UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from [ ] to [ ]
Commission file number
(Exact name of registrant as specified in its charter) |
| ||
(State or other jurisdiction of Incorporation or organization) |
| (I.R.S. Employer Identification No.) |
| ||
(Address of principal executive offices) |
| (Zip Code) |
Registrant's telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class |
| Name of Each Exchange On Which Registered |
N/A |
| N/A |
Securities registered pursuant to Section 12(g) of the Act: Common Stock
Indicate by check mark if the registrant is a well-seasoned issuer, as defined in Rule 405 of the Securities Act ☐ Yes ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15d of the Act ☐ Yes ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or such shorter period of that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by checkmark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the previous 12 months (or for such shorter period that the registrant was required to submit and post such files.)
Indicate by checkmark if disclosure of delinquent filers to Item 405 of Regulation S-K (§229.405) is not contained herein and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer," and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
☒ | Smaller reporting company | ||
|
| Emerging Growth Company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act,) Yes
The number of shares outstanding of the Company’s $.001 Par Value Common Stock as of June 28, 2024 was
DOCUMENTS INCORPORATED BY REFERENCE: None.
TABLE OF CONTENTS
|
|
| Page |
|
| ||
|
|
| 3 |
|
|
| |
|
|
|
|
| 4 | ||
| 16 | ||
| 20 | ||
| 20 | ||
| 21 | ||
| 21 | ||
| 21 | ||
|
|
|
|
PART II |
|
|
|
|
|
|
|
| 21 | ||
| 23 | ||
Management’s Discussion and Analysis of Financial Conditions and Results of Operations |
| 23 | |
| 26 | ||
| 26 | ||
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
| 26 | |
| 26 | ||
| 26 | ||
|
|
|
|
|
|
| |
|
|
|
|
| 27 | ||
| 30 | ||
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
| 32 | |
Certain Relationships and Related Transactions, and Director Independence |
| 33 | |
| 34 | ||
|
|
|
|
PART IV |
|
|
|
|
|
|
|
| 34 | ||
|
|
|
|
| 36 | ||
| 2 |
| Table of Contents |
FORWARD-LOOKING STATEMENTS
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
Various statements in this Annual Report on Form 10-K, including those that express a belief, expectation, or intention, as well as those that are not statements of historical fact, are forward-looking statements. The forward-looking statements may include projections and estimates concerning the timing and success of our business activities, our revenues, income, and capital spending. We generally identify forward-looking statements with the words “believe,” “intend,” “expect,” “seek,” “may,” “should,” “anticipate,” “could,” “estimate,” “plan,” “predict,” “project” or their negatives, and other similar expressions. All statements we make relating to our estimated timelines and commencement of operations, and our projected earnings, costs, expenditures, cash flows, and financial results or to our expectations regarding future industry trends are forward-looking statements.
These forward-looking statements are subject to risks and uncertainties that may change at any time, and, therefore, our actual results may differ materially from those that we expected. The forward-looking statements contained in this Form 10-K are largely based on our expectations, which reflect estimates and assumptions made by our management. These estimates and assumptions reflect our best judgment based on currently known market conditions and other factors. Although we believe such estimates and assumptions are reasonable, we caution that it is exceedingly difficult to predict the impact of known factors and it is impossible for us to anticipate all factors that could affect our actual results. In addition, management's assumptions about future events may prove to be inaccurate. We caution all readers that the forward-looking statements contained in this Form 10-K are not guarantees of future performance, and we cannot assure any reader that such statements will prove correct or the forward-looking events and circumstances will occur. Actual results may differ materially from those anticipated or implied in the forward-looking statements due to the numerous risks and uncertainties as described elsewhere in this Form 10-K.
The occurrence of an uncontrollable event such as the COVID-19 pandemic may negatively affect our operations. A pandemic typically results in social distancing, travel bans, and quarantine. This may limit access to our suppliers, management, support staff and professional advisors. Although the Company’s operations are virtual, we depend on numerous third party consultants and contract suppliers so we cannot measure the impact on our operations or financial condition at this point in time.
| 3 |
| Table of Contents |
PART I
ITEM 1: BUSINESS.
Overview
NASCENT BIOTECH INC., a Nevada corporation (“Nascent” or the “Company”), is actively developing its primary asset, Pritumumab, for the treatment of brain cancer and pancreatic cancer. Nascent is also actively researching other cancers that have a high probability of benefiting from the therapeutic effects of Pritumumab because they share a common molecular target. The Company continues exploring Pritumumab in its use against viruses. Nascent has completed its research with their partner, Manhattan BioSolutions developing a traditional vaccine platform to create vaccines for viral infections.
On June 8, 2021 the US Patent Office issued patent number 11028155 to the Company titled ”Enhanced Delivery of Drug to the Brain”.
On November 8, 2022 the US Patent Office issued patent number 11492394 to the Company titled “Kits and Containers for Treating Vimentin Expressing Tumors” to the Company.
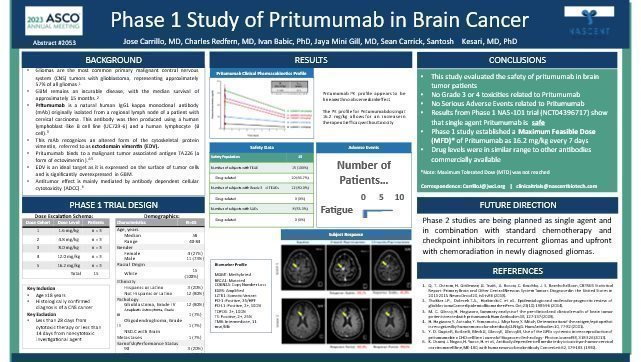
In April 2023, the Company completed phase 1 of the clinical trials and was cleared to begin phase 2 clinical trials.
On June 6, 2023, the Company mutually ended a license agreement with BioRay Pharmaceutical Co LTD and thereby regained worldwide rights for development and distribution of Pritumumab.
Current Business
We are a Phase II Clinical Stage Biopharmaceutical Company that develops monoclonal antibodies for the treatment of various cancer types. We focus on biologic drug candidates that are undergoing or have already completed initial clinical testing for the treatment of cancer and then seek to further develop those drug candidates for commercial use. We currently own drug candidate, Pritumumab, which we are developing as a treatment for epithelial cancers (which includes lung, breast, colon, brain, and pancreas).
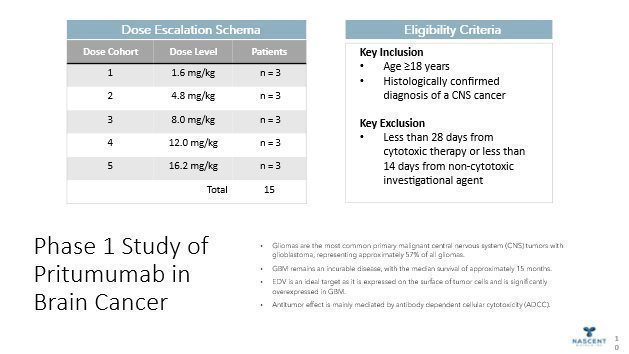
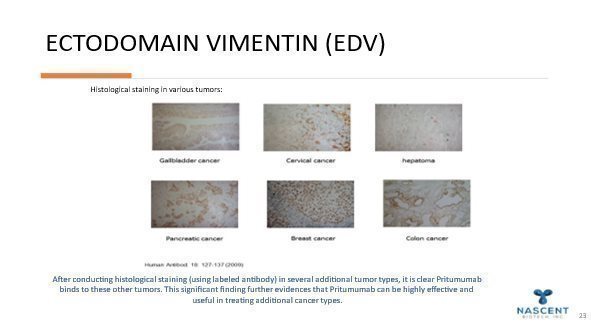
Current brain cancer therapeutic strategies include chemotherapy drug Temodar®, surgery, and/or radiation. Even when removed, most brain tumors come back within one-year post-surgery. With current standards of care, only 58% of brain cancer patients live past the first year after diagnosis with certain types, anaplastic astrocytoma and glioblastoma, the five-year survival rates are 27% and 5%, respectively. Based on clinical studies, we believe that Pritumumab may offer an advantage over existing treatments. Nascent has addressed manufacturing questions by re-engineering antibody production into the commonly used CHO cell expression system. Our initial focus is on patients with brain cancer malignancies such as glioblastoma and malignant astrocytoma. Our focus is driven primarily by two factors: (1) brain cancer is a reasonably-sized orphan market–especially including metastatic brain cancer-and (2) an unmet need in cancer treatment. Pritumumab works by binding to a target on the surface of cancer cells called ectodomain vimentin (also referred to as cell-surface vimentin). The target, generally referred to as an antigen, is prevalent in many different tumor types and is not being targeted by any other biopharmaceutical companies. By binding to this target, Pritumumab is able to make the tumor cells “known” to the body’s immune system, resulting in potentially several types of immune responses, including anti-idiotype, apoptosis, antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity, leading to death of the cancer cells and overall depletion of the tumor.
Our Phase I Clinical Trial – Performed and completed at Hoag Presbyterian Hospital-provided excellent safety outcomes and very promising therapeutic signaling.
| 4 |
| Table of Contents |
Brain Cancer Incidence and Prevalence
In the United States, the annual estimated new cases of Primary Tumor Brain Cancer each year are about 24,000, with estimated deaths about 18,000 per year. Brain cancer is the leading cause of cancer-related death in patients younger than age 20. Gliomas (a broad term which includes all tumors arising from the gluey or supportive tissue of the brain), the cancer type for which orphan drug designation has been granted by the FDA, account for 80% of all malignant brain tumors. Metastatic brain cancer (metastases to the brain from other sites) occurs in 20–40% of patients with metastatic disease and incidence increases with age. In the United States, over 100,000 cases of metastatic brain cancer are diagnosed each year with most originating from lung (50%) and breast (15-20%).
| 5 |
| Table of Contents |
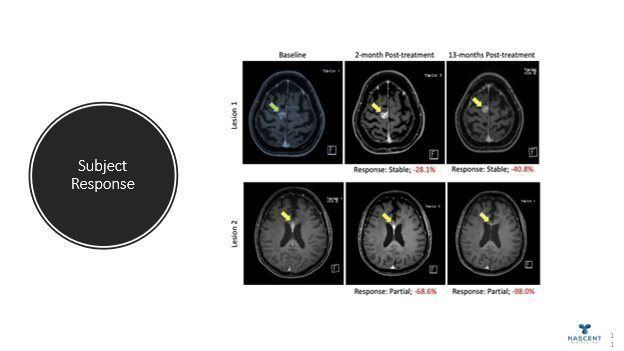

We expect to amplify on the past clinical development strategy during the next 12 months plan to:
| · | commence Phase II clinical trials in the United States with Pritumumab fourth calendar quarter 2024. |
|
|
|
| · | continue to evaluate the application of Pritumumab in the treatment of other unmet ectodomain vimentin positive cancer types such as pancreatic. |
The Company submitted its data and Phase II protocol to the USFDA and was cleared to begin Phase II Clinical trials.
| 6 |
| Table of Contents |
Our Goal and Strategy
Our goal is to become a leading oncology-focused biopharmaceutical company. The key elements of our strategy to achieve this goal are as follows:
| · | Advance Pritumumab, our drug candidate, toward regulatory approval and commercialization. In Phase I/II Clinical Trials, we are primarily focused on developing Pritumumab for the treatment of patients with primary brain tumors. |
|
|
|
| · | Expand our claims by pursuing additional indications for Pritumumab, due to the target antigen’s (ectodomain vimentin) presence in a variety of cancers apart from brain cancer. Nascent will seek to expand it into other areas. Additionally, we believe Pritumumab could be an effective conjugate (carrier protein) for other anti-cancer drugs and can be developed as a diagnostic tool. |
Pancreatic Cancer – Nascent Has been awarded Orphan Drug Designation on Pancreatic Cancer and sees it as a underserved disease That we can potentially help.
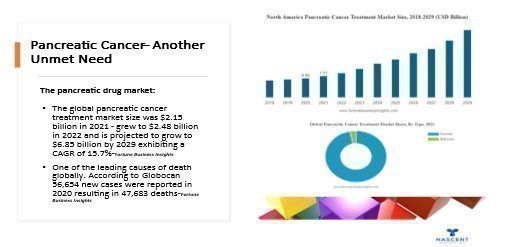
Additional Malignancies Pritumumab may be effective against:
| · | Evaluate the commercialization strategies on a product-by-product basis. As we move our drug candidates through development toward regulatory approval, we will evaluate several options for each drug candidate’s commercialization strategy. These options include entering into a joint marketing partnership with another pharmaceutical company or biotechnology company, whereby we jointly sell and market the product; and out licensing our product, whereby another pharmaceutical company or biotechnology company sells and markets our product and pays us on a developmental milestone and royalty on sales basis in North America. Our decision will be made separately for each product and will be based on several factors, including capital necessary to execute on each option, market size that needs to be addressed and terms of potential offers from other pharmaceutical and biotechnology companies. |
| 7 |
| Table of Contents |
Pritumumab in Cancer Patients
Our initial focus is on the development of Pritumumab as an intravenous treatment of patients with various types of brain cancer, most prominently gliomas and malignant astrocytoma.
Advantages of Pritumumab
Based on clinical studies to date, we believe that Pritumumab may offer an advantage over existing treatments. The antibody is fully human. This natural approach, results in the molecule posing very little toxicity to the patient, particularly in comparison to chemotherapies, radiation and cytokine approaches that currently dominate the marketplace. Further, the antibody binds to a very novel target that appears to be prevalent in a number of solid tumors. A fully human approach will mimic and harness the body’s natural defense system to fight cancer and offer an efficacious and safer approach to other protocols, which can be devastating and life threatening in and of themselves.
