UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM
(Mark One)
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _________________ to _________________
Commission File Number:
(Exact Name of Registrant as Specified in its Charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer |
|
|
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|||
|
☒ |
|
Smaller reporting company |
|
||
|
|
|
|
|
|
|
|
|
|
|
Emerging growth company |
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
As of November 5, 2021, the registrant had
Summary Risk Factors
A summary of certain risk factors affecting our business and prospects is included below. You should carefully consider the risks described below together with all of the other information in this Quarterly Report on Form 10-Q, including our financial statements and the related notes and the information included the sections titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as the “Risk Factors” included in item 1A in this Quarterly Report on From 10-Q and our Annual Report on Form 10-K for the fiscal year ended December 31, 2020. If any of the events described below actually occurs, our business, results of operations, financial conditions, cash flows or prospects could be harmed. If that were to happen, you could lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business.
i
ii
Table of Contents
|
|
Page |
PART I. |
1 |
|
Item 1. |
1 |
|
|
1 |
|
|
2 |
|
|
3 |
|
|
5 |
|
|
Notes to Unaudited Condensed Consolidated Financial Statements |
6 |
Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
16 |
Item 3. |
28 |
|
Item 4. |
28 |
|
PART II. |
29 |
|
Item 1. |
29 |
|
Item 1A. |
29 |
|
Item 2. |
47 |
|
Item 3. |
47 |
|
Item 4. |
47 |
|
Item 5. |
47 |
|
Item 6. |
47 |
|
48 |
||
iii
PART I—FINANCIAL INFORMATION
Item 1. Financial Statements
Compass Therapeutics, Inc. and Subsidiaries
Condensed Consolidated Balance Sheets
(In thousands, except per share data)
|
|
September 30, |
|
|
December 31, |
|
||
|
|
2021 |
|
|
2020 |
|
||
Assets |
|
(Unaudited) |
|
|
(Note 1) |
|
||
Current assets: |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Restricted cash |
|
|
|
|
|
|
||
Operating lease, right-of-use ("ROU") asset |
|
|
|
|
|
|
||
Other assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
Liabilities and Stockholders' Equity |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued expenses |
|
|
|
|
|
|
||
Operating lease obligations, current portion |
|
|
|
|
|
|
||
Current portion of long-term debt |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Long-term debt, net of current portion |
|
|
|
|
|
|
||
Operating lease obligations, long-term portion |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Stockholders' equity: |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Additional paid-in-capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total stockholders' equity |
|
|
|
|
|
|
||
Total liabilities and stockholders' equity |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
1
Compass Therapeutics, Inc. and Subsidiaries
Condensed Consolidated Statements of Operations (Unaudited)
(In thousands, except per share data)
|
Three Months Ended |
|
|
Nine Months Ended |
|
||||||||||
|
2021 |
|
|
2020 |
|
|
2021 |
|
|
2020 |
|
||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
||||
Research and development |
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
General and administrative |
|
|
|
|
|
|
|
|
|
|
|
||||
In-process R&D |
|
|
|
|
|
|
|
|
|
|
|
||||
Total operating expenses |
|
|
|
|
|
|
|
|
|
|
|
||||
Loss from operations |
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Other expense, net |
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Loss before income tax expense |
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Income tax expense |
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
||
Net loss |
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Net loss per share - basic and diluted |
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Basic and diluted weighted average shares outstanding |
|
|
|
|
|
|
|
|
|
|
|
||||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
2
Compass Therapeutics, Inc. and Subsidiaries
Condensed Consolidated Statements of Changes in Convertible Preferred Stock and Stockholders’ Equity (Deficit) (Unaudited)
(In thousands)
|
Convertible |
|
|
Common Stock |
|
|
Additional |
|
|
Accumulated |
|
|
Total |
|
|||||||||||||
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||||
Balance at December 31, 2020 |
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
Vesting of share-based awards |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance at March 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||||
Common shares issued for Trigr acquisition |
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Vesting of share-based awards |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance at June 30, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||||
Vesting of share-based awards |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance at September 30, 2021 |
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
Convertible |
|
|
Common Stock |
|
|
Additional |
|
|
Accumulated |
|
|
Total |
|
|||||||||||||
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity (Deficit) |
|
|||||||
Balance at December 31, 2019 |
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||
Vesting of share-based awards |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance at March 31, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|||||
Common shares issued to former shareholders |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Conversion of Compass preferred into common shares |
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Common shares issued, net of issuance costs of $ |
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Payment to non-participating Compass Therapeutics LLC members |
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
Vesting of share-based awards |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance at June 30, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||||
Adj. to costs related to June 2020 private placement |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Vesting of share-based awards |
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance at September 30, 2020 |
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||||
3
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
4
Compass Therapeutics, Inc. and Subsidiaries
Condensed Consolidated Statements of Cash Flows (Unaudited)
(In thousands)
|
|
For the Nine Months |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Cash flows from operating activities: |
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
||
Depreciation and amortization |
|
|
|
|
|
|
||
Loss (gain) on disposal of equipment |
|
|
( |
) |
|
|
|
|
Noncash interest expense |
|
|
|
|
|
|
||
Share-based compensation |
|
|
|
|
|
|
||
Write-off of in-process R&D |
|
|
|
|
|
|
||
Change in fair value of derivative liability |
|
|
|
|
|
|
||
ROU asset amortization |
|
|
|
|
|
|
||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
( |
) |
|
Other long-term assets |
|
|
|
|
|
|
||
Accounts payable |
|
|
( |
) |
|
|
|
|
Accrued expenses |
|
|
|
|
|
( |
) |
|
Operating lease liability |
|
|
( |
) |
|
|
|
|
Settlement of derivative liability |
|
|
|
|
|
( |
) |
|
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
Cash flows from investing activities: |
|
|
|
|
|
|
||
Purchases of property and equipment |
|
|
( |
) |
|
|
( |
) |
Asset acquisition costs |
|
|
( |
) |
|
|
|
|
Proceeds from sale of equipment |
|
|
|
|
|
|
||
Net cash (used in) provided by investing activities |
|
|
( |
) |
|
|
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
||
Proceeds from issuance of common stock |
|
|
|
|
|
|
||
Issuance costs from issuance of common stock |
|
|
|
|
|
( |
) |
|
Repayment of borrowings under loan |
|
|
( |
) |
|
|
( |
) |
Net cash (used in) provided by financing activities |
|
|
( |
) |
|
|
|
|
Net change in cash, cash equivalents and restricted cash |
|
|
( |
) |
|
|
|
|
Cash, cash equivalents and restricted cash at beginning of period |
|
|
|
|
|
|
||
Cash, cash equivalents and restricted cash at end of period |
|
$ |
|
|
$ |
|
||
Supplemental disclosure of cash flow information |
|
|
|
|
|
|
||
Cash paid for interest |
|
$ |
|
|
|
|
||
Supplemental disclosure of financing activities |
|
|
|
|
|
|
||
Deferred offering costs included in accrued expenses |
|
$ |
|
|
$ |
( |
) |
|
Conversion of preferred units |
|
$ |
|
|
$ |
|
||
Payment to non-participating Compass LLC investors, within accrued expenses |
|
$ |
|
|
$ |
( |
) |
|
ROU asset acquired through operating leases |
|
$ |
|
|
$ |
|
||
Acquisition of Trigr Therapeutics, Inc. |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
5
Compass Therapeutics, Inc. and Subsidiaries
Notes to Unaudited Condensed Consolidated Financial Statements
1. Nature of Business and Basis of Presentation
Compass Therapeutics, Inc. (“Compass” or the “Company”) is a clinical-stage, oncology-focused biopharmaceutical company developing proprietary antibody-based therapeutics to treat multiple human diseases. Our scientific focus is on the relationship between angiogenesis and the immune system. Our pipeline includes novel product candidates that leverage our understanding of the tumor microenvironment, including both angiogenesis-targeted agents and immune-oncology focused agents. These product candidates are designed to optimize critical components required for an effective anti-tumor response to cancer. These include modulation of the microvasculature via angiogenesis-targeted agents; induction of a potent immune response via activators on effector cells in the tumor microenvironment; and alleviation of immunosuppressive mechanisms used by tumors to evade immune surveillance. We plan to advance our product candidates through clinical development as both standalone therapies and in combination with our proprietary drug candidates as long as their continued development is supported by clinical and nonclinical data. References to Compass or the Company herein include Compass Therapeutics, Inc. and its wholly-owned subsidiaries. The Company was incorporated as Olivia Ventures, Inc. (“Olivia”) in the State of Delaware on
The Company is subject to risks and uncertainties common to companies in the biotechnology and pharmaceutical industries. There can be no assurance that the Company’s research and development will be successfully completed, that adequate protection for the Company’s technology will be obtained, that any products developed will obtain necessary government regulatory approval or that any approved products will be commercially viable. The Company operates in an environment of rapid change in technology and substantial competition from pharmaceutical and biotechnology companies. In addition, the Company is dependent upon the services of its employees and consultants.
In the opinion of management, the accompanying unaudited condensed consolidated financial statements include all normal and recurring adjustments (which consist primarily of accruals, estimates and assumptions that impact the financial statements) considered necessary to present fairly the Company’s financial position as of September 30, 2021 and its results of operations and changes in convertible preferred stock and stockholders’ equity (deficit) for the three and nine months ended September 30, 2021 and 2020 and cash flows for the nine months ended September 30, 2021 and 2020. Operating results for the nine months ended September 30, 2021 are not necessarily indicative of the results that may be expected for the year ending December 31, 2021.
The unaudited condensed consolidated financial statements include the accounts of Compass Therapeutics, Inc. and its subsidiaries, and have been prepared by the Company in conformity with accounting principles generally accepted in the United States of America (“GAAP”) and pursuant to the rules and regulations of the Securities and Exchange Commission (“SEC”) for interim financial statements. Certain information and footnote disclosures normally included in financial statements prepared in accordance with GAAP have been condensed or omitted pursuant to such rules and regulations. The condensed consolidated balance sheet at December 31, 2020 has been derived from the audited consolidated financial statements at that date, but does not include all of the information and footnotes required by GAAP for complete financial statements. Accordingly, these condensed consolidated financial statements should be read in conjunction with the Company’s audited financial statements in the Company’s Annual Report on Form 10-K for the year ended December 31, 2020 (the “Annual Report”).
Liquidity
Since our inception, we have devoted substantially all of our efforts to organizing and staffing our Company, business planning, raising capital, research and development activities, building our intellectual property portfolio and providing general and administrative support for these operations. To date, we have funded our operations primarily with proceeds from the sale of our equity securities and borrowings from debt arrangements. Through September 30, 2021, we have received $
6
development plans, we expect that such cash resources will enable us to fund our operating expenses and capital expenditure requirements through the fourth quarter of 2024.
COVID-19 Update
We have been carefully monitoring the COVID-19 pandemic and its potential impact on our business and have taken important steps to help ensure the safety of our employees and to reduce the spread of COVID-19 community-wide. We are ensuring that essential staffing levels at our operations remain in place, including maintaining key personnel in our laboratory facilities. We have implemented stringent safety measures designed to create a safe and clean environment for our employees as we continue to comply with applicable federal, state and local guidelines instituted in response to the COVID-19 pandemic.
We have been able to continue to pursue patient dosing and monitoring of our Phase 1 clinical trial of CTX-471 without significant delays. However, we have experienced increased delays in patient enrollment in several of our trial sites. In order to address the reduction in patient enrollment, we have added additional sites. In addition, there have been delays in sourcing of selected supplies required for the manufacturing of material to be used in our future clinical trials, and these delays have impacted and may continue to impact the timing of our future clinical trials. We expect that COVID-19 may continue to directly or indirectly impact (i) our employees and business operations or personnel at third-party suppliers and other vendors in the U.S. and other countries; (ii) the availability, cost or supply of materials; and (iii) the timeline for our ongoing clinical trial and potential future trials. We are continuing to assess the potential impact of the COVID-19 pandemic on our current and future business and operations, including our expenses and clinical trials, as well as on our industry and the healthcare system.
2. Summary of Significant Accounting Policies
There have been no material changes to the significant accounting policies previously disclosed in the Company’s Annual Report, except as noted below.
