UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ____________ to ____________
Commission File Number
(Exact name of Registrant as specified in its Charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
|
|
(Address of principal executive offices) |
(Zip Code) |
Registrant's telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the Registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer," "smaller reporting company," and "emerging growth company" in Rule 12b-2 of the Exchange Act.
|
☒ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|
|||
Non-accelerated filer |
|
☐ |
|
Smaller reporting company |
|
|
|
|
|
|
|
|
|
|
|
|
|
Emerging growth company |
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management's assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant's executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Act). YES
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the Registrant, based on the closing price of the shares of common stock on The Nasdaq Global Select Market on June 30, 2023, was approximately $
The number of shares of Registrant's common stock outstanding as of February 22, 2024 was
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's definitive proxy statement relating to its 2024 annual meeting of stockholders (the "2024 Proxy Statement") are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated. The 2024 Proxy Statement will be filed with the U.S. Securities and Exchange Commission within 120 days after the end of the fiscal year to which this Annual Report on Form 10-K relates.
Table of Contents
|
|
Page |
PART I |
|
|
Item 1. |
3 |
|
Item 1A. |
28 |
|
Item 1B. |
61 |
|
Item 1C. |
61 |
|
Item 2. |
62 |
|
Item 3. |
62 |
|
Item 4. |
62 |
|
|
|
|
PART II |
|
|
Item 5. |
62 |
|
Item 6. |
63 |
|
Item 7. |
Management's Discussion and Analysis of Financial Condition and Results of Operations |
64 |
Item 7A. |
74 |
|
Item 8. |
75 |
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
103 |
Item 9A. |
103 |
|
Item 9B. |
105 |
|
Item 9C. |
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
105 |
|
|
|
PART III |
|
|
Item 10. |
106 |
|
Item 11. |
106 |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
106 |
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
106 |
Item 14. |
106 |
|
|
|
|
PART IV |
|
|
Item 15. |
107 |
|
Item 16. |
109 |
|
|
110 |
i
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
As used in this Annual Report on Form 10-K ("Annual Report"), unless expressly indicated or the context otherwise requires, references to "Treace Medical Concepts," "we," "us," "our," or "the Company," refer to Treace Medical Concepts, Inc. This Annual Report contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, as codified in Section 27A of the Securities Act of 1933, as amended (the "Securities Act"), and Section 21E of the Securities Exchange Act of 1934, as amended (the "Exchange Act") concerning our business, operations and financial performance and condition, as well as our plans, objectives and expectations for our business, operations and financial performance and condition. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as "anticipate," "assume," "believe," "contemplate," "continue," "could," "due," "estimate," "expect," "goal," "intend," "may," "objective," "plan," "predict," "potential," "positioned," "seek," "should," "target," "will," "would" and other similar expressions that are predictions of or indicate future events and future trends, or the negative of these terms or other comparable terminology.
These forward-looking statements include, but are not limited to, statements about:
1
We believe that it is important to communicate our future expectations to our investors. However, there may be events in the future that we are not able to accurately predict or control and that may cause our actual results to differ materially from the expectations we describe in our forward-looking statements. These forward-looking statements are based on management's current expectations, estimates, forecasts and projections about our business and the industry in which we operate, and management's beliefs and assumptions and are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. As a result, any or all of our forward-looking statements in this Annual Report may turn out to be inaccurate. Factors that may cause actual results to differ materially from current expectations include, among other things, those set forth in this Annual Report under "Risk Factors" and elsewhere in this Annual Report. Our stockholders are urged to consider these factors carefully in evaluating the forward-looking statements.
These forward-looking statements speak only as of the date of this Annual Report. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future. You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. We undertake no obligation to update publicly any forward-looking statements for any reason after the date of this Annual Report to conform these statements to actual results or to changes in our expectations. If we update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
You should read this Annual Report and the documents that we reference in this Annual Report and have filed with the Securities and Exchange Commission ("SEC") as exhibits to this Annual Report with the understanding that our actual future results, levels of activity, performance and events and circumstances may be materially different from what we expect.
2
PART I
Item 1. Business
Overview
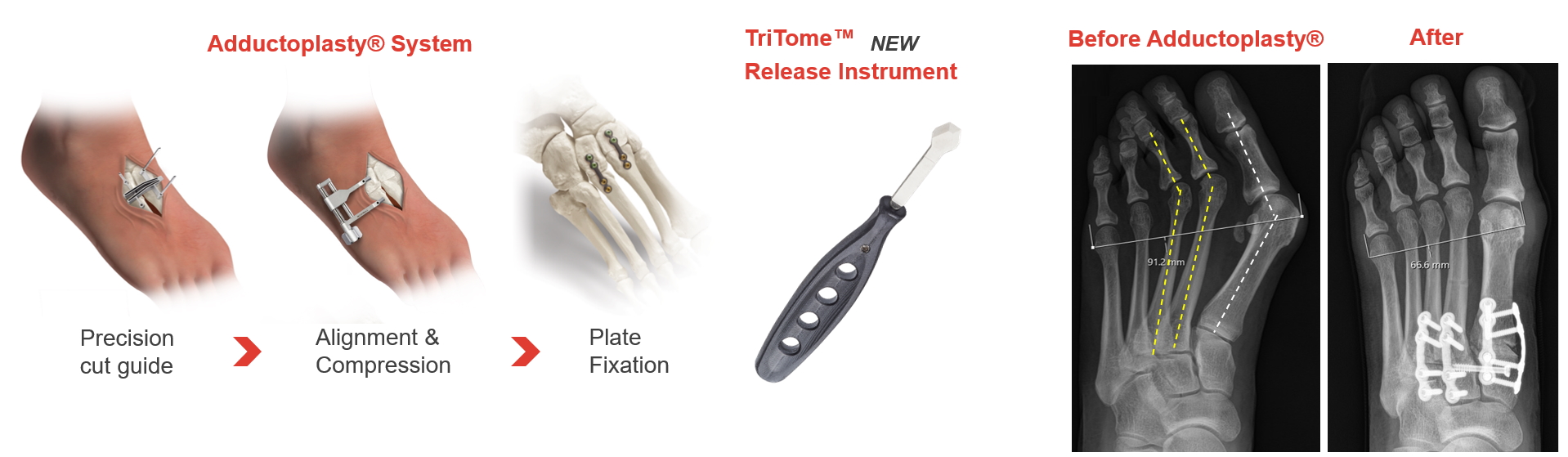
We are a medical technology company with the goal of advancing the standard of care for the surgical management of bunion and related midfoot deformities. We have pioneered and patented the Lapiplasty® 3D Bunion Correction System®—a combination of instruments, implants and surgical methods designed to surgically correct all three planes of the bunion deformity and address the root cause of the bunion, helping patients get back to their active lifestyles. To further support the needs of bunion patients, we have introduced our Adductoplasty® Midfoot Correction System, designed for reproducible surgical correction of the midfoot as well as our Hammertoe PEEK Fixation System designed to address hammertoe, claw toe and mallet toe deformities. We continue to expand our footprint in the foot and ankle market with the introduction of our SpeedPlate™ Rapid Compression Implants, an innovative fixation platform with broad versatility across Lapiplasty and Adductoplasty Procedures, as well as other common bone fusion procedures of the foot.
A bunion is a painful, disfiguring deformity characterized by a deviated position of the great toe, and easily identified visually by the "bump" at its base. Bunions affect approximately 67 million Americans, and generally increase in prevalence and severity over time. Nearly 25% of adults between the ages of 18 and 65, and over 35% of people over the age of 65, have bunions. Approximately 4.4 million patients in the United States seek medical attention for bunions annually; of these patients, an estimated 1.1 million are deemed surgical candidates, which represents a total annual addressable market opportunity of more than $5 billion. This large patient population often suffers from symptoms that worsen over time, including severe and debilitating pain, emotional burden and limited mobility, and is susceptible to further degeneration and common concomitant pathologies. Despite the significant limitations of traditional surgical treatment approaches, approximately 450,000 surgical bunion procedures are performed in the United States every year. We believe there is a significant opportunity to convert these to our Lapiplasty Procedure, representing a greater than $2.3 billion market opportunity. In addition, through better clinical outcomes and effective patient education, we believe we can increase the number of patients who seek surgical treatment, representing an incremental opportunity of $2.7 billion.
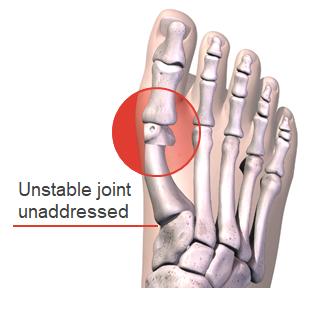
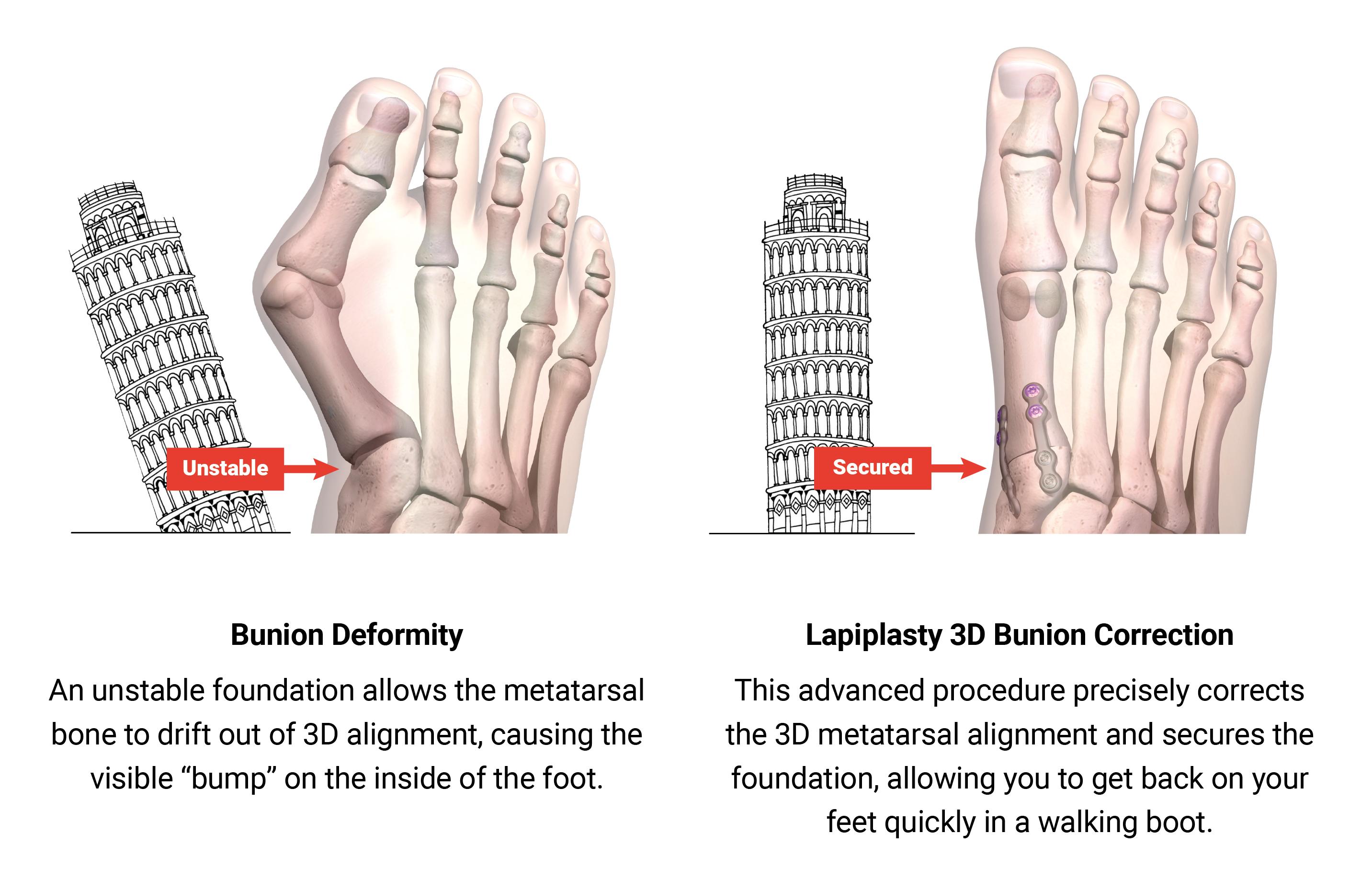
The goal of bunion surgery is to restore the normal anatomy of patients in order to return natural function and appearance in the foot and relieve pain. A common misconception is that a bunion is simply an overgrowth of bone that can be shaved off. In reality, a bunion is a complex 3D deformity caused by an unstable joint in the middle of the foot (which we may refer to as the "root cause") which causes the metatarsal bone in the foot to rotate out of alignment in all three anatomic dimensions. A 2015 study indicates that 87% of bunions have a 3D, rotational issue in addition to horizontal and vertical misalignments of the metatarsal bone. Traditional 2D approaches to bunion surgery, used in the majority of bunion surgical procedures, fail to
3
correct this third "rotational" dimension of the bunion deformity, which has been reported to result in a 10 to 12 times increase in the chance of bunion recurrence as compared to 3D surgeries.
Historically, there have been two primary approaches to the surgical treatment of bunions, which inconsistently met patient needs and physician expectations. The first and most common approach is 2D Osteotomy surgery, which involves cutting and shifting the metatarsal bone in two dimensions and mainly addressing the cosmetic bump, which may result in high variability in potential for bunion recurrence with long-term recurrence rates (up to 78%) and low patient satisfaction with the procedure. The second approach, traditionally reserved for the most advanced and severe bunion pathology, is Lapidus Fusion surgery, which fuses the unstable joint but requires a technically challenging correction through a "freehand" technique. This often results in inconsistent outcomes and has been reported to involve a protracted period of recovery, including approximately 6 to 10 weeks of non-weight-bearing. The freehand technique is highly dependent on the surgeon's skill and requires the physician to perform complex corrections without the benefits of assistive instrumentation that are standard in many other orthopaedic joint procedures and, consequently, this surgery often results in inconsistent outcomes.
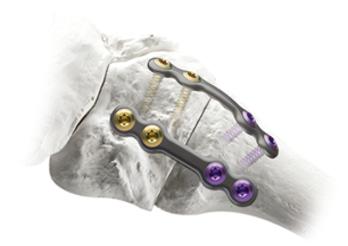
We believe our proprietary Lapiplasty System, the first of its kind, is the leading system designed to consistently and reliably correct all three dimensions of the bunion deformity, address the root cause of the bunion deformity, and allow return to weight-bearing quickly in a walking boot with low risk of recurrence (as described in more detail below in the section entitled "Key Clinical Advantages of the Lapiplasty System"). The Lapiplasty System combines our novel surgical approach, the Lapiplasty Procedure, with our procedural instrumentation and single-use implant kits. With help from our procedural instrumentation, the Lapiplasty Procedure is designed to rotate the entire metatarsal bone into normal anatomical position in all three dimensions, eliminating the bump and restoring normal anatomy. The unstable foundation in the foot is then secured with our titanium fixation implants, allowing patients to get back on their feet quickly in a walking boot. The Lapiplasty Procedure can be performed in either hospital outpatient or ambulatory surgery center settings, and utilizes existing, well-established reimbursement codes. Since receiving 510(k) clearance for the Lapiplasty System in March 2015, more than 90,000 Lapiplasty Procedures have been performed in the United States.
The safety, effectiveness and clinical advantages of the Lapiplasty System have been demonstrated in multiple post-market clinical outcome studies. We believe this portfolio of studies is unique in the bunion correction field where comprehensive outcome studies with respect to marketed bunion correction surgical products are limited. Multiple peer-reviewed publications have demonstrated the ability of the Lapiplasty System to reproducibly correct all three dimensions of the deformity and allow the patient to quickly return to weight-bearing in a walking boot while exhibiting a low rate of bunion recurrence (as described in more detail below in the section entitled "Key Clinical Advantages of the Lapiplasty System"). We have completed enrollment in our ALIGN3D™ prospective, multicenter study, which is prospectively evaluating bunion correction status through five years after the Lapiplasty Procedure and includes patient reported outcomes, range of motion
4
results and radiographic outcomes. Interim analyses from the ALIGN3D clinical study have been published in the Journal of Foot & Ankle Surgery and most recently presented at industry conferences, including at the American Orthopedic Foot and Ankle Society ("AOFAS") Annual Meeting in September 2023, and at the American College of Foot and Ankle Surgeons ("ACFAS") Annual Scientific Conference that was held in early February 2024.
As described in more detail below in the section entitled "Our Solutions," our other products include (1) the Adductoplasty System, which brings together our implants and precision instrumentation for the first comprehensive system designed for reproducible realignment, stabilization, and fusion of the midfoot, (2) the SpeedPlate Rapid Compression Implant System designed for rapid delivery of titanium compression implants through small incisions, (3) specialized osteotomes and release instruments for use in bunion and midfoot surgeries, and (4) the Hammertoe PEEK Fixation System designed to address hammertoe, claw toe and mallet toe deformities. We believe these new technologies demonstrate our commitment to rapid innovation with a focus on continually iterating the Lapiplasty System with options for reduced incision size and increased procedural efficiency and providing solutions for other procedures often performed with bunion surgeries.
We market and sell our products in the United States primarily through a direct employee sales force that is supplemented by independent sales agencies across 249 territories focused on supporting adoption and utilization of the Lapiplasty and Adductoplasty Systems among the approximately 7,400 surgical podiatrists and 2,600 orthopaedic surgeons with foot and ankle specializations in the United States. To improve clinical outcomes, we devote significant resources to training and educating physicians on the safe and effective use of the Lapiplasty and Adductoplasty Systems. Additionally, we have developed a differentiated direct-to-patient outreach program that educates patients on the benefits and risks of the Lapiplasty System. We also offer a "Find a Doctor" tool on our website that allows potential patients to search for trained Lapiplasty surgeons in their local markets. We believe our patient and surgeon education programs and specialized teams supporting surgeons in the field combined with the Lapiplasty System's differentiated clinical outcomes lead to an increase in utilization of the Lapiplasty System per physician over time.
Our employee engineering personnel and our Surgeon Advisory Board and other orthopaedic surgeon consultants help us to generate ideas and develop product innovations. Our Surgeon Advisory Board is comprised of both podiatrists and orthopaedic foot and ankle surgeons who provide us with insights for developing products that fully meet the needs of each group. Our research and development team is focused on improving clinical outcomes by designing new procedure-specific instruments and products and by developing enhanced surgical techniques.
We have experienced considerable growth since receiving 510(k) clearance for the Lapiplasty System in March 2015. The number of Lapiplasty Procedure kits sold increased from 11,113 in 2020 to 29,675 in 2023, representing a compound annual growth rate of 38.7%, despite the adverse impact of the COVID-19 pandemic on elective procedures in 2020 and 2021. Correspondingly, our revenue increased from $57.4 million in 2020 to $187.1 million in 2023, representing a compound annual growth rate of 48.3%.
Overview of Bunions
Hallux Valgus (commonly known as "bunions") is a painful, disfiguring deformity characterized by a deviated position of the great toe. Bunions are easily identified visually by the "bump" on the joint at the base of the great toe (the metatarsophalangeal ("MTP") joint). While this "bump" is widely considered to be the source of pain in bunion sufferers, a structural defect causing a 3D misalignment of the metatarsal bone is the root cause of the deformity.
Bunion deformities are most commonly considered to be the consequence of a hereditary predisposition. Prevalence increases with age, and one study found that 70% of bunion sufferers are female, and that the disorder occurs in both feet, or bilaterally, in 56% of bunion sufferers. Bunions are progressive deformities, with symptoms that typically grow in severity over time. For those with predispositions for developing bunions, constrained footwear, weight-bearing activities or occupations that aggravate the condition may accelerate progression of the joint deformity and cause symptoms to appear earlier in life. If left untreated, bunions can often have a significant long-term negative impact on sufferers, including:
5
A common misconception is that a bunion is simply an overgrowth of bone that can be shaved off. Bunions are in reality complex 3D deformities caused by deviation and rotation of the metatarsal bone in three anatomic dimensions. These three anatomic dimensions and their associated misalignments are summarized below:
The shift in the metatarsal bone causes bone or tissue at the MTP joint to move out of place, resulting in the visual "bump" associated with bunions.
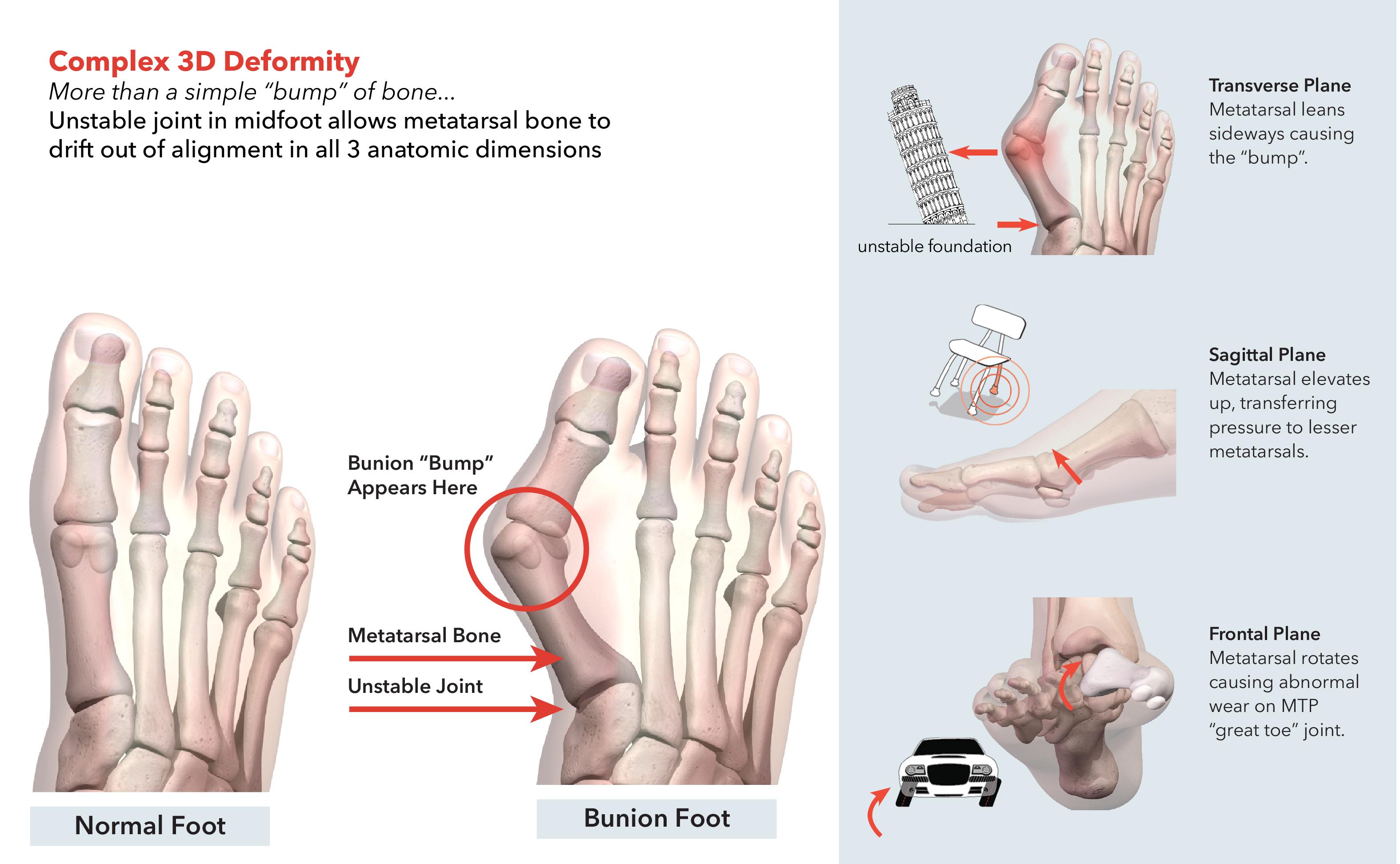
Complex 3D Deformity More than a simple bump of bone& Unstable joint in midfoot allows metatarsal bone to drift out of alignment in all 3 anatomic dimensions Transverse Plane Metatarsal leans sideways causing the bump. Saittal plane Metatarsal elevates up, transferring pressure to lesser metatarsals. Frontal Plane Metatarsal rotates causing abnormal wear on MTP great toe joint.
Traditional treatment options for bunion patients vary with the type and severity of each bunion. During the early stages of the disorder, pain can be managed but will typically worsen and additional symptoms may develop. The primary goal of most early treatment options is to relieve pressure on the bunion and halt the progression of the deformity. A physician may initially recommend various non-surgical treatments, including toe spacers, pads or splints, inserts or orthotics, medication or physical therapy. These options are prescribed to alleviate symptoms but do not address the root cause of the deformity. When these non-surgical treatments fail, or when the severity of the bunion deformity progresses past the threshold for such options, surgery is often necessary.
6
Limitations of Traditional Surgical Treatment Approaches
Historically, there have been two primary surgical approaches to bunion treatment, 2D Osteotomy and Lapidus Fusion. Between the two, approximately 450,000 bunion procedures are performed annually in the United States, of which approximately 75% are 2D Osteotomy procedures and approximately 25% are Lapidus Fusion procedures.
These traditional surgical treatment approaches are characterized by a 6% to 35% patient dissatisfaction rate for 2D Osteotomy surgery and a 7% to 13% dissatisfaction rate for Lapidus Fusion following surgery. Clinical literature has identified the primary patient expectations for bunion surgery to be pain relief, shoe fit, mobility and improvement in cosmetic appearance. Certain published long-term clinical studies have demonstrated recurrence rates as high as 78% following 2D Osteotomy surgery and 38% following Lapidus Fusion surgery. While not all patients with recurrence require a secondary surgical procedure, this high variability for potential recurrence relative to other common surgical procedures is a contributor to patient dissatisfaction.
2D Osteotomy
In a 2D Osteotomy, the bunion "bump" is shaved off and the metatarsal bone of the great toe is cut in half and shifted over to reduce the appearance of the bunion. However, by not correcting the deformity in all three dimensions, there is an increased likelihood that the metatarsal bone will continue to drift out of position over time and for the bunion to return. Additionally, the recovery time has been reported to include up to 6 weeks of non-weight bearing.
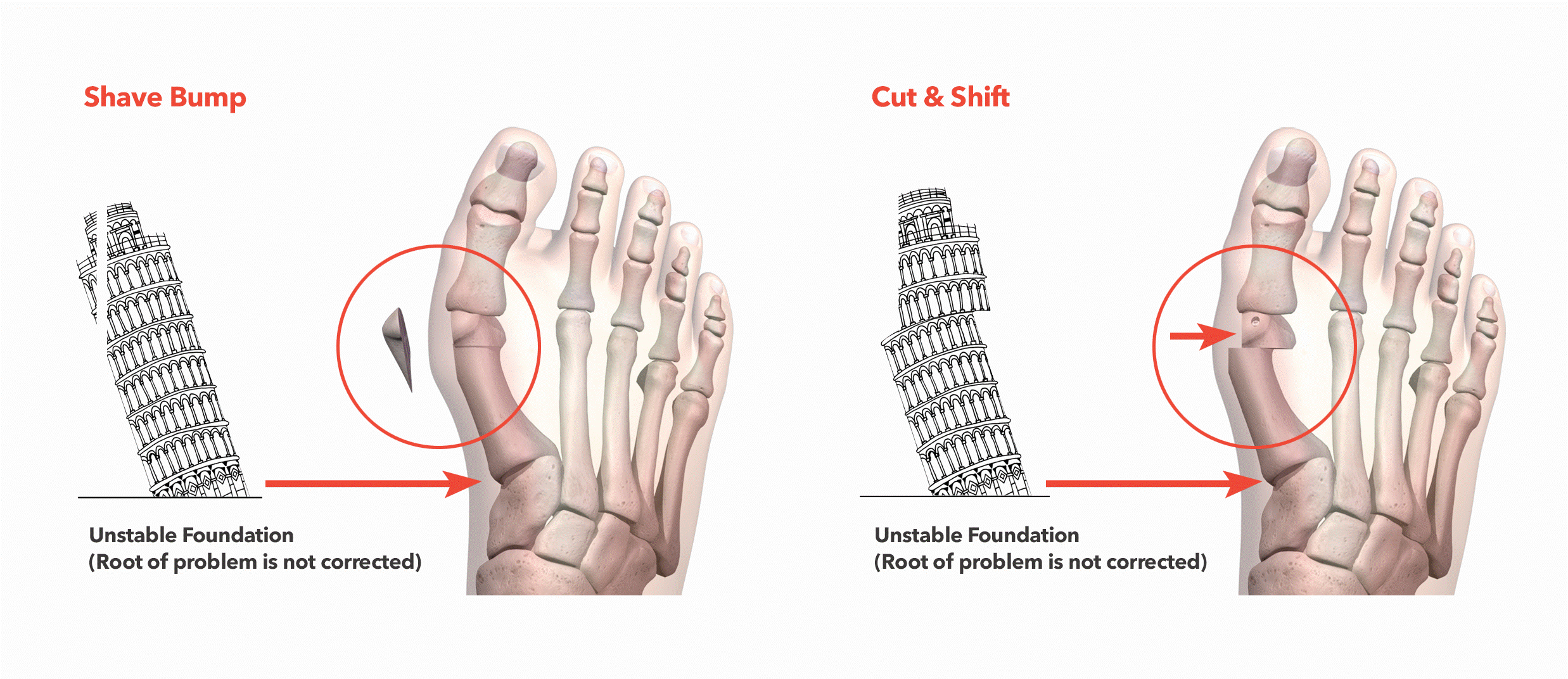 Shave Bump Unstable Foundation (Root of problem is not corrected) Cut & Shift Unstable Foundation (Root of problem is not corrected)
Shave Bump Unstable Foundation (Root of problem is not corrected) Cut & Shift Unstable Foundation (Root of problem is not corrected)
Lapidus Fusion
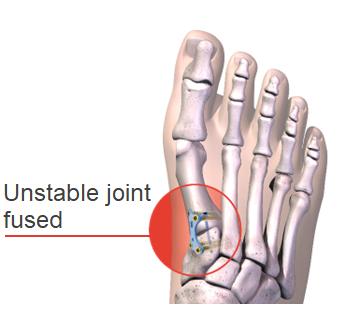
In contrast to 2D Osteotomy, the other common traditional surgical procedure, known as Lapidus Fusion, does address the root cause of the bunion and is routinely referenced in medical literature as a surgical option for bunions since the 1930s. However, even a Lapidus Fusion, as it is conventionally described and performed, still does not address the three-dimensional rotational aspect known to contribute to bunion recurrences.
A conventional Lapidus Fusion surgery fuses the unstable first tarsometatarsal ("TMT") joint but requires a technically challenging correction through a "freehand" technique and has been reported to involve a protracted period of recovery, including approximately 6 to 10 weeks of non-weight-bearing. The freehand technique is highly dependent on the surgeon's skill and requires the physician to perform complex corrections without the benefits of assistive instrumentation that are standard in many other orthopaedic joint procedures, and, consequently, this surgery often results in inconsistent outcomes. Thus, its use has been traditionally reserved for the most advanced and severe bunion pathology.
7
The table below provides a summary overview of traditional bunion surgical treatment approaches:
|
2D Osteotomy |
Lapidus Fusion |
||
% of cases |
Approximately 75% |
Approximately 25% |
||
Procedure overview |
|
Targets cosmetic bump by cutting and shifting metatarsal bone in two dimensions |
|
Fusion of the first TMT joint to realign the entire metatarsal and the toe joint and prevent the bunion from coming back |
Procedure time |
25 to 75 minutes |
40 to 120 minutes |
||
Recurrence rate |
3.6% to 78%, |
0% to 38% |
||
Reported recovery time |
1 day to 6 weeks non-weight bearing (post operative shoe or boot, some cast) |
Traditionally 6 to 10 weeks non-weight bearing (often in a cast) |
||
Non-Union Rate* |
0% to 3.3% |
2.2% to 12% |
||
Hardware Removal Rate |
0% to 12.3% |
2% to 17% |
||
Patient dissatisfaction rate |
6% to 35% |
7% to 13% |
||
Limitations |
• Does not address all 3 dimensions of the deformity reliably and leaves the unstable foundation untreated |
• Technically challenging "freehand" procedure increases inconsistency and variability of results • Primarily 2-plane procedure; does not address the frontal plane rotation problem consistently |
||
* Non-union rate is a measure of the incidence of the bone not healing together.
While bunions have traditionally been viewed as a 2D deformity, recent scientific literature has indicated that 87% of bunions have a 3D, rotational component in addition to the horizontal and vertical misalignments of the metatarsal bone. Failure to correct this third "rotational" dimension of the bunion deformity has been reported to result in a 10 to 12 times increase in the chance of bunion recurrence as compared to 3D surgeries. We believe there is a rapidly increasing awareness among surgeons of the need for 3D bunion correction based on the frequency of lectures and medical journal publications on this topic, particularly in recent years.
Our Solutions
We have pioneered our proprietary Lapiplasty 3D Bunion Correction System—a combination of innovative instruments, implants and surgical methods designed to correct all three planes of the bunion deformity and secure the unstable joint, addressing the root cause of the bunion.
Our Lapiplasty System
We believe our Lapiplasty System was the first and remains the leading system designed to consistently and reliably correct all three dimensions of the bunion deformity, stabilize the first TMT joint and allow return to weight-bearing quickly in a walking boot. In a Lapiplasty Procedure, the entire metatarsal bone is rotated and brought back into position in all three dimensions, eliminating the unsightly bump and restoring normal anatomy. The first TMT joint is secured with titanium fixation technology allowing patients to get back on their feet quickly in a walking boot. The Lapiplasty Procedure can be
8
performed on a wide range of patients with bunion deformities in the hospital outpatient or ambulatory surgery center setting and utilizes existing, well-established reimbursement codes.
The Lapiplasty System includes both procedural instrumentation and single-use, sterile-packed implant kits. Our procedural instrumentation includes innovative surgical tools designed to enable surgeons to correct all three dimensions of the bunion deformity and the root cause of bunions with accuracy and consistency. Our single-use, sterile-packed implant kits feature biplanar implants, which are two low-profile titanium fixation implants designed to stabilize the TMT joint and to allow early weight-bearing in a walking boot during the critical healing period.
