Our pipeline
We have assembled a portfolio of gene therapy product candidates with the potential to address multiple neurodegenerative diseases. Our development programs consist of:

* 8 additional CNS pipeline license options remain; 3 license options were previously exercised, and rights were subsequently returned to the University of Pennsylvania.
† US/EU prevalence per third-party sources
PBFT02 for the treatment of FTD-GRN
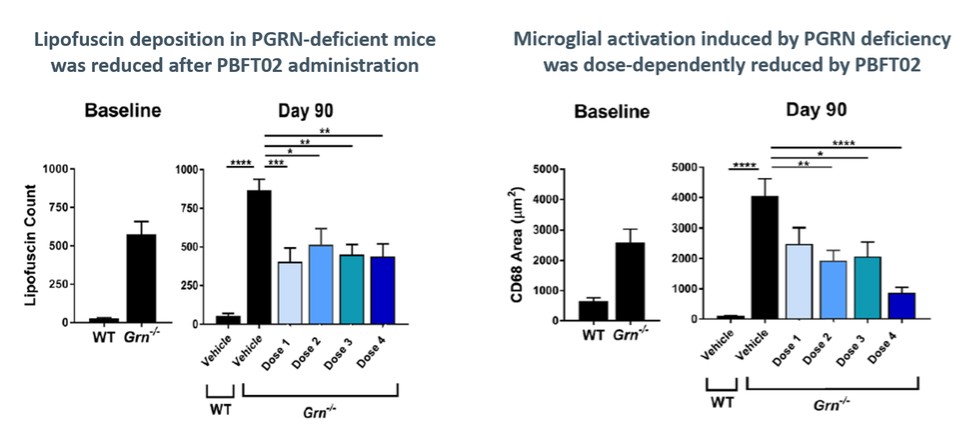
We are currently developing PBFT02, which utilizes an AAV1 capsid to deliver a functional copy of GRN encoding for PGRN, for the treatment of FTD-GRN. FTD-GRN is an inheritable form of FTD in which patients have mutations in the GRN gene, causing a deficiency in PGRN. PGRN is a complex and highly conserved protein thought to have multiple roles in cell homeostasis, neurodevelopment, and inflammation. Evidence suggests that PGRN deficiency in FTD and other neurodegenerative disorders may contribute to lysosomal dysfunction.
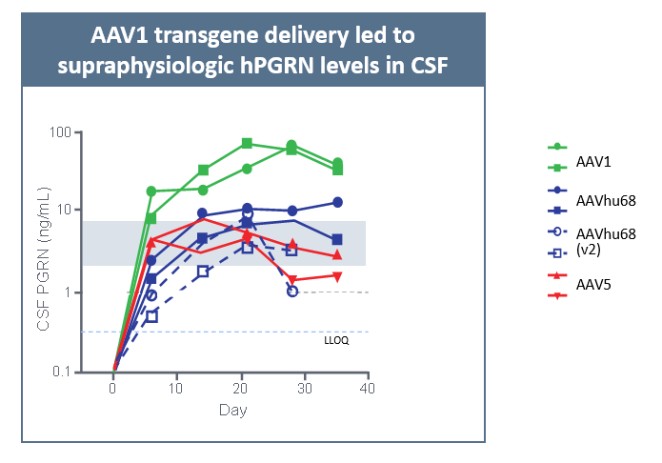
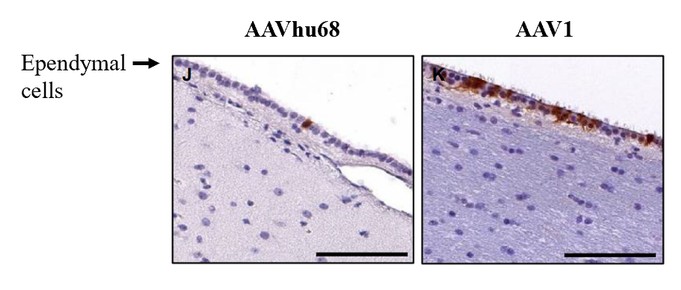
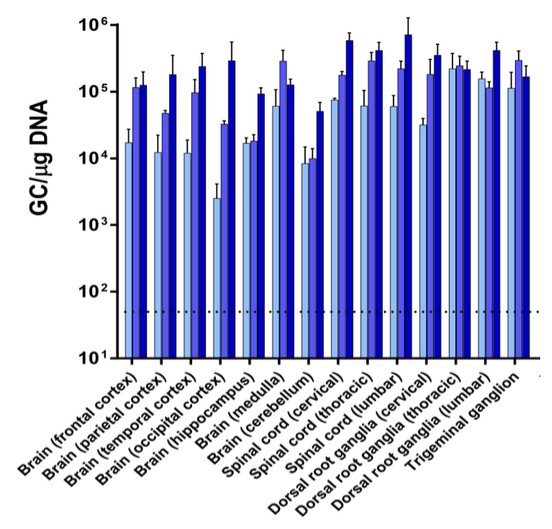