Recently Adopted Accounting Pronouncements
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016‑02, Leases, which requires a lessee to record a right‑of‑use asset and a corresponding lease liability on the balance sheet for all leases with terms longer than 12 months. A modified retrospective transition approach is required, applying the new standard to all leases existing at the date of initial application. An entity may choose to use either (1) its effective date or (2) the beginning of the earliest comparative period presented in the condensed consolidated financial statements as its date of initial application. If an entity chooses the second option, the transition requirements for existing leases also apply to leases entered into between the date of initial application and the effective date. The Company adopted this standard on January 1, 2021. See Note 7 for additional details on the Company’s accounting for leases.
Recent Accounting Pronouncements
7
3. Fair Value Measurements
The following tables present information about the Company’s financial assets and liabilities that are measured at fair value on a recurring basis and indicate the level of the fair value hierarchy utilized to determine such fair values (in thousands):
|
|
Fair Value Measurements as of September 30, 2021 Using: |
|
|||||||||||||
|
|
Quoted Prices |
|
|
Significant |
|
|
Significant |
|
|
Fair Value |
|
||||
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Cash equivalents - money market |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Total assets |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
Fair Value Measurements as of December 31, 2020 Using: |
|
|||||||||||||
|
|
Quoted Prices |
|
|
Significant |
|
|
Significant |
|
|
Fair Value |
|
||||
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Cash equivalents - money market |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Total assets |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
4. Property and Equipment
Property and equipment consist of the following (in thousands):
|
|
September 30, |
|
|
December 31, |
|
||
Equipment |
|
$ |
|
|
$ |
|
||
Software |
|
|
|
|
|
|
||
Leasehold improvements |
|
|
|
|
|
|
||
Furniture and fixtures |
|
|
|
|
|
|
||
Total property and equipment–at cost |
|
|
|
|
|
|
||
Less: Accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
Property and equipment, net |
|
$ |
|
|
$ |
|
||
8
5. Accrued Expenses
Accrued expenses consist of the following (in thousands):
|
|
September 30, |
|
|
December 31, |
|
||
|
|
2021 |
|
|
2020 |
|
||
Compensation and benefits |
|
$ |
|
|
$ |
|
||
Research and development expenses |
|
|
|
|
|
|
||
Leasehold improvements |
|
|
|
|
|
|
||
Legal and professional fees |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Total accrued expenses |
|
$ |
|
|
$ |
|
||
6. Debt
The aggregate principal amount of debt outstanding consisted of the following (in thousands):
|
|
September 30, |
|
|
December 31, |
|
||
|
|
2021 |
|
|
2020 |
|
||
Current portion of debt |
|
$ |
|
|
$ |
|
||
Less: unamortized debt discount |
|
|
( |
) |
|
|
( |
) |
Current portion of debt, net of debt |
|
$ |
|
|
$ |
|
||
Long-term debt, net of current portion |
|
$ |
|
|
$ |
|
||
Less: unamortized debt discount |
|
|
|
|
|
( |
) |
|
Long-term debt, net of current portion |
|
$ |
|
|
$ |
|
||
The Company entered into, and subsequently amended, a term loan facility with Pacific Western Bank, Inc. (the “Credit Facility”), and received $
The Credit Facility agreement contains a provision whereby the Company was obligated to pay a success fee of $
The Credit Facility contains a negative pledge on the Company’s intellectual property and also contains customary indemnification obligations and customary events of default, including, among other things, (i) non‑payment, (ii) breach of warranty, (iii) non‑performance of covenants and obligations, (iv) default on other indebtedness, (v) judgments, (iv) change of control, (vii) bankruptcy and insolvency, (viii) impairment of security, (ix) key permit events, (x) key person event, (xi) regulatory matters, and (xii) key contracts. In addition, the Company must maintain a minimum cash balance of $
The borrowings are collateralized by substantially all of the Company’s assets, excluding intellectual property, and contains affirmative and negative covenants including restrictions on the Company’s ability to incur additional indebtedness, pay dividends, encumber its property, or engage in certain fundamental business transactions, such as mergers or acquisitions of other businesses. The Company was in compliance with its covenants as of September 30, 2021.
The Company recognized interest expense of $
9
As of September 30, 2021, the aggregate minimum future principal payments due in connection with the Credit Facility, as amended, are as follows (in thousands):
Year Ending December 31, |
|
|
|
|
2021 |
|
$ |
|
|
2022 |
|
|
|
|
Total |
|
$ |
|
|
7. Leases
The Company adopted ASU 2016-02, Leases (Topic 842), or ASU 2016-02, effective January 1, 2021, using the modified retrospective transition method, in which the new standard is applied as of the date of initial adoption. The Company recognized and measured agreements executed prior to the date of initial adoption that were considered leases on January 1, 2021. No cumulative effect adjustment of initially applying the standard to the opening balance of retained earnings was made upon adoption. The Company elected the package of practical expedients permitted under the transition guidance that will retain the lease classification and initial direct costs for any leases that exist prior to adoption of the standard. In addition, the Company elected the accounting policy of not recording short-term leases with a lease term at the commencement date of 12 months or less on the condensed consolidated balance sheet as permitted by the new standard.
The Company has evaluated its leases and determined that it has one lease that is classified as an operating lease. The classification of this lease is consistent with the Company’s determination under the previous accounting standard.
When available, the Company will use the rate implicit in the lease to discount lease payments to present value; however, the Company’s current lease does not provide an implicit rate. Therefore, the Company used its incremental borrowing rate to discount the lease payments based on the date of the lease commencement.
The Company has
The table below presents the undiscounted cash flows for the lease term. The undiscounted cash flows are reconciled to the operating lease liabilities recorded on the condensed consolidated balance sheet:
|
|
(000's) |
|
|
Remainder of 2021 |
|
$ |
|
|
Years ending December 31, |
|
|
|
|
2022 |
|
|
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
Total minimum lease payments |
|
|
|
|
Less: amount of lease payments representing interest |
|
|
( |
) |
Present value of future minimum lease payments |
|
|
|
|
Less: operating lease obligations, current portion |
|
|
( |
) |
Operating lease obligations, long-term portion |
|
$ |
|
|
10
8. Stock-Based Compensation
In June 2020, the Company’s board of directors adopted the 2020 Stock Option and Incentive Plan (the “2020 Plan”) and reserved
The 2020 Plan authorizes the board of directors or a committee of the board to grant incentive stock options, nonqualified stock options and restricted stock awards to eligible officers, employees, consultants and directors of the Company. Options generally vest over a period of
Stock-based compensation expense for the nine months ended September 30, 2021 and 2020 was classified in the condensed consolidated statement of operations as follows:
|
|
Nine Months Ended September 30, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
|
|
(000’s) |
|
|||||
|
|
|
|
|
|
|
||
Research and development |
|
$ |
|
|
$ |
|
||
General and administrative |
|
|
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
||
As of September 30, 2021, remaining unrecognized compensation cost related to options and restricted stock awards to be recognized in future periods totaled $
Restricted Stock
Prior to the adoption of the 2020 Plan, the Company issued restricted stock.
|
|
Shares |
|
|
Fair Value |
|
||
Weighted Average Fair Value |
|
(000’s) |
|
|
Per Share |
|
||
Unvested, December 31, 2020 |
|
|
|
|
$ |
|
||
Granted |
|
|
|
|
$ |
|
||
Vested |
|
|
( |
) |
|
$ |
|
|
Forfeited or canceled |
|
|
( |
) |
|
$ |
|
|
Unvested, September 30, 2021 |
|
|
|
|
$ |
|
||
As of September 30, 2021, the total unrecognized compensation cost related to stock compensation expense for restricted stock is $
11
Stock Options
The following table summarizes the stock option activity for the 2020 Plan:
|
|
|
|
|
Weighted |
|
|
Weighted |
|
|||
|
|
Number of |
|
|
Average |
|
|
Average |
|
|||
|
|
Unvested |
|
|
Exercise |
|
|
Remaining |
|
|||
|
|
Options |
|
|
Price |
|
|
Contractual |
|
|||
|
|
(000’s) |
|
|
Per Share |
|
|
Life (in years) |
|
|||
Outstanding at December 31, 2020 |
|
|
|
|
$ |
|
|
|
|
|||
Granted |
|
|
|
|
$ |
|
|
|
|
|||
Exercise |
|
|
|
|
$ |
|
|
|
— |
|
||
Forfeited/cancelled |
|
|
( |
) |
|
$ |
|
|
|
— |
|
|
Outstanding at September 30, 2021 |
|
|
|
|
$ |
|
|
|
|
|||
Vested at September 30, 2021 |
|
|
|
|
$ |
|
|
|
|
|||
For the nine months ended September 30, 2021, the weighted average grant date fair value for options granted was $
For the nine months ended September 30, 2020, the weighted average grant date fair value for options granted was $
The weighted average assumptions used in the Black-Scholes pricing model to determine the fair value of stock options granted during the nine months ended September 30, 2021 and 2020 were as follows:
|
Nine Months Ended |
|
|||||
|
September 30, |
|
|||||
|
2021 |
|
|
2020 |
|
||
Expected term (in years) |
|
|
|
|
|
||
Risk-free rate |
|
% |
|
|
% |
||
Expected volatility |
|
% |
|
|
% |
||
12
9. Merger Transaction
On
To determine whether the transaction meets the definition of a business acquisition or an asset acquisition in accordance with ASC 805-10-55, we had to assess the nature of the transaction and the fair value of the assets acquired in the transaction. Our assessment suggest that the fair value of the transaction is substantially concentrated in a license to a single identifiable asset, CTX-009, and a potential financial interest (in the form of royalties) in an additional set of early-stage similar assets. The guidance further requires a business acquisition to include, at a minimum, an input and a substantive process that together significantly contribute to the ability to create outputs. Because all asset acquisitions include inputs, the existence of a substantive process is what distinguishes a business acquisition from an asset acquisition. Our assessment is that there is no process or outputs that are being acquired with the TRIGR acquisition. As a result, the TRIGR acquisition is considered to fall under the guidance of an asset acquisition rather than a business acquisition. Accordingly, the Company allocated the $
10. Related Parties and Related-Party Transactions
On October 16, 2014, the Company entered into a collaboration agreement with Adimab, LLC. The Company’s co-founder has a direct ownership interest in Adimab, LLC. The Company recorded
In connection with the acquisition of TRIGR and upon consummation of the merger agreement on June 25, 2021, Miranda Toledano, who previously served as the Chief Financial Officer and Chief Operating Officer of TRIGR, was appointed to Compass Board of Directors as an independent director. Additionally, to facilitate the transition of CTX-009 from TRIGR, the Company entered into a consulting agreement with Ms. Toledano on June 25, 2021 for a period of six months.
13
11. Other Expense
|
|
Three Months Ended |
|
|
Nine Months Ended |
|
||||||||||
|
|
September 30, |
|
|
September 30, |
|
||||||||||
|
|
2021 |
|
|
2020 |
|
|
2021 |
|
|
2020 |
|
||||
|
|
(000's) |
|
|
(000's) |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Interest income |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Interest expense |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Change in fair value of derivative liability |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
Other income (expenses) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Total other income (expenses) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
12. License, Research and Collaboration Agreements
Collaboration Agreements
ABL Bio Corporation ("ABL Bio") Agreements
In November 2018, the Company's wholly owned subsidiary TRIGR and ABL Bio, a South Korean biotechnology company, entered into an exclusive global (excluding South Korea) license agreement (the “TRIGR License Agreement”) which granted TRIGR a license to ABL001, ABL Bio’s bispecific antibody targeting DLL4 and VEGF-A (renamed CTX-009). Under the terms of the agreement, ABL Bio and TRIGR would jointly develop CTX-009, with ABL Bio responsible for development of CTX-009 throughout the end of Phase 1 clinical trials and TRIGR responsible for the development of CTX-009 from Phase 2 and onward. ABL Bio received a $
In May 2021, TRIGR and ABL Bio terminated license agreements to several preclinical assets. As a result of the return of these assets to ABL Bio and termination of the license agreements, the Company is eligible to receive royalty payments if ABL Bio develops or licensees two bispecific antibodies (ABL101 and ABL103) that were previously licensed to TRIGR.