The following table illustrates key components of the Lapiplasty System:
|
|
|
|
|
Reusable procedural instrumentation |
|
Sterile-packed implant kits |
||
Lapiplasty Positioner |
Lapiplasty Compressor |
|
Sterile Implants and Instruments |
|
|
|
|
Single-use implants and instruments used in the Lapiplasty Procedure and ancillary procedures
|
|
Engineered to quickly and reproducibly correct metatarsal alignment in all three dimensions |
Delivers controlled compression to the precision-cut joint surfaces, while maintaining the three-dimensional correction |
|
||
Lapiplasty 3-n-1® Guide Delivers precise cuts with the metatarsal held in the corrected position, ensuring optimal cut trajectory |
Lapiplasty Reusable Instrumentation
Includes the Positioner, Compressor and 3-n-1® Guide |
|
Biplanar Plating
Provides biomechanically-tested biplanar stability; designed to allow rapid return to weight-bearing in a walking boot |
SpeedPlate Implants
Designed to deliver stability of a titanium locking plate* with speed and compression of a staple |
* encompasses locking plate and screw construct
The following table illustrates our patented Lapiplasty System, with procedural instrumentation and implants used in each step of our proprietary Lapiplasty Procedure:
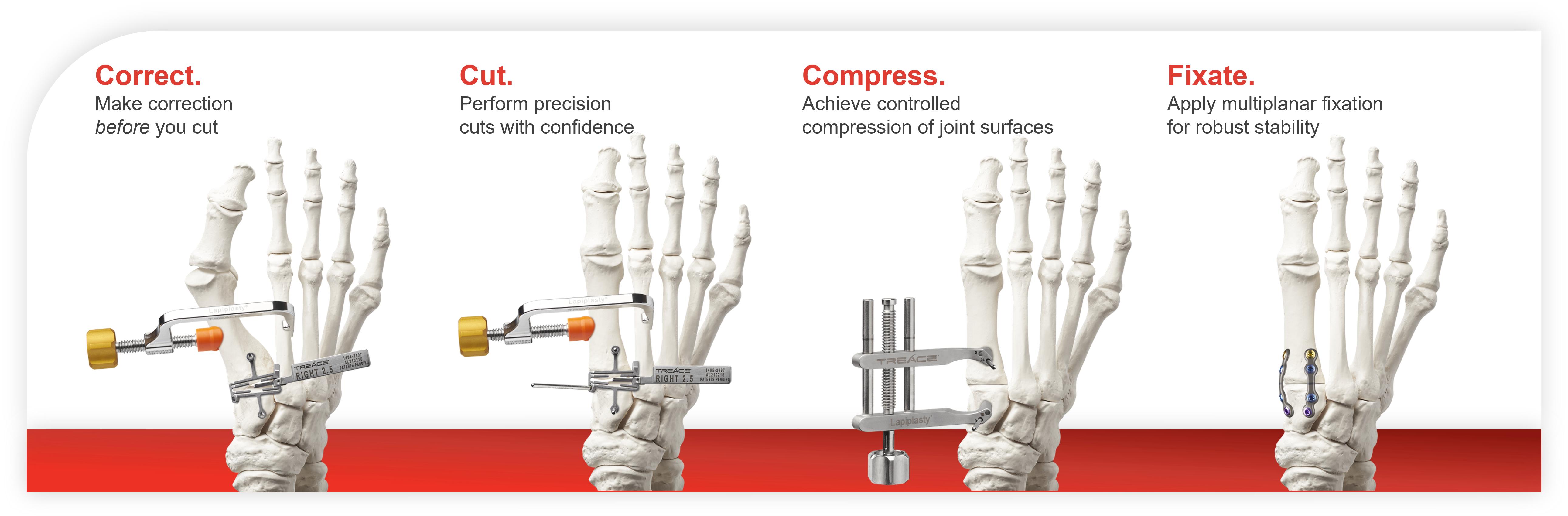
9
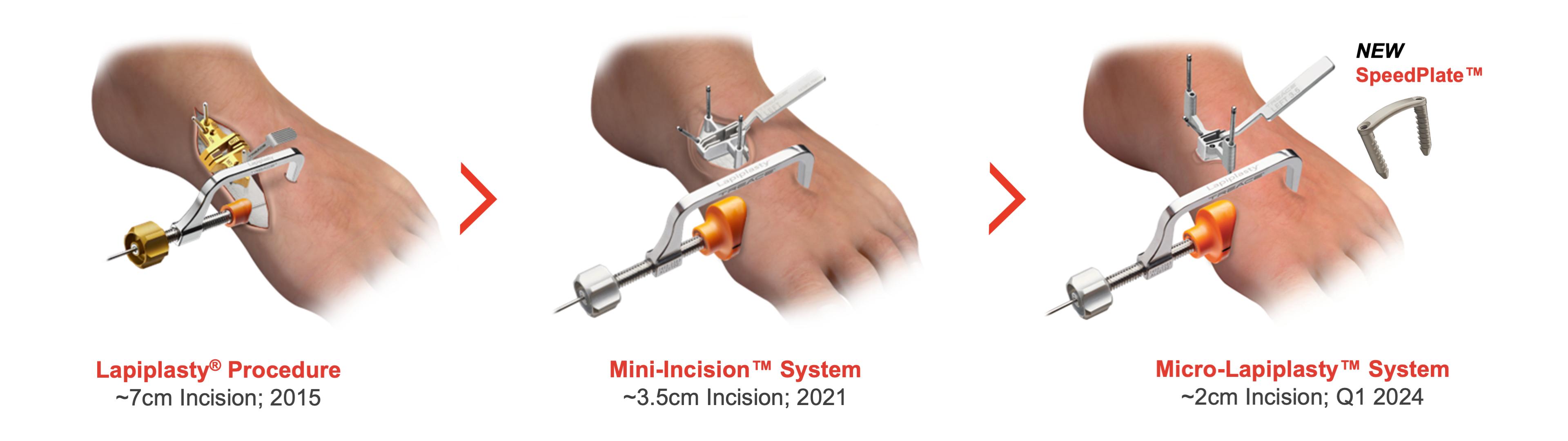
Our Minimally Invasive Approaches
To expand our Lapiplasty offerings, in 2021 we launched the Lapiplasty Mini-Incision™ System, designed to allow the Lapiplasty Procedure to be performed through a 3.5cm incision as compared to the 6cm to 8cm incision with the standard Lapiplasty Procedure. Some patients prefer smaller incisions that may leave less visible scars. The Lapiplasty Mini-Incision System includes a fixation plate known as the PlantarPower™ Plate contoured to span across the bottom half of the joint where the loads are the highest, while still providing easy access for insertion of the plate fixation screws through a small incision. Continuing with minimally invasive approaches, in the fourth quarter of 2023, we began the limited commercial launch of the Micro-Lapiplasty™ Minimally Invasive System, which is designed to enable the patented Lapiplasty Procedure to be performed through a minimally-invasive 2cm incision utilizing new SpeedPlate implant technology for fixation.
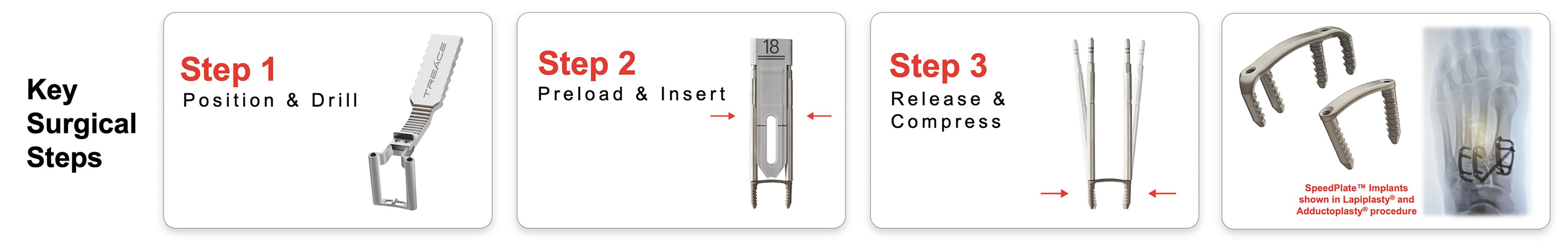
SpeedPlate Implant Fixation Platform
During 2023, we launched the SpeedPlate Rapid Compression Implant System, which is a new Lapiplasty fixation technology designed for rapid insertion while providing dynamic compression of the joint surfaces. Since it can be implanted through a small 2cm incision, the SpeedPlate technology not only offers broad applicability with our standard Lapiplasty and Adductoplasty Systems but also serves as the enabling fixation technology for our Micro-Lapiplasty System. The SpeedPlate System may also be used for other common bone fusion procedures in the foot. Full commercial launch of the SpeedPlate technology began in the fourth quarter of 2023.
Our Adductoplasty System
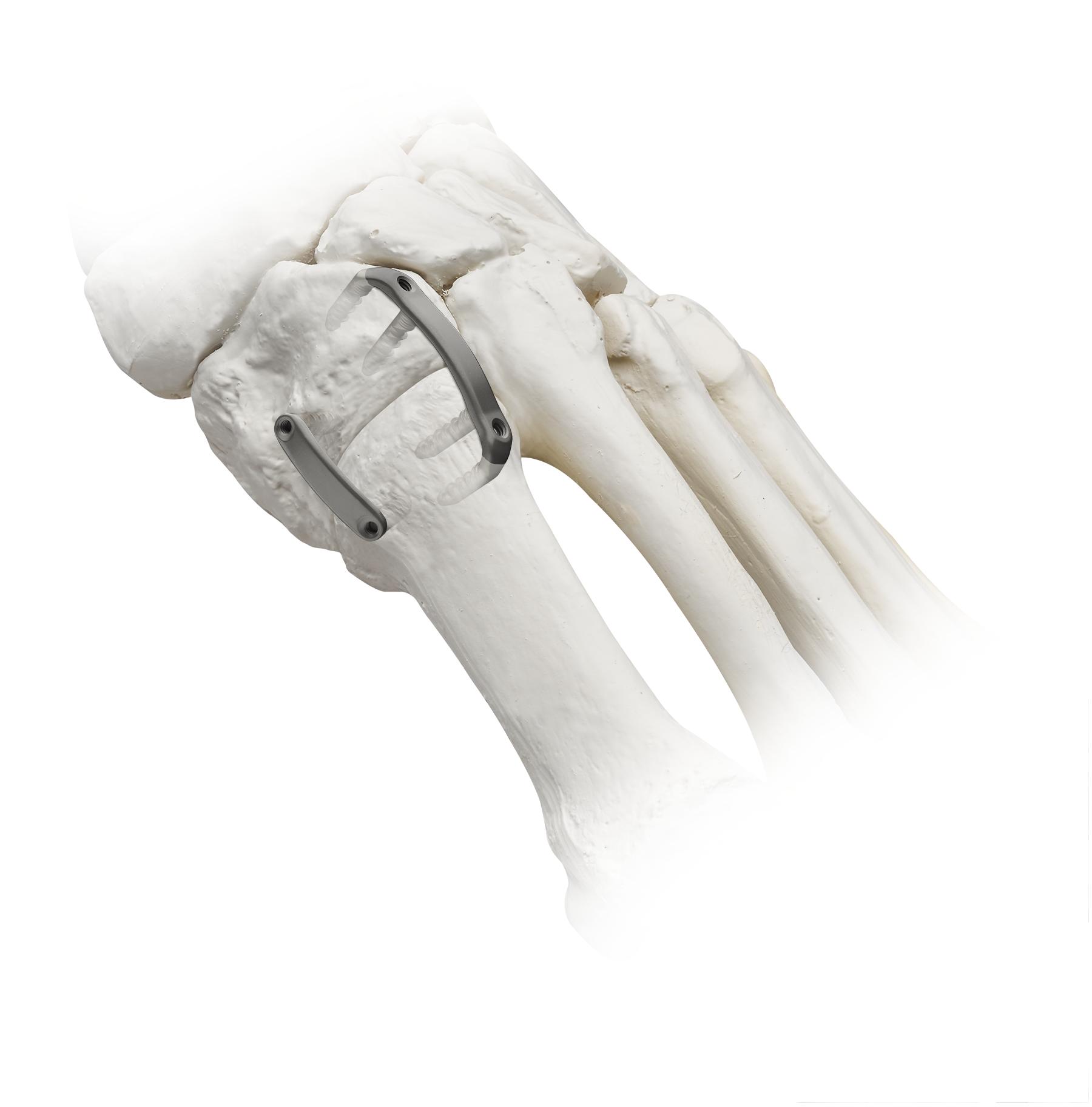
In the third quarter of 2021, we launched the Adductoplasty System, which brings together our implants and precision instrumentation for a comprehensive system designed for reproducible correction of metatarsus adductus deformities and osteoarthritis of the midfoot. Midfoot deformities may occur in up to 30% of bunion patients. The Adductoplasty System includes instruments together with our Lapiplasty fixation implants to be used for fusion of the second and third TMT joints, which may often be necessary in conjunction with bunion surgery. In 2022, we introduced the TriTome™ Release Instrument, a sterile-packed, single-use instrument with three cutting edges designed to assist with a tissue release performed in the Adductoplasty Procedure.
10
Hammertoe PEEK Fixation System
In the fourth quarter of 2023, we introduced the Hammertoe PEEK Fixation System designed to address hammertoe, claw toe and mallet toe deformities. Hammertoes often present with bunions and is one of the most prevalent deformities in the foot, resulting in approximately 700,000 surgical repairs per year in the U.S. The Hammertoe System is made with PEEK (polyether ether ketone) to offer radiolucency and mechanical properties comparable to bone, is cannulated to facilitate streamlined insertion and allow for accurate implant placement and is a sterile-packed implant and instrument kit for convenient delivery and clinical efficiency.

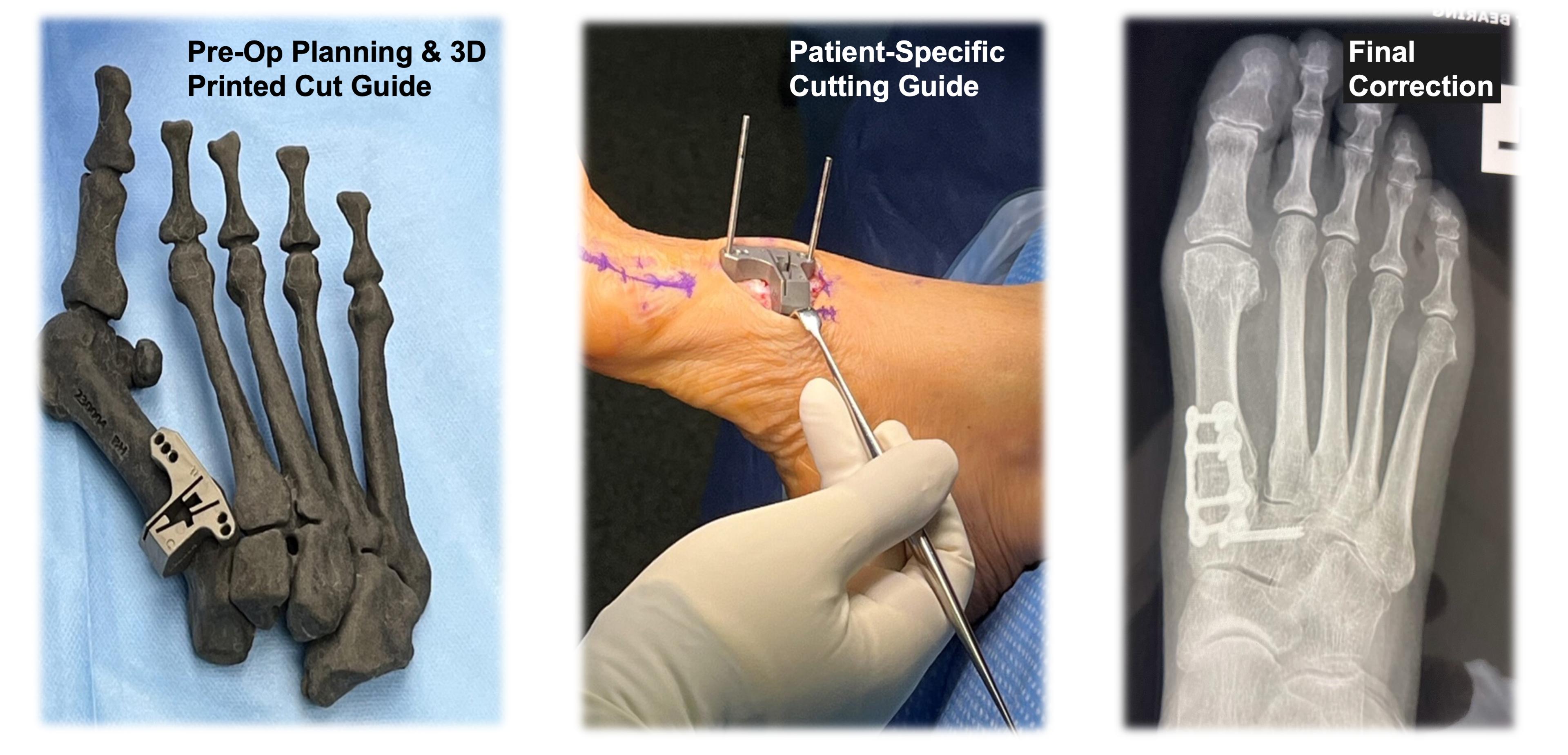
Patient Specific Instrumentation
On June 12, 2023, we acquired certain assets of MIOS Marketing, LLC d/b/a RedPoint Medical3D ("RPM-3D"). The technology acquired allows us to use patient CT scan data to which we apply software technologies to develop three-dimensional pre-operative plans for correcting a patient's deformity and produce a 3D-printed, patient-specific cut guide designed to deliver accurate surgical correction of deformities customized to the patient's unique foot anatomy. Our patient specific instrumentation is currently in limited clinical release with full commercialization planned in the second half of 2024.
Other Lapiplasty and Adductoplasty Advancements
In 2022 and 2023, we introduced additional new advancements in the Lapiplasty and Adductoplasty Procedures designed to make them faster and more efficient to perform. These include (1) the 3-n-1 Guide, which combines three separate instruments and three procedure steps into one instrument and step, (2) the S4A™ Anatomic Plating System, which features advanced 3D contours designed to accommodate variations in patient anatomy, (3) the SpeedRelease™ Release Instrument, which is a single-use instrument designed to make a challenging soft tissue release performed in the majority of Lapiplasty cases easier to perform and more reproducible for the surgeon, and (4) the LapiTome™ and RazorTome™ Osteotomes, which are sterile, single-use instruments that are designed to facilitate more efficient removal and release of bone slices and soft tissue in Lapiplasty and Adductoplasty cases.
11
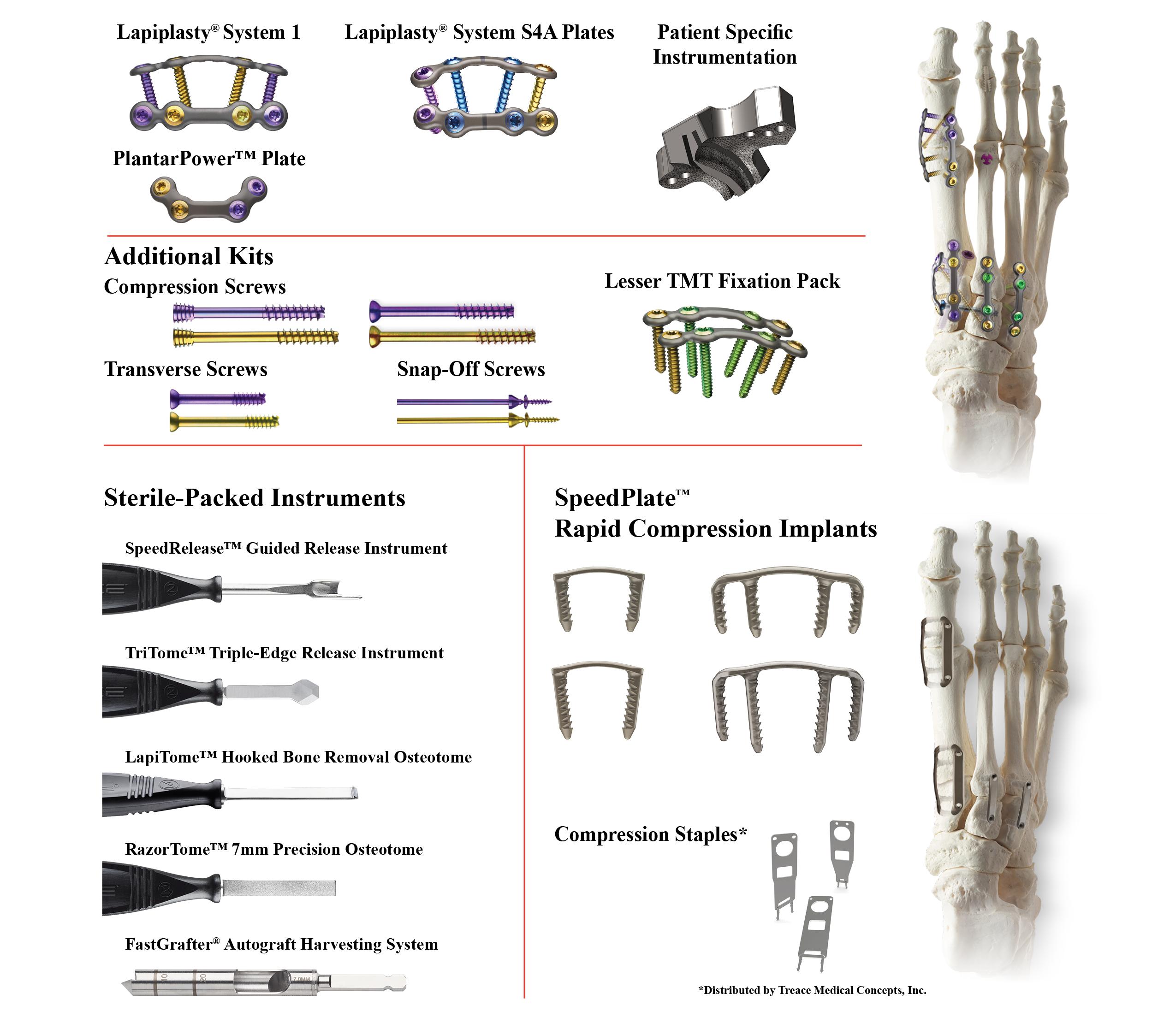
Our Product Portfolio
The following diagram depicts the implants and single use products we currently offer as part of our broader portfolio:
Key Clinical Advantages of the Lapiplasty System
We believe that the differentiated clinical advantages of the Lapiplasty Procedure support its continued clinical adoption and help establish the Lapiplasty Procedure as the standard of care for bunion surgery. We are committed to advancing the understanding of the Lapiplasty Procedure and its benefits to patients, surgeons, facilities and payors through clinical studies and publications in peer-reviewed literature. The Lapiplasty Procedure had been cited in 21 peer-reviewed journal publications as of December 2023.
Interim analyses from the ALIGN3D clinical study have been published in the Journal of Foot & Ankle Surgery and most recently presented at industry conferences, including at the AOFAS Annual Meeting in September 2023, and at the ACFAS Annual Scientific Conference in early February 2024. The Journal of Foot & Ankle Surgery publication presented an interim analysis (117 patients with at least 12 month follow-up) demonstrating early return to weight bearing in a walking boot at an average of 7.8 days, return to work at an average 25.2 days, full unrestricted activity at an average of 4 months, significant improvement in radiographic measures of 3-dimensional bunion correction with 1 recurrence reported at 12 months (0.9% recurrence rate, n=108),and significant improvement in patient-reported pain reduction on the Visual Analog Scale (VAS) and quality of life measurements on the Manchester-Oxford Foot Questionnaire (MOxFQ) and Patient-Reported Outcomes
12
Measurement Information Systems (PROMIS) scores. We have submitted our primary endpoint ALIGN3D manuscript to a top-tier, peer-reviewed foot and ankle journal at the end of 2023 and expect publication mid-year 2024.
Based on the outcomes from multiple studies, further discussed below, and our deep experience in the field of bunion surgery, we believe the key advantages of the Lapiplasty System include the system being designed for:
Our differentiated Lapiplasty System is designed to consistently and reliably correct all three dimensions of the bunion deformity and address its root cause. A traditional 2D Osteotomy performs an incomplete correction addressing the cosmetic appearance of the bunion rather than the root cause of the deformity. Alternatively, while Lapidus Fusion does seek to address the deformity's root cause, it does not address the 3D rotational aspect known to contribute to bunion recurrence and involves a technically challenging "freehand" technique, which is highly dependent on the surgeon's skill and requires the physician to perform complex corrections without the benefits of assistive instrumentation. The Lapiplasty System includes specifically engineered procedural instrumentation and implants to enable the surgeon to correct the bunion deformity with accuracy and consistency.
Clinical studies described in multiple peer-reviewed publications demonstrate the clinical benefits of the Lapiplasty System. These publications demonstrate that the Lapiplasty Procedure allows patients to quickly return to weight-bearing in a walking boot within 3 to 10 days. In addition, these publications demonstrated meaningfully low rates of recurrence (as described above). In addition, these studies indicate a low-rate in incidence of the bones not healing together (i.e., non-union rate) as well as a low rate of hardware removal. Finally, research also suggests that Lapiplasty may result in a significant decrease in post-operative bony and soft tissue width (i.e., a slimmer foot post 5-month follow up)—although not an indication for surgery, foot width reduction is often a desirable cosmetic and functional outcome and commonly associated with postoperative patient satisfaction. Given its demonstrated clinical benefits, we believe the Lapiplasty System provides a positive physician and patient experience, and through continued clinical adoption, is poised to become the standard of care for bunion surgery.
Recent Interim Data from ALIGN3D Study
Our ALIGN3D prospective, multicenter study is evaluating bunion correction status after five years and includes patient reported outcomes, range of motion results and radiographic outcomes. The study enrolled 173 patients, aged 14 to 58 years, at 7 clinical sites in the United States with 13 participating surgeons. Final patient follow-up for the primary endpoint was completed in the first half of 2023. The table below states the recent interim results of our ALIGN3D clinical study presented at the ACFAS Annual Scientific Conference held in early February 2024 demonstrating the following key outcomes, including an analysis of 173 patients with a mean follow up of 33.8 months following the Lapiplasty® Procedure:
Key outcomes |
Lapiplasty Procedure |
Recurrence rate |
0.9%1 |
Reported time to start weight-bearing |
average of 8.4 days (in a walking boot) |
Symptomatic non-union rate2 |
1.8% |
Hardware removal due to pain |
6.9% |
Patient-reported improvement in pain |
81%3 |
Patient-reported improvement in walking/standing |
86%4 |
Patient-reported improvement in social interaction |
85%4 |
13
The ACFAS presentation, which includes additional details such as patient demographics, inclusion/exclusion criteria, and complications reported in the studies, is available on Treace’s website at www.lapiplasty.com/surgeons/journal-publications/. The information found on our website, including the ACFAS presentation, is not part of this Annual Report or any other report we file with, or furnish to, the SEC.
Commercial Strategy
We are investing in and executing a five-point strategy that includes rapid product innovations, a bunion-focused direct sales channel, surgeon education programs, direct-to-patient education, and supportive clinical evidence.
We currently market and sell the Lapiplasty System through a combination of a direct employee sales force and independent sales agencies across 249 territories in the United States. As of December 31, 2023, we had 227 employee sales representatives and 22 independent sales agencies. In 2023, employee sales representatives generated approximately 81% of total revenue while approximately 19% of revenue came through independent sales agencies. In 2022, employee sales representatives generated approximately 71% of total revenue while approximately 29% came through independent sales agencies.
We have and are continuing to dedicate meaningful resources to expand our sales force and management team in the United States. We have hired and expect to continue to hire additional employee sales representatives and employee field sales management to strategically access regions with high densities of prospective patients. We believe this strategy will:
We believe our surgeon education and training programs differentiate us from our competitors. We devote significant resources to training and educating physicians on the safe and effective use of the Lapiplasty System. Our comprehensive education programs include cadaveric workshops, technical assistance in the operating room and advanced training for both new and existing surgeon customers. We believe our multiple post-market clinical outcome studies are also unique in the bunion correction field and are a key element of our medical education program.
Our practice is to require surgeons to complete a simulated surgical training program before performing the Lapiplasty Procedure. To facilitate this training, we have developed a robust curriculum including clinical and procedural details as well as hands-on surgical workshops designed to simulate a live surgical procedure. These training events incorporate
14
highly-skilled training personnel including experienced surgeon faculty and clinical specialists. Additionally, we host ongoing peer-to-peer advanced educational training programs to continue to develop the expertise of our surgeon customers, which include monthly online "Mastery Webinar" series and hands-on workshops with experienced faculty surgeons that cover more advanced Lapiplasty techniques and training on our newly developed products and procedures. Our training programs are complemented by 12 clinical specialists who assist with surgeon training and live surgery support with new surgeon users. We believe that our surgeon education programs are effective, and they are intended to result in surgeon users improving their skill and familiarity with the Lapiplasty Procedure and improved clinical outcomes for their patients.
Surgeons generally can perform their first case after they have been trained and our products have been approved by the surgical facility. Obtaining facility approval may delay surgeon access to our products for 30 to 120 days or more depending on the nature of the facility (or integrated delivery network's) approval process.
Surgeon users typically increase usage of the Lapiplasty Procedure over time as they see improved clinical outcomes for their patients relative to traditional bunion surgery approaches. The bar chart below shows as of December 31, 2023 the average number of procedures performed over the trailing twelve months by surgeons based on the number of years that the surgeons have used the Lapiplasty System.
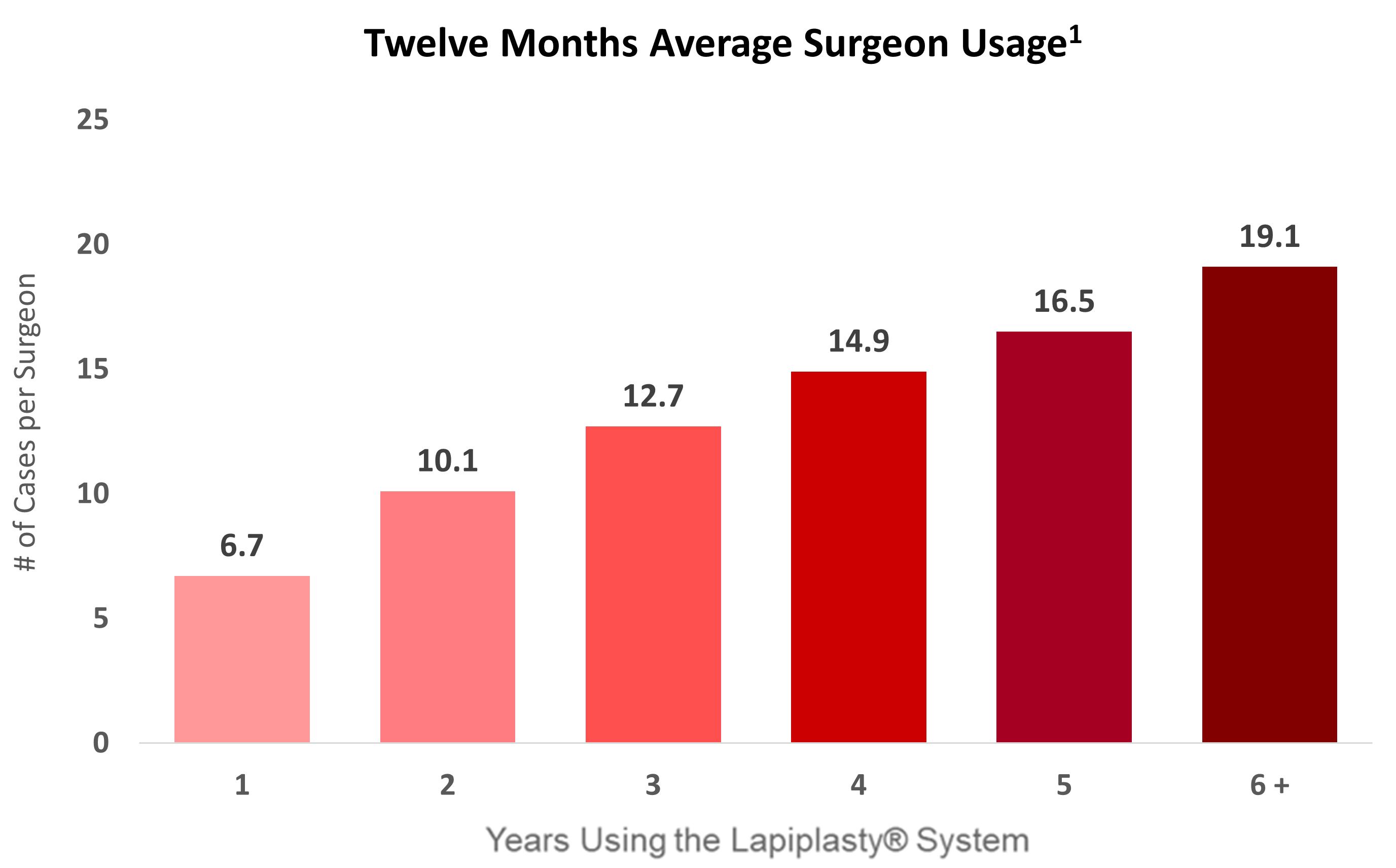
Surgeon Utilization in Last 12 Months by La2020 8.0 2019 12.0 2018 17.7 2017 and Prior Year First Case Performed
We believe our offering is differentiated by supporting surgeons with knowledgeable clinical specialists and direct sales employees who are experts in the Lapiplasty Procedure. These employees receive in-depth training to develop a thorough understanding of bunions, patient selection, procedure planning and regulatory policies to meaningfully support continued clinical adoption and existing surgeon customers. Our clinical specialists and direct sales employees participate in continuous education programs that consist of in-person foundational training, procedure observation and sales skills development. These employees are a key resource for our surgeon customers and their expertise enables them to provide meaningful clinical and technical support in the operating room and to develop strong relationships with surgeons. We believe that our approach to supporting surgeons leads to better clinical outcomes for patients.
Our direct-to-patient outreach program is a key aspect of our commercial strategy. This program is focused on educating patients on the clinical advantages of the Lapiplasty Procedure and generating brand awareness. We are working to further establish brand recognition for Lapiplasty as the leading procedure for improving bunion treatment outcomes in an industry that has traditionally not conducted significant direct-to-patient programs. We have built a sophisticated marketing infrastructure to deliver our message in a targeted manner utilizing digital and traditional marketing channels. These programs direct potential bunion surgical candidates to our educational website that further explains the Lapiplasty Procedure and its related benefits and risks. Our "Find a Doctor" tool allows them to search for trained Lapiplasty Procedure surgeons in their local markets.
15
The following diagram illustrates our patient outreach program.
Research and Development
We devote significant resources to research and development of our products. We use employee engineering personnel and our Surgeon Advisory Board and other orthopaedic surgeon consultants to generate ideas and develop product innovations. Our Surgeon Advisory Board is comprised of both podiatrists and orthopaedic foot and ankle surgeons who provide us with insights for developing products that fully meet the needs of each group. Our development team is focused on improving clinical outcomes by designing new procedure-specific products and by developing enhanced surgical techniques in attractive subspecialties within the foot and ankle market.
Our initial product development and commercial efforts have been focused on the bunion market, and our Lapiplasty System specifically. We intend to continue iterating our core Lapiplasty System instrumentation and implants to improve surgical efficiency, enhance reproducibility of outcomes and speed up surgical recovery for patients. We expanded our footprint in the foot and ankle market in 2021 with the Adductoplasty Midfoot Correction System, designed for reproducible correction of midfoot deformities, and in 2023 with SpeedPlate Rapid Compression Implants, an innovative fixation platform with broad versatility across Lapiplasty and Adductoplasty procedures, as well as other common bone fusion procedures of the foot. Also in 2023, we introduced our Hammertoe PEEK Fixation System designed to address hammertoe, claw toe and mallet toe deformities which often affect bunion patients. We are also continuing to pursue the development and potential commercialization, if cleared, of new products that we believe would leverage and expand our position in the market to treat other concomitant pathologies that occur in a high percentage of bunion surgeries. Products provided by other companies are currently utilized in some of our Lapiplasty Procedure cases to treat these concomitant conditions. Providing these ancillary products allows us to capture a higher percentage of the overall product revenue from the surgical case while providing greater efficiency and synergies to the facility and operating room staff by reducing the number of vendors needed to support the case.
Clinical Datasets
A key component of our five-point strategy is supportive clinical evidence. We devote significant time and resources to supporting clinical studies to advance the standard of care for the surgical management of bunions and to sharing these results in peer-reviewed publications. As part of our commitment to developing clinical evidence to improve the surgical treatment of bunions, we currently have three prospective, multicenter, post-market studies underway:
16
Each of these studies has a primary effectiveness endpoint that determines the maintenance of the bunion correction at 24 months after surgery. More information about the outcome of the ALIGN3D clinical study is discussed above under the heading "Key Clinical Advantages of the Lapiplasty System." We also support investigator-initiated studies conducted by surgeons seeking to study specific clinical scenarios and endpoints.
Coverage and Reimbursement
Procedures involving our products are performed by foot and ankle surgeons in both hospital outpatient facilities and ambulatory surgery centers. Hospitals, ambulatory surgery centers and surgeons that purchase or use our products generally rely on third-party payors to reimburse for all or part of the costs and fees associated with procedures using our products. As a result, sales of our products depend, in part, on the extent to which the procedures using our products are covered by third-party payors, including government programs such as Medicare and Medicaid, private insurance plans and managed care programs. Based on historical claims data from 2017, approximately 63% of Lapidus cases and 60% of all bunion surgical cases were paid by private payors.
Medicare publishes national average rates for each procedure in the hospital outpatient and ambulatory surgery center settings. Medicare rates for procedures involving our products may vary from national averages due to geographic location, the nature of facility in which the procedure is performed (i.e., teaching or community hospital) and other factors. While private payors vary in their coverage and payment policies, many use coverage and payment by Medicare as a benchmark to make their own decisions.
Coding and Reimbursement
When procedures using our products are performed in hospital outpatient or ambulatory surgery center settings, both the surgeon and the health care facility submit claims (bills) for payment to the third-party payor using established medical codes (e.g., CPT codes, diagnosis codes and HCPCS codes) that describe the patient history and medical and surgical treatments. Obtaining appropriate payment for services is dependent in part on the physician and health care facility reporting or billing the CPT code that accurately describes the procedures performed in the case.
The table below sets forth the established CPT codes that are commonly used for Lapidus-type and midfoot fusion surgeries, including the Lapiplasty and Adductoplasty Procedures, as well as for hammertoe correction.
Established CPT Codes |
|
CPT 28297 |
Correction, hallux valgus with bunionectomy, with sesamoidectomy, when performed; with first metatarsal and medial cuneiform joint arthrodesis, any method |
CPT 28730 |
Arthrodesis, midtarsal or tarsometatarsal, multiple or transverse |
CPT 28740 |
Arthrodesis, midtarsal or tarsometatarsal, single joint |
CPT 28735 |
Arthrodesis, midtarsal or tarsometatarsal, multiple or transverse; with osteotomy (e.g., flatfoot correction) |
CPT 28285 |
Correction, hammertoe (e.g., interphalangeal fusion, partial or total phalangectomy) |
CPT 20900 |
Bone graft, any donor area; minor or small (e.g., dowel or button) |
Bunion surgery also often involves multiple concomitant procedures, including Akin osteotomy, Weil osteotomy and hammertoe correction, for example. Each concomitant procedure has an applicable CPT code used for billing third-party payors, which is submitted on the same claim with the Lapiplasty Procedure for reimbursement.
Intellectual Property
We actively seek to protect the technology, inventions, and improvements that we consider important to our business using patents, trade secrets, trademarks and copyrights in the United States and foreign markets.
17
As of December 31, 2023, our patent portfolio included 52 owned U.S. patents, one licensed U.S. patent and 15 owned foreign patents. All of the registered U.S. patents are utility patents. The owned patents cover core Lapiplasty and Adductoplasty-related hardware and surgical techniques as well as other associated innovations, including the main surgical techniques used by the Lapiplasty Procedure as well as associated tools, techniques and/or implants used during the procedure. Our foreign granted patents are in Australia, the European Patent Convention and Japan. Our owned patents expire in 2035 or later.
As of December 31, 2023, we had 141 pending patent applications globally, including 81 in the United States. Outside of the United States we have patent applications pending in Australia, Canada, Europe (before the European Patent Office) and Japan as well as through the Patent Cooperation Treaty ("PCT").
