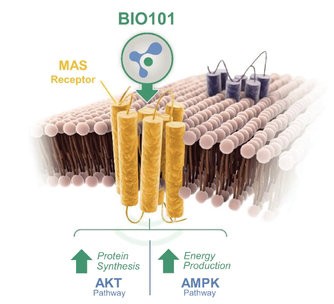
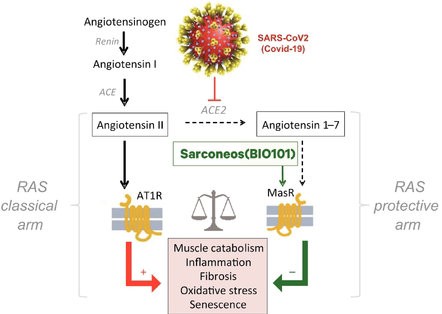
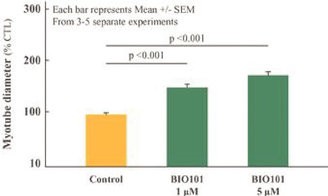
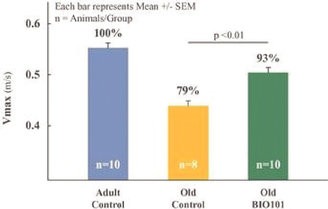
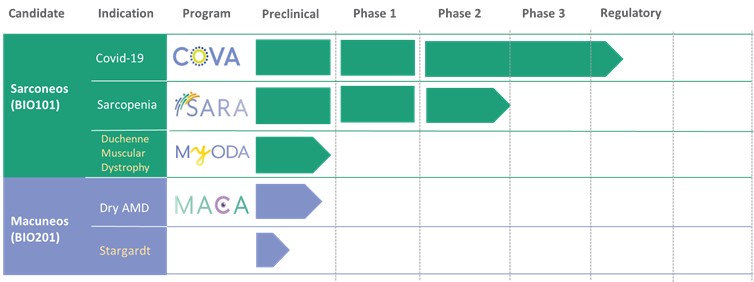
Our Clinical Pipeline
We are developing a portfolio of programs targeting biological resilience pathways that slow the degenerative processes associated with aging and improve functional outcomes for patients suffering from age-related diseases. Our current pipeline of drug candidates is illustrated below.
Our Strategy
We are focused on the development of therapeutics that improve functional outcomes for patients suffering from age-related diseases, including severe respiratory failure in patients suffering from COVID-19. Our goal is to build Biophytis into a leading biotechnology company focused on targeting biological resilience pathways that slow the degenerative processes associated with age-related disease progression in order to improve the lives of millions of patients that have limited or no treatment options. We currently plan to develop our drug candidates and then seek licensing and/or partnership opportunities for further clinical development through regulatory approval and commercialization. To achieve our goal, we are pursuing the following strategies:
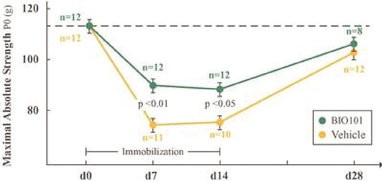
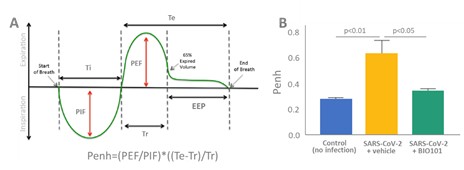
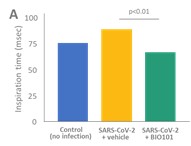
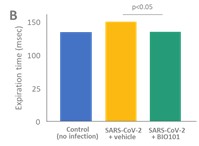
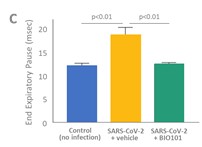
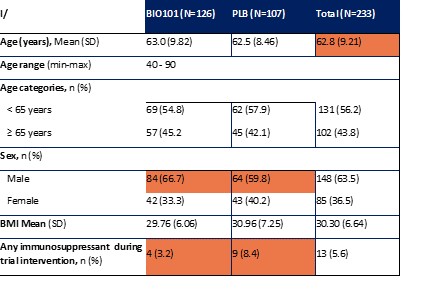
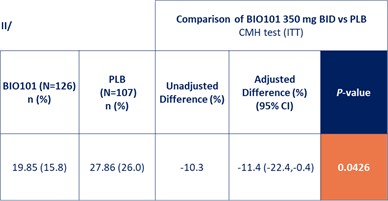
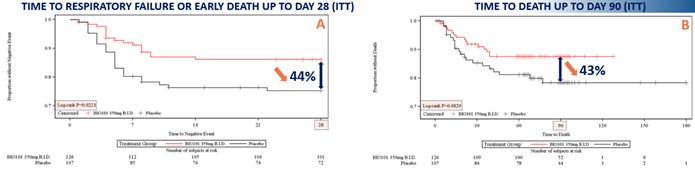
| ● | Obtain EAPs in France, Brazil and other selected countries as well as EUA (US) and conditional approval (EU) of Sarconeos (BIO101) for COVID-19 patients at risk of respiratory failure. We finalized recruitment of the Phase 2/3 COVA study in hospitalized COVID-19 patients with severe respiratory manifestations in April 2022 and reported final analysis in February 2023. The study met its pre-defined primary endpoint demonstrating a statistically significant difference between Sarconeos (BIO101) and placebo in the proportion of patients with respiratory failure or early death at day 28, representing a relative reduction of risk of 44% (p=0.043, Cochran-Mantel-Haenszel test). Based on these results, we initiated the EAP review process in France in March 2023. We also plan to apply for EAPs in Brazil and other selected countries as well as EUA in the United States and conditional approval in the EU. |