Adimab Agreement
The Company entered into a collaboration agreement with Adimab, LLC on
Other License and Research Agreements
FUJIFILM Diosynth Biotechnologies ("Fujifilm") Agreement
The Company entered into a scope of work (“SOW”) under a master services agreement with Fujifilm on July 20, 2020. The Company made cash payments of $
14
13. Subsequent events
On November 1, 2021, the Company announced that it priced an underwritten public offering (the "Offering") to sell
15
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion of the financial condition and results of operations of Compass Therapeutics, Inc. should be read in conjunction with the financial statements and the notes to those statements included in this Quarterly Report on Form 10-Q for the period ended September 30, 2021. Some of the information contained in this discussion and analysis, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risk, uncertainties and assumptions. You should read the “Risk Factors” section of this Quarterly Report on Form 10-Q and the “Risk Factors” section included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2020, for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a clinical-stage, oncology-focused biopharmaceutical company developing proprietary antibody-based therapeutics to treat multiple human diseases. Our scientific focus is on the relationship between angiogenesis, the immune system, and tumor growth. Our pipeline of novel product candidates is designed to target multiple critical biological pathways required for an effective anti-tumor response. These include modulation of the microvasculature via angiogenesis-targeted agents, induction of a potent immune response via activators on effector cells in the tumor microenvironment, and alleviation of immunosuppressive mechanisms used by tumors to evade immune surveillance. We plan to advance our product candidates through clinical development as both standalone therapies and in combination with proprietary pipeline antibodies based on supportive clinical and nonclinical data.
On June 25, 2021, we consummated a definitive merger agreement (the “Merger Agreement”) with Trigr Therapeutics, Inc. (“TRIGR”), a private biotechnology company, . Pursuant to the Merger Agreement, through our wholly owned subsidiaries and a two-step merger structure, we acquired all of the outstanding shares of TRIGR (the “TRIGR Merger”). Consideration payable to TRIGR shareholders at closing totaled an aggregate of 10,265,133 shares of our common stock (after giving effect to elimination of fractional shares that would otherwise be issued). In addition, TRIGR shareholders are eligible to receive up to $9 million, representing earnout payments which are dependent on certain events, including $5 million which is dependent on biologics license application approval of a product candidate acquired in the transaction, renamed CTX-009.
We currently have three product candidates, two of which are in clinical development:
CTX-009 (a.k.a. ABL001) - anti-DLL4 x VEGF-A bispecific antibody
CTX-009 is an investigational bispecific antibody that simultaneously blocks Delta-like ligand 4/Notch ("DLL4") and vascular endothelial growth factor A ("VEGF-A") signaling pathways, which are critical to angiogenesis and tumor vascularization. We have licensed exclusive global rights to CTX-009, outside of South Korea, from ABL Bio, Inc. (“ABL Bio”), a South Korea-based clinical-stage company focused on developing antibody therapeutics. South Korean rights are held by Handok Pharmaceuticals, Inc. (“Handok”) and China rights which were out-licensed from the Company to Elpiscience Biopharmaceuticals Co., Limited (“Elpiscience”).
CTX-009 is undergoing clinical development in patients with advanced solid tumors in South Korea. A Phase 1 dose escalation and dose expansion monotherapy study has been completed and a Phase 1b combination study of CTX-009 in combination with chemotherapy is ongoing in South Korea. In the first quarter of 2021, Handok commenced a Phase 2a study of CTX-009 in combination with paclitaxel in patients with biliary tract cancers (“BTC” or “cholangiocarcinoma”) in South Korea.
On October 8, 2021, we, along with ABL Bio, presented clinical trial data from the CTX-009 Phase 1a/1b dose-escalation and dose expansion study at a plenary oral session during the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics (Abstract Number: 4749; Session title: Plenary Session 2: New Drugs on the Horizon I). The significant findings presented are as follows:
16
Phase 1: Monotherapy Clinical Trial of CTX-009
An open-label, Phase 1 dose-escalation and expansion study designed to identify the optimal dose and to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and the anti-tumor activity of CTX-009 in patients with advanced solid tumors after failure of standard of care treatment was conducted by ABL Bio in South Korea. This trial consisted of a Phase 1a monotherapy dose escalation arm and a Phase 1b dose expansion arm. The study was initiated in September 2017 and enrollment was completed in February 2021.
The dose escalation portion of the study followed a traditional 3+3 dosing scheme where CTX-009 was administered by intravenous infusion across nine dose cohorts ranging from 0.3 to 17.5 mg/kg biweekly. Patients were enrolled in two arms: a Phase 1a dose-escalation arm and a Phase 1b dose expansion arm. The expansion cohorts were 7.5, 10, 12.5 and 15 mg/kg. Patient tumor volumes were measured using CT scans at baseline and then every eight weeks.
A total of 40 out of the 45 patients enrolled in the study are evaluable for the purpose of determination of anti-tumor activity of CTX-009 since five patients did not reach their first scan at week eight due to progressive disease or for other reasons. Sixteen of the 40 evaluable patients were dosed at the 10 or 12.5 mg/kg dose levels and represent what we project to be the efficacious dose levels. Among those 16 patients, there were three PRs confirmed by RECIST 1.1 with an ORR of 18.8% and eight patients with stable disease (“SD”), with a CBR of 68.8%. Two of the three PRs were in advanced colorectal patients and one of the three PRs was in an advanced gastric cancer patient. In addition, one of the patients with gastric cancer had a 35% decline in tumor mass relative to baseline; however, that regression was not confirmed upon a second CT scan, and hence not included in the ORR and the best response of this patient included in the data set is stable disease.
Phase 1b: Combination Clinical Trial of CTX-009 in South Korea
An open-label, combination Phase 1b clinical trial to evaluate the safety, pharmacokinetics, anti-tumor activity and the RP2D of CTX-009 in combination with paclitaxel or irinotecan chemotherapy is currently being conducted by ABL Bio and Handok in South Korea. This study was initiated in June 2020 and enrollment was completed in December 2020 (clinicaltrials.gov identifier NCT04492033).
The study includes two cohorts, each of which is divided into two groups. The first cohort was administered 10 or 12.5 mg/kg of CTX-009 on a biweekly basis, in combination with 80 mg/m2 paclitaxel administered weekly. The second cohort was administered 10 or 12.5 mg/kg of CTX-009 in combination with 150 mg/m2 irinotecan on a biweekly basis.
Of the 17 patients enrolled, there have been four PRs, including three PRs that were confirmed by RECIST 1.1. and one PR which is unconfirmed, representing a 23.5% ORR and nine patients with SD, representing a CBR of 76.5%. The unconfirmed PR was in a patient with NSCLC who has been on the study for over a year and as of October 5, 2021 remains on the study. Of the four patients with advanced cholangiocarcinoma enrolled in the study, there were two PRs confirmed by RECIST 1.1 with 41% and 62% declines in tumor burden, respectively, representing an ORR in cholangiocarcinoma of 50%. A third patient with cholangiocarcinoma had a stable disease with 28% decline in such patient’s tumor burden, and therefore the CBR observed in cholangiocarcinoma is three out of four, or 75%. The responses in cholangiocarcinoma were particularly durable with a median duration of response ("DOR") of 9.7 months as of October 5, 2021.
The observed ORR of CTX-009 at the 10 and 12.5 mg/kg doses are 18.8% (3/16) as a monotherapy and 23.5% (4/17) in combination with chemotherapy. The CBR of CTX-009 at the 10 and 12.5 mg/kg is 68.8% (11/16) as a monotherapy and 76.5% (13/17) in combination with chemotherapy.
17
Phase 2a: Interim Data from Combination Clinical Trial of CTX-009 in BTC in South Korea
A Phase 2a study of CTX-009 in combination with paclitaxel was initiated by Handok in the first quarter of 2021 in patients with BTC. The study has been enrolling patients with unresectable advanced, metastatic, or relapsed BTCs and have received one or two prior systemic therapies.
The Phase 2a study utilizes a Simon Two-Stage adaptive design where the criteria to advance to the second stage of the study is three PRs observed in 21 patients. Based on the Simon Two-Stage design, when the criteria for the first Stage is met, the study progresses to the second stage, where 45 additional patients will be enrolled. As of October 5, 2021, there had been five PRs observed among the first 17 patients evaluated (ORR=29%), and accordingly, the criteria to advance to the second part of the study has already been met. The study is being conducted at four leading medical centers in South Korea.
As of October 5, 2021, no formal safety data analysis had been completed, but CTX-009 was observed to be generally well-tolerated. AEs that were determined to be probably or possibly related to CTX-009 treatment included Grade 3 hypertension observed in 2 patients (8%). Other AEs observed were Grade 3 neutropenia (46%), Grade 3 thrombocytopenia (4%) and Grade 3 anemia (13%), which were all attributed to the concomitant chemotherapy agent (paclitaxel).
The table below depicts a preliminary summary of the drug-related AEs in 24 patients as of October 5, 2021:
|
Drug-related adverse events observed in >1 patient (preliminary as of |
Total |
Total |
Grade 3 |
Grade 3 |
10/5/2021) |
(n) |
(%) |
(n) |
(%) |
|
Neutropenia* |
11 |
46 |
11 |
46 |
|
Hypertension |
9 |
38 |
2 |
8 |
|
Thrombocytopenia* |
4 |
17 |
1 |
4 |
|
Anemia* |
3 |
13 |
3 |
13 |
|
Dyspnea |
3 |
13 |
0 |
0 |
|
General weakness |
3 |
13 |
1 |
4 |
|
Headache |
3 |
13 |
0 |
0 |
|
Hoarseness |
3 |
13 |
0 |
0 |
|
Edema, Fatigue, Fever, Pneumonia, Proteinuria |
2 each |
8 |
1 |
1** |
|
|
* Labeled Grade 3/4 cytopenia events with paclitaxel:52% neutropenia, 16% anemia, 7% thrombocytopenia ** One Grade5 pneumonia |
|
|
|
|
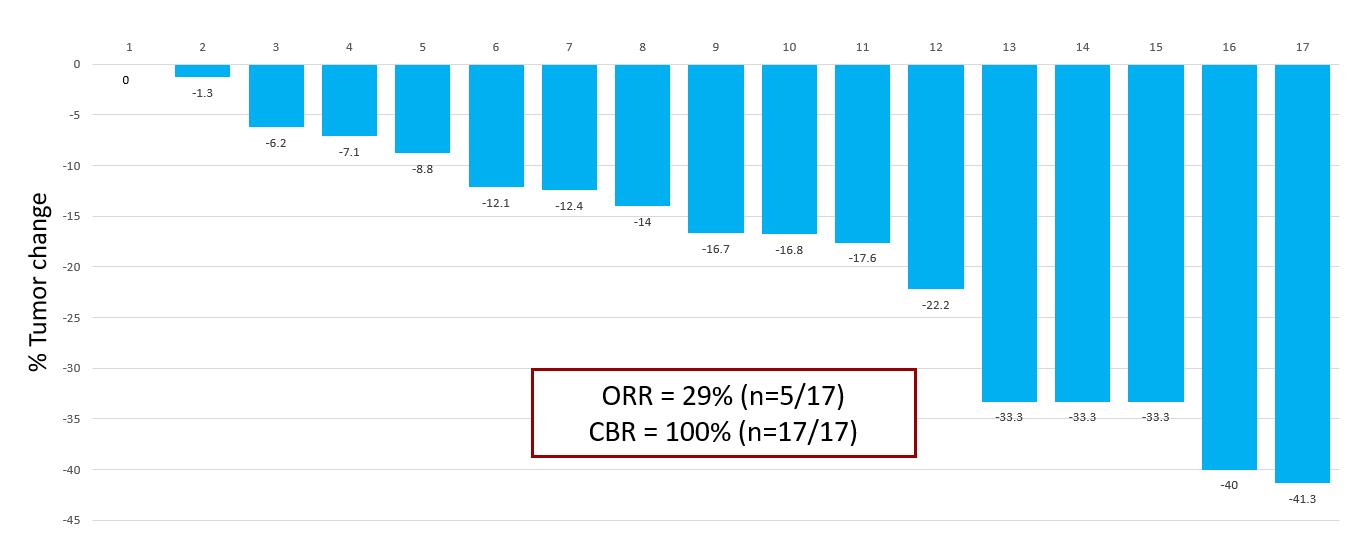
The study has enrolled 24 patients and 17 of those patients have reached their first CT scan and are considered evaluable. Five PRs confirmed by RECIST 1.1 have been observed among the first 17 evaluable patients, leading to a preliminary ORR of 29%, and all patients evaluated have had stable disease or better with a decline in tumor burden observed in 16 of the 17 patients leading to a CBR of 100%.