The licensed U.S. patent refers to our exclusive license to a U.S. patent owned by a third party that expires in 2034. Our patents are intended to exclude competitors from practicing the innovations of our currently marketed product offering and to protect potential future commercialization opportunities and to strategically block potential workarounds by competitors.
We own U.S. trademark registrations for several of our most important marks, including "Treace Medical Concepts®", the "Treace Medical Concepts®" logo, "Lapiplasty®", "Fast Grafter®", "FastPitch®", "Adductoplasty®", "3D Bunion Correction®", "Plantar Python®", "Fix It Right The First Time®", "SpeedSeeker®" and "The Leader in Hallux Valgus Surgery®". We also have pending U.S. trademark registrations on other valuable marks, including "LapiTome™", "RazorTome™", "Micro-Lapiplasty™", "SpeedPlate™", "SpeedRelease™", and "TriTome™".
The term of individual patents depends on the legal term for patents in the countries in which they are granted. In most countries, including the United States, the patent term is generally 20 years from the earliest claimed filing date of a nonprovisional patent application in the applicable country. We cannot provide assurance that patents will be issued from any of our pending applications or that, if patents are issued, they will be of sufficient scope or strength to provide meaningful protection for our technology. Notwithstanding the scope of the patent protection available to us, a competitor could develop treatment methods or devices that are not covered by our patents. Furthermore, numerous U.S. and foreign-issued patents and patent applications owned by third parties exist in the fields in which we have commercialized and are developing products. Because patent applications can take many years to issue, there may be applications unknown to us, which applications may later result in issued patents that our existing or future products or technologies may be alleged to infringe.
There has been substantial litigation regarding patent and other intellectual property rights in the medical device industry. We have initiated lawsuits in the past and may bring lawsuits in the future to enforce patents issued or licensed to us, enforce our rights in trademarks and copyright, to protect our trade secrets or know-how, to defend against claims of infringement of the intellectual property rights of others, or to determine the scope and validity of the proprietary rights of others. Litigation is costly and diverts our attention from other functions and responsibilities. Furthermore, even if our patents are found to be valid and infringed, a court may refuse to grant injunctive relief against the infringer and instead grant us monetary damages and/or ongoing royalties. Such monetary compensation may be insufficient to adequately offset the damage to our business caused by the infringer's competition in the market.
In the event we are subject to lawsuits by third parties seeking to enforce their patent rights, adverse determinations in such future litigation could subject us to significant liabilities to third parties, could require us to seek licenses from third parties and could prevent us from manufacturing, selling or using our product or techniques, any of which could severely harm our business.
Our knowledge and experience, creative product development, marketing staff and trade secret information, with respect to manufacturing processes, materials and product design, are as important as our patents in maintaining our proprietary product lines. As a condition of employment, we require all employees and key contractors to execute an agreement obligating them to maintain the confidentiality of our proprietary information and assign to us inventions and other intellectual property created during their employment. For more information, refer to Item 1A, "Risk Factors—Risks Related to Intellectual Property".
Royalty and License Agreements
We have entered into product development and fee for service agreements with members of our Surgeon Advisory Board and other surgeon consultants that specify the terms under which the consultant is compensated for his or her consulting services and grants us rights to the intellectual property created by the consultant in the course of such services. As products are commercialized with the assistance of the surgeon consultants, we may agree to enter into a royalty agreement if the consultant's contributions to the product are novel, significant and innovative.
18
We have entered into royalty agreements with surgeon consultants providing royalties based on each individual's level of contribution. Each royalty agreement: (1) confirms the irrevocable transfer to us of all pertinent intellectual property rights; (2) sets the applicable royalty rate; (3) sets the period of time during which royalties are payable; (4) is for a term of three years, renewable by the parties, and may be terminated by either party on 90 days' notice for convenience (provided that if terminated by the Company for convenience the obligation to pay royalties is not affected); and (5) prohibits the payment of royalties on products sold to entities and/or individuals with whom the surgeon advisor or any other surgeon advisor entitled to royalties is affiliated. Each of the royalty agreements may be subsequently amended to add the license of additional intellectual property covering new products, and as a result, multiple royalty rates and duration of royalty payments may be included in one royalty agreement.
For more information about royalty payments, please see Note 8, "Commitments and Contingencies," of the Notes to the Financial Statements.
Manufacturing and Supply
We currently leverage third-party manufacturing relationships to ensure low-cost production while maintaining a capital efficient business model. We generally have multiple sources of supply for critical components of the Lapiplasty and Adductoplasty Systems. Our supply agreements do not have "take or pay" commitments or financial penalties that apply if we do not meet minimum purchase obligations. Likewise, except for one supplier, our suppliers have no obligation to sell to us or to manufacture for us any given quantity of our products or components for our products. In most cases, we have redundant manufacturing capabilities for each of our products. To date, we have not experienced any significant difficulty obtaining our products or components for our products necessary to meet demand, and we have only experienced limited instances where our suppliers had difficulty supplying products by the requested delivery date. We believe manufacturing capacity is sufficient to meet market demand for our products for the foreseeable future.
The suppliers for the Lapiplasty and Adductoplasty Systems and our other products are evaluated, qualified and approved through our supplier management program, which includes various evaluations, assessments, qualifications, validations, testing and inspection to ensure the supplier can meet acceptable quality requirements. We implement a robust change control policy with our key suppliers to ensure that no component or process changes are made without our prior approval.
Order quantities and lead times for components purchased from suppliers are based on our forecasts derived from both historical demand and anticipated future demand. Lead times for components may vary depending on the size of the order, time required to fabricate, specific supplier requirements and current market demand for the components, sub-assemblies and materials.
Competition
Our industry is competitive, subject to technological change and significantly affected by new product introductions and market activities of other industry participants. Our existing products are, and any future products we commercialize will be, subject to competition. We believe the principal competitive factors in our markets include:
19
Our competition includes medical device manufacturers in the orthopaedic foot and ankle market. Stryker Corporation is currently the leader in the orthopaedic foot and ankle market and has significant market share. Additional companies operating in the orthopaedic foot and ankle market with products specifically focused on bunion surgery include CrossRoads Extremity Systems, which was acquired in 2022 by DePuy Synthes Products, Inc., a Johnson & Johnson subsidiary, Zimmer Biomet Holdings, Inc., Paragon 28, Inc., In2Bones Global, Inc., a subsidiary of CONMED Corporation, Arthrex, Inc., Enovis Corporation (formerly Colfax), Novastep Inc. (now part of Enovis Corporation), Nextremity Solutions Inc. (acquired by Medartis Holdings AG); Henry Schein, Inc. (through its newly acquired interest in TriMed and strategic relationship with Extremity Medical LLC), Gramercy Extremity Orthopedics, LLC., and Fusion Orthopedics, LLC. While foot and ankle product sales represent a relatively small percentage of our larger competitors' overall sales, many recognize the growth opportunities in this market and have been active in product additions through both internal development efforts and acquisitions.
Our competitors may have significantly greater financial resources, established presence in the market, expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified research & development, sales, marketing and management personnel, establishing clinical sites and patient registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors. In addition to competing for market share for the Lapiplasty Procedure, we also compete against these companies for personnel, including qualified personnel that are necessary to grow our business.
Finally, we may compete with medical device manufacturers outside the United States if and when we pursue plans to market our products internationally. Among other competitive advantages, such companies may have more established sales and marketing programs and networks, established relationships with health care professionals and greater name recognition in such markets.
Government Regulation
Our products and our operations are subject to extensive regulation by the U.S. Food and Drug Administration ("FDA") and other federal and state authorities in the United States, including the U.S. Department of Justice and the U.S. Department of Health and Human Services Office of Inspector General and, if we begin offering our products outside the United States, comparable authorities in foreign jurisdictions. Our products are subject to regulation as medical devices in the United States under the Federal Food, Drug, and Cosmetic Act ("FDCA") as implemented and enforced by the FDA.
United States Regulation
The FDA regulates the development, design, non-clinical and clinical research, manufacturing, safety, efficacy, labeling, packaging, storage, installation, servicing, recordkeeping, premarket clearance or approval, adverse event reporting, advertising, promotion, marketing, sale and distribution, and import and export of medical devices to ensure that medical devices distributed domestically are safe and effective for their intended uses and otherwise meet the requirements of the FDCA.
FDA Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device commercially distributed in the United States requires either FDA clearance of a 510(k) premarket notification, FDA grant of a de novo request, or FDA approval of a premarket approval application ("PMA") or FDA approval of a humanitarian device exemption. Under the FDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of manufacturer and regulatory control needed to ensure its safety and effectiveness. Class I includes devices with the lowest risk to the patient and generally require adherence to the FDA's General Controls for medical devices, which include compliance with the applicable portions of the Quality System Regulation ("QSR"), facility registration and product listing, reporting of adverse medical events, and truthful and non-misleading labeling, advertising, and promotional materials. Class II devices are subject to the FDA's General Controls, as well as special controls deemed necessary by the FDA. These special controls can include performance standards, post-market surveillance, patient registries and FDA guidance documents.
20
While many Class I and Class II devices are exempt from the 510(k) premarket notification requirement, manufacturers of certain Class I and Class II devices are required to submit to the FDA a premarket notification under Section 510(k) of the FDCA requesting permission to commercially distribute the device, also referred to as 510(k) clearance. These devices may also be exempt from Good Manufacturing Practices, which are requirements under the FDA Quality System Regulation. If a Class I or Class II device is subject to premarket notification requirements and there is no existing device previously approved by the FDA that is substantially equivalent to the device, a de novo request must be submitted and approved before the device can be marketed. Devices deemed by the FDA to pose the greatest risks, such as life sustaining, life supporting, or some implantable devices are placed in Class III. If substantial equivalence to a legally marketed device cannot be demonstrated via a Class III 510(k), a Class III device must receive PMA approval in order to be legally marketed. Our currently marketed products are Class I exempt devices and Class II devices subject to 510(k) clearance.
510(k) Clearance Marketing Pathway
Certain of our current products are subject to premarket notification and clearance under section 510(k) of the FDCA. To obtain 510(k) clearance, we must submit to the FDA a premarket notification submission demonstrating that the proposed device is "substantially equivalent" to a legally marketed predicate device, i.e., a device that was legally marketed before May 28, 1976 (pre-amendments device) and for which a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was found substantially equivalent through the 510(k) process. The FDA's 510(k) clearance process usually takes from three to twelve months but may take longer. The FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence. In addition, the FDA collects user fees for certain medical device submissions and annual fees for medical device establishments. For fiscal year 2024, the standard user fee for a 510(k) premarket notification application is $21,760.
If the FDA agrees that the device is substantially equivalent to a predicate device currently on the market, it will grant 510(k) clearance to commercially market the device. If the FDA determines that the device is "not substantially equivalent" to a previously cleared device, the applicant can resubmit the 510(k), file a reclassification petition, submit a PMA, or request a Class I or Class II designation through the "de novo" process, which is a route to market for novel medical devices that are low to moderate risk and are not substantially equivalent to a predicate device.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, will require a new 510(k) clearance or depending on the modification, PMA approval or de novo classification. The FDA requires each manufacturer to determine whether the proposed change requires submission of a 510(k), de novo classification or a PMA in the first instance, but the FDA can review any such decision and disagree with a manufacturer's determination. If the FDA disagrees with a manufacturer's determination, the FDA can require the manufacturer to cease marketing and/or request the recall of the modified device until 510(k) marketing clearance, approval of a PMA, or issuance of a de novo classification. Also, in these circumstances, the manufacturer may be subject to significant regulatory fines or penalties.
PMA Approval Pathway
Class III devices require PMA approval before they can be marketed, although some pre-amendment Class III devices for which FDA has not yet required approval of a PMA are cleared through the 510(k) process. The PMA process is more complex, costly and time consuming than the 510(k) premarket notification process. A PMA must be supported by extensive data, including, but not limited to, extensive technical information regarding device design and development, preclinical and clinical trials, manufacturing, and labeling information to demonstrate to the FDA's satisfaction the safety and effectiveness of the device for its intended use. The PMA application must provide valid scientific evidence that demonstrates to the FDA's satisfaction reasonable assurance of the safety and effectiveness of the device for its intended use. Following receipt of a PMA, the FDA determines whether the application is sufficiently complete to permit a substantive review. If the FDA accepts the application for review, it has 180 days under the FDCA to complete its review of the PMA, although in practice, the FDA's review often takes significantly longer, and can take up to several years. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant's response to deficiencies communicated by the FDA. Also, during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel's recommendation. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with quality system regulation. The PMA process can be expensive, uncertain, and lengthy, and a number of devices for which FDA approval has been sought by other companies have never been approved by the FDA for marketing. None of our products are marketed pursuant to a PMA.
21
Clinical Trials
Clinical trials are almost always required to support a PMA and are sometimes required to support a 510(k) submission. All clinical investigations of investigational devices to determine safety and effectiveness must be conducted in accordance with the FDA's investigational device exemption ("IDE") regulations which govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device is determined to present a "significant risk" to human health, the manufacturer may not begin a clinical trial until it submits an IDE application to the FDA and obtains approval of the IDE from the FDA. The IDE must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. In addition, the study must be approved by, and conducted under the oversight of, an Institutional Review Board ("IRB"), for each clinical site. The IRB is responsible for the initial and continuing review of the IDE and may pose additional requirements for the conduct of the study. If an IDE application is approved by the FDA and one or more IRBs, human clinical trials may begin at a specific number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a non-significant risk to patients, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more IRBs without separate approval from the FDA, but must still follow abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements. A clinical trial may be suspended by FDA, the sponsor, or an IRB at any time for various reasons, including a belief that the risks to the study participants outweigh the benefits of participation in the trial. Even if a clinical trial is completed, the results may not demonstrate the safety and efficacy of a device to the satisfaction of the FDA or may be equivocal or otherwise not be sufficient to obtain approval of a device. We are not currently undertaking any FDA IDE trials, as all of our existing products are FDA-cleared through the 510(k) pathway. It is possible, however, that future device development may require IDE clinical trial for approval.
Post-Market Regulation
After a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
Manufacturing processes for medical devices are required to comply with the applicable portions of the QSR, which cover the methods and the facilities and controls for the design, manufacture, testing, production, processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices intended for human use. The QSR also requires, among other things, maintenance of a device master file, device history file, and complaint files. Manufacturers
22
are subject to periodic scheduled or unscheduled inspections by the FDA. Failure to maintain compliance with the QSR requirements could result in the shut-down of, or restrictions on, manufacturing operations and the recall or seizure of marketed products. The discovery of previously unknown problems with any marketed products, including unanticipated adverse events or adverse events of increasing severity or frequency, whether resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that a manufacturer has failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
Coverage and Reimbursement
In the United States, our currently approved products are commonly treated as general supplies utilized in orthopaedic surgery and if covered by third-party payors, are paid for as part of the surgical procedure. Outside of the United States, there are many reimbursement programs through private payors as well as government programs. In some countries, government reimbursement is the predominant program available to patients and hospitals. Our commercial success depends in part on the extent to which governmental authorities, private health insurers and other third-party payors provide coverage for and establish adequate reimbursement levels for the procedures during which our products are used. Failure by physicians, hospitals, ambulatory surgery centers and other users of our products to obtain sufficient coverage and reimbursement from third-party payors for procedures in which our products are used, or adverse changes in government and private third-party payor's coverage and reimbursement policies could materially adversely affect our business, financial condition, results of operations and prospects.
Based on our experience to date, third-party payors generally reimburse for the surgical procedures in which our products are used only if the patient meets the established medical necessity criteria for surgery. Some payors are moving toward a managed care system and control their health care costs by limiting authorizations for surgical procedures, including elective procedures using our devices. Although no uniform policy of coverage and reimbursement among payors in the United States exists and coverage and reimbursement for procedures can differ significantly from payor to payor, reimbursement decisions by particular third-party payors may depend upon a number of factors, including the payor's determination that use of a product is:
Third-party payors are increasingly auditing and challenging the prices charged for medical products and services with concern for upcoding, miscoding, using inappropriate modifiers, or billing for inappropriate care settings. Some third-party payors must approve coverage for new or innovative devices or procedures before they reimburse health care providers who use the products or therapies. Even though a new product may have been cleared for commercial distribution by the FDA, we may find limited demand for the product unless and until reimbursement approval has been obtained from governmental and private third-party payors.
23
A key component in determining whether appropriate payment amounts are received for physician and other services, including those procedures using our products, is the existence of a Current Procedural Terminology ("CPT") code, which describes the procedure in which the product is used. To receive payment, health care practitioners must submit claims to insurers using these codes for payment for medical services. CPT codes are assigned, maintained and annually updated by the American Medical Association and its CPT Editorial Board. If the CPT codes that apply to the procedures performed using our products are changed or deleted, reimbursement for performance of these procedures may be adversely affected.
In the United States, some insured individuals enroll in managed care programs, which monitor and often require pre-approval of the services that a member will receive. Some managed care programs pay their providers on a per capita (patient) basis, which puts the providers at financial risk for the services provided to their patients by paying these providers a predetermined payment per member per month and, consequently, may limit the willingness of these providers to use our products.
We believe the overall escalating cost of medical products and services being paid for by the government and private health insurance has led to, and will continue to lead to, increased pressures on the health care and medical device industry to reduce the costs of products and services. All third-party reimbursement programs are developing increasingly sophisticated methods of controlling health care costs through prospective reimbursement and capitation programs, group purchasing, redesign of benefits, requiring second opinions before major surgery, careful review of bills, encouragement of healthier lifestyles and other preventative services and exploration of more cost-effective methods of delivering health care.
In addition to uncertainties surrounding coverage policies, there are periodic changes to reimbursement levels. Third-party payors regularly update reimbursement amounts and also from time to time revise the methodologies used to determine reimbursement amounts. This includes routine updates to payments to physicians, hospitals and ambulatory surgery centers for procedures during which our products are used. These updates could directly impact the demand for our products.
Health Care Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in health care systems with the stated goals of containing health care costs, improving quality or expanding access. Current and future legislative proposals to further reform health care or reduce health care costs may limit coverage of or lower reimbursement for the procedures associated with the use of our products. The cost containment measures that payors and providers are instituting and the effect of any health care reform initiative implemented in the future could impact our revenue from the sale of our products.
In the United States, the implementation of the Affordable Care Act ("ACA") for example, has changed health care financing and delivery by both governmental and private insurers substantially, and affected medical device manufacturers significantly. The ACA, among other things, provided incentives to programs that increase the federal government's comparative effectiveness research, and implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain health care services through bundled payment models. Additionally, the ACA expanded eligibility criteria for Medicaid programs and created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. We are also affected by regulatory and legislative changes affecting Medicare payments and processing and reimbursement received by our customers.
We expect additional state and federal health care reform measures to be adopted in the future, some of which could limit the amounts that federal and state governments will pay for health care products and services, which could result in reduced demand for our products or additional pricing pressure.
Federal, State and Foreign Fraud and Abuse and Physician Payment Transparency Laws
In addition to FDA restrictions on the marketing, promotion and sale of drugs and devices, other federal and state laws restrict our business practices. These laws include, without limitation, foreign, federal, and state anti-bribery, anti-kickback and false claims laws, as well as transparency laws regarding payments or other items of value provided to health care providers.
24
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal health care programs. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Several courts have interpreted the statute's intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal health care program-covered business, including the purchase of medical device products, the federal Anti-Kickback Statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Many states also have adopted anti-kickback laws which establish similar prohibitions and, in some cases, may apply more broadly to items or services covered by any third-party payor, including commercial insurers and self-pay patients.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. Under the False Claims Act, liability may be assessed against companies that cause their customers to submit false or fraudulent claims to the federal government. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes "any request or demand" for money or property presented to the U.S. government. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute may constitute a false or fraudulent claim for purposes of the federal civil False Claims Act. Private parties may initiate "qui tam" whistleblower lawsuits against any person or entity under the federal civil False Claims Act in the name of the government and share in the proceeds of the lawsuit.
In addition, the civil monetary penalties statute, subject to certain exceptions, prohibits, among other things, the offer or transfer of remuneration, including waivers of copayments and deductible amounts (or any part thereof), to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary's selection of a particular provider, practitioner or supplier of services reimbursable by Medicare or a state healthcare program.
We are subject to additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any health care benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a health care offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Also, many states have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Additionally, there has been a recent trend of increased foreign, federal, and state regulation of payments and transfers of value provided to health care professionals or entities. The federal Physician Payments Sunshine Act imposes annual reporting requirements on certain drug, biologics, medical supplies and device manufacturers for which payment is available under Medicare, Medicaid or Children's Health Insurance Program for payments and other transfers of value provided by them, directly or indirectly, to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain non-physician practitioners (physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, anesthesiologist assistants and certified nurse midwives), and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Certain foreign countries and U.S. states also mandate implementation of commercial compliance programs, impose restrictions on device manufacturer marketing practices and require tracking and reporting of gifts, compensation and other remuneration to health care professionals and entities.
Penalties for violation of any of the health care laws described above or any other governmental regulations that apply to us include, without limitation, civil, criminal and/or administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government programs, such as Medicare and Medicaid, injunctions, refusal to allow us to enter into government contracts, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of an entity's operations.
25
Data Privacy & Security
Numerous state, federal and foreign laws, regulations and standards govern the collection, use, access to, confidentiality and security of health-related and other personal information and could apply now or in the future to our operations or the operations of our service providers. In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws and consumer protection laws and regulations govern the collection, use, disclosure, and protection of health-related and other personal information. In addition, certain foreign laws govern the privacy and security of personal data, including health-related data. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
Employees and Human Capital Resources
We believe in the value of a diverse and inclusive team as an enabler for obtaining the best ideas, innovating, and creating great outcomes for our customers and their patients. To drive strong business results, we must have the right people in the right role with great leadership, culture, training and rewards, all aligned with our values and business strategies.
To achieve these goals, we are committed to developing great talent at all levels of the organization: by growing talent internally, hiring great external talent, and ensuring bench strength and succession plans at all levels throughout the organization. We have a number of initiatives in place to attract, develop and retain great talent in a rewarding and inclusive culture. Some of the highlights of our approach are as follows:
Talent Acquisition
As of December 31, 2023, we had 516 full-time employees. During 2023, we increased the number of employees by 22% to support the rapid growth of our business, with 76% of the new hires being added to our sales team. As we recruit, we are innovatively expanding external talent pools, and we actively seek talent of diverse backgrounds and strive to provide a work environment where the best ideas are welcomed from anywhere.
Talent Management and Development
On an annual basis, our leadership team participates in a talent review and succession planning exercise to identify organizational needs, development opportunities, and potential future leaders. As a result of these efforts and our commitment to providing growth opportunities for our talent, 17% of our employees were promoted or took new positions during 2023. We experienced a low undesired turnover of under 7%, despite the market's strong competition for talent. As part of our talent management process, employees participate in annual performance management to provide self-assessments and create plans to support their career objectives. We provide training and other development opportunities to help employees meet their objectives and have launched and expanded several programs that are developing technical skills, leadership skills, and helping our teams and leaders leverage the strengths of their people. In 2023, we developed and launched our Treace Leadership Development Program, created a Sales Leadership Handbook, expanded our strengths-based personal and team development programs, and expanded our sales training programs.
Total Rewards
Our human capital strategies, initiatives, and outcomes are reviewed on a regular basis with the compensation committee of our board of directors. In addition, we partner with consulting firms to regularly benchmark our peer group companies and the broader market, and as a result of this analysis, we have implemented rewards practices that we believe allow us to maintain our competitiveness in the market. We also believe strongly in providing employees the opportunity to participate as owners in the Company; this is done through broad-based equity programs granting stock options and restricted stock units, with approximately 80% of our employees having received at least one equity grant.
Culture
We are committed to delivering an inclusive culture that fosters creativity and innovation, which allows employees to be their best at work. We offer a collegial, collaborative culture supported by competitive, performance-based compensation and benefits, equity awards, career development opportunities, and access to continual growth through live and remote training. We conducted an employee engagement survey in 2022 that showed that 87% of our employees are engaged, which compares favorably with average engagement of 76% of the approximately 80 medical device and biotechnology companies
26
in the benchmark compiled by the independent third-party consulting firm that conducted the engagement survey. We have used these survey results to determine how we can continue to create work environments that energize our employees and enable them to develop and maintain a positive working culture. As part of our work to foster our outstanding culture, in 2023 we launched a Treace Culture Team of employees from diverse functions around the business. This group played a key role in promoting employee ideas and led to the development and implementation of two new recognition programs, including our first annual Treace Culture and Ethics Award. We also participate in a number of team and community-building initiatives, including where functional teams volunteer with local nonprofits and company-wide charitable activities, such as food and toy drives and staffing local charity events.
Workplace Environment and Safety
Protecting the health and safety of our colleagues, agents, visitors, and the communities in which we operate is a business priority and is central to our values. We operate from a state-of-the-art headquarters facility with many features to improve our employees' work experience, including well-equipped training and lab rooms, an onsite cafe, private health and wellness rooms, quiet areas, and collaboration zones. Our Environmental, Health and Safety ("EHS") department works to ensure that our organization complies with applicable EHS regulations. Our EHS team has implemented multiple safety programs, regularly performs safety hazard evaluations within our facility, develops and tests action plans for emergencies such as fire response, severe weather threats and shelter in place incidents, and trains our employees on maintaining safety in the workplace.
None of our employees are represented by a labor union or are a party to a collective bargaining agreement.
Facilities
As of December 31, 2023, we leased approximately 125,000 square feet for our corporate headquarters located in Ponte Vedra, Florida under a lease agreement which terminates in July 2032. We believe that this facility is sufficient to meet our current and anticipated needs in the future and that additional space can be obtained on commercially reasonable terms as needed. We also continue to lease a part of our previous corporate headquarters location until August 2026 and have entered into subleases for this space.
Available Information
We file Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, proxy statements, amendments to such documents and other information with the SEC. Our SEC filings are available to the public over the Internet at the SEC's website at http://www.sec.gov. We also make these filings available, free of charge, under the Investor Relations section of our website at http://www.treace.com as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the SEC. Our website and the information contained on or connected to that site are not incorporated into this Annual Report or any other public filing made by us with the SEC.
27
Item 1A. Risk Factors
Our business involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information in this Annual Report, including our audited financial statements and the related notes, Part II, Item 7, "Management's Discussion and Analysis of Financial Condition and Results of Operations," and "Special Note Regarding Forward-Looking Statements." The risks described below are not the only ones facing us. The occurrence of any of the following risks or additional risks and uncertainties not presently known to us or that we currently believe to be immaterial could materially and adversely affect our business, financial condition, results of operations, future prospects and stock price.
Risk Factors Summary
Below is a summary of the principal factors that make an investment in our common stock speculative or risky. The below summary is qualified in its entirety by the more complete discussion of such risks and uncertainties that follows this summary.
28
Risks Related to Our Financial Condition and Capital Requirements
We have incurred losses in the past and may be unable to achieve or sustain profitability in the future.
We incurred net losses in each period since we commenced operations. For 2023 and 2022, we incurred net losses of $49.5 million and $42.8 million, respectively. As of December 31, 2023, we had an accumulated deficit of $134.2 million and $54.0 million of principal outstanding under our term and revolving loan agreements. We expect to continue to incur significant product development, clinical and regulatory, sales and marketing, medical education, and other expenses. In addition, we expect that our general and administrative expenses will continue to increase due to the additional costs associated with being a public company. These efforts and additional expenses may be more costly than we expect, and we cannot guarantee that we will be able to increase our revenue to offset such expenses. Our revenue may decline or our revenue growth may be constrained for a number of reasons, including reduced demand for our products and services, increased competition or if we cannot capitalize on growth opportunities. We will need to generate significant additional revenue to achieve and sustain profitability and, even if we achieve profitability, we cannot be sure that we will remain profitable for any substantial period of time. Our failure to achieve or sustain profitability could negatively impact the value of our common stock.
We have a limited operating history and have grown significantly in a short period of time. If we fail to manage our growth effectively, our business could be materially and adversely affected.
We formed as a medical device consulting business in July 2013, and began focusing on the foot and ankle market in January 2014. Accordingly, we have a limited operating history, which makes it difficult to evaluate our future prospects. Our operating results have fluctuated in the past, and we expect our future quarterly and annual operating results to continue to fluctuate as we focus on increasing the demand for our products and continue to develop clinical evidence to support the safety and efficacy of our Lapiplasty and Adductoplasty Systems, as well as develop new product innovations. We may need to make business decisions that could adversely affect our operating results, such as modifications to our pricing strategy, business structure or operations.
In addition, we have experienced recent rapid growth and anticipate further growth. For example, the number of our full-time employees increased from 32 as of December 31, 2017 to 516 as of December 31, 2023. This growth has placed significant demands on our management, financial, operational, technological and other resources, and we expect that our growth will continue to place significant demands on our management and other resources and will require us to continue developing and improving our operational, financial and other internal controls. In particular, continued growth increases the challenges involved in a number of areas, including recruiting and retaining sufficient skilled personnel for our direct employee sales force, providing adequate training and supervision to maintain our high-quality standards and preserving our culture and values. We may not be able to address these challenges in a cost-effective manner, or at all. To achieve our revenue goals, we must also successfully increase our supply of products from third party manufacturers to meet expected customer demand. In the future, we may experience difficulties with quality control, component supply and shortages of qualified personnel, among other problems. These problems could result in delays in product availability and increases in expenses. Any such delay or increased expense could adversely affect our ability to generate revenue. In addition, rapid and significant growth will place a strain on our administrative and operational infrastructure. In order to manage our operations and growth, we will need to continue to improve our operational and management controls, hiring process, reporting and information technology systems and financial internal control procedures. If we do not effectively manage our growth, we may not be able to execute on our business plan, respond to competitive pressures, take advantage of market opportunities, satisfy customer requirements or maintain high-quality product offerings, which could have a material adverse effect on our business, financial condition and results of operations.
29
The terms of our credit agreements require us to meet certain operating and financial covenants and place restrictions on our operating and financial flexibility. If we raise additional capital through debt financing, the terms of any new debt could further restrict our ability to operate our business.
Under the terms of our loan agreements discussed in more detail within Note 7, "Long Term Debt," of the Notes to the Financial Statements, we are subject to certain affirmative and negative covenants, including (but not limited to), financial covenants related to minimum revenue and minimum liquidity and covenants limiting our ability to incur certain additional indebtedness, create certain liens, enter into a change of control transaction and make certain distributions and investments without our lenders' consent. Our lenders may also declare us in default for certain types of events such as non-payment of debts, inaccurate representations and warranties, failure to comply with terms of material indebtedness and material agreements, bankruptcy and insolvency, a change of control and/or a material adverse change. Upon such events, our lenders could declare an event of default, which would give them the right to declare all borrowings outstanding, together with accrued and unpaid interest and fees, to be immediately due and payable. In addition, our lenders would have the right to proceed against the assets we provided as collateral under the loan agreements. For example, under our term and revolving loan agreements with entities affiliated with MidCap Financial Trust ("MidCap"), MidCap would have the right to enforce liens and security interests in substantially all of our assets (including intellectual property) in the event of certain specified defaults under the loans with MidCap. If the debt under any of our loan agreements is accelerated, we may not have sufficient cash or be able to sell sufficient assets to repay this debt or may have to curtail our growth plans, which would harm our business and financial condition.
Additional capital, if needed, may not be available on acceptable terms, if at all.
While management believes that existing cash, cash equivalents, marketable securities, and available debt borrowings will allow us to continue our planned operations for at least the next 12 months, we may require additional capital to maintain and expand our operations. We plan to continue to invest our capital in expanding our sales force, research and development efforts, direct to consumer education programs, product offerings, and acquisitions. If we raise additional funds through the issuance of equity, equity-linked or debt securities, those securities may have rights, preferences or privileges senior to those of our common stock, and our existing stockholders may experience dilution. Any debt financing secured by us in the future could require that a substantial portion of our operating cash flow be devoted to the payment of interest and principal on such indebtedness, which may decrease available funds for other business activities, and could involve restrictive covenants relating to our capital-raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities. We cannot be certain that we will be able to obtain additional financing on favorable terms, if at all. If we cannot raise funds on acceptable terms, if and when needed, we may not be able to grow our business or respond to competitive pressures or unanticipated requirements, which could seriously harm our business.
Risks Related to Our Business and Industry
We operate in a very competitive business environment, and if we are unable to compete successfully against our existing or potential competitors, our business, financial condition, and results of operations may be adversely affected.
Our existing products and procedures are, and any new products or procedures we develop and commercialize will be subject to intense competition. The industry in which we operate is competitive, subject to change and sensitive to the introduction of new products, procedures or other market activities of industry participants. Increasingly competitors are entering into the tri-planar bunion correction market with new instruments and implants to compete with the Lapiplasty System. Our ability to compete successfully will depend on our ability to continue to train surgeons on the Lapiplasty and Adductoplasty Procedures and gain their acceptance of the procedures, develop additional products and procedures to improve these procedures and expand our product offerings that reach the market in a timely manner, receive adequate coverage and reimbursement from third-party payors and provide products that are easier to use, safer, less invasive and more effective than the products and procedures of our competitors. In addition, our ability to increase our customer base and achieve broader market acceptance of our products will depend to a significant extent on our ability to expand our marketing efforts. We have dedicated and plan to continue to dedicate significant resources to our marketing programs. It will negatively affect our business, financial condition and results of operations if our marketing efforts and expenditures do not generate a corresponding increase in revenue. In addition, we believe that developing and maintaining broad awareness of our products in a cost-effective manner is critical to achieving broad acceptance of our products and expanding domestically and internationally. Promotional activities may not generate patient or physician awareness or increase revenue, and even if they do, any increase in revenue may not offset the costs and expenses we incur in building our brand. If we fail to successfully promote, maintain and protect our brand, we may fail to attract or retain the surgeon acceptance necessary to realize a sufficient return on our brand building efforts, or to achieve the level of brand awareness that is critical for broad adoption of our products.
30
Our competition includes medical device manufacturers in the orthopaedic foot and ankle market. Stryker Corporation is currently the leader in the orthopaedic foot and ankle market and has significant market share. Additional companies operating in the orthopaedic foot and ankle market with products specifically focused on bunion surgery include CrossRoads Extremity Systems, which was acquired in 2022 by DePuy Synthes Products, Inc., a Johnson & Johnson subsidiary, Zimmer Biomet Holdings, Inc., Paragon 28, Inc., In2Bones Global, Inc., a subsidiary of CONMED Corporation, Arthrex, Inc., Enovis Corporation (formerly Colfax), Novastep Inc. (now part of Enovis), Nextremity Solutions Inc. (acquired by Medartis Holdings AG); Henry Schein, Inc. (through its newly acquired interest in TriMed and strategic relationship with Extremity Medical LLC), Gramercy Extremity Orthopedics, LLC., and Fusion Orthopedics, LLC. While foot and ankle product sales represent a relatively small percentage of our larger competitors' overall sales, many recognize the growth opportunities in this market and have been active in product additions through both internal development efforts and acquisitions. We also face potential competition from many different sources, including academic institutions, governmental agencies and public and private research institutions.
At any time, these competitors and other potential market entrants may develop new products, procedures or treatment alternatives that could render our products obsolete or uncompetitive. In addition, one or more of such competitors may gain a market advantage by developing and patenting competitive products, procedures or treatment alternatives earlier than we can, obtaining regulatory clearances or approvals more rapidly than we can or selling competitive products at prices lower than ours. If medical research were to lead to the discovery of alternative therapies or technologies that improve or cure bunions as an alternative to surgery, such as by natural correction of the unstable joint in the middle of the foot, the use of pharmaceuticals or breakthrough bio-technological innovations or therapies, our profitability could suffer through a reduction in sales or a loss in market share to a competitor. The discovery of methods of prevention or the development of other alternatives to the Lapiplasty Procedure could result in decreased demand for our products and, accordingly, could have a material adverse effect on our business, financial condition and results of operations. Many of our current and potential competitors have substantially greater sales and financial resources than we do. These competitors may also have more established distribution networks, a broader offering of products, entrenched relationships with hospitals, surgeons and distributors, or greater experience in launching, marketing, distributing and selling products or treatment alternatives.
We also compete with our competitors to hire sales representatives and engage the services of independent sales agencies. In addition, we compete with our competitors in acquiring technologies and technology licenses complementary to our products or procedures or advantageous to our business. If we are unable to compete successfully against our existing or potential competitors, our business, financial condition and results of operations may be adversely affected, and we may not be able to grow at our expected rate, if at all.
If we fail to continue to develop and retain an effective direct sales force as well as sales management and sales specialist teams, it could negatively impact our sales, and we may not generate sufficient revenue to achieve profitability.
Our revenue and profitability is directly dependent upon the sales and marketing efforts of our sales representatives, sales management and sales specialist teams. To expand our business, we have built and are continuing to build a substantial direct employee sales force supported by sales management and sales specialist teams. We have made and are continuing to make a significant investment in recruiting and training sales representatives and clinical representatives as we expand our business. There is significant competition for sales personnel experienced in relevant medical device sales. Once hired, the training process is lengthy because it requires significant education for new sales representatives and clinical specialists to achieve the level of clinical competency with our products expected by surgeons. Upon completion of the training, our sales representatives typically require lead time in the field to grow their network of accounts and achieve the productivity levels we expect them to reach in any individual territory. Furthermore, the use of our products often requires or benefits from direct support from us, including through our experienced sales representatives that provide assistance in the operating room. Our future success depends largely on our ability to continue to hire, train, retain and motivate skilled members of our sales management and sales specialist teams with significant technical knowledge in various areas. If we are unable to continue to attract, motivate, develop and retain a sufficient number of qualified sales personnel, and if our sales representatives do not achieve the productivity levels we expect them to reach, our revenue will not grow at the rate we expect, and our financial performance will suffer. Also, to the extent we hire personnel from our competitors, we may have to wait until applicable non-competition provisions have expired before deploying such personnel in restricted territories or incur costs to relocate personnel outside of such territories, and we have been in the past, and may be subject to future allegations that these new hires have been improperly solicited, and that they have divulged to us proprietary or other confidential information of their former employers. Additionally, because the market for experienced sales personnel is competitive, our competitors may try to hire our sales personnel away from us. If successful, we would be required to dedicate resources to recruiting, filling and training those vacant positions. Any of these risks may adversely affect our business.
