
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM
(Mark One)
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to ___________
Commission File Number:
(Exact Name of Registrant as Specified in its Charter)
( State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer |
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|||
|
☒ |
|
Smaller reporting company |
|
||
|
|
|
|
|
|
|
Emerging growth company |
|
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
As of May 14, 2024, the registrant had
Table of Contents
|
|
Page |
PART I. |
|
|
Item 1. |
1 |
|
|
1 |
|
|
2 |
|
|
4 |
|
|
2 |
|
|
3 |
|
Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
23 |
Item 3. |
44 |
|
Item 4. |
44 |
|
PART II. |
|
|
Item 1. |
46 |
|
Item 1A. |
46 |
|
Item 2. |
46 |
|
Item 3. |
46 |
|
Item 4. |
46 |
|
Item 5. |
46 |
|
Item 6. |
47 |
|
48 |
||
i
PART I—FINANCIAL INFORMATION
Item 1. Financial Statements.
Taysha Gene Therapies, Inc.
Condensed Consolidated Balance Sheets
(in thousands, except share and per share data)
(Unaudited)
|
|
|
|
|
|
|
||
|
|
March 31, |
|
|
December 31, |
|
||
ASSETS |
|
|
|
|
|
|
||
Current assets: |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Restricted cash |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Assets held for sale |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Restricted cash |
|
|
|
|
|
|
||
Property, plant and equipment, net |
|
|
|
|
|
|
||
Operating lease right-of-use assets |
|
|
|
|
|
|
||
Other non-current assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued expenses and other current liabilities |
|
|
|
|
|
|
||
Deferred revenue |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Term loan, net |
|
|
|
|
|
|
||
Operating lease liability, net of current portion |
|
|
|
|
|
|
||
Other non-current liabilities |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
Stockholders' equity |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated other comprehensive loss |
|
|
( |
) |
|
|
— |
|
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total stockholders’ equity |
|
|
|
|
|
|
||
Total liabilities and stockholders' equity |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
1
Taysha Gene Therapies, Inc.
Condensed Consolidated Statements of Operations
(in thousands, except share and per share data)
(Unaudited)
|
|
For the Three Months |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Revenue |
|
$ |
|
|
$ |
|
||
Operating expenses: |
|
|
|
|
|
|
||
Research and development |
|
|
|
|
|
|
||
General and administrative |
|
|
|
|
|
|
||
Total operating expenses |
|
|
|
|
|
|
||
Loss from operations |
|
|
( |
) |
|
|
( |
) |
Other income (expense): |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
( |
) |
|
|
— |
|
Change in fair value of term loan |
|
|
( |
) |
|
|
— |
|
Interest income |
|
|
|
|
|
|
||
Interest expense |
|
|
( |
) |
|
|
( |
) |
Other expense |
|
|
( |
) |
|
|
( |
) |
Total other income (expense), net |
|
|
|
|
|
( |
) |
|
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Net loss per common share, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
Weighted average common shares outstanding, basic and diluted |
|
|
|
|
|
|
||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
2
Taysha Gene Therapies, Inc.
Condensed Consolidated Statements of Comprehensive Loss
(in thousands, except share and per share data)
(Unaudited)
|
|
For the Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Other comprehensive loss: |
|
|
|
|
|
|
||
Change in fair value of terms loan attributable to instrument specific credit risk |
|
|
( |
) |
|
|
— |
|
Comprehensive loss |
|
$ |
( |
) |
|
$ |
( |
) |
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
3
Taysha Gene Therapies, Inc.
Condensed Consolidated Statements of Stockholders’ (Deficit) Equity
(in thousands, except share data)
(Unaudited)
For the Three Months Ended March 31, 2024
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
|
|
|
|
||||||
|
|
Common Stock |
|
|
Paid-in |
|
|
Accumulated |
|
|
Accumulated Other |
|
|
Total |
|
|||||||||
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Comprehensive Loss |
|
|
Stockholders' Equity |
|
||||||
Balance as of December 31, 2023 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
|
||||
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Issuance of common stock upon vesting and settlement of restricted stock units |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Issuance of common stock under ESPP |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Loss on instrument-specific credit risk |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Balance as of March 31, 2024 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
For the Three Months Ended March 31, 2023
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
|
|
Total |
|
||||||
|
|
Common Stock |
|
|
Paid-in |
|
|
Accumulated |
|
|
Accumulated Other |
|
|
Stockholders' |
|
|||||||||
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Comprehensive Loss |
|
|
Equity (Deficit) |
|
||||||
Balance as of December 31, 2022 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
|
||||
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Issuance of common stock upon vesting and settlement of restricted stock units, net |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Issuance of common stock under ESPP |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Balance as of March 31, 2023 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
( |
) |
|||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
4
Taysha Gene Therapies, Inc.
Condensed Consolidated Statements of Cash Flows
(in thousands)
(Unaudited)
|
|
For the Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Cash flows from operating activities |
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
||
Depreciation expense |
|
|
|
|
|
|
||
Stock-based compensation |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
|
|
|
— |
|
|
Non-cash change in fair value of term loan |
|
|
( |
) |
|
|
— |
|
Non-cash lease expense |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
||
Prepaid expenses and other assets |
|
|
( |
) |
|
|
( |
) |
Accounts payable |
|
|
|
|
|
|
||
Accrued expenses and other liabilities |
|
|
|
|
|
( |
) |
|
Deferred revenue |
|
|
( |
) |
|
|
( |
) |
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
Cash flows from investing activities |
|
|
|
|
|
|
||
Purchase of property, plant and equipment |
|
|
( |
) |
|
|
( |
) |
Net cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
Cash flows from financing activities |
|
|
|
|
|
|
||
Debt issuance costs for term loan |
|
|
( |
) |
|
|
— |
|
Payment of offering costs |
|
|
— |
|
|
|
( |
) |
Proceeds from common stock issuances under ESPP |
|
|
|
|
|
|
||
Other |
|
|
( |
) |
|
|
( |
) |
Net cash used in financing activities |
|
|
( |
) |
|
|
( |
) |
Net decrease in cash, cash equivalents and restricted cash |
|
|
( |
) |
|
|
( |
) |
Cash, cash equivalents and restricted cash at the beginning of the period |
|
|
|
|
|
|
||
Cash, cash equivalents and restricted cash at the end of the period |
|
$ |
|
|
$ |
|
||
Cash and cash equivalents |
|
|
|
|
|
|
||
Restricted cash |
|
|
|
|
|
|
||
Cash, cash equivalents and restricted cash at the end of the period |
|
$ |
|
|
$ |
|
||
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
||
Cash paid for interest |
|
$ |
|
|
$ |
|
||
Supplemental disclosure of noncash investing and financing activities: |
|
|
|
|
|
|
||
Property, plant and equipment in accounts payable and accrued expenses |
|
|
|
|
|
|
||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
2
Note 1—Organization and Description of Business Operations
Taysha Gene Therapies, Inc. (the “Company” or “Taysha”) was originally formed under the laws of the State of Texas on September 20, 2019. Taysha converted to a Delaware corporation on
Taysha is a clinical-stage biotechnology company focused on advancing AAV-based gene therapies for severe monogenic diseases of the central nervous system.
Sales Agreement
On October 5, 2021, the Company entered into a Sales Agreement (the “Sales Agreement”) with SVB Securities LLC (f/k/a SVB Leerink LLC) and Wells Fargo Securities, LLC (collectively, the “Sales Agents”), pursuant to which the Company may issue and sell, from time to time in its sole discretion, shares of its common stock having an aggregate offering price of up to $
Liquidity and Capital Resources
The Company has incurred operating losses since inception and expects to continue to incur significant operating losses for the foreseeable future and may never become profitable. Losses are expected to continue as the Company continues to invest in its research and development activities. As of March 31, 2024, the Company had an accumulated deficit of $
Future capital requirements will depend on many factors, including the timing and extent of spending on research and development and the market acceptance of the Company’s products. The Company will need to obtain additional financing in order to complete clinical studies and launch and commercialize any product candidates for which it receives regulatory approval. There can be no assurance that such financing will be available or will be on terms acceptable to the Company. As of March 31, 2024, the Company had cash and cash equivalents of $
3
Note 2—Summary of Significant Accounting Policies
Basis of Presentation
The unaudited condensed consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States (“GAAP”) as determined by the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) for interim financial information and the instructions to Form 10-Q and Article 10 of Regulation S-X and are consistent in all material respects with those included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2023, filed with the Securities and Exchange Commission (“SEC”) on March 19, 2024 (the “2023 Annual Report”). In the opinion of management, the unaudited condensed consolidated financial statements reflect all adjustments, which include only normal recurring adjustments necessary for the fair statement of the balances and results for the periods presented. The condensed consolidated balance sheet as of December 31, 2023 is derived from audited financial statements, however, it does not include all of the information and footnotes required by GAAP for complete financial statements. These unaudited condensed consolidated financial statements should be read in conjunction with the consolidated financial statements and related notes in the Company’s 2023 Annual Report.
Principles of Consolidation
The accompanying interim condensed consolidated financial statements include the accounts of Taysha and its wholly owned subsidiaries. All intercompany transactions and balances have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. The most significant estimates and assumptions in the Company’s financial statements relate to the determination of the fair value of the common stock prior to the initial public offering (“IPO”) (as an input into stock-based compensation), estimating manufacturing accruals and accrued or prepaid research and development expenses, the measurement of impairment of long-lived assets, the valuation of the Trinity Term Loans that are carried at fair value and the allocation of consideration received in connection with the Astellas Transactions (as defined below). These estimates and assumptions are based on current facts, historical experience and various other factors believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the recording of expenses that are not readily apparent from other sources. Actual results may differ materially from these estimates. To the extent there are material differences between the estimates and actual results, the Company’s future results of operations will be affected.
Significant Accounting Policies
There have been no changes in the Company’s significant accounting policies as disclosed in Note 2 to the audited consolidated financial statements included in the 2023 Annual Report.
Recently Adopted Accounting Pronouncements
There have been no significant changes in recently adopted accounting pronouncements from those disclosed in the section titled “Financial Statements and Supplementary Data” included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2023, as filed with the SEC.
Recently Issued Accounting Pronouncements
In November 2023, the FASB issued ASU No. 2023-07, Segment Reporting – Improvements to Reportable Segment Disclosures, to improve segment disclosure requirements under ASC 280, Segment Reporting, through enhancing disclosures about significant segment expenses. The guidance requires entities to provide significant segment expenses that are regularly provided to the chief operating decision maker and other segment expenses included in each reported measure of segment profitability. The ASU also enhances interim segment reporting requirements by aligning interim disclosures with information that must be disclosed annually in accordance with ASC 280. The guidance is effective for annual periods beginning after December 15, 2023, and interim periods
4
beginning after December 15, 2024, applied retrospectively with early adoption permitted. The Company is still evaluating the impact this ASU will have on its consolidated financial statements and related disclosures.
Note 3—Fair Value Measurements
The Company’s financial instruments that are measured at fair value on a recurring basis consist of money market funds, the Trinity Term Loans, a success fee derivative liability and certain of the Company’s warrant liabilities.
The following tables present information about the Company’s financial assets and liabilities measured at fair value on a recurring basis and indicate the level of the fair value hierarchy used to determine such fair values (in thousands):
|
March 31, 2024 |
|
|||||||||||||
|
Total |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
||||
Cash equivalents – money market funds |
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
Total assets |
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
||||
Trinity Term Loans |
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Success Fee Derivative liability |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
SSI Warrant liability |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Total liabilities |
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
|
December 31, 2023 |
|
|||||||||||||
|
Total |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
||||
Cash equivalents – money market funds
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
Total assets |
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
||||
Trinity Term Loans |
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Success Fee Derivative liability |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
SSI Warrant liability |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Total liabilities |
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
The Company classifies its money market funds, which are valued based on quoted market prices in an active market with no valuation adjustment, as Level 1 assets within the fair value hierarchy.
The Company’s Trinity Term Loans and Success Fee liability are classified as Level 3 measurements under the fair value hierarchy as the fair values were determined based on significant inputs not observable in the market. The fair values were determined utilizing a probability-weighted income approach, including variables for the timing of a success event and other probability estimates. See Note 7 for additional information on the Trinity Term Loans and Success Fee.
The Company’s SSI Warrant liability is classified as Level 3 measurements under the fair value hierarchy as the fair values were determined based on significant inputs not observable in the market. The fair values were determined using the Black-Scholes-Merton option pricing model to determine the fair value of the SSI Warrants (as defined below). See Note 10 for additional information on the SSI Warrants.
5
Note 4—Balance Sheet Components
Prepaid expenses and other current assets consisted of the following (in thousands):
|
|
March 31, |
|
|
December 31, |
|
||
Prepaid research and development |
|
$ |
|
|
$ |
|
||
Prepaid clinical trial |
|
|
|
|
|
|
||
Deferred offering costs |
|
|
|
|
|
|
||
Prepaid insurance |
|
|
|
|
|
|
||
Prepaid bonus |
|
|
|
|
|
— |
|
|
Other |
|
|
|
|
|
|
||
Total prepaid expenses and other current assets |
|
$ |
|
|
$ |
|
||
Property, plant and equipment, net consisted of the following (in thousands):
|
|
March 31, |
|
|
December 31, |
|
||
Leasehold improvements |
|
$ |
|
|
$ |
|
||
Laboratory equipment |
|
|
|
|
|
|
||
Computer equipment |
|
|
|
|
|
|
||
Furniture and fixtures |
|
|
|
|
|
|
||
Construction in progress |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
Property, plant and equipment, net |
|
$ |
|
|
$ |
|
||
Property, plant and equipment, net includes $
Depreciation expense was $
Accrued expenses and other current liabilities consisted of the following (in thousands):
|
|
March 31, |
|
|
December 31, |
|
||
Accrued research and development |
|
$ |
|
|
$ |
|
||
Lease liabilities, current portion |
|
|
|
|
|
|
||
Accrued clinical trial |
|
|
|
|
|
|
||
Accrued compensation |
|
|
|
|
|
|
||
Accrued professional and consulting fees |
|
|
|
|
|
|
||
Warrant liability |
|
|
|
|
|
|
||
Accrued severance |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Total accrued expenses and other current liabilities |
|
$ |
|
|
$ |
|
||
Note 5— Leases
The Company leases certain office, laboratory, and manufacturing space.
Dallas Lease
On January 11, 2021, the Company entered into a lease agreement (the “Dallas Lease”) with Pegasus Park, LLC, a Delaware limited liability company (the “Dallas Landlord”), pursuant to which the Company will lease approximately
6
The Dallas Lease commenced on
The Dallas Landlord has the right to terminate the Dallas Lease, or the Company’s right to possess the Office Space without terminating the Dallas Lease, upon specified events of default, including the Company’s failure to pay rent in a timely manner and upon the occurrence of certain events of insolvency with respect to the Company.
Dallas Lease Expansion
On December 14, 2021, the Company amended the Dallas Lease (the “Dallas Lease Amendment”) with the Dallas Landlord, pursuant to which the Company will lease approximately
The Dallas Lease Amendment commenced on July 1, 2022, and has a term of approximately
The Company is obligated to pay operating costs and utilities applicable to the Expansion Premises. Total future minimum lease payments under the Dallas Lease Amendment over the initial
The Company has a right of first refusal with respect to certain additional office space on the 15th floor at 3000 Pegasus Park Drive, Dallas, Texas 75247 before the Dallas Landlord accepts any offer for such space.
