
| Filed Pursuant to Rule 424(b)(4) | |
| PROSPECTUS | Registration No. 333-282231 |
SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
395,574 Shares of Common Stock
2,555,246 Pre-Funded Warrants to Purchase up to 2,555,246 Shares of Common Stock
2,950,820 Common Warrants to Purchase up to 2,950,820 Shares of Common Stock
2,555,246 Shares of Common Stock Underlying the Pre-Funded Warrants
2,950,820 Shares of Common Stock Underlying the Common Warrants
This is a reasonable best efforts offering of (i) 395,574 shares (the “Shares”) of common stock, par value $0.00001 per share (“common stock”) and 2,555,246 pre-funded warrants to purchase up to 2,555,246 shares common stock (the “Pre-Funded Warrants”) together with accompanying 2,950,820 common warrants to purchase up to 2,950,820 shares of common stock (the “Common Warrant”). Each share of common stock, or a Pre-Funded Warrant in lieu thereof, is being sold together with an accompanying Common Warrant to purchase one share of common stock.
The public offering price for each share of common stock and one accompanying Common Warrant is $1.525. The exercise price of the Common Warrants will be $1.40 per share.
We are offering Pre-Funded Warrants to certain purchasers whose purchase of shares of common stock in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if any such purchaser so chooses, Pre-Funded Warrants, in lieu of shares of common stock. The public offering price of each Pre-Funded Warrant and accompanying Common Warrant is $1.524, which equals the price at which one share of common stock and accompanying Common Warrant is sold to the public in this offering, minus $0.001, and the exercise price of each Pre-Funded Warrant will be $0.001 per share. The Pre-Funded Warrants will be immediately exercisable and may be exercised at any time until all of the Pre-Funded Warrants are exercised in full. The Pre-Funded Warrants and the Common Warrants are collectively referred to as the “Warrants.”
The shares of common stock or Pre-Funded Warrants in lieu thereof and Common Warrants are immediately separable and will be issued separately in this offering but must be purchased together in this offering.
Each Common Warrant offered hereby will be exercisable upon issuance (the “Initial Exercise Date”). The Common Warrants will expire five (5) years from the date of issuance. The exercise price and number of shares of common stock issuable under the Common Warrants are subject to adjustment as described in the Common Warrant.
This prospectus also includes the shares of common stock issuable upon exercise of the Pre-Funded Warrants and Warrants. See “Description of Securities We Are Offering” in this prospectus for more information.
Our common stock trades on the Nasdaq under the symbol “SHPH.” On October 29, 2024, the last reported sale price of our common stock was $1.80 per share. There is no established trading market for the Warrants, and we do not intend to list the Warrants on any securities exchange or nationally recognized trading system. Without an active trading market, the liquidity of the Warrants will be limited.
We have retained A.G.P./Alliance Global Partners (“A.G.P.”), who will work in conjunction with Boustead Securities, LLC (“Boustead”), as the placement agents (together, the “Placement Agents”) with respect to this Offering. The Placement Agents are not purchasing or selling any shares offered hereby, nor are they required to arrange for the purchase or sale of any specific number or dollar amount of shares, but they have agreed to use their reasonable best commercial efforts to arrange for the sale of the securities offered by this prospectus. We have agreed to pay the Placement Agents’ fees totaling 9.0% of the aggregate gross cash proceeds actually realized by the Company from the sale of the shares being offered hereby. Because there is no minimum amount of shares that must be sold as a condition to closing this offering, the placement agent fees and the proceeds to us are not presently determinable and may be substantially different than the amounts set forth below, which assume the sale of all of the shares offered hereby. See “Plan of Distribution” beginning on page 97 of this prospectus supplement for more information regarding these arrangements. Because there is no minimum number of securities or minimum aggregate amount of proceeds for this offering to close, we may sell fewer than all of the securities offered hereby, and investors in this offering will not receive a refund in the event that we do not sell an amount of securities sufficient to pursue the business goals outlined in this prospectus. Because there is no escrow account and there is no minimum offering amount, investors could be in a position where they have invested in our Company, but we are unable to fulfill our objectives due to a lack of interest in this offering. Also, any proceeds from the sale of securities offered by us will be available for our immediate use, despite uncertainty about whether we would be able to use such funds to effectively implement our business plan. We expect that the closing of the offering will occur on or about October 31, 2024 but no later than one trading day after we price the securities offered hereby. You should read this prospectus, together with additional information described under the heading “Where You Can Find More Information,” carefully before you invest in any of our securities.
We are an “emerging growth company” as defined in Section 2(a) of the Securities Act, and we have elected to comply with certain reduced public company reporting requirements.
An investment in our securities involves significant risks. You should carefully consider the risk factors beginning on page 13 of this prospectus before you make your decision to invest in our securities.
Neither the Securities and Exchange Commission, or the SEC, nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Per Share and Common Warrant | Per Pre-Funded Warrant and Common Warrant | Total(2) | ||||||||||
| Public offering price | $ | 1.525 | 1.524 | $ | 4,497,445.25 | |||||||
| Placement Agent Fees(1) | $ | 0.137 | 0.137 | $ | 404,770.07 | |||||||
| Proceeds to us, before expenses | $ | 1.388 | 1.388 | $ | 4,092,675.18 | |||||||
| (1) | We have agreed to pay the Placement Agents a cash fee equal to nine percent (9%) of the aggregate gross proceeds raised in this offering. In addition, we have agreed to reimburse the Placement Agents for certain offering-related expenses. We refer you to “Plan of Distribution” beginning on page 97 for additional information regarding compensation to be received by the Placement Agents.
|
| (2) | The amount of offering proceeds to us presented in this table does not give effect to any exercise of the Warrants. |
Delivery of the Shares and Warrants is expected to be made on or about October 31, 2024, subject to satisfaction of customary closing conditions.
Lead Placement Agent
A.G.P.
Co- Placement Agent
Boustead Securities, LLC
The date of this prospectus is October 29, 2024
TABLE OF CONTENTS
| i |
ABOUT THIS PROSPECTUS
The registration statement of which this prospectus forms a part and which we filed with the Securities and Exchange Commission (the “SEC”) includes exhibits that provide more detail of the matters discussed in this prospectus. You should read this prospectus and the related exhibits filed with the SEC, together with the additional information described under the heading “Where You Can Find More Information” before making your investment decision. You should rely only on the information provided in this prospectus, in any prospectus supplement or in a related free writing prospectus, or documents to which we otherwise refer you. In addition, this prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information.
This prospectus includes important information about us, the securities being offered and other information you should know before investing in our securities. You should not assume that the information contained in this prospectus is accurate on any date subsequent to the date set forth on the front cover of this prospectus, even though this prospectus is delivered or securities are sold or otherwise disposed of on a later date. It is important for you to read and consider all information contained in this prospectus in making your investment decision. All of the summaries in this prospectus are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described below under the heading “Where You Can Find More Information.”
We have not, and the Placement Agents have not, authorized anyone to provide any information or to make any representations other than those contained in this prospectus or in any free writing prospectuses prepared by or on behalf of us or to which we have referred you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. The information contained in this prospectus or contained in any applicable free writing prospectus is current only as of its date, regardless of its time of delivery or any sale of our securities. Our business, financial condition, results of operations and prospects may have changed since that date.
For investors outside the United States: We have not done anything that would permit a public offering of the securities or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the securities and the distribution of this prospectus outside of the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus outside of the United States.
You should rely only on the information contained in this prospectus and in any free writing prospectus prepared by or on behalf of us. We have not authorized anyone to provide you with information different from, or in addition to, that contained in this prospectus or any related free writing prospectus. This prospectus is an offer to sell only the securities offered hereby but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date. Our business, financial condition, results of operations and prospects may have changed since that date.
| ii |
We are not offering to sell or seeking offers to purchase these securities in any jurisdiction where the offer or sale is not permitted. We have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus and any free writing prospectus related to this offering in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions relating to this offering and the distribution of this prospectus and any such free writing prospectus applicable to that jurisdiction.
Unless otherwise indicated, any reference to Shuttle Pharmaceuticals Holdings, Inc., or as “we,” “us,” or “our” refers to Shuttle Pharmaceuticals Holdings, Inc. and its subsidiary (“Shuttle Pharma” or the “Company”).
Industry and Market Data
Unless otherwise indicated, information contained in this prospectus concerning our industry and the markets in which we operate or plan to operate, including our general expectations and market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry, and assumptions based on such information and knowledge which we believe to be reasonable. Our management estimates have not been verified by any independent source, and we have not independently verified any third-party information. In addition, assumptions and estimates of our company’s and our industry’s future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the section entitled “Risk Factors” beginning on page 13. These and other factors could cause our future performance to differ materially from our assumptions and estimates. See “Special Note Regarding Forward-Looking Statements” on page 42 below.
Basis of Presentation
On August 13, 2024, we filed a Certificate of Amendment to our certificate of incorporation with the Secretary of State of the State of Delaware to effect a 1-for-8 reverse stock split of our issued and outstanding shares of common stock, par value $0.00001 per share (the “2024 Reverse Stock Split”), which became effective on August 13, 2024. All historical share and per share amounts reflected throughout this prospectus have been adjusted to reflect the 2024 Reverse Stock Split. However, our periodic and current reports, and all other documents that were filed prior to August 13, 2024, do not give effect to the 2024 Reverse Stock Split.
| iii |
PROSPECTUS SUMMARY
This summary highlights information contained in greater detail elsewhere in this prospectus from our filings with the SEC. This summary is not complete and does not contain all of the information you should consider in making your investment decision. You should read the entire prospectus carefully before making an investment in our securities. You should carefully consider, among other things, our financial statements and the related notes and the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus.
Unless the context otherwise requires, references in this prospectus to “Shuttle Pharma,” “Shuttle Pharmaceuticals,” “the Company,” “we,” “our” and “us” refers to Shuttle Pharmaceuticals Holdings, Inc. and its subsidiary, Shuttle Pharmaceuticals, Inc.
Overview
Shuttle Pharma is a clinical stage pharmaceutical company leveraging our proprietary technology to develop novel therapies designed to cure cancers. Our goal is to extend the benefits of cancer treatments with surgery, radiation therapy, chemotherapy and immunotherapy. Radiation therapy (“RT”) is one of the most effective modalities for treating cancers. We are developing a pipeline of products designed to address the limitations of the current cancer therapies as well as to extend to the new applications of RT. We believe that our product candidates will enable us to deliver cancer treatments that are safer, more reliable and at a greater scale than that of the current standard of care.

The corporate structure is based on Shuttle Pharmaceuticals Holdings, Inc. (Nasdaq SHPH – a Delaware Company) with drug discovery and development performed in the wholly owned Shuttle Pharmaceuticals, Inc. (a Maryland Company) and diagnostics performed in the wholly owned Shuttle Diagnostics, Inc. (a Maryland Company). Our product candidates include Ropidoxuridine, a Phase II, clinical-stage radiation sensitizer, a platform of HDAC inhibitors (SP-1-161, SP-2-225 and SP-1-303) and two preclinical, prostate cancer-oriented diagnostics assets – the PC-RAD Test, a blood test to predict clinical response to radiation therapy and the PSMA-B ligand for potential use as a theranostic agent.
In December 2023, we submitted an Investigational New Drug (“IND”) application with the U.S. Food and Drug Administration (“FDA”) to support the next phase of development of Ropidoxuridine. In January 2024, we received the ‘Safe to Proceed’ letter from the FDA for our IND application for the Phase II study of Ropidoxuridine (IPdR) as a radiation sensitizing agent during radiotherapy in patients with newly diagnosed IDH-wildtype glioblastoma with unmethylated MGMT promoter. Receipt of the letter allows us to commence the Phase II study of Ropidoxuridine (IPdR). We have applied for and received FDA approval of Orphan designation for Ropidoxuridine and RT for treating brain cancer (glioblastoma). The Phase II clinical trial was also approved by the Institutional Review Board “IRB” on June 21, 2024. We believe our management team’s expertise in radiation therapy, combined modality cancer treatment and immuno-oncology will help drive the development and, if approved, the commercialization of these potentially curative therapies for patients with aggressive cancers.
| 1 |
Radiation Oncology has gone through transformative technological innovation over the last decade to better define tumors, allow improved shaping of radiation delivery and support dose escalation with shorter, more intensive courses of treatment. Furthermore, achieving higher dose distributions within tumor volumes has reached a practical plateau, since cancers are frequently integrated with or surrounded by more sensitive normal tissues and further dose escalation increases risks of tissue necrosis. To increase cancer cures at maximally tolerated radiation doses, pharmacological and biological modifications of cells are needed to sensitize cancers, protect normal tissues, and stimulate the immune system to react against antigens produced by irradiated, damaged cancer cells. Drugs that show sensitizing properties, or the ability to make cancer cells more sensitive to radiation, offer a solution to this problem. Currently, such drugs are chemotherapy agents used off-label, and many have inherent toxicities since they were designed for direct cancer treatments and not for sensitization. However, the clinical value of using radiation sensitizing drugs in combination with RT has been accepted in a variety of cancer types, including gynecological, gastro-intestinal, pulmonary and other malignancies. Hence, there is a critical need for new drugs that preferentially sensitize cancer cells to radiation therapy and that stimulate the innate immune response against irradiated cancer cells. Furthermore, to advance precision medicine in radiation oncology, there is a need for imaging and molecular diagnostic tests for determining the extent of cancer spread in the body and for predicting clinical responses to therapy.
We are developing our products with the goal of addressing the unmet need in cancer treatment for a commercially marketable radiation response modifier solution that leads to greater sensitivity of cancer cells to ionizing radiation therapy. The goal of our products is to increase the therapeutic index for patients receiving radiation and to decrease radiation-related toxicities in patients with solid tumors. Our products operate across three areas related to the treatment of cancer with RT:
| 1. | Sensitization of growing cancer cells, rendering them more susceptible to the effects of radiation therapy. | |
| 2. | Activation of the DNA damage response pathway to kill cancer cells and protect adjacent normal cells. | |
| 3. | Activation of the immune system to kill any remaining cells after RT. |
Our platform technology allows for the creation of an inventory of products for radiation sensitizing, immune modulation, and protection of healthy tissue.
Operations to date have focused on continuing our research and development efforts to advance Ropidoxuridine clinical testing and improved drug formulation, to advance HDAC6 inhibitor (SP-2-225) preclinical development and explore application of the PC-RAD Test, predictive biomarkers of radiation response. The clinical development of Ropidoxuridine has included completion of a Phase I clinical trial to establish drug bioavailability and a maximum tolerated dose for use in Phase II clinical trials. TCG GreenChem, Inc. (“TCG GreenChem”), with whom we have contracted for process research, development and cGMP compliant manufacture of IPdR, has manufactured the active pharmaceutical ingredient (API) of Ropidoxuridine and the University of Iowa Pharmaceuticals has formulated the drug product for use in the Company’s upcoming Phase II clinical trial in brain cancer patients undergoing radiation therapy. The drug product (capsules) were shipped to contract research organization (CRO) Theradex Oncology and distributed to clinical trial sites that are fully approved to enroll patients in the trial. Shuttle received approval from the FDA to begin the clinical trial. The FDA made recommendations to expand the clinical trial to include a randomized dose “optimization” step and we agreed with the recommendation. Meetings with engaged clinical sites to review the protocol documents have occurred and FDA required Institutional Review Board, or IRB approvals have been received. With FDA recommended changes incorporated into the revised protocol and the completion of site initiation visits, the Company has commenced its Phase II clinical study. The radiation biomarker project and the health disparities project have been completed and the Company is proceeding with plans for clinical validation and potential for commercialization of Ropidoxuridine as a radiation sensitizer.
| 2 |
Our Pipeline
We are currently developing a pipeline of small molecule radiation sensitizers and immune response regulating drugs. Our most advanced product candidate is Ropidoxuridine, an orally available halogenated pyrimidine with strong cancer radiation sensitizing properties is in preclinical studies. In addition, we have a pipeline of complimentary product candidates that we are developing to address solid tumor cancer indications by radiation sensitization or immune modification. Our therapeutic pipeline is represented in the diagram below:

Timeline for clinical phase (Ropidoxuridine) and pre-clinical phase (HDAC inhibitors) pipeline.
Our lead product candidates include:
| ● | Ropidoxuridine (IPdR) is our lead candidate radiation sensitizer for use in combination with RT to treat brain tumors (glioblastoma) and sarcomas. Phase I clinical trial results supported by a National Institutes of Health, or NIH contract to Shuttle Pharma and the NCI (CTEP) were reported in the medical journal, Clinical Cancer Research, in July 2019, by our Small Business Innovation Research, or SBIR, subcontractor. Eighteen patients completed dose escalations to 1,800 mg/day for 30 days, establishing the maximum tolerated dose (MTD) of 1,200 mg/day in combination with RT. Four partial responses, nine stable disease and one progressive disease in target lesions were reported. Four patients did not have measurable disease and, as a result, were not evaluable. These Phase I trial results demonstrate oral bioavailability and an MTD of 1,200 mg per day for 28 days for use in combination with radiation for Phase II clinical trials that we propose to perform in brain tumors and in sarcomas. The brain tumor, glioblastoma multiforme (GB) is eligible for orphan disease designations. Shuttle Pharma has advanced drug manufacture and formulation and prepared a clinical protocol of a “Phase 2 Single-Arm Study of IPdR as a Radiation Sensitizing Agent During Radiotherapy in Patients with Newly Diagnosed IDH-Wildtype MGMT Unmethylated Glioblastoma Multiforme.” In December 2023, we submitted an IND application with the FDA to support the next phase of development of Ropidoxuridine. In January 2024, we received the ‘Safe to Proceed’ letter from the FDA for our IND application for the Phase II study of Ropidoxuridine (IPdR) as a radiation sensitizing agent during radiotherapy in patients with newly diagnosed IDH-wildtype glioblastoma with unmethylated MGMT promoter. Receipt of the letter allows us to commence the Phase II study of Ropidoxuridine (IPdR). The clinical development of Ropidoxuridine has shown drug bioavailability and a maximum tolerated dose has been established for use in Phase II clinical trials. TCG GreenChem, Inc. (“TCG GreenChem”), with whom we have contracted for process research, development and cGMP compliant manufacture of IPdR, has successfully completed the manufacturing campaign for the active pharmaceutical ingredient (API) of Ropidoxuridine for use in the Company’s upcoming Phase II clinical trial in brain cancer patients undergoing radiation therapy. Shuttle also worked with University of Iowa Pharmaceuticals to develop the formulation and produce the capsules, which have been shipped to contract research organization (CRO) Theradex Oncology for distribution to clinical trial sites. Both activities have now been completed. In addition, Shuttle received approval from the FDA to begin the clinical trial. The FDA made recommendations that led to an expanded clinical trial to include randomized dose optimization and we agreed with the recommendation. We met with representatives from six candidate clinical sites to review the protocol documents and FDA required IRB approvals have been obtained. With FDA recommended changes incorporated into the revised protocol, the Company has now contractually engaged five of the planned six research centers which will be performing clinical trials and has initiated its Phase II clinical study. |
| 3 |
| ● | The Phase II clinical study is summarized below:
|
Schema for the Phase II clinical trial. The initial cohort of 40 patients will be randomized to one of two Ropidoxuridine doses. 20 patients will receive the 1200 mg dose and 20 patients will receive the 960 mg dose. The optimum dose will be determined by comparing drug bioavailability and side-effects. The optimum dose will then continue to enroll 14 additional patients to provide the required 34 patients for statistical significance in comparison to historical controls.
| ● | Ropidoxuridine and Tipiracil (IPdR/TPI) is a new combination formulation demonstrating extended bioavailability after oral administration in an animal model system. The IPdR/TPI formulation will undergo preclinical development for use as a radiation sensitizer and represents a “next generation” drug product for clinical evaluation. | |
| ● | SP-2-225 is Shuttle Pharma’s pre-clinical class IIb selective HDAC inhibitor that affects histone deacetylase HDAC6 has been identified as the candidate lead molecule for development as a post-RT innate immune system activator. The macrophage is a key target for activating the innate immune system against cancer cells. SP-2-225 has effects on the regulation of the immune system by maintaining macrophage cells in an inflammatory, anti-cancer polarization. The interactions of RT with the immune response for cancer treatment are of great current interest, offering insight into potential mechanisms for primary site and metastatic cancer treatment. For this reason, Shuttle Pharma has selected SP-2-225 as the candidate lead HDAC inhibitor for preclinical development. We have contracted with investigators at Georgetown University to perform preclinical studies of immune activation after radiation therapy in an animal tumor model. These data have been published (Noonepalle SKR, Grindrod S, Aghdam N, Li X, Gracia-Hernandez M, Zevallos-Delgado C, Jung M, Villagra A, Dritschilo A. Radiotherapy-induced Immune Response Enhanced by Selective HDAC6 Inhibition. Mol Cancer Ther. 2023 Dec 1;22(12):1376-1389. doi: 10.1158/1535-7163.MCT-23-0215. PMID: 37586844; PMCID: PMC10878032) and additional preclinical studies are in progress with our contractors. With the introduction of check-point inhibitors, CAR-T therapies and personalized medicine in cancer, regulation of the immune response following RT is of significant clinical and commercial interest. | |
| ● | SP-1-303 is Shuttle Pharma’s pre-clinical selective Class I HDAC inhibitor that preferentially affects histone deacetylases HDAC1 and HDAC3 members of the class I HDAC family of enzymes. SP-1-303 data show direct cellular toxicity in ER positive breast cancer cells. Furthermore, SP-1-303 increases PD-L1 expression. Completed preclinical in vitro studies have been published (Jung M, Nicholas N, Grindrod S, Dritschilo A. Dual-targeting class I HDAC inhibitor and ATM activator, SP-1-303, preferentially inhibits estrogen receptor positive breast cancer cell growth. PLoS One. 2024 Jul 15;19(7):e0306168. doi: 10.1371/journal.pone.0306168. PMID: 39008483; PMCID: PMC11249239.). We plan to seek university collaborations to complete SP-1-303 pre-clinical development in 2024. |
| 4 |
Our Approach
We believe that we have established a leadership position in radiation sensitizer and response modifier discovery and development. We have identified a clinical phase product candidate and discovered new pre-clinical phase molecules using our proprietary platform technologies to increase the therapeutic index for patients receiving radiation for treatment of solid tumors. Our development strategy has four key pillars: (1) to improve the efficacy of RT by demonstrating improved disease-free survival rates in patients who undergo radiation therapy, (2) reduce the amount of radiation needed for a favorable tumor response, thereby limiting the potential for radiation related toxicities to healthy cells, (3) decrease the extent of surgery needed to remove cancers and improve quality of life, and (4) leverage our next generation technologies to create drugs that regulate the immune response assisting immune checkpoint and CAR-T therapies and other personalized medicines targeting cancers.
In addition to private and public investment into our candidate therapeutic technology, we have also competed for non-dilutive funding from the NIH to support our lead sensitizer and to explore development of complimentary diagnostic products. To date, we have completed three SBIR contracts awarded to Shuttle Pharma by the NIH to:
| ● | Develop IPdR as a radiation sensitizer. This funding provided partial support for the Phase I clinical trial of Ropidoxuridine and RT. | |
| ● | Develop prostate cancer cell cultures from African-American men, with donor matched normal prostate cells, establishing 50 pairs for accelerating research to reduce prostate cancer health disparities in African-American men. This project was funded under “Moonshot” designation. Shuttle Pharma is eligible to apply for additional SBIR (Phase IIb) funding to commercialize these cells for research purposes. Currently, cells from African-American patients are distributed, on request, to investigators who are conducting health disparities research. We plan to test new small molecules using these cellular reagents for health disparities screening. | |
| ● | Develop a predictive biomarker for determining outcomes for prostate cancer patients following treatment with RT. This SBIR-funded project for blood test (PC-RAD Test) discovery and analytical validation were completed on March 15, 2022, and Shuttle Pharma intends to perform a clinical validation study. Shuttle Pharma has licensed the intellectual property for the prostate cancer predictive biomarker test from Georgetown University and will seek additional investment from the public market to advance clinical development through its Shuttle Diagnostics entity. |
All three SBIR funded projects have been completed. The Company is eligible to apply for SBIR Phase IIb funding to advance the “Moonshot” health disparities or the predictive biomarker project. The NIH SBIR program is designed to encourage small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization.
Shuttle Pharma’s scientists have also developed collaborations to invent intellectual properties for prostate cancer theranostics. From a clinical perspective, prostate-specific membrane antigen (PSMA) is a valuable target for diagnosis and therapy of prostate cancer. In a discovery project to develop a novel, boron-containing PSMA ligand to enhance proton radiation therapy of prostate cancer, we discovered PSMA-B, a molecule containing boron and demonstrating nanomolar binding activity to PSMA. Preclinical evaluations have been initiated to explore the PSMA-B ligand as a potential prostate cancer sensitizer in combination with proton therapy, as well as a PET diagnostic reagent and as a targeted prostate cancer therapeutic. By in-licensing our collaborator’s shares of the intellectual property, Shuttle Pharma has an exclusive license to the PSMA-B intellectual property and has filed a patent application. Theranostic molecules are suitable for diagnosis and therapy of cancers. The PSMA ligand is a molecule that binds to the PSMA, an enzyme that is highly expressed in prostate cancer cells. The PSMA ligand is currently used for imaging and therapy to detect and treat prostate cancer.
| ● | Develop PSMA-B as a potential diagnostic and therapeutic molecule in pre-clinical models in collaboration with academic nuclear medicine programs. Shuttle Pharma has licensed the intellectual property for the prostate cancer predictive biomarker test from inventors and will seek additional investment from NIH by applying for grant applications and from the public market to advance pre-clinical development through its Shuttle Diagnostics entity. |
| 5 |
Our Strategy
Our goal is to maintain and build upon our leadership position in radiation sensitization. We plan to develop Ropidoxuridine and the HDAC6 inhibitor (SP-2-225) and, if approved by the FDA, commercialize our product candidates for the treatment of cancers. While this process may require years to complete, we believe achieving this goal could result in new radiation sensitizer and immunotherapy products. Key elements of our strategy include:
| ● | Capitalize on Ropidoxuridine as an orally available, small molecule radiation sensitizer. To date, there is one drug (Cetuximab, a monoclonal antibody) approved by the FDA specifically as a radiation sensitizer. If we are successful in developing Ropidoxuridine and obtaining FDA approval, a small molecule sensitizer would then be enabled for clinical applications for radiation sensitization indications. | |
| ● | Expand our leadership position within radiation sensitizers. In addition to our traditional radiation sensitizers, we plan to advance our near-term pipeline to include radiation sensitizers for proton therapy. Proton Therapy is growing worldwide as a form of radiation therapy due to its unique beam shaping characteristics. As a result, this new technology offers a major opportunity for Shuttle Pharma to strive to develop an innovative and well-tolerated drug for proton therapy sensitization. | |
| ● | Execute a disciplined business development strategy to strengthen our portfolio of product candidates. We have built our current product pipeline through in-house discovery, development, partnerships with leading academic institutions and through in-licensing. We will continue to evaluate new in-licensing opportunities and collaboration agreements with leading academic institutions and other biotechnology companies around programs that seek to address areas of high unmet need and for which we believe there is a high probability of clinical success, including programs beyond our target franchise areas and current technology footprint. |
| ● | Invest in our HDAC platform technology and maximize its utility across cancer therapies. We are initially applying the platform to develop drugs for cancer radiation sensitization, normal tissue radiation protection and post radiation immune stimulation. Based on the data we have obtained thus far, these drugs are immune regulatory. We intend to invest to develop other properties of our platform technology, as well. | |
| ● | Enter into collaborations to realize the full potential of our platform. The breadth of our HDAC technology platform enables other therapeutic applications, including radiation sensitization and immune therapy. We intend to seek collaborations centered on our platform to maximize applications for cancer treatment. | |
| ● | Establish Shuttle Diagnostics, Inc. as a subsidiary of SHPH to advance development of the predictive biomarker (PC-RAD Test) and the PSMA ligand (PSMA-B) to advance prostate cancer treatment. |
Management Team
Our management team has significant experience in radiation oncology and in progressing products from early-stage research through clinical trials. Our Chief Executive Officer, or CEO, Anatoly Dritschilo, M.D., is an experienced clinician and researcher who has held senior academic and management positions including serving as Department Chairman, Hospital Medical Director and Cancer Center Director at Georgetown University Medical Center. Prior to co-founding our Company, Dr. Dritschilo was a co-founder of Oncomed, Inc., a company that became public as NeoPharm, Inc. (Nasdaq: NEOL). He has experience in providing care for patients undergoing treatment for cancers of the prostate, breast, brain, lung, sarcomas and GI systems. Dr. Dritschilo has directed basic science research supported by grants from the National Cancer Institute (“NCI”) and performed clinical trials using drugs and radiation therapy. In addition, Dr. Dritschilo served as the principal investigator of pharmaceutical industry sponsored clinical evaluations of human interferon alpha-2 (Bristol-Myers) with radiation therapy and antisense raf oligonucleotides, LErafAON (NeoPharm) with radiation therapy. He serves as a Radiation Biology and Radiation Oncology expert on committees of the NIH to review Program Project (P01) grant applications, Specialized Program of Research Excellence (SPORE) grant applications and investigator-initiated research project (R01) applications.
Dr. Dritschilo is supported in our clinical development effort by Tyvin Rich, MD, our Chief Clinical Officer and Medical Director. Dr. Rich is the former Professor and Chairman of the Department of Therapeutic Radiology and Oncology at the University of Virginia Health Sciences Center and proton radiation therapy specialist at the Hampton Proton Therapy Center in Hampton, Virginia. Dr. Rich has served as principal investigator on multi-modality clinical trials for the treatment of gastrointestinal (GI) cancers and helped to develop treatment with 5-fluorouracil (5-FU) as a radiation sensitizer for use with RT in the treatment of GI cancers. He has extensive cancer clinical trial experience in developing radiation sensitizer applications through his participation in the Radiation Therapy Oncology Group (RTOG). Dr. Rich is a co-inventor with scientists at the University of Virginia of the Proton Activated Atomic Medicine technology.
| 6 |
Our administrative services are provided by Peter Dritschilo, MBA, who has served as our President and COO since 2012. Mr. Dritschilo’s experience in hospital administration and management of medical oncology clinical services and radiation therapy facilities, including management of day-to-day operations, human resources and financial oversight. Peter Dritschilo is the son of our Chairman and CEO, Dr. Anatoly Dritschilo. Mr. Timothy J. Lorber is our Chief Financial Officer, or CFO, a position he has served in since June 2024. Mr. Lorber is a CPA with more than 40 years of professional finance experience. We believe his extensive experience and expertise in the financial industry supports his service as our CFO. Michael Vander Hoek, our former CFO, as Vice President for Operations and Regulatory expands our capability to provide the level of management needed for the proposed expansion of clinical trials. Mr. Vander Hoek served as administrative director of the Lombardi Comprehensive Cancer Center (LCC) for 12 years and has extensive experience in negotiations, management and supervision of Contract Research Organizations (CROs) and research contracts in general. As the administrative director of the Lombardi Comprehensive Cancer Center, Mr. Vander Hoek also served as the chief financial officer. Taken together, we believe our leadership team of highly qualified specialists will help us achieve the proposed milestones for the development of radiation sensitizer products.
Nasdaq Deficiency
Our common stock currently is listed for quotation on the Nasdaq Capital Market. We are required to meet Nasdaq listing rules in order to maintain such listing, including a requirement that the Company’s stockholders equity remain at or above $2.5 million.
On September 10, 2024, we received a letter from The Nasdaq notifying the Company that it is no longer in compliance with the minimum stockholders’ equity requirement for continued listing on the Nasdaq Capital Market. Nasdaq Listing Rule 5550(b)(1) requires listed companies to maintain stockholders’ equity of at least $2.5 million. In the Company’s Quarterly Report on Form 10-Q for the period ended June 30, 2024, the Company reported stockholders’ equity of $801,434, which is below the minimum stockholders’ equity required for continued listing pursuant to Nasdaq Listing Rule 5550(b)(1). In addition, at present, the Company does not meet the alternatives of market value of listed securities or net income from continuing operations.
This Nasdaq notice has no immediate effect on the listing of the Company’s securities on the Nasdaq Capital Market. Nasdaq has provided the Company with 45 calendar days, or until October 25, 2024, to submit a plan to regain compliance with the minimum stockholders’ equity standard. The Company subsequently submitted a plan of compliance to Nasdaq on October 15, 2024. If the Company’s plan to regain compliance is accepted, Nasdaq may grant an extension of up to 180 calendar days from September 10, 2024 for the Company to regain compliance.
There can be no assurance that the Company’s plan will be accepted or that, if it is, the Company will be able to regain compliance and maintain its listing on the Nasdaq Capital Market. If the Company’s plan to regain compliance is not accepted or if Nasdaq does not grant the Company a 180-day extension to regain compliance, or if the Company fails to satisfy another Nasdaq requirement for continued listing, Nasdaq could provide notice that the Company’s securities will become subject to delisting. In such event, the Company will have an opportunity to appeal Nasdaq’s decision to a hearings panel.
2024 Reverse Stock Split
On August 31, 2023 we received a letter from the Nasdaq Listing Qualifications Staff of the Nasdaq (the “Staff”) stating that for the 30 consecutive business day period the Company’s common stock had failed to maintain a minimum closing bid price of $1.00 per share, as required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”). In accordance with Nasdaq rules, the Company was provided with an initial period of 180 calendar days, or until February 27, 2024, to regain compliance with the Minimum Bid Price Requirement and a second Grace Period on February 19, 2024 through August 26, 2024.
In order to regain compliance with the Minimum Bid Price Requirement, on August 6, 2024, we filed an amendment to our certificate of incorporation to effect a 1-for-8 reverse stock split of our outstanding common stock (the “2024 Reverse Stock Split”). The 2024 Reverse Stock Split became effective on August 13, 2024, when the Company’s common stock opened for trading on Nasdaq on a post-split basis under the Company’s existing trading symbol, “SHPH.” Subsequently, on August 27, 2024, we received notice from Nasdaq that we regained compliance with the Minimum Bid Price Requirement.
All historical share and per share amounts reflected throughout this prospectus have been adjusted to reflect the 2024 Reverse Stock Split. However, our periodic and current reports, and all other documents that were filed prior to August 13, 2024 do not give effect to the 2024 Reverse Stock Split.
Going Concern Uncertainty
Our condensed consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and had a net loss of approximately $3.8 million and no revenues for the six months ended June 30, 2024 and had working capital of approximately $1.1 million as of June 30, 2024. In addition, the convertible note payable outstanding at June 30, 2024 included covenants and certain cash payment requirements. The convertible note payable outstanding at June 30, 2024, however, was paid off in full as of September 30, 2024. These conditions, and the Company’s ability to comply with such conditions, raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the condensed consolidated financial statements are issued.
| 7 |
In September 2022, the Company completed its initial public offering, or IPO, generating net proceeds of approximately $10.0 million. And in January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible note with a principal value of $4.3 million (the “January 2023 Convertible Note”), along with a four-year warrant to purchase 1,018,079 shares of common stock (127,260 shares, post Reverse Split), exercisable at $1.93 per share (post Reverse Split, as adjusted), providing the Company with approximately $3.6 million in net proceeds. While the January 2023 Note has been paid off in full, to date, the warrant has not yet been exercised.
The IPO and subsequent capital raise have supported operations leading up to the manufacture of drug product and FDA approval of the IND for the Phase II clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the Company agreed to an expansion of the clinical trial, necessitating additional capital to complete the trial as well as fund ongoing operations. The ability of the Company to continue as a going concern is dependent upon its ability to successfully raise additional equity or debt financing, including through bridge financing, to allow the Company to fund ongoing operations, conduct clinical trials and bring a drug candidate to commercialization to generate revenues.
If we fail to obtain additional material financing in the near-term, our clinical trials and targeted FDA submission timeline could be delayed, and we could be forced to abandon such activities entirely and cease operations, with the possible loss of such properties or assets. If we are unable to obtain a material quantum of financing in the imminent future or unable to continue to obtain additional financing over at least the next 12 months as we continue to generate negative cash flow, our board of directors could determine to cause the Company to undertake a process of liquidation under Chapter 7 of applicable U.S. bankruptcy laws, or otherwise seek other protection under such laws. In such event, we expect that holders of shares of our common stock would recoup little if any material value in such process.
Recent Financings – Our IPO and Post-2022 Financing
On September 2, 2022, we closed on our IPO of 1,225,888 units (each a “IPO Unit,” and collectively, the “IPO Units”), with each IPO Unit consisting of one share of the Company’s common stock and one warrant to purchase one share of common stock, at a public offering price of $8.125 per IPO Unit ($65 per IPO Unit, on a post-reverse split basis). Our IPO, which was underwritten by Boustead Securities, LLC (“Boustead”), resulted in gross proceeds of $9.96 million, before deducting underwriting discounts and commissions. On September 29, 2022, Boustead exercised its overallotment option, purchasing an additional 183,883 IPO Units, resulting in gross proceeds of $1.49 million, before deducting underwriter commissions and discounts. As a result, our IPO raised a total of $11.45 million, before deducting underwriting discounts, commissions and related IPO expenses. On a post Reverse Split basis, 352,443 shares (including exercised warrants) were issued under the IPO.
On January 11, 2023, we entered into a securities purchase agreement (the “SPA”) with Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B, a Cayman entity (the “Investor”), as amended on May 10, 2023 and June 4, 2023, pursuant to which the Company sold to the Investor a $4.3 million convertible note (the “January 2023 Convertible Note”) and warrant (the “January 2023 Warrant”) to purchase 127,260 (post Reverse Split) shares of common stock of the Company, in exchange for gross proceeds of $4.0 million (the “Investment Amount”). The January 2023 Convertible Note amortizes on a monthly basis and the Company can make such monthly amortization payments in cash or, subject to certain equity conditions, in registered shares of common stock or a combination thereof. For equity repayment, the January 2023 Convertible Note is convertible into shares of common stock at price per share equal to the lower of (i) $18.8 (post Reverse Split) (ii) 90% of the three lowest daily VWAPs of the 15 trading days prior to the payment date or (iii) 90% of the VWAP of the trading day prior to payment date. The January 2023 Convertible Note is repayable over 26 months and bears interest at the rate of 5% per annum. The January 2023 Warrant is exercisable for four years from the date of closing and is exercisable at $1.93 per share (post Reverse Split, as adjusted). In the event the Investor exercises the January 2023 Warrant in full, such exercise would result in additional gross proceeds to the Company of approximately $246 thousand. As of the date of this filing, the January 2023 Convertible Note has been paid in full.
On September 4, 2024, the Company entered into a loan agreement with our Chief Executive Officer, Dr. Anatoly Dritschilo, pursuant to which Dr. Dritschilo loaned the Company $250,000 (principal), bearing interest at the rate of 12% per annum and which is repayable in 12 substantially equal monthly installments over a one year period.
On October 14, 2024, the Company conducted the initial closing of an up to $1.3 million 5% original issue discount (“OID”) senior secured convertible note warrant offering, pursuant to which the Company issued a total of $600,000 notes and warrants to purchase 286,169 shares of common stock, exercisable at $1.53 per share. The notes bear interest at the rate of 14.5% per annum, with the interest payable quarterly in cash, mature one year after the date of issuance, are redeemable at any time by the Company at a 107% premium to the principal value of the notes and, three months after issuance, can be converted at any time by the holder at a 110% premium to the principal value of the note. Of the above investment amount, the Company’s CEO purchased $250,000 of notes and warrants in the October 2024 Bridge Offering. The closing of the offering is occurring on a rolling basis and the Company anticipates there may be additional investments in the October 2024 Bridge Offering in coming days.
| 8 |
Summary Risk Factors
Our business is subject to a number of risks you should be aware of before making an investment decision. These risks are discussed more fully in the “Risk Factors” section of this prospectus at page 13 immediately following this prospectus summary, and in Part I, Item 1A “Risk Factors” of our Annual Report on Form 10-K for the fiscal year ended December 31, 2023, as amended on September 3, 2024, and in Part II, Item 1A “Risk Factors” of our Quarterly Report on Form 10-Q for the quarter ended June 30, 2024, which are each included in this prospectus. These risks include the following:
| ● | Our ability to continue as a going concern in the near term is dependent upon us successfully raising additional equity or debt financing to fund our operations. | |
| ● | Our success is primarily dependent on achieving the development, regulatory approval and commercialization of our product candidates, both of which are in the early stages of development. | |
| ● | Our approach to the discovery and development of innovative radiation oncology drugs based on our HDAC small molecule delivery platform, which is novel, unproven and may not result in marketable products. | |
| ● | We have no product revenue, have incurred significant losses since inception, may never become profitable and may incur substantial and increasing net losses for the foreseeable future as we continue the development of, and seek regulatory approvals for our product candidates. | |
| ● | If clinical trials of our product candidates fail to demonstrate safety and efficacy, which are ongoing determinations that are solely within the authority of the FDA, we may be unable to obtain regulatory approvals to commercialize our product candidates. | |
| ● | We are subject to regulatory approval processes that are lengthy, time-consuming and unpredictable. We may not obtain approval for any of our product candidates from the FDA or foreign regulatory authorities. | |
| ● | Even if we obtain regulatory approval, the market may not be receptive to our product candidates. | |
| ● | We may not be able to establish collaborative partnerships with other pharmaceutical companies, through which we expect to complete development of, obtain marketing approval for and, if approved, manufacture and market our product candidates. | |
| ● | We may encounter difficulties satisfying the requirements of clinical trial protocols, including patient enrollment. | |
| ● | We may face competition from other companies in our field or claims from third parties alleging infringement of their intellectual property. | |
| ● | We may be unable to recruit or retain key employees, including our senior management team. | |
| ● | Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business. | |
| ● | We are a Phase I clinical stage pharmaceutical company with a limited operating history upon which you can evaluate our business and prospects. Specialty pharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. |
| 9 |
| ● | We do not currently have any product candidates in advanced clinical trials or approved for sale, and we continue to incur significant research and development and general and administrative expenses in relation to our operations. In addition, we have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the specialty pharmaceutical industry. | |
| ● | We have not submitted an application or received marketing approval for any of our product candidates. Regulatory approval of our product candidates is not guaranteed, and the approval process is expensive and may take several years. | |
| ● | It is difficult and costly to protect our intellectual property rights. | |
| ● | If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed. | |
| ● | On September 4, 2024, we restated our annual report on Form 10-K for the year ended December 31, 2023, as a result of a re-audit of our 2022 financial statements, and there may be some risk of regulatory, shareholder / investor or other actions or consequences as a result of the restatement. | |
| ● | The future issuance of equity or of debt securities that are convertible into common stock will dilute our share capital.
| |
| ● | If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed. |
Implications of Being an Emerging Growth Company
| ● | As a smaller reporting company, and as a company with less than $1.235 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the JOBS Act. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from specified disclosure and other requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include: |
| ● | being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; | |
| ● | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
| ● | not being required to comply with any mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; | |
| ● | reduced disclosure obligations regarding executive compensation; and | |
| ● | exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
| 10 |
We may take advantage of the above provisions for up to five years or until such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.235 billion in annual revenues, have more than $700 million in market value of our capital stock held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. We may also choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of some reduced reporting requirements in this prospectus. Accordingly, the information contained in this prospectus may be different than the information you receive from other public companies in which you hold stock.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an emerging growth company to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period for adopting new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Corporate Information
The Company was formed as a limited liability company in the state of Maryland in December 2012 and was converted to a C corporation in August 2016. In June 2018, Shuttle completed a share exchange with Shuttle Pharma Acquisition Corp. Inc. (“Acquisition Corp.”), pursuant to which Shuttle Pharmaceuticals, Inc. became a subsidiary of Acquisition Corp. and we subsequently changed the name of Acquisition Corp. to Shuttle Pharmaceuticals Holdings, Inc.
Our executive offices are located at 401 Professional Drive, Suite 260, Gaithersburg, MD 20879 and our telephone number is (240) 430-4212. Our corporate website is www.shuttlepharma.com. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and all amendments to those reports, if any, are available to you free of charge through the “Investor Relations” section of our website as soon as reasonably practicable after such materials have been electronically filed with or furnished to the SEC. Information contained on our websites does not form a part of this prospectus.
| 11 |
THE OFFERING
| Common stock we are offering: | 395,574 shares of common stock based on a combined public offering price of $1.525 per share of common stock and accompanying Common Warrant. |
| Pre-Funded Warrants to be offered: | We are also offering 2,555,246 Pre-Funded Warrants to purchase up to 2,555,246 shares of common stock in lieu of Shares of common stock to each purchaser whose purchase of shares of common stock in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering. The purchase price of each Pre-Funded Warrant and accompanying Common Warrant is $1.524, which equals the price at which one share of common stock and accompanying Common Warrant is sold to the public in this offering, minus $0.001, and the exercise price of each Pre-Funded Warrant will be $0.001 per share. The Pre-Funded Warrants will be exercisable immediately and may be exercised at any time until all of the Pre-Funded Warrants are exercised in full. For each Pre-Funded Warrant we sell, the number of shares of Common Stock we are offering will be decreased on a one-for-one basis.
The Pre-Funded Warrants have no stand-alone rights and will not be certificated or issued as stand-alone securities.
This prospectus also relates to the offering of our common stock issuable upon exercise of the Pre-Funded Warrants. See “Description of Securities We Are Offering – Pre-Funded Warrants.” |
| Common Warrants to be offered: | We are also offering 2,950,820 Common Warrants. Each share of common stock (or Pre-Funded Warrant to purchase one share of common stock in lieu thereof) sold includes one Common Warrant to purchase one share of common stock, exercisable at $1.40 per share. The Common Warrants will be exercisable on the Initial Exercise Date. The Common Warrants will expire five years from such date. See “Description of the Securities We Are Offering – Common Warrants.” |
| Common stock outstanding before this offering: | 2,946,099 shares of common stock(1) |
| Common stock outstanding immediately after this offering: | 3,341,673 shares (assuming no exercise of the Pre-Funded Warrants or Common Warrants issued in this offering). |
| Dividend policy: | We have never paid cash dividends on our common stock and we do not anticipate paying any cash dividends in the foreseeable future. See the section entitled “Dividend Policy.” |
| Use of proceeds: | We estimate that the net proceeds from this offering will be approximately $4.1 million, after deducting the cash fees to be paid to the Placement Agents and estimated offering expenses payable by us, and excluding the proceeds, if any, from the exercise of the Common Warrants in this offering. |
| We intend to use the net proceeds from this offering to fund IND-enabling and Phase I and II clinical trials of product candidates, including $2.3 million in payments that will be owed to Theradex Systems, Inc., the clinical research organization (CRO) supporting our Phase II clinical trials for radiation sensitizer Ropidoxuridine. We anticipate that the funds raised from this offering will allow us to complete the Phase II clinical trial, although there is no guarantee that we will not require additional funds. See “Use of Proceeds” beginning on page 43 below. | |
| Nasdaq trading symbol: | Our common stock is listed on Nasdaq under the symbol “SHPH.” There is no established trading market for the Common Warrants or Pre-Funded Warrants, and we do not expect a market to develop. In addition, we do not intend to apply for the listing of the Common Warrants or Pre-Funded Warrants on any national securities exchange or other trading market. Without an active trading market, the liquidity of the Common Warrants and Pre-Funded Warrant will be limited. |
| Risk Factors: | See “Risk Factors” beginning on page 13 of this prospectus and other information included in this prospectus for a discussion of the risk factors you should consider carefully when making an investment decision.
|
| Reasonable best efforts offering | We have agreed to offer and sell the securities offered hereby to the purchasers through the Placement Agents. The Placement Agents are not required to buy or sell any specific number or dollar amount of the securities offered hereby but will use their reasonable best efforts to solicit offers to purchase the securities offered by this prospectus. See “Plan of Distribution” on page 97 of this prospectus. |
| (1) | The number of shares of our common stock to be outstanding before this offering is based on 2,946,099 shares of our common stock outstanding as of October 29, 2024, and, unless otherwise indicated, excludes, as of that date: (i) 184,000 warrants to purchase common stock, exercisable at an average exercise price of $11.54 per share; and (ii) the 732,134 shares issuable under the convertible bridge notes and warrants to purchase 329,460 shares of common stock, exercisable at a weighted average price of $1.42 per share, issued in the October 2024 Convertible Note Offering. |
Except as otherwise indicated, the information in this prospectus assumes no exercise of any Warrants to be issued in this offering.
| 12 |
RISK FACTORS
An investment in our common stock involves a high degree of risk. You should carefully consider the following risk factors and all the other information in this registration statement before you decide to buy our common stock. If any of the following risks related to our business actually occurs, our business, financial condition, operating results, and prospects would be adversely affected. The market price of our common stock could decline due to any of these risks and uncertainties related to our business, or related to an investment in our common stock, and you may lose part or all of your investment. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial may also adversely affect our business. Certain statements below are forward-looking statements.
Risks Related to this Offering
Sales of substantial amounts of our securities in the public market could depress the market price of our common stock.
Our common stock is listed for trading on the Nasdaq Capital Market. If our stockholders sell substantial amounts of our common stock in the public market, or the market perceives that such sales may occur, the market price of our securities could fall and we may be unable to sell our securities in the future.
Our securities may experience extreme price and volume fluctuations, which could lead to costly litigation for us and make an investment in us less appealing.
The market price of our common stock may fluctuate substantially due to a variety of factors, including:
| ● | the status and results of our clinical trials for our product candidate; |
| ● | our ability to fund and complete our study and, if such study provides data supporting an FDA submission, our ability to apply for and obtain clearance from the FDA; |
| ● | our ability to remain a going concern; |
| ● | our ability to maintain our Nasdaq listing; |
| ● | our business strategy and plans; |
| ● | the potential market for our product candidate, if approved for sale in the U.S.; |
| ● | new regulatory pronouncements and changes in regulatory guidelines and timing of regulatory approvals; |
| ● | general and industry-specific economic conditions; |
| ● | variations in our quarterly financial and operating results, including the rate at which we incur negative cash flow in future periods; |
| ● | additions to or departures of our key personnel; |
| ● | changes in market valuations of other companies that operate in our business segments or in our industry; |
| ● | lack of trading liquidity; |
| ● | if our product is approved and becomes available for us to sell in the U.S., whether we ultimately achieve profitability or not; |
| ● | changes in accounting principles; and |
| ● | general market conditions, economic and other external factors. |
The market prices of the securities of early-stage companies, particularly companies like ours that are seeking to obtain regulatory approval of their product candidate and do not yet generate operating revenue, have been highly volatile and are likely to remain highly volatile in the future. This volatility has often been unrelated to the operating performance of particular companies. In the past, companies that experience volatility in the market price of their securities have often faced securities class action litigation. Whether or not meritorious, litigation brought against us could result in substantial costs, divert our management’s attention and resources and harm our financial condition and results of operations.
| 13 |
Although we have no preferred stock outstanding as of the date hereof and we have currently no intention to issue any preferred stock, our common stockholders could be adversely affected by the issuance by us of preferred stock in the future, if any.
Our certificate of incorporation does not restrict our ability to offer one or more series of preferred stock, any or all of which could rank equally with or have preferences over our common stock as to dividend payments, voting rights, rights upon liquidation or other types of rights. Our board of directors has the authority, without further action by the stockholders, to issue shares of preferred stock in one or more series and to fix the rights, preferences and the number of shares constituting any series or the designation of such series. In the case our board of directors decides to issue any preferred stock, we would have no obligation to consider the specific interests of the holders of common stock in creating any such series of preferred stock or engaging in any such offering or transaction. Our creation of any series of preferred stock or our engaging in any such offering or transaction could have a material adverse effect on holders of our common stock.
We have broad discretion in how we use the net proceeds of this offering, and we may not use these proceeds effectively or in ways with which you agree.
Our management will have broad discretion as to the application of the net proceeds of this offering and could use them for purposes other than those contemplated at the time of the offering. We currently intend to use the net proceeds from the offering to fund IND-enabling and Phase I and II clinical trials of product candidates, including radiation sensitizer Ropidoxuridine, IPdR/TPI and the HDAC inhibitor small molecule technology platform, potential acquisition or in-licensing activities and working capital and general corporate purposes. Our stockholders may not agree with the manner in which our management chooses to allocate and spend the net proceeds. Moreover, our management may use the net proceeds for corporate purposes that may not increase the market price of our common stock.
If we fail to comply with the continued listing requirements of Nasdaq, it could result in our common stock being delisted, which could adversely affect the market price and liquidity of our securities and could have other adverse effects.
On September 10, 2024, we received a letter (the “Notification”) from Nasdaq notifying the Company that it is no longer in compliance with the minimum stockholders’ equity requirement for continued listing on the Nasdaq Capital Market. Nasdaq Listing Rule 5550(b)(1) requires listed companies to maintain stockholders’ equity of at least $2,500,000. In the Company’s Quarterly Report on Form 10-Q for the period ended June 30, 2024, the Company reported stockholders’ equity of $801,434, which is below the minimum stockholders’ equity required for continued listing pursuant to Nasdaq Listing Rule 5550(b)(1). In addition, presently, the Company does not meet the alternatives of market value of listed securities or net income from continuing operations.
This Notification has no immediate effect on the listing of the Company’s securities on the Nasdaq Capital Market. Nasdaq has provided the Company with 45 calendar days, or until October 25, 2024, to submit a plan to regain compliance with the minimum stockholders’ equity standard. If the Company’s plan to regain compliance is accepted, Nasdaq may grant an extension of up to 180 calendar days from September 10, 2024 for the Company to regain compliance.
The Company is presently evaluating various courses of action to regain compliance and intends to timely submit a compliance plan to Nasdaq. However, there can be no assurance that the Company’s plan will be accepted or that, if it is, the Company will be able to regain compliance and maintain its listing on the Nasdaq Capital Market. If the Company’s plan to regain compliance is not accepted or if Nasdaq does not grant an extension and the Company does not regain compliance by October 25, 2024, or if the Company fails to satisfy another Nasdaq requirement for continued listing, Nasdaq could provide notice that the Company’s securities will become subject to delisting. In that event, the Company will have an opportunity to appeal Nasdaq’s decision to a hearings panel.