Disadvantages of Pritumumab
Given the low survival rates for current standard therapy in brain cancer, both in percentage and type, and the general safety associated with monoclonal antibodies currently approved in the United States for a variety of therapeutic indications, we believe that there are no disadvantages of Pritumumab to current therapy.
Development Plan
We initiated a US-based clinical trial of pritumumab in patients with brain cancer in March 2021. In this trial, we are investigating the efficacy of Pritumumab on metastatic brain cancer and measure the effects on the primary tumor. Our Phase I trial was a multiple ascending dose study employing significantly higher doses to test for safety and improved efficacy. We believe we can complete the Phase II studies indicated above within a period of about 24 months and for a total cost of less than $15 million dollars. This will, of course, require funding our planned capital raises as discussed elsewhere in this document.
Clinical Testing of Our Products in Development
Each of our products in development, and likely all future drug candidates, will require extensive clinical testing to determine the safety and efficacy of the product prior to seeking and obtaining regulatory approval for marketing and sale of the product. This process is expensive and time consuming. In completing this pre-clinical and clinical testing, we are dependent upon third-party consultants, consisting mainly of investigators, collaborators, and contract research organizations, who will conduct such testing.
We and our third-party consultants conducted pre-clinical testing in accordance with Good Laboratory Practices, or GLPs, and clinical testing in accordance with Good Clinical Practices, or GCPs, which are international ethical and scientific quality standards utilized for pre-clinical and clinical testing, respectively. GCP is the standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials, and is required by the FDA to be followed in conducting clinical trials. Additionally, any pre-clinical and clinical testing completed in the European Union, (EU), is conducted in accordance with applicable EU standards, such as the EU Clinical Trials Directive (Directive 2001/20/EC of April 4, 2001), or the EU Clinical Trials Directive, and the national laws of the Member States of the EU implementing its provisions.
| 8 |
| Table of Contents |
Intellectual Property
The US Food & Drug Administration has granted the Company orphan drug designation for use of Pritumumab against gliomas and pancreatic cancer. If we obtain marketing approval for Pritumumab or other drug candidates in the United States or in certain jurisdictions outside of the United States, we may be eligible for regulatory protection, such as seven years of market exclusivity under FDA orphan drug designation, up to five years of patent term extension potentially available in the United States under the Hatch-Waxman Act, 8 to 11 years of data and marketing exclusivity potentially available for new drugs in the European Union, up to five years of patent extension in Europe (Supplemental Protection Certificate), and eight years of data exclusivity potentially available in Japan. There can be no assurance that we will qualify for any such regulatory exclusivity, or that any such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies. See “Government Regulation” below. Our goal is to obtain, maintain and enforce patent protection for our products, formulation, processes, methods, and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information, and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. However, even patent protection may not always afford us with complete protection against competitors who seek to circumvent our patents. Our proprietary rights may not adequately protect our intellectual property and potential products, and if we cannot obtain adequate protection of our intellectual property and potential products, we may not be able to successfully market our potential products.”
We will depend upon the skills, knowledge, and experience of our scientific and technical personnel, as well as that of our advisors, consultants, and other contractors, none of which is patentable. To help protect our proprietary know-how, which is not patentable, and inventions for which patents may be difficult to obtain or enforce, we will in the future rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors, and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries, and inventions important to our business.
On June 8, 2021 the US Patent Office issued patent t number 11028155 to the Company titled ”Enhanced Delivery of Drug to the Brain”.
On November 8, 2022 the US Patent Office issued patent number 11492394 to the Company titled “Kits and Containers for Treating Vimentin Expressing Tumors” to the Company.
Manufacturing
We do not currently have our own manufacturing facilities. We intend to continue to use our financial resources to accelerate development of our drug candidates rather than diverting resources to establish our own manufacturing facilities. We meet our clinical trial manufacturing requirements by continuing established relationships with third-party manufacturers and other service providers to perform these services for us. We intend to continue these third-party relationships to maintain our supply of Pritumumab. Should Pritumumab obtain marketing approval, we anticipate establishing relationships with third-party manufacturers and other service providers for the commercial production and manufacture of our product. We have some flexibility in securing other manufacturers to produce our drug candidates; however, our alternatives may be limited due to proprietary technologies or methods used in the manufacture of some of our drug candidates.
Competition
The development and commercialization of new products to treat cancer is highly competitive, and we expect considerable competition from major pharmaceutical, biotechnology and specialty cancer companies. As a result, there are and will likely continue to be extensive research and substantial financial resources invested in the discovery and development of new cancer products. Our potential competitors include, but are not limited to, Genentech, Merk, Roche, Takeda, Array Biopharma and Ambit Biosciences. We are an early-stage company with no history of operations, and we acquired the rights to the drug candidates we expect to develop. Many of our competitors have substantially more resources than we do, including both financially and technically. In addition, many of our competitors have more experience than us in clinical development, manufacturing, regulatory and global commercialization. We are also competing with academic institutions, governmental agencies and private organizations that are conducting research in the field of cancer. We anticipate that we will face intense competition.
| 9 |
| Table of Contents |
We expect that our products under development and in clinical trials will address major markets within the cancer sector. Our competition will be determined in part by the indications for which drugs are developed and ultimately approved by regulatory authorities. Additionally, the timing of market introduction of some of our potential products or of competitors’ products may be an important competitive factor. Accordingly, the speed with which we can develop products, complete pre-clinical testing, clinical trials, and approval processes and supply commercial quantities to market are expected to be important competitive factors. We expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price, reimbursement, and patent position.
Government Regulation
United States—FDA Process
The research, development, testing, manufacture, labeling, promotion, advertising, distribution, and marketing, among other things, of drug products are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending New Drug Applications (NDAs) or Biologics License Applications (BLAs), warning letters, fines, civil penalties, product recalls, product seizures, total or partial suspension of production or distribution, injunctions and/or criminal prosecution. In the case of Pritumumab, it is a biologic drug and the appropriate route for approval will be to submit a BLA.
Drug Approval Process.
None of our drug product candidates may be marketed in the United States until the drug has received FDA approval. The steps required before a drug may be marketed in the United States generally include the following:
| · | completion of extensive pre-clinical laboratory tests, animal studies, and formulation studies in accordance with the FDA’s GLP regulations; |
|
|
|
| · | development of a manufacturing process in accordance with the FDA’s current Good Manufacturing Practice (cGMP) regulations; |
|
|
|
| · | submission to the FDA of an Investigational New Drug (“IND”) application for human clinical testing, which must pass FDA review before human clinical trials may begin; |
|
|
|
| · | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each proposed indication; |
|
|
|
| · | submission to the FDA of a BLA (Biologics License Application) after completion of all pivotal clinical trials; |
|
|
|
| · | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with current Good Manufacturing Practices (cGMPs); and, |
|
|
|
| · | FDA review and approval of the BLA prior to any commercial marketing or sale of the drug in the United States. |
The development and approval process requires substantial time, effort, and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. As noted above, the Company has received approval to commence human clinical trials with its currently manufactured drug product lot. Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be provided to the FDA as part of a separate submission to the IND. Further, an Institutional Review Board (IRB) for each medical center proposing to conduct the clinical trial must review and approve the study protocol and informed consent information for study subjects for any clinical trial before it commences at that center, and it must monitor the study until it is completed. Study subjects must sign an informed consent form before participating in a clinical trial.
| 10 |
| Table of Contents |
Clinical trials necessary for product approval typically are conducted in three sequential phases, but the phases may overlap. Phase 1 involved the initial introduction of the investigational drug into a limited population, typically healthy humans, to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics, and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness. Phase 2 usually involves trials in a limited patient population to (i) evaluate dosage tolerance and determine appropriate dosage; (ii) identify possible adverse effects and safety risks; and (iii) evaluate preliminarily the efficacy of the drug for specific targeted indications. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. Phase 3 trials, commonly referred to as pivotal studies, are undertaken in an expanded patient population at multiple, geographically dispersed clinical trial centers to further evaluate clinical efficacy and test further for safety by using the drug in its final form. There can be no assurance that Phase 2 or Phase 3 testing will be completed successfully within any specified period of time, if at all. Furthermore, the Company, the FDA or an IRB may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Moreover, the FDA may approve a BLA for a product candidate but require that the sponsor conduct additional clinical trials to further assess the drug after BLA approval under a post-approval commitment. Post-approval trials are typically referred to as Phase 4 clinical trials.
During the development of a new drug, sponsors are given an opportunity to meet with the FDA at certain time points. These points may be prior to submission of an IND, at the end of Phase 2, and before a BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach an agreement on the next phase of development. Sponsors typically use the end of Phase 2 meeting to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support approval of the new drug. If a Phase 3 clinical trial is the subject of discussion at an end of Phase 2 meeting with the FDA, a sponsor may be able to request a Special Protocol Assessment, the purpose of which is to reach an agreement with the FDA on the design of the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim. If such an agreement is reached, it will be documented and made part of the administrative record, and it will be binding on the FDA unless public health concerns unrecognized at the time of the protocol assessment are evident and may not be changed except under a few specific circumstances.
Concurrent with clinical trials, companies usually complete additional animal safety studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and the manufacturer must develop and validate methods for testing the quality, purity, and potency of the final drugs. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf-life. Assuming successful completion of the required clinical testing, the results of the pre-clinical studies and of the clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of a BLA requesting approval to market the product for one or more indications. A BLA must be accompanied by a significant user fee, which we anticipate will be waived for the first BLA because NBI is a “qualified small business.”
The testing and approval process requires substantial time, effort, and financial resources. The agency reviews the application and may deem it to be inadequate to support the registration, and companies cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but it typically follows such recommendations. Before approving a BLA, the FDA usually will inspect the facility or the facilities at which the drug is manufactured and will not approve the product unless the manufacturing is in compliance with cGMPs. If the FDA evaluates the BLA and the manufacturing facilities are deemed acceptable, the FDA may issue an approval letter, or in some cases, an approvable letter followed by an approval letter. Both letters usually contain a number of conditions that must be met in order to secure final approval of the BLA. When and if those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter. The approval letter authorizes commercial marketing of the drug for specific indications. As a condition of BLA approval, the FDA may require post-marketing testing and surveillance to monitor the drug’s safety or efficacy or impose other conditions.
| 11 |
| Table of Contents |
The FDA may deny approval of a BLA by issuing a Complete Response Letter if the applicable regulatory criteria are not satisfied. A Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant, expensive, and time-consuming requirements related to clinical trials, pre-clinical studies, or manufacturing. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Alternatively, approval may occur with Risk Evaluation and Mitigation Strategies, or REMS, which limit the labeling, distribution, or promotion of a drug product. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase 4 clinical trials, and surveillance programs to monitor the safety effects of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
Expedited Review and Approval
The FDA has various programs, including Orphan Drug Designation and Fast Track approval, which are intended to expedite or simplify the process for reviewing drugs, and/or provide for accelerated approval. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will be shortened. Generally, drugs that may be eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious diseases and fill an unmet medical need. Priority review is designed to give drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists and provides for an initial review within six months as compared to a standard review time of 10 months. Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track designated drug and expedite review of the application for a drug designated for priority review. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform post-marketing clinical trials.
Post-Approval Requirements
Oftentimes, even after a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. In addition, certain changes to an approved product, such as adding new indications, making certain manufacturing changes, or making certain additional labeling claims, are subject to further FDA review and approval. Before a company can market products for additional indications, it must obtain additional approvals from the FDA, typically a new BLA or NDA. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. A company cannot be sure that any additional approval for new indications for any product candidate will be approved on a timely basis, or at all. If post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved BLA are required to: (i) report certain adverse reactions to the FDA and maintain pharmacovigilance programs to proactively look for these adverse events, (ii) comply with certain requirements concerning advertising and promotional labeling for their products, and (iii) continue to have quality control and manufacturing procedures conform to cGMPs after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of ongoing compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. We intend to use third-party manufacturers to produce our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution or require substantial resources to correct. In addition, discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including recall of the product from the market or withdrawal of approval of the BLA for that drug.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials and approval of foreign countries or economic areas, such as the EU, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
| 12 |
| Table of Contents |
In the European Economic Area, or EEA (which is comprised of the 27 member states of the EU, or Member States, plus Norway, Iceland, and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
| · | The Community MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific, or technical innovation or which are in the interest of public health in the EU. |
|
|
|
| · | National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure, an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State. The competent authority of the Reference Member State prepares a draft assessment report, a draft summary of the specific product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling or packaging proposed by the Reference Member State, the product is subsequently granted a National MA in all the Member States (i.e., in the Reference Member State and the Member States Concerned). Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety, and efficacy. |
As in the United States, it may be possible in foreign countries to obtain a period of market and/or data exclusivity that would have the effect of postponing the entry into the marketplace of a competitor’s generic product. For example, if any of our products receive marketing approval in the EEA, we expect they will benefit from 8 years of data exclusivity and 10 years of marketing exclusivity. An additional non-cumulative one year period of marketing exclusivity is possible if during the data exclusivity period (the first 8 years of the 10-year marketing exclusivity period), we obtain an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies. The data exclusivity period begins on the date of the product’s first marketing authorization in the EU and prevents generics from relying on the marketing authorization holder’s pharmacological, toxicological, and clinical data for a period of 8 years. After 8 years, a generic product application may be submitted and generic companies may rely on the marketing authorization holder’s data. However, a generic cannot launch until 2 years later (or a total of 10 years after the first marketing authorization in the EU of the innovator product), or 3 years later (or a total of 11 years after the first marketing authorization in the EU of the innovator product) if the marketing authorization holder obtains marketing authorization for a new indication with significant clinical benefit within the 8 year data exclusivity period. In Japan, our products may be eligible for eight years of data exclusivity. There can be no assurance that we will qualify for such regulatory exclusivity, or that such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies. When conducting clinical trials in the EU, we must adhere to the provisions of the EU Clinical Trials Directive and the laws and regulations of the EU Member States implementing them. These provisions require, among other things, that the prior authorization of an Ethics Committee and the competent Member State authority is obtained before commencing the clinical trial.
| 13 |
| Table of Contents |
Pricing and Reimbursement
In the United States and internationally, sales of products that we market in the future, and our ability to generate revenues on such sales, are dependent, in significant part, on the availability of adequate coverage and reimbursement from third-party payers such as state and federal governments, managed care providers and private insurance plans. Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and also the out-of-pocket obligations of member patients for such products. We may need to conduct pharmacoeconomic studies to demonstrate the cost effectiveness of our products for formulary coverage and reimbursement. Even with studies, our products may be considered less safe, less effective, or less cost-effective than existing products, and third-party payers may not provide coverage and reimbursement for our product candidates, in whole or in part.