31
Our business plan relies on certain assumptions about the market for our products; however, the size and expected growth of our addressable market has not been established with precision and may be smaller than we estimate, and even if the addressable market is as large as we have estimated, we may not be able to capture additional market share.
Our estimates of the addressable market for our current products and future products are based on a number of internal and third-party estimates and assumptions, including the prevalence of bunion sufferers and the difficulty of persuading bunion suffers to undergo bunion surgery and specifically the Lapiplasty Procedure. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and our estimates may not be correct. For example, we believe that the aging of the general population and increasingly active lifestyles will continue and that these trends will increase the need for our products and that surgeons and their patients will determine that the Lapiplasty Procedure is appropriate for all severities of bunions, from mild to severe. However, the projected demand for our products could materially differ from actual demand if our assumptions regarding these trends and acceptance of our products by the medical community prove to be incorrect or do not materialize, or if non-surgical treatments or other surgical techniques gain more widespread acceptance as a viable alternative to the Lapiplasty Procedure. In addition, even if the number of bunion sufferers who elect to undergo bunion surgery, and the Lapiplasty Procedure in particular, increases as we expect, technological or medical advances could provide alternatives to address bunion deformities and reduce demand for bunion surgery. As a result, our estimates of the addressable market for our current or future products and procedures may prove to be incorrect. Further, one component of our growth strategy is our direct to patient education program, which we expect will help us educate additional bunion patients about our products and procedures; however, these patient engagements may not be as successful at educating potential surgical candidates as we expect. Thus, even if the total addressable market for our current and future products and procedures is as large as we have estimated, we may not be able to penetrate the existing market to capture additional market share for the reasons discussed in this "Risk Factors" section. If the actual number of bunion sufferers who would benefit from our products, the price at which we can sell future products or the addressable market for our products is smaller than we estimate, or if the total addressable market is as large as we have estimated but we are unable to capture additional market share, it could have a material adverse effect on our business, financial condition and results of operations.
If we cannot innovate at the pace of our competitors, we may not be able to develop or exploit new products or procedures in time to remain competitive.
For us to remain competitive, it is essential to develop and bring to market new products and procedures at an increasing speed. If we are unable to meet customer demands for new products and procedures, or if the products and procedures we introduce are viewed less favorably than our competitors' products or procedures, our results of operations and future prospects may be negatively affected. To meet our customers' needs in these areas, we must continuously design new products, update existing products and invest in and develop and enhance our procedures. Our operating results depend to a significant extent on our ability to anticipate and adapt to technological changes in the bunion and midfoot surgery markets, keep pace with developments and innovations by our competitors and maintain a strong product pipeline. Any inability to do so could have a material adverse effect on our business, financial condition and results of operations.
The seasonality of our business creates variance in our quarterly revenue, which makes it difficult to compare or forecast our financial results.
Our revenue fluctuates on a seasonal basis, which affects the comparability of our results from quarter to quarter. In particular, we have experienced and expect to continue to experience seasonality in our business, with higher sales volumes in the fourth calendar quarter followed by lower sales volumes in the subsequent first calendar quarter. Our sales volumes in fourth calendar quarters tend to be higher as many patients elect to have surgery after meeting their annual deductible and having time to recover over the winter holidays. Our sales volumes in subsequent first calendar quarters tend to be lower as a result of adverse weather and by resetting annual patient healthcare insurance plan deductibles, both of which may cause patients to delay elective procedures. The orthopaedic industry traditionally experiences lower sales volumes in the third quarter than throughout the rest of the year as elective procedures generally decline during the summer months, and in 2023, we were affected by a decline in demand for bunion surgeries during the summer months. These seasonal variations are difficult to predict accurately, may vary amongst different markets and at times may be entirely unpredictable, which introduce additional risk into our business as we rely upon forecasts of customer demand to build inventory in advance of anticipated sales. In addition, disruptions from the COVID-19 pandemic over the past few years and our limited operating history have, in part, made our seasonal patterns more difficult to discern, making it more difficult to predict future seasonal patterns.
32
Our revenue is primarily generated from sales of the Lapiplasty System, and we are, therefore, highly dependent on it for our success.
Sales of the Lapiplasty System accounted for substantially all of our revenues in prior years and are expected to continue to account for most of our revenue going forward. Our ability to execute our growth strategy and become profitable will therefore depend upon the continuing adoption by surgeons, patients, payors, hospitals, and other healthcare facilities, among others, of the Lapiplasty Procedure to correct bunions. The pace of adoption of the Lapiplasty Procedure may slow, and we may not be able to continue to penetrate the bunion surgery market for the reasons discussed in this "Risk Factors" section. We cannot ensure that the Lapiplasty Procedure will achieve broad market acceptance among surgeon, patients, payors, hospitals and healthcare facilities. Since our business has a limited product line based primarily on the Lapiplasty Procedure, any slowdown or setback in market adoption of the Lapiplasty Procedure will negatively impact our business, financial condition and results of operations.
Industry trends have resulted in increased downward pricing pressure on medical services and products, which may affect our ability to sell our products at prices necessary to support our current business strategy.
The trend toward health care cost containment through aggregating purchasing decisions and industry consolidation, along with the growth of managed care organizations, is placing increased emphasis on the delivery of more cost-effective medical therapies. For example:
More broadly, provisions of the ACA could meaningfully change the way health care is developed and delivered in the United States and may adversely affect our business and results of operations. For further discussion of these challenges, refer to "Risks Related to Regulatory Matters—Changes in health care policy and regulation may have a material adverse effect on us." We cannot predict accurately what health care programs and regulations will ultimately be implemented at the federal or state level, or the effect of any future legislation or regulation in the United States or elsewhere. However, any changes that have the effect of reducing reimbursement for procedures using our products or reducing medical procedure volumes could have a material and adverse effect on our business, financial condition and results of operations. Any decline in the amount that payors reimburse our customers for our products could make it difficult for customers to either adopt or continue to use our products, and could create additional pricing pressure for us. If we are forced to lower the price we charge for our products, or if we add more components to our systems, our gross margins will decrease, which will adversely affect our ability to invest in and grow our business. If we are unable to maintain our prices, or if our costs increase and we are unable to offset such increase with an increase in our prices, our margins could erode.
33
In addition, the largest medical device companies with multiple product franchises have increased their effort to leverage and contract broadly with customers across franchises by providing volume discounts and multi-year arrangements that could prevent our access to these customers or make it difficult, or impossible, to compete on price.
Product liability lawsuits, warranty claims and quality system problems could harm our business.
The manufacture and sale of medical devices exposes us to risk of product liability and warranty claims. While the Company offers a limited warranty and has experienced negligible returns of any products alleged to be defective, we bear the risk of warranty claims on the products we supply. We may not be successful in claiming recovery under any warranty or indemnity provided to us by our suppliers or vendors. In addition, warranty claims brought by our customers related to third-party components may arise after our ability to bring corresponding warranty claims against such suppliers expires, which could result in costs to us. Furthermore, if any of our products become the subject of a product liability claim, legal defenses are costly, regardless of the outcome. Thus, we may experience increased legal expenses as we defend any such matter, and we could incur liabilities associated with adverse outcomes that exceed our insurance coverage.
Additionally, we could experience a material design or manufacturing failure in our products, a quality system failure, other safety issues or heightened regulatory scrutiny that would warrant a recall of some of our products. Product liability lawsuits and claims, safety alerts and product recalls, regardless of their ultimate outcome, could have a material adverse effect on our business and reputation and on our ability to attract and retain customers.
Although we have product liability insurance that we believe is appropriate, this insurance is subject to deductibles and coverage limitations. Our current product liability insurance may not continue to be available to us on acceptable terms, if at all, and, if available, coverage may not be adequate to protect us against any future product liability claims. If we are unable to obtain insurance at an acceptable cost or on acceptable terms or otherwise protect against potential product liability claims, we could be exposed to significant liabilities. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could have a material adverse effect on our business, financial condition and results of operations. Further, such product liability matters may negatively impact our ability to obtain insurance coverage or cost-effective insurance coverage in future periods.
Our employees and independent contractors, including independent sales representatives and any other consultants, any future service providers and other vendors, may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have an adverse effect on our results of operations.
We are exposed to the risk that our employees and independent contractors, including independent sales agencies and any other consultants, any future commercial collaborators, and other vendors may engage in misconduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or other unauthorized activities that violate federal, state or local laws and regulations, as well as the laws, regulations and rules of regulatory bodies such as the FDA; manufacturing standards; U.S. federal and state health care fraud and abuse, data privacy laws and other similar non-U.S. laws; or laws that require the true, complete and accurate reporting of financial information or data. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. In addition, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and financial results, including, without limitation, the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid and other U.S. health care programs, other sanctions, imprisonment, contractual damages, reputational harm, diminished profits and future earnings and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
We rely in part on independent sales agencies to sell our products to our customers, and if we are unable to maintain our network of independent sales agencies, we may not achieve our anticipated revenue growth.
We utilize a hybrid sales team with a mix of employee sales personnel and independent sales agencies to sell our products to surgeons, hospitals, clinics and other end users and to assist us in promoting market acceptance of, and creating demand for, our products. If we are unable to come to commercially reasonable terms with a sales agent or agencies, we may not generate the expected level of sales and may need to spend more of our capital resources to hire sales personnel as employees. In addition, there is a risk that a sales agent that we contract with will give higher priority to the products of other medical
34
device companies, including products directly competitive with our products or may be required by larger medical devices companies to stop offering our products. There can be no assurance that a sales agent will devote the resources necessary to provide effective sales and promotional support to our products. In addition, if an independent sales agency terminates its relationship with us and is retained by one of our competitors, notwithstanding the noncompetition covenants in their contract with us, we may be unable to prevent them from helping competitors solicit business from our existing customers, which could adversely affect our sales. Furthermore, on January 5, 2023, the Federal Trade Commission proposed a rule, which if it becomes final in its current form would require the Company to rescind our non-competes, which would allow our employees and independent sales agents to terminate their engagements and compete with us, which could have a material adverse effect on the Company. While we have significantly expanded our employee sales force, we expect that in certain markets we will continue to rely on an independent sales force.
Infectious disease outbreaks, such as the COVID-19 pandemic, have adversely affected, and in the future may adversely affect, our business, financial condition and results of operations.
Our operations were adversely impacted by the COVID-19 pandemic, particularly during the government-mandated restrictions on elective procedures that occurred from March 2020 through May 2020, and continuing in 2020 and 2021, when patients continued to delay bunion surgery procedures and hospitals experienced staffing and capacity constraints that affected elective procedures. In addition to reductions in our revenue growth, we also experienced some delays in our supply chain and in study enrollment timelines and regulatory processes. If new potentially contagious and virulent variants of COVID-19 or other infectious diseases should emerge that result in delays in elective procedures or another pandemic in the United States should occur, our business, revenue growth, financial condition and results of operations could be materially adversely affected.
Risks Related to Administrative, Organizational and Commercial Operations and Growth
If hospitals, ambulatory surgery centers and other health care facilities do not approve the use of our products, our sales may not increase.
In order for surgeons to use our products at hospitals, ambulatory surgery centers and other health care facilities, we are often required to obtain approval from those hospitals, ambulatory surgery centers and health care facilities. Typically, hospitals, ambulatory surgery centers and health care facilities review the comparative effectiveness and cost of products used in the facility. The makeup and evaluation processes for health care facilities vary considerably, and it can be a lengthy, costly and time-consuming effort to obtain approval by the relevant health care facilities. Additionally, hospitals, ambulatory surgery centers, other health care facilities and GPOs, which manage purchasing for multiple facilities, may also require us to enter into a purchase agreement and satisfy numerous elements of their administrative procurement process, which can also be a lengthy, costly and time-consuming effort. If we do not obtain access to hospitals, ambulatory surgery centers and other health care facilities in a timely manner, or at all, via their approvals or purchase contract processes, or otherwise, or if we are unable to obtain approvals or secure contracts in a timely manner, or at all, our operating costs will increase, our sales may decrease, and our operating results may be adversely affected. Furthermore, we may expend significant efforts on these costly and time-consuming processes but may not be able to obtain necessary approvals or secure a purchase contract from such hospitals, ambulatory surgery centers, health care facilities or GPOs.
If adequate levels of reimbursement from third-party payors for procedures using our products are not obtained or maintained, surgeons and patients may be reluctant to use our products and our business will suffer.
In the United States, health care providers who purchase our products generally rely on third-party payors, principally federally-funded Medicare, state-funded Medicaid and private health insurance plans, to pay for all or a portion of the cost of bunion correction procedures and products utilized in those procedures. We may be unable to sell our products on a profitable basis if third-party payors deny coverage or reduce their current levels of reimbursement for procedures using products of the type we intend to offer. Our sales depend largely on governmental health care programs and private health insurers reimbursing patients' medical expenses. Surgeons, hospitals and other health care providers may not purchase our products if they do not receive appropriate reimbursement from third-party payors for procedures using our products. Payors continue to review their coverage policies for existing and new therapies and may deny coverage for treatments that include the use of our products.
In addition, some health care providers in the United States have adopted or are considering bundled payment methodologies and/or managed care systems in which the providers contract to provide comprehensive health care for a fixed cost per person. Health care providers may attempt to control costs by authorizing fewer elective surgical procedures, including bunion and midfoot correction surgeries, or by requiring the use of the least expensive procedure available. In addition,
35
third-party payors increasingly are requiring evidence that medical devices are cost-effective, and if we are unable to meet this requirement, the third-party payor may not reimburse the use of our products, which could reduce sales of our products to health care providers who depend upon reimbursement for payment. Changes in reimbursement policies or health care cost containment initiatives that limit or restrict reimbursement for procedures using our products may have an adverse effect on our business.
If we experience problems with, or are required to change, our suppliers or manufacturers, we may be unable to meet customer orders for our products in a timely manner or within our budget.
For us to be successful, our suppliers must be able to provide us with products and components in substantial quantities, in compliance with regulatory requirements, in accordance with agreed upon specifications, at acceptable costs and on a timely basis. An interruption in our commercial operations could occur if we encounter delays or difficulties in securing these components, and if we cannot then obtain an acceptable substitute. We rely on a limited number of suppliers for the components used in our products. Our suppliers may encounter manufacturing problems for a variety of reasons, including failure to follow specific protocols and procedures, failure to comply with applicable legal and regulatory requirements, equipment malfunctions and environmental factors, failure to properly conduct their own business affairs, and infringement of third-party intellectual property rights, any of which could delay or impede their ability to meet our supply requirements. Our suppliers may also not prioritize the production of our products compared to the suppliers' larger customers so we may experience longer delays in receiving our requested orders.
If we are required to transition to new third-party suppliers for certain components of our products, the use of components or materials furnished by these alternative suppliers could require us to alter our operations. Any such interruption or alteration could harm our reputation, business, financial condition and results of operations.
Furthermore, if we are required to change the manufacturer of a critical component of our Lapiplasty System or other products, we will be required to verify that the new manufacturer maintains facilities, procedures and operations that comply with our quality standards and applicable regulatory requirements, which could further impede our ability to manufacture our products in a timely manner. Transitioning to a new supplier could be time-consuming and expensive, may result in interruptions in our operations and product delivery, could affect the performance specifications of our products or could require that we modify the design of those products. A change in manufacturer could trigger the requirement to submit and obtain a new 510(k) clearance from the FDA, or similar international regulatory authorization before we implement the change, which could cause substantial delays. The occurrence of any of these events could harm our ability to meet the demand for our products in a timely manner or cost-effectively.
We cannot assure you that any need to change suppliers or manufacturers will not cause interruptions in our ability to meet customer demand for our products. For example, if we should encounter delays or difficulties in securing, reconfiguring or revalidating the equipment and components we require for our Lapiplasty Systems and other products, our reputation, business, financial condition and results of operations could be negatively impacted.
We may be unable to continue to successfully demonstrate to surgeons or key opinion leaders the merits of our products and technologies compared to those of our competitors, which may make it difficult to establish our products and technologies as a standard of care and achieve market acceptance.
Surgeons play the primary role in determining the course of treatment and, ultimately, the type of products that will be used to treat a patient. As a result, our success depends, in large part, on our ability to effectively market and demonstrate to foot and ankle surgeons the merits of our products and methodologies compared to those of our competitors. Acceptance of our products and methodologies depends on educating surgeons as to the distinctive characteristics, clinical benefits, safety and cost-effectiveness of the Lapiplasty Procedure, including the Mini-Incision and Micro-Lapiplasty Systems, the SpeedPlate Rapid Compression Implants, the Adductoplasty Procedure, and the Hammertoe PEEK Fixation System, and our other products and technologies as compared to those of our competitors, and on training surgeons in the proper use of our products. If we are not successful in convincing surgeons of the merits of our products and methodologies or educating them on the use of our products, they may not use our products or may not use them effectively, and we may be unable to increase our sales, sustain our growth or achieve and achieve profitability.
36
Also, since the Lapiplasty Procedure, including the minimally invasive variations, and the Adductoplasty Procedure, are relatively new procedures, some surgeons may be reluctant to change their surgical treatment practices for the following reasons, among others:
These reasons may affect the pace of adoption of the Lapiplasty and Adductoplasty Procedures and future products and techniques that we may offer.
In addition, we believe recommendations and support of our products and technologies by influential surgeons and key opinion leaders in our industry are essential for market acceptance and establishment of our products and procedures as a standard of care. If we do not continue to receive support from such surgeons and key opinion leaders, if long-term data does not continue to show the benefits of using our products and procedures, or if the benefits offered by our products and procedures are no longer deemed sufficient to justify their cost, surgeons, hospitals and other health care facilities may not use or continue to use our products, and we might be unable to continue to establish our products and procedures as a standard of care and continue to achieve market acceptance.
Our inability to maintain contractual relationships with health care professionals could have a negative impact on our research and development and medical education programs.
We maintain contractual relationships with respected physicians and medical personnel in hospitals, private practice and universities who assist in clinical studies, product research and development and in the training of surgeons on the safe and effective use of our products (refer to "Business — Product Development and our Surgeon Advisory Board"). We continue to place emphasis on the validation of the benefits of the Lapiplasty Procedure, including the minimally invasive variations, and Adductoplasty System, through clinical studies, the development of proprietary products and product improvements to develop our product lines as well as providing high quality training on those products. If we are unable to maintain these relationships, our ability to develop and market new and improved products and train on the use of those products could decrease, and future operating results could be unfavorably affected. At the same time, the medical device industry's relationship with physicians is under increasing scrutiny by the U.S. Department of Health and Human Services Office of Inspector General ("OIG"), the U.S. Department of Justice ("DOJ"), the state attorneys general and other foreign and domestic government agencies. Our failure to comply with requirements governing the industry's relationships with physicians or an investigation into our compliance by the OIG, the DOJ, state attorneys general and other government agencies, could negatively affect our business, financial condition, and results of operations. Refer to "Risk Factors—Risks Related to Regulatory Matters— Our relationships with customers, physicians and third-party payors are subject to federal and state health care fraud and abuse laws, false claims laws, physician payment transparency laws and other health care laws and regulations. If we or our employees, independent contractors, consultants, service providers, or vendors violate these laws, we could face substantial penalties."
If surgeons fail to use our products safely and appropriately use our products, or if we are unable to train podiatrists and orthopaedic surgeons on the safe and appropriate use of our products, we may be unable to achieve our expected sales, growth or profitability.
An important part of our sales process includes our ability to screen for and identify podiatrists and orthopaedic surgeons who have the requisite training and experience to safely and appropriately use our products and to train a sufficient number of these surgeons and to provide them with adequate instruction in use of our products. There is a training process involved for surgeons to become proficient in the safe and appropriate use of our products. This training process may take longer or be more expensive than expected and may therefore affect our ability to increase sales. Convincing surgeons to dedicate the time and energy necessary for adequate training is challenging, and we may not continue to be successful in these efforts. Evolving federal guidance regarding medical education programs under the federal Anti-Kickback Statute also could limit
37
our ability to train podiatrists and orthopaedic surgeons, and such programs could be subject to challenge under the federal Anti-Kickback Statute. Refer to "Risk Factors—Risks Related to Regulatory Matters—Our relationships with customers, physicians and third-party payors are subject to federal and state health care fraud and abuse laws, false claims laws, physician payment transparency laws and other health care laws and regulations. If we or our employees, independent contractors, consultants, service providers, or vendors violate these laws, we could face substantial penalties." Furthermore, if clinicians are not properly trained, they may misuse or ineffectively use our products. Any improper use of our products may result in unsatisfactory outcomes, patient injury, negative publicity or lawsuits against us, any of which could harm our reputation and affect future product sales. Accordingly, if surgeons fail to safely and appropriately use our products or if we are unable to train surgeons on the safe and appropriate use of our products, we may be unable to achieve our expected sales, growth or profitability.
Our brand and reputation may be diminished due to real or perceived issues with the Lapiplasty Procedure or our products, which could have an adverse effect on our business, financial condition, results of operations and prospects.
Our direct-to-patient outreach program, focused on educating patients on the clinical advantages of the Lapiplasty Procedure and generating brand awareness, is a key aspect of our commercial strategy and is important to achieving widespread acceptance of the Lapiplasty Procedure, particularly since we are seeking to change the standard of care away from traditional bunion surgical techniques. We provide direct-to-patient education through social media as well as digital and traditional marketing channels. These brand promotion activities may not yield increased sales and, even if they do, any sales increases may not offset the expenses we incur to promote our brand. Our future success depends upon increased surgeon and patient demand for our products, resulting in part from patient requests for the Lapiplasty Procedure, positive patient word-of-mouth, and patient feedback on social media that their experience with the Lapiplasty Procedure met their expectations. The Lapiplasty Procedure is surgery, and as with any surgery, patients will have a recovery period and may experience complications, and individual outcomes will vary. Patients may be dissatisfied if their expectations about the surgery, recovery process and results, among other things, are not met. Dissatisfied patients may express negative opinions to the press or through social media. Any failure to meet patient expectations and any resulting negative publicity could harm our reputation and future sales. If we fail to successfully promote and maintain our brand, our Lapiplasty solution and other products, or if we incur substantial expenses in an unsuccessful attempt to promote and maintain our brand, our Lapiplasty solution and other products may not continue to be accepted by physicians or patients, which would adversely affect our business, results of operations and financial condition.
The loss of any member on our executive management team or our inability to attract and retain highly skilled members of our sales management and marketing teams and engineers could have a material adverse effect on our business, financial condition and results of operations.
Our success depends on the skills, experience and performance of the members of our executive management team and John T. Treace, our founder and chief executive officer, in particular. The individual and collective efforts of these executives will be important as we continue to commercialize our existing products, develop new products and technologies, and expand our commercial activities. The loss or incapacity of existing members of our executive management team could have a material adverse effect on our business, financial condition and results of operations if we experience difficulties in hiring qualified successors. We do not maintain "key person' insurance for any of our executives or key employees.
Our commercial, quality and research and development programs and operations depend on our ability to attract and retain highly skilled team members. We may be unable to attract or retain qualified team members. All of our employees are at-will, which means that either we or the employee may terminate his or her employment at any time. The loss of key employees, failure of any key employee to perform, our inability to attract and retain skilled employees, as needed, or our inability to effectively plan for and implement a succession plan for key employees could have a material adverse effect on our business, financial condition and results of operations.
38
Performance issues, service interruptions or price increases by shipping carriers could adversely affect our business and harm our reputation and ability to provide our products on a timely basis.
Expedited, reliable shipping of our kits is important to our operations. We rely on providers of transport services for reliable and secure point-to-point transport of our products to our customers and sales representatives and for tracking of these shipments. Should a carrier encounter delivery performance issues such as loss, damage or destruction of our products, it would be costly to replace our products in a timely manner and could cause surgeries using our products to be delayed or canceled, and such occurrences may damage our reputation and lead to decreased demand for our products and increased cost and expense to our business. In addition, any significant increase in shipping rates could adversely affect our operating margins and results of operations. Similarly, strikes, severe weather, natural disasters or other service interruptions affecting delivery services we use would adversely affect our ability to process orders for our products on a timely basis.
Any future international expansion will subject us to additional costs and risks that may have a material adverse effect on our business, financial condition and results of operations.
Historically, all of our sales have been to customers in the United States. To the extent we enter into international markets in the future, there are significant costs and risks inherent in conducting business in international markets. If we expand, or attempt to expand, into foreign markets, we will be subject to new business risks, in addition to regulatory risks. In addition, expansion into foreign markets imposes additional burdens on our executive and administrative personnel, finance and legal teams, research and marketing teams and general managerial resources.
We have limited experience with regulatory environments and market practices internationally, and we may not be able to penetrate or successfully operate in new markets. We may also encounter difficulty expanding into international markets because of limited brand recognition in certain parts of the world, leading to delayed acceptance of our products by surgeons and their patients, hospitals, ambulatory surgery centers and payors in these international markets. If we are unable to expand internationally and manage the complexity of international operations successfully, it could have a material adverse effect on our business, financial condition and results of operations. If our efforts to introduce our products into foreign markets are not successful, we may have expended significant resources without realizing the expected benefit. Ultimately, the investment required for expansion into foreign markets could exceed the results of operations generated from this expansion.
Risks Related to Our Intellectual Property
If our patents and other intellectual property rights do not adequately protect our products, our competitors could develop and commercialize products similar or identical to ours, and we may be unable to gain significant market share and be unable to operate our business profitably.
Our success depends in large part on our ability to obtain, maintain and solidify a proprietary position for our products, which will depend on our success in obtaining and maintaining effective intellectual property protection, including through patents, trade secrets, copyrights, know-how, trademarks, license agreements and contractual provisions to establish our intellectual property rights and protect our products. These legal means, however, afford only limited protection and may not completely protect our rights. Any failure to continue to obtain or maintain patent and other intellectual property protection with respect to our products could harm our business, financial condition and results of operations.
As of December 31, 2023, our patent portfolio included 52 owned U.S. patents, one licensed U.S. patent, 81 pending U.S. patent applications, 15 granted foreign patents, 15 pending international PCT patent applications, and 45 pending foreign patent applications. We cannot assure you that our intellectual property position will not be challenged or that all patents for which we have applied will be granted. The validity and breadth of claims in patents involve complex legal and factual questions and, therefore, may be highly uncertain. Uncertainties and risks that we face include the following:
39
The patent prosecution process is expensive and time-consuming, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patent applications at a reasonable cost or in a timely manner, or in all jurisdictions. Although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, any of these parties may breach such agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek and obtain patent protection. We may choose not to seek patent protection for certain innovations and may choose not to pursue patent protection in certain jurisdictions, and under the laws of certain jurisdictions, patents or other intellectual property rights may be unavailable or limited in scope. It is also possible that we will fail to identify patentable aspects of our developments before it is too late to obtain patent protection. Furthermore, our ability to continue to obtain and maintain valid and enforceable patents depends in part on whether the differences between our inventions and the prior art allow our inventions to be patented over the prior art. Furthermore, the publication of discoveries in scientific literature often lags behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until eighteen (18) months after filing, or in some cases not at all. Therefore, we cannot be certain that we were the first to file for patent protection of such inventions.
In addition, the laws of foreign jurisdictions may not protect our rights to the same extent as the laws of the United States. For example, most countries outside of the United States do not allow patents for methods of treating the human body. This may preclude us from obtaining method patents outside of the United States having similar scope to those we have obtained or may obtain in the future in the United States. This includes certain key method patents covering the Lapiplasty Procedure. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
Moreover, we may be subject to a third-party pre-issuance submission of prior art to the U.S. Patent and Trademark Office ("USPTO") or patent offices in foreign jurisdictions, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allowing third parties to commercialize our technology and compete directly with us, without payment to us.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical products and techniques, or limit the duration of the patent protection of our technology.
We also rely on trade secrets and other unpatented proprietary technology. There can be no assurances that we can meaningfully protect our rights in our unpatented proprietary technology or that others will not independently develop substantially equivalent proprietary products or processes or otherwise gain access to our proprietary technology. We seek to protect our trade secrets and proprietary know-how, in part, with confidentiality agreements with employees and consultants that include customary intellectual property assignment obligations. There can be no assurances, however, that the agreements will not be breached, adequate remedies for any breach would be available or competitors will not discover our trade secrets or independently develop comparable intellectual property.
Continuing to obtain and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance fees, renewal fees, annuity fees and various other government fees on issued patents often must be paid to the USPTO and foreign patent agencies over the lifetime of the patent and/or applications and any patent rights we may obtain in the future. While an unintentional lapse of a patent or patent application can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our products, we may not be able to stop a competitor from marketing products that are the same as or similar to our products, which would have a material adverse effect on our business.
40
If we were to lose intellectual property lawsuits, our intellectual property rights would be impaired and, if we were found to impair the intellectual property rights of others, a court could require us to pay significant damages and/or prevent us from selling our products.
The medical device industry is litigious with respect to patents and other intellectual property rights. Companies in the medical device industry have used intellectual property litigation to gain a competitive advantage. We have brought in the past and may in the future bring lawsuits alleging that certain third parties are infringing our intellectual property rights and may in the future become a defendant in lawsuits or administrative proceedings alleging that we have infringed on another party's patents or other intellectual property or challenging our intellectual property rights. A legal proceeding, regardless of the outcome, could drain our financial resources and divert the time and effort of our management. Protracted litigation to defend or prosecute our intellectual property rights could result in our customers or potential customers deferring or limiting their purchase or use of the affected products until resolution of the litigation.
While we are aware of several third-party patents of interest, we do not believe that any of our products infringe any valid claims of patents or other proprietary rights held by others. However, there can be no assurances that we do not infringe any patents or other proprietary rights held by third parties. Litigation may also be necessary to defend infringement claims of third parties or to enforce patent rights we hold or to protect trade secrets or techniques we own. Moreover, because patent applications can take many years to issue and because publication schedules for pending applications vary by jurisdiction, there may be applications now pending of which we are unaware, and which may result in issued patents which our current or future products infringe. Also, because the claims of published patent applications can change between publication and patent grant, there may be published patent applications that may ultimately issue with claims that we infringe.
If our products were found to infringe any proprietary right of another party, we could be required to pay significant damages and/or cease production, marketing and distribution of those products to obtain a license from such third party to continue selling, developing and marketing our products and techniques. We may not be able to obtain any required license on commercially reasonable terms or at all. The acquisition or licensing of third-party intellectual property rights is a competitive area, and our competitors may pursue strategies to acquire or license third party intellectual property rights that we may consider attractive or necessary. If we are unable to successfully obtain rights to required third party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant products or redesign those products that contain the allegedly infringing intellectual property, which could harm our business, financial condition and results of operations. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys' fees if we are found to have willfully infringed a patent. A finding of infringement could force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business. Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the market price of our common shares to decline.
Because competition in our industry is intense, competitors may infringe or otherwise violate our issued patents, patents of our licensors or other intellectual property. To counter infringement or unauthorized use, we have filed and may in the future file additional infringement claims, which can be expensive and time consuming, and could distract our technical and management personnel from their normal responsibilities. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims or file administrative actions against us alleging that we infringe their patents. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent's claims narrowly or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding or administrative action could put one or more of our patents at risk of being invalidated or interpreted narrowly. Our competitors may assert invalidity on various grounds, including lack of novelty, obviousness or that we were not the first applicant to file a patent application related to our product. We may elect to enter into license agreements to settle patent infringement claims or to resolve disputes before litigation, and any such license agreements may require us to pay royalties and other fees that could be significant. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure.
Our competitors, many of which have made substantial investments in patent portfolios, trade secrets, trademarks and competing technologies, may have applied for or obtained, or may in the future apply for or obtain, patents or trademarks that may prevent, limit or otherwise interfere with our ability to make, use, sell and/or export our products or to use our technologies or product names. Moreover, individuals and groups that are non-practicing entities, commonly referred to as
41
"patent trolls," purchase patents and other intellectual property assets for the purpose of making claims of infringement in order to extract settlements. From time to time, we may receive threatening letters, notices or "invitations to license," or may be the subject of claims that our products and business operations infringe, violate or misappropriate the intellectual property rights of others. The defense of these matters can be time consuming, costly to defend in litigation, divert management's attention and resources, damage our reputation and brand and cause us to incur significant expenses or make substantial payments.
If we fail to execute invention assignment agreements with our employees and contractors involved in the development of intellectual property or are unable to protect the confidentiality of our trade secrets, the value of our products and our business and competitive position could be harmed.
In addition to patent protection, we also rely on protection of copyright, trade secrets, know-how and confidential and proprietary information. We generally enter into confidentiality and invention assignment agreements with our employees, consultants and third parties upon their commencement of a relationship with us. However, we may not enter into such agreements with all employees, consultants and third parties who have been involved in the development of our intellectual property. In addition, these agreements may not provide meaningful protection against the unauthorized use or disclosure of our trade secrets or other confidential information, and adequate remedies may not exist if unauthorized use or disclosure were to occur. The exposure of our trade secrets and other proprietary information would impair our competitive advantages and could have a material adverse effect on our business, financial condition and results of operations. In particular, a failure to protect our proprietary rights may allow competitors to copy our products and procedures, which could adversely affect our pricing and market share. Further, other parties may independently develop substantially equivalent know-how and technology.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using commonly accepted physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our products that we consider proprietary. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome is unpredictable. Even though we use commonly accepted security measures, trade secret violations are often a matter of state law, and the criteria for protection of trade secrets can vary among different jurisdictions. In addition, trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. While we have agreements with our employees, consultants and third parties that obligate them to assign their inventions to us, these agreements may not be self-executing, not all employees or consultants may enter into such agreements, or employees or consultants may breach or violate the terms of these agreements, and we may not have adequate remedies for any such breach or violation. If our intellectual property or confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, it could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest, and our competitive position may be harmed.
We rely on our trademarks, trade names and brand names to distinguish our products from the products of our competitors, and have registered or applied to register many of these trademarks. There can be no assurance that our trademark applications will be approved. Third parties may also oppose our trademark applications or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. Further, there can be no assurance that competitors will not infringe our trademarks or that we will have adequate resources to enforce our trademarks. We also license third parties to use our trademarks. In an effort to preserve our trademark rights, we enter into license agreements with these third parties, which govern the use of our trademarks and require our licensees to abide by quality control standards with respect to the goods and services that they provide under our trademarks. Although we make efforts to monitor the use of our trademarks by our licensees, there can be no assurance that these efforts will be sufficient to ensure that our licensees abide by the terms of their licenses. In the event that our licensees fail to do so, our trademark rights could be diluted. Any of the foregoing could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
42
Patent terms may not be sufficient to effectively protect our products and business for an adequate period of time.
Patents have a limited lifespan. In the United States, the natural expiration of a utility patent is generally 20 years after its first effective non-provisional filing date. Although various extensions may be available, the term of a patent, and the protection it affords, is limited. Even if patents covering our proprietary technologies and their uses are obtained, once the patent has expired, we may be open to competition. In addition, although upon issuance in the United States a patent's term can be extended based on certain delays caused by the USPTO, this extension can be reduced or eliminated based on certain delays caused by the patent applicant during patent prosecution. If we do not have sufficient patent terms to protect our products, proprietary technologies and their uses, our business would be seriously harmed.
Changes in U.S. patent laws may limit our ability to obtain, defend and/or enforce our patents.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith America Invents Act ("Leahy-Smith Act") included a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and also affect patent litigation. The USPTO has developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, which became effective in 2013. The first to file provisions limit the rights of an inventor to patent an invention if not the first to file an application for patenting that invention, even if such invention was the first invention. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the enforcement and defense of our issued patents.
The Leahy-Smith Act also includes a number of significant changes that affect the way U.S. patent applications are prosecuted and also may affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by the USPTO administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. Therefore, the Leahy-Smith Act and its implementation has increased the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. In addition, future actions by the U.S. Congress, the federal courts and the USPTO could cause the laws and regulations governing patents to change in unpredictable ways. Any of the foregoing could harm our business, financial condition and results of operations.
We may be unable to enforce our intellectual property rights throughout the world.
We have limited intellectual property rights outside the United States. Filing, prosecuting, enforcing, and defending patents or trademarks on our products and any future products in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. This could make it difficult for us to stop infringement of our foreign patents, if obtained, or the misappropriation of our other intellectual property rights. For example, some foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against third parties, including government agencies or government contractors. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and any future products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing in these jurisdictions.
Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of our intellectual property.
43
We may be subject to claims that we or our employees have misappropriated the intellectual property of a third party, including trade secrets or know-how, or are in breach of non-competition or non-solicitation agreements with our competitors and third parties may claim an ownership interest in intellectual property we regard as our own.
