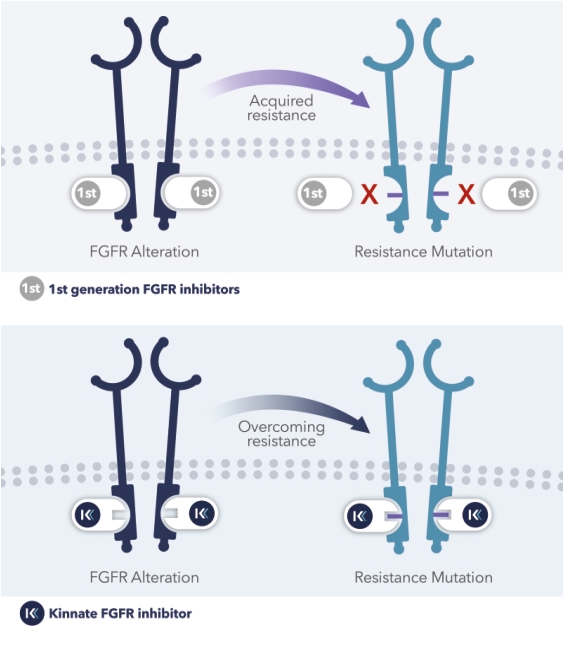
GDPR increased our responsibility and liability in relation to personal data that we process where such processing is subject to the GDPR, and we may be required to put in place additional mechanisms to ensure compliance with the GDPR, including as implemented by individual countries. Compliance with the GDPR will be a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with our European activities.
Further, the vote in the United Kingdom (UK) in favor of exiting the EU, referred to as Brexit, has created uncertainty with regard to data protection regulation in the UK. Specifically, while the Data Protection Act of 2018, which “implements” and complements the GDPR achieved Royal Assent on May 23, 2018 and is now effective in the UK, aspects of data protection in the UK, such as the transfer of data from the EEA to the UK, remain uncertain. During the period of “transition” (i.e., until December 31, 2020), EU law will continue to apply in the UK, including the GDPR, after which the GDPR will be converted into UK law. Beginning in 2021, the UK will be a “third country” under the GDPR. We may, however, incur liabilities, expenses, costs, and other operational losses under GDPR and applicable EU Member States and UK privacy laws in connection with any measures we take to comply with them.
In addition, California recently enacted the CCPA, which creates new individual privacy rights for California consumers (as defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA requires covered companies to provide new disclosure to consumers about such companies’ data collection, use and sharing practices, provide such consumers new ways to opt-out of certain sales or transfers of personal information, and provide consumers with additional causes of action in data breach situations. The CCPA went into effect on January 1, 2020, and the California Attorney General commenced enforcement actions for violations on July 1, 2020. Moreover, CPRA was recently certified by the California Secretary of State to appear on the ballot for the November 3, 2020 election. If this initiative is approved by California voters, the CPRA would significantly modify the CCPA, potentially resulting in further uncertainty and requiring us to incur additional costs and expenses in an effort to comply. The CCPA and, if it goes into effect, the CPRA, may impact our business activities and exemplifies the vulnerability of our business to the evolving regulatory environment related to personal data and protected health information.
Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Any actual or alleged failure to comply with U.S. or international laws and regulations relating to privacy, data protection, and data security could result in governmental investigations, proceedings and enforcement actions (which could include civil or criminal penalties), private litigation or adverse publicity, harm to our reputation, and could negatively affect our operating results and business. Moreover, clinical trial subjects about whom we or our potential collaborators obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information or impose other obligations or restrictions in connection with our use, retention and other processing of information, and we may otherwise face contractual restrictions applicable to our use, retention, and other processing of information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
Our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs, suppliers and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, consultants, commercial collaborators, principal investigators, CROs, suppliers and vendors may engage in misconduct or other improper activities. Misconduct by these parties could include failures to comply with FDA, EMA or comparable foreign regulatory authority regulations, provide accurate information to the FDA, EMA or comparable foreign regulatory authorities, comply with federal and state health care fraud and abuse laws and regulations, accurately report financial information or data or disclose unauthorized activities to us. In particular, research, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other