Product Pipeline
Our product candidate pipeline includes the following:
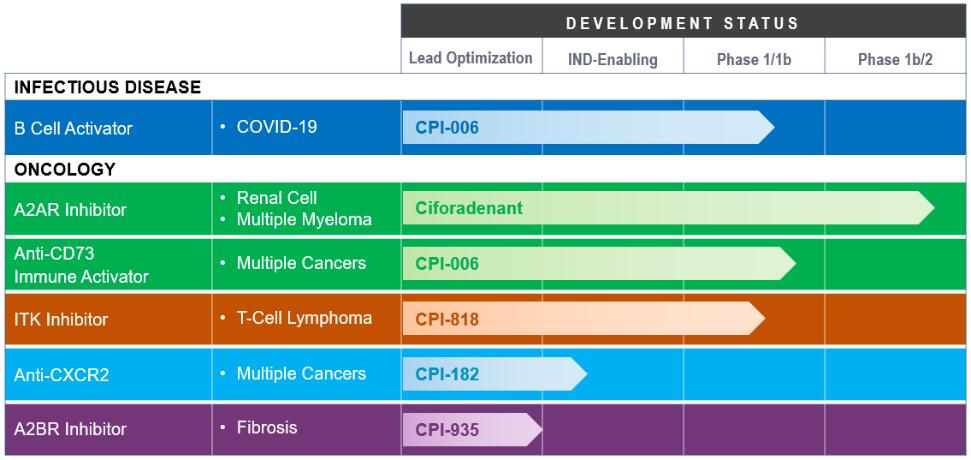
Ciforadenant Adenosine A2A Receptor Antagonist. Our initial product candidate, ciforadenant, is an oral, small molecule antagonist of the A2A receptor for adenosine that we in-licensed from Vernalis in February 2015. In January 2016, we began enrolling patients in a large expansion cohort trial for ciforadenant. This Phase 1/1b clinical trial is designed to examine safety, tolerability, biomarkers and preliminary efficacy of ciforadenant in several solid tumor types, both as a single agent and in combination with Genentech, Inc.’s cancer immunotherapy, Tecentriq, a fully humanized monoclonal antibody targeting PD-L1. In November 2016, we completed enrollment of 48 patients in the first step of the Phase 1/1b clinical trial, which was designed to determine the optimal dose of ciforadenant as both a single agent therapy and in combination with Tecentriq for use in the cohort expansion stage of the clinical trial. The expansion cohort portion of the clinical trial enrolled patients with non-small cell lung cancer (“NSCLC”), RCC, melanoma (“MEL”), triple negative breast cancer (“TNBC”) and other cancers including colorectal cancer, prostate cancer, head and neck cancer and bladder cancer at leading medical centers in the United States, Australia and Canada. We have enrolled over 300 patients in this clinical trial to date. In 2017, both the single agent and combination arms of the NSCLC and RCC cohorts met the protocol-defined criteria for expansion from 14 to 26 patients, and both arms of the RCC cohort further met the protocol-defined criteria for expansion to 48 patients. In December 2017, Genentech began enrolling patients in a Phase 1b/2 clinical trial that is evaluating ciforadenant in combination with Tecentriq in patients with NSCLC under an umbrella protocol known as Morpheus. Enrollment in this trial has been completed and the patients are being followed. In 2018, we amended our Phase 1/1b protocol to enroll patients in a Phase 1b/2 clinical trial with RCC who have failed therapies with both anti-PD-(L)1 antibodies and tyrosine kinase inhibitors (“TKI”). Based on data observed in the Phase 1b/2 clinical trial in 2019, we began enrolling patients with metastatic castration-resistant prostate cancer (“mCRPC”) in a Phase 2 expansion arm of our ongoing Phase 1/1b clinical trial with mCRPC who will receive the combination of ciforadenant with Tecentriq based on data from the Phase 1b/2 clinical trial that showed activity in this disease.
As of August 2020, the key findings from our clinical trials of ciforadenant included:
| ● | Ciforadenant has been well-tolerated at doses that achieved substantial receptor blockade; |
| ● | Ciforadenant has shown evidence of anti-tumor activity as a monotherapy and in combination with atezolizumab; |
| ● | Of cancers studied, RCC, mCRPC and NSCLC have appeared most responsive to therapy; and |
23