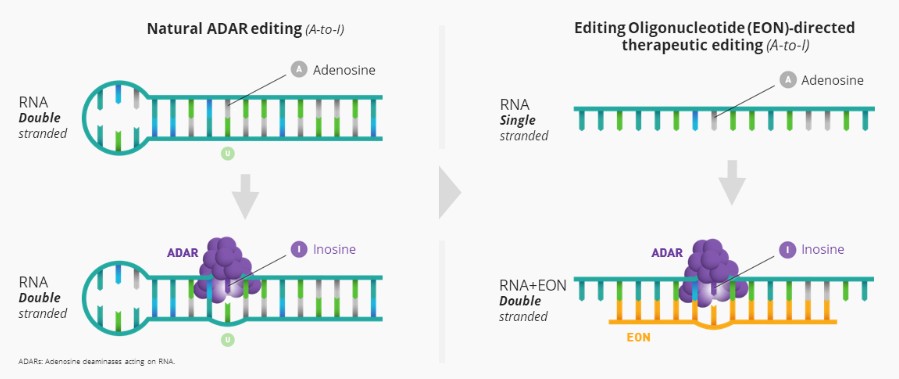
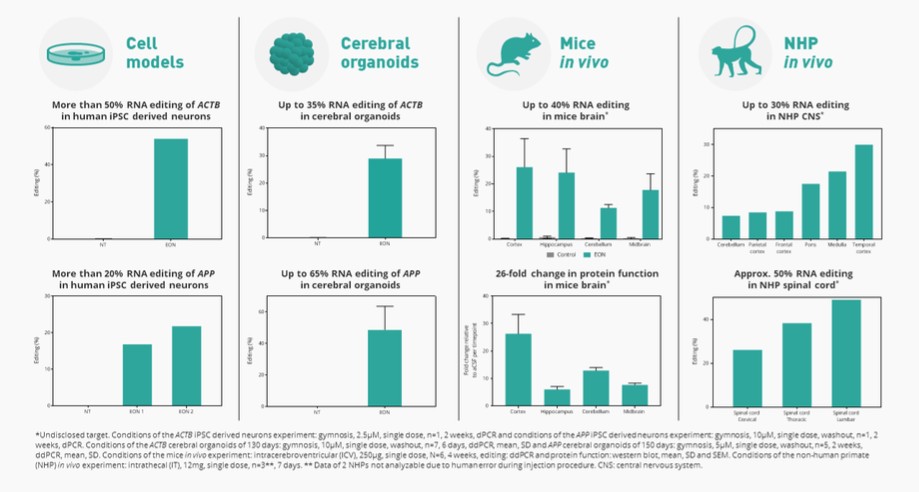
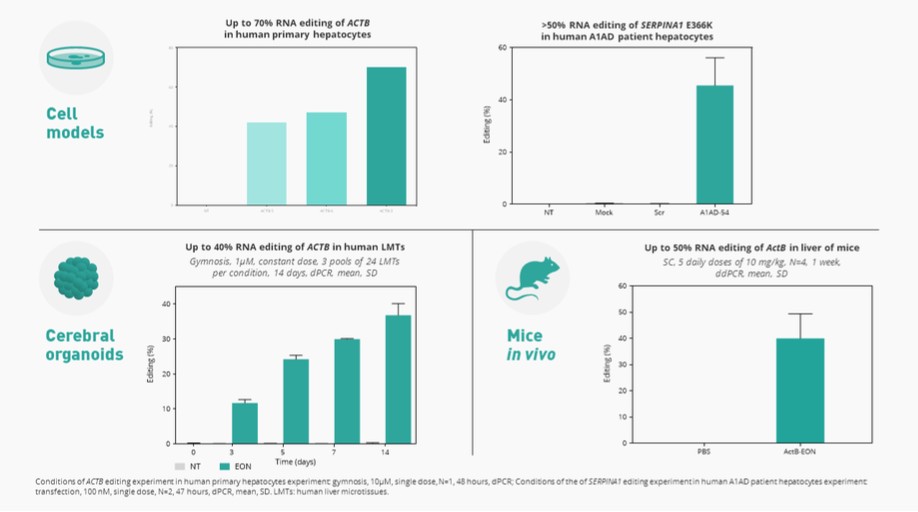
liver where research into human genetics has shown us that introduction or correction of a mutation may lead to a benefit for patients. We prioritize areas with well-established biomarkers for the assessment of early clinical activity and to establish proof of target engagement, established clinically relevant endpoints, and the ability to leverage existing proven delivery technology. We are advancing AX-0810 for cholestatic diseases targeting Na-taurocholate cotransporting polypeptide, or NTCP, and AX-1412 for cardiovascular disease targeting Beta-1,4-galactosyltransferase 1, or B4GALT1, as our initial pipeline programs.
In addition to advancing our wholly-owned pipeline programs, we entered into a global licensing and research collaboration with Eli Lilly and Company in September 2021 where our Axiomer RNA editing platform is being used to progress new drug targets for disorders toward clinical development and commercialization. Initially focused on five targets, the partnership was expanded to ten targets in December 2022, with an option for further expansion to fifteen targets.
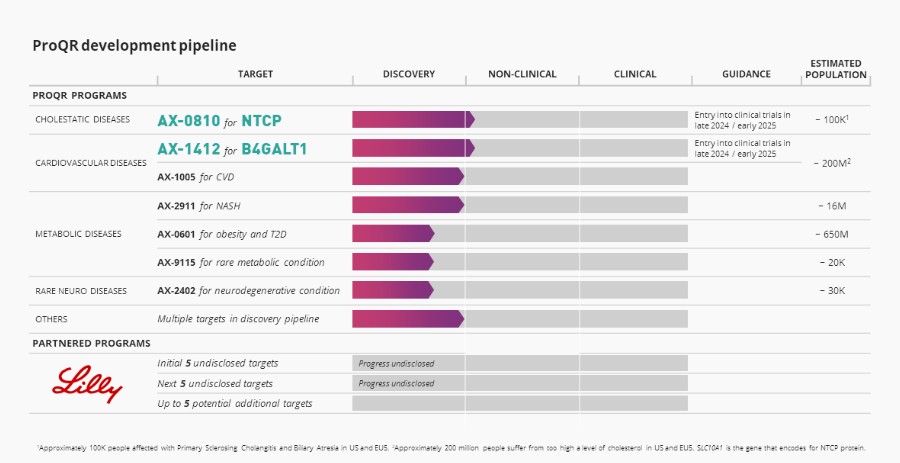
We believe the platform has significant potential to yield many additional therapeutic candidates. Thus, we continuously evaluate further opportunities for beneficial collaborations or strategic partnerships to efficiently advance product candidates with the goal of bringing medicines to patients.
We have other earlier stage RNA editing platform technologies, including our Trident platform. Our Trident RNA pseudouridylation platform is designed to enable the suppression of nonsense mutations and premature stop codons (PTC) that cause 11% of all human genetic diseases. Since all premature stop codons contain uridine, pseudouridylation of that uridine converts those nonsense codons into sense codons. The Trident technology harnesses the endogenously expressed pseudouridylation machinery with guide RNAs to inhibit nonsense messenger RNA (mRNA)-mediated decay (NMD) in a sequence-specific manner and promote PTC readthrough. The Trident technology has the potential to be applied in genetic diseases caused by PTCs.
Both the Axiomer and Trident RNA editing platforms are novel, proprietary RNA technologies invented at ProQR or with our academic collaborators. We have built a broad intellectual property estate around these technologies and together with the leading academic experts in the RNA field, we continue to advance these technologies.
52