -treated trial participants with high IL4R expression and participants with low IL4R expression that received a high dose of bizaxofusp treatment.
In September 2021, Dr. Fahar Merchant, President and Chief Executive Officer, co-authored an article related to bizaxofusp published in Lancet Oncology titled “Leveraging external data in the design and analysis of clinical trials in neuro-oncology.”
On October 2, 2021, Medicenna participated in the Virtual SNO/ASCO Conference on CNS Clinical Trials through an Oral Presentation titled: “Incorporating external control arm in bizaxofusp recurrent glioblastoma registration trial.”
On November 18, 2021, Medicenna announced that John H. Sampson, MD, PhD, MHSc, MBA, Robert H. and Gloria Wilkins Distinguished Professor of Neurosurgery at Duke University School of Medicine and member of Medicenna’s board of directors, received The Abstract Award for Excellence in Clinical Trials in connection with an oral presentation on bizaxofusp. The presentation subject to the award was delivered by Dr. Sampson at the 26th Annual Meeting of the SNO.
In January 2023, the full results of a single-arm Phase 2b trial of bizaxofusp (recently named as per WHO International Non-proprietary Names) in patients with recurrent glioblastoma were published in the peer-reviewed journal Neuro-Oncology, in an article titled “Targeting the IL4 receptor with MDNA55 in patients with recurrent glioblastoma: Results of a phase IIb trial”. Results showed the trial met its primary endpoint, with mOS in the primary and supportive analysis populations exceeding the trial’s pre-defined success criteria and the mOS historically achieved with currently approved therapies.
The Company expects the completion of a pivotal Phase 3 clinical trial of bizaxofusp to full approval to last until at least 2026, with a projected aggregate cost of up to approximately $75 million, incremental to the current cash on hand. The Company continues to work to out-license the program to one or more partners who would fund or co-fund Phase 3 clinical development of bizaxofusp as well as prepare the program for commercialization and its subsequent launch in various countries where approval has been granted.
Potential Market: bizaxofusp
The incidence of glioblastoma (GBM) multiforme in the United States and EU5 (UK, Italy, Spain, France, Germany) alone exceeded 26,000 with a market opportunity in excess of US$1 billion. Although treatment options exist, including surgery, radiation, chemotherapy, Tumor Treating Fields and targeted therapeutics, the 5-year survival rate is less than 10%.
Treatment options for rGBM are severely limited. With the exception of Avastin®, providing limited survival benefits, no universal SOC exists for rGBM. Avastin® has not been approved by the EMA for newly diagnosed GBM or rGBM, although it has been granted accelerated approval by the FDA for rGBM. Management believes that bizaxofusp is currently well positioned for the rGBM indication, when used either as monotherapy or in combination with other approved therapies. Line extension for metastatic brain cancer, newly diagnosed GBM and pediatric gliomas has the potential to increase bizaxofusp revenues.
Bizaxofusp Competition: Emerging Therapies for Adult GBM
The SOC for newly diagnosed GBM, consisting of surgery, radiotherapy and concurrent TMZ followed by adjuvant TMZ has not changed for over a decade. The lack of effective treatment options extends to a shortage of approved targeted therapies for GBM. Development of novel agents for the treatment of GBM is therefore an active area of research, and multiple agents and drug classes are being assessed for GBM.
Northwest Biotherapeutics’ DCVax-L, an autologous dendritic cell vaccine, is one of the furthest along in development for GBM. DCVax-L is being evaluated in newly diagnosed GBM patients who have received a complete surgical resection and received radiotherapy and concurrent TMZ. Northwest Biotherapeutics has completed a Phase 3 clinical trial in patients with newly diagnosed GBM for which data was announced in May 2022. It is anticipated that Northwest Biotherapeutics will seek regulatory approval for DCVax-L.
DNAtrix’s DNX-2401, an oncolytic immunotherapy, has completed enrolment in a Phase 2 clinical trial in collaboration with Merck which evaluated the efficacy and safety of DNX-2401 in combination with pembrolizumab (Keytruda®), Merck’s anti-PD-1 therapy. Positive Phase 2 data was presented in November 2020, published in May 2023 and DNAtrix has disclosed plans to initiate a Phase 3 clinical study. DNAtrix entered bankruptcy proceedings in November 2022.
Kintara Therapeutics’ product VAL-083 is a “first-in-class” small molecule chemotherapeutic and is enrolling patients. In July 2019, Kintara Therapeutics began enrolling patients in a Phase 2/3 response adaptive randomization platform trial designed to evaluate multiple regimens of VAL-083 in newly diagnosed and recurrent GBM with top-line data in the international registrational GBM AGILE Study expected before the end of calendar 2023.
Kazia Therapeutics is developing Paxalisib, a brain-penetrant inhibitor of the PI3K / Akt / mTOR pathway, which is disordered in the vast majority of patients with glioblastoma. In January 2021 Kazia Therapeutics announced that patient recruitment had commenced for Paxalisib in the GBM AGILE platform study, in August 2022, Kaxia annouced the trial had not graduated to Stage 2 of the Phase 3 GBM AGILE study, however final survival and response data from the study are expected in the second half of 2023.
CNS Pharmaceuticals drug, Berubicin, is currently being evaluating in a global potentially pivotal study that was initiated in May of 2021.The potentially pivotal trial is an adaptive, multicenter, open-label, randomized and controlled study in adult patients with recurrent glioblastoma multiforme (WHO Grade IV) after failure of standard first-line therapy. Initial data from the study is expected in Q3 2023.
Istari Oncology announced in November 2020 that it had dosed the first patient in a Phase 2 clinical trial, assessing the safety and efficacy of PVSRIPO in combination with the immune checkpoint inhibitor pembrolizumab (Keytruda®) in patients with rGBM. The study remains active but is not currently enrolling patients.
Liquidity
The Company anticipates that its current level of cash and cash equivalents and marketable securities, will be sufficient to execute its current planned expenditures for through calendar Q3 without further financing being obtained. This estimate assumes continuation of the MDNA11 Phase 1/2 ABILITY study, and that any further development of bizaxofusp will be completed by a partner.
The Company does not earn any revenues from its drug candidates and is therefore considered to be in the development stage. As required, the Company will continue to finance its operations through the sale of equity or pursue non-dilutive funding sources available to the Company in the future. The continuation of research and development activities for bizaxofusp, MDNA11 and the BiSKITs™ platform and the commercialization of bizaxofusp is dependent upon the Company’s ability to successfully finance and complete its research and development programs through a combination of equity financing and revenues from strategic partners. The Company has no current sources of revenues from strategic partners.
Environmental Regulations
We rely on third parties as for the controlled use of hazardous and radioactive materials including with respect to their manufacture, storage, handling and disposal. As such, the compliance by the Company with environmental federal, provincial, state and local laws and regulations does not and will not, to the knowledge of the Company, have any significant impact on our capital spending, profits or competitive position within the normal course of our operating activities. There can be no assurance, however, that the Company will not be required to incur significant costs to comply with environmental laws and regulations in the future or that its operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
Seasonality
Our results of operations have not been materially impacted by seasonality.
Raw Materials
We believe that sources of raw material pertinent for manufacturing bizaxofusp, MDNA11 and our BiSKIT candidates are generally available.
Intellectual Property and Partnerships
Medicenna regards its intellectual property rights as one of the foundation blocks upon which it continues to build a successful biopharmaceutical development company. Medicenna has established a strong and defensive intellectual property position to protect its proprietary technologies. To date, Medicenna has 20 patent families providing patent protection in the US and in contracting states to the Patent Corporation Treaty. The Company has a total of 104 patents issued or filed of which 50 patents have been granted and the remaining patent applications are pending in the United States and other countries.
Patent families owned or licensed by Medicenna related to bizaxofusp (granted and pending US cases listed):
| 1. |
Method for Convection Enhanced Delivery of Therapeutic Agents (U.S. Patent No. 7,371,225) |
| 2. |
Targeted Cargo Protein Combination Therapy (U.S. Patent No. 9,629,899) |
| 3. |
IL-4 Fusion Formulations for Treatment of Central Nervous System (CNS) Tumors (pending US Patent Application No. 16/753,978) |
| 4. |
ILR4 as a Biomarker in Cancer (pending US Patent Application No. 17/428,697) |
| 5. |
Combination Therapy of MDNA55 and a Vascular Endothelial Growth Factor A (VEGF-A) (pending US Patent Application No. 18/248,601) |
Expiry dates for the above patents and related family members range from 2023 to 2042 without accounting for any potential extensions.
In addition to the above patent protection, bizaxofusp has been granted Orphan Drug Designation in the United States and Europe for the treatment of GBM, which would result in seven and 10 years of orphan drug exclusivity in the U.S. and Europe, respectively. Additionally, upon approval, bizaxofusp as a biologic, is expected to be eligible for 12 years Reference Product Exclusivity in the United States, eight years data exclusivity plus two years market exclusivity in Europe, 6 years data exclusivity plus two years market exclusivity in Canada and other markets where similar means of exclusivity are available.
Patent families owned or licensed by Medicenna related to the Superkine and Empowered Superkine platforms (granted/allowed US cases listed or representative PCT listed):
| 1. |
IL-2 Superagonists in Combination with Anti-PD-1 Antibodies (Allowed US Patent Application No. 16/012,733) |
| 2. |
Interleukin-4 Receptor-Binding Fusion Proteins and Uses Thereof (Pro-apoptotic Fusions) (U.S. Patent Nos. 10,093,708 and 11,084,856) |
| 3. |
Interleukin-4 Receptor Binding Fusion Proteins and Uses Thereof (Anti-apoptotic Fusions) (U.S. Patent Nos. 10,106,592 and 11,352,402) |
| 4. |
Interleukin-2 Fusion Proteins and Uses Thereof (US Patent No. 10,781,242) |
| 5. |
Uses and Methods for Oncolytic Virus Targeting of IL-4/IL-13 and Fusions Thereof (PCT/IB2019/00759) |
| 6. |
Bifunctional Superkines and Uses Thereof (PCT/CA2021/050872) |
| 7. |
Uses and Methods For IL-2 Cytokine Fusions (unpublished) |
| 8. |
Uses and Methods for IL-2, IL-13 and IL-4 Cytokine Fusions (unpublished) |
| 9. |
Use of Immunomodulation Methods in Combination with Cytokine Fusions for Disease Treatment (unpublished) |
| 10. |
Superagonists and Antagonists of Interleukin-2 (U.S. Patent Nos. 9,428,567; 10,183,980; and 11,117,943) |
| 11. |
Superkines and Synthekines: Repurposed Cytokines with New and Enhanced Signaling Activities (U.S. Patent No. 9,738,696 and US Patent No. 10,738,096) |
| 12. |
Superagonists, Partial Agonists and Antagonists of Interleukin-2 (U.S. Patent Nos. 10,150,802 and 10,654,905; allowed US Patent Application No. 15/930,057) |
| 13. |
Therapeutic IL-13 Polypeptides (U.S. Patent Nos. 9,512,194; 9,732,133; 10,227,389 and 11,084,858) |
| 14. |
IL-13 Superkine: Immune Cell Targeting Constructs and Methods of Use Thereof (PCT/US2017/66529) |
| 15. |
IL-13/IL-4 Superkine: Immune Cell Targeting Constructs and Methods of Use Thereof (PCT/US2019/035186) |
Expiry dates for the above US patents, corresponding non-US patents and any future-issued patents claiming priority to pending patent applications filed range from 2031 to 2043, without accounting for any potential extensions. Upon approval, the above programs are expected to be eligible for 12 years Reference Product Exclusivity in the United States, 8 years data exclusivity plus 2 years market exclusivity in Europe, 6 years data exclusivity plus 2 years market exclusivity in Canada and other markets where similar means of exclusivity are available.
CPRIT Agreement
In February 2015, the Company received notice that it had been awarded a grant by CPRIT whereby the Company is eligible to receive up to US$14.1 million on eligible expenditures over a three year period related to the development of the Company’s Phase 2b clinical program for bizaxofusp. The grant from CPRIT was completed as of March 31, 2022.
If the Company is found to have used any grant proceeds for purposes other than intended, is in violation of the terms of the grant, or relocates its bizaxofusp related operations outside of the state of Texas, then the Company is required to repay any grant proceeds received.
Under the terms of the grant, the Company is also required to pay a royalty to CPRIT, comprised of 3-5% of revenues on net sales of bizaxofusp until aggregate royalty payments equal 400% of the grant funds received at which time the ongoing royalty will be 0.5% of revenues. At this time, the royalty is not probable and therefore no liability has been recorded. In addition, the Company must maintain a presence in Texas for three years following completion of the grant.
Nasdaq Listing
On October 25, 2022, the Company received the Nasdaq Notice, stating that the Company was not in compliance with the Minimum Bid Requirement per share under the Nasdaq Listing Rule 5450(a)(1) based upon the closing bid price of the Company’s common shares for the 30 consecutive business days prior to the date of the Nasdaq Notice. The Nasdaq Notice had no immediate effect on the listing or trading of the Company’s common shares on Nasdaq, and the Company’s operations currently remain unaffected by the receipt of the Nasdaq Notice.
On April 25, 2023, the Company received an Extension Notice from Nasdaq granting the Company’s request for a 180-day extension to regain compliance with the Minimum Bid Requirement of US$1.00 per share under the Nasdaq Listing Rule 5450(a)(1). The Company has until October 23, 2023 to meet the requirement. The Extension Notice had no immediate effect on the listing or trading of the Company’s Common Shares on Nasdaq, and the Company’s operations are not affected by the receipt of the Extension Notice.
The Company is closely monitoring the closing bid price of its common shares and is considering its options to regain compliance with the Minimum Bid Requirement under the Nasdaq Listing Rules. This notice does not have any impact on the Company’s TSX listing.
Business Strategy
Medicenna’s strategy is to diversify the assets in Medicenna’s pipeline based on their stage of development, mechanism of action and target product profile. To achieve this goal, we in-licensed the Superkine platform from Stanford. These candidates, namely IL-2, IL-4 and IL-13 Superkines, are expected to enable the Company to develop a library of cytokine candidates as has been demonstrated by the advancement of our lead IL-2 Superkine MDNA11 into the Phase 1/2 ABILITY study and the various candidates from our BiSKIT™ platform discussed above. The resulting product candidates derived from the Superkine and Empowered Superkine platforms have different mechanisms of action and target product profiles compared to bizaxofusp, Medicenna’s most advanced program, for the treatment of rGBM. By adopting a balanced approach, Medicenna is less reliant on a single product in Medicenna’s pipeline, with greater upside potential through opportunities to partner or develop on its own, multiple products. Medicenna believes that establishing a pipeline of drug candidates with distinct mechanisms of actions targeting multiple disease indications mitigates development risk. Medicenna intends to achieve its business strategy by focusing on the following key areas:
| 1. |
Maximize the potential clinical and commercial success of Medicenna’s drug candidates by pursuing development programs based on sound scientific rationale for multiple disease indications where there are significant unmet clinical needs. In the near-term, Medicenna’s focus will be to complete a partnership transaction for bizaxofusp as well advance MDNA11 through the Phase 1/2 ABILITY study; |
| 2. |
Develop next generation Superkines from the BiSKIT™ platform for future partnerships, collaborations or clinical development; |
| 3. |
Optimize the therapeutic potential of Medicenna’s drug candidates by selecting sub-populations of patients who stand an improved chance of responding to treatment and employing the latest technologies and strategies for optimizing drug delivery, defining relevant biomarkers, refining treatment schedules and dosing regimens and selecting appropriate combination strategies; |
| 4. |
Establish collaborations and relationships with leading scientific and clinical centres to effectively maximize the success of Medicenna’s drug development programs; and |
| 5. |
Assess strategic alliances with select pharmaceutical and/or biotechnology companies where such alliances may enable successful development and commercialization of Medicenna’s drug candidates while maximizing its return on investment. Medicenna may conduct transactions with established strategic partners on a regional or worldwide basis to accelerate product development, improve Medicenna’s marketing strength and enhance its capability of bringing products to the markets worldwide. |
Medicenna will continue to seek sources of non-dilutive funding as well as additional funds through equity financings and/or through collaborative arrangements with pharmaceutical and/or biotechnology companies for any of Medicenna’s products and technologies under development. Cash resources are carefully managed and focused on priority programs and initiatives. Accordingly, some initiatives may not be pursued or advanced in the near term as a prudent measure to preserve cash.
Regulatory Process
Government authorities in the United States, including federal, state, and local authorities, and in other countries, extensively regulate, among other things, the manufacturing, research and clinical development, marketing, labeling and packaging, storage, distribution, post-approval monitoring and reporting, advertising and promotion, and export and import of biological products, such as those Medicenna is developing. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
Securing final regulatory approval for the manufacture and sale of biological products in the United States, Europe, Canada and other commercial territories, is a long and costly process that is controlled by that particular territory’s regulatory agency. The regulatory agency in the United States is the FDA, in Canada it is Health Canada, and in Europe it is the EMA. Other regulatory agencies have similar regulatory approval processes and each regulatory agency has its own approval processes. Approval in the United States, Canada or Europe does not assure approval by other regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country.
None of Medicenna’s products have been completely developed or tested and, therefore, Medicenna is not yet in a position to seek regulatory approval to market any of Medicenna’s products. The time required to obtain approval by such regulatory authorities is unpredictable but typically takes several years following the commencement of preclinical studies and clinical trials and will require significant additional capital.
United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (“FDCA”), and its implementing regulations, and biologics under the FDCA and the Public Health Service Act (“PHSA”), and its implementing regulations. FDA approval is required before any new unapproved drug or biologic or dosage form, including a new use of a previously approved drug, can be marketed in the United States. Drugs and biologics are also subject to other federal, state, and local statutes and regulations. If Medicenna fails to comply with applicable FDA or other requirements at any time during the product development process, clinical testing, the approval process or after approval, Medicenna may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, civil monetary penalties or criminal prosecution. Any FDA enforcement action could have a material adverse effect on Medicenna.
The process required by the FDA before product candidates may be marketed in the United States generally involves the following:
| ● |
completion of extensive preclinical laboratory tests, all performed in accordance with the Good Laboratory Practices regulations; |
| ● |
completion of extensive CMC (chemistry, manufacturing and control) to produce drug in accordance with current Good Manufacturing Practices (“cGMP”); |
| ● |
submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| ● |
approval by an independent institutional review board (“IRB”) or ethics committee representing each clinical site before each clinical trial may be initiated; |
| ● |
performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; |
| ● |
preparation of and submission to the FDA of a biologics license application (“BLA”) after completion of all pivotal clinical trials; |
| ● |
potential review of the product application by an FDA advisory committee, where appropriate and if applicable; |
| ● |
a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
| ● |
satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities where the proposed product is produced to assess compliance with GMP. |
| ● |
a potential FDA audit of the preclinical research and clinical trial sites that generated the data in support of the BLA; and |
| ● |
FDA review and approval of a BLA prior to any commercial marketing or sale of the product in the United States |
The preclinical research, including production of cGMP material, clinical testing and approval process require substantial time, effort, and financial resources, and Medicenna cannot be certain that any approvals for Medicenna’s product candidates will be granted on a timely basis, if at all.
An IND is a request for authorization from the FDA to administer an investigational new drug product to humans in clinical trials. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human clinical trials. The IND also includes description of the manufacturing process and testing of the batch, results of animal studies assessing the toxicology, PK, pharmacology, and PD characteristics of the product; and any available human data or literature to support the use of the investigational new drug. An IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical trials. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical trials can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with Good Clinical Practices (“GCP”), which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the efficacy criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical trial site’s IRB or ethics committee, before the trials may be initiated, and the IRB or ethics committee must monitor the trial until completed. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries. Information related to the investigational product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is made public as part of the registration of the clinical trial on the national ClinicalTrials.gov data registry. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed in some cases for up to two years after the date of completion of the trial. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs. The Final Rule on ClinicalTrials.gov registration and reporting requirements became effective in 2017, and the government has begun enforcing those requirements against non-compliant clinical trial sponsors.
The clinical investigation of a drug is generally divided into three or four phases. Although the phases are usually conducted sequentially, they may overlap or be combined.
| ● |
Phase 1. The drug is introduced into healthy human subjects or subjects with the target disease or condition. These studies are designed to evaluate safety, dosage tolerance, metabolism and pharmacologic actions of the investigational new drug in humans, the side effects associated with increasing doses, and where possible, to gain early evidence on effectiveness. |
| ● |
Phase 2. The drug is administered to a limited patient population to evaluate dosage tolerance and optimal dosage, identify possible adverse side effects and safety risks, and preliminarily evaluate efficacy. |
| ● |
Phase 3. The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites to generate enough data to statistically evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational new drug product, and to provide an adequate basis for physician labeling. |
| ● |
Phase 4. In some cases, the FDA may condition approval of an NDA or BLA for a product candidate on the sponsor’s agreement to conduct additional clinical trials after approval. In other cases, a sponsor may voluntarily conduct additional clinical trials after approval to gain more information about the drug. Such post-approval studies are typically referred to as Phase 4 clinical trials. |
Clinical trial sponsors must also report to the FDA, within certain timeframes, serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator’s brochure, or any findings from other studies or animal testing that suggest a significant risk in humans exposed to the product candidate. The FDA, the IRB or ethics committee, or the clinical trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated check points based on access to certain data from the trial.
Congress also recently amended the FDCA, as part of the Consolidated Appropriations Act for 2023, in order to require sponsors of a Phase 3 clinical trial, or other “pivotal study” of a new drug to support marketing authorization, to design and submit a diversity action plan for such clinical trial. The action plan must include the sponsor’s diversity goals for enrollment, as well as a rationale for the goals and a description of how the sponsor will meet them. Sponsors must submit a diversity action plan to the FDA by the time the sponsor submits the relevant clinical trial protocol to the agency for review. The FDA may grant a waiver for some or all of the requirements for a diversity action plan. It is unknown at this time how the diversity action plan may affect Phase 3 trial planning and timing, but if the FDA objects to a sponsor’s diversity action plan or otherwise requires significant changes to be made, it could delay initiation of the relevant clinical trial.
The clinical trial process can take years to complete, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Results from one trial are not necessarily predictive of results from later trials. Medicenna may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Submission of an NDA or BLA to the FDA
Assuming successful completion of all required preclinical studies and clinical testing in accordance with all applicable regulatory requirements, detailed investigational new drug product information is submitted to the FDA in the form of an NDA or a BLA requesting approval to market the product for one or more indications. Under federal law, the submission of most NDAs and BLAs are subject to an application user fee, and the sponsor of an approved NDA or BLA is also subject to an annual program fee. For fiscal year 2023, the application user fee is US$3.242 million. This fee is typically increased annually. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business (fewer than 500 employees). Applications for orphan drug products are exempted from the application user fee, unless the application includes an indication for other than a rare disease or condition.
An NDA or BLA must include all relevant data available from pertinent preclinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product and may also come from a number of alternative sources, including trials initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational new drug product to the satisfaction of the FDA.
The FDA reviews all submitted NDAs and BLAs to ensure that they are sufficiently complete for substantive review before it accepts them for filing. It may refuse to file the application and request additional information rather than accept an NDA or BLA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. The FDA must make a decision on accepting an NDA or BLA for filing within 60 days of receipt and inform the sponsor by the 74th day after the FDA’s receipt of the submission whether an application is sufficiently complete to permit substantive review.
Once an NDA or BLA has been accepted for filing, the FDA begins an in-depth review of the NDA or BLA. The FDA’s current goal is to review the application for a new molecular-entity (“NME”) NDA or an original BLA within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication and is designated for priority review, six months after the FDA accepts the application for filing. The review process is often significantly extended by the FDA’s requests for additional information or clarification.
Before approving an NDA or BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
The FDA is required to refer an NDA or BLA for a novel drug (in which no active ingredient has been approved in any other application) to an advisory committee or explain why such referral was not made. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
The FDA also may require development of a risk evaluation and mitigation strategy (“REMS”) plan, if it determines that a REMS is necessary to ensure that the benefits of the drug outweigh its risks and to assure the safe use of the drug or biologic. The REMS plan could include medication guides, physician communication plans, assessment plans and/or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. The FDA determines the requirement for a REMS, as well as the specific REMS provisions, on a case-by-case basis. If the FDA concludes a REMS plan is needed, the sponsor of the NDA or BLA must submit a proposed REMS. The FDA will not approve an NDA or BLA without a REMS, if one is required.