18
The interim waterfall plot below depicts the best response for each of the 17 patients evaluated in the study as of September 28, 2021:
We plan to submit an Investigational New Drug (“IND”) application to the U.S. Food and Drug Administration (“FDA”) in the fourth quarter of 2021 and subject to the IND going into effect with the FDA, to initiate a Phase 2 study of CTX-009 in combination with paclitaxel in patients with cholangiocarcinoma in the second quarter of 2022.
The second clinical study for which we plan to submit an IND is a Phase 2/3 monotherapy clinical trial of CTX-009 in the third line setting in colorectal cancer. We believe we can advance CTX-009 into a Phase 2/3 study in the third line setting in colorectal cancer with ORR and DOR as potential endpoints for accelerated approval. We plan to submit an IND supplement to the FDA in the first half of 2022 and subject to the IND going into effect with the FDA, to initiate a Phase 2 study in patients with colorectal cancer in the second half of 2022. We have not yet discussed this plan with regulatory agencies, including the FDA.
The timing of the initiation of the clinical trials in the United States depends, among other things, on the availability of clinical drug product for the studies, communications with the FDA and FDA allowance for each of the proposed studies to proceed.
CTX-471 - a monoclonal antibody agonist of CD137, a key co-stimulatory receptor on immune cells
In July 2019, we initiated a Phase 1 trial evaluating the safety and tolerability of CTX-471 as a monotherapy in oncology patients who were previously treated with PD-1 or PD-L1 immune checkpoint inhibitors and subsequently relapsed or progressed after a period of stable disease. The design of this trial includes a dose escalation stage (Phase 1a) followed by a dose expansion stage (Phase 1b). The dose-escalation stage of the Phase 1a trial has been completed and CTX-471 was observed to be generally well-tolerated.
The dose expansion stage of the trial is currently ongoing and nearing completion. As of October 21, 2021, 36 patients have been enrolled in the study and 25 of those patients are evaluable. Of the 25 evaluable patients, 2 patients had a PR, one of which has been confirmed by RECIST 1.1 and the other PR has been seen at the first tumor evaluation at Week 9, and 11 patients have reached stable disease, leading to a preliminary ORR of 8.0% and a CBR of 52%.
The first PR observed in the study was in a patient with advanced small cell lung cancer who had a PR at week 17 and this response has been confirmed at week 25. This patient has now been dosed with CTX-471 for more than one year with a durable PR. In October 2021, a second PR was observed in a patient with metastatic melanoma who was previously treated with nivolumab and progressed on nivolumab. There has been one treatment-related serious adverse event (“SAE”) in the Phase 1b dose expansion stage of the trial. We expect to complete the Phase 1b stage of this trial during the first half of 2022. The next step for development of CTX-471 is the initiation of a Phase 1b combination study with a PD-1 or PD-L1 blocker in the first half of 2022.
19
CTX-8371 - a bispecific antibody that simultaneously targets both PD-1 and PD-L1
CTX-8371 is a bispecific antibody that simultaneously targets both PD-1 and PD-L1, the targets of well-known and widely used checkpoint inhibitor antibodies. IND-enabling studies with CTX-8371 were initiated in August 2020 and are generally progressing well except that over the last six months, our contract development manufacturing organization, Fujifilm Diosynth Biotechnologies (see Note 12 to the financial statements contained in this Form 10-Q for further description of Fujifilm agreement) has been experiencing delays with its supply chain management, leading to a delay in the GMP manufacturing campaign of CTX-8371. Based on the new timeline provided by Fujifilm for the GMP manufacturing campaign, we are currently targeting an IND submission for CTX-8371 in the second half of 2022.
We have funded our operations primarily with proceeds from the sale of our equity securities and borrowings from debt arrangements. Through September 30, 2021, we have received $132.0 million in gross proceeds from the sale of equity securities, $15.0 million in term loan borrowings under a credit facility with Pacific Western Bank ("the Credit Facility") and $60.5 million in gross proceeds from the sale of our common stock in a private placement in June 2020. In November 2021, we completed an underwritten public offering in which we received net proceeds of approximately $117.3 million after underwriting and offering expenses (See Note 13 to the financial statements contained in this Form 10-Q for further description of this transaction).
We have incurred significant operating losses since inception and have not generated any revenue from the sale of products and we do not expect to generate any revenue from the sale of products in the near future, if at all. Our ability to generate product revenue sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of our treatments and any future product candidates. Our net losses were $6.0 million and $9.2 million for the three months ended September 30, 2021 and 2020, respectively, and $69.2 million and $21.1 million for the nine months ended September 30, 2021 and 2020, respectively. The losses for the nine months ended September 30, 2021 include $50.6 million of in-process R&D expense related to the TRIGR merger, which was a stock only transaction. We had an accumulated deficit of $220.6 million at September 30, 2021. We expect to continue to incur significant expenses for at least the next several years as we advance through clinical development, develop additional product candidates and seek regulatory approval of any product candidates that complete clinical development. In addition, if we obtain marketing approval for any product candidates, we expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. We may also incur expenses in connection with the in-licensing or acquisition of additional product candidates.
Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through equity and debt financings, or other capital sources, which may include collaborations with other companies or other strategic transactions. As of September 30, 2021, we had $25.5 million in cash and cash equivalents. With the proceeds from the underwritten offering in November 2021, and based on our research and development plans, we expect that such cash resources will enable us to fund our operating expenses and capital expenditure requirements through the fourth quarter of 2024.
Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses or when, or if, we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
20
COVID-19 Update
We have been carefully monitoring the COVID-19 pandemic and its potential impact on our business and have taken important steps to help ensure the safety of our employees and to reduce the spread of COVID-19 community-wide. We are ensuring that essential staffing levels at our operations remain in place, including maintaining key personnel in our laboratory facilities. We have implemented stringent safety measures designed to create a safe and clean environment for our employees as we continue to comply with applicable federal, state and local guidelines instituted in response to the COVID-19 pandemic.
We have been able to continue to pursue patient dosing and monitoring of our Phase 1 clinical trial of CTX-471 without significant delays. However, we have experienced increased delays in patient enrollment in several of our trial sites. In order to address the reduction in patient enrollment, we have added additional sites. In addition, there have been delays in sourcing of selected supplies required for the manufacturing of material to be used in our future clinical trials, and these delays have impacted and may continue to impact the timing of our future clinical trials. We expect that COVID-19 may continue to directly or indirectly impact (i) our employees and business operations or personnel at third-party suppliers and other vendors in the U.S. and other countries; (ii) the availability, cost or supply of materials; and (iii) the timeline for our ongoing clinical trial and potential future trials. We are continuing to assess the potential impact of the COVID-19 pandemic on our current and future business and operations, including our expenses and clinical trials, as well as on our industry and the healthcare system.
Components of Results of Operations
Research and Development
Research and development expenses consist primarily of costs incurred in connection with the development of our product candidates, CTX-471, CTX-8371 and CTX-009, as well as unrelated discovery program expenses. We expense research and development costs as incurred. These expenses include:
Advance payments that we make for goods or services to be received in the future for use in research and development activities are recorded as prepaid expenses. Such amounts are recognized as an expense as the goods are delivered or the related services are performed, or until it is no longer expected that the goods will be delivered or the services rendered.
Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will increase substantially in connection with our planned clinical development activities in the future. At this time, we cannot accurately estimate or know the nature, timing and costs of the efforts that will be necessary to complete the clinical development of any future product candidates.
The successful development and commercialization of product candidates is highly uncertain. This is due to the numerous risks and uncertainties associated with product development and commercialization.
21
In-Process R&D
In-process R&D expenses consists of the acquisition of Trigr Therapeutics, Inc., whose primary asset is CTX-009, an anti-DLL4 x VEGF-A bispecific antibody. As we expense research and development costs as incurred, the cost of this acquisition was expensed. See Note 9 to the financial statements contained in this Form 10-Q for further description of the accounting of this transaction.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related costs for personnel in executive, finance, corporate and business development, and administrative functions. General and administrative expenses also include legal fees relating to patent and corporate matters; professional fees for accounting, auditing, tax and administrative consulting services; insurance costs; administrative travel expenses; marketing expenses and other operating costs.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our business operations.
Other Expense, Net
Other expense consists of interest expense, interest income and realized losses on sales of furniture and equipment.
Interest expense consists primarily of cash interest under our Credit Facility that we entered into in March 2018 and the related non-cash interest attributable to the amortization of deferred financing costs incurred in connection with this facility.
Results of Operations
Comparison of the Three Months Ended September 30, 2021 and 2020
The following table summarizes our results of operations for the three months ended September 30, 2021 and 2020:
|
|
Three Months Ended September 30, |
|
|||||||||
|
|
2021 |
|
|
2020 |
|
|
Change |
|
|||
|
|
|
|
|
(000’s) |
|
|
|
|
|||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|||
Research and development |
|
$ |
3,154 |
|
|
$ |
3,670 |
|
|
$ |
(516 |
) |
General and administrative |
|
|
2,700 |
|
|
|
5,291 |
|
|
|
(2,591 |
) |
Total operating expenses |
|
|
5,854 |
|
|
|
8,961 |
|
|
|
(3,107 |
) |
Loss from operations |
|
|
(5,854 |
) |
|
|
(8,961 |
) |
|
|
3,107 |
|
Other expense, net |
|
|
(121 |
) |
|
|
(189 |
) |
|
|
68 |
|
Loss before income tax expense |
|
|
(5,975 |
) |
|
|
(9,150 |
) |
|
|
3,175 |
|
Income tax expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
Net loss |
|
$ |
(5,975 |
) |
|
$ |
(9,150 |
) |
|
$ |
3,175 |
|
|
|
|
|
|
|
|
|
|
|
|||
Research and Development Expenses
Research and development expenses decreased by $0.5 million for the three months ended September 30, 2021 compared to the three months ended September 30, 2020. We incurred $0.3 million less in depreciation expense and $0.2 million less in program related expenses (clinical and manufacturing expense related to CTX-471 and CTX-8371, respectively) as compared to the same period in 2020.
22
We track outsourced development, outsourced personnel costs and other research and development costs of specific programs. In 2021, we began tracking our internal personnel costs on a program-by-program basis. Research and development expenses are summarized by program in the table below:
|
|
Three Months Ended September 30, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
|
|
(000’s) |
|
|||||
|
|
|
|
|
|
|
||
CTX-471 |
|
$ |
1,067 |
|
|
$ |
438 |
|
CTX-8371 |
|
|
274 |
|
|
|
599 |
|
CTX-009 |
|
|
177 |
|
|
|
— |
|
Unallocated research and development expenses |
|
|
1,636 |
|
|
|
2,633 |
|
Total research and development expenses |
|
$ |
3,154 |
|
|
$ |
3,670 |
|
General and Administrative Expenses
General and administrative expenses decreased by $2.6 million to $2.7 million for the three months ended September 30, 2021 as compared to the same period in 2020. The decrease primarily came from a decrease in stock compensation of $1.4 million, a decrease from expenses related to the reverse merger in 2020 of $0.5 million from legal expenses and professional fees and a decrease in facilities expense of $0.3 million. The stock compensation in 2020 was unusually high from immediate vesting of options issued after the reverse merger.
Other Expense, Net
We recognized interest expense of $0.1 million and $0.2 million during the three months ended September 30, 2021 and 2020, respectively. The reduction in interest is from a lower outstanding loan balance.
Income Tax Expense
During the three months ended September 30, 2021 and 2020, we recognized no income tax expense.
