antipsychotic drugs). This synergy has now been demonstrated in both laboratory rodent behavioral experiments and in multiple Phase 2 clinical trials and resulted in a Composition of Matter patent awarded in the U.S. and multiple foreign jurisdictions. Javitt subsequently observed that when patients with depression were treated with DCS, an NMDA antagonist, in combination with antidepressants, they manifested increased antidepressant effect, but did not exhibit the hallucinations and other NMDA effects previously reported with DCS. He further observed that DCS appeared to reduce some of the antidepressant side effects (akathisia) common to all known serotonin-targeted antidepressants.
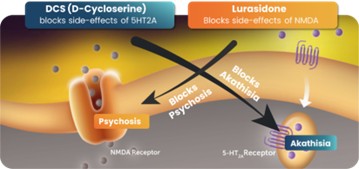
Figure 1 Synergistic composition of matter in which drugs that inhibit the NMDA receptor block the akathisia caused by serotonin-targeted drugs and serotonin-targeted drugs, in turn, block the psychedelic effects of NMDA inhibitors. Basis for US Patent 10583138. Source: NeuroRx, Inc.
These patented discoveries support NRX-101, the first investigational oral antidepressant to be granted Fast Track designation, Breakthrough Therapy designation and a Special Protocol Agreement by the FDA for severe bipolar depression in patients with ASIB. NRx is engaged in the research, development and future commercialization of this and other products for the treatment of patients suffering from suicidal ideation in the setting of bipolar depression and major depressive disorder (“MDD”) as well as PTSD and potentially chronic pain. Drugs that inhibit the brain’s NMDA receptor without ketamine’s limitations, have generated substantial interest, and have been explored for the treatment of the above conditions since the finding that ketamine has potent effects in reducing depression and suicidal ideation. It is our view that NRX-101 and our intellectual property to combine different molecules may yield a competitive advantage to use NMDA-inhibiting drugs for this purpose, as other compounds may be limited by adverse elements such as neurotoxicity (with prolonged use), hallucinations, potential habituation (i.e., addictive properties), blood pressure elevations, and/or lack of oral bioavailability.
This synergy is a key discovery underlying the patent portfolio described below. The scientific findings showed that some of the side effects of an NMDA drug can be blocked by the 5-HT2A drug and, in turn, the NMDA component can block akathisia, a known side effect of 5-HT2A-blocking drugs which is known to predispose to suicide. This dual-targeted approach is a primary basis of our worldwide patent portfolio, which currently encompasses 38 pending applications, and 48 granted patents in multiple jurisdictions covering compositions of matter and methods of use (See “NRx Patent Portfolio”). The relevant patents and patent applications in this portfolio are either owned by NeuroRx, exclusively licensed to NeuroRx by Glytech, LLC (“Glytech”), a Delaware limited liability company solely owned by Dr. Daniel Javitt (the “Glytech License”), or exclusively licensed to NeuroRx by Sarah Herzog Memorial Hospital Ezrat Nashim (“SHMH”), a non-profit organization organized under the laws of the State of Israel (the “SHMH License”).
NeuroRx owns a U.S. composition of matter patent that covers NRX-101. Patents under the Glytech License, which cover compositions of matter (including NRX-101 and pipeline therapeutic candidates) and methods of use (including methods of using NRX-101 in the treatment of bipolar depression with suicidal ideation and in treating PTSD), have been granted in the U.S., Europe (including validation by 18 members of the European Patent Convention), Japan, Australia and China.
12