Durham Lease
On December 17, 2020, the Company entered into a lease agreement (the “Durham Lease”) with Patriot Park Partners II, LLC, a Delaware limited liability company (the “Durham Landlord”), pursuant to which the Company agreed to lease approximately
The Company was not required to provide a security deposit in connection with its entry into the Durham Lease. The Company was responsible for constructing interior improvements within the Facility. The Company was required to place $
Summary of all lease costs recognized under ASC 842
The following table summarizes the lease costs recognized under ASC 842 and other information pertaining to the Company’s operating leases for the three months ended March 31, 2024 and 2023 (in thousands):
|
|
For the Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Operating lease cost |
|
$ |
|
|
$ |
|
||
Variable lease cost |
|
|
|
|
|
|
||
Total lease cost |
|
$ |
|
|
$ |
|
||
7
Supplemental information related to the remaining lease term and discount rate are as follows:
|
|
March 31, 2024 |
|
December 31, 2023 |
|
||
Weighted average remaining lease term (in years) – Finance leases |
|
|
|
|
|
||
Weighted average remaining lease term (in years) – Operating leases |
|
|
|
|
|
||
|
|
|
|
|
|
||
Weighted average discount rate – Finance leases |
|
|
% |
|
% |
||
Weighted average discount rate – Operating leases |
|
|
% |
|
% |
||
Supplemental cash flow information related to the Company’s operating leases are as follows (in thousands):
|
For the Three Months Ended March 31, |
|
|||||
|
2024 |
|
|
2023 |
|
||
Operating cash flows for operating leases |
$ |
|
|
$ |
|
||
As of March 31, 2024, future minimum commitments under ASC 842 under the Company’s operating and finance leases were as follows (in thousands):
Year Ending December 31, |
Operating |
|
Finance |
|
||
2024 |
$ |
|
$ |
|
||
2025 |
|
|
|
|
||
2026 |
|
|
|
|
||
2027 |
|
|
|
— |
|
|
2028 |
|
|
|
— |
|
|
Thereafter |
|
|
|
— |
|
|
Total lease payments |
|
|
|
|
||
Less: imputed interest |
|
( |
) |
|
( |
) |
Total lease liabilities |
$ |
|
$ |
|
||
|
|
|
|
|||
|
|
|
|
|||
$ |
|
$ |
|
|||
Note 6—Astellas Agreements
On October 21, 2022 (the “Effective Date”), the Company entered into the Option Agreement (the “Option Agreement”) with Audentes Therapeutics, Inc. (d/b/a Astellas Gene Therapy)(“Astellas”), pursuant to which the Company granted to Astellas an exclusive option to obtain an exclusive, worldwide, royalty and milestone-bearing right and license (A) to research, develop, make, have made, use, sell, offer for sale, have sold, import, export and otherwise exploit, or, collectively, exploit, the product known, as of the Effective Date, as TSHA-120 (the “120 GAN Product”), and any backup products with respect thereto for use in the treatment of Giant Axonal Neuropathy (“GAN”) or any other gene therapy product for use in the treatment of GAN that is controlled by Taysha or any of its affiliates or with respect to which the Company or any of its affiliates controls intellectual property rights covering the exploitation thereof, or a GAN Product, and (B) under any intellectual property rights controlled by Taysha or any of its affiliates with respect to such exploitation (the “GAN Option”). Subject to certain extensions, the GAN Option was exercisable from the Effective Date through a specified period of time following Astellas’ receipt of (i) the formal minutes from the Type B end-of-Phase 2 meeting between Taysha and the FDA in response to the Company’s meeting request sent to the FDA on September 19, 2022 for the 120 GAN Product (the “Type B end-of-Phase 2 Meeting”), (ii) all written feedback from the FDA with respect to the Type B end-of-Phase 2 Meeting, and (iii) all briefing documents sent by Taysha to the FDA with respect to the Type B end-of-Phase 2 Meeting. In September 2023, Astellas provided written notice of its decision not to exercise the GAN Option.
Under the Option Agreement, the Company also granted to Astellas an exclusive option to obtain an exclusive, worldwide, royalty and milestone-bearing right and license (A) to exploit any Rett Product (as defined below), and (B) under any intellectual property rights controlled by Taysha or any of its affiliates with respect to such exploitation (the “Rett Option,” and together with the GAN Option, each, an “Option”). Subject to certain extensions, the Rett Option is exercisable from the Effective Date through a specified period of time following Astellas’ receipt of (i) certain clinical data from the female pediatric trial and (ii) certain specified data with respect to TSHA-102, such period, the Rett Option Period, related to (i) the product known, as of the Effective Date, as
8
TSHA-102 and any backup products with respect thereto for use in the treatment of Rett syndrome, and (ii) any other gene therapy product for use in the treatment of Rett syndrome that is controlled by Taysha or any of its affiliates or with respect to which the Company or any of its affiliates controls intellectual property rights covering the exploitation thereof (a “Rett Product”).
The parties have agreed that, if Astellas exercises an Option, the parties will, for a specified period, negotiate a license agreement in good faith on the terms and conditions outlined in the Option Agreement, including payments by Astellas of a to be determined upfront payment, certain to be determined milestone payments, and certain to be determined royalties on net sales of GAN Products and/or Rett Products, as applicable.
During the Rett Option Period, the Company has agreed to (A) not solicit or encourage any inquiries, offers or proposals for, or that could reasonably be expected to lead to, a Change of Control (as defined in the Option Agreement), or (B) otherwise initiate a process for a potential Change of Control, in each case, without first notifying Astellas and offering Astellas the opportunity to submit an offer or proposal to the Company for a transaction that would result in a Change of Control. If Astellas fails or declines to submit any such offer within a specified period after the receipt of such notice, the Company will have the ability to solicit third party bids for a Change of Control transaction. If Astellas delivers an offer to the Company for a transaction that would result in a Change of Control, the Company and Astellas will attempt to negotiate in good faith the potential terms and conditions for such potential transaction that would result in a Change of Control for a specified period, which period may be shortened or extended by mutual agreement.
As partial consideration for the rights granted to Astellas under the Option Agreement, Astellas paid the Company an upfront payment of $
Astellas Securities Purchase Agreement
On October 21, 2022, the Company entered into a securities purchase agreement with Astellas (the “Astellas Securities Purchase Agreement”), pursuant to which the Company agreed to issue and sell to Astellas in a private placement (the “Astellas Private Placement”), an aggregate of
Accounting Treatment
In October 2022, upon closing of the Astellas Private Placement and transferring the
The Company determined that the Option Agreement falls within the scope of ASC 606, Revenue from Contracts with Customers as the development of TSHA-102 for the treatment of Rett Syndrome and TSHA-120 for the treatment of GAN are considered ordinary activities for the Company. In accordance with ASC 606, the Company evaluated the Option Agreement and identified three separate performance obligations: (1) option to obtain licensing right to GAN, (2) option to obtain licensing right to Rett and (3) performance of research and development activities in the Rett development plan. The transaction price is determined to be $
To determine the standalone selling price ("SSP") of the Rett and GAN options, which the Company concluded to be material rights, the Company utilized the probability-weighted expected return (PWERM) method. The PWERM method contemplates the
9
probability and timing of an option exercise. At contract inception, the Company estimated that the probability of exercise was
The following table summarizes the allocation of the transaction price to the three performance obligations at contract inception (amounts in thousands):
|
|
Transaction Price Allocation |
|
|
Option to obtain license for Rett |
|
$ |
|
|
Option to obtain license for GAN |
|
|
|
|
Rett research and development activities |
|
|
|
|
Total |
|
$ |
|
|
Revenue allocated to the material rights will be recognized at a point in time when each option period expires or when a decision is made by Astellas to exercise or not exercise each option. Revenue from the Rett research and development activities will be recognized as activities are performed using an input method, according to the costs incurred as related to the total costs expected to be incurred to satisfy the performance obligation. The transfer of control occurs over this time period and is a reliable measure of progress towards satisfying the performance obligation.
During the three months ended March 31, 2024, there were no significant changes to the total estimated costs to be incurred to satisfy the performance obligation associated with the Rett research and development activities.
The Company recognized revenue of $
The Company had $
Note 7 – Term Loans
Loan with Trinity Capital
On November 13, 2023 (the “Trinity Closing Date”), the Company entered into a Loan and Security Agreement (the “Trinity Term Loan Agreement”), by and among the Company, the lenders party thereto from time to time (the “Trinity Lenders”) and Trinity Capital Inc., as administrative agent and collateral agent for the Trinity Lenders (“Trinity”). The Trinity Term Loan Agreement provides for, on the Trinity Closing Date, $
Future principal debt payments on the Term Loan Agreement as of March 31, 2024 are as follows (in thousands):
Year Ending December 31, |
|
|
|
|
2024 |
|
$ |
— |
|
2025 |
|
|
— |
|
2026 |
|
|
— |
|
2027 |
|
|
|
|
2028 |
|
|
|
|
Total principal payments |
|
$ |
|
|
10
The Trinity Term Loans may be prepaid in full (i) from the Trinity Closing Date through November 13, 2024, with payment of a
The obligations under the Trinity Term Loan Agreement are secured by a perfected security interest in all of the Company’s assets except for certain customarily excluded property pursuant to the terms of the Trinity Term Loan Agreement. There are no financial covenants and
The Trinity Term Loan Agreement also contains customary representations and warranties, and also includes customary events of default, including payment default, breach of covenants, change of control, and material adverse effects. Upon the occurrence of an event of default, a default interest rate of an additional
The proceeds of the Trinity Term Loans were used to repay the Company’s obligations under the Term Loan Agreement with Silicon Valley Bank in full. The Term Loan Agreement with Silicon Valley Bank was terminated concurrently with entry into the Trinity Term Loan Agreement.
The Company assessed the terms and features of the Trinity Term Loans and determined that the Company was eligible to elect the fair value option under ASC 825, Financial Instruments. The Trinity Term Loans contain various embedded features and the election of the fair value option allowed the Company to bypass analysis of potential embedded derivatives and further analysis of bifurcation of any recognized financial liabilities. Under the fair value option, the financial liability is initially measured at its fair value on the issue date and subsequently remeasured at estimated fair value on a recurring basis at each reporting date. Changes in the fair value of the Trinity Term Loans, which include accrued interest, if any, are recorded as a component of other expense (income) in the condensed consolidated statements of operations. The Company has not elected to present interest expense separately from changes in fair value and therefore will not present interest expense associated with the Trinity Term Loans. Any changes in fair value caused by instrument-specific credit risk are presented separately in other comprehensive income or loss if material. Under the fair value option, debt issuance costs are expensed as incurred. The Company incurred $
In connection with the Trinity Term Loans, the Company entered into a Success Fee Agreement with Trinity which specifies the terms regarding a fee in the amount of
The proceeds from the Trinity Term Loans were allocated to the Success Fee and Trinity Term Loans based on their respective fair values on the Trinity Closing Date. The fair values were determined utilizing a probability-weighted income approach, including variables for the timing of a success event and other probability estimates.
The Company determined the fair value of the Trinity Term Loans and the Success Fee using a probability-weighted income approach and recorded the loan at fair value of $
11
The Company remeasured the fair value of the Trinity Term Loans and Success Fee as of March 31, 2024 using a probability-weighted income approach. The Company calculated discounted cash flows of the Trinity Term Loans using a discount rate of
The following table reconciles the change in fair value of the Trinity Term Loans during the three months ended March 31, 2024 (in thousands):
Trinity Term Loans |
|
|
|
|
Beginning fair value balance at January 1, 2024 |
|
$ |
|
|
Principal payments |
|
|
— |
|
Change in fair value reported in statements of operations |
|
|
( |
) |
Change in fair value reported in comprehensive loss |
|
|
|
|
Ending fair value balance as of March 31, 2024 |
|
$ |
|
|
During the three months ended March 31, 2024, the Company recorded $
The following table reconciles the change in fair value of the Success Fee liability during the three months ended March 31, 2024 (in thousands):
Success Fee |
|
|
|
|
Beginning fair value balance at January 1, 2024 |
|
$ |
|
|
Change in fair value of Success Fee |
|
|
|
|
Ending fair value balance as of March 31, 2024 |
|
$ |
|
|
Loan with Silicon Valley Bank
On August 12, 2021 (the “Closing Date”), the Company entered into a Loan and Security Agreement (the “Term Loan Agreement”), by and among the Company, the lenders party thereto from time to time (the “Lenders”) and Silicon Valley Bank, as administrative agent and collateral agent for the Lenders (“Agent”). The Term Loan Agreement provided for (i) on the Closing Date, $
The interest rate applicable to the Term Loans was the greater of (a) the WSJ Prime Rate plus
The Term Loans could have been prepaid in full through August 12, 2023, with payment of a
The obligations under the Term Loan Agreement were secured by a perfected security interest in all of the Company’s assets except for intellectual property and certain other customarily excluded property pursuant to the terms of the Term Loan Agreement.
Upon termination of the Term Loan Agreement with Silicon Valley Bank, the Company made a prepayment of $
12
by the Company in connection with the Term Loans included payment of the Exit Fee of $
During the three months ended March 31, 2023, the Company recognized interest expense related to the Term Loans of $
Note 8—Research, Collaboration and License Agreements
UT Southwestern Agreement
On November 19, 2019, the Company entered into a research, collaboration and license agreement (“UT Southwestern Agreement”) with the Board of Regents of the University of Texas System on behalf of The University of Texas Southwestern Medical Center (“UT Southwestern”). Under the UT Southwestern Agreement, UT Southwestern is primarily responsible for preclinical development activities with respect to licensed products for use in certain specified indications (up to investigational new drug application-enabling studies), and the Company is responsible for all subsequent clinical development and commercialization activities with respect to the licensed products. UT Southwestern will conduct such preclinical activities for a
In connection with the UT Southwestern Agreement, the Company obtained an exclusive, worldwide, royalty-free license under certain patent rights of UT Southwestern and a non-exclusive, worldwide, royalty-free license under certain know-how of UT Southwestern, in each case to make, have made, use, sell, offer for sale and import licensed products for use in certain specified indications. Additionally, the Company obtained a non-exclusive, worldwide, royalty-free license under certain patents and know-how of UT Southwestern for use in all human uses, with a right of first refusal to obtain an exclusive license under certain of such patent rights and an option to negotiate an exclusive license under other of such patent rights. The Company is required to use commercially reasonable efforts to develop, obtain regulatory approval for, and commercialize at least
On April 2, 2020, the Company amended the UT Southwestern Agreement to include the addition of another licensed product and certain indications, and a right of first refusal to the Company over certain patient dosing patents. No additional consideration was transferred in connection with this amendment. In March 2022, the Company and UT Southwestern mutually agreed to revise the payment schedules and current performance expectations of the current sponsored research agreements under the UT Southwestern Agreement and defer payments by fifteen months. In December 2023, the Company and UT Southwestern mutually agreed to terminate specific sponsored research agreements.
The UT Southwestern Agreement expires on a country-by-country and licensed product-by-licensed product basis upon the expiration of the last valid claim of a licensed patent in such country for such licensed product. After the initial research term, the Company may terminate the agreement, on an indication-by-indication and licensed product-by-licensed product basis, at any time upon specified written notice to UT Southwestern. Either party may terminate the agreement upon an uncured material breach of the agreement or insolvency of the other party. In December 2023, the Company transferred rights to specific indications back to UT Southwestern.
In November 2019, as partial consideration for the license rights granted under the UT Southwestern Agreement, the Company issued
Queen’s Agreement
On February 21, 2020, the Company entered into a license agreement with Queen’s (the “Queen’s Agreement”) to obtain the exclusive perpetual, royalty-bearing license, with the right to sublicense through multiple tiers, under certain patent rights and know-how of Queen’s, including certain improvements to such patent rights and know-how, to develop products in any field which use one or more valid claims of the patents licensed under the Queen’s Agreement (the “Licensed Patents”), or the technology, information and intellectual property related to the patents licensed under the Queen’s Agreement (together with the Licensed Patents, the “Licensed Products”), and to make, have made, use, sell, offer for sale, import and export Licensed Products and otherwise exploit such patents and know-how for use in certain specified indications. In exchange for the rights granted to the Company, the Company made a cash payment of $
13
million upon the completion of a combination of commercial milestones. In further consideration of the rights granted, beginning with the Company’s first commercial sale of the Licensed Products, the Company will also pay an annual earned royalty in the low single digits on net sales of Licensed Products, subject to certain customary reductions, and a percentage of non-royalty sublicensing revenue ranging in the low double digits. Royalties are payable, on a Licensed Products-by-Licensed Products and a country-by-country basis, until expiration of the last valid claim of a Licensed Patent covering such Licensed Products in such country and the expiration of any regulatory exclusivity for such Licensed Products in such country.
In January 2024, the Company transferred rights back to Queen’s for the Licensed Patents.
Abeona CLN1 Agreements
In August 2020, the Company entered into license and inventory purchase agreements with Abeona Therapeutics Inc. (“Abeona”) for worldwide exclusive rights to certain intellectual property rights and know-how relating to the research, development and manufacture of ABO-202, an AAV-based gene therapy for CLN1 disease (also known as infantile Batten disease). Under the terms of the agreements, the Company made initial cash payments to Abeona of $
In December 2021, a regulatory milestone was triggered in connection with this agreement and therefore the Company recorded $
Abeona Rett Agreement
On October 29, 2020, the Company entered into a license agreement (the “Abeona Rett Agreement”) with Abeona pursuant to which the Company obtained an exclusive, worldwide, royalty-bearing license, with the right to grant sublicenses under certain patents, know-how and materials originally developed by the University of North Carolina at Chapel Hill, the University of Edinburgh and Abeona to research, develop, manufacture, have manufactured, use, and commercialize licensed products for gene therapy and the use of related transgenes for Rett syndrome.
