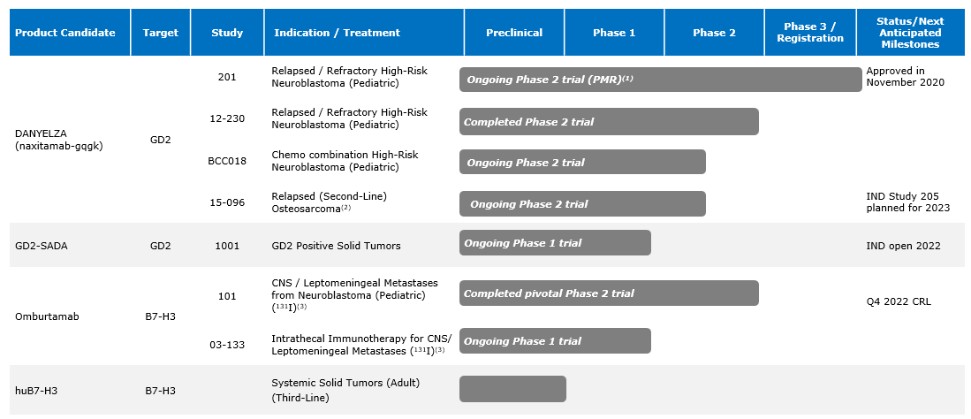
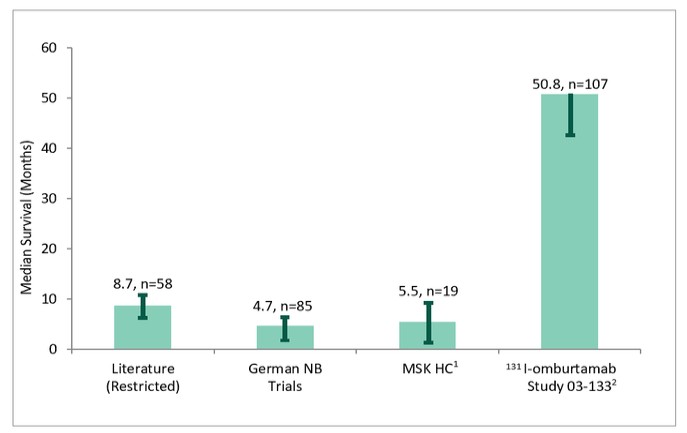
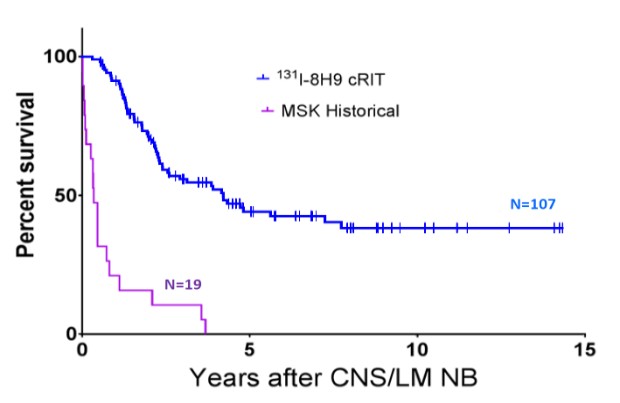
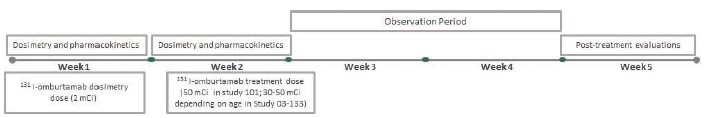
and the ODAC, who reviewed 131I-omburtamab and voted 16 to 0 that we had not provided sufficient evidence to conclude that omburtamab improves overall survival. In November 2022, we received a CRL for the BLA. In the CRL, and in our Type A meeting held subsequent to receipt of the CRL, the FDA made recommendations for us to consider in terms of trial design to demonstrate substantial evidence of effectiveness and a favorable benefit-risk profile. We are currently considering the future for our omburtamab development program and we can provide no assurance that the development of omburtamab will continue or that omburtamab will ultimately receive FDA approval.