In addition, particularly in the United States and increasingly in other countries, we are required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our products that are reimbursed by such entities. It is possible that future legislation in the United States and other jurisdictions could be enacted that could potentially impact the reimbursement rates for the products we are developing and may develop in the future and also could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market. Political, economic, and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our future business. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, the PPACA, enacted in March 2010, substantially changes the way healthcare is financed by both governmental and private insurers. Among other cost containment measures, PPACA establishes:
| · | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; |
|
|
|
| · | a Medicare Part D coverage gap discount program, in which pharmaceutical manufacturers who wish to have their drugs covered under Part D must offer discounts to eligible beneficiaries during their coverage gap period, or the donut hole; and |
|
|
|
| · | a formula that increases the rebates a manufacturer must pay under the Medicaid Drug Rebate Program. |
In the future, there may continue to be additional proposals relating to the reform of the U.S. healthcare system. Future legislation, including the current versions being considered at the federal level in the United States or regulatory actions implementing recent or future legislation, may have a significant effect on our business. Our ability to successfully commercialize products depends in part on the extent to which reimbursement for the costs of our products and related treatments will be available in the United States and worldwide from government health administration authorities, private health insurers and other organizations. Because the adoption of certain proposals could limit the prices, we are able to charge for our products, or the amounts of reimbursement available for our products, and could limit the acceptance and availability of our products, substantial uncertainty exists as to the reimbursement status of newly approved health care products by third-party payers.
Sales and Marketing
We currently have no marketing, sales, or distribution capabilities. We do, however, have worldwide commercialization rights for our drug candidates. In order to commercialize any of our drug candidates if and when they are approved for sale in the United States or elsewhere, we will need to develop the necessary marketing, sales, and distribution capabilities. The FDA regulates all advertising and promotion activities for products under its jurisdiction both prior to and after approval, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to collect additional data or conduct additional pre-clinical studies and clinical trials. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
| 14 |
| Table of Contents |
Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, and often reflect a physician’s belief that the off-label use is the best treatment for the patients. The FDA does not regulate the behavior of physicians in their choice of treatments, but FDA regulations do impose stringent restrictions on manufacturers’ communications regarding off-label uses. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA. Outside the United States, our ability to market a product is contingent upon obtaining marketing authorization from the appropriate regulatory authorities. The requirements governing marketing authorization, pricing and reimbursement vary widely from country to country.
We may also be subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations and very few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting or causing to be presented for payment to third-party payers (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws.
Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal health care programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties also can be imposed upon executive officers and employees, including criminal sanctions against executive officers under the so-called “responsible corporate officer” doctrine, even in situations where the executive officer did not intend to violate the law and was unaware of any wrongdoing. Given the penalties that can be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the government were to allege or convict us or our executive officers of violating these laws, our business could be harmed. In addition, private individuals have the ability to bring similar actions. Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. Further, there is an increasing number of state laws that require manufacturers to provide reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state authorities.
Other Laws and Regulatory Processes
We are subject to a variety of financial disclosure and securities trading regulations as a public company in the United States, including laws relating to the oversight activities of the Securities and Exchange Commission, or SEC, and, if our capital stock becomes listed on a national securities exchange, we will be subject to the regulations of such exchange on which our shares are traded. In addition, the Financial Accounting Standards Board, or FASB, the SEC, and other bodies that have jurisdiction over the form and content of our accounts, our financial statements and other public disclosure are constantly discussing and interpreting proposals and existing pronouncements designed to ensure that companies best display relevant and transparent information relating to their respective businesses.
| 15 |
| Table of Contents |
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights or acquisitions may be subject to national or supranational antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted
ITEM 1A: RISK FACTORS
There are numerous and varied risks, known and unknown, that may prevent us from achieving our goals. If any of these risks actually occur, our business, financial condition or results of operations may be materially adversely affected. In such case, the trading price of our common stock could decline and shareholders could lose all or part of their investment.
Risks Related to our Business
The occurrence of an uncontrollable event such as the COVID-19 pandemic has negatively affected our operations. The resulting social distancing, travel bans, and quarantine delayed the start of our Phase I Trial. This trial has been completed
We currently have no product revenues and no products approved for marketing and will need to raise additional capital to operate our business. We received license revenue during the year ended March 31, 2022
We have generated no product revenues but sold license agreements which had the potential to generate $5 million of license fees based on specific milestones met by the Company. To date we received $4,750,000, however by mutual agreement the licenses has been cancelled and the worldwide rights have been re-gained by the Company. Until, and unless, we receive approval from the U.S. Food and Drug Administration, or FDA, or other regulatory authorities overseas for one or more of our drug candidates, we cannot market or sell our products and will not have product revenues. We have completed our phase 1 clinical trial for Pritumumab and our submitted protocol for Phase 2 to the FDA has been accepted.
Moreover, each drug candidate will require time and capital before we can apply for IND approval from the FDA or commence clinical trials. Therefore, for the foreseeable future, we do not expect to achieve any product revenues and will have to fund all of our operations and capital expenditures from cash on hand, licensing fees and grants, and potentially, future offerings. We will need to seek additional sources of financing, which may not be available on favorable terms. If we do not succeed in timely raising additional funds on acceptable terms, we may be unable to complete planned pre-clinical and clinical trials or obtain approval of any drug candidates from the FDA and other regulatory authorities. Any additional sources of financing will likely involve the issuance of additional equity securities, which will have a dilutive effect on our stockholders.
We use the services of outside service providers to conduct our clinical trials and manufacture our products.
Development, Regulatory Approval and Marketing of Products
The outcome of the lengthy and complex process of identifying new compounds and developing new products is inherently uncertain and involves a high degree of risk and cost. Drug discovery and development is time-consuming, expensive and unpredictable. The process from early discovery or design to development to regulatory approval can take many years. Drug candidates can fail at any stage of the process, including as the result of unfavorable clinical trial results. There can be no assurance regarding our ability to meet anticipated clinical trial commencement and completion dates, regulatory submission dates, and launch dates for product candidates, or as to whether or when we will receive regulatory approval for new products or for new indications or dosage forms for existing products. Decisions by regulatory authorities regarding labeling, ingredients and other matters could adversely affect the availability or commercial potential of our products, and there is no assurance that any of our proposed products will receive regulatory approval and/or be commercially successful.
| 16 |
| Table of Contents |
Post-Approval Data
As a condition to granting marketing approval of a product, the FDA may require a company to conduct additional clinical trials. The results generated in these Phase IV trials could result in loss of marketing approval, changes in product labeling, and/or new or increased concerns about the side effects or efficacy of a product. The Food and Drug Administration Amendments Act of 2007 (the FDAAA) gave the FDA enhanced post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information, and compliance with FDA-approved risk evaluation and mitigation strategies. The FDA’s exercise of its authority under the FDAAA has in some cases resulted, and in the future, could result, in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved products. Non-U.S. regulatory agencies often have similar authority and may impose comparable costs. Post-marketing studies, whether conducted by us or by others and whether mandated by regulatory agencies or voluntary, and other emerging data about marketed products, such as adverse event reports, may also adversely affect sales of our products. Further, the discovery of significant problems with a product similar to one of our products that implicate (or are perceived to implicate) an entire class of products could have an adverse effect on sales of the affected products. Accordingly, new data about our proposed products, or products similar to our proposed products, could negatively impact demand for these products due to real or perceived side effects or uncertainty regarding efficacy and, in some cases, could result in updated labeling, restrictions on use, product withdrawal or recall. Furthermore, new data and information, including information about product misuse, may lead government agencies, professional societies, practice management groups or organizations involved with various diseases to publish guidelines or recommendations related to the use of these products or the use of related therapies or place restrictions on sales. Such guidelines or recommendations may lead to lower sales of these products if and when they reach the market.
The expiration or loss of patent protection may affect future revenues and operating income.
The Company’s proposed products may rely on patent and trademark and other intellectual property protection. To the extent any of the Company’s intellectual property are successfully challenged, invalidated, or circumvented or to the extent it does not allow the Company to compete effectively, our business may suffer.
Competitors' intellectual property may prevent the Company from selling its proposed products or have a material adverse effect on the Company’s future profitability and financial condition.
Competitors may claim that our product infringes upon their intellectual property. Resolving an intellectual property infringement claim can be costly and time consuming and may require the Company to enter into license agreements. The Company cannot guarantee that it would be able to obtain license agreements on commercially reasonable terms. A successful claim of patent or other intellectual property infringement could subject the Company to significant damages or an injunction preventing the manufacture, sale or use of affected products. Any of these events could have a material adverse effect on our profitability and financial condition.
The Company research and development efforts may not succeed in developing commercially successful products and technologies, which may cause revenue and profitability to decline.
The Company is committing substantial efforts, funds, and other resources to research and development of its proposed products. A high rate of failure is inherent in the research and development of new products and technologies. The Company will be required to make ongoing substantial expenditures without any assurance that its efforts will be commercially successful. Failure can occur at any point in the process, including after significant funds have been invested.
Promising new product candidates may fail to reach the market or may only have limited commercial success because of efficacy or safety concerns, failure to achieve positive clinical outcomes, inability to obtain necessary regulatory approvals, limited scope of approved uses, excessive costs to manufacture, the failure to establish or maintain intellectual property rights, or infringement of the intellectual property rights of others. Even if the Company successfully develops new products or enhancements, they may be quickly rendered obsolete by changing customer preferences, changing industry standards, or competitors' innovations. Innovations may not be accepted quickly in the marketplace because of, among other things, entrenched patterns of clinical practice or uncertainty over third-party reimbursement. The Company cannot state with certainty when or whether any of its products under development will be launched, whether it will be able to develop, license, or otherwise acquire compounds or products, or whether any products will be commercially successful. Failure to launch successful new products or new indications for existing products may cause the Company’s products to become obsolete, causing our revenue and operating results to suffer.
| 17 |
| Table of Contents |
New products and technological advances by our competitors may negatively affect our operations.
Any products that the Company develops will face intense competition from its competitors' products. Competitors' products may be safer, more effective; more effectively marketed or sold, or have lower prices or superior performance features than our products.
Liability claims may occur having a material adverse effect on revenues and financial condition.
The Company, once its product(s) make it to market, may be subject to product liability claims and lawsuits alleging that its products have resulted or could result in an unsafe condition for or injury to patients. Product liability claims and lawsuits, safety alerts or product recalls, and other allegations of product safety or quality issues, regardless of their validity or ultimate outcome, may have a material adverse effect on our business and reputation and on our ability to attract and retain customers. Consequences may also include additional costs, a decrease in market share for the products, lower income, or exposure to other claims. The Company has obtained product liability insurance but may not be able to obtain such coverage or obtain sufficient coverage to protect itself completely from said potential claims. Product liability claims could have a material adverse effect on our profitability and financial condition.
We have a limited operating history and are not profitable and may never become profitable.
We have a history of operating losses and no meaningful operations upon which to evaluate our business. Our accumulated deficit since inception through March 31, 2024 was $24,260,457. We expect to incur substantial losses and negative operating cash flow for the foreseeable future as we commence clinical trials of our drug candidates, which we do not expect will be commercially available for several years, if at all. Even if we succeed in developing and commercializing one or more drug candidates, we expect to incur substantial losses for the foreseeable future and may never become profitable. The successful development and commercialization of any drug candidates will require us to perform a variety of functions, including:
| · | undertaking phase 2 clinical trials; |
|
|
|
| · | hiring additional personnel; |
|
|
|
| · | participating in the regulatory approval processes; |
|
|
|
| · | manufacturing and formulating products; |
|
|
|
| · | initiating and conducting sales and marketing activities; and, |
|
|
|
| · | implementing additional internal systems and infrastructure. |
We will need to raise additional capital to fund our business and generate significant revenue to achieve and maintain profitability. Additional financing may cause dilution to current investors and there can be no assurance that any additional financing will be on terms that are favorable to the Company and our shareholders. Without ongoing revenue, our ability to stay in business is contingent on outside capital and we currently have no commitments for such capital.
