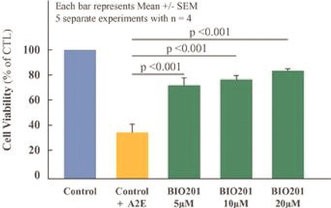
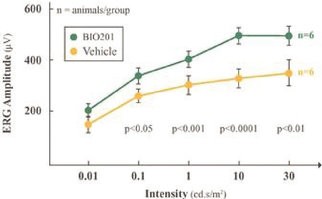
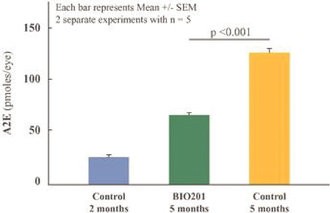
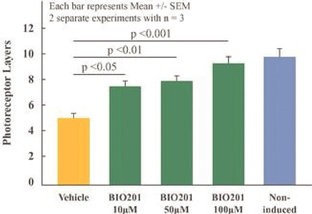
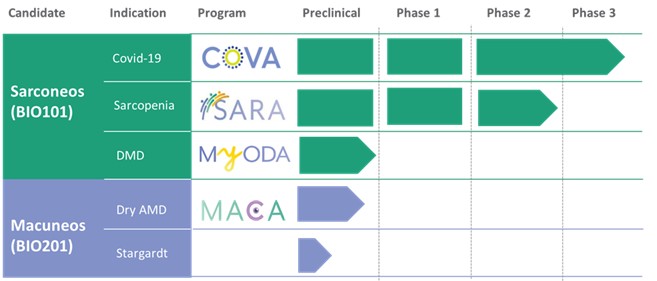
Our Clinical Pipeline
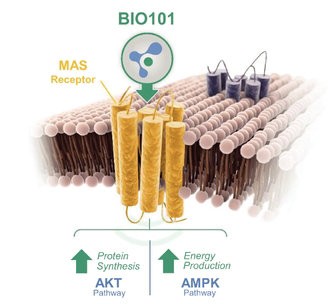
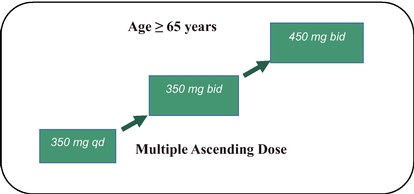
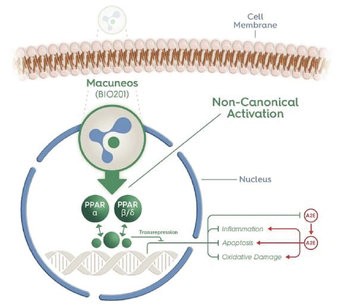
We are developing a portfolio of programs targeting biological resilience pathways that slow the degenerative processes associated with aging and improve functional outcomes for patients suffering from age-related diseases. Our current pipeline of drug candidates is illustrated below.
Our Strategy
We are focused on the development of therapeutics that improve functional outcomes for patients suffering from age-related diseases. Our goal is to build Biophytis into a leading biotechnology company focused on targeting biological resilience pathways that slow the degenerative processes associated with age-related disease progression in order to improve the lives of millions of patients that have limited or no treatment options. We currently plan to develop our drug candidates and then seek licensing and/or partnership opportunities for further clinical development through regulatory approval and commercialization. To achieve our goal, we are pursuing the following strategies:
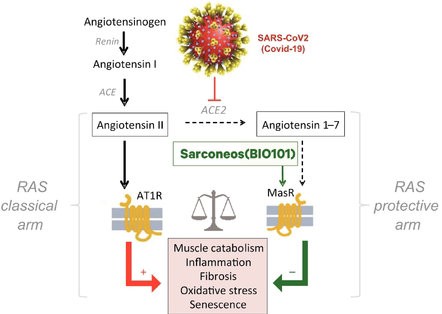
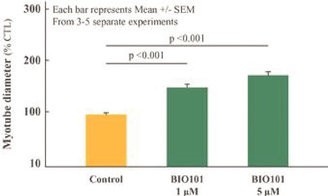
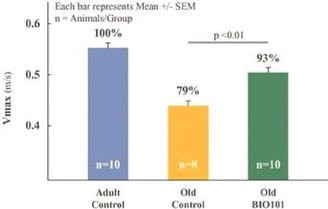
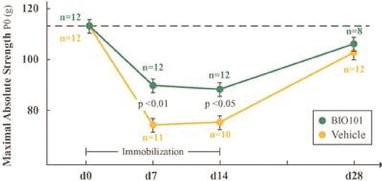
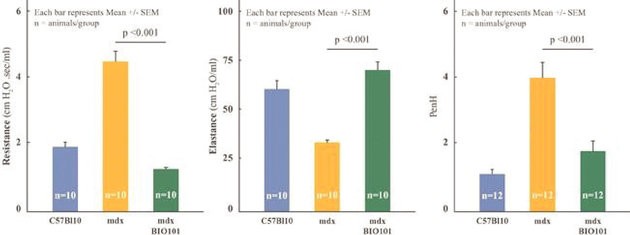
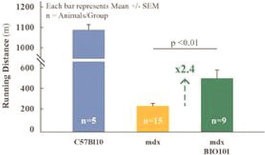
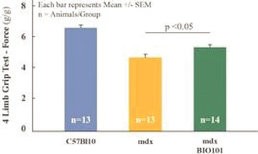
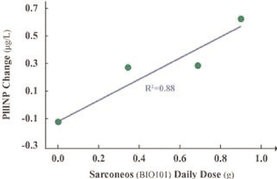
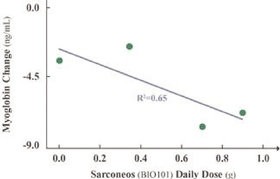
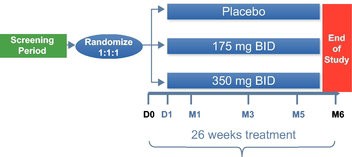
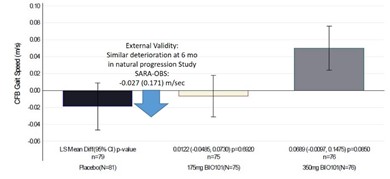
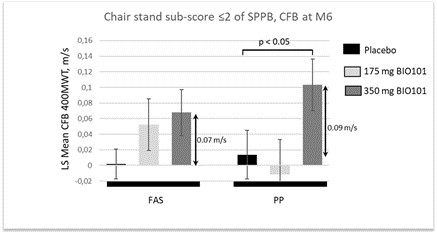