The FDA’s Decision on an NDA or a BLA
After the FDA evaluates the NDA or BLA and conducts inspections of manufacturing facilities where the product will be produced, the FDA will issue either an approval letter or a complete response letter (“Complete Response Letter”). An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. In order to satisfy deficiencies identified in a Complete Response Letter, additional clinical data and/or additional Phase 3 clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing may be required for the product candidate. Even if such additional information is submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. The FDA also may condition approval on, among other things, a REMS plan, the addition of contraindications, warnings or precautions to the product labeling, or a commitment to conduct one or more post-market studies or clinical trials. Such post-market testing may include Phase 4 clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. New government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of Medicenna’s products under development.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States for which there is no reasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years from the date of such approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety or providing a major contribution to patient care or in instances of drug supply issues. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication. Recent court cases have challenged the FDA’s approach to determining the scope of orphan drug exclusivity; however, at this time the agency continues to apply its long-standing interpretation of the governing regulations and has stated that it does not plan to change any orphan drug implementing regulations. If a drug or biological product designated as an orphan product ultimately receives marketing approval for an indication broader than what was designated in its orphan product application, it may not be entitled to exclusivity.
Expedited Development and Review Programs
Post-marketing Requirements
Following approval of a new product, the manufacturer and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and record-keeping activities, reporting of adverse experiences, and complying with promotion and advertising requirements, which include restrictions on promoting approved drugs for unapproved uses or patient populations (known as “off-label use”). Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. Prescription drug promotional materials also must be submitted to the FDA in conjunction with their first use. Further, if there are any modifications to the drug or biologic, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA/BLA or NDA/BLA supplement, which may require the applicant to develop additional data or conduct additional preclinical studies or clinical trials.
FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMPs. The cGMP regulations include requirements relating to organization of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports and returned or salvaged products. The manufacturing facilities for our drug candidates must meet cGMP requirements and satisfy the FDA or comparable foreign regulatory authorities before any product is approved and commercial products can be manufactured or distributed. Manufacturers must comply with cGMPs that require, among other things, quality control and quality assurance, the maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved drugs or biologics are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP requirements and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. The discovery of violative conditions, including failure to conform to cGMPs, could result in enforcement actions, and the discovery of problems with a product after approval may result in restrictions on a product, or on the manufacturer or holder of an approved NDA or BLA, including recall or product seizure.
Once an approval or clearance of a drug is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| ● |
restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● |
fines, warning letters or other enforcement-related letters or clinical holds on post-approval clinical trials; |
| ● |
refusal of the FDA to approve pending NDAs/BLAs or supplements to approved NDAs/BLAs, or suspension or revocation of product approvals; |
| ● |
product seizure or detention, or refusal to permit the import or export of products; |
| ● |
injunctions or the imposition of civil or criminal penalties; |
| ● |
consent decrees, corporate integrity agreements, debarment, or exclusion from federal health care programs; and/or |
| ● |
mandated modification of promotional materials and labeling and the issuance of corrective information. |
Additional Controls for Biologics
To help reduce the increased risk of the introduction of adventitious agents, the PHSA emphasizes the importance of manufacturing controls for products whose attributes cannot be precisely defined. The PHSA also provides authority to the FDA to immediately suspend biologics licenses in situations where there exists a danger to public health, to prepare or procure products in the event of shortages and critical public health needs, and to authorize the creation and enforcement of regulations to prevent the introduction or spread of communicable diseases within the United States.
After a BLA is approved, the product may also be subject to official lot release as a condition of approval. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the lot manufacturing history and the results of all of the manufacturer’s tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products, such as viral vaccines, before allowing the manufacturer to release the lots for distribution. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products. As with drugs, after approval of a BLA, biologics manufacturers must address any safety issues that arise, are subject to recalls or a halt in manufacturing, and are subject to periodic inspection after approval.
Other Healthcare Laws
Pharmaceutical manufacturers are subject to additional healthcare laws, regulation, and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation, U.S. federal anti-kickback, anti-self-referral, false claims, transparency, including the federal Physician Payments Sunshine Act, consumer fraud, pricing reporting, data privacy, data protection, and security laws and regulations. Similar state and local laws and regulations may also restrict business practices in the pharmaceutical industry, such as state anti-kickback and false claims laws, which may apply to business practices, including but not limited to, research, distribution, sales, and marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information; state and local laws which require the tracking of gifts and other remuneration and any transfer of value provided to physicians, other healthcare providers and entities; and state and local laws that require the registration of pharmaceutical sales representatives; and state and local laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Coverage and Reimbursement
Sales of any pharmaceutical product depend, in part, on the extent to which such product will be covered by third-party payers, such as federal, state, and government healthcare programs, commercial insurance, and managed healthcare organizations, and the level of reimbursement for such product by third-party payers. Significant uncertainty exists as to the coverage and reimbursement status of any newly approved product. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. One third-party payor’s decision to cover a particular product does not ensure that other payors will also provide coverage for the product. As a result, the coverage determination process can require manufacturers to provide scientific details, information on cost-effectiveness, and clinical support for the use of a product to each payor separately. This can be a time-consuming process, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
In addition, third-party payors are increasingly reducing reimbursements for pharmaceutical products and related services. The U.S. government and state legislatures have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Third-party payors are increasingly challenging the prices charged, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical products, in addition to questioning their safety and efficacy. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product.
For example, in August 2022 President Biden signed into law the Inflation Reduction Act (“IRA”). Among other things, the IRA has multiple provisions that may impact the prices of drug products that are both sold into the Medicare program and throughout the U.S. Starting in 2023, a manufacturer of drugs or biological products covered by Medicare Parts B or D must pay a rebate to the federal government if their drug product’s price increases faster than the rate of inflation. This calculation is made on a drug product by drug product basis and the amount of the rebate owed to the federal government is directly dependent on the volume of a drug product that is paid for by Medicare Parts B or D. Additionally, starting for payment year 2026, the Centers for Medicare and Medicaid Services (“CMS”) will negotiate drug prices annually for a select number of single source Part D drugs without generic or biosimilar competition. CMS will also negotiate drug prices for a select number of Part B drugs starting for payment year 2028. If a drug product is selected by CMS for negotiation, it is expected that the revenue generated from such drug will decrease. CMS has begun to implement these new authorities but their impact on the biopharmaceutical industry in the United States remains uncertain. Lawsuits against CMS alleging constitutional and other violations with respect to these aspects of the IRA have recently been initiated by industry and the U.S. Chamber of Commerce.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures. In December 2020, the U.S. Supreme Court held unanimously that federal law does not preempt the states’ ability to regulate pharmacy benefit managers (“PBMs”) and other members of the health care and pharmaceutical supply chain, an important decision that has led to further and more aggressive efforts by states in this area. The Federal Trade Commission in mid-2022 also launched sweeping investigations into the practices of the PBM industry that could lead to additional federal and state legislative or regulatory proposals targeting such entities’ operations, pharmacy networks, or financial arrangements. Significant efforts to change the PBM industry as it currently exists in the U.S. may affect the entire pharmaceutical supply chain and the business of other stakeholders, including biopharmaceutical product developers like us.
Comparable European and Other International Government Regulation
In addition to FDA regulations in the United States, we will be subject to a variety of comparable regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries.
Some countries outside of the United States have a similar process that requires the submission of a clinical trial application (“CTA”) much like the IND prior to the commencement of human clinical trials. In Europe, for example, a CTA must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed. To obtain regulatory approval to commercialize a new drug under European Union regulatory systems, we must submit a marketing authorization application (“MAA”). The MAA is similar to the NDA, with the exception of, among other things, country-specific document requirements and environmental impact assessments.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCPs and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
Specialized Skill and Knowledge
Medicenna’s business requires personnel with specialized skills and knowledge in the fields of basic and applied immunotherapy and immunology and oncology in general. Medicenna has subcontracted out several key functions to highly specialized individuals and companies to conduct the preclinical development of drug candidates from our BiSKIT’s platform, manufacturing of MDNA11 and bizaxofusp as well as certain clinical and regulatory aspects of the ABILITY study. These programs are overseen by Medicenna’s Chief Executive Officer, Chief Development Officer and Acting Chief Medical Officer, to ensure proper and timely completion of the required activities. In addition Medicenna has deep expertise available on its Clinical Advisory Board, Development Advisory Committee and Scientific Advisory Board.
4.C. Organizational Structure
MTI is the Company’s wholly-owned subsidiary. MTI has three wholly-owned subsidiaries: Medicenna Biopharma Inc., incorporated under the laws of British Columbia, Canada, Medicenna Biopharma Inc., incorporated under the laws of Delaware and Medicenna Australia PTY Ltd, incorporated under the laws of Australia. Our organizational chart is below:

4.D. Property, Plants and Equipment
Not applicable.
ITEM 4A. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
The management’s discussion and analysis of the Company for the year ended March 31, 2023 is included in this Annual Report in Exhibit 15.1.
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
6.A Directors and Senior Management
The following table sets forth the name, position, age, and functions and areas of experience in the Company of each of our directors and senior management:
| Name / Age / Province / State and Country of Residence |
Position with the Company |
Date Became a Director / Officer |
Principal Occupation Last Five Years |
|||
| Fahar Merchant, PhD Toronto, Ontario Canada Age: 65 |
President, Chief Executive Officer and Director |
Director since October 30, 2011 / Officer since 2011 |
President and Chief Executive Officer of the Company (2011 to present) |
|||
| Rosemina Merchant, MESc Toronto, Ontario, Canada Age: 67 |
Chief Development Officer and Director |
Director since April 25, 2016; Officer since October 30, 2011 |
Chief Development Officer of the Company (2011 to present) |
|||
| Elizabeth Williams, CPA, CA Georgetown, Ontario, Canada Age: 46 |
Chief Financial Officer, Corporate Secretary |
Officer since December 2016 |
Chief Financial Officer of the Company (2016 to present) |
|||
| Albert G. Beraldo, CPA, CA(1)(3) Toronto, Ontario, Canada Age: 69 |
Lead Independent Director |
Director since November 22, 2016 |
President of Idoman Ltd. (2008 to present) |
|||
| Karen Dawes, MA, MBA(1)(2) Palm Beach Gardens, Florida, USA Age: 71 |
Director |
Director since September 24, 2019 |
President, Knowledgeable Decisions, LLC (2003 to present) |
|||
| John (Jack) Geltosky, PhD(3) Portland, Oregon, USA Age: 77 |
Director |
Director since September 30, 2020 |
Managing Director of JEG and Associates, LLC (2011 to present) |
|||
| Chandrakant Panchal, PhD(1) Pierrefonds, Quebec, Canada Age: 74 |
Director |
Director since November 22, 2016 |
Chairman, Chief Executive Officer and Chief Scientific Officer of Axcelon Biopolymers Corp. (2001 to present) |
|||
| John H. Sampson, MD, PhD, MBA(2) Linville, North Carolina, USA Age: 56 |
Director |
Director since September 23, 2021 |
Robert H. and Gloria Wilkins Distinguished Professor (2009 to present), Inaugural Chair, Department of Neurosurgery, Duke University Medical Center (2015 to 2020), President, Private Diagnostic Clinic, PLLC, Duke Health (2018 to present) |
Notes:
| 1. |
Member of the Audit Committee. |
|
| 2. |
Member of the Corporate Governance and Nominating Committee. |
|
| 3. |
Member of the Compensation Committee. |
Fahar Merchant and Rosemina Merchant are married. There are no other family relationships among the directors and officers.
There are no arrangements or understandings with major shareholders, customers, suppliers or others pursuant to which any person named above was selected as a director or member of senior management.
Directors and Executive Officers
The following are short biographies of our directors and executive officers:
Albert G. Beraldo, CPA, CA
Mr. Albert G. Beraldo has been a Director of the Company since November 22, 2016. Mr. Beraldo has over 30 years’ experience in varying roles within the pharmaceutical/biotechnology industry. Mr. Beraldo has been the President of Idoman Limited since July 2008. Mr. Beraldo is the Chairman and founding shareholder of Global Transplant Solutions Inc., a US based company providing human organ preservation fluid solutions and developing products for the Human organ procurement and transplant marketplace. Mr. Beraldo was the founder and President and Chief Executive Officer of Alveda Pharmaceuticals Inc., a leading supplier of pharmaceuticals to the Canadian health care market, from 2006 until November 2015. Alveda was acquired by Teligent, Inc. (formerly IGI Laboratories, Inc., Nasdaq), a New Jersey-based specialty generic pharmaceutical company. Mr. Beraldo formerly served as President and Chief Executive Officer of Bioniche Pharma Group Limited until 2006. Mr. Beraldo has sat on the board of Pure Global Cannabis Inc. (TSXV), Helix Biopharma Corp. (January 2016 to July 2017) and was an Independent Director of Telesta Therapeutics Inc. (July 2011 to February 2014). Mr. Beraldo worked in public accounting with Ernst and Whinney until he joined Vetrepharm Canada Inc. as Financial Controller in 1983. Mr. Beraldo obtained a Bachelor of Commerce degree from the University of Windsor and a Chartered Accountant designation from the Canadian Institute of Chartered Accountants.
Karen Dawes, MA, MBA
Ms. Karen Dawes was appointed a Director of the Company in September 24, 2019. Ms. Dawes has over 20 years of commercial and executive management and has been a key player in the successful development, launch, and marketing of products in the Cardiovascular, CNS, Oncology, Metabolic, Infectious Disease, and Women’s and Men’s Health areas, including five blockbuster therapeutics. Her industry experience began with 10 years of commercial and executive management at Pfizer, where she gained increasing responsibility in product management, development, and strategy leading to her position as Vice-President, Marketing, Pratt Division. Ms. Dawes then moved to the biotech pioneer Genetics Institute (GI), where, as Chief Commercial Officer, she built the company’s initial commercial operations including strategic and operational marketing, sales, medical affairs, public relations, and market research. When GI was acquired by Wyeth, Ms. Dawes was appointed by the new parent company as Senior Vice-President, Global Strategic Marketing. Subsequently, she moved to Bayer Corporation as Division Head for the company’s U.S. Pharmaceuticals Division. Ms. Dawes is currently President of Knowledgeable Decisions, a biopharmaceutical consulting firm focusing on corporate and commercial strategy. Ms. Dawes also serves as the chairperson of the board of directors of Repligen Corporation (Nasdaq: RGEN) and is a member of the boards of directors of Vaccitech PLC (Nasdaq), JPA Health, and Medicines360. Ms. Dawes has a combined B.A. and M.A. from Simmons College and an MBA from Harvard Business School.
John (Jack) Geltosky, PhD
Dr. John (Jack) Geltosky has served as a Director of the Company since September 30, 2020. Dr. Geltosky is currently Managing Director of JEG and Associates, LLC, a business development consulting firm focused on biotech and pharmaceuticals, a position he has held since September 2011. Dr. Geltosky is an experienced pharmaceutical licensing executive with a strong R&D background. He has extensive commercial development and deals portfolio from his role as Vice President External Science, Technology & Licensing at Bristol Myers Squibb (BMS) as well as Vice President, Scientific Licensing, Worldwide Business Development at SmithKline Beecham (now GlaxoSmithKline). Dr. Geltosky also held roles of increasing responsibility within Johnson & Johnson over a 10-year period. He began his career as a research scientist at E.I. DuPont. He holds a PhD in biochemistry from the California Institute of Technology.
Fahar Merchant, PhD
Dr. Fahar Merchant has served as a Director and Officer of the Company since 2011. Dr. Merchant is a biotech veteran with 30 years’ of experience as a serial entrepreneur and co-founder of Medicenna. Previously he was President and CEO of Protox Therapeutics Inc. where he transitioned a pre-clinical start-up to a Phase 3 ready uro-oncology company in six years (2005-2011). In 1992, he co-founded IntelliGene Expressions, Inc., a biologics cGMP compliant CDMO, and built it to one of the fastest growing companies in Canada ensuring profitability during his tenure as CEO. In 2000, by strategic in-licensing, he co-founded Avicenna Medica, Inc., a clinical stage oncology company and sold it a year later to KS Biomedix (LSE) for $90 million. Dr. Merchant was CTO and Director of KS Biomedix until its acquisition by Xenova (Nasdaq and LSE) in 2003. He has raised over $150 million from public and private sources to fund development of targeted therapies for oncology and closed corporate transactions valued at over $250 million. Dr. Merchant holds a BSc in Biochemistry and Pharmacology from Aston University, MSc in Biotechnology from Birmingham University and a PhD in Biochemical Engineering from Western University.
Rosemina Merchant, MESc
Ms. Rosemina Merchant was elected a Director of the Company on April 25, 2016 and has served as an Officer of the Company since October 30, 2011. Ms. Merchant has over 30 years of experience in the development of biopharmaceuticals. Prior to co-founding Medicenna, Ms. Merchant was Senior VP of Development and Regulatory Affairs at Protox Therapeutics, Inc (TSX), and responsible for the development of PRX302 (Topsalysin) a PSA activated protoxin for localized prostate cancer and BPH. She transitioned PRX302, a discovery project to Phase 3 readiness in 6 years. During that time, she executed multiple clinical trials, managed Canadian and the United States regulatory filings and led all CMC related outsourcing activities in the United States and Europe. In 1992, Ms. Merchant co-founded, IntelliGene Expressions, Inc., a biologics cGMP compliant CDMO, where she was VP of Manufacturing and Chief Operating Officer. She also held a variety of senior level positions at KS Biomedix, GE LifeSciences, Alberta Innovates, Bioniche, and Sanofi Pasteur. Ms. Merchant holds a B.Sc in Pharmacology and Chemistry from Aston University, MSc in Applied Organic Chemistry from Birmingham University, and M.E.Sc. in Biochemical Engineering from Western University.
Chandrakant Panchal, PhD
Dr. Chandrakant Panchal has served as a Director of the Company since November 22, 2016. Dr. Panchal is the Founder of Axcelon Biopolymers Corp., a biotechnology company where he is Chairman, CEO and CSO. From 1989 to 1999 he was Co-Founder, President, and CEO of Procyon Biopharma Inc., which he took public on the TSXV in 1998 and later on the TSX in 2000. Thereafter, Dr. Panchal was CSO at Procyon until its merger with Cellpep, Inc (2006). He was then Senior Executive VP of Business Development at the merged entity, Ambrilia Biopharma Inc. During his term at Procyon and Ambrilia, he led several licensing and M&A transactions with pharmaceutical and biotechnology companies relating to cancer and HIV drugs developed by the company. Dr. Panchal sits on the boards of Avicanna Inc.(as Chairman) (TSX), and four other private corporations. Dr. Panchal obtained a PhD in biochemical engineering from Western University.
John H. Sampson, MD, PhD, MBA
Dr. John H Sampson has served as a Director of the Company since September 2021. He is the Robert H. and Gloria Wilkins Distinguished Professor of Neurosurgery at Duke University School of Medicine. He is also President of Private Diagnostic Clinic, Duke’s physician practice with revenue of over $1 billion and a member of the prestigious National Academy of Medicine. He has served on multiple Scientific and Governance Boards at publicly traded biotechnology companies and major non-profit health delivery organizations. Dr. Sampson is one of the National Institutes of Health’s top funded neurosurgeons, has helped develop various immune-based therapies, and has served as the lead investigator in dozens of early and late-stage clinical trials. He has published more than 270 peer-reviewed papers in journals such as Nature, Journal of the American Medical Association, and Proceedings of the National Academy of Sciences, and has been an editorial board member for major journals in his field. As part of his research efforts, he is actively investigating new modalities of direct brain tumor infusion and the development of novel immunotherapies. Dr. Sampson has an MD from the University of Manitoba, a PhD from Duke University, and an MBA from Duke’s Fuqua School of Business.
Elizabeth Williams, CPA, CA
Ms. Williams, CPA, CA has more than 18 years of experience in biotech, working with publicly listed entities in both Canada and the United States. Ms. Williams has extensive financing experience playing an integral role in raising more than $250 million in financing by way of public offerings, private placements, rights offerings, at-the-market facilities, warrant exercises, corporate reorganizations and debt (issuance and redemption). Prior to joining Medicenna, Ms. Williams was the Vice President of Finance and Administration at Aptose Biosciences Inc. (TSX and Nasdaq), a biotechnology company (“Aptose”). While at Aptose, Ms. Williams held several positions including acting as the Chief Financial Officer during a lengthy transition period and was responsible for a broad range of activities including financings, financial reporting and regulatory compliance. Prior to joining Aptose, Ms. Williams was an Audit Manager at Ernst & Young LLP with a focus on publicly listed multinational companies. Ms. Williams is a Director and Chair of the Audit Committee of Triumvira Immulogics Inc. Ms. Williams is a Chartered Professional Accountant and Chartered Accountant and received a Bachelor of Business Administration from Wilfrid Laurier University.
Board Diversity
The table below provides certain information regarding the diversity of our board of directors as of the date of this Annual Report.
| Board Diversity Matrix |
||||
| Country of Principal Executive Offices: |
Canada |
|||
| Foreign Private Issuer |
Yes |
|||
| Disclosure Prohibited under Home Country Law |
No |
|||
| Total Number of Directors |
7 |
|||
| Female |
Male |
Non- Binary |
Did Not |
|
| Part I: Gender Identity |
||||
| Directors |
2 |
5 |
- |
- |
| Part II: Demographic Background |
||||
| Underrepresented Individual in Home Country Jurisdiction |
3 |
|||
| LGBTQ+ |
- |
|||
| Did Not Disclose Demographic Background |
- |
|||
Corporate Cease Trade Orders, Bankruptcies, Penalties or Sanctions
Cease Trade Orders
Other than as described below, to the knowledge of the Company, no director or executive officer of the Company is, or within the 10 years prior to the date hereof has been, a director, chief executive officer, or chief financial officer, of any company (including the Company) that was subject to (a) a cease trade order; (b) an order similar to a cease trade order; or (c) an order that denied the relevant company access to any exemption under securities laws, that was in effect for a period of more than thirty consecutive days, issued while that person was acting in such capacity or issued thereafter but resulted from an event that occurred while that person was acting in such capacity.
Dr. Chandrakant Panchal is the chairman of the board of Avicanna Inc. (“Avicanna”). Avicanna announced, on March 29, 2021, that it was unable to file its audited annual financial statements for the year ended December 31, 2020, and accompanying management’s discussion and analysis, annual information form and related certifications on or before March 31, 2021, as required under applicable securities laws. On June 11, 2021, a cease trade order was issued by its principal regulator, the Ontario Securities Commission. The order was revoked on September 10, 2021, further to Avicanna filing the periodic and continuous disclosure documents required under applicable securities legislation.
Bankruptcies
Other than as described below, to the knowledge of the Company, no director or executive officer or shareholder holding a sufficient number of securities of the Company to affect materially the control of the Company is, or within the 10 years prior to the date hereof has been, a director or executive officer of any company (including the Company) that, while that person was acting in such capacity or within a year of that person ceasing to act in such capacity, became bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency or was subject to or instituted any proceedings, arrangement or compromise with creditors or had a receiver, receiver manager or trustee appointed to hold its assets.
Dr. Jack Geltosky was a director of Sophiris Bio Inc. when it decided to shut down its operations in May 2020. In connection with the shutdown, Sophiris Bio Inc. reached a compromise agreement with its senior creditor to pay an amount less than the full amount owed to the creditor.
Dr. Panchal and Mr. Albert G. Beraldo were both directors of Pure Global Cannabis Inc. when it sought and obtained, on March 19, 2020, an Order from the Ontario Superior Court of Justice (Commercial List) granting relief under the Companies’ Creditors Arrangement Act (Canada). On May 1, 2020, Dr. Panchal and Mr. Beraldo both resigned as directors of Pure Global Cannabis Inc. and a receiver and manager was appointed to hold its assets pursuant to the Bankruptcy and Insolvency Act (Canada) by Order of the Ontario Superior Court of Justice (Commercial List).