While we aim to regain compliance with Nasdaq’s stockholders equity requirement, we nonetheless run the risk that our stock may be delisted if we fail to comply with Nasdaq listing requirements. In the event our common stock is delisted, we may seek to have our common stock quoted on an over-the-counter marketplace, such as on the OTCQX. The OTCQX is not a stock exchange, and if our common stock trades on the OTCQX rather than a securities exchange, there may be significantly less trading volume and analyst coverage of, and significantly less investor interest in, our common stock, which may lead to lower trading prices for our common stock.
| 14 |
Any potential delisting of our common stock from the Nasdaq may have materially adverse consequences to our stockholders, including:
| ● | a reduced market price and liquidity with respect to our shares of common stock, which could make our ability to raise new investment capital more difficult; |
| ● | limited dissemination of market price of our common stock; |
| ● | limited news coverage; |
| ● | limited interest by investors in our common stock; |
| ● | volatility of the prices of our common stock, due to low trading volume; |
| ● | our common stock being considered a “penny stock,” which would result in broker-dealers participating in sales of our common stock being subject to the regulations set forth in Rules 15g-2 through 15g-9 promulgated under the Exchange Act |
| ● | increased difficulty in selling our common stock in certain states due to “blue sky” restrictions; and |
| ● | limited ability to issue additional securities or to secure additional financing. |
If you purchase our securities in this offering, you may experience future dilution as a result of future equity offerings or other equity issuances.
We may offer and issue additional shares of our common stock or other securities convertible into or exchangeable for our common stock in the future. We are generally not restricted from issuing additional securities, including shares of common stock, securities that are convertible into or exchangeable for, or that represent the right to receive, common stock or substantially similar securities. The issuance of securities in future offerings may cause dilution to our stockholders, including investors in this offering. We cannot assure you that we will be able to sell shares or other securities in any other offering at a price per share that is equal to or greater than the price per share paid by investors in this offering, and investors purchasing other securities in the future could have rights superior to existing stockholders. The price per share at which we sell additional shares of our common stock or other securities convertible into or exchangeable for our common stock in future transactions may be higher or lower than the price per share in this offering.
Trading of our common stock may be limited, making it difficult for our stockholders to sell their shares, and future sales of common stock could reduce our stock price.
Our common stock currently trades on Nasdaq under the ticker “SHPH.” The liquidity of our common stock may be limited, including in terms of the number of shares that can be bought and sold at a given price and reduction in security analysts’ and the media’s coverage of us, if any. These factors may result in different prices for our common stock than might otherwise be obtained in a more liquid market and could also result in a larger spread between the bid and asked prices for our common stock. In addition, in the absence of a large market capitalization, our common stock is less liquid than the stock of companies with broader public ownership, and, as a result, the trading prices of our common stock may be more volatile. In the absence of an active public trading market, an investor may be unable to liquidate his/her investment in our common stock. Trading of a relatively small volume of our common stock may have a greater impact on the trading price of our stock. We cannot predict the prices at which our common stock will trade in the future, if at all.
We do not currently intend to pay dividends on our common stock in the foreseeable future and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We do not anticipate paying any cash dividends to holders of our common stock for the foreseeable future. Consequently, investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investments. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which our stockholders have purchased their shares.
The offering price of the common stock or Pre-Funded Warrants may not be indicative of the value of our assets or the price at which securities can be resold. The offering price of the common stock or Pre-Funded Warrants may not be an indication of our actual value.
The public offering price per common stock or Pre-Funded Warrant is being determined based upon negotiations between the Company and the Placement Agents. Factors taken into consideration include the trading volume of our common stock prior to this offering, the historical prices at which our shares of common stock have recently traded, the history and prospects of our business, the stage of development of our business, our business plans for the future and the extent to which they have been implemented, an assessment of our management, general conditions of the securities markets at the time of the offering, and such other factors as we and the Placement Agents deem relevant. No assurance can be given that our common stock can be resold at the public offering price for the shares. You may not receive a positive return on your investment when you sell your shares of common stock and you may lose the entire amount of your investment in the common stock, Pre-Funded Warrants, or the Common Warrants.
| 15 |
The Common Warrants and Pre-Funded Warrants are speculative in nature.
The Common Warrants and Pre-Funded Warrants offered hereby do not confer any rights of common stock ownership on their holders, such as voting rights or the right to receive dividends, but rather merely represent the right to acquire shares of common stock at a fixed price. Specifically, commencing on the date of issuance, holders of the Pre-Funded Warrants may acquire the common stock issuable upon exercise of such warrants at an exercise price of $0.001 per share and holders of the Common Warrants may acquire the common stock issuable upon exercise of such warrants at an exercise price of $1.40 per share. Moreover, following this offering, the market value of the Pre-Funded Warrants and Common Warrants will be uncertain and there can be no assurance that the market value of such Warrants will equal or exceed their public offering price. There can be no assurance that the market price of the common stock will ever equal or exceed the exercise price of the Common Warrants, and consequently, whether it will ever be profitable for holders of the Common Warrants to exercise the Common Warrants.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock may depend in part on research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on us. If securities or industry analysts do not cover us or commence coverage of us, the trading price for our stock would likely be negatively impacted. In the event securities or industry analysts initiate coverage, if one or more of the analysts who covers us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price may decline. If one or more of these analysts ceases coverage of us or fails to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.
FINRA sales practice requirements may limit a stockholder’s ability to buy and sell our securities.
Effective June 30, 2020, the SEC implemented Regulation Best Interest requiring that “A broker, dealer, or a natural person who is an associated person of a broker or dealer, when making a recommendation of any securities transaction or investment strategy involving securities (including account recommendations) to a retail customer, shall act in the best interest of the retail customer at the time the recommendation is made, without placing the financial or other interest of the broker, dealer, or natural person who is an associated person of a broker or dealer making the recommendation ahead of the interest of the retail customer.” This is a significantly higher standard for broker-dealers to recommend securities to retail customers than before under FINRA “suitability rules.” FINRA suitability rules do still apply to institutional investors and require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending securities to their customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information, and for retail customers determine the investment is in the customer’s “best interest” and meet other SEC requirements. As a result, fewer broker-dealers may be willing to make a market in our common stock, reducing a stockholder’s ability to resell shares of our common stock.
This offering may cause the trading price of our shares of common stock to decrease.
The price per common stock or Pre-Funded Warrant, may result in an immediate decrease in the market price of our shares. This decrease may continue after the completion of this offering.
Resales of our shares of common stock in the public market by the stockholders as a result of this offering may cause the market price of our shares of common stock to decline.
Sales of substantial amounts of our shares of common stock in the public market, or the perception that such sales might occur, could adversely affect the market price of our shares of common stock. The issuance of new shares of common stock could result in resales of our shares of common stock by our current shareholders concerned about the potential ownership dilution of their holdings. Furthermore, in the future, we may issue additional shares of common stock or other equity or debt securities exercisable or convertible into shares of common stock. Any such issuance could result in substantial dilution to our existing shareholders and could cause our stock price to decline.
| 16 |
Risks Related to Our Business, Financial Condition and Capital Requirements
Our consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business; our ability to continue as a going concern is dependent upon our ability to successfully conduct clinical trials, bring a drug candidate to commercialization, generate revenues, and to raise additional equity or debt financing to fund our operations.
Our consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and had a net loss of approximately $6.6 million and no revenues for the year ended December 31, 2023, with working capital of approximately $4.6 million as of December 31, 2023. In addition, the convertible note payable outstanding at December 31, 2023 includes covenants and certain cash payment requirements. These conditions, and the Company’s ability to comply with such conditions, raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the consolidated financial statements are issued.
In September 2022, the Company completed its initial public offering of common stock, generating net proceeds of approximately $10.0 million. Additionally, in January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible note with a principal value of $4.3 million, along with a four-year warrant to purchase 127,260 shares of common stock, exercisable at $18.80 per share ($1.93 post Reverse Split, as adjusted), providing the Company with approximately $3.6 million in net proceeds. To date, the warrant has not yet been exercised. However, the Company’s existing cash resources, marketable securities and the cash received from the Company’s initial public offering and convertible note offering are not expected to provide sufficient funds to support the Company’s operations and clinical trials through the next twelve months.
The capital raise has supported operations leading up to the manufacture of drug product and FDA approval of the IND for the Phase II clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the company agreed to an expansion of the clinical trial, necessitating additional capital expenditures to complete the trial. Management intends to initiate a rights offering for $4.5 million and has submitted SBIR applications for non-dilutive NIH funding for our pre-clinical project. The ability of the Company to continue as a going concern is dependent upon our ability to successfully conduct clinical trials, bring a drug candidate to commercialization, generate revenues, and to raise additional equity or debt financing to fund our operations.
Our success is primarily dependent on the successful development, regulatory approval and commercialization of our product candidates, all of which are in the early stages of development.
We currently have one clinical stage product candidate in the early stages of development. Ropidoxuridine has undergone an SBIR funded Phase I clinical trial at Lifespan/Rhode Island Hospital. We also have an HDAC inhibitor small molecule platform. The three lead drug candidate molecules are in preclinical phases of development. None of our product candidates have gained marketing approval for sale in the United States or any other country, and we cannot guarantee that we will ever have marketable products. To date, we have invested substantially all of our efforts and financial resources in the research and development and commercial planning for our current product candidate and our HDAC small molecule delivery platform. Our near-term prospects, including our ability to finance our Company and generate revenue, as well as our future growth, will depend heavily on the development, marketing approval and commercialization of our product candidates. The clinical and commercial success of product candidates will depend on a number of factors, including the following:
| ● | building on favorable results from our Phase I clinical trial for IPdR and proceeding to Phase II and Phase III clinical trials, which may be slower or cost more than we currently anticipate; | |
| ● | our ability to demonstrate safety and efficacy of our product candidates, which are ongoing determinations that are solely within the authority of the FDA; | |
| ● | even if our clinical trials are completed, there can be no assurance that the FDA will agree that we have satisfactorily demonstrated safety or efficacy or that the FDA will not raise new issues regarding the design of our clinical trials; |
| 17 |
| ● | whether we are required by the FDA to conduct additional clinical trials to support the approval of our product candidates; | |
| ● | the acceptance by the FDA of our proposed parameters for regulatory approval, including our proposed indication, endpoints and endpoint measurement tools relating to our product candidates; | |
| ● | the incidence, duration and severity of adverse side effects; | |
| ● | the timely receipt of necessary marketing approvals from the FDA; | |
| ● | whether we are able to secure collaborations for completing the development and, if approved, commercialization of our product candidates; |
| ● | the effectiveness of our and our potential collaborators’ marketing, sales and distribution strategy and operations of product candidates that are approved; | |
| ● | our success in educating physicians and patients about the benefits, administration and use of our product candidates; | |
| ● | the ability of our third-party manufacturers and potential collaborators to manufacture clinical trial and commercial supplies of our product candidates to remain in good standing with regulatory bodies, and to develop, validate and maintain commercially viable manufacturing processes that are compliant with current Good Manufacturing Practices (“cGMP”) regulations; | |
| ● | our ability to commercialize our product candidates, if approved for marketing; | |
| ● | our ability to enforce our intellectual property rights; | |
| ● | our ability to avoid third-party patent interference or patent infringement claims; | |
| ● | acceptance of our product candidates as safe and effective by patients and the medical community; and | |
| ● | a continued acceptable quality profile of our product candidates following approval. |
Many of the above-listed risk factors are beyond our control. Accordingly, we cannot assure you that we will ever be able to generate revenue through the sale of our product candidates. Any one of these factors or other factors discussed in this Annual Report could affect our ability to commercialize product candidates, which could impact our ability to earn sufficient revenues to transition from a developmental stage company and continue our business. If we do not obtain marketing approval of and commercialization of our product candidates, or are significantly delayed in doing so, our business will be materially harmed. We have a limited operating history and have incurred significant losses since our inception, and we anticipate that we will continue to incur losses for the foreseeable future and may never achieve or maintain profitability.
We are a clinical stage pharmaceutical company, preparing to commence Phase II clinical trials of our lead drug candidate, with a limited operating history upon which you can evaluate our business and prospects. Specialty pharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We do not currently have any product candidates in advanced clinical trials or approved for sale, and we continue to incur significant research and development and general and administrative expenses related to our operations. In addition, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the specialty pharmaceutical industry. We have not generated any revenue and have incurred losses in each year since our founding in December 2012. Our accumulated deficit as of December 31, 2023 was $25.4 million. We expect to continue to incur significant losses for the foreseeable future. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
| 18 |
We currently have no source of product sales revenue.
We have not yet completed clinical trials and thus do not yet have commercial sales of our products and have not yet not generated any revenues from commercial sales of our product candidates. Our ability to generate product revenue depends upon our ability to develop and commercialize products, including any of our current product candidates or other product candidates that we may develop, in-license or acquire in the future. We do not anticipate generating revenue from the sale of products for the foreseeable future. Our ability to generate future product revenue from our current or future product candidates also depends on a number of additional factors, including our ability to:
| ● | complete research and clinical development of current and future product candidates, either directly or through collaborative relationships; | |
| ● | establish and maintain supply and manufacturing relationships with third parties, and ensure adequate and legally compliant manufacturing of bulk drug substances and drug products to maintain that supply; | |
| ● | obtain regulatory approval from relevant regulatory authorities in jurisdictions where we intend to market our product candidates, either directly or through collaborative relationships; | |
| ● | launch and commercialize future product candidates for which we obtain marketing approval, if any, through collaborative partners; | |
| ● | obtain coverage and adequate product reimbursement from third-party payors, including government payors; | |
| ● | achieve market acceptance for our products, if any; | |
| ● | establish, maintain and protect our intellectual property rights; and | |
| ● | attract, hire and retain qualified personnel. |
In addition, because of the numerous risks and uncertainties associated with clinical product development, including that our product candidates may not advance through development or achieve the endpoints of applicable clinical trials, we are unable to predict the timing or amount of any potential future product sales revenues. Our expenses also could increase beyond expectations if we decide to or are required by the FDA, or comparable foreign regulatory authorities, to perform studies or trials in addition to those that we currently anticipate. Even if we complete the development and regulatory processes described above, we anticipate incurring significant costs associated with launching and commercializing these products.
The market may not be receptive to our product candidates based on our novel therapeutic modality, and we may not generate any future revenue from the sale or licensing of product candidates.
Even if approval is obtained for a product candidate, we may not generate or sustain revenue from sales of the product due to factors such as whether the product can be sold at a competitive cost and otherwise accepted in the market. The product candidates that we are developing are based on new delivery platform therapeutic approaches (there currently is no drug which has FDA approval for indications of radiation sensitization). Market participants with significant influence over acceptance of new treatments, such as physicians and third-party payors, may not accept our delivery platform, and we may not be able to convince the medical community and third-party payors to accept and use, or to provide favorable reimbursement for, any product candidates developed by us. Market acceptance of our product candidates will depend on, among other factors:
| ● | timing of our receipt of any marketing and commercialization approvals; |
| ● | terms of any approvals and the countries in which approvals are obtained; | |
| ● | safety and efficacy of our product candidates, which are determinations solely within the authority of the FDA; |
| 19 |
| ● | prevalence and severity of any adverse side effects associated with our product candidates; | |
| ● | warnings contained in any labeling approved by the FDA or other regulatory authority; | |
| ● | convenience and ease of administration of our product candidates; | |
| ● | success of our physician education programs; | |
| ● | availability of adequate government and third-party payor reimbursement; | |
| ● | pricing of our products, particularly as compared to alternative treatments; and | |
| ● | availability of alternative effective products for indications our product candidates are intended to treat. |
We will require substantial additional financing in order to obtain marketing approval of our product candidates and commercialize our product candidates; a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development, other operations or commercialization efforts.
Since our inception, substantially all of our resources have been dedicated to the preclinical and clinical development of our HDAC small molecule delivery platform and our initial product candidate, Ropidoxuridine. Our capital needs to date have been met by contributions from existing stockholders, as well as through private offerings and IPO of our securities and our SBIR contracts. We believe that we will continue to expend substantial resources for the foreseeable future on the completion of clinical development and regulatory preparedness of our product candidates, preparations for a commercial launch of our product candidates, if approved, and development of any other current or future product candidates we may choose to further develop. These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, obtaining marketing approvals, and, if we are not able to enter into planned collaborations, manufacturing and supply as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any drug development process is highly uncertain, we cannot reasonably estimate the actual amounts necessary to complete the development and commercialization of our current product candidates, if approved, or future product candidates, if any.
We believe that the proceeds we received in our IPO and subsequent $4.0 million follow on convertible note offering, for which we received net proceeds of $3.6 million, along with our existing capital resources, will not be sufficient to fund our operations one year after the date that the consolidated financial statements are issued without additional capital infusion. We anticipate we will need to seek additional funding through public or private equity or debt financings or other sources, such as through a rights offering, strategic collaborations or grants and contracts. Such financing may result in dilution to stockholders, imposition of debt covenants and repayment obligations, or other restrictions that may adversely affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements depend on many factors, including:
| ● | the scope, progress, results and costs of researching and developing our current product candidates, future product candidates and conducting preclinical and clinical trials; | |
| ● | the cost of commercialization activities if our current product candidates and future product candidates are approved for sale, including securing collaborative ventures for completing development of, securing marketing approval for and ultimately marketing, selling and distributing our product candidates, if approved or building a corporate infrastructure if we have to undertake these activities directly; |
| ● | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; | |
| ● | the number and characteristics of any additional product candidates we may develop or acquire; | |
| ● | any product liability or other lawsuits related to our products or commenced against us; | |
| ● | the expenses needed to attract and retain skilled personnel; |
| ● | the costs associated with being a public company; | |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; and | |
| ● | the timing, receipt and amount of sales of, or royalties on, any future approved products, if any. |
| 20 |
Additional funds may not be available when we need them, on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to:
| ● | delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for our current product candidates or future product candidates, if any; | |
| ● | delay, limit, reduce or terminate our research and development activities; or | |
| ● | delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize our current or future product candidates. |
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic collaborations and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. The incurrence of indebtedness would result in increased fixed payment obligations and could involve certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through strategic collaborations and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates or grant licenses on terms unfavorable to us.
Unfavorable and/or unstable global market and economic conditions, including those caused by the ongoing conflict between the Ukraine and Russia and the ongoing COVID-19 pandemic, could have serious adverse consequences on our business, financial condition and results of operations.
The global economy, including credit and financial markets, has experienced extreme volatility and disruptions as a result of the ongoing conflict between the Ukraine and Russia and challenges arising from the ongoing COVID-19 pandemic, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, increases in inflation rates and uncertainty about economic stability. Our results of operations could be adversely affected by the general conditions of the global economy and the global financial markets. In addition, any such volatility and disruptions may have adverse consequences on us or the third parties upon whom we rely. For example, in 2008, the global financial crisis caused extreme volatility and disruptions in the capital and credit markets and the current COVID-19 pandemic has caused significant volatility and uncertainty in U.S. and international markets. Inflation rates, particularly in the United States, have increased recently to levels not seen in years. Increased inflation may result in increased operating costs (including our labor costs), reduced liquidity, and limitations on our ability to access credit or otherwise raise debt and equity capital. In addition, the United States Federal Reserve has raised, and may again raise, interest rates in response to concerns about inflation. Increases in interest rates, especially if coupled with reduced government spending and volatility in financial markets, may have the effect of further increasing economic uncertainty and heightening these risks, which may impact our ability to raise additional capital in the future. The March 2023 failure of Silicon Valley Bank, the pressure such failure has placed on other mid-sized banks, and its potential near- and long-term effects on the biotechnology industry and its participants such as our vendors, suppliers and investors, may also adversely affect our operations and stock price. In addition, U.S. and global markets are experiencing volatility and disruption following the escalation of geopolitical tensions and the start of the military conflict between Russia and Ukraine. On February 24, 2022, a full-scale military invasion of Ukraine by Russian troops began. Although the length and impact of the ongoing military conflict is highly unpredictable, the conflict in Ukraine has led to market disruptions, including significant volatility in commodity prices, credit and capital markets, as well as supply chain disruptions. Various of Russia’s actions have led to sanctions and other penalties being levied by the United States, Australia, the European Union, and other countries, as well as other public and private actors and companies, against Russia and certain other geographic areas, including agreement to remove certain Russian financial institutions from the Society for Worldwide Interbank Financial Telecommunication payment system and restrictions on imports of Russian oil, liquified natural gas and coal. Additional potential sanctions and penalties have also been proposed and/or threatened. Russian military actions and the resulting sanctions could disrupt or otherwise adversely impact our operations and the operations of third parties upon which we rely, as well as the global economy and financial markets, and lead to instability and lack of liquidity in capital markets, potentially making it more difficult for us to obtain additional funds. Related sanctions, export controls or other actions that may be initiated by nations including the United States, the European Union or Russia (e.g., potential cyberattacks, disruption of energy flows, etc.), which could adversely affect our business and/or our supply chain, our CROs, CMOs and other third parties with which we conduct business. A severe or prolonged economic downturn, inflationary environment, rising interest rates, or political unrest could result in a variety of risks to our business, including, weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services. The extent and duration of the military action, sanctions, and resulting market disruptions are impossible to predict, but could be substantial. Any such disruptions may also magnify the impact of other risks described in this Annual Report on Form 10-K and elsewhere herein.
| 21 |
Our product candidates are in the early stages of development and may fail in development or suffer delays that materially adversely affect their commercial viability.
We have no products on the market and all of our product candidates are in the early stages of development. Our ability to achieve and sustain profitability depends on obtaining regulatory approvals, including IRB approval, for and commercializing our product candidates, either alone or with third parties. Before obtaining regulatory approval for the commercial distribution of our product candidates, we or one of our collaborators must conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates, the final determination of which rests solely in the authority of the FDA. Preclinical testing and clinical trials are expensive, difficult to design and implement, can take many years to complete and are uncertain as to outcome. The start or end of a clinical study is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative drug or required prior therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, the age and condition of the patients, the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments for the relevant disease.
A product candidate can unexpectedly fail at any stage of preclinical and clinical development. The historical failure rate for product candidates is high due to scientific feasibility, lack of quality and effectiveness, changing standards of medical care and other variables. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects. We may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
| ● | negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program; | |
| ● | serious and unexpected drug-related side effects experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates; | |
| ● | delays in submitting an Investigational New Drug application (“IND”) or delays or failure in obtaining the necessary approvals from regulators to commence a clinical trial, or a suspension or termination of a clinical trial once commenced; | |
| ● | conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; | |
| ● | delays in enrolling research subjects in clinical trials; | |
| ● | high drop-out rates of research subjects; | |
| ● | greater than anticipated clinical trial costs; | |
| ● | poor effectiveness of our product candidates during clinical trials; | |
| ● | unfavorable FDA or other regulatory agency inspection and review of a clinical trial site; | |
| ● | failure of our third-party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all; | |
| ● | delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular; or | |
| ● | varying interpretations of data by the FDA and similar foreign regulatory agencies. |
| 22 |
If third parties on which we depend to conduct our preclinical studies, or any future clinical trials, do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our development program could be delayed with materially adverse effects on our business, financial condition, results of operations and prospects.
We are relying on third party collaborators to conduct our efficacy clinical trials for Ropidoxuridine and plan to rely on third party clinical investigators, CROs, clinical data management organizations and consultants to design, conduct, supervise and monitor preclinical studies of our product candidates and will do the same for any clinical trials. Because we plan to largely rely on third parties and do not have the ability to conduct preclinical studies or clinical trials independently, we have less control over the timing, quality and other aspects of preclinical studies and clinical trials than we would if we conducted them on our own. These investigators, CROs, and consultants are not our employees and we have limited control over the amount of time and resources that they dedicate to our programs. These third parties may have contractual relationships with other entities, some of which may be our competitors, which may draw time and resources from our programs. The third parties with whom we contract might not be diligent, careful or timely in conducting our preclinical studies or clinical trials, resulting in the preclinical studies or clinical trials being delayed or unsuccessful.
If we cannot contract with acceptable third parties on commercially reasonable terms, or at all, or if these third parties do not carry out their contractual duties, satisfy legal and regulatory requirements for the conduct of preclinical studies or clinical trials or meet expected deadlines, our clinical development programs could be delayed and otherwise adversely affected. In all events, we are responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. The FDA requires clinical trials to be conducted in accordance with good clinical practices, including for conducting, recording and reporting the results of preclinical studies and clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Any such event could have a material adverse effect on our business, financial condition, results of operations and/or prospects.
Because we rely on third party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality.
We rely on third party supply and manufacturing partners to supply the materials and components for, and manufacture, our research and development, preclinical and clinical trial drug supplies. We do not own manufacturing facilities or supply sources for such components and materials. There can be no assurance that our supply of research and development, preclinical and clinical development drugs and other materials will not be limited, interrupted, restricted in certain geographic regions or of satisfactory quality or continue to be available at acceptable prices. In particular, any replacement of any drug product formulation manufacturer we may use could require significant effort and expertise in the event there are a limited number of qualified replacements for a particular product candidate.
The manufacturing process for a product candidate is subject to FDA and foreign regulatory authority review. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards, such as Current Good Manufacturing Practice (or CGMP). In the event that any of our suppliers or manufacturers fail to comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on reasonable terms, if at all. In some cases, the technical skills or technology required to manufacture our product candidates may be unique or proprietary to the original manufacturer, and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills or technology to another third party and a feasible alternative may not exist. These factors would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our product candidates. If we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.
We expect to continue to rely on third party manufacturers if we receive regulatory approval for any product candidate. To the extent that we have existing or future manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner consistent with contractual and regulatory requirements, including those related to quality control and assurance. If we are unable to obtain or maintain third-party manufacturing for product candidates, or to do so on commercially reasonable terms, we may not be able to fully develop and commercialize our product candidates. Our or a third party’s failure to execute on our manufacturing requirements could adversely affect our business in a number of ways, including:
| ● | an inability to initiate or continue clinical trials of product candidates under development; | |
| ● | delay in submitting regulatory applications, or receiving regulatory approvals, for product candidates; |
| 23 |
| ● | loss of the cooperation of a collaborator; | |
| ● | subjecting our product candidates to additional inspections by regulatory authorities; | |
| ● | requirements to cease distribution or to recall batches of our product candidates; and | |
| ● | in the event of approval to market and commercialize a product candidate, an inability to meet commercial demands for our products. |
We may be unsuccessful in engaging in strategic transactions which could adversely affect our ability to develop and commercialize product candidates, impact our cash position, increase our expense and present significant distractions to our management.
From time to time, we may consider strategic transactions, such as collaborations, acquisitions of companies, asset purchases and out- or in- licensing of product candidates or technologies. In particular, we will evaluate and, if strategically attractive, seek to enter into additional collaborations, including with major biotechnology or pharmaceutical companies to complete development and marketing of our product candidates, if approved. The competition for collaborators is intense, and the negotiation process is time-consuming and complex. Any proposed collaboration may be on terms that are not optimal for us, and we may not be able to maintain any new or existing collaboration if, for example, development or approval of a product candidate is delayed, sales of an approved product candidate do not meet expectations or the collaborator terminates the collaboration. Any such collaboration, or other strategic transaction, may require us to incur non-recurring or other charges, increase our near- and long-term expenditures and pose significant integration or implementation challenges or disrupt our management or business. These transactions would entail numerous operational and financial risks, including exposure to unknown liabilities, disruption of our business and diversion of our management’s time and attention in order to manage a collaboration or develop acquired products, product candidates or technologies, incurrence of substantial debt or dilutive issuances of equity securities to pay transaction consideration or costs, higher than expected collaboration, acquisition or integration costs, write-downs of assets or goodwill or impairment charges, increased amortization expenses, difficulty and cost in facilitating the collaboration or combining the operations and personnel of any acquired business, impairment of relationships with key suppliers, manufacturers or customers of any acquired business due to changes in management and ownership and the inability to retain key employees of any acquired business. Accordingly, although there can be no assurance that we will undertake or successfully complete any transactions of the nature described above, any transactions that we do complete may be subject to the foregoing or other risks and have a material adverse effect on our business, results of operations, financial condition and prospects. Conversely, any failure to enter into any collaboration or other strategic transaction that would be beneficial to us could delay the development and potential commercialization of our product candidates and have a negative impact on the competitiveness of any product candidate that reaches market.
We face competition from entities that have developed or may develop product candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to ours. If these companies develop technologies or product candidates more rapidly than we do or their technologies, including delivery technologies, are more effective, our ability to develop and commercialize product candidates may be adversely affected.
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized biotechnology companies, as well as with universities and other research institutions which are developing new technology. Our competitors have developed, are developing or will develop product candidates and processes competitive with our product candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community and any new treatments that enter the market. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may try to develop product candidates.
| 24 |
Many of our competitors have significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than we have. If we obtain approval for any product candidate, we will face competition based on many different factors, including the quality and effectiveness of our products, the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these products, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position. Competing products could present superior treatment alternatives, including by being more effective, safer, less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
Any inability to attract and retain qualified key management and technical personnel would impair our ability to implement our business plan.
Our success largely depends on the continued service of certain key management and other specialized personnel, including Anatoly Dritschilo, M.D., our Chief Executive Officer, Mira Jung, Ph.D., our Chief Scientific Officer, Michael Vander Hoek, our Chief Financial Officer and Vice President Operations and Regulatory, and Peter Dritschilo, our President and Chief Operating Officer. The loss of one or more members of our management team or other key employees or advisors could delay our research and development programs and materially harm our business, financial condition, results of operations and prospects. The relationships that our key managers have cultivated within our industry make us particularly dependent upon their continued employment with us. We are dependent on the continued service of our technical personnel because of the highly technical nature of our product candidates and technologies and the specialized nature of the regulatory approval process. Because our management team and key employees are not obligated to provide us with continued service, they could terminate their employment with us at any time without penalty. We do not maintain key person life insurance policies on any of our management team members or key employees. Our future success will depend in large part on our continued ability to attract and retain other highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing, manufacturing, governmental regulation and commercialization. We face competition for personnel from other companies, universities, public and private research institutions, government entities and other organizations.
If our product candidates advance into Phase II and Phase III clinical trials, we may experience difficulties in managing our growth and expanding our operations.
As our product candidates enter and advance through preclinical studies and any clinical trials, we will need to expand our development, regulatory and manufacturing capabilities or contract with other organizations to provide these capabilities for us. In the future, we expect to have to manage additional relationships with collaborators or partners, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
If any of our product candidates are approved for marketing and commercialization and we are unable to develop sales, marketing and distribution capabilities on our own or enter into agreements with third parties to perform these functions on acceptable terms, we will be unable to commercialize any such future products.
We currently have no sales, marketing or distribution capabilities or experience. If any of our product candidates is approved, we plan to enter into collaborations with third parties to sell, market and distribute our products. In the alternative, we would have to develop internal sales, marketing and distribution capabilities to commercialize any approved product, which would be expensive and time-consuming, or, as is more likely, enter into collaborations with third parties to perform these services. If we rely on third parties with sales, marketing and distribution capabilities to market our products or decide to co-promote products with collaborators, we will need to establish and maintain marketing and distribution arrangements with third parties, and there can be no assurance that we will be able to enter into such arrangements on acceptable terms, if, at all. In entering into third-party marketing or distribution arrangements, any revenue we receive will depend upon the efforts of the third parties and there can be no assurance that such third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance of any approved product. If we decide to market our products directly, we will need to commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and supporting distribution, administration and compliance capabilities. If we are not able to commercialize any product approved in the future, either on our own or through third parties, our business, financial condition, results of operations and prospects could be materially adversely affected.
| 25 |
If we fail to comply with U.S. and foreign regulatory requirements, regulatory authorities could limit or withdraw any marketing or commercialization approvals we may receive and subject us to other penalties that could materially harm our business.
Even if we receive marketing and commercialization approval of a product candidate, there can be no assurance we will not be subject to future or continuing regulatory review, including in relation to adverse patient experiences with the product and clinical results that are reported after a product is made commercially available, both in the U.S. and any foreign jurisdiction in which we seek regulatory approval. The FDA has significant post-market authority, including the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate safety risks related to the use of a product or to require withdrawal of the product from the market. The FDA also has the authority to require a risk evaluation and mitigation strategies (“REMS”) plan after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug. The manufacturer and manufacturing facilities we use to make a future product, if any, will also be subject to periodic review and inspection by the FDA and other regulatory agencies, including for continued compliance with CGMP requirements. The discovery of any new or previously unknown problems with our third-party manufacturers, manufacturing processes or facilities may result in restrictions on the product, manufacturer or facility, including withdrawal of the product from the market. If we rely on third-party manufacturers, we will not have control over compliance with applicable rules and regulations by such manufacturers. Any product promotion and advertising will also be subject to regulatory requirements and continuing regulatory review. If we or our collaborators, manufacturers or service providers fail to comply with applicable continuing regulatory requirements in the U.S. or foreign jurisdictions in which we seek to market our products, we or they may be subject to, among other things, fines, warning letters, holds on clinical trials, refusal by the FDA to approve pending applications or supplements to approved applications, suspension or withdrawal of regulatory approval, product recalls and seizures, refusal to permit the import or export of products, operating restrictions, injunction, civil penalties and criminal prosecution.
Our business entails a significant risk of product liability and our ability to obtain sufficient insurance coverage could have a material effect on our business, financial condition, results of operations or prospects.
Our business exposes us to significant product liability risks inherent in the development, testing, manufacturing and marketing of therapeutic treatments. Product liability claims could delay or prevent completion of our development programs. If we succeed in marketing products, such claims could result in an FDA investigation of the quality and effectiveness of our products, our manufacturing processes and facilities or our marketing programs and potentially a recall of our products or more serious enforcement action, limitations on the approved indications for which they may be used or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to trial participants or patients and a decline in our stock price. We currently have product liability insurance that we believe is appropriate for our stage of development and may need to obtain higher levels prior to marketing any of our product candidates. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
Our employees, principal investigators, CROs and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, principal investigators, CROs and consultants may engage in fraud, other misconduct or illegal activity. Misconduct by these parties could include intentional failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing standards we may establish, comply with federal and state healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. While we make an effort to maintain strict work processes and oversight of our employees, contractors and consultants, any misconduct could expose us to liability through the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. Furthermore, it is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
| 26 |
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of cyber security measures, our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Such events could cause interruptions of our operations. For example, the loss of preclinical data or data from any future clinical trial involving our product candidates could result in delays in our development and regulatory filing efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the development of our product candidates could be delayed.
Our proprietary information, or that of our customers, suppliers and business partners, may be lost or we may suffer security breaches.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, clinical trial data, our proprietary business information and that of our customers, suppliers and business partners, and personally identifiable information of our customers, clinical trial subjects and employees, in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Although to our knowledge we have not experienced any such material security breach to date, any such breach could compromise our network, or the networks of our CROs or other third-party service providers, and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals for our drugs. Although we maintain business interruption insurance coverage, our insurance might not cover all losses from any future breaches of our systems.
Failure of our information technology systems could significantly disrupt the operation of our business.
Our business increasingly depends on the use of information technologies, which means that certain key areas such as research and development, production and sales are to a large extent dependent on our information systems or those of third-party providers. Our ability to execute our business plan and to comply with regulatory requirements with respect to data control and data integrity, depends, in part, on the continued and uninterrupted performance of our information technology systems, or IT systems and the IT systems supplied by third-party service providers. These systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and backup measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we and our third-party service providers have taken to prevent unanticipated problems that could affect our IT systems, sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data, and in particular to operate our proprietary technology platform, could adversely affect our ability to operate our business.
| 27 |
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research, development and manufacturing involve the use of hazardous materials and various chemicals. We maintain quantities of various flammable and toxic chemicals in our facilities in Gaithersburg, Maryland that are required for our research, development and manufacturing activities. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We believe our procedures for storing, handling and disposing these materials in our Gaithersburg facilities comply with the relevant guidelines of Gaithersburg, the State of Maryland and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of animals and biohazardous materials. Although we maintain workers’ compensation insurance to cover us for costs and expenses, we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological or hazardous materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate any of these laws or regulations.
Our information technology systems could face serious disruptions that could adversely affect our business.
Our information technology and other internal infrastructure systems, including corporate firewalls, servers, leased lines and connection to the Internet, face the risk of systemic failure that could disrupt our operations. A significant disruption in the availability of our information technology and other internal infrastructure systems could cause interruptions in our collaborations with our partners and delays in our research and development work.
Changes in accounting rules and regulations, or interpretations thereof, could result in unfavorable accounting charges or require us to change our compensation policies.
Accounting methods and policies for pharmaceutical companies, including policies governing revenue recognition, research and development and related expenses and accounting for stock-based compensation are subject to review, interpretation and guidance from relevant accounting authorities, including the SEC. Changes to accounting methods or policies, or interpretations thereof, may require us to reclassify, restate or otherwise change or revise our financial statements, including those contained in this Annual Report.
Risks Related to Our Intellectual Property
If we are not able to obtain and enforce patent protection for our technologies or product candidates, development and commercialization of our product candidates may be adversely affected.
Our success depends in part on our ability to obtain and maintain patents and other forms of intellectual property rights, including in-licenses of intellectual property rights of others, for our product candidates, methods used to manufacture our product candidates and methods for treating patients using our product candidates, as well as our ability to preserve our trade secrets, to prevent third parties from infringing upon our proprietary rights and to operate without infringing upon the proprietary rights of others. As of the date of this registration statement, we have filed 6 patent applications with the U.S. Patent and Trademark Office (the “USPTO”) with respect to various aspects of our HDAC inhibitor small molecule delivery platforms and Ropidoxuridine, our lead product candidate. However, we may not be able to apply for patents on certain aspects of our product candidates or delivery technologies in a timely fashion or at all. To date, four U.S. patents and eighteen European patents have been granted. There is no guarantee that any of our pending patent applications will result in issued or granted patents, that any of our issued, granted or licensed patents will not later be found to be invalid or unenforceable or that any issued, granted or licensed patents will include claims that are sufficiently broad to cover our product candidates or delivery technologies or to provide meaningful protection from our competitors. Moreover, the patent position of specialty pharmaceutical companies can be highly uncertain because it involves complex legal and factual questions. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our current and future proprietary technology and product candidates are covered by valid and enforceable patents or are effectively maintained as trade secrets. If third parties disclose or misappropriate our proprietary rights, it may materially and adversely impact our position in the market.
| 28 |
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other requirements during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case. The standards applied by the USPTO and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in pharmaceutical patents. As such, we do not know the degree of future protection that we will have on our proprietary products and technology. While we will endeavor to try to protect our product candidates with intellectual property rights such as patents, as appropriate, the process of obtaining patents is time-consuming, expensive and sometimes unpredictable.
We may decide for business reasons to no longer pursue or to abandon certain intellectual property rights in the U.S. or elsewhere, including due to non-cooperation of inventors or owners of such intellectual property, prior art, or scope of protection, or for other reasons.
Once granted, patents may remain open to opposition, interference, re-examination, post-grant review, inter partes review, nullification or derivation action in court or before patent offices or similar proceedings for a given period after allowance or grant, during which time third parties can raise objections against such initial grant. In the course of such proceedings, which may continue for a protracted period of time, the patent owner may be compelled to limit the scope of the allowed or granted claims thus attacked, or may lose the allowed or granted claims altogether. In addition, there can be no assurance that:
| ● | others will not or may not be able to make, use or sell compounds that are the same as or similar to our product candidates but that are not covered by the claims of the patents that we own or license; | |
| ● | we or our licensors, collaborators or any future collaborators are the first to make the inventions covered by each of our issued patents and pending patent applications that we own or license; | |
| ● | we or our licensors, collaborators or any future collaborators are the first to file patent applications covering certain aspects of our inventions; | |
| ● | others will not independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; | |
| ● | A third party may not challenge our patents and, if challenged, a court may not hold that our patents are valid, enforceable and infringed; |
| ● | any issued patents that we own or have licensed will provide us with any competitive advantages, or will not be challenged by third parties; | |
| ● | we may develop additional proprietary technologies that are patentable; | |
| ● | the patents of others will not have an adverse effect on our business; and | |
| ● | our competitors do not conduct research and development activities in countries where we do not have enforceable patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets. |
| 29 |
We intend to license patent rights from third-party owners or licensees. If such owners or licensees do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, or if they retain or license to others any competing rights, our competitive position and business prospects may be adversely affected. We may not be able to protect our intellectual property rights throughout the world.
Obtaining a valid and enforceable issued or granted patent covering our technology in the U.S. and worldwide can be extremely costly. In jurisdictions where we have not obtained patent protection, competitors may use our technology to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but where it is more difficult to enforce a patent as compared to the U.S. Competitor products may compete with our future products in jurisdictions where we do not have issued or granted patents or where our issued or granted patent claims or other intellectual property rights are not sufficient to prevent competitor activities in these jurisdictions. The legal systems of certain countries, particularly certain developing countries, make it difficult to enforce patents and such countries may not recognize other types of intellectual property protection, particularly that relating to biopharmaceuticals. This could make it difficult for us to prevent the infringement of patents or marketing of competing products in violation of our proprietary rights generally in certain jurisdictions. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We generally file a provisional patent application first (a priority filing) at the USPTO. A U.S. utility application and international application under the Patent Cooperation Treaty (PCT) are usually filed within twelve months after the priority filing. Based on the PCT filing, national and regional patent applications may be filed in the European Union, Japan, Australia and Canada and other countries. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. In addition, we may decide to abandon national and regional patent applications before grant. Finally, the grant proceeding of each national or regional patent is an independent proceeding which may lead to situations in which applications might in some jurisdictions be refused by the relevant registration authorities, while granted by others. It is also quite common that depending on the country, various scopes of patent protection may be granted on the same product candidate or technology. The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the U.S., and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition from others in those jurisdictions. Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position in the relevant jurisdiction may be impaired and our business and results of operations may be adversely affected.
We or our licensors, or any future collaborators or a strategic partners may become subject to third party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development and commercialization of our product candidates, or put our patents and other proprietary rights at risk.
We or our licensors, or any future collaborators or strategic partners may be subject to third-party claims for infringement or misappropriation of patent or other proprietary rights. We are generally obligated under our license or collaboration agreements to indemnify and hold harmless our licensors or collaborator for damages arising from intellectual property infringement by us. If we or our licensors, or any future collaborators or strategic partners are found to infringe a third-party patent or other intellectual property rights, we could be required to pay damages, potentially including treble damages, if we are found to have willfully infringed. In addition, we or our licensors, collaborators or any future strategic partners may choose to seek, or be required to seek, a license from a third party, which may not be available on acceptable terms, if at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to us. If we fail to obtain a required license, we or our collaborator, or any future collaborator, may be unable to effectively market product candidates based on our technology, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. In addition, we may find it necessary to pursue claims or initiate lawsuits to protect or enforce our patent or other intellectual property rights. The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
| 30 |
If we were to initiate legal proceedings against a third party to enforce a patent covering one of our products or our technology, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the U.S., defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one or more of our products or certain aspects of our platform technology. Such a loss of patent protection could have a material adverse impact on our business. Patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without legally infringing our patents or other intellectual property rights.
Intellectual property rights of third parties could adversely affect our ability to commercialize our product candidates, and we might be required to litigate or obtain licenses from third parties in order to develop or market our product candidates. Such litigation or licenses could be costly or not available on commercially reasonable terms.
Our competitive position may suffer if patents issued to third parties or other third-party intellectual property rights cover our products or elements thereof, or our manufacture or uses relevant to our development plans. In such cases, we may not be in a position to develop or commercialize products or product candidates unless we successfully pursue litigation to nullify or invalidate the third-party intellectual property right concerned, or enter into a license agreement with the intellectual property right holder, if available on commercially reasonable terms.
Third party intellectual property right holders may also actively bring infringement claims against us. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and time-consuming litigation and may be prevented from or experience substantial delays in marketing our products. If we fail in any such dispute, in addition to being forced to pay damages, we may be temporarily or permanently prohibited from commercializing any of our product candidates that are held to be infringing. We might, if possible, also be forced to redesign product candidates so that we no longer infringe the third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
If we fail to comply with our obligations under any license, collaboration or other agreements, we may be required to pay damages and could lose intellectual property rights that are necessary for developing and protecting our product candidates and delivery technologies or we could lose certain rights to grant sublicenses.
Our current licenses impose, and any future licenses we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology. Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we would be required to pay on sales of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in products that we aim to develop and commercialize, if any. Therefore, even if we are able to develop and commercialize products, we may be unable to achieve or maintain profitability.
| 31 |
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patent protection for certain aspects of our product candidates and delivery technologies, we also consider trade secrets, including confidential and unpatented know-how important to the maintenance of our competitive position. We protect trade secrets and confidential and unpatented know-how, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to such knowledge, such as our employees, corporate collaborators, outside scientific collaborators, CROs, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants that obligate them to maintain confidentiality and assign their inventions to us. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time- consuming, and the outcome is unpredictable. In addition, some courts in the U.S. and certain foreign jurisdictions are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
We may be subject to claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages and may lose valuable intellectual property rights or personnel.
Many of our employees were previously employed at universities or biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper our ability to commercialize, or prevent us from commercializing, our product candidates, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to effectively compete and our business may be adversely affected.
Risks Related to Government Regulation and Product Approvals
We may be unable to obtain U.S. or foreign regulatory approval and, as a result, unable to commercialize our product candidates.
Our product candidates are subject to extensive governmental regulations relating to, among other things, research, testing, development, manufacturing, safety, efficacy, approval, recordkeeping, reporting, labeling, storage, packaging, advertising and promotion, pricing, marketing and distribution of drugs. Rigorous preclinical testing and clinical trials and an extensive regulatory approval process are required to be completed in the U.S. and in many foreign jurisdictions before a new drug can be marketed. Satisfaction of these and other regulatory requirements is costly, time-consuming, uncertain and subject to unanticipated delays. It is possible that none of the product candidates we may develop will obtain the regulatory approvals necessary for us or our collaborators to begin selling them.
| 32 |
We have very limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA. The time required to obtain FDA and other approvals is unpredictable but typically takes many years following the commencement of clinical trials, depending upon the type, complexity and novelty of the product candidate. The standards that the FDA and its foreign counterparts use when regulating us are not always applied predictably or uniformly and can change. Any analysis we perform of data from preclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations, for example, from future legislation or administrative action, or from changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. It is impossible to predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be.
Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues from the particular product candidate for which we are seeking approval. Furthermore, any regulatory approval to market a product may be subject to limitations on the approved uses for which we may market the product or the labeling or other restrictions. In addition, the FDA has the authority to require a Risk Evaluation and Mitigation Strategy (REMS) plan as part of an NDA or biologics license application (BLA) or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug or biologic, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. These limitations and restrictions may limit the size of the market for the product and affect reimbursement by third-party payors.
If we or our collaborators, manufacturers or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions, which could affect our ability to develop, market and sell our products and may harm our reputation.
We and our collaborators are subject to federal, state, and foreign healthcare laws and regulations pertaining to fraud and abuse and patients’ rights. These laws and regulations include:
| ● | the U.S. federal healthcare program anti-kickback law, which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual for a healthcare item or service, or the purchasing or ordering of an item or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid; | |
| ● | the U.S. federal false claims law, which prohibits, among other things, individuals or entities from knowingly presenting or causing to be presented, claims for payment by government funded programs such as Medicare or Medicaid that are false or fraudulent, and which may apply to us by virtue of statements and representations made to customers or third parties; | |
| ● | the U.S. federal Health Insurance Portability and Accountability Act (HIPAA) and Health Information Technology for Economic and Clinical Health (HITECH) Act, which prohibit executing a scheme to defraud healthcare programs, impose requirements relating to the privacy, security, and transmission of individually identifiable health information, and require notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information; | |
| ● | the federal Open Payments regulations under the National Physician Payment Transparency Program have been issued under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, and will require that manufacturers of pharmaceutical and biological drugs covered by Medicare, Medicaid, and Children’s Health Insurance Programs report all consulting fees, travel reimbursements, research grants, and other payments or gifts with values over $10 made to physicians and teaching hospitals; and | |
| ● | state laws comparable to each of the above federal laws, such as, for example, anti-kickback and false claims laws applicable to commercial insurers and other non-federal payors, requirements for mandatory corporate regulatory compliance programs, and laws relating to patient data privacy and security. |
| 33 |
If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, monetary damages, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, any of which could adversely our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time, and resources.
If we or our collaborators, manufacturers or service providers fail to comply with applicable federal, state, or foreign laws or regulations, we could be subject to enforcement actions, which could affect our ability to develop, market and sell our products successfully and could harm our reputation and lead to reduced acceptance of our products by the market. These enforcement actions include, among others:
| ● | adverse regulatory inspection findings; | |
| ● | warning letters; |
| ● | voluntary or mandatory product recalls or public notification or medical product safety alerts to healthcare professionals; | |
| ● | restrictions on, or prohibitions against, marketing our products; |
| ● | restrictions on, or prohibitions against, importation or exportation of our products; | |
| ● | suspension of review or refusal to approve pending applications or supplements to approved applications; | |
| ● | exclusion from participation in government-funded healthcare programs; | |
| ● | exclusion from eligibility for the award of government contracts for our products; | |
| ● | suspension or withdrawal of product approvals; | |
| ● | product seizures; | |
| ● | injunctions; and | |
| ● | civil and criminal penalties and fines. |
Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drugs vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. Although we intend to monitor these regulations, our programs are currently in the early stages of development and we will not be able to assess the impact of price regulations for a number of years. As a result, we might obtain regulatory approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product and negatively impact the revenues we are able to generate from the sale of the product in that country.
| 34 |
Our ability to commercialize any products also will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if we succeed in bringing one or more products to the market, these products may not be considered cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. Because our programs are in the early stages of development, we are unable at this time to determine their cost effectiveness or the likely level or method of reimbursement. Increasingly, the third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are requiring that drug companies provide them with predetermined discounts from list prices and are seeking to reduce the prices charged or the amounts reimbursed for pharmaceutical products. If the price we are able to charge for any products we develop, or the reimbursement provided for such products, is inadequate in light of our development and other costs, our return on investment could be adversely affected.
Our current product candidates will need to be administered under the supervision of a physician on an outpatient basis. Under currently applicable U.S. law, certain drugs that are not usually self-administered (including injectable drugs) may be eligible for coverage under the Medicare Part B program if:
| ● | they are incident to a physician’s services; | |
| ● | they are reasonable and necessary for the diagnosis or treatment of the illness or injury for which they are administered according to accepted standards of medical practice; and | |
| ● | they have been approved by the FDA and meet other requirements of the statute. |
There may be significant delays in obtaining coverage for newly-approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA. Moreover, eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement may be based on payments allowed for lower- cost drugs that are already reimbursed, may be incorporated into existing payments for other services and may reflect budgetary constraints or imperfections in Medicare data. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for new drugs that we develop and for which we obtain regulatory approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our financial condition.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the healthcare system in the U.S. and other major healthcare markets have been proposed in recent years, and such efforts have expanded substantially in recent years. These developments have included prescription drug benefit legislation that was enacted and took effect in January 2006, healthcare reform legislation enacted by certain states, and Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (the “ACA”), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending and enhance remedies against fraud and abuse. The ACA also contains provisions that will affect companies in the pharmaceutical industry and other healthcare related industries by imposing additional costs and changes to business practices. Provisions affecting pharmaceutical companies include the following:
| ● | mandatory rebates for drugs sold into the Medicaid program have been increased, and the rebate requirement has been extended to drugs used in risk-based Medicaid managed care plans; |
| 35 |
| ● | the 340B Drug Pricing Program under the Public Health Services Act has been extended to require mandatory discounts for drug products sold to certain critical access hospitals, cancer hospitals and other covered entities; | |
| ● | pharmaceutical companies are required to offer discounts on brand-name drugs to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “Donut Hole”; and | |
| ● | pharmaceutical companies are required to pay an annual non-tax deductible fee to the federal government based on each company’s market share of prior year total sales of branded products to certain federal healthcare programs, such as Medicare, Medicaid, Department of Veterans Affairs and Department of Defense. Since we expect our branded pharmaceutical sales to constitute a small portion of the total federal health program pharmaceutical market, we do not expect this annual assessment to have a material impact on our financial condition. |
Moreover, we cannot predict what healthcare reform initiatives may be adopted in the future. Further federal and state legislative and regulatory developments are likely, and we expect ongoing initiatives in the U.S. to increase pressure on drug pricing. Such reforms could have an adverse effect on anticipated revenues from product candidates that we may develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates.
Our ability to obtain services, reimbursement or funding from the federal government may be impacted by possible reductions in federal spending.
U.S. federal government agencies currently face potentially significant spending reductions. Under the Budget Control Act of 2011, the failure of Congress to enact deficit reduction measures of at least $1.2 trillion for the years 2013 through 2021 triggered automatic cuts to most federal programs. These cuts would include aggregate reductions to Medicare payments to providers of up to two percent per fiscal year, starting in 2013. Under the American Taxpayer Relief Act of 2012, which was enacted on January 1, 2013, the imposition of these automatic cuts was delayed until March 1, 2013. Certain of these automatic cuts have been implemented. The full impact on our business of these automatic cuts is uncertain. If federal spending is reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or the NIH to continue to function at current levels. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop.
If any of our product candidates receives marketing approval and we or others later identify undesirable side effects caused by the product candidate, our ability to market and derive revenue from the product candidates could be compromised.
In the event that any of our product candidates receive regulatory approval and we or others identify undesirable side effects caused by one of our products, any of the following adverse events could occur, which could result in the loss of significant revenue to us and materially and adversely affect our results of operations and business:
| ● | regulatory authorities may withdraw their approval of the product or seize the product; | |
| ● | we may be required to recall the product or change the way the product is administered to patients; | |
| ● | additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof; | |
| ● | we may be subject to fines, injunctions or the imposition of civil or criminal penalties; |
| ● | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; | |
| ● | we may be required to create a Medication Guide outlining the risks of such side effects for distribution to patients; |
| 36 |
| ● | we could be sued and held liable for harm caused to patients; | |
| ● | the product may become less competitive; and | |
| ● | our reputation. |
Risks Related to our Common Stock
The financial covenants of the convertible note could result in default and could have a material adverse effect on our liquidity.
Despite our ability to settle debt on the convertible note through cash or issuing shares of common stock, we may be required to repay the convertible note and interest thereon in cash if we do not meet certain equity conditions, including but not limited to volume or price failure on each trading day during the equity conditions measuring period, authorized shares failure, or occurrence of default as defined in the note. Events that could trigger a default include failure to pay the note holder any amount, bankruptcy, insolvency, or violation of the Cash Burn Covenant.