Many of our employees and consultants were previously employed at or engaged by other medical device companies, including our competitors or potential competitors. Some of these employees, consultants and contractors may have executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees and consultants do not use the intellectual property, proprietary information, know-how or trade secrets of others in their work for us, we may be subject to claims that we or these individuals have, inadvertently or otherwise, misappropriated the intellectual property or disclosed the alleged trade secrets or other proprietary information, of these former employers, competitors or other third parties. Additionally, we may be subject to claims from third parties challenging our ownership interest in or inventorship of intellectual property we regard as our own, for example, based on claims that our agreements with employees or consultants obligating them to assign intellectual property to us are ineffective or in conflict with prior or competing contractual obligations to assign inventions to another employer, to a former employer, or to another person or entity. Litigation may be necessary to defend against claims, and it may be necessary or we may desire to enter into a license to settle any such claim; however, there can be no assurance that we would be able to obtain a license on commercially reasonable terms, if at all. If our defense to those claims fails, in addition to paying monetary damages or a settlement payment, a court could prohibit us from using technologies, features or other intellectual property that are essential to our products, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. In addition, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. Any litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our products, which could materially and adversely affect our business, financial condition, operating results, cash flows and prospects.
Risks Related to Regulatory Matters
We are subject to substantial government regulation that could have a material adverse effect on our business.
Our products are regulated as medical devices. The development, testing, manufacture, distribution, sale, and marketing of our products are subject to extensive regulation and review by numerous governmental authorities. U.S. regulations govern the design, development, testing, clinical trials, premarket clearance and approval, safety, marketing, sale, and registration of new medical devices, in addition to regulating manufacturing practices, reporting, labeling, relationships with health care professionals and recordkeeping procedures. State laws also regulate the sale and distribution of our products and may require registration or licensure. Our failure to comply with applicable regulatory requirements could result in these governmental authorities:
Any adverse regulatory action, depending on its magnitude, may restrict us from effectively marketing and selling our products and limit our ability to obtain future pre-market clearances or approvals, and could result in a substantial modification to our business practices and operations.
44
Our relationships with customers, physicians, other health care providers and third-party payors are subject to federal and state health care fraud and abuse laws, false claims laws, physician payment transparency laws and other health care laws and regulations. If we or our sales representatives or other employees, independent contractors, consultants, or vendors violate these laws, we could face substantial penalties.
Our relationships with customers, physicians, other health care providers and third-party payors are subject to federal and state health care fraud and abuse laws, false claims laws, physician payment transparency laws and other health care laws and regulations. In particular, the promotion, sale and marketing of health care items and services is subject to extensive laws and regulations designed to prevent fraud, kickbacks, overutilization, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales practices, customer incentive and other business arrangements. The U.S. health care laws and regulations that may affect our ability to operate include, but are not limited to:
45
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities, patient outreach programs or our arrangements with physicians, other health care providers, independent sales agencies and customers could be subject to challenge and expose us to liability under one or more of such laws. It is not always possible to identify and deter employee misconduct or business noncompliance, and the precautions we take to detect and prevent inappropriate conduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Efforts to ensure that our business arrangements will comply with applicable health care laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other health care laws and regulations.
If we or our sales representatives, other employees, agents, independent contractors, consultants, and vendors violate these laws, we may be subject to investigations, enforcement actions and/or significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, disgorgement, monetary fines, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and/or oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
We may not receive, or may be delayed in receiving, the necessary clearances or approvals for our future products or modifications to our current products, and failure to timely obtain necessary clearances or approvals for our future products or modifications to our current products would adversely affect our ability to grow our business.
In the United States, before we can market a new medical device, or a new use of, new claim for or significant modification to an existing product, we must first receive either clearance under Section 510(k) of the FDCA or approval of a PMA or de novo request, from the FDA, unless an exemption applies. In the 510(k) clearance process, before a device may be marketed, the FDA must determine that a proposed device is "substantially equivalent" to a legally-marketed "predicate" device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed before May 28, 1976 (pre-amendments device), a device that was originally on the U.S. market pursuant to an approved PMA and later down-classified, or a 510(k)-exempt device. To be "substantially equivalent," the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data is sometimes required to support substantial equivalence. In the process of obtaining PMA approval, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. To date, our Class II devices have received marketing authorization pursuant to the 510(k) clearance process.
Modifications to products that are approved through a PMA application generally require FDA approval. Similarly, certain modifications made to products cleared through a 510(k) may require a new 510(k) clearance. Both the PMA and the 510(k) clearance process can be expensive, lengthy and uncertain. The FDA's 510(k) clearance process usually takes from three to 12 months, but can last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is submitted to the FDA, and generally requires the performance of one or more clinical trials. Despite the time, effort and cost, a device may not be approved or cleared by the FDA. Any delay or failure to obtain necessary regulatory clearances or approvals could harm our business. Furthermore, even if we are granted regulatory clearances or approvals, they may include significant limitations on the indicated uses for the device, which may limit the market for the device.
46
We have made modifications to 510(k)-cleared products in the past and have determined based on our review of the applicable FDA regulations and guidance that in certain instances new 510(k) clearances or PMA approvals were not required. We may make modifications or add additional features in the future that we believe do not require a new 510(k) clearance or approval of a PMA. If the FDA disagrees with our determination and requires us to submit new 510(k) notifications or PMA applications for modifications to our previously cleared products for which we have concluded that new clearances or approvals are unnecessary, we may be required to cease marketing or to recall the modified product until we obtain clearance or approval, and we may be subject to significant regulatory fines or penalties. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to existing products than we had expected, product introductions or modifications could be delayed or canceled, which could adversely affect our ability to grow our business. The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
Failure to comply with post-marketing regulatory requirements could subject us to enforcement actions, including substantial penalties, and might require us to recall or withdraw a product from the market.
Even though we have obtained clearance for our Lapiplasty and Adductoplasty Systems and other products in the United States, we are subject to ongoing and pervasive regulatory requirements governing, among other things, the manufacture, marketing, advertising, medical device reporting, sale, promotion, import, export, registration and listing of devices. For example, we must submit periodic reports to the FDA as a condition of 510(k) clearance. These reports include information about failures and certain adverse events associated with the device after its clearance. Failure to submit such reports, or failure to submit the reports in a timely manner, could result in enforcement action by the FDA. Following its review of the periodic reports, the FDA might ask for additional information or initiate further investigation. In addition, any marketing authorizations we are granted are limited to the cleared indications for use. Further, the manufacturing facilities for a product are subject to periodic review and inspection. Subsequent discovery of problems with a product, manufacturer, or manufacturing facility may result in restrictions on the product, manufacturer or manufacturing facility, withdrawal of the product from the market or other enforcement actions.
The regulations to which we are subject are complex and have become more stringent over time. Regulatory changes could result in restrictions on our ability to continue or expand our operations, higher than anticipated costs, or lower than anticipated sales. Plus regulators such as the FDA and other state and foreign regulatory authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by federal, state or foreign regulatory authorities, which could result in any of the following sanctions:
47
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and have a material adverse effect on our reputation, business, financial condition, and results of operations.
Legislative or regulatory reforms may have a material adverse effect on us.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulation of medical devices. In addition to new government regulations, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations or take other actions, which may prevent or delay approval or clearance of our future products under development or impact our ability to modify our currently cleared products on a timely basis. Furthermore, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new statutes, regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of any future products or make it more difficult to obtain clearance or approval for, manufacture, market or distribute our products. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require additional testing before obtaining clearance or approval; changes to manufacturing methods; recall, replacement or discontinuance of our products; or additional record keeping. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may be subject to enforcement action, and we may not achieve or sustain profitability.
In addition, in response to perceived increases in health care costs, in recent years there have been and continue to be proposals by the federal government, state governments, regulators and third-party payors to control these costs and, more generally, to reform the U.S. health care system. Certain of these proposals could limit the prices we will be able to charge for our products or the amount of reimbursement available for our products and could limit the acceptance and availability of our products. See Part I, Item 1. "Business — Government Regulation." The expansion in the government's role in the U.S. healthcare industry may result in decreased profits to us, lower reimbursement by payors for procedures using our products, and/or reduced medical procedure volumes, all of which may have a material adverse effect on our business, financial condition, and results of operations.
Our products must be manufactured in accordance with federal and state quality regulations and are subject to FDA inspection, and our failure to comply with these regulations could result in fines, product recalls, product liability claims, limits on future product clearances, reputational damage and other adverse impacts.
The methods used in, and the facilities used for, the manufacture of our products must comply with the FDA's QSR which is a complex regulatory scheme that covers the procedures and documentation of the design, testing, production, process controls, quality assurance, labeling, packaging, handling, storage, distribution, installation, servicing and shipping of medical devices. Furthermore, we are required to verify that our suppliers maintain facilities, procedures and operations that comply with our quality standards and applicable regulatory requirements. The FDA enforces the QSR through periodic announced or unannounced inspections of medical device manufacturing facilities, which may include the facilities of subcontractors. Our products are also subject to similar state regulations.
We or our third-party manufacturers may not take the necessary steps to comply with applicable regulations, including the QSR, which could cause delays in the delivery of our products or result in the production of products that cannot be sold or distributed. In addition, failure to comply with applicable FDA requirements or later discovery of previously unknown problems with our products or manufacturing processes could result in, among other things: (i) warning letters or untitled letters; (ii) fines, injunctions or civil penalties; (iii) suspension or withdrawal of approvals or clearances; (iv) customer notifications or repair, replacement, refunds, detention, seizures or recalls of our products; (v) total or partial suspension of production or distribution; (vi) administrative or judicially imposed sanctions; (vii) the FDA's refusal to grant pending or future clearances or approvals for our products; (viii) clinical holds; (ix) refusal to permit the import or export of our products; and (x) criminal prosecution of us or our employees. Any of these actions could significantly and negatively impact supply of our products. If any of these events occur, our reputation could be harmed, we could be exposed to tort or product liability claims, and we could lose customers and suffer reduced revenue and increased costs.
48
The misuse or off-label use of our products may harm our reputation in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.
Our currently marketed products are either Class II medical devices cleared by the FDA for specific indications or they are Class I exempt for general orthopaedic use. For example, our Lapiplasty plating system has been cleared by the FDA for use in stabilization of fresh fractures, revision procedures, joint fusion and reconstruction of small bones of the feet. We train our marketing personnel and sales representatives to not promote our devices for uses outside of the FDA-authorized indications for use, known as "off-label uses." We cannot, however, prevent a physician from using our devices off-label, when in the physician's independent professional medical judgment, he or she deems it appropriate. There may be increased risk of injury to patients if physicians attempt to use our devices off-label. Furthermore, the use of our devices for indications other than those cleared by the FDA or approved by any foreign regulatory body (to the extent our products are cleared for use outside the United States in the future) may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients.
If the FDA determines that our promotional materials, sales and marketing activities or health care provider training constitute promotion of an off-label use (e.g., misbranding), it could request that we modify our training, sales, marketing or promotional materials and/or subject us to regulatory or enforcement actions, including the issuance or imposition of an untitled letter, a warning letter, an injunction, a debarment, seizure of the products, recalls, a civil fine or criminal penalties. We could face similar consequences from actions by foreign regulatory bodies if we should offer our products outside the United States. It is also possible that other federal, state or foreign enforcement authorities might take action under other regulatory authority, such as false claims laws, if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government health care programs and the curtailment of our operation.
In addition, physicians may misuse our products or use improper techniques if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our devices are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. As described above, product liability claims could divert management's attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance.
Our products may cause or contribute to adverse medical events or be subject to failures or malfunctions that we are required to report to the FDA, and if we fail to do so, we could be subject to sanctions that could harm our reputation, business, financial condition and results of operations. The discovery of serious safety or efficacy issues with our products, or a recall or a market withdrawal of our products either voluntarily or at the direction of the FDA or another governmental authority, could have a negative impact on us.
We are subject to the FDA's medical device reporting regulations, which require us to report to the FDA when we receive or become aware of information that reasonably suggests that one or more of our products may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. We are also required to maintain records under the medical device reporting regulations evidencing our decisions regarding whether incidents are medical device reportable events. If we fail to comply with our reporting obligations, the FDA could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearance or approval, seizure of our products or delay in clearance or approval of future products.
The FDA and foreign regulatory bodies have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA's authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury, adverse health consequences, or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government-mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects or other deficiencies or failures to comply with applicable regulations. Product defects or other errors may occur in the future.
49
Depending on the corrective action we take to address a product's deficiencies or defects, the FDA may require, or we may decide, that we will need to obtain new clearances or approvals for the device before we may market or distribute the corrected device. Seeking such clearances or approvals may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement actions, including FDA warning letters, cease distribution and notification orders, product seizure, injunctions, administrative penalties or civil or criminal fines.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA. We may initiate voluntary withdrawals or corrections for our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, it could require us to report those actions as recalls, and we may be subject to enforcement action. A future recall announcement could harm our reputation with customers, potentially lead to product liability claims against us and negatively affect our sales. In addition, we have had in the past, and may in the future, have reports of adverse events associated with the Lapiplasty Procedure. While adverse events are inherent in the medical device and surgical industry, frequent adverse events can lead to reputational harm and negatively affect our sales. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
The clinical study process is lengthy and expensive with uncertain outcomes. We have limited long-term data regarding the safety and efficacy of our products. Results of earlier studies may not be predictive of future clinical trial results, or the safety or efficacy profile for such products.
Clinical studies can be difficult to design and implement, can take many years, can be expensive and carry uncertain outcomes. We have conducted multiple post-market clinical outcome studies that we believe are unique in the bunion correction field and are a key element of our medical education program. Among other studies, we currently have three prospective, multicenter, post-market studies underway: (1) the ALIGN3D clinical study designed to evaluate outcomes of the Lapiplasty Procedure, which has completed enrollment with 173 patients; (2) the Mini3D™ study designed to evaluate outcomes of the Lapiplasty Procedure using Lapiplasty® Mini-Incision System, which has completed enrollment with 105 patients; and (3) the MTA3D™ Clinical Study designed to evaluate outcomes of the combined Adductoplasty and Lapiplasty Procedures for patients in need of metatarsus adductus and hallux valgus corrective surgery, which is still enrolling patients. Each of these studies has a primary effectiveness endpoint of twenty-four months. It takes time to enroll patients and to evaluate outcomes 24 months after surgery. Furthermore, after these studies are completed at the 24-month endpoint, data about outcomes for periods longer than 24 months after the Lapiplasty surgery will not yet be available, limiting comparability with traditional surgical approaches for bunions that have longer term data about their outcomes.
While we have reported positive interim results in our ALIGN3D post-market clinical studies, we cannot guarantee our post-market clinical studies, or any other clinical study we may conduct or sponsor in the future, will be successful or will otherwise generate data that supports the performance of our products. Our post-market clinical studies are subject to oversight by the institutional review board at the medical institutions where the clinical studies are conducted. We rely also on, and in the future may continue to rely on, contract research organizations ("CROs") and clinical trial sites to ensure the proper and timely conduct of our clinical studies. While we have agreements governing their committed activities, we have limited influence over their actual performance. To the extent our collaborators or the CROs fail to enroll participants in our clinical studies, fail to conduct the studies in accordance with good clinical practice standards and their protocols, or are delayed for a significant time in the execution of our studies, including achieving full enrollment, we may be affected by increased costs and/or program delays, and the utility, reliability, and acceptance of the studies by the FDA or other regulatory authorities could be impaired.
The initiation and completion of any clinical studies may be prevented, delayed or halted for numerous reasons, which could adversely affect the costs, timing or successful completion of our clinical studies. Patient enrollment in clinical studies and completion of patient follow-up depend on many factors, including the size of the patient population, the nature of the study protocol, the proximity of patients to clinical sites, the eligibility criteria for the clinical study, patient compliance, and clinicians' and patients' perceptions as to the potential advantages of the product being studied in relation to other available therapies, including any new treatments that may be approved for the indications we are investigating. If the clinical studies do not continue to show positive outcomes from our products, adoption of our products may slow, and our business, financial condition and prospects may be significantly harmed.
50
Actual or perceived failures to comply with applicable data protection, privacy and security laws, regulations, standards and other requirements could adversely affect our business, results of operations and financial condition.
We and our service providers may be subject to federal, state and foreign data protection laws and regulations (i.e., laws and regulations that address data privacy and security). In the United States, numerous federal and state laws and regulations, including HIPAA, state data breach notification laws, state health information privacy laws and federal and state consumer protection laws and regulations (e.g., Section 5 of the Federal Trade Commission Act, which is discussed below), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our services providers. We may also be subject to U.S. federal rules, regulations and guidance concerning data security for medical devices, including guidance from the FDA. In addition, we may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under HIPAA. Depending on the facts and circumstances, we could be subject to significant civil and criminal penalties if we obtain, use or disclose individually identifiable health information maintained by a HIPAA-covered entity or business associate in a manner that is not authorized or permitted by HIPAA. The regulatory framework for data privacy and security worldwide is continuously evolving and developing and, as a result, interpretation and implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future and may be subject to change.
Certain states have also adopted comparable privacy and security laws and regulations, some of which may be more stringent than HIPAA. Such laws and regulations will be subject to interpretation by various courts and other governmental authorities, thus creating potentially complex compliance issues for us and our future customers and strategic partners. In addition, California enacted the California Consumer Privacy Act ("CCPA") on June 28, 2018, which went into effect on January 1, 2020. The CCPA creates individual privacy rights for California consumers and increases the privacy and security obligations of entities handling certain personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. Further, the California Privacy Rights Act ("CPRA") recently passed in California. The CPRA significantly amends the CCPA and imposes additional data protection obligations on covered businesses, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk data, and opt outs for certain uses of sensitive data. It also creates a new California data protection agency authorized to issue substantive regulations and could result in increased privacy and information security enforcement. The majority of the provisions went into effect on January 1, 2023, and additional compliance investment and potential business process changes may be required. Similar laws have been adopted in other states, including Virginia, Colorado, Connecticut and Utah, and have been proposed in other states and at the federal level, reflecting a trend toward more stringent privacy legislation in the United States. The enactment of such laws could have potentially conflicting requirements that would make compliance challenging. In the event that we are subject to or affected by HIPAA, the CCPA, the CPRA or other domestic privacy and data protection laws, any liability from failure to comply with the requirements of these laws could adversely affect our financial condition.
Furthermore, the Federal Trade Commission ("FTC") and many state Attorneys General continue to enforce federal and state consumer protection laws against companies for online collection, use, dissemination and security practices that appear to be unfair or deceptive. For example, according to the FTC, failing to take appropriate steps to keep consumers' personal information secure can constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act. The FTC expects a company's data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities.
As our operations and business grow, we may become subject to or affected by rapidly evolving data protection laws, rules and regulations in foreign jurisdictions. For example, there are also extensive data protection laws and regulations in effect in the European Economic Area and the United Kingdom that are actively enforced in those jurisdictions. To the extent we are obligated to comply with such data protection laws and regulations and we fail to do so, we may be the subject of litigation, government investigations and enforcement actions, claims by third parties and/or adverse publicity, which could adversely affect our business, results of operations and financial condition.
In addition to government regulation, privacy advocates and industry groups have and may in the future propose self-regulatory standards from time to time. These and other industry standards may legally or contractually apply to us, or we may elect to comply with such standards. We expect that there will continue to be new proposed laws and regulations concerning data privacy and security, and we cannot yet determine the impact such future laws, regulations and standards may have on our business. New laws, amendments to or re-interpretations of existing laws, regulations, standards and other obligations may require us to incur additional costs and restrict our business operations. Because the interpretation and application of laws, regulations, standards and other obligations relating to data privacy and security are still uncertain, it is possible that these laws, regulations, standards and other obligations may be interpreted and applied in a manner that is inconsistent with our data processing and storage practices and policies or the features of our products. If so, in addition to
51
the possibility of fines, lawsuits, regulatory investigations, public censure, other claims and penalties, and significant costs for remediation and damage to our reputation, we could be materially and adversely affected if legislation or regulations are expanded to require changes in our data processing and storage practices and policies or if governing jurisdictions interpret or implement their legislation or regulations in ways that negatively impact our business, financial condition and results of operations. We may be unable to make such changes and modifications in a commercially reasonable manner, or at all. Any inability to adequately address data privacy or security-related concerns, even if unfounded, or to comply with applicable laws, regulations, standards and other obligations relating to data privacy and security, could result in additional cost and liability to us, harm our reputation and brand, damage our relationships with consumers and harm our business, financial condition and results of operations.
We make public statements about our use and disclosure of personal information through our privacy policies, information provided on our website and press statements. Although we endeavor to comply with our public statements and documentation, we may at times fail to do so or be alleged to have failed to do so. The publication of our privacy policies and other statements that provide promises and assurances about data privacy and security can subject us to potential government or legal action if they are found to be deceptive, unfair or misrepresentative of our actual practices. Any concerns about our data privacy and security practices, even if unfounded, could damage the reputation of our business and harm our business, financial condition and results of operations.
Although we work to comply with applicable laws, regulations and standards, our contractual obligations and other legal obligations, these requirements are evolving and may be modified, interpreted and applied in an inconsistent manner from one jurisdiction to another, and may conflict with one another or other legal obligations with which we must comply. Any failure or perceived failure by us or our employees, representatives, contractors, consultants, collaborators or other third parties to comply with such requirements or adequately address privacy and security concerns, even if unfounded, could result in additional cost and liability to us, damage our reputation and adversely affect our business and results of operations. In addition, if our practices are not consistent, or viewed as not consistent, with legal and regulatory requirements, including changes in laws, regulations and standards or new interpretations or applications of existing laws, regulations and standards, we may also become subject to audits, inquiries, whistleblower complaints, adverse media coverage, investigations, criminal or civil sanctions, all of which may harm our business, financial condition and results of operations.
Our information technology systems may be subject to breaches, cyber-attacks or other disruptions that could, among other adverse consequences, cause us to violate laws, regulations and contractual obligations, could impair our ability to operate our business and could adversely affect our business, results of operations, financial condition, cash flows, reputation or competitive position.
We utilize our own and third party information technology systems to operate our business, and some of these systems are used to process, transmit and store sensitive data, including patient information, consumer information, and other information subject to data protection, privacy and security laws, regulations, standards and other requirements. The breach of these systems, ransomware attacks, cyber-attacks, malicious intrusions or significant disruptions, the wrongful use or disclosure of such sensitive data, and security failures in these systems, including a failure to maintain the security of these systems, as well as through diverse attack vectors, such as social engineering/phishing, company insiders, suppliers or providers, and as a result of human or technological error, including misconfigurations, bugs, or other vulnerabilities in software and hardware, may result in a violation of laws, regulations, standards and other requirements and could expose us to substantial liability, reputational harm, civil and criminal penalties and fines, litigation and enforcement actions and materially and adversely affect our business. Information technology systems and the confidential and sensitive information, including intellectual property, business financial information, trade secrets, and other confidential information, are vulnerable to a range of cybersecurity risks and threats, including malicious code embedded in open-source software, or misconfigurations, "bugs" or other vulnerabilities in commercial software that is integrated into our (or our suppliers’ or service providers’) IT systems, products or services. IT system incidents may disrupt our ability to access important systems and records and could prevent us from running our business for a period of time. We cannot guarantee that we will be immune from an incident or be able to respond rapidly enough to prevent a negative impact on our business. Furthermore, cyber-attacks are expected to accelerate on a global basis in frequency and magnitude as threat actors are becoming increasingly sophisticated in using techniques and tools – including generative and other artificial intelligence – that circumvent security controls, evade detection and remove forensic evidence. As a result, we may be unable to detect, investigate, remediate or recover from future attacks or incidents, or to avoid a material adverse impact to our IT systems, confidential information or business. There can be no assurance that our cybersecurity risk management program and processes, including our policies, controls or procedures, will be fully implemented, complied with or effective in protecting our systems and information. Although we maintain insurance policies, we cannot be certain that any or all of the costs and liabilities incurred in relation any cybersecurity attack or incident will be covered or that applicable insurance will be available to us in the future on economically reasonable terms or at all.
52
For more information, see the risk factor entitled "We, along with our suppliers, are dependent on various information technology systems, and failures of, interruptions to, or unauthorized tampering of those systems could have a material adverse effect on our business."
Risks Related to Ownership of Our Common Stock
The market price of our common stock may be volatile and fluctuate substantially.
The market price of our common stock may be highly volatile and could fluctuate or decline substantially as a result of a variety of factors, some of which are beyond our control or are related in complex ways, including:
In recent years, the stock market in general, and the market for medical device companies in particular, has experienced significant price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Further, the stock market in general has been highly volatile due to inflation, interest rate increases, supply chain disruptions, general economic conditions, and the COVID-19 pandemic in the United States. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance. These fluctuations may be even more pronounced in the trading market for our stock in the early years after our initial public offering. Following periods of such volatility in the market price of a company's securities, securities class action litigation has often been brought against that company. Because of the potential volatility of our stock price, we may become the target of securities litigation in the future. Securities litigation could result in substantial costs and divert management's attention and resources from our business.
If our operating and financial performance in any given period does not meet any guidance that we provide to the public, the market price of our common stock may decline.
We may, but are not obligated to, provide public guidance on our expected operating and financial results for future periods. Any such guidance will be comprised of forward-looking statements subject to the risks and uncertainties described in this Annual Report and in our other public filings and public statements. Our actual results may not always be in line with or
53
exceed any guidance we have provided, especially in times of economic uncertainty. If, in the future, our operating or financial results for a particular period do not meet any guidance we provide or the expectations of investment analysts, or if we reduce our guidance for future periods, the market price of our common stock may decline. Even if we do issue public guidance, there can be no assurance that we will continue to do so in the future.
If we do raise additional capital, stockholders may be subject to dilution.
If we issue additional shares of our common stock or other equity securities convertible into common stock to fund operations, develop new products, accelerate other strategies, make acquisitions or support other activities, the ownership interests of our stockholders will be diluted. Because our decision to issue debt or equity securities in any future offering will depend on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing or nature of any future offerings. To the extent that we raise additional capital through the sale of equity securities, stockholders' ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect stockholders' rights. The incurrence of indebtedness would result in increased fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Additionally, any future collaborations we enter into with third parties may provide capital in the near term but limit our potential cash flow and revenue in the future. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
A significant portion of our total outstanding shares may be sold into the market in the near future, which could cause the market price of our common stock to decline significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. As of December 31, 2023, we had outstanding 61,749,654 shares of common stock. Sale of these shares, or the perception in the market that the holders of a large number of shares of our common stock intend to sell shares, could reduce the market price of our common stock.
Furthermore, we have registered all shares of our common stock that we may issue under our equity compensation plans. Shares issued upon the exercise of stock options and upon vesting of restricted stock units that are outstanding under our equity incentive plans, or pursuant to future awards granted under those plans, have and will continue to become available for sale in the public market to the extent permitted by the provisions of applicable vesting schedules, and Rule 144 and Rule 701 under the Securities Act. Refer to Note 11, "Stockholders' Equity," of the Notes to the Financial Statements for information about outstanding options, restricted stock units and performance stock units. If any of these additional shares are sold, or if it is perceived that they will be sold, in the public market, the market price of our common stock could decline.
We may seek to grow our business through acquisitions or investments in new or complementary businesses, products or technologies, through the licensing of products or technologies from third parties or other strategic alliances, and the failure to manage acquisitions, investments, licenses or other strategic alliances, or the failure to integrate them with our existing business, could have a material adverse effect on our operating results, dilute our stockholders' ownership, increase our debt or cause us to incur significant expense.
Our success depends on our ability to continually enhance and broaden our product offerings in response to changing clinician and patients' needs, competitive technologies and market pressures. Accordingly, from time to time, as we did in 2023 with the acquisition of certain assets of RPM-3D, we may consider opportunities to acquire, make investments in or license other technologies, products and businesses that may enhance our capabilities, complement our existing products and technologies, or expand the breadth of our markets or customer base. Potential and completed acquisitions, strategic investments, licenses and other alliances involve numerous risks, including:
54
We do not know if we will be able to identify acquisitions or strategic relationships we deem suitable, whether we will be able to successfully complete any such transactions on favorable terms, if at all, or whether we will be able to successfully integrate any acquired business, product or technology into our business or retain any key personnel, suppliers, sales agent, or customer relationships. Our ability to successfully grow through strategic transactions depends upon our ability to identify, negotiate, complete, and integrate suitable target businesses, technologies or products and to obtain any necessary financing. These efforts could be expensive and time-consuming and may disrupt our ongoing business and prevent management from focusing on our operations.
If we pursue any foreign acquisitions, they typically involve unique risks in addition to those mentioned above, including those related to integration of operations across different cultures, languages and legal and regulatory environments, currency risks and the particular economic, political and regulatory risks associated with specific countries.
To finance any acquisitions, investments or strategic alliances, we may choose to issue shares of our common stock as consideration, which could dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may be unable to consummate any acquisitions, investments or strategic alliances using our common stock as consideration. Additional funds may not be available on terms that are favorable to us, or at all.
If securities analysts do not publish research or reports about our business or if they publish negative evaluations of our stock, the price of our stock could decline.
The trading market for our common stock relies in part on the research and reports that industry or financial analysts publish about us or our business. We have a limited number of analysts who cover us. If one or more of the analysts covering our business downgrade their evaluations of our stock, the price of our stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our stock, which in turn could cause our stock price to decline.
We have in the past and may in the future be subject to short selling strategies that may drive down the market price of our common stock and negatively affect our reputation.
Short sellers have in the past and may attempt in the future to drive down the market price of our common stock and damage our reputation. Short selling is the practice of selling securities that the seller does not own but may have borrowed with the intention of buying identical securities back at a later date. The short seller hopes to profit from a decline in the value of the securities between the time the securities are borrowed and the time they are replaced. As it is in the short seller's best interests for the price of the stock to decline, many short sellers publish, or arrange for the publication of, negative opinions regarding the relevant issuer and its business prospects to create negative market momentum. These short seller publications are not regulated by any governmental, self-regulatory organization or other official authority in the U.S., are not subject to certification requirements imposed by the SEC, and may express opinions based on distortions, omissions or fabrications. Issuers, like us, whose securities have historically had limited trading history or volumes and/or have been susceptible to relatively high volatility levels can be vulnerable to such short seller attacks. Short selling reports can cause increased volatility in an issuer's stock price, and result in regulatory and governmental inquiries, stockholder lawsuits, and negative market reactions. Furthermore, when subject to unfavorable allegations, even if untrue, we may have to expend a significant amount of resources to investigate such allegations, respond to questions from customers and other stakeholders, and defend ourselves, including against inquiries or investigations from governmental authorities, lawsuits on behalf of stockholders, and negative selling tactics from competitors, that may be prompted by such allegations.
55
Our business could be negatively impacted by corporate citizenship and ESG matters and/or our reporting of such matters.
Institutional, individual, and other investors, proxy advisory services, regulatory authorities, consumers and other stakeholders are increasingly focused on environmental, social and governance ("ESG") practices of companies. If we are perceived as lagging with respect to ESG initiatives, certain investors may engage with us to improve ESG disclosures or performance, make voting decisions against our directors, reduce or eliminate our stock from their holdings, or exclude our stock from consideration. In addition, the criteria by which our corporate responsibility practices are assessed may change, which could result in greater expectations of us and cause us to undertake costly initiatives to identify, measure and report ESG metrics and satisfy such new criteria. Any failure or perceived failure by us to meet investor or other stakeholder expectations and evolving standards or regulatory requirements may negatively impact our financial results, our reputation, our ability to attract or retain employees, our attractiveness as a service provider, investment, or business partner, or expose us to government enforcement actions, private litigation, and actions by stockholders or other stakeholders.
We have not paid dividends in the past and do not expect to pay dividends in the future, and, as a result, any return on investment may be limited to appreciation in the price of our stock.
We have never paid cash dividends and do not anticipate paying cash dividends on our common stock in the foreseeable future. The payment of dividends will depend on our earnings, capital requirements, financial condition, prospects for future earnings and other factors our board of directors may deem relevant. There is no guarantee that our common stock will appreciate or even maintain the price at which our holders have purchased it. In addition, our loan agreements limit our ability to, among other things, pay dividends or make other distributions or payments on account of our common stock, in each case subject to certain exceptions. If we do not pay dividends, our stock may be less valuable because a return on stockholders' investment will only occur if our stock price appreciates and the stockholder then sells our common stock.
Insiders will continue to have substantial influence over us, which could limit your ability to affect the outcome of key transactions, including a change of control.
As of December 31, 2023, officers and their respective affiliates beneficially owned shares (not including any vested and exercisable options and unvested restricted stock units) representing approximately 22% of our outstanding common stock. Further, while there is no voting agreement or other similar arrangements between them, our CEO and one other director are family members. As a result, these stockholders, if they act together, will be able to influence our management and affairs and all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control of our company and might affect the market price of our common stock.
Anti-takeover provisions in our amended and restated certificate of incorporation and bylaws, and Delaware law, could discourage a change in control of our company or a change in our management.
Our amended and restated certificate of incorporation and bylaws as currently in effect contain provisions that might enable our management to resist a takeover. These provisions include:
56
These provisions might discourage, delay or prevent a change in control of our company or a change in our management. The existence of these provisions could adversely affect the voting power of holders of common stock and limit the price that investors might be willing to pay in the future for shares of our common stock. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law (the "DGCL"), which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with any "interested" stockholder for a period of three years following the date on which the stockholder became an "interested" stockholder.
Our amended and restated certificate of incorporation and amended and restated bylaws provide that the Court of Chancery of the State of Delaware is the sole and exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders' abilities to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation and amended and restated bylaws provide that the Court of Chancery of the State of Delaware (or, in the event that the Court of Chancery does not have jurisdiction, the federal district court for the District of Delaware or other state courts of the State of Delaware) is the exclusive forum for any derivative action or proceeding brought on our behalf, any action asserting a claim of breach of fiduciary duty, any action asserting a claim against us arising pursuant to the DGCL, our amended and restated certificate of incorporation or our amended and restated bylaws, or any action asserting a claim against us that is governed by the internal affairs doctrine; provided that, the exclusive forum provision does not apply to suits brought to enforce any liability or duty created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction; and provided further that, if and only if the Court of Chancery of the State of Delaware dismisses any such action for lack of subject matter jurisdiction, such action may be brought in another state or federal court sitting in the State of Delaware. Our amended and restated certificate of incorporation and amended and restated bylaws also provide that the federal district courts of the United States of America will be the exclusive forum for the resolution of any complaint asserting a cause of action against any defendant arising under the Securities Act. Such provision is intended to benefit and may be enforced by us, our officers and directors, employees, and agents, including the underwriters and any other professional or entity who has prepared or certified any prospectus, including the prospectuses for our initial public offering in 2021 and our offering in 2023.
We believe these provisions may benefit us by providing increased consistency in the application of Delaware law and federal securities laws by chancellors and judges, as applicable, particularly experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums and protection against the burdens of multi-forum litigation. This choice of forum provision may limit a stockholder's ability to bring a claim in a judicial forum that it finds favorable for disputes with us or any of our directors, officers, other employees or stockholders, which may discourage lawsuits with respect to such claims or make such lawsuits more costly for stockholders, although our stockholders will not be deemed to have waived our compliance with federal securities laws and the rules and regulations thereunder. Furthermore, the enforceability of similar choice of forum provisions in other companies' certificates of incorporation has been challenged in legal proceedings, and it is possible that a court could find these types of provisions to be inapplicable or unenforceable. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions, and there can be no assurance that such provisions will be enforced by a court in those other jurisdictions. If a court were to find the choice of forum provision that contained in our amended and restated certificate of incorporation and amended and restated bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could seriously harm our business.
We are a large accelerated filer and, subject to certain grace periods, may no longer provide scaled disclosures as an emerging growth company or as a smaller reporting company, which will increase our costs and demands on management.
As of December 31, 2023, we became a large accelerated filer, and are no longer an emerging growth company or a smaller reporting company. As an emerging growth company and a smaller reporting company, we had the option to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies, including, but not
57
limited to, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. Subject to certain grace periods, we may no longer provide scaled disclosure as an "emerging growth company" or a "smaller reporting company" as defined under the Exchange Act, which will increase our costs and demands on management.
In addition, as a non-accelerated filer and emerging growth company, we have availed ourselves of the exemption from the requirement that our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting under Section 404(b) of the Sarbanes Oxley Act. However, we may no longer avail ourselves of this exemption as a large accelerated filer, which will increase our expenses and require a significant amount of management time.
General Risk Factors
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. Furthermore, a severe or prolonged economic downturn, including a recession or depression resulting from the inflation reduction initiatives, higher interest rates, wars, invasions, political disruption or pandemics could result in a variety of risks to our business, including weakened demand for our procedures or products and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy, political disruption and inflation, including those resulting from pandemics, wars, invasions, energy supply interruptions or international trade disputes, staffing shortages, or similar events, could also strain our manufacturers or suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our products. Any of the foregoing could seriously harm our business, and we cannot anticipate all of the ways in which the political or economic climate and financial market conditions could seriously harm our business.
We, along with our suppliers, are dependent on various information technology systems, and failures of, interruptions to, or unauthorized tampering of those systems could have a material adverse effect on our business.
We and our suppliers rely extensively on information technology systems, networks and services, including internet sites, data hosting and processing facilities and tools, physical security system and other hardware, software and technical applications and platforms, some of which are managed, hosted, provided or used by third-parties or their vendors, to conduct business. These systems include, but are not limited to, ordering and managing materials from suppliers, converting materials to finished products (suppliers), shipping products to customers, processing transactions, accessing documents, receiving payments, summarizing and reporting results of operations, complying with regulatory, legal or tax requirements, providing data security, maintaining databases of consumer information, storing patient information, and other processes necessary to manage our business.