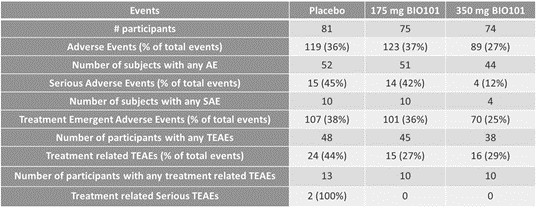
| ● | Demonstrate efficacy of Sarconeos (BIO101) in sarcopenia. Our resources and business efforts are significantly focused on advancing the clinical development of Sarconeos (BIO101) for the treatment of neuromuscular disorders, with an initial focus on sarcopenia. The topline data for our SARA-INT Phase 2b clinical trial was published in October 2021. Due to the effect of the pandemic on patient population, only 45 percent of the study subjects were able to complete the study with end-of-treatment efficacy assessments and the study was underpowered to observe the hypothesized effect size, and the primary and secondary endpoints were not met. Additional Phase 2 dose-finding studies are planned to identify an optimal dose or dosing regimens, and further inform the safety profile for higher dosing regimens and related safety information as well as pharmacokinetics data are planned. |
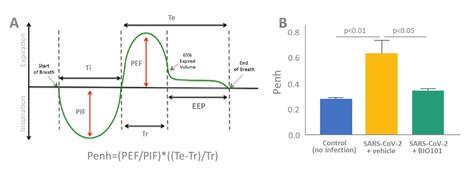
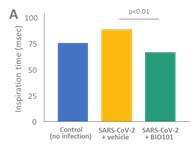
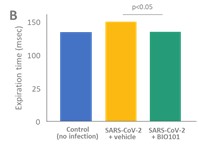
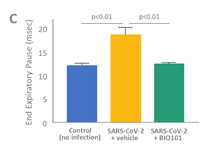
| ● | Demonstrate the therapeutic benefit and obtain an EUA (US, Brazil) and conditional approval (EU) of Sarconeos (BIO101) for COVID-19 patients. We are concluding the enrollment part of the two-part Phase 2/3 COVA study in hospitalized COVID-19 patients with severe respiratory manifestations earlier than planned due to the progression of the pandemic and the difficulty we experienced in enrollment. Since April 2020, 237 patients have been enrolled in France, the United States, Belgium, and Brazil, in approximately 35 clinical centers. The initial target for enrollment was 310 pateints. We plan to conclude our data analysis by the end of the third quarter of 2022. Based on these results, we will further determine development and regulatory strategies. |
60