While our Company’s management is working to improve our internal controls and procedures, at present management has determined that our internal controls were deemed to be inadequate, which could cause our financial reporting to be unreliable and lead to misinformation being disseminated to the public.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. As defined in Rule 13a-15(f) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), internal control over financial reporting is a process designed by, or under the supervision of, the principal executive and principal financial officer and effected by the board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| ● | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and/or directors of the Company; and | |
| ● | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
We are required to include a report of management on the effectiveness of our internal control over financial reporting. We expect to incur additional expenses and diversion of management’s time as a result of performing the system and process evaluation, testing and remediation required in order to comply with the management certification requirements.
Presently, we do not have a sufficient number of employees to segregate responsibilities and may be unable to afford increasing our staff or engaging outside consultants or professionals to overcome our lack of employees. During the course of our testing, we may identify other deficiencies that we may not be able to timely remediate. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock, if a market ever develops, could drop significantly.
The JOBS Act has reduced the information that we are required to disclose.
Under the JOBS Act, the information that we will be required to disclose has been reduced in a number of ways.
As a company that had gross revenues of less than $1.235 billion during the Company’s last fiscal year, the Company is an “emerging growth company,” as defined in the JOBS Act (an “EGC”). We will retain that status until the earliest of (a) the last day of the fiscal year which we have total annual gross revenues of $1.235 billion (as indexed for inflation in the manner set forth in the JOBS Act) or more; (b) the last day of the fiscal year of following the fifth anniversary of the date of the first sale of the common stock pursuant to an effective registration statement under the Securities Act of 1933, as amended (the “Securities Act”); (c) the date on which we have, during the previous three year period, issued more than $1.0 billion in non-convertible debt; or (d) the date on which we are deemed to be a “large accelerated filer,” as defined in Rule 12b-2 under the Exchange Act or any successor thereto. As an EGC, the Company is relieved from the following:
| ● | The Company is excluded from Section 404(b) of Sarbanes-Oxley Act (“Sarbanes-Oxley”), which otherwise would have required the Company’s auditors to attest to and report on the Company’s internal control over financial reporting. The JOBS Act also amended Section 103(a)(3) of Sarbanes-Oxley to provide that (i) any new rules adopted by the PCAOB requiring mandatory audit firm rotation or changes to the auditor’s report to include auditor discussion and analysis (in the event the PCAOB adopts an auditor rotation requirement) will not apply to an audit of an EGC; and (ii) any other future rules adopted by the PCAOB will not apply to the Company’s audits unless the SEC determines otherwise. |
| ● | The JOBS Act amended Section 7(a) of the Securities Act to provide that the Company need not present more than two years of audited financial statements in an initial public offering registration statement and in any other registration statement, need not present selected financial data pursuant to Item 301 of Regulation S-K for any period prior to the earliest audited period presented in connection with such initial public offering. In addition, the Company is not required to comply with any new or revised financial accounting standard until such date as a private company (i.e., a company that is not an “issuer” as defined by Section 2(a) of Sarbanes-Oxley) is required to comply with such new or revised accounting standard. Corresponding changes have been made to the Exchange Act, which relates to periodic reporting requirements, which would be applicable if the Company were required to comply with them. |
| ● | As long as we are an EGC, we may comply with Item 402 of Regulation S-K, which requires extensive quantitative and qualitative disclosure regarding executive compensation, by disclosing the more limited information required of a “smaller reporting company.” |
| 37 |
| ● | The JOBS Act will also exempt us from the following additional compensation-related disclosure provisions that were imposed on U.S. public companies pursuant to the Dodd-Frank Act: (i) the advisory vote on executive compensation required by Section 14A(a) of the Exchange Act; (ii) the requirements of Section 14A(b) of the Exchange Act relating to stockholders advisory votes on “golden parachute” compensation; (iii) the requirements of Section 14(i) of the Exchange Act as to disclosure relating to the relationship between executive compensation and our financial performance; and (iv) the requirement of Section 953(b)(1) of the Dodd-Frank Act, which requires disclosure as to the relationship between the compensation of our chief executive officer and median employee pay. |
Our stock price may be volatile, and purchasers of our common stock could incur substantial losses.
Since commencement of trading on Nasdaq Stock Market LLC or Nasdaq, on August 29, 2022, our stock price has been extremely volatile, having traded as high as $1,010.08 and as low as $1.03. As a result of this volatility, investors may not be able to sell their common stock at or above the price when they purchased our common stock. The market price for our common stock may be influenced by many factors, including the other risks described in this section of this Annual Report entitled “Risk Factors” and the following:
| ● | the success of competitive products or technologies; | |
| ● | results of preclinical and clinical studies of our product candidates, or those of our competitors, our existing collaborator or any future collaborators; | |
| ● | regulatory or legal developments in the U.S. and other countries, especially changes in laws or regulations applicable to our products; | |
| ● | introductions and announcements of new products by us, our commercialization partners, or our competitors, and the timing of these introductions or announcements; | |
| ● | actions taken by regulatory agencies with respect to our products, clinical studies, manufacturing process or sales and marketing terms; | |
| ● | actual or anticipated variations in our financial results or those of companies that are perceived to be similar to us; | |
| ● | the success of our efforts to acquire or in-license additional technologies, products or product candidates; | |
| ● | developments concerning our collaborations, including but not limited to those with our sources of manufacturing supply and our commercialization partners; | |
| ● | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; | |
| ● | developments or disputes concerning patents or other proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our products; | |
| ● | our ability or inability to raise additional capital and the terms on which we raise it; | |
| ● | the recruitment or departure of key personnel; | |
| ● | changes in the structure of healthcare payment systems; | |
| ● | market conditions in the pharmaceutical and biotechnology sectors; |
| ● | actual or anticipated changes in earnings estimates or changes in stock market analyst recommendations regarding our common stock, other comparable companies or our industry generally; |
| ● | our failure or the failure of our competitors to meet analysts’ projections or guidance that we or our competitors may give to the market; | |
| ● | fluctuations in the valuation of companies perceived by investors to be comparable to us; | |
| ● | announcement and expectation of additional financing efforts; | |
| ● | speculation in the press or investment community; | |
| ● | trading volume of our common stock; | |
| ● | sales of our common stock by us or our stockholders; | |
| ● | the concentrated ownership of our common stock; | |
| ● | changes in accounting principles; | |
| ● | terrorist acts, acts of war or periods of widespread civil unrest; | |
| ● | natural disasters and other calamities; and | |
| ● | general economic, industry and market conditions. |
| 38 |
In addition, the stock markets in general, and the markets for pharmaceutical stocks, in particular, have experienced extreme volatility that has been often unrelated to the operating performance of the issuer. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance.
The future issuance of equity or of debt securities that are convertible into common stock will dilute our share capital.
We may choose to raise additional capital in the future, depending on market conditions, strategic considerations and operational requirements. To the extent that additional capital is raised through the issuance of shares or other securities convertible into shares of our common stock, our stockholders will be diluted. Future issuances of our common stock or other equity securities, or the perception that such sales may occur, could adversely affect the trading price of our common stock and impair our ability to raise capital through future offerings of shares or equity securities. No prediction can be made as to the effect, if any, that future sales of common stock or the availability of common stock for future sales will have on the trading price of our common stock.
If we fail to maintain applicable listing requirements, Nasdaq may delist our common stock from trading, in which case the liquidity and market price of our common stock could decline.
We cannot assure you that we will be able to meet the continued listing standards of Nasdaq in the future. If we fail to comply with the applicable listing standards and Nasdaq delists our common stock, we and our stockholders could face significant material adverse consequences, including:
| ● | a limited availability of market quotations for shares of our common stock; | |
| ● | reduced liquidity for our common stock; | |
| ● | a determination that our common stock is “penny stock,” which would require brokers trading in our common stock to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for shares of our common stock; | |
| ● | a limited amount of news about us and analyst coverage of us; and | |
| ● | a decreased ability for us to issue additional equity securities or obtain additional equity or debt financing in the future. |
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our common stock is listed on Nasdaq, such securities will be deemed covered securities. Although the states will be preempted from regulating the sale of our securities, the federal statute does allow states to investigate companies if there is a suspicion of fraud and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Further, if we were no longer listed on Nasdaq, our securities would not be covered securities and we would be subject to regulations in each state in which we offer our securities.
Because our management has broad discretion over the use of the net proceeds we received from our IPO and any follow-on offerings, you may not agree with how we use them and the proceeds may not be invested successfully.
We intend to use the net proceeds to us from our IPO and any follow-on offerings to fund preclinical and clinical trials of product candidates, Ropidoxuridine and new formulations of Ropidoxuridine with Tipiracil, O-18 containing molecules for proton radiation sensitization, continued HDAC technology platform development, working capital and general corporate purposes, including the costs of operating as a public company, as well as potential acquisition or in-licensing activities. Therefore, our management has broad discretion as to the use of the IPO proceeds and proceeds from our subsequent private placements or follow-on offerings. Accordingly, you will be relying on the judgment of our management with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that the proceeds will be invested in a way that does not yield a favorable, or any, return for our Company.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our stock, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. We do not currently have and may never obtain research coverage by securities and industry analysts. If no or few securities or industry analysts commence coverage of us, the trading price for our stock would be negatively impacted. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our target studies and operating results fail to meet the expectations of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
Our board of directors has the authority, without stockholder approval, to issue preferred stock with terms that may not be beneficial to holders of our common stock and such issuance could potentially adversely affect stockholders’ voting power and perpetuate their control over us.
Our Certificate of Incorporation, as amended to date, allows us to issue shares of preferred stock without any vote or further action by our stockholders. Our board of directors has the authority to fix and determine the relative rights and preferences of any preferred stock. As a result, our board of directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of shares of our common stock. These rights and preferences could negatively affect the holders of our common stock.
The ability of our executive officers and directors, who are our principal stockholders, to control our business may limit or eliminate the ability of minority stockholders to influence corporate affairs.
Our executive officers and directors, who are our principal stockholders, own approximately 39.4% of our issued and outstanding common stock. Accordingly, they may be able to effectively control the election of directors, as well as all other matters requiring stockholder approval. The interests of our principal stockholders may differ from the interests of other stockholders with respect to the issuance of shares, business transactions with or sales to other companies, selection of other directors and other business decisions. The minority stockholders have no way of overriding decisions made by our principal stockholders. This level of control may also have an adverse impact on the market value of our shares because our principal stockholders may institute or undertake transactions, policies or programs that result in losses and may not take any steps to increase our visibility in the financial community and/or may sell sufficient numbers of shares to significantly decrease our price per share.
| 39 |
Our Certificate of Incorporation and Bylaws, each as amended to date, provide for indemnification of officers and directors at the expense of the Company and limit their liability that may result in a major cost to us and hurt the interests of our stockholders because corporate resources may be expended for the benefit of officers and/or directors.
Our Certificate of Incorporation and Bylaws, each as amended to date, provide for the indemnification of our officers and directors. We have been advised that, in the opinion of the SEC, indemnification for liabilities arising under federal securities laws is against public policy as expressed in the Securities Act and is therefore, unenforceable.
Our Certificate of Incorporation, as amended to date, provides that disputes must be resolved in the Court of Chancery of the State of Delaware, except for cases brought under the Securities Act or Exchange Act.
Our Certificate of Incorporation, as amended to date, provides that the Court of Chancery in the State of Delaware will be the exclusive forum for dispute resolution for certain enumerated actions, excluding any actions brought under the Securities Act or Exchange Act, or unless the Company consents in writing to an alternative jurisdiction. This exclusive forum selection clause may cause inconvenience of our stockholders or other stakeholders, should they need to bring suit against the Company for an action other than one arising under the Securities Act or Exchange Act.
We do not expect to pay cash dividends in the foreseeable future.
We have never paid cash dividends on our common stock. We do not expect to pay cash dividends on our common stock at any time in the foreseeable future. The future payment of dividends on our common stock directly depends upon our future earnings, capital requirements, financial requirements and other factors that our board of directors will consider. Since we do not anticipate paying cash dividends on our common stock, return on your investment, if any, will depend solely on an increase, if any, in the market value of our common stock.
Provisions in our amended and restated certificate of incorporation, as amended, and bylaws, as amended, as well as Delaware law, might discourage, delay or prevent a change of control of our company or changes in our management and, therefore, depress the market price of our common stock.
Our Certificate of Incorporation and Bylaws, each as amended to date, and bylaws contain provisions that could depress the market price of our common stock by acting to discourage, delay, or prevent a change in control of our company or changes in our management that the stockholders of our company may deem advantageous. These provisions, among other things:
| ● | permit the board of directors to establish the number of directors; | |
| ● | provide that directors may only be removed “for cause” and only with the approval of 66 2/3 percent of our stockholders; |
| ● | require super-majority voting to amend some provisions in our Certificate of Incorporation and Bylaws; | |
| ● | authorize the issuance of “blank check” preferred stock that our board of directors could use to implement a stockholder rights plan (also known as a “poison pill”); | |
| ● | eliminate the ability of our stockholders to call special meetings of stockholders; | |
| ● | prohibit stockholder action by written consent, which requires all stockholder actions to be taken at a meeting of our stockholders; | |
| ● | provide that the board of directors is expressly authorized to make, alter or repeal our bylaws; and | |
| ● | establish advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted upon by stockholders at annual stockholder meetings. |
| 40 |
In addition, Section 203 of the Delaware General Corporation Law may discourage, delay or prevent a change in control of our company. Section 203 imposes certain restrictions on merger, business combinations and other transactions between us and holders of 15% or more of our common stock.
The restatement of our consolidated financial statements and identification of material weaknesses in our internal controls may result in additional risks and uncertainties, including regulatory, stockholder or other actions, loss of investor and counterparty confidence and negative impacts on our stock price.
On July 10, 2024, the Audit Committee and Company management, after consultation and discussion with our independent registered public accounting firm, concluded that certain of our previously issued audited consolidated financial statements and unaudited consolidated financial statements need to be restated and should no longer be relied on for the reasons described in the Current Report on Form 8-K dated July 10, 2024, and in “Explanatory Note” preceding Part I, Item 1 of this Form 10-K/A and Note 2 to the Consolidated Financial Statements under Part II, Item 8 of this Form 10-K/A.
The accounting errors impacted the Company’s consolidated financial statements for the year ended December 31, 2022. These errors further resulted in an impact to the Company’s consolidated balance sheet for the year ended December 31, 2023 and the unaudited condensed consolidated balance sheet as of and for the periods ended March 31, 2023, June 30, 2023, and September 30, 2023, specific to Additional Paid In Capital and Accumulated Deficit. Additionally, the restatement constituted an event of default under the terms of one of the Company’s financing arrangements that was subsequently waived, but required additional cash collateral from the Company.
In addition, in connection with the restatement of the previously issued financial statements for the fiscal year ended December 31, 2023 and December 31, 2022, and fiscal quarter ended March 31, 2024 (the “Affected Periods”), management identified certain material weaknesses in our internal control over financial reporting. Although we are taking remediation measures, including having hired a new Chief Financial Officer during the second quarter of 2024 to bolster the Company’s internal technical accounting and financial reporting experience and provide bandwidth for the prior CFO to focus on the Company’s expanding clinical trials, we can give no assurance that the measures we have taken, are taking and plan to take in the future will remediate the material weaknesses identified or that any additional material weaknesses or restatements of financial results will not arise in the future due to a failure to implement and maintain adequate internal control over financial reporting. In addition, even if we are successful in strengthening our controls and procedures, in the future those controls and procedures may not be adequate to prevent or identify irregularities or errors or to facilitate the fair presentation of our consolidated financial statements.
As a result of the restatement and associated non-reliance on the previously issued financial statements for the Affected Periods and the identified material weakness, we have become subject to a number of additional costs and risks, including unanticipated costs for accounting and legal fees in connection with or related to the restatement and the ongoing process of remediating the material weaknesses. We could also be subject to regulatory, stockholder or other actions in connection with the restatement and/or the material weaknesses, which would, regardless of the outcome, consume management’s time and attention and may result in additional legal, accounting and other costs. If such proceedings arise and we do not prevail in such proceedings, we could be required to pay damages or settlement costs. In addition, the restatement and related matters could impair our reputation or could cause our stockholders or other counterparties to lose confidence in us. Any of these occurrences could adversely affect our business, financial condition, liquidity and results of operations.
| 41 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents included herein contain forward-looking statements that involve risks and uncertainties. All statements other than statements of historical facts contained in this prospectus and the documents included herein are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “may,” “could,” “will,” “would,” “should,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “intend,” “predict,” “seek,” “contemplate,” “project,” “continue,” “potential,” “ongoing” or the negative of these terms or other comparable terminology. These forward-looking statements include, but are not limited to, statements about:
| ● | the initiation, timing, progress and results of our research and development programs, preclinical studies, any clinical trials and INDs, NDAs other regulatory submissions; | |
| ● | our expected dependence on third party collaborators for developing, obtaining regulatory approval for and commercializing product candidates; | |
| ● | our receipt and timing of any milestone payments or royalties under any research collaboration and license agreement we enter into; | |
| ● | our ability to identify and develop product candidates; | |
| ● | our or a collaborator’s ability to obtain and maintain regulatory approval of any of our product candidates; | |
| ● | the rate and degree of market acceptance of any approved products candidates; | |
| ● | the commercialization of any approved product candidates; | |
| ● | our ability to establish and maintain additional collaborations and retain commercial rights for our product candidates subject to collaborations; | |
| ● | the implementation of our business model and strategic plans for our business, technologies and product candidates; | |
| ● | our estimates of our expenses, ongoing losses, future revenue and capital requirements; | |
| ● | our ability to obtain additional funds for our operations; | |
| ● | our ability to obtain and maintain intellectual property protection for our technologies and product candidates and our ability to operate our business without infringing the intellectual property rights of others; | |
| ● | our reliance on third parties to conduct our preclinical studies or any future clinical trials; | |
| ● | our reliance on third party supply and manufacturing partners to supply the materials and components for, and manufacture, our research and development, preclinical and clinical trial drug supplies; | |
| ● | our ability to attract and retain qualified key management and technical personnel; | |
| ● | our use of net proceeds to us from this offering; | |
| ● | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act; | |
| ● | our financial performance; and | |
| ● | developments relating to our competitors or our industry. |
These statements relate to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance, or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Factors that may cause actual results to differ materially from current expectations include, among other things, those described in the section entitled “Risk Factors” in this prospectus and contained in our Annual Report on Form 10-K for the fiscal year ended December 31, 2023, as amended, our Quarterly Report on Form 10-Q for the six months ended June 30, 2024, which are included in this prospectus, together with the information included in this prospectus, and in any free writing prospectus that we have authorized for use in connection with this offering.
| 42 |
Any forward-looking statement in this prospectus and the documents included herein reflects our current view with respect to future events and is subject to these and other risks, uncertainties and assumptions relating to our operations, results of operations, industry, and future growth. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by U.S. federal securities law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
MARKET AND INDUSTRY DATA AND FORECASTS
We obtained the industry and market data used throughout this prospectus from our own internal estimates and research, as well as from independent market research, industry and general publications and surveys, governmental agencies, publicly available information and research, surveys and studies conducted by third parties. Internal estimates are derived from publicly available information released by industry analysts and third- party sources, our internal research and our industry experience, and are based on assumptions made by us based on such data and our knowledge of our industry and market, which we believe to be reliable and reasonable. In some cases, we do not expressly refer to the sources from which this data is derived. In addition, while we believe the industry and market data included in this prospectus is reliable and based on reasonable assumptions, such data involve material risks and other uncertainties and are subject to change based on various factors, including those discussed in the section entitled “Risk Factors” and “Special Note Regarding Forward-Looking Statements.” These and other factors could cause results to differ materially from those expressed in the estimates made by the independent parties or by us.
SELECTED FINANCIAL DATA
The following selected financial data has been derived from our audited financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2023, as amended, filed with the SEC on September 4, 2024, and our unaudited financial statements included in our Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2024, filed with the SEC on September 4, 2024.
All historical share and per share amounts reflected throughout this prospectus have been adjusted to reflect the 2024 Reverse Stock Split. However, our periodic and current reports, and all other documents included in this prospectus that were filed prior to August 13, 2024 do not give effect to the 2024 Reverse Stock Split.
Our historical results are not indicative of the results that may be expected in the future, and results of interim periods are not indicative of the results for the entire year.
Statements of Operations Data:
| For the Years Ended | For the Six Months Ended | |||||||||||||||
| December 31, | June 30, | |||||||||||||||
| 2023 | 2022 | 2024 | 2023 | |||||||||||||
| (As Restated) | ||||||||||||||||
| Revenue | $ | - | $ | - | $ | - | $ | - | ||||||||
| Operating expenses | $ | 5,892,382 | $ | 2,343,826 | $ | 2,867,481 | $ | 3,010,411 | ||||||||
| Loss from operations | $ | (5,892,382 | ) | $ | (2,343,826 | ) | $ | (2,867,481 | ) | $ | (3,010,411 | ) | ||||
| Other income (expense) | $ | (700,341 | ) | $ | (2,652,243 | ) | $ | (894,765 | ) | $ | (149,057 | ) | ||||
| Net loss | $ | (6,592,723 | ) | $ | (4,996,069 | ) | $ | (3,762,246 | ) | $ | (3,159,468 | ) | ||||
| Weighted average common shares outstanding – basic and diluted | 1,886,275 | 1,337,742 | 2,090,890 | 1,742,520 | ||||||||||||
| Net loss per share – basic and diluted | $ | (3.53 | ) | $ | (5.04 | ) | $ | (1.80 | ) | $ | (1.81 | ) | ||||
USE OF PROCEEDS
We estimate that the net proceeds from this offering, after deducting Placement Agents’ fees and estimated offering expenses payable by us, will be approximately $4.1 million assuming no exercise of the Common Warrants. We intend to use the net proceeds from this offering to fund our Phase II clinical trial for our product candidate, including $2.3 million in payments that will be owed to Theradex Systems, Inc., the clinical research organization (CRO) supporting our Phase II clinical trials for our radiation sensitizer Ropidoxuridine, and for working capital and general corporate purposes.
The expected use of net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. While we anticipate that the majority of the net proceeds will be used to support our Phase II clinical trial for Ropidoxuridine, and the general corporate efforts required to support them, we cannot currently allocate specific percentages of the net proceeds to us from this offering that we may use for the purposes specified above. Our management will have broad discretion in the application of the net proceeds from this offering and could use them for purposes other than those contemplated at the time of this offering. Our stockholders may not agree with the manner in which our management chooses to allocate and spend the net proceeds. Moreover, our management may use the net proceeds for corporate purposes that may not result in our being profitable or that increases our market value.
Pending our use of the net proceeds from this offering, we intend to invest the net proceeds in a variety of capital preservation investments, including short-term, investment-grade, interest-bearing instruments and U.S. government securities.
| 43 |
BUSINESS
Shuttle Pharma is a clinical stage pharmaceutical company leveraging our proprietary technology to develop novel therapies designed to cure cancers. Our goal is to extend the benefits of cancer treatments with surgery, radiation therapy, chemotherapy and immunotherapy. Radiation therapy (“RT”) is one of the most effective modalities for treating cancers. We are developing a pipeline of products designed to address the limitations of the current cancer therapies as well as to extend to the new applications of RT. We believe that our product candidates will enable us to deliver cancer treatments that are safer, more reliable and at a greater scale than that of the current standard of care.

The corporate structure is based on Shuttle Pharmaceuticals Holdings, Inc. (Nasdaq SHPH – a Delaware Company) with drug discovery and development performed in the wholly owned Shuttle Pharmaceuticals, Inc. (a Maryland Company) and diagnostics performed in the wholly owned Shuttle Diagnostics, Inc. (a Maryland Company). Our product candidates include Ropidoxuridine, a Phase II, clinical-stage radiation sensitizer, a platform of HDAC inhibitors (SP-1-161, SP-2-225 and SP-1-303), and two preclinical, prostate cancer-oriented diagnostics assets – the PC-RAD Test, a blood test to predict clinical response to radiation therapy and the PSMA-B ligand for potential use as a theranostic agent.
In December 2023, we submitted an Investigational New Drug (“IND”) application with the U.S. Food and Drug Administration (“FDA”) to support the next phase of development of Ropidoxuridine. In January 2024, we received the ‘Safe to Proceed’ letter from the FDA for our IND application for the Phase II study of Ropidoxuridine (IPdR) as a radiation sensitizing agent during radiotherapy in patients with newly diagnosed IDH-wildtype glioblastoma with unmethylated MGMT promoter. Receipt of the letter allows us to commence the Phase II study of Ropidoxuridine (IPdR). We have applied for and received FDA approval of Orphan designation for Ropidoxuridine and RT for treating brain cancer (glioblastoma). The Phase II clinical trial was also approved by the Institutional Review Board “IRB” on June 21, 2024. We believe our management team’s expertise in radiation therapy, combined modality cancer treatment and immuno-oncology will help drive the development and, if approved, the commercialization of these potentially curative therapies for patients with aggressive cancers.
Radiation Oncology has gone through transformative technological innovation over the last decade to better define tumors, allow improved shaping of radiation delivery and support dose escalation with shorter, more intensive courses of treatment. Furthermore, achieving higher dose distributions within tumor volumes has reached a practical plateau, since cancers are frequently integrated with or surrounded by more sensitive normal tissues and further dose escalation increases risks of tissue necrosis. To increase cancer cures at maximally tolerated radiation doses, pharmacological and biological modifications of cells are needed to sensitize cancers, protect normal tissues, and stimulate the immune system to react against antigens produced by irradiated, damaged cancer cells. Drugs that show sensitizing properties, or the ability to make cancer cells more sensitive to radiation, offer a solution to this problem. Currently, such drugs are chemotherapy agents used off-label, and many have inherent toxicities since they were designed for direct cancer treatments and not for sensitization. However, the clinical value of using radiation sensitizing drugs in combination with RT has been accepted in a variety of cancer types, including gynecological, gastro-intestinal, pulmonary and other malignancies. Hence, there is a critical need for new drugs that preferentially sensitize cancer cells to radiation therapy and that stimulate the innate immune response against irradiated cancer cells. Furthermore, to advance precision medicine in radiation oncology, there is a need for imaging and molecular diagnostic tests for determining the extent of cancer spread in the body and for predicting clinical responses to therapy.
| 44 |
We are developing our products with the goal of addressing the unmet need in cancer treatment for a commercially marketable radiation response modifier solution that leads to greater sensitivity of cancer cells to ionizing radiation therapy. The goal of our products is to increase the therapeutic index for patients receiving radiation and to decrease radiation-related toxicities in patients with solid tumors. Our products operate across three areas related to the treatment of cancer with RT:
| 1. | Sensitization of growing cancer cells, rendering them more susceptible to the effects of radiation therapy. | |
| 2. | Activation of the DNA damage response pathway to kill cancer cells and protect adjacent normal cells. | |
| 3. | Activation of the immune system to kill any remaining cells after RT. |
Our platform technology allows for the creation of an inventory of products for radiation sensitizing, immune modulation, and protection of healthy tissue.
Operations to date have focused on continuing our research and development efforts to advance Ropidoxuridine clinical testing and improved drug formulation, to advance HDAC6 inhibitor (SP-2-225) preclinical development and explore application of the PC-RAD Test, predictive biomarkers of radiation response. The clinical development of Ropidoxuridine has included completion of a Phase I clinical trial to establish drug bioavailability and a maximum tolerated dose for use in Phase II clinical trials. TCG GreenChem, Inc. (“TCG GreenChem”), with whom we have contracted for process research, development and cGMP compliant manufacture of IPdR, has manufactured the active pharmaceutical ingredient (API) of Ropidoxuridine and the University of Iowa Pharmaceuticals has formulated the drug product for use in the Company’s upcoming Phase II clinical trial in brain cancer patients undergoing radiation therapy. The drug product (capsules) were shipped to contract research organization (CRO) Theradex Oncology and distributed to clinical trial sites that are fully approved to enroll patients in the trial. Shuttle received approval from the FDA to begin the clinical trial. The FDA made recommendations to expand the clinical trial to include a randomized dose “optimization” step and we agreed with the recommendation. Meetings with engaged clinical sites to review the protocol documents have occurred and FDA required Institutional Review Board, or IRB approvals have been received. With FDA recommended changes incorporated into the revised protocol and the completion of site initiation visits, the Company has commenced its Phase II clinical study. The radiation biomarker project and the health disparities project have been completed and the Company is proceeding with plans for clinical validation and potential for commercialization of Ropidoxuridine as a radiation sensitizer.
Our Pipeline
We are currently developing a pipeline of small molecule radiation sensitizers and immune response regulating drugs. Our most advanced product candidate is Ropidoxuridine, an orally available halogenated pyrimidine with strong cancer radiation sensitizing properties is in preclinical studies. In addition, we have a pipeline of complimentary product candidates that we are developing to address solid tumor cancer indications by radiation sensitization or immune modification. Our therapeutic pipeline is represented in the diagram below:
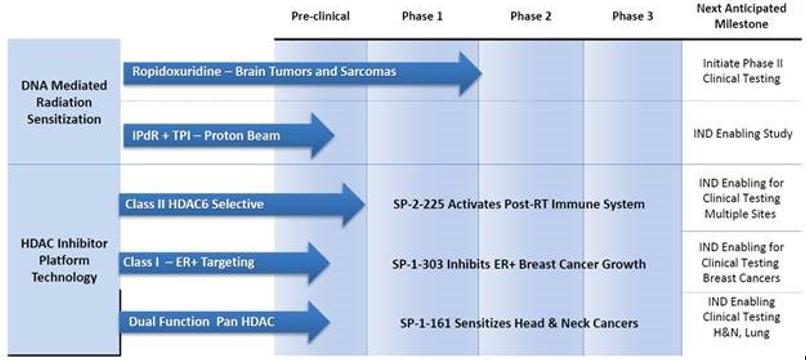
Timeline for clinical phase (Ropidoxuridine) and pre-clinical phase (HDAC inhibitors) pipeline.
| 45 |
Our lead product candidates include:
| ● | Ropidoxuridine (IPdR) is our lead candidate radiation sensitizer for use in combination with RT to treat brain tumors (glioblastoma) and sarcomas. Phase I clinical trial results supported by an NIH contract to Shuttle Pharma and the NCI (CTEP) were reported in the medical journal, Clinical Cancer Research, in July 2019, by our SBIR subcontractor. Eighteen patients completed dose escalations to 1,800 mg/day for 30 days, establishing the maximum tolerated dose (MTD) of 1,200 mg/day in combination with RT. Four partial responses, nine stable disease and one progressive disease in target lesions were reported. Four patients did not have measurable disease and, as a result, were not evaluable. These Phase I trial results demonstrate oral bioavailability and an MTD of 1,200 mg per day for 28 days for use in combination with radiation for Phase II clinical trials that we propose to perform in brain tumors and in sarcomas. The brain tumor, glioblastoma multiforme (GB) is eligible for orphan disease designations. Shuttle Pharma has advanced drug manufacture and formulation and prepared a clinical protocol of a “Phase 2 Single-Arm Study of IPdR as a Radiation Sensitizing Agent During Radiotherapy in Patients with Newly Diagnosed IDH-Wildtype MGMT Unmethylated Glioblastoma Multiforme.” In December 2023, we submitted an IND application with the FDA to support the next phase of development of Ropidoxuridine. In January 2024, we received the ‘Safe to Proceed’ letter from the FDA for our IND application for the Phase II study of Ropidoxuridine (IPdR) as a radiation sensitizing agent during radiotherapy in patients with newly diagnosed IDH-wildtype glioblastoma with unmethylated MGMT promoter. Receipt of the letter allows us to commence the Phase II study of Ropidoxuridine (IPdR). The clinical development of Ropidoxuridine has shown drug bioavailability and a maximum tolerated dose has been established for use in Phase II clinical trials. TCG GreenChem, Inc. (“TCG GreenChem”), with whom we have contracted for process research, development and cGMP compliant manufacture of IPdR, has successfully completed the manufacturing campaign for the active pharmaceutical ingredient (API) of Ropidoxuridine for use in the Company’s upcoming Phase II clinical trial in brain cancer patients undergoing radiation therapy. Shuttle also worked with University of Iowa Pharmaceuticals to develop the formulation and produce the capsules, which have been shipped to contract research organization (CRO) Theradex Oncology for distribution to clinical trial sites. Both activities have now been completed. In addition, Shuttle received approval from the FDA to begin the clinical trial. The FDA made recommendations that led to an expanded clinical trial to include randomized dose optimization and we agreed with the recommendation. We met with representatives from six candidate clinical sites to review the protocol documents and FDA required IRB approvals have been obtained. With FDA recommended changes incorporated into the revised protocol, the Company has now contractually engaged all of the planned six research centers which will be performing clinical trials and has initiated its Phase II clinical study. |
| ● | The Phase II clinical study is summarized below:
|
| 46 |
Schema for the Phase II clinical trial. The initial cohort of 40 patients will be randomized to one of two Ropidoxuridine doses. 20 patients will receive the 1200 mg dose and 20 patients will receive the 960 mg dose. The optimum dose will be determined by comparing drug bioavailability and side-effects. The optimum dose will then continue to enroll 14 additional patients to provide the required 34 patients for statistical significance in comparison to historical controls.
| ● | Ropidoxuridine and Tipiracil (IPdR/TPI) is a new combination formulation demonstrating extended bioavailability after oral administration in an animal model system. The IPdR/TPI formulation will undergo preclinical development for use as a radiation sensitizer and represents a “next generation” drug product for clinical evaluation. | |
| ● | SP-2-225 is Shuttle Pharma’s pre-clinical class IIb selective HDAC inhibitor that affects histone deacetylase HDAC6 has been identified as the candidate lead molecule for development as a post-RT innate immune system activator. The macrophage is a key target for activating the innate immune system against cancer cells. SP-2-225 has effects on the regulation of the immune system by maintaining macrophage cells in an inflammatory, anti-cancer polarization. The interactions of RT with the immune response for cancer treatment are of great current interest, offering insight into potential mechanisms for primary site and metastatic cancer treatment. For this reason, Shuttle Pharma has selected SP-2-225 as the candidate lead HDAC inhibitor for preclinical development. We have contracted with investigators at Georgetown University to perform preclinical studies of immune activation after radiation therapy in an animal tumor model. These data have been published (Noonepalle SKR, Grindrod S, Aghdam N, Li X, Gracia-Hernandez M, Zevallos-Delgado C, Jung M, Villagra A, Dritschilo A. Radiotherapy-induced Immune Response Enhanced by Selective HDAC6 Inhibition. Mol Cancer Ther. 2023 Dec 1;22(12):1376-1389. doi: 10.1158/1535-7163.MCT-23-0215. PMID: 37586844; PMCID: PMC10878032) and additional preclinical studies are in progress with our contractors. With the introduction of check-point inhibitors, CAR-T therapies and personalized medicine in cancer, regulation of the immune response following RT is of significant clinical and commercial interest. | |
| ● | SP-1-303 is Shuttle Pharma’s pre-clinical selective Class I HDAC inhibitor that preferentially affects histone deacetylases HDAC1 and HDAC3 members of the class I HDAC family of enzymes. SP-1-303 data show direct cellular toxicity in ER positive breast cancer cells. Furthermore, SP-1-303 increases PD-L1 expression. Completed preclinical in vitro studies have been published (Jung M, Nicholas N, Grindrod S, Dritschilo A. Dual-targeting class I HDAC inhibitor and ATM activator, SP-1-303, preferentially inhibits estrogen receptor positive breast cancer cell growth. PLoS One. 2024 Jul 15;19(7):e0306168. doi: 10.1371/journal.pone.0306168. PMID: 39008483; PMCID: PMC11249239.). We plan to seek university collaborations to complete SP-1-303 pre-clinical development in 2024. |
Our Approach
We believe that we have established a leadership position in radiation sensitizer and response modifier discovery and development. We have identified a clinical phase product candidate and discovered new pre-clinical phase molecules using our proprietary platform technologies to increase the therapeutic index for patients receiving radiation for treatment of solid tumors. Our development strategy has four key pillars: (1) to improve the efficacy of RT by demonstrating improved disease-free survival rates in patients who undergo radiation therapy, (2) reduce the amount of radiation needed for a favorable tumor response, thereby limiting the potential for radiation related toxicities to healthy cells, (3) decrease the extent of surgery needed to remove cancers and improve quality of life, and (4) leverage our next generation technologies to create drugs that regulate the immune response assisting immune checkpoint and CAR-T therapies and other personalized medicines targeting cancers.
In addition to private and public investment into our candidate therapeutic technology, we have also competed for non-dilutive funding from the NIH to support our lead sensitizer and to explore development of complimentary diagnostic products. To date, we have completed three SBIR contracts awarded to Shuttle Pharma by the NIH to:
| ● | Develop IPdR as a radiation sensitizer. This funding provided partial support for the Phase I clinical trial of Ropidoxuridine and RT. | |
| ● | Develop prostate cancer cell cultures from African-American men, with donor matched normal prostate cells, establishing 50 pairs for accelerating research to reduce prostate cancer health disparities in African-American men. This project was funded under “Moonshot” designation. Shuttle Pharma is eligible to apply for additional SBIR (Phase IIb) funding to commercialize these cells for research purposes. Currently, cells from African-American patients are distributed, on request, to investigators who are conducting health disparities research. We plan to test new small molecules using these cellular reagents for health disparities screening. |
| 47 |
| ● | Develop a predictive biomarker for determining outcomes for prostate cancer patients following treatment with RT. This SBIR-funded project for blood test (PC-RAD Test) discovery and analytical validation were completed on March 15, 2022, and Shuttle Pharma intends to perform a clinical validation study. Shuttle Pharma has licensed the intellectual property for the prostate cancer predictive biomarker test from Georgetown University and will seek additional investment from the public market to advance clinical development through its Shuttle Diagnostics entity. |
All three SBIR funded projects have been completed. The Company is eligible to apply for SBIR Phase IIb funding to advance the “Moonshot” health disparities or the predictive biomarker project. The NIH SBIR program is designed to encourage small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization.
Shuttle Pharma’s scientists have also developed collaborations to invent intellectual properties for prostate cancer theranostics. From a clinical perspective, prostate-specific membrane antigen (PSMA) is a valuable target for diagnosis and therapy of prostate cancer. In a discovery project to develop a novel, boron-containing PSMA ligand to enhance proton radiation therapy of prostate cancer, we discovered PSMA-B, a molecule containing boron and demonstrating nanomolar binding activity to PSMA. Preclinical evaluations have been initiated to explore the PSMA-B ligand as a potential prostate cancer sensitizer in combination with proton therapy, as well as a PET diagnostic reagent and as a targeted prostate cancer therapeutic. By in-licensing our collaborator’s shares of the intellectual property, Shuttle Pharma has an exclusive license to the PSMA-B intellectual property and has filed a patent application. Theranostic molecules are suitable for diagnosis and therapy of cancers. The PSMA ligand is a molecule that binds to the PSMA, an enzyme that is highly expressed in prostate cancer cells. The PSMA ligand is currently used for imaging and therapy to detect and treat prostate cancer.
| ● | Develop PSMA-B as a potential diagnostic and therapeutic molecule in pre-clinical models in collaboration with academic nuclear medicine programs. Shuttle Pharma has licensed the intellectual property for the prostate cancer predictive biomarker test from inventors and will seek additional investment from NIH by applying for grant applications and from the public market to advance clinical pre-development through its Shuttle Diagnostics entity. |
Our Strategy
Our goal is to maintain and build upon our leadership position in radiation sensitization. We plan to develop Ropidoxuridine and the HDAC6 inhibitor (SP-2-225) and, if approved by the FDA, commercialize our product candidates for the treatment of cancers. While this process may require years to complete, we believe achieving this goal could result in new radiation sensitizer and immunotherapy products. Key elements of our strategy include:
| ● | Capitalize on Ropidoxuridine as an orally available, small molecule radiation sensitizer. To date, there is one drug (Cetuximab, a monoclonal antibody) approved by the FDA specifically as a radiation sensitizer. If we are successful in developing Ropidoxuridine and obtaining FDA approval, a small molecule sensitizer would then be enabled for clinical applications for radiation sensitization indications. | |
| ● | Expand our leadership position within radiation sensitizers. In addition to our traditional radiation sensitizers, we plan to advance our near-term pipeline to include radiation sensitizers for proton therapy. Proton Therapy is growing worldwide as a form of radiation therapy due to its unique beam shaping characteristics. As a result, this new technology offers a major opportunity for Shuttle Pharma to strive to develop an innovative and well-tolerated drug for proton therapy sensitization. | |
| ● | Execute a disciplined business development strategy to strengthen our portfolio of product candidates. We have built our current product pipeline through in-house discovery, development, partnerships with leading academic institutions and through in-licensing. We will continue to evaluate new in-licensing opportunities and collaboration agreements with leading academic institutions and other biotechnology companies around programs that seek to address areas of high unmet need and for which we believe there is a high probability of clinical success, including programs beyond our target franchise areas and current technology footprint. |
| 48 |
| ● | Invest in our HDAC platform technology and maximize its utility across cancer therapies. We are initially applying the platform to develop drugs for cancer radiation sensitization, normal tissue radiation protection and post radiation immune stimulation. Based on the data we have obtained thus far, these drugs are immune regulatory. We intend to invest to develop other properties of our platform technology, as well. | |
| ● | Enter into collaborations to realize the full potential of our platform. The breadth of our HDAC technology platform enables other therapeutic applications, including radiation sensitization and immune therapy. We intend to seek collaborations centered on our platform to maximize applications for cancer treatment. | |
| ● | Establish Shuttle Diagnostics, Inc. as a subsidiary of SHPH to advance development of the predictive biomarker (PC-RAD Test) and the PSMA ligand (PSMA-B) to advance prostate cancer treatment. |
Radiation Therapy
Radiation Oncologists use Radiation Therapy (RT) to treat cancers that cannot be completely removed by surgery but have not yet spread to distant sites within the body. RT has been a mainstay for the treatment of cancer malignancies for more than half a century. The combination treatment of radiation therapy and chemotherapy has involved the use of cytotoxic drugs, targeted biologic agents and targeted external beam radiation to increase the destruction of tumor cells and cure or delay cancer progression. The low number of drugs and biologic agents under investigation as radiation sensitizing agents highlights an unmet need for new approaches and agents that provide greater effectiveness, increased quality and better tolerability for patients.
Currently, “chemo-radiation” treatments are established in cancers of the head and neck, esophagus, lung, stomach, breast, brain, pancreas, rectum and uterine cervix. The ideal radiation sensitizer would reach the tumor in adequate concentrations and act selectively in the tumor, as compared to surrounding normal tissues. It would have predictable pharmacokinetics for timing with radiation therapy and could be administered with every radiation treatment approach. The ideal radiation sensitizer would have minimal toxicity or manageable enhancement of radiation toxicity.
The U.S. market for radiation sensitizing agents is experiencing dynamic growth through development of new radiation technology, the introduction of new agents, growth in the number of diagnosed patients in a variety of cancers and changes in treatment patterns. New agents have been introduced, including bevacizumab (Avastin®, Roche), panitumumab (Vectibix®, Amgen), temozolomide (Temodar®, Merck) and cetuximab (Erbitux®, Eli Lilly/Imclone), with potential as radiation sensitizing agents (though all but cetuximab are used off label); and all are recommended by the NCCN® (National Comprehensive Cancer Network) in clinical practice guidelines for use in combination with established therapies such as FOLFOX (leucovorin, 5-FU, oxaliplatin), CapeOX (capecitabine, oxaliplatin) and FOLFIRI (leucovorin, 5-FU, irinotecan).
The growth in the number of patients with cancers is being driven by an aging population and improved diagnostic tools. According to the National Cancer Institute (NCI), more than half (~50 - 60%) of all cancer patients undergo some type of radiotherapy during the course of their treatment. Confirming the patient estimate from the NCI, the American Society for Therapeutic Radiology and Oncology (ASTRO) factsheet states approximately 67% of approximately 1.25 million cancer patients are treated with radiation therapy annually, either one or more times. In addition, in a study published by the Journal of Clinical Oncology in 2016, it is estimated that the number of cancer patients needing radiation therapy will increase by 22% in the next 10 years. (See “The Future of Radiation Oncology in the United States From 2010 to 2020: Will Supply Keep Pace With Demand?” Benjamin D. Smith, Bruce G. Haffty, Lynn D. Wilson, Grace L. Smith, Akshar N. Patel, and Thomas A. Buchholz Journal of Clinical Oncology 2010 28:35, 5160-5165).
The American Society of Clinical Oncology (ASCO) estimates more than 80% of cancers in the U.S. occur in people in the age group of 50 and above with over 60% of cancers occurring in those 65 and over. (See, 2018 Clinical Cancer Advances Report, American College of Clinical Oncology, 2018). For example, according to the American Cancer Society (ACS), more than 90% of colorectal cancer patients are individuals aged 50 years and older, with approximately 40% of all cases occurring in patients aged 75 years and over. The Colon Cancer Alliance estimates that 90% of new cases and 95% of deaths from colorectal cancers occur in people aged 50 or older. Also, the U.S. Census estimates that the age group of 65-84 will grow by 23% within the next five years, indicating a likely increase in the overall number of cancer patients in the U.S.
| 49 |
The table below details the number of cancers estimated in the United States in 2024:
Estimated New Cancer Cases in the U.S.
| Male | Female | |||||||||||||||||
| Prostate | 299,010 | 27 | % | Breast | 310,720 | 31 | % | |||||||||||
| Lung & bronchus | 116,310 | 12 | % | Lung & bronchus | 118,270 | 13 | % | |||||||||||
| Colon & rectum | 81,540 | 8 | % | Colon & rectum | 71,270 | 8 | % | |||||||||||
| Urinary bladder | 118,330 | 6 | % | Uterine corpus | 67,880 | 7 | % | |||||||||||
| Melanoma of the skin | 59,170 | 6 | % | Melanoma of the skin | 41,470 | 5 | % | |||||||||||
| Kidney & renal pelvis | 52,380 | 5 | % | Thyroid | 31,520 | 3 | % | |||||||||||
| Non-Hodgkin lymphoma | 44,590 | 4 | % | Non-Hodgkin lymphoma | 36,030 | 4 | % | |||||||||||
| Oral cavity & pharynx | 41,510 | 4 | % | Kidney & renal pelvis | 29,230 | 3 | % | |||||||||||
| Leukemia | 36,450 | 4 | % | Pancreas | 31,910 | 3 | % | |||||||||||
| Pancreas | 34,530 | 3 | % | Leukemia | 26,320 | 3 | % | |||||||||||
| All sites | 983,160 | All sites | 934,870 | |||||||||||||||
ACS Facts & Figures, 2024
The U.S. estimated incidence, deaths and five-year survival rate of cancer patients responsive to radiation therapy is significant (ACS Facts & Figures, 2024). The top cancers responsive to radiation are shown, based on the number of newly diagnosed patients. The incidence rates for some cancers are increasing by approximately 1-2% per year in the U.S. The number of newly diagnosed patients is significant and growing due to the aging of the population and improved diagnostic techniques.
The cancers listed above illustrate the opportunity presented for radiation sensitizers. Of note is the low five-year survival of pancreas, brain, lung and esophagus cancers—all are candidates for Shuttle Pharma’s pipeline of radiation sensitizing compounds. Cancers with low survival rates are of interest since they show a high unmet need for new therapeutics and an opportunity for Shuttle Pharma to gain significant uptake of their pipeline compounds.
Factors that present challenges and may restrict growth in the radiation sensitizer market include the safety and tolerability of many of the newer agents with radiation sensitizing properties; a regulatory environment that engenders greater levels of scrutiny of clinical practice issues; the high cost of newer agents; and the changing (and more restrictive) reimbursement environment in radiation oncology through CMS (Center for Medicare and Medicaid Services) and private payors. These factors may negatively impact the potential for growth in the U.S. market.
Many of the drugs used “off-label” as radiation sensitizers currently require close scrutiny of their potential for side effects that can affect the safety and tolerability of their use with patients. All of the current agents carry significant potential for side effects that can affect patients’ therapies and quality of life. Radiation sensitizing agents can cause both acute and chronic side effects in patients. Side effects can vary from person to person depending on age, sex, type of cancer, dose given per day, total dose given, and the patient’s general medical condition. Some common side effects of currently used radiation sensitizers include leukopenia, skin damage, hair loss, fatigue, bladder problems, nausea, fibrosis, memory loss, infertility, and enhanced risk of developing a second cancer, which may arise as a result of the patient’s weakened immune system due to cytotoxic drugs used in treatment or when newer biologic agents cause the over-production of specific cytokines or proteins, which can lead to developing secondary cancers.
Over the past five years, the FDA has taken an increasingly conservative approach to the approval of new agents for oncology treatment. There is greater scrutiny of results from clinical trials regarding progression free survival, overall survival, and safety and tolerability of new agents. Restrictions such as black box warnings and REMS (Risk Evaluation and Migration Strategies) are being applied to more new products over the past five years compared to the previous five years. These restrictions require physicians to be more careful in evaluating the use of newer agents and newer diagnostic tools to select the most appropriate patients for newer approved agents.
Many of the new agents are molecularly targeted therapies that are biologic in their development and manufacturing. The cost of the newer agents can be significant. For example, the cost for Avastin for one treatment course as a radiation sensitizer is estimated at $9,000-12,000 according to one Key Opinion Leader in the U.S. (Carl Schmidt, Consultant, Shuttle Pharmaceuticals Holdings, Inc., Business Plan 2018). Recently, a CAR-T gene therapy from Novartis was launched with a yearly cost of $475,000. Further, as many private payors scrutinize the cost and appropriate use of newer drugs, they require physicians to provide justification through prior authorization requests, use of step therapy and guidelines that delay treatment, increase administrative costs and limit the therapeutic choices for physicians and hospitals.
| 50 |
Public payors for radiation oncology therapies such as CMS have instituted reimbursement reductions that potentially affect the overall cost of therapy and can limit the acceptance of newer agents. With CMS announced reductions in reimbursement for radiation oncology, there is increased pressure to find a more potent radiation sensitizer agent with reduced side effects, and greater cost-effectiveness.
Escalating healthcare spending is adding pressure on government and commercial payors to contain drug costs. While the oncology space is arguably not as tightly managed by payors as other therapeutic areas, utilization management of costly cancer therapeutics has become an increasing priority for U.S. payors, especially with the advent of biologics. Payors (and market access agencies in the EU) will most often restrict high-cost drugs, drugs with limited or no survival benefits, and drugs deemed to be at high risk for widespread off-label use.
Beyond efforts at cost containment by insurers (which often require patients to first be prescribed lower cost drugs to determine effectiveness prior to allowing for reimbursements for more expensive (or less cost effective) drugs), payors are also looking toward implementing clinical pathways as a way to maintain or improve health outcomes while lowering costs. Clinical pathways are designed to address the limitations of prior authorization and of reduced fee schedules, offering more durable cost containment to payors. These pathways may lead to cost savings by encouraging the use of generics, streamlining treatment choices, and reducing side effects while maintaining outcomes.
Engineered Radiation Sensitizers
The market for radiation sensitizers in selected cancer types is defined by the need to improve local-regional tumor control. Treatment regimens have been developed to address patient needs for tumor control and quality of life. Since the initial applications of Ropidoxuridine and selective HDAC inhibitors are as adjuncts to the standard of care for the treatment of radiation responsive cancers, the unmet needs of the market lie in the potential for the following:
| ● | Improvement in efficacy of radiation treatments as determined by overall survival, progression free survival and response rates in comparison to currently used “off-label” sensitizer drugs. | |
| ● | Reduction in radiation doses needed to affect a positive clinical response for the patient. | |
| ● | Reduction in the surgical extent that is needed to remove residual cancer. | |
| ● | Improvement in quality-of-life outcomes. |
Various sources have estimated that more than 800,000 patients in the U.S. are treated with radiation therapy for their cancers. According to the American Cancer Society, about 50% are treated for curative purposes and the balance for palliative care. The market opportunity for radiation sensitizers lies with the 400,000 patients treated for curative purposes. The number of patients being treated with RT is expected to grow by more than 22% over the next five years. Based on a rough estimate of a course of radiation sensitizing brand drug therapy (off label at this time) of $12,000 per patient—the market size would exceed $4.0 billion. This would represent about 4% of the annual cost of cancer care in the U.S.
In the past two decades, developments in the field of oncology have resulted in an increase in the number of clinical trials of marketed products that exhibit radiation sensitizing properties. The following are a few examples of recently approved products that exhibit radiation sensitizing properties: topotecan (Hycamtin®) was approved for ovarian and small-cell lung cancer and also in cervical cancer when used in combination with cisplatin. Irinotecan (Camptosar®) is used for metastatic colorectal carcinoma, trastuzumab (Hercepetin®) for breast cancer, and gefitinib (Iressa®) for locally advanced non-small-cell lung cancer. However, the claims on radiation sensitization are anecdotal in the scientific literature.
In addition, clinical trials are in progress to develop novel molecules (such as poly (ADP-ribose) polymerase (PARP) inhibitors (such as Lynparza (olaparib)), histone deacetylase (HDAC) inhibitors (such Zolinza® (vorinostat) and heat-shock protein 90 (hsp90) inhibitors with potential to increase the therapeutic use of compounds with radiation sensitizing properties for other cancers. Several drugs with radiation sensitizing properties are currently in Phase III clinical trials, such as nimorazole (for head and neck cancer), motexafin gadolinium (for brain metastases), and cisplatin (for cervical cancer); though none are likely to apply for a radiosensitizing claim with the FDA since the radiosensitizing element in their clinical trials are not primary endpoints. While additional drugs with radiation sensitizing properties are expected to be launched in the future, thereby driving the radiation sensitizers market further, to date, there is no indication that any drug in development is expected to be approved specifically as a radiation sensitizer.
| 51 |
The competitive environment for “off-label” radiation sensitizers for solid tumor cancers is anticipated to become predominantly generic. Avastin, Erbitux, Camptosar and Xeloda have or will lose patent protection in the next three years. Newer products under investigation or approved, such as Vectibix® (panitumumab) from Amgen will be promoted as having radiation sensitizing properties, along with indications for treatment for specific cancers. The high cost of these new therapies coupled with limited efficacy compared to current standard of care will be constrained by both public and private payors. Other new agents are in development but will face similar challenges.