Subject to certain obligations of Abeona, the Company is required to use commercially reasonable efforts to develop at least one licensed product and commercialize at least one licensed product in the United States.
In connection with the Abeona Rett Agreement, the Company paid Abeona a one-time upfront license fee of $
The Abeona Rett Agreement expires on a country-by-country and licensed product-by-licensed product basis upon the expiration of the last royalty term of a licensed product. Either party may terminate the agreement upon an uncured material breach of the agreement or insolvency of the other party. The Company may terminate the agreement for convenience upon specified prior written notice to Abeona.
In March 2022, the Company’s clinical trial application, (“CTA”) filing for TSHA-102 for the treatment of Rett Syndrome was approved by Health Canada and therefore triggered a regulatory milestone payment in connection with this agreement. The
14
Company recorded $
Acquisition of Worldwide Rights for TSHA-120 for the treatment of GAN
In March 2021, the Company acquired the exclusive worldwide rights to a clinical-stage AAV9 gene therapy program, now known as TSHA-120, for the treatment of GAN pursuant to a license agreement with Hannah’s Hope Fund (“HHF”) for Giant Axonal Neuropathy, Inc. TSHA-120 is an intrathecally dosed AAV9 gene therapy currently being evaluated in a clinical trial for the treatment of GAN.
Under the terms of the GAN Agreement, in exchange for granting the Company the exclusive worldwide rights to TSHA-120, HHF received an upfront payment of $
License Agreement for CLN7
In March 2022, the Company entered into a license agreement with UT Southwestern (the “CLN7 Agreement”) pursuant to which the Company obtained an exclusive worldwide, royalty-bearing license with right to grant sublicenses to develop, manufacture, use, and commercialize licensed products for gene therapy for CLN7, a form of Batten Disease. In connection with the CLN7 Agreement, the Company paid a one-time upfront license fee of $
Note 9—Stock-Based Compensation
On July 1, 2020, the Company’s board of directors approved the 2020 Equity Incentive Plan (“Previous Plan”) which permits the granting of incentive stock options, non-statutory stock options, stock appreciation rights, restricted stock awards (“RSAs”), restricted stock units (“RSUs”) and other stock-based awards to employees, directors, officers and consultants. As of September 16, 2020, the approval date of the New Plan (as defined below), no additional awards will be granted under the Previous Plan. The terms of the Previous Plan will continue to govern the terms of outstanding equity awards that were granted prior to approval of the New Plan.
On September 16, 2020, the Company’s stockholders approved the 2020 Stock Incentive Plan (“New Plan”), which became effective upon the execution of the underwriting agreement in connection with the IPO.
Furthermore, on September 16, 2020, the Company’s stockholders approved the Employee Stock Purchase Plan (“ESPP”), which became effective upon the execution of the underwriting agreement in connection with the IPO. The maximum number of shares of common stock that may be issued under the ESPP will not exceed
15
the Company increased the number of shares of common stock reserved for issuance under the ESPP by
On December 15, 2023, the Company’s board of directors adopted the Taysha Gene Therapies, Inc. 2023 Inducement Plan (the “Inducement Plan”). The Inducement Plan was adopted without stockholder approval pursuant to Nasdaq Listing Rule 5635(c)(4). The Board reserved
The only persons eligible to receive grants of Inducement Awards (as defined below) under the Inducement Plan are individuals who satisfy the standards for inducement grants under Nasdaq Listing Rule 5635(c)(4). The Inducement Plan will be administered by the Board and the Company’s Compensation Committee. Inducement Awards may only be granted by: (i) the Compensation Committee, provided such committee is comprised solely of “independent directors” (as defined by Nasdaq Listing Rule 5605(a)(2)) or (ii) a majority of the Company’s “independent directors.” An “Inducement Award” means any right to receive the Company’s common stock, cash or other property granted under the Inducement Plan (including nonstatutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights, performance stock awards, performance cash awards or other stock-based awards).
The number of shares available for grant under the Company’s incentive plans were as follows:
|
|
New |
|
|
Inducement |
|
|
|
|
|||
|
|
Plan |
|
|
Plan |
|
|
Total |
|
|||
Available for grant - January 1, 2024 |
|
|
|
|
|
|
|
|
|
|||
Plan adjustments and amendments |
|
|
|
|
|
— |
|
|
|
|
||
Grants |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Forfeitures |
|
|
|
|
|
— |
|
|
|
|
||
Available for grant - March 31, 2024 |
|
|
|
|
|
|
|
|
|
|||
Stock Options
For the three months ended March 31, 2024,
The following weighted-average assumptions were used to estimate the fair value of time-based vesting stock options that were granted during the three months ended March 31, 2024 and 2023:
|
|
Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Risk-free interest rate |
|
|
% |
|
|
% |
||
Expected dividend yield |
|
|
— |
|
|
|
— |
|
Expected term (in years) |
|
|
|
|
|
|
||
Expected volatility |
|
|
% |
|
|
% |
||
The following table summarizes time-based vesting stock option activity, during the three months ended March 31, 2024:
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
||||
|
|
|
|
|
Weighted |
|
|
Average |
|
|
Aggregate |
|
||||
|
|
|
|
|
Average |
|
|
Remaining |
|
|
Intrinsic |
|
||||
|
|
Stock |
|
|
Exercise |
|
|
Contractual |
|
|
Value |
|
||||
|
|
Options |
|
|
Price |
|
|
Life (in years) |
|
|
(in thousands) |
|
||||
Outstanding at January 1, 2024 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Options granted |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Options cancelled or forfeited |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Options expired |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Outstanding at March 31, 2024 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Options exercisable at March 31, 2024 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
The aggregate intrinsic value in the above table is calculated as the difference between the fair value of the Company’s common stock at the respective reporting date and the exercise price of the stock options. As of March 31, 2024, the total unrecognized compensation related to unvested stock option awards granted was $
16
recognize over a weighted-average period of approximately
Performance Stock Options
In February 2023, the Company issued options to purchase
In May 2023, the Company issued options to purchase
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
|
% |
|
Expected dividend yield |
|
|
— |
|
Expected term (in years) |
|
|
|
|
Expected volatility |
|
|
% |
|
In December 2023, the Company modified all of the Original Options to amend the clinical and regulatory performance conditions and decreased the number of options granted to
|
|
|
|
|
Risk-free interest rate |
|
|
% |
|
Expected dividend yield |
|
|
— |
|
Expected term (in years) |
|
|
|
|
Expected volatility |
|
|
% |
|
The Modified Options will vest over
Market-based Stock Options
In February 2023, the Company issued options to purchase
17
Restricted Stock Units
For the three months ended March 31, 2024, the Company issued
The Company’s default tax withholding method for RSUs granted prior to 2023 is the sell-to-cover method, in which shares with a market value equivalent to the tax withholding obligation are sold on behalf of the holder of the RSUs upon vesting and settlement to cover the tax withholding liability and the cash proceeds from such sales are remitted by the Company to taxing authorities. For RSUs granted in 2023, the Company’s tax withholding policy allows the RSU holder to choose to either pay cash to the Company for the tax withholding obligation or elect the net withholding method, in which shares with a market equivalent to the tax withholding obligation are withheld and the net shares are issued to the RSU holder.
The Company’s RSU activity for the three months ended March 31, 2024 was as follows:
|
|
|
|
|
Weighted |
|
||
|
|
|
|
|
Average |
|
||
|
|
|
|
|
Grant Date |
|
||
|
|
Number |
|
|
Fair Value |
|
||
|
|
of Shares |
|
|
per Share |
|
||
Nonvested at January 1, 2024 |
|
|
|
|
$ |
|
||
Restricted units granted |
|
|
|
|
|
|
||
Vested |
|
|
( |
) |
|
|
|
|
Cancelled or forfeited |
|
|
— |
|
|
|
— |
|
Nonvested at March 31, 2024 |
|
|
|
|
$ |
|
||
As of March 31, 2024, the total unrecognized compensation cost related to the unvested RSU's was $
Performance and Market-based Restricted Stock Units
In February 2023, the Company issued
Employee Stock Purchase Plan
In February 2022, the Company’s board of directors authorized the first offering under the ESPP. Under the ESPP, eligible employees may purchase shares of Taysha common stock through payroll deductions at a price equal to
The following table summarizes the total stock-based compensation expense for the stock options, ESPP, RSAs and RSUs recorded in the condensed consolidated statements of operations for the three months ended March 31, 2024 and 2023 (in thousands):
|
|
For the Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Research and development expense |
|
$ |
|
|
$ |
( |
) |
|
General and administrative expense |
|
|
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
||
18
Note 10—Warrants
Pre-Funded Warrants
On August 14, 2023, the Company entered into a Securities Purchase Agreement (the “August 2023 Purchase Agreement”) with certain institutional and other accredited investors (the “Purchasers”), pursuant to which the Company agreed to sell and issue to the Purchasers in a private placement transaction (the “August 2023 Private Placement”) (i)
The Pre-Funded Warrants have a per share exercise price of $
The closing of the August 2023 Private Placement occurred on August 16, 2023 (the “Closing”). The total gross proceeds to the Company at the Closing were $
The Company concluded that at the closing of the Private Placement in August 2023, the Pre-Funded Warrants did not meet the criteria for equity classification under the guidance of ASC 815 as the Company did not have sufficient authorized and unissued shares to satisfy the warrants if exercised. The Company recorded the Pre-Funded Warrants as liabilities at their fair value. This liability is subject to remeasurement at each balance sheet date and any change in fair value is recognized in the Company’s consolidated statements of operations. The Company incurred $
The Company measured the fair value of the PIPE Shares and Pre-Funded Warrants based on the $
|
|
Purchase Price Allocation |
|
|
PIPE Shares |
|
$ |
|
|
Pre-Funded Warrants |
|
|
|
|
Total |
|
$ |
|
|
The Company remeasured the fair value of the Pre-Funded Warrants using the closing price of the Company’s common stock on the Nasdaq Global Market as of November 15, 2023 of $
SSI Warrants
In April 2023, the Company entered into a securities purchase agreement (the “SSI Securities Purchase Agreement”), with two affiliates of SSI Strategy Holdings LLC (“SSI”), named therein (the “SSI Investors”) pursuant to which the Company agreed to issue and sell to the SSI Investors in a private placement (the “SSI Private Placement”),
19
Warrant Share, which was the closing price of the Company’s common stock on the Nasdaq Global Market on April 4, 2023. The SSI Warrants issued in the SSI Private Placement provide that the holder of the SSI Warrants will not have the right to exercise any portion of its SSI Warrants until the achievement of certain clinical and regulatory milestones related to the Company’s clinical programs. The SSI Private Placement closed on April 5, 2023. Gross proceeds of the SSI Private Placement were $
The Company concluded that the SSI Warrants do not meet the criteria for equity classification under the guidance of ASC 815 due to settlement provisions that permit the holder to receive a variable number of shares in the event of a specified fundamental transaction as well as provisions that permit the holder to participate in dividends. As the SSI Warrants do not meet the criteria for equity classification, the Company recorded the warrants as liabilities at their fair value. This liability is subject to remeasurement at each balance sheet date until the warrants are exercised or expire, and any change in fair value is recognized in the Company’s condensed consolidated statements of operations.
The Company determined the fair value of the SSI Warrants at issuance was $
Risk-free interest rate |
|
|
% |
|
Expected dividend yield |
|
— |
|
|
Expected term (in years) |
|
|
|
|
Expected volatility |
|
|
% |
|
Market value of common stock (per share) |
|
$ |
|
|
The fair value adjustment as of March 31, 2024 was $
The Company estimated the fair value of the SSI Warrant liability using the following assumptions as of March 31, 2024:
Risk-free interest rate |
|
|
% |
|
Expected dividend yield |
|
— |
|
|
Expected term (in years) |
|
|
|
|
Expected volatility |
|
|
% |
|
Market value of common stock (per share) |
|
$ |
|
|
The following table summarizes changes in the Company’s warrant liability during the year ended March 31, 2024 (in thousands):
|
|
Warrant Liability |
|
|
Balance at January 1, 2024 |
|
$ |
|
|
Change in fair value |
|
|
|
|
Balance at March 31, 2024 |
|
$ |
|
|
Note 11—Net Loss Per Common Share
Basic net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted-average number of shares of common stock outstanding for the period. Since the Company had a net loss in all periods presented, basic and diluted net loss per common share are the same.
In August 2023, the Company issued liability-classified Pre-Funded Warrants with a nominal exercise price of $
20
The following table represents the calculation of basic and diluted net loss per common share (in thousands, except share and per share data):
|
|
For the Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Weighted-average shares of common stock outstanding used to compute net loss per common share, basic and diluted |
|
|
|
|
|
|
||
Net loss per common share, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
The following common stock equivalents outstanding as of March 31, 2024 and 2023 were excluded from the computation of diluted net loss per share attributable to common stockholders for the periods presented because including them would have been anti‑dilutive:
|
|
March 31, |
|
|
March 31, |
|
||
Unvested RSUs |
|
|
|
|
|
|
||
Stock options |
|
|
|
|
|
|
||
SSI Warrants |
|
|
|
|
|
— |
|
|
Total |
|
|
|
|
|
|
||
Note 12—Income Taxes
Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Valuation allowances are provided if based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. The Company has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets. There is
As of March 31, 2024, there were no material changes to either the nature or the amounts of the uncertain tax positions previously determined for the year ended December 31, 2023.
Note 13—Commitments and Contingencies
Litigation
From time to time, the Company may be subject to various legal proceedings and claims that arise in the ordinary course of its business activities. The Company records a liability when a particular contingency is probable and estimable.
In January 2024 and April 2024, the Company was named a nominal defendant in two putative stockholder derivative actions filed by stockholders of the Company in the Court of Chancery of the State of Delaware. Shortly after filing suit, the plaintiff in the second-filed action voluntarily dismissed his lawsuit and filed a substantially similar action except with fewer named defendants. The complaints assert claims relating to the Company’s August 2023 Private Placement against (i) certain of the Company’s current and former directors and officers for breach of fiduciary duty and unjust enrichment; and (ii) against certain participants in the Company’s August 2023 Private Placement for aiding and abetting breach of fiduciary duty and unjust enrichment. The complaints seek an unspecified award of damages in the Company’s favor, plus pre-judgment and post-judgement interest, and an award to the plaintiffs for the costs and disbursement of the action, including fees for their attorneys, experts, and accountants. The Company has not recorded a liability related to these lawsuits because, at this time, the Company is unable to reasonably estimate possible losses or gains or determine whether an unfavorable outcome is either probable or remote.
Commitments
In the normal course of business, the Company enters into contracts that contain a variety of indemnifications with its directors, officers, employees, licensors, suppliers and service providers. The Company’s maximum exposure under these arrangements is unknown at March 31, 2024. The Company does not anticipate recognizing any significant losses relating to these arrangements.
21
Note 14 – Strategic Reprioritization
In March 2022, the Company implemented changes to the Company’s organizational structure as well as a broader operational cost reduction plan to enable the Company to focus on specific clinical-stage programs for GAN and Rett syndrome. Substantially all other research and development activities have been paused to increase operational efficiency.
In connection with prioritization of programs, the Company reduced headcount by approximately
Payment of these costs are substantially complete as of March 31, 2024. The amount of accrued severance recorded as of March 31, 2024 is as follows (amounts in thousands):
|
|
Accrued Severance |
|
|
Accrued severance as of January 1, 2024 |
|
$ |
|
|
Severance recorded |
|
|
— |
|
Severance paid |
|
|
( |
) |
Accrued severance as of March 31, 2024 |
|
$ |
|
|
Note 15 – Retirement Plan
In July 2021, the Company adopted a 401(k) retirement savings plan that provides retirement benefits to all full-time employees. Eligible employees may contribute a percentage of their annual compensation, subject to Internal Revenue Service limitations. The Company contributed $
22
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited condensed consolidated financial statements and related notes included in this Quarterly Report on Form 10-Q and the audited financial statements and notes thereto as of and for the year ended December 31, 2023 and the related Management’s Discussion and Analysis of Financial Condition and Results of Operations, included in our Annual Report on Form 10-K for the year ended December 31, 2023, or Annual Report, filed with the Securities and Exchange Commission, or the SEC, on March 19, 2024. Unless the context requires otherwise, references in this Quarterly Report on Form 10-Q to “we,” “us,” and “our” refer to Taysha Gene Therapies, Inc. together with its consolidated subsidiaries.
