Three months ended March 31, 2024 and 2023
The Company incurred a comprehensive loss of $8,990,284 for the three months ended March 31, 2024 compared to a comprehensive loss of $7,119,354 for the three months ended March 31, 2023. The detailed changes in research and development and general and administration expenses for the three months ended March 31, 2024 and 2023 are included in the tables above.
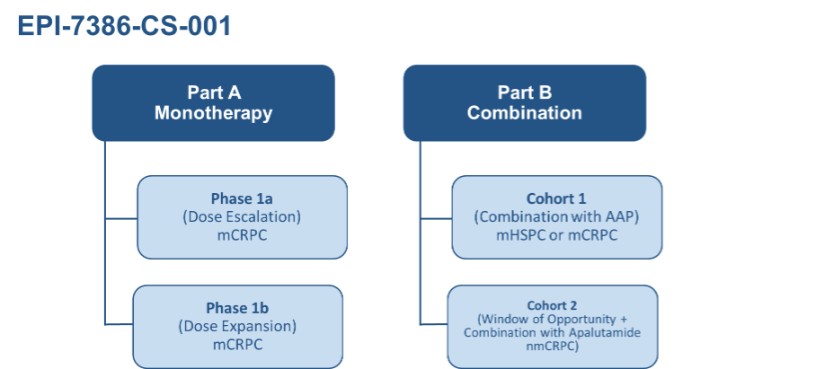
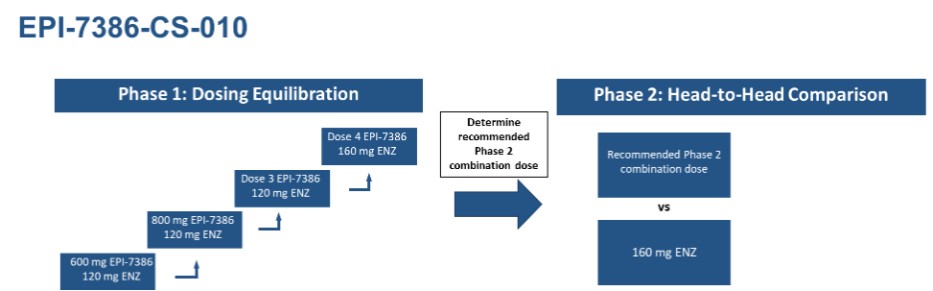
Research and development expenses were $6,177,987 (2023 - $4,480,863) for the three months ended March 31, 2024; the Company’s research and development investment continued with the ongoing clinical trial of masofaniten (EPI-7386). Clinical costs of $3,013,161 (2023 - $623,805) and ongoing preclinical costs and data analysis costs of $1,150,869 (2023 - $1,714,345) support the clinical trial and associated analysis. Manufacturing costs of $332,772 (2023 - $269,748) reflect the ongoing production of masofaniten (EPI-7386) to support the ongoing clinical trial.
General and administration expenses were $4,315,502 (2023 - $3,730,692) for the three months ended March 31, 2024. Professional fees of $477,757 (2023 - $159,318) have increased for legal and accounting services in conjunction with ongoing corporate activities. Salaries and benefits of $2,448,855 (2023 - $2,026,935) for the three months ended March 31, 2024 include annual bonuses paid to senior management and employees during the period. Directors’ fees of $104,000 (2023 - $95,250) are paid to directors for their membership on the Board based on an annual fee structure.
Share-based payments for research and development team members were $459,141 (2023 - $750,159) and share-based payments allocated for general and administrative expenses were $673,460 (2023 – $686,932) for directors, key management and employees of the Company. Share-based payments expense is non-cash and is estimated under the Black-Scholes method and expensed relative to vesting conditions for the underlying stock options.
Liquidity and Capital Resources
ESSA is a clinical stage company and does not currently generate revenue.
As of March 31, 2024, the Company has working capital of $133,123,568 (September 30, 2023 - $145,301,807). Operational activities during the six months ended March 31, 2024 were financed mainly by proceeds from the financings completed in July 2020 and February 2021. At March 31, 2024, the Company had available cash reserves and short-term investments of $135,888,666 (September 30, 2023 - $148,076,401) to settle current liabilities of $4,174,304 (September 30, 2023 - $3,495,071). At March 31, 2024, the Company believed that it had sufficient capital to satisfy its obligations as they became due and execute its planned expenditures for more than twelve months.
ESSA’s future cash requirements may vary materially from those now expected due to a number of factors, including the costs associated with future preclinical work and to take advantage of strategic opportunities, such as partnering collaborations or mergers and acquisitions activities. In the future, it may be necessary to raise additional funds. These funds may come from sources such as entering into strategic collaboration arrangements, the issuance of shares from treasury, or alternative sources of financing. However, there can be no assurance that ESSA will successfully raise funds to continue its operational activities. See “Risk Factors” in our Annual Report on Form 10-K.
Critical Accounting Policies and Estimates
The Company makes estimates and assumptions about the future that affect the reported amounts of assets and liabilities. Estimates and judgments are continually evaluated based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. In the future, actual experience may differ from these estimates and assumptions.
The effect of a change in an accounting estimate is recognized prospectively by including it in comprehensive income in the period of the change, if the change affects that period only, or in the period of the change and future periods, if the change affects both.