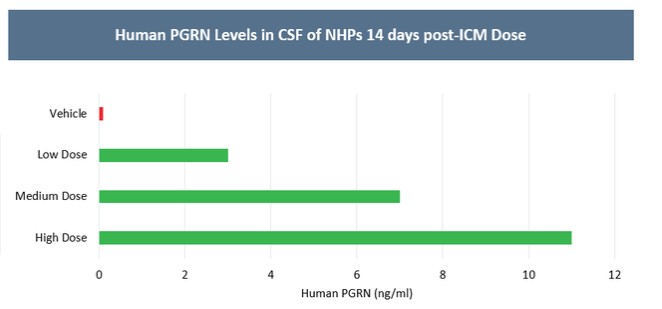
Currently, there are no disease-modifying therapies approved for the treatment of FTD-GRN. Based on findings in preclinical studies, we believe that PBFT02 may provide FTD-GRN patients with significantly improved outcomes. We selected the AAV1 capsid and ICM administration for PBFT02 because this approach led to extensive and robust expression of human PGRN throughout the brain and spinal cord of non-human primates, or NHPs, and due to the higher PGRN levels in cerebral spinal fluid, or CSF, achieved using AAV1 as compared with other serotypes tested. ICM administration of AAV1 to NHPs resulted in supraphysiologic CSF levels of human PGRN when compared with CSF levels in healthy human subjects, and in excess of levels achieved in NHPs with AAVhu68 or AAV5. We have an active IND from the U.S. Food and Drug Administration, or FDA, and approved clinical trial authorizations, or CTAs, in multiple countries for PBFT02. We are conducting our upliFT-D trial, an international, multi-center, open-label, single-arm Phase 1/2 clinical trial of PBFT02 in patients with a diagnosis of symptomatic FTD-GRN.