Despite the implementation of security measures, our internal computer systems and those of our contractors, consultants and collaborators are vulnerable to damage from cyberattacks, including ransomware and "phishing" attacks, intentional or accidental actions or omissions to act that cause vulnerabilities, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Attacks upon information technology systems are increasing in their frequency, levels of persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives and expertise. Geopolitical events or issues may increase cybersecurity risks on a global basis. Furthermore, because the techniques used to obtain unauthorized access to, or to sabotage, systems change frequently and often are not recognized until launched against a target, we may be unable to anticipate these techniques or implement adequate preventative measures. We may also experience security breaches that may remain undetected for an extended period, and we may not anticipate these acts or mitigate them adequately or timely, which may compound damages before the incident is discovered or remediated. The extent of a particular cyber incident and the steps that we may need to take to investigate the incident may not be immediately clear, and it may take a significant amount of time before such investigation can be completed and full and reliable information about the incident is known. New regulations may require us to disclose information about a material cybersecurity incident before it has been resolved or fully investigated.
While we do not believe that we have experienced any significant system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our or our critical third parties' operations, it could result in material disruptions of our operations and ultimately, our financial results. Consequences of data breaches and information technology disruptions include, but are not limited, to patients or employees being exposed to financial or medical identity theft, losing existing customers or have difficulty attracting new customers, experiencing difficulty preventing, detecting, and controlling
58
fraud, being exposed to the loss or misuse of confidential information, having disputes with customers, physicians, and other health care professionals, suffering regulatory sanctions or penalties under federal laws, state laws, or the laws of other jurisdictions, experiencing increases in operating expenses or an impairment in our ability to conduct our operations, delays in conducting business transactions and reporting financial results, incurring expenses or losing revenues as a result of a data privacy breach, product failure, information technology outages or disruptions, or suffering other adverse consequences including lawsuits or other legal action and damage to our reputation. Further, if our systems are damaged or cease to function properly due to any number of causes, ranging from catastrophic events to power outages to security breaches, and our business continuity plans do not effectively and timely compensate, we may suffer interruptions in our ability to manage operations, conduct business, and report financial results and would also be exposed to a risk of loss, including financial assets or litigation and potential liability, which could materially adversely affect our business, financial condition, results of operations and prospects.
We cannot assure you that any limitations of liability provisions in our contracts would be enforceable or adequate or would otherwise protect us from any liabilities or damages with respect to any particular claim relating to a security lapse or breach. While we maintain certain insurance coverage, our insurance may be insufficient or may not cover all liabilities incurred by such attacks. We also cannot be certain that our insurance coverage will be adequate for data handling or data security liabilities actually incurred, that insurance will continue to be available to us on economically reasonable terms, or at all, or that any insurer will not deny coverage as to any future claim. The successful assertion of one or more large claims against us that exceeds available insurance coverage, or the occurrence of changes in our insurance policies, including premium increases or the imposition of large deductible or co-insurance requirements, could have a material adverse effect on our business, including our financial condition, operating results and reputation.
Our operations are vulnerable to interruption or loss due to natural or other disasters, power loss, strikes and other events beyond our control.
A major hurricane, fire or other disaster (such as a major flood, earthquake or terrorist attack) affecting our headquarters or our other facilities, or facilities of our suppliers and manufacturers, could significantly disrupt our operations, and delay or prevent product shipment or installation during the time required to repair, rebuild or replace our suppliers' and manufacturers' damaged facilities, which delays could be lengthy and costly. If any of our customers' facilities are negatively impacted by a disaster, shipments of our products could be delayed. Additionally, customers may delay purchases of our products until operations return to normal. Even if we are able to quickly respond to a disaster, the ongoing effects of the disaster could create some uncertainty in the operations of our business. Concerns about terrorism, the effects of a terrorist attack or political turmoil could also have a negative effect on our operations, those of our suppliers and manufacturers and our customers.
We have incurred and will continue to incur significant costs as a result of operating as a public company, and our executive management team expects to devote substantial time to public company compliance programs.
As a public company, we have and will continue to incur significant legal, accounting and other expenses due to our compliance with regulations and disclosure obligations applicable to us, including compliance with the Sarbanes-Oxley Act, as well as rules implemented by the SEC and the Nasdaq Stock Market. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact, in ways we cannot currently anticipate, the manner in which we operate our business. Our executive management team and other personnel has and will continue to devote a substantial amount of time to these compliance programs and monitoring of public company reporting obligations and as a result of the corporate governance and executive compensation related rules, regulations and guidelines prompted by the Dodd-Frank Wall Street Reform and Consumer Protection Act, and further regulations and disclosure obligations expected in the future, we must devote additional time and costs to comply with such compliance programs and rules. These rules and regulations cause us to incur significant legal and financial compliance costs and make some activities more time-consuming and costly.
Failure to establish and maintain an effective system of internal controls could result in material misstatements of our financial statements or cause us to fail to meet our reporting obligations or fail to prevent fraud in which case, our stockholders could lose confidence in our financial reporting and the market price of our common stock could decline.
We are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act and the rules and regulations of the Nasdaq Stock Market. Under Section 404 of the Sarbanes-Oxley Act and these other rules and regulations, we must furnish a report by our management on our internal control over financial reporting, which must include disclosure of any material weaknesses identified by our management in our internal control over financial reporting, and our independent
59
registered public accounting firm must attest to the effectiveness of our internal control over financial reporting. To achieve compliance with Section 404 for 2023, we have been engaged in a process to document and evaluate our internal control over financial reporting, which has been both costly and extensive. In this regard, we have and will need to continue to dedicate internal resources, including employing financial and accounting personnel, engaging outside consultants and adopting a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. While we have not identified any material weakness in our internal controls over financial reporting for 2023, if in any future period we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective and would receive an adverse opinion regarding our internal control over financial reporting from our independent registered accounting firm.
We may in the future discover that material weaknesses in our system of internal financial and accounting controls and procedures could result or has resulted in a misstatement of our financial statements. If we are unable to remediate future material weaknesses, or otherwise maintain effective internal control over financial reporting, we may not be able to report our financial results accurately, prevent fraud or file our periodic reports in a timely manner, which may adversely affect investor confidence in us and, as a result, our stock price and ability to access the capital markets in the future.
In addition, our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system's objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.
Furthermore, in connection with the future attestation process by our independent registered public accounting firm, we may encounter problems or delays in completing the implementation of any requested improvements and receiving a favorable attestation. If we cannot favorably assess the effectiveness of our internal control over financial reporting, or if our independent registered public accounting firm is unable to provide an unqualified attestation report on our internal controls, our stockholders could lose confidence in our reporting and the market price of our common stock could decline. In addition, we could be subject to sanctions or investigations by the Nasdaq Stock Market, the SEC or other regulatory authorities.
We are subject to U.S. anti-corruption, export control, sanctions and other trade laws and regulations (collectively, the "Trade Laws"). We can face serious consequences for violations.
We are subject to anti-corruption laws, including the U.S. domestic bribery statute contained in 18 U.S.C. 201, the U.S. Travel Act, and the U.S. Foreign Corrupt Practices Act of 1977, as amended. These anti-corruption laws generally prohibit companies and their employees, agents and intermediaries from authorizing, promising, offering or providing, directly or indirectly, corrupt or improper payments or anything else of value to recipients in the public or private sector. We can be held liable for the corrupt or illegal activities of our agents and intermediaries, even if we do not explicitly authorize or have actual knowledge of such activities. We are also subject to other U.S. laws and regulations governing export controls, as well as economic sanctions and embargoes on certain countries and persons.
Violations of Trade Laws can result in substantial criminal fines and civil penalties, imprisonment, the loss of trade privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences. Likewise, any investigation of potential violations of Trade Laws could also have an adverse impact on our reputation, our business, results of operations and financial condition.
Changes in tax laws or regulations that are applied adversely to us or our customers may seriously harm our business.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of any of our future domestic and foreign earnings. Any new taxes could adversely affect our domestic and any future international business operations, and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us.
Our ability to use net operating losses to offset future taxable income may be subject to certain limitations.
Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an "ownership change," generally defined as a cumulative change of more than 50 percentage points (by value) in its equity ownership by certain stockholders over a three-year period, the corporation's ability to use its pre-change net operating loss ("NOL")
60
carryforwards and other pre-change NOLs tax attributes (such as research tax credits) to offset its post-change income or taxes may be limited. Based upon our analysis as of December 31, 2023, we have determined that we do not expect these limitations to impair our ability to use our NOLs prior to expiration. However, if changes in our ownership occur in the future, our ability to use our NOLs may be further limited. For these reasons, we may not be able to utilize a material portion of the NOLs, even if we achieve profitability.
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
Cybersecurity Risk Management and Strategy
We are committed to protecting the privacy and security of our information assets and the data entrusted to us. Our cybersecurity program comprises multiple levels of physical, technical, and administrative safeguards. Our cybersecurity program is informed by industry standards, including the Center for Information Security (CIS) framework for security controls and benchmarks, the National Institute of Standards and Technology (NIST) standards, and the ISO 27000 framework. This does not imply that we meet any particular technical standards, specifications, or requirements at all times, only that we use CIS, NIST, and ISO 27000 as a guide to help us identify, assess, and manage cybersecurity risks relevant to our business.
We have integrated cybersecurity risk management into our broader risk management framework to promote a company-wide culture of cybersecurity risk management. This integration results in cybersecurity considerations as a key part of our decision-making processes. Our cybersecurity team considers emerging threats and new vectors of cyberattack and pursues a deliberate risk-avoidance approach. We also maintain our written security incident response runbook detailing the response and notifications involved with various security events.
Our cybersecurity training and education emphasizes periodic phishing tests and a mandatory training curriculum to assist our employees' awareness of common types of attacks. This includes awareness of phishing, malware, social engineering, and overall security best practices for new and existing employees. We also perform periodic, independent risk assessments that consider four primary areas of risk: physical, digital, social, and administrative/governance.
Since we understand that cybersecurity threats are complex and evolving, we have a dedicated team of both internal and external cybersecurity experts, which is led by our Chief Information Officer (CIO), Security Officer. This team is responsible for publishing information technology and security policies, promoting compliance with those policies, implementing a program to mitigate potential threats, and performing periodic risk and maturity assessments. Our risk mitigation measures include network segmentation, cyber protection and containment, detection and response, and recovery. The primary goal of this team is to decrease the risk of cyber incidents having a material impact.
We also have plans in place to respond to cybersecurity incidents. These plans address issues relating to preparation for and detection of incidents, as well as responding to and recovering from incidents. We have procedures designed to assess, investigate, contain, remediate, and mitigate cybersecurity incidents, as well as procedures that seek to comply with legal obligations and regulatory reporting requirements. We periodically engage with assessors, consultants, auditors, and other third parties to review our cybersecurity processes.
Recognizing the risks associated with third-party service providers, we implement processes to manage these risks. We conduct assessments of critical third-party providers before engagement and maintain ongoing monitoring to assess compliance with our cybersecurity standards. In addition, we require SOC 1 Type II attestations from those IT vendors whose applications or cloud infrastructure handle sensitive information.
In the past three years, we have not experienced any cybersecurity incidents that have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations, or financial condition. We face certain ongoing risks from cybersecurity threats that, if realized, are reasonably likely to materially affect us, including our operations, business strategy, results of operations, or financial condition. See the risk factor entitled "Our information technology systems may be subject to breaches, cyber-attacks or other disruptions that could, among other adverse consequences, cause us to violate laws and regulations and could adversely affect our business, results of operations, financial condition, cash flows, reputation or competitive position."
61
Cybersecurity Governance
Our Board of Directors considers cybersecurity risk as part of its risk oversight function and has delegated to the Audit Committee responsibility for oversight of risk assessment and risk management, including cybersecurity, and the Company's policies and controls relating to information technology, management information systems, and cybersecurity. The Audit Committee receives quarterly reports from management on our cybersecurity risk management activities. In addition, management updates the Audit Committee, as necessary, regarding any material cybersecurity incidents, as well as any incidents with lesser impact potential. The Audit Committee reports to the full Board of Directors regarding its activities, including those related to cybersecurity.
The CIO, Security Officer has over three decades of technology experience, working for leading technology and consulting companies and previously served as CIO, Security Officer for both public and private medical device and healthcare organizations. Our cybersecurity team includes a former chief information security officer for large healthcare organizations, a former head of global security for a major enterprise cybersecurity platform, and other similarly credentialed professionals. Our CIO, Security Officer reports to the Chief Financial Officer and provides regular reports to the Audit Committee on cybersecurity policies, procedures, and risk and remediation efforts. Our CIO, Security Officer also serves on our Disclosure Committee and has regular dialogue with the senior management team on information security matters and risk management practices.
The CIO, Security Officer is regularly informed about the latest developments in cybersecurity, including potential threats and innovative risk management techniques. We believe this ongoing knowledge acquisition is crucial for the effective prevention, detection, mitigation, and remediation of cybersecurity incidents. The cybersecurity team implements and oversees processes for the regular monitoring of our information systems. In the event of a cybersecurity incident, the cybersecurity team follows a written security incident response runbook, which includes procedures to, among other things, respond to the incident, mitigate its impact, and evaluate and satisfy applicable obligations.
Item 2. Properties
As of December 31, 2023, we leased approximately 125,000 square feet for our corporate headquarters located in Ponte Vedra, Florida under a lease agreement which terminates in July 2032. We believe that this facility is sufficient to meet our current and anticipated needs in the future and that additional space can be obtained on commercially reasonable terms as needed. We also continue to lease a part of our previous corporate headquarters location until August 2026 and have entered into subleases for this space.
Item 3. Legal Proceedings
We are not currently party to any material legal proceedings. We may, however, in the ordinary course of business, face various claims brought against us by third parties and we may, from time to time, make claims or take legal actions to assert our rights, including our intellectual property rights, restrictive covenants and contractual claims. For more information, see the discussion of legal matters in Part II, Item 8, Note 8, "Commitments and Contingencies," of the Notes to the Financial Statements.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock began trading on the NASDAQ Global Select Market under the symbol "TMCI" on April 23, 2021. Prior to that time, there was no public market for our common stock.
62
Holders of Record
At February 22, 2024, there were approximately 24 stockholders of record of our common stock. Since many of our shares of common stock are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividends
We have never declared or paid, and do not anticipate declaring or paying, any cash dividends on any of our capital stock for the foreseeable future. We currently intend to retain all available funds and any future earnings for use in the operation of our business, to finance the growth and development of our business and for future repayment of debt. Future determinations as to the declaration and payment of dividends, if any, will be at the discretion of our board of directors and will depend on then-existing conditions, including our operating results, financial condition, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant. In addition, our term loan agreement limits our ability to pay dividends or make other distributions or payments on account of our common stock, in each case subject to certain exceptions.
Recent Sales of Unregistered Securities
None.
Share Repurchases
The following table presents the information with respect to purchases made by us of our common stock during the three months ended December 31, 2023:
Period |
|
Total Number of Shares Purchased |
|
|
Average Price Paid Per Share |
|
|
Total Number of Shares Purchased as part of Publicly Announced Plans or Programs |
|
|
Approximate Dollar Value of Shares that May Yet Be Purchased Under the Plans or Programs |
|
||||
October 1 to October 31, 2023 (1)(2) |
|
|
1,218 |
|
|
$ |
10.52 |
|
|
|
- |
|
|
|
- |
|
November 1 to November 30, 2023 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
December 1 to December 31, 2023 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
Totals |
|
|
1,218 |
|
|
$ |
10.52 |
|
|
|
- |
|
|
|
- |
|
Performance Graph
We previously qualified as a "smaller reporting company," as defined in Rule 12b-2 under the Exchange Act, and have been permitted to rely, and have relied, on the reduced disclosure requirements available to smaller reporting companies, including not being required to provide the information required by this item pursuant to Item 201(e) of Regulation S-K. Our ability to rely on the reduced disclosure requirements available to smaller reporting companies will cease after the filing of this Annual Report and our definitive proxy statement related to our 2024 Annual Meeting of Stockholders (the "2024 Proxy Statement"), including those portions of the 2024 Proxy Statement that will be incorporated by reference into Part III of this Annual Report.
Item 6. [Reserved]
63
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and related notes thereto included elsewhere in this Annual Report. This discussion and other parts of this Annual Report contain forward-looking statements that involve risks and uncertainties, such as statements of our plans, objectives, expectations and intentions that are based on the beliefs of our management, as well as assumptions made by, and information currently available to, our management. Our actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in the section titled "Risk Factors." Please also see the section titled "Special Note Regarding Forward-Looking Statements."
Overview
We are a medical technology company with the goal of advancing the standard of care for the surgical management of bunion and related midfoot deformities. We have pioneered our proprietary Lapiplasty 3D Bunion Correction System—a combination of instruments, implants and surgical methods designed to surgically correct all three planes of the bunion deformity and secure the unstable joint, addressing the root cause of the bunion and helping patients get back to their active lifestyles. Although bunions are deformities typically caused by an unstable joint in the middle of the foot that leads to a three-dimensional ("3D") misalignment in the foot's anatomical structure, the majority of traditional surgical approaches focus on correcting the deformity from a two-dimensional ("2D") perspective and therefore fail to address the root cause of the disorder. To effectively restore the normal anatomy of bunion patients and improve clinical outcomes, we believe addressing the root cause of the bunion is critical and have developed the Lapiplasty System to correct the deformity across all three anatomic dimensions. Our other products often used in conjunction with bunion surgery include the Adductoplasty System, the Hammertoe PEEK Fixation System, the SpeedPlate Rapid Compression Implant System, and specialized osteotomes and release instruments.
We were formed in 2013, and since receiving 510(k) clearance for the Lapiplasty System in March 2015, we have sold more than 90,000 Lapiplasty Procedure kits in the United States. We market and sell our Lapiplasty Systems to physicians, surgeons, ambulatory surgery centers and hospitals. The Lapiplasty Procedure can be performed in either hospital outpatient or ambulatory surgery centers settings, and utilizes existing, well-established reimbursement codes. We currently market and sell the Lapiplasty System and other products through a combination of a direct employee sales force and independent sales agencies across 249 territories in the United States. As of December 31, 2023, we had 227 direct sales representatives and 22 independent sales agencies. In 2023, employee sales representatives generated approximately 81% of revenues while approximately 19% of revenues came through independent sales agencies.
On February 10, 2023, we completed a follow-on public offering of 5,476,190 shares of our common stock, which included the exercise in full of the underwriters' option to purchase additional shares, at a price to the public of $21.00 per share. This offering resulted in net proceeds of approximately $107.5 million after deducting underwriting discounts and commissions of $6.9 million and offering expenses payable by us of approximately $0.6 million.
On June 12, 2023, we acquired certain assets of RPM-3D used in providing pre-operative planning and patient-specific guides for the surgical correction of foot and ankle deformities for $20.0 million in cash. In addition, we are obligated to pay additional amounts of up to $10.0 million in cash upon completion of certain milestones. This acquisition adds FDA-cleared patient specific instrumentation ("PSI") technologies and capabilities to our portfolio, building upon our pioneering 3D bunion correction and related midfoot solutions, as well as 22 additional patent applications that further expand and reinforce our global intellectual property portfolio covering technologies for the correction of bunion and related deformities.
As of December 31, 2023, we had cash and cash equivalents of $13.0 million and marketable securities of $110.2 million available for sale to fund operations, an accumulated deficit of $134.2 million and $54.0 million of principal outstanding under our term loan and revolving loan agreements.
64
Economic Environment
There is continuing uncertainty in the macro-economic environment. Inflationary pressures, interest rates, recession fears and reduced consumer confidence, and ongoing supply chain challenges have resulted, and may continue to result, in higher costs and longer lead times from suppliers and potentially reduced demand for our Lapiplasty Procedure kits and other products. General economic conditions may also negatively impact demand for elective surgeries. While we continuously work with suppliers to mitigate higher costs and longer lead times and continue to invest in our direct sales channel, patient education initiatives, clinical evidence and product innovations to build demand for our products, we expect these macro-economic challenges to continue for the foreseeable future, which likely will impact our results of operations.
Key Business Metrics
We regularly review a number of operating and financial metrics, including the number of Lapiplasty Procedure kits sold, blended average revenue per Lapiplasty Procedure kit sold, the number of active surgeons using the Lapiplasty System and the surgeon utilization rate, to evaluate our business, measure our performance, identify trends affecting our business, formulate our business plans and make strategic decisions. The number of Lapiplasty Procedure kits sold during the twelve months ended December 31, 2023 increased by 5,019, or 20%, over the twelve months ended December 31, 2022. The blended average sales price per Lapiplasty Procedure kits sold was $6,306 during the twelve months ended December 31, 2023, a 10% increase over the same period in 2022. We define the blended average sales price as revenue divided by Lapiplasty Procedure kit sold that includes the revenue for all products sold from our expanding product line. The number of active surgeons as of December 31, 2023 was 2,855, an increase of 20% from the prior year. We define the number of active surgeons as the number of surgeons that performed at least one procedure using the Lapiplasty System in the trailing twelve-month period. The surgeon utilization rate for the twelve months ended December 31, 2023 increased 0.6% over the twelve months ended December 31, 2022 to an average of 10.4 Lapiplasty Procedure kits per active surgeon.
We believe that the number of Lapiplasty Procedure kits sold, blended average revenue per Lapiplasty Procedure kit sold, number of active surgeons using the Lapiplasty System and the surgeon utilization rate are useful indicators of our ability to drive adoption of the Lapiplasty System and generate revenue and are helpful in tracking the progress of our business. While we believe these metrics are representative of our current business, we anticipate these metrics may be substituted by additional or different metrics as our business grows.
Factors Affecting Our Business
We believe that our financial performance has been and in the foreseeable future, will continue to depend on many factors, including macro-economic conditions as described above, those described below, those noted in the section titled "Special Note Regarding Forward-Looking Statements" and in the section titled "Risk Factors".
Adoption of the Lapiplasty System
The growth of our business depends on our ability to gain broader acceptance of the Lapiplasty System by successfully marketing and distributing the Lapiplasty System and ancillary products. We currently have approval at over 2,200 facilities across the United States and plan to continue to increase access by marketing to more surgeons and facility administrators that our products are alternatives to traditional products used in bunion surgical procedures. While surgeon adoption of the Lapiplasty Procedure remains critical to supporting procedure growth, hospital and ambulatory surgery center facility approvals are necessary for both existing and future surgeon customers to access our products. To facilitate greater access to our products and support future sales growth, we intend to continue educating hospitals and facility administrators on the differentiated benefits associated with the Lapiplasty System, supported by our robust portfolio of clinical data. If we are unable to successfully continue to commercialize our Lapiplasty System, we may not be able to generate sufficient revenue to achieve or sustain profitability. In the near term, we expect we will continue to operate at a loss, and we anticipate we will finance our operations principally through the use of our cash and cash equivalents, marketable securities, and expected revenues. We may also raise funds by incurring debt and through offerings of our capital stock.
Investments in Innovation and Growth
We expect to continue to focus on long-term revenue growth through investments in our business. In sales and marketing, we are dedicating meaningful resources to continue to expand our sales force and management team in the United States, as well as our patient focused outreach and education campaigns. We have approximately 340 employee sales representatives, associate sales representatives, clinical specialists, and sales managers at December 31, 2023. We plan to grow this specialized team in 2024 to ensure strong surgeon support and continued market penetration.
65
In research and development, our team and surgeon consultants are continually working on next-generation innovations for the surgical correction of bunions and other conditions that often present with bunions. In 2021, we launched (1) the Lapiplasty Mini-Incision System, which is designed to allow the Lapiplasty Procedure to be performed through a miniature, 3.5cm incision as compared to the current 6cm to 8cm incision, and (2) the Adductoplasty System, which brings together our implants and instrumentation to provide a comprehensive system designed for reproducible realignment, stabilization, and fusion of the midfoot to address midfoot deformities that can occur in up to 30% of bunion patients. In 2022, we introduced (1) the 3-n-1 Guide, which combines three separate instruments and three procedure steps into one instrument and step, (2) the S4A plating system, which features advanced 3D contours designed to accommodate variations in patient anatomy, and (3) the SpeedRelease Instrument, which is a single-use instrument designed to make a challenging soft tissue release performed in the majority of Lapiplasty cases easier to perform and more reproducible for the surgeon. In 2023, in addition to the acquisition of the assets of RPM-3D discussed above, we began the market release of (1) the SpeedPlate fixation platform, which can be used in the Lapiplasty and Adductoplasty Procedures, as well as other common bone fusion procedures of the foot, (2) the Hammertoe PEEK Fixation System designed to address hammertoe, claw toe and mallet toe deformities, which often present concomitantly with bunions, and (3) LapiTome and RazorTome Osteotomes, which are sterile, single-use instruments that are designed to facilitate more efficient removal and release of bone slices and soft tissue in Lapiplasty and Adductoplasty cases.
In our general and administrative functions, we expect to continue to hire personnel and expand our infrastructure to both drive and support our anticipated growth and operations as a public company. Accordingly, in the near term, we expect to have net losses from these activities, but in the longer term we anticipate they will positively impact our business and results of operations.
Seasonality
We have experienced and expect to continue to experience seasonality in our business, with higher sales volumes in the fourth calendar quarter, historically accounting for approximately 30 to 40% of full year revenues, and lower sales volumes in the subsequent first calendar quarter. Our sales volumes in the fourth quarter tend to be higher as many patients elect to have surgery after meeting their annual deductible and having time to recover over the winter holidays. Our sales volumes in subsequent first calendar quarters also tend to be lower as a result of adverse weather and by resetting annual patient healthcare insurance plan deductibles, both of which may cause patients to delay elective procedures; however, in some years the first quarter may benefit from additional sales volumes when high patient demand for surgeries in the fourth quarter cannot be fully accommodated and those surgical procedures are rolled over into the first quarter. Similar to the rest of the orthopaedic industry, we have experienced and expect to continue to experience lower sales volumes in the third quarter than throughout the rest of the year as elective procedures generally decline during the summer months.
Coverage and Reimbursement
Hospitals, ambulatory surgery centers and surgeons that purchase or use our products generally rely on third-party payors to reimburse for all or part of the costs and fees associated with procedures using our products. As a result, sales of our products depend, in part, on the extent to which the procedures using our products are covered by third-party payors, including government programs such as Medicare and Medicaid, private insurance plans and managed care programs. Based on historical claims data from 2017, approximately 63% of Lapidus cases and 60% of all bunion surgical cases were paid by private payors.
Medicare payment rates to hospital outpatient departments are set under the Medicare hospital outpatient prospective payment system, which groups clinically similar hospital outpatient procedures and services with similar costs to ambulatory payment classifications ("APCs"). Each APC is assigned a single lump sum payment rate, which includes payment for the primary procedure as well as any integral, ancillary, and adjunctive services. The primary CPT codes for the Lapiplasty Procedure, CPT 28297 and CPT 28740, are grouped together under APC 5114. For Lapiplasty Procedures in which fusion is performed on multiple TMT joints, CPT 28730 applies and is classified under APC 5115. For Adductoplasty Procedures in which fusion is performed on multiple TMT joints, either CPT 28730 or CPT 27835 applies and are classified under APC 5115.
66
Components of Our Results of Operations
Revenue
We currently generate revenue from the sale of our proprietary Lapiplasty System and the minimally invasive variations, Adductoplasty System, Hammertoe PEEK Fixation System, SpeedPlate Implant Fixation Platform, single use osteotomes and release instruments, and other ancillary products. These systems bring together single-use implant kits, reusable instrument trays, and surgical techniques. We sell the kits and single use instruments and other products to physicians, surgeons, hospitals and ambulatory surgery centers in the United States through a network of employee sales representatives and independent sales agencies.
No single customer accounted for 10% or more of our revenue during 2023. We expect our revenue to increase in absolute dollars in the foreseeable future as we expand our product offerings, sales territories, new accounts and trained physician base and as existing physician customers perform more Lapiplasty Procedures, though it may fluctuate from quarter to quarter due to a variety of factors, including seasonality and the macro-economic environment.
Cost of Goods Sold
Cost of goods sold consists primarily of costs for the purchase of our products from third-party manufacturers. Direct costs from our third-party manufacturers include costs for raw materials plus the markup for the assembly of the components. Cost of goods sold also includes royalties, allocated overhead for indirect labor, certain direct costs such as those incurred for shipping our products, and personnel costs. We expense all inventory provisions for excess, obsolete and field losses as cost of goods sold. We record adjustments to our inventory valuation for estimated excess, obsolete and non-sellable inventories based on assumptions about future demand, past usage, changes to manufacturing processes and overall market conditions. We expect our cost of goods sold to increase in absolute dollars in the foreseeable future to the extent more of our products are sold, though it may fluctuate from quarter to quarter.
Gross Profit and Gross Margin
We calculate gross profit as revenue less cost of goods sold, and gross margin as gross profit divided by revenue. Our gross margin has been and will continue to be affected by a variety of factors, primarily average selling prices, production and ordering volumes, change in mix of customers and products, third-party manufacturing costs and cost-reduction strategies. We expect our gross profit to increase in the foreseeable future as our revenue grows, though our gross margin may fluctuate from quarter to quarter due to changes in average selling prices as we introduce new products, and as we adopt new manufacturing processes and technologies.
Operating Expenses
Sales and Marketing
Sales and marketing expenses consist primarily of compensation for personnel, including salaries, bonuses, benefits, sales commissions and share-based compensation, related to selling and marketing functions, surgical instrument expense, physician education programs, training, shipping costs related to sending products to our sales representatives, travel expenses, marketing initiatives including our direct-to-patient outreach program and advertising, market research and analysis and conferences and trade shows. We expect sales and marketing expenses to continue to increase in absolute dollars in the foreseeable future as we continue to invest in our direct sales force and expand our marketing efforts, and as we continue to expand our sales and marketing infrastructure to both drive and support anticipated sales growth, though it may fluctuate from quarter to quarter.
Research and Development
Research and development ("R&D") expenses consist primarily of engineering, product development, clinical studies to develop and support our products, regulatory expenses, and other costs associated with products and technologies that are in development. These expenses include compensation for personnel, including salaries, bonuses, benefits and share-based compensation, supplies, consulting, prototyping, testing, materials, travel expenses, depreciation, and an allocation of facility overhead expenses. We expect R&D expenses to continue to increase in absolute dollars in the foreseeable future as we continue to hire personnel and invest in next-generation innovations of the Lapiplasty System and other products, though it may fluctuate from quarter to quarter due to a variety of factors, including the level and timing of our new product development efforts, as well as our clinical development, clinical studies, and other related activities.
67
General and Administrative
General and administrative expenses consist primarily of compensation for personnel, including salaries, bonuses, benefits and share-based compensation, related to finance, information technology, legal and human resource functions, as well as professional services fees (including legal, audit and tax fees), insurance costs, general corporate expenses and allocated facilities-related expenses. We expect general and administrative expenses to continue to increase in absolute dollars in the foreseeable future as we hire personnel and expand our infrastructure to drive and support the anticipated growth in our organization.
Interest Income
Interest income consists of interest earned on our money market funds and marketable securities.
Interest Expense
Interest expense consists of interest incurred and amortization of debt discount and issuance costs related to outstanding borrowings.
Results of Operations
Comparison of the years ended December 31, 2023 and 2022
The following table summarizes our results of operations for the periods indicated ($ in thousands):
|
|
Year Ended December 31, |
|
|
Change |
|||||||||
|
|
2023 |
|
|
2022 |
|
|
Amount |
|
|
% |
|||
Revenue |
|
$ |
187,118 |
|
|
$ |
141,838 |
|
|
$ |
45,280 |
|
|
31.9% |
Cost of goods sold |
|
|
35,181 |
|
|
|
25,532 |
|
|
|
9,649 |
|
|
37.8% |
Gross profit |
|
|
151,937 |
|
|
|
116,306 |
|
|
|
35,631 |
|
|
30.6% |
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|||
Sales and marketing |
|
|
140,894 |
|
|
|
104,567 |
|
|
|
36,327 |
|
|
34.7% |
Research and development |
|
|
15,440 |
|
|
|
13,584 |
|
|
|
1,856 |
|
|
13.7% |
General and administrative |
|
|
47,031 |
|
|
|
32,999 |
|
|
|
14,032 |
|
|
42.5% |
Total operating expenses |
|
|
203,365 |
|
|
|
151,150 |
|
|
|
52,215 |
|
|
34.5% |
Loss from operations |
|
|
(51,428 |
) |
|
|
(34,844 |
) |
|
|
(16,584 |
) |
|
47.6% |
Interest income |
|
|
6,726 |
|
|
|
1,313 |
|
|
|
5,413 |
|
|
* |
Interest expense |
|
|
(5,167 |
) |
|
|
(4,398 |
) |
|
|
(769 |
) |
|
17.5% |
Debt extinguishment loss |
|
|
— |
|
|
|
(4,483 |
) |
|
|
4,483 |
|
|
* |
Other income, net |
|
|
342 |
|
|
|
(403 |
) |
|
|
745 |
|
|
* |
Other non-operating income (expense), net |
|
|
1,901 |
|
|
|
(7,971 |
) |
|
|
9,872 |
|
|
(123.8)% |
Net loss |
|
$ |
(49,527 |
) |
|
$ |
(42,815 |
) |
|
$ |
(6,712 |
) |
|
15.7% |
* Not meaningful
Revenue. Revenue increased by $45.3 million, or 31.9%, in the year ended December 31, 2023, as compared to 2022. The increase was partially driven by a 20% increase in the number of Lapiplasty Procedure kits sold in 2023 compared to 2022 as the result of an expanded surgeon customer base. The increase in the volume of Lapiplasty Procedure kits sold resulted in 50.9% of the revenue growth from 2022 to 2023. The remaining revenue growth was a result of increased adoption of our newer technologies and selling more products used in bunion cases resulting in a 10% increase in average blended revenue per Lapiplasty Procedure kit sold compared to the same period in 2022.
Cost of Goods Sold, Gross Profit and Gross Margin. Cost of goods sold increased by $9.6 million, or 37.8%, in the year ended December 31, 2023, as compared to 2022. The increase in cost of goods sold was primarily due to a $6.7 million increase in direct costs of goods sold and a $0.5 million increase in royalty expense both resulting from increased sales, $1.8 million increase in overhead expenses resulting from an increase in our headcount to support our growing business, and a $0.7 million increase in inventory provisions. Gross profit increased $35.6 million from the year ended December 31, 2023 as compared to 2022, primarily due to sales growth. Gross profit margin for the year end December 31, 2023 decreased from 82.0% to 81.2%, as compared to the same period in 2022, primarily due to changes in product mix, an increase in overhead, and an increase in inventory provisions, partially offset by lower royalty rates on newer products.
68
Sales and Marketing Expenses. Sales and marketing expenses increased by $36.3 million, or 34.7%, in the year ended December 31, 2023, as compared to 2022. The increase in sales and marketing expenses was due to growth in our overall business. Sales and marketing expenses increased due to an increase of $15.7 million in payroll and related expenses from increased headcount of sales and marketing personnel, an increase of $11.6 million for higher commissions from increased sales by our employee sales representatives and independent sales agencies, $2.5 million in surgeon training and clinical-related expenses, an increase of $2.2 million in surgical instrument expense, an increase of $1.9 million primarily due to higher advertising fees for direct to consumer campaigns and sales meeting costs, an increase of $1.2 million in professional services related to marketing, an increase of $0.5 million in shipping costs due to increased sales, and an increase of $0.3 million in rent expense and occupancy costs related to moving into our new headquarters building in third quarter of 2022.
Research and Development Expenses. Research and development expenses increased by $1.9 million, or 13.7%, in the year ended December 31, 2023, as compared to 2022. The increase in research and development expenses was due to increases of $0.9 million in payroll and payroll-related costs resulting from increased headcount of research and development personnel, an increase of $0.6 million in product testing and validation, and an increase of $0.3 million in rent expense and occupancy costs related to moving into our new headquarters building in third quarter of 2022.
General and Administrative Expenses. General and administrative expenses increased by $14.0 million, or 42.5%, in the year ended December 31, 2023, as compared to 2022. The increase in general and administrative expenses was primarily due to an increase of $8.9 million in payroll and payroll-related costs from increased headcount to support our growing business, an increase of $4.2 million in professional services, primarily consisting of a $3.3 million increase in accrued compensation expense related to the milestone payments for our acquisition of RPM-3D in second quarter of 2023 and a $0.9 million increase in legal expenses, an increase of $0.7 million in rent expense and occupancy costs related to moving into our new headquarters building in third quarter of 2022, and a $0.5 million increase in amortization of finite-lived intangible assets from our acquisition of RPM-3D.
Interest Income. Interest income increased by $5.4 million in 2023 as compared to 2022. The increase in interest income was due to higher cash balances invested in marketable securities during 2023 due to our equity offering in the first quarter of 2023 and higher interest rates compared to 2022.
Interest Expense. Interest expense increased by $0.8 million, or 17.5%, from the year ended December 31, 2023 as compared to 2022. The increase in interest expense was due to a higher average debt balance in 2023 compared to 2022 and slightly higher interest rates on our outstanding debt in 2023 compared to 2022.
Debt Extinguishment Loss. Debt extinguishment loss decreased by $4.5 million, for the year ended December 31, 2023 as compared to the same period of 2022 due to our debt refinancing in 2022.
For the comparison of the results of operations for the years ended December 31, 2022 and 2021, refer to our Annual Report on Form 10-K, for the year ended December 31, 2022, as filed with the U.S Securities and Exchange Commission on March 8, 2023, in Part II Item 7 "Management's Discussion and Analysis of Financial Condition and Results of Operations."
