UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
(Mark One)
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For
the fiscal year ended
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period from [●] to [●]
Commission
File Number:
(Exact name of registrant as specified in its charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| (Address of principal executive offices) | (Zip Code) |
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| The | ||
| The Capital Market | ||
| Securities registered pursuant to Section 12(g) of the Act: None | ||
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data
File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding
12 months (or for such shorter period that the registrant was required to submit and post such files).
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | Smaller reporting
company |
Emerging growth
company |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐
The
aggregate market value of the voting and non-voting common equity held by non-affiliates based on a closing sale price of $6.32 per share,
which was the last sale price of the common shares as of January 31, 2022, the last business day of the registrant’s most recently
completed second fiscal quarter, was $
As of October 27, 2022, shares of the registrant’s common shares, no par value per share, were issued and outstanding.
TABLE OF CONTENTS
| 2 |
Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements which are made pursuant to the safe harbor provisions of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These statements may be identified by such forward-looking terminology as “may,” “should,” “expects,” “intends,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue” or the negative of these terms or other comparable terminology. Our forward-looking statements are based on a series of expectations, assumptions, estimates and projections about our company, are not guarantees of future results or performance and involve substantial risks and uncertainty. We may not actually achieve the plans, intentions or expectations disclosed in these forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in these forward-looking statements. Our business and our forward-looking statements involve substantial known and unknown risks and uncertainties, including the risks in the section titled “Risk Factors” beginning on page 40, that may cause our or our industry’s actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. In addition, you are directed to factors discussed in the “Business” section beginning on page 5 and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section beginning on page 59, as well as those discussed elsewhere in this Annual Report on Form 10-K.
All of our forward-looking statements are as of the date of this Annual Report on Form 10-K only. In each case, actual results may differ materially from such forward-looking information. We can give no assurance that such expectations or forward-looking statements will prove to be correct. An occurrence of, or any material adverse change in, one or more of the risk factors or risks and uncertainties referred to in this Annual Report on Form 10-K or included in our other public disclosures or our other periodic reports or other documents or filings filed with or furnished to the U.S. Securities and Exchange Commission (the “SEC”) could materially and adversely affect our business, prospects, financial condition and results of operations. Except as required by law, we do not undertake or plan to update or revise any such forward-looking statements to reflect actual results, changes in plans, assumptions, estimates or projections or other circumstances affecting such forward-looking statements occurring after the date of this Annual Report on Form 10-K, even if such results, changes or circumstances make it clear that any forward-looking information will not be realized. Any public statements or disclosures by us following this Annual Report on Form 10-K that modify or impact any of the forward-looking statements contained in this Annual Report on Form 10-K will be deemed to modify or supersede such statements in this Annual Report on Form 10-K.
| 3 |
This Annual Report on Form 10-K may include market data and certain industry data and forecasts, which we may obtain from internal company surveys, market research, consultant surveys, publicly available information, reports of governmental agencies and industry publications, articles and surveys. Industry surveys, publications, consultant surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but the accuracy and completeness of such information is not guaranteed. While we believe that such studies, clinical trials and publications are reliable, we have not independently verified market and industry data from third-party sources.
Risk Factor Summary
Our business is subject to significant risks and uncertainties that make an investment in us speculative and risky. Below we summarize what we believe are the principal risk factors but these risks are not the only ones we face, and you should carefully review and consider the full discussion of our risk factors in the section titled “Risk Factors”, together with the other information in this Annual Report on Form 10-K. If any of the following risks actually occurs (or if any of those listed elsewhere in this Annual Report on Form 10-K occur), our business, reputation, financial condition, results of operations, revenue, and future prospects could be seriously harmed. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that adversely affect our business.
| ● | We have a history of losses, may incur future losses and may not achieve profitability; |
| ● | We are an early stage development company; |
● |
We are developing novel technologies which may not be effective or safe; |
| ● | We have an unproven market for our product candidates; |
| ● | We are heavily reliant on third-parties to carry out a large portion of our business; |
| ● | Pre-clinical studies and initial clinical trials are not necessarily predictive of future results; |
| ● | We must obtain additional capital to continue our operations; |
| ● | We are highly dependent on our key personnel; |
| ● | We may not succeed in completing the development of our products, commercializing our products or generating significant revenues; |
| ● | We may not successfully develop, maintain and protect our proprietary products and technologies; |
| ● | Changes in legislation and regulations may affect our revenue and profitability; |
| ● | If we or our licensees are unable to obtain U.S., Canadian and/or foreign regulatory approval for our product candidates, we will be unable to commercialize our therapeutic candidates; |
| ● | Clinical trials involve a lengthy and expensive process with uncertain outcomes, and results of earlier studies and trials may not be predictive of future trial results; |
| ● | Future issuance of our common shares could dilute the interests of existing shareholders; and |
| ● | We have a significant number of options and warrants outstanding, and while these options and warrants are outstanding, it may be more difficult to raise additional equity capital. |
| 4 |
PART I
ITEM 1. BUSINESS
BUSINESS
Overview of the Company
BriaCell (the “Company”) is an immuno-oncology biotechnology company with a strong focus on cancer immunotherapy. Immunotherapies have come to the forefront in the fight against cancer since they harness the body’s own immune system to recognize and destroy cancer cells. BriaCell owns the U.S. patent to SV-BR-1-GM (“Bria-IMT™”), a whole-cell targeted immunotherapy for cancer (U.S. Patent No. 7,674,456), as well as patents related to PKCδ inhibitors (U.S. Patent Nos. 9,364,460 and 9,572,793). The Company is currently advancing our targeted immunotherapy program by prioritizing a Phase I/IIa clinical trial with Bria-IMT™ in combination with an immune checkpoint inhibitor and a companion diagnostic test, BriaDx™, to identify patients most likely to benefit from Bria-IMT™. The Bria-IMT™ regimen was evaluated in four patients in a prior study in 2004-2006 by Dr. Charles Wiseman, the scientific founder, former member of the board of directors of the Company (the “Board”) and principal scientific advisor. Encouraging results were obtained, especially in a patient who matched Bria-IMT™ at HLA-DR alleles and had a grade II tumor. In 2017-2018 BriaCell evaluated 23 patients with advanced breast cancer with the Bria-IMT™ regimen and obtained confirmation of the ability of the Bria-IMT™ regimen to induce regression of metastatic breast cancer in patients who match Bria-IMT™ at least at one HLA allele and/or if they had grade I or grade II tumors. A combination study with the immune checkpoint inhibitor pembrolizumab (KEYTRUDA®) was initiated and the first patient dosing in the “combination therapy” clinical trial occurred in September 2018. BriaCell purchased the KEYTRUDA® for this study as BriaCell does not have an agreement with Merck & Co., Inc. for the supply of KEYTRUDA®. Eleven patients were dosed in the combination therapy trial with Bria-IMT™ and the immune checkpoint inhibitor KEYTRUDA® and subsequently dosing with this combination was discontinued. The study was modified under an amended protocol which evaluates the combination of the Bria-IMT™ regimen with Incyte Corporation experimental drugs retifanlimab (anti-PD-1 antibody similar to pembrolizumab). The study is ongoing.
Market
It is estimated by the National Cancer Institute that in 2022, approximately 287,500 women will be diagnosed with breast cancer in the United States. That means that every two minutes an American woman is diagnosed with breast cancer and more than 43,000 are projected to die in 2022. Although about 100 times less common than in women, breast cancer also affects men. It is estimated that the lifetime risk of men getting breast cancer is about 1 in 1,000, and the American Cancer Society estimates that approximately 2,710 new cases of invasive male breast cancer will be diagnosed and approximately 530 men will die from breast cancer in 2022.
According to the May 2019 “Global Oncology Trends 2021” report by the IQVIA Institute, the global market for cancer drugs (including immunotherapy drugs) is expected to reach nearly $269 billion by the end of 2025, growing at a compound annual growth rate (“CAGR”) of 10% between 2021 and 2025, of which about 20% is expected to be immuno-oncology drugs.
| 5 |
About 12.9% percent of women will be diagnosed with breast cancer at some point during their lifetime. In 2018, there were an estimated 3,676,262 women living with female breast cancer in the United States. Approximately 81% of cases present as invasive breast cancer. Approximately 6% of new breast cancer diagnoses are Stage IV (metastatic breast cancer (“MBC”), which has already spread to other organs). Twenty to thirty percent of all women diagnosed with breast cancer will develop MBC. Breast cancer can be subdivided based on receptor status – the hormone receptors for estrogen (ER) and progesterone (PR), collectively referred to as hormone receptors (HR), and the Her2/neu growth factor receptor (HER2). Based on the latest SEER statistics, 74.6% were found to be HR+/HER2−, 10.8% were triple-negative (HR−/HER2−), 10.5% were HR+/HER2+, and 4.0% were HR−/HER2+.1
It is estimated that over 150,000 women in the US are living with MBC.2 For those with metastatic disease at diagnosis, their 5-year survival rate is 27%.3 For patients who develop MBC after initially having localized disease, if they had a good response to treatment (i.e. a disease-free interval of more than 24 months), their survival rate is similar to that of patients with MBC at initial diagnosis, but if their disease-free interval is less than 24 months, their prognosis is worse.4 We currently propose that Bria-IMT’s™ indication will be for the treatment of patients with MBC who have failed at least two lines of therapy. Similarly, another study showed that the median overall survival among patients with de novo stage IV MBC was 39.2 months, while for patients with relapsed disease it was 27.2 months.5 Median progression free survival after first-line therapy is only 9 months and the survival benefit decreases with subsequent lines of therapy.6 One study showed that of 386 patients with MBC, 374 (97%) received first-line therapy, 254 (66%) received second-line therapy, 175 (45%) received third-line therapy, and 105 (27%) received therapy beyond third-line.7
1 See https://seer.cancer.gov/statfacts/html/breast.html
2 Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2017 Jun;26(6):809-815.
3 Breast Cancer Facts & Figures 2017-2018. Atlanta: American Cancer Society, Inc. 2017.
4 Lobbezoo, D. J. A. et al. Prognosis of metastatic breast cancer subtypes: the hormone receptor/HER2-positive subtype is associated with the most favorable outcome. Breast Cancer Res. Treat. 141, 507–514 (2013).
5 Dawood S, Broglio K, Ensor J, Hortobagyi GN, Giordano SH. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol. 2010 Nov; 21(11):2169–74.
6 Bonotto M, Gerratana L, Iacono D, Minisini AM, Rihawi K, Fasola G, Puglisi F. Treatment of Metastatic Breast Cancer in a Real-World Scenario: Is Progression-Free Survival With First Line Predictive of Benefit From Second and Later Lines? Oncologist.
7 Kotsakis A, Ardavanis A, Koumakis G, Samantas E, Psyrri A, Papadimitriou C. Epidemiological characteristics, clinical outcomes and management patterns of metastatic breast cancer patients in routine clinical care settings of Greece: Results
| 6 |
Figure A: Overview of current drugs for breast cancer, demonstrating the pattern of novel therapeutic introductions and significant market uptake. These precedents demonstrate a strong market pull for Bria-IMT™.
| Drug | Technology | Company | Indication | 2018 Sales US (Mil $US) | 2018 Sales Ex-US (Mil $US) | 2018 Sales WW (Mil $US) | ||||||||||||
| HERCEPTIN® (trastuzumab) | Monoclonal antibody | Roche | HER2+BC & HER2+ metastatic gastric cancer | 2,955 | 4,140 | 7,096 | ||||||||||||
| IBRANCE® (palbociclib) in combination with fluvestrant or aromatase inhibitor | CDK 4/6 inhibitor | Pfizer | HR+/HER2- MBC | 2,922 | 1,196 | 4,118 | ||||||||||||
| PERJETA® (pertuzumab) in combination with Herceptin® (trastuzumab) and chemotherapy | HER2/neu receptor antagonist | Roche | HER2+ early BC that has a high likelihood of recurrence | 1,347 | 1,499 | 2,846 | ||||||||||||
| FASLODEX® (fulvestrant) | Estrogen receptor antagonist | AstraZeneca | HR+/HER2- MBC | 537 | 491 | 1,028 | ||||||||||||
| KADCYLA® (ado-trastuzumab emtansine) | HER2 targeted antibody & microtubule inhibitor conjugate | Roche | HER2+BC | 365 | 630 | 995 | ||||||||||||
| LYNPARZA® (olaparib) | Poly (ADP-ribose) polymerase (PARP) inhibitor | AstraZeneca | BC & Ovarian cancer | 345 | 302 | 647 | ||||||||||||
| Verzenio® (abemaciclib) monotherapy or in combination with fulvestrant or aromatase inhibitor | CDK 4/6 inhibitor | Eli Lilly | HR+/HER2- MBC | 255 | - | 255 | ||||||||||||
| KISQALI® (ribociclib) in combination with fluvestrant or aromatase inhibitor | CDK 4/6 inhibitor | Novartis | HR+/HER2- MBC | 235 | - | 235 | ||||||||||||
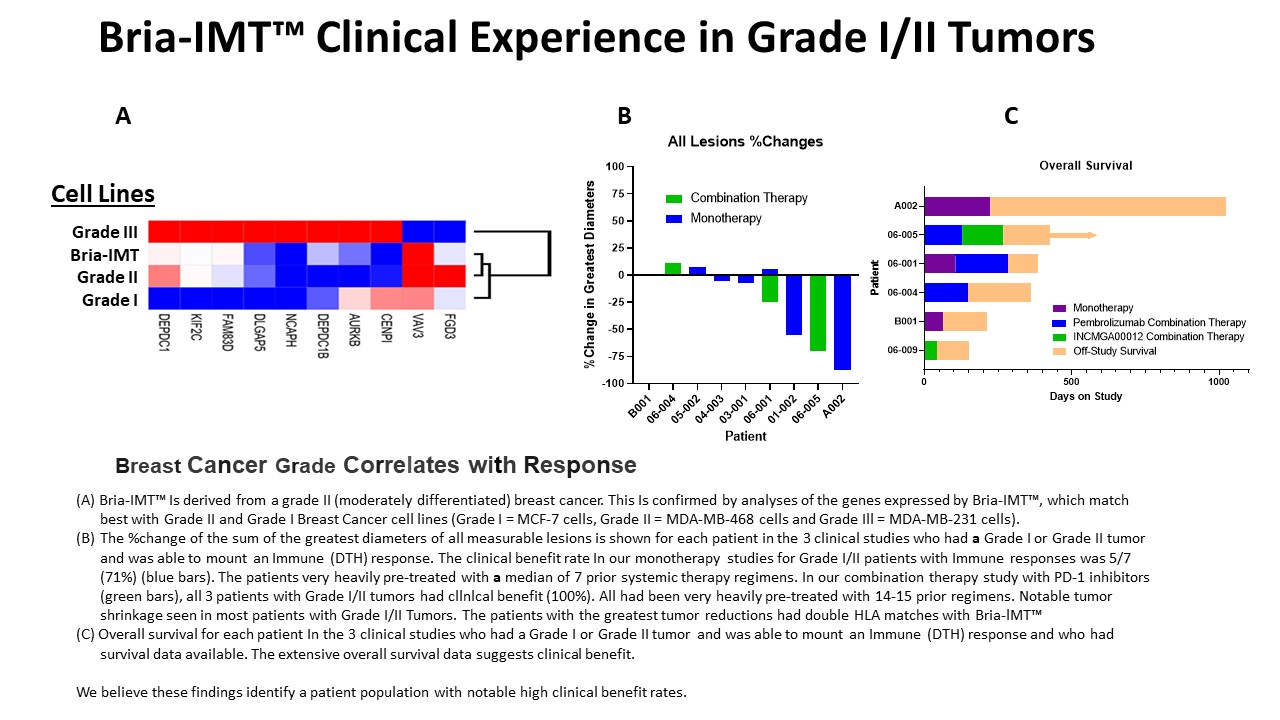
The best response to Bria-IMT™ to date is in patients who matched Bria-IMT™ at one or more HLA alleles, with higher response rates for patients with 2+ HLA allele matches. If one HLA allele match is found to be sufficient, we will be able to treat ~50-60% of the patient population, while patients with 2+ HLA matches constitutes ~15-35% of cases.8 We also saw higher clinical benefit rates for patients with grade I/II tumors. Tumor differentiation in breast cancer cell lines is often described by their classification as Luminal, Basal A and Basal B subtypes, with Luminal representing well differentiated tumors, Basal B poorly differentiated tumors, and Basal A an intermediate stage tumor (“moderately” differentiated).2 Yao and colleagues in 2005 identified a 9-gene signature (AURKB, CENPI, DEPDC1, DEPDC1B, FAM83D, FGD3, NCAPH, TNFRSF18, FCGR1A) discriminating poorly (grade 3) from moderately (grade 2) differentiated tumors.3 To understand the place of SV-BR-1-GM in this model, we compared its RNA expression profile with those of three other cell lines representing Luminal (MCF-7), Basal A (MDA-MB-468) and Basal B (MDA-MB-231), using a 10-gene signature (AURKB, CENPI, DEPDC1, DEPDC1B, FAM83D, FGD3, NCAPH, DLGAP, KIF2C, VAV3) derived from those by Yao and colleagues. The results, shown in the figure below, demonstrate that Bria-IMT™ most closely clusters with MDA-MB-468 and as such is considered a grade II “moderately differentiated” cell line.
Greece: Results from the EMERGE multicenter retrospective chart review study. BMC Cancer. 2019 Jan 18;19(1):88.
8 Gragert, Loren, Abeer Madbouly, John Freeman, and Martin Maiers. 2013. “Six-Locus High Resolution HLA Haplotype Frequencies Derived from Mixed-Resolution DNA Typing for the Entire US Donor Registry.” Human Immunology.
2 Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515-527. doi:10.1016/j.ccr.2006.10.008)
3 Yao F, Zhang C, Du W, Liu C, Xu Y. Identification of gene-expression signatures and protein markers for breast cancer grading and staging. PLoS One. 2015;10(9). doi:10.1371/journal.pone.0138213)
| 7 |
Based on a recent publication of patients with relapsed breast cancer, we estimate that this will account for ~40% of relapsed metastatic breast cancer cases (33% grade II and 7% grade I) (Sundquist M, Brudin L, Tejler G. Improved survival in metastatic breast cancer 1985-2016. Breast. 2017 Feb;31:46-50. doi: 10.1016/j.breast.2016.10.005. Epub 2016 Nov 2). In patients with relapsed disease, the overall survival following relapse appears similar for those with grade II and grade III tumors.9 The market for breast cancer drugs is a multibillion-dollar market with new drugs being approved on an ongoing basis, indicating the shortage of safe and effective treatments for this deadly disease. Figure A summarizes current drugs on the market utilized in combination therapy along with their reported market sales, which further supports market potential for Bria-IMT™ to be used for combination therapy for breast cancer patients.
We propose the following calculation in order to show the rationale behind the number of patients that we anticipate can be currently treated by SV-BR-1-GM:
| ● | There are 150,000 women with metastatic breast cancer in the U.S.10 | |
| ● | ~45% will receive third line therapy11 = 68,000 patients available | |
| ● | 68,000 x 50% (matched for 1 HLA allele group)12 = 34,000 patients available for treatment13 | |
| ● | 40% have grade I/II tumors14 = 13,600 patients available for treatment |
9 See note 5, above.
10 Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2017 Jun;26(6):809-815.
11 Kotsakis A, Ardavanis A, Koumakis G, Samantas E, Psyrri A, Papadimitriou C. Epidemiological characteristics, clinical outcomes and management patterns of metastatic breast cancer patients in routine clinical care settings of Greece: Results from the EMERGE multicenter retrospective chart review study. BMC Cancer. 2019 Jan 18;19(1):88.
12 Gragert, Loren, Abeer Madbouly, John Freeman, and Martin Maiers. 2013. “Six-Locus High Resolution HLA Haplotype Frequencies Derived from Mixed-Resolution DNA Typing for the Entire US Donor Registry.” Human Immunology.
13 Momenimovahed Z, Salehiniya H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer (Dove Med Press). 2019 Apr 10;11:151-164. SEER Cancer Statistics Factsheets: Female Breast Cancer. National Cancer Institute. Bethesda, MD; American Cancer Society. Breast Cancer Facts & Figures 2017-2018. Atlanta: American Cancer Society, Inc. 2017.
14 See note 5, above.
| 8 |
Competition
Currently available therapeutic options for breast cancer offer some hope for patients, but there is much room for improvement. Comparable studies looking primarily at second line or later treatment are shown in Table “A”, below. Evaluating response rates (partial and complete responses = ORR), progression free survival (“PFS”) and overall survival (“OS”) from clinical trials in similar subjects with metastatic or recurrent breast cancer indicate that response rates range from 6.9% up to 59%, depending on the population studied and the intervention (median 24%). PFS ranges from 8 weeks to 12 months (median 5 months) and OS from 6 months to 31 months (median 13 months).
Table A: Studies evaluating second-line or later treatment options. Data depict an unpredictable response rate to treatment ranging from 6.9-59%, therefore establishing and confirming the opportunity for Bria-IMT™.
| Study | Treatment & Design | # of Pts | ORR | PFS/TTP | OS | |||||||||||||
| Perez15 | Paclitaxel Monotherapy | 212 | 21.5 | % | 4.7 mo | 12.8 mo | ||||||||||||
| Seidman16 | Gemcitabine Monotherapy | 160 | 26 | % | ||||||||||||||
| Zelek17 | Vinorelbine Monotherapy | 40 | 25 | % | 6 mo | |||||||||||||
| Licchetta18 | Cyclophosphamide and megestrol acetate | 29 | 31 | % | 7.4 mo | 13.4 mo | ||||||||||||
| Harvey19 | Docetaxel Monotherapy 60 mg/m2 | 122 | 22.1 | % | 12.7 wk | 10.6 mo | ||||||||||||
| Docetaxel Monotherapy 75 mg/m2 | 146 | 23.3 | % | 15.0 wk | 10.3 mo | |||||||||||||
| Docetaxel Monotherapy 100 mg/m2 | 139 | 36.0 | % | 16.6 wk | 12.3 mo | |||||||||||||
| Rivera20 | Docetaxel Monotherapy q3wk | 59 | 35.6 | % | 5.7 mo | 18.3 mo | ||||||||||||
| Docetaxel Monotherapy qwk | 59 | 20.3 | % | 5.5 mo | 18.6 mo | |||||||||||||
| Gradishar21 | ABI-007 (Nab paclitaxel) | 229 | 33 | % | 23.0 wk | 65.0 wk | ||||||||||||
| Paclitaxel Monotherapy | 225 | 19 | % | 16.9 wk | 55.7 wk | |||||||||||||
| ABI-007 (Nab paclitaxel) 2nd line | 132 | 27 | % | 20.9 wk | 56.4 wk | |||||||||||||
| Paclitaxel Monotherapy 2nd line | 136 | 13 | % | 16.1 wk | 46.7 wk | |||||||||||||
| Perez22 | Ixabepilone Monotherapy | 126 | 11.5 | % | 3.1 mo | 8.6 mo | ||||||||||||
| Leyland-Jones23 | Trastuzumab with paclitaxel | 32 | 59 | % | 12.2 mo | |||||||||||||
| von Minckwitz24 | Trastuzumab with capecitabine | 78 | 48.1 | % | 8.2 mo | 25.5 mo | ||||||||||||
| Capecitabine Monotherapy | 78 | 27.0 | % | 5.6 mo | 20.4 mo | |||||||||||||
| Verma25 | Trastuzumab emtansine | 495 | 43.6 | % | 9.6 mo | 30.9 mo | ||||||||||||
| lapatinib plus capecitabine | 496 | 30.8 | % | 6.4 mo | 25.1 mo | |||||||||||||
| Geyer26 | Lapatinib plus capecitabine | 163 | 22 | % | 8.4 mo | |||||||||||||
| Capecitabine Monotherapy | 161 | 14 | % | 4.4 mo | ||||||||||||||
| Bartsch27 | Capecitabine and trastuzumab | 40 | 20 | % | 8 mo | 24 mo | ||||||||||||
| Blackwell28 | Lapatinib Monotherapy | 148 | 6.9 | % | 8.1 wk | 39.0 wk | ||||||||||||
| Lapatinib with trastuzumab | 148 | 10.3 | % | 12.0 wk | 51.6 wk | |||||||||||||
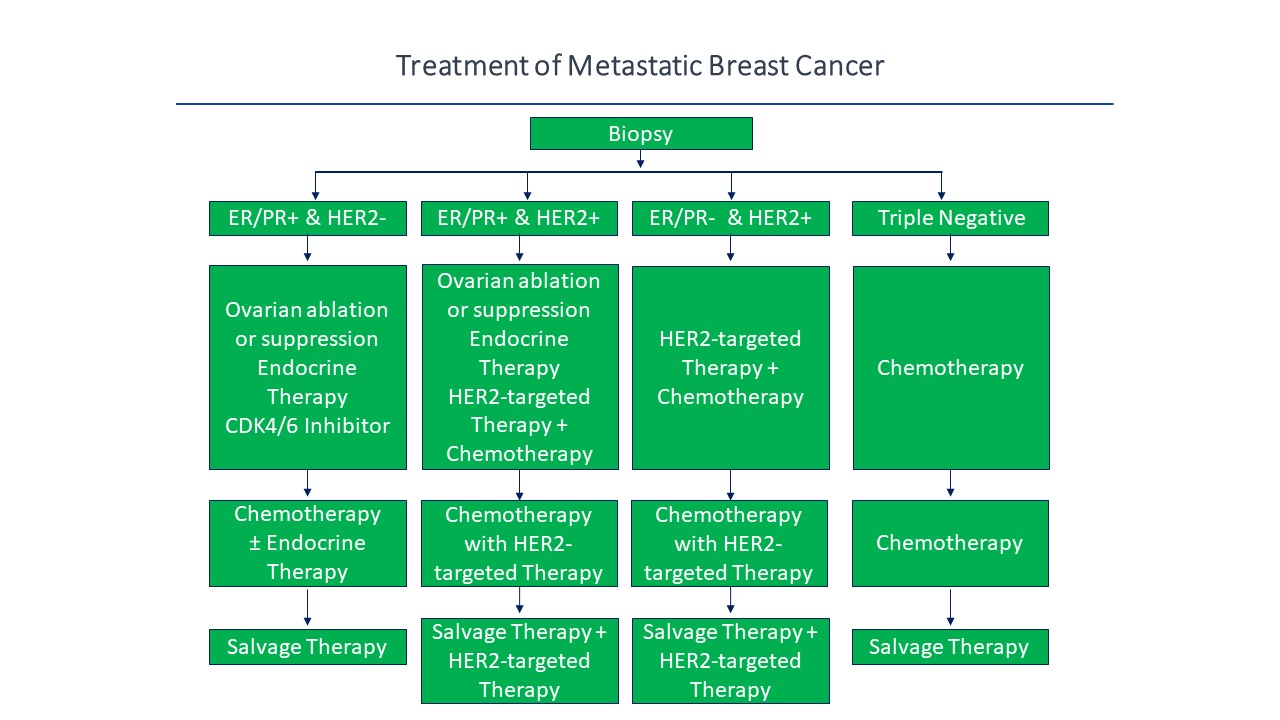
MBC treated with second or higher lines of therapy has a very poor prognosis and few effective therapies that consistently induce long-term remission,29 which indicates the market demand and clinical need for new and improved therapeutic drugs and treatment options in order to improve these response outcomes and patient survival rates. Thus, Bria-IMT™ has the potential to induce long-term remission, especially in combination with immunotherapies. Current treatment of MBC is outlined in Figure “B”, below, which illustrates different therapeutic treatment options and drugs used upon diagnoses from biopsy and identification of breast cancer biomarkers.30
15 Perez, E. A., Vogel, C. L., Irwin, D. H., Kirshner, J. J. & Patel, R. Multicenter Phase II Trial of Weekly Paclitaxel in Women With Metastatic Breast Cancer. J. Clin. Oncol. 19, 4216–4223 (2001).
