PART I
ITEM 1. BUSINESS.
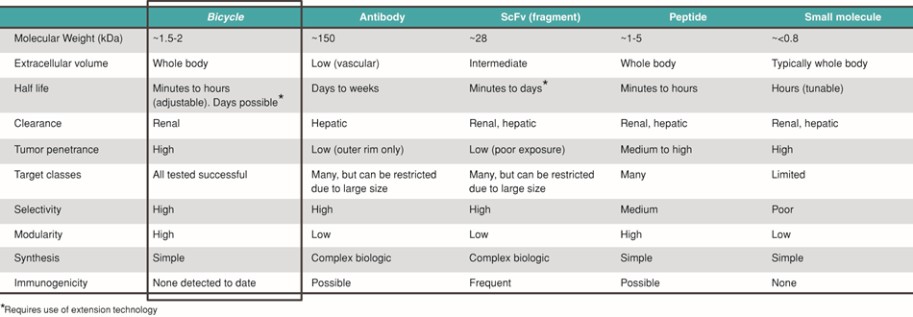
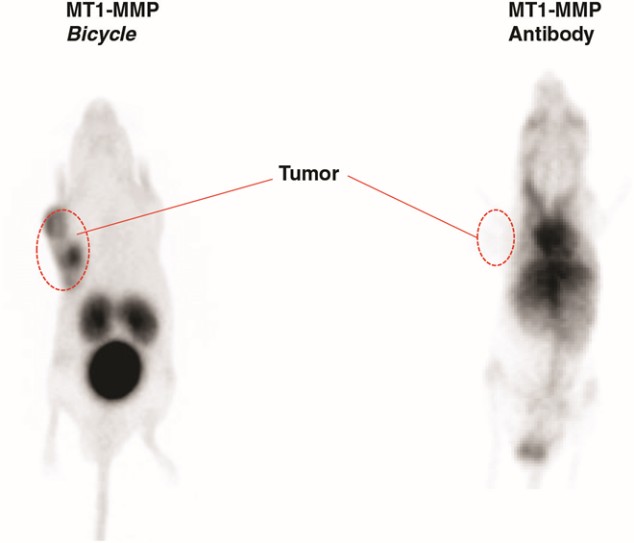
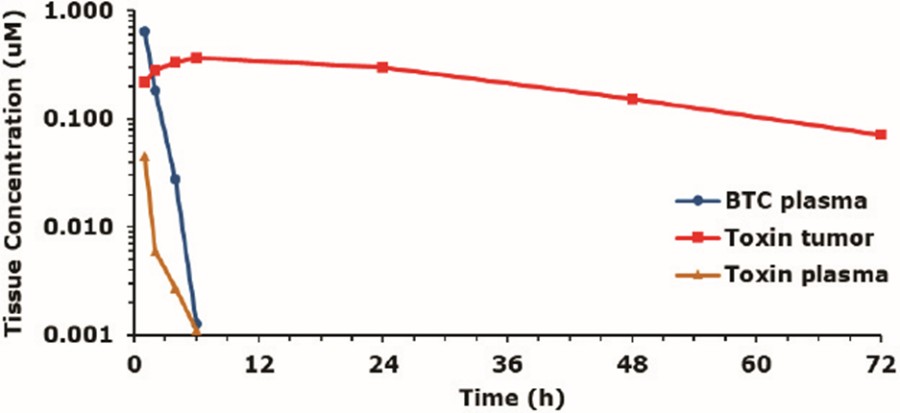
We are a clinical-stage biopharmaceutical company developing a novel class of medicines, which we refer to as Bicycles, for diseases that are underserved by existing therapeutics. Bicycles are fully synthetic short peptides constrained to form two loops which stabilize their structural geometry. This constraint facilitates target binding with high affinity and selectivity, making Bicycles attractive candidates for drug development. Bicycles are a unique therapeutic modality combining the pharmacology usually associated with a biologic with the manufacturing and pharmacokinetic, or PK, properties of a small molecule. The relatively large surface area presented by Bicycles allows targets to be drugged that have historically been intractable to non-biological approaches. Bicycles are excreted by the kidney rather than the liver and have shown no signs of immunogenicity to date, qualities which we believe explain the molecules’ favorable toxicological profile.
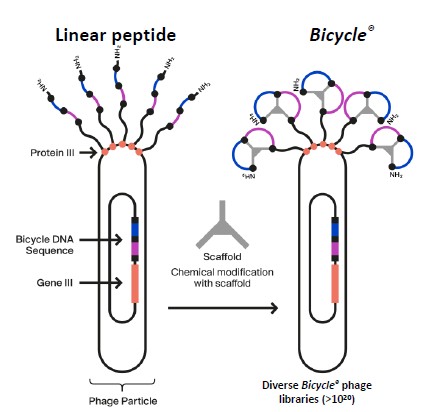
We have a novel and proprietary phage display screening platform which we use to identify Bicycles in an efficient manner. The platform initially displays linear peptides on the surface of engineered bacteriophages, or phages, before “on-phage” cyclization with a range of small molecule scaffolds which can confer differentiated physicochemical and structural properties. Our platform encodes quadrillions of potential Bicycles which can be screened to identify molecules for optimization to potential product candidates. We have used this powerful screening technology to identify our current portfolio of candidates in oncology and intend to use it in conjunction with our collaborators to seek to develop additional future candidates across a range of other disease areas.