The departure of key personnel could affect the Company due to the loss of their expertise.
Our business plan was developed by our officers and will depend on their ability to develop pharmaceutical products. Without their expertise, it is unlikely we will be able to complete the development, testing and FDA approval process. We do not have the funds, at this time, to hire additional personnel and without our current management team; it is unlikely we would be able to obtain further funding. The loss of any member of management would severely hinder our ability to develop our proposed products. A failure on our part to retain the services of these key personnel could have a material adverse effect on our operating results and financial conditions. We do not maintain key man life insurance on any of our officers or employees.
| 18 |
| Table of Contents |
Our auditors have expressed substantial doubt about our ability to continue as a going concern.
Our audited financial statements for the year ended March 31, 2024 were prepared using the assumption that we will continue our operations as a going concern. We were incorporated in 2014 and do not have a history of earnings except for the years ended March 31, 2017 and 2022. As a result, our independent registered public accounting firm, in their audit report, has expressed substantial doubt about our ability to continue as a going concern. Continued operations are dependent on our ability to complete equity or debt financing activities or to generate profitable operations. Such activities may not be available or may not be available on reasonable terms. We believe that if we do not have sufficient funding, we may have to suspend or cease operations within twelve months. Therefore, we may be unable to continue operations in the future as a going concern. If we cannot continue as a viable entity, our stockholders may lose some or all their investment in the Company.
We may be exposed to risks relating to management’s conclusion that our disclosure controls and procedures and internal controls over financial reporting are ineffective.
Currently, we do not have an independent audit committee. Our independent Director along with the other Directors functions as our audit committee and is comprised of three directors, two of whom are not considered to be "independent" in accordance with the requirements of Rule 10A-3 under the Securities Exchange Act of 1934. An independent audit committee plays a crucial role in the corporate governance process, assessment of the Company's processes relating to its risks and control environment, oversight of financial reporting, and evaluation of internal and independent audit processes. The lack of an independent audit committee may prevent the Board of Directors from being independent in its judgments and its ability to pursue the committee's responsibilities, this could compromise management of our business.
Risks Relating to Our Common Stock
The Company’s stock price may be volatile.
The market price of the Company’s common stock is likely to be highly volatile and could fluctuate widely in price in response to various factors, many of which are beyond the Company’s control, including the following:
| · | technological innovations or new products and services by the Company or its competitors. |
| · | additions or departures of key personnel. |
| · | the Company’s ability to execute its business plan. |
| · | operating results that fall below expectations. |
| · | loss of any strategic relationship. |
| · | industry developments. |
| · | economic and other external factors; and, |
| · | period-to-period fluctuations in the Company’s financial results. |
In addition, the securities markets have from time-to-time experienced significant price and volume fluctuations that are unrelated to the operating performance of companies. These market fluctuations may also materially and adversely affect the market price of the Company’s common stock.
We may in the future issue additional shares of our common stock which would reduce investors’ ownership interests in the Company, and which may dilute our share value.
Our Articles of Incorporation were amended to increase the authorized shares to 550,000,000 of which 500,000,000 shares are common Shares, par value $0.001 per share and 50,000,000 shares are preferred shares, $0.001 par value. The future issuance of all or part of our remaining authorized common stock may result in substantial dilution in the percentage of our common stock held by our then existing stockholders. We may value any common stock issued in the future on an arbitrary basis. The issuance of common stock for future services or acquisitions or other corporate actions may have the effect of diluting the value of the shares held by our investors and might have an adverse effect on any trading market for our common stock.
The Company authorized a class of Series A convertible non-voting preferred stock with rights and preferences superior to those of the common stockholders and which might contain provisions giving them priority over the rights of the common stockholders. Any such class of preferred stock may result in substantial dilution to our common stockholders and have an adverse effect on any trading market for our common stock.
| 19 |
| Table of Contents |
FINRA sales practice requirements may limit a stockholders’ ability to buy and sell our stock.
The Financial Industry Regulatory Authority (“FINRA”) has adopted rules that relate to the application of the SEC’s penny stock rules in trading our securities and require that a broker/dealer have reasonable grounds for believing that the investment is suitable for that customer, prior to recommending the investment. Prior to recommending speculative, low priced securities to their non-institutional customers, broker/dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information.
Under interpretations of these rules, FINRA believes that there is a high probability that speculative, low priced securities will not be suitable for at least some customers. FINRA’s requirements make it more difficult for broker/dealers to recommend that their customers buy our common stock, which may have the effect of reducing the level of trading activity and liquidity of our common stock. Further, many brokers charge higher transactional fees for penny stock transactions. As a result, fewer broker/dealers may be willing to make a market in our common stock, reducing a shareholder’s ability to resell shares of our common stock.
The Company’s common stock is currently deemed to be “penny stock,” which makes it more difficult for investors to sell their shares.
The Company’s common stock is and will be subject to the “penny stock” rules adopted under section 15(g) of the Exchange Act. The penny stock rules apply to companies whose common stock is not listed on the NASDAQ Stock Market or other national securities exchange and trades at less than $5.00 per share or that have tangible net worth of less than $5,000,000 ($2,000,000 if the company has been operating for three or more years). These rules require, among other things, that brokers who trade penny stock to persons other than “established customers” complete certain documentation, make suitability inquiries of investors and provide investors with certain information concerning trading in the security, including a risk disclosure document and quote information under certain circumstances. Many brokers have decided not to trade penny stocks because of the requirements of the penny stock rules and, as a result, the number of broker-dealers willing to act as market makers in such securities is limited. If the Company remains subject to the penny stock rules for any significant period, it could have an adverse effect on the market, if any, for the Company’s securities. If the Company’s securities are subject to the penny stock rules, investors will find it more difficult to dispose of the Company’s securities.
ITEM 1B: UNSOLVED STAFF COMMENTS
As a “smaller reporting company”, we are not required to provide the information required by this Item.
ITEM 1C: CYBERSECURITY.
We are highly dependent on third-party provided software applications to conduct key operations. We depend on both our own systems as well as the systems, networks and technology of our contractors, consultants, vendors and other business partners.
Our cybersecurity evaluation identifies various risks and issues that we continue to mitigate to further improve our program. This includes:
· | Establishing a cybersecurity training program.. |
· | Setting up and implementing a third-party risk management program to support a Third-Party Risk Management Policy and process to assess the risks associated with our critical third-party vendor engagements. |
· | Setting up and testing a Cybersecurity Incident Response Plan. |
· | Establishing additional processes for identifying cybersecurity threats and vulnerabilities within the environment in which we operate. |
· | Enhancing our technical security safeguards and configurations. |
| 20 |
| Table of Contents |
Process for Assessing, Identifying and Managing Material Risks from Cybersecurity Threats
In the event of a cybersecurity incident designated personnel are responsible for assessing the severity of an incident and associated threat, containing the threat, remediating the threat, including recovery of data and access to systems, analyzing any reporting obligations associated with the incident, and performing post-incident analysis and program enhancements. Our Company maintains an insurance policy through an independent insurance company for claims pursuant to a cybersecurity claim for damages.
ITEM 2: PROPERTIES
Our principal executive offices are located at 631 US Hwy 1 Suite 407, N Palm Beach, FL 33408, which we receive rent free on a month to month basis. Our telephone number is (612) 961-5656. We have no policies with respect to investments in real estate or interests in real estate, real estate mortgages, or securities of or interests in persons primarily engaged in real estate activities.
ITEM 3: LEGAL PROCEEDINGS
None
ITEM 4: MINE SAFETY DISCLOSURE
Not applicable
ITEM 5: MARKET FOR REGISTRANT’S COMMON EQUITY AND RELATED SHAREHOLDER MATTERS
Our shares of common stock are currently trading on the Over the Counter Market: Bulletin Board under the Symbol “NBIO”.
The following quotations, obtained from www.nasdaq.com, reflect the high and low bids for our common shares.
The high and low bid prices of our common stock for the periods indicated below are as follows:
Quarter Ended |
| High |
|
| Low |
| ||
March 31, 2024 |
| $ | 0.18 |
|
| $ | 0.08 |
|
December 31, 2023 |
| $ | 0.28 |
|
| $ | 0.07 |
|
September 30, 2023 |
| $ | 0.17 |
|
| $ | 0.04 |
|
June 30, 2023 |
| $ | 0.08 |
|
| $ | 0.03 |
|
March 31, 2023 |
| $ | 0.20 |
|
| $ | 0.04 |
|
December 31, 2022 |
| $ | 0.31 |
|
| $ | 0.14 |
|
September 30, 2022 |
| $ | 0.45 |
|
| $ | 0.15 |
|
June 30, 2022 |
| $ | 0.32 |
|
| $ | 0.05 |
|
(1) Over-the-counter market quotations reflect inter-dealer prices without retail mark-up, mark-down, or commission, and may not represent actual transactions.
Holders
As of March 31, 2024, there were approximately 164 holders of record of our common stock and 170,594,165 shares of common stock issued and outstanding.
Our transfer agent is Transfer Online, Inc. Its mailing address is 512 SE Salmon Street, Portland, OR, 97214 and its telephone number is (503) 227-2950.
| 21 |
| Table of Contents |
Equity Compensation Plans
We have adopted a 2015 Stock Option Plan (the “2015 Plan”) which was amended in the year ending March 31, 2020 under which we are authorized to issue up to a maximum of 10,000,000 incentive stock options and non-qualified stock options to our directors, officers, employees and consultants. The 2015 Plan authorizes the Board of Directors or a committee thereof, to grant awards of incentive stock options and non-qualified stock options upon such terms and conditions as the Board may determine.
We currently have 930,000 options issued and outstanding under our 2015 Stock Option Plan (amended March 9, 2020) which have been granted to two key officers and directors and plus three consultants.
Purchase of Equity Securities by the Issuer and Affiliated Purchasers
We did not purchase any of our shares of common stock or other securities during our fiscal year ended March 31, 2024.
Recent Sales of Unregistered Securities; Use of Proceeds from Registered Securities
On July 9, 2020, the Company increased its authorized shares to 550,000,000 consisting of 50,000,000 preferred shares and 500,000,000 common shares all at par value $0.001.
Preferred Stock
On July 10, 2019, the Company filed an amended articles of Incorporation designating 1,500,000 shares of preferred stock as Series A non-voting Convertible preferred shares convertible into common stock at four shares of common for each one share of preferred.
As of March 31, 2024 there were no preferred shares outstanding.
Common Stock
During the year ended March 31, 2023 the Company issued 37,700 shares of common stock with a value of $9,275 for accounts payable.
During the year ended March 31, 2023, the Company issued 1,000,000 shares of common stock for clinical consulting with a value of $60,000.
During the year ended March 31, 2023, the Company 4,200,000 shares of common stock for the conversion of 4,200,000 warrants with a value of $208,000 received in cash.
During the year ended March 31, 2023 , the Company issued 3,815,414 shares of common stock with a value of $624,320 to three related parties for service.
During the year ended March 31, 2023 the Company issued 11,398,059 shares of common stock with a value of $1,023,975 for the conversion of debt and accrued interest.
During the year ended March 31, 2024 , the Company issued 7,186,385 shares of common stock with a value of $773,243 to three related parties for service.
During the year ended March 31, 2024 the Company issued 10,787,180 shares of common stock with a value of $684,054 for the conversion of debt and accrued interest.
During the year ended March 31, 2024 the Company issued 19,970,000 shares of common stock with a value of $1,561,000 for cash.
| 22 |
| Table of Contents |
During the year ended March 31, 2024 the Company issued 886,250 shares of common stock with a value of $60,850 for service. The Company issued 500,000 shares of these as conversion of warrants paid by the accounts payable of one party of $25,000.
The Company relied upon exemptions from registration of these securities issuances pursuant to Regulation D and Section 4(2) of the Securities Act of 1933, as amended.
ITEM 6: SELECTED FINANCIAL DATA.
As a “smaller reporting company”, we are not required to provide the information required by this Item.
ITEM 7: MANAGEMENT’S DISCUSSION AND ANALYSIS OR PLAN OF OPERATION
OVERVIEW
The following discussion should be read in conjunction with our audited financial statements and the related notes for the years ended March 31, 2024 and March 31, 2023 that appear elsewhere in this annual report. The following discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to such differences include but are not limited to those discussed below and elsewhere in this annual report, particularly in the section entitled "Risk Factors".
Our consolidated financial statements are stated in United States Dollars and are prepared in accordance with United States Generally Accepted Accounting Principles.
Results of Operations
The following summary of our results of operations should be read in conjunction with our consolidated financial statements for the years ended March 31, 2024 and 2023, which are included herein.
Expenses
Operating expenses for the year ended March 31, 2024 were $2,229,879, including research and development costs of $110,975, clinical trials of $92,466, manufacturing and vial filling $319,507 general and administrative expenses of $403,589 and consulting of $1,303,342 compared to the year ended March 31, 2023 in which our operating expenses were $2,155,757 with research and development costs of $270,529, phase 1 clinical trials of $383,688, consulting of $1,096,245 and general and administrative costs of $405,295. Operating expenses increased slightly in the year ended March 31, 2024 over the same period in 2023 by $74,122 due primarily to higher product manufacturing and vialing costs in 2024 verses 2023.
