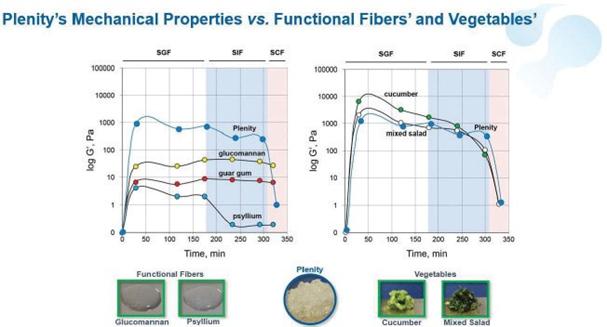
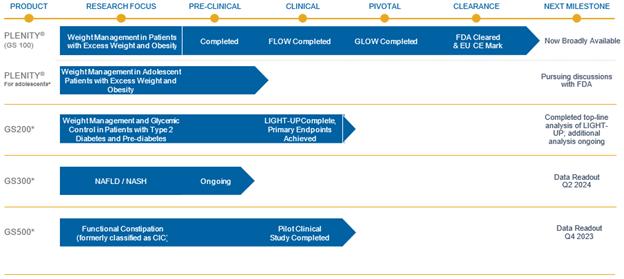
Results of Operations
Our only activities through December 31, 2021 were organizational activities, those necessary to prepare for the Initial Public Offering, described below, and after the Initial Public Offering, finding a target company for a Business Combination and activities in connection with the acquisition of Gelesis Holdings, Inc. We do not expect to generate any operating revenues until after the completion of our Business Combination. We generate non-operating income in the form of interest income on marketable securities held in the Trust Account. We incur expenses as a result of being a public company (for legal, financial reporting, accounting and auditing compliance), as well as for due diligence expenses in connection with completing a Business Combination.
For the year ended December 31, 2021, we had net loss of $12,898,082, which consisted of operating costs of $20,674,209 and a change in fair value of warrant liabilities of $7,602,367, offset by interest income on marketable securities held in the Trust Account of $167,045 and an unrealized gain on marketable securities held in our Trust Account of $6,715.
For the period from February 14, 2020 (inception) through December 31, 2020, we had net loss of $15,294,860, which consisted of transaction cost related to IPO of $671,901, operating costs of $2,426,204 and a change in fair value of warrant liabilities of $12,406,208, offset by interest income on marketable securities held in the Trust Account of $201,441 and an unrealized gain on marketable securities held in our Trust Account of $8,012.
Liquidity and Capital Resources
On July 7, 2020, we consummated the Initial Public Offering of 27,600,000 Units at a price of $10.00 per Unit, which includes the full exercise by the underwriters of the over-allotment option to purchase an additional 3,600,000, generating gross proceeds of $276,000,000. Simultaneously with the closing of the Initial Public Offering, we consummated the sale of 7,520,000 Private Placement Warrants at a price of $1.00 per Private Placement Warrant in a private placement to our stockholders, generating gross proceeds of $7,520,000.
Following the Initial Public Offering, the full exercise of the over-allotment option by the underwriters’ and the sale of the Private Placement Warrants, a total of $276,000,000 was placed in the Trust Account and we had $1,389,212 of cash held outside of the Trust Account, after payment of costs related to the Initial Public Offering, and available for working capital purposes. We incurred $15,851,828 in transaction costs, including $5,520,000 of underwriting fees, $9,660,000 of deferred underwriting fees and $671,828 of other offering costs.
For the year ended December 31, 2021, cash used in operating activities was $446,644, which consisted of our net loss of $12,898,082, interest earned on marketable securities held in the Trust Account of $167,045, an unrealized gain on marketable securities held in our Trust Account of $6,715 and changes in fair value of warrant liabilities of $7,602,367. Changes in operating assets and liabilities provided $20,227,565 of cash.
For the period from February 14, 2020 (inception) through December 31, 2020, cash used in operating activities was $861,345. Net loss of $15,294,860 was affected by interest earned on marketable securities held in the Trust Account of $201,441, an unrealized gain on marketable securities of $8,012, transaction costs related to the Initial Public Offering of $671,901, changes in fair value or warrants of $12,406,208 and changes in operating assets and liabilities, which provided $1,564,859 of cash from operating activities.
As of December 31, 2021, we had cash and marketable securities held in the Trust Account of $276,207,207. We may withdraw interest to pay franchise and income taxes. During the year ended December 31, 2021, we have withdrawn $176,006 of interest earned on the Trust Account for the payment of franchise taxes.
Until the consummation of the Business Combination, we used the funds held outside the Trust Account primarily to identify and evaluate target businesses, perform business due diligence on prospective target businesses, travel to and from the offices, plants or similar locations of prospective target businesses or their representatives or owners, review corporate documents and material agreements of prospective target businesses, and structure, negotiate and complete a Business Combination.
Upon the close of the Business Combination, we received approximately $105 million of gross proceeds to fund our operations. We believe that the cash available from the consummation of the Business Combination and related transactions will be sufficient to fund our short-term liquidity needs and the execution of our business plan through at least the twelve month-period from the date of the Business Combination.