16 Seidman, A. D. Gemcitabine as single-agent therapy in the management of advanced breast cancer. Oncology (Williston Park). 15, 11–4 (2001).
17 Zelek, L. et al. Weekly vinorelbine is an effective palliative regimen after failure with anthracyclines and taxanes in metastatic breast carcinoma. Cancer 92, 2267–72 (2001).
18 Licchetta A, Correale P, Migali C, Remondo C, Francini E, Pascucci A, Magliocca A, Guarnieri A, Savelli V, Piccolomini A, Carli AF, Francini G. Oral metronomic chemo-hormonal-therapy of metastatic breast cancer with cyclophosphamide and megestrol acetate. J Chemother. 2010 Jun;22(3):201-4.
19 Harvey, V. et al. Phase III Trial Comparing Three Doses of Docetaxel for Second-Line Treatment of Advanced Breast Cancer. J. Clin. Oncol. 24, 4963–4970 (2006).
20 Rivera, E. et al. Phase 3 study comparing the use of docetaxel on an every-3-week versus weekly schedule in the treatment of metastatic breast cancer. Cancer 112, 1455–1461 (2008).
21 Gradishar WJ. Taxanes for the treatment of metastatic breast cancer. Breast Cancer (Auckl). 2012;6:159-71.
22 Perez, E. A. et al. Efficacy and Safety of Ixabepilone (BMS-247550) in a Phase II Study of Patients With Advanced Breast Cancer Resistant to an Anthracycline, a Taxane, and Capecitabine. J. Clin. Oncol. 25, 3407–3414 (2007).
23 Leyland-Jones, B. et al. Pharmacokinetics, Safety, and Efficacy of Trastuzumab Administered Every Three Weeks in Combination With Paclitaxel. J. Clin. Oncol. 21, 3965–3971 (2003). Only 41% of patients had prior systemic chemotherapy.
24 von Minckwitz G et el. Trastuzumab beyond progression: overall survival analysis of the GBG 26/BIG 3-05 phase III study in HER2-positive breast cancer. Eur J Cancer. 2011 Oct;47(15):2273-81. Prior therapy limited to trastuzamab alone or in combination with a taxane.
25 Verma, S. et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 367, 1783–1791 (2012).
26 Geyer, C. E. et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 355, 2733–2743 (2006).
27 Bartsch, R. et al. Capecitabine and Trastuzumab in Heavily Pretreated Metastatic Breast Cancer. J. Clin. Oncol. 25, 3853–3858 (2007).
28 Blackwell, K. L. et al. Randomized Study of Lapatinib Alone or in Combination With Trastuzumab in Women With ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. J. Clin. Oncol. 28, 1124–1130 (2010).
29 Dawood S, Broglio K, Ensor J, Hortobagyi GN, Giordano SH. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol. 2010 Nov; 21(11):2169–74; Bonotto M, Gerratana L, Iacono D, Minisini AM, Rihawi K, Fasola G, Puglisi F. Treatment of Metastatic Breast Cancer in a Real-World Scenario: Is Progression-Free Survival With First Line Predictive of Benefit From Second and Later Lines? Oncologist. 2015 Jul;20(7):719-24; Kotsakis A, Ardavanis A, Koumakis G, Samantas E, Psyrri A, Papadimitriou C. Epidemiological characteristics, clinical outcomes and management patterns of metastatic breast cancer patients in routine clinical care settings of Greece: Results from the EMERGE multicenter retrospective chart review study. BMC Cancer. 2019 Jan 18;19(1):88.
30 NCCN Guidelines Version 2.2019, 07/02/2019 © 2019 National Comprehensive Cancer Network (NCCN®).
| 9 |
Figure B: Current treatment paradigm for metastatic breast cancer including between different treatment strategies and combination therapies dependent upon biomarker identification and activity within the breast cancer signaling pathway.
Of patients treated with trastuzumab for MBC, one study showed that 241/331 (72%) progressed within 27 months (32% per year) with median survival of 13-14 months (CI 10-15 months).31 This indicates the high unmet need in this patient population which should facilitate regulatory review of novel therapies such as Bria-IMT™.
While there are approximately 36 different biotech companies working to create an effective breast cancer vaccine, a significant gap remains in the effectiveness and safety of second or higher lines of therapy. The most studied targeted immunotherapy, Neuvax (Galena), a HER2 peptide vaccine, failed a Phase III trial, but there is encouraging data to support at least three ongoing clinical trials combining trastuzumab with HER2 epitope immunogens.32 The National Cancer Institute (“NCI”) randomized trial adding PANVAC (a poxviral-based immunogen) to docetaxel increased the median PFS from 3.9 months to 7.9 months and is to be used as a basis for larger, more sophisticated clinical trials.33 An immunogen targeting a carbohydrate antigen, globo-H, was associated with improved PFS, but only in the subset able to mount antibody responses.34 A Johns Hopkins breast cancer trial using a breast cancer cell line transfected with the gene for GM-CSF has not been positive but, using the same cell line with trastuzumab, 40% of patients enjoyed clinical benefit (CR+PR+stable) at one year.35 Finally, the study of targeted cancer immunotherapies in combination with other therapies is receiving much attention, particularly combination with checkpoint inhibitors.36
31 Rossi, V.; Nole, F.; Redana, S.; Adamoli, L.; Martinello, R.; Aurilio, G.; Verri, E.; Sapino, A.; Viale, G.; Aglietta, M.; Montemurro, F., Clinical outcome in women with HER2-positive de novo or recurring stage IV breast cancer receiving trastuzumab-based therapy. Breast 2014, 23 (1), 44-9.
32 Mittendorf, E. A.; Peoples, G. E., Injecting Hope—A Review of Breast Cancer Vaccines. Oncology (Williston Park) 2016, 30 (5), 475-81, 485.
33 Heery, C. R.; Ibrahim, N. K.; Arlen, P. M.; Mohebtash, M.; Murray, J. L.; Koenig, K.; Madan, R. A.; McMahon, S.; Marte, J. L.; Steinberg, S. M.; Donahue, R. N.; Grenga, I.; Jochems, C.; Farsaci, B.; Folio, L. R.; Schlom, J.; Gulley, J. L., Docetaxel Alone or in Combination With a Therapeutic Cancer Vaccine (PANVAC) in Patients With Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol 2015, 1 (8), 1087-95.
34 Huang, C.; Yu, A.; Tseng, L., Randomized phase II/III trial of active immunotherapy with OPT-822/OPT-821 in patients with metastatic breast cancer. J Clin Oncol 2016, 34 (15).
35 Chen, G.; Gupta, R.; Petrik, S.; Laiko, M.; Leatherman, J. M.; Asquith, J. M.; Daphtary, M. M.; Garrett-Mayer, E.; Davidson, N. E.; Hirt, K.; Berg, M.; Uram, J. N.; Dauses, T.; Fetting, J.; Duus, E. M.; Atay-Rosenthal, S.; Ye, X.; Wolff, A. C.; Stearns, V.; Jaffee, E. M.; Emens, L. A., A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol Res 2014, 2 (10), 949-61.
36 McArthur, H. L.; Page, D. B., Immunotherapy for the treatment of breast cancer: checkpoint blockade, cancer vaccines, and future directions in combination immunotherapy. Clin Adv Hematol Oncol 2016, 14 (11), 922-933.
| 10 |
There are several other approaches to developing targeted breast cancer immunotherapies. These include using peptide cocktails, a triple peptide regimen, recombinant HER2, antigen-pulsed dendritic cells, DNA immunogens, whole cell allogeneic GM-CSF secreting SKBR3 or T47D cells, an (HLA)-A2/A3-restricted immunogenic peptide derived from the HER2 protein, oxidized mannan-MUC1, and personalized peptide immunogens.
Among the most promising results in patients with advanced disease have been using whole-cell preparations, particularly if the cells are engineered to express GM-CSF. We are taking this approach and capitalizing on positive initial results with Bria-IMT™ monotherapy in difficult to treat patients using a regimen that both limits regulatory T cell activity (using low dose cyclophosphamide pre-treatment) and boosts the immune response (using post-dose alpha interferon in the inoculation sites). The combination with PD-1 inhibitors is a logical extension of our findings where 21 of 23 MBC patients had demonstrable PD-L1 expression on the circulating tumor cells (“CTCs”) and/or circulating cancer-associated macrophage-like cells (“CAMLs”). The overall strategy, once the initial milestones have been met, to enroll additional patients for product registration, will allow rapid progression of the best therapeutic option to a Biologics License Application (“BLA”).
Products/Pipeline
Bria-IMT™
Bria-IMT™, BriaCell’s lead candidate, is a whole-cell immunotherapy undergoing clinical testing in patients with MBC who have failed prior lines of therapy. BriaCell has been conducting a Phase I/IIa clinical trial of Bria-IMT™, in combination with immune checkpoint inhibitors such as pembrolizumab (KEYTRUDA®; manufactured by Merck & Co., Inc.). The combination study is listed in ClinicalTrials.gov as NCT03328026 under FDA-approved BB-IND 10312 under protocol BRI-ROL-001 at three clinical sites: St. Joseph Heritage Healthcare in Santa Rosa, California, United States; University of Miami/Sylvester at Plantation, in Plantation, Florida, USA; Cancer Center of Kansas, in Wichita, Kansas, USA. Subsequent to the establishment of a collaboration with Incyte Corporation, this study has been modified to evaluate the combination of the Bria-IMT™ with retifanlimab (also referred to as INCMGA00012 ,a PD-1 inhibitor).
BriaCell has achieved proof of concept based on data from a Phase I/IIa study of Bria-IMT™ in advanced breast cancer patients. In essence, BriaCell obtained evidence that patients with certain HLA molecules also present in Bria-IMT™ have a higher likelihood of responding to the Bria-IMT™ regimen with tumor regression (“shrinkage”), which is consistent with results from a molecular analysis of Bria-IMT™ conducted by BriaCell.
Positive Proof of Concept
| ● | Bria-IMT™ has been evaluated in a regimen including pre-dose low-dose cyclophosphamide (to reduce immune suppression), intradermal inoculation with 20-50 million irradiated Bria-IMT™ cells between two and three days later, with subsequent intradermal inoculation with interferon-α2 approximately two and four days later. This is known as the Bria-IMT™ regimen. Both were single arm studies, so there were no untreated patients for comparison. | |
| ● | BriaCell has evaluated the Bria-IMT™ regimen in two Phase I/IIa studies of Bria-IMT™ in advanced breast cancer patients. |
X.; Wolff, A. C.; Stearns, V.; Jaffee, E. M.; Emens, L. A., A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol Res 2014, 2 (10), 949-61.
| 11 |
| ● | There were four evaluable patients treated in one study (Study SVMC #01-026) and 23 evaluable patients treated in another study (Study WRI-GEV-007) with this regimen with cycles every two weeks for the first month and then monthly. They were heavily pre-treated with a median of four prior systemic therapy regimens. | |
| ● | The data shows an outstanding safety and tolerability profile for Bria-IMT™ in advanced breast cancer patients. | |
| ● | In the SVMC #01-026 study, treatment was limited to six cycles over five months. Four post-menopausal white women were enrolled aged between 58.7 and 73 years. Three had breast cancer and one had Her2+ ovarian cancer. All had failed at least one prior systemic therapy. |
| ● | These patients received between four and six cycles of treatment on protocol. One patient had an additional 13 cycles off protocol. | |
| ● | The only adverse events that occurred in more than one patient were itch and rash at the inoculation sites. No deaths were reported during this study. There were four serious adverse events (“SAEs”) in 3 patients with one (transient urticaria, grade 3) judged probably related to treatment. All SAEs were manageable with community practice therapies. | |
| ● | The Bria-IMT™ regimen was able to elicit delayed-type hypersensitivity (“DTH”) responses in all patients. DTH is a measure of cell-mediated immunity. This response involves the interaction of T-cells, monocytes, and macrophages. This reaction is caused when CD4+ Th1 helper T cells recognize foreign antigen in a complex with the Class II HLA molecule on the surface of antigen-presenting cells. These can be macrophages or dendritic cells that secrete monokines such as IL-12 and IL-15, which stimulates the proliferation of additional CD4+ Th1 cells. CD4+ T cells secrete other cytokines including IL-2 and interferon gamma, inducing the further release of other Th1 cytokines, thus mediating the immune response. This results also in the activation of CD8+ T cells which destroy target cells on contact, and activated macrophages which produce hydrolytic enzymes. | |
| ● | The DTH response involves the interaction of T-cells, monocytes, and macrophages. This reaction is caused when CD4+ Th1 helper T cells recognize foreign antigen in a complex with the Class II HLA molecule on the surface of antigen-presenting cells. These can be macrophages or dendritic cells that secrete monokines such as IL-12 and IL-15, which stimulates the proliferation of additional CD4+ Th1 cells. CD4+ T cells secrete other cytokines including IL-2 and interferon gamma, inducing the further release of other Th1 cytokines, thus mediating the immune response. This results also in the activation of CD8+ T cells which destroy target cells on contact and activated macrophages which produce hydrolytic enzymes. |
| 12 |
| ● | One patient (A002) had a partial response with regression of breast lesions, resolution of lung and soft tissue lesions, and improvement of stability of bone lesions. She completed therapy and 3 months after her last Bria-IMT™ inoculation, imaging studies identified regrowth of tumor notably in the breast, lung, and brain. After consultation with the FDA, the patient was treated off-protocol which also produced tumor regression, including the resolution of brain metastases. The HLA-DRB3 allele of patient A002 matched with that of SV-BR-1-GM and the HLA-DRB1 allele of patient A002 also matched that of SV-BR-1-GM. Her tumor was grade II (moderately differentiated). One other patient on this study (B001) with a grade II tumor had disease limited to bony metastases. She did not have measurable disease but was felt to progress on study. | |
| ● | Median time to tumor progression was 144 days (range 64 – 223 days) for the initial round of treatment. Overall survival was more than 33 months in all patients except B001 (7 months). |
| ● | In the WRI-GEV-007 study, patients were treated with a median of three cycles of therapy (range 1-8). |
| ● | The Bria-IMT™ regimen was able to elicit both cellular immune responses (as evidenced by DTH responses in 85% of patients evaluated) and antibody responses (present in 58% of patients evaluated). |
| ● | The most common adverse events seen were local irritation at the inoculation sites. There was one serious adverse event of gastrointestinal reflux disease possibly related to Bria-IMT™. | |
| ● | Several patients showed evidence of anti-tumor activity of the Bria-IMT™ regimen in spite of their being heavily pre-treated advanced breast cancer patients. Specifically, one patient (designated 01-002) had regression or disappearance of 20 lung metastases, but stable disease in liver metastases (as the liver metastases were the target lesions, she did not qualify as a partial response). She displayed a robust DTH response, had a grade I tumor and matched Bria-IMT™ at 2 HLA loci. One patient (05-002) had a reduction in the size of a breast lesion but progression of a liver lesion and did not meet criteria for a partial response. She also displayed a robust DTH response, had a grade II tumor and matched Bria-IMT™ at 2 HLA loci. One patient (01-005) had a marked reduction in cutaneous involvement but developed restrictive cardiomyopathy (unrelated to study drug) with subsequent mortality. She had a grade III (poorly differentiated) tumor and matched Bria-IMT™ at one HLA locus. She was not on study long enough to be evaluated for her response. | |
| ● | Patients 01-002, 05-002 and 01-005 who showed objective evidence of tumor shrinkage all matched the Bria-IMT™ cell line at least at one HLA locus and all had evidence of DTH responses to Bria-IMT™ and/or the parent cell line (SV-BR-1 – the breast cancer cell line from which Bria-IMT™ was derived). Patients who did not develop a DTH response did not show evidence of tumor shrinkage. | |
| ● | Patients 01-002 and 05-002 had grade I/II tumors. Both of them also had two HLA matches with Bria-IMT™. Two other patients with grade II tumors (patient 03-001 and 06-001) had stable disease on the study and were also considered to have received clinical benefit from the treatment. (Clinical benefit was defined as some evidence of tumor shrinkage (including a mixed response with shrinkage of some tumors but progression of others, as for 05-002) with over 90 days on study; or as stable disease, a partial response or a complete response as per RECIST criteria). Neither 03-001 or 06-001 had HLA matches with Bria-IMT™, suggesting that HLA matching may not be required for clinical benefit in patients with grade I/II tumors. Thus, four of the six patients with grade I/II tumors exhibited clinical benefit. One of the remaining patients showed no evidence of an immune response as evaluated by DTH. Thus, four of the five grade I/II patients able to develop an immune response, as noted by DTH, exhibited clinical benefit. | |
| ● | These preliminary data indicate that the Bria-IMT™ regimen in advanced breast cancer patients is well tolerated, able to elicit an immune response and able to induce reduction in tumor burden. |
| 13 |
| ● | Another phase I/IIa study (BRI-ROL-001) was initiated evaluating the combination of the Bria-IMT™ regimen with KEYTRUDA® (pembrolizumab). This combination combines the induction of an immune response by Bria-IMT™ (i.e. “putting the foot on the gas” of the immune response) with the ability of KEYTRUDA® to block the PD-1 – PD-L1 immune checkpoint (i.e. to “take the foot off the brakes” of the immune response). | |
| ● | Eleven patients with advanced breast cancer (median of four prior systemic therapy regimens) have been treated with this regimen with cycles every three weeks for a median of three cycles (range 1 – 9 cycles). | |
| ● | Two patients had evidence of tumor regression, both of whom had robust immune responses (as measured by DTH) to Bria-IMT™. Both of them had grade II tumors. One matched Bria-IMT™ at two HLA types (06-005) while the other did not match Bria-IMT™ at any HLA types (06-001, who “rolled over” from the WRI-GEV-007 study where she had stable disease), suggesting that the Bria-IMT™ regimen, when given in combination with a PD-1 inhibitor, may be able to induce tumor regression without an HLA match especially in patients with grade I/II tumors. One additional patient (06-004) in this study had a grade II tumor and was noted to have stable disease. The other seven patients treated had grade III tumors (poorly differentiated). Thus, all three of the patients with grade I/II tumors showed evidence of clinical benefit. | |
| ● | Following the establishment of a collaboration with Incyte Corporation, this study is being altered to evaluate the combination of the Bria-IMT™ regimen with INCMGA00012 (anti-PD-1 antibody similar to KEYTRUDA®) and epacadostat (inhibitor of IDO, which suppresses the immune response). The combination with KEYTRUDA® has been discontinued but may be resumed in other studies. | |
| ● | The data confirms the “HLA Matching Hypothesis” and supports BriaCell’s strategy for the development of Bria-OTS™, BriaCell’s first personalized off-the-shelf immunotherapy for advanced breast cancer. |
About Bria-IMT™
Developed and characterized by a team of dedicated scientists and clinicians, Bria-IMT™ (SV-BR-1-GM) is a targeted immunotherapy being developed for the treatment of breast cancer. Bria-IMT™ is a genetically engineered human breast cancer cell line with features of immune cells and clinically applied as a targeted immunotherapy.
In short, Bria-IMT™ immunotherapy is a genetically engineered human breast cancer cell line derived from a grade II tumor which activates the immune system to attack and destroy breast cancer tumors.
Mechanism of Action of Bria-IMT™
The mechanism of action of Bria-IMT™ is currently under investigation. It is likely that the expression of certain breast cancer antigens (proteins expressed in breast cancer cells) in Bria-IMT™ generates strong T cell and potentially antibody responses – resulting in recognition and destruction of cancerous cells.37
Bria-IMT™ is designed to secrete GM-CSF, a factor that stimulates components of the immune system. Specifically, GM-CSF activates dendritic cells, the cells that start immune responses. These activated dendritic cells then activate T cells, a key component of the immune system, to recognize the tumor cells as foreign, and eliminate them. To amplify this action, we have combined Bria-IMT™ with other immune system activators including cyclophosphamide (used in low doses to reduce immune suppression), and interferon-α, a cytokine that further activates the immune system. We believe this approach of simultaneous activation of the immune system via different pathways will improve the immune system response to attack and destroy cancer cells.
| 14 |
Bria-OTS™
Using BriaCell’s novel technology platform and our strong research and development capabilities, BriaCell plans to develop Bria-OTS™, a personalized off-the-shelf immunotherapy for breast cancer, and similar immunotherapy cell lines for other cancer indications.
| ● | Bria-OTS™ is under development as an off-the-shelf personalized immunotherapy for advanced breast cancer. | |
| ● | The concept for Bria-OTS™ comes from BriaCell’s work with Bria-IMT™, where BriaCell noted that if a patient “matches” Bria-IMT™ in their HLA type, they were more likely to respond. | |
| ● | HLA molecules are the molecules that start immune responses but are polymorphic – i.e. they are different in different people, although some people will share the same HLA molecules (referred to as HLA alleles or HLA types). | |
| ● | Bria-OTS™ is made from cell lines that are genetically engineered to expresses the immune boosters GM-CSF and interferon-α, as well as specific HLA types (a.k.a. alleles). | |
| ● | Different cell lines are being pre-manufactured to express different HLA types covering >99% of the overall breast cancer patient population. |
| ● | Using the BriaDX™, a companion diagnostic test performed on the patient’s saliva, the suitable personalized treatment will be selected for each patient for administration. | |
| ● | This approach allows personalized treatment without the need for personalized manufacturing. Additionally, it saves time, and skips expensive and complicated manufacturing procedures associated with other personalized treatments. | |
| ● | Bria-OTS™ cell lines are being engineered and transferred to good manufacturing practice (“GMP”) production in 2022 and commencing clinical evaluation in 2022 (expected authorization by FDA and expected first patient to be dosed in 2022) with safety and efficacy data expected to be released during 2022 and 2023. |
BriaDx™
BriaDx™ is a diagnostic test that BriaCell is developing to identify the patients most likely to respond to Bria-IMT™. Currently, BriaDx™ includes HLA typing of the patients, as patients having HLA alleles also present in Bria-IMT™ appear to have a higher likelihood of responding to the Bria-IMT™ regimen with tumor shrinkage. Additional markers of potential diagnostic use are being developed based on the expression of specific biomarkers in the responder (i.e. biomarkers which identify the patients for which Bria-IMT™ immunotherapy appears more effective) vs the non-responder patients from clinical studies of Bria-IMT™ in advanced breast cancer patients.
Blood and tumor samples from the patients are analyzed using cutting-edge technologies including gene expression analysis and assessment of the levels of antibodies predicted to bind to Bria-IMTTM.
The insights gained from biomarker studies conducted to date have provided us with a solid basis for the development of Bria-OTS™, an off-the-shelf personalized immunotherapy which would match over 99% of patients with advanced breast cancer.
37 Lacher M.D., Bauer G. Fury B., Graeve S., Fledderman E.L., Petrie T.D., Coleal-Bergum D.P., Hackett T., Perotti N.H., Kong Y.Y., Kwok W.W., Wagner J.P., Wiseman C.L., and Williams W.V. SV-BR-1-GM, a Clinically Effective GM-CSF- Secreting Breast Cancer Cell Line, Expresses an Immune Signature and Directly Activates CD4+ T Lymphocytes. Frontiers in Immunology 2018; 9: Article 776.
| 15 |
BriaDx™ is being developed to help understand which patients are most likely to respond to Bria-IMT™ targeted immunotherapy. Based on the proposed mechanism of action of Bria-IMT™, HLA molecules play a key role inducing cellular immune responses to Bria-IMT™ which boosts the patient’s immune response to their cancer.