Forward-Looking Statements
The information in this discussion contains forward-looking statements and information within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, which are subject to the “safe harbor” created by those sections. These forward-looking statements include, but are not limited to, statements concerning our strategy, future operations, future financial position, future revenues, projected costs, prospects and plans and objectives of management. The words “anticipates,” “believes,” “estimates,” “expects,” “intends,” “may,” “plans,” “projects,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We may not actually achieve the plans, intentions, or expectations disclosed in our forward-looking statements and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements that we make. These forward-looking statements involve risks and uncertainties that could cause our actual results to differ materially from those in the forward-looking statements, including, without limitation, the risks set forth in Part I, Item 1A, “Risk Factors” in our Annual Report. The forward-looking statements are applicable only as of the date on which they are made, and we do not assume any obligation to update any forward-looking statements.
Note Regarding Trademarks
All brand names or trademarks appearing in this report are the property of their respective holders. Unless the context requires otherwise, references in this report to the “Company,” “we,” “us,” and “our” refer to Taysha Gene Therapies, Inc.
Overview
We are a clinical-stage biotechnology company focused on advancing AAV-based gene therapies for the treatment of severe monogenic diseases of the central nervous system, or CNS. Our lead clinical program TSHA-102 is in development for the treatment of Rett syndrome, a rare neurodevelopmental disorder with no approved disease-modifying therapies that address the genetic root cause of the disease. With a singular focus on developing transformative medicines, we aim to address severe unmet medical needs and dramatically improve the lives of patients and their caregivers. Our management team has proven experience in gene therapy development and commercialization. We leverage this experience, our manufacturing process and a clinically and commercially proven AAV9 capsid in an effort to rapidly translate treatments from bench to bedside.
We are evaluating TSHA-102 in the REVEAL Phase 1/2 adolescent and adult trial, which is a first-in-human, open-label, randomized, dose-escalation and dose-expansion study evaluating the safety and preliminary efficacy of TSHA-102 in adolescent and adult females aged 12 years and older with Rett syndrome due to MECP2 loss-of-function mutation. The trial is taking place in Canada and the United States. We dosed the first two adult patients with Rett syndrome in 2023. There have been no treatment-emergent serious adverse events as of the 35-week assessment post-treatment for the first adult patient treated. In addition, there have been no treatment-emergent serious adverse events as of the 19-week assessment post-treatment for the second adult patient treated. The independent data monitoring committee, or IDMC, meeting to review the clinical data from the first two adult patients and the first pediatric patient took place in February 2024. The IDMC approved our request to proceed to earlier dose escalation in the adolescent and adult trial, enabling early advancement to cohort 2. The first patient in cohort 2 (high dose, 1x1015 total vg) has been enrolled, and dosing has been scheduled for the second quarter of 2024. We expect to provide an update on available safety and efficacy data from completed cohort 1 (low dose, 5.7x1014 total vg) in mid-2024. We expect to report initial available safety and efficacy data from cohort 2 (high dose, 1x1015 total vg) in the second half of 2024.
We are also evaluating TSHA-102 in the REVEAL Phase 1/2 pediatric trial, which is an open-label, randomized, dose-escalation and dose-expansion study evaluating the safety and preliminary efficacy of TSHA-102 in pediatric females with Rett syndrome due to MECP2 loss-of-function mutation. The trial is taking place in the United States. We submitted a CTA to the United Kingdom’s Medicines and Healthcare Products Regulatory Agency, or MHRA, for pediatric patients with Rett syndrome and submitted an IND application for pediatric patients with Rett syndrome to the FDA for TSHA-102 early in the third quarter of 2023.
23
In August 2023, we received clearance from the FDA on our IND for TSHA-102 in pediatric patients with Rett syndrome and dosed the first Rett syndrome pediatric patient in December 2023. In February 2024, the IDMC provided clearance to dose the second pediatric patient following review of initial clinical data from the six-week post-treatment assessment from the first pediatric patient dosed. We dosed the second patient in cohort 1 in the first quarter of 2024. We expect to report initial available safety and efficacy data from cohort 1 (low dose, 5.7x1014 total vg) in mid-2024. We expect to report initial available safety and efficacy data from cohort 2 (high dose, 1x1015 total vg) in the second half of 2024.
We have received orphan drug designation and rare pediatric disease designation from the FDA and orphan drug designation from the European Commission for TSHA-102 for the treatment of Rett syndrome. We also received Fast Track Designation from the FDA for TSHA-102 for the treatment of Rett syndrome. We also received CTA clearance from the MHRA in early 2024. In February 2024, we received Innovative Licensing and Access Pathway, or ILAP, designation for TSHA-102 from the U.K. MHRA. The ILAP aims to facilitate patient access to novel treatments by accelerating time to market through opportunities for enhanced engagements with U.K. regulatory authorities and other stakeholders. In April 2024, the FDA granted Regenerative Medicine Advanced Therapy (RMAT) designation for TSHA-102 in Rett syndrome. RMAT designation follows the FDA’s review of available safety and efficacy data from the first three patients with Rett syndrome dosed with the low dose of TSHA-102 (5.7x1014 total vg) across the REVEAL Phase 1/2 adolescent and adult trial and the REVEAL Phase 1/2 pediatric trial. RMAT designation was designed to expedite the development and review of regenerative medicine therapies. A regenerative medicine therapy is eligible for RMAT designation if it is intended to treat, modify, reverse or cure a serious condition, and preliminary clinical evidence indicates the therapy has the potential to address unmet medical needs for such condition. Sponsor companies receiving RMAT designation can benefit from increased interactions with the FDA involving senior managers, with the goal of expediting drug development.
We have a limited operating history. Since our inception, our operations have focused on organizing and staffing our company, business planning, raising capital and entering into collaboration agreements for conducting preclinical and clinical development activities for our product candidates. Our lead product candidate is still in the clinical stage. We do not have any product candidates approved for sale and have not generated any revenue from product sales. Through March 31, 2024, we have funded our operations primarily through: (i) the sale of equity, raising an aggregate of $589.0 million of gross proceeds from our initial public offering, or the IPO, sales of common stock pursuant to our Sales Agreement (as defined below), our October 2022 follow-on offering and our 2023 private placements; (ii) pre-IPO private placements of our convertible preferred stock; (iii) our Term Loan Agreement (as defined below) and subsequently the Trinity Term Loan Agreement (as defined below); and (iv) the Astellas Transactions (as defined below).
On November 13, 2023, or the Trinity Closing Date, we entered into a Loan and Security Agreement, or the Trinity Term Loan Agreement, by and among us, the lenders party thereto from time to time, or the Trinity Lenders, and Trinity Capital Inc., as administrative agent and collateral agent for the Trinity Lenders, or Trinity. The Trinity Term Loan Agreement provides for, on the Trinity Closing Date, $40.0 million aggregate principal amount of term loans, or, collectively, the Trinity Term Loans. We drew the Trinity Term Loans in full on the Trinity Closing Date. The proceeds of the Trinity Term Loans were used to repay our obligations under the Loan and Security Agreement, or the Term Loan Agreement, with the lenders party thereto from time to time, or the Lenders and Silicon Valley Bank, as administrative agent and collateral agent for the Lenders, or the Agent, in full. The Term Loan Agreement with Silicon Valley Bank was terminated concurrently with entry into the Trinity Term Loan Agreement.
Since our inception, we have incurred significant operating losses. Our net losses were $24.1 million for the three months ended March 31, 2024 and $17.6 million for the three months ended March 31, 2023. As of March 31, 2024, we had an accumulated deficit of $537.1 million. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate that our expenses will increase significantly in connection with our ongoing activities, as we:
24
Our Pipeline
We are focused on discovering, developing and commercializing gene therapies for the treatment of monogenic diseases of the CNS, in both rare and large patient populations. Our primary focus is advancing our lead TSHA-102 clinical program in Rett syndrome, while our pipeline of CNS programs offers the potential for additional development opportunities in the future. The stage of development of our Rett syndrome program, including the progress in our ongoing clinical trials, is represented in the below table:
As of the date of this report, we have deprioritized the company-sponsored evaluation of certain clinical-stage programs, including TSHA-120 for GAN, TSHA-105 for SLC13A5, TSHA-118 for CLN1 and TSHA-121 for CLN7, and are seeking external strategic options to potentially enable further development of these programs.
TSHA-102 for Rett Syndrome
TSHA-102 is a self-complementary intrathecally delivered AAV9 gene transfer therapy in clinical evaluation for Rett syndrome, a rare progressive neurodevelopmental disorder caused by mutations in the X-linked MECP2 gene encoding methyl CpG-binding protein 2, or MeCP2, which is essential for regulating neuronal and synaptic function in the brain. The disorder is characterized by loss of communication and hand function, slowing and/or regression of development, motor and respiratory impairment, seizures, intellectual disabilities and shortened life expectancy. Rett syndrome progression is divided into four key stages, beginning with early onset stagnation at 6 to 18 months of age followed by rapid regression, plateau and late motor deterioration. Rett syndrome primarily occurs in females and is one of the most common genetic causes of severe intellectual disability.
Designed as a one-time treatment, TSHA-102 aims to address the genetic root cause of the disease by delivering a functional form of MECP2 to cells in the CNS. The vector is delivered directly to the cerebrospinal fluid via intrathecal administration, which facilitates optimal biodistribution and cell transduction within key regions of the CNS. Because of the risks associated with both under- and over-expression of MeCP2, we have combined high-throughput microRNA, or miRNA, profiling and genome mining to create miRNA-Responsive Auto-Regulatory Element, or miRARE, our novel miRNA target panel. The miRARE element includes binding sites for endogenous miRNA, which are responsive to MeCP2 levels to prevent overexpression. By utilizing the miRARE technology, TSHA-102 is designed to mediate levels of MeCP2 in the CNS on a cell-by-cell basis without risk of overexpression. By increasing MECP2 levels in MECP2 deficient cells and maintaining healthy levels of MECP2 output of healthy cells, TSHA-102 has demonstrated the ability to produce and maintain safe transgene expression levels in the CNS. (Sinnet, SE, et al. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain. 2021 awab182.)
Currently, there are no approved disease-modifying therapies that treat the genetic root cause of Rett syndrome, and there is a significant unmet medical need. According to the Rett Syndrome Research Trust, Rett syndrome affects more than 350,000 patients
25
worldwide. The estimated addressable patient population with typical Rett syndrome caused by a pathogenic/likely pathogenic MECP2 mutation is between 15,000 and 20,000 patients in the United States, European Union and United Kingdom.
Phase 1/2 REVEAL Clinical Trials
We currently have two Phase 1/2 clinical trials ongoing for TSHA-102: an adolescent/adult study in the United States and Canada and a pediatric study in the United States. In addition, approval has been granted to open a pediatric study in the U.K. The trials are described below:
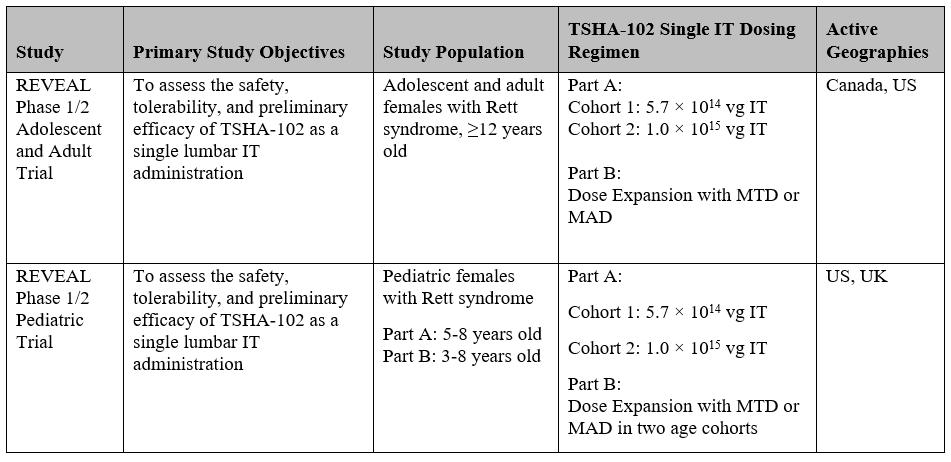
IT – Intrathecal; MTD – Maximum Tolerated Dose; MAD – Maximum Administered Dose.
We dosed the first adult patient with Rett syndrome in May 2023. The second adult patient was dosed in September 2023. We dosed the first pediatric patient with Rett syndrome in the Phase 1/2 REVEAL pediatric trial in December 2023 and the second pediatric patient was dosed in the first quarter of 2024. In early 2024, we announced that the U.K. MHRA authorized the CTA for TSHA-102 in pediatric patients with Rett syndrome, enabling expansion of our ongoing pediatric trial into the United Kingdom. In February 2024, we announced the expansion of the ongoing REVEAL Phase 1/2 adolescent and adult trial in Canada into the United States following submission of the adolescent and adult trial protocol to the FDA.
Cohort 1 of both trials is evaluating the low dose of TSHA-102 of 5.7x1014 total vg. Two adult patients have been dosed in cohort 1 in the adolescent and adult trial, and TSHA-102 showed a well-tolerated safety profile with no treatment-emergent serious adverse events as of the week 35 post-treatment assessment for the first adult patient and as of the week 19 post-treatment assessment for the second adult patient. Following review of available clinical data from the first two dosed adult patients and first dosed pediatric patient showing that TSHA-102 was generally well-tolerated, and in light of the potential for improved benefit at the higher dose (1x1015 total vg), in February 2024, the IDMC approved our request to proceed to earlier dose escalation in the adolescent and adult trial, enabling earlier advancement to cohort 2 evaluating 1x1015 total vg. Dosing in cohort 1 of the adolescent and adult trial is now considered complete. We have enrolled the first patient in cohort 2 of the adolescent and adult trial and scheduled dosing for the second quarter of 2024. The IDMC also approved the dosing of the second patient in cohort 1 of the pediatric trial, and the second pediatric patient was dosed in the first quarter of 2024.
We expect to report initial available safety and efficacy data from cohort 1 of the pediatric trial and an update on available safety and efficacy data from completed cohort 1 (low dose, 5.7x1014 total vg) of the adolescent and adult trial in mid-2024. We expect to report initial available safety and efficacy data from cohort 2 (high dose, 1x1015 total vg) of both the adolescent and adult trial and the pediatric trial in the second half of 2024.
26
The maximum tolerated dose or maximum administered dose established in Part A will be administered during dose expansion in Part B. Data from Part A will be assessed by regulatory agencies and the IDMC to determine key elements of Part B of the study, including efficacy endpoints, study duration and the MTD or MAD.
TSHA-102 REVEAL Adolescent / Adult Clinical Trial TSHA-102-CL-101 Safety and Efficacy Summary
Efficacy endpoints include patient assessments performed by clinicians using the Clinical Global Impressions Scale – Improvement, or CGI-I, the Clinical Global Impressions Scale – Severity, or CGI-S, Rett Syndrome Hand Function Scale, or RSHFS, Vineland Adaptive Behavior Scales Third Edition, and Revised Motor Behavior Assessment, or R-MBA. Additional efficacy endpoints also include patient assessments by caregivers using Parental Global Impressions Improvement, or PGI-I, the Rett Syndrome Behavior Questionnaire, or RSBQ, Seizure Diaries and other clinical assessment scales.
The first adult patient has the most advanced stage of Rett syndrome, Stage IV, with a genetic change consisting of a large deletion within the MECP2 gene that is known to cause Rett syndrome. This patient's phenotypic manifestation is severe, having lost all abilities to: walk, stand, and sit without support around age eight (non-ambulatory, wheelchair bound, limited movements of her lower extremities), use her hands around age six (unable to grasp and hold objects of any size) and speak around age two (non-verbal, minimal vocalizations). Per the Principal Investigator, or PI, the first adult patient’s baseline reported seizure frequency was approximately two to four seizures per year. After TSHA-102 administration, the first adult patient has showed a well-tolerated safety profile with no treatment-emergent serious adverse events as of the 35-week safety assessment post-treatment. Per the protocol, prophylactic immunosuppressant therapy began seven days prior to TSHA-102 administration. The first adult patient’s steroid taper was initiated on week 17 and was completed by week 33. At week 25, the patient demonstrated sustained and new improvements across key efficacy assessments at decreased steroid levels compared to earlier post-treatment assessments and the patient was subsequently at physiologic levels within a few days post the week 25 assessment.