As part of the restructuring plan, we also deprioritized 124I-omburtamab , which is omburtamab radiolabeled with Iodine-124, that was being studied for the treatment of Diffuse Intrinsic Pontine Glioma, or DIPG, and 131I-omburtamab that was being studied for the treatment of Desmoplastic Small Round Cell Tumors, or DSRCT.
Our Phase 1 multicenter study for 177Lu-omburtamab-DTPA, for the treatment of medulloblastoma, and our Phase 1 multicenter study with 177Lu-omburtamab-DTPA targeting B7-H3 positive CNS/LM tumors in adults were also deprioritized in 2022.
The SADA Technology
On April 15, 2020, we entered into a license agreement, or the SADA License Agreement, with MSK and Massachusetts Institute of Technology, or MIT, that grants us an exclusive, worldwide, sublicensable license to certain patent and intellectual property rights developed by MSK and MIT to develop, make, and commercialize licensed products and to perform services for all therapeutic and diagnostic uses in the field of cancer diagnostics and cancer treatments the SADA Technology. The patents and patent applications covered by the SADA License Agreement are directed, in part, to the SADA Technology, as well as a number of SADA constructs developed by MSK.
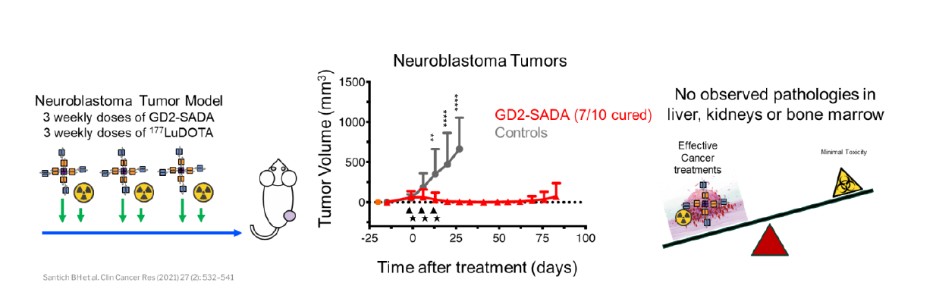
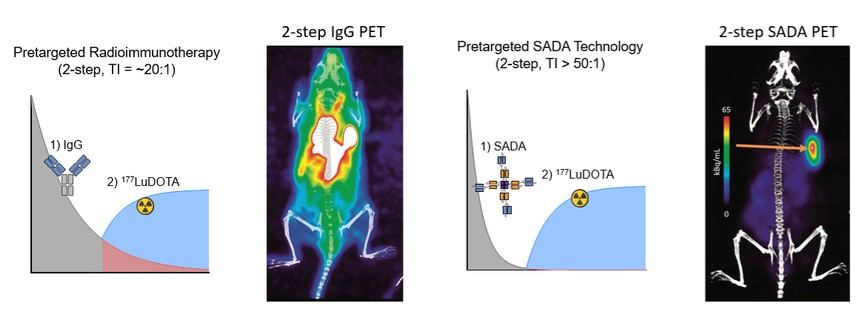
We are using the SADA Technology to advance a series of antibody constructs based on the SADA technology, where bispecific antibody fragments bind to the tumor before a radioactive payload is injected in a two-step approach. We have designated GD2-SADA for potential use in GD2-positive solid tumors as our first SADA construct and filed an IND for GD2-SADA in December 2021. We obtained clearance for the IND in July 2022. The first clinical sites were activated in November 2022, and MSK opened in March 2023. We believe the SADA technology could potentially improve the efficacy of radiolabeled therapeutics in tumors that have not historically demonstrated meaningful responses to radiolabeled agents.
8