HLA molecules are polymorphic, in that they are different in different individuals, but shared by some individuals (similar to eye color). Based on our clinical data to date, we hypothesize that patients with HLA alleles also present in Bria-IMT™ have a higher likelihood of responding to the Bria-IMT™ regimen with tumor regression. Therefore, BriaDx™, a companion diagnostic test, determines the patients’ HLA types.
Available Clinical Data for Treatment with the Bria-IMT™ Regimen
BriaCell conducted three Proof of Concept clinical trials, one using parental SV-BR-1 cells and the other two using Bria-IMT™ (i.e. genetically engineered SV-BR-1 cells – producing GM-CSF also called SV-BR-1-GM), in metastatic (i.e. Stage IV) breast cancer patients who had failed prior treatments. The patients were treated with the Bria-IMT™ regimen according to the following schedule, and the results are summarized below.
Treatment schedule:
| ● | Cyclophosphamide
300 mg/m2 intravenously 2-3 days prior to Bria-IMT™ inoculation 
| |
| ● | Bria-IMT™
20 million irradiated cells given intradermally split into 4 inoculations (2 in the upper
back and 2 in the thighs) | |
| ● | Interferon alpha-2b 10,000 units into each inoculation site 2 and 4 days after the Bria-IMT™ inoculations | |
| ● | Treatment cycles every 2 weeks for four weeks (3 inoculations) then every month. |
| 16 |

First Proof of Concept Trial
| ● | Used unmodified cell line (parental SV-BR-1 cells) + GM-CSF + cyclophosphamide. | |
| ● | N = 14 late stage, treatment-refractory breast cancer patients. | |
| ● | No significant adverse treatment-associated events, well tolerated. | |
| ● | Median Overall Survival = 12.1 months. |
Second Proof of Concept Trial
| ● | Used Bria-IMT™ (i.e. genetically engineered SV-BR-1 cells – producing GM-CSF) with pre-dose, low dose cyclophosphamide and post-dose local interferon-α to boost the response (the Bria-IMT™ regimen) with cycles every two weeks for four weeks (three inoculations) then monthly up to a total of six cycles. | |
| ● | N = 4 late stage, treatment-refractory (3 breast cancer (2 grade II and 1 grade III), and 1 ovarian cancer) patients. | |
| ● | No significant adverse treatment-associated events, well tolerated. | |
| ● | Median Overall Survival = 35 months. | |
| ● | One robust responder with greater than 90% regression during treatment and a subsequent relapse (upon halting treatment) responded to re-treatment. | |
| ● | This patient matched Bria-IMT™ at a key HLA type (HLA-DR) and had a grade II tumor. |
Third Proof of Concept Trial
Thirty patients were screened, 24 enrolled and 23 dosed in the Phase I/IIa study (2017-2018).
| ● | The Bria-IMT™ regimen included pre-dose low-dose cyclophosphamide (to reduce immune suppression), intradermal inoculation with 20-50 million irradiated Bria-IMT™ cells between two and three days later, with subsequent intradermal inoculation with interferon-α2b approximately two and four days later. The majority of adverse events (“AEs”) were limited to expected minor local irritation at the injection sites. | |
| ● | The 23 patients treated with this regimen received cycles every two weeks for the first month and then monthly. They were heavily pre-treated with a median of four prior systemic therapy regimens. | |
| ● | Patients were treated with a median of three cycles of therapy (range 1-8). | |
| ● | The Bria-IMT™ regimen was able to elicit both cellular immune responses (as evidenced by DTH responses in 85% of patients evaluated) and antibody responses (present in 58% of patients evaluated). | |
| ● | There were no serious, unexpected, drug-related AEs. |
Most patients who withdrew from the trial did so due to the worsening of their underlying disease. Specifically, 14 patients terminated participation due to progressive disease, four withdrew, three terminated participation due to mortality (unrelated to study drug), and two terminated participation due to adverse events (both judged unrelated to study drug).
In the combined experience of the second and third studies (which both use the same Bria-IMT™ regimen), disease control (i.e. stable disease or partial response) was evaluated.
| 17 |
| ● | Disease control was seen in four of twenty patients who match with Bria-IMT™ at one or more HLA locus, including in three of six patients who match Bria-IMT™ at two or more HLA loci, further supporting our “HLA Matching Hypothesis”, and the development of Bria-OTS ™ to single match over 99% and double match approximately 90% of the patient population. | |
| ● | Effectiveness also depends on the ability of the patient to develop an immune response to Bria-IMT™, as measured by DTH to the Bria-IMT™ or to the parental cell line (SV-BR-1). Across both “monotherapy” studies (SVMC #01-026 and WRI-GEV-007), a positive DTH response was noted in 22 patients while five were not responsive. | |
| ● | Results are shown in the tables below, combining the second and third proof of concept studies, both of which used Bria-IMT™ in an identical regimen. |
Disease Control* in Studies SVMC #01-026 and WRI-GEV-007 Based on HLA Matching to Bria-IMT™ and Immune Response to Treatment
| Patients | HLA Match | Disease Control | Disease Control in Immune Responders | |||||||||
| N=6 | ≥2 | 50 | % | 75 | % | |||||||
| N=20 | ≥1 | 25 | % | 33 | % | |||||||
| N=7 | 0 | 29 | % | 29 | % | |||||||
| All Patients N=27 | 26 | % | 32 | % | ||||||||
Disease Control in Studies SVMC #01-026 and WRI-GEV-007 Based on Tumor Grade
| Patients | Grade I/II | Disease Control | ||||||
| All Patients N=27 | 8 | 63 | % | |||||
| Immune Responders (as measured by DTH) | ||||||||
| Immune Responders N=22 | 7 | 71 | % | |||||
| ● | Bria-IMT™ was dosed in 27 patients (four in 2004-2006, 23 in 2017-2018) as the Bria-IMT™ regimen alone. | |
| ● | Bria-IMT™ has been very well tolerated (over 100 doses given to date). | |
| ● | Tumor regression was seen in patients who were able to mount an immune response and matched Bria-IMT™ at HLA types, confirming our main hypothesis and supporting using HLA typing as a marker to predict who is most likely to respond. | |
| ● | BriaCell continues to monitor their clinical trials, proposing that BriaDx™ would include HLA typing as well as other potential biomarkers (such as tumour grade or the ability to mount a DTH response) to identify the patients most likely to respond to the Bria-IMT™ regimen. |
Development of Additional Immunotherapy Cell Lines
| ● | Based on these observations, BriaCell is extending this technology to other types of cancer by developing additional immunotherapy cell lines. | |
| ● | Cell lines currently being genetically engineered include a breast cancer cell line, a prostate cancer cell line, a non-small cell lung cancer cell line and a melanoma cell line. | |
| ● | Initial steps in the genetic engineering have been completed with subsequent steps planned for 2022 and 2023. | |
| ● | IND filings for these immunotherapy cell lines are anticipated starting in 2022. |
| 18 |
Protein Kinase C Delta (PKCδ) Inhibitors
Overview
The delta isoform of the Protein Kinase C family (PKCδ) is implicated in a multitude of cellular responses to external and internal stimuli, playing both pro- and anti-tumorigenic roles. In contrast to PKCα, PKCδ does not seem to be required for survival of normal cells. In PKCδ knockout mice, mild lymphoproliferation was observed, but overall, PKCδ inhibition is well tolerated at the organismal level. BriaCell scientists develop small-molecule PKCδ inhibitors for use in those situations where PKCδ carries out pro-tumorigenic functions. Preliminary data suggest that PKCδ inhibition may be particularly beneficial in a subset of cancers with oncogenic Ras or with otherwise activated Ras signaling, for instance in endometrial cancers with estrogen-induced K-Ras stabilization. In particular, PKCδ inhibition may be of therapeutic use in cancers dependent on Ras signaling for proliferation, as shown in vitro for lung cancer. BriaCell, through our subsidiary Sapientia, uses structural information of Rottlerin, a PKCδ inhibitor with modest activity, and Staurosporine, a potent but nonspecific PKC inhibitor, to develop a series of “hybrid” compounds. This rational design approach is envisioned to yield molecules with, compared to Rottlerin, enhanced activity yet retained PKC δ-selectivity.
Strategy and Results
PKCδ inhibition was achieved with small molecules using a pharmacophore model based on Staurosporine and Rottlerin. One of the most promising molecules based on this approach, BC106 (BJE6-106), presents an IC50 for PKCδ inhibition of approximately 50 nM and is approximately 1000-fold more selective for PKCδ than for PKCα. In cellular and animal model studies, BC106 shows effective anti-proliferative and anti-tumor activity, but this molecule is not water soluble, hence not appropriate as a drug candidate. Efforts to improve water solubility have been initiated, with a series of compounds undergoing testing in in vitro kinase and cell-based assays.
To develop PKCδ inhibitors, BriaCell affiliates started with two molecules known to have PKC-inhibitory properties: Staurosporine and Rottlerin. Multiple chemical manipulations and testing resulted in BC106, one of the Company’s most effective compounds to-date. Staurosporine is a well-known PKC inhibitor with anti-cancer activity, while Rottlerin, also known as Mallotoxin, opens potassium channels that have been used to induce apoptosis. Rottlerin has also been shown to be an immunosuppressive agent, affecting multiple oncogenic pathways. Although some reports claim that Rottlerin does not act primarily via PKCδ inhibition, BriaCell’s data supports Rottlerin-derived molecules as viable tumor suppressors.
The Company’s strategy for compound synthesis is based on a hitherto unexplored design concept, wherein functional moieties of two natural products known to strongly inhibit PKCδ – Rottlerin and Staurosporine – have been “intellectually cut” from each natural product and then covalently joined to make a novel, chimeric scaffold. The Company’s synthetic analogs, in essence, combine the bottom benzopyran moiety of Rottlerin and chemically join that to the indolyl carbazole moiety of Staurosporine. Further, new chimeric scaffolds are synthesized in a novel, convergent modular fashion, allowing for the rapid assembly and testing of many derivatives.
Rottlerin was initially used because this molecule inhibits purified PKCδ at an IC50 of 3-5 μM in vitro, and in cultured cells with an IC50 of 5 μM. Rottlerin is relatively more selective for PKCδ than for PKCα (PKCδ IC50:PKCα IC50 ≈ 1:30). BriaCell further advanced our pharmacophore model using the Rottlerin-based prototype chimeric structure in combination with Staurosporine by incorporating protein structural data for the novel class PKCs. This strategy produced a second generation of PKCδ inhibitors with the “head” group resembling that of Staurosporine and the other domains conserved from the Rottlerin scaffold to preserve isozyme specificity. A second generation successful product is represented by BC128, which has an IC50 of 4 μM for PKCδ (similar to Rottlerin), and better isozyme selectivity (IC50 of >120 μM for PKCα). BC128 showed anti-tumor cell activity in vitro and in vivo.
| 19 |
BC106, BriaCell’s most-recent “lead” compound, produces substantial cytotoxicity against multiple human tumor lines at nM concentrations (10-40 times lower than Rottlerin or BC128). BC106 dramatically inhibited the clonogenic capacity of RAS-mut tumor cell lines after as little as 12 hours of exposure. BC106 is 1000-fold more selective for PKCδ than for PKCα. The latter is an important finding because inhibition of PKCα is generally toxic to all cells (normal and malignant) and would make BC106 non-tumor-targeted.
Approximately 40% of melanomas harbor NRAS mutations and there is no effective RAS-targeted treatment available for this subgroup. BriaCell affiliates have demonstrated that NRAS-mutant melanoma cells were highly sensitive to PKCδ siRNA knock-down and to BC106 at nM concentrations. Clonogenic assays demonstrated that irreversible inhibition of proliferation required as little as 12 hours of exposure to Rottlerin or BC106.
BriaCell affiliates also assessed the effects of PKCδ inhibition on breast tumor growth and survival in a xenograft human breast cancer stem cell model. PKCδ inhibition prevented tumor grown and promoted the survival of the animals evaluated over the course of 300 days (note that the vehicle treated animals all died within the first 20 days of the study).
Furthermore, PKCδ inhibition also inhibited the growth of neuroendocrine cells.
Summary and Outlook – Early Stage Preclinical Program
| ● | Thirty percent of all human malignancies display activating RAS mutations, with another 60% showing over-activity of Ras-signaling pathways.38 | |
| ● | BriaCell’s novel, proprietary PKCδ inhibitors have shown activity against multiple RAS transformed tumors.39 | |
| ● | This target has an attractive safety profile based on in vivo studies and knock out mouse studies.40 | |
| ● | PKCδ also has potential activity as an immunotherapeutic by blocking TGFβ signaling.41 | |
| ● | PKCδ inhibitors are applicable to specific niche tumor types which provide an accelerated clinical development plan. | |
| ● | Structural aspects of first-generation inhibitor rottlerin and staurosporine ([a] pan-PKC inhibitor) were combined to create second generation inhibitor KAM1. | |
| ● | Third generation inhibitors such as BC-106 have improved potency and selectivity. | |
| ● | Fourth generation inhibitors are under development to optimize their drug-like characteristics. | |
| ● | PKCδ inhibitors lack endothelial cell cytotoxicity, while PKCδ deficient mice develop normally and are fertile. This shows that there is no marked intrinsic toxicity as a result of inhibiting PKCδ. | |
| ● | Candidate selection is anticipated in 2022. |
38 Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012 May 15; 72(10): 2457–2467.
39 Xia, S., Forman, L. W. & Faller, D. V. Protein Kinase Cδ Is Required for Survival of Cells Expressing Activated p21RAS. J. Biol. Chem. 282, 13199–13210 (2007); Chen, Z. et al. Protein kinase Cδ inactivation inhibits cellular proliferation and decreases survival in human neuroendocrine tumors. Endocr. Relat. Cancer 18, 759–71 (2011); Xia, S., Chen, Z., Forman, L. W. & Faller, D. V. PKCδ survival signaling in cells containing an activated p21Ras protein requires PDK1. Cell. Signal. 21, 502–508 (2009); Liou, J. S., Chen, C.-Y., Chen, J. S. & Faller, D. V. Oncogenic Ras Mediates Apoptosis in Response to Protein Kinase C Inhibition through the Generation of Reactive Oxygen Species. J. Biol. Chem. 275, 39001–39011 (2000); Liou, J. S., Chen, J. S. & Faller, D. V. Characterization of p21Ras-mediated apoptosis induced by protein kinase C inhibition and application to human tumor cell lines. J. Cell. Physiol. 198, 277–294 (2004); Chen, C. Y., Liou, J., Forman, L. W. & Faller, D. V. Differential regulation of discrete apoptotic pathways by Ras. J. Biol. Chem. 273, 16700–9 (1998); Chen, C. Y. & Faller, D. V. Direction of p21ras-generated signals towards cell growth or apoptosis is determined by protein kinase C and Bcl-2. Oncogene 11, 1487–98 (1995); Chen, C. Y. & Faller, D. V. Phosphorylation of Bcl-2 protein and association with p21Ras in Ras-induced apoptosis. J. Biol. Chem. 271, 2376–9 (1996); Chen, C.-Y., Liou, J., Forman, L. W. & Faller, D. V. Correlation of genetic instability and apoptosis in the presence of oncogenic Ki-Ras. Cell Death Differ. 5, 984–995 (1998); Chen, C. Y. et al. The recruitment of Fas-associated death domain/caspase-8 in Ras-induced apoptosis. Cell Growth Differ. 12, 297–306 (2001).
40 Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, Tsukiyama T, Nagahama H, Ohno S, Hatakeyama S, Nakayama KI. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002 Apr 25;416(6883):865-9.
41 Wermuth PJ, Addya S, Jimenez SA. Effect of Protein Kinase C delta (PKC-δ) Inhibition on the Transcriptome of Normal and Systemic Sclerosis Human Dermal Fibroblasts In Vitro. PLoS ONE, November 2011, Volume 6, Issue 11, e27110; PMCID: PMC3214051; Li Z, Jimenez SA. Protein Kinase C δ and c-Abl Kinase Are Required for Transforming Growth Factor β Induction of Endothelial–Mesenchymal Transition In Vitro. Arthritis and Rheumatism, Vol. 63, No. 8, August 2011, pp 2473–2483 PMCID: PMC3134600; Bujor AM, Asano Y, Haines P, Lafyatis R, Trojanowska M. The c-Abl Tyrosine Kinase Controls Protein Kinase C δ –Induced Fli-1 Phosphorylation in Human Dermal Fibroblasts. Arthritis & Rheumatism, Vol. 63, No. 6, June 2011, pp 1729–1737. PMCID: PMC3381734.
| 20 |
Early Phase Programs
BriaCell is developing multi-specific binding reagents that simultaneously bind to an immune cell and a cancer cell, or just to a cancer cell, and activate the immune system against the cancer cells. The novel binding reagents are designed to act, among others, as potent immune cell activators/immune checkpoint inhibitors without the toxicity of current checkpoint inhibitors. The expected effect is a highly targeted therapy envisioned to selectively destroy cancer cells without affecting normal (i.e. non-cancerous) cells. This may mean less severe side effects for the treated cancer patients compared to alternative therapies. The Company cautions that these novel therapeutics are still in early-stage research and development and is not making any express or implied claims as to their success in cancer treatment or commercial viability. The patent application seeks protection for, among others, the design of new therapeutics and methods for their use. These are designated “Bria-TILs-Rx”. IND filings for Bria-TILs-Rx for the treatment of prostate cancer and epithelial and glandular cancer, respectively, are anticipated to be made in 2022 and require an additional cost of approximately US$1,000,000 each.
On October 28, 2020, BriaCell entered into a Cooperative Research and Development Agreement (“CRADA”) with the U.S. Department of Health and Human Services, as represented by the NCI. Under the CRADA, NCI and BriaCell will work together to conduct preclinical studies to develop and test BriaCell’s proprietary Bria-OTS cellular immunotherapy as a treatment for cancer, to improve and broaden applicability of this therapeutic strategy. Under the terms of the CRADA, BriaCell will provide funding (totaling $433,400 over three years) to support the project. The NCI estimates that 1.3 person-years of effort per year will be required to complete the CRADA research, which includes the development of a mouse model of this therapeutic strategy. BriaCell and NCI will be using their combined expertise in tumor immunology, molecular biology and development of cellular therapies to design studies which are intended to trigger the immunologic pathways necessary to create potent immune responses against cancer. The goal of the collaboration is to develop novel therapeutics for future clinical collaborations, allowing cancer patients to potentially benefit from potent and personalized cancer immunotherapy.
Mechanism of Action of Bria-IMT™ and Bria-OTS™4
The mechanism of action of Bria-IMT™/Bria-OTS™ is currently under investigation.
| 21 |
We believe that Bria-IMT™/Bria-OTS™ activates the patient’s immune system to recognize tumor cells and destroy them. We hypothesize that Bria-IMT™/Bria-OTS™ exerts its action via the patient’s antigen-presentation system (i.e. the system that presents antigen material on the surface of cells for recognition by the T cells of the immune system as either self (i.e. safe) or foreign (i.e. to be destroyed)). Specifically, Bria-IMT™/Bria-OTS is thought to stimulate dendritic cells, a key component of the antigen-presenting system, to display certain immunogenic (i.e. immune response-generating) protein fragments to T cells, which activates the T cells to destroy the tumor cells either directly, or indirectly by inducing a humoral (i.e. antibody-generating) immune response. In addition, we also have shown that Bria-IMT™ is capable of directly stimulating T cells, thereby potentially adding additional therapeutic benefits. The latter property of Bria-IMT™ is the basis of the Bria-OTS™ project as it requires HLA matching between the therapeutic cells and the patient.42
BriaCell’s preliminary analyses have shown several up-regulated genes in Bria-IMT™ that encode proteins known to be immunogenic (i.e. immune response-generating), suggesting that Bria-IMT™ can stimulate the immune system against the cancer cells.
Bria-IMT™ is a human breast cancer cell line which expresses Her2/neu (a protein well known for its overexpression in breast cancer but also associated other epithelial malignancies, including ovarian, pancreatic, colon, bladder and prostate cancers). Bria-IMT™ has been engineered to produce and secrete GM-CSF, a protein that promotes dendritic cell function, a key component of the immune system, and hence activates the immune system.
Potential Mechanisms of Specific Immune Activation in Advanced Breast Cancer
| 1. | Bria-IMT/OTS™ produces breast cancer antigens (i.e. proteins made by breast cancer cells). |
| 2. | Bria-IMT/OTS™ secretes GM-CSF, which further promotes dendritic cell-based antigen presentation (i.e. boosts the response). |
| 3. | Breast cancer antigens are taken up by dendritic cells and “presented” to CD4+ and CD8+ T cells implicated in tumor destruction. |
| 4. | Bria-IMT/OTS™ directly stimulates cancer fighting CD4+ and CD8+ T cells (i.e. further boosts the response). |
| 5. | Bria-IMT/OTS™ biological activity depends on HLA matching of Bria-IMT/OTS™ and the patient. |
Clinical Trials
Phase I/IIA Combination Study of Bria-IMT™ with Immune Checkpoint Inhibitors in Advanced Breast Cancer
The FDA approved the combination study of Bria-IMT™ with immune checkpoint inhibitors in advanced breast cancer (third line or later). The initial study used pembrolizumab (KEYTRUDA®, purchased by the Company as the Company does not have an agreement with Merck for the supply of KEYTRUDA®). The Company dosed 11 patients with this combination and no dose limiting toxicities were observed. Additionally, evidence of additive or synergistic activity was observed.
The combination with KEYTRUDA® was discontinued and the study was subsequently modified to use a combination of Bria-IMT with the Incyte PD-1 inhibitor retifanlimab (INCMGA00012). The Company anticipates additional safety and efficacy data for the combination of Bria-IMT™ with INCMGA00012 to be released throughout 2022 and 2023.
42 Lacher M.D., Bauer G. Fury B., Graeve S., Fledderman E.L., Petrie T.D., Coleal-Bergum D.P., Hackett T., Perotti N.H., Kong Y.Y., Kwok W.W., Wagner J.P., Wiseman C.L., and Williams W.V. SV-BR-1-GM, a Clinically Effective GM-CSF- Secreting Breast Cancer Cell Line, Expresses an Immune Signature and Directly Activates CD4+ T Lymphocytes. Frontiers in Immunology 2018; 9: Article 776.
| 22 |
For the year ended July 31, 2022, research costs amounted to $8,021,489 as compared to $2,020,899 for the year ended July 31, 2021. The increase is attributed to the recommencing of the Company’s clinical trials and the increased activity in the lab, including the hiring of additional lab employees.
We may face difficulties recruiting or retaining patients in our ongoing and planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to our clinical trial sites because of the outbreak of COVID-19. In the event that clinical trial sites are slowed down or closed to enrollment in our trials, this could have a material adverse impact on our clinical trial plans and timelines. We are continuing to assess our business plans and the impact COVID-19 is having on our clinical trial timelines and our ability to recruit candidates for clinical trials, but there can be no assurance that this analysis will enable us to avoid part or all of any impact from the spread of COVID-19 or its consequences, including downturns in business sentiment generally or in our sector in particular. The extent to which COVID-19 and global efforts to contain its spread will impact our operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scope of the outbreak and the actions taken to contain or treat the coronavirus outbreak. We currently believe that the execution of our clinical trials and research programs will be delayed by at least one quarter due to COVID-19.
One patient transitioned from combined treatment of the Bria-IMT™ regimen with KEYTRUDA® to combination treatment with retifanlimab. She has continued to have stable disease, with further reduction in the size of some of her breast cancer nodules around the brain, including disappearance of one nodule behind the left eye which was causing proptosis (i.e. pushing the eye forward). This nodule has completely disappeared and her eye has gone back into place.
Rationale for the Combination Study of Bria-IMT™ with Immune Checkpoint Inhibitors
Immune checkpoint inhibitors, such as anti-PD-1 antibodies, have come to the forefront in the fight against cancer, with substantial benefits for some patients. Recently, the significance of immune checkpoint inhibitors was recognized by the Nobel committee by awarding Dr. Tasuku Honjo and Dr. James P. Allison with the 2018 Nobel Prize in Physiology or Medicine (scientists behind game-changing cancer immunotherapies win Nobel medicine prize), validating the Company’s decision to initiate a combination therapy with immune checkpoint inhibitors.
Drs. Allison and Honjo independently, using different strategies, showed a new approach of treating patients by awakening certain cells of the immune system, T cells, to attack tumors. This new approach of treating patients with immune checkpoint inhibitors (such as anti-PD-1 antibodies), designed to overcome immune suppression in cancer patients, is revolutionizing the fight against cancer.
In 2010, a pre-clinical study by Dr. Allison’s research group showed that combination with anti-PD-1 antibodies potentiated the tumor-destroying effect of melanoma cells engineered to produce GM-CSF, a substance that activates the immune system, compared to the treatment with the GM-CSF producing cells alone. Bria-IMT™, a breast cancer cell line, also produces GM-CSF. Bria-IMT™ has been shown to indirectly and directly stimulate T cells, and hence has displayed immune-activating properties. BriaCell has published these findings in a leading immunology journal. It is important to note that anti-PD-1 antibodies have not been shown to work on their own in breast cancer.