Comparison of the Nine Months Ended September 30, 2021 and 2020
The following table summarizes our results of operations for the nine months ended September 30, 2021 and 2020:
|
|
Nine Months Ended September 30, |
|
|||||||||
|
|
2021 |
|
|
2020 |
|
|
Change |
|
|||
|
|
|
|
|
(000’s) |
|
|
|
|
|||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|||
Research and development |
|
$ |
10,763 |
|
|
$ |
10,498 |
|
|
$ |
265 |
|
General and administrative |
|
|
7,500 |
|
|
|
9,364 |
|
|
|
(1,864 |
) |
In-process R&D |
|
|
50,618 |
|
|
|
— |
|
|
|
50,618 |
|
Total operating expenses |
|
|
68,881 |
|
|
|
19,862 |
|
|
|
(1,599 |
) |
Loss from operations |
|
|
(68,881 |
) |
|
|
(19,862 |
) |
|
|
1,599 |
|
Other expense, net |
|
|
(306 |
) |
|
|
(1,215 |
) |
|
|
909 |
|
Loss before income tax expense |
|
|
(69,187 |
) |
|
|
(21,077 |
) |
|
|
2,508 |
|
Income tax expense |
|
|
(13 |
) |
|
|
(32 |
) |
|
|
19 |
|
Net loss |
|
$ |
(69,200 |
) |
|
$ |
(21,109 |
) |
|
$ |
2,527 |
|
Research and Development Expenses
Research and development expenses increased by $0.3 million from $10.5 million for the nine months ended September 30, 2020 to $10.8 million for the nine months ended September 30, 2021. The increase was primarily attributable to an increase in lab supply costs of $0.5 million, manufacturing costs related to CTX-8371 of $0.4 million and increased facilities expenses of $0.3 million. These increases were partially offset by decreases in depreciation expense of $0.8 million.
23
We track outsourced development, outsourced personnel costs and other research and development costs of specific programs. In 2021, we began tracking our internal personnel costs on a program-by-program basis. Research and development expenses are summarized by program in the table below:
|
|
Nine Months Ended September 30, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
|
|
(000’s) |
|
|||||
|
|
|
|
|
|
|
||
CTX-471 |
|
$ |
2,763 |
|
|
$ |
2,250 |
|
CTX-8371 |
|
|
2,083 |
|
|
|
685 |
|
NKP30 cell engagement platform |
|
|
130 |
|
|
|
22 |
|
CTX-009 |
|
|
282 |
|
|
|
— |
|
Unallocated research and development expenses |
|
|
5,505 |
|
|
|
7,541 |
|
Total research and development expenses |
|
$ |
10,763 |
|
|
$ |
10,498 |
|
In-process R&D
In-process R&D was $50.6 million for the nine months ended September 30, 2021 and consisted of costs related to the TRIGR acquisition. There were no in-process R&D expenses in 2020. See Note 9 to the financial statements contained in this Form 10-Q for further description.
General and Administrative Expenses
General and administrative expenses decreased by $1.8 million to $7.5 million for the nine months ended September 30, 2021 from $9.4 million for the same period in 2020. The decrease was primarily attributable to a decrease in legal expenses of $0.9 million and a decrease in facilities expenses of $0.6 million.
Other Expense, net
We recognized interest expense of $0.3 million and $0.7 million during the nine months ended September 30, 2021 and 2020, respectively. The reduction in interest is from a lower outstanding loan balance.
A fair value of the derivative related to the Credit Facility was increased by $0.6 million as of September 30, 2020. Following our reverse merger with Compass Therapeutics LLC in June 2020, the derivative was settled.
Income Tax Expense
During the nine months ended September 30, 2021, we recognized income tax expenses of $13 thousand. During the nine months ended September 30, 2020, we recognized income tax expenses of $32 thousand.
Liquidity and Capital Resources
Since our inception, we have devoted substantially all of our efforts to organizing and staffing our Company, business planning, raising capital, research and development activities, building our intellectual property portfolio and providing general and administrative support for these operations. We have funded our operations primarily with proceeds from the sale of our equity securities and borrowings from debt arrangements. Through September 30, 2021, we have received $132.0 million in gross proceeds from the sale of equity securities and $15.0 million in term loan borrowings under the Credit Facility. Following the completion of the TRIGR Merger, we completed a private placement of our common stock and received gross proceeds of $60.5 million. As of September 30, 2021, we had cash and cash equivalents of $25.5 million.
In November 2021, we completed an underwritten public offering in which it received net proceeds of approximately $117.3 million after underwriting and offering fees. Following this offering, we paid off the balance of its outstanding loan. See Note 13 to the financial statements contained in this Form 10-Q for further description of these transactions.
Funding Requirements
Our primary use of cash is to fund operating expenses, primarily research and development expenditures. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in
24
our outstanding accounts payable, accrued expenses and prepaid expenses. Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amount of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our operations through a combination of equity offerings, debt financings, collaborations, strategic alliances and marketing, distribution or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, current stockholders’ interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect rights of common stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making acquisitions or capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or drug candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or other arrangements when needed, we may be required to delay, limit, reduce or terminate our research, product development or future commercialization efforts, or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Cash Flows
The following table shows a summary of our cash flows for the periods indicated:
|
|
Nine Months Ended September 30, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
|
|
(000’s) |
|
|||||
|
|
|
|
|
|
|
||
Cash used in operating activities |
|
$ |
(15,001 |
) |
|
$ |
(20,230 |
) |
Cash provided by (used in) investing activities |
|
|
(994 |
) |
|
|
32 |
|
Cash provided by (used in) financing activities |
|
|
(5,625 |
) |
|
|
50,413 |
|
Net increase (decrease) in cash, cash equivalents |
|
$ |
(21,620 |
) |
|
$ |
30,215 |
|
25
Operating Activities
During the nine months ended September 30, 2021, we used $15.0 million of cash in operating activities, resulting from our net loss of $69.2 million, offset by non-cash charges of $54.7 million. Our non-cash charges are from the TRIGR acquisition expense of in-process R&D of $50.6 million, share-based compensation expense of $2.8 million and depreciation and amortization of $0.4 million.
During the nine months ended September 30, 2020, we used $20.2 million of cash in operating activities, resulting from our net loss of $21.1 million and the change in operating assets and liabilities of $4.0 million, offset by non‑cash charges of $4.9 million. Our non‑cash charges were comprised of depreciation and amortization of $1.3 million, share-based compensation expense of $3.0 million, non-cash interest expense of $72 thousand, and a change in fair value of our derivative liability of $0.6 million. The change in our operating assets was primarily related to the settlement of a derivative liability and a decrease in our accrued expenses due to the timing in which we pay our vendors.
Investing Activities
During the nine months ended September 30, 2021, cash used in investing activities was $1.0 million attributed to $0.8 million in leasehold improvements and purchases of equipment and $0.3 million for acquisition costs related to TRIGR, partially offset by the sale of property and equipment. During the nine months ended September 30, 2020, cash provided by investing activities was $32 thousand attributable to the sale of property and equipment.
Financing Activities
During the nine months ended September 30, 2021, we had $5.6 million in principal payments under the Credit Facility. During the nine months ended September 30, 2020 we received net cash proceeds of $50.4 million from financing activities. This was primarily due to the closing of a private placement in June 2020, which resulted in net proceeds of $54.2 million that were partially offset by $3.8 in principal payments under the Credit Facility.
Indebtedness
In March 2018, we entered into the Credit Facility which matures on March 1, 2022 and consists of $15.0 million in term loans. The term loans bear interest at the greater of (i) 6.25% and (ii) the prime rate plus an applicable margin of 2.0%. As of September 30, 2021, the interest rate was 6.25%. We made interest-only payments through June 30, 2020, and beginning in April 2020, we began to make equal monthly principal payments of $625 thousand. Payments are scheduled through March 2022. As of September 30, 2021, $3.7 million was outstanding.
Future Funding Requirements
We expect our expenses to increase substantially in connection with our ongoing activities. The timing and amount of our operating expenditures will depend largely on:
26
We believe that our existing cash and cash equivalents as of filing of the form 10-Q will enable us to fund our operating expenses and capital expenditure requirements through the fourth quarter of 2024 based on our current plans, which may change based on clinical or pre-clinical results. These plans include: initiation of two Phase 2 clinical trials of CTX-009, completion of Phase 1 clinical trial of CTX-471, initiation of a Phase 1b combination trial for CTX-471 and commencement the planned Phase 1 development of CTX-8371, subject to satisfactory completion of IND-enabling activities for that product candidate. We expect that we will require additional funding to complete the clinical development of CTX-009, CTX-471 and CTX-8371, commercialize our product candidates, if we receive regulatory approval, and pursue in-licenses or acquisitions of other product candidates. If we receive regulatory approval for CTX-009, CTX-471 or CTX-8371 or other product candidates, we expect to incur significant commercialization expenses related to product manufacturing, sales, marketing and distribution, depending on where we choose to commercialize ourselves.
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity and debt financings, collaborations, strategic alliances, and marketing, distribution or licensing arrangements with third parties. To the extent that we raise additional capital through the sale of equity or convertible debt securities, ownership interest may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or other arrangements when needed, we may be required to delay, reduce or eliminate our product development or future commercialization efforts, or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
27
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
There have been no material changes from the quantitative and qualitative disclosures about market risk previously disclosed in Item 7A of our Annual Report on Form 10-K for the fiscal year ended December 31, 2020.
Critical Accounting Policies and Significant Judgments and Estimates
See Note 2 of the financial statements included in this Quarterly Report on Form 10-Q for the period ended September 30, 2021 and Part II, Item 7 “Critical Accounting Policies and Significant Judgements and Estimates” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2020 for our critical accounting policies and estimates.
Off‑Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off‑balance sheet arrangements, as defined in the rules and regulations of the SEC.
Recently Issued and Adopted Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our consolidated financial statements.
Item 4. Controls and Procedures.
Management’s Evaluation of our Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as of September 30, 2021. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of September 30, 2021, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act, that occurred during the quarter ended September 30, 2021 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
28
PART II—OTHER INFORMATION
Item 1. Legal Proceedings.
As of the date of this Quarterly Report on Form 10-Q, we are not involved in any material legal proceedings. However, from time to time, we could be subject to various legal proceedings and claims that arise in the ordinary course of our business activities. Regardless of the outcome, legal proceedings can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Item 1A. Risk Factors
In addition to the other information set forth in this report, you should carefully consider the risk factors discussed in Part I, Item 1A “Risk Factors” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2020, which could materially affect our business, financial condition, or results of operations.
Additional risk factors from the Annual Report on Form 10-K are as follows:
We may experience increased costs, disruptions or other difficulties with the integration of TRIGR.
On June 25, 2021, we consummated the acquisition of all outstanding shares of TRIGR Therapeutics, Inc., or TRIGR. As a result of this acquisition, we also acquired certain assets of TRIGR, including CTX-009 (formerly designated TR009/ABL001/NOV1501), an anti-DLL4 x VEGF-A bispecific antibody that is undergoing clinical development in patients with advanced solid tumors in South Korea. A Phase 1 dose escalation study and a Phase 1b dose expansion monotherapy study have been completed, and a Phase 1b combination study is ongoing. We plan to file an IND in the United States and continue the development of this product candidate across multiple indications. While we have invested, and continue to invest, significant resources in due diligence, planning and integration of TRIGR, it is possible that significant issues and potential unknown liabilities may arise during the course of the integration and ongoing development of CTX-009, which may result in increased costs, delays in the initiation of clinical trials and other difficulties that are not presently contemplated.
We have not historically developed the product candidate we acquired in our recent acquisition of TRIGR and are relying on TRIGR’s prior research to advance this product candidate and intellectual property obtained by TRIGR.
The product candidate that was acquired in the TRIGR acquisition, CTX-009, was initially developed by TRIGR and its licensor ABL Bio. We have not yet demonstrated an ability to develop, advance, or run clinical trials with this product candidate. In addition, our intellectual property rights covering the product candidate that was acquired in the TRIGR acquisition, CTX-009, were developed by TRIGR and its licensor, ABL Bio. We were not responsible for obtaining, maintaining or enforcing such intellectual property rights and we are relying on the previous work of TRIGR and its licensor to have adequately protected such intellectual property. We are relying on TRIGR’s previous work to continue our development of this product candidate. As a result, we cannot ensure that we will be able to successfully advance this product candidate going forward or that the intellectual property rights were protected at a level comparable to our previously disclosed intellectual property.
Certain of our clinical trials are conducted in overseas jurisdictions, which may subject us to delays and expenses.
We are conducting certain clinical trials in overseas jurisdictions. For example, clinical trials for CTX-009 are currently being conducted in South Korea. Regulators in the United States, such as the FDA, or in other foreign jurisdictions, may not support our trial design and protocol, which would delay our clinical development plans and increase our expenses.