To the knowledge of the Company, no director or executive officer or shareholder holding a sufficient number of securities of the Company to affect materially the control of the Company has, within the ten years prior to the date hereof, become bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency, or become subject to or instituted any proceedings, arrangement, or compromise with creditors, or had a receiver, receiver manager or trustee appointed to hold that person’s assets.
Penalties and Sanctions
No director or executive officer of the Company, or a shareholder holding a sufficient number of securities of Medicenna to affect materially the control of the Company has been subject to (a) any penalties or sanctions imposed by a court relating to securities laws or by a securities regulatory authority or has entered into a settlement agreement with a securities regulatory authority; or (b) any other penalties or sanctions imposed by a court or regulatory body that would likely be considered important to a reasonable investor in making an investment decision.
All of the above disclosure also applies to any personal holding companies of any of the persons referred to above.
Conflicts of Interest
Certain of the Company’s officers and directors are also officers and/or directors of other, or may otherwise be involved with or consulted by, companies engaged in the biotechnology industry and research business generally and may be presented from time to time with situations or opportunities which give rise to apparent conflicts of interest which cannot be resolved by arm’s length negotiations but only through exercise by the officers and directors of such judgment as is consistent with their fiduciary duties to the Company which arise under applicable corporate law, especially insofar as taking advantage, directly or indirectly, of information or opportunities acquired in their capacities as directors or officers of the Company. Any such conflict is governed by applicable corporate laws, which require that directors act honestly, in good faith and with a view to the best interests of the Company. It is expected that any transactions with officers and directors will be on terms consistent with industry standards and sound business practice in accordance with the fiduciary duties of those persons to the Company, and, depending upon the magnitude of the transactions and the absence of any disinterested board members, may be submitted to the shareholders for their approval.
In addition, the CBCA requires officers and directors to disclose any personal interest which they may have in any material contract or transaction which is proposed to be entered into with the Company and, in the case of directors, to abstain from voting as a director for the approval of any such contract or transaction, unless otherwise permitted under the CBCA.
6.B. Compensation
STATEMENT OF EXECUTIVE COMPENSATION
Compensation Discussion and Analysis
Objectives
The Company has historically relied on the experience of its Board and independent compensation consultants in setting executive compensation. In considering compensation awards, the Board has considered the skill level of its executives as well as comparable levels of compensation for individuals with similar capabilities and experience. In regard to the Company’s current executive compensation arrangements, the Board has also considered such factors as the Company’s current financial situation, the estimated financial situation of the Company in the mid‑term and the need to attract and retain the key executives necessary for the Company’s long-term success.
On March 28, 2017, the Board established a Compensation Committee to, among other things, (i) consider the overall remuneration strategy and, where information is available, verifying the appropriateness of existing remuneration levels using external sources for comparison; (ii) compare the nature and amount of directors’ and executive officers’ compensation to performance against goals set for the year while considering relevant comparative information, independent expert advice and the Company’s financial position, and (iii) make recommendations to the Board in respect of director and executive officer remuneration matters, with the overall objective of ensuring maximum Shareholder benefit from the retention of high quality board and executive team members.
Medicenna’s executive compensation program is designed to:
| ● |
attract and retain qualified, motivated and achievement-oriented individuals by offering compensation that is competitive in the industry and marketplace; |
| ● |
align executive interests with the interests of Shareholders; and |
| ● |
ensure that individuals continue to be compensated in accordance with their personal performance and responsibilities and their contribution to the overall objectives of the Company. |
These objectives are achieved by offering executives and employees a compensation package that is competitive and rewards the achievement of both short-term and long-term objectives of the Company. As such, our compensation package consists of three key elements:
| ● |
base salary and initial Options; |
| ● |
short-term compensation incentives to reward corporate and personal performance through potential annual cash bonuses; and |
| ● |
long-term compensation incentives related to long-term increase in Common Share value through participation in the Stock Option Plan. |
The Compensation Committee reviews each of these items on a stand-alone basis and also reviews compensation as a total package. Adjustments to compensation are made as appropriate following a review of the compensation package as a whole.
Benchmarking
In February 2023, the Compensation Committee retained the services of Gallagher Benefit Services (Canada) Group Inc. (“Gallagher”) to perform an analysis of Executive and Director compensation with respect to the Company’s executive compensation program.
Gallagher was hired directly by the Compensation Committee and may not receive other mandates from the Company unless said Committee gives its prior consent.
The following table presents the fees paid by the Company to Gallagher:
| March 31, 2023 |
March 31, 2022 |
|
| Executive compensation related fees |
$23,500 |
Nil |
| Other fees |
Nil |
Nil |
Named Executive Officers - Compensation Comparator Group
In order to perform its analysis, Gallagher compared Medicenna’s executive compensation against the following named peer companies (“Peer Group”) approved by the Compensation Committee. All criteria were assessed as of February 2023.
| Company |
Industry (Biotech) |
Exchange (Nasdaq) |
Geography (Canada) |
Market Capitalization1 ($0-100M) |
Market Capitalization1 (>$200M) |
| Aptose Biosciences Inc. |
X |
X |
X |
X |
|
| Bolt Biotherapeutics Inc. |
X |
X |
X |
||
| BriaCell Therapeutics Corp. |
X |
X |
X |
X |
|
| Cue Biopharma, Inc. |
X |
X |
X |
||
| Essa Pharma Inc. |
X |
X |
X |
X |
|
| IMV Inc. |
X |
X |
X |
X |
|
| Neoleukin Therapeutics, Inc. |
X |
X |
X |
||
| Oncolytics Biotech Inc. |
X |
X |
X |
X |
|
| Werewolf Therapeutics, Inc. |
X |
X |
X |
||
| Xilio Therapeutics, Inc. |
X |
X |
X |
||
| Medicenna |
X |
X |
X |
X |
| 1. |
All financial data has been extracted from S&P Global Market Intelligence’s S&P Capital IQ platform. Market Capitalization data is as of February 1, 2023. |
In addition to proxy data, Gallagher gathered competitive market data from its proprietary databases to:
| ● |
arrive at competitive market compensation; |
| ● |
evaluate market data at the 25th, 50th, and 75th percentiles for all pay elements; and |
| ● |
assess compensation based on base salary, target total cash compensation (base salary + target short term incentive opportunity), long-term incentives (annual and total stock option Black-Scholes value + full-value share face value) and target total direct compensation (target total cash compensation + long-term incentives). |
Gallagher’s findings included that Medicenna’s target total direct compensation to the executive team was well below the 25th percentile for each of the Named Executive Officers.
Base Salary
In establishing base salaries, the objective of the Board is to establish levels that will enable Medicenna to attract and retain executive officers who can effectively contribute to the long-term success of the Company. Base salary for each executive officer is determined by the individual’s skills, abilities, experience, past performance and anticipated future contributions to the success of Medicenna.
Short-Term Compensation Incentives
The role of short-term compensation incentives at Medicenna is to motivate our executive officers to achieve specified performance objectives for fiscal 2023 and to reward them for their achievement in the event that those objectives are met. The Board sets annual corporate objectives encompassing scientific, clinical, regulatory, business and corporate development and financial criteria. The annual cash bonus for the executive officers is based, at least in part, on the level of achievement of these annual objectives, assuming these objectives are still relevant at the time of evaluation. All current corporate and executive officer objectives are reviewed by the Compensation Committee and approved by the Board. The Compensation Committee recommends to the Board the awarding of bonuses, payable in cash, stock or share options if warranted by individual performance.
Cash bonuses are determined as soon as practicable after the end of the fiscal year and, for the Named Executive Officers (as defined hereinafter), are included in the Summary Compensation Table in the year in respect of which they are earned.
Long-Term Incentive Plans
Long-term incentives, in the form of Options, are intended to align the interests of the Company’s directors and its executive officers with those of its shareholders, to provide a long-term incentive that rewards these individuals for their contribution to the creation of shareholder value and to reduce the cash compensation that the Company would otherwise have to pay. In determining the size and terms of individual grants, the Board takes into account, among other things (i) the level of effort, time, responsibility, ability, experience and level of commitment of the executive officer and (ii) market comparatives for similarly situated executives.
Hedge or Offset Instruments
Named Executive Officers or directors are not permitted to purchase financial instruments that are designed to hedge or offset a decrease in market value of equity securities granted as compensation or held, directly or indirectly, by Named Executive Officers or directors, including, for greater certainty, prepaid variable forward contracts, equity swaps, collars, or units of exchange funds.
Risk Assessment of Compensation
The implications of the risks associated with the Company’s compensation practices were not considered by the Board or a committee of the Board.
Performance Graph
The following graph compares the total shareholder return of $100 invested in our Common Shares since the Company’s initial public offering with the total return of the S&P/TSX Capped Health Care Index:
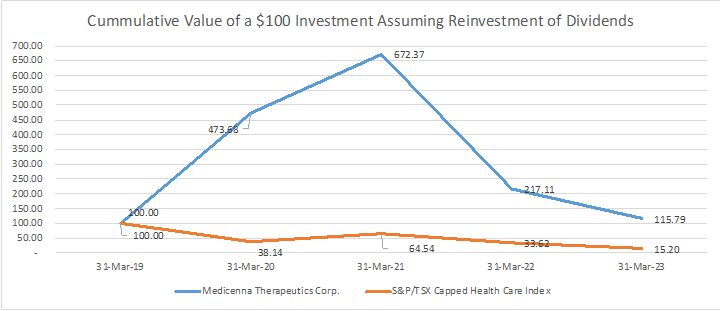
The performance trend shown by the above graph does not necessarily reflect the trend in our compensation to Named Executive Officers reported over the same period. The market price of the Common Shares, similar to the share prices of many publicly-traded biotechnology companies, has historically been highly volatile. Our approach to compensation is designed to attract and retain quality executives while promoting long-term profitability and maximizing shareholder value. Our Named Executive Officers are compensated on the basis of individual and corporate performance rather than on factors strictly tied to the short-term performance of our Common Shares in the market.
Summary Compensation Table
The following table details the compensation information for the three fiscal years ended March 31, 2023 of the Company, for the Chairman, President and Chief Executive Officer, the Chief Financial Officer and the Chief Development Officer (each, an “NEO” and, collectively the “Named Executive Officers”).
| Name and Principal Position |
Year Ended |
Salary ($) |
Share-based awards ($) |
Option-based awards ($) |
Non-equity incentive |
Pension value ($) |
All other compensation ($) |
Total Compensation ($) |
|
| Annual incentive plan ($) |
Long-term incentive plans ($) |
||||||||
| Dr. Fahar Merchant Chairman, President and Chief Executive Officer |
March 31, 2023 March 31, 2022 March 31, 2021 |
428,365(1) 436,154(1) 543,383(1) |
N/A N/A N/A |
422,820(2) 432,305(3) 298,915(4) |
Nil Nil Nil |
Nil Nil Nil |
N/A N/A N/A |
26,000(5) 26,000(5) 26,000(5) |
877,185 894,459 868,298 |
| Ms. Elizabeth Williams, Chief Financial Officer |
March 31, 2023 March 31, 2022 March 31, 2021 |
285,000 285,000 260,000 |
N/A N/A N/A |
186,732(2) 148,236(3) 118,052(4) |
Nil Nil 74,880 |
Nil Nil Nil |
N/A N/A N/A |
26,000(5) 26,000(5) 26,000(5) |
497,732 459,236 478,932 |
| Ms. Rosemina Merchant Chief Development Officer |
March 31, 2023 March 31, 2022 March 31, 2021 |
338,462(1) 344,615(1) 401,418(1) |
N/A N/A N/A |
249,251(2) 193,071(3) 133,941(4) |
Nil Nil 38,350 |
Nil Nil Nil |
N/A N/A N/A |
26,000(5) 26,000(5) 26,000(5) |
613,713 563,686 599,709 |
| Dr. Mann Muhsin Former Chief Medical Officer |
March 31, 2023 March 31, 2022 |
Nil 331,846(6) |
N/A N/A |
Nil 572,205(3)(8) |
Nil Nil |
Nil Nil |
N/A N/A |
Nil Nil |
Nil 904,051 |
| Dr. Kevin Moulder Former Chief Scientific Officer |
March 31, 2023 March 31, 2022 |
Nil 99,525(7) |
N/A N/A |
Nil 370,040(3)(8) |
Nil Nil |
Nil Nil |
N/A N/A |
Nil Nil |
Nil 469,565 |
| (1) |
Includes amounts paid to the Executive for vacation pay accrued but unused. For Dr. Merchant, an amount of $23,365 was paid for unused vacation (base salary $405,000) in the year ended March 31, 2023, $31,154 (base salary $405,000) in the year ended March 31, 2022 and $148,383 (base salary $395,000) in the year ended March 31, 2021. For Ms. Merchant, an amount of $18,462 (base salary $320,000) was paid for unused vacation in the year ended March 31, 2023, $24,615 (base salary $320,000) in the year ended March 31, 2021 and $106,418 (base salary $295,000) for the year ended March 31, 2021. |
| (2) |
In determining the fair value of these option-based awards, the Black-Scholes valuation methodology was used with the following assumptions: (i) expected life of five years; (ii) volatility 90%; (iii) risk-free interest rate of 4.50%; and (iv) no dividend yield. The Company has decided to use the Black-Scholes valuation methodology because it is equivalent to the option value reported in the Company’s consolidated financial statements. |
| (3) |
In determining the fair value of these option-based awards, the Black-Scholes valuation methodology was used with the following assumptions: (i) expected life of five years; (ii) volatility 90%; (iii) risk-free interest rate of 1.00%; and (iv) no dividend yield. The Company has decided to use the Black-Scholes valuation methodology because it is equivalent to the option value reported in the Company’s consolidated financial statements. |
| (4) |
In determining the fair value of these option-based awards, the Black-Scholes valuation methodology was used with the following assumptions: (i) expected life of five years; (ii) volatility 103%; (iii) risk-free interest rate of 1.00%; and (iv) no dividend yield. The Company has decided to use the Black-Scholes valuation methodology because it is equivalent to the option value reported in the Company’s consolidated financial statements. |
| (5) |
Represents amount paid into an RRSP by the Company on the NEOs’ behalf. |
| (6) |
Dr. Mann Muhsin was appointed as Chief Medical Officer on May 10, 2021 and resigned from his position effective January 21, 2022. Dr. Muhsin was paid in US dollars, the amounts presented have been converted to Cdn dollars at a US/Cdn exchange rate of $/$1.2536. |
| (7) |
Dr. Keven Moulder was appointed as Chief Scientific Officer on April 20, 2022 and resigned from his position effective February 28, 2022. Dr. Moulder was paid in UK Pounds, the amounts presented have been converted to Cdn dollars at a UK Pound/Cdn exchange rate of £1/$1.7131. |
| (8) |
Options granted to Dr. Muhsin and Dr. Moulder were forfeited unvested upon their resignation. |
Incentive Plan Award – Named Executive Officers
Outstanding Share-Based Awards and Option-Based Awards
The following tables show all awards outstanding to each NEO as at March 31, 2023:
| Option-based Awards | Share-based Awards | ||||||
| Name and Principal Position |
Number of securities underlying unexercised options (#) |
Option exercise price ($) |
Option expiration date |
Value of unexercised in-the-money options ($) (1) |
Number of shares or units of shares that have not vested (#) |
Market or payout value of share-based awards that have not vested ($) |
Market or payout value of vested share-based awards not paid out or distributed ($) |
| Dr. Fahar Merchant Chairman, President and Chief Executive Officer |
198,487 77,299 300,000 300,000 350,000 350,000 405,000 |
3.14 5.11 1.30 1.00 2.00 2.01 1.45 |
Sep 23, 2031 Nov 3, 2030 Nov 8, 2029 Feb 14, 2029 Feb 13, 2027 Sept 21, 2027 June 24, 2032 |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
| Ms. Elizabeth Williams Chief Financial Officer |
68,073 30,528 150,000 200,000 125,000 75,000 178,862 |
3.14 5.11 1.30 1.00 2.00 2.01 1.45 |
Sep 23, 2031 Nov 3, 2030 Nov 8, 2029 Feb 14, 2029 Feb 13, 2027 Sept 21, 2027 June 24, 2032 |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
| Ms. Rosemina Merchant Chief Development Officer |
88,646 34,637 200,000 200,000 250,000 150,000 238,746 |
3.14 5.11 1.30 1.00 2.00 2.01 1.45 |
Sep 23, 2031 Nov 3, 2030 Nov 8, 2029 Feb 14, 2029 Feb 13, 2027 Sept 21, 2027 June 24, 2032 |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
Nil Nil Nil Nil Nil Nil Nil |
| (1) |
These amounts are calculated based on the difference between the market value of the securities underlying the Options on March 31, 2023 at the end of the fiscal year ($0.88), and the exercise price of the Options. |
Value Vested or Earned During the Year
The following table sets forth for each NEO the value vested or earned on all option-based awards, share-based awards and non-equity incentive plan compensation during the year ended March 31, 2023:
| Name and Principal Position |
Option-based awards – Value vested during the year ($) |
Share-based awards – Value vested during the year ($) |
Non-equity incentive plan compensation – Value earned during the year ($) |
| Dr. Fahar Merchant Chairman, President and Chief Executive Officer |
Nil |
N/A |
Nil |
| Ms. Elizabeth Williams Chief Financial Officer |
Nil |
N/A |
Nil |
| Ms. Rosemina Merchant Chief Development Officer |
Nil |
N/A |
Nil |
Pension Plan Benefits
The Company does not provide pension plan benefits to its NEOs or employees of the Company.
Director Compensation Table
The following table details the compensation received by each director for the year ended March 31, 2023 (other than directors who were also Named Executive Officers and for whom information is shown in the table under the heading “Summary Compensation Table” above):
| Name |
Fees earned ($) |
Share-based awards ($) |
Option-based awards(1) ($) |
Non-equity incentive plan compensation ($) |
Pension value ($) |
All other Compensation ($) |
Total ($) |
| Mr. Albert G. Beraldo |
81,750 |
Nil |
61,200 |
Nil |
N/A |
Nil |
142,950 |
| Dr. Chandrakant Panchal |
51,875 |
Nil |
61,200 |
Nil |
N/A |
Nil |
113,075 |
| Dr. John (Jack) Geltosky |
56,250 |
Nil |
61,200 |
Nil |
N/A |
Nil |
117,450 |
| Ms. Karen Dawes |
62,500 |
Nil |
61,200 |
Nil |
N/A |
Nil |
123,700 |
| Dr. John Sampson |
50,625 |
Nil |
61,200 |
Nil |
N/A |
Nil |
111,825 |
| (1) |
In determining the fair value of these option-based awards, the Black-Scholes valuation methodology was used with the following assumptions: (i) expected life of 5 years; (ii) volatility 90%; (iii) risk-free interest rate of 4.50%; and (iv) no dividend yield. The Company has decided to use the Black-Scholes valuation methodology because it is equivalent to the option value reported in the Company’s consolidated financial statements. |
Since April 1, 2021, the directors are entitled to an annual fee of $45,000 with no per meeting fees. The lead director is entitled to an additional annual fee of $18,000. The chair of the Audit Committee is entitled to an annual fee of $15,000, with each committee member receiving an annual fee of $7,500. The respective chairs of the Governance Committee and of the Compensation Committee are entitled to an annual fee of $10,000, with each committee member receiving an annual fee of $5,000 per committee.
Non-executive directors are reimbursed for any out-of-pocket travel expenses incurred in order to attend meetings. Executive directors are not entitled to directors’ compensation.
Dr. Merchant and Ms. Merchant did not receive any compensation for their role as directors of the Company.
Incentive Plan Awards – Directors
Outstanding Share-Based Awards and Option Based Awards
The following table sets forth for each director, other than the Named Executive Officers who are directors, all option-based and share-based awards outstanding at March 31, 2023:
| Option-based Awards |
Share-based Awards |
||||||
| Name |
Number of securities underlying unexercised options (#) |
Option exercise price ($) |
Option expiration date |
Value of unexercised in-the-money options ($) (1) |
Number of shares or units of shares that have not vested (#) |
Market or payout value of share-based awards that have not vested ($) |
Market or payout value of vested share-based awards not paid out or distributed ($) |
| Mr. Albert G. Beraldo |
27,070 15,655 75,000 50,000 50,000 50,000 58,621 |
3.14 5.11 1.30 1.00 2.00 2.88 1.45 |
Sep 23, 2026 Nov 3, 2025 Nov 8, 2024 Feb 14, 2024 Feb 13, 2027 Nov 10, 2022 June 24, 2027 |
Nil Nil Nil Nil Nil Nil Nil |
N/A |
N/A |
N/A |
| Dr. Chandrakant Panchal |
27,070 15,655 75,000 50,000 50,000 50,000 58,621 |
3.14 5.11 1.30 1.00 2.00 2.88 1.45 |
Sep 23, 2026 Nov 3, 2025 Nov 8, 2024 Feb 14, 2024 Feb 13, 2027 Nov 10, 2022 June 24, 2027 |
Nil Nil Nil Nil Nil Nil Nil |
N/A |
N/A |
N/A |
| Dr. John (Jack) Geltosky |
27,070 15,655 58,621 |
3.14 5.11 1.45 |
Sep 23, 2026 Nov 3, 2025 June 24, 2027 |
Nil Nil Nil |
N/A |
N/A |
N/A |
| Karen Dawes |
27,070 15,655 75,000 58,621 |
3.14 5.11 1.30 1.45 |
Sep 23, 2026 Nov 3, 2025 Nov 8, 2024 June 24, 2027 |
Nil Nil Nil Nil |
N/A |
N/A |
N/A |
| Dr. John Sampson |
27,070 58,621 |
3.14 1.45 |
Sep 23, 2026 June 24, 2027 |
Nil Nil |
N/A |
N/A |
N/A |
| (1) |
These amounts are calculated based on the difference between the market value of the securities underlying the Options on March 31, 2023 at the end of the fiscal year ($0.88), and the exercise price of the Options. |
Value Vested or Earned During the Year
The following table sets forth for each director the value vested or earned on all option-based awards, share-based awards, and non-equity incentive plan compensation during the year ended March 31, 2023.
| Name |
Option-based awards – Value vested during the year ($) |
Share-based awards – Value vested during the year ($) |
Non-equity incentive plan compensation – Value earned during the year ($) |
| Mr. Albert G. Beraldo |
3,810 |
N/A |
Nil |
| Dr. Chandrakant Panchal |
3,810 |
N/A |
Nil |
| Dr. Jack Geltosky |
3,810 |
N/A |
Nil |
| Ms. Karen Dawes |
3,810 |
N/A |
Nil |
| Dr. John Sampson |
3,810 |
N/A |
Nil |
Securities Authorized for Issuance Under Equity Compensation Plans
The following table sets forth certain details as at the end of the year ended March 31, 2023 with respect to compensation plans pursuant to which equity securities of the Company are authorized for issuance.
| Plan Category |
Number of Shares to be |
Weighted-average |
Number of Shares remaining |
| Equity compensation plans approved by Shareholders |
5,610,353 |
$1.84 |
4,832,840 |
| Equity compensation plans not approved by Shareholders |
Nil |
Nil |
Nil |
| Total |
5,610,353 |
$1.84 |
4,832,840 |
Employment Agreements
We have entered into employment agreements with Fahar Merchant, Rosemina Merchant and Elizabeth Williams.
Fahar Merchant
On October 1, 2016, Fahar Merchant entered into an amended and restated employment agreement with MTI and MBI. Pursuant to this agreement, as amended, both MTI and MBI employ Dr. Merchant as an executive officer for a base annual salary of $405,000, a $26,000 annual retirement contribution and an annual bonus of up to 50% of his base annual salary. Dr. Merchant is also entitled to executive benefits comparable to those provided by MBI and MTI to other senior executives, including but not limited to executive level health insurance and benefits. MBI and MTI may also grant Dr. Merchant stock options pursuant to the terms outlined in the Company’s Stock Option Plan. See “6.E Share Ownership” below for a description of the Company’s Stock Option Plan. This employment agreement can be terminated by any party for convenience or for cause, and in the event of termination, MTI and MBI will pay Dr. Merchant a severance fee pursuant to the terms set forth in the agreement and described below. The foregoing description is qualified in its entirety by reference to Dr. Merchant’s employment agreement, which is included as Exhibits 10.8, 10.9, 10.10 and 10.11 hereto and incorporated by reference herein.