We anticipate that new products launching into the cancer market with anecdotal claims for use as radiation sensitizers with improved effectiveness, quality and tolerability will initially be limited in their growth until they have been added to established clinical pathways and guidelines. If their effectiveness, quality and tolerability are demonstrated clinically, as determined by the FDA, it is anticipated the National Comprehensive Cancer Network (NCCN), the leading authority in oncology drug evaluation for treatment guidelines, would issue a recommendation and addition to standard of care within approximately six to twelve months after launch. An NCCN recommendation would positively impact the growth potential for a new product entering the market. Also, payors, both public and private, would add the new product to their approved drug lists and provide reimbursement giving providers incentive to use the product as neoadjuvant and adjuvant therapy to standard of care.
As with many cancer therapies, side effects can often have a distinct impact on quality of life and influence the potential for market growth. Patients increasingly have a stronger voice in the decision-making process for the appropriate therapies and costs to treat their cancers. As payors are increasingly placing more of the financial burden of the cost of therapy directly on patients, patients are voicing their opinions to their physicians and payors which have a direct effect on which products are selected. Many of the current therapies have significant side effects:
Private insurers are expected to have more restrictive formularies and medical benefits in which patients will be expected to carry more of the burden of the cost of drugs. Also, it is anticipated that increased application of third party developed treatment guidelines, such as those from the NCCN (National Comprehensive Cancer Network), are expected to be used by private payors to limit the access to products for specific conditions through prior authorizations and implementation of step therapy or increased out of pocket cost approaches. As many of the current drugs used as radiation sensitizers are expensive and not approved for use as radiation sensitizers (thus, such treatment is “off label”), and as many of the products in clinical trials are expected to be at the current or higher price levels, new products that may be specifically approved for an indication as the only approved product as a radiation sensitizer will have increased consideration for reimbursement.
CMS is increasingly moving many patients to private insurance through Medicare Advantage and ACOs. Medicare Advantage plans are capitation HMO and PPO plans offered through private insurers to Medicare patients. ACOs are being developed to increase quality of care for their patients. Most of the new ACOs are initially positioned for Medicare patients with over 400 approved by CMS. Several studies from the Center for Health Strategies, 2017, Journal of American Medical Association, 2018 and the Brookings Institute, 2015 estimated that almost 1000 ACOs for Medicare and non-Medicare patient populations would be approved by CMS or developed by a variety of healthcare entities to begin operating under the ACA in 2017. We expect the growth in ACOs to continue, regardless of any changes that may be made to the ACA going forward. In early 2017, Health Affairs, a magazine tracking ACOs, estimated that over 22 million patients are enrolled in Medicare and private ACOs. To address the quality of care measures designated by CMS and to gain additional incentives, use of clinical pathways or treatment guidelines is anticipated to be increasingly instituted to manage patient care. The impact on the uptake of new products in this environment can be profound if the new product is first in class and is included in national guidelines from organizations such as the NCCN and/or approval by the regional CMS contracting groups.
ROPIDOXURIDINE
The halogenated thymidine (TdR) analogs, bromodeoxyuridine (BUdR) and iododeoxyuridine (IUdR), are a class of pyrimidine analogs that have been recognized as potent radiosensitizing agents since the early 1960s. (See Kinsella TJ. An Approach to the Radiosensitization of Human Tumors. Cancer J Sci Am. Jul-Aug 1996:2(4); 184-193). Their cellular uptake and metabolism are dependent on the TdR salvage pathway where they are initially phosphorylated to the monophosphate derivative by the rate-limiting enzyme, thymidine kinase (TK). (See Shewach DS, Lawrence TS. Antimetabolite radiosensitizers. J Clin Oncol, Sep 10 2007; 25(26):4043-4050). After sequential phosphorylation to triphosphates, they are then used in DNA replication, in competition with deoxythymidine triphosphate (dTTP), by DNA polymerase. DNA incorporation is a prerequisite for radiosensitization of human tumors by the halogenated TdR analogs, and the extent of radiosensitization correlates directly with the percentage TdR replacement in DNA. (See Lawrence TS, Davis MA, Maybaum J, Stetson PL, Ensminger WD. The Dependence of Halogenated Pyrimidine Incorporation and Radiosensitization on the Duration of Drug Exposure. International Journal of radiation oncology, biology, physics. Jun 1990; 18(6);1393-1398). The molecular mechanisms of radiosensitization are most likely the result of increased susceptibility of TdR analog-substituted DNA to the generation of highly reactive uracil free radicals by ionizing radiation (IR), which may also damage unsubstituted complementary-strand DNA. Repair of IR damage may also be reduced by pre-IR exposure to these analogs.
| 52 |
The rationale for using Ropidoxuridine as a radiation sensitizer is based on prior clinical studies with the active metabolite IUdR; identified in NIH laboratories as a potent radiation sensitizer. Ropidoxuridine is an orally available prodrug of IUdR. In the body, Ropidoxuridine is metabolized in the liver into IUdR. IUdR is incorporated into the DNA of actively growing cells and when cells are exposed to ionizing radiation, DNA strand breaks are generated, resulting in more cell death and radiation sensitization. (See Gurkan E, Schupp JE, Aziz MA, Kinsella TJ, Loparo KA. Probabilistic modeling of DNA mismatch repair effects on cell cycle dynamics and iododeoxyuridine-DNA incorporation. Cancer Res. Nov 15 2007; 67(22):10993-11000).
Most of the clinical efficacy data were obtained from NIH supported studies performed with IUdR, the active metabolite of Ropidoxuridine. However, IUdR requires constant infusion over six weeks of therapy which creates a significant compliance issue for patients. Ropidoxuridine can be given as a capsule for oral administration, resulting in greater ease of medication delivery and potentially improved compliance and fewer complications.
Over the last 20 years, there has been renewed interest in these halogenated TdR analogs as experimental radiation sensitizers in selected cancer patient groups. These analogs are rapidly metabolized in both rodents and humans, principally with cleavage of deoxyribose and subsequent dehalogenation by hepatic and extrahepatic metabolism, when given as a bolus infusion with a plasma half-life of <5 min. Consequently, prolonged continuous or repeated intermittent drug infusions over several weeks before and during irradiation are necessary, based on in vivo human tumor kinetics, to maximize the proportion of tumor cells that incorporate these analogs into DNA during the S phase of the cell cycle. (See Fowler JF, Kinsella TJ. The Limiting Radiosensitization of Tumors by S-phase Sensitizers. Br J Cancer. 1996;74 (Suppl)(27):294-296). Phase I and Phase II trials using prolonged continuous or repeated intermittent intravenous infusions of BUdR or IUdR before and during radiation therapy (RT) have focused principally on patients with high-grade brain tumors. These clinically radiation resistant tumors can have a rapid proliferation rate (potential tumor doubling times of 5–15 days) and are surrounded by non-proliferating normal brain tissues that show little to no DNA incorporation of the TdR analogs. As such, high-grade brain tumors are ideal targets for this approach to radiation sensitization. In Phase I/Phase II clinical trials, prolonged survival outcomes were observed compared to RT alone in patients with anaplastic astrocytomas and in patients with glioblastoma multiforme IUdR continuous IV infusion (1000 mg/m2/ day/ 14 days), Total 39 patients (F. Sullivan, et al. Int J Radiat Oncol Biol Phys. 1994; 30(3):583-90.) A therapeutic gain in clinical radiation sensitization using these halogenated TdR analogs was proposed for other types of poorly responsive (radiation resistant) cancers, including locally advanced cervical cancer, head and neck cancers, unresectable hepatic metastases from colorectal cancers, and locally advanced sarcomas, based on the results of other Phase I/Phase II clinical trials.
Target Indication: Glioblastoma, Sarcomas and Rectal Cancers
After completion of the Phase I clinical trial of Ropidoxuridine and RT in advanced GI cancers, we proposed to perform Phase II efficacy clinical trials in brain tumors (glioblastoma), soft tissue sarcomas, and rectal cancers. Glioblastoma multiforme is a deadly malignancy of the brain with no known cure. Radiation therapy provides delay of disease progression and is standard of care following surgical resection or biopsy. Radiation therapy is combined with Temodar, a drug that has shown activity (~ four months survival benefit) in treating brain tumors. Preliminary data using radiation therapy in combination with IUdR resulted in a delay of disease progression of up to six months. We propose to test IPdR in combination with radiation therapy in the Phase II clinical trial. Similarly, delay in disease progression has been observed following treatment of sarcomas by the combination of IUdR and RT. Based on the Phase I data of our clinical trial we know that therapeutic levels of IUdR are reached by administering the orally available prodrug, IPdR.
Clinical Data
The Phase I results of the clinical trial supported by an SBIR contract to Shuttle Pharma and a sub-contract to the Brown University Oncology Group (BrUOG) at the LifeSpan/Rhode Island Hospital were reported by the subcontractor at the 30th EORTC-NCI-AACR Symposium in November 2018 and in the medical journal, Clinical Cancer Research, in 2019. Eighteen patients completed dose escalation to 1800 mg/day for 30 days, establishing the maximum tolerated dose (MTD) of 1,200 mg/day in combination with RT. Therapeutic blood levels of IUdR were achieved. Four patients were scored as partial responses, nine patients had stable disease and one patient progressed in the target lesions. These data support advancing IPdR and RT to clinical trials for the FDA to determine efficacy.
| 53 |
Development Plan
A key to driving the Ropidoxuridine product forward is the development of a clinical plan with aggressive timelines and support within the radiation oncology community to participate in clinical trials with the appropriate patients to ensure a comprehensive NDA dossier for each product. Initially, the plan is focused on the Phase I and Phase II clinical trials. Upon completion of Phase II studies, we will determine whether to extend the Phase II study to a randomized Phase II, or to perform a randomized Phase III clinical trial. Such determination will be based on results of the initial clinical trials and the end of a Phase II meeting with the FDA. Shuttle Pharmaceuticals requested and received FDA orphan drug status for Ropidoxuridine as a clinical radiation sensitizer for treatment of glioblastoma and pre-operative treatment of soft tissue sarcomas. As a result, the application for “orphan” designation for Ropidoxuridine with RT for glioblastoma has been approved. The application for sarcomas, however, was not approved and will require addressing certain FDA comments and resubmission. The IPdR/TPI formulation clinical plan will focus on resectable stage II and III rectal cancer patients.
Our clinical plan for Ropidoxuridine development as implemented to date, includes:
| ● | GMP manufacture and formulation of 24 kg of Ropidoxuridine for use in clinical trials has been completed. | |
| ● | The IND for a Phase II clinical trial of Ropidoxuridine and RT in glioblastoma has been approved for the study to proceed. | |
| ● | The contract research organizations (CRO), Theradex Oncology has been engaged to assist in the performance the Phase II clinical trial. | |
| ● | Completion of the Phase II clinical trial in glioblastomas to determine appropriate dosing, effectiveness and tolerability of the treatment. |
The data obtained from the NIH/NCI SBIR funded Phase I clinical trial supported efforts to raise capital to enable performing the Phase II clinical trials of Ropidoxuridine. We aim to conduct and complete the Phase II clinical trial so that we may present data to the FDA for its determination of efficacy. We believe this will support our efforts to raise the additional required capital to complete Phase II and to fund the Phase III clinical trials and seek FDA approval of an NDA with “orphan” designation.
The clinical plan for the IPdR/TPI formulation will focus on resectable Stage II and Stage III rectal cancer patients and on recurrent glioblastoma treated with proton therapy. Nonetheless, we cannot guarantee the successful completion of any of these trials. Our inability to meet any of the aforementioned milestones in the Phase II or Phase III clinical trials will cause us to be unable to proceed with our present efforts and will likely cause us to be unable to raise additional funds.
Our HDAC Small Molecule Delivery Platform
General
Since the founding of Shuttle Pharma, our discovery research and development efforts have been focused on our small molecule technology delivery platform which uses HDAC inhibitors, designed to target cancer cells, while protecting healthy tissue.
HDACs are a class of enzymes that regulate gene expression through chemical modification of histones and non-histone proteins. Increased HDAC activity leads to a more condensed chromatin (which is a protein complex consisting of DNA and other proteins), decreased gene expression and loss of key gene products, including tumor suppressor gene function. Inhibition of HDAC activity leads to a more open chromatin and increased expression of the key gene products. This chromatin modification underlies the epigenetic cellular regulatory system and is an area of intense investigation.
Our research and development efforts to date have focused on the discovery of novel, dual functional molecules for potential use in cancer treatment as radiation sensitizers of cancers, protectors of normal tissues, and activators of the immune responses to antigens expressed by irradiated cancer cells. To date, we have produced three candidate molecules:
| ● | SP-1-161, a candidate lead of compounds demonstrating activation of the “ATM” gene product (mutated in Ataxia-Telangiectasia). Ataxia-Telangiectasia is a human genetic disease characterized by neurological, immunological and radiobiological clinical features. |
| 54 |
| ● | SP-2-225, a candidate lead of compounds demonstrating Class II (HDAC6) selective inhibition. HDAC6 is a molecule integral to the presentation of antigens by macrophages to T-lymphocytes. | |
| ● | SP-1-303 is a candidate Class I HDAC inhibitor with preferential efficacy against ER positive cancers. |
SP-1-161 - A Dual Functional Agent
SP-1-161 is an HDAC inhibitor of the hydroxamate chemical class of compounds and an ATM activator of the indole chemical class. HDACs modify histones and non-histone proteins, which are key components of the chromatin structure, gene expression regulation, and cell growth. HDAC inhibitors inhibit cell proliferation, angiogenesis and immunity. Eighteen human HDACs have been identified, subdivided into four classes based on sequence and functional homology. In cancer cells, HDAC activity silences tumor suppressor genes important for cell growth regulation and to chromosomal instability. Abnormal HDAC activity is also associated with tumor cell growth, invasion, metastasis and resistance to therapy. Therefore, inhibitors of HDACs have emerged as anti-cancer agents for cancer therapy. Vorinostat and romidepsin have been approved by the FDA for treatment of patients with relapsed or refractory T-cell lymphomas. In addition, panobinostat received FDA approval for treatment of recurrent multiple myeloma in combination with bortezomib and dexamethasone.
In preclinical studies, SP-1-161 inhibited the activity of pan-HDACs and activated the ATM gene product. ATM is a critical protein for the activation of the cell stress response for cellular recovery from radiation exposure in normal cells, but not in cancer cells. ATM activates the P53 protein, referred to as the “guardian of the genome,” and serves as a tumor suppressor critical for normal cell function and activation of programmed cell death in cancer cells.
In preclinical studies, SP-1-161 protected normal breast epithelial cells (184A1) following exposure to ionizing radiation while increasing sensitivity of breast cancer cells (MCF7). SP-1-161 provides this dual function in a single molecule and this molecule is differentiated from other HDAC inhibitors by treatment of cancers while protecting normal cells. (See Grindrod S, Brown M, Jung M. “Development of dual Function Small Molecules as Therapeutic Agents for Cancer Research,” Poster presentation #A178, American Association of Cancer Research Oct 2017).
SP-2-225
SP-2-225 is a selective HDAC inhibitor that affects histone deacetylase (HDAC6) and is a member of the class IIb HDAC family. Class II HDACs play important roles in cancer motility, invasion, neurological diseases, and immune checkpoint. HDAC6 inhibition has been most extensively studied for its role in the treatment of hematological cancers. HDAC6 is unique among HDAC enzymes in having two active catalytic domains and a unique physiological function. In addition to the modification of histones, HDAC6 targets specific substrates including α-tubulin and HSP90, and are involved in protein trafficking and degradation, cell shape and migration. Selective HDAC6 inhibitors are an emerging class of pharmaceuticals due to the involvement of HDAC6 in pathways related to neurodegenerative diseases, cancer and immunology. Specifically, its potential to affect regulation of the immune system and enhance the immune response in cancer is of great interest. With the introduction of check-point inhibitors, CAR-T therapies and personalized medicine in cancer, regulation of the immune response to this therapy is of significant clinical and commercial interest. (See Noonepalle SKR, Grindrod S, Aghdam N, Li X, Gracia-Hernandez M, Zevallos-Delgado C, Jung M, Villagra A, Dritschilo A. Radiotherapy-induced Immune Response Enhanced by Selective HDAC6 Inhibition. Mol Cancer Ther. 2023 Dec 1;22(12):1376-1389. doi: 10.1158/1535-7163.)
Selective inhibition of HCAC6 reduces dose limiting side effects associated with non-selective HDAC inhibitors. Selective HDAC6 inhibitors may be combined with other cytotoxic agents. Shuttle’s discovery of selective HDAC inhibitors has yielded several HDAC6 selective candidate molecules including SP-2-225. HDAC6 inhibitors are under investigation for roles in the treatment of diseases such as multiple myeloma.
| 55 |
SP-1-303 - Target Indication: Breast Cancer
Histone deacetylase inhibitors sensitize cancers to the effects of radiation, protect normal tissues from radiation injury and activate the immune system. SP-1-303 is a selective Class I HDAC inhibitor that inhibits HDAC1, 3 and 6 and has direct cellular toxicity in ER positive breast cancer cells. Furthermore, SP-1-303 increases the PD-L1 expression level in a time-dependent manner, support combination of SP-1-303 with an immune checkpoint blocker to enhance the therapeutic benefits. We are currently conducting preclinical efficacy studies of these molecules.
Development Plan
The HDAC inhibitor platform of candidate molecules will require pre-clinical evaluation, completion of IND-enabling studies and the lead drug candidates will be tested in Phase I clinical trials for pharmacokinetics and MTD determination. We have three lead candidates for potential development for the treatment of solid tumors, including breast cancer, lung cancer and multiple myeloma.
The results of Phase I and Phase II clinical trials will determine further drug development and Shuttle will seek to establish collaborative partnerships with other pharmaceutical companies to complete pre-clinical and clinical development, drug manufacturing and marketing of our product candidates. In the event we are unsuccessful in completing our clinical trials at any stage, or in the event we obtain negative results, we will likely be unable to raise additional funding related to our HDAC studies or will have to change direction of our research efforts regarding the HDAC inhibitor platform of candidate molecules.
Our Manufacturing Strategy
We have no manufacturing facilities that are owned or operated by us. We have performed laboratory scale synthesis and testing in our research laboratories in Gaithersburg, Maryland. GMP synthesis of API, drug formulation and human dosage preparation will be performed under contracts with third-party manufacturers.
Strategic Agreements
We have developed important strategic agreements with academic institutions for access to resources such as intellectual property, core facilities and contracting relationships. In addition, we have established an agreement with Propagenix for intellectual property in-licensing. Our current and ongoing relationships include:
| ● | Georgetown University |
| ○ | Sub-contractor for the SBIR supported African American prostate cancer patient health disparities project (completed). The conditional reprogramming of cells (CRC) technology was invented at Georgetown University and Georgetown University owns the intellectual property. Propagenix holds the license for the intellectual property for the CRC technology from Georgetown University. The intellectual property for cells derived from African American patients under the Georgetown University subcontract belong to Shuttle Pharmaceuticals, Inc. based on our sub-licensing agreement with Propagenix. | |
| ○ | Sub-contractor for the SBIR supported metabolomic predictive biomarker project (completed). The metabolomic biomarker intellectual property belongs to Georgetown University and Shuttle Pharma holds an exclusive option to license the intellectual property. | |
| ○ | Exclusive licensing agreement with Georgetown University pursuant to which Georgetown University agreed to license the intellectual property known as “Predictive Biomarkers for Adverse Effects of Radiation Therapy” (U.S. Patent Application No. 17/476,184, filed on September 15, 2021) (the “Patent Rights”), which was developed by Dr. Anatoly Dritschilo, the Company’s Chief Executive Officer, Dr. Scott Grindrod, the Company’s Principal Scientist, and Drs. Amrita Cheema and Yaoxiang Li, employees of Georgetown. The Patent Rights will be available for the Company’s use worldwide. | |
| ○ | Shuttle Pharma entered into a research agreement (the “Research Agreement”) with Georgetown University for testing small molecule radiation sensitizers and immune activation candidates discovered and developed by Shuttle Pharma in cell-based and animal xenograft models. | |
| ○ | In conjunction with the Research Agreement, Shuttle Pharma also entered into a material transfer agreement (the “MTA”), dated March 21, 2023, with Georgetown University. Under the MTA, Shuttle Pharma agreed to transfer research quantities of candidate drug molecules to Georgetown University, which materials will be used by Georgetown University solely to carry out additional research for Shuttle Pharma and which materials shall at all times remain the property of Shuttle Pharma. |
| ● | Brown University |
| ○ | Sub-contractor of the SBIR supported Phase I clinical trial of IPdR and RT (completed). |
| ● | University of Virginia |
| ○ | Research collaboration to develop heavy oxygen molecules for proton radiation sensitizer applications. |
| ● | George Washington University |
| ○ | Material transfer agreement for testing HDAC inhibitor effects in immune model systems | |
| ○ | The material transfer agreement that protects our HDAC inhibitor intellectual property is with George Washington University, transferring drugs for research purposes and sharing authorship on publications. There is no transfer of funds related to such activities. |
| ● | Propagenix, Inc. |
| ○ | License agreement for “conditional re-programmed cell” (CRC) technology. The cells established by Shuttle Pharma scientists at Georgetown University belong to us, based on the sublicense from Propagenix, Inc. An up-front licensing fee of $25,000 was paid to Propagenix. No other future milestone or royalty payments owed related to the Propagenix agreement. |
Competition “Off-Label” Use

Drugs with radiation sensitizing properties.
Our Product Candidates
We are advancing a clinical stage product candidate, Ropidoxuridine, that we believe will target cancer cells while protecting healthy tissue when used in conjunction with RT.
| 56 |
Ropidoxuridine
Ropidoxuridine, an orally available halogenated pyrimidine with strong cancer radiation sensitizing properties, is our lead “clinical phase” product candidate. Halogenated pyrimidines are incorporated into DNA by rapidly growing cancer cells and become more sensitive to the effects of RT. We have received an SBIR contract from the NIH to fund a Phase I clinical trial in collaboration with Brown University at the Lifespan/Rhode Island Hospital to determine the maximum tolerated dose in patients with advanced gastrointestinal cancers. In connection with the trial, NCI approved the Phase I clinical protocol and provided drug and clinical data management support to Rhode Island Hospital. The Phase I clinical trial has been completed and the results support advancing Ropidoxuridine to Phase II clinical trials of brain tumors, sarcomas and other tumors.
The following tables provide data from reported clinical trials of Iododeoxyuridine and RT therapy in brain cancers (glioblastoma multiforme) and high-grade sarcomas. Our primary strategy for Ropidoxuridine and RT therapy is to provide oral drug delivery to effect radiation sensitization of cancers and validate effectiveness in glioblastoma and sarcoma, potential “Orphan” indications.
Brain Cancer Treatment
Efficacy compared to historical RT-alone controls for treatment
of high-grade primary brain tumors (RTOG*, NCI** trials)

| ** | IUdR continuous IV infusion (1000 mg/m2/ day/ 14 days), Total of 39 patients (F. Sullivan, et al. Int J Radiat Oncol Biol Phys. 1994; 30(3):583-90) |
| * | IUdR continuous IV infusion (2000 mg/m2/ 4 day infusion/ 6 week treatment), Total of 21 patients (R. Urtasun, et al. Int J Radiat Oncol Biol Phys. 1996;36(5):1163-7.) |
Sarcoma Treatment
Efficacy compared to historical RT-alone controls for treatment
of high-grade sarcomas (University of Michigan*** trials)
| *** | 16 patients were treated with continuous infusion (1000-1600 mg/m2/day) plus RT (J.M. Robertson, et al. Int J Radiat Oncol Biol Phys. 1995; 31(1):87-92). |
In addition to our primary product candidate, we are developing and planning to develop other cancer radiation sensitizers and radiation protectors, which target protecting normal tissue during the administration of RT, and other products utilizing our HDAC small molecule technology platform.
SBIR Contracts
The SBIR Program
The Small Business Innovation Research program, as developed by Congress under the Small Business Innovation Development Act of 1982, is designed to encourage domestic small businesses to engage in Federal Research/Research and Development (“R/R&D”) that has the potential for commercialization. Through a competitive awards-based program, SBIR enables small businesses to explore their technological potential and provides the incentive to profit from its commercialization. Some of the SBIR’s program goals include stimulating technological innovation, meeting Federal research and development needs and encouraging participation in innovation and entrepreneurship.
The SBIR program is a three-phase program. Phase 1 is to establish the technical merit and commercial potential of the proposed R/R&D efforts. Phase 2 is to continue the R/R&D efforts initiated in Phase 1 and funding is based on the results achieved in Phase 1. Phase 3 allows for the small business to pursue commercialization objectives resulting from the Phase 1 and 2 R/R&D activities. In addition, companies that have successfully completed Phases I and II are also eligible to apply for Phase IIb funding.
In addition to the SBIR contract to fund our Phase I clinical study on Ropidoxuridine in combination with RT for treatment of advanced gastrointestinal cancers, we have also received awards of SBIR contracts from the NIH to address prostate cancer health disparities and prostate cancer radiation biomarker development.
As of the date of this registration statement, all SBIR contracts received by the Company have been completed. The Company submitted a final report for SBIR contract # 75N81018C00031 on March 28, 2022. The following summary of terms for the three Phase II SBIR contracts is provided below.
Summary of SBIR Contracts
| ● | SBIR contract #261201400013C: Phase I ($191,971) and Phase II ($1,428,117) for Clinical Development of IPdR for Radiosensitization, dates September 19, 2014 through August 3, 2017. Subcontract to Brown University/LifeSpan Rhode Island Hospital. No related intellectual property. | |
| ● | SBIR contracts # HHSN261201600038C; Phase I ($224,687) and #261201800016C: Cell-Based Models for Prostate Cancer Health Disparity Research - Moonshot Project (Phase II), award amount $1,484,350, dates September 19, 2016 through September 16, 2021. Subcontract to Georgetown University. Intellectual property consists of cell cultures and is property of Shuttle Pharmaceuticals, Inc. via licensing agreement. | |
| ● | SBIR contracts #HHSN261201600027C ($299,502) and #75N81018C00031: Predictive Biomarkers of Prostate Cancer Patient Sensitivity for Radiation Late Effects, award amount $1,903,095, dates September 16, 2019 through March 15, 2022. Subcontract to Georgetown University. Intellectual property is owned by subcontractor Georgetown University with option to license to Shuttle Pharmaceuticals, Inc. |
Prostate Cancer Studies to Address Health Disparities
Prostate cancer health disparities studies have shown that African American men are at higher risk for developing prostate cancer, as well as at higher risk of cancer specific death rates as compared to Caucasian American men. The causes of disparities have been attributed to socioeconomic differences, environmental exposures and biological factors. Most disparities studies have been population based, in part, due to the lack of relevant in vitro and in vivo models to support biological studies.
| 57 |
Shuttle Pharma has been awarded Phase I and II SBIR contracts entitled “Cell-based models for prostate cancer health disparity research” to develop African American prostate cancer cell lines with donor matched normal prostate epithelial cell lines from African American men.
The commercialization of the prostate cells will require additional support through the SBIR funding mechanism. Companies that have completed Phase I and II SBIR awards are eligible to apply for Phase IIb SBIR funding. These awards are intended to de-risk a project by providing up to $4 million of matching funding for product development to commercialization. We intend to apply for such government funding to advance laboratory facilities and to expand the availability of the cell cultures. We did not raise capital through our IPO for the health disparities project. Should we not be successful with SBIR IIb funding, we will pause and may have to terminate this project.
Prostate Cancer Biomarker Development
Patients treated for prostate cancer may experience treatment related late effects that adversely affect quality of life and may prove life-threatening. Shuttle Pharma has been awarded a Phase I SBIR contract entitled “Predictive biomarkers for prostate cancer patient sensitivity for radiation late effects” to determine the technical and commercial feasibility of a biomarker panel predictive of radiation mediated late effects in patients treated for prostate cancer.
Through collaboration with Georgetown University, patients treated with SBRT for prostate cancers were analyzed for urinary and rectal symptoms and their blood was analyzed by mass spectroscopy for predictive biomarkers. The discovery and validation of metabolite panels to serve as a predictive biomarker of patient outcomes following radiation therapy and supports future development and commercialization of a diagnostic product through a Phase 2 SBIR effort.
The development to commercialization of the metabolite predictive biomarker panel requires additional support through the SBIR funding mechanism. We will be eligible to apply for Phase IIb SBIR funding the next round of solicitation. A Phase IIb will help de-risk the project by providing up to $4 million of matching funds for performing the clinical validation trial for product development to commercialization. We intend to apply for such government funding to advance this project. We also intend to raise capital through the public market for predictive biomarker development through the Shuttle Diagnostics entity. We do not intend to use the funds raised through our IPO for the health disparities project. Should we not be successful for SBIR IIb funding, we will terminate this project.
Collaborative Arrangements
While we intend to enter into selective collaborative arrangements to further develop our drug candidates in the future, at present we have not entered into any collaborative arrangements with third parties to develop our drug candidates as we are still completing clinical trials and, as a result, there can be no assurance that we will be able to do so on commercially reasonable terms or otherwise.
Intellectual Property
We invest significant amounts of funds in research and development. Our research and development expenses before contract reimbursements were $3,517,093 and $1,258,840 for the fiscal years ended December 31, 2023 and 2022 respectively. After reimbursements for contracts of $0 and $211,455 for the fiscal years ended December 31, 2023 and 2022, net research and development expenses were $3,517,093 and $1,047,385, respectively.
We are seeking multifaceted protection for our intellectual property that includes licenses, confidentiality and non-disclosure agreements, copyrights, patents, trademarks and common law rights, such as trade secrets. We enter into confidentiality and proprietary rights agreements with our employees, consultants, collaborators, subcontractors and other third parties and generally control access to our documentation and proprietary information.
| 58 |
As of the date of this registration statement, we have filed five patent applications with the USPTO with respect to various aspects of our HDAC small molecule delivery platform and Ropidoxuridine, our lead product candidate. The following is the status of the patent applications Shuttle has filed to date:
Summary of Shuttle Pharma’s Intellectual Property Portfolio

Morgan, Lewis & Bockius LLP prepared patent applications related to Ropidoxuridine (IPdR) and HDAC inhibitors, and, in the fourth quarter of 2018, found no freedom to operate (FTO) issue for Ropidoxuridine used as radiosensitizer and used with tipiracil, and HDAC inhibitors SP-1-161 and SP-2-225.
Our strategy around protection of our proprietary technology, including any innovations and improvements, is to obtain worldwide patent coverage with a focus on jurisdictions that represent significant global pharmaceutical markets. Generally, patents have a term of twenty years from the earliest priority date, assuming that all maintenance fees are paid, no portion of the patent has been terminally disclaimed and the patent has not been invalidated. In certain jurisdictions, and in certain circumstances, patent terms can be extended or shortened. We are obtaining worldwide patent protection for at least novel molecules, composition of matter, pharmaceutical formulations, methods of use, including treatment of disease, methods of manufacture and other novel uses for the inventive molecules originating from our research and development efforts. We continuously assess whether it is strategically more favorable to maintain confidentiality for the “know-how” regarding a novel invention rather than pursue patent protection. For each patent application that is filed we strategically tailor our claims in accordance with the existing patent landscape around a particular technology.
There can be no assurance that an issued patent will remain valid and enforceable in a court of law through the entire patent term. Should the validity of a patent be challenged, the legal process associated with defending the patent can be costly and time consuming. Issued patents can be subject to oppositions, interferences and other third-party challenges that can result in the revocation of the patent limit patent claims such that patent coverage lacks sufficient breadth to protect subject matter that is commercially relevant. Competitors may be able to circumvent our patents. Development and commercialization of pharmaceutical products can be subject to substantial delays and it is possible that at the time of commercialization any patent covering the product has expired or will be in force for only a short period of time following commercialization. We cannot predict with any certainty if any third-party U.S. or foreign patent rights or other proprietary rights will be deemed infringed by the use of our technology. Nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. Should we need to defend ourselves and our partners against any such claims, substantial costs may be incurred. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability to develop or commercialize some or all of our products in the U.S. and abroad and could result in the award of substantial damages. In the event of a claim of infringement, we or our partners may be required to obtain one or more licenses from a third party. There can be no assurance that we can obtain a license on a reasonable basis should we deem it necessary to obtain rights to an alternative technology that meets our needs. The failure to obtain a license may have a material adverse effect on our business, results of operations and financial condition.
| 59 |
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that we can meaningfully protect our trade secrets on a continuing basis. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to our trade secrets.
It is our policy to require our employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from us to execute confidentiality agreements upon the commencement of employment or consulting relationships. These agreements provide that all confidential information developed or made known to these individuals during the course of the individual’s relationship with the company is to be kept confidential and is not to be disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee will be the property of our company. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Our success will depend in part on our ability to obtain and maintain patent protection, preserve trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others, both in the U.S. and other territories worldwide.
Manufacturing and Supply
We do not currently own or operate manufacturing facilities for the production of preclinical, clinical or commercial quantities of any of our product candidates. We currently use a number of our suppliers for the raw materials and formulation to meet the preclinical and any clinical requirements of our product candidates. We do not have a long-term agreement with any of these parties and we believe alternative sources of supply exist.
We intend to enter into collaborations for the manufacture of our product candidates, with our collaborators assuming responsibility for such manufacturing. Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Any collaborator or third-party contract manufacturer we use would need to be compliant with cGMP. cGMP is a regulatory standard for the production of pharmaceuticals that will be used in humans.
Sales and Marketing
Our current focus is on the development of our existing portfolio, the completion of clinical trials and, if and where appropriate, the registration of our product candidates. We currently do not have marketing, sales and distribution capabilities. If we receive marketing and commercialization approval for any of our product candidates, we intend to market the product either directly or through collaborations, strategic alliances and distribution agreements with third parties. The ultimate implementation of our strategy for realizing the financial value of our product candidates is dependent on the results of clinical trials for our product candidates, the availability of funds and the ability to negotiate acceptable commercial terms with third parties.
Employees
As of the date of this registration statement, we have nine full-time employees, including our six executive officers, and three engaged in research and development. We consider our relationship with our employees to be good.
Nasdaq Deficiency and 2024 Reverse Stock Split
Our common stock currently is listed for quotation on the Nasdaq Capital Market. We are required to meet Nasdaq listing rules in order to maintain such listing, including a requirement that the Company maintain stockholders’ equity of at least $2.5 million.
| 60 |
Our common stock currently is listed for quotation on the Nasdaq Capital Market. We are required to meet Nasdaq listing rules in order to maintain such listing, including a requirement that the Company’s stockholders equity remain at or above $2.5 million.
On September 10, 2024, we received a letter from The Nasdaq notifying the Company that it is no longer in compliance with the minimum stockholders’ equity requirement for continued listing on the Nasdaq Capital Market. Nasdaq Listing Rule 5550(b)(1) requires listed companies to maintain stockholders’ equity of at least $2.5 million. In the Company’s Quarterly Report on Form 10-Q for the period ended June 30, 2024, the Company reported stockholders’ equity of $801,434, which is below the minimum stockholders’ equity required for continued listing pursuant to Nasdaq Listing Rule 5550(b)(1). In addition, at present, the Company does not meet the alternatives of market value of listed securities or net income from continuing operations.
This Nasdaq notice has no immediate effect on the listing of the Company’s securities on the Nasdaq Capital Market. Nasdaq has provided the Company with 45 calendar days, or until October 25, 2024, to submit a plan to regain compliance with the minimum stockholders’ equity standard. If the Company’s plan to regain compliance is accepted, Nasdaq may grant an extension of up to 180 calendar days from September 10, 2024 for the Company to regain compliance.
The Company is presently evaluating various courses of action to regain compliance and intends to timely submit a compliance plan to Nasdaq. However, there can be no assurance that the Company’s plan will be accepted or that, if it is, the Company will be able to regain compliance and maintain its listing on the Nasdaq Capital Market. If the Company’s plan to regain compliance is not accepted or if Nasdaq does not grant an extension and the Company does not regain compliance by October 25, 2024, or if the Company fails to satisfy another Nasdaq requirement for continued listing, Nasdaq could provide notice that the Company’s securities will become subject to delisting. In that event, the Company will have an opportunity to appeal Nasdaq’s decision to a hearings panel.
On August 31, 2023 we received a letter from the Nasdaq Listing Qualifications Staff of the Nasdaq (the “Staff”) stating that for the 30 consecutive business day period the Company’s common stock had failed to maintain a minimum closing bid price of $1.00 per share, as required for continued listing on The Nasdaq Capital Market pursuant to Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”). In accordance with Nasdaq rules, the Company was provided with an initial period of 180 calendar days, or until February 27, 2024, to regain compliance with the Minimum Bid Price Requirement (the “Initial Grace Period”) and a second Grace Period on February 19, 2024 through August 26, 2024.
In order to regain compliance with the Minimum Bid Price Requirement, on August 6, 2024, we filed an amendment to our certificate or incorporation to effect a 1-for-8 reverse stock split of our outstanding common stock (the “2024 Reverse Stock Split”). The 2024 Reverse Stock Split became effective on August 13, 2024, when the Company’s common stock opened for trading on Nasdaq on a post-split basis under the Company’s existing trading symbol, “SHPH.” Subsequently, on August 27, 2024, we received notice from Nasdaq that we regained compliance with the Minimum Bid Price Requirement.
All historical share and per share amounts reflected throughout this prospectus have been adjusted to reflect the 2024 Reverse Stock Split. However, our periodic and current reports, and all other documents included in this prospectus that were filed prior to August 13, 2024 do not give effect to the 2024 Reverse Stock Split.
Going Concern Uncertainty
Our condensed consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and had a net loss of approximately $3.8 million and no revenues for the six months ended June 30, 2024 and has working capital of approximately $1.1 million as of June 30, 2024. In addition, the convertible note, which was paid in full as of September 30, 2024, includes covenants and certain cash payment requirements. These conditions, and the Company’s ability to comply with such conditions, raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the condensed consolidated financial statements are issued.
In September 2022, the Company completed its initial public offering, or IPO, generating net proceeds of approximately $10.0 million. And in January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible note with a principal value of $4.3 million, along with a four-year warrant to purchase 1,018,079 shares (127,260 shares post Reverse Split) of common stock, exercisable at $1.93 per share (post Reverse Split, as adjusted), providing the Company with approximately $3.6 million in net proceeds. To date, the warrant has not yet been exercised. In September 2024, the investor converted $1.2 million of principal and interest outstanding on the note, leaving a balance remaining on the note of $14 thousand, including principal and interest as of September 6, 2024, and collateral of $600 thousand of Company cash was released.
The IPO and subsequent capital raise have supported operations leading up to the manufacture of drug product and FDA approval of the IND for the Phase II clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the Company agreed to an expansion of the clinical trial, necessitating additional capital to complete the trial as well as fund ongoing operations. The ability of the Company to continue as a going concern is dependent upon its ability to successfully raise additional equity or debt financing, including through bridge financing, to allow the Company to fund ongoing operations, conduct clinical trials and bring a drug candidate to commercialization to generate revenues.
If we fail to obtain additional material financing in the near-term, our clinical trials and targeted FDA submission timeline could be delayed, and we could be forced to abandon such activities entirely and cease operations, with the possible loss of such properties or assets. If we are unable to obtain a material quantum of financing in the imminent future or unable to continue to obtain additional financing over at least the next 12 months as we continue to generate negative cash flow, our board of directors could determine to cause the Company to undertake a process of liquidation under Chapter 7 of applicable U.S. bankruptcy laws, or otherwise seek other protection under such laws. In such event, we expect that holders of shares of our common stock would recoup little if any material value in such process.
| 61 |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management’s Discussion and Analysis should be read in conjunction with our financial statements and the related notes thereto included elsewhere in this registration statement. The discussion for the fiscal year ended December 31, 2023 has been impacted by the restatement described in the Explanatory Note and in Note 2 of the consolidated financial statements of this registration statement. Certain of the financial and other information provided in this Management’s Discussion and Analysis has been updated to reflect the restatement adjustments.
The Management’s Discussion and Analysis contains forward-looking statements that involve risks and uncertainties, such as statements of our plans, objectives, expectations, and intentions. Any statements that are not statements of historical fact are forward-looking statements. When used, the words “believe,” “plan,” “intend,” “anticipate,” “target,” “estimate,” “expect,” and the like, and/or future-tense or conditional constructions (“will,” “may,” “could,” “should,” etc.), or similar expressions, identify certain of these forward-looking statements. These forward-looking statements are subject to risks and uncertainties that could cause actual results or events to differ materially from those expressed or implied by the forward-looking statements in this registration statement. Our actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of several factors including, but not limited to, those noted under “Risk Factors” in this registration statement.
We do not undertake any obligation to update forward-looking statements to reflect events or circumstances occurring after the date of this registration statement, except as required by U.S. federal securities laws.
Overview
Founded by Georgetown University Medical School faculty members, Shuttle Pharmaceuticals Holdings, Inc. (the “Company”) is a discovery and development stage pharmaceutical company leveraging our proprietary technology to develop novel therapies that are designed to cure cancer. Originally formed as Shuttle Pharmaceuticals, LLC in 2012, our goal is to extend the benefits of cancer treatments by leveraging insights into cancer therapy with surgery, radiation therapy, chemotherapy and immunotherapy. While there are several therapies being developed with the goal of curing cancer, one of the most effective and proven approaches to this is radiation therapy (RT). We are developing a pipeline of products designed to address the limitations of the current standard of cancer therapies. We believe that our product candidates will enable us to deliver cancer treatments that are safer, more reliable and at a greater scale than that of the current standard of care.
Operations to date have focused on continuing our research and development efforts to advance Ropidoxuridine clinical testing and improved drug formulation, to advance HDAC6 inhibitor (SP-2-225) preclinical development and explore new SBIR contract work on predictive biomarkers of radiation response, as well as prostate cell lines for health disparities research. We received Small Business Innovation Research (“SBIR”) contract funding from the NIH for the aforementioned projects. The clinical development of Ropidoxuridine has shown drug bioavailability and a maximum tolerated dose has been established for use in Phase II clinical trials. TCG GreenChem, Inc. (“TCG GreenChem”), with whom we have contracted for process research, development and cGMP compliant manufacture of IPdR, has successfully completed the manufacturing campaign for the active pharmaceutical ingredient (API) of Ropidoxuridine for use in the Company’s upcoming Phase II clinical trial in brain cancer patients undergoing radiation therapy. Shuttle also worked with University of Iowa Pharmaceuticals to develop the formulation, produce the capsules, and which have been shipped to Contract Research Organization (CRO) Theradex Oncology for distribution to clinical trial sites. Both activities have now been completed. In addition, Shuttle received approval from the FDA to begin the clinical trial. The FDA made recommendations to expand the clinical trial and the company agreed with the recommendation. With this change incorporated into the revised protocol, the Company believes it remains on track to commence its Phase II clinical study in the second quarter of 2024. The radiation biomarker project and the health disparities project have been completed and the company is following up with plans for clinical validation and potential commercialization. Changes in operational, administrative, legal and professional expenses related to our operations are set forth in more detail in the discussion below.
Restatement of Previously Issued Consolidated Financial Statements
As previously disclosed in the Company’s Quarterly Report on Form 10-Q for the period ended March 31, 2024, filed with the Securities and Exchange Commission (“SEC”) on May 13, 2024, following the entry of a cease-and-desist order by the SEC against our former auditor, B.F. Borgers CPA PC (“BF Borgers”), we commenced the re-audit (the “Re-audit”) of our consolidated financial statements for the year ended December 31, 2022, which had been audited by BF Borgers. Since that time and as a result of the Re-audit, as of July 10, 2024, the Company and the Audit Committee have concluded and informed our current auditor, Forvis Mazars, LLP, that our audited consolidated financial statements for the year ended December 31, 2022 (the “2022 Financial Statements”), our audited financial statements for the year ended December 31, 2023 (the “2023 Financial Statements”), and the quarterly periods included in the Company’s Annual Report for the year ended December 31, 2023, and the quarterly report for the period ended March 31, 2024 require restatement and are not reliable.
| 62 |
The accounting errors impacting our 2022 Financial Statements include (i) correcting historical and fiscal 2022 stock compensation transactions; (ii) correcting the initial and subsequent accounting for certain convertible notes, promissory notes and warrants to purchase shares of common stock in fiscal 2022; (iii) correcting the historical and subsequent accounting for the Series A Convertible Preferred Stock in fiscal 2022; (iv) correcting the initial and subsequent accounting for warrants issued with the Series A Convertible Preferred Stock, and (v) correcting the recognition of issuance costs related to the initial public offering in fiscal 2022.
The impact of the accounting errors from the 2022 Financial Statements also impacts the Company’s 2023 Financial Statements specific to opening balances of additional paid in capital and accumulated deficit.
The discussion of financial results presented here is reflective of the restatement adjustments. Refer to Note 2 of the consolidated financial statements included elsewhere in this Annual Report for more information on the restatement.
Reverse Stock Split
On August 13, 2024, in order to meet the Minimum Bid Price Requirement, the Company effectuated a 1-for-8 reverse stock split of its issued and outstanding common stock, rounding up to account for any fractional shares (the “Reverse Stock Split”). The Reverse Stock Split had no effect on the Company’s authorized shares of common stock or preferred stock. Following the reverse stock split on August 13, 2024, the Company had a total of 2,111,235 shares issued and outstanding (or 2,008,689 on a post-split basis as of December 31, 2023). All share, option, warrant and per share amounts (except our authorized outstanding) have been retroactively restated in these financial statements and related disclosures.
Results of Operations
Comparison of the three months ended June 30, 2024 and 2023
The following table summarizes the results of our operations:
| Three Months Ended | ||||||||||||||||
| June 30, | ||||||||||||||||
| 2024 | 2023 | Change | % | |||||||||||||
| Revenue | $ | - | $ | - | $ | - | - | |||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 645,719 | 839,665 | (193,946 | ) | (23 | )% | ||||||||||
| General and administrative | 310,038 | 209,279 | 100,759 | 48 | % | |||||||||||
| Legal and professional | 526,877 | 416,688 | 110,189 | 26 | % | |||||||||||
| Total operating expenses and loss from operations | 1,482,634 | 1,465,632 | 17,002 | 1 | % | |||||||||||
| Other income (expense): | ||||||||||||||||
| Interest expense - related parties | - | (2,588 | ) | 2,588 | (100 | )% | ||||||||||
| Interest expense | (430,685 | ) | (729,351 | ) | 298,666 | (41 | )% | |||||||||
| Gain on sale of marketable securities | 39,683 | 1,744 | 37,939 | 2,175 | % | |||||||||||
| Change in fair value of marketable securities | (27,964 | ) | (26,534 | ) | (1,430 | ) | 5 | % | ||||||||
| Interest income | 14,158 | 19,267 | (5,109 | ) | (27 | )% | ||||||||||
| Change in fair value of derivative liabilities | (143,773 | ) | 434,275 | (578,048 | ) | (133 | )% | |||||||||
| Loss on settlement of convertible debt | - | (415,553 | ) | 415,553 | (100 | )% | ||||||||||
| Total other expense | (548,581 | ) | (718,740 | ) | 170,159 | (24 | )% | |||||||||
| Net loss | $ | (2,031,215 | ) | $ | (2,184,372 | ) | $ | 153,157 | (7 | )% | ||||||
Research and Development. Research and development (“R&D”) expense was $0.6 million for the three months ended June 30, 2024, as compared to $0.8 million for three months ended June 30, 2023. The decrease of $0.2 million, or 23%, is primarily related to the Company having completed production of the drug product and waiting for the initiation of trials.
| 63 |
R&D compensation related expenses were $0.3 million in the three months ended June 30, 2024 as compared to $0.3 million in the three months ended June 30, 2023. Compensation related expenses were 48% for the three months ended June 30, 2024, representing an increase from 39% of total R&D in the three months ended June 30, 2023. Subcontract work made up 42% of total R&D expenses in the three months ended June 30, 2024 and 56% of total R&D expenses during the three months ended June 30, 2023.
General and Administrative Expenses. General and Administrative expenses in the three months ended June 30, 2024 increased by $0.1 million, or 48% to $0.3 million in the three months ended June 30, 2024, compared to $0.2 million in the three months ended June 30, 2023.
Legal and Professional Expenses. During the three months ended June 30, 2024, legal and professional expenses increased by $0.1 million or 26% to $0.5 million compared to $0.4 million in the same period in the prior year. The increase in legal and professional fees was primarily due to increases in our expenses related to our public filing requirements, contracts and financing related work.
Other Income Expense. Other expense was $0.5 million for the three months ended June 30, 2024, which consisted of $0.4 million in interest expense on convertible loans, interest income of $14 thousand, change in marketable securities of $28 thousand, and a loss on change in fair value of derivative liabilities of $0.1 million. Other income was $0.7 million for the three months ended June 30, 2023, which consisted of $0.7 million in interest expense on convertible loans, $3 thousand in interest expense on related party loans, interest income of $19 thousand, loss on settlement of convertible debt of $0.4 million, unrealized loss on marketable securities of $26 thousand, and a gain on change in fair value of derivative liability of $0.4 million. The $0.4 million gain in fair value of derivative liability resulted from the fair value measurement using the Monte Carlo pricing model and the impact of the decrease in the market price of warrant shares.
Comparison of the six months ended June 30, 2024 and 2023
The following table summarizes the results of our operations:
| Six Months Ended | ||||||||||||||||
| June 30, | ||||||||||||||||
| 2024 | 2023 | Change | % | |||||||||||||
| Revenue | $ | - | $ | - | $ | - | - | |||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 1,231,823 | 1,761,466 | (529,643 | ) | (30 | %) | ||||||||||
| General and administrative | 634,647 | 464,633 | 170,014 | 37 | % | |||||||||||
| Legal and professional | 1,001,011 | 784,312 | 216,699 | 28 | % | |||||||||||
| Total operating expenses and loss of operations | 2,867,481 | 3,010,411 | (142,930 | ) | (5 | %) | ||||||||||
| Other income (expense): | ||||||||||||||||
| Interest expense - related parties | - | (6,825 | ) | 6,825 | (100 | %) | ||||||||||
| Interest expense | (928,200 | ) | (1,328,682 | ) | 400,482 | (30 | %) | |||||||||
| Interest income | 35,611 | 35,955 | (344 | ) | (1 | %) | ||||||||||
| Finance fee | - | (104,245 | ) | 104,245 | (100 | %) | ||||||||||
| Change in fair value of derivative liabilities | 54,089 | 1,675,275 | (1,621,186 | ) | (97 | %) | ||||||||||
| Gain on sale of marketable securities | 43,720 | 1,744 | 41,976 | 2,407 | % | |||||||||||
| Change in fair value of marketable securities | (28,670 | ) | 11,528 | (40,198 | ) | (349 | %) | |||||||||
| Loss on settlement of convertible debt | (71,315 | ) | (433,807 | ) | 362,492 | (84 | %) | |||||||||
| Total other expense | (894,765 | ) | (149,057 | ) | (745,708 | ) | 500 | % | ||||||||
| Net loss | $ | (3,762,246 | ) | $ | (3,159,468 | ) | $ | (602,778 | ) | 19 | % | |||||
Research and Development. Research and development (“R&D”) expense was $1.2 million for the six months ended June 30, 2024, as compared to $1.8 million for six months ended June 30, 2023. The decrease of $0.5 million, or 30%, is primarily related to the Company having completed production of the drug product and waiting for the initiation of trials.
| 64 |
R&D compensation related expenses were $0.6 million in the six months ended June 30, 2024 as compared to $0.9 million in the six months ended June 30, 2023. Compensation related expenses were 52% for the six months ended June 30, 2024, representing an increase from 51% of total R&D in the six months ended June 30, 2023. Subcontract work made up 38% of total R&D expenses in the six months ended June 30, 2024 and 56% of total R&D expenses during the six months ended June 30, 2023.
General and Administrative Expenses. General and Administrative expenses in the six months ended June 30, 2024 increased by $0.2 million, or 37% to $0.6 million in the six months ended June 30, 2024. The increase in general and administrative expenses was primarily due to a restricted stock-based grant to an officer of the Company valued at $42 thousand, and increases in marketing, advertising and filing expenses.
Legal and Professional Expenses. During the six months ended June 30, 2024, legal and professional expenses increased by $0.2 million or 28%. The increase in legal and professional fees was primarily due to increases in our expenses related to our public filing requirements, contracts and financing related work.
Other Income Expense. Other expense was $0.9 million for the six months ended June 30, 2024, which consisted of $0.9 million in interest expense on convertible loans, interest income of $36 thousand, loss on settlement of convertible debt of $71 thousand, change in marketable securities of $29 thousand, and a gain on change in fair value of derivative liabilities of $54 thousand. Other expense was $0.1 million for the six months ended June 30, 2023, which consisted of $1.3 million in interest expense on convertible loans, $7 thousand in interest expense on related party loans, interest income of $36 thousand, finance fee on convertible loans of $0.1 million, loss on settlement of convertible debt of $0.4 million, gain on sale of marketable securities of $2 thousand, unrealized gain on marketable securities of $12 thousand, and a gain on change in fair value of derivative liabilities of $1.7 million. The $1.7 million gain in fair value of derivative liability resulted from the fair value measurement using the Monte Carlo pricing model and the impact of the decrease in the market price of warrant shares.
Liquidity and Capital Resources
Our unaudited condensed consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and has a net loss of approximately $3.8 million and no revenues for the six months ended June 30, 2024 and has working capital of approximately $1.1 million as of June 30, 2024. In addition, the convertible note payable outstanding at June 30, 2024 includes covenants and certain cash payment requirements. On July 12, 2024, the Company informed the investor of its convertible note that the Company’s expected restatement of its financial statements for the years ended December 31, 2022 and 2023 constituted an event of default under the terms of the convertible note. On August 6, 2024, the Company paid $0.6 million to the investor of the convertible note and received a waiver from the investor related to the default. The funds paid are restricted and will be held as collateral to the balance owed under the convertible note (see Note 9). These conditions, and the Company’s ability to comply with such conditions, raise substantial doubt about the Company’s ability to continue as a going concern.
| 65 |
In January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible note with a principal value of $4.3 million, along with a four-year warrant to purchase 127,260 shares of common stock, exercisable at $18.80 per share ($1.93 post Reverse Split, as adjusted), providing the Company with approximately $3.6 million in net proceeds. To date, the warrant has not yet been exercised. However, the Company’s existing cash resources, marketable securities and the cash received from the equity offering and convertible note are not expected to provide sufficient funds to carry out the Company’s operations and clinical trials through the next twelve months.