As of 35 weeks post-treatment, following completion of the steroid taper, the PI noted that the first adult patient’s improvements observed across multiple clinical domains had been sustained with new improvements as well. These include sustained improvements in motor function, with the gained ability to kick legs against gravity and sit unassisted for the first time in over one decade, including after the steroids have been fully tapered from the patient at week 35 post-treatment, as supported by video evidence. Further sustained improvements from the first adult patient were also observed in motor function, including improved hand function as the patient’s hands were more open, and the improved ability to grasp and hold objects. The PI also observed new improvements in the patient’s socialization/communication skills at week 35 post-treatment, as the patient was more alert and interactive during the day, made more vocalizations, and showed the enhanced ability to use her eye-driven communication device, which caregivers reported she had not expressed interest in before treatment. Specifically, at week 35 post-treatment, the patient was able to use the eye-driven device much more efficiently, with the gained ability to activate functions on the screen of the device. Difficulty in communication, including loss of speech, is one of the most prominent symptoms of Rett syndrome and a key area of concern for caregivers as it interferes with patients’ ability to communicate their needs and express their interests. Further, the first adult patient showed sustained improvements in autonomic function at week 35 post-treatment, including sustained normalized sleep/night-time behaviors for the first time in twenty years, improved breathing patterns with fewer breath holding spells and infrequent hyperventilation compared to before treatment, and improved circulation with hands and feet at a more normal temperature and color. Finally, the PI observed that the patient’s seizures had been overall well controlled through week 35 following treatment at lower levels of anti-seizure medication, relative to baseline, and the patient no longer experienced unprovoked seizures. These observations are supported by data from the Seizure diaries. The PI’s clinical observations are supported by clinical and video evidence as well as caregiver-reported seizure diaries.
The second adult patient has the most advanced stage of Rett syndrome, Stage IV, with a missense mutation in the MECP2 gene, which has been reported in over 25 publications to cause Rett syndrome. This patient's phenotypic manifestation is milder than the first adult patient, with partial loss of ambulation (able to walk/stand without support, wide based, slow, unsteady gait) and hand function (with significant stereotypies that emerged by age three, inability to transfer objects between hands). She has been nonverbal since the age of two years old. Per the PI, the second adult patient’s baseline reported seizure frequency was approximately two to four seizures per week. After TSHA-102 administration, the patient has showed a well-tolerated safety profile with no treatment-emergent serious adverse events as of the 19-week safety assessment post-treatment. Tapering of the steroids was initiated on week 17 and is expected to be complete by week 25.
As of 19-weeks post-treatment, the PI noted that the second adult patient’s improvements observed across multiple clinical domains had been maintained with new improvements at decreased steroid levels relative to earlier post-treatment assessments. These include sustained improvement in motor function, with the patient’s hands more open and relaxed, and improved hand stereotypies, with less forceful hand wringing. These observations from the PI are supported by video evidence. The second adult patient also showed sustained improvements in social/communication skills as she was more interested, engaged and alert, including showing increased response to spoken words and eye contact, as supported by improved social skills on the clinician administered R-MBA scale. The second adult patient also showed sustained improvements in autonomic function, including improved breathing patterns with fewer breath holding spells and infrequent hyperventilation compared to before treatment, and improved circulation, with hands and feet at a more normal temperature and color. Finally, the second adult patient showed pronounced improvements in seizure
27
frequency at week 19 post-treatment, with a significant reduction in seizures at 25% lower levels of anti-seizure medication, relative to baseline. The PI noted that the patient’s epilepsy had been much better controlled following treatment at a lower dose of anti-seizure medication, and has been seizure-free for 17 weeks as of the week 19 post-treatment time point, despite a pre-treatment seizure frequency of approximately two to four seizures per week. The PI’s clinical observations are supported by clinical and video evidence as well as caregiver-reported seizure diaries.
TSHA-102-CL-101 Trial Adult Patient 1 Efficacy Data
The first adult patient showed clinically significant improvement in CGI-S from score of six (severely ill) at baseline to score of five (markedly ill) in this measure four weeks post-TSHA-102 administration and this improvement was sustained through week 25. Similarly, the patient demonstrated sustained improvement in CGI-I and PGI-I as of week 25 assessment post-TSHA-102 administration with scores of two (much improved and much better for CGI-I and PGI-I, respectively).
The first adult patient dosed with TSHA-102 demonstrated a sustained clinical improvement in RSBQ Total Score at week 25 post-TSHA-102 administration as depicted in the chart below.
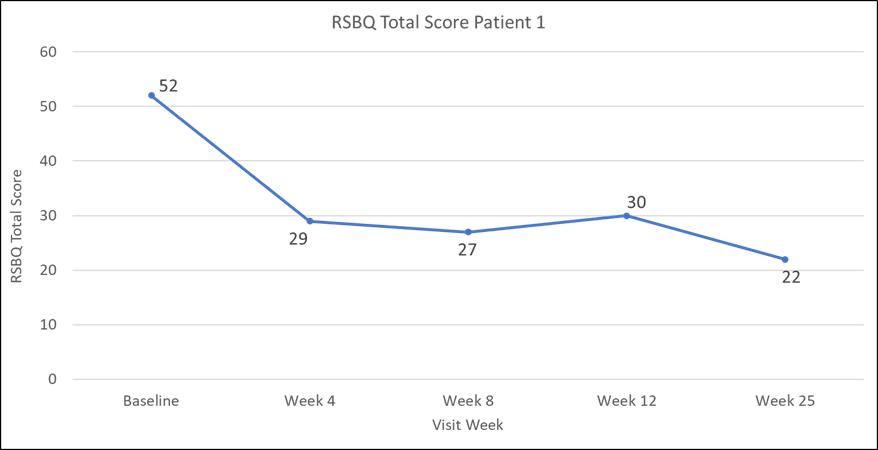
Compared to baseline, a 30-point improvement was observed in RSBQ total score at week 25 post TSHA-102 administration. RSBQ changes at week 25 were driven by improvements in hand behaviors, breathing problems, general mood, repetitive face movements, night-time behaviors, fear/anxiety, and body rocking/expressionless face.
28
As shown in the diagram below, the first adult patient started to show notable overall improvements in R-MBA total scores at the week-12 visit. However, the total R-MBA score increased closer to baseline at the week 25 visit where improvements were demonstrated in motor dysfunction and respiratory behaviors.
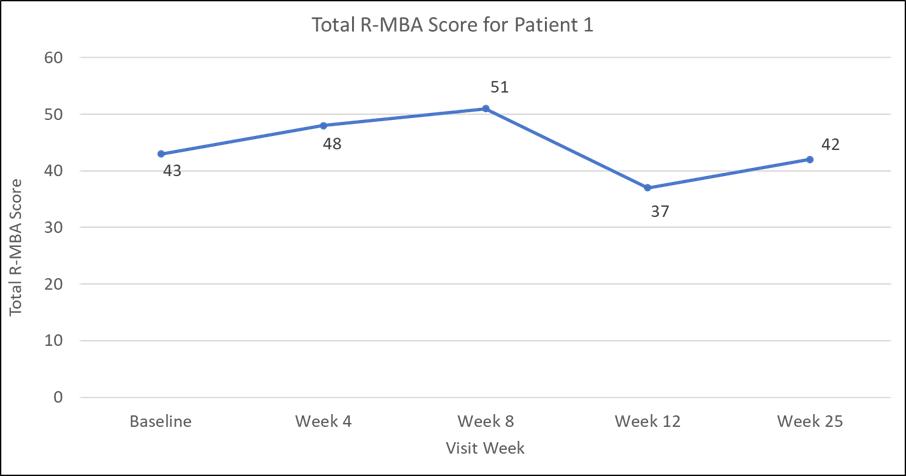
The first adult patient has demonstrated stable seizure events relative to baseline through week 35 post-treatment, based on caregiver-reported medical history, and seizures are confined to periods where phenytoin level declines to <50 μmol/L (previously <100 μmol/L). The first adult patient has been on phenytoin as antiepileptic therapy, which she has continued following treatment with TSHA-102. Prior to treatment and per medical history, the patient required phenytoin levels of >100 µmol/L to control her seizures. The first adult patient had seizures prior to TSHA-102 administration on Day –8 and Day –7, and post-administration she had seizures on Days 45-49 and Day 82 associated with lower than target phenytoin levels. Specifically, the seizures on Days 45-49 corresponded with a phenytoin level of 45.9 µmol/L, and the seizure on Day 82 corresponded with a phenytoin level was 35.9 umol/L.
Loss of hand function is a hallmark characteristic of Rett syndrome and a key area of concern for caregivers. It impacts a patient’s ability to communicate and impedes daily activities, which ultimately limits independence. The RSHFS is a scale designed to evaluate hand function in patients with Rett syndrome. Hand function is evaluated by an experienced independent physical therapist with expertise in evaluating hand function in patients with Rett syndrome. Sessions are videotaped in which the patient’s caregiver offers the patient both large (e.g. a toy, cup, or spoon) and small (e.g. a grape or small piece of sandwich) objects so that she may demonstrate her ability to grasp, pick up, and hold the objects. The independent physical therapist then codes the demonstrated hand function in each video at one of four levels of hand function, ranging from no active grasping of any objects to independent grasping, for the best level for large objects assessment.
The first adult patient showed an improvement in RSHFS at 25 weeks post-TSHA-102 administration as depicted in the tables below. As of week 25 following treatment, the first adult patient is using her non-dominant hand for some basic grasping whereas before treatment, she was not able to grasp at all. As of the week 25 assessment, her dominant hand function improved from
29
baseline with the demonstrated ability to grasp of two different objects (spoon and toy) rather than just one object (spoon). These clinical observations reported by the independent physical therapist are supported by video evidence.
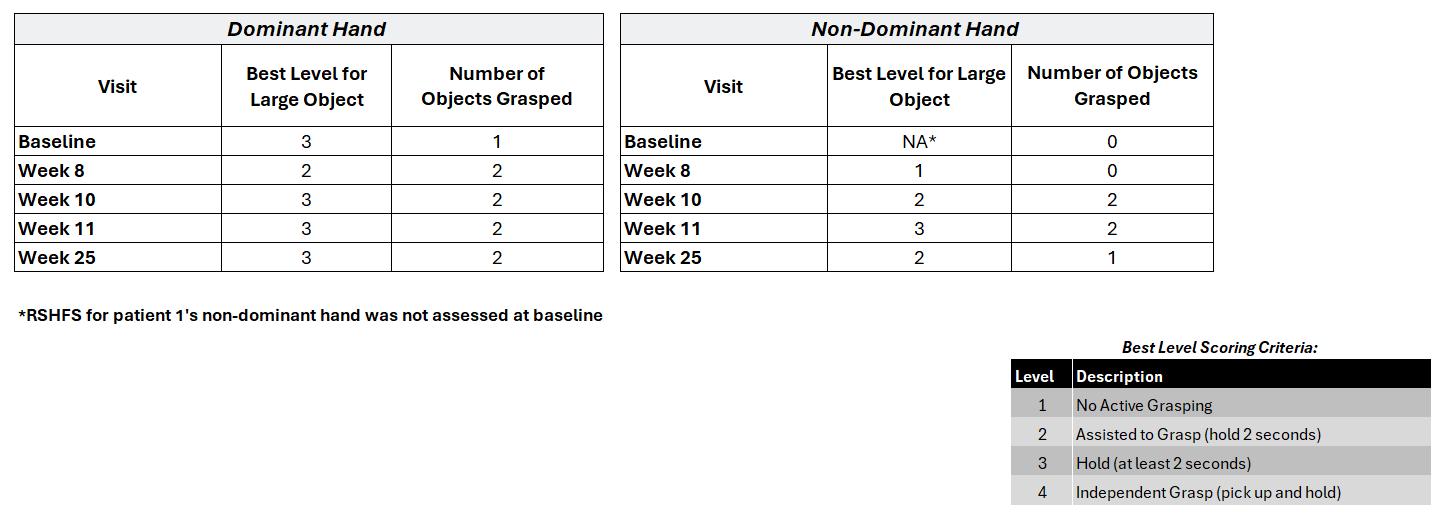
TSHA-102-CL-101 Trial Adult Patient 2 Efficacy Data
While the two adult patients dosed to date in our REVEAL trial both have the most advanced stage of Rett syndrome, Stage, IV, they possess different genetic backgrounds and mutation types, which manifest in different phenotypes and clinical severity.
While there was no change at 12 weeks post TSHA-102 administration in the second adult patient’s CGI-S score of four (moderately ill) at baseline, her CGI-I and PGI-I scores show sustained improvement (score of three, minimally better and a little better, respectively) at week 12.
The second adult patient showed a sustained clinical improvement in RSBQ Total Score 12 weeks post-TSHA-102 administration as depicted in the chart below.
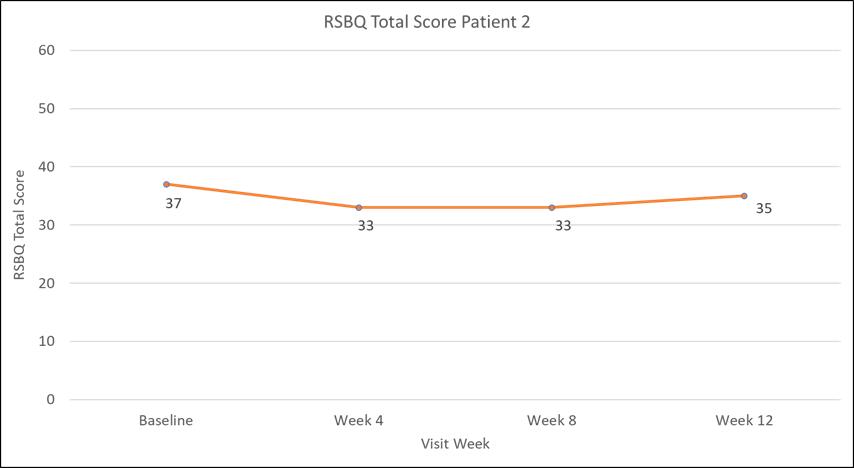
Improvements were documented in several subdomains. Specifically, compared to baseline, improvements were noted in breathing, body rocking, facial expressions, general mood, and repetitive face movements.
30
The second adult patient showed continued improvement in the R-MBA Total Score at 12 weeks post-TSHA-102 administration as depicted in the graph below.
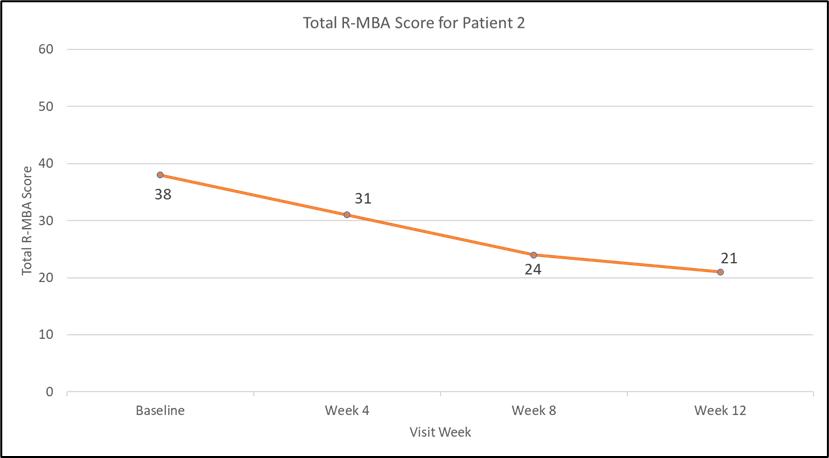
A 17-point improvement at week 12 was demonstrated in the R-MBA Total Score. Most notable improvements were documented in the following subscales: social skills, respiratory behaviors, seizures, truncal rocking and stereotypic hand movements and/or mouthing, and aberrant behaviors.
Seizure diary demonstrated reduced seizure events relative to baseline through 19-weeks post-treatment for the second adult patient at lower levels of anti-seizure medication, based on caregiver-reported medical history. Pre-treatment, the second adult patient had approximately two to four seizures per week, and there has been a significant reduction in seizures post-treatment with TSHA-102 at 25% lower levels of anti-seizure medication, relative to baseline. Post-treatment, the second adult patient had a single seizure event on day 13 as of week 19 post-treatment. The seizure was an unknown type, with motor manifestations and lasted less than one minute duration. The patient has been seizure-free for 17 weeks as of the week 19 post-treatment time point.
Hand function in the dominant hand for the second adult patient is challenging to interpret due to inconsistency in the video recording. At the week 8 post-treatment assessment, the second adult patient’s dominant hand received a hand function score of four, an independent grasp (pick up and hold) and was able to grasp three objects. Hand function in the second adult patient remained unchanged in the non-dominant hand on the RSHFS 12 weeks post-TSHA-102 treatment. These clinical observations reported by the independent physical therapist are supported by video evidence.