KEYTRUDA® (pembrolizumab)
Manufactured by Merck & Co., Inc., KEYTRUDA® (pembrolizumab) is a prescription medicine that may treat certain cancers by working with the immune system. It has been approved for the treatment of a number of cancer indications, excluding breast cancer. The company is not a party to any agreements with Merck for the supply of KEYTRUDA®.
| 23 |
| ● | A phase I/IIa study was performed evaluating the combination of the Bria-IMT™ regimen with KEYTRUDA® (pembrolizumab). This combination combines the induction of an immune response by Bria-IMT™ (putting the foot on the gas of the immune response) with the ability of KEYTRUDA® to block the PD-1 – PD-L1 immune checkpoint (i.e. taking the foot off the brakes of the immune response). | |
| ● | Eleven patients with advanced breast cancer (with a median of four prior systemic therapy regimens) were treated with this regimen, with cycles every three weeks for a median of three cycles each (range: 1 – 9 cycles). | |
| ● | Two patients had evidence of tumor regression, both of whom had grade II tumors and also had robust immune responses (as measured by DTH) to Bria-IMT™. One matched Bria-IMT™ at two HLA types while the other did not match Bria-IMT™ at any HLA types, suggesting that the Bria-IMT™ regimen, when given in combination with a PD-1 inhibitor, may be able to induce tumor regression without an HLA match, especially in patients with grade I/II tumors. One other patient in this cohort had a grade II tumor and that patient had stable disease. Thus, of the three patients with grade I/II tumors treated with the combination of the Bria-IMT™ regimen with pembrolizumab, all three showed evidence of a clinical benefit, as typically defined in clinical research. BriaCell purchased the KEYTRUDA® for this study without a collaboration with Merck while pursuing other avenues to collaborate with a company that has an anti-PD-1 antibody and/or other immune checkpoint inhibitors to use in combination with the Bria-IMT™ regimen. BriaCell has obtained such an agreement with Incyte Corporation as noted below. Based on this, the combination therapy study (BRI-ROL-001) had been amended to evaluate combination of the Bria-IMT™ regimen with Incyte’s PD-1 inhibitor as noted below. The combination with KEYTRUDA® has been discontinued. |
BriaCell & Incyte Collaboration and Supply Agreement
The following summarizes the non-exclusive clinical trial collaboration to evaluate the effects of combinations of novel clinical candidates:
| ● | The clinical study focuses on BriaCell’s lead candidate, Bria-IMT™, in combination with Incyte’s retifanlimab, for treatment of advanced breast cancer. |
| ● | Incyte is providing retifanlimab, an anti-PD-1 monoclonal antibody, for use in combination studies with BriaCell’s lead candidate, Bria-IMT™. | |
| ● | Incyte is a global biopharmaceutical company focused on discovering and developing novel therapeutics in oncology and other serious diseases. | |
| ● | The first twelve patients will receive the Bria-IMT™ regimen in combination with retifanlimab. Once safety of the combination has been established, an expansion cohort of 24 subjects will be evaluated. This expansion cohort will be limited to patients who match Bria-IMT™ at one or more HLA alleles and/or have Grade I/II disease. | |
| ● | Dosing of the novel combinations commenced in the fourth quarter of 2019 and is ongoing. | |
| ● | The Company anticipates safety and efficacy data to be released during 2022 and 2023. | |
| ● | Pending discussions with the U.S. Food and Drug Administration (“FDA”), a registration study focused on, but not limited to, Bria-IMT™, in combination with Incyte’s selected compounds for advanced breast cancer, is planned to commence in late 2022 or early 2023 with the Bria-OTS™ program following by approximately 8-10 quarters. |
| 24 |
Additional data was presented at the American Association for Cancer Research San Antonio Breast Cancer Meeting on December 10-11, 2020. The data presented summarized the clinical and pathological data of the Bria-IMT™ monotherapy (i.e. the Bria-IMT™ regimen alone) study and the Phase I/IIa clinical study of Bria-IMT™ in combination with immune checkpoint inhibitors, including pembrolizumab (KEYTRUDA®; manufactured by Merck & Co., Inc.), and more recently, Incyte’s retifanlimab (developed by Incyte Corporation), in advanced breast cancer. Thirty patients were treated with the Bria-IMT™ regimen (19 with the Bria-IMT™ regimen alone, four who began on the Bria-IMT™ regimen and transitioned to a combination of Bria-IMT™ with Incyte’s retifanlimab, and seven with a combination of Bria-IMT™ with KEYTRUDA®). Eleven of those patients had moderately-well differentiated tumors. Seventy percent of these patients who were able to develop an immune response showed disease control, suggesting that Bria-IMT™, with a molecular signature most closely related to moderately-well differentiated tumors, may result in disease control, especially in patients with moderately-well differentiated tumors. These patients were very heavily pre-treated with a median of seven prior systemic therapy regimens (including chemotherapy, biological and “targeted” therapy). The median PFS of this cohort was 5.7 months in the monotherapy study, and 6.9 months in combination therapy. Of the group, there were nine patients with evaluable lesions including six with stable disease and two with partial responses according to RECIST criteria. One patient with stable disease had a marked reduction in numerous non-target lesions. The data suggests clinical and survival benefits for patients with moderately-well differentiated tumors who were treated with the Bria-IMT™ regimen with or without check point inhibitors. Notably, the survival benefit was higher in the group that received the Bria-IMT™ regimen with check point inhibitors, suggesting an additive or synergistic effect.
The median OS for the combined monotherapy and combination therapy was 12.5 months (data on six patients with moderately-well differentiated tumors). An OS of 7.2-9.8 months in similar patients with metastatic breast cancer in the third line setting has recently been published (Kazmi S, et al. “Overall survival analysis in patients with metastatic breast cancer and liver or lung metastases treated with eribulin, gemcitabine, or capecitabine.” Breast Cancer Res Treat. 2020). This suggests a potentially significant survival benefit for the patients treated with the Bria-IMT™ regimen alone or in combination with check point inhibitors.
Marketing and Sales Strategy
The product will initially be marketed to oncologists who are well-versed in the use of immunotherapy for cancer. Partnering with other pharmaceutical companies in order to market combinations with a number of drugs is also an option that we intend to pursue. This study will utilize a frozen formulation which consists of irradiated SV-BR-1-GM cells in viable freezing media. This formulation will permit stockpiling of the immunotherapy so that it can be sent on demand to clinical sites. The eventual goal is to reach all oncologists who treat late stage breast cancer, either by direct outreach or by partnering with another company that has an established presence in the oncology space.
Other Commercial Considerations
There is a high unmet medical need in late-stage breast cancer, providing potential for accelerated approval of Bria-IMT™. The FDA is interested in facilitating the availability of novel therapies of patients with unmet medical needs, especially those that can target the population most likely to respond. In addition, the FDA has granted “Fast Track” status to BriaCell’s lead candidate, Bria-IMT™, for the treatment of metastatic breast cancer. These two facts may help facilitate the accelerated approval of Bria-IMT™.
Production and Marketing Plan
Bria-IMT™ cells grow in simple tissue culture media and are irradiated prior to inoculation. Bria-IMT™ manufacturing will be performed by Contract Manufacturing Organizations. We have been working with KBI Biopharma, Inc. (“KBI”) and the University of California, Davis Health System (“UC Davis”) GMP facility, who have developed a frozen formulation where the cells are grown, harvested and irradiated, followed by cryopreservation in a viable state. The cells are stockpiled and shipped directly to clinical sites for inoculation. Each lot of Bria-IMT™ is tested for potency (i.e. GM-CSF production), identity (i.e. HER2+ and ER/PR-) and adventitious agents to rule out contamination with infectious agents. To date, there have been no issues with these tests. Additional manufacturing facilities have been evaluated and may be enlisted as demand grows.
| 25 |
Marketing will target oncologists who are well-versed in the use of immunotherapy and especially breast cancer treatment centers. The initial target will be patients with metastatic or recurrent breast cancer who have failed at least two prior treatment regimens. We plan to develop the clinical data for Bria-IMT™ and to use this information to reach out to oncologists seeking additional therapeutic options for their patients. We will include in this effort a physician education campaign targeting the oncologists most likely to treat metastatic breast cancer. As these physicians become more aware of the data regarding Bria-IMT™ in breast cancer, we will make sure they also understand how best to use Bria-IMT™ in combination with other therapies that have complementary synergistic mechanisms of action. This will also come from the clinical studies described above focusing on combination therapy. Partnering with other pharmaceutical companies in order to market a number of drugs is also an option that we intend to pursue. Our eventual goal is to reach all oncologists who treat late stage breast cancer, either by direct outreach or by partnering with another company that has an established presence in the oncology space.
License Agreements
On July 24, 2017, the Company entered into a Share Exchange Agreement with its wholly-owned subsidiary, BriaCell Therapeutics Corp., Sapientia, and all the shareholders of Sapientia. Sapientia, a biotechnology company based in Havertown, PA, is developing novel targeted therapeutics for multiple indications, including several cancers and fibrotic diseases.
Pursuant to the terms of the Share Exchange Agreement, BriaCell Therapeutics Corp. agreed to acquire from the Sapientia shareholders all of the issued and outstanding shares in the capital of Sapientia in consideration to the Sapientia shareholders, pro rata, of an aggregate of 8,333 common shares in the capital of BriaCell (the “Transaction”), which were issued on September 5, 2017.
As part of the Transaction, BriaCell acquired the license agreement Sapientia entered into with Faller-Williams Technology (“FWT”), dated March 16, 2017, (the “License Agreement”), pursuant to which BriaCell acquired all rights, including composition of matter patents (the “PKCδ Patents”), and preclinical study data to a novel therapeutic technology platform, PKCδ inhibitors, which represents a unique, highly-targeted approach to treat cancer and to boost the immune system.
Pursuant to the License Agreement, FWT is eligible to receive certain milestone payments, including i) $5,000,000 upon the filing of each New Drug Application with the FDA with respect to products disclosed and/or described in the PKCδ Patents (the “PKCδ Products”); ii) $25,000,000 upon final approval of each New Drug Application by the FDA for the marketing of a PKCδ Product; iii) $1,000,000 upon the filing of each Marketing Authorization Application (“MAA”) with the Medicines and Healthcare Products Regulatory Agency of United Kingdom or the Committee for Medicinal Products for Human Use of the European Commission with respect to a PKCδ Product; and iv) $5,000,000 upon the final approval of each MAA with the Medicines and Healthcare Products Regulatory Agency of United Kingdom or the Committee for Medicinal Products for Human Use of the European Commission for the marketing of a PKCδ Product.
FWT is eligible to receive certain royalty payments under the License Agreement. Following the first commercial sale of a PKCδ Product in the United States, FWT shall receive i) 5% of worldwide net sales of PKCδ Products encompassed by one or more valid claims of the PKCδ Patents and/or improvements thereto, and ii) 2.5% of worldwide net sales from PKCδ Products not encompassed within one or more valid claims of the PKCδ Patents. Additionally, upon BriaCell’s receipt of marketing approval for a PKCδ Product from the FDA, the Medicines and Healthcare Products Regulatory Agency of United Kingdom, the Committee for Medicinal Products for Human Use of the European Commission or an equivalent authority, FWT shall receive minimum royalty payments of $250,000 per year.
Unless terminated earlier pursuant to the provisions therein, the License Agreement shall expire ten years after the last PKCδ Patent expires.
Intellectual Property
The proprietary nature of, and protection for, the Company’s current and/or any future product candidates, processes and know-how are important to its business, as is its ability to operate without infringing on the proprietary rights of others, and to prevent others from infringing its proprietary rights. The Company seeks patent protection in the U.S. and internationally for its current and future product candidates it may develop through other technology. In order to protect its proprietary technologies, the Company relies on combinations of applications for patent and trade secret protection, as well as confidentiality agreements with employees, consultants, and third parties.
| 26 |
The Company has filed and own or have licensed all rights in the following pending patent applications and issued patents:
Filed with the United States Patent and Trademark Office (“USPTO”) on June 14, 2004, U.S. Patent No. 7,674,456 B2, includes claims to the following:
| 1. | Compositions comprising SV-BR cells |
| 2. | Therapeutic methods of using said compositions |
On February 27, 2017, BriaCell filed an international patent application under the Patent Cooperation Treaty (“PCT”) to further expand its intellectual property portfolio underlying the Company’s current and anticipated pipeline of whole-cell cancer immunotherapeutics, including Bria-IMT™ and Bria-OTS™. The PCT application (PCT/US2017/019757) claims priority to two provisional patent applications filed by the Company with the USPTO in 2016. In essence, it provides the framework for additional whole-cell cancer immunotherapeutics beyond Bria-IMT™ and strategies for patient-specific selection of the most likely effective whole-cell immunotherapeutic (BriaDx™). The PCT application entered the National Phase in the second half of 2018.
On July 24, 2017, BriaCell obtained the exclusive license to certain patents related to PKCδ inhibitor technology, including patents to specific compounds, methods of using the compounds, and methods of assessing patients regarding the compounds. These patents include U.S. Patent No. 9,364,460, which was issued on June 14, 2016; U.S. Patent No. 9,572,793, which issued on February 21, 2017; U.S. Patent No. 9,844,534, which was issued December 19, 2017; and EP Patent No. 2897610, which was issued on January 10, 2018.
To the knowledge of the Company’s management, there are no contested proceedings or third-party claims over any of our patent applications. Our success depends upon our ability to protect our technologies through intellectual property agreements including patents, trademarks, know-how, and confidentiality agreements. However, there can be no assurance that the above-mentioned patent applications will be approved by the appropriate agencies.
All of the technology for which patents are currently sought is owned by the Company. Our patents are entirely owned or exclusively licensed by the Company.
Competition
Cancer immunotherapy has become a significant growth area for the biopharmaceutical industry, attracting large pharmaceutical companies as well as small niche players. Generally, our principal competitors in the cancer immunotherapy market comprise both companies with currently approved products for various indications, such as manufacturers of approved bispecific antibodies, CAR-T cells, and checkpoint inhibitors, as well as companies currently engaged in cancer immunotherapy clinical development. The large and medium-size players who have successfully obtained approval for cancer immunotherapy products include Bristol-Myers Squib Company, Merck & Co., Inc., Genentech, Inc. (a subsidiary of Roche Holding AG), AstraZeneca PLC, Celgene Corporation, Johnson & Johnson/Janssen Pharmaceuticals, Amgen, Novartis, Acerta Pharmaceuticals (a subsidiary of AstraZeneca), Juno Therapeutics, Inc. (a subsidiary of Celgene), Kite Pharma, Inc., a wholly-owned subsidiary of Gilead Sciences, Inc. and Pfizer, Inc./EMD Serono, Inc. Most of these companies, either alone or together with their collaborative partners, have substantially greater financial resources than does BriaCell.
Companies developing novel products with similar indications to those we are pursuing are expected to influence our ability to penetrate and maintain market share. For patients with early stage breast cancer, adjuvant therapy is often given to prevent recurrence and increase the chance of long-term disease-free survival. Adjuvant therapy for breast cancer can include chemotherapy, hormonal therapy, radiation therapy, or combinations thereof. In addition, the HER2 targeted drug trastuzumab (HERCEPTIN), alone or in combination with pertuzumab (PERJETA), both manufactured and marketed by Roche/Genentech, may be given to patients with tumors with high expression of HER2 (IHC 3+), as well as other novel targets such as MUC1, which may be useful in treating breast cancer. In addition, the FDA approved the first ever immunotherapy regimen for breast cancer to the Roche/Genentech PD-L1 checkpoint inhibitor atezolizumab (TECENTRIQ), combined with Celgene’s nab-paclitaxel (ABRAXANE) for TNBC that cannot be removed with surgery and is locally advanced or metastatic.
| 27 |
There are a number of cancer vaccines in development for breast cancer, including but not limited to TPIV200 (Marker Therapeutics, Inc.), AE-37 (Antigen Express), and Stimuvax (Merck KgA). While these development candidates are aimed at a number of different targets, and AE-37 has published data in the HER2 breast cancer patient population, there is no guarantee that any of these compounds will not in the future be indicated for treatment of low-to-intermediate HER2 breast cancer patients and become directly competitive with NPS.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do, and also have greater experience in obtaining FDA and other regulatory approvals of treatments and commercializing those treatments. Accordingly, our competitors may be more successful than us in obtaining approval for cancer immunotherapy products and achieving widespread market acceptance. Our competitors’ treatments may be more effectively marketed and sold than any products we may commercialize, thus causing limited market share before we can recover the expenses of developing and commercializing of our cancer immunotherapy product candidate.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These activities may lead to consolidated efforts that allow for more rapid development of cancer immunotherapy product candidates.
These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, the ability to work with specific clinical contract organizations due to conflicts of interest, and the conduct of trials in the ability to recruit clinical trial sites and subjects for our clinical trials.
We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, price and the availability of reimbursement from government and other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are viewed as safer, more convenient or less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our current product candidates or any other future product candidate, which could result in our competitors establishing a strong market position before we are able to enter the market.
Employees
As of the date of this filing, we had eleven full-time employees and one part-time employee, located in various US states including: NY, CA, PA, SC, HI, and NJ. We also have international employees located in Canada and Israel.
For the year ended July 31, 2022, the average number of employees has been eight, of whom four were executive management.
Research and Development Activities and Costs
For information regarding our clinical studies, please see above under the caption “Description of the Business – Clinical Trials.”
For the years ended July 31, 2022 and 2021, we incurred $7,585,926 and $1,315,496, respectively, of net research and development expenses (excluding share based compensation allocated to research and development employees)
| 28 |
Manufacturing
We do not own or operate manufacturing facilities for the production of our product candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently depend on third-party contract manufacturers for all of our required raw materials, active pharmaceutical ingredients, and finished product candidate for our clinical trials. We currently employ internal resources and third-party consultants to manage our manufacturing contractors.
Bria-IMT™ is currently manufactured under current Good Manufacturing Practices (“cGMP”) pursuant to agreements with UC Davis and with KBI, which is located in The Woodlands, Texas.
On June 11, 2015, the Company entered into an Agreement for Services with The Regents of the University of California, acting for and on behalf of UC Davis, pursuant to which UC Davis manufactures Bria-IMT™ (previously known as BriaVax) at its GMP facility. The Company pays UC Davis certain hourly rates depending on the specific services provided by UC Davis in connection with its manufacturing of Bria-IMT™.
Pursuant to the Company’s master services agreement with KBI, dated March 17, 2017, KBI has conducted developmental studies to derive and optimize a cryopreserved formulation of Bria-IMT™ (previously known as BriaVax) as a research working cell bank of final drug product doses suitable for cold chain shipment (the “KBI Services”). The Company pays for the cost of materials, consumables, and third party services, plus an additional 5% fee to compensate KBI for the cost of purchasing, material handling, inventory and administration and management of third party services necessary for KBI to perform the KBI Services. The master services agreement with KBI terminates on May 4, 2027.
On July 5, 2022, BriaCell announced that it had entered into a manufacturing service agreement with Waisman Biomanufacturing at the University of Wisconsin–Madison (“Waisman”), to manufacture Bria-Pros™, BriaCell’s off-the-shelf personalized immunotherapy for prostate cancer, for anticipated use in clinical studies. Waisman is a leading contract manufacturing organization with experience in the manufacturing of cellular therapies for clinical trials. Under the terms of the agreement, Waisman will be responsible for GMP manufacturing of Bria-Pros™ for anticipated use in clinical studies. Waisman’s expert team will be working closely with BriaCell’s scientific and product development teams to ensure timely production of Bria-Pros™ in compliance with applicable regulatory requirements by the FDA.
Sales and Marketing
Our future commercial strategy may include the use of strategic partners, distributors, a contract sale force, or the establishment of our own commercial and specialty sales force, as well as similar strategies for regions and territories outside the United States. We plan to further evaluate these alternatives as we approach approval for the use of our product candidates for one or more indications.
Property, Plant and Equipment
The Company does not own any real property. BriaCell’s corporate offices in Canada are located at Suite 300, Bellevue Centre, 235-15th Street, West Vancouver, BC V7T 2XI, and its corporate and research offices in the United States are located at 2929 Arch Street 3rd Floor, Philadelphia, PA 19104.
We consider our current office space sufficient to meet our anticipated needs for the foreseeable future and suitable for the conduct of our business.
Government Regulation
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of biologics such as those we are developing. Along with third-party contractors, we will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our current or future product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label.
| 29 |
The process required by the FDA before biologic product candidates may be marketed in the United States generally involves the following:
| ● | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices (“GLP”) regulations; |
| ● | submission to the FDA of an Investigational New Drug Application (“IND”), which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| ● | approval by an independent Institutional Review Board (“IRB”) or ethics committee at each clinical site before the trial is begun; |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed biologic product candidate for its intended purpose; |
| ● | preparation of and submission to the FDA of a Biologics License Application (“BLA”), after completion of all pivotal clinical trials; |
| ● | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| ● | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with cGMP, and to assure that the facilities, methods and controls are adequate to preserve the biological product’s continued safety, purity and potency, and of selected clinical investigations to assess compliance with current Good Clinical Practices (“GCP”); and |
| ● | FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States, which must be updated annually when significant changes are made. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our current or future product candidates will be granted on a timely basis, if at all. Prior to beginning the first clinical trial with a product candidate, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
| 30 |
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site and must monitor the clinical trial until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by a Data & Safety Monitoring Board (“DSMB”) organized by the clinical trial sponsor, which provides authorization for whether or not a clinical trial may move forward at designated check points based on access to certain data from the clinical trial, and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects, or based on other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical trial results to public registries.
For purposes of BLA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| ● | Phase 1-The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. |
| ● | Phase 2-The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. In some cases, FDA will grant preliminary marketing authorization for drugs treating areas of high unmet medical need based on Phase 2 clinical trials. If granted, they will also require confirmatory Phase 3 evaluation post-marketing. BriaCell is evaluating Bria-IMT in patients with breast cancer who have failed at least two prior lines of therapy. In this population there is no approved therapy. Therefore, the development plan for Bria-IMT is an area of high unmet medical need. It is anticipated that BriaCell will not need to complete Phase 3 clinical trials prior to submitting the marketing application for Bria-IMT in patients with advanced breast cancer who have failed at least two prior lines of therapy. In this case, a confirmatory Phase 3 evaluation post-marketing will be required. It is anticipated that this would consist of a randomized, controlled clinical trial of Bria-IMT in combination with immune checkpoint inhibitors compared with best available therapy. However, this design is subject to negotiation with the FDA. |
| ● | Phase 3-The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. |
| ● | Phase 4-In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be made a condition to approval of the BLA. |
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product, or for biologics, the safety, purity and potency. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
| 31 |
BLA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a BLA requires payment of a substantial user fee to FDA, and the sponsor of an approved BLA is also subject to annual product and establishment user fees. These fees are typically increased annually. A waiver of user fees may be obtained under certain limited circumstances.
Once a BLA has been submitted, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and whether the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and we may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may request additional information or clarification. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
| 32 |
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a product candidate with Fast Track designation, the FDA may consider sections of the BLA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A Fast Track designated product candidate may also qualify for priority review, under which the FDA sets the target date for FDA action on the BLA at six months after the FDA accepts the application for filing. Priority review is granted when there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the Accelerated Approval program, the FDA may approve a BLA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or a clinical endpoint that can be measured earlier than irreversible morbidity or mortality and that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the biologic’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit.
In addition, a sponsor may seek FDA designation of its product candidate as a Breakthrough Therapy, if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application. Breakthrough designation also allows the sponsor to file sections of the BLA for review on a rolling basis.
Fast Track, Priority Review and Breakthrough Therapy designations do not change the standards for approval but may expedite the development or approval process.
Other Healthcare Laws and Compliance Requirements
Our sales, promotion, medical education and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to the FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services, other divisions of the Department of Health and Human Services, and state and local governments. Our promotional and scientific/educational programs must comply with the federal Anti-Kickback Statute, the Foreign Corrupt Practices Act, the False Claims Act (“FCA”), the Veterans Health Care Act, physician payment transparency laws, privacy laws, security laws, and additional state laws similar to the foregoing.
The federal Anti-Kickback Statute prohibits, among other things, the offer, receipt, or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham research or consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. Many states have similar laws that apply to their state health care programs as well as private payors.
| 33 |
The FCA imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. Actions under the FCA may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the FCA can result in significant monetary penalties and treble damages. For example, the federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multibillion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, companies have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or corporate integrity agreements, restricting the manner in which they conduct their business. The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) also created federal criminal statutes that prohibit, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “Affordable Care Act”), among other things, imposed new reporting requirements on drug manufacturers for payments or other transfers of value made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties. Certain states also mandate implementation of commercial compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare professionals.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act (“HITECH”) and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increases the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gives state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect.
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to it, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results. Also, the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to foreign officials for the purpose of obtaining or retaining business. We cannot assure you that our internal control policies and procedures will protect us from reckless or negligent acts committed by our employees, future distributors, partners, collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on our business, results of operations and reputation.
Coverage and Reimbursement
Sales of pharmaceutical products depend significantly on the availability of third-party coverage and reimbursement. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. Although we currently believe that third-party payors will provide coverage and reimbursement for our product candidates, if approved, these third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of our product candidates. Seeking coverage and reimbursement from third-party payors can be time consuming and expensive. Moreover, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis.
| 34 |
Foreign Regulation
In addition to regulations in the United States, we are and will be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and commercial sales and distribution of our products, if approved.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-U.S. countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have processes that require the submission of a clinical trial application much like an IND prior to the commencement of human clinical trials. In Europe, for example, a clinical trial application (“CTA”) must be submitted to the competent national health authority and to independent ethics committees in each country in which a company plans to conduct clinical trials. Once the CTA is approved in accordance with a country’s requirements, clinical trials may proceed in that country.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country, even though there is already some degree of legal harmonization in the European Union (the “E.U.”) member states resulting from the national implementation of underlying E.U. legislation. In all cases, the clinical trials are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of a new drug or medicinal product in the E.U., a sponsor must obtain approval of a marketing authorization application. The way in which a medicinal product can be approved in the E.U. depends on the nature of the medicinal product.
The centralized procedure results in a single marketing authorization granted by the European Commission that is valid across the E.U., as well as in Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human drugs that are: (i) derived from biotechnology processes, such as genetic engineering, (ii) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions and viral diseases, (iii) officially designated as “orphan drugs” and (iv) advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines. The centralized procedure may at the request of the applicant also be used for human drugs which do not fall within the above mentioned categories if the human drug (a) contains a new active substance which was not authorized in the European Community; or (b) the applicant shows that the medicinal product constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization in the centralized procedure is in the interests of patients or animal health at the European Community level.