The IPO and subsequent capital raise have supported operations, the manufacture of drug product and FDA approval of the IND for the Phase 2 clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the Company agreed to an expansion of the clinical trial, necessitating additional capital to complete the trial as well as fund ongoing operations. As a result, management has submitted an application for a SBIR grant for non-dilutive funding for pre-clinical project through the NIH. The Phase II clinical trial was also approved by the Institutional Review Board “IRB” in June, 2024. In August 2024, the Company took action to pursue a separate capital raise of up to $10.0 million and entered into a work order with a contract research organization (“CRO”) for purposes of supporting the Company’s Phase II Study of Ropidoxuridine. Under the terms of the work order, the CRO will oversee the studies for a period of 53 months in exchange for a fee of approximately $2.3 million, payable in stages and based upon services performed during the study. The ability of the Company to continue as a going concern is dependent upon its ability to successfully raise additional equity or debt financing to allow it to fund ongoing operations, conduct clinical trials and bring a drug candidate to commercialization to generate revenues.
The accompanying unaudited condensed consolidated financial statements do not include any adjustments to reflect the future effects on the recoverability and classification of assets or the amounts and classification of liabilities if the Company is unable to continue as a going concern.
Balance Sheet Data:
| June 30, | December 31, | |||||||||||||||
| 2024 | 2023 | Change | % | |||||||||||||
| Current assets | $ | 2,485,333 | $ | 5,593,005 | $ | (3,107,672 | ) | (56 | )% | |||||||
| Current liabilities | 1,384,620 | 1,042,237 | 342,383 | 33 | % | |||||||||||
| Working capital | $ | 1,100,713 | $ | 4,550,768 | $ | (3,450,055 | ) | (76 | )% | |||||||
As of June 30, 2024, total current assets were $2.5 million and total current liabilities were $1.4 million, resulting in working capital of $1.1 million. As of December 31, 2023, total current assets were $5.6 million and total current liabilities were $1.0 million, resulting in a working capital of $4.6 million. As of June 30, 2024, the current assets primarily resulted from $0.7 million cash and $1.6 million marketable securities, with the decrease from December 31, 2023 being primarily due to ongoing cash burn from our R&D programs, filing expenses and general operations. The increase in current liabilities is primarily due to the current portion of the $1.0 million convertible note and accrued interest that increased $0.3 million and an increase in accounts payable of $82 thousand.
Cash Flows from Operating Activities
| Six Months Ended | ||||||||||||||||
| June 30, | ||||||||||||||||
| 2024 | 2023 | Change | % | |||||||||||||
| Cash used in operating activities | $ | (2,638,425 | ) | $ | (2,984,039 | ) | $ | 345,614 | (12 | )% | ||||||
| Cash provided by (used in) investing activities | $ | 1,259,270 | $ | (2,890,905 | ) | $ | 4,150,175 | (144 | )% | |||||||
| Cash provided by (used in) financing activities | $ | (501,667 | ) | $ | 2,904,527 | $ | (3,406,194 | ) | (117 | )% | ||||||
| Cash on hand | $ | 695,594 | $ | 5,446,786 | $ | (4,751,192 | ) | (87 | )% | |||||||
| 66 |
To date, we have not generated positive cash flows from operating activities. For the six months ended June 30, 2024, net cash flows used in operating activities was $2.6 million, consisting of a net loss of $3.8 million, increased by a gain on change in derivative liabilities of $54 thousand, offset by amortization of debt discount of $0.8 million, loss on settlement of convertible debt of $71 thousand, accrued interest settled with common stock of $19 thousand, stock-based compensation of $183 thousand and increased by a net change in working capital of $85 thousand. For the six months ended June 30, 2023, net cash flows used in operating activities was $3.0 million, consisting of a net loss of $3.2 million, increased by a gain on change in derivative liabilities of $1.7 million, offset by amortization of debt discount of $1.1 million, loss on settlement of convertible debt of $0.4 million, accrued interest settled with common stock of $0.2 million, stock-based compensation of $58 thousand and further reduced by a net change in working capital of $12 thousand.
Cash Flows from Investing Activities
For the six months ended June 30, 2024, we invested in trading marketable securities for $36 thousand and received $1.3 million in proceeds from disposition of marketable securities. For the six months ended June 30, 2023, we invested in trading marketable securities for $3.0 million and received $80 thousand in proceeds from disposition of marketable securities.
Cash Flows from Financing Activities
For the six months ended June 30, 2024, we paid $0.5 million related to payments on a convertible note. For the six months ended June 30, 2023, we received a net of $3.6 million from the sale and issuance of a convertible note payable and warrants and repaid $0.7 million in related party notes payable.
Results of Operations
Comparison of the year ended December 31, 2023 and 2022 (as restated)
The following table summarizes the results of our operations:
| Years Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
| 2023 | 2022 | Change | % | |||||||||||||
| (as restated) | ||||||||||||||||
| Revenue | $ | — | $ | — | $ | — | — | |||||||||
| Operating expenses: | ||||||||||||||||
| Research and development, net of contract expense reimbursements, as restated | 3,517,093 | 1,047,385 | 2,469,708 | 236 | % | |||||||||||
| General and administrative, as restated | 1,046,854 | 473,714 | 573,140 | 121 | % | |||||||||||
| Legal and professional, as restated | 1,328,435 | 822,727 | 505,708 | 61 | % | |||||||||||
| Total operating expenses and loss of operations, as restated | 5,892,382 | 2,343,826 | 3,548,556 | 151 | % | |||||||||||
| Other income (expense): | ||||||||||||||||
| Interest expense - related parties | (6,825 | ) | (52,010 | ) | 45,185 | (87 | )% | |||||||||
| Interest expense, as restated | (2,484,193 | ) | (428,688 | ) | (2,055,505 | ) | 479 | % | ||||||||
| Interest income | 79,117 | — | 79,117 | 100 | % | |||||||||||
| Finance fee, as restated | (104,245 | ) | (189,912 | ) | 85,667 | (45 | )% | |||||||||
| Change in fair value of derivative liabilities, as restated | 2,216,488 | (189,000 | ) | 2,405,488 | 1,273 | % | ||||||||||
| Change in fair value of warrant liabilities, as restated | — | (398,675 | ) | 398,675 | 100 | % | ||||||||||
| Gain on sale of marketable securities | 4,970 | — | 4,970 | 100 | % | |||||||||||
| Change in fair value of marketable securities | 71,568 | — | 71,568 | 100 | % | |||||||||||
| Loss on settlement of convertible debt, as restated | (477,221 | ) | (1,795,652 | ) | 1,318,431 | (73 | )% | |||||||||
| Gain on settlement of accounts payable | — | 328,687 | (328,687 | ) | (100 | )% | ||||||||||
| Gain on forgiveness of Paycheck Protection Program note payable | — | 73,007 | (73,007 | ) | (100 | )% | ||||||||||
| Total other expense, as restated | (700,341 | ) | (2,652,243 | ) | 1,951,902 | (74 | )% | |||||||||
| Net loss, as restated | $ | (6,592,723 | ) | $ | (4,996,069 | ) | (1,596,654 | ) | 32 | % | ||||||
Research and Development, net of contract expense reimbursements. Total research and development (“R&D”) expense was $3.5 million for the year ended December 31, 2023, as compared to $1.0 million for the year ended December 31, 2022 (as restated), which included R&D expense reimbursements of $0 and $211,455, respectively. The increase of $2.5 million, or 236%, is primarily related to the Company increasing R&D spending as a result of having received funding from the Company’s initial public offering in the third quarter of fiscal 2022 and, the convertible note issued during the period ended March 31, 2023.
| 67 |
R&D compensation related expenses were $1.5 million in the year ended December 31, 2023 as compared to $0.7 million in the year ended December 31, 2022 (as restated). Compensation related expenses, excluding reimbursements, for the year ended December 31, 2023 was 43% as a percent of R&D expense, representing a decrease from the 56% of total R&D incurred in the year ended December 31, 2022. Subcontract work, excluding reimbursements, made up 52% of total R&D expenses in the year ended December 31, 2023 and 35% of total R&D expenses during the year ended December 31, 2022.
General and Administrative Expenses. General and Administrative expenses in the year ended December 31, 2023 increased by $0.6 million, or 121%, from $0.5 million in the year ended December 31, 2022 (as restated) to $1.1 million in the year ended December 31, 2023. The increase in general and administrative expenses was primarily due to increases in insurance expenses of $0.1 million, compensation of $0.2 million and advertising costs of $0.1 million.
Legal and Professional Expenses. During the year ended December 31, 2023, legal and professional expenses increased by $0.5 million or 61%. The increase in legal and professional fees was primarily due to increases in our expenses related to our public filing requirements, contracts and financing related work.
Other Income (expense). Other expense was $0.7 million for the year ended December 31, 2023, which mainly consisted of $2.5 million in interest expense on convertible notes, loss on settlement of convertible debt of $0.5 million, and finance fees of $0.1 million, which were partially offset by a gain on change in fair value of derivative liabilities of $2.2 million, change in fair value of marketable securities of $0.1 million and interest income of $0.1 million. Other expense was $2.7 million for the year ended December 31, 2022 (as restated), which mainly consisted of $0.4 million in interest expense on convertible notes (as restated), loss on settlement of convertible debt of $1.8 million (as restated), finance fees of $0.2 million (as restated) a loss on change in fair value of derivative liability of $0.2 million (as restated), and a loss on change in fair value of warrant liabilities of $0.4 million (as restated), partially offset by a gain on settlement of accounts payable of $0.3 million and a gain on forgiveness of Paycheck Protection Program note payable of $0.1 million.
Liquidity and Capital Resources
Our consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and had a net loss of $6.6 million and no revenues during the year ended December 31, 2023 and working capital of approximately $4.6 million as of December 31, 2023. In addition, the convertible notes payable outstanding at December 31, 2023 includes covenants and certain cash payment requirements. On July 12, 2024, the Company informed the investor of its convertible note that the Company’s expected restatement of its financial statements for the years ended December 31, 2022 and 2023 constituted an event of default under the terms of the convertible note. On August 6, 2024, the Company paid $0.6 million to the investor of the convertible note and received a waiver from the investor related to the default. The funds paid are restricted and will be held as collateral to the balance owed under the convertible note (see Note 12). These conditions, and the Company’s ability to comply with such conditions, raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that these financial statements are issued.
| 68 |
In January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible note with a principal value of $4.3 million, along with a four-year warrant to purchase 127,260 shares of common stock, exercisable at $18.80 per share ($1.93 post-reverse split, as adjusted), providing the Company with approximately $3.6 million in net proceeds. To date, the warrant has not yet been exercised. However, the Company’s existing cash resources, marketable securities and the cash received from the equity offering and convertible note are not expected to provide sufficient funds to carry out the Company’s operations and clinical trials through the next twelve months.
The IPO and subsequent capital raise have supported operations, the manufacture of drug product and FDA approval of the IND for the Phase 2 clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the Company agreed to an expansion of the clinical trial, necessitating additional capital to complete the trial as well as fund ongoing operations. As a result, management had initiated a $4.5 million rights offering and submitted an application for a SBIR grant for non-dilutive funding for pre-clinical project through the NIH. The Phase II clinical trial was also approved by the Institutional Review Board “IRB” in June, 2024. In August 2024, the Company took actions to pursue a separate capital raise of up to $10 million and entered into a work order with a contract research organization (“CRO”) for purposes of supporting the Company’s Phase II Study of Ropidoxuridine. Under the terms of the work order, the CRO will oversee the studies for a period of 53 months in exchange for a fee of approximately $2.3 million, payable in stages and based upon services performed during the study. The ability of the Company to continue as a going concern is dependent upon its ability to successfully raise additional equity or debt financing to allow it to fund ongoing operations, conduct clinical trials and bring a drug candidate to commercialization to generate revenues.
The accompanying financial statements do not include any adjustments to reflect the future effects on the recoverability and classification of assets or the amounts and classification of liabilities if the Company is unable to continue as a going concern.
Balance Sheet Data:
| December 31, | December 31, | |||||||||||||||
| 2023 | 2022 | Change | % | |||||||||||||
| Current assets | $ | 5,593,005 | $ | 8,578,351 | $ | (2,985,346 | ) | (35 | )% | |||||||
| Current liabilities | 1,042,237 | 975,676 | 66,561 | 7 | % | |||||||||||
| Working capital | $ | 4,550,768 | $ | 7,602,675 | $ | (3,051,907 | ) | (40 | )% | |||||||
As of December 31, 2023, total current assets were $5.6 million and total current liabilities were $1.0 million, resulting in working capital of $4.6 million. As of December 31, 2022 (as restated), total current assets were $8.6 million and total current liabilities were $1.0 million, resulting in a working capital of $7.6 million. The Company’s current assets as of December 31, 2023 primarily resulted from $3.6 million in net cash received from the issuance of a convertible note payable, offset by $0.8 million repaid for related party notes ($685,473 in principal and $98,135 of accrued interest), and $0.4 million repaid in cash for a convertible note ($334,444 in principal and $59,846 in accrued interest). In addition, we continued progress on our R&D programs during the year ended December 31, 2023 that resulted in increased cash expenditures. The increase in current liabilities is primarily due to the current portion of the $4.3 million convertible note which is $0.7 million of convertible note payable and accrued interest, an increase in accounts payable of $0.2 million, offset by the reduction of $0.8 million in related party notes payable and accrued interest.
Cash Flows
| Years Ended December 31, | ||||||||||||||||
| 2023 | 2022 | Change | % | |||||||||||||
| (as restated) | ||||||||||||||||
| Cash used in operating activities, as restated | (5,581,147 | ) | (2,666,411 | ) | (2,914,736 | ) | 109 | % | ||||||||
| Cash used in investing activities | (2,829,723 | ) | — | (2,829,723 | ) | 100 | % | |||||||||
| Cash provided by financing activities, as restated | 2,570,083 | 10,578,865 | (8,008,782 | ) | (76 | )% | ||||||||||
| Cash and cash equivalents on hand | 2,576,416 | 8,417,203 | (5,840,787 | ) | (69 | )% | ||||||||||
| 69 |
Cash Flows from Operating Activities
To date, we have not generated positive cash flows from operating activities. For the year ended December 31, 2023, net cash flows used in operating activities was $5.6 million, primarily consisting of a net loss of $6.6 million, increased by a gain on change in derivative liabilities of $2.2 million, offset by amortization of debt discount of $2.1 million, loss on settlement of convertible debt of $0.5 million, accrued interest settled with common stock of $0.3 million, stock-based compensation of $0.2 million and further reduced by a net change in operating assets and liabilities of $0.2 million. For the year ended December 31, 2022, net cash flows used in operating activities was $2.7 million (as restated), primarily consisting of a net loss of $5.0 million (as restated), decreased by a loss on change in derivative liability of $0.2 million, amortization of debt discount and finance fees of $0.4 million, a loss settlement of convertible debt of $1.7 million (as restated), a change in fair value of freestanding instruments in excess of proceeds of $0.2 million (as restated), and stock-based compensation of $0.2 million (as restated), partially offset by a gain on settlement of accounts payable of $0.3 million, a gain on forgiveness of Paycheck Protection Program of $0.1 million, a change in fair value of warrant liabilities of $0.4 million (as restated), and a net change in in operating assets and liabilities of $0.5 million.
Cash Flows from Investing Activities
For the year ended December 31, 2023, we invested in trading marketable securities for $3.0 million and received $0.2 million in proceeds from disposition of marketable securities and purchased less than $0.1 million of equipment. For the year ended December 31, 2022, we had no investing activities.
Cash Flows from Financing Activities
For the year ended December 31, 2023, we received net proceeds of $3.9 million from the sale and issuance of convertible notes payable and warrants and repaid $0.3 million in convertible notes, $0.3 million for finance costs related to convertible note payable, and $0.7 million in related party notes payable. For the year ended December 31, 2022, we received $10.0 million from the issuance of shares of common stock and exercise of warrants and $0.6 million from the issuance of convertible notes and warrants.
Recent Financings – Our IPO and Post-2022 Financing
On September 2, 2022, we closed on our IPO of 1,225,888 units (each a “IPO Unit,” and collectively, the “IPO Units”), with each IPO Unit consisting of one share of the Company’s common stock and one warrant to purchase one share of common stock, at a public offering price of $8.125 per IPO Unit ($65 per IPO Unit on a post Reverse Split basis). Our IPO, which was underwritten by Boustead Securities, LLC (“Boustead”), resulted in gross proceeds of $9.96 million, before deducting underwriting discounts and commissions. On September 29, 2022, Boustead exercised its overallotment option, purchasing an additional 183,883 IPO Units, resulting in gross proceeds of $1.49 million, before deducting underwriter commissions and discounts. As a result, our IPO raised a total of $11.45 million, before deducting underwriting discounts, commissions and related IPO expenses. On a post Reverse Split basis, 352,443 shares (including exercised warrants) were issued under the IPO.
On January 11, 2023, we entered into a securities purchase agreement (the “SPA”) with Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B, a Cayman entity (the “Investor”), pursuant to which the Company sold to the Investor a $4.3 million convertible note (the “January 2023 Convertible Note”) and warrant (the “January 2023 Warrant”) to purchase 127,260 (post Reverse Split) shares of common stock of the Company, in exchange for gross proceeds of $4.0 million (the “Investment Amount”). The January 2023 Convertible Note amortizes on a monthly basis and the Company can make such monthly amortization payments in cash or, subject to certain equity conditions, in registered shares of common stock or a combination thereof. For equity repayment, the January 2023 Convertible Note is convertible into shares of common stock at price per share equal to the lower of (i) $18.8 (post Reverse Split) (ii) 90% of the three lowest daily VWAPs of the 15 trading days prior to the payment date or (iii) 90% of the VWAP of the trading day prior to payment date. The January 2023 Convertible Note is repayable over 26 months and bears interest at the rate of 5% per annum. The January 2023 Warrant is exercisable for four years from the date of closing and is exercisable at $1.93 per share (post Reverse Split, as adjusted). In the event the Investor exercises the January 2023 Warrant in full, such exercise would result in additional gross proceeds to the Company of approximately $246 thousand.
| 70 |
On May 10, 2023, we entered into an amendment agreement (the “Amendment Agreement”) to the SPA. Under the Amendment Agreement, the Company and the Investor amended the transaction documents as follows: (i) amended and restated Section 2 of the January 2023 Warrant so as to remove a provision that would have potentially required an adjustment to the number of warrant shares exercisable under the January 2023 Warrant, (ii) stipulated that the Company would obtain majority shareholder approval to issue up to an additional $10 million in convertible notes (the “Subsequent Notes”) and warrants (the “Subsequent Warrants”) equal to 42.5% of the outstanding principal value of the Subsequent Notes, which Subsequent Note and Subsequent Warrant would be sold to the Investor on substantially the same terms as the existing Convertible Note and Warrant (each as amended by the Amendment Agreement) and upon conversion and/or exercise would cause the potential issuance of in excess of 19.9% of the Company’s issued and outstanding stock, (iii) that, upon obtaining majority stockholder approval, the Company would file a Schedule 14C related to such potential issuance of the shares of common stock related to the potential sale of the Subsequent Notes and Subsequent Warrants to the Investor within 30 calendar days of entry into the Amendment Agreement, and (iv) stipulated that the Investor would release $1.5 million in cash collateral to the Company, with $1.0 million to be released to the Company immediately upon singing of the Amendment Agreement and $0.5 million to be released upon the Company’s filing of the Schedule 14C. The Company obtained majority stockholder consent to the potential sale of the Subsequent Notes and Subsequent Warrants to the Investor in advance of entry into the Amendment Agreement.
On June 4, 2023, we entered into amendment no. 1 (“Amendment No. 1”) to the Amendment Agreement dated May 11, 2023, for purposes of amending the terms of the SPA. Under Amendment No. 1 to the Amendment Agreement, the Company and the Investor agreed as follows: (i) that Section 15(q) to the January 2023 Convertible Note, which required the Company to hold the Cash Collateral in a Controlled Account Agreement (as defined in the Convertible Note), would no longer be applicable, (ii) that the Investor would stipulate the release to the Company of the remaining Cash Collateral totaling $2.92 million (thus releasing the full amount of the Cash Collateral to the Company), and (iii) that, should the Investor exercise its option to purchase the Subsequent Notes and Subsequent Warrants, that such Subsequent Notes would omit Section 15(q) and that the Company would not be required to maintain any controlled accounts or otherwise be subject to any controlled account agreements. In August 2024, we entered into an additional Amendment Agreement pursuant to which the Investor waived certain defaults in exchange for the Company’s payment of $0.6 million in collateral to be held against the Investment Amount pending the January 2023 Convertible Note’s maturity. As of September 30, 2024, the January 2023 Convertible Note had been paid off in full.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
Critical Accounting Policies and Significant Judgments and Estimates
This discussion and analysis of our financial condition and results of operations is based on our unaudited condensed consolidated financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”). The preparation of these unaudited condensed consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the unaudited condensed consolidated financial statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our unaudited condensed consolidated financial statements included elsewhere in this report, we believe that the following accounting policies are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
| 71 |
Our most critical accounting policies and estimates relate to the following:
| ● | Research and Development Expenses | |
| ● | Fair Value of Common Stock (Common Stock Valuations) | |
| ● | Fair Value of Warrants to Purchase Common Stock | |
| ● | Fair Value of Bifurcated Derivative Liabilities and other Financial Instruments | |
| ● | Fair Value of Preferred Stock (upon extinguishment) |
Research and Development Expenses
Research and development expenses are expensed as incurred and, prior to our initial public offering in September 2022, have historically been offset by contract receivable payments from an NIH SBIR contract that has supported our scientific research. This is stated in the consolidated financial statements as research and development, net of contract expense reimbursements.
Fair Value of Common Stock (Common Stock Valuations)
For all periods prior to our initial public offering, there was no public market for our common stock, and, as a result, the fair value of the shares of common stock underlying our share-based awards, warrants and other financial instruments was estimated on each grant or measurement date by our board of directors. To determine the fair value of our common stock underlying these grants and financial instrument, our board of directors considered, among other things, input from management, input from investment banks and financial advisors, valuations of our common stock prepared by unrelated third-party valuation firms in accordance with the guidance provided by the American Institute of Certified Public Accountants 2013 Practice Aid, Valuation of Privately Held Company Equity Securities Issued as Compensation, and our board of directors’ assessment of additional objective and subjective factors that it believe were relevant and factors that may have changed from the date of the most recent valuation through the date of the grant. These factors included, but were not limited to:
| ● | Our results of operations and financial position, including our levels of available capital resources; | |
| ● | Our stage of development and material risk related to our business; | |
| ● | Our business conditions and projections; | |
| ● | The valuation of publicly traded companies in the life sciences and pharmaceuticals sectors, as well as recently completed mergers and acquisitions of peer companies; | |
| ● | The lack of marketability of our common stock as a private company; | |
| ● | The prices at which we sold shares of our Series A Preferred Stock to outside investors in arms-length transactions; | |
| ● | The rights, preferences and privileges of our Series A Preferred Stock relative to those of our common stock; | |
| ● | The likelihood of achieving a liquidity event for our securityholders, such as an initial public offering or a sale of our Company, given prevailing market conditions; | |
| ● | Trends and developments in our industry; and | |
| ● | External market conditions affecting the life sciences and pharmaceutical industry sectors. |
As a result of the initial public offering in September 2022, it is not necessary to determine the fair value of our common stock for any fair value measurements subsequent to this date, as our shares are traded in the public market.
During the year ended December 31, 2022, and prior to the closing of our initial public offering, the estimated fair value of our common stock was utilized to:
| ● | Measure stock compensation expense | |
| ● | Estimate the fair value of certain warrants to purchase common stock | |
| ● | Estimate the fair value of certain bifurcated derivative features in the Convertible Notes | |
| ● | Estimate the deemed dividends recorded related to the extinguishment of the Series A Preferred Stock and the modification of the December 2021 Warrants | |
| ● | Measure any final fair value adjustments upon the settlement, exercise or reclassification of certain financial instruments upon our initial public offering |
In developing its fair value estimates, we performed certain sensitivity analysis around the estimated fair value of the common stock and how changes in this estimate could impact accounting conclusions. For most of these fair value measurements, we determined that $1.00 to $2.00 changes in the estimated fair value per share would not have material impacts on the measurements or ultimate accounting conclusions or entries.
| 72 |
In performing this sensitivity analysis, we identified two measurements that are the most sensitive to changes in our estimated stock price fair value measurement is the valuation of the Series A Preferred Stock post-extinguishment in April 2022 (upon the modification of terms that was determined to be substantive and represent an extinguishment) and the estimate of the incremental fair value provided to the holders of the December 2021 Warrants upon their modification in April 2022 (to allow the warrants to remain exercisable for same number of shares and at same exercise price as per the contract pre-April 2022 reverse stock split). We estimated that a $1.00 per share increase or decrease in our estimated fair value of common stock could result in an increase or decrease in these measurements of approximately $250,000.
The accounting for both transactions was to reflect the determined amounts as a deemed dividend, which would reduce the net loss attributable to common stockholders and increase the net loss per share available to common stockholders. These measurements are non-recurring fair value measurements in nature and did not impact any other balance sheet accounts or operating or non-operating income.
Fair Value of Warrants to Purchase Common Stock
We have issued warrants to investors in our debt and equity offerings. We have also issued warrants to service providers in relation to our financing offerings.
We evaluate all warrants issued to determine the appropriate classification under ASC 480 and ASC 815 (as well as under ASC 718 for warrants issued as share-based payments). In addition to determining classification, we evaluate these instruments to determine if such instruments meet the definition of a derivative.
For warrants that are determined to be equity-classified, we estimate the fair value at issuance and record the amounts to additional paid in capital (potentially on a relative fair value basis if issued in a basket transaction with other financial instruments). Warrants that are equity-classified are not subsequently remeasured unless modified or required to be reclassified as liabilities. For warrants that are determined to be liability-classified, we estimate the fair value at issuance and each subsequent reporting date, with changes in the fair value reported in the consolidated statements of operations. The classification of all outstanding warrants, including whether such instruments should be recorded as equity, is evaluated at the end of each reporting period.
For warrants issued with ‘vanilla’ or less complex terms, we use the Black Scholes valuation model to estimate fair value. Key inputs to the Black Scholes valuation model include the fair value of our stock, the exercise price and number of warrant shares of the contract, the remaining contractual term, stock price volatility (based on our stock price history as well as an analysis of the historical volatility of the common stock of a group of comparable entities) and a risk-free rate.
For warrants with uncertain or more complex terms (such as variability in the warrant shares or exercise price), we may utilize more complex models to address such provisions. These may include probability-weighted scenario models or Monte Carlo simulation models. Monte Carlo simulation models require the use of simulations that are weighted based on significant unobservable inputs including stock prices, the average volatility of a population set and probabilities assigned. Each simulation is based on the range of inputs in a scenario with the mean of the output on each simulation calculated as an average.
The use of these valuation models requires the input of highly subjective assumptions. Any change to these inputs could produce significantly higher or lower fair value measurements.
Fair Value of Bifurcated Derivative Liabilities and Other Financial Instruments
We evaluate all of our financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the consolidated statements of operations. The classification of derivative instruments, including whether such instruments should be recorded as liabilities are evaluated at the end of each reporting period.
For our derivative financial instruments classified as a liability, the Company will select a valuation model that best aligns with the type of instrument and economic profile of the embedded feature being valued. This may include the use of Monte Carlo or other appropriate valuation models to value the derivative instruments at inception and on subsequent valuation dates. Monte Carlo simulation models require the use of simulations that are weighted based on significant unobservable inputs including the average volatility of a population set and probabilities assigned. Each simulation is based on the range of inputs in a scenario with the mean of the output on each simulation calculated as an average. For other embed features, the Company may utilize a ‘with and without’ income-based valuation approach, designed to compare the fair value of a financial instrument with the embedded feature and the fair value of the financial instrument without the embedded feature, in order to isolate the fair value of the embedded feature, or certain probability-weighted scenario analyses to compare various potential settlement amounts. These complex valuations require the use of key inputs, some of which are based on estimates and judgements by management and/or external consultants, such as the fair value of the Company’s stock price, volatility, estimates of the occurrence or non-occurrence of certain events and estimates of the probability of default or other external impacts.
The use of these valuation models requires the input of highly subjective assumptions. Any change to these inputs could produce significantly higher or lower fair value measurements.
Fair Value of Preferred Stock (upon extinguishment)
During the year ended December 31, 2022, we agreed to modify the terms of the Series A Preferred Stock, resulting in the conclusion that the modification was a substantial change in fair value of the Series A Preferred Stock and that extinguishment accounting should be applied. In order to account for the extinguishment, we estimated the fair value of the Series A Preferred Stock based on the modified terms. The difference between the estimated fair value of the Series A Preferred Stock post-modification and its carrying value was recorded as a deemed dividend adjusting net loss available to common stockholders during the year ended December 31, 2022.
For this fair value measurement, we utilized a model that includes a Monte Carlo simulation to estimate the fair value of the Series A Preferred Stock in an initial public offering or other listing scenario and a discounted cash flow approach to estimate the fair value of the Series A Preferred Stock in a redemption (or non-conversion) scenario. The Monte Carlo simulation includes management inputs around the fair value of our common stock and the estimated initial public offering date. The simulation determines whether the estimated future initial public offering would be a “Qualified IPO”, as defined, or a “Listing” conversion, as these two conversion types represented different economic outcomes. The present value calculation for the redemption (or non-conversion scenario) assumes the future repayment of the Series A Preferred Stock stated value and accrued cumulative dividends, discounted to the date of the modification. The estimated fair values of these scenarios were then applied a probability of each event occurring (probability of initial public offering or redemption/non-conversion). These complex valuations require the use of key inputs, some of which are based on estimates and judgements by management assisted by external consultants, such as the fair value of the Company’s stock price, volatility, estimates of the occurrence or non-occurrence of certain events and estimates of the probability of default or other external impacts.
The use of these valuation models requires the input of highly subjective assumptions. Any change to these inputs could produce significantly higher or lower fair value measurements that could materially impact the financial statements.
| 73 |
MANAGEMENT
Our directors and executive officers and their respective ages and titles are as follows:
| Name | Age | Position(s) and Office(s) Held | ||
| Anatoly Dritschilo, M.D. | 80 | Chairman of the board of directors and Chief Executive Officer | ||
| Timothy J. Lorber | 66 | Chief Financial Officer* | ||
| Michael Vander Hoek | 65 | VP for Operations and Regulatory, former Chief Financial Officer* | ||
| Peter Dritschilo | 55 | President and Chief Operating Officer | ||
| Mira Jung, Ph.D. | 74 | Chief Scientific Officer | ||
| Tyvin Rich, M.D. | 76 | Chief Clinical Officer | ||
| Milton Brown, M.D., Ph.D. | 59 | Director | ||
| Steven Richards | 55 | Independent Director (1)(2)(3) | ||
| Joshua Schafer | 53 | Independent Director (2)(3) | ||
| Chris H. Senanayake, Ph.D. | 66 | Independent Director (1) | ||
| Bette Jacobs, Ph.D. | 73 | Independent Director (1)(3) |
| (1) | Member of the Audit Committee |
| (2) | Member of the Compensation Committee |
| (3) | Member of the Nominating and Corporate Governance Committee |
*Timothy Lorber became our Chief Financial Officer on June 13, 2024. Prior to that time, Michael Vander Hoek served as our Chief Financial Officer.
Set forth below is a description of the background and business experience of our directors and executive officers.
Anatoly Dritschilo, M.D. Dr. Dritschilo is a co-founder of the Company and has served as Chief Executive Officer and Chairman of the Board of Directors since the Company’s formation in December 2012. Dr. Dritschilo is a radiation oncologist by training and has held multiple leadership positions in health care. At Georgetown University Medical School in Washington, D.C., he served principally as Department Chair from 1980 to 2022; Chief of Radiation Oncology at MedStar-Georgetown University Hospital from 2005 to 2022; Medical Director of Georgetown University Hospital from 1994 to 1997; and Interim Director of the NCI-funded Lombardi Comprehensive Cancer Center from 2005 to 2007. He has also served on the boards of directors of MedStar-Georgetown University Hospital, the National Capital Rehabilitation Hospital, and the MedStar Health Research Institute. Previously, he was a founding director of Oncomed, Inc. and a member of the board of directors of Neopharm, Inc. His 250+ scientific publications and 12 issued patents have earned him election as a Fellow of the National Academy of Inventors. Dr. Dritschilo holds a Bachelor of Science degree in Chemical Engineering from the University of Pennsylvania, a medical degree from the College of Medicine of New Jersey and residency training from the Harvard Joint Center for Radiation Therapy. His qualifications support his service as our Chief Executive Officer and Chairman of the Board of Directors.
Timothy J. Lorber, CPA serves as the Company’s Chief Financial Officer, a position he assumed on a part-time basis commencing June 13, 2024. He will assume the full-time position of Chief Financial Officer on September 9, 2024. Mr. Lorber has more than 40 years of professional finance experience, including Legg Mason, Inc. (“Legg Mason”), one of the world’s larger public global asset management firms, where he worked from March 2006 to July 2020 serving in various finance leadership roles, including Managing Director and Chief Accounting Officer until its sale in 2020. From August 2021 until June 2024, Mr. Lorber has served in leadership roles with several privately held businesses, overseeing finance, technology and human resources functions. Prior to Legg Mason, Mr. Lorber served as Internal Audit Director of Freddie Mac from August 2003 to March 2006 and has also worked for several international public accounting firms. Mr. Lorber has extensive experience with mergers and acquisitions, valuations and complex accounting and financial reporting matters and holds a Bachelor of Arts in Accounting from Loyola University, Maryland, and is a licensed CPA.
| 74 |
Michael P. Vander Hoek serves as the Company’s Vice President, Operations and Regulatory, a position he has held since 2019, and served as the Company’s Chief Financial Officer from 2019 until June 13, 2024 . From November 2019 until April 2021, Mr. Vander Hoek served as Director, Finance and Business Development at Georgetown Lombardi Comprehensive Cancer Center (“LCCC”), where he directed a new five-year $221.9 million institutional commitment for cancer center research under a new NCI-approved cancer consortium arrangement and recruited scientists to fulfill strategic objectives with senior leaders to improve cancer research and treatment. From 2007 until November 2019, Mr. Vander Hoek served as Associate Director, Administration, at Georgetown’s LCCC, where he was responsible for direct administrative operations for more than 400 faculty and staff in the department of oncology, radiation medicine, pathology and biostatistics, bioinformatics and biomathematics, including managing $216.9 million in institutional commitments to LCCC from Medstar Health, John Theurer Cancer Center (“JTCC”), and Georgetown University. and implementing an enterprise-wide clinical trial management system for Georgetown University and Medstar Health. From 2004 until 2007, Mr. Vander Hoek served as Chief Financial Officer at Georgetown’s LCCC. During his time at Georgetown, Mr. Vander Hoek negotiated a series of 12 research integration agreements between LCCC and the JTCC that resulted in the approval of an NCI recognized Consortium in 2019. From 2001 until 2004, Mr. Vander Hoek served as Vice-Chair, Planning and Administration, at MedStar Georgetown University Hospital, where he was responsible for managing administrative and financial operations for some 440 staff, physicians, residents and fellows in the departments of Medicine and Neurology. From 1996 until 2001, Mr. Vander Hoek served as Senior Associate Administrator, Finance and Information Systems, for the Department of Medicine, Georgetown University Medical Center, where he designed and managed the faculty compensation system, while managing the finances and information systems for the department. His financial management experience in publicly held companies includes Director of Managed Care Reimbursement for Critical Care America from 1990 to 1993 and Regional Controller for Laboratory Corporation of America (LabCorp) from 1993 to 1996. His responsibilities at both companies included extensive financial management related to mergers, acquisitions, and start-up operations. Mr. Vander Hoek holds a Master’s in Health Services Administration from The George Washington University and a Bachelor of Arts in Biology and Psychology from Hope College.
Peter Dritschilo has served as our President and Chief Operating Officer since Shuttle was formed in December 2012. He also served as our Chief Financial Officer until 2019. Mr. Dritschilo has more than 25 years of business management experience in medical services and cancer treatment. He has held administrative positions with Medstar-Rad America from 2001 to 2005, Georgetown University 2005 to 2006, Prince William Hospital and the Fauquier Hospital Cancer Center 2006 to 2011 and Inova Health System’s Schar Cancer Institute from 2011 to 2018. In 2014, Mr. Dritschilo filed for Chapter 7 bankruptcy protection due to the failure of a personal business venture. Mr. Dritschilo graduated from Georgetown University and received his MBA from the George Washington University.
Mira Jung, Ph.D., a co-founder of our company, has served as our Chief Scientific Officer for Biology since December 2012, and was a member of our board of directors from our formation in December 2012 until 2019. Since 2004, Dr. Jung has served as Professor of Radiation Medicine and Microbiology at Georgetown University Medical School. With over 30 years of experience in molecular radiation biology research, she is an expert in mechanisms of radiation resistance and on the roles of HDAC inhibitors in modifying the radiation response. Dr. Jung’s research has been funded by NIH and the DOD leading to 100+ publications and nine patents granted by the USPTO, including the first reports of HDAC inhibitor drug classes modifying cancer cell radiation resistance and protecting normal tissues from radiation damage. Dr. Jung holds an MA degree and a PhD in Microbiology and Molecular Virology from the University of Kansas, Lawrence.
Tyvin A. Rich, M.D. serves as our company’s Chief Medical Officer and is responsible for the clinical development of novel radiation sensitizers. Since 2010, Dr. Rich has served as a Staff Radiation Oncologist at the Hampton University Proton Therapy Institute in Hampton Virginia and Professor Emeritus at University of Virginia Health Sciences Center, Department of Radiation Oncology. From 1995 until 2010, Dr. Rich was a Professor and Chairman of the Department of Therapeutic Radiology and Oncology at the University of Virginia Health Sciences Center. Prior to that, from 1984 through 1995, Dr. Rich was a Professor of Radiotherapy and Director of Clinics in the Department of Radiotherapy of the University of Texas M. D. Anderson Cancer Center. He has served as the protocol chair for RTOG clinical trials that advanced the use of chemoradiation for the treatment of rectal and pancreatic cancers. He is an expert in the applications of infusional 5-Fluorouracil for chemoradiation therapy of gastro-intestinal cancers and has authored more than 200 scientific articles, reviews and book chapters. Dr. Rich received his undergraduate degree at Rutgers University, his medical degree at the University of Virginia, and completed residencies in internal medicine at Georgetown University Medical Center and radiation therapy at Massachusetts General Hospital, Harvard Medical School.
| 75 |
Milton Brown, M.D., Ph.D., FNAI. Dr. Brown is a co-founder of our Company, previously served as our Chief Scientific Officer for Chemistry, and has served a member of our Board of Directors since the Company’s formation in December 2012. Since August 2022, Dr. Brown has also served as Vice Dean of Research, Professor of Internal Medicine and the Prudence and Louis Ryan endowed chair in translational research at Eastern Virginia Medical School. Previously, he was Director of the Center for Drug Discovery at the George Mason University from 2020 to 2022 and Director of the Inova Center for Drug Discovery and Development from 2016 to 2020. Dr. Brown was a founder of Rivanna Pharmaceuticals in 2004 and co-founder, Chairman and CEO of Trocar Pharma in 2020, both of which are Virginia-based biopharmaceutical companies engaged in the discovery and development of novel small molecule therapeutics for the treatment of neurological, oncological, and infectious diseases. Dr. Brown has also served as Director of the Drug Discovery Center at Georgetown University Medical School from 2012 to 2016 and principal investigator of the NIH/NCI funded Chemical Diversity Center from 2010 to 2015. Dr. Brown brings to Shuttle Pharma 25 years of experience in drug discovery with over 100 publications and 67 issued patents, including discovery of novel HDAC inhibitors. Dr. Brown was a 2015 recipient of the Percy Julian Award by the National Organization of Black Chemists and Chemical Engineers for significant contributions in pure and/or applied research in science. He has served on government committees including the NIH Experimental Therapeutics Study Section, the NIH Drug Discovery and Molecular Pharmacology Study Section and was a scientific counselor to the U.S. Secretary of Health. Dr. Brown holds a Ph.D. in synthetic chemistry from the University of Alabama, and an M.D. from the University of Virginia. He is an elected fellow of the National Academy of Inventors (FNAI). His extensive experience and expertise in drug discovery makes him uniquely qualified to guide the company’s drug discovery program as a member of our Board of Directors.
Steven Richards. Mr. Richards was appointed to be a member of our Board of Directors in 2019. He is CEO and Founder of Endurance Media, a media finance company based in Santa Monica, California, that launched in 2014 with a strategic alliance with eOne Entertainment and a mandate to produce and finance commercially driven feature films. From 2006 to 2014, Mr. Richards served as Co-President and Chief Operating Officer of Silver Pictures where he oversaw all business activities and managed a team of more than 20 people responsible for film development, production, and financial information. From 2000 to 2006, he served as Chief Financial Officer at Silver Pictures and from 1995 to 2000 as Vice President, Finance, at Silver Pictures. Mr. Richards holds an MBA in Finance from UCLA, a BBA in accounting from Temple University, and holds his CPA license. We believe his experience as a chief financial officer and his knowledge of accounting will assist in providing guidance and oversight to our Board of Directors as we grow our Company.
Joshua Schafer. Mr. Schafer was appointed to be a member of our Board of Directors in 2019. From January 2023 until present, Mr. Schafer has been serving as the Chief Commercial Officer, and EVP Business Development at Zevra Therapeutics, a rare disease company. From November 2022 until January 2023, Mr. Schafer was interim CEO and Chair of the board of directors at PHARNEXT, an entity he has served on the board of directors since July 2020. From December 2020 until November 2022, Mr. Schafer served as Senior Vice President and General Manager, Autoimmune and Rare Disease Business for Mallinckrodt Pharmaceuticals Incorporated. In addition, he served as Chief Strategy and Business Development Officer from September 2019 until December 2020, and from 2015 to September 2019 he was SCP of Business Development and General Manager of International Operations at Mallinckrodt Pharmaceuticals. From 2009 until 2015, he served as Vice President and Oncology Therapeutic Area Head at Astellas Pharmaceuticals Incorporated, where he was responsible for building the company’s global oncology franchise. From 2000 until 2009, Mr. Schafer served in positions of increasing seniority at Takeda Pharmaceuticals North America, including Manager and Senior Manager, New Product and New Business Development; Senior Product Manager, Gastrointestinal Marketing; and Director, Oncology and Renal Marketing and Commercial Development. He began working in the healthcare and pharmaceutical industry in 1998 and has served in various positions including management consulting at Accenture (formerly Anderson Consulting), G.D. Searle & Co. (later acquired by Pfizer) and Cognia Corporation. He received his Bachelor of Arts in Biology and German at the University of Notre Dame, his MS in Biotechnology from Northwestern University and his MBA from Northwestern University. We believe Mr. Schafer’s extensive experience in pharmaceutical strategy, marketing and business development will assist our Board of Directors’ oversight role as we build and develop our Company.
| 76 |
Chris H. Senanayake, Ph.D. Dr. Senanayake was appointed to be a member of the Company’s Board of Directors in 2021. In 2019, Dr. Senanayake founded TCG GreenChem, Inc., a U.S. subsidiary of TCG Lifesciences Pvt. Ltd., a leading global Contract Research and Manufacturing Services (CRAMS) company in the area of drug discovery, development and commercialization, where he serves as chief executive officer. Dr. Senanayake has more than 30 years of pharmaceutical industry experience, making him an invaluable asset to Shuttle Pharma’s mission as the Company advances its pharmaceutical candidates in clinical trials. He has held positions of Senior Scientist at Dow Chemical, and Research Fellow at Merck & Co, Inc. (from 1990 to 1996), Director and Executive Director of Process Research at Sepracor, Inc. (1996 to 2002), Director of Chemical Development and Vice President of Chemical Development for Boehringer Ingelheim Pharmaceuticals, Inc. In 2018, he was appointed as the CEO of Asta GreenChem, Inc in Richmond VA and Astatech (Chengdu) Biopharmaceuticals Corp. in China. He has a record of leading and delivering high complexity APIs for manufacturing. Dr. Senanayake participated in development activities of many drugs, including multi-billion-dollar blockbuster drugs, such as Crixivan, Lunesta, Jardiance, Formotorol, Desvenlafaxine and other drug candidates. He is co-author of 425 scientific publications and is co-inventor of more than 150 patents. Dr. Senanayake received his Ph.D. in synthetic organic chemistry at Wayne State University, where he developed the total synthesis of complex natural products and completed the first total synthesis of grosshemin in the guaianolide family. In his postdoctoral fellowship, he conducted total synthesis of polyol systems such as amphotericin B, compactin and C-nucleosides. We believe Dr. Senanayake’s detailed and in-depth experience as an executive and developer of pharmaceuticals will enable him to provide value to us by introducing potential joint venture partners, as well as enhancing our oversight through his in-depth understanding of and experience in the pharmaceuticals industry.
Bette Jacobs, Ph.D. Dr. Jacobs was appointed to be a member of the Company’s Board of Directors in October 2022. Dr. Jacobs is an experienced researcher, administrator, and businesswoman currently serving as a professor in the department of health systems administration at Georgetown University and as a distinguished scholar at the O’Neill Institute for National and Global Health Law. Dr. Jacobs holds her Ph.D. from the University of Texas and is noted for her groundbreaking transdisciplinary and cross-sector work in systems design. As a voting member of the Cherokee Nation, she has lifetime involvement in equity programs and has testified before Congress. In addition to serving on several start-up boards, Dr. Jacobs founded the National Coalition of Ethnic Minority Nurse Associations funded by the NIH National Institute of General Medical Sciences. Prior to her current role at Georgetown, she served as dean at the Georgetown School of Nursing and Health Studies, vice president for Honda of America Manufacturing, associate director of applied research at UAB Civitan International Research Center and acting dean of graduate studies and research at California State University. She has been a fellow and visiting professor at the University of Oxford and an academic guest scholar and lecturer at several acclaimed universities worldwide. Her wealth of experience in research, administration and serving on boards coupled with her unique background and perspectives makes her ideally suited to serving as a member of our Board of Directors.
Scientific Advisory Committee
Theodore L. Phillips, M.D. has served as the Chair of our Scientific Advisory Committee since 2018. He held the position of Chief Medical Officer and Clinical Director at Shuttle Pharmaceuticals from 2014 until 2018. Dr. Phillips’ distinguished career has included positions of Chair of the Department of Radiation Oncology (from1978 to 1998) and Associate Director (from 1996 to 1999) of the UCSF Cancer Center at the University of California at San Francisco. He is highly experienced in radiation oncology clinical trials of hypoxic radiation sensitizers. Dr. Phillips served as the principal investigator of the SBIR contract for the Phase I clinical trial of Ropidoxuridine. He previously served as Associate Director of the Northern California Oncology Group from 1983-1990, president of the American Society of Therapeutic Radiation Oncologists from 1984 to 1985 and is an elected member of the Institute of Medicine of the National Academy of Science. Dr. Phillips holds a BS degree from Dickinson College in Carlisle, Pennsylvania and a MD from the University of Pennsylvania. He provides advice to the leadership team to help design and implement clinical trials of radiation therapy and radiation response modifying drugs.
Ralph R. Weichselbaum, M.D. has served as Scientific Advisor to Shuttle Pharmaceuticals for translational research for the discovery and development of radiation response modifiers since 2013. Dr. Weichselbaum is the Daniel K. Ludwig Professor and Chairman of the Department of Radiation and Cellular Oncology, the University of Chicago, a position he has held since 1985. He is also an elected member of the Institute of Medicine, National Academy of Sciences. He has devoted his career to translational research in cancer with combined radiotherapy and chemotherapy. Dr. Weichselbaum and his colleagues conceived “genetic radiotherapy” and developed viral constructs for use in clinical tumor radiation sensitization. These were commercialized as TNFerade (GenVec, Inc.) and tested in a Phase I clinical trial in prostate cancer and a Phase III clinical trial for pancreatic cancer.
| 77 |
J. Martin Brown, Ph.D. has served as a Scientific Advisor to Shuttle Pharmaceuticals for translational research for the development of hypoxic radiation sensitizers since 2017. Dr. Brown received his Ph.D.in Cancer Biology from Oxford University in 1968 and was Director of the Division of Radiation and Cancer Biology at Stanford University from 1984 to 2004. He is an expert in the radiation biology of hypoxia in cancers and has more than 300 peer-reviewed published articles. He has received awards in recognition of his work, including the Gold Medal, American Society for Therapeutic Radiology and Oncology (1999, the Failla Memorial Award, Radiation Research Society (2000), the Weiss Medal, Association for Radiation Research (2001) and the Henry S. Kaplan Distinguished Scientist Award, International Association for Radiation Research (2007). He developed etanidazole, a hypoxic radiation sensitizer, and tirapazamine, a hypoxic cytotoxic drug, from bench to clinical trials.
Alejandro Villagra, Ph.D. has served as a Scientific Advisor to Shuttle Pharmaceuticals with expertise in cellular signaling pathways, epigenetics and immunology since 2017. Dr. Villagra received his Ph.D. in Molecular Biology from the University of Concepcion, in Chile in 2004 and completed post-graduate training at the H. Lee Moffitt Cancer Center and Research Institute in Tampa, Florida in Molecular Immunology in 2009, in the Laboratory of Eduardo Sotomayor, MD. He joined the faculty of the Moffitt Cancer Center and Research Institute, as a research scientist from 2009 through 2015 and advanced to Assistant Professor of Oncologic Sciences. He became an Assistant Professor in the Department of Biochemistry and Molecular Medicine at the George Washington University (GWU) School of Medicine and Health Sciences in 2015, as a member of the GWU Cancer Center. His research is focused on molecular and cellular roles of histone deacetylases (HDACs) in tumor immunology and as adjuvants for immunotherapy of cancers.
Joseph Armstrong, III, Ph.D. joined as a Scientific Advisor to Shuttle Pharmaceuticals in 2021, He received his Ph.D. from the University of Colorado in 1988, completed his post-doctoral work at the University of Virginia at Charlottesville and holds the position of Chief Operating Officer at and Global Head of Business Development TCG GreenChem, Inc. He provides industry experience in chemistry, drug development and process research, having previously held positions at Merck & Co. Inc. in Rahway, N.J and in the U.K. for two pharmaceutical companies in the areas of Pharmaceutical Research and Development. His primary areas of focus have been in the design and implementation of efficient synthesis of drug candidates amenable to large scale production. Dr. Armstrong led the development team that designed, developed and implemented the manufacturing process for the new treatment for Type II diabetes, Januvia TM. His team was awarded the Solvias Prize in 2004 (Basel, Switzerland), the IChemE Aztra-Zeneca Award for Green Chemistry and Engineering in 2005 (London, England), Dr. Armstrong has more than 40 publications and holds 10 patents.
Family Relationships
Dr. Anatoly Dritschilo and Peter Dritschilo are father and son. There are no other family relationships among our directors and executive officers.
Involvement in Certain Legal Proceedings
During the past ten years, other than Peter Dritschilo, our President, who filed for personal bankruptcy under Chapter 7 of the U.S. Bankruptcy Code in 2014, no director, executive officer, promoter, or control person of the Company has been involved in the following:
| (1) | A petition under the Federal bankruptcy laws or any state insolvency law which was filed by or against, or a receiver, fiscal agent or similar officer was appointed by a court for the business or property of such person, or any partnership in which he was a general partner at or within two years before the time of such filing, or any corporation or business association of which he was an executive officer at or within two years before the time of such filing; | |
| (2) | Such person was convicted in a criminal proceeding or is a named subject of a pending criminal proceeding (excluding traffic violations and other minor offenses); | |
| (3) | Such person was the subject of any order, judgment, or decree, not subsequently reversed, suspended, or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from, or otherwise limiting, the following activities: |
| i. | Acting as a futures commission merchant, introducing broker, commodity trading advisor, commodity pool operator, floor broker, leverage transaction merchant, any other person regulated by the Commodity Futures Trading Commission, or an associated person of any of the foregoing, or as an investment adviser, underwriter, broker or dealer in securities, or as an affiliated person, director or employee of any investment company, bank, savings and loan association or insurance company, or engaging in or continuing any conduct or practice in connection with such activity; |
| 78 |
| ii. | Engaging in any type of business practice; or | |
| iii. | Engaging in any activity in connection with the purchase or sale of any security or commodity or in connection with any violation of Federal or State securities laws or Federal commodities laws; |
| (4) | Such person was the subject of any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any Federal or State authority barring, suspending or otherwise limiting for more than 60 days the right of such person to engage in any activity described in paragraph (f)(3)(i) below, or to be associated with persons engaged in any such activity; | |
| (5) | Such person was found by a court of competent jurisdiction in a civil action or by the Commission to have violated any Federal or State securities law, and the judgment in such civil action or finding by the Commission has not been subsequently reversed, suspended, or vacated; | |
| (6) | Such person was found by a court of competent jurisdiction in a civil action or by the Commodity Futures Trading Commission to have violated any Federal commodities law, and the judgment in such civil action or finding by the Commodity Futures Trading Commission has not been subsequently reversed, suspended, or vacated; | |
| (7) | Such person was the subject of, or a party to, any Federal or State judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended, or vacated, relating to an alleged violation of: |
| i. | Any Federal or State securities or commodities law or regulation; or | |
| ii. | Any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order; or | |
| iii. | Any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| (8) | Such person was the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act (15 U.S.C. 78c(a)(26))), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act (7 U.S.C. 1(a)(29))), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
Board of Directors
Our board of directors is responsible for overseeing the Company’s business consistent with its fiduciary duty to the stockholders. This significant responsibility requires highly skilled individuals with various qualities, attributes and professional experience. There are general requirements for service on the board of directors that are applicable to directors and there are other skills and experience that should be represented on the board of directors as a whole but not necessarily by each director. Our Corporate Governance and Nominating Committee, detailed below, considers the qualifications of director candidates individually and in the broader context of the board of directors’ overall composition and the Company’s current and future needs.
Terms of Office
All of our directors are elected to one-year terms to hold office until the next annual meeting of our stockholders and until a successor is appointed and qualified, or until their removal, resignation, or death. Executive officers serve at the pleasure of the board of directors.
