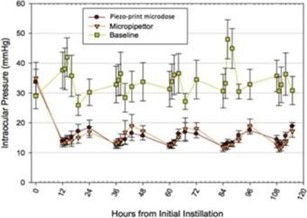
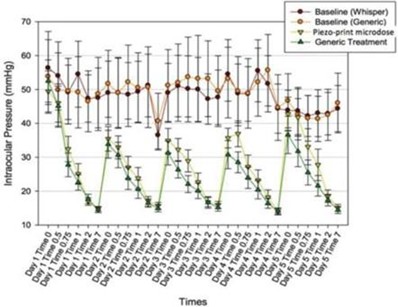
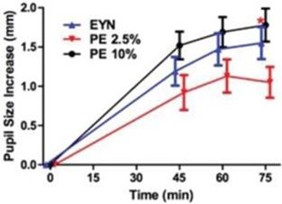
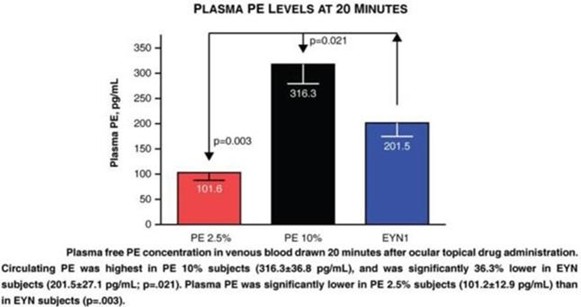
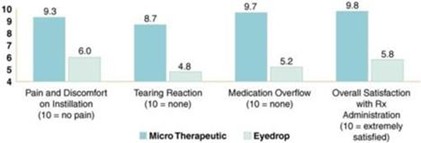
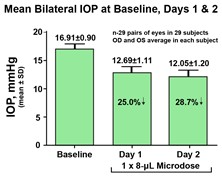
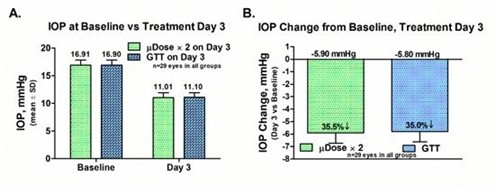
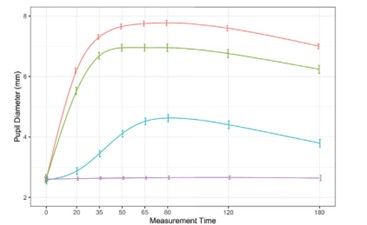
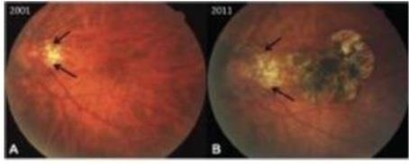
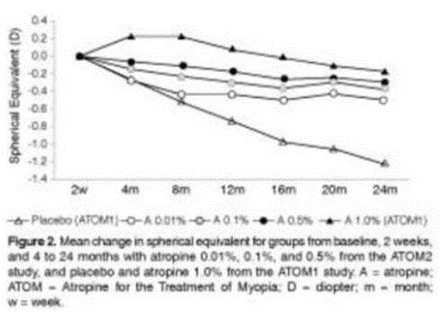
paid during the year, of $150,000, subject to certain exceptions for a non-executive chair of the Board. As of December 31, 2023, the number of securities remaining available for future issuance under equity compensation plans was 2,054,409.
At-The-Market Offering
December 2021 Sales Agreement
On December 14, 2021, the Company entered into a Sales Agreement, (the “December 2021 Sales Agreement”), with SVB Securities under which the Company may offer and sell, from time to time at its sole discretion, shares of common stock for gross proceeds of up to $50.0 million through SVB Securities as its sales agent, or the Offering. The issuance and sale of shares, if any, of common stock by the Company under the December 2021 Sales Agreement will be pursuant to the Company’s Registration Statement on Form S-3 (File No. 333-261638) filed with the SEC on December 14, 2021, or the Registration Statement, and the prospectus relating to the Offering filed therewith that forms a part of the Registration Statement.
Subject to the terms and conditions of the December 2021 Sales Agreement, SVB Securities may sell the common stock by any method permitted by law deemed to be an “at the market offering” as defined in Rule 415(a)(4) of the Securities Act of 1933, as amended. SVB Securities will use commercially reasonable efforts to sell the common stock from time to time, based upon instructions from the Company (including any price, time or size limits or other customary parameters or conditions the Company may impose). The Company will pay SVB Securities a commission equal to three percent (3.0)% of the gross sales proceeds of any common stock sold through SVB Securities under the December 2021 Sales Agreement, and also has provided SVB Securities with certain indemnification rights.
During the year ended December 31, 2022, the Company received approximately $5.4 million in gross proceeds and $5.3 million in net proceeds from the sale of 2,716,061 shares of its common stock under the December 2021 Sales Agreement. During the year ended December 31, 2023, the Company received approximately $4.7 million in gross proceeds and $4.6 million in net proceeds from the sale of 1,866,147 shares of its common stock.
Securities Purchase Agreement
On March 3, 2022, the Company entered into a securities purchase agreement, (the “Purchase Agreement”) with a certain institutional and accredited investor, or the Purchaser, pursuant to which the Company issued (i) 3,000,000 shares of common stock, (ii) pre-funded warrants, (the “Pre-Funded Warrants”), to purchase an aggregate of 1,870,130 shares of common stock and (iii) warrants to purchase an aggregate of 4,870,130 shares of common stock, (the “Investor Warrants”), (together, the “the March 2022 Offering”). The Company determined that the warrants qualified for equity classification.
The offering price for the shares was $3.08 per share and the offering price for the Pre-Funded Warrants was $3.07 per Pre-Funded Warrant, which represents the per share public offering price less $0.01 per share exercise price for each Pre-Funded Warrant. The Investor Warrants will have an exercise price of $3.54 per share and each Investor Warrant became exercisable for one share of Common Stock. The Investor Warrants became exercisable six months from the date of issuance and the Pre-Funded Warrants were exercisable immediately upon issuance. The Pre-Funded Warrants shall terminate when fully exercised and the Investor Warrants will terminate five years from the initial exercisability date. The aggregate gross proceeds to the Company from the March 2022 Offering were approximately $15 million, excluding the proceeds, if any, from the exercise of the Pre-Funded Warrants and the Investor Warrants. No underwriter or placement agent participated in the March 2022 Offering. The Company incurred issuance costs in the amount of $83,391 in connection with the March 2022 offering.
The March 2022 Offering was made pursuant to an effective registration statement on Form S-3 (Registration Statement No. 333-261638), as previously filed with and declared effective by the Securities and Exchange Commission and a related prospectus.
Registered Direct Offering
On August 24, 2023, the Company entered into a securities purchase agreement with a certain institutional and accredited investor (the “Purchaser”), pursuant to which the Company agreed to sell, in a registered direct offering by the Company directly to the Purchaser (the “August 2023 Offering”), 4,198,633 shares of common stock, pre-funded warrants to purchase up to 2,252,979 shares of common stock and warrants to purchase up to 4,838,709 shares of common stock (the “Common Warrants” and, together with the Pre-Funded