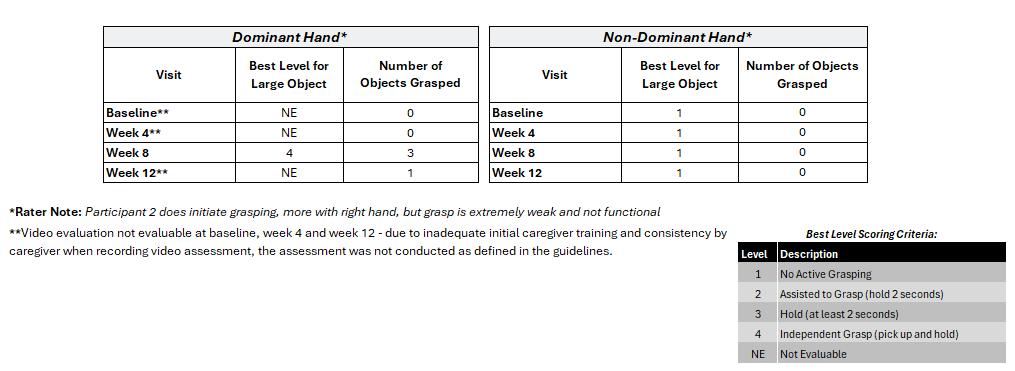
31
Deprioritized Programs
We have previously deprioritized the evaluation of our clinical product candidates TSHA-120 for GAN, TSHA-105 for SLC13A5, TSHA-118 for CLN1 and TSHA-121 for CLN7. Although we are not currently evaluating the potential of TSHA-105, TSHA-118 and TSHA-121, we may again evaluate any of these in the future as a product candidate as a component of our pipeline expansion plans, or pursue partnerships to advance these programs.
TSHA-120 for Giant Axonal Neuropathy (GAN)
GAN is an ultra-rare autosomal recessive, progressive neurodegenerative disease of the central, peripheral and autonomic nervous systems caused by deficiency or complete loss-of-function of gigaxonin and the accumulation of intermediate filaments. Epidemiology studies indicate there are between 1,000 and 1,500 treatable GAN patients in the United States, European Union and United Kingdom.
There is an early (classical) and late-onset (non-classical) phenotype associated with the disease, with shared pathophysiology due to accumulation of intermediate filaments. Symptoms and features of children with classical GAN usually develop before the age of five years with distal muscle weakness and sensory loss due to axonal sensory motor neuropathy, manifesting as bilateral foot drop and difficulties with fine motor coordination. An abnormal, wide based, unsteady gait due to CNS and cerebellar involvement is also a common initial clinical manifestation. Children with the classical phenotype typically have dull, tightly curled, coarse hair (“kinky” hair), “giant” axons pathognomonic on a nerve biopsy due to accumulation of intermediate filaments, and progressive spinal cord atrophy and white matter abnormalities, initially around the cerebellar dentate nucleus, on MRI images. Symptoms progress and, as the children grow older, they develop progressive proximal muscle weakness, resulting in difficulties raising their arms and standing from the floor or a chair, scoliosis, distal contractures, progressive gait and limb ataxia, leading to loss of ambulation by the second decade. Progressive optic nerve atrophy, seen early in the disease, results in increasing deterioration of visual acuity in later stages and has been more recently described. Indeed, decreased visual acuity was seen at baseline in approximately half of GAN patients aged 3-21 years, enrolled in a natural history study [Brain. 2021 Nov 29;144(10):3239-3250]. Due to increased respiratory muscle weakness and restrictive respiratory failure as a result of severe scoliosis, assisted ventilation is required in adolescents. GAN patients often die during their late teens or early twenties, typically due to respiratory failure.
The late-onset, or non-classical, phenotype is often categorized as Charcot-Marie-Tooth Type 2, or CMT2, as it presents as a typical early onset axonal sensory motor neuropathy without the typical kinky hair and CNS involvement of the classical phenotype and has a relatively slow progression. This phenotype might represent up to 6% of all CMT2 diagnosis. In the late-onset population, patients have poor quality of life and significantly compromised activities of daily living. The disease is life limiting but not as severely as classic GAN. In classic GAN, symptomatic treatments attempt to maximize physical development and minimize the rate of deterioration. Currently, there are no approved disease-modifying therapies available, only palliative treatments.
In March 2021, we acquired the exclusive worldwide rights to a clinical-stage, intrathecally dosed AAV9 gene therapy program, now known as TSHA-120, for the treatment of GAN, pursuant to a license agreement with Hannah’s Hope Fund for Giant Axonal Neuropathy, Inc., or HHF. Under the terms of the agreement, HHF received an upfront payment of $5.5 million and will be eligible to receive clinical, regulatory and commercial milestones totaling up to $19.3 million, as well as a low, single-digit royalty on net sales upon commercialization of TSHA-120. We received orphan drug designation and rare pediatric disease designation from the FDA for TSHA-120 for the treatment of GAN. In April 2022, we received orphan drug designation from the European Commission for TSHA-120 for the treatment of GAN.
In September 2023, subsequent to the receipt of Type C meeting feedback from the FDA regarding a registrational path for TSHA-120, we announced that we discontinued the development of our TSHA-120 program in the evaluation for the treatment of GAN. In January 2024, we initiated the transfer of the FDA IND application and investigational clinical trial material for TSHA-120 in GAN to clinical trial collaborator NINDS, creating an opportunity for continued clinical evaluation of TSHA-120 in GAN. Additionally, we have entered discussions with the originating advocacy organization regarding TSHA-120 in an effort to transfer rights back to the advocacy organization to move the program forward.
TSHA-118 for CLN1 Disease
CLN1 disease (one of the forms of Batten disease), a lysosomal storage disorder, is a progressive, fatal neurodegenerative disease with early childhood onset that has an estimated incidence of approximately 1 in 138,000 live births worldwide. The estimated prevalence of CLN1 disease is 1,000 patients in the United States and European Union. CLN1 disease is caused by loss-of-function mutations in the CLN1 gene that encodes the enzyme palmitoyl-protein thioesterase-1, a small glycoprotein involved in the
32
degradation of certain lipid-modified proteins. Loss of function mutations in the CLN1 gene causes accumulation of these lipid-modified proteins in cells, eventually leading to aggregation, neuronal cellular dysfunction and ultimately neuronal cell death.
In the infantile-onset form of CLN1 disease, clinical symptoms appear between six to 24 months and include rapid deterioration of speech and motor function, refractory epilepsy, ataxia and visual failure. Infantile-onset CLN1 patients are typically poorly responsive by five years of age and remain noncommunicative until their death, which usually occurs by seven years of age. Late-infantile-onset CLN1 disease begins between two to four years of age with initial visual and cognitive decline followed by the development of ataxia and myoclonus, or quick, involuntary muscle jerks. Juvenile-onset CLN1 disease patients present between the ages of five to ten years old, with vision loss as a first symptom followed by cognitive decline, seizures and motor decline. Approximately 60% of the children diagnosed with CLN1 disease in the United States present with early-onset infantile forms, with the remaining 40% experiencing later-onset childhood forms.
All currently available therapeutic approaches for patients with CLN1 disease are targeted towards the treatment of symptoms, and no disease-modifying therapies have been approved. Gene therapy has shown promise in correcting forms of neuronal ceroid lipofuscinoses diseases that involve mutations in soluble enzymes, in part, due to cross-correction of neighboring non-transduced cells.
We believe that the introduction of a functional CLN1 gene using an AAV9 vector delivered intrathecally to the CNS offers the potential of a disease-modifying therapeutic approach for this disease. TSHA-118 is a self-complementary AAV9 viral vector that expresses human codon-optimized CLN1 complementary deoxyribonucleic acid under control of the chicken ß-actin hybrid promoter. We acquired exclusive worldwide rights to certain intellectual property rights and know-how relating to the research, development and manufacture of TSHA-118 (formerly ABO-202) in August 2020 pursuant to a license agreement with Abeona Therapeutics Inc., or Abeona.
TSHA-118 has been granted orphan drug designation, rare pediatric disease designation and fast track designation from the FDA and orphan drug designation from the European Medicines Agency for the treatment of CLN1 disease.
There is currently an open IND for the CLN1 program. We submitted a CTA filing for TSHA-118 which was approved by Health Canada in 2021. Clinical trial material has been manufactured and released and is now ready for use in a clinical trial setting. We provided investigational clinical trial material for TSHA-118 in CLN1 to support an individual-patient investigator-initiated IND request from RUSH University Medical Center for the treatment of a patient with CLN1 disease.
TSHA-105 for SLC13A5 Deficiency
TSHA-105 is a gene replacement therapy in development for the treatment of SLC13A5 deficiency, a rare autosomal recessive epileptic encephalopathy characterized by the onset of seizures within the first few days of life. SLC13A5 deficiency is caused by bi-allelic loss-of function mutations in the SLC13A5 gene, which codes for a sodium dependent citrate transporter, or NaCT, that is largely expressed in the brain and liver. To date, all tested mutations result in no or a greatly reduced amount of the citrate in the cells. Diminished NaCT function leads to loss of neuronal uptake of citrate and other metabolites such as succinate that are critical to brain energy metabolism and function. Affected children have impairments in gross motor function and speech production with relative preservation of fine motor skills and receptive speech. Currently, there are no approved therapies for SLC13A5 deficiency, and treatment is largely to address symptoms. The estimated prevalence of SLC13A5 deficiency is 1,900 patients in the United States and European Union.
TSHA-105 is constructed from a codon-optimized human SLC13A5 gene packaged in a self-complementary AAV9 capsid.
We have received orphan drug designation and rare pediatric disease designation from the FDA and orphan drug designation from the European Commission for TSHA-105 for the treatment of epilepsy caused by SLC13A5 deficiency. Clinical trial material has been manufactured and released and is now ready for use in a clinical trial setting.
Other Programs
We have a pipeline of early-stage gene therapy programs targeting CNS diseases that we may progress in the future or advance through potential partnerships.
TSHA-113 for Tauopathies
We are developing TSHA-113 for the treatment of tauopathies. Tauopathies comprise a large subset of neurodegenerative diseases involving the aggregation of microtubule associated protein tau, or MAPT, protein into neurofibrillary or gliofibrillary tangles in the human brain. These include MAPT-associated frontotemporal dementia, or FTD, progressive supranuclear palsy, or PSP,
33
corticobasal degeneration, or CD, and Alzheimer’s disease. There are an estimated 11,000 patients in United States and Europe affected by MAPT mediated FTD and 2,000 to 2,500 are affected with MAPT-mediated PSP. and CD, and Alzheimer’s disease affects an estimated 6.2 million Americans and 7.8 million Europeans.
Intrathecal delivery of an antisense oligonucleotide, or ASO, targeting Tau mRNA by Biogen/Ionis in a Phase 1 study demonstrated durable, robust, time and dose dependent lowering of tau protein and phospho-tau in cerebrospinal fluid of Alzheimer’s disease patients. Buoyed by these results, in August 2022, Biogen started a Phase 2 trial in people with mild cognitive impairment or mild dementia due to Alzheimer’s disease. This ASO target validation paved the way for other approaches targeting intercellular tau mRNA (reduce tau protein production), for treating Tauopathies.
Unlike an ASO treatment, which would require repeat lifelong administration, we are developing a one-time treatment for Tauopathies. TSHA-113 is an AAV9 capsid that packages a tau-specific miRNA and is delivered in the cerebrospinal fluid for the treatment of tauopathies. This miRNA targets all six isoforms of tau mRNA.
We tested the efficacy of TSHA-113 in PS19 mice, a validated mouse model for tauopathies. These mice express human MAPT, and they exhibit significant tau pathology, neurodegeneration, loss of body weight and progressive hind-limb paralysis around nine to 12 months of age. We tested efficacy of our treatment by delivering TSHA-113 to PS19 mice at three months, six months and nine months of age via intracisterna magna injection. We found that the tau mRNA and protein levels were significantly reduced by TSHA-113 treatment. Consistently, the tau seeding assay showed reduced levels of pathological tau in brains from PS19 mice treated with TSHA-113. In addition, TSHA-113 treatment was able to rescue the survival rate, loss in body weight, and the hind limb clasping phenotype in the PS19 mice when treated at three months, six months and nine months of age. Taken together, these results demonstrate that a one-time, vectorized delivery of a tau-specific miRNA is a promising approach for treatment for tauopathies. Ongoing and future work is focused on optimal dose determination for IND-enabling studies.
TSHA-106 for Angelman syndrome
We are developing TSHA-106 for the treatment of Angelman syndrome, a neurodevelopmental disorder caused by a maternal deficiency of the UBE3A gene. Angelman syndrome is characterized by profound developmental delay, ataxia and gait disturbance, sleep disorder, seizures, heightened anxiety, aggression and severe speech impairments. Angelman syndrome affects approximately one per 12,000 to 20,000 patients worldwide.
Angelman syndrome is an imprinting disorder in which the maternal gene is deficient and the paternal copy of UBE3A is intact but silenced by a long non-coding RNA, UBE3A antisense transcript, or UBE3A-ATS. Delivery of an ASO targeting UBE3A-ATS showed promising results in ameliorating Angelman syndrome symptoms in a transgenic mouse model.
We have in-licensed a novel gene replacement therapy from University of North Carolina. This novel construct is designed to express two isoforms of UBE3A mRNA from the same codon optimized transgene cassette and could potentially be a one-time treatment for the disease. The unique design feature allows short and long hUBE3A isoforms expression at a near-endogenous 3:1 (short/long) ratio, a feature that could help to support optimal therapeutic outcomes. Additionally, this construct uses human Synapsin 1 promoter, to limit UBE3A expression primarily in neurons, the primary therapeutic target for treating Angelman syndrome.
In a published study, this dual isoform expressing cassette was packaged into PHP.B capsids and administered by intracerebroventricular injections in neonatal mice models. This treatment significantly improved motor learning and innate behaviors in Angelman syndrome mice (PMID: 34676830). It rendered Angelman syndrome mice resilient to epileptogenesis and associated hippocampal neuropathologies induced by seizure kindling. These results demonstrated the feasibility, tolerability, and therapeutic potential for dual-isoform hUBE3A gene transfer in the treatment of AS in mice.
To advance these findings into translatable interventions, our collaborators packaged the dual isoform expressing cassette into AAV9 capsids and undertook animal proof of concept studies. Overall, these results are highly consistent with the published data describing neonatal ICV delivery of a similar dose of the PHP.B/hUBE3Aopt vector (PMID: 34676830) and support continued development. Ongoing and future work is focused on optimal dose and route of administration determination for IND enabling studies.
There are an estimated 55,000 patients with Angelman syndrome in the United States and Europe.
TSHA-114 for Fragile X Syndrome
We are developing TSHA-114 for the treatment of Fragile X syndrome, the most common single gene cause of autism and cognitive impairment, affecting about one in 6,000 individuals worldwide. Fragile X syndrome is diagnosed around three years of age and characterized by anxiety, aggression, hyperactivity, attention deficits and sleep and communication disruption.
34
Fragile X syndrome is caused by a pathological expansion of a CGG triplet repeat in the 5’ untranslated region of the FMR1 gene. Expansion of the triplet above the normal 5–55 repeats to 200 or more causes hypermethylation of the gene promoter, and shutdown of transcription and translation of the encoded protein, fragile X mental retardation protein, or FMRP. The expanded repeat also induces formation of RNA: DNA heteroduplexes that induces epigenetic gene silencing. Although most patients with Fragile X syndrome do not express FMRP, some individuals with the full mutation produce low amounts of the protein (less than 10% of normal levels). FMRP expression in unaffected persons varies greatly from person to person. Current pharmacotherapeutic treatments for Fragile X syndrome are solely directed towards symptom relief.
We conducted proof of concept studies in animal models of Fragile X (Fmr1 KO) with TSHA-114. No significant adverse effects were observed in behavioral, serological or pathohistological markers up to 12 months after intrathecal administration of TSHA-114 in wild-type mice. TSHA-114 treated FMRKO showed widespread FMRP expression was observed throughout brain post administration. TSHA-114 treated FMRKO mice showed robust suppression of audiogenic seizures and normalization of fear conditioning behavior. In addition, assessment of circadian locomotor activity revealed restoration of hyperactivity and sleep. Assessment of transgene expression and behavioral responses in individual mice demonstrated correlations between the level of FMRP expression and potential drug efficacy.
The results from the study support continued development. Ongoing and future work is focused on optimal dose and route of administration determination for IND enabling studies.
There are an estimated 75,000 patients with Fragile X syndrome in the United States and Europe.
License Agreements
Research, Collaboration and License Agreement with The University of Texas Southwestern Medical Center
In November 2019, we entered into a research, collaboration and license agreement, or the UT Southwestern Agreement, with The Board of Regents of the University of Texas System on behalf of UT Southwestern, as amended in April 2020.