Director Independence
In order to qualify for continued listing on Nasdaq, our board of directors must consist of a majority of “independent” directors, as defined under Nasdaq listing standards and Rule 10A-3(b)(1) under the Exchange Act. At present, four of the six directors serving on our board of directors qualify as “independent.” Our independent directors consist of Messrs. Richards and Schafer, Dr. Senanayake and Dr. Jacobs.
| 79 |
Board Committees
General
Our board of directors has established three committees consisting of an audit committee, a compensation committee, and a nominating and corporate governance committee. The members of each committee qualify as “independent” as defined under Nasdaq listing standards and Rule 10A-3(b)(1). Moreover, at least one member of the audit committee qualifies as an “audit committee financial expert” as the term is defined under Nasdaq listing standards and applicable rules and regulations of the SEC, based on their respective business professional experience in the financial and accounting fields.
Audit Committee
The audit committee, which consists of Steve Richards, MBA, CPA (Chair), Bette Jacobs and Chris H. Senanayake, assists our board of directors in its oversight of the Company’s accounting and financial reporting processes and the audits of the Company’s financial statements, including (a) the quality and integrity of the Company’s financial statements (b) the Company’s compliance with legal and regulatory requirements, (c) the independent auditors’ qualifications and independence and (d) the performance of the Company’s internal audit functions and independent auditors, as well as other matters which may come before it as directed by the board of directors. Further, the audit committee, to the extent it deems necessary or appropriate, among its several other responsibilities, will:
| ● | be responsible for the appointment, compensation, retention, termination and oversight of the work of any independent auditor engaged for the purpose of preparing or issuing an audit report or performing other audit, review or attest services for the Company; | |
| ● | discuss the annual audited financial statements and the quarterly unaudited financial statements with management and the independent auditor prior to their filing with the SEC in our Annual Report on Form 10-K and Quarterly Reports on Form 10-Q; | |
| ● | review with the Company’s management on a periodic basis (i) issues regarding accounting principles and financial statement presentations, including any significant changes in our company’s selection or application of accounting principles; and (ii) the effect of any regulatory and accounting initiatives, as well as off-balance sheet structures, on the financial statements of the company; | |
| ● | monitor the Company’s policies for compliance with federal, state, local and foreign laws and regulations and the Company’s policies on corporate conduct; | |
| ● | maintain open, continuing and direct communication between the board of directors, the audit committee and our independent auditors; and | |
| ● | monitor our compliance with legal and regulatory requirements and will have the authority to initiate any special investigations of conflicts of interest, and compliance with federal, state and local laws and regulations, including the Foreign Corrupt Practices Act, as may be warranted. |
Compensation Committee
The compensation committee, which consists of Steve Richards (Chair) and Joshua Schafer, aids our board of directors in meeting its responsibilities relating to the compensation of the Company’s executive officers and to administer all incentive compensation plans and equity-based plans of the Company, including the plans under which Company securities may be acquired by directors, executive officers, employees and consultants. Further, the compensation committee, to the extent it deems necessary or appropriate, among its several other responsibilities, will:
| ● | review periodically our Company’s philosophy regarding executive compensation to (i) ensure the attraction and retention of corporate officers; (ii) ensure the motivation of corporate officers to achieve the Company’s business objectives; and (iii) align the interests of key management with the long-term interests of the Company’s stockholders; |
| 80 |
| ● | review and approve corporate goals and objectives relating to chief executive officer compensation and other executive officers of Shuttle; | |
| ● | make recommendations to the board of directors regarding compensation for non-employee directors, and review periodically non- employee director compensation in relation to other comparable companies and in light of such factors as the compensation committee may deem appropriate; and | |
| ● | review periodically reports from management regarding funding the Company’s pension, retirement, long-term disability and other management welfare and benefit plans. |
Nominating and Corporate Governance Committee
The nominating and corporate governance committee, which consists of Joshua Schafer (Chair), Steve Richards and Bette Jacobs, recommends to the board of directors individuals qualified to serve as directors and on committees of the board of directors to advise the board of directors with respect to the board of directors composition, procedures and committees to develop and recommend to the board of directors a set of corporate governance principles applicable to the Company, and to oversee the evaluation of the board of directors and Shuttle’s management. In addition, the nominating and corporate governance committee will consider diversity of background including diversity of race, ethnicity, international background, gender and age when evaluating candidates for board of directors membership.
Further, the nominating and corporate governance committee, to the extent it deems necessary or appropriate, among its several other responsibilities will:
| ● | recommend to the board of directors and for approval by a majority of independent directors for election by stockholders or appointment by the board of directors as the case may be, pursuant to our bylaws and consistent with the board of director’s evidence for selecting new directors; | |
| ● | review the suitability for continued service as a director of each member of the board of directors when his or her term expires or when he or she has a significant change in status; | |
| ● | review annually the composition of the board of directors and to review periodically the size of the board of directors; | |
| ● | make recommendations on the frequency and structure of board of directors meetings or any other aspect of procedures of the board of directors; | |
| ● | make recommendations regarding the chairmanship and composition of standing committees and monitor their functions; | |
| ● | review annually committee assignments and chairmanships; | |
| ● | recommend the establishment of special committees as may be necessary or desirable from time to time; and | |
| ● | develop and periodically review corporate governance procedures and consider any other corporate governance issue. |
Code of Ethics
We have adopted a code of ethics that applies to all of our executive officers, directors and employees. The code of ethics codifies the business and ethical principles that govern all aspects of our business. This document will be made available in print, free of charge, to any stockholder requesting a copy in writing from our secretary at our executive offices in Gaithersburg, Maryland. A copy of our code of ethics is available on our website at www.shuttlepharma.com.
| 81 |
Insider Trading Policies and Procedures
The Company has adopted an insider trading policy, as amended and restated on March 10, 2023 (the “Insider Trading Policy”), overseen by the Company’s corporate secretary, that applies to all (i) directors, (ii) executive officers and (iii) employees who are exposed to insider information (together, the “Covered Persons”). The Insider Trading Policy prohibits the use of material non-public information obtained by Covered Persons through their involvement with the Company when making decisions to purchase, sell, give away or otherwise trade in the Company’s securities or to provide such information to others outside the organization. Under the Insider Trading Policy, material non-public information includes, among other things, significant changes in the Company’s prospects, significant write-downs, liquidity problems, changes in management, extraordinary borrowings, changes in debt, planned public offerings or any other information that may be deemed material to the Company or the Company’s prospects. Further, we have established black-out periods to which all Covered Persons are subject, including quarterly black-out periods, which commence three weeks before the end of each quarter and continue until the quarterly results are disclosed by filing the Company’s Quarterly Report on Form 10-Q or Annual Report on Form 10-K. The Company may impose black-out periods from time to time as other types of material non-public information occur when material non-public events or disclosures are pending. If the Company imposes a special black-out period, the Company will notify Covered Persons accordingly. Covered Persons are permitted to trade in the Company’s securities only when there is no black-out period in effect and such trade has been pre-cleared by the Company’s corporate secretary, or when a qualified 10b5-1 plan has been established in accordance with federal securities laws.
Clawback Policy
While the Company does not presently have in place any significant incentive compensation agreements or awards related to the Company’s overall financial performance, the Company’s board of directors has adopted a clawback policy in order to comply with federal securities laws. As such, we have adopted a clawback policy in which we may seek the recovery or forfeiture of incentive compensation paid by us, including cash, equity or equity-based compensation, in the event we restate our financial statements under certain circumstances. The clawback policy applies to our Section 16 officers, any employee who was eligible to receive incentive compensation and whose conduct contributed to the need for a restatement, and any other former Section 16 officer or other employee who contributed to the need for such restatement.
Board of Directors Role in Risk Oversight
Members of the board of directors have periodic meetings with management and the Company’s independent auditors to perform risk oversight with respect to the Company’s internal control processes. The Company believes that the board of directors’ role in risk oversight does not materially affect the leadership structure of the Company. The Company believes that its founders, leadership team and members of the board of directors exemplify diversity and inclusivity with respect to race, sex and ethnic origin. The board of directors presently has two diverse directors and is in the process of reviewing and vetting a female candidate to serve as a director. As such, the Company anticipates being in full compliance with Nasdaq’s newly adopted diversity requirements by the end of its first year of listing.
| 82 |
EXECUTIVE AND DIRECTOR COMPENSATION
Summary Compensation Table
The table below summarizes all compensation awarded to, earned by, or paid to our Chief Executive Officer and Chief Financial Officer and certain of our other executive officers for 2023 and 2022.
| Name and principal position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards ($) | Non-Equity Incentive Plan Compensation ($) | Nonqualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | ||||||||||||||||||||||||||
| Anatoly Dritschilo M.D., CEO | 2023 | 287,175 | 112,000 | - | - | - | - | - | 399,175 | ||||||||||||||||||||||||||
| 2022 | 97,253 | - | 83,717 | - | - | - | - | 268,921 | |||||||||||||||||||||||||||
| Michael Vander Hoek, CFO, VP | 2023 | 230,530 | 72,000 | - | - | - | - | 302,530 | |||||||||||||||||||||||||||
| 2022 | 82,792 | - | 34,115 | - | - | - | - | 128,792 | |||||||||||||||||||||||||||
| Peter Dritschilo, President and COO | 2023 | 242,012 | 72,000 | - | - | - | - | 314,012 | |||||||||||||||||||||||||||
| 2022 | 100,308 | - | 31,977 | - | - | - | - | 178,641 | |||||||||||||||||||||||||||
| Tyvin Rich, Chief Medical Officer | 2023 | 220,226 | 43,000 | - | - | - | - | - | 263,226 | ||||||||||||||||||||||||||
| 2022 | 67,077 | - | 11,918 | - | - | - | - | 96,077 | |||||||||||||||||||||||||||
Employment Agreements
Each of our executive officers has entered into an employment agreement with us. The employees each will receive compensation on an annual basis in cash, payable in monthly installments commencing at the completion of our IPO, as well as restricted stock units (“RSUs”) subject to achieving certain key performance indicators. Certain of our executive officers are entitled to various target bonuses, upon achievement of certain milestones. The terms of the employment agreements are as follows:
Employment Agreement with Anatoly Dritschilo, MD
On June 28, 2019, we entered into an employment agreement with our Chief Executive Officer and Chairman of the board of directors, Anatoly Dritschilo, M.D. Under Dr. Dritschilo’s employment agreement, Dr. Dritschilo will receive base compensation of $274,000 per year. Dr. Dritschilo also received an initial RSU grant of 5,687 RSUs (2,844 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vested over three years in substantially equal one-third installments on each one year anniversary of the agreement. Under his employment agreement, if Dr. Dritschilo terminates his employment for “Good Reason,” as defined in the agreement, Dr. Dritschilo will be entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain requirements of his employment agreement. Dr. Dritschilo accepted a reduced salary prior to the Company’s completion of its initial public offering in September 2022.
| 83 |
Employment Agreement with Timothy J. Lorber
On June 10, 2024, we entered into an employment agreement with our Chief Financial Officer, Timothy J. Lorber. Under Mr. Lorber’s employment agreement, he will receive base compensation of $227,000 and is entitled to a target bonus of $72,000 upon achievement of certain milestones. Mr. Lorber also received an initial RSU grant of $100,000 worth of RSU issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest annually in one-third increments commencing on the first anniversary date of the grant of RSU, in accordance with the terms of the RSUs award agreement.
Employment Agreement with Michael Vander Hoek
On September 1, 2019, we entered into an amended employment agreement with our pervious Chief Financial Officer and Vice President for Operations and Regulatory, Michael Vander Hoek. Under Mr. Vander Hoek’s employment agreement, he will receive base compensation of $227,000 and is entitled to a target bonus of $72,000 upon achievement of certain milestones. Mr. Vander Hoek also received an initial RSU grant of 762 RSUs (381 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one year anniversary of the agreement. Under Mr. Vander Hoek’s employment agreement, if he terminates his employment for “Good Reason,” as defined in the agreement, he will be entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain requirements of his employment agreement. Mr. Vander Hoek accepted a reduced salary prior to the Company’s completion of its initial public offering in September 2022. Effective September 10, 2024, Mr. Vander Hoek assumes the Vice President, Regulatory position on a full-time basis and stops being our Chief Financial Officer.
Employment Agreement with Peter Dritschilo
On May 30, 2019, we entered into an employment agreement with our President and Chief Operating Officer, Peter Dritschilo. Under Mr. Dritschilo’s employment agreement, Mr. Dritschilo will receive base compensation of $236,000 and is entitled to a target bonus of $72,000 upon achievement of certain milestones. Mr. Dritschilo also received an initial RSU grant of 2,595 RSUs (1,298 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one-year anniversary of the agreement. Under Mr. Dritschilo’s employment agreement, if Mr. Dritschilo terminates his employment for “Good Reason,” as defined in the agreement, he will be entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain requirements of his employment agreement. Mr. Dritschilo accepted a reduced salary prior to the Company’s completion of its initial public offering in September 2022.
Employment Agreement with Tyvin Rich, M.D.
On May 31, 2019, we entered into an employment agreement with our Chief Clinical Officer, Tyvin Rich, M.D. Under Dr. Rich’s employment agreement, Dr. Rich receives base compensation of $218,000 per year and is entitled to a target bonus of $43,000 upon achievement of certain milestones. Dr. Rich also received an initial RSU grant of 481 RSUs (241 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one year anniversary of the agreement. Under Dr. Rich’s employment agreement, if Dr. Rich terminates his employment for “Good Reason,” as defined in the agreement, he is entitled to his then applicable base salary for period of 12 months, subject to his continued compliance with certain provisions of his employment agreement. Dr. Rich accepted a reduced salary prior to the Company’s completion of its initial public offering in September 2022.
| 84 |
Employment Agreement with Mira Jung, Ph.D.
On May 1, 2023, we entered into an employment agreement with Mira Jung, Ph.D., our Chief Scientific Officer, replacing the May 30, 2019 employment agreement. Under Dr. Jung’s current agreement, Dr. Jung receives a base salary of $46,000 (20%) per year and is entitled to a target bonus of $15,620. She also received an additional grant worth $20,200 worth of RSUs, vesting annually in one-third increments commencing on the first anniversary date of the grant of RSUs. Under Dr. Jung’s previous agreement, Dr. Jung received base compensation of $46,800 and was entitled to a target bonus of $14,200 upon achievement of certain milestones. Dr. Jung also received an initial RSU grant of 112 RSUs (56 on a post-reverse split basis) issuable under the Company’s 2018 Equity Incentive Plan, which RSUs vest over three years in substantially equal installments on each one-year anniversary of the agreement. Under Dr. Jung’s employment agreement, if Dr. Jung terminates her employment for “Good Reason,” as defined in the agreement, Dr. Jung is then entitled to her then applicable base salary for period of 12 months, subject to her continued compliance with certain requirements of her employment agreement. Dr. Jung accepted a reduced salary prior to the Company’s completion of its initial public offering in September 2022.
Outstanding Equity Awards at Fiscal Year-End
As of December 31, 2023, on a post-reverse split basis, a total of 32,416 RSUs have been granted to our executive officers and Directors under our 2018 Equity Incentive Plan (the “Plan”), 12,415 RSUs were outstanding on December 31, 2022, 20,100 RSUs vested, of which 24,731 remain subject to vesting. The Company has filed a registration statement on Form S-8 (SEC File No. 333-268758) to register the shares granted under the Plan.
The following table sets forth information concerning the number of shares of common stock underlying outstanding equity incentive awards for each of our executive officers as of December 31, 2023:
| Option Awards | Stock Awards | ||||||||||||||||||||||||||
| Name | Grant Date | Number of Securities Underlying Unexercised Options Exercisable (#) | Number of Securities Underlying Unexercised Options Unexercisable (#) | Option Exercise Price ($) | Option Expiration Date | Number of Shares or Units of Stock not yet Vested (#)) | Market Value of Shares or Units not yet Vested ($) | ||||||||||||||||||||
| Bette Jacobs | 10/28/2022 | - | - | - | - | 1,483 | (1) | $ | 5,457 | ||||||||||||||||||
| Milton Brown | 02/15/2023 | 2,084 | (1) | $ | 7,667 | ||||||||||||||||||||||
| (1) | These RSUs vest in two installments on the anniversary of the grant date. |
2018 Equity Incentive Plan
Our 2018 Equity Incentive Plan provides for equity incentives to be granted to our employees, executive officers or directors and to key advisers and consultants. Equity incentives may be in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuant to the 2018 Equity Incentive Plan, restricted stock awards, other stock- based awards, or any combination of the foregoing. The 2018 Equity Incentive Plan is administered by the Company’s compensation committee or, alternatively, if there is no compensation committee, the Company’s board of directors. We have reserved 3,000,000 shares of our common stock for issuance under the Plan, of which 84,779 shares have been granted under the Plan as of the date of this Annual Report.
Director Compensation
Each of our non-employee directors, pursuant to the terms of director agreements (the “Director Agreements”), between each of the directors and the Company, receives compensation on an annual basis consisting of $25,000 in cash, payable in quarterly installments commencing 90 days after completion of our initial public offering, and received $100,000 in RSUs upon their respective dates of election. The RSUs vest over a two-year period in one third increments, with one-third vesting immediately upon signing and one-third vesting on each of the first and second anniversary of election. In addition, non-employee directors will also be reimbursed for out-of-pocket costs incurred in connection with attending meetings.
| 85 |
BENEFICIAL OWNERSHIP OF SECURITIES
The following table sets forth, as of the date of this registration statement, the beneficial ownership of our common stock by each director and executive officer, by each person known by us to beneficially own 5% or more of our common stock and by directors and executive officers as a group. Unless otherwise stated, the address of the persons set forth in the table is c/o Shuttle Pharmaceuticals Holdings, Inc., 401 Professional Drive, Suite 260, Gaithersburg, MD 20879.
Beneficial ownership is determined in accordance with SEC rules and generally includes voting or investment power with respect to securities. Unless otherwise indicated, the stockholders listed in the table below have sole voting and investment power with respect to the shares indicated.
All share ownership figures include shares of our commons stock issuable upon securities convertible or exchangeable into shares of our common stock, whether or not convertible or exchangeable within 60 days of the effective date of this Annual Report. Such shares are deemed outstanding and beneficially owned by such person only for purposes of computing his or her percentage ownership, but not for purposes of computing the percentage ownership for any other person.
As of October 29, 2024, there were issued and outstanding 2,946,099 shares of common stock.
| Names and addresses | Number of shares of common stock beneficially owned (#) | Percentage of shares of common stock beneficially owned (%) | ||||||
| Directors and Named Executive Officers: | ||||||||
| Anatoly Dritschilo, M.D.(1) | 538,701 | 18.3 | % | |||||
| Timothy Lorber(2) | - | - | ||||||
| Milton Brown, M.D., Ph.D.(3) | 137,869 | 4.7 | % | |||||
| Mira Jung, Ph.D. | 135,715 | 4.6 | % | |||||
| Michael Vander Hoek | 12,982 | - | ||||||
| Peter Dritschilo | 820 | - | ||||||
| Tyvin A. Rich, M.D. | 304 | - | ||||||
| Steve Richards | 214 | - | ||||||
| Joshua Schafer | 214 | - | ||||||
| Chris H. Senanayake | 349 | - | ||||||
| Bette Jacobs | 1,901 | - | ||||||
| All directors and officers as a group (ten persons) | 829,070 | 28.1 | % | |||||
| Other 5% beneficial owners: | ||||||||
| None. | ||||||||
| - | Denotes the holder owns less than one percent of the outstanding common stock. |
| ± | The persons named above have full voting and investment power with respect to the shares indicated. Under the rules of the SEC, a person (or group of persons) is deemed to be a “beneficial owner” of a security if he or she, directly or indirectly, has or shares the power to vote or to direct the voting of such security, or the power to dispose of or to direct the disposition of such security. Accordingly, more than one person may be deemed to be a beneficial owner of the same security. |
| (1) | Consists of (i) 135,650 shares of common stock held of record by Dr. Anatoly Dritschilo, (ii) 138,051 shares of common stock and warrants to purchase 2,500 shares of commons stock, each held of record by Joy Dritschilo, his spouse, and (iii) 262,500 shares of common stock held by PAL Trust, a trust formed for the benefit of Dr. and Mrs. Dritschilo’s adult children and for which a third party serves as external trustee and two of their children serve as co-trustees . Dr. Dritschilo disclaims beneficial ownership over all securities held by Mrs. Dritschilo and PAL Trust. |
(2) |
Does not include 28,455 RSUs that remain subject to vesting. |
| (3) | Does not include 1,042 RSUs which remain subject to vesting. |
| 86 |
Change of Control
The Company is not aware of any arrangements which may at a subsequent date result in a change of control of the Company.
Securities authorized for issuance under equity compensation plans
2018 Equity Incentive Plan
Our 2018 Equity Incentive Plan (the “Plan”) provides for equity incentives to be granted to our employees, executive officers or directors and to key advisers and consultants. Equity incentives may be in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuant to the Plan, restricted stock awards, other stock- based awards, or any combination of the foregoing. The Plan is administered by the Company’s compensation committee or, alternatively, if there is no compensation committee, the Company’s board of directors. We have reserved 3,000,000 shares of our common stock for issuance under the Plan, of which 84,779 shares have been granted under the Plan as of the date of this Annual Report, of which 24,731 remain subject to vesting.
The following table provides information as of December 31, 2023 about our equity compensation plans and arrangements.
| Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted- average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans | |||||||||
| Equity compensation plans approved by security holders | 24,731 | $ | 11.76 | 2,321,820 | ||||||||
| Equity compensation plans not approved by security holders | – | – | – | |||||||||
| Total | 24,731 | * | $ | 2,321,820 | ||||||||
*Outstanding equity incentive grants consist entirely of RSUs which automatically vest over time into an equal number of shares of common stock at no additional cost to the holder.
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Unless described below, during the last two fiscal years, there were no transactions or series of similar transactions to which we were a party or will be a party, in which:
| ● | the amounts involved exceed or will exceed $120,000; and |
| ● | any of our directors, executive officers or holders of more than 5% of our capital stock, or any member of the immediate family of any of the foregoing had, or will have, a direct or indirect material interest. |
| 87 |
On December 1, 2020, the Company consolidated two loans obtained in 2018 for a total of $350,000 from Joy Dritschilo, the wife our Chief Executive Officer, which loans accrued interest at 7.5% since the date of inception, into a single loan between Mrs. Dritschilo and the Company (the “2018 Consolidated Loan”) such that, with accrued interest, the 2018 Consolidated Loan had a principal balance of $424,005.65, bore interest at a rate of 7.5% per annum, and had a maturity date of December 31, 2021. The 2018 Consolidated Loan were then extended until June 30, 2022, pursuant to an amendment to the 2018 Consolidated Loan agreement dated January 24, 2022. On July 29, 2022, the Company and Mrs. Dritschilo entered into an amendment to the 2018 Consolidated Loan, pursuant to which repayment was extended through June 30, 2023. On January 15, 2023, following closing on the Convertible Note and Warrant offering to Ayrton Capital, the 2018 Consolidated Loan was paid off in full.
On December 1, 2020, the Company consolidated the May 2018 Loan and the September 2019 Loan with our Chief Executive Officer (the “2019 Consolidated Loan”), such that, with accrued interest, the 2019 Consolidated Loan had a principal balance of $138,448.20, bears interest at the rate of 7.5% per annum, and has a maturity date of December 31, 2021. The 2019 Consolidated Loan was extended until June 30, 2022, pursuant to an amendment to the 2019 Consolidated Loan agreement dated January 24, 2022. On July 29, 2022, the Company and our Chief Executive Officer entered into an amendment to the 2019 Consolidated Loan, pursuant to which repayment was extended through June 30, 2023.
On June 21, 2021, the Company entered into a loan agreement with Mrs. Dritschilo in the amount of $120,000 (principal), bearing interest at the rate of 7.5% per annum, with a single balloon payment due at maturity on June 21, 2022 (the “June 2021 Loan Agreement”). On July 29, 2022, the Company and Mrs. Dritschilo entered into an amendment to the June 2021 Loan Agreement, pursuant to which repayment was extended through June 30, 2023.
On August 1, 2022, in conjunction with our private placement of $125,000 of units consisting of 10% notes and warrants to purchase common stock, which were sold to three accredited investors in total, Mrs. Dritschilo purchased a $50,000 note and received warrants to purchase 2,500 shares of common stock at $20.00 per share. The notes and warrants were sold pursuant to an exemption from registration pursuant to Rule 506(b) of Regulation D of the Securities Act.
On September 14, 2022, we entered into a manufacturing agreement with TCG GreenChem, Inc. (“TCG GreenChem”), the U.S. subsidiary of TCG Lifesciences Pvt Ltd., a global contract research and manufacturing services company located in India. Dr. Chis Senanayake, one of our independent directors, is CEO and CSO of TCG GreenChem and CSO of TCG Lifesciences Pvt Ltd. TCG GreenChem was contracted for process research, development and cGMP compliant manufacture of IPdR, The Company paid TCG GreenChem $450,000 during the year ended December 31, 2022 and a total of $1,096,370 during the year ended December 31, 2023, completing the contract.
On September 4, 2024, the Company entered into a loan agreement with our Chief Executive Officer, Dr. Anatoly Dritschilo, pursuant to which Dr. Dritschilo loaned the Company of $250,000 (principal), bearing interest at the rate of 12% per annum and which is repayable in 12 substantially equal monthly installments over a one year period.
On October 14, 2024, the Company conducted the initial closing of an up to $1.3 million 5% OID senior secured convertible note warrant offering, pursuant to which the Company issued a total of $600,000 notes and warrants to purchase 286,169 shares of common stock, exercisable at $1.53 per share. The notes bear interest at the rate of 14.5% per annum, with the interest payable quarterly in cash, mature one year after the date of issuance, are redeemable at any time by the Company at a 107% premium to the principal value of the notes and, three months after issuance, can be converted at any time by the holder at a 110% premium to the principal value of the note. The Company’s CEO purchased $250,000 of notes and warrants in the October 2024 Bridge Offering.
Review, Approval and Ratification of Related Party Transactions
All related party transactions are subject to the review, approval, or ratification of our board of directors or an appropriate committee thereof.
CAPITALIZATION
The following table sets forth our cash and cash equivalents and capitalization as of June 30, 2024:
| ● | on an actual basis; | |
| ● | on a pro forma basis to reflect the issuance of a $250,000 note payable to Dr. Dritschilo on September 4, 2024, and the conversion of $1,172,850 of principal outstanding under the Alto Convertible Note for 719,148 shares of common stock. The Alto Convertible Note has been paid in full as of the date of this prospectus; and the $732,134 in convertible bridge notes to related parties (including $250,000 to Dr. Dritschilo) in October 2024, excluding proceeds from any exercise of 329,460 warrants issued in conjunction with the Convertible Bridge Notes; and; | |
| ● | on a pro forma as adjusted basis to reflect the sale of 395,574 shares of common stock, 2,555,246 Pre-Funded Warrants and 2,950,820 Common Warrants in this offering at the combined offering price of $1.525 per share of Common Stock (or less $0.001 per Pre-Funded Warrant) and one accompanying Common Warrant, resulting in net proceeds of $4.1 million, after deducting approximately $405,000 in expenses, but excluding proceeds from any exercise of the Common Warrants being offered in this offering, as well as the payment in full of the Alto Convertible Note. |
Our capitalization following the closing of this offering will depend on the actual public offering price and other terms of this offering determined at pricing. Cash and cash equivalents are not components of our total capitalization. You should read this table together with the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and related notes included elsewhere in this prospectus.
| 88 |
| As of June 30, 2024 | As of June 30, 2024 | As of June 30, 2024 Pro Forma As | ||||||||||
| Actual | Pro Forma | Adjusted | ||||||||||
| Cash and cash equivalents | $ | 695,594 | 1,390,456 | $ | 5,483,131 | |||||||
| Capitalization: | ||||||||||||
| Notes payable | ||||||||||||
| Alto Convertible Note | $ | 1,517,989 | - | $ | - | |||||||
| Related party | - | 250,000 | 250,000 | |||||||||
Bridge Convertible Notes | 831,579 | 831,579 | ||||||||||
| Stockholders’ equity: | ||||||||||||
| Series A convertible preferred stock, $0.00001 par value; $1,000 per share liquidation value; 20,000,000 shares authorized; no shares outstanding | - | |||||||||||
| Common stock, $0.00001 par value; 100,000,000 shares authorized; 2,106,223 shares issued and outstanding at June 30, 2024, actual, 2,825,371 shares pro forma, and 3,220,945 shares pro forma as adjusted | 21 | 28 | 68 | |||||||||
| Additional paid-in capital | 29,996,963 | 31,205,707 | 35,298,343 | |||||||||
| Accumulated deficit | (29,195,550 | ) | (29,195,550 | ) | (29,195,550 | ) | ||||||
| Total stockholders’ equity (deficit) | 801,434 | 2,010,185 | 6,102,860 | |||||||||
| Total capitalization | $ | 2,319,423 | 3,091,764 | $ | 7,184,439 | |||||||
The number of shares of common stock issued and outstanding actual, pro forma and pro forma as adjusted in the table above excludes 2,321,820 shares reserved for issuance under our 2018 Equity Incentive Plan and 513,460 warrants which remain subject to exercise.
DILUTION
If you invest in our common stock in this offering, your ownership interest will be diluted immediately to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock immediately after this offering. As of June 30, 2024, our historical net tangible book value was $801,434, or $0.38 per share. Our historical net tangible book value is the amount of our total tangible assets less our total liabilities. Historical net tangible book value per share represents historical net tangible book value divided by the 2,106,223 shares of our common stock outstanding as of June 30, 2024.
After giving effect to our issuance and sale of common stock, Pre-Funded Warrants and Common Warrants in this offering at the public offering price of $1.525 per share of Common Stock (or less $0.001 per Pre-Funded Warrant) and one accompanying Common Warrant, and after deducting estimated Placement Agents’ fees payable by us, our pro forma as adjusted net tangible book value as of June 30, 2024 would have been $6.1 million, or $1.06 per share, after including 2,555,246 of Pre-Funded Warrant shares. This represents an immediate increase in as adjusted net tangible book value per share of $1.15 to existing stockholders and immediate dilution of $0.47 in pro forma as adjusted net tangible book value per share to new investors purchasing common stock in this offering. Dilution per share to new investors is determined by subtracting as adjusted net tangible book value per share after this offering from the public offering price per share paid by new investors.
The following table illustrates this dilution on a per share basis:
| Public offering price per share | $ | 1.525 | ||
| Historical net tangible book value per share as of June 30, 2024 | $ | 0.38 | ||
| Increase in as adjusted net tangible book value per share attributable to this offering | $ | 1.15 | ||
| As adjusted net tangible book value per share after giving effect to this offering | $ | 1.06 | ||
| Dilution in as adjusted net tangible book value per share to new investors in this offering | $ | 0.47 |
The information above is as of June 30, 2024 and excludes:
| ● | Up to 732,134 shares of our common stock issuable from time to time upon the conversion of outstanding Convertible Bridge Notes and the exercise of 329,460 related warrants, with a weighted-average exercise price of $1.42 per share; | |
| ● | 184,000 shares of our common stock issuable upon the exercise of outstanding warrants, with a weighted-average exercise price of $11.54 per share; | |
| ● | 2,321,820 other shares of our common stock reserved for future issuance from time to time under our 2018 Equity Incentive Plan. |
| 89 |
DIVIDEND POLICY
We have not paid any dividends on our common stock since inception and we currently expect that, in the foreseeable future, all earnings, if any, will be retained for the development of our business and no dividends will be declared or paid. Any future dividends will be subject to the discretion of our board of directors and will depend upon, among other things, our earnings, if any, operating results, financial condition and capital requirements, general business conditions and other pertinent facts.
DESCRIPTION OF CAPITAL STOCK
Capital Stock
Our authorized capital stock consists of 100,000,000 shares of common stock, par value $0.00001 per share, and 20,000,000 shares of preferred stock, par value $0.00001 per share.
Common Stock
As of the date of this prospectus, we had 2,946,099 shares of common stock issued and outstanding. Each holder of common stock is entitled to one vote for each share owned on all matters voted upon by stockholders, and a majority vote is required for all actions to be taken by stockholders. In the event we liquidate, dissolve or wind-up our operations, the holders of the common stock are entitled to share equally and ratably in our assets, if any, remaining after the payment of all our debts and liabilities and the liquidation preference of any shares of preferred stock that may then be outstanding. The common stock has no preemptive rights, no cumulative voting rights, and no redemption, sinking fund, or conversion provisions.
Holders of common stock are entitled to receive dividends, if and when declared by the board of directors, out of funds legally available for such purpose, subject to the dividend and liquidation rights of any preferred stock that may then be outstanding.
Preferred Stock
Our board of directors has the authority, without further action by the stockholders, to issue shares of preferred stock in one or more series and to fix the rights, preferences and the number of shares constituting any series or the designation of such series. While our certificate of incorporation and bylaws, each as amended to date, do not contain any provisions that may delay, defer or prevent a change in control, the issuance of preferred stock may have the effect of delaying or preventing a change in control or make removal of our management more difficult. At present, our board of directors has authorized the issuance of up to 10,000 shares of Series A convertible preferred stock, of which 1,212.5 shares were issued and subsequently converted into common stock following completion of our IPO. As of the date of this prospectus, no preferred stock is issued or outstanding.
| 90 |
Series A Convertible Preferred Stock
Our board of directors previously designated and authorized the issuance of up to 10,000 shares of Series A convertible preferred stock, par value $0.00001 per share (the “Series A Convertible Preferred Stock”), of which there were 1,212.5 shares issued, which were subsequently converted into common stock following completion of our IPO. A total of $1,212,500 was raised during 2018 and 2019 from the sale of our Series A Convertible Preferred Stock. The Series A Convertible Preferred Stock had a stated value of $1,000 per share, was entitled to receive a dividend at the rate of 8.5% per annum, which dividend was cumulative and payable at our option in shares of common stock or cash upon the date of conversion or redemption, as so determined by the Company. The Series A Convertible Preferred Stock, pursuant to its terms, were automatically convertible upon the earlier of (a) the closing of the sale of shares of common stock to the public at an offering price of at least $32.00 per share (on a post-Reverse Split Basis) in a firm commitment underwritten public offering pursuant to an effective registration statement under the Securities Act, resulting in gross proceeds to us (before underwriter’s discounts, commissions and expenses) of an amount of at least $10 million (a “Qualified IPO”), or (b) listing of the common stock on the NYSE or Nasdaq (a “Qualified Listing”). All shares of Series A Convertible Preferred Stock were then convertible at either (i) 90% of the gross public offering price per share of the Qualified IPO (before deducting underwriter’s discounts, commissions and expenses) or (ii) in the case of (b) above, $5.00 per share. Following consummation of our IPO, the Series A Convertible Preferred Stock was converted into a total of 336,810 shares of common stock (42,101 shares on a post-reverse split basis), plus an additional 100,517 shares of common stock (12,565 shares on a post-reverse split basis) paid to the Series A Convertible Shareholders as dividends payable. As of the date of this prospectus, there are no shares of Series A Convertible Preferred Stock outstanding.
Series A Warrants
In conjunction with our sale of Series A Convertible Preferred Stock, our board of directors authorized the issuance of warrants to purchase up to approximately 42,113 shares of common stock (5,265 shares on a post-reverse split basis) (the “Series A Warrants”), to the holders of Series A Preferred Stock. The Series A Warrants were issued following consummation of our IPO, are exercisable for a period of three years following issuance and have an exercise price of $32.00 per share.
January 2023 Note and Warrant Offering
On January 11, 2023, we entered into a securities purchase agreement (the “SPA”) with Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B, a Cayman entity (the “Investor”) and consummated the sale to such Investor of a senior secured convertible note (the “January 2023 Convertible Note”) with an initial principal amount of $4,300,000, bearing interest at 5% per annum, and a warrant (the “January 2023 Warrant”) to purchase 1,018,079 shares (127,260 shares on a post-2024 Reverse Split Basis) of our common stock at a fixed exercise price of $2.35 per share ($18.80 per share on a post-2024 Reverse Split Basis, and subsequently reset to $1.93).
The January 2023 Convertible Note was sold with an original issue discount of $300,000. As such, the Investor paid for the January 2023 Convertible Note by delivering $4,000,000 in cash consideration to the Company. Boustead served as the sole placement agent for the private placement (the “January 2023 Private Placement”) of the January 2023 Convertible Note and January 2023 Warrant. Boustead received a placement agent fee of $320,000 at the closing of the January 2023 Private Placement, representing 7.0% of the gross cash proceeds at the closing plus 1% in non-accountable expenses. In addition, Boustead received a placement agent warrant to purchase 71,266 shares (8,909 shares on a post-2024 Reverse Split basis) of common stock, representing 7.0% of the warrant shares issued in the January 2023 Private Placement. After deducting the placement agent fee and our estimated expenses associated with the January 2023 Private Placement, our estimated net cash proceeds at the closing were approximately $3,493,423. As of September 6, 2024, the majority of the January 2023 Convertible Note had been converted and there remained just over $14,000 outstanding under the note. The Alto Convertible Note has been paid in full as of the date of this prospectus.
The SPA
The SPA contains certain representations and warranties, covenants and indemnities customary for similar transactions. Under the SPA, we also agreed to the following additional covenants:
| ● | So long as the January 2023 Convertible Note remains outstanding, we will not effect or enter into an agreement to effect any variable rate transaction other than a bona fide at-the-market offering or equity line of credit. |
| ● | We obtained majority stockholder consent approving the offering and the issuance of common stock upon conversion of the January 2023 Convertible Note and exercise of the January 2023 Warrant, as is required under the rules of the Nasdaq, and subsequently filed an information statement on Schedule 14C (the “Information Statement”) informing all stockholders of the action and mailed such Information Statement to all stockholders within 70 days of closing. |
| 91 |
In addition, we granted the Investor participation rights in future equity and equity-linked offerings of securities, subject to certain limited exceptions, during the two years after the later of (a) the closing or (b) the date the January 2023 Convertible Note no longer remains outstanding, in an amount of up to 30% of the securities being sold in such offerings.
January 2023 Convertible Note
General
The January 2023 Convertible Note was issued to the Investor on January 11, 2023 and matures on March 11, 2025 (subject to extension in certain circumstances, including bankruptcy and outstanding events of default).
Amortization
Starting on the earlier of (x) the date of a registration statement which registers the shares underlying the January 2023 Convertible Note is declared effective by the SEC and (y) February 28, 2023, then on the first trading day of the month for each month thereafter, and on the maturity date (each, an “Installment Date”), subject to certain exceptions and unless deferred as described below, the Company is required to make monthly amortization payments equal to 1/26th of the unrestricted principal (and related unrestricted original issue discount) and interest of the January 2023 Convertible Note payable (the “Installment Amount”), which must be satisfied in cash at a redemption price equal to 105% of such Installment Amount (each, an “Installment Redemption”).
Notwithstanding the foregoing, the noteholder may, at its sole option, elect to defer any Installment Amount until a subsequent Installment Date selected by the noteholder, provided that any such deferred amounts shall not continue to accrue interest unless the deferral is at the request of the Company.
Interest
The January 2023 Convertible Note bears interest at a rate of 5% per annum and, upon any conversion or redemption, shall include a make-whole of interest from such date of determination through the maturity date. After the occurrence and during the continuance of an Event of Default (as defined in the January 2023 Convertible Note), the January 2023 Convertible Note will accrue interest at the rate of 15% per annum. See “Events of Default” below.
Conversion; Alternate Conversion upon Event of Default
The January 2023 Convertible Note is convertible, at the option of the noteholder, into shares of our common stock at the lower of (i) $18.80 per share (post Reverse Split), (ii) 90% of the three lowest VWAPs in the 15 trading days prior to the payment date or (iii) 90% of the VWAP on the trading day prior to the payment date. The conversion price is subject to full ratchet antidilution protection upon any subsequent transaction at a fixed price lower than the conversion price then in effect and standard adjustments in the event of any stock split, stock dividend, stock combination, recapitalization or other similar transaction. If we enter into any agreement to issue (or issue) any variable rate securities, other than a bona fide at-the-market offering or equity line of credit, the noteholder has the additional right to substitute such variable price (or formula) for the conversion price.
If an Event of Default has occurred under the January 2023 Convertible Note, the noteholder may elect to alternatively convert the January 2023 Convertible Note at a redemption premium of 115% of the conversion amount. As of September 30, 2024, the January 2023 Convertible Note was paid in full.
Subsequent Placement Optional Redemption and Future Right of Participation
At any time after the earlier of the date a noteholder becomes aware of any placement by us of equity or equity-linked securities or the date of consummation of such a placement, subject to certain limited exceptions, the noteholder will have the right to have us redeem a portion of each January 2023 Convertible Note not in excess of 30% of the net proceeds from such placement at a redemption price of 100% of the portion of the January 2023 Convertible Note subject to redemption. In addition, through March 11, 2026, the noteholder has the right to be notified of any future offering of securities and to participate in up to 30% of the total offering amount.
| 92 |
Covenants
We are subject to certain customary affirmative and negative covenants regarding the incurrence of certain indebtedness, the existence of liens, the repayment of indebtedness, the payment of cash in respect of dividends, distributions or redemptions, and the transfer of assets, among other matters.
January 2023 Warrant
In addition to the January 2023 Convertible Note, we issued a January 2023 Warrant exercisable for four (4) years for the purchase of an aggregate of up to 127,260 shares of common stock (the “January 2023 Warrant Shares”), at an adjusted exercise price of $1.93 per share. The number of January 2023 Warrant Shares and exercise price are each subject to adjustment as provided under the terms of the January 2023 Warrant. If, at the time of exercise of the January 2023 Warrant, there is no effective registration statement registering, or no current prospectus available for, the issuance of the January 2023 Warrant Shares to the Investor, and the registration statement is not subject to SEC review and the Company has otherwise affirmatively failed to maintain such registration statement’s effectiveness, then the January 2023 Warrant may also be exercised, in whole or in part, by means of a “cashless exercise.” The January 2023 Warrant may not be exercised if, after giving effect to the exercise the Investor, would beneficially own in excess of 4.99% of the number of shares of common stock outstanding immediately after giving effect to the issuance of the January 2023 Warrant Shares. At the Investor’s option, the ownership limitation blocker may be raised or lowered to any other percentage not in excess of 9.99%, as applicable, except that any raise will only be effective upon 61-days’ prior notice to the Company.
If the Company issues or sells, or the Company publicly announces the issuance or sale of, any shares of common stock, or convertible securities or options issuable or exchangeable into common stock (a “New Issuance”), under which such common stock is sold for a consideration per share less than the exercise price then in effect, the exercise price of the January 2023 Warrant will be adjusted to the New Issuance price in accordance with the formulas provided in the January 2023 Warrant. Any such adjustment will not apply with respect to the issuance of Excluded Securities (as defined in the January 2023 Warrant). Upon any adjustment to the exercise price, the number of January 2023 Warrant Shares that may be purchased upon exercise of the January 2023 Warrant will be increased or decreased proportionately, so that after such adjustment the aggregate exercise price payable for the adjusted number of January 2023 Warrant Shares will be the same as the aggregate exercise price in effect immediately prior to such adjustment. In addition, if the Company enters into a Fundamental Transaction (as defined in the January 2023 Warrants) at any time that a January 2023 Warrant is outstanding, then, upon any subsequent exercise of the January 2023 Warrant, the Investor will have the right to receive, for each January 2023 Warrant Share that would have been issuable upon such exercise immediately prior to the occurrence of such Fundamental Transaction, the number of shares of common stock of the successor or acquiring corporation or of the Company, if it is the surviving corporation, and any additional consideration receivable as a result of such Fundamental Transaction by a holder of the number of shares of common stock for which the January 2023 Warrant is exercisable immediately prior to such Fundamental Transaction, provided, further, that if holders of common stock are not offered or paid any consideration in such Fundamental Transaction, such holder of common stock will be deemed to have received common stock of the successor entity (which entity may be the Company following such Fundamental Transaction) in such Fundamental Transaction.
Registration Rights Agreement
Pursuant to a Registration Rights Agreement, dated as of January 11, 2023, between the Investor and the Company, we have granted certain registration rights to the noteholder and January 2023 Warrant holder. The Registration Rights Agreement requires us to have a registration statement declared effective by the 60th day following the date of the Registration Rights Agreement (of March 11, 2023), or the 90th day (or April 10, 2023) following the date of the Registration Rights Agreement in the event such registration statement is subject to SEC review. It also grants the Investor customary “piggyback” registration rights.
| 93 |
Additional Information
The foregoing is only a summary of the material terms of the SPA, the January 2023 Convertible Note, the January 2023 Warrant, the Registration Rights Agreement, and the other ancillary transaction documents, and does not purport to be a complete description of the rights and obligations of the parties thereunder.
Anti-Takeover Effects of Provisions of our Certificate of Incorporation, our Bylaws and Delaware Law
Some provisions of Delaware law, our certificate of incorporation and our bylaws contain provisions that could make the following transactions more difficult: acquisition of us by means of a tender offer; acquisition of us by means of a proxy contest or otherwise; or removal of our incumbent officers and directors. It is possible that these provisions could make it more difficult to accomplish or could deter transactions that stockholders may otherwise consider to be in their best interest or in our best interests, including transactions that might result in a premium over the market price for our shares.
These provisions, summarized below, are expected to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to encourage persons seeking to acquire control of us to first negotiate with our Board of Directors. We believe that the benefits of increased protection of our potential ability to negotiate with the proponent of a non-friendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging these proposals because negotiation of these proposals could result in an improvement of their terms.
Delaware Anti-Takeover Statute
In general, Delaware corporations are subject to Section 203 of the DGCL, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| ● | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested holder; | |
| ● | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; |
| ● | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock that is not owned by the interested stockholder; or | |
| ● | the corporation does not have a class of voting stock that is: (i) listed on a national securities exchange; or (ii) held of record by more than 2,000 stockholders, unless any of the foregoing results from action taken, directly or indirectly, by an interested stockholder or from a transaction in which a person becomes an interested stockholder |
In general, Section 203 defines business combination to include the following:
| ● | any merger or consolidation involving the corporation and the interested stockholder; | |
| ● | any sale, transfer, pledge, or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; | |
| ● | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
| 94 |
| ● | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or | |
| ● | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges or other financial benefits by or through the corporation. |
Section 203 defines interested stockholder as an entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation or any entity or person affiliated with or controlling or controlled by such entity or person.
Nasdaq Listing
Our shares of common stock are traded on The Nasdaq Capital Market under the symbol “SHPH.” We do not intend to apply for listing of the Warrants on any securities exchange or other nationally recognized trading system.
Transfer Agent
The transfer agent and registrar of our common stock is VStock Transfer, LLC, of Woodmere, New York. Our transfer agent’s telephone number is (212) 828-8436.
DESCRIPTION OF SECURITIES WE ARE OFFERING
We are offering, on a reasonable best-efforts basis (i) 395,574 Shares of common stock and 2,555,246 Pre-Funded Warrants, and (ii) 2,950,820 Common Warrants to purchase up to 2,950,820 shares of common stock. Each share of common stock (or one Pre-Funded Warrant to purchase one share of our common stock in lieu thereof) will be sold with one Common Warrant to purchase one share of common stock. The shares of common stock, or Pre-Funded Warrants in lieu thereof, and Common Warrants are immediately separable and will be issued separately in this offering but must be purchased together in this offering.
The public offering price for each share of common stock and one accompanying Common Warrant is $1.525. The public offering price for each Pre-Funded Warrant and one accompanying Common Warrant is $1.524, which equals the price at which one share of common stock and one accompanying Common Warrant is sold to the public in this offering, minus $0.001. Each Common Warrant offered hereby will be a warrant to purchase one share of common stock and will have an initial exercise price equal to $1.40.
We have agreed to offer and sell the securities offered hereby to the purchasers through the Placement Agents. The Placement Agents are not required to buy or sell any specific number or dollar amount of the securities offered hereby but will use their reasonable best efforts to solicit offers to purchase the securities offered by this prospectus.
Common Stock
The material terms and provisions of our common stock are described under the caption “Description of Capital Stock” in this prospectus.
Common Warrants
The following summary of certain terms and provisions of the Common Warrants offered hereby is not complete and is subject to, and qualified in its entirety by the provisions of the forms of Common Warrants which is filed as an exhibit to the registration statement of which this prospectus is a part. Prospective investors should carefully review the terms and provisions set forth in the form of Common Warrants.
Exercisability. The Common Warrants are exercisable upon issuance and at any time up to the date that is five (5) years after its issuance. The Common Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and by payment in full in immediately available funds for the number of shares of common stock purchased upon such exercise. No fractional shares of common stock will be issued in connection with the exercise of the Common Warrants. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price.
Exercise Limitation. A holder will not have the right to exercise any portion of the Common Warrants if the holder (together with its affiliates) would beneficially own in excess of 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Common Warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99%, provided that any increase in such percentage shall not be effective until 61 days following notice from the holder to us.
| 95 |
Exercise Price. The exercise price per whole share of common stock purchasable upon exercise of one Common Warrant is $1.40. The exercise price is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock and also upon any distributions of assets, including cash, stock or other property to our stockholders.
Transferability. Subject to applicable laws, the Common Warrants may be offered for sale, sold, transferred or assigned without our consent.
Fundamental Transactions. In the event of a fundamental transaction, as described in the Common Warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding common stock, or any person or group becoming the beneficial owner of 50% of the voting power represented by our outstanding common stock, the holders of the Common Warrants will be entitled to receive upon exercise of the Common Warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the Common Warrants immediately prior to such fundamental transaction.
No Rights as a Stockholder. Except by virtue of such holder’s ownership of shares of our common stock, the holder of Common Warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the Common Warrants.
Pre-Funded Warrants
The following summary of certain terms and provisions of the Pre-Funded Warrants that are being offered hereby is not complete and is subject to and qualified in its entirety by the provisions of the forms of Pre-Funded Warrants which is filed as an exhibit to the registration statement of which this prospectus is a part. Prospective investors should carefully review the terms and provisions of the form of Pre-Funded Warrant for a complete description of the terms and conditions of the Pre-Funded Warrants.
The term “prefunded” refers to the fact that the purchase price of our common stock in this offering includes almost the entire exercise price that will be paid under the Pre-Funded Warrants, except for a nominal remaining exercise price of $0.001 per share. The purpose of the Pre-Funded Warrants is to enable investors that may have restrictions on their ability to beneficially own more than 4.99% (or, upon election of the holder, 9.99%) of our outstanding shares of common stock following the consummation of this offering the opportunity to make an investment in the Company without triggering their ownership restrictions, by receiving Pre-Funded Warrants in lieu of our common stock which would result in such ownership of more than 4.99% (or 9.99%), and receive the ability to exercise their option to purchase the shares underlying the Pre-Funded Warrants at such nominal price at a later date.
Duration and Exercise Price
Each Pre-Funded Warrant offered hereby will have an initial exercise price of $0.001 per share. The Pre-Funded Warrants will be immediately exercisable and may be exercised at any time until the Pre-Funded Warrants are exercised in full. The exercise price and number of shares of common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting our common stock and the exercise price.
Exercise Limitation.
The Pre-Funded Warrants will be exercisable, at the option of each holder, in whole or in part, by delivering a duly executed exercise notice accompanied by payment in full for the number of purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of the Pre-Funded Warrant to the extent that the holder would own more than 4.99% (or, at the election of a purchaser, 9.99%) of the outstanding common stock immediately after exercise, except that upon at least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder’s Pre-Funded Warrants up to 9.99% of the number of shares of common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Pre-Funded Warrants. No fractional shares of common stock will be issued in connection with the exercise of a Pre-Funded Warrant. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price.
Fundamental Transaction
In the event of a fundamental transaction, as described in the Pre-Funded Warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding voting securities, the holders of the Pre-Funded Warrants will be entitled to receive upon exercise of the Pre-Funded Warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the Pre-Funded Warrants immediately prior to such fundamental transaction, other than one in which a successor entity that is a publicly traded corporation (whose stock is quoted or listed for trading on a national securities exchange, including, but not limited to, the New York Stock Exchange, the NYSE American, the Nasdaq Global Select Market, the Nasdaq Global Market or the Nasdaq Capital Market) assumes the common warrant such that the warrant shall be exercisable for the publicly traded common stock of such successor entity.
Transferability
Subject to applicable laws, a Pre-Funded Warrant may be transferred at the option of the holder upon surrender of the Pre-Funded Warrant to us together with the appropriate instruments of transfer.
Exchange Listing
We do not intend to list the Pre-Funded Warrants on any securities exchange or nationally recognized trading system.
No Rights as a Stockholder
Except as otherwise provided in the Pre-Funded Warrants or by virtue of such holder’s ownership of, the holders of the Pre-Funded Warrants do not have the rights or privileges of holders of our common stock, including any voting rights, until they exercise their Pre-Funded Warrants.
| 96 |
PLAN OF DISTRIBUTION
A.G.P./Alliance Global Partners (“A.G.P.”), in conjunction with Boustead, have agreed to act as Placement Agents with respect to this offering subject to the terms and conditions contained in the placement agent agreement, dated October 29, 2024 (the “Placement Agent Agreement”).
The Placement Agents are not purchasing or selling any shares offered hereby, nor are they required to arrange for the purchase or sale of any specific number or dollar amount of shares but have agreed to use their reasonable commercial efforts to arrange for the sale of all of the shares. Because there is no minimum amount of shares of common stock that must be sold as a condition to closing this offering, the actual number of shares sold in this offering is not presently determinable and may be substantially less than the shares noted herein above. We have entered into a securities purchase agreement directly with one of the investors. Investors that did not enter into a securities purchase agreement are relying solely on this prospectus in connection with the purchase of our securities in this offering.
We negotiated the offering price for the shares in this offering with prospective investors. The factors considered in determining the price included the recent market price of our common stock, the general condition of the securities market at the time of this offering, the history of, and the prospects for the industry in which we compete, our past and present operations and our prospects for future revenues.
We have agreed to indemnify the Placement Agents against specified liabilities, including liabilities under the Securities Act, and to contribute to payments the Placement Agents may be required to make in respect thereof.
Placement Agents Fees, Commissions and Expenses
This offering is being conducted on a reasonable best-efforts basis and the Placement Agents have no obligation to buy any of the securities from us or to arrange for the purchase or sale of any specific number or dollar amount of securities. Upon the closing of this offering, we will pay the Placement Agents a placement agents’ fee equal to 9% of the aggregate gross cash proceeds to us from the sale of the securities in the offering, of which A.G.P. will receive 60% and Boustead will receive 40%. With respect to gross proceeds raised from the sale of securities to certain identified purchasers introduced by to us by Boustead, Boustead will receive 60% and 40% shall be paid to A.G.P. In addition, we have also agreed to reimburse the Placement Agents at closing for legal expenses incurred by them in connection with the offering in an amount not to exceed $100,000 and up to $50,000 for certain reasonable non-accountable fees and expenses.