Liquidity and Capital Resources
Overview
Before our IPO, our primary sources of capital were private placements of common stock and convertible preferred stock, debt financing agreements and revenue from the sale of our products. In April 2021, we received net proceeds of $107.6 million from our IPO. On February 10, 2023, we completed a follow-on public offering of 5,476,190 shares of our common stock, which included the exercise in full of the underwriters' option to purchase additional shares, at a price to the public of $21.00 per share. This offering resulted in net proceeds of approximately $107.5 million after deducting underwriting discounts and commissions of $6.9 million and offering expenses payable by us of approximately $0.6 million.
In April 2022, we entered into a five-year $150.0 million loan arrangement, initially consisting of up to $120.0 million in term loans over four tranches and up to $30.0 million in a revolving loan facility with entities affiliated with MidCap. As of December 31, 2023, we have $50.0 million under the term loan and $4.0 million under the revolving loan facility outstanding. As of December 31, 2023, tranche two ($30.0 million) of the term loan expired and is no longer available. Tranches three and four provide up to an additional $40.0 million in borrowing capacity in the aggregate, subject to the achievement of certain revenue targets.
69
As of December 31, 2023, we had cash and cash equivalents of $13.0 million and marketable securities of $110.2 million available for sale, an accumulated deficit of $134.2 million and $54.0 million of principal outstanding under our term loan and revolving loan agreements. We believe that our existing cash and cash equivalents, marketable securities, available debt borrowings, and expected revenues will be sufficient to meet our capital requirements and fund our operations for at least the next 12 months from the date of the issuance of these financial statements. We may be required or decide to raise additional financing to support further growth of our operations.
Funding Requirements
We use our cash to fund our operations, which primarily includes product costs as well as our sales and marketing and R&D expenses and related personnel costs. We expect our sales and marketing expenses to increase for the foreseeable future as we continue to invest in our direct sales force and expand our marketing efforts, and as we continue to expand our sales and marketing infrastructure to both drive and support anticipated sales growth. We also expect R&D expenses to increase for the foreseeable future as we continue to hire personnel and invest in next-generation innovations of our products. In addition, we expect our general and administrative expenses to increase for the foreseeable future as we hire personnel and expand our infrastructure to both drive and support the anticipated growth in our organization. We also incur and will continue to incur additional expenses as a result of operating as a public company. In the second quarter of 2023, funds were used for the acquisition of RPM-3D, and from time to time in the future, we may also consider additional investments in technologies, assets and businesses to expand or enhance our product offerings. The timing and amount of our operating and capital expenditures will depend on many factors, including:
70
Based upon our current operating plan, we believe that our existing cash, cash equivalents, marketable securities, and available debt borrowings will enable us to fund our operating expenses and capital expenditure requirements for at least the next twelve months. We have based this estimate on assumptions that may prove to be wrong or that may change in the future, and we could utilize our available capital resources sooner than we expect. We may seek to raise any necessary additional capital through public or private equity offerings or debt financings, credit or loan facilities or a combination of one or more of these or other funding sources. Additional funds may not be available to us on acceptable terms or at all. If we fail to obtain necessary capital when needed on acceptable terms, or at all, we could be forced to delay, limit, reduce or terminate our product development programs, commercialization efforts, sales and marketing initiatives, or other operations. If we raise additional funds by issuing equity securities, our stockholders will suffer dilution and the terms of any financing may adversely affect the rights of our stockholders. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, is likely to involve restrictive covenants limiting our flexibility in conducting future business activities, and, in the event of insolvency, debt holders would be repaid before holders of our equity securities received any distribution of our corporate assets.
Cash Flows
The following table sets forth the primary sources and uses of cash and cash equivalents for the periods presented below (in thousands):
|
|
Year Ended December 31, |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Net cash (used in) provided by: |
|
|
|
|
|
|
|
|
|
|||
Operating activities |
|
$ |
(34,575 |
) |
|
$ |
(30,648 |
) |
|
$ |
(17,193 |
) |
Investing activities |
|
|
(81,299 |
) |
|
|
(76,518 |
) |
|
|
(2,705 |
) |
Financing activities |
|
|
109,383 |
|
|
|
20,806 |
|
|
|
107,652 |
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
(6,491 |
) |
|
$ |
(86,360 |
) |
|
$ |
87,754 |
|
Net Cash Used in Operating Activities
Net cash used in operating activities for the year ended December 31, 2023 was $34.6 million, consisting primarily of a net loss of $49.5 million and an increase in net operating assets of $9.7 million, which were partially offset by non-cash charges of $24.7 million. The non-cash charges consist primarily of share-based compensation expense of $17.4 million, depreciation and amortization expense of $5.4 million and non-cash lease expense of $2.5 million, offset by amortization and accretion of marketable securities of $1.4 million. The increase in net operating assets was primarily due to an increase of $9.8 million in inventories to meet demand for new products and safety stock, an increase of $9.3 million in accounts receivable due to sales growth in 2023, and an increase of $1.2 million in prepaid expenses and other assets (excluding unsettled securities transactions), which were partially offset by a $7.5 million increase to accrued liabilities and a $3.2 million increase to accounts payable due to timing of payments and growth of our operations. The increase of $7.5 million in accrued liabilities consisted of an increase of $4.2 million due to timing of payments, and an increase of $3.3 million due to increased accrued compensation expense related to our acquisition of RPM-3D in the second quarter 2023.
Net cash used in operating activities for the year ended December 31, 2022 was $30.6 million, consisting primarily of a net loss of $42.8 million and an increase in net operating assets of $5.8 million, which were partially offset by non-cash charges of $18.0 million. The increase in net operating assets was primarily due to an increase in accounts receivable resulting from higher sales revenue in 2022, higher inventories resulting from higher purchases in anticipation of growing demand in 2023 and to guard against supply chain risks, and an increase in prepaid expenses and other assets due to timing of payments, which were offset by increases in accounts payable and accrued liabilities due to timing of payments and growth of our operations. The non-cash charges primarily consisted of share-based compensation expense of $8.1 million, a loss on extinguishment of debt of $4.5 million, non-cash lease amortization of $2.5 million, depreciation and amortization expense of $2.1 million, and $0.4 million in provision for doubtful accounts primarily driven by higher account receivable balances at year end.
71
Net cash used in operating activities for the year ended December 31, 2021 was $17.2 million, consisting primarily of a net loss of $20.6 million and an increase in net operating assets of $1.0 million, which were partially offset by non-cash charges of $4.3 million. The increase in net operating assets was primarily due to an increase in accounts receivable resulting from higher revenues in 2021, higher inventories resulting from higher purchases in anticipation of growing demand in 2022, and an increase in prepaid expenses and other assets due to timing of payments and growth of our operations, which were offset by increases in accounts payable and accrued liabilities due to timing of payments and growth of our operations. The non-cash charges primarily consisted of depreciation and amortization expense of $0.7 million, share-based compensation expense of $3.4 million, and amortization of debt issuance costs of $0.2 million.
Net Cash Used in Investing Activities
Net cash used in investing activities for the year ended December 31, 2023 was $81.3 million consisting of $169.9 million in purchases of marketable securities available for sale, $20.0 million for the acquisition of the RPM-3D assets, and $11.5 million in purchases of property and equipment, partially offset by $120.0 million in sales and maturities of marketable securities available for sale. The purchases of marketable securities were the result of cash invested from our public offering of common stock during the first quarter of 2023. The purchases of property and equipment consist of $5.0 million in purchases of capitalized surgical instruments for our reusable instrument trays driven by higher numbers of employee sales representatives and sales growth and $6.5 million of purchases of fixed assets and leasehold improvements primarily for our new corporate headquarters building.
Net cash used in investing activities for the year ended December 31, 2022 was $76.5 million consisting of $63.4 million in purchases of marketable securities available for sale, partially offset by $1.7 million in sales and maturities of marketable securities available for sale, and $14.8 million in purchases of property and equipment. The purchases of property and equipment consist of $5.1 million in purchases of capitalized surgical instruments for our reusable instrument trays driven by higher numbers of employee sales representatives and sales growth and $8.0 million of purchases of fixed assets and leasehold improvements primarily for our new corporate headquarters building.
Net cash used in investing activities for the year ended December 31, 2021 was $2.7 million, consisting of purchases of capitalized surgical instruments for our reusable instrument trays.
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the year ended December 31, 2023 was $109.4 million, consisting of $107.5 million of net cash proceeds from our public offering of common stock in the first quarter of 2023 and $1.9 million from the exercise of stock options.
Net cash provided by financing activities for the year ended December 31, 2022 was $20.8 million, consisting of $53.5 million of net cash proceeds from the term loan agreement and revolving loan facility with MidCap and $2.2 million from the exercise of stock options, partially offset by the $33.9 million repayment of the CRG Group L.P. term loan and $1.0 million of third party debt issuance costs paid to related to the new MidCap borrowings.
Net cash provided by financing activities for the year ended December 31, 2021 was $107.7 million, consisting primarily of net proceeds from our IPO of $107.6 million and $1.8 million proceeds from exercise of stock options, partially offset by repayment of our Paycheck Protection Program loan of $1.8 million.
Surgeon Advisory Board Royalty Agreements
We recognized royalties expense of $6.9 million and $6.5 million for the years ended December 31, 2023 and 2022, respectively. For the years ended December 31, 2023 and 2022, the aggregate royalty rate was 3.7% and 4.6%, respectively. Each of the royalty agreements with our surgeon consultants prohibits the payment of royalties on products sold to entities and/or individuals with whom any of the surgeon advisors is affiliated.
Operating Lease
We have commitments for future payments related to our new corporate headquarters office located in Ponte Vedra, Florida. We entered into a 10-year lease in February 2022 for our new location which expires in July 2032. Lease payments comprise the base rent plus operating costs which includes taxes, insurance, and common area maintenance. We also have commitments for future payments related to our former headquarters which expire in April 2026. We have obtained subleases for this space. The remaining lease obligations are $25.7 million under these leases.
72
Critical Accounting Policies and Estimates
Management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenue, expenses and related disclosures. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
While our significant accounting policies are more fully described in Note 2, "Summary of Significant Accounting Policies," of the Notes to the Financial Statements included in this Annual Report, we believe the following discussion addresses our most critical accounting policies, which are those that are most important to our financial condition and our results of operations and require our most difficult, subjective and complex judgments.
Stock-Based Compensation
We account for stock-based compensation arrangements at fair value. We determine the fair value on the grant date for stock options using the Black-Scholes model and performance-based restricted stock unit ("PSU") awards using a Monte Carlo valuation model.
The fair value of these awards is recognized over the period during which an award holder is required to provide services in exchange for the award, known as the requisite service period, which is typically the vesting period using the straight-line method. Stock-based compensation expense is recorded net of estimated forfeitures in our Statements of Operations and are adjusted to actuals as they occur. The estimated forfeiture rate is based on historical analysis of actual forfeiture rates of similar awards.
The fair value of the stock options and PSUs are locked at grant date and will not fluctuate after grant date. However, the underlying inputs to the Black-Scholes option model and the Monte Carlo valuation model may change in the future for new grants which could impact the fair value of stock-based awards granted each year and could cause compensation expense to vary in future periods.
Goodwill and Other Intangible Assets
Our goodwill represents the excess of the cost over the fair value of net assets acquired. The determination of the value of goodwill and intangibles assets arising from acquisitions requires extensive use of accounting estimates and judgments to allocate the purchase price to the fair value of net tangible and intangible assets acquired. Goodwill is not amortized and is assessed for impairment using fair value measurement techniques on an annual basis or more frequently if facts and circumstances warrant such a review. Goodwill is considered to be impaired if we determine that the carrying value of our reporting unit exceeds its respective fair value.
We have a definite lived intangible technology asset that is reviewed for impairment upon triggering events that indicate the carrying value of the asset may not be recoverable. Recoverability is measured by a comparison of the carrying amount to future net undiscounted cash flows expected to be generated by the associated asset. If the asset's carrying value is determined to not be recoverable, the impairment to be recognized is measured by the amount by which the carry amount exceeds the fair market value of the intangible asset. Calculating cash flows for this measurement requires us to make significant estimates and assumptions related to forecasts of future revenues, expenses and discount rates. Changes in these assumptions could have a significant impact on the fair value of the technology intangible. If such assets are determined to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount exceeds the fair market value of the assets. The calculation of the fair value of the intangible assets involves Level 3 fair value measurements. Any impairment recognized could significantly impact our results of operations in the period of impairment.
Recently Issued Accounting Pronouncements
For information regarding recent accounting pronouncements and their expected impact on our future results of operations and financial condition, refer to Note 3, "Recent Accounting Pronouncements," of the Notes to the Financial Statements.
73
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We previously qualified as a "smaller reporting company," as defined in Rule 12b-2 under the Exchange Act, and have been permitted to rely, and have relied, on the reduced disclosure requirements available to smaller reporting companies, including not being required to provide the information required by this item pursuant to Item 305 of Regulation S-K. Our ability to rely on the reduced disclosure requirements available to smaller reporting companies will cease after the filing of this Annual Report and our definitive proxy statement related to our 2024 Annual Meeting of Stockholders (the "2024 Proxy Statement"), including those portions of the 2024 Proxy Statement that will be incorporated by reference into Part III of this Annual Report.
74
Item 8. Financial Statements and Supplementary Data
INDEX TO FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm (PCAOB ID Number |
76 |
77 |
|
78 |
|
79 |
|
80 |
|
81 |
75
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Treace Medical Concepts, Inc.
Opinion on the financial statements
We have audited the accompanying balance sheets of Treace Medical Concepts, Inc. (a Delaware corporation) (the "Company") as of December 31, 2023 and 2022, the related statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2023, and the related notes and financial statement schedules included under Item 15(a) (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) ("PCAOB"), the Company’s internal control over financial reporting as of December 31, 2023, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"), and our report dated February 27, 2024 expressed "an unqualified opinion".
Basis for opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical audit matters
Critical audit matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/
We have served as the Company's auditor since 2018.
February 27, 2024
76
TREACE MEDICAL CONCEPTS, INC.
Balance Sheets
(in thousands, except share and per share amounts)
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
Assets |
|
|
|
|
|
|
||
Current assets |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Marketable securities, short-term |
|
|
|
|
|
|
||
Accounts receivable, net of allowance for doubtful accounts of $ |
|
|
|
|
|
|
||
Inventories |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Intangible assets, net of accumulated amortization of $ |
|
|
|
|
|
|
||
Goodwill |
|
|
|
|
|
|
||
Operating lease right-of-use assets |
|
|
|
|
|
|
||
Other non-current assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
||
Current liabilities |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued liabilities |
|
|
|
|
|
|
||
Accrued commissions |
|
|
|
|
|
|
||
Accrued compensation |
|
|
|
|
|
|
||
Other liabilities |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Long-term debt, net of discount of $ |
|
|
|
|
|
|
||
Operating lease liabilities, net of current portion |
|
|
|
|
|
|
||
Other long-term liabilities |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
Commitments and contingencies (Note 8) |
|
|
|
|
|
|
||
Stockholders’ equity |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Accumulated other comprehensive (loss) income |
|
|
|
|
|
( |
) |
|
Treasury stock, at cost; |
|
|
( |
) |
|
|
|
|
Total stockholders’ equity |
|
|
|
|
|
|
||
Total liabilities and stockholders’ equity |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these financial statements.
77
P TREACE MEDICAL CONCEPTS, INC.
Statements of Operations and Comprehensive Loss
(in thousands, except share and per share amounts)
|
|
Year Ended December 31, |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Revenue |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Cost of goods sold |
|
|
|
|
|
|
|
|
|
|||
Gross profit |
|
|
|
|
|
|
|
|
|
|||
Operating expenses |
|
|
|
|
|
|
|
|
|
|||
Sales and marketing |
|
|
|
|
|
|
|
|
|
|||
Research and development |
|
|
|
|
|
|
|
|
|
|||
General and administrative |
|
|
|
|
|
|
|
|
|
|||
Total operating expenses |
|
|
|
|
|
|
|
|
|
|||
Loss from operations |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Interest income |
|
|
|
|
|
|
|
|
|
|||
Interest expense |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Debt extinguishment loss |
|
|
|
|
|
( |
) |
|
|
|
||
Other income, net |
|
|
|
|
|
( |
) |
|
|
|
||
Other non-operating income (expense), net |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Net loss |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Convertible preferred stock cumulative and undeclared dividends |
|
|
|
|
|
|
|
|
( |
) |
||
Net loss attributable to common stockholders |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Other comprehensive income (loss) |
|
|
|
|
|
|
|
|
|
|||
Unrealized gain (loss) on marketable securities |
|
|
|
|
|
( |
) |
|
|
|
||
Comprehensive loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Net loss per share attributable to common stockholders, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Weighted-average shares used in computing net loss per share attributable to common stockholders, basic and diluted |
|
|
|
|
|
|
|
|
|
|||
The accompanying notes are an integral part of these financial statements.
78
TREACE MEDICAL CONCEPTS, INC.
Statements of Stockholders' Equity
(in thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|||||||||
|
|
Convertible |
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
Other |
|
|
|
|
|
Total |
|
||||||||||||
|
|
Preferred Stock |
|
|
Common Stock |
|
|
Paid-In |
|
|
Accumulated |
|
|
Comprehensive |
|
|
Treasury |
|
|
Stockholders’ |
|
|||||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Loss |
|
|
Stock |
|
|
Equity |
|
|||||||||
Balances at January 1, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
||||||||
Issuance of common stock upon exercise of stock options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
||||||
Issuance of restricted stock awards |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Share-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Issuance of common stock from public offering, net of issuance costs and underwriting discount of $ |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of common stock upon net exercise of warrants |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
Conversion of convertible preferred stock and accrued dividends on convertible preferred stock into common stock |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
|
|
|
( |
) |
||
Balances at December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
||||||||
Issuance of common stock upon exercise of stock options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||||
Issuance of common stock for vesting of restricted stock units |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Share-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
Unrealized loss on available-for-sale marketable securities |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
Balances at December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|||||||
Issuance of common stock upon exercise of stock options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of common stock for vesting of restricted stock units |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Share-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Issuance of common stock from public offering, net of issuance costs and underwriting discount of $ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
Unrealized loss on available-for-sale marketable securities |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Shares directly withheld from employees for tax payment |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
||
Balances at December 31, 2023 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||||
The accompanying notes are an integral part of these financial statements.
79
TREACE MEDICAL CONCEPTS, INC.
Statements of Cash Flows
(in thousands)
|
|
Year Ended December 31, |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Cash flows from operating activities |
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating |
|
|
|
|
|
|
|
|
|
|||
Depreciation and amortization expense |
|
|
|
|
|
|
|
|
|
|||
Provision for allowance for doubtful accounts |
|
|
|
|
|
|
|
|
|
|||
Share-based compensation expense |
|
|
|
|
|
|
|
|
|
|||
Non-cash lease expense |
|
|
|
|
|
|
|
|
|
|||
Amortization of debt issuance costs |
|
|
|
|
|
|
|
|
|
|||
Gain on fair value adjustment to derivative liability |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Debt extinguishment loss |
|
|
|
|
|
|
|
|
|
|||
Loss on impairment of long-lived assets |
|
|
|
|
|
|
|
|
|
|||
Accretion (amortization) of discount (premium) on marketable securities, net |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Other, net |
|
|
|
|
|
|
|
|
|
|||
Net changes in operating assets and liabilities, net of acquisitions: |
|
|
|
|
|
|
|
|
|
|||
Accounts Receivable |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Inventory |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Prepaid expenses and other assets |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Other non-current assets |
|
|
|
|
|
( |
) |
|
|
|
||
Other liabilities |
|
|
( |
) |
|
|
|
|
|
|
||
Accounts payable |
|
|
|
|
|
|
|
|
|
|||
Accrued liabilities |
|
|
|
|
|
|
|
|
|
|||
Other, net |
|
|
|
|
|
|
|
|
|
|||
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Cash flows from investing activities |
|
|
|
|
|
|
|
|
|
|||
Purchases of available-for-sale marketable securities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Sales and maturities of available-for-sale marketable securities |
|
|
|
|
|
|
|
|
|
|||
Purchases of property and equipment |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Acquisition, net of cash acquired |
|
|
( |
) |
|
|
|
|
|
|
||
Net cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Cash flows from financing activities |
|
|
|
|
|
|
|
|
|
|||
Proceeds from interest bearing term debt |
|
|
|
|
|
|
|
|
|
|||
Proceeds from interest bearing revolving debt |
|
|
|
|
|
|
|
|
|
|||
Debt issuance costs |
|
|
|
|
|
( |
) |
|
|
|
||
Payments on interest bearing debt |
|
|
|
|
|
( |
) |
|
|
|
||
Payments on PPP Loan |
|
|
|
|
|
|
|
|
( |
) |
||
Proceeds from issuance of common stock from public offering, net of issuance costs and underwriting discount of $ |
|
|
|
|
|
|
|
|
|
|||
Proceeds from exercise of employee stock options |
|
|
|
|
|
|
|
|
|
|||
Taxes from withheld shares |
|
|
( |
) |
|
|
|
|
|
|
||
Net cash provided by financing activities |
|
|
|
|
|
|
|
|
|
|||
Net decrease in cash and cash equivalents |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Cash and cash equivalents at beginning of period |
|
|
|
|
|
|
|
|
|
|||
Cash and cash equivalents at end of period |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|||
Cash paid for interest |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Operating lease right-of-use assets obtained in exchange for new lease liabilities |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Operating lease right-of-use asset and lease liability adjustment due to lease incentive |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
||
Noncash investing activities: |
|
|
|
|
|
|
|
|
|
|||
Unrealized (gains) losses on marketable securities |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
||
Unsettled matured marketable security and receivable from broker |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Noncash portion of internally developed software |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
||
Noncash financing activities: |
|
|
|
|
|
|
|
|
|
|||
Issuance of common stock upon exercise of warrants |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Conversion of convertible preferred stock and accrued dividends on convertible preferred stock into common stock |
|
$ |
|
|
$ |
|
|
$ |
|
|||
The accompanying notes are an integral part of these financial statements.
80
1. Formation and Business of the Company
The Company
Treace Medical Concepts, LLC was formed on
Initial Public Offering and Follow-on Offering
On April 27, 2021, the Company completed its initial public offering ("IPO"). The Company received net proceeds of approximately $
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying financial statements are prepared in accordance with U.S. generally accepted accounting principles ("GAAP").
The financial statements presented have been prepared on a consistent basis with the exception of the following item. An adjustment has been made to the Statement of Operations and Comprehensive Loss for the years ended 2022 and 2021 for $
The Company evaluated events or transactions that may have occurred after the balance sheet date for potential recognition or disclosure through the date the financial statements were issued. No subsequent events or transactions requiring recognition or disclosure were identified.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Although these estimates are based on the Company’s knowledge of current events and actions it may undertake in the future, actual results may ultimately materially differ from these estimates and assumptions.
Significant estimates and assumptions include write-downs related to accounts receivable, reserves and inventories, valuation of intangible assets and goodwill, the recoverability of long-term assets, deferred tax assets and related valuation allowances, contingencies, earn-out liabilities, and stock-based compensation.
Business Combinations
The Company allocates the purchase consideration to the identifiable assets and liabilities acquired, including intangible assets at fair value on the date of the acquisition. The excess of the fair value of the purchase consideration over the fair value of the identifiable assets and liabilities, if any, is recorded as goodwill. During the measurement period, which is up to one year from the acquisition date, the Company may adjust initial amounts that were recognized at the acquisition date to reflect new information obtained about facts and circumstances that existed as of the acquisition date.
81
Determining the fair value of assets acquired and liabilities assumed requires significant judgment, including the selection of valuation methodologies that may include the income approach, the cost approach, or the market approach. Significant assumptions used in those methodologies include the timing and amounts of cash flow projections, including revenue growth rates, obsolescence rates, margins, royalty rates, counterparty risk rates, and other discount rates.
Intangibles
Definite-life intangible assets are assessed for impairment upon triggering events that indicate that the carrying value of an asset may not be recoverable. Recoverability is measured by a comparison of the carrying amount to future net undiscounted cash flows expected to be generated by the associated asset. If the asset's carrying value is determined to not be recoverable, the impairment to be recognized is measured by the amount by which the carrying amount exceeds the fair market value of the intangible assets.
Goodwill
Goodwill represents the excess of the purchase price as compared to the fair value of net assets acquired and liabilities assumed. Goodwill is not amortized but is tested for impairment annually or when indications of impairment exist. The Company can elect to qualitatively assess goodwill for impairment if it is more likely than not that the fair value of a reporting unit exceeds its carrying value.
Impairment exists when the carrying amount, including goodwill, of the reporting unit exceeds its fair value, resulting in an impairment charge for this excess (not to exceed the carrying amount of the goodwill). The Company's annual impairment testing date is July 1. The impairment, if determined, is recorded within operating expenses in the Statements of Operations and Comprehensive Loss in the period the determination is made. There were no impairments recorded during the periods presented.
Contingent Consideration
Business combinations may include contingent consideration as part of the purchase price under which the Company will make future payments to the seller upon the achievement of certain milestones. The fair value of the contingent consideration is estimated as of the acquisition date at the present value of the expected contingent payments and is subsequently remeasured at each balance sheet date. The scenario-based model was used and relies on multiple outcomes to estimate the likelihood of future payout of the contingent consideration. The resulting earnout payout is then probability-weighted and discounted at an appropriate risk adjusted rate in order to arrive at the present value of the expected payment.
The Company reviews the probabilities of achievement of the earnout milestones to determine the impact on the fair value of the contingent consideration on a quarterly basis over the earn-out period. Actual results are compared to the estimates and probabilities of achievement used in its forecasts. The estimated fair value of the contingent consideration liability will increase or decrease, up to the contractual limit, as applicable. Changes in the estimated fair value of the contingent consideration are recorded in operating expenses in the Statement of Operations and Comprehensive Loss and are reflected in the period in which they are identified. Changes in the estimated fair value of the Company's contingent consideration may materially impact or cause volatility in its operating results.
Segments
The Company operates and manages its business as
82
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original or remaining maturity at the time of purchase of 90 days or less to be cash equivalents. All of the Company's cash equivalents have liquid markets and high credit ratings. The Company maintains its cash in money market funds and bank deposits, the balances of which at times may exceed federally insured limits.
Marketable Securities
The Company considers its debt securities and Yankee certificate of deposits ("Yankee CDs") to be available-for-sale securities. Available-for-sale securities are classified as cash equivalents or short-term marketable securities.
Marketable securities classified as available-for-sale are measured at fair value with temporary unrealized gains and losses reported in other comprehensive loss, and as a component of stockholders' equity (deficit) until their disposition or maturity. The Company reviews all available-for-sale securities at each period end to determine if they remain available-for-sale based on the Company's current intent and ability to sell the security if it is required to do so. Realized gains and losses from the sale of marketable securities, if any, are calculated using the specific-identification method. Premiums and discounts are amortized and accreted, respectively, using the effective interest method. Refer to Note 4, "Fair Value Measurements," for fair value disclosures for cash equivalents and short-term marketable securities.
Available-for-sale securities are subject to a periodic impairment review. The Company may recognize an impairment charge when a decline in the fair value of investments below the cost basis is determined to be other-than-temporary. In determining whether a decline in market value is other-than-temporary, various factors are considered, including the cause, duration of time and severity of the impairment, any adverse changes in the investees' financial condition and the Company's intent and ability to hold the security for a period of time sufficient to allow for an anticipated recovery in market value. Declines in fair value judged to be other-than-temporary are included in the Company's Statements of Operations and Comprehensive Loss. The Company did not record any other-than-temporary impairments related to marketable securities in the Company's Statements of Operations and Comprehensive Loss for the years ended December 31, 2023 or 2022.
Accounts Receivable and Allowances
Accounts receivable are generally from hospitals and ambulatory surgery centers and are stated at amounts billed less allowances for doubtful accounts. The Company continually monitors customer payments and maintains an allowance for losses resulting from a customer's inability to make required payments. The Company considers factors such as historical experience, credit quality, age of the accounts receivable balances, geographic related risks and economic conditions that may affect a customer's ability to pay. Accounts receivable are written off when individual balances are no longer collectible. As of December 31, 2023 and 2022, accounts receivable are presented net of an allowance for doubtful accounts of $
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of risk consist principally of cash, cash equivalents, marketable securities, and accounts receivable. The Company maintains its cash with established financial institutions and, at times, such balances with any one financial institution may be in excess of the Federal Deposit Insurance Corporation ("FDIC") insured limits. The Company's available-for-sale securities portfolio primarily consists of U.S. treasury and agency securities, money market funds, commercial paper, Yankee CDs, high credit quality asset-backed securities and corporate debt securities. The Company's investment policy requires its available for sale securities to meet certain criteria including investment type, credit ratings, and a maximum portfolio duration of one year.
The Company earns revenue from the sale of its products to customers such as hospitals and ambulatory surgery centers. The Company's accounts receivable is derived from revenue earned from customers. The Company performs ongoing credit evaluations of its customers' financial condition and generally requires no collateral from its customers. At December 31, 2023 and 2022, no customer accounted for more than
83
Leases
The Company determines whether an arrangement is or contains a lease at the inception of the arrangement and whether such a lease is classified as a financing lease or an operating lease at the commencement date of the lease. Lease liabilities and their corresponding right-of-use assets are recorded based on the present value of lease payments over the expected lease term. The Company determines the commencement date of a lease to be the date on which a lessor makes an underlying asset available for use by the Company. Leases with a term greater than one year are recognized on the Balance Sheets as operating lease right-of-use assets, Operating lease liabilities and Operating lease liabilities, net of current portion. The Company elected not to recognize right-of-use assets and lease liabilities for leases with terms of 12 months or less (short-term leases). As the interest rates implicit in our lease contracts are not readily determinable, the Company utilizes its incremental borrowing rate based on the information available at the commencement date to determine the present value of lease payments. Certain adjustments to the right-of-use asset may be required for items such as initial direct costs paid, incentives received, or impairment charges if the Company determines the right-of-use asset is impaired. In the fourth quarter of 2022, the Company recorded an impairment to the right-of-use asset associated with the lease on its previous corporate headquarters. See Note 6, "Balance Sheet Components," for further details of the impairment.
The Company considers the lease term to be the noncancelable period that the Company has the right to use the underlying asset, together with any periods where it is reasonably certain the Company will exercise an option to extend (or not terminate) the lease.
Rent expense for operating leases is recognized on a straight-line basis over the lease term and is presented in operating expenses on the Statements of Operations and Comprehensive Loss. The Company has elected to not separate lease and non-lease components for its real estate leases and instead accounts for each separate lease component and the non-lease components associated with that lease component as a single lease component. Variable lease payments are recognized as lease expense as incurred and are recorded in operating expenses on the Statements of Operations and Comprehensive Loss.
The Company has no finance leases.
Inventories
Inventories consist primarily of surgical kits and components as finished goods and are stated at the lower of cost or net realizable value. Cost is determined based on an average cost method which approximates the first-in, first-out basis and includes primarily outsourced manufacturing costs and direct manufacturing overhead costs. The Company reviews inventory for excess, obsolescence, and field losses and writes down inventory, as necessary. For the years ended December 31, 2023, 2022, and 2021, the Company recorded a provision to cost of goods sold of $
Property and Equipment, Net
Property and equipment are recorded at cost. Depreciation of property and equipment is recorded using the straight-line method over the following estimated useful lives as follows:
|
|
Years |
Furniture, fixtures and equipment |
|
|
Machinery and equipment |
|
|
Capitalized surgical instruments |
|
|
Computer equipment |
|
|
Leasehold improvements |
|
|
Software |
|
Long-lived assets are evaluated whenever a change in circumstances indicates that the carrying amount of an asset may not be recoverable. If assets are considered to be impaired, a charge is recorded for an amount that the carrying value exceeds the fair value. The Company capitalized $
84
Revenue Recognition
The Company generates revenue from the sale of its proprietary Lapiplasty System and its minimally invasive variations, Adductoplasty System, Hammertoe PEEK Fixation System, SpeedPlate Implant Fixation Platform, single use osteotomes and release instruments, and other implanted ancillary products. The Company receives payment for all these products that are consumed during the surgery and does not receive separate consideration for the use of the instrument tray furnished by the Company for the surgeon's use. The Company identifies the instrument trays as a lease component and the implants and other single use products as a non-lease component in its arrangements with its customers. The Company concluded that the non-lease component is predominant, and as such, elected the practical expedient to not separate the lease and non-lease components. Therefore, the overall arrangement is accounted for under ASC 606.
Implant kits, single use instruments, and other ancillary products are sold in the United States through a combination of a direct employee sales force and independent sales agencies. The Company invoices hospitals and ambulatory surgery centers for the implant kits and other products and pays commissions to the sales representatives and independent sales agencies. The Company has
Under ASC 606, revenue is recognized when the customer obtains control of promised goods or services, in an amount that reflects consideration which the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that an entity determines are within the scope of ASC 606, the Company performs the following five steps as prescribed by ASC 606:
A contract with a customer exists when (i) the Company enters into a legally enforceable contract with a customer that defines each party's rights regarding the products to be transferred and identifies the payment terms related to these products, (ii) the contract has commercial substance, and (iii) the Company determines that collection of substantially all consideration for its products that are transferred is probable based on the customer's intent and ability to pay the promised consideration. The Company considers signed agreements and purchase orders as a customer's contract. Sales prices are specified in either the customer contract or agreed price list, which is executed prior to the transfer of control to the customer.
The Company identifies performance obligations based on the terms of the contract and customary business practices, which include products that are distinct, or a series of distinct goods that are substantially the same and that have the same pattern of transfer to the customer. The transfer of the Company's products to the customer are distinct performance obligations. The Company does not have any material performance obligations other than the transfer of the products to the customer.
The transaction price in the Company's customer contracts includes fixed consideration to be contractually billed to the customer while variable consideration includes the right of return. The Company does not allocate the transaction price or any variable consideration to the right of return. The Company did
For
Revenue for products is recognized when a customer obtains control of the promised products, which is generally when the customer has the ability to (i) direct its use and (ii) obtain substantially all of the remaining benefits from it. Revenue recognition occurs for the majority of our sales when control of the product transfers to the customer, which is generally at the time the product is used in surgery. The Company also generates a small percentage of total revenues from the sale of
85
products when the products are ordered in advance of a surgery procedure. The performance obligation is the delivery of the products and therefore, revenue is recognized either at shipment or upon receipt by the customer depending upon the contractual terms of the purchase and when the risk of ownership transfers to the customer.
Contract Costs
The Company applies the practical expedient to recognize the incremental costs of obtaining a contract as expense when
Cost of Goods Sold
Cost of goods sold consists primarily of costs for the purchase of the Company's products from third-party manufacturers. Direct costs from the Company's third-party manufacturers include costs for raw materials plus the markup for the assembly of the components. Cost of goods sold also includes royalties, allocated overhead for indirect labor, certain direct costs such as those incurred for shipping our products and personnel costs. The Company expenses all inventory provisions for excess and obsolete inventories as cost of goods sold. The Company records adjustments to its inventory valuation for estimated excess, obsolete, field loss rate, and non-sellable inventories based on assumptions about future demand, past usage, changes to manufacturing processes and overall market conditions.
Research and Development Expenses
Research and development ("R&D") expenses consist primarily of engineering, product development, clinical studies to develop and support the Company's products, regulatory expenses, and other costs associated with products and technologies that are in development. These expenses include compensation for personnel, including salaries, bonuses, benefits and stock-based compensation, supplies, consulting, prototyping, testing, materials, travel expenses, depreciation and an allocation of facility overhead expenses.
Shipping and Handling
The Company has elected to account for shipping and handling activities as fulfillment activities. As such, the Company does not evaluate shipping and handling as promised services to its customers. The Company may bill customers for shipping and handling costs. Amounts billed for shipping and handling are included in revenue. Shipping and handling costs incurred by the Company are included in sales and marketing expense. Shipping and handling costs totaled $
Advertising Costs
Advertising costs are expensed as incurred and are included as a component of marketing and sales expenses. Advertising expense includes the cost of advertising across the various mediums we employ, including print, digital, radio and television. Advertising costs totaled approximately $
Income Taxes
The Company accounts for income taxes under the liability method, whereby deferred tax assets and liabilities are determined based on the difference between the financial statements and tax bases of assets and liabilities using the enacted tax rates in effect for the year in which the differences are expected to affect taxable income. A valuation allowance is established when necessary to reduce deferred tax assets when management estimates, based on available objective evidence, that it is more likely than not that the benefit will not be realized for the deferred tax assets.
The Company also follows the provisions of ASC 740-10, Accounting for Uncertainty in Income Taxes. ASC 740-10, which prescribes a comprehensive model for the recognition, measurement, presentation and disclosure in financial statements of any uncertain tax positions that have been taken or expected to be taken on a tax return.
86
Product Liability
The Company believes it carries adequate insurance for possible product liability claims. Accruals for product liability claims and legal defense costs in excess of insured amounts are recorded if it is probable that a liability has been incurred and the amount of any liability can be reasonably estimated.
Debt
Debt issuance costs and discount are amortized to interest expense using the effective interest method.
Stock-Based Compensation
The Company accounts for stock-based compensation arrangements with employees in accordance with ASC 718, Compensation-Stock Compensation, using a fair-value based method. The Company determines the fair value of stock options and performance-based restricted stock awards units ("PSUs") on the date of grant. The fair value for restricted stock and restricted stock units awards is the fair value of the stock at the grant date.
The fair value of time-based awards is recognized over the period during which an award holder is required to provide services in exchange for the award, known as the requisite service period, which is typically the vesting period using the straight-line method. The Company accrues for estimated forfeitures on share-based awards and adjusts stock-based compensation cost to actual as forfeitures occur. The estimated forfeitures are based on a historical analysis of actual forfeitures of awards.