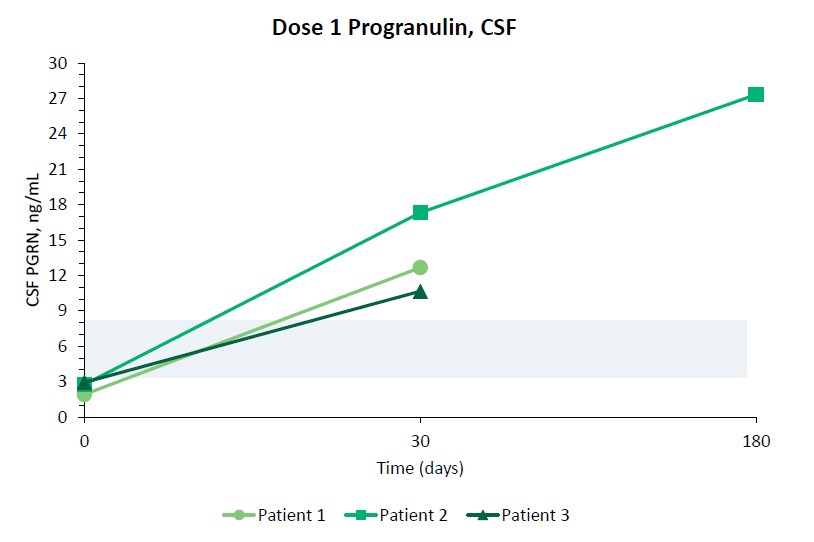
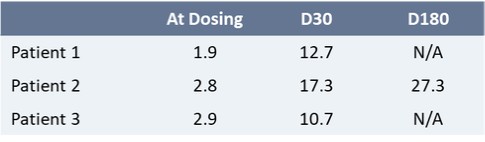
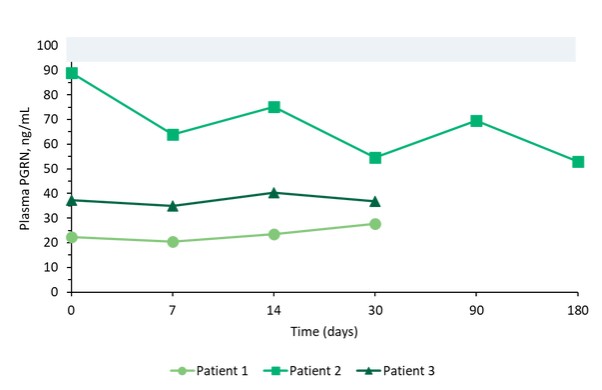
We reported initial safety and biomarker data from three patients in Cohort 1 of our upliFT-D trial in December of 2023. In this trial, Dose 1 of PBFT02 treatment resulted in supraphysiologic levels of CSF PGRN with concentrations ranging from 10.7 to 17.3 ng/mL at 30 days post-treatment (n=3), exceeding the range found in healthy adult controls of 3.3 to 8.2 ng/mL (n=61). In the first patient to reach 6-months post PBFT02 administration, CSF PGRN remained at supraphysiologic levels with a concentration of 27.3 ng/mL. By contrast, following PBFT02 treatment plasma PGRN levels were unaltered, remaining similar to baseline concentrations and below levels found in healthy adult controls throughout the available follow-up period across all three patients.
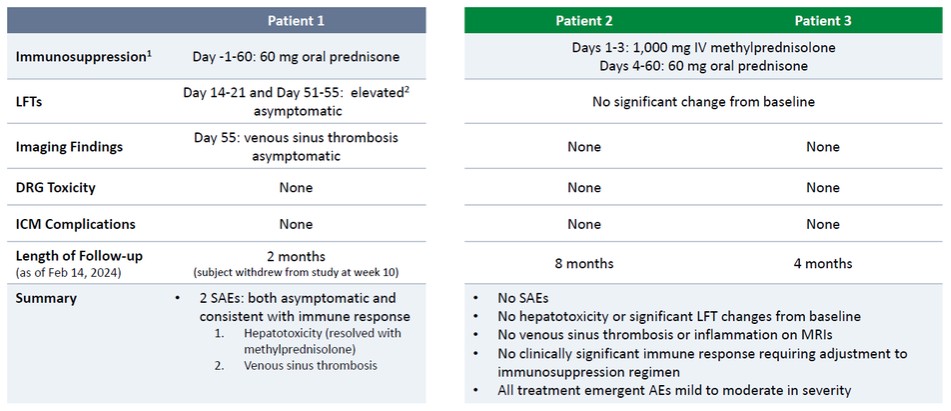
As previously reported, Patient 1, who received a low level of immunosuppression (60 mg oral prednisone daily for 60 days), per the initial trial protocol, experienced two serious adverse events, or SAEs, that were both asymptomatic and
7