Rosemina Merchant
On October 1, 2016, Rosemina Merchant entered into an amended and restated employment agreement with MTI and MBI. Pursuant to this agreement, as amended, both MTI and MBI employ Ms. Merchant as an executive officer for a base annual salary of $320,000, a $26,000 annual retirement contribution and an annual bonus of up to 40% of her base annual salary. Ms. Merchant is also entitled to executive benefits comparable to those provided by MTI and MBI to other senior executives, including but not limited to executive level health insurance and benefits. MBI and MTI may also grant Ms. Merchant stock options pursuant to the terms defined in the Company’s Stock Option Plan. See “6.E Share Ownership” below for a description of the Company’s Stock Option Plan. This agreement can be terminated by any party for convenience or for cause, and in the event of termination, MTI and MBI will pay Ms. Merchant a severance fee pursuant to the terms set forth in the Agreement and described below. The foregoing description is qualified in its entirety by reference to Ms. Merchant’s employment agreement, which is included as Exhibits 10.12, 10.13, 10.14 and 10.15 hereto and incorporated by reference herein.
Elizabeth Williams
On December 12, 2016, Elizabeth Williams entered into an employment agreement with MTI. Pursuant to this Agreement, as amended, MTI employs Elizabeth Williams as an executive officer for a base annual salary of $285,000, with a $26,000 annual retirement contribution and an annual bonus of up to 40% of her annual base salary based on MTI’s achievement of certain milestones agreed upon by the parties. Ms. Williams is also entitled to dental and extended health benefits coverage in accordance with the policies and procedures of MTI. MTI may also grant Ms. Williams stock options pursuant to the terms defined in the Company’s Stock Option Plan. See “6.E Share Ownership” below for a description of the Company’s Stock Option Plan. The agreement can be terminated by any party for convenience or for cause, and in the event of termination, MTI will provide a severance fee pursuant to the terms set forth in the Agreement and described below. The foregoing description is qualified in its entirety by reference to Ms. Williams’ employment agreement, which is included as Exhibits 10.16, 10.17, 10.18 and 10.19 hereto and incorporated by reference herein.
Termination and Change of Control Benefits
The employment agreements of Dr. Merchant, Ms. Williams and Ms. Merchant provide that if their employment is terminated by the Company other than for cause, they will be entitled to the following benefits:
| Name |
Termination Without Cause |
Change of Control |
||||||||||
| Dr. Fahar Merchant |
$ | 607,500 | (1) | $ | 607,500 | (1) | ||||||
| Ms. Elizabeth Williams |
$ | 285,000 | (2) | $ | 285,000 | (2) | ||||||
| Ms. Rosemina Merchant |
$ | 320,000 | (3) | $ | 320,000 | (3) | ||||||
| (1) |
This amount represents 18 months of Dr. Merchant’s annual base salary as of March 31, 2023. |
| (2) (3) |
This amount represents 12 months of Ms. Williams annual base salary as of March 31, 2023. This amount represents 12 months of Ms. Merchant’s annual base salary as of March 31, 2023. |
Fahar Merchant
In the event that Dr. Merchant’s employment is terminated by Medicenna other than for cause, Dr. Merchant shall be entitled to receive a lump sum payment equal to one and one half times his then annual base salary (less applicable source deductions) as well as any bonus eligible but not yet paid as of the time of termination. As at March 31, 2023, this obligation would have been $607,500. In addition, all unvested Options will become immediately vested and exercisable. In the event of termination without cause or for good reason either in connection with or twelve months following a change of control, Dr. Merchant shall be entitled to severance pay equivalent to one and one half times his then annual base salary (less applicable source deductions) as well as any bonus eligible but not yet paid as of the time of termination. As at March 31, 2023, this obligation would have been $607,500. In addition, all unvested Options will become immediately vested and exercisable.
Elizabeth Williams
In the event that Ms. Williams’s employment is terminated by Medicenna other than for cause, Ms. Williams shall be entitled to receive a lump sum payment equal to twelve months of her then annual base salary. As at March 31, 2023, this obligation would have been $285,000.
In the event of termination without cause or for good reason either in connection with or twelve months following a Change of Control, Ms. Williams shall be entitled to severance pay equivalent to be entitled to receive a lump sum payment of twelve months of her then annual base salary as well as any bonus eligible but not yet paid as of the time of termination. As at March 31, 2023, this obligation would have been $285,000.
Rosemina Merchant
In the event that Ms. Merchant’s employment is terminated by Medicenna other than for cause, Ms. Merchant shall be entitled to receive a lump sum payment equal to one times her then annual base salary (less applicable source deductions). As at March 31, 2023, this obligation would have been $320,000.
In the event of termination without cause or for good reason either in connection with or twelve months following a change of control, Ms. Merchant shall be entitled to severance pay equivalent to one times her then annual base salary (less applicable source deductions) as well as any bonus eligible but not yet paid as of the time of termination. As at March 31, 2023, this obligation would have been $320,000. In addition, all unvested Options will become immediately vested and exercisable.
6.C. Board Practices
All of our directors are elected at the annual meeting of our shareholders, or at any special meeting of shareholders if one of the purposes for which a special meeting was called was the election of directors, and each holds such office until his or her successor is elected or appointed, unless his or her office is earlier vacated by way of the director’s resignation or death or under any of the relevant provisions of our Articles or the CBCA.
Employment, Consulting and Directors’ Service Contracts
For information on employment agreements with Dr. Merchant and Ms. Merchant, see Item 6.B. above.
Audit Committee
The Audit Committee is a committee of the Board that assists the Board in fulfilling its oversight of, and recommend appropriate actions with respect to:
| the integrity of the Company’s financial statements, accounting and financial reporting processes, system of internal controls over financial reporting and audit process; |
| the Company’s compliance with, and process for monitoring compliance with, legal and regulatory requirements so far as they relate to matters of financial reporting; |
| the independent auditor’s qualifications, independence and performance; and |
| the design, implementation and performance of the Company’s internal audit function. |
Audit Committee Terms of Reference
The Company has a written charter which sets out the duties and responsibilities of its Audit Committee. The Audit Committee Charter is attached hereto as Exhibit 15.2.
Audit Committee Composition
The Company’s Audit Committee is comprised of three directors: Albert G. Beraldo (Chair), Karen Dawes and Chandrakant Panchal.
Relevant Education and Experience
All of the Audit Committee members are independent of management of the Company as required by the TSX and the Nasdaq and each member is financially literate in that he or she has the ability to read and understand a set of financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of the issues that can reasonably be expected to be raised by the Company’s financial statements.
| Albert G. Beraldo, CPA, CA (Chair) – Mr. Beraldo worked in public accounting with Ernst and Whinney until he joined Vetrepharm Canada Inc. as Financial Controller in 1983. Mr. Beraldo is the Financial Expert of the Audit Committee and has many years of experience as the Chief Financial Officer of both private and public companies. Mr. Beraldo obtained a Bachelor of Commerce degree from the University of Windsor and a Chartered Accountant designation from the Canadian Institute of Chartered Accountants. Mr. Beraldo is financially literate and an independent director of the Company for the purpose of NI 52-110. | |
| Karen Dawes, MA, MBA – Ms. Dawes worked as Chief Commercial Officer of biotech pioneer Genetics Institute (GI), where she built that company’s initial commercial operations including strategic and operational marketing, sales, medical affairs, public relations, and market research. When GI was acquired by Wyeth, Karen was appointed by the new parent company as Senior Vice-President, Global Strategic Marketing. Subsequently, Karen moved to Bayer Corporation as Division Head for the company’s U.S. Pharmaceuticals Division. Ms. Dawes is currently President of Knowledgeable Decisions, a biopharmaceutical consulting firm focusing on corporate and commercial strategy. Ms. Dawes also serves as the chairperson of the board of directors of RepliGen (Nasdaq: RGEN) and is a member of the board of directors of Medicines360. Ms. Dawes has a combined B.A. and M.A. from Simmons College and an MBA from Harvard Business School. Ms. Dawes is financially literate and an independent director of the Company for the purpose of NI 52-110. | |
| Chandrakant Panchal, PhD - Dr. Panchal is the Founder of Axcelon Biopolymers Corp., a biotechnology company where he is Chairman, CEO and CSO. From 1989 to 1999 he was Co-Founder, President, and CEO of Procyon Biopharma Inc., which he took public on the TSXV in 1998 and later on the TSX in 2000. Thereafter, Dr. Panchal was CSO at Procyon until its merger with Cellpep, Inc (2006). He was then Senior Executive VP of Business Development at the merged entity, Ambrilia Biopharma Inc. During his term at Procyon and Ambrilia, he led several licensing and M&A transactions with pharmaceutical and biotechnology companies relating to cancer and HIV drugs developed by the company. Dr. Panchal sits on the boards of Avicanna Inc.(as Chairman) (TSX), and four other private corporations. Dr. Panchal obtained a PhD in biochemical engineering from Western University. Dr. Panchal is financially literate and an independent director of the Company for the purpose of NI 52-110. |
Pre-Approval Policies and Procedures
The Audit Committee has adopted specific policies and procedures for the engagement of non-audit services, as described in the Audit Committee Charter attached hereto as Exhibit 15.2. All non-audit services performed by our auditors for the twelve-month period ended March 31, 2023 were pre-approved by our Audit Committee. It is our policy that all non-audit services performed by our auditors will continue to be pre-approved by our Audit Committee.
Compensation Committee
The Compensation Committee has the responsibility of assisting Board oversight of executive and director compensation. Without limiting the generality of the foregoing, the Compensation Committee has the following responsibilities:
| a. |
review and approve corporate goals and objectives relevant to the compensation of the Company’s CEO, evaluate the CEO’s performance in light of those goals and objectives and determine and approve the CEO’s compensation level; |
|
| b. |
grant options under the Company’s Stock Option Plan, as amended from time to time; |
|
| c. | review and approve the cash and non-cash compensation of the executive officers; | |
| d. |
recommend to the Board the cash and non-cash compensation policies for the non-employee directors; |
|
| e. |
make recommendations to the Board with respect to amendments to the Company’s Stock Option Plan or implementing other equity-based plans; |
|
| f. |
assist the Board in evaluating potential candidates for executive officer positions with the Company; and |
|
| g. |
produce a compensation committee report on executive officer compensation as required by the applicable securities laws. |
The Compensation Committee is composed of independent directors, including John (Jack) Geltosky and Albert G. Beraldo. The Chair of the Compensation Committee is John (Jack) Geltosky. In discharging its responsibilities, the Compensation Committee shall meet as often as it determines necessary or advisable, but not less frequently than annually. The Compensation Committee may also hold special meetings or act by unanimous written consent as the Compensation Committee may decide.
Corporate Governance and Nomination Committee
The Corporate Governance and Nomination Committee has the responsibility of assisting the Board in fulfilling its corporate governance responsibilities. Without limiting the generality of the foregoing, the Corporate Governance and Nomination Committee has the following responsibilities:
| a. |
identify qualified individuals to become Board members, consistent with criteria approved by the Board; |
| b. |
determine the composition of the Board and its committees; |
| c. |
select the director nominees for the next annual meeting of shareholders; |
| d. |
monitoring a process to assess Board, committee and management effectiveness; |
| e. |
aid and monitor management succession planning; and |
| f. |
developing, recommending to the Board, implementing and monitoring policies and processes related to the Company’s corporate governance guidelines consistent with applicable securities laws and applicable rules and guidelines of any stock exchange on which the securities of the Company are listed and any other laws applicable to the Company. |
The Corporate Governance and Nomination Committee is composed of independent directors, including Karen Dawes and John Sampson. The Chair of the Corporate Governance and Nomination Committee is Karen Dawes. In discharging its responsibilities, the Corporate Governance and Nomination Committee shall meet as often as it determines necessary or advisable, but not less than twice a year. The Corporate Governance and Nomination Committee may also hold special meetings or act by unanimous written consent as the Corporate Governance and Nomination Committee may decide.
6.D. Employees
As at March 31, 2023, Medicenna had 16 full-time employees and one part-time consultant, including seven holding PhD degrees, two with an MBBS and two employees holding CPA designations.
Medicenna depends on certain key members of its management and scientific staff and the loss of services of one or more of these persons could adversely affect the Company.
Medicenna also uses consultants and outside contractors to carry on many of Medicenna’s activities, including preclinical testing and validation, formulation, assay development, manufacturing, clinical and regulatory affairs, toxicology and clinical trials.
None of our employees or consultants are represented by a labor organization or are party to a collective bargaining arrangement. We consider our relationships with our employees to be good.
6.E. Share Ownership
The following table indicates information as of June 10, 2022, regarding the beneficial ownership of our Common Shares, for:
| ● |
each person who is known by us to beneficially own more than 5% of our Common Shares; |
| ● |
each named executive officer; |
| ● |
each of our directors; and |
| ● |
all of our directors and executive officers as a group. |
Unless otherwise indicated in the footnotes to the table, and subject to community property laws where applicable, the following persons have sole voting and investment control with respect to the shares beneficially owned by them. In accordance with SEC rules, if a person has a right to acquire beneficial ownership of any Common Shares on or within 60 days of June 20, 2023, upon conversion or exercise of outstanding securities or otherwise, the shares are deemed beneficially owned by that person and are deemed to be outstanding solely for the purpose of determining the percentage of our shares that person beneficially owns. These shares are not included in the computations of percentage ownership for any other person. As of June 9, 2023, we had 11 record holders of our Common Shares, with 6 record holders in Canada, representing 67% of our outstanding Common Shares, and 5 record holders in the United States, representing 33% of our outstanding Common Shares.
Except as otherwise indicated, the address of each of the persons in this table is 2 Bloor St., 7th Floor, Toronto, Ontario M4W 3E2.
| Name and Address of Beneficial Owner |
Shares Beneficially Owned |
Percentage of Shares Beneficially Owned |
||||||
| 5% and Greater Shareholders: |
||||||||
| Fahar Merchant, PhD(1) |
18,782,468 | 27.0 | % |
|||||
| Rosemina Merchant(2) |
18,782,468 | 27.0 | % |
|||||
| Aries Biologics Inc.(3) |
5,500,000 | 7.9 | % |
|||||
| AIGH Capital Management, LLC (4) |
5,429,983 | 7.8 | % | |||||
| Directors and Named Executive Officers: |
||||||||
| Elizabeth Williams, CPA, CA(5) |
666,937 | 1.0 | % |
|||||
| Albert G. Beraldo, CPA, CA(6) |
651,346 | * | % |
|||||
| Karen Dawes, MA(7) |
201,346 | * | % |
|||||
| John (Jack) Geltosky, PhD(8) |
101,346 | * | % |
|||||
| Chandrakant Panchal, PhD(9) |
329,346 | * | % |
|||||
| John H. Sampson, MD, PhD, MBA(10) |
85,691 | * | % |
|||||
| All executive officers and directors as a group (8 persons)(11) |
20,818,480 | 29.9 | % |
|||||
| * |
Indicates beneficial ownership of less than 1%. |
| (1) |
Includes 100,000 Common Shares underlying warrants and 1,550,168 Common Shares underlying options held directly by Dr. Merchant. Also includes (i) 5,250,000 Common Shares, 100,000 Common Shares underlying warrants and 930,900 Common Shares underlying warrants held by Rosemina Merchant who is married to Dr. Merchant; and (ii) 5,500,000 Common Shares held by Aries Biologics Inc., the voting shares of which are held 50% by Dr. Merchant and 50% by Ms. Merchant. |
| (2) |
Includes 100,000 Common Shares underlying warrants and 1,550,168 Common Shares underlying options held directly by Dr. Merchant. Also includes (i) 5,250,000 Common Shares, 100,000 Common Shares underlying warrants and 930,900 Common Shares underlying warrants held by Rosemina Merchant who is married to Dr. Merchant; and (ii) 5,500,000 Common Shares held by Aries Biologics Inc., the voting shares of which are held 50% by Dr. Merchant and 50% by Ms. Merchant. |
| (3) |
The voting shares of Aries Biologics, Inc. are held 50% by Dr. Merchant and 50% by Ms. Merchant. |
|
| (4) |
Includes 1,000,000 warrants to purchase Common Shares. Based solely on the Form 13G/A filed on February 15, 2023, with respect to securities jointly held by AIGH Capital Management, LLC, AIGH Inventory Partners, LLC, and Mr. Orin Hirschman with a principal address of 6006 Berkeley Ave., Baltimore, MD 21209. |
| (5) |
Includes 651,637 Common Shares underlying options held by Ms. Williams. |
| (6) |
Includes 326,346 Common Shares underlying options. |
| (7) |
Includes 176,346 Common Shares underlying options. |
| (8) |
Consists of 101,346 Common Shares underlying options. |
| (9) |
Includes 326,346 Common Shares underlying options. |
| (10) |
Consists of 85,691 Common Shares underlying options. |
| (11) |
Includes 200,000 Common Shares underlying warrants and 4,148,780 Common Shares underlying options. |
Stock Option Plan
The Company’s Stock Option Plan was approved for adoption by shareholders on September 21, 2017 to amend, restate and supersede the previous stock option plan adopted by the Company in 2015.
The Stock Option Plan does not have a fixed number of Shares issuable thereunder, but permits the issuance of up to an aggregate of 15% of the outstanding Shares from time to time. As at March 31, 2023, the Company had Options outstanding under the Stock Option Plan to purchase up to 5,610,353 Shares (representing approximately 8.06% of the then 69,637,469 issued and outstanding Shares). Accordingly, unallocated options with respect to an aggregate of 4,832,840 Shares were available for future grants (representing approximately 6.94% of the then 69,637,469 issued and outstanding Shares).
The Company’s annual “burn rate” for stock options granted under the Stock Option Plan (including predecessor plans), calculated as described in Section 613(p) of the TSX Company Manual with respect to the number of issued and outstanding Shares (total number of stock options issued in a fiscal year, divided by the weighted average number of outstanding Shares for that year) was 0.9% in the fiscal year ended March 31, 2021, 2.02% in the fiscal year ended March 31, 2022 and 1.99% in the fiscal year ended March 31, 2023.
Summary of Material Terms
The Stock Option Plan is intended to attract, retain and motivate persons of training, experience and leadership as key service providers to the Company and its subsidiaries, including their directors, officers and employees, and to advance the interests of the Company. Options may be granted to a director, officer, employee or service provider of the Company or any related entity (being a person that controls or is controlled by the Company or that is controlled by the same person that controls the Company) (each, an “Eligible Person”).
The aggregate number of Shares issuable upon the exercise of all Options granted under the Stock Option Plan and under all other share compensation arrangements will not exceed 15% of the issued and outstanding Shares as at the date of grant of each Option under the Stock Option Plan. If any Option granted under the Stock Option Plan expires, terminates for any reason in accordance with the terms of the Stock Option Plan or is exercised, Shares subject thereto shall again be available for the purpose of the Stock Option Plan. Accordingly, the Stock Option Plan is considered an “evergreen” plan and unallocated options under the Stock Option Plan must be submitted for approval by the Shareholders every three years.
Subject to the terms and conditions of the Stock Option Plan, the number of Shares subject to each Option, the price of each Option, the expiration date of each Option, the extent to which each Option is exercisable from time to time during the term of the Option and other terms and conditions relating to each such Option shall be determined by the Compensation Committee and recommended to the Board.
Option and other terms and conditions relating to each such Option shall be determined by the Compensation Committee and recommended to the Board.
The exercise price for any Option issued under the Stock Option Plan may not be less than the Market Price of the Shares on the date of which the grant of the Option is approved by the Board. For these purposes, “Market Price” at any date in respect of the Shares means the closing sale price of the Shares on the TSX on the trading date immediately preceding such date; provided that, (i) in the event that such Shares did not trade on such trading day, the Market Price shall be the average of the bid and ask prices in respect of such Shares at the close of trading on such trading day, (ii) if no quotation is made for the applicable day, the Market Price on such day shall be determined in the manner set forth in the preceding clause for the next preceding trading day, and (iii) notwithstanding the foregoing, if there is no reported closing price or high bid/low asked price that satisfies the preceding clauses, the Market Price on any day shall be determined by such methods and procedures as shall be established from time to time by the Compensation Committee.
Options issued under the Stock Option Plan may be exercised during a period determined under the Stock Option Plan, which may not exceed ten years. Unless otherwise determined by the Board, Options will vest as follows: 50% on the first anniversary of the grant, 25% on the second anniversary of the grant and 25% on the third anniversary of the grant. Any or all Shares that have vested may be purchased during the term of the Options.
In addition to the restrictions on maximum issuances set forth above for all security based compensation arrangements, the number Shares which may be issued pursuant to Options granted under the Stock Option Plan to any one person may not exceed 5% of the then aggregate issued and outstanding Shares at the date of the grant.
The following insider participation limits also apply under the Stock Option Plan: (i) the number of Shares issuable to insiders, at any time, pursuant to the Stock Option Plan and other share compensation arrangements shall not exceed 10% of the issued and outstanding Shares (on a non-diluted basis); and (ii) the number of Shares issued to insiders, within a one-year period, pursuant to the Stock Option Plan and other share compensation arrangements shall not exceed 10% of the issued and outstanding Shares (on a non-diluted basis).
An Option is personal to the optionholder and non-assignable (whether by operation of law or otherwise); provided, however, that Options may be transferred or assigned to certain permitted assignees which include a spouse, a trustee acting on behalf of the optionholder or spouse, a holding entity or an RRSP, RRIF or TFSA of the optionholder or spouse. If the optionholder resigns, is terminated for cause or fails to be re-elected as a director, the Options terminate immediately. If the optionholder dies or ceases to be eligible under the Stock Option Plan for any other reason, Options that are entitled to be exercised may generally be exercised (subject to certain extensions at the discretion of the Board or a committee thereof) until the earlier of (i) one year or three months, respectively, of the applicable date, or (ii) the expiry date of the Option.
The Option Plan also provides for the cashless exercise of Options which allows for the option holder to receive, without cash payment (other than taxes), a number of Shares based on the following formula:

In the event that the expiry of an Option occurs during a blackout period imposed by management or the Board in accordance with the Company’s insider trading policy, the expiry date of such Option shall be deemed to be amended to that date which is ten business days following the end of such blackout period.
In the event of a Change of Control (as such term is defined in the Stock Option Plan) with respect to the Company or a Corporate Group entity (which, under the Stock Option Plan, means the Company and any subsidiary or related or affiliated business entities of the Company and includes any successor corporations or entities thereto), notwithstanding anything in the Stock Option Plan to the contrary:
| ● |
if the employment of an optionee is terminated by the Company or a Corporate Group entity without cause or if the optionee resigns in circumstances constituting constructive dismissal by the Company or the Corporate Group entity, respectively, in each case, within twelve months (or such other period as determined by the Board in its sole discretion) following a Change of Control with respect to the Company or the Corporate Group entity, respectively (such date being the “Termination Date”), all or any of the optionee’s Options will vest immediately prior to the Termination Date (or such later period as determined by the Board in its sole discretion), subject to any performance conditions which shall be dealt with at the discretion of the Board. All vested Options may be exercised until 90 days (or such other period as may be determined by the Board in its sole discretion) following the Termination Date (but until the normal expiry date of the Option rights of such optionee, if earlier). Upon the expiration of such period, all unexercised Option rights of that optionee shall immediately become terminated and shall lapse notwithstanding the original term of the Option granted to such optionee under the Stock Option Plan; and |
| ● |
any surviving, successor or acquiring entity will assume any outstanding Options or will substitute similar awards for the outstanding Options. If the surviving, successor or acquiring entity is a “private issuer” or does not have any securities listed on an established securities exchange, does not assume the outstanding Options or substitute similar awards for the outstanding Options, or if the Board otherwise determines in its sole discretion and subject to the rules of the TSX, the Company will give written notice to all optionees advising that the Stock Option Plan will be terminated effective immediately prior to the Change of Control and all Options will be deemed to be vested Options, and may provide for the exercise of Options and tender of Shares in connection with the Change of Control and may otherwise provide for the cash out or termination of Options that are not exercised within a specified period of time. |
The Stock Option Plan contains certain customary adjustment provisions, including in connection with a subdivision, redivision, consolidation, reclassification, reorganization or other change of, or involving, the Shares.
