Our Pipeline
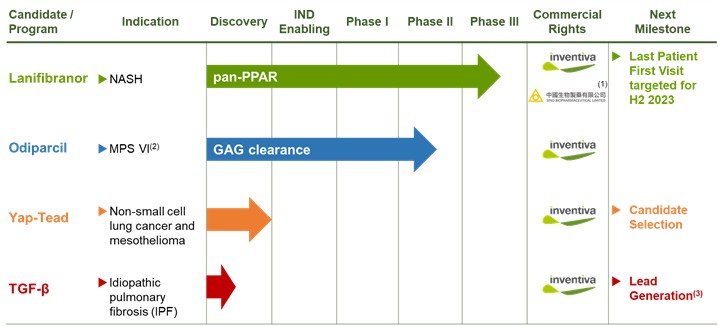
(1) Licensing agreement giving Chia Tai Tianqing Pharmaceutical Group, Co., LTD. ("CTTQ"), an affiliate of Sino Biopharm, exclusive rights to develop and commercialize in China, Hong-Kong, Macao and Taiwan; (2) We suspended all MPS-related research and development activities in 2020, and continue to evaluate our options for potential further development of odiparcil for the treatment of MPS; (3) Lead generation means identifying molecules in anticipation of selecting candidates.
Our goal is to rapidly deliver multiple, novel and differentiated oral small molecule therapies to patients suffering from NASH, MPS, cancer and other diseases with significant unmet medical need. To work towards achieving our goal, we are pursuing the following strategies:
| ● | Demonstrate the Safety and Efficacy of Lanifibranor in the Treatment of NASH with Two Pivotal Clinical Trials. In June 2020, we announced positive topline results from our NATIVE Phase IIb clinical trial of lanifibranor in patients with NASH. In this trial, treatment with lanifibranor at a dose of 1,200 mg met the primary endpoint of a reduction in inflammation and ballooning with no worsening of fibrosis after 24 weeks of treatment, while continuing to show the favorable tolerability profile observed in prior clinical trials of lanifibranor. Treatment with lanifibranor at doses of 800 mg and 1,200 mg also met the key secondary endpoints of resolution of NASH with no worsening of fibrosis and, at the 1,200 mg does, improvement in liver fibrosis without worsening NASH, which are the primary endpoints relevant for seeking accelerated approval from the FDA and conditional approval from the EMA after the Phase III clinical trial is completed, if successful. |
We have initiated LEGEND, a Phase IIa combination trial with lanifibranor and SGLT2 inhibitor empagliflozin, in patients with NASH and Type-2 Diabetes. The primary efficacy endpoint of the trial is a change in Hemoglobin A1c (HbA1c) at the end of the 24-week treatment compared to baseline. Secondary endpoints include changes in liver enzymes, glycaemic and lipids parameters, inflammatory markers and body fat composition
We are also supporting a Phase II investigator-initiated clinical trial studying lanifibranor for the treatment of non-alcoholic fatty liver disease, or NAFLD, the most common liver disorder in developed countries and a precursor to NASH. If positive, we expect that these data would support our registrational filings for lanifibranor for the treatment of NASH.
Lanifibranor has received fast track designation from the FDA for the treatment of NASH. Based on the broad anti-fibrotic and anti-inflammatory properties, as well as beneficial vascular and metabolic effects, of lanifibranor observed in pre-clinical and clinical development to date, we may also pursue development of lanifibranor for the treatment of NASH patients with stage 4
71