In connection with the UT Southwestern Agreement, we obtained an exclusive, worldwide, royalty-free license under certain patent rights of UT Southwestern and a non-exclusive, worldwide, royalty-free license under certain know-how of UT Southwestern, in each case to make, have made, use, sell, offer for sale and import licensed products for use in certain specified indications. Additionally, we obtained a non-exclusive, worldwide, royalty-free license under certain patents and know-how of UT Southwestern for use in all human uses, with a right of first refusal to obtain an exclusive license under certain of such patent rights and an option to negotiate an exclusive license under other of such patent rights. We are required to use commercially reasonable efforts to develop, obtain regulatory approval for, and commercialize at least one licensed product.
On April 2, 2020, we amended the UT Southwestern Agreement to include the addition of another licensed product and certain indications, and a right of first refusal to us over certain patient dosing patents. No additional consideration was transferred in connection with this amendment. In March 2022, we and UT Southwestern mutually agreed to revise the payment schedules and current performance expectations of the current sponsored research agreements under the UT Southwestern Agreement and defer payments by fifteen months. In December 2023, we and UT Southwestern mutually agreed to terminate specific sponsored research agreements. There are no outstanding payments due for these terminated programs as of March 31, 2024.
In connection with the UT Southwestern Agreement, we issued to UT Southwestern 2,179,000 shares of our common stock. We do not have any future milestone or royalty obligations to UT Southwestern under the UT Southwestern Agreement, other than costs related to the maintenance of patents.
The UT Southwestern Agreement expires on a country-by-country and licensed product-by-licensed product basis upon the expiration of the last valid claim of a licensed patent in such country for such licensed product. After the initial research term, we may terminate the agreement, on an indication-by-indication and licensed product-by-licensed product basis, at any time upon specified written notice to UT Southwestern. Either party may terminate the agreement upon an uncured material breach of the agreement or insolvency of the other party. In December 2023, we transferred rights to specific indications back to UT Southwestern.
License Agreement with Abeona (CLN1 Disease)
In August 2020, we entered into a license agreement, or the Abeona CLN1 Agreement, with Abeona Therapeutics Inc., or Abeona. In connection with the Abeona CLN1 Agreement, we obtained an exclusive, worldwide, royalty-bearing license, with the right to grant sublicenses under certain patents, know-how and materials originally developed by the University of North Carolina at Chapel Hill and Abeona to research, develop, manufacture, have manufactured, use, and commercialize licensed products for gene therapy for the prevention, treatment, or diagnosis of CLN1 Disease (one of the forms of Batten disease) in humans.
35
Subject to certain obligations of Abeona, we are obligated to use commercially reasonable efforts to develop at least one product and commercialize at least one product in the United States.
In connection with the license grant, we paid Abeona a one-time upfront license fee of $3.0 million during fiscal year 2020. We are obligated to pay Abeona up to $26.0 million in regulatory-related milestones and up to $30.0 million in sales-related milestones per licensed product and high single-digit royalties on net sales of licensed products. Royalties are payable on a licensed product-by-licensed product and country-by-country basis until the latest of the expiration or revocation or complete rejection of the last licensed patent covering such licensed product in the country where the licensed product is sold, the loss of market exclusivity in such country where the product is sold, or, if no licensed product exists in such country and no market exclusivity exists in such country, ten years from first commercial sale of such licensed product in such country. In addition, concurrent with the Abeona CLN1 Agreement, we entered into a purchase and reimbursement agreement with Abeona, pursuant to which we purchased specified inventory from Abeona and reimbursed Abeona for certain research and development costs previously incurred for total consideration of $4.0 million paid in fiscal year 2020.
In December 2021, our CTA filing for TSHA-118 for the treatment of CLN1 disease was approved by Health Canada and therefore triggered a regulatory milestone payment in connection with the Abeona CLN1 Agreement. We recorded $3.0 million within research and development expenses in the consolidated statements of operations for the year ended December 31, 2021. The milestone fee was paid in January 2022 and has been classified as an investing outflow in the condensed consolidated statements of cash flows for the three months ended March 31, 2022. No additional milestone payments were made or triggered during the three months ended March 31, 2024.
The Abeona CLN1 Agreement expires on a country-by-country and licensed product-by-licensed product basis upon the expiration of the last royalty term of a licensed product in such country. Either party may terminate the agreement upon an uncured material breach of the agreement or insolvency of the other party. We may terminate the agreement for convenience upon specified prior written notice to Abeona.
License Agreement with Abeona (Rett Syndrome)
In October 2020, we entered into a license agreement, or the Abeona Rett Agreement, with Abeona pursuant to which we obtained an exclusive, worldwide, royalty-bearing license, with the right to grant sublicenses under certain patents, know-how and materials originally developed by the University of North Carolina at Chapel Hill, the University of Edinburgh and Abeona to research, develop, manufacture, have manufactured, use, and commercialize licensed products for gene therapy and the use of related transgenes for Rett syndrome.
Subject to certain obligations of Abeona, we are required to use commercially reasonable efforts to develop at least one licensed product and commercialize at least one licensed product in the United States.
In connection with the Abeona Rett Agreement, we paid Abeona a one-time upfront license fee of $3.0 million during fiscal year 2020. We are obligated to pay Abeona up to $26.5 million in regulatory-related milestones and up to $30.0 million in sales-related milestones per licensed product and high single-digit royalties on net sales of licensed products. Royalties are payable on a licensed product-by-licensed product and country-by-country basis until the latest of the expiration or revocation or complete rejection of the last licensed patent covering such licensed product in the country where the licensed product is sold, the loss of market exclusivity in such country where the product is sold, or, if no licensed product exists in such country and no market exclusivity exists in such country, ten years from first commercial sale of such licensed product in such country.
In March 2022, our CTA filing for TSHA-102 for the treatment of Rett Syndrome was approved by Health Canada and therefore triggered a regulatory milestone payment of $1.0 million in connection with the Rett Agreement, which was paid in July 2022. In May 2023, we dosed the first patient with TSHA-102 in the Phase 1/2 REVEAL trial evaluating the safety and preliminary efficacy of TSHA-102 in adult patients with Rett syndrome and therefore triggered a milestone payment of $3.5 million in connection with the Rett Agreement, which was paid in August 2023. No additional milestone payments were made during the three months ended March 31, 2024.
The Abeona Rett Agreement expires on a country-by-country and licensed product-by-licensed product basis upon the expiration of the last royalty term of a licensed product in such country. Either party may terminate the agreement upon an uncured material breach of the agreement or insolvency of the other party. We may terminate the agreement for convenience upon specified prior written notice to Abeona.
Option Agreement with Astellas
36
On October 21, 2022, or the Effective Date, we entered into an Option Agreement, or the Option Agreement, with Audentes Therapeutics, Inc. (d/b/a Astellas Gene Therapy), or Astellas.
TSHA-120 Giant Axonal Neuropathy
Under the Option Agreement, we granted to Astellas an exclusive option to obtain an exclusive, worldwide, royalty and milestone-bearing right and license (A) to research, develop, make, have made, use, sell, offer for sale, have sold, import, export and otherwise exploit, or, collectively, Exploit or the Exploitation, the product known, as of the Effective Date, as TSHA-120, or the 120 GAN Product, and any backup products with respect thereto for use in the treatment of GAN or any other gene therapy product for use in the treatment of GAN that is controlled by us or any of our affiliates or with respect to which we or any of our affiliates controls intellectual property rights covering the Exploitation thereof, or a GAN Product, and (B) under any intellectual property rights controlled by us or any of our affiliates with respect to such Exploitation, or the GAN Option. Subject to certain extensions, the GAN Option was exercisable from the Effective Date through a specified period of time following Astellas’ receipt of (i) the formal minutes from the Type B end-of-Phase 2 meeting between us and the FDA in response to our meeting request sent to the FDA on September 19, 2022 for the 120 GAN Product, (ii) all written feedback from the FDA with respect to the Type B end-of-Phase 2 Meeting, and (iii) all briefing documents sent by us to the FDA with respect to the Type B end-of-Phase 2 Meeting. Following the receipt of Type C meeting feedback from the FDA regarding a registrational path for TSHA-120 in September 2023, Astellas elected not to exercise the GAN Option, and we recognized revenue related to this expiration during the third quarter of 2023.
TSHA-102 Rett Syndrome
Under the Option Agreement, we also granted to Astellas an exclusive option to obtain an exclusive, worldwide, royalty and milestone-bearing right and license (A) to Exploit any Rett Product (as defined below), and (B) under any intellectual property rights controlled by us or any of our affiliates with respect to such Exploitation, or the Rett Option. Subject to certain extensions, the Rett Option is exercisable from the Effective Date through a specified period of time following Astellas’ receipt of (1) certain clinical data from the female pediatric trial and (2) certain specified data with respect to TSHA-102, or the Rett Option Period related to (i) the product known, as of the Effective Date, as TSHA-102 and any backup products with respect thereto for use in the treatment of Rett syndrome, and (ii) any other gene therapy product for use in the treatment of Rett syndrome that is controlled by us or any of our affiliates or with respect to which we or any of our affiliates controls intellectual property rights covering the Exploitation thereof, or a Rett Product.
The parties have agreed that, if Astellas exercises an Option, the parties will, for a specified period, negotiate a license agreement in good faith on the terms and conditions outlined in the Option Agreement, including payments by Astellas of a to-be-determined upfront payment, certain to-be-determined milestone payments, and certain to-be-determined royalties on net sales of GAN Products and/or Rett Products, as applicable.
Components of Results of Operations
Revenue
Revenue for the three months ended March 31, 2024 was derived from the Astellas Transactions. We recognize revenue as research and development activities related to our Rett program are performed. Revenue related to the material rights associated with the Rett Option and the GAN Option must be recognized at a point in time when the options are exercised or the option period expires. In September 2023, Astellas elected not to exercise the GAN Option, therefore we recognized revenue related to the GAN Option during the year ended December 31, 2023.
To date, we have not recognized any revenue from product sales, and we do not expect to generate any revenue from the sale of products, if approved, in the foreseeable future. If our development efforts for our product candidates are successful and result in regulatory approval, or license agreements with third parties, we may generate revenue in the future from product sales. However, there can be no assurance as to when we will generate such revenue, if at all.
Operating Expenses
Research and Development Expenses
Research and development expenses primarily consist of clinical and preclinical development of our product candidates and discovery efforts, including conducting preclinical studies, manufacturing development efforts, preparing for and conducting clinical trials and activities related to regulatory filings for our product candidates. Research and development expenses are recognized as incurred and payments made prior to the receipt of goods or services to be used in research and development are capitalized until the goods or services are received. Costs incurred in obtaining technology licenses through asset acquisitions are charged to research and
37
development expense if the licensed technology has not reached technological feasibility and has no alternative future use. Research and development expenses include or could include:
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We reduced our research and development and general and administrative spend from 2022 to 2023 but have increased and expect to continue to increase our research and development expenses with respect to the Rett clinical trials for the foreseeable future as we continue the development of TSHA-102 and manufacturing processes and conduct discovery and research activities for our preclinical programs. We cannot determine with certainty the timing of initiation, the duration or the completion costs of current or future preclinical studies and clinical trials of our product candidates due to the inherently unpredictable nature of preclinical and clinical development. Clinical and preclinical development timelines, the probability of success and development costs can differ materially from expectations. We anticipate that we will make determinations as to which product candidates to pursue and how much funding to direct to each product candidate on an ongoing basis in response to the results of ongoing and future preclinical studies and clinical trials, regulatory developments and our ongoing assessments as to each product candidate’s commercial potential. We will need to raise substantial additional capital in the future. Our clinical development costs are expected to increase significantly as we commence clinical trials. Our future expenses may vary significantly each period based on factors such as:
38
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive and administrative functions, including stock-based compensation, severance costs, travel expenses and recruiting expenses. Other general and administrative expenses include professional fees for legal, consulting, accounting and audit and tax-related services and insurance costs.
We anticipate that certain of our general and administrative expenses will decrease in the future as a result of the reductions in our headcount in 2022 and 2023 to support our infrastructure and focus on our Rett program. We also anticipate that our general and administrative expenses may increase in the future as a result of payments for accounting, audit, legal, consulting services, costs associated with maintaining compliance with Nasdaq listing rules and SEC requirements, director and officer liability insurance, investor and public relations activities and other expenses associated with operating as a public company to support planned future Rett program development.
Other Income (Expense)
Other income (expense) consists primarily of dividends earned from our money market fund and interest income on our cash and cash equivalents, interest expense on borrowings under the Trinity Term Loan, and non-cash changes in the fair value of our outstanding warrant liability and the Trinity Term Loan.
Results of Operations
Results of Operations for the Three Months ended March 31, 2024 and 2023
The following table summarizes our results of operations for the three months ended March 31, 2024 and 2023 (in thousands):
|
|
For the Three Months |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Revenue |
|
$ |
3,411 |
|
|
$ |
4,706 |
|
Operating expenses: |
|
|
|
|
|
|
||
Research and development |
|
|
20,657 |
|
|
|
12,514 |
|
General and administrative |
|
|
7,084 |
|
|
|
8,751 |
|
Total operating expenses |
|
|
27,741 |
|
|
|
21,265 |
|
Loss from operations |
|
|
(24,330 |
) |
|
|
(16,559 |
) |
Other income (expense): |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
(337 |
) |
|
|
— |
|
Change in fair value of term loan |
|
|
(1,053 |
) |
|
|
— |
|
Interest income |
|
|
1,693 |
|
|
|
319 |
|
Interest expense |
|
|
(29 |
) |
|
|
(1,374 |
) |
Other expense |
|
|
(5 |
) |
|
|
(8 |
) |
Total other income (expense), net |
|
|
269 |
|
|
|
(1,063 |
) |
Net loss |
|
$ |
(24,061 |
) |
|
$ |
(17,622 |
) |
Revenue
Revenue related to the Astellas Transactions was $3.4 million for the three months ended March 31, 2024, compared to $4.7 million for the three months ended March 31, 2023. The revenue recorded is the result of Rett research and development activities performed during the respective first quarter of 2024 and 2023.
Research and Development Expenses
Research and development expenses were $20.7 million for the three months ended March 31, 2024, compared to $12.5 million for the three months ended March 31, 2023. The $8.2 million increase was primarily driven by a $7.6 million increase in GMP batch activities during the three months ended March 31, 2024, which is representative of the intended commercial manufacturing process for TSHA-102 in Rett syndrome. Additionally, clinical trial expenses increased due to ongoing activities in the REVEAL adolescent/adult and pediatric trials.
39
General and Administrative Expenses
General and administrative expenses were $7.1 million for the three months ended March 31, 2024, compared to $8.8 million for the three months ended March 31, 2023. The decrease of $1.7 million was due to reduced general and administrative compensation as a result of lower headcount and reduced consulting and professional fees.
Other Income (Expense)
Change in fair value of warrant liability
Change in fair value of warrant liability was a non-cash expense totaling $0.3 million for the three months ended March 31, 2024 related to the SSI Warrants (as defined below).
Change in fair value of term loan
We elected the fair value option for the Trinity Term Loan and changes to fair value, other than changes that are directly attributable to instrument-specific credit risk, are recorded as a component of other income (expense). The change in fair value was $1.1 million for the three months ended March 31, 2024.
Interest Income
Interest income was $1.7 million for the three months ended March 31, 2024 compared to $0.3 million for the three months ended March 31, 2023. The increase in income is primarily attributable to dividends earned from our money market fund and interest earned on our savings account following the investment of proceeds raised in our August 2023 Private Placement (as defined below).
Interest Expense
Interest expense was less than $0.1 million for the three months ended March 31, 2024, compared to $1.4 million for the three months ended March 31, 2023. The decrease of approximately $1.3 million was primarily attributable to interest expense incurred under the Term Loan Agreement for the three months ended March 31, 2023.
Liquidity and Capital Resources
Overview
Since our inception, we have not generated any revenue from product sales and have incurred significant operating losses. As of March 31, 2024, we had cash and cash equivalents of $124.0 million. We have funded our operations primarily through equity financings, raising an aggregate of $589.0 million in gross proceeds from equity financings, including from pre-IPO private placements of convertible preferred stock, our IPO, and subsequent sales of common stock in public and private securities offerings, our term loans and the Astellas Transactions.