Under the centralized procedure in the E.U., the maximum timeframe for the evaluation of a marketing authorization application by the EMA is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Committee for Medicinal Products for Human Use (“CHMP”)), with adoption of the actual marketing authorization by the European Commission thereafter. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest from the point of view of therapeutic innovation, defined by three cumulative criteria: (i) the seriousness of the disease to be treated, (ii) the absence of an appropriate alternative therapeutic approach, and (iii) anticipation of exceptional high therapeutic benefit. In this circumstance, EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days and the opinion issued thereafter.
The Mutual Recognition Procedure (“MRP”) for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the E.U. The MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is applicable to the majority of conventional medicinal products, and is based on the principle of recognition of an already existing national marketing authorization by one or more member states.
| 35 |
The characteristic of the MRP is that the procedure builds on an already existing marketing authorization in a member state of the E.U. that is used as reference in order to obtain marketing authorizations in other E.U. member states. In the MRP, a marketing authorization for a drug already exists in one or more member states of the E.U. and subsequently marketing authorization applications are made in other E.U. member states by referring to the initial marketing authorization. The member state in which the marketing authorization was first granted will then act as the reference member state. The member states where the marketing authorization is subsequently applied for act as concerned member states.
The MRP is based on the principle of the mutual recognition by E.U. member states of their respective national marketing authorizations. Based on a marketing authorization in the reference member state, the applicant may apply for marketing authorizations in other member states. In such case, the reference member state shall update its existing assessment report about the drug in 90 days. After the assessment is completed, copies of the report are sent to all member states, together with the approved summary of product characteristics, labeling and package leaflet. The concerned member states then have 90 days to recognize the decision of the reference member state and the summary of product characteristics, labeling and package leaflet. National marketing authorizations shall be granted within 30 days after acknowledgement of the agreement.
Should any E.U. member state refuse to recognize the marketing authorization by the reference member state, on the grounds of potential serious risk to public health, the issue will be referred to a coordination group. Within a timeframe of 60 days, member states shall, within the coordination group, make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the Commission, for the start of the decision-making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products or Veterinary Medicinal Products, as appropriate.
For other countries outside of the E.U., such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the other applicable regulatory requirements.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Recent Developments
BriaCell Appoints Renowned Oncologist, Giuseppe Del Priore, MD, MPH, as Chief Medical Officer
On February 16, 2022, the Company announced its appointment of Giuseppe Del Priore, MD, MPH, as the Company’s Chief Medical Officer (“CMO”). Dr. Del Priore will oversee the clinical and regulatory aspects of BriaCell’s current and upcoming clinical trials, including the ongoing Phase I/IIa combination study of BriaCell’s lead candidate, Bria-IMT™, with Incyte’s checkpoint inhibitor, retifanlimab, in advanced breast cancer.
Dr. Del Priore is a seasoned healthcare executive with over 25 years of experience in research, drug development, and clinical trials management. Dr. Del Priore’s prior work experience includes serving as a C-Suite biotechnology executive, a National Director at the Cancer Treatment Centers of America, and faculty at Indiana University School of Medicine, Weill Cornell Medicine, and New York University School of Medicine. Dr. Del Priore completed his MPH degree in Biostatistics and Epidemiology at the University of Illinois Chicago School of Public Health, his medical degree with Distinction at The State University of New York, and his BA, magna cum laude, in Philosophy, at The City University of New York, with additional training at Memorial Sloan Kettering Cancer Center, The University of Chicago, Northwestern University, and the University of Rochester. He has authored numerous publications, was named on several patents, and was listed as the “Best Doctors” by the U.S. News & World Report. He regularly appears in various media outlets as a Key Opinion Leader in oncology. His work has been reported on by CNN, NY Times, Bloomberg, NPR, and countless worldwide outlets and has earned editorial comments from leaders at MSKCC just in the past year.
| 36 |
BriaCell Appoints Leading Immunologist Dr. Alexander Kharazi to its Scientific Advisory Board
On February 23, 2022, the Company announced its appointment of leading immunologist, Alexander Kharazi, M.D., Ph.D., to its Scientific Advisory Board. Dr. Kharazi co-invented Bria-IMT™, BriaCell’s lead clinical candidate, in collaboration with Dr. Charles L. Wiseman, BriaCell’s Founder and Principal Research Advisor. Dr. Kharazi currently serves as Chief Technology Officer at Stemedica Cell Technologies, Inc. His experience includes roles as Chief Scientist of the Immunotherapy laboratory at St. Vincent Medical Center in Los Angeles (1998-2006) and Chief Pathologist of a large good laboratory practice animal study at the University of California, Los Angeles (UCLA) (1991-1998) reporting results to the U.S. Congress. Additionally, he has worked as a Research Fellow in the department of Pathology at the Tokyo Metropolitan Institute of Gerontology in Japan from 1989 to 1991. Dr. Kharazi earned his Ph.D. in immunology and his medical degree in internal medicine and pathology in Kiev, Ukraine. He is a named inventor on eight U.S. patents and several foreign patents. He is the author of numerous U.S. and international publications and has been an invited speaker/chairman/panelist on several scientific meetings.
BriaCell Adds Two Clinical Trial Sites to Accelerate Patient Enrollment
On February 28, 2022, the Company announced it has recruited two additional clinical sites for screening and enrolling advanced breast cancer patients in the Phase I/IIa combination study of BriaCell’s lead candidate, Bria-IMT™, with Incyte’s checkpoint inhibitor, retifanlimab. The additional clinical sites include the following: 1) Atlantic Health System, Morristown, New Jersey, and 2) Tranquil Clinical Research, Webster, Texas.
BriaCell advances preparatory work for new Bria-OTS™ breast cancer clinical trial
On April 7, 2022, the Company announced that its novel off-the-shelf (“OTS”) personalized immunotherapy, Bria-OTS™, is currently being manufactured at a cGMP facility and undergoing quality control testing for the potential upcoming clinical trial for patients with advanced breast cancer.
BriaCell Receives FDA Fast Track Approval for Targeted Breast Cancer Immunotherapy
On April 13, 2022, the Company announced that the FDA has granted Fast Track status to BriaCell’s lead candidate, Bria-IMT™, for the treatment of metastatic breast cancer (i.e. breast cancer that has spread beyond the breast). The Fast Track designation will apply to patients with metastatic breast cancer. BriaCell is developing Bria-IMT™ in combination with immune checkpoint inhibitors in a clinical trial listed in ClinicalTrials.gov as NCT03328026. BriaCell is currently enrolling and dosing advanced breast cancer patients in its Phase I/IIa combination study of Bria-IMT™ with Incyte’s checkpoint inhibitor, retifanlimab under corporate collaboration with Incyte.
Initial data on patient survival in this study was first presented at the San Antonio Breast Cancer Symposium in December 2021 and was over 12 months in spite of the patients being very heavily pre-treated having failed on average 9 prior regimens) compared with 7-10 months in a study in 3rd line breast cancer patients (i.e. those who failed 2 prior regimens for metastatic breast cancer) 1. Other patient subsets with possible survival benefit included those who match Bria-IMT™ at 1 or more HLA type and those with grade I (i.e. well differentiated) or grade II (i.e. moderately differentiated) breast cancer.
BriaCell Adds Additional Clinical Sites to Broaden Patient Access and Further Boost Enrollment
On May 18, 2022, the Company announced that it has activated Hoag Memorial Hospital Presbyterian and re-engaged Sylvester Comprehensive Cancer Center, part of the University of Miami Health System, as two additional clinical sites for the screening and enrollment of advanced breast cancer patients in the Phase I/IIa combination study of BriaCell’s lead candidate, Bria-IMT™, with Incyte’s checkpoint inhibitor, retifanlimab.
| 37 |
BriaCell Anti-Cancer Technology Published in Leading Cancer Drug Discovery Journal
On June 24, 2022, the Company announced the publication of novel scientific data and clinical data (previously reported) on BriaCell’s lead candidate, Bria-IMT™. The abstract of the paper was published on-line in Recent Patents on Anti-Cancer Drug Discovery, a publication focused on research (where patents have been registered) in leading therapeutic areas, targets, and agents related to anti-cancer drug discovery. The publication highlights BriaCell’s approach to developing novel cellular immunotherapies for cancer and the safety and efficacy of Bria-IMT™ against advanced breast cancer in a prior clinical study through a potentially unique mechanism of action. The full text of the article will be made available.
BriaCell Enters Research Agreement to Identify Novel Targets for Cancer Treatment
On June 28, 2022, the Company announced a research collaboration agreement with Harvard Medical School in support of a project led by Joan S. Brugge, PhD, a faculty member. The project aims to discover new targets that may lead to the development of novel anti-cancer treatments.
The research collaboration will focus on the discovery and development of novel targets to enhance tumor cell responsiveness to chemotherapy and immunotherapies in specific cancers including lung, head and neck, cervical, and bladder cancers. The research team at Harvard Medical School is led by Joan S. Brugge, PhD, who is the Louise Foote Pfeiffer Professor of Cell Biology and Co-Director of the Ludwig Cancer Center.
The Company will have the option to negotiate a license to innovations owned by Harvard University that arise under the one-year collaboration. The research agreement was coordinated by Harvard’s Office of Technology Development.
BriaCell Partners with Waisman Biomanufacturing to Manufacture and Supply Prostate Cancer Immunotherapy
On July 5, 2022, the Company announced a clinical-stage biotechnology company specializing in targeted immunotherapies for cancer, has entered a manufacturing service agreement with Waisman Biomanufacturing at the University of Wisconsin–Madison (“Waisman”), to manufacture Bria-Pros™, the Company’s off-the-shelf personalized immunotherapy for prostate cancer, for anticipated use in clinical studies. Waisman is a leading contract manufacturing organization with experience in the manufacturing of cellular therapies for clinical trials.
Under the terms of the agreement, Waisman will be responsible for good manufacturing practice in the manufacturing of Bria-Pros™ for anticipated use in clinical studies. Waisman’s expert team will be working closely with the Company’s scientific and product development teams to ensure timely production of Bria-Pros™ in compliance with applicable regulatory requirements by the FDA.
BriaCell Secures License for a Promising Novel Anti-Cancer Agent
On August 2, 2022, the Company secured an exclusive license from University of Maryland, Baltimore County (“UMBC”) to develop and commercialize Soluble CD80 (“sCD80”) as a biologic agent for the treatment of cancer. The novel technology, originally developed by Suzanne Ostrand-Rosenberg, PhD, Faculty at UMBC, and BriaCell’s scientific advisory board member, is entitled “Soluble CD80 as a Therapeutic to Reverse Immune Suppression in Cancer Patients” (Patent No. US 9,650,429 B2). In animal models, sCD80 has been shown to be safe and effective in stopping the tumor growth in animal models by potentially restoring natural anti-tumor immunity. Importantly, sCD80’s unique actions may involve both awakening and boosting the immune system to recognize and destroy tumor cells.
Under the terms of the agreement, BriaCell gains the worldwide rights to develop and commercialize sCD80 as a therapeutic agent for the treatment of cancer, while UMBC holds all rights, title and interest in the inventions and the patent, except for certain rights retained by the United States Government. BriaCell will pay 2% royalties to UMBC upon the commercialization of the product plus other development costs. The licensing agreement was coordinated by UMBC.
| 38 |
BriaCell Partners with Caris Life Sciences® to Expand Patient Outreach and Molecular Profiling
On September 14, 2022, the Company signed an agreement with Caris Life Sciences ® (Caris), a leading molecular science and technology company actively developing and delivering innovative solutions to revolutionize healthcare.
Under the terms of the agreement, Caris will help BriaCell with efficient patient identification, accelerating enrollment for its current Phase I/II clinical trial in advanced metastatic breast cancer of certain genetically defined subgroups. The partnership between BriaCell and Caris leverages Caris’ Right-In-Time (RIT) Clinical Trial Network, a group of over 495 oncology sites that are able to quickly identify and enroll eligible patients in biomarker-directed clinical trials. This service offers patients and physicians access to the most cutting-edge precision medicine in development. Additionally, through Caris’ comprehensive molecular profiling (Whole Exome and Whole Transcriptome Sequencing), Caris will perform tumor profiling for the patients enrolled in the clinical trial.
BriaCell Announces New Clinical Trial Site to Bring Novel Cancer Treatments to Advanced Breast Cancer Patients
On October 12, 2022, the Company announced it has added Mayo Clinic, Jacksonville, Florida as a clinical site in the Phase I/II study of BriaCell’s lead candidate, Bria-IMT™, with Incyte’s PD-1 inhibitor, retifanlimab, in advanced breast cancer.
BriaCell Announces Successful Completion of Phase I Portion of Clinical Study in Advanced Breast Cancer; Randomized Phase II Efficacy Evaluation Progressing
On October 21, 2022, the Company announced the completion of the Phase I part of the clinical trial of its lead candidate, Bria-IMT™, in combination with Incyte’s PD-1 inhibitor, retifanlimab, in advanced breast cancer. The efficacy and survival data of the treated patients is being evaluated in the Phase II part of the study which was recently awarded the FDA’s fast track designation. Under an FDA approved protocol, another arm has recently been added to the Phase II study to evaluate the effects of dosing schedules for patients in the study.
The Phase I portion of the trial, with the primary goal of assessing safety and tolerability of the combination, enrolled 12 subjects who had previously failed at least two prior lines of therapy, characterized as a difficult-to-treat patient population. The combination treatment had a favorable safety profile and appeared well-tolerated with no dose-limiting toxicities.
Progressing through the Phase II part of the clinical trial, a randomized controlled design will be used to allow comparison of the effectiveness of the treatment regimens between the two arms of the study with different dosing schedules.
BriaCell noted it is on schedule to meet with the FDA later this year to discuss the design of a key registration study.
| 39 |
ITEM 1A. RISK FACTORS
An investment in our securities involves a high degree of risk. An investor should carefully consider the risks described below as well as other information contained in this Annual Report on Form 10-K and our other reports filed with the U.S. Securities and Exchange Commission (“SEC”). The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our securities could decline, and investors in our company may lose all or part of their investment.
Risks Related to Our Business
We have a history of losses, may incur future losses and may not achieve profitability
BriaCell is a development stage immune-oncology biotechnology corporation that to date has not recorded any revenues from the sale of diagnostic or therapeutic products. Since incorporation, BriaCell has accumulated net losses and expects such losses to continue as it commences product and pre-clinical development and eventually enters into license agreements for its technology. We incurred net losses of $26,838,903 and $13,816,200 in the fiscal years ended 2022 and 2021, respectively. Management expects to continue to incur substantial operating losses unless and until such time as product sales generate sufficient revenues to fund continuing operations. BriaCell has neither a history of earnings nor has it paid any dividends, and it is unlikely to pay dividends or enjoy earnings in the immediate or foreseeable future.
We are an early stage development company
The Company is developing novel technologies that may not be efficacious or safe. The Company expects to spend a significant amount of capital to fund research and development. As a result, the Company expects that its operating expenses will increase significantly and, consequently, it will need to generate significant revenues to become profitable. Even if the Company does become profitable, it may not be able to sustain or increase profitability on a quarterly or annual basis. The Company cannot predict when, if ever, it will be profitable. There can be no assurances that the intellectual property of BriaCell, or other technologies it may acquire, will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs, or be successfully marketed. The Company will be undertaking additional laboratory studies or trials with respect to the intellectual property of BriaCell, and there can be no assurance that the results from such studies or trials will result in a commercially viable product or will not identify unwanted side effects.
There is a lack of supporting clinical data.
The clinical effectiveness and safety of any of the Company’s developmental products is not yet supported by extensive clinical data and the medical community has not yet developed a large body of peer reviewed literature that supports the safety and efficacy of the Company’s products. If future studies call into question the safety or efficacy of the Company’s products, the Company’s business, financial condition, and results of operations could be adversely affected.
We have an unproven market for our product candidates
The Company believes that the anticipated market for its potential products and technologies if successfully developed will continue to exist and expand. These assumptions may prove to be incorrect for a variety of reasons, including competition from other products and the degree of commercial viability of the potential product.
We may not succeed in adapting to and meeting the business needs associated with our anticipated growth
Anticipated growth in all areas of BriaCell’s business is expected to continue to place a significant strain on its managerial, operational and technical resources. The Company expects operating expenses and staffing levels to increase in the future. To manage such growth, the Company must expand its operational and technical capabilities and manage its employee base while effectively administering multiple relationships with various third parties. There can be no assurance that the Company will be able to manage its expanding operations effectively. Any failure to implement cohesive management and operating systems, to add resources on a cost-effective basis or to properly manage the Company’s expansion could have a material adverse effect on its business and results of operations.
| 40 |
We are heavily reliant on third-parties to carry out a large portion of our business
The Company does not expect to have any in-house manufacturing, pharmaceutical development or marketing capability. To be successful, a product must be manufactured and packaged in commercial quantities in compliance with regulatory requirements and in reasonable time frames and at accepted costs. The Company intends to contract with third parties to develop its products. No assurance can be given that the Company or its suppliers will be able to meet the supply requirements in respect of the product development or commercial sales.
Production of therapeutic products may require raw materials for which the sources and amount of supply are limited, or may be hindered by quality or scheduling issues in respect of the third party suppliers over which the Company has limited control. An inability to obtain adequate supplies of raw materials could significantly delay the development, regulatory approval and marketing of a product. The Company has limited in-house personnel to internally manage all aspects of product development, including the management of multi-center clinical trials. The Company is significantly reliant on third-party consultants and contractors to provide the requisite advice and management. There can be no assurance that the clinical trials and product development will not encounter delays which could adversely affect prospects for the Company’s success.
To be successful, an approved product must also be successfully marketed. The market for the Company’s product being developed by the Company may be large and will require substantial sales and marketing capability. At the present time, the Company does not have any internal capability to market pharmaceutical products. The Company intends to enter into one or more strategic partnerships or collaborative arrangements with pharmaceutical companies or other companies with marketing and distribution expertise to address this need. If necessary, the Company will establish arrangements with various partners for geographical areas. There can be no assurance that the Company can market, or can enter into a satisfactory arrangement with a third party to market a product in a manner that would assure its acceptance in the marketplace. However, if a satisfactory arrangement with a third party to market and/or distribute a product is obtained; the Company will be dependent on the corporate collaborator(s) who may not devote sufficient time, resources and attention to the Company’s programs, which may hinder efforts to market the products.
Should the Company not establish marketing and distribution strategic partnerships and collaborative arrangements on acceptable terms, and undertake some or all of those functions, the Company will require significant additional human and financial resources and expertise to undertake these activities, the availability of which is not guaranteed. The Company will rely on third parties for the timely supply of raw materials, equipment, contract manufacturing, and formulation or packaging services. Although the Company intends to manage these third-party relationships to ensure continuity and quality, some events beyond the Company’s control could result in complete or partial failure of these goods and services. Any such failure could have a material adverse effect on the financial conditions and result of operation of the Company.
Due to the complexity of the process of developing pharmaceutical products, the Company’s business may depend on arrangements with pharmaceutical and biotechnology companies, corporate and academic collaborators, licensors, licensees and others for the research, development, clinical testing, technology rights, manufacturing, marketing and commercialization of its products. Such agreements could obligate the Company to diligently bring potential products to market, make milestone payments and royalties that, in some instances, could be substantial, and incur the costs of filing and prosecuting patent applications. There can be no assurance that the Company will be able to establish or maintain collaborations that are important to its business on favorable terms, or at all.
A number of risks arise from the Company’s potential dependence on collaborative agreements with third parties. Product development and commercialization efforts could be adversely affected if any collaborative partner terminates or suspends its agreement with the Company, causes delays, fails to on a timely basis develop or manufacture in adequate quantities a substance needed in order to conduct clinical trials, fails to adequately perform clinical trials, determines not to develop, manufacture or commercialize a product to which it has rights, or otherwise fails to meet its contractual obligations. The Company’s collaborative partners could pursue other technologies or develop alternative products that could compete with the products the Company is developing.
| 41 |
The Company has signed Non-Disclosure Agreements (“NDA”) with many different third parties. as is customary in the industry. There is no guarantee that, despite the terms of the NDA which bind third parties, the Company will ultimately be able to prevent from such third parties from breaching their obligations under the NDA. Use of the Company’s confidential information in an unauthorized manner is likely to negatively affect the Company.
Pre-clinical studies and initial clinical trials are not necessarily predictive of future results
Pre-clinical tests and Phase I/II clinical trials are primarily designed to test safety, to study pharmacokinetics and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Success in pre-clinical and early clinical trials does not ensure that later large-scale efficacy trials will be successful, nor does it predict final results. Favorable results in early trials may not be repeated in later trials.
A number of companies in the life sciences industry have suffered significant setbacks in advanced clinical trials, even after positive results in earlier trials. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated. Any pre-clinical data and the clinical results obtained for BriaCell’s technology may not predict results from studies in larger numbers of subjects drawn from more diverse populations or in the commercial setting, and also may not predict the ability of our products to achieve their intended goals, or to do so safely.
An inability to obtain raw materials or product supply could have a material adverse effect on the Company’s business, financial condition and results of operations
Raw materials and supplies are generally available in quantities to meet the needs of the Company’s business. The Company will be dependent on third-party manufacturers for the pharmaceutical products that it markets An inability to obtain raw materials or product supply could have a material adverse impact on the Company’s business, financial condition and results of operations.
We must obtain additional capital to continue our operations
The Company anticipates that additional capital will be required to complete its current research and development programs. It is anticipated that future research, additional pre-clinical and toxicology studies and manufacturing initiatives, including to prepare for market approval and successful product market launch, will require additional funds. Further financing may dilute the current holdings of shareholders and may thereby result in a loss for the shareholders. There can be no assurance that the Company will be able to obtain adequate financing, or financing on terms that are reasonable or acceptable for these or other purposes, or to fulfill the Company’s obligations under various license agreements. Failure to obtain such additional financing could result in delay or indefinite postponement of further research and development of the Company’s technologies with the possible loss of license rights to these technologies.
We are highly dependent on our key personnel
Although the Company is expected to have experienced senior management and personnel, the Company will be substantially dependent upon the services of a few key personnel, particularly Dr. William V. Williams, Dr. Giuseppe Del Priore, Dr. Miguel Lopez-Lago and other professionals for the successful operation of its business. Phase I of the Company’s research and development is planned to be completed by qualified professionals and is expected to concentrate on treatment of advanced breast cancer. The loss of the services of any of these personnel could have a material adverse effect on the business of the Company. The Company may not be able to attract and retain personnel on acceptable terms given the intense competition for such personnel among high technology enterprises, including biotechnology and healthcare companies, universities and non-profit research institutions. If we lose any of these persons, or are unable to attract and retain qualified personnel, our business, financial condition and results of operations may be materially and adversely affected.
| 42 |
BriaCell in the future may acquire businesses, products or technologies that it believes complement or expand its existing business.
Acquisitions of this type involve a number of risks, including the possibility that the operations of the acquired business will not be profitable or that the attention of the Company’s management will be diverted from the day-to-day operation of its business. An unsuccessful acquisition could reduce the Company’s margins or otherwise harm its financial condition.
If the Company experiences a data security breach and confidential information is disclosed, the Company may be subject to penalties and experience negative publicity
The Company and its customers could suffer harm if personal and health information were accessed by third parties due to a system security failure. The collection of data requires the Company to receive and store a large amount of personally identifiable data. Recently, data security breaches suffered by well-known companies and institutions have attracted a substantial amount of media attention, prompting legislative proposals addressing data privacy and security. The Company may become exposed to potential liabilities with respect to the data that it collects, manages and processes, and may incur legal costs if information security policies and procedures are not effective or if the Company is required to defend its methods of collection, processing and storage of personal data. Future investigations, lawsuits or adverse publicity relating to its methods of handling such information could have a material adverse effect on the Company’s business, financial condition and results of operations due to the costs and negative market reaction relating to such developments.
We may not succeed in completing the development of our products, commercializing our products or generating significant revenues
Since commencing our operations, we have focused on the research and development and limited clinical trials of our product candidates. Our ability to generate revenues and achieve profitability depends on our ability to successfully complete the development of our products, obtain market approval and generate significant revenues. The future success of our business cannot be determined at this time, and we do not anticipate generating revenues from product sales for the foreseeable future. In addition, we face a number of challenges with respect to our future commercialization efforts, including, among others, that:
| ● | we may not have adequate financial or other resources to complete the development of our product, including two stages of clinical development that are necessary in order to commercialize our products; | |
| ● | we may not be able to manufacture our products in commercial quantities, at an adequate quality or at an acceptable cost; | |
| ● | we may not be able to maintain our CE mark due to regulatory changes; | |
| ● | we may never receive FDA or Health Canada approval for our intended development plans; | |
| ● | we may not be able to establish adequate sales, marketing and distribution channels; | |
| ● | healthcare professionals and patients may not accept our product candidates; | |
| ● | technological breakthroughs in cancer detection, treatment and prevention may reduce the demand for our product candidates; | |
| ● | changes in the market for cancer treatment, new alliances between existing market participants and the entrance of new market participants may interfere with our market penetration efforts; | |
| ● | third-party payors may not agree to reimburse patients for any or all of the purchase price of our products, which may adversely affect patients’ willingness to purchase our product candidates; | |
| ● | uncertainty as to market demand may result in inefficient pricing of our product candidates; | |
| ● | we may face third-party claims of intellectual property infringement; | |
| 43 |
| ● | we may fail to obtain or maintain regulatory approvals for our products candidates in our target markets or may face adverse regulatory or legal actions relating to our product candidates even if regulatory approval is obtained; and | |
| ● | we are dependent upon the results of ongoing clinical studies relating to our product candidates and the products of our competitors. We may fail in obtaining positive results. |
If we are unable to meet any one or more of these challenges successfully, our ability to effectively commercialize our product candidates could be limited, which in turn could have a material adverse effect on our business, financial condition and results of operations.