Subject to applicable regulatory requirements, including the rules of the TSX, and except as provided below, the Board may, in its sole and absolute discretion and without Shareholder approval, amend, suspend, terminate or discontinue the Stock Option Plan and may amend the terms and conditions of Options granted pursuant to the Stock Option Plan. Without limiting the generality of the foregoing, the Board may make the following amendments to the Stock Option Plan, without obtaining Shareholder approval: (i) amendments to the terms and conditions of the Stock Option Plan necessary to ensure that the Stock Option Plan complies with the applicable regulatory requirements, including the rules of the TSX, in place from time to time; (ii) amendments to the provisions of the Stock Option Plan respecting administration of the Stock Option Plan and eligibility for participation under the Stock Option Plan; (iii) amendments to the provisions of the Stock Option Plan respecting the terms and conditions on which Options may be granted pursuant to the Stock Option Plan, including the provisions relating to the term of the Option and the vesting schedule; and (iv) amendments to the Stock Option Plan that are of a “housekeeping” nature.
However, the Board may not, without the approval of the Shareholders, make amendments with respect to the following: (i) an increase to the Stock Option Plan maximum or the number of securities issuable under the Stock Option Plan; (ii) a reduction in the option price of an Option benefitting an insider; (iii) an extension to the term of Options (other than as a result of a blackout period extension) benefitting an insider; (iv) any amendment which would permit Options granted under the Stock Option Plan to be transferable or assignable other than to a permitted assignee and for normal estate settlement purposes; (v) changes to the insider participation limits; and (vi) amendments to the Stock Option Plan amendment provisions.
The Company does not currently have any other security-based compensation arrangement.
6.F. Disclosure of a registrant’s action to recover erroneously awarded compensation
Not applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
7.A. Major Shareholders
See Item 6.E. above.
7.B. Related Party Transactions
Except as otherwise set out herein, there are no material interests, direct or indirect, of any director, executive officer, person who beneficially owns, or controls or directs, directly or indirectly, more than 10% of the outstanding Common Shares, or any known associates or affiliates of such persons, in any transaction within the last three completed financial years or during the current financial year which has materially affected or is reasonably expected to materially affect the Company.
7.C. Interests of Experts and Counsel
Not applicable.
8.A. Consolidated Statements and Other Financial Information
The audited consolidated financial statements for the years ended March 31, 2021, 2022 and 2023 can be found under “Item 17. Financial Statements”.
8.B. Significant Changes
We are not aware of any significant change that has occurred since March 31, 2023, the date of the audited consolidated financial statements included in this Annual Report, and that has not been disclosed elsewhere in this Annual Report.
ITEM 9. THE OFFER AND LISTING.
9.A. Offer and Listing Details
The Common Shares are listed and posted for trading on each of the TSX and Nasdaq under the trading symbol “MDNA”.
The following tables shows the price ranges and volumes traded on the TSX and Nasdaq for the periods noted:
| Month |
TSX |
|||
| High ($) |
Low ($) |
Volume (#) |
||
| April 2022 |
$1.97 |
$1.35 |
759,096 |
|
| May 2022 |
$1.50 |
$1.18 |
337,182 |
|
| June 2022 |
$1.70 |
$0.98 |
649,295 |
|
| July 2022 |
$2.14 |
$1.40 |
626,848 |
|
| August 2022 |
$2.38 |
$1.13 |
3,115,804 |
|
| September 2022 |
$1.35 |
$1.06 |
880,743 |
|
| October 2022 |
$1.21 |
$0.89 |
830,546 |
|
| November 2022 |
$1.00 |
$0.55 |
1,131,966 |
|
| December 2022 |
$0.88 |
$0.54 |
792,462 |
|
| January 2023 |
$1.04 |
$0.64 |
565,931 |
|
| February 2023 |
$1.05 |
$0.84 |
514,248 |
|
| March 2023 |
$0.85 |
$0.68 |
400,629 |
|
| Month |
Nasdaq |
|||
| High ($) |
Low ($) |
Volume (#) |
||
| April 2022 |
US$1.59 |
US$1.07 |
3,531,700 |
|
| May 2022 |
US$1.24 |
US$0.91 |
1,828,600 |
|
| June 2022 |
US$1.33 |
US$0.76 |
1,416,700 |
|
| July 2022 |
US$1.64 |
US$1.10 |
996,400 |
|
| August 2022 |
US$1.88 |
US$0.86 |
14,694,300 |
|
| September 2022 |
US$1.06 |
US$0.77 |
4,702,500 |
|
| October 2022 |
US$0.95 |
US$0.65 |
3,111,500 |
|
| November 2022 |
US$0.77 |
US$0.40 |
4,325,100 |
|
| December 2022 |
US$0.62 |
US$0.40 |
3,225,900 |
|
| January 2023 |
US$0.79 |
US$0.47 |
3,360,200 |
|
| February 2023 |
US$0.79 |
US$0.57 |
1,776,800 |
|
| March 2023 |
US$0.63 |
US$0.50 |
1,341,200 |
|
Convertible Securities
As at the date hereof, there are issued and outstanding the following convertible securities of the Company, details of which are outlined in the table below:
| Security |
Number |
Exercise or Conversion Price |
Expiry Date (dd/mm/yyyy) |
| Stock options |
5,610,353 |
$1.00 to $5.19 |
24/02/2024 to 12/08/2032 |
| Warrants |
13,333,333 |
US$1.85 |
10/08/2027 |
| Warrants |
1,303,000 |
$1.20 |
21/12/2023 |
| Warrants |
1,549,042 |
$1.75 |
17/07/2023 |
The following securities of the Company (other than Common Shares) were issued during the fiscal year ended March 31, 2023:
| Date of Issue |
Security |
Number |
Exercise Price |
| June 24, 2022 |
Stock options |
1,115,713 |
$1.45 |
| August 12, 2022 |
Stock options |
175,000 |
$1.36 |
9.B. Plan of Distribution
Not applicable.
9.C. Markets
A discussion of all stock exchanges and other regulated markets on which our securities are listed is provided under “Item 9.A. Offer and Listing Details.”
9.D. Selling Shareholders
Not applicable.
9.E. Dilution
Not applicable.
9.F. Expenses of the Issue
Not applicable.
ITEM 10. ADDITIONAL INFORMATION
10.A. Share Capital
Not applicable.
10.B. Memorandum and Articles of Association
Articles of Incorporation and By-laws
We are governed by our articles of continuance dated November 13, 2017 (the “Articles”) under the CBCA and by our by-law no. 2 dated July 31, 2020 (the “By-law”). Our Articles are on file with Corporations Canada under Corporation Number 1049266-3.
Purposes of the Company
Our Articles and By-law do not include a stated purpose and do not place any restrictions on the business that the Company may carry on.
Directors
Our Articles provide that the minimum number of directors we must have is one (1) and the maximum number is eleven (11). In accordance with the CBCA, at least 25% of our directors must be residents of Canada. In order to serve as a director, a person must be a natural person at least 18 years of age, capable and not bankrupt. Neither the Articles nor the By-law contain an age limit requirement for the retirement of non-retirement of directors. Our Articles provide that the directors may, between annual general meetings of the shareholders, appoint one (1) or more additional directors of the Company to serve until the next annual general meeting, but the number of additional directors shall not at any time exceed 1/3 of the number of directors who held office at the expiration of the last annual general meeting of the Company.
The directors are elected by a majority of the votes cast at the annual general meeting at which an election of directors is required or at any special meeting of shareholders, to hold office until the election of their successors, except in the case of resignations or if their offices become vacant by death or otherwise.
Neither the Articles nor the By-law require directors to hold a minimum number of shares of the Company to qualify as a director.
The directors are entitled to remuneration determined by the Board or by a committee to which the Board may delegate the power to do so from time to time. There is no requirement for an independent quorum. Under the mandate of our Compensation Committee, comprised of a minimum of two directors all of whom shall be independent directors, such committee is tasked with making recommendations to the Board concerning directors’ remuneration.
Our By-law provide that no director or officer shall be disqualified by his office from contracting with the Company nor shall any contract or arrangement entered into by or on behalf of the Company with any director or officer or in which any director or officer is in any way interested be liable to be voided nor shall any director or officer so contracting or being so interested be liable to account to the Company for any profit realized by any such contract or arrangement by reason of such director or officer holding that office or of the fiduciary relationship thereby established; provided that the director or officer shall have complied with the provisions of the CBCA. The CBCA provides that a director who is a party to, or who is a director or officer of, or has a material interest in, any person who is a party to a material contract or transaction or proposed material contract or transaction with us must disclose to us the nature and extent of his or her interest at the time and in the manner provided by the CBCA, or request that same be entered in the minutes of the meetings of the Board, even if such contract, in connection with our normal business activity, does not require the approval of either the directors or the shareholders. At the request of the president or any director, the director placed in a situation of conflict of interest must leave the meeting while the Board discusses the matter. The CBCA prohibits such a director from voting on any resolution to approve the contract or transaction unless the contract or transaction:
| - |
relates primarily to his or her remuneration as our director, officer, employee or agent or as a director, officer, employee or agent of an affiliate of us; |
| - |
is for indemnity or insurance for director’s liability as permitted by the CBCA; or |
| - |
is with our affiliate. |
The CBCA provides that the Board may, on our behalf and without authorization of our shareholders:
| - |
borrow money upon our credit; |
| - |
issue, reissue, sell or pledge our debt obligations; |
| - |
give a guarantee on our behalf to secure performance of an obligation of any person; and |
| - |
mortgage, hypothecate, pledge or otherwise create a security interest in all or any of our property, owned or subsequently acquired, to secure any of our obligations. |
The shareholders have the ability to restrict such powers through our Articles or By-law (or through a unanimous shareholder agreement), but no such restrictions are in place.
Pursuant to the CBCA, our directors manage and administer our business and affairs and exercise all such powers and authority as we are authorized to exercise pursuant to the CBCA, the Articles and the By-law. The general duties of our directors and officers under the CBCA are to act honestly and in good faith with a view to our best interests and to exercise the care, diligence and skill that a reasonably prudent person would exercise in comparable circumstances. Any breach of these duties may lead to liability to us and our shareholders for breach of fiduciary duty. In addition, a breach of certain provisions of the CBCA, including the improper payment of dividends or the improper purchase or redemption of shares, will render the directors who authorized such action liable to account to us for any amounts improperly paid or distributed.
Our By-law provide that we shall, to the full extent provided by law, indemnify a director or an officer, a former director or officer of the Company or another individual who acts or acted at the Company’s request as a director or officer, or in a similar capacity, of another entity, and his heirs and legal representatives to the extent permitted by the CBCA against all expenses (including legal fees), judgments, fines and any amount actually and reasonably incurred by him in respect of any civil, criminal, administrative or investigative (other than an action by or in the right of the Company) by reason of the fact that he is or was an employee or agent of the Company, or is or was serving at the request of the Company as a director, officer, employee, agent of or participant in another entity, provide he acted honestly and in good faith with a view to the best interests of the Company or, as the case may be, to the best interests of the other entity for which he served at the Company’s request and, with respect to any criminal or administrative action or proceeding that is enforced by a monetary penalty, had reasonable grounds for believing that his conduct was lawful.
Share Capitalization
The authorized share capital of the Company consists of an unlimited number of Common shares, and an unlimited number of Preferred shares. As of the date hereof, our authorized share capital consists of (i) an unlimited number of Common Shares, of which 69,637,469 are issued and outstanding, and (ii) an unlimited number of Preferred shares, of which nil are issued and outstanding. In addition, we have 5,610,353 Common Shares issuable pursuant to outstanding stock options and 16,185,386 issuable upon the exercise of outstanding warrants. We had nil beneficial owners of our Preferred Shares as of March 31, 2023.
Common Shares
The holders of the common shares are entitled to receive notice of and attend all meetings of shareholders and have one vote for each common shares held by them, except meetings at which only shareholders of a specified class of shares are entitled to vote, provided that they were shareholders as of the record date. In addition, the holders are entitled to receive dividends if, as and when declared by our Board on the common shares, provided that the Company is entitled to declare dividends on the preferred shares, or on any of such classes of shares without being obliged to declare any dividends on the common shares. Finally, the holders of the common shares are entitled, subject to the rights, privileges, restrictions and conditions attaching to any other class of shares of the Company, to receive our remaining property upon any liquidation, dissolution or winding-up of our affairs, whether voluntary or involuntary in equal rank with the holders of all common shares of the Company. Shareholders have no liability to further capital calls as all shares issued and outstanding are fully paid and non-assessable.
Shareholder Actions
The CBCA provides that our shareholders may, with leave of a court, bring an action in our name and on our behalf for the purpose of prosecuting, defending or discontinuing an action on our behalf. In order to grant leave to permit such an action, the CBCA provides that the court must be satisfied that our directors were given adequate notice of the application, the shareholder is acting in good faith and that it appears to be in our best interests that the action be brought.
Action Necessary to Change Rights of Shareholders
In order to change the rights of our shareholders, we would need to amend our Articles to effect the change. Such an amendment would require the approval of holders of two-thirds of the issued and outstanding shares cast at a duly called special meeting and, for certain amendments, the holders of shares of a class or of a series are entitled to vote separately as a class or series on a proposal to amend the articles. For certain amendments, a shareholder is entitled under the CBCA to dissent in respect of such a resolution amending the Articles and, if the resolution is adopted and we implement such changes, demand payment of the fair value of its shares.
Meetings of Shareholders
An annual meeting of shareholders is held each year for the purpose of considering the financial statements and reports, electing directors, appointing auditors and for the transaction of other business as may be brought before the meeting. The board of directors has the power to call a special meeting of shareholders at any time. A quorum at any meeting of shareholders shall be persons present not being less than two in number and holding or representing more than twenty-five percent (25%) of the total number of issued and outstanding common shares of the Company.
Notice of the time and place of each meeting of shareholders must be given not less than 21 days, nor more than 60 days, before the date of each meeting to each director, to the auditor and to each shareholder who at the close of business on the record date for notice is entered in the securities register as the holder of one or more shares carrying the right to vote at the meeting. Notice of meeting of shareholders called for any other purpose other than consideration of the minutes of an earlier meeting, financial statements and auditor's report, election of directors and reappointment of the incumbent auditor, must state the nature of the business in sufficient detail to permit the shareholder to form a reasoned judgment on and must state the text of any special resolution or by-law to be submitted to the meeting.
The only persons entitled to be present at a meeting of shareholders are those entitled to vote, the directors of the Company and the auditor of the Company. Any other person may be admitted only on the invitation of the chairman of the meeting or with the consent of the meeting. In circumstances where a court orders a meeting of shareholders, the court may direct how the meeting may be held, including who may attend the meeting.
The CBCA provides that the holders of not less than 5% of our outstanding voting shares may requisition our directors to call a meeting of shareholders for the purpose stated in the requisition. Except in limited circumstances, including where a meeting of shareholders has already been called and a notice of meeting already given or where it is clear that the primary purpose of the requisition is to redress a personal grievance against us or our directors, officers or shareholders, our directors, on receipt of such requisition, must call a meeting of shareholders. If the directors fail to call a meeting of shareholders within twenty-one days after receiving the requisition, any shareholder who signed the requisition may call the meeting of shareholders and, unless the shareholders resolve otherwise at the meeting, we shall reimburse the shareholders for the expenses reasonably incurred by them in requisitioning, calling and holding the meeting of shareholders.
The CBCA also provides that, except in limited circumstances, a resolution in writing signed by all of the shareholders entitled to vote on that resolution at a meeting of shareholders is as valid as if it had been passed at a meeting of shareholders.
Our By-law include an advance notice provision (the “Advance Notice Requirement”). The Advance Notice Requirement applies in certain circumstances where nominations of persons for election to the Board are made by our shareholders other than pursuant to: (a) a requisition of a meeting made pursuant to the provisions of the CBCA; or (b) a shareholder proposal made pursuant to the provisions of the CBCA. Among other things, the Advance Notice Requirement fixes a deadline by which shareholders must submit a notice of director nominations to us prior to any annual or special meeting of shareholders where directors are to be elected and sets forth the information that a shareholder must include in the notice for it to be valid. In the case of an annual meeting of shareholders, we must be given not less than 30 days’ notice prior to the date of the annual meeting; provided, however, that in the event that the annual meeting is to be held on a date that is less than 50 days after the date on which the first public announcement of the date of the annual meeting was made, notice may be made not later than the close of business on the 10th day following such public announcement. In the case of a special meeting of shareholders (which is not also an annual meeting) called for the purpose of electing directors, we must be given notice not later than the close of business on the 15th day following the day on which the first public announcement of the date of the special meeting was made. In the case of an annual meeting of shareholders or a special meeting of shareholders (which is not also an annual meeting of shareholders) called for the purpose of electing directors where notice-and-access is used for delivery of proxy-related materials, must be given notice not later than the close of business on the 40th day prior to the date of the meeting of shareholders; provided, however, that if the shareholders’ meeting is to be held on a date that is less 50 days after the notice date or the special meeting notice date, as applicable, the notice shall be made, in the case of an annual meeting of shareholders, not later than the close of business on the 10th day following the notice date and, in the case of a special meeting (which is not also an annual meeting of shareholders) called for the purpose of electing directors (whether or not called for other purposes), not later than the close of business on the 15th day following the special meeting notice date.
The Board may, in its sole discretion, waive any requirement of the Advance Notice Requirement.
Limitations on Right to Own Securities
There is no limitation imposed by the laws of Canada or by the Articles or By-law on the right of a non-resident to hold or vote the common shares, other than as provided in the Investment Canada Act (Canada). The Investment Canada Act (Canada) may require review and approval by the Minister of Industry (Canada) of certain acquisitions of “control” of the Company by a “non-Canadian”. The threshold for acquisitions of control is generally defined as being at least one-third or more of the voting shares of the Company. “Non-Canadian” generally means an individual who is not a Canadian citizen, or a corporation, partnership, trust or joint venture that is ultimately controlled by non-Canadian.
Change of Control
There are no provisions in our By-law or Articles that would have an effect of delaying, deferring or preventing a change in control of the Company and that would operate only with respect to a merger, acquisition or corporate restructuring involving the Company. However, certain types of change of control transactions will require shareholder approval of the Company’s Shareholders and calling the necessary shareholder meeting for such transaction would delay the completion of the transaction.
Disclosure of Share Ownership
In general, under applicable securities regulation in Canada, a person or company who beneficially owns, or who directly or indirectly exercises control or direction over voting securities of a reporting issuer, voting securities of an issuer or a combination of both, carrying more than ten percent of the voting rights attached to all the issuer’s outstanding voting securities is an insider and must, within ten days of becoming an insider, file a report in the required form effective the date on which the person became an insider, disclosing any direct or indirect beneficial ownership of, or control or direction over, securities of the reporting issuer.
Additionally, securities regulation in Canada provides for the filing of a report by an insider of a reporting issuer whose holdings change, which report must be filed within five days from the day on which the change takes place.
Our By-law do not contain a provision governing the ownership threshold above which shareholder ownership must be disclosed.
10.C. Material Contracts
| ● |
2017 Stock Option Plan, effective as of September 21, 2017, pursuant to which the Company may grant stock options of the Company to Eligible Persons on terms determined by the Compensation Committee and approved by the Board; |
| ● |
Exclusive (Equity) Agreement, by and between the Board of Trustees of the Leland Stanford Junior University (“Stanford”) and MTI, effective as of August 21, 2015, as amended by that certain Amendment to Exclusive (Equity) Agreement, effective as of August 21, 2019, pursuant to which Stanford granted a license to the Company to any rights that Stanford has in certain patent applications related to IL-2 superagonists and antagonists; |
| ● |
Exclusive (Equity) Agreement, by and between Stanford and the Company, effective as of August 21, 2015, as amended by that certain Amendment to Exclusive (Equity) Agreement, effective as of August 21, 2019, pursuant to which Stanford granted a license to the Company to any rights that Stanford has in certain patent applications related to therapeutic IL-13 and IL-4 polypeptides; |
| ● |
Start-Up Patent License Agreement – Exclusive, by and between the National Institutes of Health (“NIH”) and the Company, pursuant to which NIH transferred certain inventions related to biomedical and behavioral research to the Company to facilitate the commercial development of products and processes for public use and benefit; |
| ● |
Cancer Research Grant Contract, by and between the Cancer Prevention and Research Institute of Texas (“CPRIT”) and the Company, effective as of March 1, 2015, pursuant to which CPRIT has granted funding to the Company to assist in the development of treatments for cancers; |
| ● |
Employment Agreement, by and among MBI, MTI and Fahar Merchant, effective as of October 1, 2016, as amended by that certain Amendment Letter, effective as of April 1, 2017, that certain Amendment Letter, effective as of April 1, 2020 and that certain Amendment Letter, effective as of April 1, 2021, pursuant to which both MBI and MTI employed Fahar Merchant as an executive officer for a base annual salary of $405,000; |
| ● |
Employment Agreement, by and among MBI, MTI and Rosemina Merchant, effective as of October 1, 2016, as amended by that certain Amendment Letter, effective as of April 1, 2017 and that Amendment Letter, effective as of April 1, 2020, pursuant to which both MBI and MTI employed Rosemina Merchant as an executive officer for a base annual salary of $295,000; and |
| ● |
Employment Agreement, by and between MTI and Elizabeth Williams, effective as of December 12, 2016, as amended by that certain Amendment Letter, effective as of April 1, 2017, that certain Amendment Letter, effective as of April 1, 2020 and that certain Amendment Letter, effective as of April 1, 2021, pursuant to which MTI employed Elizabeth Williams as an executive officer for a base annual salary of $285,000. |
10.D. Exchange Controls
There are currently no government laws, decrees, regulations or other legislation of Canada or the United States that restrict the export or import of capital (including the availability of cash and cash equivalents) or that affect the remittance of dividends, distributions, interest or other payments to non-residents of Canada or the United States holding our Common Shares. Any remittances of dividends to United States residents and to other non-residents are, however, subject to withholding tax. See “Taxation” below.
10.E. Taxation
Material U.S. Federal Income Tax Considerations for U.S. Holders
Subject to the limitations and qualifications stated herein, this discussion sets forth material U.S. federal income tax considerations relating to the acquisition, ownership and disposition by U.S. Holders (as hereinafter defined) of the Common Shares. The discussion is based on the Internal Revenue Code of 1986, as amended (the “Code”), its legislative history, existing and proposed regulations thereunder, published rulings and court decisions, and the Canada-United States Income Tax Convention (1980) as amended (the “Treaty”) all as currently in effect and all subject to change at any time, possibly with retroactive effect. This summary applies only to U.S. Holders. This discussion of a U.S. Holder’s tax consequences addresses only those persons that acquire Common Shares in an offering and that hold those Common Shares as capital assets (generally, property held for investment). In addition, it does not describe all of the tax consequences that may be relevant in light of a U.S. Holder’s particular circumstances, including state and local tax consequences, estate and gift tax consequences, alternative minimum tax consequences, and tax consequences applicable to U.S. Holders subject to special rules, such as:
| ● |
banks, insurance companies, and certain other financial institutions; |
| ● |
U.S. expatriates and certain former citizens or long-term residents of the United States; |
| ● |
dealers or traders in securities who use a mark-to-market method of tax accounting; |
| ● |
persons holding Common Shares as part of a hedging transaction, “straddle,” wash sale, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to Common Shares; |
| ● |
persons whose “functional currency” for U.S. federal income tax purposes is not the U.S. dollar; |
| ● |
brokers, dealers or traders in securities, commodities or currencies; |
| ● |
tax-exempt entities or government organizations; |
| ● |
S corporations, partnerships, or other entities or arrangements classified as partnerships for U.S. federal income tax purposes; |
| ● |
regulated investment companies or real estate investment trusts; |
| ● |
persons who acquired the Common Shares pursuant to the exercise of any employee stock option or otherwise as compensation; |
| ● |
persons required to accelerate the recognition of any item of gross income with respect to the Common Shares as a result of such income being recognized on an applicable financial statement; |
| ● |
persons holding the Common Shares in connection with a trade or business, permanent establishment, or fixed base outside the United States; and |
| ● |
persons who own (directly or through attribution) 10% or more (by vote or value) of the outstanding Common Shares. |
If an entity that is classified as a partnership for U.S. federal income tax purposes holds Common Shares, the U.S. federal income tax treatment of a partner will generally depend on the status of the partner, the activities of the partnership and certain determinations made at the partner level. Partnerships holding Common Shares and partners in such partnerships are encouraged to consult their tax advisers as to the particular U.S. federal income tax consequences of holding and disposing of Common Shares.