On the Trinity Closing Date, we entered into the Trinity Term Loan Agreement, by and among us, the Trinity Lenders, and Trinity. The Trinity Term Loan Agreement provided for, on the Trinity Closing Date, $40.0 million aggregate principal amount of term loans, or, collectively, the Trinity Term Loans. We drew the Trinity Term Loans in full on the Trinity Closing Date. The interest rate applicable to the Trinity Term Loans is the greater of (a) the Wall Street Journal Prime Rate plus 4.50% or (b) 12.75% per annum. The Trinity Term Loans are interest only from the Trinity Closing Date through 36 months from the Trinity Closing Date, which may be extended to 48 months from the Trinity Closing Date upon the satisfaction of certain milestones set forth in the Trinity Term Loan Agreement, after which we are required to pay equal monthly installments of principal through November 13, 2028, or the Maturity Date. The Trinity Term Loans may be prepaid in full (i) from the Trinity Closing Date through November 13, 2024, with payment of a 3.00% prepayment premium, (ii) from November 13, 2024 through November 13, 2025, with payment of a 2% prepayment premium, and (iii) from November 13, 2025 through, but excluding, the Maturity Date, with payment of a 1% prepayment premium. On the Trinity Closing Date, we paid Trinity a commitment fee of 1.00% of the original principal amount of the Trinity Term Loans. Upon repayment in full of the Trinity Term Loans, we will pay Trinity an end of term payment equal to 5.00% of the original principal amount of the Trinity Term Loans.
The obligations under the Trinity Term Loan Agreement are secured by a perfected security interest in all of our assets except for certain customarily excluded property pursuant to the terms of the Trinity Term Loan Agreement. There are no financial covenants and no warrants associated with the Trinity Term Loan Agreement. The Trinity Term Loan Agreement contains various covenants that limit our ability to engage in specified types of transactions without the consent of Trinity and the Trinity Lenders which include, among others, incurring or assuming certain debt; merging, consolidating or acquiring all or substantially all of the capital stock or property of another entity; changing the nature of our business; changing our organizational structure or type; licensing, transferring or
40
disposing of certain assets; granting certain types of liens on our assets; making certain investments; and paying cash dividends. The Trinity Term Loan Agreement also contains customary representations and warranties, and also includes customary events of default, including payment default, breach of covenants, change of control, and material adverse effects. Upon the occurrence of an event of default, a default interest rate of an additional 5% per annum may be applied to the outstanding loan balances, and the Trinity Lenders may declare all outstanding obligations immediately due and payable and exercise all of its rights and remedies as set forth in the Trinity Term Loan Agreement and under applicable law. The proceeds of the Trinity Term Loans were used to repay our obligations under the Term Loan Agreement with Silicon Valley Bank in full. The Term Loan Agreement with Silicon Valley Bank was terminated concurrently with entry into the Trinity Term Loan Agreement.
On October 5, 2021, we filed a shelf registration statement on Form S-3 with the SEC in relation to the registration of common stock, preferred stock, debt securities, warrants and units or any combination thereof up to a total aggregate offering price of $350.0 million. We also simultaneously entered into a Sales Agreement, or the Sales Agreement with SVB Leerink LLC and Wells Fargo Securities, LLC, or the Sales Agents, pursuant to which we may issue and sell, from time to time at our discretion, shares of our common stock having an aggregate offering price of up to $150.0 million through the Sales Agents. In March 2022, we amended the Sales Agreement to, among other things, include Goldman Sachs & Co. LLC as an additional Sales Agent. In April 2022, we sold 2,000,000 shares of common stock pursuant to the Sales Agreement and received net proceeds of $11.6 million. No other shares of common stock have been issued and sold pursuant to the Sales Agreement as of March 31, 2024.
On October 21, 2022, we entered into the Option Agreement with Astellas granting Astellas an exclusive option to obtain exclusive, worldwide, royalty and milestone-bearing rights and licenses related to TSHA-120 and TSHA-102. As partial consideration for the rights granted to Astellas under the Option Agreement, Astellas paid us a one-time payment in the amount of $20.0 million, or the Upfront Payment, in November 2022.
Also on October 21, 2022, we entered into a securities Purchase Agreement with Astellas, or the Astellas Securities Purchase Agreement, and together with the Option Agreement, the Astellas Transactions, pursuant to which we agreed to issue and sell to Astellas in a private placement, or the Astellas Private Placement, an aggregate of 7,266,342 shares of our common stock, or the Astellas Private Placement Shares, for aggregate proceeds of approximately $30.0 million. The Astellas Private Placement closed on October 24, 2022. Pursuant to the Astellas Securities Purchase Agreement, in connection with the Astellas Private Placement, Astellas has the right to designate one individual to attend all meetings of the Board in a non-voting observer capacity. We also granted Astellas certain registration rights with respect to the Astellas Private Placement Shares.
On October 26, 2022, we entered into an underwriting agreement, or the Underwriting Agreement, with Goldman Sachs & Co. LLC, or the Underwriter, to issue and sell 14,000,000 shares of our common stock, par value $0.00001 per share, in an underwritten public offering, or the Follow-on Offering, pursuant to an effective registration statement on Form S-3 and a related prospectus and prospectus supplement. The offering price to the public was $2.00 per share and the Underwriter purchased the shares from us pursuant to the Underwriting Agreement at a price of $1.88 per share. In addition, we granted the Underwriter an option to purchase, for a period of 30 days, up to an additional 2,100,000 shares of our common stock. The Follow-on Offering closed on October 31, 2022 and we received net proceeds of $26.0 million after deducting underwriting discounts, commissions and offering expenses. On November 10, 2022, the Underwriter exercised their option to purchase an additional 765,226 shares of our common stock and we received net proceeds of $1.4 million after deducting underwriting discounts and commissions.
In April 2023, we entered into a securities purchase agreement, or the SSI Securities Purchase Agreement, with two affiliates of SSI Strategy Holdings LLC, or SSI, named therein, or the SSI Investors, pursuant to which we agreed to issue and sell to the SSI Investors in a private placement, or the SSI Private Placement, 705,218 shares of our common stock, or the SSI Shares, and warrants, or the SSI Warrants, to purchase an aggregate of 525,000 shares of our common stock, or the Warrant Shares. SSI provides certain consulting services to us. Each SSI Warrant has an exercise price of $0.7090 per Warrant Share, which was the closing price of our common stock on the Nasdaq Global Market on April 4, 2023. The SSI Warrants issued in the SSI Private Placement provide that the holder of the SSI Warrants will not have the right to exercise any portion of its SSI Warrants until the achievement of certain clinical and regulatory milestones related to our clinical programs. The SSI Private Placement closed on April 5, 2023. Gross proceeds of the SSI Private Placement were $0.5 million.
On August 14, 2023, we entered into a securities purchase agreement, or the August 2023 Securities Purchase Agreement, with certain institutional and other accredited investors, or the Purchasers, pursuant to which we agreed to sell and issue to the Purchasers in a private placement transaction, or the August 2023 Private Placement, that closed on August 16, 2023: (i) 122,412,376 shares of our common stock and (ii) with respect to certain Purchasers, pre-funded warrants, or the Pre-Funded Warrants, to purchase 44,250,978 shares of common stock in lieu of shares of common stock. The closing of the August 2023 Private Placement, or the Closing, occurred on August 16, 2023. The total gross proceeds to us at the Closing were $150.0 million, and after deducting placement agent commissions and offering expenses payable by us, net proceeds were $140.3 million.
41
Funding Requirements
To date, we have not generated any revenues from the commercial sale of approved drug products, and we do not expect to generate substantial revenue for at least the next few years. If we fail to complete the development of our product candidates in a timely manner or fail to obtain their regulatory approval, our ability to generate future revenue will be compromised. We do not know when, or if, we will generate any revenue from our product candidates, and we do not expect to generate significant revenue unless and until we obtain regulatory approval of, and commercialize, our product candidates. We reduced spending in 2023, and anticipate such reductions will continue in 2024, as a result of our decision to discontinue development of our GAN clinical program. We have increased and expect to continue to increase our research and development expenses, particularly with respect to the Rett clinical trials, for the foreseeable future as we continue the development of our product candidates and manufacturing processes and conduct discovery and research activities for our preclinical programs. If we obtain approval for any of our product candidates, we expect to incur significant commercialization expenses related to sales, marketing, manufacturing and distribution. We anticipate that we will need substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts.
As of March 31, 2024, our material cash requirements consisted of $31.0 million in total lease payments under our noncancelable leases for equipment, laboratory space and office space. These leases are described in further detail in Note 5 to our unaudited condensed consolidated financial statements located in “Part I – Financial Information, Item 1. Financial Statements” in this Quarterly Report on Form 10-Q. Our most significant purchase commitments consist of approximately $9.6 million in cancellable purchase obligations to our CROs and other clinical trial vendors.
We believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital requirements into 2026. We will require additional capital to fund the research and development of our product candidates, to fund our manufacturing activities, to fund precommercial activities of our programs and for working capital and general corporate purposes. The assessment of our ability to meet our future obligations is inherently judgmental, subjective and susceptible to change.
Because of the numerous risks and uncertainties associated with research, development and commercialization of biological products, we are unable to estimate the exact amount of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time-consuming, expensive and uncertain process that takes many years to complete, and we may never generate the necessary data or results required to obtain
42
marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our commercial revenues, if any, will be derived from sales of product candidates that we do not expect to be commercially available in the near term, if at all. Accordingly, we will need to continue to rely on additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the terms of these equity securities or this debt may restrict our ability to operate. The Trinity Term Loan Agreement contains negative covenants, including, among other things, restrictions on indebtedness, liens investments, mergers, dispositions, prepayment of other indebtedness and dividends and other distributions. Any future additional debt financing and equity financing, if available, may involve agreements that include covenants limiting and restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, entering into profit-sharing or other arrangements or declaring dividends. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may be required to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
Cash Flows
The following table shows a summary of our cash flows for the three months ended March 31, 2024 and 2023 (in thousands):
|
|
For the Three Months Ended March 31, |
|
|||||
|
|
2024 |
|
|
2023 |
|
||
Net cash used in operating activities |
|
$ |
(19,798 |
) |
|
$ |
(20,185 |
) |
Net cash used in investing activities |
|
|
(140 |
) |
|
|
(3,900 |
) |
Net cash used in financing activities |
|
|
(22 |
) |
|
|
(370 |
) |
Net change in cash, cash equivalents and restricted cash |
|
$ |
(19,960 |
) |
|
$ |
(24,455 |
) |
Operating Activities
For the three months ended March 31, 2024, our net cash used in operating activities of $19.8 million primarily consisted of a net loss of $24.1 million, primarily attributable to our spending on research and development expenses. The net loss of $24.1 million was partially offset by adjustments for non-cash items, primarily stock-based compensation expense of $3.2 million and other non-cash items of $1.0 million, net. Additional cash provided by operating assets and liabilities of $0.3 million was primarily attributable to an increase in accounts payable of $4.0 million and offset by a decrease in deferred revenue of $3.4 million.
For the three months ended March 31, 2023, our net cash used in operating activities of $20.2 million primarily consisted of a net loss of $17.6 million, primarily attributable to our spending on research and development expenses. The net loss of $17.6 million was partially offset by adjustments for non-cash items, primarily stock-based compensation expense of $1.7 million. Additional cash used in operating activities of $5.0 million was primarily due to a decrease in deferred revenue.
Investing Activities
During the three months ended March 31, 2024, investing activities used $0.1 million of cash primarily attributable to the purchase of lab equipment. During the three months ended March 31, 2023, investing activities used $3.9 million of cash primarily attributable to capital expenditures related to the close-out of our in-house manufacturing facility project.
Financing Activities
During the three months ended March 31, 2024, financing activities used less than $0.1 million of cash, which is primarily attributable to the payment of lease financing obligations which were partially offset by ESPP contributions. During the three months ended March 31, 2023, financing activities used $0.4 million of cash, which is primarily attributable to the payment of shelf registration costs and other financing transactions.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Critical Accounting Policies and Significant Judgments and Estimates
There were no material changes to our critical accounting policies that are disclosed in our audited consolidated financial statements for the year ended December 31, 2023 filed with the SEC on March 19, 2024.
43
Recent Accounting Pronouncements
See Note 2 to our unaudited condensed consolidated financial statements located in “Part I – Financial Information, Item 1. Financial Statements” in this Quarterly Report on Form 10-Q for a description of recent accounting pronouncements applicable to our condensed consolidated financial statements.
Emerging Growth Company and Smaller Reporting Company Status
In April 2012, the Jumpstart Our Business Startups Act of 2012, or JOBS Act, was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We elected the extended transition period for complying with new or revised accounting standards, which delays the adoption of these accounting standards until they would apply to private companies.
In addition, as an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include:
We may take advantage of these provisions until we no longer qualify as an emerging growth company. We will cease to qualify as an emerging growth company on the date that is the earliest of: (i) December 31, 2025, (ii) the last day of the fiscal year in which we have more than $1.235 billion in total annual gross revenues, (iii) the date on which we are deemed to be a “large accelerated filer” under the rules of the SEC, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th, or (iv) the date on which we have issued more than $1.0 billion of non-convertible debt over the prior three-year period. We may choose to take advantage of some but not all of these reduced reporting burdens. We have taken advantage of certain reduced reporting requirements in this Quarterly Report on Form 10-Q and our other filings with the SEC. Accordingly, the information contained herein may be different than you might obtain from other public companies in which you hold equity interests.
We are also a “smaller reporting company,” meaning that the market value of our shares held by non-affiliates is less than $700 million and our annual revenue was less than $100 million during the most recently completed fiscal year. We may continue to be a smaller reporting company if either (i) the market value of our shares held by non-affiliates is less than $250 million or (ii) our annual revenue was less than $100 million during the most recently completed fiscal year and the market value of our shares held by non-affiliates is less than $700 million. If we are a smaller reporting company at the time we cease to be an emerging growth company, we may continue to rely on exemptions from certain disclosure requirements that are available to smaller reporting companies. Specifically, as a smaller reporting company, we may choose to present only the two most recent fiscal years of audited financial statements in our Annual Report on Form 10-K and, similar to emerging growth companies, smaller reporting companies have reduced disclosure obligations regarding executive compensation.
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this Item.
Item 4. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act ), as of
44
the end of the period covered by this Form 10-Q. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that as of March 31, 2024, our disclosure controls and procedures were effective to provide reasonable assurance that the information required to be disclosed by us in this Form 10-Q was (a) reported within the time periods specified by SEC rules and regulations, and (b) communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding any required disclosure.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting identified in management’s evaluation pursuant to Rules 13a-15(d) or 15d-15(d) of the Exchange Act during the period covered by this Quarterly Report on Form 10-Q that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Internal Controls
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable, not absolute, assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs. Our management, including our Chief Executive Officer and Chief Financial Officer, believes that our disclosure controls and procedures and internal control over financial reporting are designed to provide reasonable assurance of achieving their objectives and are effective at the reasonable assurance level. However, our management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud.
45
PART II—OTHER INFORMATION
Item 1. Legal Proceedings.
We are not subject to any material legal proceedings. From time to time, we may be involved in various claims and legal proceedings relating to claims arising out of our operations. We are not currently a party to any legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Item 1A. Risk Factors.
Our business is subject to risks and events that, if they occur, could adversely affect our financial condition and results of operations and the trading price of our securities. In addition to the other information set forth in this quarterly report on Form 10-Q, you should carefully consider the factors described in Part I, Item 1A. “Risk Factors” of our Annual Report on Form 10-K for the fiscal year ended December 31, 2023, filed with the Securities and Exchange Commission on March 19, 2024.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
(a) Recent Sales of Unregistered Equity Securities
None.
(b) Use of Proceeds
None.
(c) Issuer Purchases of Equity Securities
None.
Item 3. Defaults Upon Senior Securities.
Not applicable.
Item 4. Mine Safety Disclosures.
Not applicable.
Item 5. Other Information.
None.
46
Item 6. Exhibits.
The exhibits listed on the Exhibit Index are either filed or furnished with this report or incorporated herein by reference.
Exhibit Number |
|
Description |
3.1 |
|
|
3.2 |
|
|
3.3 |
|
|
31.1* |
|
|
31.2* |
|
|
32.1# |
|
|
32.2# |
|
|
101.INS |
|
XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document |
101.SCH |
|
Inline XBRL Taxonomy Extension Schema Document |
101.CAL |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
101.DEF |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
101.PRE |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
104 |
|
Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101). |
* Filed herewith.
# These certifications are being furnished solely to accompany this quarterly report pursuant to 18 U.S.C. Section 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and are not to be incorporated by reference into any filing of the registrant, whether made before or after the date hereof, regardless of any general incorporation language in such filing.
47
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
|
Taysha Gene Therapies, Inc. |
|
|
|
|
|
Date: May 14, 2024 |
|
By: |
/s/ Sean Nolan |
|
|
|
Sean Nolan |
|
|
|
Chief Executive Officer (Principal Executive Officer) |
|
|
|
|
Date: May 14, 2024 |
|
By: |
/s/ Kamran Alam |
|
|
|
Kamran Alam |
|
|
|
Chief Financial Officer (Principal Financial and Accounting Officer) |
48