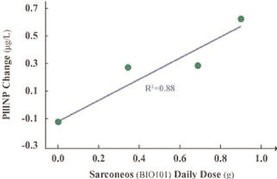
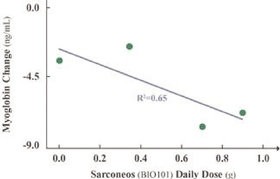
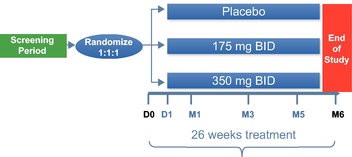
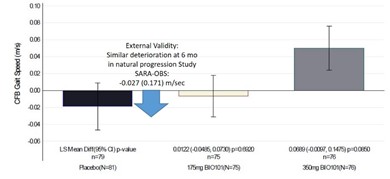
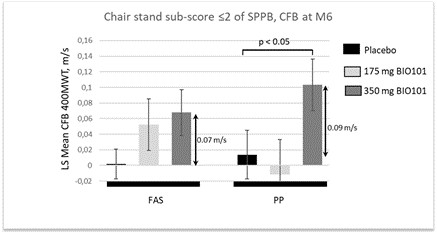
| ● | Demonstrate efficacy of Sarconeos (BIO101) in sarcopenia. Our resources and business efforts have been significantly focused on advancing the clinical development of Sarconeos (BIO101) from the treatment of neuromuscular disorders, with an initial focus on sarcopenia. The topline data for our SARA-INT Phase 2b clinical trial was published in October 2021. Due to the effect of the pandemic on the patient population, only 45 percent of the study subjects were able to complete the study with end-of-treatment efficacy assessments and the study was underpowered to observe the hypothesized effect size, and the primary and secondary endpoints were not met. We have extensively interacted with the regulatory authorities in Europe (EMA) and the United States (FDA) throughout 2022 to define the design of a Phase 3 clinical trial in this indication while in parallel holding discussions with potential partners to be part of this development process. |
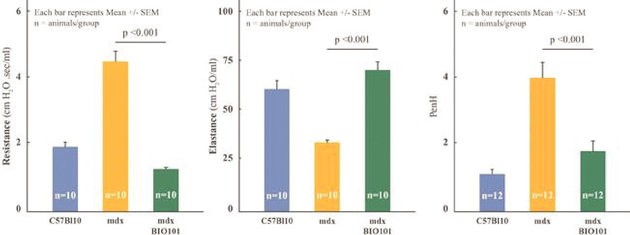
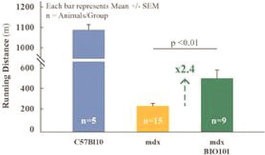
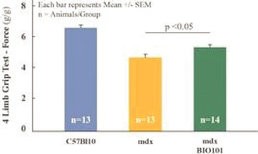
| ● | Initiate clinical development of Sarconeos (BIO101) in DMD. Our efforts are also focused on leveraging our knowledge and the development of Sarconeos (BIO101) in sarcopenia and COVID-19 to commence and advance the clinical development of Sarconeos (BIO101) for the treatment of non-ambulatory DMD patients with signs of respiratory deterioration, independent of genetic mutation and across the disease spectrum. We have already received an IND “may proceed” letter from the FDA in the United States and a CTA approval from FAMHP in Belgium. In the “may proceed” letter from the FDA, the FDA noted that it had significant concerns with the design of the study, and that the results of the study, as originally designed to enroll ambulatory and non-ambulatory patients and measure muscle function deterioration through a |
56