If product liability lawsuits are brought against us, we may incur substantial liabilities and the commercialization of our drug candidates may be affected
As our drug candidates enter clinical trials, we will face an inherent risk of product liability suits and will face an even greater risk if we obtain approval to commercialize any drugs. For example, we may be sued if our drug candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the drug, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our drug candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our drugs; | |
| ● | injury to our reputation; | |
| ● | withdrawal of clinical trial participants and inability to continue clinical trials; | |
| ● | initiation of investigations by regulators; | |
| ● | costs to defend the related litigation; | |
| ● | diversion of management’s time and our resources; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
| ● | loss of revenue; | |
| ● | exhaustion of any available insurance and our capital resources; | |
| ● | the inability to commercialize any drug candidate; and | |
| ● | a decline in the price of our common shares. |
| 44 |
We shall seek to obtain the appropriate insurance once our candidates are ready for clinical trial. However, our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of drugs we develop, alone or with collaborators. We currently do not have in place product liability insurance and although we plan to have in place such insurance as and when the products are ready for commercialization, as well as insurance covering clinical trials, the amount of such insurance coverage may not be adequate, we may be unable to maintain such insurance, or we may not be able to obtain additional or replacement insurance at a reasonable cost, if at all. Our insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
Additionally, we may be sued if the products that we commercialize, market or sell cause or are perceived to cause injury or are found to be otherwise unsuitable, and may result in:
| ● | decreased demand for those products; | |
| ● | damage to our reputation; | |
| ● | costs incurred related to product recalls; | |
| ● | limiting our opportunities to enter into future commercial partnerships; and | |
| ● | a decline in the price of our common shares. |
We face business disruption and related risks resulting from the recent outbreak of COVID-19, which could have a material adverse effect on our business plan.
The development of our product candidates could be disrupted and materially adversely affected by the outbreak of COVID-19. As a result of measures imposed by the governments in affected regions, businesses and schools have been suspended due to quarantines intended to contain this outbreak. We have enrolled, and will seek to enroll, subject to funding constraints, cancer patients in our clinical trials. In the event that clinical trial sites are slowed down or closed to enrollment in our trials, this could have a material adverse impact on our clinical trial plans and timelines. We may face difficulties recruiting or retaining patients in our ongoing and planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to our clinical trial sites because of the outbreak. We are continuing to assess our business plans and the impact COVID-19 is having on our clinical trial timelines and our ability to recruit candidates for clinical trials, but there can be no assurance that this analysis will enable us to avoid part or all of any impact from the spread of COVID-19 or its consequences, including downturns in business sentiment generally or in our sector in particular. The extent to which COVID-19 and global efforts to contain its spread will impact our operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scope of the outbreak and the actions taken to contain or treat the coronavirus outbreak. We currently believe that the execution of our clinical trials and research programs are delayed by at least one quarter due to COVID-19.
Global economic uncertainty and financial market volatility caused by political instability, changes in international trade relationships and conflicts, such as the conflict between Russia and Ukraine, could make it more difficult for us to access financing and could adversely affect our business and operations.
Our ability to raise capital is subject to the risk of adverse changes in the market value of our stock. Periods of macroeconomic weakness or recession and heightened market volatility caused by adverse geopolitical developments could increase these risks, potentially resulting in adverse impacts on our ability to raise further capital on favorable terms. The impact of geopolitical tension, such as a deterioration in the bilateral relationship between the US and China or an escalation in conflict between Russia and Ukraine, including any resulting sanctions, export controls or other restrictive actions that may be imposed by the US and/or other countries against governmental or other entities in, for example, Russia, also could lead to disruption, instability and volatility in global trade patterns, which may in turn impact our ability to source necessary reagents, raw materials and other inputs for our research and development operations.
| 45 |
We may be adversely affected by the effects of inflation.
Inflation has the potential to adversely affect our business, results of operations, financial position and liquidity by increasing our overall cost structure, particularly if we are unable to achieve commensurate increases in the prices we charge our customers. The existence of inflation in the economy has the potential to result in higher interest rates and capital costs, supply shortages, increased costs of labor and other similar effects. As a result of inflation, we may experience increases in the costs of labor, materials, and other inputs, such as engineering consultants. Although we may take measures to mitigate the impact of this inflation, if these measures are not effective our business, results of operations, financial position and liquidity could be materially adversely affected. Even if such measures are effective, there could be a difference between the timing of when these beneficial actions impact our results of operations and when the cost inflation is incurred.
Failure to remediate material weaknesses in internal accounting controls could result in material misstatements in our financial statements.
Our management has identified material weaknesses in our internal control over financial reporting related to lack of segregation of duties within account processes, and systems, inadequate documentation to evidence the operation of controls, inconsistent procedures and approvals, lack of periodic user access reviews, lack of assessment of controls of financially significant vendors and insufficient written policies and procedures for accounting, IT and financial reporting and record keeping. Our management has concluded that, due to such material weakness, our disclosure controls and procedures were not effective as of July 31, 2022. We have implemented remediation efforts including independent review and approval of transactions and reconciliations in some processes by hiring personnel and segregating duties amongst the team. Management is implementing processes to document and retain evidence to support reviews and reconciliations. Such changes may not, however, be effective in establishing the adequacy of our internal control over financial reporting. If the material weaknesses are not adequately remedied, or if we identify further material weaknesses in our internal controls, our failure to establish and maintain effective disclosure controls and procedures and internal control over financial reporting could result in material misstatements in our financial statements and a failure to meet our reporting and financial obligations, each of which could have a material adverse effect on our financial condition and the trading price of our securities. In addition, investors’ perceptions that our internal control over financial reporting is inadequate or that we are unable to produce accurate financial statements may materially adversely affect the price of our securities.
Risks Related to Our Intellectual Property
We may not successfully develop, maintain and protect our proprietary products and technologies
BriaCell’s success depends to a significant degree upon its ability to develop, maintain and protect proprietary products and technologies. BriaCell files patent applications in the United States and other countries as part of its global strategy to protect its intellectual property and maintains certain U.S. and Non-U.S. patents in its intellectual property portfolio. However, patents provide only limited protection of BriaCell’s intellectual property. The assertion of patent protection involves complex legal and factual determinations and is therefore uncertain and can be expensive. BriaCell cannot provide assurances that patents will be granted with respect to any of its pending patent applications, or that the scope of any of its granted patents, or any patents granted in the future, will be sufficiently broad to offer meaningful protection, or that it will develop and file patent applications on additional proprietary technologies that are patentable, or, if patentable, that any patents will be granted from such patent applications. BriaCell’s current or future patents could be successfully challenged, invalidated or circumvented. This could result in BriaCell’s patent rights failing to create an effective competitive barrier. Losing a significant patent or failing to get a patent to issue from a pending patent application that BriaCell considers significant could have a material adverse effect on BriaCell’s business. The laws governing the scope of patent coverage in various countries continue to evolve. The laws of some foreign countries may not protect BriaCell’s intellectual property rights to the same extent as the laws of the United States. BriaCell has applied for patent protection only in selected countries. Therefore, third parties may be able to replicate BriaCell technologies covered by BriaCell’s patent portfolio in countries in which it does not have patent protection.
BriaCell’s future success and competitive position depends in part upon its ability to maintain its intellectual property portfolio. There can be no assurance that any patents will be issued on any existing or future patent applications.
We are susceptible to intellectual property suits that could cause us to incur substantial costs or pay substantial damages or prohibit us from selling our product candidates
There is a substantial amount of litigation over patent and other intellectual property rights in the biotechnology industry. Whether or not a product infringes a patent involves complex legal and factual considerations, the determination of which is often uncertain. Our management is presently unaware of any other parties’ patents and proprietary rights which our products under development would infringe. Searches typically performed to identify potentially infringed patents of third parties are often not conclusive and, because patent applications can take many years to issue, there may be applications now pending, which may later result in issued patents which our current or future products may infringe or be alleged to infringe. In addition, our competitors or other parties may assert that our product candidates and the methods employed may be covered by patents held by them. If any of our products infringes a valid patent, we could be prevented from manufacturing or selling such product unless we are able to obtain a license or able to redesign the product in such a manner as to avoid infringement. A license may not always be available or may require us to pay substantial royalties. We also may not be successful in any attempt to redesign our product to avoid infringement, nor does a later redesign protect BriaCell from prior infringement. Infringement and other intellectual property claims, with or without merit, can be expensive and time-consuming to litigate and can divert our management’s attention from operating our business.
| 46 |
The steps we have taken to protect our intellectual property may not be adequate, which could have a material adverse effect on our ability to compete in the market
BriaCell’s ability to establish and maintain a competitive position may be achieved in part by prosecuting claims against others who it believes to be infringing its rights. In addition, enforcement of BriaCell’s patents in foreign jurisdictions will depend on the legal procedures in those jurisdictions. In addition to filing patent applications, we rely on confidentiality, non-compete, non-disclosure and assignment of inventions provisions, as appropriate, in our agreements with our employees, consultants, and service providers, to protect and otherwise seek to control access to, and distribution of, our proprietary information. These measures may not be adequate to protect our intellectual property from unauthorized disclosure, third-party infringement or misappropriation, for the following reasons:
| ● | the agreements may be breached, may not provide the scope of protection we believe they provide or may be determined to be unenforceable; | |
| ● | we may have inadequate remedies for any breach; | |
| ● | proprietary information could be disclosed to our competitors; or | |
| ● | others may independently develop substantially equivalent or superior proprietary information and techniques or otherwise gain access to our trade secrets or disclose such technologies. |
Specifically, with respect to non-compete agreements, both state law and precedent varies greatly from state to state and we may be unable to enforce these agreements, in whole or in part, and it may be difficult for us to restrict our competitors from gaining the expertise that our former employees gained while working for us. If our intellectual property is disclosed or misappropriated, it could harm our ability to protect our rights and could have a material adverse effect on our business, financial condition and results of operations.
We may need to initiate lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive and, if we lose, could cause us to lose some of our intellectual property rights, which would harm our ability to compete in the market
We rely on patents, confidentiality and trade secrets to protect a portion of our intellectual property and our competitive position. Patent law relating to the scope of claims in the technology fields in which we operate is still evolving and, consequently, patent positions in the biotechnology/pharmaceutical industry can be uncertain. In order to protect or enforce our patent rights, we may initiate patent and related litigation against third parties, such as infringement suits or requests for injunctive relief. BriaCell’s ability to establish and maintain a competitive position may be achieved in part by prosecuting claims against others who it believes to be infringing its rights. In addition, enforcement of BriaCell’s patents in foreign jurisdictions will depend on the legal procedures in those jurisdictions. Any lawsuits that we initiate could be expensive, take significant time and divert our management’s attention from other business concerns and the outcome of litigation to enforce our intellectual property rights in patents, copyrights, trade secrets or trademarks is highly unpredictable. Litigation also puts our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, or adversely affect its ability to distribute any products that are subject to such litigation. In addition, we may provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, including attorney fees, if any, may not be commercially valuable. The occurrence of any of these events could have a material adverse effect on our business, financial condition and results of operations.
We may be subject to damages resulting from claims that we or our employees or contractors have wrongfully used or disclosed alleged trade secrets of their former employers
Many of our employees and contractors were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that we or any employee or contractor have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of his or her former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize certain therapeutic candidates, which could severely harm our business, financial condition and results of operations. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
| 47 |
If the FDA or comparable foreign regulatory authorities approve generic versions of any of our products that receive marketing approval, or such authorities do not grant our products appropriate periods of exclusivity before approving generic versions of our products, the sales of our products could be adversely affected.
Once a new drug application is approved, the product covered thereby becomes a “reference listed drug” in the FDA’s publication, “Approved Drug Products with Therapeutic Equivalence Evaluations,” commonly known as the Orange Book. Manufacturers may seek approval of generic versions of reference listed drugs through submission of abbreviated new drug applications in the United States. In support of an abbreviated new drug applications, a generic manufacturer need not conduct clinical trials. Rather, the applicant generally must show that its product has the same active ingredient(s), dosage form, strength, route of administration and conditions of use or labeling as the reference listed drug and that the generic version is bioequivalent to the reference listed drug, meaning it is absorbed in the body at the same rate and to the same extent. Generic products may be significantly less costly to bring to market than the reference listed drug and companies that produce generic products are generally able to offer them at lower prices. Thus, following the introduction of a generic drug, a significant percentage of the sales of any branded product or reference listed drug is typically lost to the generic product.
The FDA may not approve abbreviated new drug applications for a generic product until any applicable period of non-patent exclusivity for the reference listed drug has expired. The United States Federal Food, Drug, and Cosmetic Act provides a period of five years of non-patent exclusivity for a new drug containing a new chemical entity (“NCE”). Specifically, in cases where such exclusivity has been granted, abbreviated new drug applications may not be submitted to the FDA until the expiration of five years, unless the submission is accompanied by a Paragraph IV certification that a patent covering the reference listed drug is either invalid or will not be infringed by the generic product, in which case the applicant may submit its application four years following approval of the reference listed drug.
While we believe that our products contain active ingredients that would be treated as NCEs by the FDA and, therefore, if approved, should be afforded five years of data exclusivity, the FDA may disagree with that conclusion and may approve generic products after a period that is less than five years. If the FDA were to award NCE exclusivity to someone other than us, we believe that we would still be awarded three year “Other” exclusivity protection from generic competition, which is awarded when an application or supplement contains reports of new clinical investigations (not bioavailability studies) conducted or sponsored by an applicant and essential for approval. Manufacturers may seek to launch these generic products following the expiration of the applicable marketing exclusivity period, even if we still have patent protection for our product. If we do not maintain patent protection and data exclusivity for our product candidates, our business may be materially harmed.
Competition that our products may face from generic versions of our products could materially and adversely impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on the investments we have made in those product candidates.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest United States non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products, including generics or biosimilars. Given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
| 48 |
Risks Related to Regulations
Changes in legislation and regulations may affect our revenue and profitability
Existing and proposed changes in the laws and regulations affecting public companies may cause the Company to incur increased costs as the Company evaluates the implications of new rules and responds to new requirements. Failure to comply with new rules and regulations could result in enforcement actions or the assessment of other penalties. New laws and regulations could make it more difficult to obtain certain types of insurance, including director’s and officer’s liability insurance, and the Company may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage, to the extent that such coverage remains available.
The impact of these events could also make it more difficult for the Company to attract and retain qualified persons to serve on the Board, or as executive officers. The Company may be required to hire additional personnel and utilize additional outside legal, accounting and advisory services, all of which could cause the Company’s general and administrative costs to increase beyond what the Company currently has planned. Although the Company evaluates and monitors developments with respect to new rules and laws, the Company cannot predict or estimate the amount of the additional costs the Company may incur or the timing of such costs with respect to such evaluations and/or compliance and cannot provide assurances that such additional costs will render the Company compliant with such new rules and laws.
If we or our licensees are unable to obtain U.S., Canadian and/or foreign regulatory approval for our product candidates, we will be unable to commercialize our therapeutic candidates
To date, we have not marketed, distributed or sold an approved product. Our therapeutic candidates are subject to extensive governmental regulations relating to development, clinical trials, manufacturing and commercialization of drugs. We may not obtain marketing approval for any of our therapeutic candidates in a timely manner or at all. In connection with the clinical trials for our product candidates and other therapeutic candidates that we may seek to develop in the future, either on our own or throughout licensing arrangements, we face the risk that:
| ● | a product candidate may not prove safe or efficacious; | |
| ● | the results with respect to any product candidate may not confirm the positive results from earlier preclinical studies or clinical trials; | |
| ● | the results may not meet the level of statistical significance required by the FDA, Health Canada or other regulatory authorities; and | |
| ● | the results will justify only limited and/or restrictive uses, including the inclusion of warnings and contraindications, which could significantly limit the marketability and profitability of the therapeutic candidate. |
Any delay or failure in obtaining the required regulatory approvals will materially and adversely affect our ability to generate future revenues from a particular product candidate. Any regulatory approval to market a product may be subject to limitations on the indicated uses for which we may market the product or may impose restrictive conditions of use, including cautionary information, thereby limiting the size of the market for the product. We and our licensees, as applicable, also are, and will be, subject to numerous foreign regulatory requirements that govern the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process includes all of the risks associated with the FDA approval process that we describe above, as well as risks attributable to the satisfaction of foreign requirements. Approval by the FDA does not ensure approval by regulatory authorities outside the United States. Foreign jurisdictions may have different approval processes than those required by the FDA and may impose additional testing requirements for our therapeutic candidates.
| 49 |
If the third parties on which we rely to conduct our clinical trials and clinical development do not perform as contractually required or expected, we may not be able to obtain regulatory clearance or approval for, or commercialize, our product candidates
We do not have the ability to independently conduct our clinical trials for our product candidates and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct such trials. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory clearance for, or successfully commercialize, our product candidates on a timely basis, if at all, and our business, operating results and prospects may be adversely affected. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control.
Modifications to our product candidates, or to any other product candidates that we may develop in the future, may require new regulatory clearances or approvals or may require us or our licensees, as applicable, to recall or cease marketing these therapeutic candidates until clearances are obtained
Modifications to our product candidates, after they have been approved for marketing, if at all, or to any other pharmaceutical product that we may develop in the future, may require new regulatory clearance, or approvals, and, if necessitated by a problem with a marketed product, may result in the recall or suspension of marketing of the previously approved and marketed product until clearances or approvals of the modified product are obtained. The FDA requires pharmaceutical products manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance. A manufacturer may determine in conformity with applicable regulations and guidelines that a modification may be implemented without pre-clearance by the FDA; however, the FDA can review a manufacturer’s decision and may disagree. The FDA may also on its own initiative determine that a new clearance or approval is required. If the FDA requires new clearances or approvals of any pharmaceutical product or medical device for which we or our licensees receive marketing approval, if any, we or our licensees may be required to recall such product and to stop marketing the product as modified, which could require us or our licensees to redesign the product and will have a material adverse effect on our business, financial condition and results of operations. In these circumstances, we may be subject to significant enforcement actions.
The results of our clinical trials may not support our product claims or may result in the discovery of adverse side effects
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product claims or that any regulatory authority whose approval we will require in order to market and sell our products in any territory will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that clinical trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our regulatory submissions and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results
We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including FDA approval. Clinical trials are expensive and complex, can take many years and have uncertain outcomes. We cannot predict whether we or our licensees will encounter problems with any of the completed, ongoing or planned clinical trials that will cause us, our licensees or regulatory authorities to delay or suspend clinical trials, or delay the analysis of data from completed or ongoing clinical trials. We estimate that clinical trials of our most advanced therapeutic candidates will continue for several years, but they may take significantly longer to complete. Failure can occur at any stage of the testing and we may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent commercialization of our current or future therapeutic candidates, including but not limited to:
| ● | delays in securing clinical investigators or trial sites for the clinical trials; |
| 50 |
| ● | delays in obtaining institutional review board and other regulatory approvals to commence a clinical trial; | |
| ● | slower than anticipated patient recruitment and enrollment; | |
| ● | negative or inconclusive results from clinical trials; | |
| ● | unforeseen safety issues; | |
| ● | uncertain dosing issues; | |
| ● | an inability to monitor patients adequately during or after treatment; and | |
| ● | problems with investigator or patient compliance with the trial protocols. |
A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience than us, have suffered significant setbacks in advanced clinical trials, even after seeing promising results in earlier clinical trials. Despite the results reported in earlier clinical trials for our therapeutic candidates, we do not know whether any phase 3 or other clinical trials we or our licensees may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market our therapeutic candidates. If later-stage clinical trials of any therapeutic candidate do not produce favorable results, our ability to obtain regulatory approval for the therapeutic candidate may be adversely impacted, which will have a material adverse effect on our business, financial condition and results of operations.
The pharmaceutical business is subject to increasing government price controls and other restrictions on pricing, reimbursement and access to drugs, which could adversely affect our future revenues and profitability
To the extent our products are developed, commercialized, and successfully introduced to market, they may not be considered cost-effective and third-party or government reimbursement might not be available or sufficient. Globally, governmental and other third-party payors are becoming increasingly aggressive in attempting to contain health care costs by strictly controlling, directly or indirectly, pricing and reimbursement and, in some cases, limiting or denying coverage altogether on the basis of a variety of justifications, and we expect pressures on pricing and reimbursement from both governments and private payors inside and outside the U.S. to continue.
In the U.S., we are subject to substantial pricing, reimbursement, and access pressures from state Medicaid programs, private insurance programs and pharmacy benefit managers, and implementation of U.S. health care reform legislation is increasing these pricing pressures. The Affordable Care Act instituted comprehensive health care reform, and includes provisions that, among other things, reduce and/or limit Medicare reimbursement, require all individuals to have health insurance (with limited exceptions), and impose new and/or increased taxes. The future of the Affordable Care Act and its constituent parts are uncertain at this time.
In almost all markets, pricing and choice of prescription pharmaceuticals are subject to governmental control. Therefore, the price of our products and their reimbursement in Europe and in other countries is and will be determined by national regulatory authorities. Reimbursement decisions from one or more of the European markets may impact reimbursement decisions in other European markets. A variety of factors are considered in making reimbursement decisions, including whether there is sufficient evidence to show that treatment with the product is more effective than current treatments, that the product represents good value for money for the health service it provides, and that treatment with the product works at least as well as currently available treatments.
The continuing efforts of government and insurance companies, health maintenance organizations, and other payors of health care costs to contain or reduce costs of health care may affect our future revenues and profitability or those of our potential customers, suppliers, and collaborative partners, as well as the availability of capital.
| 51 |
United States federal and state privacy laws, and equivalent laws of other nations, may increase our costs of operation and expose us to civil and criminal sanctions
HIPPA, and the regulations that have been issued under it, and similar laws outside the United States, contains substantial restrictions and requirements with respect to the use and disclosure of individuals’ protected health information. The HIPAA privacy rules prohibit “covered entities,” such as healthcare providers and health plans, from using or disclosing an individual’s protected health information, unless the use or disclosure is authorized by the individual or is specifically required or permitted under the privacy rules. Under the HIPAA security rules, covered entities must establish administrative, physical and technical safeguards to protect the confidentiality, integrity and availability of electronic protected health information maintained or transmitted by them or by others on their behalf. While we do not believe that we will be a covered entity under HIPAA, we believe many of our customers will be covered entities subject to HIPAA. Such customers may require us to enter into business associate agreements, which will obligate us to safeguard certain health information we obtain in the course of our relationship with them, restrict the manner in which we use and disclose such information and impose liability on us for failure to meet our contractual obligations.
In addition, under HITECH, which was signed into law as part of the U.S. stimulus package in February 2009, certain of HIPAA’s privacy and security requirements are now also directly applicable to “business associates” of covered entities and subject them to direct governmental enforcement for failure to comply with these requirements. We may be deemed as a “business associate” of some of our customers. As a result, we may be subject as a “business associate” to civil and criminal penalties for failure to comply with applicable privacy and security rule requirements. Moreover, HITECH created a new requirement obligating “business associates” to report any breach of unsecured, individually identifiable health information to their covered entity customers and imposes penalties for failing to do so.
In addition to HIPAA, most U.S. states have enacted patient confidentiality laws that protect against the disclosure of confidential medical information, and many U.S. states have adopted or are considering adopting further legislation in this area, including privacy safeguards, security standards, and data security breach notification requirements. These U.S. state laws, which may be even more stringent than the HIPAA requirements, are not supplanted by the federal requirements, and we are therefore required to comply with them to the extent they are applicable to our operations.
These and other possible changes to HIPAA or other U.S. federal or state laws or regulations, or comparable laws and regulations in countries where we conduct business, could affect our business and the costs of compliance could be significant. Failure by us to comply with any of the standards regarding patient privacy, identity theft prevention and detection, and data security may subject us to penalties, including civil monetary penalties and in some circumstances, criminal penalties. In addition, such failure may damage our reputation and adversely affect our ability to retain customers and attract new customers.
The protection of personal data, particularly patient data, is subject to strict laws and regulations in many countries. The collection and use of personal health data in the E.U. is governed by the provisions of Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data (the “Data Protection Directive”). The Data Protection Directive imposes a number of requirements, including an obligation to seek the consent of individuals to whom the personal data relates, the information that must be provided to the individuals, notification of data processing obligations to the competent national data protection authorities of individual E.U. member states and the security and confidentiality of the personal data. The Data Protection Directive also imposes strict rules on the transfer of personal data out of the E.U. to the U.S.. Failure to comply with the requirements of the Data Protection Directive and the related national data protection laws of the E.U. member states may result in fines and other administrative penalties and harm our business. We may incur extensive costs in ensuring compliance with these laws and regulations, particularly if we are considered to be a data controller within the meaning of the Data Protection Directive.
| 52 |
If we fail to comply with the U.S. federal Anti-Kickback Statute and similar state and foreign country laws, we could be subject to criminal and civil penalties and exclusion from federally funded healthcare programs including the Medicare and Medicaid programs and equivalent third country programs, which would have a material adverse effect on our business and results of operations
A provision of the Social Security Act, commonly referred to as the federal Anti-Kickback Statute, prohibits the knowing and willful offer, payment, solicitation or receipt of any form of remuneration, directly or indirectly, in cash or in kind, to induce or reward the referring, ordering, leasing, purchasing or arranging for, or recommending the ordering, purchasing or leasing of, items or services payable, in whole or in part, by Medicare, Medicaid or any other federal healthcare program. Although there are a number of statutory exemptions and regulatory safe harbors to the federal Anti-Kickback Statute protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exemption or safe harbor may be subject to scrutiny. The federal Anti-Kickback Statute is very broad in scope and many of its provisions have not been uniformly or definitively interpreted by existing case law or regulations. In addition, most of the states have adopted laws similar to the federal Anti-Kickback Statute, and some of these laws are even broader than the federal Anti-Kickback Statute in that their prohibitions may apply to items or services reimbursed under Medicaid and other state programs or, in several states, apply regardless of the source of payment. Violations of the federal Anti-Kickback Statute may result in substantial criminal, civil or administrative penalties, damages, fines and exclusion from participation in federal healthcare programs.