In addition, there are risks inherent in conducting clinical trials in overseas jurisdictions, which may subject us to delays and expenses, such as:
29
The following risk factors from the Annual Report on Form 10-K have been modified:
We have a limited operating history and no products approved for commercial sale. We have a history of significant losses, expect to continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability.
We are a clinical-stage biopharmaceutical company with a limited operating history. Since our founding in 2014, we have incurred significant net losses. Our net losses were $29.5 million and $34.7 million for the years ended December 31, 2020 and 2019, respectively, and as of December 31, 2020 and September 30, 2021, we had an accumulated deficit of $151.4 million and $220.6 million, respectively. In addition, as of December 31, 2020 and September 30, 2021, we had stockholders’ equity of $39.9 million and $23.9 million, respectively. We have funded our operations to date primarily with proceeds from private placements of preferred and common equity and borrowings under the Credit Facility. Since commencing operations, we have devoted substantially all of our efforts and financial resources to organizing and staffing our company, identifying business development opportunities, raising capital, securing intellectual property rights related to our product candidates, conducting discovery, and research and development activities for our product candidates.
We expect that it will be several years, if ever, before we have a commercialized product. We expect to continue to incur significant expenses and operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially if, and as, we:
To become and remain profitable, we must develop and eventually commercialize products with significant market potential. This will require us to be successful in a range of challenging activities, including completing preclinical studies and clinical trials, obtaining marketing approval for product candidates, manufacturing, marketing and selling products and satisfying any post-marketing requirements. We may never succeed in any or all of these activities and, even if we do, we may never generate revenue that is significant or large enough to achieve profitability. If we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
30
We have never generated revenue from product sales and may never be profitable.
Our ability to generate revenue from product sales and achieve profitability depends on our ability, alone or with our collaboration partners, to successfully complete the development of, and obtain the regulatory approvals necessary to commercialize, our product candidates. We do not anticipate generating revenue from product sales for the next several years, if ever. Our ability to generate future revenue from product sales depends heavily on our, or our existing or future collaborators’, success in:
We anticipate incurring significant costs associated with commercializing any product candidate that is approved for commercial sale. Our expenses could increase beyond expectations if we are required by the FDA or other regulatory agencies to perform clinical trials or studies in addition to those that we currently anticipate, or if there are any delays in establishing appropriate manufacturing arrangements for or in completing our clinical trials for the development of any of our product candidates, for example, as a result of any setbacks or delays due to the COVID-19 pandemic. Even if we are able to generate revenue from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations.
We will require substantial additional financing to pursue our business objectives, which may not be available on acceptable terms, or at all. A failure to obtain this necessary capital when needed could force us to delay, limit, reduce or terminate our product development, commercialization efforts or other operations.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to continue the preclinical and clinical development of our current and future programs. If we receive marketing approval for any product candidates, we will require significant additional amounts of cash in order to launch and commercialize such product candidates. In addition, other unanticipated costs may arise. Because the designs and outcomes of our planned and anticipated clinical trials are highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development of and commercialize any product candidate we develop. Additionally, any COVID-19 related program setbacks or delays due to changes in federal, state, or local laws and regulations or clinical site policies could impact our programs and increase our expenditures.
31
Our future capital requirements depend on many factors, including:
Until we can generate sufficient product revenue to finance our cash requirements, which we may never do, we expect to finance our future cash needs through a combination of public or private equity and debt financings, marketing and distribution arrangements, other collaborations, strategic alliances and licensing arrangements. Based on our research and development plans, we expect that our cash resources will enable us to fund our operating expenses and capital expenditure requirements over the next 12 months. This estimate is based on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we expect. Changes may occur beyond our control that would cause us to consume our available capital before that time, including changes in, and progress of, our development activities, acquisitions of additional product candidates and changes in regulation.
If we raise additional capital through marketing, sales and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish certain valuable rights to our product candidates, future revenue streams or research programs, technologies or grant licenses on terms that may not be favorable to us. If we raise additional capital through public or private equity offerings, the terms of these securities may include liquidation or other preferences that adversely affect our stockholders’ rights. Further, to the extent that we raise additional capital through additional sales of common stock or securities convertible or exchangeable into common stock, investors’ ownership interest will be diluted. If we raise additional capital through debt financing, we would be subject to fixed payment obligations and may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
Adequate additional financing may not be available to us on acceptable terms, or at all. If we are unable to obtain additional financing on favorable terms when needed, we may be required to delay, limit, reduce or terminate preclinical studies, clinical trials, or other research and development activities or one or more of our development programs.
32
Risks related to the discovery and development of our product candidates
Our business is dependent on our ability to advance our current and future product candidates through clinical trials, obtain marketing approval and ultimately commercialize them.
We are early in our development efforts. We will be conducting our first clinical trials since acquiring CTX-009, currently our lead product candidate, and are currently conducting clinical trials for CTX-471. Our ability to generate product revenues, which we do not expect will occur for several years, if ever, will depend heavily on the successful development and eventual commercialization of CTX-009, CTX-471, CTX-8371 and any other current or future product candidates we develop, which may never occur. Our current product candidates, including CTX-009, CTX-471, CTX-8371 and any future product candidates we develop will require additional preclinical or clinical development, management of clinical, preclinical and manufacturing activities, marketing approval in the United States and other jurisdictions, demonstration of effectiveness to pricing and reimbursement authorities, sufficient cGMP manufacturing supply for both preclinical and clinical development and commercial production, building of a commercial organization and substantial investment and significant marketing efforts before we generate any revenues from product sales.
The clinical and commercial success of our current and future product candidates will depend on several factors, including the following:
33
These factors, many of which are beyond our control, could cause us to experience significant delays or an inability to obtain regulatory approvals or commercialize our current or future product candidates, and could otherwise materially harm our business. Successful completion of preclinical studies and clinical trials does not mean that CTX-009, CTX-471, CTX-8371 or any other current or future product candidates we develop will receive regulatory approval. Even if regulatory approvals are obtained, we could experience significant delays or an inability to successfully commercialize our current and any future product candidates we develop, which would materially harm our business. If we are not able to generate sufficient revenue through the sale of any current or future product candidate, we may not be able to continue our business operations or achieve profitability.
Clinical development involves a lengthy and expensive process with uncertain outcomes. We may incur additional costs and experience delays in developing and commercializing or be unable to develop or commercialize our current and future product candidates.
To obtain the requisite regulatory approvals to commercialize any of our product candidates, we must demonstrate through extensive preclinical studies and clinical trials that our product candidates are safe, pure and potent in humans. Clinical testing is expensive and can take many years to complete, and its outcome is highly uncertain. Failure can occur at any time during the clinical trial process and our future clinical trial results may not be successful. We may experience delays in completing our clinical trials or preclinical studies and initiating or completing additional clinical trials. Although certain trials (or portions thereof) of CTX-009 and CTX-471 have been completed, we may experience delays in completing ongoing or future trials or in initiating any planned clinical trials and development efforts. Additionally, we cannot be certain the ongoing and planned preclinical studies or clinical trials for CTX-009, CTX-471, CTX-8371 or any other future product candidates will begin on time, not require redesign, enroll an adequate number of subjects on time or be completed on schedule, if at all. We may also experience numerous unforeseen events during our clinical trials that could delay or prevent our ability to receive marketing approval or commercialize the product candidates we develop, including:
34
We could encounter delays if a clinical trial is suspended or terminated by us, or by the IRBs of the institutions in which such trials are being conducted, ethics committees or the Data Safety Monitoring Board, or DSMB, for such trial or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of marketing approval of our product candidates. The FDA or other regulatory authorities may change the requirements for approval even after they have reviewed and commented on the design for our clinical trials. Further, the FDA or other regulatory authorities may disagree with our clinical trial design and our interpretation of data from clinical trials. For example, we are conducting and may in the future conduct additional “open-label” clinical trials. An “open-label” clinical trial is one where both the patient and investigator know whether the patient is receiving the investigational product candidate or either an existing approved drug or placebo. Most typically, open-label clinical trials test only the investigational product candidate and sometimes may do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients may be subject to a “patient bias” where patients perceive their symptoms to have improved merely due to their awareness of receiving an experimental treatment. Moreover, patients selected for early clinical studies often include the most severe sufferers and their symptoms may have been bound to improve notwithstanding the new treatment. In addition, open-label clinical trials may be subject to an “investigator bias” where those assessing and reviewing the physiological outcomes of the clinical trials
35
are aware of which patients have received treatment and may interpret the information of the treated group more favorably given this knowledge.
Principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or a regulatory authority concludes that the financial relationship may have affected the interpretation of the trial, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection of the marketing application we submit. Any such delay or rejection could prevent or delay us from commercializing our current or future product candidates.
If we experience delays in the completion, or termination, of any clinical trial of our product candidates, including as a result of the COVID-19 pandemic, the commercial prospects of our product candidates will be harmed and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down the development and approval process for our product candidates and jeopardize our ability to commence product sales and generate revenues. Significant clinical trial delays could also allow our competitors to bring products to market before we do or shorten any periods during which we have the exclusive right to commercialize our product candidates.
Any such events would impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates or result in the development of our product candidates stopping early.
Preclinical development is uncertain. Our preclinical programs may experience delays or may never advance to clinical trials, which would adversely affect our ability to obtain regulatory approvals or commercialize these programs on a timely basis or at all.
With the exception of CTX-009, which is undergoing clinical trials in South Korea, and CTX-471, which is currently being tested in a Phase 1 clinical trial, all of our product candidates are still in the discovery or preclinical stage, and the risk of failure for such product candidates is high. In addition, any one or more of our product candidates that have not yet entered the clinic may never advance into clinical development. For instance, in early 2021, we conducted a review of our pipeline and made the strategic decision to deprioritize the development efforts for our NKp30 innate cell engager platform and to refrain from advancing CTX-8573 to IND-enabling studies. In order to obtain FDA approval to market a new biologic we must demonstrate proof of safety, purity and potency, including efficacy, in humans. To meet these requirements we will have to conduct adequate and well-controlled clinical trials. Before we can commence clinical trials for a product candidate, we must complete extensive preclinical testing and studies that support our planned clinical trials in humans. We cannot be certain of the timely completion or outcome of our preclinical testing and studies and cannot predict if the FDA will accept our proposed clinical programs or if the outcome of our preclinical testing and studies will ultimately support the further development of our current or future product candidates. As a result, we cannot be sure that we will be able to submit INDs or similar applications for our preclinical programs on the timelines we expect, if at all, and we cannot be sure that submission of INDs or similar applications will result in the FDA or other regulatory authorities allowing clinical trials to begin.
Conducting preclinical testing is a lengthy, time-consuming and expensive process. The length of time of such testing may vary substantially according to the type, complexity and novelty of the program, and often can be several years or more per program. Delays associated with programs for which we are conducting preclinical testing and studies may cause us to incur additional operating expenses. The commencement and rate of completion of preclinical studies and clinical trials for a product candidate may be delayed by many factors, including but not limited to:
36
Our current or future product candidates may cause undesirable side effects or have other properties when used alone or in combination with other approved products or investigational new drugs that could halt their clinical development, delay or prevent their regulatory approval, limit their commercial potential or result in significant negative consequences.
Before obtaining regulatory approvals for the commercial sale of our product candidates, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that our product candidates are safe, pure and potent for use in each target indication, and failures can occur at any stage of testing. As with most biological products, use of our current or future product candidates could be associated with side effects or adverse events which can vary in severity from minor reactions to death and in frequency from infrequent to prevalent. There have been serious adverse side effects reported in response to antibody therapeutics and bispecifics in oncology.
Immuno-oncology drugs have been observed to cause side effects, generally related to over-activation of the immune system. These include colitis, diabetes, pituitary inflammation, thyroiditis, myocarditis, liver inflammation, thrombocytopenia, among others. Our immuno-oncology product candidates, including CTX-471, may have similar or additional side effects.
We are developing CTX-8371 as a potential bispecific antibody that simultaneously targets both PD-1 and PD-L1, the targets of well-known and widely used checkpoint inhibitor antibodies. While we have observed so far in preclinical testing that simultaneous targeting of both PD-1 and PD-L1 has been associated with less toxicity than targeting either PD-1 alone or PD-L1 alone, there can be no assurance that CTX-8371 will not demonstrate unacceptable toxicities in later testing that may render it unsafe or intolerable.