Other income totaled $142,197 for the year ended March 31, 2024 compared to other expense of $645,212 for the same period in 2023. Other income consisted of change in fair value of $472,168, original debt discount to interest of $25,000, interest expense of $385,236, other income of $5,265 and gain on debt forgiveness of $50,000 in the year ended March 31, 2024. This compared to interest expense of $1,566,919, gain in debt forgiveness of $28,000, finance costs of $5,124, original debt discount to interest of $156,229 and a change in fair value of $1,054,786 and other income of $274 for the same period in 2023.
| 23 |
| Table of Contents |
Revenue, Net Income and Loss
The Company recorded no revenue for the years ended March 31, 2024 and 2023.
Our net loss for the year ended March 31, 2024 was $2,087,682 compared to a net loss of $2,800,969 during the year ended March 31, 2023. The decrease in net loss for the year ended March 31, 2024 is due to lower clinical trials expense and lower interest expense in 2024 compared to the same period in 2023.
Our operations to date have been financed by the sale of our common stock and initial payments of a licenses sale. Our largest expenses to date have been research and development, IND filings and tests required for the filing and consulting fees.
We do not anticipate generating revenues in the foreseeable future , and any revenues that we generate may not be sufficient to cover our operating expenses. If we do not succeed in raising additional capital, we may have to cease operations and you may lose your entire investment.
Liquidity and Capital Resources
At March 31, 2024 we had cash of $372,120 and prepaid of $180,000 compared to cash of $172,186 and prepaid of $82,816 in the same period in 2023. Our accounts payable and accrued expense at March 31, 2024 was $686,294 consisting of $684,115 in accounts payable and accrued expense as compared to $498,122 in accounts payable and accrued expense as of March 31, 2023. Other liabilities include a convertible notes of $125,000
We had no revenues to help satisfy some of our ongoing liabilities.. Our auditors have issued a going concern opinion. Unless we secure additional equity or debt financing, of which there can be no assurance, we may not be able to continue any operations.
Working Capital
Our total current assets, as of March 31, 2024, consisted of $372,120 in cash and $180,000 in prepaid as compared to total current assets of $172,186 consisting of cash and prepaid of $82,816 in the same period in 2023.
Our total current liabilities as of March 31, 2024 were $808,794 as compared to total current liabilities of $1,551,139 as of March 31, 2023. The decrease in current liabilities in the year ended March 31, 2024 over the same period in 2023 was primarily attributed to the reduction of convertible notes to $125,000 and derivative liability of zero in the year ended March 31, 2024 compared to convertible notes of $389,591 and derivative liability of $ 663,426 in the same period in 2023.
As of March 31, 2024 the Company’s negative working capital was $256,674 compared to negative working capital of $1,296,137 as of March 31, 2023. The lower negative working capital from 2024 over 2023 can be attributed to a reduction of the convertible notes and no derivative liability offset by higher accounts payable and accrued expenses in 2024 over 2023.
Cash Flows
Operating Activities
Cash used in operating activities was $1,186,066 for the fiscal year ended March 31, 2024 compared to cash used in operating activities of $1,787,228 in the year ended March 31, 2023. The decrease in cash used in operating activities as of March 31, 2024 is attributed to lower change in fair value on derivatives and lower debt discount amortization in 2024 over the same period in 2023.
Investing Activities
Cash used in investing activities was zero for the fiscal years ended March 31, 2024 and 2023.
| 24 |
| Table of Contents |
Financing Activities
Cash provided by financing activities during the fiscal year ended March 31, 2024 was $1,386,066 compared to cash provided by financing activities of $1,865,000 for the fiscal year ended March 31, 2023. The cash provided by financing activities in 2024 was due to proceeds common stock issued for cash of $1,561,000, convertible notes of $225,000 offset by repayment of convertible notes of $400,000, compared to proceeds from convertible notes of $1,932,000, conversion of warrants of $208,000 and repayment of convertible notes of $275,000 in the year ended March 31, 2023
Cash Requirements
We will require additional funds to fund our budgeted expenses over the next 12 months. These funds may be raised through equity financing, debt financing, or other sources, which may result in further dilution in the equity ownership of our shares. There is still no assurance that we will be able to maintain operations at a level sufficient for investors to obtain returns on their investments in our common stock. Further, we may continue to be unprofitable. We need to raise additional funds in the immediate future in order to proceed with our budgeted expenses including the clinical trials of our product.
Going Concern
The financial statements accompanying this report have been prepared on a going concern basis, which implies that our company will continue to realize its assets and discharge its liabilities and commitments in the normal course of business. The Company has no revenues and has not paid any dividends and is unlikely to pay dividends or generate earnings in the immediate or foreseeable future. The continuation of our company as a going concern is dependent upon the continued financial support from our shareholders, the ability of our company to obtain necessary equity financing to achieve our operating objectives, and the attainment of profitable operations. As of March 31, 2024, our company has accumulated deficit of $24,260,457 and a working capital deficit of $256,674. We do not have sufficient working capital to enable us to carry out our stated plan of operation for the next twelve months.
Due to the uncertainty of our ability to meet our current operating expenses and the capital requirements noted below in their report on the financial statements for the year ended March 31, 2024 our independent auditors, included an explanatory paragraph regarding concerns about our ability to continue as a going concern. Our financial statements contain additional note disclosures describing the circumstances that lead to this disclosure by our independent auditors. The continuation of our business is dependent upon us raising additional financial support. The issuance of additional equity securities by us could result in a significant dilution in the equity interests of our current stockholders. Obtaining commercial loans, assuming those loans would be available, will increase our liabilities and future cash commitments.
Future Financings
We will require additional financing of an estimated $15,000,000 to enable us to proceed with our plan of operations, including completion of phase 2 clinical trials plus presentation of data to the FDA to proceed to phase 3 clinical trials. These cash requirements are more than our current cash and working capital resources. Accordingly, we will require additional financing to continue operations and to repay our liabilities. There is no assurance that any party will advance additional funds to us to enable us to sustain our plan of operations or to repay our liabilities.
We anticipate continuing to rely on equity sales of our common stock to continue to fund our business operations. Issuances of additional shares will result in dilution to our existing stockholders. There is no assurance that we will achieve any additional sales of our equity securities or arrange for debt or other financing to fund our planned business activities.
We presently do not have any arrangements for additional financing for the expansion of our exploration operations, and no potential lines of credit or sources of financing are currently available for proceeding with our plan of operations.
If we are unable to raise the funds that we require to execute our plan of operation, we intend to scale back our operations commensurately with the funds available to us.
| 25 |
| Table of Contents |
Off-Balance Sheet Arrangements
We currently have no off-balance sheet arrangements.
Item 7A: Quantitative and Qualitative Disclosures About Market Risk
As a “smaller reporting company”, we are not required to provide the information required by this Item.
ITEM 8: FINANCIAL STATEMENTS and SUPPLEMENTARY DATA.
Financial statements are audited and included in this Form 10-K as an exhibit and are incorporated herein by this reference.
ITEM 9: CHANGE IN AND DISAGREEMENTS WITH ACCOUNTANTS AND FINANCIAL DISCLOSURE.
None
ITEM 9A: CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
For purposes of this section, the term disclosure controls and procedures means controls and other procedures of an issuer that are designed to ensure that information required to be disclosed by the issuer in the reports that it files or submits under the Securities Exchange Act of 1934, as amended (the “Act”) (15 U.S.C. 78a et seq.) is recorded, processed, summarized and reported, within the time periods specified in the Commission's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Act is accumulated and communicated to the issuer's management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. It should be noted that the design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote. As of the end of the period covered by this Annual Report, we carried out an evaluation, under the supervision and with the participation of our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures. Based on this evaluation, our CEO and CFO has concluded that the Company’s disclosure controls and procedures are not effective because of the identification of a material weakness in our internal control over financial reporting, which is identified below, which we view as an integral part of our disclosure controls and procedures.
Changes in Internal Controls over Financial Reporting
We have not made any changes in our internal controls over financial reporting that occurred during the period covered by this Form 10-K that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act of 1934. Our internal control system was designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes, in accordance with generally accepted accounting principles. Because of inherent limitations, a system of internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate due to change in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management conducted an evaluation of the effectiveness of our internal control over financial reporting as of March 31, 2024 using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework (2013). Based on its evaluation, our management concluded that our internal control over financial reporting was not effective as of March 31, 2024 and there are material weaknesses in our internal control over financial reporting. We lack full time personnel in accounting and financial staff to sufficiently monitor and process financial transactions in an efficient and timely manner. This allows for insufficient segregation of duties and a lack of multiple levels of supervision and review. Our history of losses has severely limited our budget to hire and train enough accounting and financial personnel needed to adequately provide this function. Consequently, we lacked sufficient technical expertise, reporting standards and written policies and procedures. A material weakness is a deficiency, or a combination of control deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.
This Form 10-K does not include an attestation report of the Company’s registered public accounting firm regarding internal control over financial reporting because the attestation report requirement has been removed for “smaller reporting companies” under the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010.
ITEM 9B: OTHER INFORMATION
None
| 26 |
| Table of Contents |
PART III
ITEM 10: DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
The following individuals serve as the directors and executive officers of our company as of the date of this annual report. All directors of our company hold office until the next annual meeting of our shareholders or until their successors have been elected and qualified. The executive officers of our company are appointed by our board of directors and hold office until their death, resignation, or removal from office.
Name | Position Held with Our Company |
Age | Date First Elected or Appointed |
Sean Carrick | President, Secretary, and Director | 56 | July, 2014 |
Doug Karas | Independent Director | 61 | April, 2016 |
Lowell Holden | Chief Financial Officer and Director | 81 | July, 2014 |
Business Experience
The following is a brief account of the education and business experience during at least the past five years of each director, executive officer and key employee of our company, indicating the person’s principal occupation during that period, and the name and principal business of the organization in which such occupation and employment were carried out.
The business experience during the past five years of the person presently listed above as an Officer or Director of the Company is as follows:
Lowell Holden – Chief Financial Officer and Director - Lowell Holden has been the Chief Financial Officer and Chief Accounting Officer of the Company since May of 2014. Since 1983, Mr. Holden has owned and operated his own consulting firm, LS Enterprises, Inc., which provides business consulting, accounting, and other services to businesses. Mr. Holden has a broad range of business experience including managing, securing financing, structuring of transactions, and is experienced and knowledgeable in managing relationships with customers, financing institutions and stockholders. Presently Mr. Holden serves as the Chief Financial Officer of Skkynet Cloud Systems, Inc (SKKY) and director of EMR Technology, and director of PTS, Inc (PTSH). Mr. Holden also has a background in assisting companies in fulfilling their financial auditing and SEC reporting requirements. Mr. Lowell Holden has a Bachelor of Science degree from Iowa State University.
| 27 |
| Table of Contents |
Sean Carrick – President and Director - Sean Carrick brings to Nascent a career that spans more than 25 years of experience building and leading successful medical device, Pharmaceutical and Biotech companies in large, mid-cap and venture-backed stages. Previously, Mr. Carrick served as President of Silver Star Mining Corporation from January 1, 2013 to November 2013 where he was responsible for all business management and strategic direction. Prior to Silverstar, Mr. Carrick served as Director of Sales, Southern US, from August 2010 through November 2012 at Maquet Medical Systems and Florida Director of Sales at the Linvatec Division of Conmed Corporation (December 2007 through July 2010). Mr. Carrick holds a BS Degree in Economics and Business Administration from Duquesne University and strategic leadership and management certificates from the Cogency Group, Eckerd College and Maquet Medical Systems.
Douglas J. Karas – Independent Director - Douglas J. Karas served as Vice President, Performance Analysis, and Investment Risk for Franklin Templeton Companies LLC. Mr. Karas is responsible for investment Regulatory Governance, GIPS® Compliance, Fund Board, Sales and Marketing support. Mr. Karas brings cross-functional leadership and collaboration with a strong customer service focus at all organizational levels. Mr. Karas is an Affiliate member of the Tampa Bay CFA Society. Mr. Karas has been with Franklin Templeton Investments for 18 years. Prior to this position, he was the director- Financial Business Processes & Systems and was responsible for the firm’s corporate finance applications including treasury, accounting, corporate and fund taxation and corporate development and planning. He has extensive operations management, consulting, risk and regulatory compliance, re-engineering, project management, and system development experience. Previous positions with Franklin Templeton include senior manager financial operations, manager financial program management office and manager treasury accounting control compliance. Mr. Karas’ experience cuts across operations and applications and he has exposure to treasury cash, deal and risk; revenue calculations and recognition; external and internal financial reporting (including 10-K/10-Q, Business Line Profitability, Transfer Pricing and Fund Profitability), financial instruments (dollar rolls, derivatives, ABS and MBS- Security set-up and Maintenance, Pricing, Revenue Recognition, etc.); and Procedures of Portfolio Managers and Investment Advisors in accordance with the Securities Act of 1933 and 1934, Investment Company and Investment Advisor Acts of 1940. Prior to joining Franklin Templeton, Mr. Karas held positions with Transamerica Life Companies and Federated Investors. Mr. Karas graduated with a B.S. in business, accounting from the University of Pittsburgh and is a Certified Public Accountant.
Identification of Significant Employees
As of the date of this Report, other than our current directors and officers, we have no other full-time or part-time employees.
Family Relationships
There are no family relationships among any of our directors, executive officers and proposed directors or executive officers.
Compliance with Section 16(a) of the Securities Exchange Act of 1934
Section 16(a) of the Securities Exchange Act of 1934, as amended, and the rules of the Securities and Exchange Commission (SEC) require our directors, executive officers and persons who own more than 10% of our Class A common stock to file reports of their ownership and changes in ownership of our Class A common stock with the SEC. Based solely upon a review of copies of such forms filed on Forms 3, 4, and 5, and amendments thereto furnished to us, we determined that none of the directors, executive officers or beneficial owners of more than 10% of our common stock failed to file a report on a timely basis during the year ended March 31, 2024.