All of our future financial relationships with U.S. healthcare providers, purchasers, formulary managers, and others who provide products or services to federal healthcare program beneficiaries will potentially be governed by the federal Anti-Kickback Statute and similar state laws. We believe our operations will be in compliance with the federal Anti-Kickback Statute and similar state laws. However, we cannot be certain that we will not be subject to investigations or litigation alleging violations of these laws, which could be time-consuming and costly to us and could divert management’s attention from operating our business, which in turn could have a material adverse effect on our business. In addition, if our arrangements were found to violate the federal Anti-Kickback Statute or similar state laws, the consequences of such violations would likely have a material adverse effect on our business, results of operations and financial condition.
There are other federal and state laws that may affect our ability to operate, including the federal civil False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. Moreover, we may be subject to other federal false claim laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs. Moreover, there are analogous state laws. Violations of these laws can result in substantial criminal, civil or administrative penalties, damages, fines and exclusion from participation in federal healthcare programs.
Moreover, the provisions of the Foreign Corrupt Practices Act of 1997 and other similar anti-bribery laws in other jurisdictions generally prohibit companies and their intermediaries from providing money or anything of value to officials of foreign governments, foreign political parties, or international organizations with the intent to obtain or retain business or seek a business advantage. Recently, there has been a substantial increase in anti-bribery law enforcement activity by U.S. regulators, with more aggressive and frequent investigations and enforcement by both the SEC and the Department of Justice. A determination that our operations or activities violated U.S. or foreign laws or regulations could result in imposition of substantial fines, interruption of business, loss of supplier, vendor or other third-party relationships, termination of necessary licenses and permits, and other legal or equitable sanctions. In addition, lawsuits brought by private litigants may also follow as a consequence.
| 53 |
In both domestic and foreign markets, the development, formulation, manufacturing, packaging, labeling, handling, distribution, import, export, licensing, sale and storage of pharmaceuticals and medical devices are affected by a body of laws, governmental regulations, administrative determinations, including those by Health Canada and the FDA, court decisions and similar constraints.
Such laws, regulations and other constraints can exist at the federal, provincial or local levels in Canada and at all levels of government in foreign jurisdictions. There can be no assurance that the Company and the Company’s partners are in compliance with all of these laws, regulations and other constraints. The Company and its partners may be required to incur significant costs to comply with such laws and regulations in the future, and such laws and regulations may have an adverse effect on the business. The failure of the Company or its partners to comply with current or future regulatory requirements could lead to the imposition of significant penalties or claims and may have a material adverse effect on the business. In addition, the adoption of new laws, regulations or other constraints or changes in the interpretations of such requirements might result in significant compliance costs or lead the Company and its partners to discontinue product development and could have an adverse effect on the business.
The Company’s international operations expose it and its representatives, agents and distributors to risks inherent to operating in foreign jurisdictions that could materially adversely affect its operations and financial position.
These risks include:
| ● | country specific taxation policies; | |
| ● | imposition of additional foreign governmental controls or regulations; | |
| ● | export license requirements; | |
| ● | changes in tariffs and other trade restrictions; and | |
| ● | complexity of collecting receivables in a foreign jurisdiction. |
Moreover, applicable agreements relating to business in foreign jurisdictions are governed by foreign laws and are subject to dispute resolution in the courts of, or through arbitration proceedings in, the country or region in which the parties are located or another jurisdiction agreed upon by the parties. The Company cannot accurately predict whether such jurisdictions will provide an effective and efficient means of resolving disputes that may arise in the future. Even if it obtains a satisfactory decision through arbitration or a court proceeding, the Company could have difficulty in enforcing any award or judgment on a timely basis or at all.
Risks Related to Our Securities
If we are not able to comply with the applicable continued listing requirements or standards of the TSX Exchange or Nasdaq, TSX Exchange or Nasdaq could delist our common shares
In order to maintain the listing of our common shares on the TSX Exchange and the Nasdaq Capital Market, we must satisfy minimum financial and other continued listing requirements and standards, including those regarding director independence and independent committee requirements, minimum stockholders’ equity, minimum share price, and certain corporate governance requirements. There can be no assurances that we will be able to comply with such applicable listing standards.
Future issuance of our common shares could dilute the interests of existing shareholders
We may issue additional common shares in the future. The issuance of a substantial number of common shares could have the effect of substantially diluting the interests of our shareholders. In addition, the sale of a substantial amount of common shares in the public market, in the initial issuance, in a situation in which we acquire a company and the acquired company receives common shares as consideration and the acquired company subsequently sells its common shares, or by investors who acquired such common shares in a private placement, could have an adverse effect on the market price of our common shares.
| 54 |
We have a significant number of options and warrants outstanding, and while these options and warrants are outstanding, it may be more difficult to raise additional equity capital
As of October 27, 2022 we had outstanding options and warrants to purchase 9,674,638 common shares, respectively. The holders of these options and warrants are given the opportunity to profit from a rise in the market price of our common shares. We may find it more difficult to raise additional equity capital while these options and warrants are outstanding. At any time during which these warrants are likely to be exercised, we may be unable to obtain additional equity capital on more favorable terms from other sources. Additionally, the exercise of these options and warrants will cause the increase of our outstanding Common shares, which could have the effect of substantially diluting the interests of our current shareholders.
Sales of a substantial number of our common shares in the public market by our existing shareholders could cause our share price to fall
Sales of a substantial number of our common shares in the public market, or the perception that these sales might occur, could depress the market price of our common shares and could impair our ability to raise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our common shares. As of October 27, 2022 we have 8,184,338 shares issuable upon exercise of outstanding warrants. Sales of shares by these shareholders could have a material adverse effect on the trading price of our common shares. We intend to register the offering, issuance, and sale of all common shares that we may issue under our equity compensation plans. Once we register these shares, they can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates and the lock-up agreements.
We are an Emerging Growth Company, which may reduce the amount of information available to investors
The Jumpstart Our Business Start-ups Act (the “JOBS Act”), and our status as a foreign private issuer will allow us to postpone the date by which we must comply with some of the laws and regulations intended to protect investors and to reduce the amount of information we provide in our reports filed with the SEC, which could undermine investor confidence in our company and adversely affect the market price of our Common shares.
For as long as we remain an “emerging growth company” as defined in the JOBS Act, we intend to take advantage of certain exemptions from various requirements that are applicable to public companies that are not emerging growth companies including:
| ● | the provisions of the Sarbanes-Oxley Act requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting; | |
| ● | any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements. |
We intend to take advantage of these exemptions until we are no longer an “emerging growth company.” We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year of the fifth anniversary of our initial public offering in the United States, (b) in which we have total annual gross revenue of at least US$1.07 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our Common shares that is held by non-affiliates exceeds US$700 million as of the prior June 30; and (2) the date on which we have issued more than US$1.0 billion in non-convertible debt during the prior three-year period.
We cannot predict if investors will find our common shares or listed warrants (“Warrants”) less attractive because we may rely on these exemptions. If some investors find our common shares or Warrants less attractive as a result, there may be a less active trading market for our common shares or Warrants, and our common share or Warrant price may be more volatile and may decline.
We have never paid cash dividends on our capital stock and we do not anticipate paying any dividends in the foreseeable future. Consequently, any gains from an investment in our common shares will likely depend on whether the price of our Common shares increases, which may not occur
We have not paid cash dividends on any capital stock to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. Consequently, in the foreseeable future, you will likely only experience a gain from your investment in our Common shares if the price of our Common shares increases beyond the price in which you originally acquired the Common shares.
| 55 |
In the event a market develops for our common shares or Warrants, the market price of our common shares or Warrants may be volatile
In the event a market develops for our common shares or Warrants, the market price of our common shares or Warrants may be highly volatile. Some of the factors that may materially affect the market price of our common shares or Warrants are beyond our control, such as changes in financial estimates by industry and securities analysts, conditions or trends in the industry in which we operate or sales of our common shares or Warrants. These factors may materially adversely affect the market price of our common shares or Warrants, regardless of our performance. In addition, the public stock markets have experienced extreme price and trading volume volatility. This volatility has significantly affected the market prices of securities of many companies for reasons frequently unrelated to the operating performance of the specific companies. These broad market fluctuations may adversely affect the market price of our Common shares.
Our executive officers, directors and principal shareholders will maintain the ability to exert significant control over matters submitted to our shareholders for approval
Our executive officers, directors and principal shareholders who owned more than 5% of our outstanding common shares will, in the aggregate, beneficially own shares representing approximately 19.14% of our share capital. As a result, if these shareholders were to act together, they would be able to control all matters submitted to our shareholders for approval, as well as our management and affairs. For example, these persons, if they act together, would control the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of voting power could delay or prevent an acquisition of our company on terms that other shareholders may desire or result in management of our company that our public shareholders disagree with.
If we are or become classified as a passive foreign investment company, our U.S. shareholders may suffer adverse tax consequences as a result
Generally, for any taxable year, if at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company (“PFIC”) for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income (including amounts derived by reason of the temporary investment of funds raised in offerings of our shares) and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. If we are characterized as a PFIC, our U.S. shareholders may suffer adverse tax consequences, including having gains realized on the sale of our common shares treated as ordinary income, rather than capital gains, the loss of the preferential rate applicable to dividends received on our common shares by individuals who are U.S. holders, and having interest charges apply to distributions by us and gains from the sales of our shares.
Our status as a PFIC will depend on the nature and composition of our income and the nature, composition and value of our assets. Asset value is based on which the fair market value of each asset, including goodwill and going concern value (which may be determined by reference to the market value of our common shares, which may be volatile). Our status will also depend, in part, on when and how we utilize the cash proceeds from any securities offerings our business. Based upon the value of our assets, including any goodwill, and the nature and composition of our income and assets, we believe that we will be classified as a PFIC for the taxable year ending July 31, 2022 and possibly for succeeding years. However, even if we are classified as a PFIC for the year ending July 31, 2022, under an exception to the PFIC classification rules, we may be able to avoid such classification altogether if we can meet certain conditions set forth in the exception. (See the discussion of PFIC status under “Taxation, U.S. Federal Income Taxation”, below. Because the determination of whether we are a PFIC for any taxable year is a factual determination made annually after the end of each taxable year, there can be no assurance as to our status as a PFIC in any taxable year.
| 56 |
The tax consequences that would apply if we are classified as a PFIC would also be different from those described above if a U.S. shareholder were able to make a valid qualified electing fund (“QEF”) election. If we are classified as a PFIC, then we expect to provide U.S. shareholders with the information necessary for a U.S. shareholder to make a QEF election but there is no assurance that we will do so. See the discussion of PFIC status under “Taxation, U.S. Federal Income Taxation”, below.
If estimates of revenue, expenses, or capital or liquidity requirements change or are inaccurate, or if cash generated from operations is insufficient to satisfy liquidity requirements, the Company may arrange additional financings
BriaCell expects that its current cash and cash equivalent reserves will be sufficient to meet its anticipated needs for working capital and capital expenditures for the near future. In the future, the Company may also arrange financings to give it the financial flexibility to pursue attractive acquisition or investment opportunities that may arise. The Company may pursue additional financing through various means, including equity investments, issuances of debt, joint venture projects, licensing arrangements or through other means. The Company cannot be certain that it will be able to obtain additional financing on commercially reasonable terms or at all. The Company’s ability to obtain additional financing may be impaired by such factors as the status of capital markets, both generally and specifically in the pharmaceutical and medical device industries, and by the fact that it is a new enterprise without a proven operating history. If the amount of capital raised from additional financing activities, together with revenues from operations (if any), is not sufficient to satisfy the Company’s capital needs, it may not be able to develop or advance its products, execute its business and growth plans, take advantage of future opportunities, or respond to competitive pressures or unanticipated customer or partner requirements. If any of these events occur, the Company’s business, financial condition, and results of operations could be adversely affected. Any future equity financings undertaken are likely to be dilutive to existing shareholders. Finally, the terms of securities issued in future capital transactions may include preferences that are more favourable to new investors.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they adversely change their recommendations or publish negative reports regarding our business or our shares, our share price and trading volume could decline
The trading market for our securities will be influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. We do not have any control over these analysts and we cannot provide any assurance that analysts will cover us or provide favorable coverage. If any of the analysts who may cover us adversely change their recommendation regarding our shares, or provide more favorable relative recommendations about our competitors, the market value of our securities would likely decline. If any analyst who may cover us were to cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause the price of our common shares and Warrants and our trading volume to decline.
Certain Canadian legislation contains provisions that may have the effect of delaying or preventing a change in control
Canadian legislation could discourage potential acquisition proposals, delay or prevent a change in control and limit the price that certain investors may be willing to pay for our subordinate voting shares. For instance, a non-Canadian must file an application for review with the Minister responsible for the Investment Canada Act and obtain approval of the Minister prior to acquiring control of a “Canadian business” within the meaning of the Investment Canada Act, where prescribed financial thresholds are exceeded. Furthermore, limitations on the ability to acquire and hold our subordinate voting shares and multiple voting shares may be imposed by the Competition Act (Canada). This legislation permits the Commissioner of Competition to review any acquisition or establishment, directly or indirectly, including through the acquisition of shares, of control over or of a significant interest in us. Otherwise, there are no limitations either under the laws of Canada or British Columbia, or in our articles on the rights of non-Canadians to hold or vote our subordinate voting shares and multiple voting shares. Any of these provisions may discourage a potential acquirer from proposing or completing a transaction that may have otherwise presented a premium to our shareholders.
| 57 |
Because we are a corporation incorporated in British Columbia and some of our directors and officers are resident in Canada or other countries, it may be difficult for investors in the United States to enforce civil liabilities against us based solely upon the federal securities laws of the United States. Similarly, it may be difficult for Canadian investors to enforce civil liabilities against our directors and officers residing outside of Canada
We are a corporation incorporated under the laws of British Columbia with our principal place of business in West Vancouver. Some of our directors and officers and the auditors or other experts named herein are residents of Canada and all or a substantial portion of our assets and those of such persons are located outside the United States. Consequently, it may be difficult for U.S. investors to effect service of process within the United States upon us or our directors or officers or such auditors who are not residents of the United States, or to realize in the United States upon judgments of courts of the United States predicated upon civil liabilities under the Securities Act. Investors should not assume that Canadian courts: (1) would enforce judgments of U.S. courts obtained in actions against us or such persons predicated upon the civil liability provisions of the U.S. federal securities laws or the securities or blue sky laws of any state within the United States, or (2) would enforce, in original actions, liabilities against us or such persons predicated upon the U.S. federal securities laws or any such state securities or blue sky laws.
Similarly, some of our directors and officers are residents of countries other than Canada and all or a substantial portion of the assets of such persons are located outside Canada. As a result, it may be difficult for Canadian investors to initiate a lawsuit within Canada against these non-Canadian residents. In addition, it may not be possible for Canadian investors to collect from these non-Canadian residents judgments obtained in courts in Canada predicated on the civil liability provisions of securities legislation of certain of the provinces and territories of Canada. It may also be difficult for Canadian investors to succeed in a lawsuit in the United States, based solely on violations of Canadian securities laws.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
In July 2021, the Company ended its lease agreement in Berkeley, California. During the same time, the Company started a month-to-month lease arrangement for office and lab space in New York, New York, in the amount of approximately $8,600 per month. This lease was terminated in March 2022. As of April 2022, the Company commenced a month-to-month lease arrangement for office and lab space in Philadelphia, Pennsylvania, in the amount of approximately $16,000 per month.
ITEM 3. LEGAL PROCEEDINGS
We may be involved from time to time in ordinary litigation, negotiation, and settlement matters that will not have a material effect on our operations or finances.
On May 19, 2021, Alpha Capital Anstalt (“Alpha”) filed a lawsuit in the New York State Supreme Court, Commercial Division, New York County against BriaCell Therapeutics Corp. (“BriaCell”), alleging that BriaCell breached a loan contract when it refused to reprice and extend the term of warrants purported held by Alpha in spring 2021, seeking monetary and injunctive relief for delivery of those amended warrants. Counterclaiming and defending against Alpha’s complaint, BriaCell alleges that Alpha’s loan to BriaCell is unenforceable both because the loan is criminally usurious under New York law and because Alpha acted as an unregistered securities dealer in violation of American securities law. BriaCell also has alleged that Canadian securities law, regulation, and rules prohibited it from amending the warrants to comply with Alpha’s spring 2021 demands. On May 11, 2022, Alpha moved to dismiss BriaCell’s operative Amended Counterclaim. The parties have fully briefed that motion, and the Court has calendared oral argument on that motion for February 7, 2023. Expert discovery is ongoing and may affect the value of the parties’ respective claims and damages.
The Company disagrees with Alpha’s claims, is defending these claims, and has filed a counter claim. At this time, while it is impossible to provide any guarantee as to the outcome of the lawsuit, it is the Company’s assessment, based on advice from the Company’s legal counsel at this time, and based on the information known by the Company, that it’s more likely than not that BriaCell will not have to pay Alpha in the litigation.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 58 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market information
Our common shares and Warrants to purchase common shares trade on The Nasdaq Capital Market under the symbols “BCTX” and “BCTXW”, respectively, since February 24, 2021 and on the Toronto Stock Exchange (“TSX”) under the symbol “BCT” since December 31, 2021, and prior to that, on the TSX Venture Exchange from December 3, 2014.
Number of Shareholders
As of October 27, 2022, we have approximately 24 shareholders of record of our common shares.
Dividend Policy
Historically, we have not paid any dividends to the holders of shares of our common shares and we do not expect to pay any such dividends in the foreseeable future as we expect to retain our future earnings for use in the operation and expansion of our business.
Issuer Purchases of Equity Securities
On September 9, 2021, the Board authorized a securities repurchase program through September 27, 2022 of (i) up to 1,341,515 common shares and (ii) up to 411,962 Warrants in total, representing 10% of the 13,415,154 common shares and 10% of the 4,119,622 Warrants comprising the “public float” as of September 8, 2021. The program expired in September 2022.
We expect to execute the securities repurchase program primarily in open market transactions on the TSX or Nasdaq, subject to market conditions.
The Company most recently repurchased its common shares in January 2022 and did not repurchase any common shares during its fourth fiscal quarter.
The following table contains information for the Warrants repurchased during the three months ended July 31, 2022.
| Month | Total Number of Warrants Purchased | Average Price Paid Per Warrant | Total Number of Warrants Purchased as Part of Publicly Announced Plans or Programs | Approximate Dollar Value of Warrants that May Yet Be Purchased Under the Plans or Programs | ||||||||||||
| May 2022 | 14,821 | $ | 2.52 | 219,261 | $ | 485,607 | ||||||||||
| June 2022 | 9,550 | $ | 2.65 | 228,881 | 485,165 | |||||||||||
| July 2022 | 14,512 | $ | 2.66 | 243,323 | 438,461 | |||||||||||
| Total | 38,883 | $ | 2.60 | 243,323 | $ | 438,461 | ||||||||||
ITEM 6.
Not applicable.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with our financial statements and related notes included elsewhere in this Annual Report. This discussion and other parts of this Annual Report contain forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under “Risk Factors” and elsewhere in this Annual Report.
The preparation of financial statements in conformity with these accounting principles requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent liabilities at the financial statement date and reported amounts of revenue and expenses during the reporting period. On an on-going basis, we review our estimates and assumptions. The estimates were based on historical experience and other assumptions that we believe to be reasonable under the circumstances. Actual results are likely to differ from those estimates or other forward-looking statements under different assumptions or conditions, but we do not believe such differences will materially affect our financial position or results of operations. Our actual results may differ materially as a result of many factors, including those set forth under the headings entitled “Special Note Regarding Forward-Looking Statements” and “Risk Factors”.
| 59 |
Recent Developments
BriaCell (the “Company”) is an immuno-oncology biotechnology company with a strong focus on cancer immunotherapy. Immunotherapies have come to the forefront in the fight against cancer since they harness the body’s own immune system to recognize and destroy cancer cells. BriaCell owns the U.S. patent to SV-BR-1-GM (“Bria-IMT™”), a whole-cell targeted immunotherapy for cancer (U.S. Patent No. 7,674,456), as well as patents related to PKCδ inhibitors (U.S. Patent Nos. 9,364,460 and 9,572,793). The Company is currently advancing our targeted immunotherapy program by prioritizing a Phase I/IIa clinical trial with Bria-IMT™ in combination with an immune checkpoint inhibitor and a companion diagnostic test, BriaDx™, to identify patients most likely to benefit from Bria-IMT™. The Bria-IMT™ regimen was evaluated in four patients in a prior study in 2004-2006 by Dr. Charles Wiseman, the scientific founder, former member of the board of directors of the Company (the “Board”) and principal scientific advisor. Encouraging results were obtained, especially in a patient who matched Bria-IMT™ at HLA-DR alleles and had a grade II tumor. In 2017-2018 BriaCell evaluated 23 patients with advanced breast cancer with the Bria-IMT™ regimen and obtained confirmation of the ability of the Bria-IMT™ regimen to induce regression of metastatic breast cancer in patients who match Bria-IMT™ at least at one HLA allele and/or if they had grade I or grade II tumors. A combination study with the immune checkpoint inhibitor pembrolizumab (KEYTRUDA®) was initiated and the first patient dosing in the “combination therapy” clinical trial occurred in September 2018. BriaCell purchased the KEYTRUDA® for this study as BriaCell does not have an agreement with Merck & Co., Inc. for the supply of KEYTRUDA®. Eleven patients were dosed in the combination therapy trial with Bria-IMT™ and the immune checkpoint inhibitor KEYTRUDA® and subsequently dosing with this combination was discontinued. The study was modified under an amended protocol which evaluates the combination of the Bria-IMT™ regimen with Incyte Corporation experimental drugs retifanlimab (anti-PD-1 antibody similar to pembrolizumab). The study is ongoing.
It is estimated by the National Cancer Institute that in 2022, approximately 287,500 women will be diagnosed with breast cancer in the United States. That means that every two minutes an American woman is diagnosed with breast cancer and more than 43,000 are projected to die in 2022. Although about 100 times less common than in women, breast cancer also affects men. It is estimated that the lifetime risk of men getting breast cancer is about 1 in 1,000, and the American Cancer Society estimates that approximately 2,710 new cases of invasive male breast cancer will be diagnosed and approximately 530 men will die from breast cancer in 2022.
According to the May 2019 “Global Oncology Trends 2021” report by the IQVIA Institute, the global market for cancer drugs (including immunotherapy drugs) is expected to reach nearly $269 billion by the end of 2025, growing at a compound annual growth rate (“CAGR”) of 10% between 2021 and 2025, of which about 20% is expected to be immuno-oncology drugs.
About 12.9% percent of women will be diagnosed with breast cancer at some point during their lifetime. In 2018, there were an estimated 3,676,262 women living with female breast cancer in the United States. Approximately 81% of cases present as invasive breast cancer. Approximately 6% of new breast cancer diagnoses are Stage IV (metastatic breast cancer (“MBC”), which has already spread to other organs). Twenty to thirty percent of all women diagnosed with breast cancer will develop MBC. Breast cancer can be subdivided based on receptor status – the hormone receptors for estrogen (ER) and progesterone (PR), collectively referred to as hormone receptors (HR), and the Her2/neu growth factor receptor (HER2). Based on the latest SEER statistics, 74.6% were found to be HR+/HER2−, 10.8% were triple-negative (HR−/HER2−), 10.5% were HR+/HER2+, and 4.0% were HR−/HER2+.1
It is estimated that over 150,000 women in the US are living with MBC. For those with metastatic disease at diagnosis, their 5-year survival rate is 27%. For patients who develop MBC after initially having localized disease, if they had a good response to treatment (i.e. a disease-free interval of more than 24 months), their survival rate is similar to that of patients with MBC at initial diagnosis, but if their disease-free interval is less than 24 months, their prognosis is worse.4 We currently propose that Bria-IMT’s™ indication will be for the treatment of patients with MBC who have failed at least two lines of therapy. Similarly, another study showed that the median overall survival among patients with de novo stage IV MBC was 39.2 months, while for patients with relapsed disease it was 27.2 months. Median progression free survival after first-line therapy is only 9 months and the survival benefit decreases with subsequent lines of therapy. One study showed that of 386 patients with MBC, 374 (97%) received first-line therapy, 254 (66%) received second-line therapy, 175 (45%) received third-line therapy, and 105 (27%) received therapy beyond third-line.
On September 14, 2022, the Company signed an agreement with Caris Life Sciences ® (Caris), a leading molecular science and technology company actively developing and delivering innovative solutions to revolutionize healthcare.
| 60 |
Under the terms of the agreement, Caris will help BriaCell with efficient patient identification, accelerating enrollment for its current Phase I/II clinical trial in advanced metastatic breast cancer of certain genetically defined subgroups. The partnership between BriaCell and Caris leverages Caris’ Right-In-Time (RIT) Clinical Trial Network, a group of over 495 oncology sites that are able to quickly identify and enroll eligible patients in biomarker-directed clinical trials. This service offers patients and physicians access to the most cutting-edge precision medicine in development. Additionally, through Caris’ comprehensive molecular profiling (Whole Exome and Whole Transcriptome Sequencing), Caris will perform tumor profiling for the patients enrolled in the clinical trial.