As further consideration for the Placement Agents’ services, we have also agreed and paid the Placement Agents a refundable, to the extent not incurred, up-front cash fee (the “Retainer Fee”) of $50,000 upon the engagement of the Placement Agents. The Retainer Fee will be applied against the Placement Agents’ accountable out-of-pocket expenses (in compliance with FINRA Rule 5110(g)(4)) that are payable by us in connection with this offering).
The following table shows the per share and total cash placement agents’ fee we will pay to the Placement Agents in connection with the sale of the securities offered pursuant to this prospectus supplement and the accompanying prospectus, assuming the purchase of all of the securities hereby on a best-efforts basis:
Per
Share and | Per
Pre-Funded Warrant and Accompanying Common Warrant | Total | ||||||||||
| Public offering price | $ | 1.525 | $ | 1.524 | $ | 4,497,445.25 | ||||||
| Placement Agents fees to be paid by us (9%) | $ | 0.137 | $ | 0.137 | $ | 404,770.07 | ||||||
| Proceeds, before expenses, to us | $ | 1.388 | $ | 1.388 | $ | 4,092,675.18 | ||||||
Lock-Up Agreements
Pursuant to certain “lock-up” agreements, our executive officers, directors and holders of at least 5% of our common stock and securities exercisable for or convertible into common stock outstanding immediately upon the closing of this offering, have agreed, subject to certain exceptions, not to offer, sell, assign, transfer, pledge, contract to sell, or otherwise dispose of or announce the intention to otherwise dispose of, or enter into any swap, hedge or similar agreement or arrangement that transfers, in whole or in part, the economic risk of ownership of, directly or indirectly, engage in any short selling of any shares of common stock or securities convertible into or exchangeable or exercisable for any shares of common stock, whether currently owned or subsequently acquired, without the prior written consent of the Placement Agents, for a period of sixty (60) days after the closing date of the offering.
The Placement Agents, in their sole discretion, may release the common stock and other securities subject to the lock-up agreements described above in whole or in part at any time. When determining whether or not to release common stock and other securities from lock-up agreements, the Placement Agents will consider, among other factors, the holder’s reasons for requesting the release, the number of shares of common stock and other securities for which the release is being requested and market conditions at the time.
In addition, we have agreed that, subject to certain exceptions, we will not (i) conduct any issuances of our common stock for a period of 60 days following closing of this offering or (ii) enter into a variable rate transaction (as defined in the securities purchase agreement) for a period of 120 days following closing of this offering; provided that following the closing date of this offering we will be permitted to enter into an agreement with A.G.P. in connection with an “at the market” offering under Rule 415(a)(4) under the Securities Act and make sales thereunder.
| 97 |
Other Relationships
We retained Boustead to act as underwriter in our IPO, which we closed on September 2, 2022, pursuant to which we sold IPO Units consisting of shares of our common stock and one warrant to purchase one share of common stock, at a public offering price of $8.125 per unit, resulting in gross proceeds of $9,960,430, before deducting underwriting discounts and commissions. On September 29, 2022, Boustead exercised its overallotment option, purchasing an additional 183,883 IPO Units (22,986 shares on a post-reverse split basis), resulting in gross proceeds of $1,494,041, before deducting underwriter commissions and discounts. As a result, our IPO raised a total of $11,454,474, before deducting underwriting discounts, commissions and related IPO expenses.
On January 11, 2023, we entered into a securities purchase agreement with the Investor and consummated the sale to such investor of the January 2023 Convertible Note with an initial principal amount of $4,300,000, bearing interest at 5% per annum, and a warrant to purchase 1,018,079 shares (127,260 shares on a post-reverse split basis) of our common stock at a fixed exercise price of $2.35 per share ($1.93 per share on a post-2024 Reverse Split Basis, as adjusted). Boustead served as our placement agent in the January 2023 Convertible Note offering and received a placement agent fee of $320,000 at the closing, representing 7.0% of the gross cash proceeds at the closing plus 1% in non-accountable expenses. In addition, Boustead received a placement agent warrant to purchase 71,266 shares of common stock, representing 7.0% of the warrant shares issued in the offering.
On February 8, 2022 and March 11, 2022, we completed private placement offerings pursuant to which we sold to several accredited investors an aggregate of $365,000 and $225,000, respectively, of our 2022 Convertible Notes. Boustead acted as placement agent in the 2022 Convertible Note and warrant offering and received commissions and non-accountable reimbursements of 10% of the gross proceeds received and warrants to purchase 10% of the common stock.
On August 1, 2022, we completed the August 2022 Note and Warrant Offering, pursuant to which we sold to three accredited investors an aggregate of $125,000 of our 10% promissory notes, which were due upon completion of our IPO, and warrants to purchase 50,000 shares of common stock, at an exercise price of $2.50 per share (6,250 shares at $20.00 per share on a post-reverse split basis). Boustead acted as placement agent in the August 2022 Note and warrant offering but deferred its cash compensation in relation to such offering. At the time of the IPO, 30,000 of the warrants sold in August 2022 Note and Warrant Offering were registered for resale pursuant to a separate resale prospectus and were subsequently exercised.
The Placement Agents are full-service financial institutions engaged in various activities, which may include sales and trading, commercial and investment banking, advisory, investment management, investment research, principal investment, hedging, market making, brokerage and other financial and non-financial activities and services. The Placement Agents may in the future provide various investment banking, commercial banking and other financial services for us and our affiliates for which they may in the future receive customary fees.
In the ordinary course of their various business activities, the Placement Agents and certain of their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments issued by us and our affiliates. If the Placement Agents or their respective affiliates have a lending relationship with us, they routinely hedge their credit exposure to us consistent with their customary risk management policies. The Placement Agents and their respective affiliates may hedge such exposure by entering into transactions that consist of either the purchase of credit default swaps or the creation of short positions in our securities or the securities of our affiliates, including potentially the common stock offered hereby. Any such short positions could adversely affect future trading prices of the common stock offered hereby. The Placement Agents and certain of their respective affiliates may also communicate independent investment recommendations, market color or trading ideas and/or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
| 98 |
Regulation M
Each Placement Agent may be deemed to be an underwriter within the meaning of Section 2(a)(11) of the Securities Act, and any commissions received by it and any profit realized on the resale of the shares sold by it while acting as principal might be deemed to be underwriting discounts or commissions under the Securities Act. As an underwriter, each Placement Agent would be required to comply with the requirements of the Securities Act and the Exchange Act, including, without limitation, Rule 415(a)(4) under the Securities Act and Rule 10b-5 and Regulation M under the Exchange Act. These rules and regulations may limit the timing of purchases and sales of shares by the Placement Agents acting as principal. Under these rules and regulations, the Placement Agents:
● may not engage in any stabilization activity in connection with our securities; and
● may not bid for or purchase any of our securities or attempt to induce any person to purchase any of our securities, other than as permitted under the Exchange Act, until it has completed its participation in the distribution.
Trading Market
Our common stock is listed on the Nasdaq Capital Market under the symbol “SHPH.” We do not intend to apply for listing of the Warrants on any securities exchange or other nationally recognized trading system.
LEGAL MATTERS
The validity of the securities offered hereby and certain legal matters in connection with this offering relating to U.S. law will be passed upon for us by Dorsey & Whitney LLP, New York, NY. The Placement Agents are being represented by Sullivan & Worcester LLP, New York, New York, in connection with this offering.
EXPERTS
The consolidated financial statements of Shuttle Pharmaceuticals Holdings, Inc. (the “Company”) as of December 31, 2023 and 2022 and for the fiscal years then ended, have been audited by Forvis Mazars LLP, independent registered public accounting firm, as set forth in their report thereon, included in the Company’s Annual Report on Form 10-K/A for the fiscal year ended December 31, 2023, and included herein. Such consolidated financial statements have been included herein in reliance upon such report given on the authority of such firm as experts in accounting and auditing. The report of Forvis Mazars LLP contains an explanatory paragraph regarding substantial doubt about the Company’s ability to continue as a going concern and an explanatory paragraph regarding a restatement of previously issued consolidated financial statements to correct misstatements.
WHERE YOU CAN FIND MORE INFORMATION
We filed with the SEC a registration statement under the Securities Act for the securities offered by this prospectus. This prospectus does not contain all of the information in the registration statement and the exhibits and schedule that were filed with the registration statement. For further information with respect to us and our securities, we refer you to the registration statement and the exhibits and schedule that were filed with the registration statement. Statements contained in this prospectus about the contents of any contract or any other document that is filed as an exhibit to the registration statement are not necessarily complete, and we refer you to the full text of the contract or other document filed as an exhibit to the registration statement. The SEC maintains an Internet website at http://www.sec.gov that contains reports, proxy and information statements, and other information regarding registrants that file electronically with the SEC.
We file periodic reports and current reports under the Exchange Act, including Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K, and other information with the SEC. These periodic reports and other information are available for inspection and copying at the SEC regional offices, public reference facilities and on the website of the SEC referred to above.
We make available free of charge on or through our internet website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. The information found on our website, https://shuttlepharma.com, other than as specifically included in this prospectus, is not part of this prospectus.
| 99 |
SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
Consolidated Financial Statements
Contents
| F-1 |
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Balance Sheets
(Unaudited)
| June 30, | December 31, | |||||||
| 2024 | 2023 | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Prepaid expenses | ||||||||
| Marketable securities | ||||||||
| Accrued interest income | ||||||||
| Total Current Assets | ||||||||
| Property and equipment, net | ||||||||
| Operating lease right-of-use asset | ||||||||
| Total Assets | $ | $ | ||||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current Liabilities | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Accounts payable and accrued expenses related party | ||||||||
| Accrued interest payable | ||||||||
| Convertible notes payable, net | ||||||||
| Derivative liability | ||||||||
| Operating lease liability | ||||||||
| Total Current Liabilities | ||||||||
| Convertible notes payable non-current, net | ||||||||
| Derivative liability | ||||||||
| Operating lease liability non-current | ||||||||
| Total Liabilities | ||||||||
| Stockholders’ Equity | ||||||||
| Series A Convertible Preferred Stock, $ par value; $ | ||||||||
| Common stock, $ par value; shares authorized; and shares issued and outstanding as of June 30, 2024 and December 31, 2023, respectively | ||||||||
| Additional paid in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ Equity | ||||||||
| Total Liabilities and Stockholders’ Equity | $ | $ | ||||||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
| F-2 |
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Statements of Operations
(Unaudited)
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2024 | 2023 | 2024 | 2023 | |||||||||||||
| Revenue | $ | $ | $ | $ | ||||||||||||
| Operating expenses | ||||||||||||||||
| Research and development | ||||||||||||||||
| General and administrative | ||||||||||||||||
| Legal and professional | ||||||||||||||||
| Total operating expenses | ||||||||||||||||
| Loss from operations | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Other income (expense) | ||||||||||||||||
| Interest expense - related parties | ( | ) | ( | ) | ||||||||||||
| Interest expense | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Interest income | ||||||||||||||||
| Finance fee | ( | ) | ||||||||||||||
| Change in fair value of derivative liabilities | ( | ) | ||||||||||||||
| Gain on sale of marketable securities | ||||||||||||||||
| Change in fair value of marketable securities | ( | ) | ( | ) | ( | ) | ||||||||||
| Loss on settlement of convertible debt | ( | ) | ( | ) | ( | ) | ||||||||||
| Total other expense | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Net loss attributable to common stockholders | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | ( | ) | ||||
| Weighted average common shares outstanding - basic and diluted | ||||||||||||||||
| Net loss per shares - basic and diluted | $ | ) | $ | ) | $ | ) | $ | ) | ||||||||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
| F-3 |
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Statements of Changes in Stockholders’ Equity
(Unaudited)
For the Six Months Ended June 30, 2024
| Additional | Total | |||||||||||||||||||
| Common Stock | Paid in | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Equity | ||||||||||||||||
| Balance - December 31, 2023 | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Common stock issued for conversion of accrued interest and principal | ||||||||||||||||||||
| Common stock issued for restricted stock units | ||||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance - March 31, 2024 | ( | ) | ||||||||||||||||||
| Common stock issued for restricted stock units | ||||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance - June 30, 2024 | $ | $ | $ | ( | ) | $ | ||||||||||||||
For the Six months ended June 30, 2023
| Total | ||||||||||||||||||||
| Common Stock | Additional Paid in | Accumulated | Stockholders’ Equity | |||||||||||||||||
| Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||
| Balance - December 31, 2022 | $ | $ | $ | ( | ) | $ | | |||||||||||||
| Warrants issued for financing costs, net of issuance fees of $ | - | |||||||||||||||||||
| Common stock issued for conversion of accrued interest and principal | ||||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance - March 31, 2023 | ( | ) | ||||||||||||||||||
| Common stock issued for conversion of accrued interest and principal | ||||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance - June 30, 2023 | $ | $ | $ | ( | ) | $ | ||||||||||||||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
| F-4 |
Shuttle Pharmaceuticals Holdings, Inc.
Condensed Consolidated Statements of Cash Flows
(Unaudited)
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2024 | 2023 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | ||||||||
| Change in fair value of derivative liabilities | ( | ) | ( | ) | ||||
| Amortization of debt discount and finance fees | ||||||||
| Gain on sale of marketable securities | ( | ) | ( | ) | ||||
| Change in fair value of marketable securities | ( | ) | ||||||
| Accrued interest settled with common stock | ||||||||
| Loss on settlement of convertible debt | ||||||||
| Stock-based compensation | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Accrued interest income | ( | ) | ||||||
| Prepaid expenses | ( | ) | ||||||
| Accounts payable and accrued expenses | ( | ) | ||||||
| Accounts payable and accrued expenses - related parties | ( | ) | ( | ) | ||||
| Accrued interest payable | ||||||||
| Accrued interest payable - related parties | ( | ) | ||||||
| Change in operating lease asset and liability | ( | ) | ||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Investment in marketable securities | ( | ) | ( | ) | ||||
| Proceeds from disposition of marketable securities | ||||||||
| Net cash provided by (used in) investing activities | ( | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Repayment of note payable-related party | ( | ) | ||||||
| Proceeds from convertible notes payable and warrants | ||||||||
| Payment for finance costs related to convertible note payable | ( | ) | ||||||
| Payment of convertible note payable | ( | ) | ||||||
| Net cash provided by (used in) financing activities | ( | ) | ||||||
| Net change in cash and cash equivalents | ( | ) | ( | ) | ||||
| Cash and cash equivalents, beginning of period | ||||||||
| Cash and cash equivalents, end of period | $ | $ | ||||||
| Cash paid for: | ||||||||
| Interest | $ | $ | ||||||
| Income taxes | $ | $ | ||||||
| Supplemental non-cash financing activities: | ||||||||
| Common stock issued for settlement of debt | $ | $ | ||||||
| Warrants issued for financing fees, net of issuance fees of $ | $ | $ | ||||||
| Initial recognition of right of use asset and liability | $ | $ | ||||||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
| F-5 |
Shuttle Pharmaceuticals Holdings, Inc.
Notes to Unaudited Condensed Consolidated Financial Statements
June 30, 2024
Note 1 – Organization and Liquidity
Organization and Line of Business
Shuttle
Pharmaceuticals Holdings, Inc. (“we”, “us”, “our”, the “Company”) was originally formed
as Shuttle Pharmaceuticals, LLC in the State of Maryland on December 18, 2012. On August 12, 2016, the Company filed articles of conversion
with the State of Maryland to convert from an LLC to a C corporation, at which time the Company changed its name to Shuttle Pharmaceuticals,
Inc. (“Shuttle”). In connection with the conversion the Company issued shares of common stock in exchange for
The Company’s primary purpose is to develop and commercialize unique drugs for the sensitization of cancers and protection of normal tissues, with the goal of improving outcomes for cancer patients receiving radiation therapy. Shuttle has deployed its proprietary technology to develop novel cancer immunotherapies, producing a pipeline of selective HDAC inhibitors for cancer and immunotherapy applications. The Company’s HDAC platform is designed to target candidate molecules with potential roles in therapeutics beyond cancer, including autoimmune, inflammatory, metabolic, neurological and infectious diseases. The Company’s Ropidoxuridine product, which is used with radiation therapy to sensitize cancer cells, was initially funded by a Small Business Innovation Research (“SBIR”) contract provided by the National Cancer Institute (“NCI”), a unit of the National Institutes of Health (“NIH”). Ropidoxuridine has been further developed though the Company’s collaborations with scientists at the University of Virginia for use in combination with proton therapy to improve patient survival. Historically, and prior to the Company’s initial public offering in September 2022, the Company has obtained funding to develop products through NIH grants, including a product to predict late effects of radiation with metabolite biomarkers and develop prostate cancer cell lines in health disparities research.
The production and marketing of the Company’s products and its ongoing research and development activities will be and are subject to extensive regulation by numerous governmental authorities in the United States. Prior to marketing in the United States, any products or combination of products developed by the Company must undergo rigorous preclinical (animal) and clinical (human) testing and an extensive regulatory approval process implemented by the Food and Drug Administration (“FDA”) under the Food, Drug and Cosmetic Act. There can be no assurance that the Company will not encounter problems in its clinical trials that will cause the Company or the FDA to delay or suspend the clinical trials.
The Company’s success will depend in part on its ability to obtain patents and product license rights, maintain trade secrets, and operate without infringing on the proprietary rights of others, both in the United States and in other countries. There can be no assurance that patents issued to or licensed by the Company will not be challenged, invalidated or circumvented, or that the rights granted thereunder will provide proprietary protection or competitive advantages to the Company now or in the future.
Liquidity and Going Concern
Our
unaudited condensed consolidated financial statements are prepared on a going concern basis, which contemplates the realization of
assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since
inception and has a net loss of approximately $
| F-6 |
In January 2023,
the Company entered into a securities purchase agreement with an institutional investor through which the Company sold a convertible
note with a principal value of $
The
IPO and subsequent capital raise have supported operations, the manufacture of drug product and FDA approval of the IND
for the Phase II clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and the Company agreed
to an expansion of the clinical trial, necessitating additional capital to complete the trial as well as fund ongoing operations. As
a result, management had initiated a $
The accompanying unaudited condensed consolidated financial statements do not include any adjustments to reflect the future effects on the recoverability and classification of assets or the amounts and classification of liabilities if the Company is unable to continue as a going concern.
Note 2 – Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”) for interim financial statements and with the instructions to Form 10-Q and Rule 8-03 of Regulation S-X of the United States Securities and Exchange Commission (“SEC”). Accordingly, they do not contain all information and notes required by GAAP for annual financial statements. A complete discussion of the Company’s significant accounting policies is included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2023.
In the opinion of the Company’s management, the accompanying unaudited condensed consolidated financial statements contain all of the adjustments necessary to present the financial position of the Company as of June 30, 2024 and the results of operations and cash flows for the periods presented. The accompanying condensed consolidated financial statements of the Company have not been audited by the Company’s independent registered public accounting firm, except that the year-end consolidated balance sheet was derived from audited financial statements. The results of operations for the six months ended June 30, 2024 are not necessarily indicative of the operating results for the full fiscal year or any future period. These unaudited condensed consolidated financial statements should be read in conjunction with the restated financial statements and related notes thereto for the year ended December 31, 2023 included in the Company’s Annual Report on Form 10-K/A filed with the SEC on September 3, 2024.
Reverse Stock Split
On
August 13, 2024, in order to meet Nasdaq’s minimum bid price requirement of $
per share (the “Minimum Bid Price Requirement”), the Company effectuated a
| F-7 |
Basis of Consolidation
The unaudited condensed consolidated financial statements have been prepared on a consolidated basis with those of the Company’s wholly-owned subsidiaries, Shuttle Pharmaceuticals, Inc. and Shuttle Diagnostics Inc. All intercompany transactions and balances have been eliminated.
Correction of an Immaterial Error in the Prior Period Financial Statements
During
the fourth quarter of 2023, the Company determined that the prior year consolidated financial statements had a misstatement caused
by an immaterial classification error of certain research and development expenses in accordance with Accounting Standards
Codification (“ASC”) 730. As a result, certain prior year amounts have been corrected for consistency with the current
year presentation. The Company assessed the materiality of this change in presentation on prior period consolidated financial
statements in accordance with SEC Staff Accounting Bulletin No. 99, “Materiality,” (ASC Topic 250, Accounting Changes
and Error Corrections). Based on this assessment, the Company concluded that these error corrections in its unaudited condensed
consolidated statements of operations are not material to any previously presented financial statements based upon overall
considerations of both quantitative and qualitative factors. The corrections had no impact on the unaudited condensed consolidated
balance sheet, unaudited condensed consolidated statements of cash flows, or unaudited condensed consolidated statement of changes
in stockholders’ equity, to these financial statements, or for any previously presented interim or annual financial
statements. Further, the corrections did not result in a change in quarterly or year-to-date operating losses, basic or diluted
earnings per share, or working capital. The quarterly correction required for the three and six months ended June 30, 2023 was
$
| June 30, 2023 | Correction | Corrected June 30, 2023 | ||||||||||
| Research and development expense | $ | $ | ( | ) | $ | |||||||
| General and administrative expense | ||||||||||||
| $ | $ | $ | ||||||||||
Use of Estimates
The preparation of unaudited condensed consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the unaudited condensed consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The Company regularly evaluates estimates and assumptions. The Company bases its estimates and assumptions on current facts, historical experience, and various other factors that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the accrual of costs and expenses that are not readily apparent from other sources. The actual results experienced by the Company may differ materially and adversely from the Company’s estimates. To the extent there are material differences between the estimates and the actual results, future results of operations will be affected. Significant estimates are contained in the accompanying unaudited condensed consolidated financial statements for the recognition of research and development expenses, valuation of warrants and valuation of bifurcated derivative liabilities and other financial instruments.
Cash and Cash Equivalents
Cash and cash equivalents include cash in bank accounts and money market funds with maturities of less than three months from inception, which are readily convertible to known amounts of cash and which, in the opinion of management, are subject to an insignificant risk of loss in value. As of June 30, 2024 and December 31, 2023, cash and cash equivalents consisted of the following:
| June 30, | December 31, | |||||||
| 2024 | 2023 | |||||||
| Cash | $ | $ | ||||||
| Money market funds | ||||||||
| $ | $ | |||||||
| F-8 |
Periodically,
the Company may carry cash balances at financial institutions in excess of the federally insured limit of $
Marketable Securities
Our investments in debt securities are carried at fair value. Investments in debt securities that are not classified as held-to-maturity are carried at fair value and classified as either trading or available-for-sale. Realized and unrealized gains and losses on trading of debt securities are charged to income.
The
marketable securities held by the Company, which are classified as trading marketable securities, consisted of an outstanding
balance of $
Fair Value of Financial Instruments
The Company follows accounting guidelines on fair value measurements for financial instruments measured on a recurring basis, as well as for certain assets and liabilities that are initially recorded at their estimated fair values. Fair Value is defined as the exit price, or the amount that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The Company uses the following three-level hierarchy that maximizes the use of observable inputs and minimizes the use of unobservable inputs to value its financial instruments:
| ● | Level 1: Observable inputs such as unadjusted quoted prices in active markets for identical instruments. | |
| ● | Level 2: Quoted prices for similar instruments that are directly or indirectly observable in the marketplace. | |
| ● | Level 3: Significant unobservable inputs which are supported by little or no market activity and that are financial instruments whose values are determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires a significant judgment or estimation. |
Financial instruments measured at fair value are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires the Company to make judgments and consider factors specific to the asset or liability. The use of different assumptions and/or estimation methodologies may have a material effect on estimated fair values. Accordingly, the fair value estimates disclosed, or initial amounts recorded, may not be indicative of the amount that the Company or holders of the instruments could realize in a current market exchange.
The carrying amounts of the Company’s financial instruments including cash and cash equivalents, prepaid expenses, accounts payable and accrued liabilities approximate fair value due to the short-term maturities of these instruments.
| F-9 |
Set out below are the Company’s financial instruments that are required to be remeasured at fair value on a recurring basis and their fair value hierarchy as of June 30, 2024 and December 31, 2023:
| June 30, 2024 | Level 1 | Level 2 | Level 3 | Carrying Value | ||||||||||||
| Assets | ||||||||||||||||
| Marketable Securities: | ||||||||||||||||
| United States Treasury Bonds | $ | $ | $ | $ | ||||||||||||
| Total Assets | $ | $ | $ | $ | ||||||||||||
| Liabilities | ||||||||||||||||
| Derivative Liability - Warrants | $ | $ | $ | $ | ||||||||||||
| Derivative Liability - Accelerated feature | ||||||||||||||||
| Total Liabilities | $ | $ | $ | $ | ||||||||||||
| December 31, 2023 | Level 1 | Level 2 | Level 3 | Carrying Value | ||||||||||||
| Assets | ||||||||||||||||
| Marketable Securities: | ||||||||||||||||
| United States Treasury Bonds | $ | $ | $ | $ | ||||||||||||
| Total Assets | $ | $ | $ | $ | ||||||||||||
| Liabilities | ||||||||||||||||
| Derivative Liability - Warrants | $ | $ | $ | $ | ||||||||||||
| Derivative Liability - Accelerated feature | ||||||||||||||||
| Total Liabilities | $ | $ | $ | $ | ||||||||||||
Derivative Financial Instruments
The Company does not use derivative instruments to hedge exposures to cash flow, market or foreign currency risks. We evaluate all of our financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the unaudited condensed consolidated statements of operations. For our liability classified derivative financial instruments, the Company used a Monte Carlo valuation model to value the derivative instruments at inception and on subsequent valuation dates. For our equity classified derivative financial instruments, we used a Black-Scholes option-pricing model at the grant date to value the derivative instrument. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is evaluated at the end of each reporting period. Derivative liabilities are classified in the unaudited condensed consolidated balance sheets as current or non-current based on whether or not net-cash settlement or conversion of the instrument could be required within twelve (12) months of the balance sheet date.
Warrants
The Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance in FASB ASC 480, Distinguishing Liabilities from Equity (“ASC 480”) and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own ordinary shares and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of warrant issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
For issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all the criteria for equity classification, the warrants are required to be recorded at their initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the estimated fair value of the warrants are recognized as a non-cash gain or loss on the unaudited condensed consolidated statements of operations. The fair value of the warrants is estimated using a Black-Scholes pricing model or a Monte Carlo simulation.
| F-10 |
Research and Development Expenses
Research and development expenses are charged to expense as incurred. Research and development expenses include, but are not limited to, product development, clinical and regulatory expenses, payroll and other personnel expenses, which include a certain portion of our chief executive officer, chief operating officer, chief financial officer and directors’ compensation. For the three and six months ended June 30, 2024 and 2023, a portion of personnel-related expenses and stock-based compensation expense for these individuals totaling $ million and $ million, respectively, and $ million and $ million, respectively, was included within research and development due to their active involvement in the research and development activities, materials, supplies, related subcontract expenses, and consulting costs.
Regarding the accounting treatment for reimbursements, GAAP provides limited guidance on the accounting for government grants received by for-profit companies. In accordance with ASC Topic 832, Government Assistance, as adopted January 1, 2022, we disclose certain types of government assistance received in the notes to the consolidated financial statements that includes: a) the nature of the transaction including the nature of the assistance being given, b) the accounting policies being used to account for the transaction and c) other provisions of relevance, where required. Depending on the type of grant or contract, we understand there is more than one acceptable alternative for the accounting treatment – a reduction of costs, a deferred credit to be amortized, revenue or other income. The Company has concluded that reimbursements received for R&D expenses incurred, are more akin to a reduction of costs and applies reimbursements against incurred research costs. There were no reimbursements received for the six months ended June 30, 2024 and 2023.
Net loss per share of common stock requires presentation of basic earnings per share on the face of the unaudited condensed consolidated statements of operations for all entities with complex capital structures and requires a reconciliation of the numerator and denominator of the basic earnings per share computation. In the accompanying unaudited condensed consolidated financial statements, basic loss per share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the year. Diluted earnings per share is computed by dividing net income by the weighted average number of shares of common stock and potentially dilutive outstanding shares of common stock during the period to reflect the potential dilution that could occur from common shares issuable through contingent share arrangements, stock options and warrants unless the result would be antidilutive.
The dilutive effect of restricted stock units subject to vesting and other stock-based payment awards is calculated using the “treasury stock method,” which assumes that the “proceeds” from the exercise of these instruments are used to purchase common shares at the average market price for the period. The dilutive effect of convertible securities is calculated using the “if-converted method.” Under the if-converted method, securities are assumed to be converted at the beginning of the period, and the resulting shares of common stock are included in the denominator of the diluted calculation for the entire period being presented.
| June 30, | June 30, | |||||||
| 2024 | 2023 | |||||||
| Convertible notes (Note 5) | ||||||||
| Warrant (Note 6) | ||||||||
| Restricted stock units (Note 6) | ||||||||
Deferred Offering Costs
Pursuant to ASC 340-10-S99-1, costs directly attributable to an offering of equity securities are deferred and would be charged against the gross proceeds of an offering as a reduction of additional paid-in capital. Deferred offering costs may consist of underwriting, legal, accounting, and other expenses incurred through the balance sheet date that are directly related to a proposed public offering. Should the proposed public offering prove to be unsuccessful, any deferred costs, as well as additional expenses to be incurred, will be expensed.
| F-11 |
Recent Accounting Pronouncements
In November 2023, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2023-07, Segment Reporting Topic 280, “Segment Reporting-Improvements to Reportable Segment Disclosures,” which allows disclosure of one or more measures of segment profit or loss used by the chief operating decision maker to allocate resources and assess performance. Additionally, the standard requires enhanced disclosures of significant segment expenses and other segment items, as well as incremental qualitative disclosures on both an annual and interim basis. This guidance is effective for annual reporting periods beginning after December 15, 2023, and interim reporting periods after December 15, 2024. Early adoption is permitted and retrospective application is required for all periods presented. The adoption of this new guidance is not expected to have a significant impact on our consolidated financial statements.
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes Topic 740, “Income Tax-Improvements to Income Tax Disclosures,” which requires enhanced disclosures, including specific categories and disaggregation of information in the effective tax rate reconciliation, disaggregated information related to income taxes paid, income or loss from continuing operations before income tax expense or benefit, and income tax expense or benefit from continuing operations. This guidance is effective for annual reporting periods beginning after December 15, 2024. Early adoption is permitted and should be applied on a prospective basis, however retrospective application is permitted. The Company is currently evaluating the impact of adopting this guidance on its consolidated and condensed financial statements and disclosures included within notes to consolidated and condensed financial statements.
Note 3 – Leases
Operating lease right-of-use (“ROU”) assets and liabilities are recognized at the present value of the future lease payments as of the lease commencement date. Operating lease expense is recognized on a straight-line basis over the lease term.
During
the six months ended June 30, 2023, the Company had a lease agreement which allowed for the use of a laboratory facility for a monthly
payment of $
The
Company currently has a lease agreement which allows for the use of a laboratory facility, entered into on February 16, 2023, with base
rent of $
The following summarizes the ROU lease expense components and cash flow information for the Company’s operating leases:
| Three Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2024 | 2023 | 2024 | 2023 | |||||||||||||
| Operating lease cost | $ | $ | $ | $ | ||||||||||||
| Variable lease cost | ||||||||||||||||
| Sublease income | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Total lease cost | $ | $ | $ | $ | ||||||||||||
| Cash paid for operating cash flows from operating leases | $ | $ | $ | $ | ||||||||||||
| Right-of-use assets obtained in exchange for new operating lease liability | $ | $ | $ | $ | ||||||||||||
Supplemental balance sheet information related to operating leases was as follows:
| June 30, | December 31, | |||||||
| 2024 | 2023 | |||||||
Weighted-average remaining lease term (year) | ||||||||
| Weighted-average discount rate | % | % | ||||||
| F-12 |
Future non-cancelable minimum lease payments under the operating lease liability as of June 30, 2024, are as follows:
| Years Ended December 31, | ||||
| 2024 (excluding the six months ended June 30, 2024) | $ | |||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 2028 | ||||
| Total future minimum lease payments | ||||
| Less: imputed interest | ( | ) | ||
| Present value of payments | $ | |||
Note 4 – Notes Payable-Related Party
During
the three and six months ended June 30, 2023 the Company incurred $
Note 5 – Convertible Notes and Notes Payable
Alto Opportunity Master Fund, SPC
On
January 11, 2023, the Company entered into a securities purchase agreement (the “SPA”) with Alto Opportunity Master
Fund, SPC – Segregated Master Portfolio B, a Cayman entity (the “Investor”), pursuant to which the Company sold to
the Investor a $
| F-13 |
In
conjunction with entry into the SPA, the Company entered into a series of related agreements, including a security agreement (the “Security
Agreement”), an intellectual property security agreement (the “IP Security Agreement”) and a subsidiary guaranty (the
“Subsidiary Guaranty”). The security agreements and guaranty allow, among other things, for the Investor to have a security
interest in and place a lien on all of the Company’s assets and intellectual property until such time as the Alto Convertible Note
is paid off. In addition, the SPA called for the Company to enter into a springing deposit account control agreement (the “Springing
DACA”), which, in the event the Company defaults on its repayment of the Alto Convertible Note, would allow the Investor to assume
control of the Company’s bank account only with regard to any funds remaining outstanding under the Alto Convertible Note. As such,
in conjunction with entry into the SPA, the Company established a separate bank account in which it deposited the Investment Amount and
pursuant to which the Company, the Investor and the bank holding the Investment Amount, First Republic Bank, entered into the Springing
DACA agreement. As the Investment Amount had been held at First Republic Bank, in light of certain banking crises then affecting smaller
banks, on March 12, 2023, the Company and the Investor moved the Investment Amount from First Republic Bank, after which time the Springing
DACA was no longer in effect. Further, pursuant to amendments to the SPA entered into in May and June of 2023, the Company and the Investor
agreed that all of the Investment Amount would be released to the Company and the relevant provision of the SPA which required the Springing
DACA would no longer be deemed applicable. In addition, the Company granted the Investor the option to purchase up to an additional $
Boustead
Securities, LLC (“Boustead”) served as a placement agent for the Alto Convertible Note and Warrant offering and received
$
The
Company allocated the finance costs related to the Boustead placement agent fee of $
The
Company allocated to the debt component of the note an original discount of $
During
the three and six months ended June 30, 2024, the Company recorded interest expense of $
During
the three and six months ended June 30, 2023, the Company recorded interest expense of $
As
of June 30, 2024, the outstanding principal for the convertible note was $
| F-14 |
Note 6 – Stockholders’ Equity
Common Stock
During the three and six months ended June 30, 2024, the Company issued:
| ● |
and shares of common stock to settle $ | |
| ● | and shares of common stock issued for vesting of restricted stock units, respectively. |
During the three and six months ended June 30, 2023, the Company issued:
| ● |
and shares of common stock to settle $ |
Warrants
In
connection with the January 2023 Alto Convertible Note, Boustead was granted warrants to purchase
A summary of activity regarding all warrants issued for the six months ended June 30, 2024 were as follows:
| Number of | Weighted Average | Average | ||||||||||
| Warrants Shares | Exercise Price | Life (years) | ||||||||||
| Outstanding, December 31, 2023 | $ | |||||||||||
| Granted | - | |||||||||||
| Outstanding, June 30, 2024 | $ | |||||||||||
The intrinsic value of the warrants as of June 30, 2024 is $. All of the outstanding warrants are exercisable as of June 30, 2024.
Equity Incentive Plan
Our 2018 Equity Incentive Plan (the “2018 Plan”) provides for equity incentives to be granted to our employees, executive officers, directors and key advisers and consultants. Equity incentive grants may be made in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuant to the 2018 Equity Incentive Plan, restricted stock awards, other stock-based awards, or any combination of the foregoing. The 2018 Equity Incentive Plan is administered by the Company’s compensation committee. We have reserved shares of our common stock for issuance under the 2018 Equity Incentive Plan. As of June 30, 2024, shares have been granted under the 2018 Equity Incentive Plan, of which shares have vested.
Restricted Stock Units
We may grant restricted stock units (“RSU”) under our 2018 Plan. RSUs are bookkeeping entries representing an amount equal to the fair market value of one share of our common stock. Subject to the provisions of our 2018 Plan, the administrator determines the terms and conditions of RSUs, including the vesting criteria and the form and timing of payment. Notwithstanding the foregoing, the administrator, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed. RSUs granted typically vest annually in one third increments from the date of appointment.
| F-15 |
During
the three and six months ended June 30, 2024 and 2023, pursuant to agreements with officers and consultants, and ,
respectively and and
RSUs, respectively with a value of $
As of June 30, 2024, there was $ of unrecognized RSU compensation cost related to non-vested stock-based compensation arrangements which is expected to be recognized over a weighted-average period of years.
| Number of RSUs | Weighted Average Fair Value Per RSU | |||||||
| Outstanding, December 31, 2023 | $ | |||||||
| Granted | ||||||||
| Vested | ( | ) | ||||||
| Outstanding, June 30, 2024 | $ | |||||||
Rights Offering and Financing Commitment
On
February 7, 2024, the Company and its wholly-owned subsidiary, Shuttle Diagnostics, Inc., entered into a securities purchase agreement
(the “Purchase Agreement”) with SRO, LLC, a Nevada limited liability company, pursuant to which SRO LLC agreed to commit
to purchasing from the Company $
The
Company filed an initial registration statement on Form S-1 (the “Form S-1”) with the SEC in April 2024 related to the
registration of subscription rights to purchase the Units to be sold in the Rights Offering. The Form S-1 has not been declared
effective as of the date these unaudited condensed consolidated financial statements were issued. Upon the Form S-1 being declared
effective, the Purchase Agreement allows SRO LLC up to 60 days to raise the initial $
In
conjunction with its entry into the Purchase Agreement, on February 7, 2024, the Company entered into a placement agent and advisory
services agreement (the “Placement Agent Agreement”) with BSL, pursuant to which BSL and BSL’s affiliates will provide
the Company with regular and customary financial consulting advice and will act as placement agent, on a best efforts basis, for the
Rights Offering. In exchange for its services, BSL will receive a commitment fee equal to $
| F-16 |
As
of June 30, 2024, the Company has incurred $
Note 7 – Derivative Liabilities
Fair Value Assumptions Used in Accounting for Derivative Liabilities
ASC 815 requires us to assess the fair market value of derivative liabilities at the end of each reporting period and recognize any change in the fair market value as other income or expense.
In
January 2023, in connection with the Alto Convertible Note, the Company issued warrants to purchase
The
Company has determined the Acceleration Option is an embedded derivative within the host instrument and has bifurcated it from the host
instrument and recorded it as a derivative liability valued at $
The Company
classified these derivative liabilities as a Level 3 fair value measurement and used the Monte Carlo pricing model to calculate the fair
value as of January 11, 2023 ($
The key inputs for the Monte Carlo simulation as of June 30, 2024, were as follows:
| Net cash settlement and down round key valuation inputs – warrants* | ||||
| Annualized volatility | % | |||
| Risk-free interest rate | % | |||
| Quoted VWAP | $ | |||
| Exercise price | $ | |||
| Probability assessment | % | |||
| Illiquidity discount | - | % | ||
| Time period (years) | ||||
| * | Based on a Monte Carlo simulation analysis of 250,000 iterations |
| Acceleration option key valuation inputs* | ||||
| Annualized volatility | % | |||
| Risk-free interest rate | % | |||
| Quoted VWAP | $ | |||
| Illiquidity discount | - | % | ||
| Time period (years) | ||||
| * |
| F-17 |
The following table summarizes the changes in the derivative liabilities:
| Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | ||||||||
| Warrants | Accelerated Feature | |||||||
| Balance - December 31, 2023 | $ | $ | ||||||
| Gain on change in fair value | ( | ) | ( | ) | ||||
| Balance - March 31, 2024 | $ | $ | ||||||
| (Gain) loss on change in fair value | ( | ) | ||||||
| Balance - June 30, 2024 | $ | $ | ||||||
Note 8 – Related Party transactions
On
September 14, 2022, we entered into a manufacturing agreement with TCG GreenChem, Inc. (“TCG GreenChem”), the U.S. subsidiary
of TCG Lifesciences Pvt Ltd., a global contract research and manufacturing services company located in India. Dr. Chis Senanayake, one
of our independent directors, is CEO and CSO of TCG GreenChem and CSO of TCG Lifesciences Pvt Ltd. TCG GreenChem was contracted for process
research, development and cGMP compliant manufacture of IPdR. During the six months ended June 30, 2024 and 2023, the Company expensed
$
Note 9 – Subsequent Events
See Note 2, Summary of Significant Accounting Policies, Reverse Stock Split, above.
Alto Opportunity Master Fund, SPC
On
July 12, 2024, the Company informed the Investor in the Alto Convertible Note that the Company’s expected restatement of its consolidated
financial statements for the years ended December 31, 2022 and 2023 constituted an event of default under the terms of the Alto Convertible
Note. On August 6, 2024, the Company paid $
Alliance Global Partners
On
July 30, 2024, the Company engaged A.G.P./Alliance Global Partners (“AGP”) to serve as exclusive underwriter, placement
agent or advisor in any public or private offering or financing (as defined, the “Offering”) of up to $
Phase II study of Ropidoxuridine
On August 8, 2024, the Company entered into a work order (the “Work Order”) with Theradex Systems, Inc., a New Jersey contract research organization (“CRO”) for purposes of supporting the Company’s Phase II Study of Ropidoxuridine as a Radiation Sensitizing Agent During Radiotherapy in Patients with Newly Diagnosed IDH-Wildtype Glioblastoma with Unmethylated MGMT Promotor.” As such, Shuttle Pharma is now in the process of signing up six clinical sites where the clinical studies will be performed – two of which have completed initial site initiation visits and one of which is ready to enroll patients – and where the CRO will oversee such studies.
Under
the terms of the Work Order, the CRO will oversee the studies for a period of 53 months (the “Term”), including overseeing
and monitoring the regulatory aspects of the Phase II clinical trial, and managing the documentation surrounding the clinical trial in
exchange for a fee of approximately $
| F-18 |
Index to Financial Statements
| F-19 |
Report of Independent Registered Public Accounting Firm
To the Shareholders, Board of Directors, and Audit Committee of
Shuttle Pharmaceuticals Holdings, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Shuttle Pharmaceuticals Holdings, Inc. and subsidiaries (the “Company”) as of December 31, 2023 and 2022, the related consolidated statements of operations, changes in stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2023, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”).
Restatement of Previously Issued Financial Statements
As discussed in Note 2 to the financial statements, the 2023 and 2022 financial statements have been restated to correct misstatements.
Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses and negative cash flows from operations since inception. These conditions, along with other matters as set forth in Note 1, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits.
We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures include examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Forvis Mazars, LLP
We have served as the Company’s auditor since 2023.
Atlanta, Georgia
September 3, 2024
| F-20 |
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Balance Sheets
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| (as restated) | (as restated) | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Prepaid expenses | ||||||||
| Marketable securities | ||||||||
| Accrued interest income | ||||||||
| Total current assets | ||||||||
| Property and equipment, net of accumulated depreciation of $ | ||||||||
| Other assets | ||||||||
| Operating lease right-of-use asset | ||||||||
| Total Assets | ||||||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current Liabilities | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Accounts payable and accrued expenses related party | ||||||||
| Accrued interest payable | ||||||||
| Accrued interest payable - related parties | ||||||||
| Notes payable to related parties | ||||||||
| Convertible notes payable, net | ||||||||
| Operating lease liability | ||||||||
| Total Current Liabilities | ||||||||
| Convertible notes payable non-current, net | ||||||||
| Derivative liabilities | ||||||||
| Operating lease liability non-current | ||||||||
| Total Liabilities | ||||||||
| Stockholders’ Equity | ||||||||
| Series A Convertible Preferred Stock, $ par value; $ | ||||||||
| Common stock, $ par value; shares authorized; and shares issued and outstanding, respectively | ||||||||
| Additional paid in capital, as restated | ||||||||
| Accumulated deficit, as restated | ( | ) | ( | ) | ||||
| Total Stockholders’ Equity | ||||||||
| Total Liabilities and Stockholders’ Equity | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-21 |
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Operations
| Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (as restated) | ||||||||
| Revenue | $ | |||||||
| Operating expenses | ||||||||
| Research and development, net of contract expense reimbursements, as restated | ||||||||
| General and administrative, as restated | ||||||||
| Legal and professional, as restated | ||||||||
| Total operating expenses, as restated | ||||||||
| Net loss from operations | ( | ) | ( | ) | ||||
| Other income (expense) | ||||||||
| Interest expense - related parties | ( | ) | ( | ) | ||||
| Interest expense, as restated | ( | ) | ( | ) | ||||
| Interest income | ||||||||
| Finance fee, as restated | ( | ) | ( | ) | ||||
| Change in fair value of derivative liabilities, as restated | ( | ) | ||||||
| Change in fair value of warrant liabilities, as restated | ( | ) | ||||||
| Gain on sale of marketable securities | ||||||||
| Change in fair value of marketable securities | ||||||||
| Loss on settlement of convertible debt, as restated | ( | ) | ( | ) | ||||
| Gain on settlement of accounts payable | ||||||||
| Gain on forgiveness of Paycheck Protection Program note payable | ||||||||
| Total other expense, as restated | ( | ) | ( | ) | ||||
| Net loss, as restated | ( | ) | ( | ) | ||||
| Dividend on Series A Convertible Preferred Stock, as restated | ( | ) | ||||||
| Deemed dividend on extinguishment of Series A Convertible Preferred Stock, as restated | ( | ) | ||||||
| Deemed dividend on modification of equity-classified warrants, as restated | ( | ) | ||||||
| Deemed contribution on conversion of preferred stock and settlement of preferred stock dividends, as restated | ||||||||
| Net loss attributable to common stockholders | ( | ) | ( | ) | ||||
| Weighted average common shares outstanding - basic and diluted | ||||||||
| Net loss per share - basic and diluted | ) | ) | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-22 |
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Changes in Stockholders’ Equity
For the Years Ended December 31, 2023 (as restated) and 2022 (as restated)
| Series A Preferred Stock | Common Stock | Paid-In | Common Stock to be | Accumulated | Total Stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Issued | Deficit | Equity | |||||||||||||||||||||||||
| Balance at December 31, 2021 (as restated) | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||
| Common stock issued for cash upon initial public offering and exercise of warrants, net of issuance costs, as restated | — | |||||||||||||||||||||||||||||||
| Warrants issued for financing costs, as restated | — | — | ||||||||||||||||||||||||||||||
| Common stock issued for conversion of accrued interest, as restated | — | ( | ) | |||||||||||||||||||||||||||||
| Common stock issued upon conversion of convertible debt and exercise of related warrants, as restated | — | |||||||||||||||||||||||||||||||
| Common stock issued for exercise of warrants with settlement of notes payable, as restated | — | |||||||||||||||||||||||||||||||
| Common stock issued upon exercise of warrants, as restated | — | |||||||||||||||||||||||||||||||
| Common stock issued for restricted stock units, as restated | — | |||||||||||||||||||||||||||||||
| Stock-based compensation, as restated | — | — | ||||||||||||||||||||||||||||||
| Dividends on Series A convertible preferred stock, as restated | — | — | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Deemed dividend on extinguishment of Series A Preferred Stock | — | — | ||||||||||||||||||||||||||||||
| Deemed dividend on modification of equity-classified warrants | — | — | ||||||||||||||||||||||||||||||
| Deemed contribution on conversion of preferred stock and settlement of preferred stock dividend | — | — | ||||||||||||||||||||||||||||||
| Common shares issued for dividends on and conversion of Series A convertible preferred stock, as restated | ( | ) | ||||||||||||||||||||||||||||||
| Reclassification of fair value of warrant liabilities to equity, as restated | — | — | ||||||||||||||||||||||||||||||
| Net loss, as restated | — | — | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2022 (as restated) | $ | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||||||||
| Warrants issued for financing
costs, net of issuance fees of $ | — | — | ||||||||||||||||||||||||||||||
| Common stock issued for conversion of accrued interest and principal | — | |||||||||||||||||||||||||||||||
| Common stock issued for restricted stock units | — | |||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | ||||||||||||||||||||||||||||||
| Net loss | — | — | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2023 (as restated) | $ | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-23 |
Shuttle Pharmaceuticals Holdings, Inc.
Consolidated Statements of Cash Flows
| Years Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
(as restated) | ||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | ||||||||
| Change in fair value of derivative liability, as restated | ( | ) | ||||||
| Change in fair value of warrant liabilities, as restated | ||||||||
| Amortization of debt discount and finance fees, as restated | ||||||||
| Gain on marketable securities | ( | ) | ||||||
| Change in fair value of marketable securities | ( | ) | ||||||
| Accrued interest settled with common stock | ||||||||
| Loss on settlement of notes payable and convertible notes payable, as restated | ||||||||
Fair value of freestanding instruments in excess of proceeds, as restated | ||||||||
| Gain on settlement on accounts payable | ( | ) | ||||||
| Gain on forgiveness of Paycheck Protection Program | ( | ) | ||||||
| Stock-based compensation, as restated | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Accrued interest income | ( | ) | ||||||
| Prepaid expenses | ( | ) | ||||||
| Accounts payable and accrued expenses | ( | ) | ||||||
| Accounts payable and accrued expenses - related parties | ( | ) | ||||||
| Accrued interest payable, as restated | ||||||||
| Accrued interest payable - related parties | ( | ) | ||||||
| Other assets | ||||||||
| Change in operating lease asset and liabilities | ( | ) | ||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Investment in marketable securities | ( | ) | ||||||
| Proceeds from disposition of marketable securities | ||||||||
| Purchase of equipment | ( | ) | ||||||
| Net cash used in investing activities | ( | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from issuance of common shares and exercise of warrants, as restated | ||||||||
| Proceeds from note payable-related party | ||||||||
| Repayment of note payable-related party | ( | ) | ( | ) | ||||
| Proceeds from convertible notes payable, as restated | ||||||||
| Proceeds from notes payable and warrants, as restated | ||||||||
| Payment for finance costs related to convertible note payable, as restated | ( | ) | ( | ) | ||||
| Payment of convertible note payable | ( | ) | ||||||
| Net cash provided by financing activities | ||||||||
| Net change in cash and cash equivalents | ( | ) | ||||||
| Cash and cash equivalents, beginning of period | ||||||||
| Cash and cash equivalents, end of period | $ | $ | ||||||
| Cash paid for: | ||||||||
| Interest | $ | $ | ||||||
| Income taxes | $ | $ | ||||||
| Supplemental non-cash financing activities: | ||||||||
| Common stock issued for conversion of accrued interest, as restated | $ | $ | ||||||
| Common stock issued upon conversion of convertible debt and exercise of related warrants, as restated | $ | $ | ||||||
| Common stock issued for exercise of warrants with settlement of notes payable, as restated | $ | $ | ||||||
| Common shares issued for dividends on and conversion of Series A convertible preferred stock, as restated | $ | $ | ||||||
| Warrants issued for financing fees, net of issuance fees of $ | $ | $ | ||||||
| Reclassification of fair value of warrant liabilities to equity, as restated | $ | $ | ||||||
| Initial recognition of right of use asset and liability | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-24 |
Shuttle Pharmaceuticals Holdings, Inc.
Notes to Consolidated Financial Statements
December 31, 2023 and 2022
Note 1 – Organization and Liquidity
Organization and Line of Business
The
Company was formed as Shuttle Pharmaceuticals, LLC, in the State of Maryland on December 18, 2012. On August 12, 2016, the Company filed
articles of conversion with the State of Maryland to convert from an LLC to a C corporation, at which time the Company changed its name
to Shuttle Pharmaceuticals, Inc. (“Shuttle”). In connection with the conversion the Company issued shares of common
stock in exchange for
The Company’s primary purpose is to develop and commercialize unique drugs for the sensitization of cancers and protection of normal tissues, with the goal of improving outcomes for cancer patients receiving radiation therapy. Shuttle has deployed its proprietary technology to develop novel cancer immunotherapies, producing a pipeline of selective HDAC inhibitors for cancer and immunotherapy applications. The Company’s HDAC platform is designed to target candidate molecules with potential roles in therapeutics beyond cancer, including autoimmune, inflammatory, metabolic, neurological and infectious diseases. The Company’s Ropidoxuridine product, which is used with radiation therapy to sensitize cancer cells, was funded by a Small Business Innovation Research (“SBIR”) contract provided by the National Cancer Institute (“NCI”), a unit of the National Institutes of Health (“NIH”). Ropidoxuridine has been further developed though the Company’s collaborations with scientists at the University of Virginia for use in combination with proton therapy to improve patient survival. Historically, and prior to the Company’s initial public offering, the Company has obtained funding to develop products through NIH grants, including a product to predict late effects of radiation with metabolite biomarkers and develop prostate cancer cell lines in health disparities research.
The production and marketing of the Company’s products and its ongoing research and development activities will be and are subject to extensive regulation by numerous governmental authorities in the United States. Prior to marketing in the United States, any products or combination of products developed by the Company must undergo rigorous preclinical (animal) and clinical (human) testing and an extensive regulatory approval process implemented by the Food and Drug Administration (“FDA”) under the Food, Drug and Cosmetic Act. There can be no assurance that the Company will not encounter problems in clinical trials that will cause the Company or the FDA to delay or suspend clinical trials.
The Company’s success will depend in part on its ability to obtain patents and product license rights, maintain trade secrets, and operate without infringing on the proprietary rights of others, both in the United States and in other countries. There can be no assurance that patents issued to or licensed by the Company will not be challenged, invalidated or circumvented, or that the rights granted thereunder will provide proprietary protection or competitive advantages to the Company now or in the future.
Liquidity and Going Concern
Our
consolidated financial statements are prepared on a going concern basis, which contemplates the realization of assets and the
satisfaction of liabilities and commitments in the normal course of business. The Company has incurred losses since inception and
has a net loss of approximately $
In
January 2023, the Company entered into a securities purchase agreement with an institutional investor through which the Company sold
a convertible note with a principal value of $
The
IPO and subsequent capital raise have supported operations, the manufacture of drug product and FDA
approval of the IND for the Phase II clinical trial of Ropidoxuridine and radiation therapy in glioblastoma. The FDA recommended and
the company agreed to an expansion of the clinical trial, necessitating additional capital to complete the trial, as well as fund ongoing operations. As a result, management had initiated
a $
| F-25 |
The accompanying consolidated financial statements do not include any adjustments to reflect the future effects on the recoverability and classification of assets or the amounts and classification of liabilities if the Company is unable to continue as a going concern.