Name | Position | Filed Reports Timely |
Sean Carrick | Officer, Director | Yes |
Douglas Karas | Director | Yes |
Lowell Holden | Officer, Director | Yes |
| 28 |
| Table of Contents |
Committees of the Board
All proceedings of our board of directors were conducted by resolutions consented to in writing by all the directors and filed with the minutes of the proceedings of the directors. Such resolutions consented to in writing by the directors entitled to vote on that resolution at a meeting of the directors are, according to the Incorporation laws of the state of Nevada and the bylaws of our company, as valid and effective as if they had been passed at a meeting of the directors duly called and held.
Our company currently does not have nominating, compensation committees or committees performing similar functions nor does our company have a written nominating, compensation, or audit committee charter. Our board of directors does not believe that it is necessary to have such committees because it believes that the functions of such committees can be adequately performed by our directors.
Our company does not have any defined policy or procedure requirements for shareholders to submit recommendations or nominations for directors. The directors believe that, given the early stage of our development, a specific nominating policy would be premature and of little assistance until our business operations develop to a more advanced level. Our company does not currently have any specific or minimum criteria for the election of nominees to the board of directors and we do not have any specific process or procedure for evaluating such nominees. Our directors assess all candidates, whether submitted by management or shareholders, and make recommendations for election or appointment.
Audit Committee and Audit Committee Financial Expert
Our audit committee consists of our outside director Douglas Karas who serves as Chairman of the committee and sole member. Mr. Karas is a Certified Public Account. Our board of directors has determined that none of the members of our audit committee qualifies as an "audit committee financial expert" as defined in Item 407(d)(5)(ii) of Regulation S-K and is "independent" as the term is used in Item 7(d)(3)(iv) of Schedule 14A under the Securities Exchange Act of 1934, as amended.
The audit committee along with the board of directors approves the retaining of the Independent Public Account Firm. When annual meets are held the firm is approved by the shareholders. At such meeting, representatives of the firm are invited to attend and interact with shareholders present through their independent statements plus answer appropriate questions from the shareholders in attendance. Continued engagement of the appointed Independent Registered Accounting Firm continues until the audit committee along with the board of directors makes a recommendation for change.
We believe that the members of our board of directors, which includes the audit committee chairman, are collectively capable of analyzing and evaluating our financial statements and understanding internal controls and procedures for financial reporting. In addition the audit committee and the board of directors reviews all correspondence from the audit firm along with qualifications and any changes in the personnel on the audit. The audit committee meets and or corresponds though the board members with the audit firm as needed.
Code of Ethics
We adopted a Code of Ethics applicable to all of our directors, officers, employees, and consultants, which is a “code of ethics” as defined by applicable rules of the SEC. If we make any amendments to our Code of Ethics other than technical, administrative, or other non-substantive amendments, or grant any waivers, including implicit waivers, from a provision of our Code of Ethics to our chief executive officer, chief financial officer, or certain other finance executives, we will disclose the nature of the amendment or waiver, its effective date and to whom it applies in a Current Report on Form 8-K filed with the SEC.
| 29 |
| Table of Contents |
ITEM 11: EXECUTIVE COMPENSATION.
The following tables sets for the compensation cash and accrual for all officers and directors during the past three years:
DIRECTORS and OFFICERS - COMPENSATION
Name and Principal Position |
| Annual compensation | Long-term compensation |
| |||||
| Salary ($) | Bonus ($) | Other annual compen -sation ($) | Awards | Payouts | All other compen- sation ($) |
| ||
Year | Restricted stock award(s) ($) | Securities under- lying options/ SARs (#) | LTIP payouts ($) |
Total Compensation | |||||
Sean Carrick President-Director(2) | 2024 2023
| 252,000 252,000
| --
| - -
| -- --
| - -
| - -
| - -
| 252,000 252,000
|
Lowell Holden(1) CFO, Director
| 2024 2023
| 180,000 180,000
| --
| - -
| -- -
| - -
| - - | - --
| 180,000 180,000
|
Douglas (3) Karas, Director | 2024 2023 |
| -- -- | -- -- | 20,000 20,000 | -- -- | -- -- | -- -- | 20,000 20,000 |
For the year ended March 31, 2024:
| (1) | The Chief Financial Officers was paid $180,000 in cash. |
| (2) | The President and Chief Executive Officer was paid $252,000 in cash. |
| (3) | The director of the Company was paid $20,000 in director fees which was paid in stock. |
As of March 31, 2024, the Company has no other Executive Compensation issues which would require the inclusion of other mandated table disclosures.
Narrative Disclosure to Summary Compensation Table
There are employment contracts, with our executive officers, that could result in payments to such person because of his or her resignation, retirement or other termination of employment with our company, or its subsidiaries, or any change in control, or a change in the person’s responsibilities following a change in control of our company.
Stock Option Plan
We have adopted a 2015 Stock Option Plan (the “2015 Plan”) under which we are authorized to issue up to a maximum of 1,500,000 incentive stock options and non-qualified stock options to our directors, officers, employees, and consultants. The 2015 Plan was approved by our stockholders. On March 8, 2020, the Board of Director under their authorization of the Bylaws of the Company increased the authorized options to 10,000,000. The 2015 Plan authorizes the Board of Directors or a committee thereof, to grant awards of incentive stock options and non-qualified stock options upon such terms and conditions as the Board may determine. The Board of Directors administer the 2015 Plan.
| 30 |
| Table of Contents |
As of March 31, 2024, the Company has 930,000 options issued and outstanding under our Stock Option Plan which have been granted to two officers and directors and two consultants. Each of the foregoing individuals has been awarded options which will vest in equal annual installments over a four year period with the first 20% vesting at the date of grant. All the options are exercisable between $0.18 and $0.50 per share.
The following table sets forth outstanding options and holders as of March 31, 2024:
Recipient |
| Title |
|
| Number Options |
| ||
Sean Carrick |
| Officer and Director |
|
|
| 375,000 |
| |
Lowell Holden |
| Officer and Director |
|
|
| 375,000 |
| |
Employees and consultants as a group |
|
| -- |
|
|
| 180,000 |
|
Total |
|
|
|
|
|
| 930,000 |
|
Equity Compensation Plan Information
Plan Category |
| Number of securities to be issued upon exercise of outstanding options, warrants and rights |
|
| Weighted-average exercise price of outstanding options, warrants and rights |
|
| Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a) |
| |||
|
| (a) |
|
| (b) |
|
| (c) |
| |||
Equity compensation plans approved by security holders |
|
| 10,000,000 |
|
|
| -- |
|
|
| 9,070,000 |
|
Equity compensation plans not approved by security holders |
|
| -- |
|
|
| -- |
|
|
| -- |
|
Total |
|
| 10,000,000 |
|
|
| -- |
|
|
| 9,070,000 |
|
The Company utilizes the shares available under the Plan described above to issue shares of stock as compensation to employees, consultants and officers and directors. At the end of each quarter, the Board of Directors of the Company determines the number of shares to be issued pursuant to the Plan.
Compensation of Directors
We reimburse our directors for expenses incurred in connection with attending board meetings. We pay our independent director $5,000 per quarter either in cash or stock for being a director. Directors who are officers of the Company do not receive compensation for being directors.
We have no formal plan for compensating our directors for their service in their capacity as directors, although such directors have received stock options to purchase common shares as awarded by our board of directors. Directors are entitled to reimbursement for reasonable travel and other out-of-pocket expenses incurred in connection with attendance at meetings of our board of directors. No director received and/or accrued any cash compensation for their services as a director, including committee participation and/or special assignments. The directors have been awarded an aggregate of 750,000 options for the efforts as directors.
| 31 |
| Table of Contents |
Pension, Retirement or Similar Benefit Plans
There are no arrangements or plans in which we provide pension, retirement or similar benefits for directors or executive officers. We have no material bonus or profit sharing plans pursuant to which cash or non-cash compensation is or may be paid to our directors or executive officers, except that stock options may be granted at the discretion of the board of directors or a committee thereof.
ITEM 12: SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS and MANAGEMENT and RELATED STOCKHOLDER MATTERS
The following table sets forth certain information concerning the number of shares of common stock owned beneficially as of March 31, 2024 by: (i) each of our directors; (ii) each of our named executive officers; and (iii) each person or group known by us to beneficially own more than 5% of our outstanding shares of common stock. Unless otherwise indicated, the shareholders listed below possess sole voting and investment power with respect to the shares of common stock they own.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS | ||||||||||
Title of Class |
| Name and Address of Beneficial Owner |
| Amount and Nature of Beneficial Ownership |
|
| Percentage of Class (1) |
| ||
Common |
| Douglas Karas- Director 631 US Hwy 1 Suite 407 N Palm Beach, FL 33408 |
|
| 1,744,552 |
|
|
| 1.02 | % |
Common |
| Lowell Holden – CFO & Director (1) 631 US Hwy 1 Suite 407 N Palm Beach, FL 33408 |
|
| 12,029,997 |
|
|
| 7.05 | % |
Common |
| Dr Robert D Burke – 5% shareholder 631 US Hwy 1 Suite 407 N Palm Beach, FL 33408 |
|
| 25,768,464 |
|
|
| 15.17 | % |
Common |
| Sean Carrick President, Secretary & Director 6631 US Hwy 1 Suite 407 N Palm Beach, FL 33408 |
|
| 22,934,007 |
|
|
| 13.44 | % |
Common |
| Sid Lorio 5% shareholder 631 US Hwy 1 Suite 407 North Palm Beach, FL 33408 |
|
| 10,281,249 |
|
|
| 6.02 | % |
|
| All directors and executive officers as a group (3 persons) |
|
| 36,708,556 |
|
|
| 21.51 | % |
(1) Mr. Holden holds 10,029,997shares directly and 2, 000,000 shares indirectly
We are unaware of any contract or other arrangement or provisions of our Articles or Bylaws the operation of which may at a subsequent date result in a change of control of our company. There are not any provisions in our Articles or Bylaws, the operation of which would delay, defer, or prevent a change in control of our company.
| 32 |
| Table of Contents |
ITEM 13: CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
On September 1, 2020, the Company entered five-year compensation agreements with two of its officers and directors. Under the terms of the agreements the Company issued shares of common stock to the officers and directors equaling 18% of the outstanding shares of the Company as additional future shares are issued. The officers and directors are entitled to additional future shares, so their aggregate ownership percentage remains at 18% of the future outstanding shares of the Company. In addition, the officers ae entitled to future bonuses including a signing bonus totaling $170,000 which was has not been accrued during the period ending March 31, 2024 plus additional bonuses based on their performance.
Officer and Director |
| Fiscal Year Annualized Compensation Base Being Paid |
|
| Non-dilutive shares percentage |
| ||
President and CEO (2) |
| $ | 252,000 |
|
|
| 12 | % |
Chief Financial Officer (1) |
| $ | 180,000 |
|
|
| 6 | % |
Director (3) |
| $ | 20,000 |
|
|
| -- |
|
Total |
| $ | 592,000 |
|
|
| 18 | % |
For the year ended March 31, 2024:
| (1) | The Chief Financial Officers was paid $180,000 in cash. |
| (2) | The President and Chief Executive Officer was paid $252,000 in cash. |
| (3) | The director of the Company was paid $20,000 in director fees which was paid in common stock of the Company. |
The following table sets forth the shares earned under these contracts from inception through year ended March 31, 2024:
Officer and Director |
| Initial Share Awards Under the Contracts |
|
| Additional Shares Earned to Maintain Ownership Percentage |
|
| Total Shares Earned |
| |||
President |
|
| 1,028,910 |
|
|
| 14,712,669 |
|
|
| 15,741,579 |
|
Chief Financial Officer |
|
| 617,346 |
|
|
| 7,588,673 |
|
|
| 8,206,019 |
|
Total |
|
| 1,646,256 |
|
| 22.301.342 |
|
|
| 23,947,598 |
| |
During the year ended March 31, 2024 the Company issued 7,186,387 shares of common stock with a value of $773,241 to three related parties for service.
Except as described above, none of the following persons has any direct or indirect material interest in any transaction to which we are a party during the past two years, or in any proposed transaction to which our company is proposed to be a party:
A. | any director or officer; |
B. | any proposed nominee for election as a director; |
C. | any person who beneficially owns, directly or indirectly, shares carrying more than 5% of the voting rights attached to our common stock; or |
D. | any relative or spouse of any of the foregoing persons, or any relative of such spouse, who has the same house as such person or who is a director or officer of any parent or subsidiary. |
Board Independence
As currently constituted and applying the rules of NASDAQ one of the members of our Board of Directors is independent. We are committed to eventually establishing a board in which a majority of our members consist of independent directors, as defined under the NASDAQ rules. Our ability to implement this goal will depend upon the growth of the Company and our ability to attract and compensate strategic persons willing to serve in that function.
We currently act with three directors, consisting of Lowell Holden, Doug Karas and Sean Carrick. We have a standing but not independent audit committee except for our one independent director, Doug Karas, but not a standing compensation or nominating committee, for which our entire board of director’s act in such capacities. We believe that our members of our board of directors are capable of analyzing and evaluating our financial statements and understanding internal controls and procedures for financial reporting.
| 33 |
| Table of Contents |
ITEM 14: PRINCIPAL ACCOUNTING FEES AND SERVICE.