If unacceptable side effects arise in the development of our product candidates, we, the FDA, the IRBs at the institutions in which our studies are conducted or the DSMB could suspend or terminate our clinical trials or the FDA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete any of our clinical trials or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel using our product candidates to understand the side effect profiles for our clinical trials and upon any commercialization of any of our product candidates. Inadequate training in recognizing or managing the potential side effects of our product candidates could result in patient injury or death. Any of these occurrences may harm our business, financial condition and prospects significantly.
Although our current and future product candidates have undergone and will undergo safety testing to the extent possible and, where applicable, under such conditions discussed with regulatory authorities, not all adverse effects of drugs can be predicted or anticipated. Antibody therapeutics and bispecifics and their method of action of harnessing the body’s immune system are powerful and could lead to serious side effects that we only discover in clinical trials or during commercial marketing. Unforeseen side effects could arise either during clinical development or after our product candidates have been approved by regulatory authorities and the approved product has been marketed, resulting in the exposure of additional patients. So far, we have not demonstrated that CTX-009, CTX-471, CTX-8371 or any other product candidate is safe in humans, and we cannot predict if ongoing or future clinical trials will do so. If any of our current or future product candidates fail to demonstrate safety and efficacy in clinical trials or do not gain marketing approval, we will not be able to generate revenue and our business will be harmed.
In addition, we intend to pursue CTX-009 and CTX-471 in part in combination with other therapies and may develop CTX-8371 and future product candidates in combination with other therapies, which exposes us to additional risks relating to undesirable side effects or other properties. For example, the other therapies may lead to toxicities that are improperly attributed to our product candidates or the combination of our product candidates with other therapies may result in toxicities that the product candidate or other therapy does not produce when used alone. The other therapies we are using in combination may be removed from the market, or we may not be able to secure adequate quantities of such materials for which we have no guaranteed supply contract, and thus be unavailable for testing or commercial use with any of our approved products. The other therapies we may use in combination with our product candidates may also be supplanted in the market by newer, safer or more efficacious products or combinations of products.
37
Even if we successfully advance one of our product candidates through clinical trials, such trials will likely only include a limited number of subjects and limited duration of exposure to our product candidates. As a result, we cannot be assured that adverse effects of our product candidates will not be uncovered when a significantly larger number of patients are exposed to the product candidate. Further, any clinical trial may not be sufficient to determine the effect and safety consequences of taking our product candidates over a multi-year period.
If any of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
Any of the foregoing events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and result in the loss of significant revenues, which would materially harm our business. In addition, if one or more of our product candidates or our antibody therapeutic development approach generally prove to be unsafe, our entire technology platform and pipeline could be affected, which would also materially harm our business.
As an organization, we have limited experience designing and implementing clinical trials and we have never conducted pivotal clinical trials. Failure to adequately design a trial, or incorrect assumptions about the design of the trial, could adversely affect the ability to initiate the trial, enroll patients, complete the trial, or obtain regulatory approval on the basis of the trial results, as well as lead to increased or unexpected costs and in delayed timelines.
The design and implementation of clinical trials is a complex process. We have limited experience designing and implementing clinical trials, and we may not successfully or cost-effectively design and implement clinical trials that achieve our desired clinical endpoints efficiently, or at all. A clinical trial that is not well designed may delay or even prevent initiation of the trial, can lead to increased difficulty in enrolling patients, may make it more difficult to obtain regulatory approval for the product candidate on the basis of the study results, or, even if a product candidate is approved, could make it more difficult to commercialize the product successfully or obtain reimbursement from third-party payors. Additionally, a trial that is not well-designed could be inefficient or more expensive than it otherwise would have been, or we may incorrectly estimate the costs to implement the clinical trial, which could lead to a shortfall in funding. We also expect to continue to rely on third parties to conduct our clinical trials.
See “—Risks Related to Reliance on Third Parties—We rely or will rely on third parties to help conduct our ongoing and planned preclinical studies and clinical trials for CTX-009, CTX-471, CTX-8371 and any future product candidates we develop. If these third parties do not successfully carry out their contractual duties, comply with regulatory requirements or meet expected deadlines, we may not be able to obtain marketing approval for or commercialize CTX-009, CTX-471, CTX-8371 and any current or future product candidates we develop and our business could be materially harmed.”
Consequently, we may be unable to successfully and efficiently execute and complete clinical trials that are required for BLA submission and FDA approval of CTX-009, CTX-471, CTX-8371 or current or future product candidates. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we develop.
38
We have chosen to prioritize development of CTX-009, CTX-471 and CTX-8371. We may expend our limited resources on product candidates or indications that do not yield a successful product and fail to capitalize on other candidates or indications for which there may be a greater likelihood of success or may be more profitable.
Because we have limited resources, we have strategically determined to prioritize development of CTX-009, CTX-471 and CTX-8371 rather than other product candidates. This decision is based, in part, on the significant resources required for developing and manufacturing antibody therapeutics and bispecifics. To date, no regulatory authority has granted approval for an antibody therapeutic targeting CD137, also known as 4-1BB, as well as the target of CTX-471. Of note, several drugs targeting CD137 have been tested in early-stage clinical trials, and at least one of these drugs had severe side effects. It is possible that CTX-471 may have similar adverse effects, including toxicity, in humans. As a result, we may be foregoing other potentially more profitable antibody therapies or drugs with a greater likelihood of success. Our decisions concerning the allocation of research, development, collaboration, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of any viable commercial product and may divert resources away from better opportunities.
Similarly, our potential decisions to delay, terminate or collaborate with third parties with respect to certain programs may subsequently also prove to be suboptimal and could cause us to miss valuable opportunities. If we make incorrect determinations regarding the viability or market potential of any of our current or future product candidates or misread trends in the oncology, autoimmunology or biopharmaceutical industry, our business, financial condition and results of operations could be materially adversely affected. As a result, we may fail to capitalize on viable commercial products or profitable market opportunities, be required to forego or delay pursuit of opportunities with other product candidates or other diseases and disease pathways that may later prove to have greater commercial potential than those we choose to pursue, or relinquish valuable rights to such product candidates through collaboration, licensing or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain development and commercialization rights.
Risks related to regulatory approval of our product candidates
Regulatory agencies may not agree with the design of our clinical development programs for one or more of our product candidates that we intend to utilize to support an expedited development process.
We intend to pursue an accelerated approval pathway for one or more of our product candidates, such as CTX-009, where we believe our proposed adaptive design of our clinical development program for this product candidate could support expedited development. However, we have not yet reached any agreement with the FDA or comparable foreign regulatory authorities on the availability of such a strategy, nor have we initiated discussions to seek such agencies’ feedback on such pathway. There can be no assurance that we will be successful in reaching alignment with regulatory authorities with the effect and purpose of shortening the development path for any of our product candidates.
We intend to develop CTX-009 and CTX-471 in part in combination with other therapies and may develop CTX-8371 and future product candidates in combination with other therapies, which exposes us to additional regulatory risks.
We intend to develop CTX-009 in combination with other therapies, such as chemotherapy, and CTX-471 in part in combination with other therapies, such as trastuzumab, and may develop CTX-8371 and future product candidates in combination with one or more currently approved cancer therapies. These combinations have not been previously tested in the clinic and may, among other things, fail to demonstrate synergistic activity, may fail to achieve superior outcomes relative to the use of single agents or other combination therapies, or may fail to demonstrate sufficient safety or efficacy traits in clinical trials to enable us to complete those clinical trials or obtain marketing approval for the combination therapy.
In addition, we did not develop or obtain regulatory approval for, and we do not manufacture or sell, any of these approved therapeutics. Therefore, even if any product candidate we develop were to receive marketing approval or be commercialized for use in combination with other existing therapies, we would continue to be subject to the risk that the FDA or comparable foreign regulatory authorities could revoke approval of the therapy used in combination with our product candidate or that safety, efficacy, manufacturing or supply issues could arise with these existing therapies. This could result in our own products being removed from the market or being less successful commercially. Combination
39
therapies are commonly used for the treatment of cancer diseases, and we would be subject to similar risks if we develop any of our product candidates for use in combination with other drugs or for indications other than cancer.
We may also evaluate CTX-009, CTX-471, CTX-8371 or any future product candidate in combination with one or more other cancer therapies that have not yet been approved for marketing by the FDA or comparable foreign regulatory authorities. We will not be able to market and sell CTX-009, CTX-471, CTX-8371 or any product candidate we develop in combination with any such unapproved cancer therapies that do not ultimately obtain marketing approval.
If the FDA or comparable foreign regulatory authorities do not approve these other biological products or revoke their approval of, or if safety, efficacy, manufacturing or supply issues arise with, the biological products we choose to evaluate in combination with CTX-009, CTX-471, CTX-8371 or any product candidate we develop, we may be unable to obtain approval of or market any such product candidate.
Risks related to the commercialization of our product candidates
The market opportunities for any current or future product candidate we develop, if approved, may be limited to those patients who are ineligible for established therapies or for whom prior therapies have failed, and may be small.
Any revenue we are able to generate in the future from product sales will be dependent, in part, upon the size of the market in the United States and any other jurisdiction for which we gain regulatory approval and have commercial rights. If the markets or patient subsets that we are targeting are not as significant as we estimate, we may not generate significant revenues from sales of such products, even if approved.
Cancer therapies are sometimes characterized as first-line, second-line or third-line, and the FDA often approves new therapies initially only for third-line use. When cancer is detected early enough, first-line therapy, usually chemotherapy, hormone therapy, surgery, radiation therapy or a combination of these, is sometimes adequate to cure the cancer or prolong life without a cure. Second- and third-line therapies are administered to patients when prior therapy is not effective. The number of patients who receive second- and third-line treatment is significantly smaller than the number of patients who receive first-line treatment, and the prognosis of patients who receive second- or third-line treatment is often poorer than that of patients who receive first-line treatment.
We may initially seek approval for CTX-009, CTX-471, CTX-8371 and any other product candidates we develop as second- or third-line therapies. If we do so, for those products that prove to be sufficiently beneficial, if any, we would expect potentially to seek approval as a first-line therapy, but there is no guarantee that any product candidate we develop, even if approved, would be approved for first-line therapy, and, prior to any such approvals, we may have to conduct additional clinical trials.
The number of patients who have the types of cancer or autoimmune diseases we are targeting may turn out to be lower than expected. Additionally, the potentially addressable patient population for our current or future product candidates may be limited, if and when approved. Even if we obtain significant market share for any product candidate, if and when approved, if the potential target populations are small, we may never achieve profitability without obtaining marketing approval for additional indications, including to be used as first- or second-line therapy.
If we are unable to establish marketing, sales and distribution capabilities for CTX-009, CTX-471, CTX-8371 or any other product candidate that may receive regulatory approval, we may not be successful in commercializing those product candidates if and when they are approved.
We do not have sales or marketing infrastructure. To achieve commercial success for CTX-009, CTX-471, CTX-8371 and any other product candidate for which we may obtain marketing approval, we will need to establish a sales and marketing organization. In the future, we expect to build a focused sales and marketing infrastructure to market some of our product candidates in the United States, if and when they are approved. There are risks involved with establishing our own marketing, sales and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
40
Factors that may inhibit our efforts to market our products on our own include:
If we are unable to establish our own marketing, sales and distribution capabilities and are forced to enter into arrangements with, and rely on, third parties to perform these services, our revenue and our profitability, if any, are likely to be lower than if we had developed such capabilities ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute our product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish marketing, sales and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
Risks related to healthcare, insurance and legal matters
Enacted healthcare legislation, changes in healthcare law and implementation of regulations, as well as changes in healthcare policy, may increase the difficulty and cost for us to commercialize our product candidates, may impact our business in ways that we cannot currently predict, could affect the prices we may set, and could have a material adverse effect on our business and financial condition.
In the United States and in some foreign jurisdictions, there have been and likely will continue to be a number of legislative and regulatory changes regarding the healthcare system directed at broadening the availability of healthcare, improving the quality of healthcare, and containing or lowering the cost of healthcare. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or, collectively, the ACA, was passed, which substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA, among other things, subjects biological products to potential competition by lower-cost biosimilars, expands the types of entities eligible for the 340B drug discount program, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program, extends the rebate program to individuals enrolled in Medicaid managed care organizations, establishes annual fees and taxes on manufacturers of certain branded prescription drugs, and creates a new Medicare Part D coverage gap discount program in which, as a condition of coverage of its products under Medicare Part D, manufacturers must now agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, or BBA, effective as of 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period.
