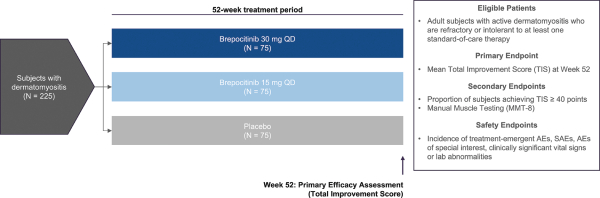
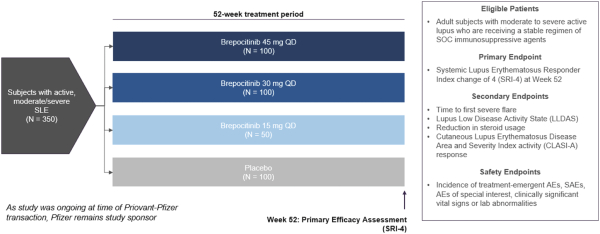
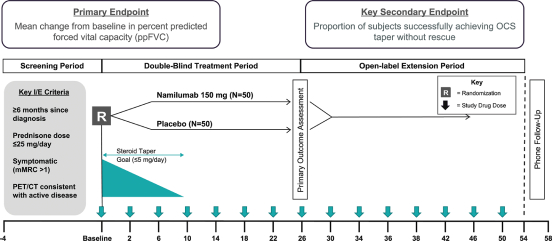
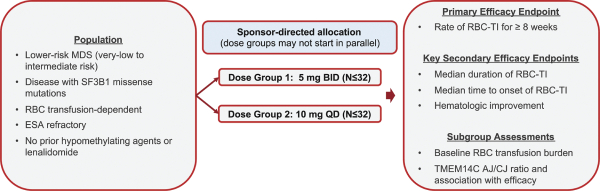
have no alternative future use. Accordingly, the Company recorded $
399.6 million as acquired
in-process
research and development expense in the accompanying consolidated statement of operations for the year ended March 31, 2021.
In connection with the transaction, the vesting of certain outstanding Silicon Therapeutics share-based compensation awards held by employees of Silicon Therapeutics was discretionarily accelerated at closing. As a result, the Company recorded share-based compensation expense of $23.5 million in the accompanying consolidated statements of operations for the year ended March 31, 2021.
In addition, certain share-based compensation awards of Silicon Therapeutics were exchanged with restricted
of the Company, subject to certain service-based vesting requirements, with a fair value of $22.6 million. Of this amount, $15.6 million was attributed to precombination service and therefore included in the total fair value of consideration transferred.
In September 2021, Priovant Therapeutics, Inc. (“Priovant”) entered into a license and collaboration agreement with Pfizer, Inc. (“Pfizer”) (the “Pfizer License Agreement”). The transaction was accounted for as an asset acquisition as the acquired assets did not meet the definition of a business. The fair value of consideration transferred was $
82.1 million, consisting of $
70.0 million of preferred stock issued to Pfizer, representing a dilution-protected minority ownership interest in Priovant; a $
10.0 million upfront cash payment; and $
2.1 million relating to other obligations. The acquired rights, which included the licensed rights, starting materials and
in-process
inventory for each drug candidate, represent
in-process
research and development assets, which were determined to have no alternative future use. Accordingly, the Company recorded $
82.1 million as acquired
in-process
research and development expense in the accompanying consolidated statements of operations for the year ended March 31, 2022.
Priovant is obligated to pay Pfizer a mid tens-of-millions sales milestone payment if aggregate net sales of its licensed products in Priovant’s territory in a given year exceed a mid hundreds-of-millions amount. Pfizer is obligated to pay Priovant a low tens-of-millions sales milestone payment if aggregate net sales of its licensed products outside of Priovant’s territory in a given year exceed a mid hundreds-of-millions amount.
Priovant is obligated to pay Pfizer a tiered,
sub-teens
royalty, on aggregate net sales of its licensed products in Priovant’s territory. Pfizer is obligated to pay Priovant a tiered high single-digit to
sub-teens
royalty, on aggregate net sales of its licensed products outside of Priovant’s territory.
In November 2021, Hemavant Sciences GmbH (“Hemavant”), a wholly owned subsidiary of the Company, entered into a license agreement with Eisai Co., Ltd. (“Eisai”) (the “Eisai License Agreement”). Pursuant to the Eisai License Agreement, Eisai granted Hemavant (i) an exclusive, worldwide, sublicensable, royalty-bearing license under certain patents and
know-how
and (ii) a
non-exclusive,
worldwide, sublicensable, royalty-bearing license under certain additional patents,
know-how
and inventions, in each case, to develop, manufacture and commercialize the compound known as
RVT-2001
and products incorporating
RVT-2001
for all human and animal uses. In exchange for the rights, the Company made an upfront payment to Eisai consisting of $
8.0 million in cash and the issuance of $
7.0 million in shares of the Company’s
at an agreed price of $
8.00 per share. Hemavant may also be obligated to pay up to a maximum of $
65.0 million in development and regulatory milestone payments (with respect the product for the first indication) and up to a maximum of $
18.0 million in payments (with respect to the product for each additional indication) and up to a maximum of $
295.0 million in commercial milestone payments. Hemavant may also be obligated to pay a tiered high single-digit to
sub-teens
royalty, subject to certain customary reductions, on net sales of licensed products.