The following table presents for the fiscal year March 31, 2024, $38,389, the aggregate fees billed in connection with the audits of our financial statements and other professional services rendered by our independent registered public accounting Fruci & Associates II, PLLC.
|
| 2024 |
|
| 2023 |
| ||
Audit fees |
| $ | 37,300 |
|
| $ | 32,250 |
|
Audit related fees |
|
| 1,089 |
|
|
| -- |
|
Tax fees |
|
| -- |
|
|
| -- |
|
All other fees |
|
| -- |
|
|
| -- |
|
Audit fees represent the professional services rendered for the audit of our annual financial statements and the review of our financial statements included in quarterly reports, along with services normally provided by the accounting firm in connection with statutory and regulatory filings or engagements. Audit-related fees represent professional services rendered for assurance and related services by the accounting firm that are reasonably related to the performance of the audit or review of our financial statements that are not reported under audit fees.
Tax fees represent professional services rendered by the accounting firm for tax compliance, tax advice, and tax planning. All other fees represent fees billed for products and services provided by the accounting firm, other than the services reported for in the other categories.
ITEM 15: EXHIBITS, FINANCIAL STATEMENT SECHEDULES.
(a) | Financial Statements | |
|
|
|
(1) | Financial statements for our company are listed in the index under Item 8 of this document | |
|
|
|
(2) | All financial statement schedules are omitted because they are not applicable, not material or the required information is shown in the financial statements or notes thereto. | |
|
|
|
(b) | Exhibits | |
Exhibit Number |
| Description |
(31) |
| Section 302 Certifications |
| ||
(32) |
| Section 906 Certification |
| ||
101** |
| Interactive Data Files |
101.INS |
| Inline XBRL Instance Document |
101.SCH |
| Inline XBRL Taxonomy Extension Schema Document. |
101.CAL |
| Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
101.DEF |
| Inline XBRL Taxonomy Extension Definition Linkbase Document. |
101.LAB |
| Inline XBRL Taxonomy Extension Label Linkbase Document. |
101.PRE |
| Inline XBRL Taxonomy Extension Presentation Linkbase Document. |
104 |
| Cover Page Interactive Data File (Formatted as inline XBRL and contained in Exhibit 101). |
| 34 |
| Table of Contents |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereto duly authorized.
|
| NASCENT BIOTECH INC. |
|
|
| (Registrant) |
|
|
|
|
|
Dated: July 1, 2024 | By: | /s/ Sean Carrick |
|
| Sean Carrick, President |
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Dated: July 1, 2024 | By: | /s/ Sean Carrick |
|
|
| Sean Carrick, President, Secretary and Director |
|
|
|
|
|
Dated: July 1, 2024 | By: | /s/ Douglas Karas |
|
|
| Douglas Karas, Independent Director |
|
|
|
|
|
Dated: July 1, 2024 | By: | /s/ Lowell Holden |
|
|
| Lowell Holden, Chief Financial Officer, and Director |
|
| 35 |
| Table of Contents |

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Nascent Biotech, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Nascent Biotech, Inc.(“the Company”) as of March 31, 2024 and 2023, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the years in the two-year period ended March 31, 2024, and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2024 and 2023 and the results of its operations and its cash flows for each of the years in the two-year period ended March 31, 2024, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 3 to the financial statements, the Company has an accumulated deficit, net losses, and negative cash flows from operations. These factors, among others, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 3. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there were no critical audit matters.
We have served as the Company’s auditor since 2019.
| |
July 1, 2024 |
|
| 36 |
| Table of Contents |
NASCENT BIOTECH, INC.
CONSOLIDATED BALANCE SHEETS
AS OF MARCH 31,
|
| 2024 |
|
| 2023 |
| ||
ASSETS |
|
|
|
|
|
| ||
Current assets: |
|
|
|
|
|
| ||
Cash |
| $ |
|
| $ |
| ||
Prepaid |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Total current assets |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Total assets |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable and accrued expense |
| $ |
|
| $ |
| ||
Convertible note- net of discount |
|
|
|
|
|
| ||
Derivative liability |
|
|
|
|
|
| ||
Total current liabilities |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Total liabilities |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Commitments and Contingencies |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
Preferred stock, $ |
|
|
|
|
|
| ||
Common stock, $ |
|
|
|
|
|
| ||
Additional paid-in capital |
|
|
|
|
|
| ||
Accumulated deficit |
|
| ( | ) |
|
| ( | ) |
Total stockholders’ equity (deficit) |
|
| ( | ) |
|
| ( | ) |
Total liabilities and stockholders’ equity |
| $ |
|
| $ |
| ||
The accompanying notes are an integral part of these consolidated financial statements.
| 37 |
| Table of Contents |
NASCENT BIOTECH, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
FOR YEARS ENDED MARCH 31,
|
| 2024 |
|
| 2023 |
| ||
|
|
|
|
|
|
| ||
Operating expenses: |
|
|
|
|
|
| ||
Consulting expense |
| $ |
|
| $ |
| ||
General and administrative expense |
|
|
|
|
|
| ||
Clinical trials |
|
|
|
|
|
| ||
Product manufacturing and vial filling |
|
|
|
|
|
| ||
Research and development |
|
|
|
|
|
| ||
Total operating expense |
|
|
|
|
|
| ||
Income (loss) from operations |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
Interest income |
|
|
|
|
|
| ||
Interest expense |
|
| ( | ) |
|
| ( | ) |
Finance costs |
|
|
|
|
| ( | ) | |
Gain on debt forgiveness |
|
|
|
|
|
| ||
Original interest debt discount |
|
|
|
|
| ( | ) | |
Gain on change in fair value of derivative liabilities |
|
|
|
|
|
| ||
Total other income (expense) |
|
|
|
|
| ( | ) | |
|
|
|
|
|
|
|
|
|
Net income (loss) before income tax |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Income tax |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Net income (loss) |
| $ | ( | ) |
| $ | ( | ) |
|
|
|
|
|
|
|
|
|
Net income (loss) per share, basic and diluted |
| $ | ( | ) |
| $ | ( | ) |
|
|
|
|
|
|
|
|
|
Weighted average number of shares outstanding, basic and diluted |
|
|
|
|
|
| ||
The accompanying notes are an integral part of these consolidated financial statements.
| 38 |
| Table of Contents |
NASCENT BIOTECH, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
YEARS ENDED MARCH 31, 2024 AND 2023
|
|
|
|
|
|
|
| Total |
| |||||||||||
|
| Common Stock |
|
| Additional Paid-In |
|
| Accumulated |
|
| Stockholders’ Equity |
| ||||||||
|
| Shares |
|
| Amount |
|
| Capital |
|
| Deficit |
|
| (Deficit) |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Balance at March 31, 2022 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ | ( | ) | |||
Common stock issued for service |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued for conversion of payables |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued to related party for service |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued for the conversion of debt |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued for the conversion of warrants |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Warrants issued with convertible notes |
|
| - |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Stock option expense |
|
| - |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
| - |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at March 31, 2023 |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Common stock issued for cash |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued for service |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued for the conversion of stock option |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued to related parties for service |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Common stock issued for conversion of debt |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Change in derivative at conversion |
|
| - |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Stock option expense |
|
| - |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Down round of warrants |
|
| - |
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| |||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
| - |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at March 31, 2024 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ | ( | ) | |||
The accompanying notes are an integral part of these consolidated financial statements.
| 39 |
| Table of Contents |
NASCENT BIOTECH, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR YEARS ENDED MARCH 31,
|
| 2024 |
|
| 2023 |
| ||
|
|
|
|
|
|
| ||
Cash flows from operating activities: |
|
|
|
|
|
| ||
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
Stock based compensation |
|
|
|
|
|
| ||
Stock based compensation- related parties |
|
|
|
|
|
| ||
(Gain) loss on fair value of derivative liabilities |
|
| ( | ) |
|
| ( | ) |
Debt discount amortization |
|
|
|
|
|
| ||
Loss on notes |
|
| ( | ) |
|
|
| |
Stock option expense |
|
|
|
|
|
| ||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
|
|
|
| ||
Prepaid |
|
| ( | ) |
|
| ( | ) |
Accounts payable and accrued expenses |
|
|
|
|
| ( | ) | |
Net provided by (cash used) in operating activities |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
Common stock issued for cash |
|
|
|
|
|
| ||
Proceeds from convertible note |
|
|
|
|
|
| ||
Common stock issued for the exercise of warrants |
|
|
|
|
|
| ||
Repayment of convertible notes |
|
| ( | ) |
|
| ( | ) |
Net cash provided by ( used in) financing activities |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash |
|
|
|
|
|
| ||
Cash – beginning of year |
|
|
|
|
|
| ||
Cash – end of year |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
SUPPLEMENT DISCLOSURES: |
|
|
|
|
|
|
|
|
Interest paid |
| $ |
|
| $ |
| ||
Income taxes paid |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
NON MONETARY TRANSACTIONS |
|
|
|
|
|
|
|
|
Common stock issued for convertible debt |
| $ |
|
| $ |
| ||
Common stock issued for accounts payable |
| $ |
|
| $ |
| ||
Initial discount from derivatives |
| $ |
|
| $ |
| ||
The accompanying notes are an integral part of these consolidated financial statements.
| 40 |
| Table of Contents |
NASCENT BIOTECH, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 – ORGANIZATION AND NATURE OF OPERATIONS
Nascent Biotech, Inc (“Nascent “the Company”) was incorporated on March 3, 2014 under the laws of the State of Nevada.
On July 15, 2014 Biotech entered into a reverse merger with Jin-En Group International Holding Company (Jin-En). Jin-En issued
The Company is actively developing its primary asset Pritumumab for the treatment of brain cancer and pancreatic cancer. Nascent is also actively researching other cancers that have a high probability of benefiting from the therapeutic effects of Pritumumab because they share a common target. Nascent is a clinical stage biopharmaceutical company that focuses on biologic drug candidates that are preparing for initial clinical testing for the treatment of brain and pancreatic cancer. The Company has completed its phase one clinical trials and clearance was received from the FDA for the Company to commence its phase 2 clinical trials.
NOTE 2 – SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles. The Company has elected a fiscal year ending on March 31.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Nascent Biotech, Inc. and its wholly-owned subsidiary Nascent Biologics, Inc, which has been inactive since March 2015. All intercompany accounts and transactions have been eliminated.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents. The Company as of March 31, 2024 did not have any cash equivalents.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the dates of the financial statements and the reported amounts of revenues and expenses during the periods. The extent to which the COVID-19 pandemic may directly or indirectly impact our business, financial condition, and results of operations is highly uncertain and subject to change. We considered the potential impact of the COVID-19 pandemic on our estimates and assumptions and there was not a material impact to our consolidated financial statements as of and for the year ended March 31, 2024. Actual results could differ from estimates making it reasonably possible that a change in the estimates could occur in the near term.
| 41 |
| Table of Contents |
Revenue recognition
In April 2016, the FASB issued ASU 2016–10 Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing. The amendments in this Update do not change the core principle of the guidance in Topic 606. Rather, the amendments in this Update clarify the following two aspects of Topic 606: identifying performance obligations and the licensing implementation guidance, while retaining the related principles for those areas. Topic 606 includes implementation guidance on (a) contracts with customers to transfer goods and services in exchange for consideration and (b) determining whether an entity’s promise to grant a license provides a customer with either a right to use the entity’s intellectual property (which is satisfied at a point in time) or a right to access the entity’s intellectual property (which is satisfied over time). The amendments are intended to render more detailed implementation guidance with the expectation to reduce the degree of judgement necessary to comply with Topic 606.
ASC Topic 606 prescribes a new five-step model entities should follow in order to recognize revenue in accordance with the core principle. These five steps are:
| 1. | Identify the contract(s) with a customer. |
| 2. | Identify the performance obligations in the contract. |
| 3. | Determine the transaction price. |
| 4. | Allocate the transaction price to the performance obligations in the contract. |
| 5. | Recognize revenue when (or as) the entity satisfied the performance obligations. |
The Company implemented the transition using the modified retrospective method of transition. The funds are not earned on milestones that have not been reached per the contract. Based on the cut off treatment of the recognition of revenue per the milestones specific to the license agreements, the Company has determined that there are no adjustments in the value of the revenue recognized from these contracts.
The Company had one revenue stream, which was the milestone payments of the license agreement with BioRay Pharmaceutical which is not earned or billed until the milestone per the agreement is met. The license agreement for this revenue stream was terminated. The Company did not receive any revenue for the year ended March 31, 2024.
Stock-Based Compensation
The Company accounts for stock-based compensation in accordance with the fair value recognition provision of the Financial Accounting Standards Board(“FASB”) Accounting Standards Codification (“ASC”) No 718. The Company issues restricted stock to employees and consultants for their services. Cost of these transactions are measured at fair value of the equity instrument issued at the date of grant. These shares are considered fully vested and the fair market value is recognized as the expense in the period granted. The Company recognized consulting expense and a corresponding increase to the additional paid in capital related to the stock issued for services. For agreements requiring future services the consulting expense is to be recognized ratably over the requisite service period.