Since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future. Various portions of the ACA are currently undergoing legal and constitutional challenges in the United States Supreme Court; the previous Trump Administration issued various Executive Orders which eliminated cost sharing subsidies and various provisions that would impose a fiscal burden on states or a cost, fee, tax, penalty or regulatory burden on individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices; and Congress has introduced several pieces of legislation aimed at significantly revising or repealing the ACA. Also, in December 2018, the Centers for Medicare and Medicaid Services, or CMS, issued a final rule permitting further collections and payments to and from certain ACA qualified health plans and health insurance issuers under the ACA risk adjustment program. It is unclear whether the ACA will be overturned, repealed, replaced, or further amended. We cannot predict what affect further changes to the ACA would have on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example, on August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit
41
reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030 unless additional congressional action is taken. However, the Medicare sequester reductions under the BCA will be suspended from May 1, 2020 through December 31, 2020 due to the COVID-19 pandemic. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Additionally, the BBA, among other things, amended the ACA, effective January 1, 2019, to increase the point-of-sale discount (from 50% under the ACA to 70%) that is owed by pharmaceutical manufacturers who participate in Medicare Part D and, closed the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”.
Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, CMS may develop new payment and delivery models, such as bundled payment models. Recently, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Such scrutiny has resulted in several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the cost of drugs under Medicare and reform government program reimbursement methodologies for drug products. For example, at the federal level, the previous Trump administration’s budget proposal for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the previous Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Additionally, the previous Trump Administration previously released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out-of-pocket costs of drug products paid by consumers. HHS has started soliciting feedback on some of these measures and, at the same, is implementing others under its existing authority. For example, in May 2019, CMS issued a final rule to allow Medicare Advantage Plans the option of using step therapy, a type of prior authorization, for Part B drugs beginning January 1, 2020. This final rule codified CMS’s policy change that was effective January 1, 2019. Although a number of these and other proposed measures may require additional authorization to become effective, Congress has indicated that they will continue to seek new legislative and/or administrative measures to control drug costs.
On July 24, 2020 and September 13, 2020, then-President Trump announced several executive orders related to prescription drug pricing that seek to implement several of the administration's proposals. In response, the FDA released a final rule on September 24, 2020, which went into effect on November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, CMS issued an Interim Final Rule implementing the Most Favored Nation (“MFN”) Model under which Medicare Part B reimbursement rates will be calculated for certain drugs and biologicals based on the lowest price drug manufacturers receive in Organization for Economic Cooperation and Development countries with a similar gross domestic product per capita. The MFN Model regulations mandate participation by identified Part B providers and will apply in all U.S. states and territories for a seven-year period beginning January 1, 2021 and ending December 31, 2027. On December 28, 2020, a judge in the U.S. District Court for the Northern District of California granted a preliminary injunction prohibiting CMS from implementing the MFN rule. On August 6, 2021, CMS released a proposed rule that would rescind the MFN rule, although the timing of its adoption remains uncertain as of the date hereof.
At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions on coverage or access could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our product candidates that we successfully commercialize or put pressure on our product pricing.
42
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the extent to which state and federal governments cover particular healthcare products and services and could limit the amounts that the federal and state governments will pay for healthcare products and services. This could result in reduced demand for any product candidate or complementary or companion diagnostics we develop or could result in additional pricing pressures.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the United States. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
Risks related to manufacturing of our product candidates
The loss of our third-party manufacturing partners or our, or our partners’, failure to comply with applicable regulatory requirements or to supply sufficient quantities at acceptable quality levels or prices, or at all, would materially and adversely affect our business.
We have contracted with qualified third-party contract manufacturing organizations, or CMOs, to manufacture our product candidates for preclinical and clinical trials. If approved, commercial supply of CTX-009, CTX-471, CTX-8371 and any future product candidates may also be manufactured at one or more CMOs.
The facilities used by our CMOs to manufacture our product candidates are subject to various regulatory requirements and may be subject to the inspection of the FDA or other regulatory authorities. We do not control the manufacturing process at our CMOs, and are completely dependent on them for compliance with current regulatory requirements. If we or our CMOs cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or comparable regulatory authorities in foreign jurisdictions, we may not be able to rely on their manufacturing facilities for the manufacture of elements of our product candidates. In addition, we have limited control over the ability of our CMOs to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority finds our facilities or those of our CMOs inadequate for the manufacture of our product candidates or if such facilities are subject to enforcement action in the future or are otherwise inadequate, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates.
Additionally, our CMOs may experience manufacturing difficulties due to resource constraints or as a result of labor disputes or unstable political environments or on account of global pandemics or similar events, including the COVID-19 pandemic. If our CMOs were to encounter any of these difficulties, our ability to provide our product candidate to patients in clinical trials, or to provide product for the treatment of patients once approved, would be jeopardized.
Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates proceed through preclinical studies to late-stage clinical trials towards potential approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and results. Therefore, we will likely need to change our CMOs for manufacturing of any product candidates that advance to later-stage trials to those that can support commercial-scale manufacturing. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the materials manufactured using altered processes. Such changes may also require additional testing, FDA notification or FDA approval. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commence sales and generate revenue.
43
We are subject to multiple manufacturing risks, any of which could substantially increase our costs and limit supply of our product candidates.
The process of manufacturing antibody therapeutics and bispecifics, including our product candidates, is complex, time-consuming, highly regulated and subject to several risks, including:
We have experienced, and may in the future experience challenges with our third party manufacturers that could have an adverse impact on the development of our product candidates. For example, the expected timing of submission of our IND for our product candidate CTX-8371 was shifted to mid-2022 due to delays at our outside manufacturer for that candidate.
We may also make changes to our manufacturing processes at various points during development, for a number of reasons, such as controlling costs, achieving scale, decreasing processing time, increasing manufacturing success rate or other reasons. Such changes carry the risk that they will not achieve their intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of our ongoing or future clinical trials. In some circumstances, changes in the manufacturing process may require us to perform ex vivo comparability studies and to collect additional data from patients prior to undertaking more advanced clinical trials. For instance, changes in our process during the course of clinical development may require us to show the comparability of the product used in earlier clinical phases or at earlier portions of a trial to the product used in later clinical phases or later portions of the trial.
Risks related to intellectual property
Our intellectual property agreements with third parties may be subject to disagreements over contract interpretation, which could narrow the scope of our rights to the relevant intellectual property or technology, resulting in termination of our access to such intellectual property or increase our financial or other obligations to our licensors.
The agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in our intellectual property agreements may be susceptible to multiple interpretations. Such licenses include those with ABL Bio Inc. with respect to our CTX-009 product candidate and with Adimab, LLC, with respect to our CTX-471 product candidate. The resolution of any contract interpretation disagreement that may arise could affect the scope of our rights to the relevant intellectual property or technology, or affect financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, or we otherwise are unable to maintain our licenses for our product candidates, we may be unable to successfully develop and commercialize the affected product candidates.
44
Risks related to our work with third parties
We rely or will rely on third parties to help conduct our ongoing and planned preclinical studies and clinical trials for CTX-009, CTX-471, CTX-8371 and any future product candidates we develop. If these third parties do not successfully carry out their contractual duties, comply with regulatory requirements or meet expected deadlines, we may not be able to obtain marketing approval for or commercialize CTX-009, CTX-471, CTX-8371 and any current or future product candidates we develop and our business could be materially harmed.
We currently do not have the ability to independently conduct preclinical studies that comply with the regulatory requirements known as current good laboratory practice, or GLP, requirements. We also do not currently have the ability to independently conduct any clinical trials. The FDA and regulatory authorities in other jurisdictions require us to comply with regulations and standards, including cGCP, or requirements for conducting, monitoring, recording and reporting the results of clinical trials, in order to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. We rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, to conduct GLP-compliant preclinical studies and cGCP-compliant clinical trials on our product candidates properly and on time. While we have agreements governing their activities, we control only certain aspects of their activities and have limited influence over their actual performance. The third parties with whom we contract for execution of our GLP-compliant preclinical studies and our cGCP-compliant clinical trials play a significant role in the conduct of these studies and trials and the subsequent collection and analysis of data. These third parties are not our employees and, except for restrictions imposed by our contracts with such third parties, we have limited ability to control the amount or timing of resources that they devote to our current or future product candidates. Although we rely on these third parties to conduct our GLP-compliant preclinical studies and cGCP-compliant clinical trials, we remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol and applicable laws and regulations, and our reliance on the CROs does not relieve us of our regulatory responsibilities.
Many of the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position. Further, under certain circumstances, these third parties may unilaterally terminate their agreements with us. If the third parties conducting our preclinical studies or our clinical trials do not adequately perform their contractual duties or obligations, experience significant business challenges, disruptions or failures, do not meet expected deadlines, including on account of the COVID-19 pandemic, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our protocols or to GLP and cGCP, or for any other reason, we may need to enter into new arrangements with alternative third parties. This could be difficult, costly or impossible, and our preclinical studies or clinical trials may need to be extended, delayed, terminated or repeated. As a result, we may not be able to obtain regulatory approval in a timely fashion, or at all, for the applicable product candidate, our financial results and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
Risks related to our business
We will need to grow the size of our organization, and we may experience difficulties in managing this growth.
As our development plans and strategies develop, and as we transition into operating as a public company, we expect to need additional managerial, operational, marketing, sales, financial and other personnel. Future growth would impose significant added responsibilities on members of management, including:
Our future financial performance and our ability to advance development of and, if approved, commercialize CTX-009, CTX-471, CTX-8371 and any current or future product candidates we develop will depend, in part, on our ability to
45
effectively manage any future growth, and our management may have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors and consultants to provide certain services. We cannot assure you that the services of independent organizations, advisors and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain marketing approval of any current or future product candidates or otherwise advance our business. We cannot assure you that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, or at all.
If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop and commercialize CTX-009, CTX-471, CTX-8371 and any current or future product candidates we develop and, accordingly, may not achieve our research, development and commercialization goals.
46
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
None.
Item 3. Defaults Upon Senior Securities.
None.
Item 4. Mine Safety Disclosures.
Not applicable.
Item 5. Other Information.
On November 8, 2021, we paid off the balance of the notes and terminated the Credit Facility. After this pay off, we have no additional debt facilities.
On November 12, 2021, we issued a press release announcing the Company's financial results for the quarter ended September 30, 2021. A copy of this press release is attached as Exhibit 99.1 to this Quarterly Report. The information regarding this press release in this Item 5 (including Exhibit 99.1) shall not be deemed "filed" for purposes of Section 18 of the Exchange Act or otherwise subject to the liabilities of that section, nor shall it be deemed incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as expressly set forth by specific reference in such a filing.
Item 6. Exhibits.
Exhibit Number |
|
Description |
|
|
|
31.1* |
|
|
|
|
|
31.2* |
|
|
|
|
|
32.1** |
|
|
|
|
|
32.2** |
|
|
99.1 |
|
|
|
|
|
101.INS |
|
Inline XBRL Instance Document – the instance document does not appear in Interactive Data File because its XBRL tags are embedded within the Inline XBRL Document |
|
|
|
101.SCH |
|
Inline XBRL Taxonomy Extension Schema Document |
|
|
|
101.CAL |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
101.DEF |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
101.LAB |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
101.PRE |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
104 |
|
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
* Filed herewith.
** These exhibits are being furnished and shall not be deemed to be “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, nor shall such exhibits be deemed to be incorporated by reference in any registration statement or other document filed under the Securities Act or the Exchange Act, except as otherwise stated in such filing.
47
SIGNATURES
Pursuant to the requirements of the Exchange Act, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
|
Company Name |
|
|
|
|
|
Date: November 12, 2021 |
|
By: |
/s/ Thomas Schuetz |
|
|
|
Thomas Schuetz, MD |
|
|
|
Co-Founder and Chief Executive Officer |
|
|
|
|
Date: November 12, 2021 |
|
By: |
/s/ Vered Bisker-Leib |
|
|
|
Vered Bisker-Leib, PhD |
|
|
|
President and Chief Operating Officer
|
Date: November 12, 2021 |
|
By: |
/s/ Neil Lerner |
|
|
|
Neil Lerner, CPA |
|
|
|
Vice President - Finance
|
48