Reverse Stock Split
On August 13, 2024, in order to meet
Nasdaq’s minimum bid price requirement of $
per share (the “Minimum Bid Price Requirement”), the Company effectuated a
Note 2 – Restatement of Previously Issued Financial Statements
As previously disclosed in the Company’s Quarterly Report on Form 10-Q for the period ended March 31, 2024, filed with the Securities and Exchange Commission (“SEC”) on May 13, 2024, following the entry of a cease-and-desist order by the SEC against our former auditor, B.F. Borgers CPA PC (“BF Borgers”), we commenced the re-audit (the “Re-audit”) of our consolidated financial statements for the year ended December 31, 2022, which had been audited by BF Borgers. Since that time and as a result of the Re-audit, and as initially disclosed on Form 8-K dated July 10, 2024, the Company and the Audit Committee have concluded and informed our current auditor, Forvis Mazars, LLP, that our audited consolidated financial statements for the year ended December 31, 2022 (the “2022 Financial Statements”), our audited consolidated financial statements for the year ended December 31, 2023 (the “2023 Financial Statements”), and the quarterly periods included in the Company’s Annual Report for the year ended December 31, 2023, and the quarterly report for the period ended March 31, 2024 require restatement and are not reliable.
The accounting errors impacting our 2022 Financial Statements include:
Stock Compensation
| ● | Application of the incorrect grant date fair values in determining the value of certain equity awards granted during the years ended December 31, 2019 and 2020; | |
| ● | Miscalculation of the pattern of recognition of stock-based compensation over the requisite service periods for certain equity awards granted during the years ended December 31, 2019 and 2020; |
Series A Convertible Preferred Stock
| ● | Utilization of incorrect inputs to the fair value estimates for the warrant obligation related to the Series A Convertible Preferred Stock; incorrectly recording the reclassification of the warrant obligation upon issuance as a gain and not as a reclassification to stockholders’ equity; | |
| ● | Recording of accrued dividends on the Series A Convertible Preferred Stock as a liability prior to the dividends being declared; | |
| ● | Failure to recognize the accounting impact for the amendment to the terms of the Series A Convertible Preferred Stock in April 2022, which the Company subsequently concluded should have been recognized as an extinguishment; | |
| ● | Failure to recognize deemed contribution related to the settlement of the Series A Convertible Preferred Stock and accrued dividends; |
Notes Payable
| ● | Misidentification of certain debt and equity issuance costs and incorrect association with certain of the Company’s financings during the years ended December 31, 2022 and 2021; | |
| ● | Application of the incorrect amortization period and pattern of recognition for debt discounts recorded in relation to the Company’s debt financings during the years ended December 31, 2022 and 2021; | |
| ● | Incorrect recognition of bifurcated derivative liabilities, as well as incorrect initial and subsequent fair value measurements, in relation to the December 2021 Notes, 2022 Convertible Notes and August 2022 Notes; | |
| ● | Misclassification of certain warrants issued with debt transactions during the year ended December 31, 2022; | |
| ● | Failure to recognize certain liability-classified warrants issued in exchange for services provided related to the Company’s financing activities during the year ended December 31, 2022; | |
| ● | Incorrect application of extinguishment or conversion models in accounting for certain debt instruments upon their settlement; and |
Initial Public Offering
| ● | Incorrect identification of deferred issuance costs incurred in relation to the initial public offering; Incorrect identification and classification of issuance costs incurred in relation to the initial public offering. |
In addition to the restatement of the consolidated financial statements, the Company has also restated the following notes to reflect the errors described above:
| ● | Note 2 - Summary of Significant Accounting Policies (now Note 3) | |
| ● | Note 5 – Notes Payable-Related Party | |
| ● | Note 6 – Convertible Notes (now labeled Notes Payable) | |
| ● | Note 7 – Stockholders’ Equity | |
| ● | Note 8 – Derivative Liabilities | |
| ● | Note 10 – Income Taxes |
| F-26 |
The impact of the errors described above also impact the Company’s Annual Report on Form 10-K for the years ended December 31, 2023 and December 31, 2022 within additional paid in capital and accumulated deficit, resulting in the preparation and filing of this Annual Report on Form 10-K/A.
| Consolidated Balance Sheet as of December 31, 2023 | As Reported | Adjustment | As Restated | |||||||||
| Additional paid in capital | ||||||||||||
| Accumulated deficit | ( | ) | ( | ) | ( | ) | ||||||
Additionally, the Company has provided Note 11 – Quarterly Financial Data (unaudited and restated) to present the impact of the above restatements on the unaudited quarterly financial information for the period ended September 30, 2023.
The Company’s prior and updated accounting for the errors described above do not have any effect on the Company’s previously reported cash or cash flows.
The following tables summarize the effect of the restatement on each financial statement line item as of the dates indicated:
| Consolidated Balance Sheet as of December 31, 2022 | As Reported | Adjustment | As Restated | |||||||||
| Additional paid in capital | (1) | |||||||||||
| Accumulated deficit | ( | ) | ( | )(2) | ( | ) | ||||||
| (1) | ||
| (2) |
| Consolidated Statement of Operations for the year ended December 31, 2022 | As Reported | Adjustment | As Restated | |||||||||
| Research and development, net of contract expense reimbursements | $ | $ | ( | ) | $ | |||||||
| General and administrative | ( | ) | ||||||||||
| Legal and professional | ( | ) | ||||||||||
| Net loss from operations | ( | ) | ( | ) | ||||||||
| Interest expenses | ( | ) | ( | ) | ||||||||
| Financing fee | ( | ) | ( | ) | ||||||||
| Change in fair value of derivative liabilities | ( | ) | ( | ) | ||||||||
| Change in fair value of warrant liabilities | ( | ) | ( | ) | ||||||||
| Loss on extinguishment of debt | ( | ) | ( | ) | ||||||||
| Net loss | ( | ) | ( | ) | ( | ) | ||||||
| Dividend on Series A Preferred Stock | ( | ) | ( | ) | ( | ) | ||||||
| Deemed dividend on extinguishment of Series A Preferred Stock | ( | ) | ( | ) | ||||||||
| Deemed dividend on modification of equity-classified warrants, as restated | ( | ) | ( | ) | ||||||||
| Deemed contribution on conversion of preferred stock and settlement of preferred stock dividends, as restated | ||||||||||||
| Net loss attributable to common stockholders | ( | ) | ( | ) | ( | ) | ||||||
| Net loss per shares - basic and diluted | $ | ) | $ | ) | $ | ) | ||||||
| Consolidated Statement of Stockholders’ Equity as of December 31, 2022 | As Reported | Adjustment | As Restated | |||||||||
| Common stock issued for cash upon initial public offering and exercise of warrants, net of issuance costs | $ | $ | $ | |||||||||
| Common stock issued for cash | ( | ) | ||||||||||
| Warrants exercised for cash | ( | ) | ||||||||||
| Warrants issued for financing costs | ( | ) | ||||||||||
| Common stock issued for conversion of accrued interest | ( | ) | ||||||||||
| Common stock issued upon conversion of convertible debt and exercise of related warrants | ||||||||||||
| Common stock issued for exercise of warrants with settlement of notes payable | ||||||||||||
| Stock-based compensation | ( | ) | ||||||||||
| Dividends on Series A preferred stock | ( | ) | ( | ) | ( | ) | ||||||
| Common shares issued for dividends on and conversion of Series A preferred stock | ( | ) | ||||||||||
| Reclassification of fair value of warrant liability to equity upon initial public offering | ||||||||||||
| Net loss | ( | ) | ( | ) | ( | ) | ||||||
| F-27 |
| Consolidated Statement of Cash Flows for the year ended December 31, 2022 | As Reported | Adjustment | As Restated | |||||||||
| Net loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Change in fair value of derivative liabilities | ||||||||||||
| Change in fair value of warrant liabilities | ( | ) | ||||||||||
| Amortization of debt discount and finance fees | ( | ) | ||||||||||
| Loss on settlement of convertible debt | ||||||||||||
| Fair value of freestanding instruments in excess of proceeds | ||||||||||||
| Gain on interest relief on conversion of notes payable | ( | ) | ||||||||||
| Stock-based compensation | ( | ) | ||||||||||
| Accrued interest payable | ( | ) | ||||||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||||||
| Proceeds from issuance of common shares and warrants | ( | ) | ||||||||||
| Proceeds from convertible notes payable | ( | ) | ||||||||||
| Proceeds from notes payable and warrants | ||||||||||||
| Payment for finance costs related to convertible notes payable | ( | ) | ( | ) | ||||||||
| Net cash provided by financing activities | ( | ) | ||||||||||
| Supplemental non-cash financing activities: | ||||||||||||
| Common stock issued upon conversion of convertible debt | $ | $ | $ | |||||||||
| Common stock issued for exercise of warrants with settlement of notes payable | $ | $ | $ | |||||||||
| Common shares issued for dividends on and conversion of Series A preferred stock | $ | $ | ( | ) | $ | |||||||
| Warrants issued for financing fees, net of issuance fees of $ | $ | $ | ( | ) | $ | |||||||
| Reclassification of fair value of warrant liability to equity upon initial public offering | $ | $ | $ | |||||||||
| Consolidated Balance Sheet as of December 31, 2021 | As Reported | Adjustment | As Restated | |||||||||
| Additional paid in capital | (1) | |||||||||||
| Accumulated Deficit | ( | ) | ( | )(2) | ( | ) | ||||||
| (1) | |
| (2) |
Note 3 – Summary of Significant Accounting Policies
Basis of Presentation
These consolidated financial statements and related disclosures have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”). The consolidated financial statements and disclosures have been prepared using the accrual basis of accounting in accordance with U.S. generally accepted accounting principles (“GAAP”).
Basis of Consolidation
The consolidated financial statements have been prepared on a consolidated basis with those of the Company’s wholly-owned subsidiaries, Shuttle Pharmaceuticals, Inc. and Shuttle Diagnostics, Inc. All intercompany transactions and balances have been eliminated.
Reclassifications
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The Company regularly evaluates estimates and assumptions. The Company bases its estimates and assumptions on current facts, historical experience, and various other factors that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the accrual of costs and expenses that are not readily apparent from other sources. The actual results experienced by the Company may differ materially and adversely from the Company’s estimates. To the extent there are material differences between the estimates and the actual results, future results of operations will be affected. Significant estimates are contained in the accompanying consolidated financial statements for the recognition of research and development expenses, valuation of warrants and valuation of bifurcated derivative liabilities and other financial instruments.
| F-28 |
Cash and Cash Equivalents
Cash and cash equivalents include cash in bank accounts and money market funds with maturities of less than three months from inception, which are readily convertible to known amounts of cash and which, in the opinion of management, are subject to an insignificant risk of loss in value. As of December 31, 2023 and 2022, cash and cash equivalents consisted of the following:
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| Cash | $ | $ | ||||||
| Money market funds | ||||||||
| $ | $ | |||||||
Periodically,
the Company may carry cash balances at financial institutions in excess of the federally insured limit of $
Marketable Securities
Our investments in debt securities are carried at fair value. Investments in debt securities that are not classified as held-to-maturity are carried at fair value and classified as either trading or available-for-sale. Realized and unrealized gains and losses on trading of debt securities are charged to income.
As
of December 31, 2023, the marketable securities held by the Company, which are classified as trading marketable securities, consisted
of an outstanding balance of $
Fair Value of Financial Instruments
The Company follows accounting guidelines on fair value measurements for financial instruments measured on a recurring basis, as well as for certain assets and liabilities that are initially recorded at their estimated fair values. Fair value is defined as the exit price, or the amount that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The Company uses the following three-level hierarchy that maximizes the use of observable inputs and minimizes the use of unobservable inputs to value its financial instruments:
| ● | Level 1: Observable inputs such as unadjusted quoted prices in active markets for identical instruments. | |
| ● | Level 2: Quoted prices for similar instruments that are directly or indirectly observable in the marketplace. | |
| ● | Level 3: Significant unobservable inputs which are supported by little or no market activity and that are financial instruments whose values are determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires a significant judgment or estimation. |
Financial instruments measured at fair value are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires the Company to make judgments and consider factors specific to the asset or liability. The use of different assumptions and/or estimation methodologies may have a material effect on estimated fair values. Accordingly, the fair value estimates disclosed, or initial amounts recorded, may not be indicative of the amount that the Company or holders of the instruments could realize in a current market exchange.
The carrying amounts of the Company’s financial instruments including cash and cash equivalents, prepaid expenses, accounts payable and accrued liabilities approximate fair value due to the short-term maturities of these instruments.
Set out below are the Company’s financial instruments that are required to be remeasured at fair value on a recurring basis and their fair value hierarchy as of December 31, 2023:
| December 31, 2023 | Level 1 | Level 2 | Level 3 | Carrying Value | ||||||||||||
| Assets | ||||||||||||||||
| Marketable Securities: | ||||||||||||||||
| United States Treasury Bonds | $ | $ | $ | $ | ||||||||||||
| Total Assets | $ | $ | $ | $ | ||||||||||||
| Liabilities | ||||||||||||||||
| Derivative Liability - Warrants | $ | $ | $ | $ | ||||||||||||
| Derivative Liability - Accelerated feature | ||||||||||||||||
| Total Liabilities | $ | $ | $ | $ | ||||||||||||
| F-29 |
During the year ended December 31, 2022, the Company had non-derivative warrant liabilities that were measured at fair value on a recurring basis. Each of these fair value measurements was considered to be a Level 3 measurement by the Company as they used significant unobservable inputs, such as the Company’s pre-IPO stock price, probability of event occurrence and other management estimates. The key inputs for each of these warrant liabilities were as follows:
| Warrant Liability – Series A Preferred Stock Obligation | December 31, 2021 | Reclassification Date | ||||||
| Estimated fair value of Company’s stock | $ | $ | ||||||
| Exercise price (based on estimated IPO or Listing terms at issuance) | $ | $ | ||||||
| Number of warrants (based on estimated Series A Preferred Stock conversion shares upon IPO or Listing at issuance) | ||||||||
| Annualized volatility | % | % | ||||||
| Risk-free interest rate | % | % | ||||||
| Estimated IPO date | ||||||||
| Estimated Term | ||||||||
| Probability of IPO or Listing Occurring | % | |||||||
| Probability of IPO as “Qualified IPO” | % | |||||||
| Probability of IPO as “Listing” | % | |||||||
| Estimated fair value | $ | $ | ||||||
| Warrant Liability – August 2022 Warrants | Issuance Dates | Reclassification/Settlement Date | ||||||
| Estimated fair value of Company’s stock | $ | $ | ||||||
| Exercise price | $ | $ | ||||||
| Number of warrants | ||||||||
| Remaining term | ||||||||
| Volatility | % | % | ||||||
| Risk-free interest rate | % | % | ||||||
| Estimated fair value | $ | $ | ||||||
| Warrant Liability – Boustead Placement Agent Warrants | Issuance Dates | Reclassification Date | ||||||
| Estimated fair value of Company’s stock | $ | $ | ||||||
| Exercise price (based on estimated Convertible Note conversion terms at issuance) | $ | $ | ||||||
| Number of warrants (based on estimated Convertible Note conversion terms at issuance) | ||||||||
| Remaining term | ||||||||
| Volatility | % | % | ||||||
| Risk-free interest rate | % | |||||||
| Estimated fair value | $ | $ | ||||||
A rollforward of these warrant liabilities for the year ended December 31, 2022 is as follows:
| Warrant Liability – Series A Preferred Stock Obligation | ||||
| Balance, December 31, 2021 (as restated) | ||||
| Issued | ||||
| Exercised (settled) | ||||
| Loss on change in fair value | ||||
| Reclassifications to stockholders’ equity | ( | ) | ||
| Balance, December 31, 2022 | ||||
| Warrant Liability – August 2022 Warrants | ||||
| Balance, December 31, 2021 (as restated) | ||||
| Issued | ||||
| Exercised (settled) | ( | ) | ||
| Loss on change in fair value | ||||
| Reclassifications to stockholders’ equity | ( | ) | ||
| Balance, December 31, 2022 | ||||
| Warrant Liability – Boustead Placement Agent Warrants | ||||
| Balance, December 31, 2021 (as restated) | ||||
| Issued | ||||
| Exercised (settled) | ||||
| Gain on change in fair value | ( | ) | ||
| Reclassification to stockholders’ equity | ( | ) | ||
| Balance, December 31, 2022 | ||||
Refer to Note 7 for additional background on the reclassification of warrant liabilities to equity. See Note 8 for additional disclosures related to the fair value of the Company’s derivative liabilities.
| F-30 |
Leases
We determine if an arrangement is a lease at inception. Operating leases are included in operating lease right-of-use asset (“ROU”), operating lease liability - current, and operating lease liability - noncurrent on the consolidated balance sheets. Finance leases are included in property and equipment, other current liabilities, and other long-term liabilities in our consolidated balance sheets. The Company did not utilize any financing that required recognition of finance leases during the years ended December 31, 2023 and 2022.
ROU assets represent our right to use an underlying asset for the lease term and lease liabilities represent our obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As our leases do not provide an implicit rate, we use our incremental borrowing rate based on the estimated rate of interest for collateralized borrowing, over a similar term of the lease payments at commencement date. The operating lease ROU asset also includes any lease payments made and excludes lease incentives. Our lease terms may include options to extend or terminate the lease when it is reasonably certain that we will exercise that option. Lease expense for lease payments is recognized on a straight-line basis over the lease term.
Leases with a lease term of 12 months or less at inception are not recorded on our consolidated balance sheets and are expensed on a straight-line basis over the lease term in our statement of operations. We have lease agreements with lease and non-lease components, which are generally accounted for separately.
The Company determines the present value of minimum future lease payments for operating leases by estimating a rate of interest that it would have to pay to borrow on a collateralized basis over a similar term, an amount equal to the lease payments and a similar economic environment (the “incremental borrowing rate” or “IBR”). The Company determines the appropriate IBR by identifying a reference rate and making adjustments that take into consideration financing options and certain lease-specific circumstances. For the reference rate, the Company used an equity built up, risk adjusted rate, as the implicit interest rate.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Expenditures for maintenance and repairs are charged to expense as incurred; additions, renewals and betterments are capitalized. When property and equipment are retired or otherwise disposed of, the related cost and accumulated depreciation are removed from the respective accounts, and any gain or loss is included in operations. Depreciation of property and equipment is provided using the straight-line method for substantially all assets with estimated lives as follows:
| Furniture | ||||
| Computers and equipment | ||||
| Research Equipment |
Derivative Financial Instruments
The Company does not use derivative instruments to hedge exposures to cash flow, market or foreign currency risks. We evaluate all of our financial instruments to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the consolidated statements of operations.
For our derivative financial instruments, the Company utilizes the most appropriate valuation model (such as Monte Carlo simulations or probability-weighted expected return models, based on the nature of the terms of the instrument) to value the derivative instruments at inception and on subsequent valuation dates. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is evaluated at the end of each reporting period. Derivative liabilities are classified in the consolidated balance sheet sheets as current or non-current based on whether or not net-cash settlement or conversion of the instrument could be required within twelve (12) months of the balance sheet date.
Convertible Notes
The Company evaluates its convertible notes for embedded features and bifurcates these features (such as conversion options and redemption options) from their host instruments and accounts for them as free standing derivative financial instruments if certain criteria are met. The criteria include circumstances in which (a) the economic characteristics and risks of the embedded derivative instrument are not clearly and closely related to the economic characteristics and risks of the host contract, (b) the hybrid instrument that embodies both the embedded derivative instrument and the host contract is not re-measured at fair value under otherwise applicable generally accepted accounting principles with changes in fair value reported in earnings as they occur and (c) a separate instrument with the same terms as the embedded derivative instrument would be considered a derivative instrument.
| F-31 |
Warrants
The Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance in FASB ASC 480, Distinguishing Liabilities from Equity (“ASC 480”) and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own ordinary shares and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. Finally, the Company determines if the warrants meet the definition of a derivative based on their contractual terms. This assessment, which requires the use of professional judgment, is conducted at the time of warrant issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
For issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all the criteria for equity classification, the warrants are required to be recorded at their initial fair value on the date of issuance, and at each balance sheet date thereafter. Changes in the estimated fair value of the warrants are recognized as a non-cash gain or loss on the consolidated statements of operations. The Company also evaluates if changes in contractual terms or other considerations would result in the reclassification of outstanding warrants from liabilities to stockholders’ equity (or vice versa).
The fair value of the warrants is estimated using a Black-Scholes pricing model or a Monte Carlo simulation.
Impairment of Long-Lived Assets
The Company reviews its long-lived assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of the asset may not be fully recoverable. Recoverability of assets is measured by a comparison of the carrying amount of an asset to the estimated undiscounted cash flows expected to be generated by the asset. If the carrying amount of the asset exceeds its estimated future cash flows, an impairment charge will be recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. There were no impairments of long-lived assets during the periods presented.
Compensation cost for stock awards, which include restricted stock units (“RSUs”), is measured at the fair value on the grant date and recognized as expense, over the related service or performance period. The fair value of stock awards is based on the quoted price of our common stock on the grant date. Compensation expense related to the RSUs is reduced by the fair value of the units that are forfeited by employees that leave the Company prior to vesting as they occur. Compensation cost for RSUs are recognized using the straight-line method over the requisite service period.
Research and Development Expenses
Research and development expenses are charged to expense as incurred. Research and development expenses include, but are not limited to, product development, clinical and regulatory expenses, payroll and other personnel expenses, which include a certain portion of our chief executive officer, chief operating officer, chief financial officer and directors’ compensation. For the year ended December 31, 2023 and 2022, a portion of personnel-related expenses and stock-based compensation expense for these individuals totaling $ million and $ million, respectively, was included within research and development due to their active involvement in the research and development activities, materials, supplies, related subcontract expenses, and consulting costs.
Regarding
the accounting treatment for reimbursements, GAAP provides limited guidance on the accounting for government grants received by for-profit
companies. In accordance with ASC Topic 832, Government Assistance, as adopted January 1, 2022, we disclose certain types of
government assistance received in the notes to the consolidated financial statements that includes: a) the nature of the transaction
including the nature of the assistance being given, b) the accounting policies being used to account for the transaction and c) other
provisions of relevance, where required. Depending on the type of grant or contract, we understand there is more than one acceptable
alternative for the accounting treatment – a reduction of costs, a deferred credit to be amortized, revenue or other income. The
Company has concluded that reimbursements received for R&D expenses incurred, are more akin to a reduction of costs and applies reimbursements
against incurred research costs. For the years ended December 31, 2023 and 2022, we recorded $
Income Taxes
The Company accounts for income taxes in accordance with ASC Topic 740, Income Taxes. ASC 740 requires a company to use the asset and liability method of accounting for income taxes, whereby deferred tax assets are recognized for deductible temporary differences, and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, the Company does not foresee generating taxable income in the near future and utilizing its deferred tax asset, therefore, it is more likely than not that some portion, or all of, the deferred tax assets will not be realized. Deferred tax assets and liabilities are adjusted for the effects of changes in tax laws and rates on the date of enactment.
Under ASC 740, a tax position is recognized as a benefit only if it is “more likely than not” that the tax position would be sustained in a tax examination, with a tax examination being presumed to occur. The amount recognized is the largest amount of tax benefit that is greater than 50% likely of being realized on examination. For tax positions not meeting the “more likely than not” test, no tax benefit is recorded. The Company has no material uncertain tax positions for any of the reporting periods presented.
| F-32 |
Segment Information
Operating segments are defined as components of an enterprise about which separate and discrete information is available for evaluation by the chief operating decision-maker (“CODM”) in deciding how to allocate resources and assess performance. The Company and the Company’s CODM, the Company’s chief executive officer, evaluates the Company’s operations and manages its business as a operating segment. All of the Company’s long-lived assets are held in the United States.
Net loss per share of common stock requires presentation of basic earnings per common share on the face of the statements of operations for all entities with complex capital structures and requires a reconciliation of the numerator and denominator of the basic earnings per share computation. In the accompanying consolidated financial statements, basic loss per common share is computed by dividing net loss attributable to common stockholders by the weighted average number of shares of common stock outstanding during the year. Diluted earnings per share is computed by dividing net income attributable to common stockholders by the weighted average number of shares of common stock outstanding and potentially dilutive shares of common stock during the period to reflect the potential dilution that could occur from common shares issuable through convertible securities, contingent share arrangements, stock options and warrants unless the result would be antidilutive.
The dilutive effect of restricted stock units and other stock-based payment awards subject to vesting and common stock warrants is calculated using the “treasury stock method,” which assumes that the “proceeds” from the exercise of these instruments are used to purchase common shares at the average market price for the period. The dilutive effect of convertible securities is calculated using the “if-converted method.” Under the if-converted method, securities are assumed to be converted at the beginning of the period, and the resulting shares of common stock are included in the denominator of the diluted calculation for the entire period being presented.
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
(as restated) | ||||||||
| Convertible notes (Note 6) | ||||||||
| Warrants (Note 7) | ||||||||
| Restricted stock units (Note 7) | ||||||||
Recent Accounting Pronouncements
In December 2023, the FASB issued ASU 2023-09, “Income Taxes (Topic 740): Improvements to Income Tax Disclosures,” which requires disaggregated information about a reporting entity’s effective tax rate reconciliation as well as information on income taxes paid. The guidance is effective for the Company’s fiscal years beginning after February 1, 2025, with early adoption permitted. The Company does not expect the adoption of this standard to have any material impact on its financial statements.
There have been no other recent accounting pronouncements, changes in accounting pronouncements or recently adopted accounting guidance during the year ended December 31, 2023 that are of significance or potential significance to the Company.
Note 4 - Leases
The
Company had two lease agreements during the year which allowed for the use of laboratory facilities.
On
February 16, 2023, the Company entered into a new lease agreement for new office and laboratory space, with base rent of $
The following summarizes the right-of use asset and lease information for the Company’s operating leases:
| Years Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Operating lease cost | ||||||||
| Variable lease cost | ||||||||
| Sublease income | ( | ) | ( | ) | ||||
| Total lease cost | ||||||||
| Other information | ||||||||
| Operating cash flows from operating leases | ||||||||
| Right-of-use assets obtained in exchange for new operating lease liabilities | ||||||||
| Weighted-average remaining lease term - operating leases (year) | ||||||||
| Weighted-average discount rate - operating leases | % | % | ||||||
| F-33 |
Future non-cancelable minimum lease payments under the operating lease liability as of December 31, 2023, are as follows:
| Years ended December 31, | ||||
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 2028 | ||||
| Total future minimum lease payments | ||||
| Less: imputed interest | ( | ) | ||
| Present value of payments | $ | |||
Note 5 – Notes Payable-Related Party
On
December 1, 2020, the Company consolidated all of the outstanding loans owed to an officer of the Company and to his spouse, resulting
in the following two loans: (i) a single loan from the spouse of an officer of the Company, dated December 1, 2020, with a principal
balance of $
On
June 21, 2021, the Company entered into a loan from the spouse of an officer of the Company in the amount of $
The Company accounted for these loan extensions during the year ended December 31, 2022, as a troubled debt restructuring as the Company was experiencing financial difficulties and the extensions represented a concession by the lenders. As the future undiscounted cash flows of these related party notes exceeded their carrying value, no gain or loss was recognized upon the extensions.
As
of June 30, 2023 principal payments of $
On August 1, 2022, in conjunction with a private placement of notes and
warrants (as detailed in Note 6 below), in exchange for a $
Note 6 - Notes Payable
Alto Opportunity Master Fund, SPC
On
January 11, 2023, the Company entered into a securities purchase agreement (the “SPA”) with Alto Opportunity Master
Fund, SPC – Segregated Master Portfolio B, a Cayman entity (the “Investor”), pursuant to which the Company sold to
the Investor a $
| F-34 |
In
conjunction with entry into the SPA, the Company entered into a series of related agreements, including a security agreement (the
“Security Agreement”), an intellectual property security agreement (the “IP Security Agreement”) and a
subsidiary guaranty (the “Subsidiary Guaranty”). The security agreements and guaranty allow, among other things, for the
Investor to have a security interest in and place a lien on all of the Company’s assets and intellectual property until such
time as the Alto Convertible Note is paid off. In addition, the SPA called for the Company to enter into a springing deposit account
control agreement (the “Springing DACA”), which, in the event the Company defaults on its repayment of the Alto
Convertible Note, would allow the Investor to assume control of the Company’s bank account only with regard to any funds
remaining outstanding under the Alto Convertible Note. As such, in conjunction with entry into the SPA, the Company established a
separate bank account in which it deposited the Investment Amount and pursuant to which the Company, the Investor and the bank
holding the Investment Amount, First Republic Bank, entered into the Springing DACA agreement. As the Investment Amount had been
held at First Republic Bank, in light of certain banking crises then affecting smaller banks, on March 12, 2023, the Company and the
Investor moved the Investment Amount from First Republic Bank, after which time the Springing DACA was no longer in effect. Further,
pursuant to amendments to the SPA entered into in May and June of 2023, the Company and the Investor agreed that all of the
Investment Amount would be released to the Company and the relevant provision of the SPA which required the Springing DACA would no
longer be deemed applicable. In addition, the Company granted the Investor the option to purchase up to an additional $
Boustead
Securities, LLC (“Boustead”) served as a placement agent for the Alto Convertible Note and Warrant offering and received
$
The
Company allocated the finance costs related to the Boustead placement agent fee of $
The
Company allocated to the debt component of the note an original discount of $
During
the year ended December 31, 2023, the Company recorded interest expense of $
As
of December 31, 2023, the outstanding principal for the convertible note was $
| F-35 |
Subsequent to December 31, 2023, the Company and the Investor agreed to certain changes in terms of the Alto Convertible Note as well as the waiver of certain covenant violations. See Subsequent Events for further details.
Paycheck Protection Program Term Notes
On
March 9, 2021, the Company obtained a $
December 2021 Promissory Notes
On
December 28, 2021, the Company issued promissory note units, consisting of an aggregate $
The
Company determined that the warrants issued with the December 2021 Promissory Notes should be classified as equity. The Company
measured the fair value of these warrants using a Black-Scholes model and allocated fair value to these warrants on a relative fair
value basis. The proceeds from the December 2021 Promissory Notes allocated to the warrants at issuance was $
The
discount on the December 2021 Promissory Notes was being amortized using the straight-line method as the initial proceeds received were
fully allocated to the warrants and bifurcated derivative, through the maturity date of the notes. The Company recognized amortization
expense of $
In April 2022, the warrants were modified by the Company
to nullify the effect of the April 2022 reverse stock split, allowing the warrant to remain outstanding and exercisable for
In
September 2022, the warrants were exercised by the lenders in exchange for tendering the outstanding principal balance ($
2022 Convertible Bridge Notes
On
February 8, 2022 and March 11, 2022, the Company sold $
The
Company assessed the embedded features of the 2022 Convertible Notes and determined that the automatic conversion feature with a
| F-36 |
For
services provided in the offering, Boustead Securities LLC also received warrants to purchase
During
the year ended December 31, 2022, the 2022 Convertible Notes were automatically converted into the securities issued by the Company
to the investors in the initial public offering, at a discounted price of $
August 2022 Notes
On
August 1, 2022, the Company issued promissory note units, consisting of an aggregate $
The Company assessed the embedded features of the August 2022 Promissory Notes and determined that the accelerated repayment upon an initial
public offering represented a derivative feature requiring bifurcation. The Company estimated the fair value of the derivative to be $
The
Company determined that the warrants issued with the August 2022 Notes should be classified as liabilities under ASC 815, as the
settlement amount could vary based on the occurrence of the initial public offering. The Company measured the fair value of these
warrants using a Black-Scholes model with inputs that included an estimated fair value of the Company’s common stock of
$
The
fair value of these warrants, the fair value of the bifurcated derivative liabilities and the other debt issuance costs incurred (totaling $
Boustead
Securities LLC acted as placement agent for the August 2022 Notes and received warrants to purchase shares of common stock at an
exercise price of $ per share. The fair value of the warrants was estimated as $
During
the year ended December 31, 2022, the lender under the August 2022 Promissory Notes and one lender under the August 2022 Convertible
Notes exercised warrants to purchase
Note 7 - Stockholders’ Equity
Common Stock
During the year ended December 31, 2023, the Company issued:
| ● |
shares of common stock to settle $ | |
| ● | shares of common stock issued for vesting of restricted stock units. |
| F-37 |
During the year ended December 31, 2022, the Company issued:
| ● | shares of common stock for conversion of $of accrued interest. | |
| ● | ||
| ● |
shares of common stock upon completion of the Company’s initial public offering (including the exercise of the underwriter’s overallotment option),
and the immediate exercise of warrants issued as part of the IPO Units, for net proceeds of $ | |
| ● |
shares of common stock upon the automatic conversion of preferred shares and
shares of common stock to settle cumulative dividends on the preferred shares totaling $ | |
| ● | A total of
shares of common stock issued to the lenders upon conversion of the 2022 Convertible Notes, including
shares of common stock issued upon the conversion of the 2022 Convertible Notes into units with an estimated fair value of
$ | |
| ● | A total of | |
| ● |
Series A Preferred Shares
The
Series A Preferred Stock, in accordance with its terms, is automatically convertible into a number of shares of the Company’s
common stock upon the closing of the sale of shares of common stock to the public in a qualified offering (at a 10% discount to the
initial public offering price, as set forth in the Series A certificate of designation) or upon listing of the Company’s
common stock on a national securities exchange at a fixed price. The Series A Preferred Stock, while outstanding, accrued cumulative dividends at a
rate of
In
April 2022, the Company and holders of the Series A Preferred Stock agreed to modifications of the Series A certificate of designation
which changed the definition of a “Qualified IPO” by lowering the minimum required proceeds to $and removing the minimum per share IPO price,
and changed the conversion price upon the listing of the Company’s common stock from $
During
the year ended December 31, 2022 and upon completion of its initial public offering, the Company converted
shares of Series A Preferred Stock into
shares of common stock, based on a conversion price of
During
the year ended December 31, 2022, the Company accrued $
As of December 31, 2023 and 2022, the Company had no shares of Series A Preferred Stock outstanding.
Warrants
In April 2022, the Company modified the terms of the Series A Preferred
Stock as discussed above. These modifications impacted the potential terms of these contingently issuable warrants (potential warrant
shares and exercise price). The impact of this modification was reflected in the fair value of the warrant obligation recorded during
the interim periods within the year ended December 31, 2022. Upon
completion of our initial public offering, the Series A Preferred Stock was converted, and warrants were issued in relation to the conversion,
with each warrant then exercisable at the equivalent of the per share initial public offering price. In total, the Company issued warrants
to purchase
The
warrant liability related to the Series A Preferred Stock was $
| F-38 |
Warrants Issued to Boustead as Placement Agent or Underwriter
For services provided in relation to the 2022 Convertible
Notes and August 2022 Notes offerings, Boustead received warrants to purchase
Boustead acted as placement agent for the August 2022
Notes and received warrants to purchase shares of common stock at an exercise price of $
Boustead additionally received a warrant to purchase
Alto Opportunity Master Fund, SPC
In
connection with the January 2023 Alto Convertible Note, Boustead was granted warrants to purchase
A summary of activity regarding all warrants issued for the year ended December 31, 2023 and 2022 were as follows:
| Number of | Weighted Average | Weighted Average | ||||||||||
| Warrants | Exercise Price | Life (years) | ||||||||||
| (as restated) | (as restated) | (as restated) | ||||||||||
| Outstanding, December 31, 2021 | $ | |||||||||||
| Granted(1) | ||||||||||||
| Exercised(2) | ( | ) | — | |||||||||
| Outstanding, December 31, 2022 | ||||||||||||
| Granted - Boustead | ||||||||||||
| Granted - Ayrton | ||||||||||||
| Outstanding, December 31, 2023 | ||||||||||||
| (1) | ||
| (2) | Exercised warrants include the exercise of the warrants issued with the December 2021 Notes (warrants); the exercise of the warrants issued with the August 2022 Notes (warrants); the exercise of the warrants issued upon conversion of the Convertible Notes ( warrants); and the exercise of the warrants issued during the IPO, including overallotment option ( warrants), and warrants exercised on a cashless basis ( warrants). |
The intrinsic value of the warrants as of December 31, 2023 is $. All of the outstanding warrants are exercisable as of December 31, 2023.
Equity Incentive Plan
Our 2018 Equity Incentive Plan (the “2018 Plan”) provides for equity incentives to be granted to our employees, executive officers, directors and key advisers and consultants. Equity incentive grants may be made in the form of stock options with an exercise price of not less than the fair market value of the underlying shares as determined pursuant to the 2018 Equity Incentive Plan, restricted stock awards, other stock-based awards, or any combination of the foregoing. The 2018 Equity Incentive Plan is administered by the Company’s compensation committee. We have reserved shares of our common stock for issuance under the 2018 Equity Incentive Plan. As of December 31, 2023, shares have been granted under the 2018 Equity Incentive Plan, of which shares have vested.
Restricted Stock Units
We may grant restricted stock units (“RSU”) under our 2018 Plan. RSUs are bookkeeping entries representing an amount equal to the fair market value of one share of our common stock. Subject to the provisions of our 2018 Plan, the administrator determines the terms and conditions of RSUs, including the vesting criteria and the form and timing of payment. Notwithstanding the foregoing, the administrator, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed. RSUs granted typically vest annually in one third increments from the date of appointment.
During
the years ended December 31, 2023 and 2022, pursuant to agreements with directors, officers and consultants, and RSUs
with a value of $
| Year ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (as restated) | ||||||||
| Recognized in general and administrative expense | $ | $ | ||||||
| Recognized in research and development expense | ||||||||
| Total | $ | $ | ||||||
As of December 31, 2023 and 2022, there was $ and $, respectively, of unrecognized stock-based compensation expense related to unvested RSUs, which is expected to be recognized over a weighted-average period of years and years, respectively.
| F-39 |
| Number of Shares | Weighted Average Grant Date Fair Value Per Share | |||||||
| Outstanding, December 31, 2021 (as restated) | $ | |||||||
| Granted | ||||||||
| Vested (as restated) | ( | ) | ||||||
| Outstanding, December 31, 2022 (as restated) | $ | |||||||
| Granted | ||||||||
Vested (as restated) | ( | ) | ||||||
| Outstanding, December 31, 2023 | $ | |||||||
Note 8 – Derivative Liabilities
Fair Value Assumptions Used in Accounting for Derivative Liabilities
ASC 815 requires us to assess the fair market value of derivative liabilities at the end of each reporting period and recognize any change in the fair market value as other income or expense.
Alto Opportunity Master Fund, SPC
In
January 2023, in connection with the Alto Convertible Note, the Company issued warrants to purchase
The Company determined our derivative liability from the noteholder’s Acceleration Option for the Alto Convertible Note is not clearly and closely related to the host and should be thus accounted for as a bifurcated derivative liability.
We
classified these derivative liabilities as a Level 3 fair value measurement and used the Monte Carlo pricing model to calculate the fair
value as of January 11, 2023, and for each reporting period. Key inputs for the simulation are summarized below. The Monte Carlo simulation
uses an implied VWAP for each valuation date. The implied VWAP was backsolved by setting the summation of the parts (e.g., derivatives
and debt without derivatives) equal to the cash proceeds. The simulation was then iterated and manipulated to solve for the implied share
price, which was approximately $per share (or an approximate
The range of key inputs for the Monte Carlo simulation for the year ended December 31, 2023, were as follows:
| Net cash settlement and down round key valuation inputs – Alto Warrants* | ||||
| Annualized volatility | % | |||
| Risk-free interest rate | % | |||
| Quoted VWAP* | $ | |||
| Exercise price | $ | |||
| Probability assessment | % | |||
| Illiquidity discount | ( | )% | ||
| Time period (years) | ||||
| Estimated fair value (issuance) | $ | |||
| Estimated fair value (December 31, 2023) | $ | |||
| * | Based on a Monte Carlo simulation analysis of 50,000 iterations |
| F-40 |
| Alto Acceleration Option key valuation inputs* | ||||
| Annualized volatility | % | |||
| Risk-free interest rate | % | |||
| Quoted VWAP* | $ | |||
| Illiquidity discount | ( | )% | ||
| Time period (years) | ||||
| Estimated fair value (issuance) | $ | |||
| Estimated fair value (December 31, 2023) | $ |
| * |
December 2021 Notes
In connection with the issuance of the December 2021 Notes, the Company identified an accelerated repayment upon IPO feature that required bifurcation. We classified this derivative liability as a Level 3 fair value measurement and used a probability weighted scenario model to estimate the fair value of the derivative just prior to the settlement of the December 2021 Notes. Key inputs for this valuation model are summarized below.
| December 2021 Notes derivative key valuation inputs | Issuance Date | Settlement Date | ||||||
| Amount due upon acceleration | $ | $ | ||||||
| Carrying value of December 2021 Notes at measurement date | $ | |||||||
| IPO date | ||||||||
| Probability of IPO acceleration | % | % | ||||||
| Probability of maturity | % | % | ||||||
| Time period to maturity (years) | ||||||||
| Discount rate | % | % | ||||||
| Estimated fair value | $ | |||||||
2022 Convertible Bridge Notes
In connection with the issuance of the 2022 Convertible Notes in February and March 2022, the Company identified certain embedded features that required bifurcation as a combined derivative liability, including the share-settled redemption of the 2022 Convertible Notes at a 50% discount and the automatic increase in principal of 10% if conversion does not occur within 12 months.
We classified this derivative liability as a Level 3 fair value measurement and used the a with and without probability weighted scenario model to estimate the fair value as of each issuance date of the 2022 Convertible Notes, as well as just prior to their settlement upon the initial public offering. Key inputs for this valuation model are summarized below.
| 2022 Convertible Notes derivative key valuation inputs* | Issuance Dates | Settlement Date | ||
| Annualized volatility | ||||
| Risk-free interest rate | ||||
| Exercise price (implied) | $ - $ | |||
| Estimated IPO date | ||||
| Probability of default | % | |||
| Probability of IPO or Alternative Liquidity Event | % | % | ||
| Probability of Maturity | % | % | ||
| Time period (years) | ||||
| Estimated fair value | $ |
$ |
| F-41 |
August 2022 Notes
In connection with the issuance of the August 2022 Promissory Note, the Company identified an accelerated repayment upon IPO feature that required bifurcation. We classified this derivative liability as a Level 3 fair value measurement and used a probability weighted scenario model to estimate the fair value of the derivative just prior to the settlement of the August 2022 Promissory Notes. Key inputs for this valuation model are summarized below.
| August 2022 Promissory Notes derivative key valuation inputs | Issuance Dates | Settlement Date | ||
| Amount due upon acceleration (estimated at issuance) | $ | $ | ||
| Carrying value of August 2022 Promissory Notes at measurement date | $ | $ | ||
| IPO date | ||||
| Probability of IPO acceleration | ||||
| Probability of maturity | % | |||
| Time period to maturity (years) | ||||
| Discount rate | ||||
| Estimated fair value | $ |
$ |
In connection with the issuance of the August 2022 Convertible Notes, the Company identified certain embedded features that required bifurcation as a combined derivative liability, including the share-settled redemption of the 2022 Convertible Notes at a 50% discount and the automatic increase in principal of 10% if conversion does not occur within 12 months. Key inputs for this valuation model are summarized below:
| 2022 Convertible Notes derivative key valuation inputs* | Issuance Dates | Settlement Date | ||||||
| Estimated IPO date | ||||||||
| Probability of default | % | % | ||||||
| Probability of IPO or Alternative Liquidity Event | % | % | ||||||
| Probability of Maturity | % | % | ||||||
| Time period (years) | ||||||||
| Estimated fair value | $ | $ | ||||||
The following table summarizes the changes in the derivative liabilities:
| Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | ||||||||
| Alto Warrants | Alto Acceleration Option | |||||||
| Balance - December 31, 2021 | $ | $ | ||||||
| Addition of new derivative | ||||||||
| Loss on change in fair value | ||||||||
| Settlements | ||||||||
| Balance - December 31, 2022 | ||||||||
| Addition of new derivatives | ||||||||
| Gain on change in fair value | ( | ) | ( | ) | ||||
| Balance - December 31, 2023 | $ | $ | ||||||
| Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | ||||||||||||
| December 2021 Notes Derivative | August 2022 Notes Derivative | 2022 Convertible Notes Derivative | ||||||||||
| Balance - December 31, 2021 | $ | |||||||||||
| Addition of new derivative | ||||||||||||
| Loss (gain) on change in fair value | ( | ) | ||||||||||
| Settlements | ( | ) | ( | ) | ( | ) | ||||||
| Balance - December 31, 2022 | ||||||||||||
| Addition of new derivatives | ||||||||||||
| Gain on change in fair value | ||||||||||||
| Balance - December 31, 2023 | $ | $ | ||||||||||
Note 9 – Related Party Transactions
On
September 14, 2022, we entered into a manufacturing agreement with TCG GreenChem, Inc. (“TCG GreenChem”), the U.S. subsidiary
of TCG Lifesciences Pvt Ltd., a global contract research and manufacturing services company located in India. Dr. Chis Senanayake, one
of our independent directors, is CEO and CSO of TCG GreenChem and CSO of TCG Lifesciences Pvt Ltd. TCG GreenChem was contracted for process
research, development and cGMP compliant manufacture of IPdR. The Company paid TCG GreenChem $
| F-42 |
Note 10 – Income Taxes
The Company has no income tax expense or benefit for the years ended December 31, 2023 and 2022, since the Company has a full valuation allowance for the net operating loss carryforwards in these periods.
We recognize tax benefits from uncertain tax positions only if it is more likely than not that the tax position will be sustained upon examination by the taxing authorities based on the technical merits of the position. The tax benefits recognized from such positions are estimated based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. There are no uncertain tax positions to be reported for the tax years 2023 and 2022.
The
Tax Cuts and Jobs Act of 2017 (“TCJA”) amended IRC Section 174 to require capitalization of all research and development
(“R&D”) costs incurred in tax years beginning after December 31, 2021. These costs are required to be amortized over
five years if the R&D activities are performed in the U.S., or over 15 years if the activities were performed outside the U.S. For
tax reporting purposes, the Company capitalized $
The
reconciliation of income tax benefit at the U.S. statutory rate of
| December 31, | December 31 | |||||||
| 2023 | 2022 (as restated) | |||||||
| Federal tax benefit at statutory rate | $ | ( | ) | $ | ( | ) | ||
| State income taxes, net of federal tax effect | ( | ) | ( | ) | ||||
| Rate change | ( | ) | ( | ) | ||||
| R & D tax credits | ( | ) | ( | ) | ||||
| Return to provision adjustments | ||||||||
| Permanent differences | ||||||||
| Derivative Debt Discount Amortization | ||||||||
| Change in FMV of Warrant Liability | ( | ) | ||||||
| Loss on convertible note conversion | ||||||||
| Other | ||||||||
| Change in valuation allowance | ||||||||
| Shortfall of stock compensation expense | ||||||||
| Other adjustments | ||||||||
| Income tax expense | $ | $ | ||||||
The principal components of deferred tax assets consist of the following:
| December 31, | December 31, | |||||||
| 2023 | 2022 (as restated) | |||||||
| Deferred tax asset: | ||||||||
| Net operating loss carryforwards | $ | $ | ||||||
| Fixed assets | ||||||||
| Intangibles (includes Section 174 capitalization) | ||||||||
| R&D tax credits | ||||||||
| Equity based compensation | ||||||||
| Interest & other accrued expenses | ||||||||
| Lease asset/(liability) | ||||||||
| Total | $ | $ | ||||||
| December 31, | December 31, | |||||||
| Deferred tax liabilities: | 2023 | 2022
(as restated) | ||||||
| Change in fair market value of securities | $ | ( | ) | $ | ||||
| Prepaid expenses | ( | ) | ( | ) | ||||
| State income tax deferred | ( | ) | ||||||
| Fixed assets | ( | ) | ||||||
| Total | $ | ( | ) | $ | ( | ) | ||
| Total deferred tax asset | $ | $ | ||||||
| Less: valuation allowance | ( | ) | ( | ) | ||||
| Net deferred tax asset | $ | $ | ||||||
| F-43 |
A reconciliation of the U.S. federal income tax rate to the Company’s effective tax rate is as follows:
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| Federal income tax benefit at statutory rate | % | % | ||||||
| State income tax benefit, net of federal tax effect | % | % | ||||||
| Change in tax rate | % | % | ||||||
| R&D tax credits | % | % | ||||||
| Return to provision adjustments | ( | )% | ( | )% | ||||
| Permanent differences | ||||||||
| Derivative Debt Discount Amortization | ( | )% | % | |||||
| Change in FMV of Warrant Liability | % | ( | )% | |||||
| Loss on convertible note conversion | ( | )% | ( | )% | ||||
| Other | ( | )% | ( | )% | ||||
| Change in valuation allowance | ( | )% | ( | )% | ||||
| Shortfall of stock compensation expense | ( | )% | % | |||||
| Other adjustments | ( | )% | % | |||||
| Total income tax expense | % | % | ||||||
As
of December 31, 2023, the Company had approximately $
NOLs created prior to
2018 could be carried back two years and carried forward
As of December 31, 2023, the Company has $
Note 11 – Quarterly Financial Data (Unaudited and Restated)
The Company is providing restated quarterly unaudited consolidated financial information as of September 30, 2023 in the table below. See Note 2, Restatement of Previously Issued Consolidated Financial Statements, for further background concerning the events preceding the restatement of financial information in this Comprehensive Form 10-K/A.
The restated consolidated balance sheet line items for the third fiscal quarter of 2023 are as follows:
| As Reported | Adjustments | Restated | ||||||||||
| September 30, 2023 | September 30, 2023 | September 30, 2023 | ||||||||||
| Additional paid in capital (1) | ||||||||||||
| Accumulated deficit | ( | ) | ( | ) | ( | ) | ||||||
| (1) |
The adjustments did not have an effect on the consolidated statement of operations or cash flows for the three fiscal quarters of 2023.
The Company has included its restated quarterly unaudited consolidated financial information for the interim periods ended March 31, 2023 and June 30, 2023 within Form 10-Q/A for the period ended March 31, 2024 and Form 10-Q for the period ended June 30, 2024, respectively, filed concurrent with this Form 10-K/A.
| F-44 |
Note 12 – Subsequent Events
Management evaluated all additional events subsequent to the balance sheet date through the date the consolidated financial statements were issued and determined that the following items are required to be disclosed.
Restatement of Previously Issued Financial Statements
See Note 2.
Nasdaq Bid Price Requirement Extension Granted until August 26, 2024
On
August 31, 2023, after the Company’s common stock had traded below $per share for
FDA Approval to Proceed with Phase II study of Ropidoxuridine (IPdR)
On January 8, 2024, Shuttle Pharmaceuticals Holdings, Inc., a Delaware corporation (the “Company”), issued a press release announcing that the Company had received the “Safe to Proceed” letter from the U.S. Food and Drug Administration for the Company’s investigational new drug (IND) application for its Phase II study of Ropidoxuridine (IPdR) as a radiation sensitizing agent during radiotherapy in patients with newly diagnosed IDH-wildtype glioblastoma with unmethylated MGMT promoter. Receipt of the letter allows Shuttle to commence the Phase II study and, as a result, the Company is currently finalizing site enrollment with “first patient, first dose” expected in the coming months. The Phase II clinical trial was also approved by the Institutional Review Board “IRB” on June 21, 2024.
On August 8, 2024, the Company entered into a work order (the “Work Order”) with Theradex Systems, Inc., a New Jersey contract research organization (“CRO”) for purposes of supporting the Company’s Phase II Study of Ropidoxuridine as a Radiation Sensitizing Agent During Radiotherapy in Patients with Newly Diagnosed IDH-Wildtype Glioblastoma with Unmethylated MGMT Promotor.” As such, Shuttle Pharma is now in the process of signing up six clinical sites where the clinical studies will be performed – two of which have completed initial site initiation visits and one of which is ready to enroll patients – and where the CRO will oversee such studies.
Under the terms of the Work Order, the CRO will oversee the studies for a period of 53 months (the “Term”), including overseeing and monitoring the regulatory aspects of the Phase II clinical trial, and managing the documentation surrounding the clinical trial in exchange for a fee of approximately $2.3 million, payable in stages and based upon services performed during the Term of the study.
Rights Offering
Effective
February 7, 2024, the Company and its wholly-owned subsidiary, Shuttle Diagnostics, Inc., entered into a securities purchase
agreement (the “Purchase Agreement”) with SRO, LLC, a Nevada limited liability company, pursuant to which SRO LLC agreed
to commit to purchasing from the Company $
The
Purchase Agreement allows SRO LLC up to 60 days to raise the initial $
Restricted Stock Awarded to Chief Financial Officer
On March 8, 2024, the Company’s then CFO
was awarded
restricted stock units (“RSUs”), with a value of $
On
June 13, 2024, the Company’s newly appointed CFO was awarded
restricted stock units (“RSUs”), with a value of $
Alto Opportunity Master Fund, SPC
On
July 12, 2024, the Company informed the Investor in the Alto Convertible Note that the Company’s expected restatement of its
consolidated financial statements for the years ended December 31, 2022 and 2023 constituted an event of default under the terms of
the Alto Convertible Note. On August 6, 2024, the Company paid $
Alliance Global Partners
On July 30, 2024, the Company
engaged A.G.P./Alliance Global Partners (“AGP”) to serve as exclusive underwriter, placement agent or advisor in
any public or private offering or financing (as defined, the “Offering”) of up to $
| F-45 |
395,574 Shares of Common Stock
2,555,246 Pre-Funded Warrants to Purchase up to 2,555,246 Shares of Common Stock
2,950,820 Common Warrants to Purchase up to 2,950,820 Shares of Common Stock
2,555,246 Shares of Common Stock Underlying such Pre-Funded Warrants
2,950,820 Shares of Common Stock Underlying the Common Warrants
SHUTTLE PHARMACEUTICALS HOLDINGS, INC.
Lead Placement Agent
A.G.P.
Co- Placement Agent
Boustead Securities, LLC
PROSPECTUS
October 29, 2024