A “U.S. Holder” is a holder who, for U.S. federal income tax purposes, is a beneficial owner of Common Shares and is:
| ● |
An individual who is a citizen or individual resident of United States; |
| ● |
a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia; |
| ● |
an estate the income of which is subject to U.S. federal income taxation regardless of its source; or |
| ● |
a trust if (1) a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust or (2) the trust has a valid election in effect to be treated as a U.S. person under applicable U.S. Treasury Regulations. |
PERSONS CONSIDERING AN INVESTMENT IN COMMON SHARES SHOULD CONSULT THEIR OWN TAX ADVISORS AS TO THE PARTICULAR TAX CONSEQUENCES APPLICABLE TO THEM RELATING TO THE ACQUISITION, OWNERSHIP AND DISPOSITION OF THE COMMON SHARES, INCLUDING THE APPLICABILITY OF U.S. FEDERAL, STATE AND LOCAL TAX LAWS.
Passive Foreign Investment Company Rules
If the Company is classified as a PFIC in any taxable year, a U.S. Holder will be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. Holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
A non-U.S. corporation will be classified as a PFIC for any taxable year in which, after applying certain look-through rules, either:
| ● |
at least 75% of its gross income is passive income (such as interest income); or |
| ● |
at least 50% of its gross assets (determined on the basis of a quarterly average) is attributable to assets that produce passive income or are held for the production of passive income. |
The Company will be treated as owning its proportionate share of the assets and earning its proportionate share of the income of any other corporation, the equity of which it owns, directly or indirectly, 25% or more (by value).
Based on the Company’s interpretation of the law, the Company’s recent financial statements, and taking into account expectations about the Company’s income, assets and activities, the Company believes that it may have been a PFIC for the taxable year ended March 31, 2023 and expects that it will be a PFIC for the current taxable year. The determination of whether the Company is a PFIC for the taxable year ended March 31, 2023 and the current taxable year will depend, in part, on whether the Company receives government grants (including certain grants similar to those previously awarded by CPRIT) during the taxable year ended March 31, 2024, and the Company’s determination of whether such grants (if received) constitute passive income for PFIC testing purposes. A separate determination must be made after the close of each taxable year as to whether the Company is a PFIC for that year, and as a result, its PFIC status may change from year to year. The total value of the Company’s assets for purposes of the asset test generally will be calculated using the market price of the Common Shares, which may fluctuate considerably. Fluctuations in the market price of the Common Shares may result in the Company’s being a PFIC for any taxable year. Because of the uncertainties involved in establishing the Company’s PFIC status, there can be no assurance regarding if the Company currently is treated as a PFIC, or may be treated as a PFIC in the future.
If the Company is classified as a PFIC in any year with respect to which a U.S. Holder owns the Common Shares, the Company will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding years during which the U.S. Holder owns the Common Shares, regardless of whether the Company continues to meet the tests described above unless (i) the Company ceases to be a PFIC and the U.S. Holder has made a “deemed sale” election under the PFIC rules, or (ii) the U.S. Holder makes a Qualified Electing Fund Election (“QEF Election”) with respect to all taxable years during such U.S. Holders’ holding period in which the Company is a PFIC. If the “deemed sale” election is made, a U.S. Holder will be deemed to have sold the Common Shares the U.S. Holder holds at their fair market value and any gain from such deemed sale would be subject to the rules described below. After the deemed sale election, so long as the Company does not become a PFIC in a subsequent taxable year, the U.S. Holder’s Common Shares with respect to which such election was made will not be treated as shares in a PFIC and the U.S. Holder will not be subject to the rules described below with respect to any “excess distribution” the U.S. Holder receives from us or any gain from an actual sale or other disposition of the Common Shares. U.S. Holders should consult their tax advisors as to the possibility and consequences of making a deemed sale election if the Company ceases to be a PFIC and such election becomes available.
For each taxable year the Company is treated as a PFIC with respect to a U.S. Holder, such U.S. Holder will be subject to special tax rules with respect to any “excess distribution” such U.S. Holder receives and any gain such U.S. Holder recognizes from a sale or other disposition (including, under certain circumstances, a pledge) of Common Shares, unless (i) such U.S. Holder makes a QEF Election or (ii) the Common Shares constitute “marketable” securities, and such U.S. Holder makes a mark-to-market election as discussed below. Absent the making of a QEF Election or a mark-to-market election, distributions a U.S. Holder receives in a taxable year that are greater than 125% of the average annual distributions a U.S. Holder received during the shorter of the three preceding taxable years or the U.S. Holder’s holding period for the Common Shares will be treated as an excess distribution. Under these special tax rules:
| ● |
the excess distribution or gain will be allocated ratably over a U.S. Holder’s holding period for the Common Shares; |
| ● |
the amount allocated to the current taxable year, and any taxable year prior to the first taxable year in which the Company became a PFIC, will be treated as ordinary income; and |
| ● |
the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such year. |
The tax liability for amounts allocated to years prior to the year of disposition or “excess distribution” cannot be offset by any net operating losses for such years, and gains (but not losses) realized on the sale of the Common Shares cannot be treated as capital, even if a U.S. Holder holds the Common Shares as capital assets.
In addition, if the Company is a PFIC, a U.S. Holder will generally be subject to similar rules with respect to distributions the Company receives from, and the Company’s dispositions of the stock of, any of the Company’s direct or indirect subsidiaries that also are PFICs, as if such distributions were indirectly received by, and/or dispositions were indirectly carried out by, such U.S. Holder. U.S. Holders should consult their tax advisors regarding the application of the PFIC rules to the Company’s subsidiaries.
If a U.S. Holder makes an effective QEF Election, the U.S. Holder will be required to include in gross income each year, whether or not the Company makes distributions, as capital gains, such U.S. Holder’s pro rata share of the Company’s net capital gains and, as ordinary income, such U.S. Holder’s pro rata share of the Company’s earnings in excess of the Company’s net capital gains. If the Company determines that it is a PFIC for this year or any future taxable year, the Company currently expects that it would provide the information necessary for U.S. Holders to make a QEF Election.
U.S. Holders also can avoid the interest charge on excess distributions or gain relating to the Common Shares by making a mark-to-market election with respect to the Common Shares, provided that the Common Shares are “marketable.” Common Shares will be marketable if they are “regularly traded” on certain U.S. stock exchanges or on a foreign stock exchange that meets certain conditions. For these purposes, the Common Shares will be considered regularly traded during any calendar year during which they are traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. Any trades that have as their principal purpose meeting this requirement will be disregarded. The Common Shares are listed on the Nasdaq and the TSX, which are qualified exchanges for these purposes. Consequently, if the Common Shares remain listed on the Nasdaq or the TSX and are regularly traded, and you are a holder of Common Shares, the Company expects the mark-to-market election would be available to U.S. Holders if the Company is a PFIC. Each U.S. Holder should consult its tax advisor as to the whether a mark-to-market election is available or advisable with respect to the Common Shares.
A U.S. Holder that makes a mark-to-market election must include in ordinary income for each year an amount equal to the excess, if any, of the fair market value of the Common Shares at the close of the taxable year over the U.S. Holder’s adjusted tax basis in the Common Shares. An electing holder may also claim an ordinary loss deduction for the excess, if any, of the U.S. Holder’s adjusted basis in the Common Shares over the fair market value of the Common Shares at the close of the taxable year, but this deduction is allowable only to the extent of any net mark-to-market gains for prior years. Gains from an actual sale or other disposition of the Common Shares will be treated as ordinary income, and any losses incurred on a sale or other disposition of the shares will be treated as an ordinary loss to the extent of any net mark-to-market gains for prior years. Once made, the election cannot be revoked without the consent of the Internal Revenue Service (the “IRS”), unless the Common Shares cease to be marketable.
However, a mark-to-market election generally cannot be made for equity interests in any lower-tier PFICs that we own, unless shares of such lower-tier PFIC are themselves “marketable.” As a result, even if a U.S. Holder validly makes a mark-to-market election with respect to the Common Shares, the U.S. Holder may continue to be subject to the PFIC rules (described above) with respect to its indirect interest in any of the Company’s investments that are treated as an equity interest in a PFIC for U.S. federal income tax purposes. U.S. Holders should consult their tax advisors to determine whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
Unless otherwise provided by the United States Treasury Department (the “U.S. Treasury”), each U.S. shareholder of a PFIC is required to file an annual report containing such information as the U.S. Treasury may require. A U.S. Holder’s failure to file the annual report will cause the statute of limitations for such U.S. Holder’s U.S. federal income tax return to remain open with regard to the items required to be included in such report until three years after the U.S. Holder files the annual report, and, unless such failure is due to reasonable cause and not willful neglect, the statute of limitations for the U.S. Holder’s entire U.S. federal income tax return will remain open during such period. U.S. Holders should consult their tax advisors regarding the requirements of filing such information returns under these rules.
WE STRONGLY URGE YOU TO CONSULT YOUR TAX ADVISOR REGARDING THE IMPACT OF THE COMPANY’S PFIC STATUS ON YOUR INVESTMENT IN THE COMMON SHARES AS WELL AS THE APPLICATION OF THE PFIC RULES TO YOUR INVESTMENT IN THE COMMON SHARES.
Cash Dividends and Other Distributions
Subject to the discussion under “Passive Foreign Investment Company Rules” above, to the extent there are any distributions made with respect to the Common Shares, a U.S. Holder generally will be required to include in its gross income distributions received with respect to its Common Shares (including the amount of Canadian taxes withheld, if any) as dividend income, but only to the extent that the distribution is paid out of the Company’s current or accumulated earnings and profits (computed using U.S. federal income tax principles), with the excess treated first as a non-taxable return of capital to the extent of the holder’s adjusted tax basis in its Common Shares and, thereafter, as capital gain recognized on a sale or exchange on the day actually or constructively received by the holder (as described below under “Sale or Disposition of Common Shares”). There can be no assurance that the Company will maintain calculations of the Company’s earnings and profits in accordance with U.S. federal income tax accounting principles. U.S. Holders should therefore assume that any distribution with respect to the Common Shares will constitute ordinary dividend income. Dividends paid on the Common Shares will not be eligible for the dividends received deduction allowed to U.S. corporations.
Dividends paid to a non-corporate U.S. Holder by a “qualified foreign corporation” may be subject to reduced rates of taxation if certain holding period and other requirements are met. A qualified foreign corporation generally includes a foreign corporation if (i) its Common Shares are readily tradable on an established securities market in the United States or it is eligible for benefits under a comprehensive U.S. income tax treaty that includes an exchange of information program and which the U.S. Treasury has determined is satisfactory for these purposes and (ii) if such foreign corporation is not a PFIC (as discussed above) for either the taxable year in which the dividend is paid or the preceding taxable year. The Common Shares are readily tradable on the Nasdaq, an established securities market in the United States, and the Company may be eligible for the benefits of the Treaty. Accordingly, subject to the PFIC rules discussed above, a non-corporate U.S. Holder may qualify for the reduced rate on dividends so long as the applicable holding period requirements are met. U.S. Holders should consult their own tax advisors regarding the availability of the reduced tax rate on dividends in light of their particular circumstances.
Distributions paid in a currency other than U.S. dollars will be included in a U.S. Holder’s gross income in a U.S. dollar amount based on the spot exchange rate in effect on the date of actual or constructive receipt, whether or not the payment is converted into U.S. dollars at that time. The U.S. Holder will have a tax basis in such currency equal to such U.S. dollar amount, and any gain or loss recognized upon a subsequent sale or conversion of the foreign currency for a different U.S. dollar amount will generally be U.S. source ordinary income or loss.
If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder generally should generally not be required to recognize foreign currency gain or loss in respect of the dividend income.
If a U.S. Holder is subject to Canadian withholding taxes (at the rate applicable to such U.S. Holder) with respect to dividends paid on the Common Shares, such U.S. Holder may be entitled to receive either a deduction or a foreign tax credit for such Canadian taxes paid. Complex limitations apply to the foreign tax credit. Dividends paid by us generally will constitute “foreign source” income and generally will be categorized as “passive category income.” Because the foreign tax credit rules are complex, each U.S. Holder should consult its own tax advisor regarding the foreign tax credit rules.
Sale or Disposition of Common Shares
A U.S. Holder generally will recognize gain or loss on the taxable sale or exchange of the Common Shares in an amount equal to the difference between the U.S. dollar amount realized on such sale or exchange (determined in the case of the Common Shares sold or exchanged for currencies other than U.S. dollars by reference to the spot exchange rate in effect on the date of the sale or exchange or, if the Common Shares sold or exchanged are traded on an established securities market and the U.S. Holder is a cash basis taxpayer or an electing accrual basis taxpayer, which election must be applied consistently from year to year and cannot be changed without the consent of the IRS, the spot exchange rate in effect on the settlement date) and the U.S. Holder’s adjusted tax basis in the Common Shares determined in U.S. dollars. The initial tax basis of the Common Shares to a U.S. Holder will be the U.S. Holder’s U.S. dollar purchase price for the Common Shares (determined by reference to the spot exchange rate in effect on the date of the purchase, or if the Common Shares purchased are traded on an established securities market and the U.S. Holder is a cash basis taxpayer or an electing accrual basis taxpayer, which election must be applied consistently from year to year and cannot be changed without the consent of the IRS, the spot exchange rate in effect on the settlement date). An accrual basis U.S. Holder that does not make the special election will recognize exchange gain or loss to the extent attributable to the difference between the exchange rates on the sale date and the settlement date, and such exchange gain or loss generally will constitute ordinary income or loss.
Subject to the discussion under “Passive Foreign Investment Company Rules” above, such gain or loss will be capital gain or loss and will be long-term gain or loss if the Common Shares have been held for more than one year. Under current law, long-term capital gains of non-corporate U.S. Holders generally are eligible for reduced rates of taxation. The deductibility of capital losses is subject to limitations. Capital gain or loss, if any, recognized by a U.S. Holder generally will be treated as U.S. source income or loss for U.S. foreign tax credit purposes. U.S. Holders are encouraged to consult their own tax advisors regarding the availability of the U.S. foreign tax credit in their particular circumstances.
Medicare Contribution Tax
Certain U.S. Holders that are individuals, estates or certain trusts must pay a 3.8% tax, or “Medicare contribution tax”, on their “net investment income.” Net investment income generally includes, among other things, dividend income and net gains from the disposition of stock. A U.S. Holder that is an individual, estate or trust should consult its tax advisor regarding the applicability of the Medicare contribution tax to its income and gains in respect of its investment in the Common Shares.
Information Reporting and Backup Withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.-related financial intermediaries generally are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding on a duly executed IRS Form W-9 or otherwise establishes an exemption.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against the U.S. Holder’s U.S. federal income tax liability and may entitle the U.S. Holder to a refund, provided that the required information is timely furnished to the IRS.
Certain Reporting Requirements
U.S. Holders paying more than US$100,000 for the Common Shares generally may be required to file IRS Form 926 reporting the payment of the offer price for the Common Shares to us. Substantial penalties may be imposed upon a U.S. Holder that fails to comply. Each U.S. Holder should consult its own tax advisor as to the possible obligation to file IRS Form 926.
Information with Respect to Foreign Financial Assets
Certain U.S. Holders who are individuals (and, under regulations, certain entities) may be required to report information relating to the Common Shares, subject to certain exceptions (including an exception for Common Shares held in accounts maintained by certain U.S. financial institutions), by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. Such U.S. Holders who fail to timely furnish the required information may be subject to a penalty. Additionally, if a U.S. Holder does not file the required information, the statute of limitations with respect to tax returns of the U.S. Holder to which the information relates may not close until three years after such information is filed. U.S. Holders should consult their tax advisers regarding their reporting obligations with respect to their ownership and disposition of the Common Shares.
10.F. Dividends and Paying Agents
Not applicable.
10.G. Statement by Experts
Not applicable.
10.H. Documents on Display
Documents concerning our company referred to in this Annual Report may be viewed by appointment during normal business hours at our registered and records office at 2 Bloor St. W., 7th Floor, Toronto, Ontario M4W 3E2.
10.I. Subsidiary Information
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We have exposure to credit risk, liquidity risk and market risk. Our Board of Directors has the overall responsibility for the oversight of these risks and reviews our policies on an ongoing basis to ensure that these risks are appropriately managed.
i. Credit risk
Credit risk arises from the potential that a counterparty will fail to perform its obligations. The financial instruments that are exposed to concentrations of credit risk consist of cash and cash equivalents and marketable securities.
The Company manages credit risk associated with its cash and cash equivalents and marketable securities by maintaining minimum standards of R1-med or A-high investments.
ii. Interest rate risk
Interest rate risk is the risk that the fair values and future cash flows of the Company will fluctuate because of changes in market interest rates. We believe our exposure to interest rate risk is not significant.
iii. Liquidity risk
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they fall due. The Company currently settles all of its financial obligations out of cash and cash equivalents. The ability to do so relies on the Company maintaining sufficient cash in excess of anticipated needs. As at March 31, 2023, the Company’s liabilities consist of accounts payable and accrued liabilities that have contracted maturities of less than one year.
iv. Currency risk
Currency risk is the risk that future cash flows of a financial instrument will fluctuate because of changes in foreign exchange rates. The Company is exposed to currency risk from employee costs as well as the purchase of goods and services primarily in the United States and the cash balances held in foreign currencies. Fluctuations in the US dollar exchange rate could have a significant impact on the Company’s results. Assuming all other variables remain constant, a 10% depreciation or appreciation of the Canadian dollar against the US dollar would result in an increase or decrease in loss and comprehensive loss for the year ended March 31, 2023 or $2.3 million (March 31, 2022 - $0.6 million).
Balances in thousands of US dollars are as follows:
| March 31, 2023 |
March 31, 2022 |
|||||||
| US$ |
US$ |
|||||||
| Cash and cash equivalents |
18,250 | 5,456 | ||||||
| Accounts payable and accrued liabilities |
(1,598 | ) |
(1,269 | ) |
||||
| 16,652 | 4,187 | |||||||
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
12.A. Debt Securities
Not applicable.
12.B. Warrants and Rights
Not applicable.
12.C. Other Securities
Not applicable.
12.D. American Depositary Shares
Not applicable.
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
Not applicable.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
Not applicable.
14.E. Use of Proceeds
On December 30, 2020, the Company entered into a sales agreement with SVB acting as a sales agent, pursuant to which the Company may, from time to time sell, at-the-market on the Nasdaq such number of common shares as would have an aggregate offering price of up to US$25.0 million (the “2020 ATM Offering”), which expired December 30, 2022.
During the year ended March 31, 2023, the Company issued 656,656 common shares (March 31, 2022 – 1,748,600) for gross proceeds of US$0.8 million (March 31, 2022 - US$3.1 million) at an average price of US$1.20 (March 31, 2022 - US$1.77) on the 2020 ATM Offering. The Company received; net of commissions US$0.8 million (March 31, 2022 - US$2.8 million). In total, we incurred share issuance costs (including commissions) of US$0.1 million (March 31, 2022 - US$0.1 million).
On August 11, 2022, pursuant to an underwritten public offering, 13,333,334 units were sold by the Company at a purchase price of US$1.50 per unit for gross proceeds of US$20.0 million ($25.6 million). Each unit included one common share with a fair value of US$1.06 and one common share purchase warrant with a fair value of US$0.44 (see Note 6). Each common share purchase warrant entitles the holder to purchase one common share at an exercise price of US$1.85 until August 9, 2027. We incurred transaction costs of $2.2 million (US1.7 million) of which $1.6 million (US$1.2 million) were allocated to share issue costs and $0.6 million (US$0.5 million) were allocated to operating expenses, based on their relative fair values.
On February 17, 2023, the Company entered into a sales agreement with Oppenheimer & Co. Inc. acting as a sales agent, pursuant to which the Company may, from time to time sell, through at-the-market on the NASDAQ such number of common shares as would have an aggregate offering price of up to US10.0 million (the “2023 ATM Offering”). During the year ended March 31, 2023, the Company has issued no common shares on the 2023 ATM Offering.
ITEM 15. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
At the end of the period covered by this Annual Report, an evaluation of the effectiveness of the design and operation of the Company’s “disclosure controls and procedures” (as such term is defined in Rules 13a-15(e) of the Exchange Act) was carried out by the Company’s principal executive officer and principal financial officer. Based upon that evaluation, the Company’s CEO and CFO have concluded that, as of the end of the period covered by this report, the design and operation of the Company’s disclosure controls and procedures are effective to ensure that (i) information required to be disclosed in reports that the Company files or submits to regulatory authorities is recorded, processed, summarized and reported within the time periods specified by regulation, and (ii) is accumulated and communicated to management, including the Company’s CEO and CFO, to allow timely decisions regarding required disclosure.
It should be noted that while the Company’s CEO and CFO believe that the Company’s disclosure controls and procedures provide a reasonable level of assurance that they are effective, they do not expect that the Company’s disclosure controls and procedures will prevent all errors and fraud. A control system, no matter how well conceived or operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
Management Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Rule 13a-15(f) and Rule 15d-15(f) of the Exchange Act) and has designed such internal controls over financial reporting to provide reasonable assurance regarding the reliability of financial reporting and preparation of financial statements for external purposes in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board.
In designing and evaluating the Company’s internal control over financial reporting, the Company’s management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its reasonable judgment in evaluating the cost-benefit relationship of possible controls and procedures. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies and procedures may deteriorate.
Management conducted an evaluation of the effectiveness of the Company’s internal control over financial reporting as of March 31, 2023. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this evaluation, management concluded that the Company’s internal control over financial reporting was effective as of March 31, 2023, based on those criteria.
Attestation Report of Independent Auditor
In accordance with the JOBS Act enacted on April 5, 2012, the Company qualifies as an “emerging growth company” ( “EGC”), which entitles the Company to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not EGCs. Specifically, the JOBS Act defers the requirement to have the Company’s independent auditor assess the Company’s internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act. As such, the Company is exempted from the requirement to include an auditor attestation report in this Annual Report for so long as the Company remains an EGC, which may be for as long as five years following its initial registration in the United States.
Changes in Internal Control over Financial Reporting
During the year ended March 31, 2023, there were no changes in the Company’s internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERT
The Company’s Audit Committee, which consists exclusively of independent directors in accordance with Nasdaq listing requirements, is comprised of Albert G. Beraldo, Karen Dawes and Chandrakant Panchal. Albert G. Beraldo is the Chair of the Audit Committee. The Board of Directors has determined that Albert G. Beraldo, Karen Dawes and Chandrakant Panchal each meet the independence requirements for directors, including the heightened independence standards for members of the audit committee under Rule 10A-3 of the Exchange Act. The Board has determined that Albert G. Beraldo is financially literate as required by Nasdaq listing requirements and an “audit committee financial expert” as defined by Item 407(d)(5) of Regulation S-K. For a description of the education and experience of each member of the Audit Committee, see “Item 6A. Directors, Senior Management and Employees.”
The Company has adopted a Code of Business Conduct and Ethics, attached hereto as Exhibit 11.1, applicable to all of its directors, officers and employees, including its CEO and CFO, which is a “code of ethics” as defined in Section 406(c) of the Sarbanes-Oxley Act. The Code of Business Conduct and Ethics sets out the fundamental values and standards of behavior that the Company expects from our directors, officers and employees with respect to all aspects of its business.
If the Company grants any waiver of the Code of Business Conduct and Ethics, whether explicit or implicit, to a director or executive officer, it will be promptly disclosed as required by any applicable law or applicable rules and guidelines of any stock exchange on which the securities of the Company are listed
The full text of the Code of Business Conduct and Ethics is posted on the Company’s website at www.medicenna.com. The information on or accessible through the website is not part of and is not incorporated by reference into this Annual Report, and the inclusion of the website address in this Annual Report is only for reference.
