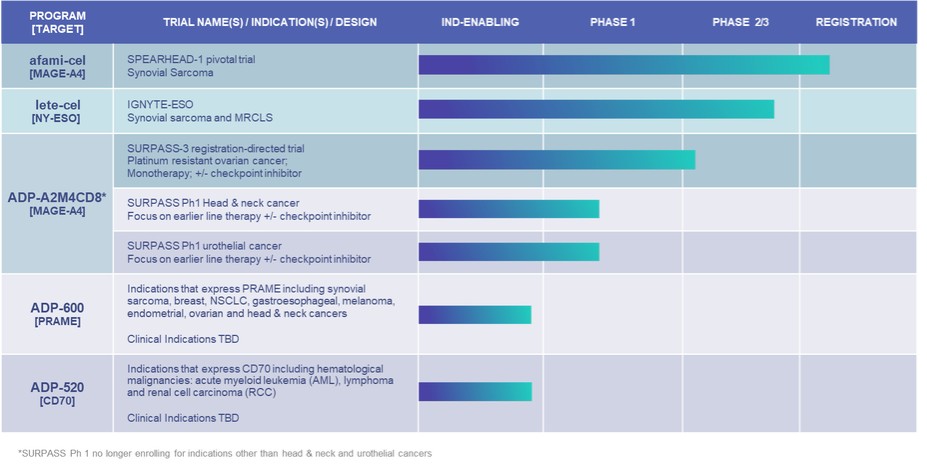
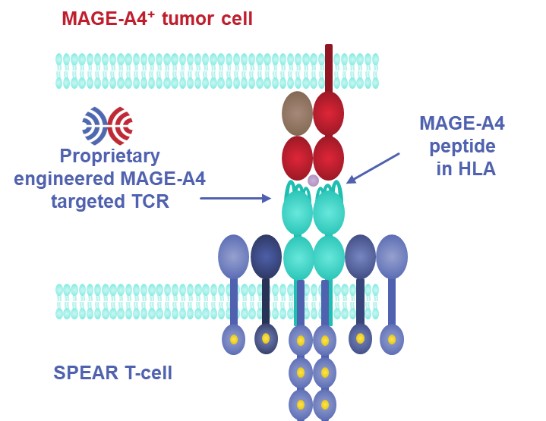
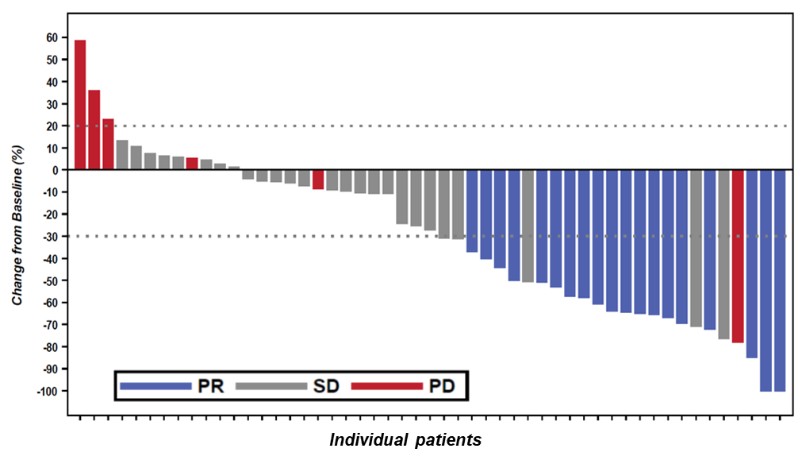
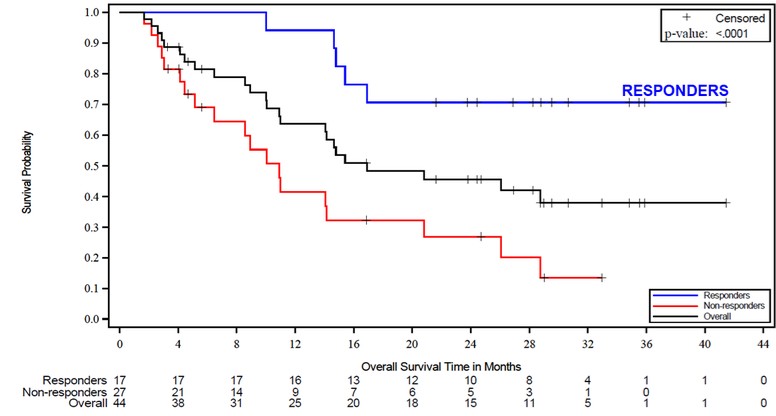
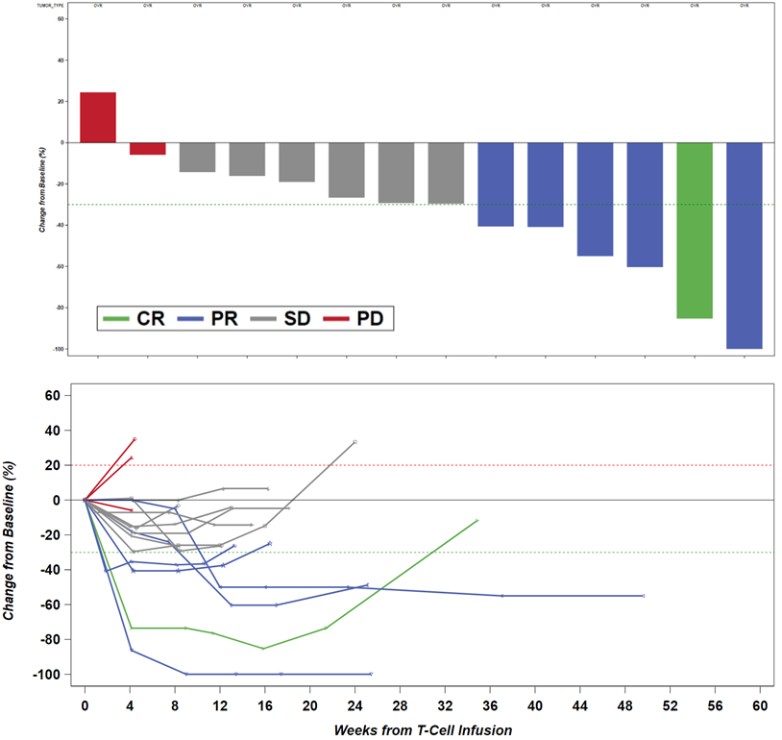
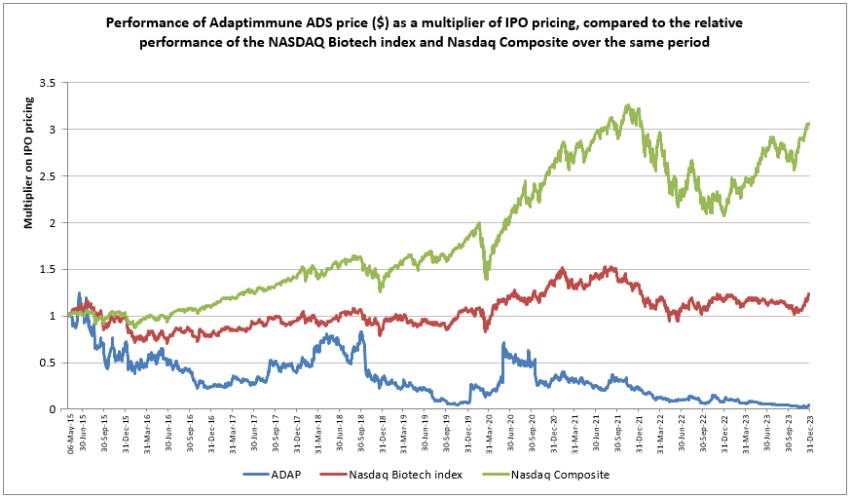
Government Regulation and Product Approvals
Government authorities in the U.S., at the federal, state and local level, and in other countries and jurisdictions, including the EU and U.K, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products. The processes for obtaining regulatory approvals in the U.S. and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources. Failure to comply with the various federal, state and local level laws and requirements can also result in severe penalties and restrictions to the business.
FDA Approval Process
In the U.S., therapeutic products, including drugs, biologics, and medical devices are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act (the “FDCA”), and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products, including biological products. Biological products are subject to regulation under the FDC Act, and are approved for marketing under provisions of the Public Health Service Act (“PHSA”) via a BLA. The application process and requirements for approval of BLAs are generally similar to those for new drug applications (“NDAs”), and biologics are associated with generally similar, if not greater, approval risks and costs as drugs. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs or BLAs, clinical holds, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
In the U.S., the development of a new biological product and certain changes to an approved biological product typically involves:
| ● | preclinical studies, including laboratory and animal tests, |
| ● | the submission to the FDA of an Investigational New Drug application (“IND”), which must become effective before human clinical testing may commence, and |
| ● | adequate and well-controlled clinical trials to establish the safety and effectiveness of the biological product for each indication for which FDA approval is sought. |
Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
Preclinical Studies
Preclinical studies include laboratory evaluation of product biochemistry, formulation and stability, as well as in vitro and animal studies to assess the potential for toxicity and to establish a rationale for therapeutic use for supporting subsequent clinical testing. In the United States, certain preclinical trials must comply with the FDA's Good Laboratory Practice requirements ("GLPs") and the U.S. Department of Agriculture's Animal Welfare Act, as well as other federal regulations and requirements.
IND Submission
In order to begin clinical testing of an investigational biologic product, the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, among other things, are submitted to the FDA as part of an IND application. An IND application is a request for authorization from the FDA to administer an investigational product to humans and must become effective before human clinical trials may begin. Some long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after