Research and Development Expense
Research and development costs are expensed in the period they are incurred in accordance with ASC 730, Research and Development unless they meet specific criteria related to technical, market and financial feasibility, as determined by management, including but not limited to the establishment of a clearly defined future alternative use for the product, and the availability of adequate resources to complete the project. If all criteria are met, the costs are deferred and amortized over the expected useful life of the project or expensed as research and development as the material are consumed or written off if a product is abandoned. At March 31, 2024 and 2023, the Company had zero capitalization associated with materials held with a future alternative use. The cost of these materials was expensed as research and development as the materials are consumed or designated for usage. As the Company is preparing to begin clinical studies using a dose escalation method, it is not feasible to determine if the additional product will be needed for the brain cancer studies.
| 42 |
| Table of Contents |
Income Taxes
Deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is established when necessary to reduce deferred tax assets to the amounts expected to be realized.
The Company accounts for income taxes under the provisions of FASB ASC 740, Accounting for Income Taxes. It prescribes a recognition threshold and measurement attributes for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. As a result, the Company has applied a more-likely-than-not recognition threshold for all tax uncertainties. The guidance only allows the recognition of those tax benefits that have a greater than 50% likelihood of being sustained upon examination by the various taxing authorities.
Basic and Diluted Net Income (Loss) per Share
Basic net income (loss) per share calculations are calculated on the basis of the weighted average number of common shares outstanding during the year. During the year ended March 31, 2024 the Company had a net loss so the options and warrants outstanding were not part the loss per share calculation as they would be antidilutive. Diluted income (loss) per share calculations includes the dilutive effect of warrants and options on the weighted average of the per share calculation.
Fair Value of Financial Instruments
The Company’s financial instruments consist of cash and cash equivalents, accounts payable and accrued expenses and shareholder loans. The carrying amount of these financial instruments approximates fair value due either to length of maturity or interest rates that approximate prevailing market rates unless otherwise disclosed in these financial statements.
As defined in (Financial Accounting Standards Board ASC 820), fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date (exit price). The Company utilized the market data of similar entities in its industry or assumptions that market participants would use in pricing the asset or liability, including assumptions about risk and the risks inherent in the inputs to the valuation technique. These inputs can be readily observable, market corroborated, or generally unobservable. The Company classifies fair value balances based on the observability of those inputs. FASB ASC 820 establishes a fair value hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (level 1 measurement) and the lowest priority to unobservable inputs (level 3 measurement).
The three levels of the fair value hierarchy are as follows:
Level 1 – | Quoted prices are available in active markets for identical assets or liabilities as of the reporting date. Active markets are those in which transactions for the asset or liability occur in sufficient frequency and volume to provide pricing information on an ongoing basis. Level 1 primarily consists of financial instruments such as exchange-traded derivatives, marketable securities, and listed equities. |
|
|
Level 2 - | Pricing inputs are other than quoted prices in active markets included in level 1, which are either directly or indirectly observable as of the reported date and includes those financial instruments that are valued using models or other valuation methodologies. These models are primarily industry-standard models that consider various assumptions, including quoted forward prices for commodities, time value, volatility factors, and current market and contractual prices for the underlying instruments, as well as other relevant economic measures. Substantially all of these assumptions are observable in the marketplace throughout the full term of the instrument, can be derived from observable data or are supported by observable levels at which transactions are executed in the marketplace. Instruments in this category generally include non-exchange-traded derivatives such as commodity swaps, interest rate swaps, options, and collars. |
|
|
Level 3 – | Pricing inputs include significant inputs that are generally less observable from objective sources. These inputs may be used with internally developed methodologies that result in management’s best estimate of fair value |
| 43 |
| Table of Contents |
As of March 31, 2024 the Company believes the amounts reported for cash, payables, accrued liabilities and amounts due to related parties approximate their fair values due to the nature or duration of these instruments.
The following table represents the change in the fair value of the derivative liabilities during the year ended March 31, 2024:
|
| Level 1 |
|
| Level 2 |
|
| Level 3 |
| |||
Balance at March 31, 2022 |
| $ |
|
| $ |
|
| $ |
| |||
Change in fair value at initial issuance |
|
|
|
|
|
|
|
|
| |||
Change in fair value at conversion |
|
|
|
|
|
|
|
|
|
| ( | ) |
Change in fair value of the derivative |
|
|
|
|
|
|
|
|
| |||
Balance at March 31, 2023 |
| $ |
|
| $ |
|
| $ |
| |||
Change in fair value at initial issuance |
|
|
|
|
|
|
|
|
|
|
| |
Change in fair value at conversion |
|
|
|
|
|
|
|
|
|
| ( | ) |
Change in fair value of the derivative |
|
|
|
|
|
|
|
|
|
| ( | ) |
Balance at March 31, 2024 |
| $ |
|
| $ |
|
| $ |
| |||
NOTE 3 – GOING CONCERN
The Company’s consolidated financial statements are prepared using accounting principles generally accepted in the United States of America applicable to a going concern that contemplates the realization of assets and liquidation of liabilities in the normal course of business. The Company, as shown in the accompanying consolidated balance sheets, has working capital deficit of $
The Company will engage in research and development activities that must be satisfied in cash secured through outside funding. The Company will offer noncash consideration and seek equity lines as a means of financing its operations. If the Company is unable to obtain revenue producing contracts or financing or if the revenue or financing it does obtain is insufficient to cover any operating losses it may incur, it may substantially curtail or terminate its operations or seek other business opportunities through strategic alliances, acquisitions or other arrangements that may dilute the interests of existing stockholders.
NOTE 4 – RELATED PARTY TRANSACTIONS
On September 1, 2020, the Company entered five-year compensation agreements with two of its officers and an independent director.
Officer and Director |
| Fiscal Year Annualized Compensation Base Being Paid |
|
| Non-dilutive shares percentage |
| ||
President and CEO (2) |
| $ |
|
|
| % | ||
Chief Financial Officer (1) |
| $ |
|
|
| % | ||
Director (3) |
| $ |
|
|
| - |
| |
Total |
| $ |
|
|
| % | ||
| 44 |
| Table of Contents |
For the year ended March 31, 2024:
| (1) | The Chief Financial Officers was paid $ |
| (2) | The President and Chief Executive Officer was paid $ |
| (3) | The director of the Company was paid $ |
The following table sets forth the shares earned under these contracts from inception through year ended March 31, 2024:
24,000Officer and Director |
| Initial Share Awards Under the Contracts |
|
| Additional Shares Earned to Maintain Ownership Percentage |
|
| Total Shares Earned |
| |||
President |
|
|
|
|
|
|
|
|
| |||
Chief Financial Officer |
|
|
|
|
|
|
|
|
| |||
Total |
|
|
|
|
|
|
|
|
| |||
During the year ended March 31, 2023 the Company issued
During the year ended March 31, 2024 the Company issued
NOTE 5 – LICENSE SETTLEMENT
On October 12, 2017, the Company signed a consulting agreement with a former license holder. Under the terms of the agreement the Company, commencing February 1, 2018 will pay the consultant $
NOTE 6 – EQUITY
On July 9, 2020, the Company increased its authorized shares to
Preferred Stock
On July 10, 2019, the Company filed an amended articles of Incorporation designating
As of March 31, 2024 and 2023 there we no outstanding preferred shares.
Common Stock
During the year ended March 31, 2023 the Company issued
During the year ended March 31, 2023, the Company issued
| 45 |
| Table of Contents |
During the year ended March 31, 2023, the Company
During the year ended March 31, 2023 , the Company issued
During the year ended March 31, 2023 the Company issued
During the year ended March 31, 2024 , the Company issued
During the year ended March 31, 2024 the Company issued
During the year ended March 31, 2024 the Company issued
During the year ended March 31, 2024 the Company issued
NOTE 7 – OPTIONS
The Company under its 2015 option plan issues options to various officers, directors, and consultants.
On April 1, 2016, the Company issued
On April 1, 2016, the Company entered a consulting agreement under which the consultant was granted
On July 20, 2016, the Company granted from the 2015 Option Program
On April 1, 2018, the Company granted from the 2015 Option Program
On November 17, 2022 the Company issued
| 46 |
| Table of Contents |
During the years ended March 31, 2024 and 2023, the Company expensed $
The following sets forth the options granted and outstanding during the years ended March 31, 2024 and 2023:
|
| Options |
|
| Weighted Average Exercise Price |
|
| Weighted Average Remaining Contract Life |
|
| Number of Options Exercisable |
|
| Intrinsic Value |
| |||||
Outstanding at March 31, 2022 |
|
|
|
| $ |
|
|
|
|
|
|
|
| $ | -- |
| ||||
Granted |
|
|
|
|
|
|
|
|
|
|
| -- |
|
|
| -- |
| |||
Exercised |
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
Forfeited |
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
Outstanding at March 31, 2023 |
|
|
|
| $ |
|
|
|
|
|
|
|
| $ | -- |
| ||||
Granted |
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
Exercised |
|
| ( | ) |
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
Forfeited |
|
|
|
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
|
| -- |
|
Outstanding at March 31, 2024 |
|
|
|
| $ |
|
|
|
|
|
|
|
| $ | - |
| ||||
NOTE 8 – WARRANTS
During the year ended March 31, 2021, the Company granted
The Company used the Black Scholes Pricing model to estimate the fair value of the warrants as of grant date, using the following key inputs: market prices of the Company’s common stock at dates of grant between $
On April 7, 2022, the Company issued
On August 31, 2022 the Company issued
On September 2, 2022 the Company issued
| 47 |
| Table of Contents |
The
On January 30, 2024 the Company issued
The weighted average remaining life and intrinsic value of the warrants as of March 31, 2024 was.
|
| Warrants |
|
| Weighted Average Exercise Price |
|
| Weighted Average Remaining Contract Life |
|
| Intrinsic Value |
| ||||
Outstanding at March 31, 2022 |
|
|
|
| $ |
|
|
|
|
| $ |
| ||||
Granted |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Exercised |
|
| ( | ) |
|
|
|
|
| -- |
|
|
|
| ||
Expired |
|
| -- |
|
|
|
|
|
| -- |
|
|
|
| ||
Outstanding at March 31, 2023 |
|
|
|
| $ |
|
|
|
|
| $ |
| ||||
Granted |
|
|
|
|
|
|
|
|
|
|
|
|
| |||
Exercised |
|
| -- |
|
|
|
|
|
| -- |
|
|
|
| ||
Expired |
|
| -- |
|
|
|
|
|
| -- |
|
|
|
| ||
Outstanding at March 31, 2024 |
|
|
|
| $ |
|
|
| .95 |
|
| $ |
| |||
As of March 31, 2024 there were
NOTE 9 – INCOME TAXES
At March 31, 2024 and 2023 the Company had federal net operating loss carry forwards of approximately $
Components of net deferred tax assets, including a valuation allowance, are as follows at March 31, 2024 and 2023:
|
| March 31, 2024 |
|
| March 31, 2023 |
| ||
Deferred tax assets: |
|
|
|
|
|
| ||
Net operating loss |
| $ |
|
| $ |
| ||
Less: Valuation allowance |
|
| ( | ) |
|
| ( | ) |
Net deferred tax assets |
| $ |
|
| $ |
| ||
In assessing the recovery of the deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income in the periods in which those temporary differences become deductible. Management considers the scheduled reversals of future deferred tax assets, projected future taxable income, and tax planning strategies in making this assessment. As a result, management determined it was more likely than not the deferred tax assets would not be realized as of March 31, 2024.
Based on the corporation tax rates the Company calculated the deferred tax asset for the years ended March 31, 2024 and 2023 at
The Company, due to its losses, has not filed US Corporate tax returns and is subject to examination back to March 31, 2015.
| 48 |
| Table of Contents |
NOTE 10 – CONVERTIBLE DEBT
On August 31, 2022 the Company entered into an agreement with an unrelated third party for convertible debentures totaling $
The initial derivatives were calculated for each debenture as follows:
| 1. | Debenture 1- risk free interest of |
| 2. | Debenture 2- risk free interest of |
| 3. | Debenture 3- risk free interest of |
During the period ended March 31, 2023, the Company issued
On June 29, 2023 the Company signed a redemption agreement, amended on August 31, 2023, with the debentures owner pertaining to the outstanding principal balance of $
On September 2, 2022 the Company entered into an agreement with an unrelated third party for convertible debentures totaling $
| 49 |
| Table of Contents |
On December 1, 2023 the Company issued an amended convertible debenture to an unrelated third party under the agreement dated September 2, 2022 (noted in the paragraph above), totaling $
During the year ended March 31, 2024, the Company issued an aggregate of
NOTE 11 – LICENSE AGREEMENT TERMINATION
On March 31, 2021, the Company issued a license agreement for US $
NOTE 12 – COMMITMENTS AND CONTINGENCIES
On June 30, 2016, the Company entered into a cell line sales agreement with the product manufacture. Under the terms of the agreement the company is obligated to make future payments based on the milestones of its achievements. These future payments may be as followed;
| 1. | $ |
| 2. | $ |
| 3. | $ |
| 4. | Annual maintenance fee upon completion of phase I manufacturing or the transfer of the cell line from Catalent’s control of $ |
| 5. |
As of March 31, 2024 the outstanding amount due the manufacture was zero
On March 9, 2020, the Board of Directors of the Company adapted an expense bonus program.
On September 1, 2020, the Company entered five-year compensation agreements with two of its officers and directors.
NOTE 13 – SUBSEQUENT EVENTS
On March 29, 2024 the Company file an S-1 registering
On June 26, 2024, the Company issued
On June 27, 2024, the Company issued
The Company has evaluated subsequent events to determine events occurring after March 31, 2024 through the date this report was issued and determined there are no events that would have a material impact on the Company’s financial results or require disclosure and have determined none exist other than those noted above in this footnote.
| 50 |