The Audit Committee is responsible for reviewing and evaluating the Code of Business Conduct and Ethics periodically and will recommend any necessary or appropriate changes thereto to the Board for consideration. The Audit Committee will also assist the Board of Directors with the monitoring of compliance with the Code of Business Conduct and Ethics.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following table sets forth information regarding the amount billed and accrued to the Company by PricewaterhouseCoopers LLP, for the fiscal years ended March 31, 2022 and 2023:
| Year Ended March 31, |
||||||||
| Services |
2022 |
2023 |
||||||
| Audit Fees(1) |
$ | 153,000 | $ | 179,000 | ||||
| Audit-Related Fees(2) |
$ | 72,500 | $ | 50,000 | ||||
| Tax Fees(3) |
$ | 29,095 | $ | 24,308 | ||||
| Other Fees(4) |
$ | 5,202 | $ | 9,588 | ||||
Notes:
| (1) |
“Audit fees” means the aggregate fees billed for professional services rendered by our principal accounting firm for the audit of the Company’s annual financial statements and the review of its comparative interim financial statements. |
| (2) |
“Audit-related fees” means the aggregate fees billed for professional services rendered by the Company’s principal accounting firm for the assurance and related services, which mainly included the audit and review of financial statements and are not reported under “Audit fees” above. |
| (3) |
“Tax fees” means the aggregate fees billed for professional services rendered by the Company’s principal accounting firm for tax compliance, tax advice and tax planning. |
| (4) |
“Other fees” means the aggregate fees incurred in each of the fiscal years listed for the professional tax services rendered by the Company’s principal accounting firm other than services reported under “Audit fees,” “Audit-related fees” and “Tax fees”. |
The policy of the Company’s Audit Committee is to pre-approve all non-audit services provided by PricewaterhouseCoopers LLP, its independent registered public accounting firm, including audit services, audit-related services, tax services, and other services as described above.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not Applicable.
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
Not Applicable.
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
None.
ITEM 16G. CORPORATE GOVERNANCE
The Company is a foreign private issuer and its Common Shares are listed on the Nasdaq Capital Market. Rule 5615(a)(3) of the Nasdaq Rules permits a foreign private issuer to follow its home country’s practices in lieu of certain requirements of the 5600 Series of the Nasdaq Rules, which set forth corporate governance requirements. In order to claim such an exemption, the Company must disclose the significant differences between its corporate governance practices and those required to be followed by U.S. domestic issuers under the Nasdaq Rules. Set forth below is a brief summary of such differences.
As a Canadian corporation listed on Nasdaq, we are not required to comply with certain Nasdaq corporate governance standards. Section 5615(a)(3) of the Nasdaq Stock Market Rules permits Nasdaq to grant exemptions to a foreign private issuer for certain provisions of the Rule 5600 series, Rule 5250(b)(3) and Rule 5250(d). We are organized under the laws of Canada and our Common Shares are listed for trading on the TSX. We comply with the applicable laws of Canada and rules and regulations of the TSX, including rules related to corporate governance practices. A description of the significant ways in which our corporate governance practices differ from those followed by U.S. domestic companies pursuant to the Nasdaq Stock Market Rules is as follow: Quorum Requirements: Rule 5620(c) of the Nasdaq Stock Market Rules requires that the minimum quorum requirement for any meeting of a Company’s shareholders is 33 1/3% of the outstanding Common Shares. In addition, Rule 5620(c) requires that an issuer listed on Nasdaq state its quorum requirement in its bylaws. Our quorum requirement for a meeting of shareholders is set forth in our by-laws, which require at least two (2) persons holding or representing by proxy not less than twenty-five (25%) percent of the outstanding shares of the Company entitled to vote at the meeting. The foregoing is consistent with the applicable laws in Canada and the rules of the TSX.
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
Pursuant to applicable SEC transition guidance, the disclosure required by Item 16J will be applicable to the Company from the fiscal year ending March 31, 2024.
Financial Statements Filed as Part of this Annual Report:
Audited Annual Financial Statements for the years ended March 31, 2021, 2022 and 2023:
Independent Auditor's Report of PricewaterhouseCoopers LLP, dated June 26, 2023;
Consolidated Statements of Financial Position for the years ended March 31, 2022 and 2023;
Consolidated Statements of Loss and Comprehensive Loss for the years ended March 31, 2021, 2022 and 2023;
Consolidated Statements of Cash Flows for the years ended March 31, 2021, 2022 and 2023;
Consolidated Statements of Changes in Shareholders' Equity for the years ended March 31, 2021, 2022 and 2023;
Notes to the Consolidated financial statements.
Refer to Item 17. Financial Statements.
The following Exhibits are being filed as part of this Annual Report, or are incorporated by reference where indicated:
| Exhibit Number |
Description |
| 1.1(a) |
|
| 1.2 |
|
| 1.3(a) |
|
| 2.1(a)% |
Warrant indenture dated December 21, 2018 between the Company and TSX Trust Company |
| 2.2(a) |
Warrant indenture dated October 17, 2019 between the Company and TSX Trust Company |
| 2.3 |
|
| 2.4 |
|
| 4.1(a)# |
|
| 4.2(a)% |
|
| 4.3(a)% |
|
| 4.4(a) |
|
| 4.5(a) |
|
| 4.6(a)% |
|
| 4.7(a)% |
|
| 4.8(a)#% |
|
| 4.9(a)# |
|
| 4.10(a)# |
|
| 4.11(a)# |
| 4.12(a)#% |
|
| 4.13(a)# |
|
| 4.14(a)# |
|
| 4.15(a)# |
|
| 4.16(a)#% |
|
| 4.17(a)# |
|
| 4.18(a)# |
|
| 4.19(a)# |
|
| 8.1(a) |
|
| 11.1(a) |
|
| 12.1* |
Rule 13a-14(a)/15d-14(a) Certification of Chief Executive Officer |
| 12.2* |
Rule 13a-14(a)/15d-14(a) Certification of Chief Financial Officer |
| 13.1* |
|
| 13.2* |
|
| 15.1* |
Management Discussion and Analysis of the Company for the year ended March 31, 2023. |
| 15.2(a) |
|
| 15.3* |
|
| 101 |
The following materials from the Company’s Annual Report on Form 20-F for the fiscal year ended March 31, 2023, formatted in Inline eXtensible Business Reporting Language (iXBRL): |
| (i) Consolidated Balance Sheets as of March 31, 2021, 2022 and 2023; (ii) Consolidated Statements of Operations for the years ended March 31, 2021, 2022 and 2023; (iii) Consolidated Statements of Comprehensive Loss for the years ended March 31, 2021, 2022 and 2023; (iv) Consolidated Statements of Changes in Shareholders’ Equity for the years ended March 31, 2021, 2022 and 2023; (v) Consolidated Statements of Cash Flows for the years ended March 31, 2021, 2022 and 2023; and (vi) Notes to Consolidated Financial Statements |
| 104 |
Cover Page Interactive Data File (formatted as Inline eXtensible Business Reporting Language (iXBRL) and contained in Exhibit 101) |
* Filed herewith
# Indicates management contract or compensatory plan.
% Portions of this exhibit (indicated by asterisks) have been omitted as the Company has determined that (a) the omitted information is not material and (b) the omitted information is of the type that the Company customarily and actually treats as private or confidential.
(a) Previously filed with the SEC on Form 20-F dated June 22, 2022, and incorporated by reference.
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| MEDICENNA THERAPEUTICS CORP. |
||
| /s/ Elizabeth Williams |
||
| By: Elizabeth Williams |
||
| Title: Chief Financial Officer |
Date: June 27, 2023
![]()
Consolidated financial statements of
Medicenna Therapeutics Corp.
(Expressed in Canadian Dollars)
For the years ended March 31, 2023, 2022 and 2021

Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors of Medicenna Therapeutics Corp.
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of Medicenna Therapeutics Corp. and its subsidiaries (together, the Company) as of March 31, 2023 and 2022, and the related consolidated statements of loss and comprehensive loss, of shareholders’ equity and of cash flows for the years then ended, including the related notes (collectively referred to as the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2023 and 2022, and its financial performance and its cash flows for the years then ended in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
PricewaterhouseCoopers LLP
PwC Centre, 354 Davis Road, Suite 600, Oakville, Ontario, Canada L6J 0C5
T: +1 905 815 6300, F: +1 905 815 6499, ca_oakville_main_fax@pwc.com
“PwC” refers to PricewaterhouseCoopers LLP, an Ontario limited liability partnership.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
Chartered Professional Accountants, Licensed Public Accountants
June 26, 2023
We have served as the Company’s auditor since
Medicenna Therapeutics Corp.
Consolidated Statements of Financial Position
(Expressed in thousands of Canadian Dollars, except for share and per share amounts)
| March 31, 2023 |
March 31, 2022 |
|||||||
| Assets |
$ | $ | ||||||
| Current assets | ||||||||
| Cash and cash equivalents (Note 2e) |
||||||||
| Prepaids and deposits |
||||||||
| Other receivables (Note 7) |
||||||||
| Intangible assets (Note 14) |
||||||||
| Liabilities |
||||||||
| Current liabilities |
||||||||
| Accounts payable and accrued liabilities (Note 8) |
||||||||
| Warrant derivative (Note 12) |
||||||||
| Shareholders' Equity |
||||||||
| Common shares (Note 9) |
||||||||
| Contributed surplus (Notes 10 and 11) |
||||||||
| Accumulated other comprehensive income |
||||||||
| Deficit | ( |
) | ( |
) | ||||
| Approved by the Board | ||
| /s/ Albert Beraldo | Director | |
| /s/ Karen Dawes | Director |
The accompanying notes are an integral part of these Consolidated financial statements.
Medicenna Therapeutics Corp.
Consolidated Statements of Loss and Comprehensive Loss
(Expressed in thousands of Canadian Dollars, except for share and per share amounts)
| Year ended March 31, 2023 | Year ended March 31, 2022 | Year ended March 31, 2021 | ||||||||||
| $ | $ | $ | ||||||||||
| Operating expenses | ||||||||||||
| General and administration (Note 17) | ||||||||||||
| Research and development (Note 17) | ||||||||||||
| Total operating expenses | ||||||||||||
| Finance income (Note 2d) | ( | ) | ( | ) | ( | ) | ||||||
| Transaction costs on derivative warrant liability | ||||||||||||
| Change in fair value of warrant derivative (Note 12) | ( | ) | ||||||||||
| Foreign exchange (gain) loss | ( | ) | ||||||||||
| ( | ) | ( | ) | |||||||||
| Net loss for the year | ( | ) | ( | ) | ( | ) | ||||||
| Cumulative translation adjustment | ( | ) | ( | ) | ( | ) | ||||||
| Comprehensive loss for the year | ( | ) | ( | ) | ( | ) | ||||||
| Basic and diluted loss per share for the year | ( | ) | ( | ) | ( | ) | ||||||
| Weighted average number of common shares outstanding (Note 9) | ||||||||||||
The accompanying notes are an integral part of these Consolidated financial statements.
Medicenna Therapeutics Corp.
Consolidated Statements of Cash Flows
(Expressed in thousands of Canadian Dollars)
| Year ended March 31, 2023 |
Year ended March 31, 2022 |
Year ended March 31, 2021 |
||||||||||
| $ | $ | $ | ||||||||||
| Operating activities |
||||||||||||
| Net loss for the year |
( |
) | ( |
) | ( |
) | ||||||
| Items not involving cash |
||||||||||||
| Depreciation |
||||||||||||
| Stock based compensation |
||||||||||||
| Government grant expense recoveries (Note 7) |
( |
) | ( |
) | ||||||||
| Warrant amendment (Note 12) |
||||||||||||
| Unrealized foreign exchange |
( |
) | ||||||||||
| Accrued interest |
( |
) | ( |
) | ( |
) | ||||||
| Change in fair value of warrant derivative (Note 12) |
( |
) | ||||||||||
| Changes in non-cash working capital |
||||||||||||
| Other receivables and deposits |
( |
) | ( |
) | ||||||||
| Accounts payable and accrued liabilities |
( |
) | ||||||||||
| ( |
) | ( |
) | ( |
) | |||||||
| Investing activities |
||||||||||||
| Acquisition of marketable securities |
( |
) | ( |
) | ||||||||
| Disposition of marketable securities |
||||||||||||
| Financing activities |
||||||||||||
| Repayment of lease liabilities | ( |
) | ( |
) | ||||||||
| Issuance of share capital and warrants on public offering, net of issuance costs (Note 9) |
||||||||||||
| Issuance of share capital, net of issuance costs (Note 9) |
||||||||||||
| Warrant and option exercises (Notes 10 and 11) |
||||||||||||
| Effect of foreign exchange on cash |
( |
) | ( |
) | ||||||||
| Net increase (decrease) in cash |
( |
) | ||||||||||
| Cash, beginning of year |
||||||||||||
| Cash, end of year |
||||||||||||
| Other non-cash transactions |
||||||||||||
| Broker warrants issued |
$ | $ | $ | |||||||||
The accompanying notes are an integral part of these Consolidated financial statements.
Medicenna Therapeutics Corp.
Consolidated Statements of Changes in Shareholders' Equity
(Expressed in thousands Canadian Dollars, except for share and per share amounts)
| Common shares issued and outstanding |
Contributed surplus |
Accumulated other comprehensive income |
Deficit |
Total shareholders' equity |
||||||||||||||||||||
| Number |
Amount |
|||||||||||||||||||||||
| $ | $ | $ | $ | $ | ||||||||||||||||||||
| Balance, March 31, 2020 |
( |
) | ||||||||||||||||||||||
| Stock based compensation |
- | |||||||||||||||||||||||
| Warrant and option exercises |
( |
) | ||||||||||||||||||||||
| Issued on April 2020 overallotment |
||||||||||||||||||||||||
| Issued on ATM financing |
||||||||||||||||||||||||
| Cumulative translation adjustment |
- | ( |
) | ( |
) | |||||||||||||||||||
| Net loss for the year |
- | ( |
) | ( |
) | |||||||||||||||||||
| Balance, March 31, 2021 |
( |
) | ||||||||||||||||||||||
| Balance, March 31, 2021 |
( |
) | ||||||||||||||||||||||
| Stock based compensation |
- | |||||||||||||||||||||||
| Warrant and option exercises |
( |
) | ||||||||||||||||||||||
| Issued on April 2020 overallotment |
||||||||||||||||||||||||
| Cumulative translation adjustment |
- | ( |
) | ( |
) | |||||||||||||||||||
| Net loss for the year |
- | ( |
) | ( |
) | |||||||||||||||||||
| Balance, March 31, 2022 |
( |
) | ||||||||||||||||||||||
| Balance, March 31, 2022 |
( |
) | ||||||||||||||||||||||
| Stock based compensation |
- | |||||||||||||||||||||||
| Issued on ATM financing (Note 9) |
||||||||||||||||||||||||
| Issued pursuant to public offering, net of warrant derivative (Note 9) |
||||||||||||||||||||||||
| Warrant amendment (Note 12) |
- | |||||||||||||||||||||||
| Cumulative translation adjustment |
- | ( |
) | ( |
) | |||||||||||||||||||
| Net loss for the year |
- | ( |
) | ( |
) | |||||||||||||||||||
| Balance, March 31, 2023 |
( |
) | ||||||||||||||||||||||
The accompanying notes are an integral part of these Consolidated financial statements.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 1. | Nature of business and liquidity |
The Company's principal business activity is the development and commercialization of IL-2, IL-4 and IL-13 Superkines and Empowered Superkines for the treatment of cancer, inflammation and immune-mediated diseases. Medicenna has
As at March 31, 2023, the head and registered office is located at 2 Bloor St W, 7th Floor, Toronto, Ontario, Canada.
Since inception, the Company has devoted its resources to funding R&D programs, including securing intellectual property rights and licenses, conducting discovery research, manufacturing drug supplies, initiating preclinical and clinical studies, submitting regulatory dossiers and providing administrative support to R&D activities, which has resulted in an accumulated deficit of $
We currently do not earn any revenues from our product candidates and are therefore considered to be in the development stage. As required, the Company will continue to finance its operations through the sale of equity or pursue non-dilutive funding sources available to the Company in the future. The continuation of our research and development activities for bizaxofusp (formerly MDNA55), MDNA11 and the BiSKITs™ platform and the commercialization of bizaxofusp is dependent upon our ability to successfully finance and complete our research and development programs through a combination of equity financing and revenues from strategic partners. There is no guarantee of future financing and that our research and development activities associated with bizaxofusp, MDNA11 and the BiSKITs platform will be successful which may require a change in plans of the Company. We have no current sources of revenues from strategic partners.
Management has forecasted that the Company’s current level of cash will be sufficient to execute its current planned expenditures through calendar Q3 2024.
| 2. | Basis of presentation and significant accounting policies |
| a) | Statement of compliance |
These consolidated financial statements have been prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board (“IASB”) (“IFRS”) and the Interpretations of the International Financial Reporting and Interpretations Committee (“IFRIC”).
The Consolidated financial statements were approved by the Company’s Board of Directors and authorized for issue on June 26, 2023.
| b) | Principles of Consolidation |
These consolidated financial statements include the accounts of the Company and its wholly owned Subsidiaries MTI, MBI, MAL, and MBIBC (British Columbia, Inactive). Subsidiaries are fully consolidated from the date at which control is determined to have occurred and are deconsolidated from the date that the Company no longer controls the entity. The financial statements of the subsidiaries are prepared for the same reporting period as the Company using consistent accounting policies. Intercompany transactions, balances, and gains and losses on transactions between subsidiaries are eliminated.
| c) | Functional and presentation currency |
The functional currency of an entity and its subsidiary is the currency of the primary economic environment in which the entity operates. The functional currency of the parent company is the Canadian dollar and the functional currency of MBI is the US dollar, the functional currency of MTI and MBI BC is the Canadian dollar, the functional currency of MAL is the Australian dollar, and the presentation currency of the parent company is the Canadian dollar.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 2. | Basis of presentation and significant accounting policies cont’d |
|
| d) | Foreign currency translation |
Foreign currency translation of monetary assets and liabilities, denominated in currencies other than the Company’s functional currency, are converted at the rate of exchange in effect at the consolidated statements of financial position date. Revenue and expense items are translated at the rate of exchange in effect at the transaction date. Translation gains or losses are included in determining income or loss for the year.
| e) | Cash and cash equivalents and marketable securities |
Cash and cash equivalents
Cash equivalents include guaranteed investment certificates ( March 31, 2023 – $
Marketable securities
Marketable securities consist of guaranteed investment certificates with a maturity of greater than 90 days and less than one year. The Company has classified its marketable securities at fair value through profit or loss.
| f) | Research and development costs |
Expenditures on research and development activities, undertaken with the prospect of gaining new scientific or technical knowledge and understanding, are recognized in profit or loss as incurred. Investment tax credits related to current expenditures are included in the determination of net income as the expenditures are incurred when there is reasonable assurance they will be realized.
Development activities involve a plan or design for the production of new or substantially improved products and processes. Development expenditures are capitalized only if development costs can be measured reliably, the product or process is technically and commercially feasible, future economic benefits are probable, and the Company intends to and has sufficient resources to complete development and to use or sell the asset. These criteria will be deemed by the Company to have been met when revenue is received by the Company and a determination that it has sufficient resources to market and sell its product offerings. Upon a determination that the criteria to capitalize development expenditures have been met, the expenditures capitalized will include the cost of materials, direct labour and overhead costs that are directly attributable to preparing the asset for its intended use.
Other development expenditures will be expensed as incurred.
Capitalized development expenditures will be measured at cost less accumulated amortization and accumulated impairment losses.
| g) | Government assistance |
Government grants, including grants from similar bodies, consisting of investment tax credits are recorded as a reduction of the related expense or cost of the asset acquired. Government grants are recognized when there is reasonable assurance that the Company has met the requirements of the approved grant program and there is reasonable assurance that the grant will be received.
Research grants that compensate the Company for expenses incurred are recognized in profit, or loss in reduction thereof on a systematic basis in the same years in which the expenses are recognized.
Grants that compensate the Company for the cost of an asset are recognized in profit or loss on a systematic basis over the useful life of the asset.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 2. | Basis of presentation and significant accounting policies cont’d |
|
| h) | Intangible assets |
The Company owns certain patents, intellectual property licenses and options to acquire intellectual property. The Company expenses patent costs, including license fees and other maintenance costs, until such time as the Company has certainty over the future recoverability of the intellectual property at which time it capitalizes the costs incurred. The Company capitalizes costs directly related to the acquisition of existing license patents. The Company does hold any intangible asset with an indefinite life.
Intangible assets with finite lives are amortized over the useful economic life and assessed for impairment whenever there is an indication that the intangible asset may be impaired. The amortization method and amortization period of an intangible asset with a finite life is reviewed at least annually. Changes in the expected useful life or the expected pattern of consumption of future economic benefits embodied in the asset is accounted for by changing the amortization period or method, as appropriate, and are treated as changes in accounting estimates. The amortization expense on intangible assets with finite lives is recognized in general and administrative expenses.
Amortization is recognized in profit or loss on a straight-line basis over the estimated useful lives of intangible assets from the date they are available for use to August 31, 2035.
| i) | Income taxes |
Current tax and deferred tax are recognized in the Company’s profit and loss, except to the extent that it relates to a business combination or items recognized directly in equity or in net loss and comprehensive loss.
Current income taxes are recognized for the estimated taxes payable or receivable on taxable income or loss for the current year and any adjustment to income taxes payable in respect of previous years. Current income taxes are determined using tax rates and tax laws that have been enacted or substantively enacted by the period end date.
Deferred tax assets and liabilities are recognized where the carrying amount of an asset or liability differs from its tax base, except for taxable temporary differences arising on the initial recognition of goodwill and temporary differences arising on the initial recognition of an asset or liability in a transaction which is not a business combination and at the time of the transaction affects neither accounting nor taxable profit or loss.
Recognition of deferred tax assets for unused tax losses, tax credits and deductible temporary differences is restricted to those instances where it is probable that future taxable profit will be available against which the deferred tax assets can be utilized. At the end of each reporting period, the Company reassesses unrecognized deferred tax assets. The Company recognizes a previously unrecognized deferred tax asset to the extent that it has been probable that future taxable profit will allow the deferred tax asset to be recovered.
| j) | Basic and diluted loss per common share |
Basic loss per share is computed by dividing the loss available to common shareholders by the weighted average number of common shares outstanding during the year. The computation of diluted earnings per share assumes the conversion, exercise or contingent issuance of securities only when such conversion, exercise or issuance would have a dilutive effect on earnings per share. The dilutive effect of convertible securities is reflected in diluted earnings per share by application of the “if converted” method. The dilutive effect of outstanding options and warrants and their equivalents is reflected in diluted earnings per share. Since the Company has losses, the exercise of outstanding options and warrants have not been included in this calculation as it would be anti-dilutive.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 2. | Basis of presentation and significant accounting policies cont’d |
|
| k) | Equipment |
The Company’s fixed assets comprise of computer equipment for use in general and administrative and research activities.
Depreciation is recognized using the straight-line method based on an expected life of the assets:
| Computer equipment | |
Impairment of long-lived assets:
The Company’s long-lived assets are reviewed for indications of impairment at the date of preparing each statement of financial position. If indication of impairment exists, the asset’s recoverable amount is estimated.
An impairment loss is recognized when the carrying value of an asset, or its cash-generating unit, exceeds its recoverable amount. A cash-generating unit is the smallest identifiable group of assets that generates cash inflows that are largely independent of cash inflows from other assets or groups of assets. For the purpose of impairment testing, the Company determined it has
| l) | Warrant derivative |
When a warrant exercise price is denominated in a currency which differs from the Company’s functional currency, the financial instruments are treated as liabilities and measured at fair value. The fair value of the warrants are calculated using the Black-Scholes model. The change in the liability has been recorded in the consolidated statement of loss and comprehensive loss.
| m) | Stock-based compensation |
The Company has a stock-based compensation plan (the "Plan") available to officers, directors, employees and consultants with grants under the Plan approved by the Company's Board of Directors. Under the Plan, the exercise price of each option equals the closing trading price of the Company's stock on the day prior to the grant or a higher price as determined by the Board of Directors. Vesting is provided for at the discretion of the Board of Directors and the expiration of options is to be not greater than
Stock options awarded to non-employees are accounted for at the fair value of the goods received or the services rendered. The fair value is measured at the date the Company obtains the goods or the date the counterparty renders the service. If the fair value of the goods or services cannot be reliably measured, the fair value of the options granted will be used.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 2. | Basis of presentation and significant accounting policies cont’d |
|
| n) | Share Capital |
Common shares are classified as equity. Incremental costs directly attributable to the issue of common shares are recognized as a reduction of equity.