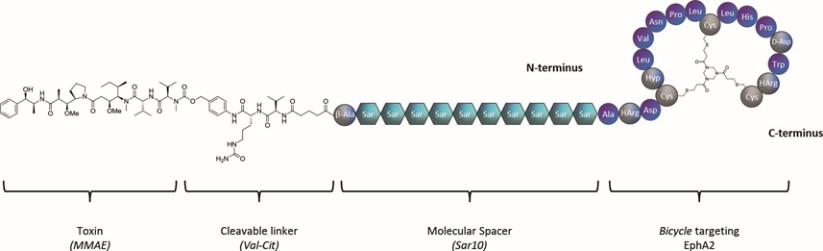
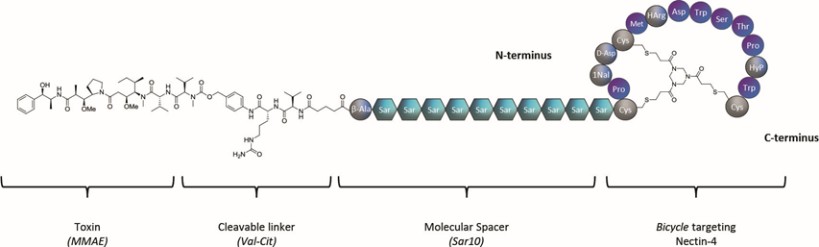
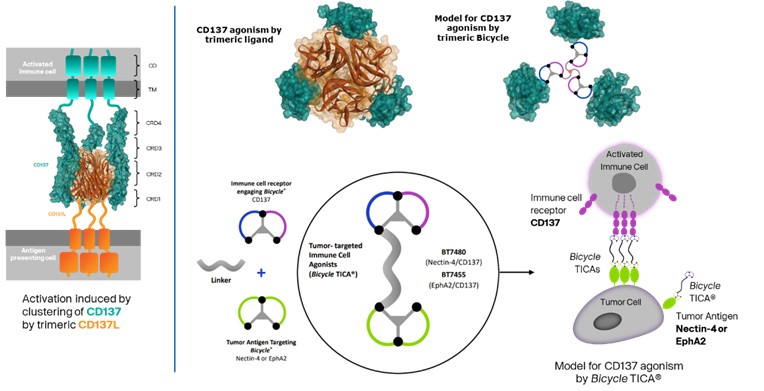
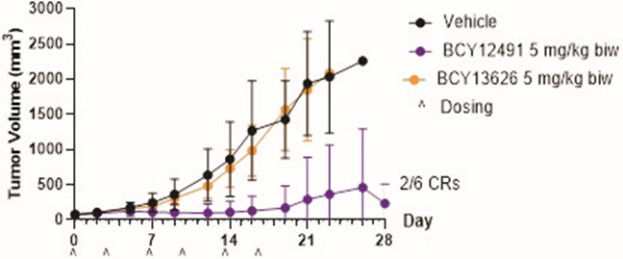
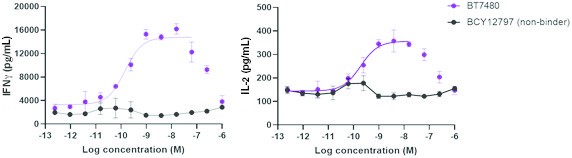
Our product candidates, BT5528, BT8009, and BT1718, are each a Bicycle® Toxin Conjugate, or BTCTM. These Bicycles are chemically attached to a toxin that when administered is cleaved from the Bicycle and kills the tumor cells. We are evaluating BT5528, a second-generation BTC targeting Ephrin type A receptor 2, or EphA2, in a company-sponsored Phase I/II clinical trial and BT8009, a second-generation BTC targeting Nectin-4, in a company-sponsored Phase I/II clinical trial. In addition, BT1718 is being developed to target tumors that express Membrane Type 1 matrix metalloproteinase, or MT1 MMP, and is being investigated for safety, tolerability and efficacy in an ongoing Phase I/IIa clinical trial sponsored and fully funded by the Cancer Research UK Centre for Drug Development, or Cancer Research UK. In addition, our other product candidates, BT7480 and BT7455, are each a Bicycle tumor-targeted immune cell agonist®, or Bicycle TICATM. A Bicycle TICA links immune cell receptor binding Bicycles to tumor antigen binding Bicycles. We are evaluating BT7480, a Bicycle TICA targeting Nectin-4 and agonizing CD137, in a company-sponsored Phase I/II clinical trial, and we are conducting IND-enabling studies for BT7455, an EphA2/CD137 Bicycle TICA. Our discovery pipeline in oncology includes Bicycle-based systemic immune cell agonists and Bicycle TICAs.
Beyond our wholly owned oncology portfolio, we are collaborating with biopharmaceutical companies and organizations in additional therapeutic areas in which we believe our proprietary Bicycle screening platform can identify therapies to treat diseases with significant unmet medical need. Our partnered programs include collaborations in immuno-oncology, or I-O, anti-infective, cardiovascular, ophthalmology, dementia, central nervous system, neuromuscular and respiratory indications.
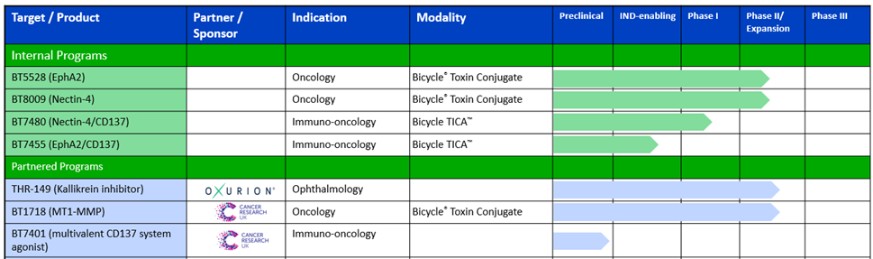
The following table summarizes key information about our programs:
2