The Company estimates the fair value of the stock-based awards using the Black-Scholes option-pricing model, which requires the input of highly subjective assumptions. The assumptions are as follows:
The Company continues to use judgment in evaluating the expected volatility and expected terms utilized for the fair value of stock options on a prospective basis. The Company used the simplified method to determine the expected term for options due to the lack employee exercise history since our IPO. As the Company continues to accumulate additional data, the Company may make refinements to the assumptions, which could materially impact the future stock-based compensation expense.
The Company estimates the fair value of PSUs awards using the Monte Carlo model, which requires the input of several assumptions. The assumptions are as follows:
87
Comprehensive Loss
The Company is required to report all components of comprehensive loss, including net loss, in the financial statements in the period in which they are recognized. Comprehensive loss is defined as a change in equity of a business enterprise during a period, resulting from transactions and other events and circumstances from non-owner sources. Comprehensive loss equaled $
Net Loss Per Share Attributable to Common Stockholders
Basic net loss per share attributable to common stockholders is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration for potentially dilutive securities. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common shares and potentially dilutive securities outstanding for the period. For purposes of the diluted net loss per share calculation, common stock options, unvested restricted stock units and restricted stock awards are considered to be potentially dilutive securities. Because the Company has reported a net loss for the years ended December 31, 2023, 2022, and 2021, diluted net losses per common share were the same as basic net losses per common share for these periods.
3. Recent Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842) ("ASC 842"). The update establishes principles for recognition, measurement, presentation, and disclosure intended to increase transparency and comparability for the accounting of lease transactions. The standard required lessees to recognize leases with terms greater than
In June 2016, the FASB issued ASU 2016-13, Financial Instruments – Credit Losses. This guidance required financial instruments to be measured at amortized cost, and trade accounts receivable to be presented at the net amount expected to be collected. The new model requires an entity to estimate credit losses based on historical information, current information, and reasonable and supportable forecasts, including estimates of prepayments. In November 2019, the FASB issued ASU 2019-10, that was effective for fiscal years beginning after December 15, 2022, and interim periods within that fiscal year. The Company adopted the standard on January 1, 2023. The adoption of this guidance did not have a material impact on the Company's financial statements.
Recent Accounting Pronouncements Not Yet Adopted
In November 2023, the FASB issued ASU 2023-07, Segment Reporting (Topic 280) ("ASC 280"). The update requires all public business entities to identify their reportable segments, including the basis of organization, types of products and services from which each reportable segment derives its revenues, and the title and position of the individual or the name of the group or committee identified as the chief operating decision maker ("CODM") and an explanation of how the CODM uses the reported measure(s) of segment profit or loss in assessing segment performance and deciding how to allocate
88
resources. Public entities shall disclose on an annual and interim basis for each reportable segment including entities that only have one reportable segment, certain significant expense categories and amounts that are regularly provided to the CODM and included in reported segment profit or loss. ASC 280 is applied retrospectively to all prior periods presented in the financial statements. This new guidance is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024. Early adoption is permitted. The Company is currently evaluating the impact of the new standard on its financial statements and related disclosures.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740) ("ASC 740"). The update requires all public business entities on an annual basis to (1) disclose specific categories in the rate reconciliation and (2) provide additional information for reconciling items that meet a quantitative threshold and an explanation, if not otherwise evident, of the individual reconciling items disclosed, such as the nature, effect, and underlying causes of the reconciling items and the judgment used in categorizing the reconciling items. In addition, the update requires certain new disclosures of the amount of income taxes paid (net of refunds received) disaggregated by federal, state, and foreign taxes and the amount of income taxes paid (net of refunds received) disaggregated by individual jurisdictions in which income taxes paid is equal to or greater than five percent of total income taxes paid (net of refunds received). Other new disclosures required include income (or loss) from continuing operations before income tax expense (or benefit) disaggregated between domestic and foreign and income tax expense (or benefit) from continuing operations disaggregated by federal, state, and foreign. The new guidance is effective for annual periods beginning after December 15, 2024. Early adoption is permitted. The amendments are to be applied on a prospective basis, with retrospective application permitted. The Company is currently evaluating the impact of the new standard on its financial statements and related disclosures.
4. Fair Value Measurements
Assets and liabilities recorded at fair value in the financial statements are categorized based upon the level of judgment associated with the inputs used to measure their fair value. Hierarchical levels which are directly related to the amount of subjectivity associated with the inputs to the valuation of these assets or liabilities are as follows:
Level 1—Inputs are unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access as of the measurement date.
Level 2—Inputs are observable, unadjusted quoted prices in active markets for similar assets or liabilities, unadjusted quoted prices for identical or similar assets or liabilities in markets that are not active or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the related assets or liabilities.
Level 3—Unobservable inputs for the asset or liability only used when there is little, if any, market activity for the asset or liability at the measurement date. This hierarchy requires the Company to use observable market data, when available, and to minimize the use of unobservable inputs when determining fair value.
Assets and Liabilities Measured and Recorded at Fair Value on a Recurring Basis – The following assets and liabilities are measured at fair value on a recurring basis as of December 31, 2023 and December 31, 2022 (in thousands):
|
|
December 31, 2023 |
|
|||||||||||||
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Cash equivalents |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Money market funds |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Short-term marketable securities at fair value |
|
|
|
|
|
|
|
|
|
|
|
|
||||
U.S. treasury and government agencies |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Commercial paper |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Corporate debt |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Asset-backed securities |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Yankee CD |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Contingent consideration |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
Total liabilities |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
89
|
|
December 31, 2022 |
|
|||||||||||||
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Cash and cash equivalents |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Money market funds |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Commercial paper |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Corporate debt |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Yankee CD |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Short-term marketable securities at fair value |
|
|
|
|
|
|
|
|
|
|
|
|
||||
U.S. treasury and government agencies |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
Corporate debt |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Asset-backed securities |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Yankee CD |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
The carrying amounts of the Company's money market funds classified as cash and cash equivalents, accounts receivable, accounts payable, and accrued liabilities, approximate their fair value due to the short-term nature of these assets and liabilities. Based on the borrowing rates currently available to the Company for debt with similar terms and consideration of default and credit risk, the carrying value of the term loan approximates their fair value.
The Company's available-for-sale securities portfolio consists of investments in U.S. treasury and government agency securities, commercial paper, corporate debt securities, asset-backed securities, and Yankee CDs. Yankee CDs are certificates of deposit issued in the United States by a branch of a foreign bank and are denominated in U.S. dollars. The fair value of Level 1 securities is determined on trade prices in active markets for identical assets. The fair value of Level 2 securities is determined using valuation models using inputs that are observable either directly or indirectly, such as quoted prices for similar assets, interest rates, yield curves, credit spreads, default rates, loss severity, broker and dealer quotes, as well as other relevant economic measures. The Level 3 contingent consideration was recorded at fair value on the date of the acquisition and thereafter based on the consideration expected to be transferred on the projected payment date estimated as the probability weighted future cash flows, discounted back to the present value. This calculation uses unobservable inputs that reflect the Company's own assumptions as to the ability of the acquired business to meet the targeted benchmarks and the discount rate used in the determination of fair value.
The following table sets forth a summary of the changes in the fair value of the Company's Level 3 financial instruments (in thousands):
Fair value as of December 31, 2022 |
|
$ |
|
|
Contingent consideration from acquisition |
|
|
|
|
Change in fair value |
|
|
|
|
Fair value as of December 31, 2023 |
|
$ |
|
Contingent consideration is included in other liabilities on the Balance Sheets. As of December 31, 2023, the entire balance is classified as current due to the timing of the expected payment. The change in fair value for the contingent consideration related to the technological advancements milestone payment is classified as research and development expense within the Statements of Operations and Comprehensive Loss. We made
There were no assets or liabilities measured at fair value on a nonrecurring basis as of December 31, 2023 and December 31, 2022.
5. Business Combination
On June 12, 2023 (the "closing date"), the Company acquired certain assets of MIOS Marketing, LLC d/b/a RedPoint Medical3D ("RPM-3D"), a medical technology company offering pre-operative planning and patient-specific guides designed to deliver accurate surgical correction of deformities tailored to the patient's unique foot anatomy. RPM-3D's 22 patent applications further expand and reinforce the Company's global intellectual property portfolio covering technologies for the correction of bunion and related deformities.
90
The Company paid $
The following table summarizes the components of the acquisition-date fair value of consideration transferred (in thousands):
Cash |
$ |
|
|
Contingent consideration |
|
|
|
Total |
$ |
|
The following table summarizes the preliminary components of the acquisition-date fair value of assets acquired (in thousands):
Prepaid expenses and other current assets |
$ |
|
|
Inventory |
|
|
|
Property, plant and equipment |
|
|
|
Intangible assets |
|
|
|
Total identifiable assets acquired |
|
|
|
Goodwill |
|
|
|
Total assets acquired |
$ |
|
Identified intangible assets consist of developed technology. The fair value was determined with the assistance of an external valuation specialist using an income approach, in accordance with ASC Topic 805 - Business Combinations. The developed technology is a finite-lived intangible asset with a useful life of ten years that is amortized on straight-line basis. There were no other material intangibles from the RPM-3D acquisition.
The purchase consideration was allocated to the identifiable net assets acquired based on estimated fair values at the date of the acquisition. The excess of the fair value of the purchase consideration over the fair value of the identifiable assets and liabilities was recorded as goodwill. The goodwill is attributable to the expected synergies with the Company's existing operations. The purchase price allocated to goodwill will be deductible for income tax purposes over a 15-year period.
The intangible assets balance as of December 31, 2023 consisted of the following (in thousands):
|
Gross Carrying Amount |
|
Accumulated Amortization |
|
Net Carrying Amount |
|
|||
Developed technology |
$ |
|
$ |
|
$ |
|
|||
The intangible amortization for the twelve months ended December 31, 2023 and 2022 was $
Estimated intangible amortization expense as of December 31, 2023 for the next five years is as follows (in thousands):
2024 |
$ |
|
|
2025 |
|
|
|
2026 |
|
|
|
2027 |
|
|
|
2028 |
|
|
91
The goodwill balance as of December 31, 2023 is as follows (in thousands):
Balance as of December 31, 2022 |
$ |
|
|
Acquisitions |
|
|
|
Balance as of December 31, 2023 |
$ |
|
There is no supplemental proforma presentation of operating results of the acquisition of the RPM-3D assets due to the immaterial impact on the Company's operations for the twelve months ended December 31, 2023 and 2022.
6. Balance Sheet Components
Cash and Cash Equivalents
The Company's cash and cash equivalents consisted of the following (in thousands):
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
Cash |
|
$ |
|
|
$ |
|
||
Cash equivalents: |
|
|
|
|
|
|
||
Money market funds |
|
|
|
|
|
|
||
Commercial paper |
|
|
|
|
|
|
||
Corporate debt |
|
|
|
|
|
|
||
Yankee CD |
|
|
|
|
|
|
||
Total cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Included in cash as of December 31, 2022 is $
Marketable Securities
The Company's available-for-sale marketable securities consisted of the following (in thousands):
|
|
December 31, 2023 |
|
|||||||||||||
|
|
Amortized Cost |
|
|
Gross Unrealized Gains |
|
|
Gross Unrealized Losses |
|
|
Fair Value |
|
||||
Marketable securities - short-term |
|
|
|
|
|
|
|
|
|
|
|
|
||||
U.S. treasury and government agencies |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Commercial paper |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Corporate debt |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Asset-backed securities |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Yankee CD |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Total marketable securities - short-term |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
December 31, 2022 |
|
|||||||||||||
|
|
Amortized Cost |
|
|
Gross Unrealized Gains |
|
|
Gross Unrealized Losses |
|
|
Fair Value |
|
||||
Marketable securities - short-term |
|
|
|
|
|
|
|
|
|
|
|
|
||||
U.S. treasury and government agencies |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Corporate debt |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Asset-backed securities |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Yankee CD |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
Total marketable securities - short-term |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
92
As of December 31, 2023, there were no available-for-sale securities with unrealized losses greater than 12 months. There was not an allowance for credit losses required as of December 31, 2023 and 2022.
As of December 31, 2023, the Company had no plans to sell securities with unrealized losses, and believes it is more likely than not that it would not be required to sell such securities before recovery of their amortized cost. As of December 31, 2023 and 2022, there were no material gains or losses from sales of available-for-sale securities.
As of December 31, 2023 and 2022, accrued interest of $
As of December 31, 2023 all marketable securities mature within two years, except for asset-backed securities. Asset-backed securities are not due at a single maturity date. As such, these securities were not included.
Property and Equipment, Net
The Company's property and equipment, net consisted of the following (in thousands):
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
Furniture and fixtures, and equipment |
|
$ |
|
|
$ |
|
||
Construction in progress |
|
|
|
|
|
|
||
Machinery and equipment |
|
|
|
|
|
|
||
Capitalized surgical equipment |
|
|
|
|
|
|
||
Computer equipment |
|
|
|
|
|
|
||
Leasehold improvements |
|
|
|
|
|
|
||
Software |
|
|
|
|
|
|
||
Total property and equipment |
|
|
|
|
|
|
||
Less: accumulated depreciation and amortization |
|
|
( |
) |
|
|
( |
) |
Property and equipment, net |
|
$ |
|
|
$ |
|
||
Depreciation and amortization expense for property and equipment was $
In the fourth quarter of 2022, the Company recognized a $
Accrued Liabilities
Accrued liabilities consist of the following (in thousands):
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
Accrued royalties expense |
|
$ |
|
|
$ |
|
||
Accrued interest |
|
|
|
|
|
|
||
Accrued professional services |
|
|
|
|
|
|
||
Accrued compensation expense for RPM-3D earn-out |
|
|
|
|
|
|
||
Other accrued expense |
|
|
|
|
|
|
||
Total accrued liabilities |
|
$ |
|
|
$ |
|
||
93
Other liabilities
Other liabilities consist of the following (in thousands):
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
|
$ |
|
|
$ |
|
|||
Contingent consideration |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Total other liabilities |
|
$ |
|
|
$ |
|
||
7. Long Term Debt
The Company's debt consisted of the following (in thousands):
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
Revolving line of credit |
|
|
|
|
|
|
||
MidCap revolving loan facility |
|
$ |
|
|
$ |
|
||
Term loans |
|
|
|
|
|
|
||
MidCap term loan facility |
|
|
|
|
|
|
||
Total term and revolving loans |
|
|
|
|
|
|
||
Less: debt discount and issuance costs |
|
|
( |
) |
|
|
( |
) |
Total long-term debt, net |
|
$ |
|
|
$ |
|
||
As of December 31, 2023, future payments of long-term debt were as follows (in thousands):
Fiscal Year |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
Total principal payments |
|
|
|
|
Less: Unamortized debt discount and debt issuance costs |
|
|
( |
) |
Total long-term debt, net |
|
$ |
|
|
MidCap Loan and Revolving Loan Facility
On April 29, 2022, the Company entered into a
The term loan facility provides for a 60-month term loan up to $
The revolving loan facility provides up to $
94
The loans are secured by all of the Company's assets, including intellectual property. The loan agreements and other ancillary loan documents contain customary representations and warranties and affirmative and negative covenants. Under the loan agreements, the Company is not required to meet any minimum level of revenue if liquidity (defined as unrestricted cash plus undrawn availability under the revolving loan agreement) is greater than the outstanding balance under the term loan. If liquidity falls below such outstanding balance, then the Company is subject to a minimum trailing twelve-month revenue covenant. The Company is not subject to this covenant at December 31, 2023.
During the years ended December 31, 2023, 2022, and 2021, the Company recognized $
8. Commitments and Contingencies
License and Royalty Commitments
The Company has entered into product development and fee for service agreements with members of its Surgeon Advisory Board and other surgeon consultants that specify the terms under which the consultant is compensated for his or her consulting services and grants the Company rights to the intellectual property created by the consultant in the course of such services. As products are commercialized with the assistance of members of the Surgeon Advisory Board and other surgeon consultants, the Company may agree to enter into a royalty agreement if the consultant's contributions to the product are novel, significant and innovative.
As of December 31, 2023 and 2022, the Company has royalty agreements with certain members of its Surgeon Advisory Board and other surgeon consultants providing for royalties based on each individual's level of contribution. Each royalty agreement: (i) confirms the irrevocable transfer to the Company of all pertinent intellectual property rights; (ii) sets the applicable royalty rate; (iii) sets the period of time during which royalties are payable; (iv) is for a term of
As of December 31, 2023 and 2022, the Company's royalty agreements provide for (i) royalty payments for 10 years from first commercial sale of the relevant product and (ii) a royalty rate for each such agreement ranging from
The Company recognized royalties expense of $
95
Contingencies
From time to time, the Company may be a party to various litigation claims in the normal course of business. Legal fees and other costs associated with such actions are expensed as incurred. The Company assesses, in conjunction with legal counsel, the need to record a liability for litigation and contingencies. Accrual estimates are recorded when and if it is determinable that such a liability for litigation and contingencies are both probable and reasonably estimable. There were
9. Operating Leases
The Company's leases consist of real estate leases in Ponte Vedra, Florida.
On February 9, 2022, the Company entered into a
The Company's leases contain options to renew, none of which the Company is reasonably certain to exercise. The lease agreements do not contain any residual value guarantees or restrictive covenants. For the new headquarters lease, the Company is provided a tenant improvement allowance for the construction of leasehold improvements, of which $
In addition to base rent, the Company pays variable costs related to its share of operating expenses under certain of its lease arrangements. These variable costs are recorded as lease expense as incurred and presented as operating expenses in the Statements of Operations and Comprehensive Loss. Variable lease costs were $
Rent expense was $
Operating lease cost was $
Additional information related to operating leases is as follows:
|
|
December 31, 2023 |
|
|
Weighted average remaining lease term (years) |
|
|
|
|
Weighted average discount rate |
|
|
% |
|
The following table summarizes a maturity analysis of operating lease liabilities showing the aggregate lease payments as of December 31, 2023 (in thousands):
Fiscal Year |
|
|
|
|
2024 |
|
$ |
|
|
2025* |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
2028 |
|
|
|
|
Thereafter |
|
|
|
|
Total undiscounted lease payments |
|
|
|
|
Less: imputed interest |
|
|
( |
) |
Total discounted lease payments |
|
|
|
|
|
|
( |
) |
|
Noncurrent portion of lease liability |
|
$ |
|
|
*
In 2023, the Company entered into subleases for a portion of its former headquarter space as a lessor. The lease is classified as an operating lease. The Company recorded rental income of $
96
10. Income Taxes
The Company has
Reconciliation of the statutory federal income tax to the Company's effective tax is as follows:
|
|
December 31, |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Income tax at the statutory rate |
|
|
( |
)% |
|
|
( |
)% |
|
|
( |
)% |
Stock-based and other compensation |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
State taxes, net of federal benefit |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Research and development credits |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Change in valuation allowance |
|
|
|
|
|
|
|
|
|
|||
Other |
|
|
|
|
|
|
|
|
|
|||
Effective tax rate |
|
|
% |
|
|
% |
|
|
% |
|||
The tax effects of temporary differences and carryforwards that give rise to significant portions of deferred tax assets and deferred tax liabilities were as follows (in thousands):
|
|
December 31, |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
Deferred income tax assets |
|
|
|
|
|
|
||
Net operating loss carryforwards |
|
$ |
|
|
$ |
|
||
Interest expense |
|
|
|
|
|
|
||
Stock-based compensation expense |
|
|
|
|
|
|
||
Accrued bonus |
|
|
|
|
|
|
||
Research and development expenses |
|
|
|
|
|
|
||
Research and development credits |
|
|
|
|
|
|
||
Operating lease liabilities |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Gross deferred income tax assets |
|
|
|
|
|
|
||
Less: valuation allowance |
|
|
( |
) |
|
|
( |
) |
Net deferred tax assets |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Deferred income tax liabilities |
|
|
|
|
|
|
||
Property and equipment |
|
$ |
( |
) |
|
$ |
( |
) |
Operating lease right-of-use assets |
|
|
( |
) |
|
|
( |
) |
Other |
|
|
( |
) |
|
|
( |
) |
Gross deferred income tax liabilities |
|
|
( |
) |
|
|
( |
) |
Net deferred tax liabilities |
|
$ |
|
|
$ |
|
||
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax asset will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax assets, projected future taxable income, and tax planning strategies in making this assessment. Due to the Company's history of net losses, the deferred tax assets have been fully offset by a full valuation allowance of $
The Company had unused federal and state net operating loss carryforwards of approximately $
97
The federal and state net operating loss carryforwards and credits may be subject to significant limitations under Section 382 and Section 383 of the Internal Revenue Code and similar provisions under state law. The Tax Reform Act contains provisions that limit the federal net operating loss carryforwards that may be used in any given year in the event of special occurrences, including significant ownership changes. A Section 382 "ownership change" generally occurs if one or more stockholders or groups of stockholders, who own at least 5% of the Company's stock, increase their ownership by more than 50 percentage points over their lowest ownership percentage within a rolling three-year period. The Company may have previously experienced, and may in the future experience, one or more Section 382 "ownership changes," including in connection with the Company's initial public offering. If so, the Company may lose some or all of the tax benefits of its carryforwards and credits.
Management has reviewed and evaluated the relevant technical merits of each of its tax positions in accordance with accounting principles generally accepted in the United States of America for accounting for uncertainty in income taxes and determined that there are
11. Stockholders' Equity
Preferred Stock
The Company is authorized to issue
Common Stock
On April 27, 2021, the Company amended and restated its Certificate of Incorporation, which became effective upon the closing of the IPO. The Company is authorized to issue
Shares Reserved for Future Issuance
The Company has reserved shares of common stock for future issuances as follows:
|
|
December 31, |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
Common stock options issued and outstanding |
|
|
|
|
|
|
||
Shares available for future awards granted under the 2021 Plan |
|
|
|
|
|
|
||
Unvested restricted stock units under the 2021 Plan |
|
|
|
|
|
|
||
Common stock available for Employee Stock Purchase Plan |
|
|
|
|
|
|
||
Total |
|
|
|
|
|
|
||
Share-based Incentive Plans
In April 2021, prior to the IPO closing, the Company's board of directors and stockholders approved the 2021 Incentive Award Plan ("2021 Plan"), which became effective upon the IPO closing. The Company initially reserved
Prior to the IPO, the Company was authorized to issue stock purchase rights and to grant options to purchase Class A common stock to employees, directors, and consultants under the 2014 Plan. Stock options under the 2014 Plan have a term of no more than
98
(i) to the extent that such an option award terminates, expires or lapses for any reason or is settled in cash without the delivery of shares, any shares subject to the award at such time will be available for future grants under the 2021 Plan; (ii) to the extent shares are tendered or withheld to satisfy the grant, exercise price or tax withholding obligation with respect to any award under the 2014 Plan, such tendered or withheld shares will be available for future grants under the 2021 Plan; and (iii) to the extent that the Company repurchases the shares prior to vesting so that shares are returned to the Company, such shares will be available for future grants under the 2021 Plan.
At December 31, 2023,
Stock Options
Options under the 2021 Plan may be granted for periods of up to
The Company uses the Black-Scholes option pricing model to determine the fair value of stock options at the grant dates with the following assumptions for options granted during 2023, 2022, and 2021 fiscal years:
|
|
December 31, |
||||
|
|
2023 |
|
2022 |
|
2021 |
Expected term (in years) |
|
|
|
|||
Expected volatility |
|
|
|
|||
Risk-free interest rate |
|
|
|
|||
Expected dividend yield |
|
|
|
|||
Stock option activity for 2023 under the Stock Plans is set forth below:
|
|
Number of |
|
|
Weighted- |
|
|
Weighted- |
|
|
Aggregate Intrinsic Value |
|
||||
Outstanding as of December 31, 2022 |
|
|
|
|
|
|
|
$ |
|
|
|
|
||||
Options granted |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Options exercised |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Options canceled |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Outstanding as of December 31, 2023 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
||||
Options vested and expected to vest at December 31, 2023 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
||||
Options vested and exercisable at December 31, 2023 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
||||
The aggregate intrinsic value of options exercised during the years ended December 31, 2023, 2022, and 2021 was $
99
Restricted Stock Units and Awards
Full value award activity for 2023 under the Stock Plans is set forth below:
|
|
Full Value Awards |
|
|||||||||||||
|
|
RSUs |
|
|
Weighted-Average Grant Date Fair Value |
|
|
RSAs |
|
|
Weighted-Average Grant Date Fair Value |
|
||||
Unvested as of December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Shares or units granted |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Shares or units vested or released |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
||
Shares or units forfeited |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Unvested as of December 31, 2023 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
RSUs and RSAs granted under the 2021 Plan generally vest annually over
Performance Share Units
The Company granted performance-based restricted stock unit ("PSU") awards in the third quarter of 2023 subject to market and service vesting conditions to certain executives under the Company's 2021 Plan. The actual number of PSUs that will vest at the end of the measurement period is determined based on the Company's total stockholder return ("TSR") ranking relative to the TSR of a published index of the Company's peers. The measurement period is
The table below summarizes the assumptions used to estimate the grant date fair value of the PSUs granted:
|
|
Year Ended December 31, |
|
|
2023 |
Weighted average expected volatility of common stock |
|
|
Expected volatility of peer index |
|
|
Correlation coefficient of peer index |
|
|
Weighted average risk-free interest rate |
|
|
Dividend yield |
|
PSU activity for 2023 under the Stock Plans is set forth below:
|
|
Number of Shares |
|
|
Weighted Average Granted Date Fair Value |
|
||
Unvested as of December 31, 2022 |
|
|
|
|
$ |
|
||
Granted |
|
|
|
|
|
|
||
Vested |
|
|
|
|
|
|
||
Canceled or forfeited |
|
|
|
|
|
|
||
Unvested as of December 31, 2023 |
|
|
|
|
$ |
|
||
Share-Based Compensation Expense
Stock-based compensation expense is reflected in the Statements of Operations and Comprehensive Loss as follows (in thousands):
|
Year Ended December 31, |
|
|||||||||
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Cost of goods sold |
$ |
|
|
$ |
|
|
$ |
|
|||
Sales and marketing expense |
|
|
|
|
|
|
|
|
|||
Research and development expense |
|
|
|
|
|
|
|
|
|||
General and administrative expense |
|
|
|
|
|
|
|
|
|||
Total |
$ |
|
|
$ |
|
|
$ |
|
|||
100
The weighted-average grant date fair values of the stock options granted were $
As of December 31, 2023, there was $
Employee Share Purchase Plan
In April 2021, the Company's board of directors and stockholders approved the 2021 Employee Stock Purchase Plan ("ESPP"). The Company initially reserved
In January 2024, the number of shares of common stock available for issuance under the ESPP was increased by
12. Employee Benefit Plan
The Company matches employee contributions to the 401(k) plan at a rate equal to
Employer contributions under this plan were $
101
13. Net Loss Per Share Attributable to Common Stockholders
The following table sets forth the computation of basic and diluted net loss per share attributable to common stockholders which is computed by dividing the net loss attributable to common stockholders by the weighted-average number of shares of common stock outstanding for the period.
|
|
Year Ended December 31, |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
Numerator |
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Adjust: Convertible preferred stock cumulative |
|
|
|
|
|
|
|
|
( |
) |
||
Net loss attributable to common stockholders |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Denominator |
|
|
|
|
|
|
|
|
|
|||
Weighted-average common stock outstanding, |
|
|
|
|
|
|
|
|
|
|||
Net loss per share attributable to common |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
The following potentially dilutive securities outstanding have been excluded from the computation of diluted weighted average shares outstanding because such securities have an antidilutive impact due to the Company's net loss, in common stock equivalent shares:
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2023 |
|
|
2022 |
|
||
Common stock options issued and outstanding |
|
|
|
|
|
|
||
Unvested full value awards |
|
|
|
|
|
|
||
Total |
|
|
|
|
|
|
||
102
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation and supervision of our Chief Executive Officer and our Chief Financial Officer, have evaluated our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) before filing this Annual Report. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2023, the end of the period covered by this Annual Report, our disclosure controls and procedures were effective to provide reasonable assurance that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Management's Annual Report on Internal Control Over Financial Reporting
The Company's management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) for the Company. Internal control over financial reporting is a process designed under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. Management conducted an assessment of the effectiveness of the Company's internal control over financial reporting based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control–Integrated Framework (2013 Framework). Based on this assessment, our management concluded that, as of December 31, 2023, our internal control over financial reporting was effective based on those criteria.
Changes in internal control over financial reporting
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the quarter ended December 31, 2023 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Inherent limitation on the effectiveness of internal control
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
103
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Treace Medical Concepts, Inc.
Opinion on internal control over financial reporting
We have audited the internal control over financial reporting of Treace Medical Concepts, Inc. (a Delaware corporation) (the "Company") as of December 31, 2023, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023, based on criteria established in the 2013 Internal Control—Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) ("PCAOB"), the financial statements of the Company as of and for the year ended December 31, 2023, and our report dated February 27, 2024 expressed "an unqualified opinion" on those financial statements.
Basis for opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and limitations of internal control over financial reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ GRANT THORNTON LLP
Jacksonville, FL
February 27, 2024
104
Item 9B. Other Information.
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
105
PART III
Item 10. Directors, Executive Officers and Corporate Governance
We have adopted a code of conduct applicable to our principal executive, financial and accounting officers and all persons performing similar functions. A copy of our code of conduct is available on our principal corporate website at www.treace.com in the Investors section under "Corporate Governance". We intend to post any required disclosures regarding an amendment to, or waiver from, a provision of our code of conduct on the same website. The information found on our website is not part of this Annual Report or any other report we file with, or furnish to, the SEC.
The other information required by this item is incorporated by reference from our definitive Proxy Statement to be filed with the SEC in connection with our 2024 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2023.
Item 11. Executive Compensation
The information required by this item is incorporated by reference from our definitive Proxy Statement to be filed with the SEC in connection with our 2024 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2023.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item is incorporated by reference from our definitive Proxy Statement to be filed with the SEC in connection with our 2024 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2023.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item is incorporated by reference from our definitive Proxy Statement to be filed with the SEC in connection with our 2024 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2023.
Item 14. Principal Accounting Fees and Services
Our independent registered public accounting firm is GRANT THORNTON LLP, Jacksonville, FL, Auditor Firm ID: 248.
The other information required by this item is incorporated by reference from our definitive Proxy Statement to be filed with the SEC in connection with our 2024 Annual Meeting of Stockholders within 120 days after the end of the fiscal year ended December 31, 2023.
106
PART IV
Item 15. Exhibits, Financial Statement Schedules
Our financial statements are listed in the "Index to the Financial Statements" under Part II, Item 8, "Financial Statements and Supplementary Data" of this Annual Report.
Schedule II. Valuation and Qualifying Accounts (in thousands):
|
|
|
Additions |
|
|
|
|
|
|||||||
|
Balance at beginning of period |
|
Charged to expenses |
|
Charged to other accounts |
|
Write-offs and deductions |
|
Balance at end of period |
|
|||||
Allowance for doubtful accounts: |
|
|
|
|
|
|
|
|
|
|
|||||
Year ended December 31, 2023 |
$ |
735 |
|
$ |
434 |
|
$ |
- |
|
$ |
(189 |
) |
$ |
980 |
|
Year ended December 31, 2022 |
|
414 |
|
|
411 |
|
|
- |
|
|
(90 |
) |
|
735 |
|
Year ended December 31, 2021 |
|
446 |
|
|
144 |
|
|
- |
|
|
(176 |
) |
|
414 |
|
Deferred tax asset valuation allowance |
|
|
|
|
|
|
|
|
|
|
|||||
Year ended December 31, 2023 |
$ |
22,864 |
|
$ |
12,347 |
|
$ |
- |
|
$ |
- |
|
$ |
35,211 |
|
Year ended December 31, 2022 |
|
11,941 |
|
|
10,923 |
|
|
- |
|
|
- |
|
|
22,864 |
|
Year ended December 31, 2021 |
|
5,501 |
|
|
6,440 |
|
|
- |
|
|
- |
|
|
11,941 |
|
The documents listed in the Exhibit Index of this Annual Report are incorporated by reference or are filed with this Annual Report, in each case as indicated therein.
EXHIBIT INDEX
|
Incorporated by Reference |
Exhibit Number |
Description |
Form |
File No. |
Exhibit |
Filing Date |
Filed Herewith |
3.1 |
Amended and Restated Certificate of Incorporation of Treace Medical Concepts, Inc. |
8-K |
001-40355 |
3.1 |
4-27-21 |
|
3.2 |
Amended and Restated Bylaws of Treace Medical Concepts, Inc. |
8-K |
001-40355 |
3.2 |
4-27-21 |
|
4.1 |
S-1/A |
333-254863 |
4.2 |
4-19-21 |
|
|
4.2 |
Description of common stock of Treace Medical Concepts, Inc. |
10-K |
001-40355 |
4.2 |
3-4-22 |
|
10.1+ |
S-1/A |
333-254863 |
10.13 |
4-19-21 |
|
|
10.2+ |
Change in Control Severance Agreement by and between Treace Medical Concepts, Inc. and Mark L. Hair |
S-1/A |
333-254863 |
10.6 |
4-19-21 |
|
10.3+ |
Change in Control Severance Agreement by and between Treace Medical Concepts, Inc. and Aaron Berutti |
10-Q |
001-40355 |
10.2 |
11-4-21 |
|
107
10.4+
|
Change in Control Severance Agreement by and between Treace Medical Concepts, Inc. and Scot M. Elder |
10-K |
001-40355 |
10.7 |
3-4-22 |
|
10.5+ |
10-Q |
001-40355 |
10.7 |
8-5-21 |
|
|
10.6+ |
Form of Indemnification Agreement for directors and executive officers |
S-1/A |
333-254863 |
10.1 |
4-19-21 |
|
10.7+ |
S-1 |
333-254863 |
10.2(a) |
3-30-21 |
|
|
10.8+ |
Form of Stock Option Agreement for Directors under 2014 Stock Plan |
S-1 |
333-254863 |
10.2(b) |
3-30-21 |
|
10.9+ |
Form of Stock Option Agreement for Employees under 2014 Stock Plan |
S-1 |
333-254863 |
10.2(c) |
3-30-21 |
|
10.10+ |
S-1 |
333-254863 |
10.2(d) |
3-30-21 |
|
|
10.11+ |
S-1/A |
333-254863 |
10.3 |
4-19-21 |
|
|
10.12+ |
Form of Performance Stock Unit Award For Employees under 2021 Incentive Award Plan |
10-Q |
001-40355 |
10.1 |
11-9-23 |
|
10.13+ |
S-1/A |
333-254863 |
10.11 |
4-19-21 |
|
|
10.14+ |
|
|
|
|
X |
|
10.15 |
S-1 |
333-254863 |
10.12 |
3-30-21 |
|
|
10.16 |
10-Q |
001-40355 |
10.1 |
8-10-22 |
|
|
10.17 |
10-Q |
001-40355 |
10.2 |
8-10-22 |
|
|
10.18 |
10-Q |
001-40355 |
10.1 |
11-9-22 |
|
|
10.19 |
10-Q |
001-40355 |
10.2 |
11-9-22 |
|
|
23.1 |
Consent of Grant Thornton LLP, Independent Registered Public Accounting Firm |
|
|
|
|
X |
31.1 |
|
|
|
|
X |
|
31.2 |
|
|
|
|
X |
|
32.1# |
|
|
|
|
X |
|
32.2# |
Certification of Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as |
|
|
|
|
X |
108
|
Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
|
|
|
|
|
97.1 |
|
|
|
|
X |
|
101.INS |
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
|
|
|
|
|
101.SCH |
Inline XBRL Taxonomy Extension Schema With Embedded Linkbase Documents |
|
|
|
|
|
104 |
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
|
|
|
|
|
+ Indicates management contract or compensatory plan.
________________________________________________________________________________________________
# The certifications attached as Exhibit 32.1 and 32.2 that accompany this Annual Report are deemed furnished and not filed with the U.S. Securities and Exchange Commission and are not to be incorporated by reference into any filing of Treace Medical Concepts, Inc. under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report, irrespective of any general incorporation language contained in such filing.
Item 16. Form 10-K Summary
None.
109
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934 the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
Treace Medical Concepts, Inc. |
|
|||
|
|
|
|
|
|
|
Date: February 27, 2024 |
|
By: |
|
/s/ Mark L. Hair |
|
|
|
|
|
|
Name: |
Mark L. Hair |
|
|
|
|
|
Title: |
Chief Financial Officer |
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature
/s/ John T. Treace |
Title
Chief Executive Officer, Founder and Director (Principal Executive Officer) |
Date
February 27, 2024 |
John T. Treace |
|
|
/s/ Mark L. Hair Mark L. Hair |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer)
|
February 27, 2024 |
/s/ James T. Treace James T. Treace
|
Chairman of the Board of Directors |
February 27, 2024 |
/s/ John K. Bakewell John K. Bakewell
|
Director
|
February 27, 2024 |
/s/ Lance A. Berry Lance A. Berry
|
Director |
February 27, 2024 |
/s/ Lawrence W. Hamilton Lawrence W. Hamilton
|
Director |
February 27, 2024 |
/s/ Elizabeth S. Hanna |
Director |
February 27, 2024 |
Elizabeth S. Hanna |
|
|
|
|
|
/s/ Deepti Jain |
Director |
February 27, 2024 |
Deepti Jain |
|
|
|
|
|
/s/ Jane E. Kiernan |
Director |
February 27, 2024 |
Jane E. Kiernan |
|
|
|
|
|
/s/ Richard W. Mott Richard W. Mott
|
Director |
February 27, 2024 |
110