The Company has adopted a relative fair value method with respect to the measurement of shares and warrants issued as private placement units. The relative fair value method allocates value to each component on a pro-rata basis, based on the fair value of the components calculated independently of one another. The Company measures the fair value of the warrant component of the unit using the Black-Scholes option pricing model. The unit value is then allocated, pro-rata, between the two components, with the fair value attributed to the warrants being recorded to contributed surplus.
| o) | Financial Instruments |
Financial assets and liabilities are recognized when the Company becomes a party to the contractual provisions of the instrument. Financial assets are derecognized when the rights to receive cash flows from the assets have expired or have been transferred and the Company has transferred substantially all risks and rewards of ownership.
Financial assets and liabilities are offset and the net amount is reported in the consolidated statement of financial position when there is a legally enforceable right to offset the recognized amounts and there is an intention to settle on a net basis, or realize the asset and settle the liability simultaneously.
The Company recognizes financial instruments based on their classification. Depending on the financial instruments’ classification, changes in subsequent measurements are recognized in net loss and comprehensive loss.
The Company has implemented the following classifications:
| ● | Cash, cash equivalents and marketable securities are classified at fair value through profit or loss. |
| ● | Other receivables, prepaid and deposits are classified as amortized cost. After their initial fair value measurement, they are measured at amortized cost using the effective interest method; and |
| ● | Accounts payable and accrued liabilities are classified as other amortized cost. After their initial fair value measurement, they are measured at amortized cost using the effective interest method. |
| p) | Impairment of financial assets |
The Company applies the simplified method of the expected credit loss model required under IFRS 9. Under this method, the Company estimates a lifetime expected loss allowance for all receivables. Receivables are written off when there is no reasonable expectation of recovery.
If there is objective evidence that an impairment loss has been incurred, the amount of the loss is measured as the difference between the asset’s carrying amount and the present value of estimated future cash flows. The present value of the estimated future cash flows is discounted at the financial asset’s original effective interest rate.
| q) | Employee benefits |
Short-term employee benefit obligations are measured on an undiscounted basis and are expensed as the related service is provided. A liability is recognized for the amount expected to be paid in short-term cash bonuses if the Company expects to pay these amounts as approved by the Board of Directors as a result of past services provided by the employee and the obligation can be estimated reliably.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 2. | Basis of presentation and significant accounting policies cont’d |
|
| r) | Provisions |
A provision is recognized if, as a result of a past event, the Company has a present legal or constructive obligation that can be estimated reliably, and it is probable that an outflow of economic benefits will be required to settle the obligation. Provisions are assessed by discounting the expected future cash flows at a pre-tax rate that reflects current market assessments of the time value of money and the risks specific to the liability. The unwinding of the discount on provisions is recognized in finance costs. A provision for onerous contracts is recognized when the unavoidable costs of meeting the obligations under the contract exceed the economic benefits expected to be received under it. The provision is measured at the present value of the lower of the expected cost of terminating the contract and the expected net cost of continuing with the contract.
| s) | Research and development tax credits |
Research and development tax credits Refundable investment tax credits relating to Research and Development Tax Incentive (“RDTI”) are recorded in the accounts in the fiscal period in which the qualifying expenditures are incurred provided there is reasonable assurance that the tax credits will be realized. Refundable investment tax credits, in connection with RDTI activities, are accounted for using the cost reduction method and included in government assistance on the statements of loss and comprehensive loss. Amounts recorded for refundable investment tax credits are calculated based on the expected eligibility and tax treatment of qualifying RDTI expenditures recorded in the Company’s consolidated financial statements.
| t) | COVID-19 Pandemic |
The continued evolution of COVID-19 and its variants, as well as periodic spikes in infection rates and local outbreaks, in spite of safety measures or vaccinations, could cause disruptions to our operations or those of third parties with whom we engage. The COVID-19 pandemic has led to global supply chain challenges, which could adversely impact our ability to conduct business in the manner and timelines presently planned. As new variants of the virus appear, especially variants that are more easily spread, cause more serious outcomes, or are resistant to existing vaccines, new health orders and safety protocols could further impact our operations. The Company will continue to monitor developments of the pandemic and continuously assess its potential further impact on its operations to prevent any disruptions to the conduct of its business and clinical trials. In the event of a prolonged continuation of the pandemic, it is not clear what the potential impact may be on the Company's business, financial position and financial performance.
| 3. | New standards and interpretations not yet adopted |
In January 2020, the IASB issued amendments to Presentation of financial statements (“IAS 1”) to provide a more general approach to the classification of liabilities under IAS 1 based on the contractual arrangements in place at the reporting date. The amendments to IAS 1 are effective for annual reporting periods beginning on or after January 1, 2023. The Company does not anticipate adoption of this standard to have a material impact on the consolidated financial statements.
There are no other standards, interpretations or amendments to existing standards that are not yet effective that are expected to have a material impact on the consolidated financial statements of the Company.
| 4. | Key sources of estimation uncertainty and judgement |
The preparation of consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates. Estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are accounted for prospectively.
The key sources of estimation uncertainty that have a significant risk of causing material adjustment to the carrying amounts of assets and liabilities are discussed below:
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 4. | Key sources of estimation uncertainty and judgement cont’d |
|
Valuation of stock-based compensation and warrants
Management measures the costs for stock-based compensation and warrants using market-based option valuation techniques. Assumptions are made and estimates are used in applying the valuation techniques. These include estimating the future volatility of the share price, expected dividend yield, expected risk-free interest rate, future employee turnover rates, future exercise behaviors and corporate performance. Such estimates and assumptions are inherently uncertain. Changes in these assumptions affect the fair value estimates of stock-based compensation and warrants.
Estimate of prepaid expenses and accruals for research and development expenses
The Company records an estimate of accrued expenses for research and development. The period-end process involves assessing the status of research and development activities by analyzing open purchase orders and having discussions with the Company’s personnel and service providers. This information is used to identify services that have been performed and estimate the level of service performed on the Company’s behalf and the associated cost incurred for the service when the Company has not yet been invoiced or otherwise notified of the actual cost.
For research and development activities, the majority of service providers invoice the Company in arrears for services performed, either on a pre-determined schedule or when contractual milestones are met; however, some require advanced payments, such that payments to the service providers exceed the level of services provided. This impacts the amount of accrued expenses and prepaid balances related to research and developments costs as of period end.
The Company estimates its accrued expenses and prepaid expenses as of each statement of financial position date in its financial statements based on facts and circumstances known at that time. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjust research and development expenses in subsequent periods.
Valuation of warrant derivative
Estimating the fair value of the warrant derivative at initial measurement, at each exercise date and at each reporting period requires determining the most appropriate valuation model. This estimate also requires determining the most appropriate inputs to the valuation model, including the expected life, share price volatility, and dividend yield, and making assumptions about them. Changes in the inputs and assumptions used affect the fair value estimate of the warrant derivative.
| 5. | Capital disclosures |
The Company’s objectives, when managing capital, are to safeguard cash and cash equivalents as well as maintain financial liquidity and flexibility in order to preserve its ability to meet financial obligations and deploy capital to grow its businesses.
The Company’s financial strategy is designed to maintain a flexible capital structure consistent with the objectives stated above and to respond to business growth opportunities and changes in economic conditions. In order to maintain or adjust its capital structure, the Company may issue shares or issue debt (secured, unsecured, convertible and/or other types of available debt instruments).
There were no changes to the Company’s capital management policy during the year. The Company is not subject to any externally imposed capital requirements.
| 6. | Financial risk management |
| (a) | Fair value |
The Company’s financial instruments recognized on the Consolidated statements of financial position consist of cash and cash equivalents, marketable securities, government and other receivables, and accounts payable and accrued liabilities. The fair value of these instruments, approximate their carrying values due to their short-term maturity.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 6. | Financial risk management cont’d |
|
| (a) | Fair value |
Classification of financial instruments
Financial instruments measured at fair value on the statements of financial position are summarized into the following fair value hierarchy levels:
Level 1: quoted prices (unadjusted) in active markets for identical assets or liabilities.
Level 2: inputs other than quoted prices included within Level 1 that are observable for the asset or liability.
Level 3: inputs for the asset or liability that are not based on observable market data (unobservable inputs).
The Company classifies its financial assets and liabilities depending on the purpose for which the financial instruments were acquired, their characteristics, and management intent as outlined below:
| ● | Cash and cash equivalents and marketable securities are measured using Level 1 inputs and changes in fair value are recognized through profit or loss, with changes in fair value being recorded in net income at each year-end. |
| ● | Other receivables, prepaids and deposits are measured at amortized cost less impairments. |
| ● | Accounts payable and accrued liabilities are measured at amortized cost. |
The Company has exposure to the following risks from its use of financial instruments: credit, interest rate, currency, and liquidity risk. The Company reviews its risk management framework on a quarterly basis and makes adjustments as necessary.
| (b) | Credit risk |
Credit risk arises from the potential that a counterparty will fail to perform its obligations. The financial instruments that are exposed to concentrations of credit risk consist of cash and cash equivalents and marketable securities.
The Company manages credit risk associated with its cash and cash equivalents and marketable securities by maintaining minimum standards of R1-med or A-high investments.
| (c) | Interest rate risk |
Interest rate risk is the risk that the fair values and future cash flows of the Company will fluctuate because of changes in market interest rates. The Company believes that its exposure to interest rate risk is not significant.
| (d) | Liquidity risk |
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they fall due. The Company currently settles all of its financial obligations out of cash and cash equivalents. The ability to do so relies on the Company maintaining sufficient cash in excess of anticipated needs. As at March 31, 2023, the Company’s liabilities consist of accounts payable and accrued liabilities that have contracted maturities of less than one year.
| (e) | Currency risk |
Currency risk is the risk that future cash flows of a financial instrument will fluctuate because of changes in foreign exchange rates. The Company is exposed to currency risk from employee costs as well as the purchase of goods and services primarily in the United States and cash and cash equivalent balances held in foreign currencies. Fluctuations in the US dollar exchange rate could have a significant impact on the Company’s results. Assuming all other variables remain constant, a 10% depreciation or appreciation of the Canadian dollar against the US dollar would result in an increase or decrease in loss and comprehensive loss for the year ended March 31, 2023 of $
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 6. | Financial risk management cont’d |
|
| |
(e) | Currency risk (cont’d) |
|
Balances in US dollars are as follows:
| March 31, 2023 | March 31, 2022 | |||||||
| $ | $ | |||||||
| Cash and cash equivalents | ||||||||
| Accounts payable and accrued liabilities | ( | ) | ( | ) | ||||
| 7. | Other receivables |
| March 31, 2023 | March 31, 2022 | |||||||
| $ | $ | |||||||
| Investment tax credits receivable | ||||||||
| Sales tax receivable | ||||||||
Refundable Tax
The Company is entitled to receive $
| 8. | Accounts payable and accrued liabilities |
| March 31, 2023 | March 31, 2022 | |||||||
| $ | $ | |||||||
| Trade payables | ||||||||
| Accrued liabilities | ||||||||
| 9. | Share capital |
Authorized
Unlimited common shares
Equity Issuances
August 2022 Public Offering
On August 11, 2022, pursuant to an underwritten public offering,
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 9. | Share capital cont’d |
|
April 2020 Financing
On April 15, 2020, the Company announced the full exercise of the overallotment option, issuing an additional
At-The-Market Facilities
On December 30, 2020, the Company entered into a sales agreement with SVB Leerink acting as a sales agent, pursuant to which the Company may, from time to time sell, through at-the-market on the Nasdaq such number of common shares as would have an aggregate offering price of up to million (the “2020 ATM Offering”), which expired December 30, 2022.
During the year ended March 31, 2023, the Company issued
On February 17, 2023, the Company entered into a sales agreement with Oppenheimer & Co. Inc. acting as a sales agent, pursuant to which the Company may, from time to time sell, through at-the-market on the Nasdaq such number of common shares as would have an aggregate offering price of up to million (the 2023 ATM Offering). During the year ended March 31, 2023, the Company has issued
Calculation of loss per share
Loss per common share is calculated using the weighted average number of common shares outstanding. For the year ended March 31, 2022, 2021 and 2020, the calculation was as follows:
| 2023 | 2022 | 2021 | ||||||||||
| Common shares issued and outstanding, beginning of year | ||||||||||||
| Shares issued in March/April 2020 financing | ||||||||||||
| Shares issued on 2022 Public Offering | ||||||||||||
| ATM issuances | ||||||||||||
| Effect of warrants and options exercised | ||||||||||||
| Weighted average common shares issued and outstanding, end of year | ||||||||||||
| Common shares issued and outstanding, end of year | ||||||||||||
The effect of any potential exercise of the Company’s stock options and warrants outstanding during the year has been excluded from the calculation of diluted loss per common share as it would be anti- dilutive.
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 10. | Warrants |
Warrant continuity:
| Number of Warrants | Weighted average exercise price | |||||||
| Balance outstanding at March 31, 2020 | $ | |||||||
| Broker warrants issued in overallotment | ||||||||
| Warrants exercised during the year | ( | ) | ||||||
| Warrants outstanding at March 31, 2021 | $ | |||||||
| Warrants expired during the year | ( | ) | ||||||
| Warrants exercised during the year | ( | ) | ||||||
| Warrants outstanding at March 31, 2022 | $ | |||||||
| Common share purchase warrants issued in the 2022 Public Offering | ||||||||
| Warrants expired during the year | ( | ) | ||||||
| Warrants outstanding at March 31, 2023 | $ | |||||||
There were
At March 31, 2023, warrants were outstanding and exercisable, enabling holders to acquire common shares as follows:
| Number of Warrants | Exercise Price | Expiry Date | ||||
| $ | ||||||
| July 17, 2023 | ||||||
| December 31, 2023 | ||||||
|
| August 9, 2027 | |||||
Warrants outstanding and exercisable, totaling
| 11. | Stock options |
Year ended March 31, 2023
During the year ended March 31, 2023, the Company granted
Year ended March 31, 2022
During the year ended March 31, 2022, the Company granted
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 11. | Stock options cont’d |
|
Year ended March 31, 2021
During the year ended March 31, 2021, the Company granted
Stock option transactions for the years ended March 31, 2023, 2022, and 2021 are set forth below:
| Number of options | Weighted average exercise price | |||||||
| Balance outstanding at March 31, 2020 | $ | |||||||
| Granted | ||||||||
| Exercised | ( | ) | ||||||
| Forfeited | ( | ) | ||||||
| Balance outstanding at March 31, 2021 | $ | |||||||
| Granted | ||||||||
| Exercised | ( | ) | ||||||
| Forfeited | ( | ) | ||||||
| Balance outstanding at March 31, 2022 | $ | |||||||
| Granted | ||||||||
| Expired | ( | ) | ||||||
| Forfeited | ( | ) | ||||||
| Balance outstanding at March 31, 2023 | ||||||||
The following table summarizes information about stock options outstanding at March 31, 2023:
| Options Outstanding | Options Exercisable | ||||
| Exercise Prices | Options | Weighted average remaining contractual life | Weighted average | Options | Weighted average |
| $ | Years | $ | $ | ||
| - | | | | | |
| - | | | | | |
| - | | | | | |
| | | | | | |
The following assumptions were used in the Black-Scholes option-pricing model to determine the fair value of stock options granted during the year:
| March 31, 2023 | March 31, 2022 | March 31, 2021 | ||||||||||
| Exercise price | $ | $ | $ | |||||||||
| Grant date share price | $ | $ | $ | |||||||||
| Risk free interest rate | % | % | % | |||||||||
| Expected life of options (years) | | | | |||||||||
| Expected volatility | % | % | % | |||||||||
| Expected dividend yield | - | - | - | |||||||||
| Forfeiture rate | % | % | % | |||||||||
| Weighted average fair value of options granted during the year | $ | $ | $ | |||||||||
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 12. | Warrant Derivative |
On August 11, 2022, pursuant to an underwritten public offering,
We incurred transaction costs of $
Under IFRS 9 Financial Instruments and IAS 32 Financial Instruments: Presentation, warrants with an exercise price denominated in a currency that differs from an entity's functional currency are treated as a derivative measured at fair value with subsequent changes in fair value accounted for through the consolidated statement of loss. Our warrants with an exercise price of meet this requirement and we have presented the value of these warrants as a non-current liability on the consolidated statement of financial position. Upon exercise, the recorded liability will be included in our share capital along with the proceeds from the exercise. If these warrants expire, the related liability is reversed through the consolidated statement of loss. There is no cash flow impact as a result of the accounting treatment for changes in the fair value of the warrant derivative or when warrants expire unexercised.
Estimating the fair value for our warrant derivative requires determining the most appropriate valuation model which is dependent on the terms and conditions of the issuance. This estimate also requires determining the most appropriate inputs to the valuation model, including the expected life of the warrant derivative, expected share price volatility and expected dividend yield and making assumptions about them.
A reconciliation of the change in fair value of the warrant derivative is as follows:
| Fair value of Warrant Derivatives | ||||
| $ | ||||
| Balance, August 11, 2022 | ||||
| Change in fair value of warrant derivative | ( | ) | ||
| Foreign exchange loss | ||||
| Balance, March 31, 2023 | ||||
We use historical data to estimate the expected dividend yield and expected volatility of our stock in determining the fair value of the warrants. The risk-free interest rate is based on U.S. Department of Treasury benchmark treasury yield rates in effect at the time of valuation and the expected life of the warrants represents the estimated length of time the warrants are expected to remain outstanding.
The following table summarizes the key assumptions used in the Black-Scholes valuation of the warrant derivative at March 31, 2023:
| March 31, 2023 | August 11, 2022 | |||||||
| Fair value of warrants | $ | $ | ||||||
| Underlying share price | $ | $ | ||||||
| Risk free interest rate | % | % | ||||||
| Expected hold period to exercise (years) | | | ||||||
| Expected share price volatility | % | % | ||||||
| Expected dividend yield | Nil | Nil | ||||||
The following table summarizes our outstanding warrant derivative for the year ended ending March 31, 2023:
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 12. | Warrant Derivative cont’d |
|
| Exercise Price | Outstanding Beginning of the Period | Granted during the Period | Outstanding, End of the Period | Weighted Average Remaining Contractual Life (years) |
|
| - | | | |
| 13. | Government assistance |
CPRIT assistance
In February 2015, the Company received notice that it had been awarded a grant by the Cancer Prevention Research Institute of Texas (“CPRIT”) whereby the Company was eligible to receive up to million on eligible expenditures over a -year period related to the development of the Company’s phase 2b clinical program for bizaxofusp. As of March 31, 2022, the grant with CPRIT was complete.
Of the US$14.1 million grant approved by CPRIT, Medicenna had received million from CPRIT at March 31, 2022.
Under the terms of the grant, the Company is required to pay a royalty to CPRIT, comprised of
| 14. | Commitments |
Intellectual property
On August 21, 2015, the Company exercised its right to enter into two license agreements (the “Stanford License Agreements”) with the Board of Trustees of the Leland Stanford Junior University (“Stanford”). In connection with this licensing agreement, the Company issued
The Company has entered into various license agreements with respect to accessing patented technology. In order to maintain these agreements, the Company is obligated to pay certain costs based on timing or certain milestones within the agreements, the timing of which is uncertain. These costs include ongoing license fees, patent prosecution and maintenance costs, royalty and other milestone payments. As at March 31, 2023, the Company is obligated to pay the following:
| • | Given the current development plans and expected timelines of the Company it is assumed that project milestones of million will be due in the next five years. |
| • | Project milestone payments, assuming continued success in the development programs, of uncertain timing totaling million and an additional million in sales milestones. |
| Contractual obligations | Less than 1 year | 1-3 years | 3-5 years | Total | ||||||||||||
| $ | $ | $ | $ | |||||||||||||
| Patent licensing costs | ||||||||||||||||
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 15. | Related party disclosures |
| (a) | Key management personnel |
Key management personnel, which consists of the Company’s officers (President and Chief Executive Officer, Chief Financial Officer, Chief Development Officer, former Chief Medical Officer and former Chief Scientific Officer) and directors, earned the following compensation for the following years:
| 2023 | 2022 | 2021 | ||||||||||
| $ | $ | $ | ||||||||||
| Salaries and wages | ||||||||||||
| Board fees | ||||||||||||
| Stock option expense | ||||||||||||
| (b) | Amounts payable to related parties |
As at March 31, 2023, the Company had trade and other payables in the normal course of business, owing to directors and officers of $
| 16. | Income taxes |
a) Provision for Income Tax
A reconciliation of income taxes at statutory rates with the reported taxes is as follows:
| 2023 | 2022 | 2021 | ||||||||||
| $ | $ | $ | ||||||||||
| Loss before income taxes | ( | ) | ( | ) | ( | ) | ||||||
| Tax rate | % | % | % | |||||||||
| Expected tax recovery | ( | ) | ( | ) | ( | ) | ||||||
| Change in statutory rates and foreign exchange rates | ( | ) | ( | ) | ||||||||
| Permanent differences | ( | ) | ||||||||||
| Share issuance costs | ( | ) | ( | ) | ( | ) | ||||||
| Change in unrecognized deductible temporary difference | ||||||||||||
| Total income tax expense (recovery) | ||||||||||||
b) Deferred Income Tax
The significant components of the Company’s deferred tax assets that have not been included on the consolidated statement of financial position are as follows:
| 2023 | 2022 | 2021 | ||||||||||
| $ | $ | $ | ||||||||||
| Non-capital losses carry-forward | ||||||||||||
| Property and equipment | ||||||||||||
| Share issuance costs | ||||||||||||
| Unrecognized deferred tax asset | ( | ) | ( | ) | ( | ) | ||||||
| Net deferred tax assets | ||||||||||||
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 16. | Income taxes cont’d |
|
The significant components of the Company’s temporary differences, unused tax credits and unused tax losses that have not been included in the consolidated statements of financial position are as follows:
| Type | Amount | Expiry | ||||||
| Non-capital losses carry-forward | $ | 2037-2043 | ||||||
| Property and equipment | N/A | |||||||
| Share issuance costs | 2041-2044 | |||||||
| 17. | Components of Expenses |
| 2023 | 2022 | 2021 | ||||||||||
| $ | $ | $ | ||||||||||
| General and Administration Expenses | ||||||||||||
| Depreciation expense | ||||||||||||
| Stock based compensation | ||||||||||||
| Facilities and operations | ||||||||||||
| Public company expenses | ||||||||||||
| Salaries and benefits | ||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| $ | $ | $ | ||||||||||
| Research and Development Expenses | ||||||||||||
| Chemistry, manufacturing, and controls | ||||||||||||
| Regulatory | ||||||||||||
| Discovery and pre-clinical | ||||||||||||
| Clinical | ||||||||||||
| Salaries and benefits | ||||||||||||
| Licensing, patent, legal fees and royalties | ||||||||||||
| Stock based compensation | ||||||||||||
| CPRIT grant claimed in eligible expenses (Note 13) | ( | ) | ||||||||||
| Australian R&D refund claimed in eligible expenses (Note 7) | ( | ) | ( | ) | ||||||||
| Other research and development expenses | ||||||||||||
Medicenna Therapeutics Corp.
Notes to the Consolidated financial statements
For the Years Ended March 31, 2023, 2022 and 2021
(Tabular amounts expressed in thousands of Canadian Dollars, except for share and per share amounts)
| 18. | Subsequent events |
On April 25, 2023, the Company received an extension notice (“Extension Notice”) from the Nasdaq Stock Market LLC granting the Company’s request for a 180-day extension to regain compliance with the minimum bid price requirement (“Minimum Bid Requirement”) of US$1.00 per share under the Nasdaq Listing Rule 5450(a)(1). The Company was first notified by Nasdaq of its failure to comply with the Minimum Bid Requirement on October 25, 2022, and was given until April 24, 2023 to regain compliance. The Company now has until October 23, 2023 to meet the requirement. The Extension Notice has no immediate effect on the listing or trading of the Company’s common stock on Nasdaq, and the Company’s operations are not affected by the receipt of the Extension Notice.