On August 2, 2022, the Company secured an exclusive license from University of Maryland, Baltimore County (“UMBC”) to develop and commercialize Soluble CD80 (“sCD80”) as a biologic agent for the treatment of cancer. The novel technology, originally developed by Suzanne Ostrand-Rosenberg, PhD, Faculty at UMBC, and BriaCell’s scientific advisory board member, is entitled “Soluble CD80 as a Therapeutic to Reverse Immune Suppression in Cancer Patients” (Patent No. US 9,650,429 B2). In animal models, sCD80 has been shown to be safe and effective in stopping the tumor growth in animal models by potentially restoring natural anti-tumor immunity. Importantly, sCD80’s unique actions may involve both awakening and boosting the immune system to recognize and destroy tumor cells.
Under the terms of the agreement, BriaCell gains the worldwide rights to develop and commercialize sCD80 as a therapeutic agent for the treatment of cancer, while UMBC holds all rights, title and interest in the inventions and the patent, except for certain rights retained by the United States Government. BriaCell will pay 2% royalties to UMBC upon the commercialization of the product plus other development costs. The licensing agreement was coordinated by UMBC.
On October 12, 2022, the Company announced it has added Mayo Clinic, Jacksonville, Florida as a clinical site in the Phase I/II study of BriaCell’s lead candidate, Bria-IMT™, with Incyte’s PD-1 inhibitor, retifanlimab, in advanced breast cancer.
On October 21, 2022, the Company announced the completion of the Phase I part of the clinical trial of its lead candidate, Bria-IMT™, in combination with Incyte’s PD-1 inhibitor, retifanlimab, in advanced breast cancer. The efficacy and survival data of the treated patients is being evaluated in the Phase II part of the study which was recently awarded the FDA’s fast track designation. Under an FDA approved protocol, another arm has recently been added to the Phase II study to evaluate the effects of dosing schedules for patients in the study.
The Phase I portion of the trial, with the primary goal of assessing safety and tolerability of the combination, enrolled 12 subjects who had previously failed at least two prior lines of therapy, characterized as a difficult-to-treat patient population. The combination treatment had a favorable safety profile and appeared well-tolerated with no dose-limiting toxicities.
Progressing through the Phase II part of the clinical trial, a randomized controlled design will be used to allow comparison of the effectiveness of the treatment regimens between the two arms of the study with different dosing schedules.
BriaCell noted it is on schedule to meet with the FDA later this year to discuss the design of a key registration study.
| 61 |
Approval of Omnibus Incentive Plan
On August 2, 2022, the Company approved an omnibus equity incentive plan (“Omnibus Plan), which will permit the Company to grant incentive stock options, preferred share units, restricted share units (“RSU’s”), and deferred share units (collectively, the “Awards”) for the benefit of any employee, officer, director, or consultant of the Company or any subsidiary of the Company. The maximum number of Shares available for issuance under the Omnibus Plan shall not exceed 15% of the issued and outstanding Shares, from time to time, less the number of Shares reserved for issuance under all other security-based compensation arrangements of the Company, including the existing Stock Option Plan. The Omnibus Plan remains subject to approval by the shareholders of the Company (the “Shareholders”) and final approval of the Toronto Stock Exchange (“Exchange”) and will replace the Company’s existing Stock Option Plan upon receipt of such approvals (“Approvals”).
The Company may make grants under the Omnibus Plan, however, the grants cannot be settled until the Approvals have been received.
Stock Option and RSU Grants
On September 1, 2021, the Company issued 100,000 options to a consultant with an exercise price of $5.74, which vest immediately and expire on September 1, 2026.
On November 1, 2021, the Company issued 12,600 options with an exercise price of $7.94, and expire on November 1, 2026. 10,000 of the options were issued to a director and vest immediately, and 2,600 options were issued to members of the Company’s scientific advisory board and vest in five equal instalments every six months, with the first instalment vesting immediately.
On January 13, 2022, the Company issued 524,700 options to directors, officers, and employees with an exercise price of $8.47 and expire on January 13, 2027. 482,300 of the options were granted to Insiders, as such term is defined in the Securities Act (British Columbia) and vest in four equal instalments every 90 days, with the first instalment vesting immediately. The remaining 42,400 options vest in eight equal instalments every 90 days, with the first installment vesting immediately.
On February 16, 2022, the Company issued 150,000 options to an officer with an exercise price of $7.51 and expire on February 16, 2027. The options vest in eight equal instalments every 90 days, with the first instalment vesting immediately.
On May 20, 2022, the Company issued 31,000 options with an exercise price of $4.71 and expire on May 20, 2027. The options vest in eight equal instalments every 90 days, with the first instalment vesting immediately. 20,000 options were issued to the Company’s CFO.
On August 2, 2022, the Company issued 180,100 options with an exercise price of C$8.38 and expire on August 2, 2027. The options vest in eight equal instalments every 90 days, with the first instalment vesting immediately. 142,100 of the options were issued to the Company’s officers. In addition, the Company issued RSU
On August 2, 2022, the Company also granted, under the Omnibus Plan, 19,200 RSU’s to the CEO. The RSU’s vested immediately.
Exercise of warrants
During the year ended July 2022, 1,615,645 warrants with an weighted aggregate exercise price of $5.98 were exercised for gross proceeds of $6,509,767.
Securities Repurchase Program
As noted in a press release dated September 9, 2021, BriaCell announced that the Board has authorized the Company’s securities repurchase program whereby the Company may purchase through the facilities of the TSX Venture Exchange (“TSXV”) or The NASDAQ Capital Market (“NASDAQ”) (i) up to 1,341,515 common shares (the “Common Shares”) and (ii) up to 411,962 publicly traded BCTXW warrants (the “Listed Warrants”) in total, representing 10% of the 13,415,154 Common Shares and 10% of the 4,119,622 Listed Warrants comprising the “public float” as of September 8, 2021, over the next 12 months (the “Buyback”). Independent Trading Group (ITG), Inc. will act as the Company’s advisor and dealer manager in respect of the Buyback. The Company received final regulatory approval on September 22, 2021.
The repurchase program will in no way interfere with BriaCell’s ambitious growth plans to expand into previously-announced areas of cancer immunotherapy and/or advance its current breast cancer clinical trials. BriaCell’s proposed repurchases may be conducted through open market transactions at prevailing market prices, in privately negotiated transactions, in block trades, and/or through other legally permissible means, subject to the market conditions and in compliance with applicable rules and regulations. The timing and dollar amount of repurchase transactions will be subject to the SEC’s Rule 10b-18 and/or Rule 10b5-1 requirements. Purchases of Common Shares or Listed Warrants through the NASDAQ will not, during the 12-month period, exceed 5% of the outstanding Common Shares or Listed Warrants in the aggregate, as at the commencement of the Buyback. BriaCell’s Board of Directors will be reviewing the program periodically and may revise the terms and/or size or suspend or discontinue the program.
As of October 27, 2022, the company has repurchased 1,031,672 common shares and 259,059 publicly traded warrants. All of the warrants and shares repurchased have been cancelled.
| 62 |
Changes in the Board of Directors
On September 1, 2021, Mr. Marc Lustig was appointed to the Company’s Board of Directors. Mr. Lustig is a highly regarded investor, entrepreneur, and corporate finance veteran with a deep understanding of the life sciences industry, including biotechnology and pharmaceuticals, as well as the legal cannabis industry. Marc holds MSc and MBA degrees from McGill University. His professional experience includes working at Merck & Co., and his capital markets career includes roles in biotechnology equity research and corporate finance. Mr. Lustig was the founder and CEO of Origin House, which was sold to Cresco Labs Inc. (CSE: CL; OTCQX: CRLBF) in 2020, where he currently serves as a director and as Head of Capital Markets. In addition to being a director of a number of public companies, Marc founded the Lustig Family Medical Cannabis Research & Care Fund of the Cedars Cancer Foundation that provides cannabis to palliative cancer patients.
Shareholder Meeting
On May 19, 2021, BriaCell announced the results of its annual general and special meeting of shareholders of the Company (the “Shareholders”) for the years ended July 31, 2019 and July 31, 2020, held on May 18, 2021 (the “Meeting”). A total of 1,685,180 common shares of the Company (the “Common Shares”) were voted, representing 22.36% of the Company’s issued and outstanding Common Shares. At the Meeting, the Shareholders overwhelmingly voted in favor of all proposed resolutions that consisted of the following:
| ● | The number of directors set at six; | |
| ● | Election of Dr. William V. Williams, Mr. Jamieson Bondarenko, Dr. Charles Wiseman, Dr. Rebecca Taub, Mr. Vaughn C. Embro-Pantalony, and Mr. Martin Schmieg as directors of the Company; | |
| ● | Appointment of MNP LLP as auditors of the Company for the ensuing year and authorizing the directors to fix their remuneration; | |
| ● | Renewal of the Company’s stock option plan; | |
| ● | Ratification of the number of directors set at six for the prior year ended July 31, 2019; | |
| ● | Ratification of the election of Dr. William V. Williams, Mr. Jamieson Bondarenko, Mr. Richard Berman, Mr. Vaughn C. Embro-Pantalony, Dr. Rebecca Taub, and Dr. Charles Wiseman as directors of the Company for the prior year ended July 31, 2019; | |
| ● | Ratification of the appointment of MNP LLP as the auditors of the Company for the prior year ended July 31, 2019 and ratifying the directors authorization to fix their remuneration; | |
| ● | Ratification of the Company’s stock option plan for the prior year ended July 31, 2019; and | |
| ● | Ratification of holding the Company’s annual general and special meeting for the year ended July 31, 2019 on May 18, 2021. |
Having received shareholder approval, the Company’s stock option plan remains subject to approval from the TSX Venture Exchange. The formal report on voting results with respect to all matters voted upon during the Meeting will be filed on the Company’s SEDAR profile at www.sedar.com and will be filed with the SEC at www.sec.gov.
Overview
Critical Accounting Policies and Estimates
1. Critical Estimates and Judgements
The preparation of these consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and reported amounts of expenses during the reporting period. Actual outcomes could differ from these estimates. The financial statements include estimates which, by their nature, are uncertain. The impacts of such estimates are pervasive throughout the financial statements, and may require accounting adjustments based on future occurrences. Revisions to accounting estimates are recognized in the period in which the estimate is revised and also in future periods when the revision affects both current and future periods.
The critical judgments and significant estimates in applying accounting policies that have the most significant effect on the amounts recognized in the consolidated financial statements are:
| ● | Intangible assets are tested for impairment annually or more frequently if there is an indication of impairment. The carrying value of intangibles with definite lives is reviewed each reporting period to determine whether there is any indication of impairment. If there are indications of impairment the impairment analysis is completed and if the carrying amount of an asset exceeds its recoverable amount, the asset is impaired and impairment loss is recognized. |
| 63 |
| ● | The Company uses the Black-Scholes option-pricing model to estimate fair value of options and the warrant liability at each reporting date. The key assumptions used in the model are the expected future volatility in the price of the Company’s shares and the expected life of the warrants. | |
| ● | The financial statements of each company within the consolidated group are measured using their functional currency which is the currency of the primary economic environment in which an entity operates. The Company changed its functional currency from the Canadian dollar (C$) to the United States dollar (US$) as of May 1, 2021. The change in presentation currency is a voluntary change which is accounted for retrospectively. For comparative reporting purposes, historical financial information has been translated to United States dollars using the exchange rate as of May 1, 2021, which is the date of the change in the functional and presentation currency. |
2. New Accounting Policies Adopted
No new accounting policies were adopted during the year ended July 31, 2022.
Results of Operations
Comparison of the year ended July 31, 2022, compared to the year ended July 31, 2021
Research Costs
Research costs are comprised primarily of (i) Salaries and wages to Company employees at our laboratory; and (ii) Clinical trials and investigational drug costs, which include the testing and manufacture of our investigational drugs and costs of our clinical trials.
For the year ended July 31, 2022, research costs amounted to $8,021,489 as compared to $2,020,899 for the year ended July 31, 2021. The increase is attributed to the recommencing of the Company’s clinical trials and the increased activity in the lab, including the hiring of additional lab employees.
General and Administrative Expenses
For the year ended July 31, 2022, general and administrative expenses amounted to $7,267,452 as compared to $ 4,955,136 for the year ended July 31, 2021. The increase in 2022 is mainly due to a significant ramp up of activity in the Company, following the financings completed in 2021. These increases relate primarily to share based compensation (i.e. non-cash), increase in salaries due to hiring more personnel, and consulting and professional fees incurred by the Company.
Financial expenses, net
For the year ended July 31, 2022, financial expense, net amounted to $11,549,962 as compared to $6,840,165 for the year ended July 31, 2021. Financial expense, net in 2022 is the result of the revaluation of warrant liability at period end offset slightly by interest income earned during the period on funds held in interest bearing accounts. The higher expense in 2021 can be attributed to a larger adjustment to the warrant liability from the issuance of warrants and the revaluation of warrants at period end.
Loss for the period
The Company reported a loss for the year ended July 31, 2022, of $26,838,903 as compared to $13,816,200 for the year ended July 31, 2021. The primary reason for reduced losses in 2022 is due to the decrease in fair value of the warrant liability.
| 64 |
Going Concern Uncertainty
The financial statements have been prepared on a going concern basis, which assumes that the Company will be able to realize its assets and discharge its liabilities in the normal course of business for the foreseeable future. The continuing operations of the Company are dependent upon its ability to continue to raise adequate financing and to commence profitable operations in the future.
As of July 31, 2022, the Company has total assets of $42,577,041 (July 31, 2021 - $58,043,762) and a positive working capital balance of $41,405,614 (July 31, 2021 –$57,241,355).
The Company is planning to finance its research and developmental activities from its existing and future working capital resources and to continue to evaluate additional sources of capital and financing. The Company believes that its existing capital resources will be adequate to satisfy its expected liquidity requirements for at least twelve months from the issuance of the consolidated financial statements.
Liquidity and Capital Resources
As of July 31, 2022, the Company has a working capital of $41,405,614 (July 31, 2021 – $57,241,355) and an accumulated deficit of $60,349,837 (July 31, 2021 - $29,141,897). In June 2021, the Company completed a private placement of gross proceeds of $27.2 million.
As of July 31, 2022, the Company’s capital resources consist primarily of cash and cash equivalents, comprising mostly of cash on deposit with banks, investments in money market funds, investments in U.S. government securities, U.S. government agency securities, and investment grade corporate debt securities. Our investment policy and strategy are focused on preservation of capital and supporting our liquidity requirements.
| 65 |
Historically, the Company has financed its operation through private and public placement of equity securities, as well as debt financing. The Company’s ability to fund its longer-term cash requirements is subject to multiple risks, many of which are beyond its control. The Company intends to raise additional capital, either through debt or equity financings in order to achieve its business plan objectives. Management believes that it can be successful in obtaining additional capital; however, no assurance can be provided that the Company will be able to do so. There is no assurance that any funds raised will be sufficient to enable the Company to attain profitable operations or continue as a going concern. To the extent that the Company is unsuccessful, the Company may need to curtail or cease its operations and implement a plan to extend payables or reduce overhead until sufficient additional capital is raised to support further operations. There can be no assurance that such a plan will be successful
During the year ended July 31, 2022, the Company’s overall position of cash and cash equivalents decreased by $16,227,033 from the year ended July 31, 2021 (including effects of foreign exchange). This decrease in cash can be attributed to the following:
The Company’s net cash used in operating activities during the year ended July 31, 2022 was $12,484,376 as compared to $7,750,188 for year ended July 31, 2021. This increase is mostly due to company growth and increased expenditures during the period.
Cash used in financing activities for the year ended July 31, 2022 was $3,742,657 as compared to $64,997,624 for the year ended July 31, 2021. Cash used in 2022 is attributed to the money spent on the buyback program offset by warrant exercise proceeds. Cash provided in 2021 was mainly from the Nasdaq Financing in February 2021, the private placement proceeds in June 2021, and the exercise of warrants offset by the repayment of these loans.
Off-balance Sheet Arrangements
None.
Tabular Disclosure of Contractual Obligations
None.
| 66 |
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are a smaller reporting company, as defined by Rule 12b-2 of the Securities Exchange Act of 1934, as amended, and are not required to provide the information required under this Item 7A.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The report of independent registered public accounting firm with PCAOB ID:
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as defined in Rule 13a-15(e) and Rule 15d-15(e) under the Exchange Act that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure.
Our management, with the participation of our principal executive officer and principal accounting and financial officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of the end of the period covered by this Annual Report on Form 10-K. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on such evaluation, our principal executive officer and principal accounting and financial officer have concluded that as of July 31, 2022, our disclosure controls and procedures were not effective as of such date as a result of material weaknesses in our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as such term is defined in Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the U.S.. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
As of July 31, 2022, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this assessment, our management concluded that, as of July 31, 2022, our internal control over financial reporting lacked adequate segregation of duties within account processes, and systems, inadequate documentation to evidence the operation of controls, inconsistent procedures and approvals, lack of periodic user access reviews, lack of assessment of controls of financially significant vendors and insufficient written policies and procedures for accounting, IT and financial reporting and record keeping. We are implementing plans to improve such internal control.
Changes in Internal Control Over Financial Reporting
There has been material changes in our internal control over financial reporting during the quarter ended July 31, 2022 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. Independent review and approval of transactions and reconciliations has been implemented in some processes by hiring personnel and segregating duties amongst the team. Management is implementing processes to document and retain evidence to support reviews and reconciliations.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
| 67 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Executive Officers, Directors and Key Employees
The following table sets forth the name, age and position of each of our executive officers, key employees and directors as of October 27, 2022. All directors hold office until the next annual meeting of shareholders and the election and qualification of their successors. Officers serve at the discretion of the board.
| Name | Age | Position | ||
| William V. Williams, MD, FRCP | 67 | President, Chief Executive Officer, and Director | ||
| Gadi Levin, CA, MBA | 49 | Chief Financial Officer and Corporate Secretary | ||
| Giuseppe Del Priore, MD, MPH | 60 | Chief Medical Officer | ||
| Miguel A. Lopez-Lago, PhD | 53 | Chief Scientific Officer | ||
| Jamieson Bondarenko, CFA, CMT | 38 | Chairman of the Board of Directors | ||
| Vaughn C. Embro-Pantalony, MBA, FCPA, FCMA, CDIR, ACC | 65 | Director | ||
| Marc Lustig, MSC, MBA | 50 | Director | ||
| Martin E. Schmieg | 60 | Director | ||
| Rebecca Taub, MD | 70 | Director | ||
| Jane A. Gross, PhD | 66 | Director |
Biographies
William V. Williams, MD, President, Chief Executive Officer and Director, is a seasoned biopharmaceutical executive with over 35 years of industry and academic expertise, including significant clinical management in multinational pharmaceutical companies. Dr. Williams has served as President, Chief Executive Officer and Director of the Company since November 1, 2016. Dr. Williams served as Vice President of Exploratory Development at Incyte Corporation from March 2005 through November 2016. There he facilitated entry of over 20 compounds into the clinic, including ruxolitinib (Jakafi), baricitinib (Olumiant), and epacadostat. Dr. Williams held several positions at GlaxoSmithKline Pharmaceuticals, including Head of Experimental Medicine and Vice President of Clinical Pharmacology from December 2000 through March 2002; Director and Head of Clinical Pharmacology, Oncology, Musculoskeletal and Inflammation from March 2002 through December 2004 and Director and Head of Clinical Pharmacology, Musculoskeletal, Inflammation, Gastrointestinal and Urology from December 2004 through March 2005. He has also served as Assistant Professor of Medicine and the Director of Rheumatology Research at the University of Pennsylvania from July 1991 through January 1998. Dr. Williams earned his BSc in Chemistry and Biotechnology from Massachusetts Institute of Technology and Medical Doctorate from Tufts University School of Medicine. We believe that Dr. Williams is qualified to serve as a member of our Board because of his experience as our President and Chief Executive Officer, as well as his depth of academic and industry experience.
Gadi Levin, CA, MBA, Chief Financial Officer and Secretary, was appointed Chief Financial Officer and Secretary of the Company on February 1, 2016. Mr. Levin has also served as Chief Financial Officer and Director of Vaxil Bio Ltd since March 1, 2016, and as the Finance Director of Eco (Atlantic) Oil & Gas Ltd. since December 1, 2016. Mr. Levin has over 15 years of experience working with public U.S., Canadian and multi-jurisdictional public companies. Previously, Mr. Levin served as Chief Financial Officer of DarioHeath Corp from November 2013 through January 2015. Mr. Levin also served as the Vice President of Finance and Chief Financial Officer for two Israeli investment firms specializing in private equity, hedge funds and real estate. Mr. Levin began his CPA career at the accounting firm Arthur Andersen, where he worked for nine years, specializing in U.S. listed companies involved in initial public offerings. Mr. Levin has a Bachelor of Commerce degree in Accounting and Information Systems from the University of Cape Town, South Africa, and a post graduate diploma in Accounting from the University of South Africa. He received his Chartered Accountant designation in South Africa and has an MBA from Bar Ilan University in Israel.
| 68 |
Giuseppe Del Priore, MD, MPH, Chief Medical Officer, was appointed Chief Medical Officer on February 16, 2022. Dr. Del Priore is a seasoned healthcare executive with over 25 years of experience in research, drug development, and clinical trial management. Dr. Del Priore’s prior work experience includes serving as a biotechnology company Chief Medical Officer, a National Director at the Cancer Treatment Centers of America, and faculty at Indiana University School of Medicine, Weill Cornell Medicine, and New York University School of Medicine. Dr. Del Priore completed his MPH degree in Biostatistics and Epidemiology at the University of Illinois Chicago School of Public Health, his medical degree with Distinction at The State University of New York, and his BA, magna cum laude, in Philosophy, at The City University of New York, with additional training at Memorial Sloan Kettering Cancer Center, The University of Chicago, Northwestern University, and the University of Rochester. He has authored numerous publications, was named on several patents, and was listed as the “Best Doctors” by the U.S. News & World Report. He regularly appears in various media outlets as a Key Opinion Leader in oncology. We believe that Dr. Del Priore is qualified to serve as Chief Medical Officer because of his medical and clinical trial experience.
Miguel A. Lopez-Lago, PhD, Chief Scientific Officer, was appointed Chief Scientific Officer on May 26, 2022, a promotion from his prior title of Senior Director, Research and Development. Since 2000, Dr. Lopez-Lago has been working as a cancer scientist at Memorial Sloan Kettering Cancer Center, New York. Specifically, he has investigated various aspects of tumor biology, including the development of targeted therapies for mesothelioma and the characterization of the biological mechanisms underlying cancer metastasis. More recently, Dr. Lopez-Lago has been interested in the study of the tumor immune-microenvironment and in the development of immunotherapies for thoracic cancers using chimeric antigen receptor T cell technologies. Since 2013, Dr. Lopez-Lago has been working as Senior Research Scientist at MSKCC. Dr. Lopez-Lago received his Bachelor of Science in Bio-Sciences and his doctorate in Molecular Biology from Santiago of Compostela University, Spain. We believe that Dr. Lopez-Lago is qualified to serve as Chief Scientific Officer because of his scientific training, especially in immunology and cellular therapies.
Jamieson Bondarenko, CFA, CMT, Chairman of the Board, was appointed as a Director of the Company on February 12, 2019 and elected as Chairman on April 24, 2019. Mr. Bondarenko provides strategic capital markets & corporate development advice to early-stage life sciences companies through his merchant capital company, JGRNT Capital Corp., a company he founded in November 2016. From December 2016 through October 2017, he served as Principal and Managing Director of the Equity Capital Markets group of Eight Capital. He also held several positions in the Capital Markets division of Dundee Securities Ltd., including Managing Director from July 2016 through December 2016, Director from October 2015 through July 2016, Vice President from December 2012 through October 2015 and Associate from February 2010 through December 2012. We believe that Mr. Bondarenko is qualified to serve as a member of our Board because of his industry-specific and capital markets experience.
Vaughn C. Embro-Pantalony, MBA, FCPA, FCMA, CDIR, ACC, Director, has been a Director of the Company since his appointment on March 18, 2019. In February 2018, he joined the Board of Directors of Soricimed Biopharma Inc., a private clinical-stage biopharma company developing targeted cancer therapies, and in August 2018 he was appointed Chairman of the Board of Soricimed, where he continues to serve in this capacity. He is also a Director of Microbix Biosystems Inc., a public company and leading manufacturer of viral and bacterial antigens and reagents for the global diagnostics industry. He originally joined the Microbix Board in February 2007, and he also served as its President and Chief Executive Officer from November 2012 to July 2017. He is President of Stratpath Management Inc., consulting on strategy and governance to the life sciences sector. He has held other executive positions in life sciences with responsibility for finance, business development, strategic planning and information technology, including Vice President, Finance, and Chief Financial Officer of Novopharm Limited from May 2003 through April 2006; Vice President, Information Technology, and Chief Information Officer of Bayer Inc. from July 1999 through April 2003; Vice President, Finance and Administration of Bayer Healthcare from October 1996 through June 1999; and Director, Finance and Administration and Chief Financial Officer of Zeneca Pharma Inc. from March 1995 through August 1996. He received his bachelor’s degree from Wilfrid Laurier University and his master of business administration degree from University of Windsor. He is a Fellow Chartered Professional Accountant and a Chartered Director (C. Dir.) and is Audit Committee Certified (A.C.C.) through the Directors College, McMaster University. We believe that Mr. Embro-Pantalony is qualified to serve as a member of our Board due to his extensive experience as a pharmaceutical and life sciences executive.
| 69 |
Marc Lustig, Director, was appointed to the Company’s Board on September 1, 2021. Mr. Lustig is a highly regarded investor, entrepreneur, and corporate finance veteran with a deep understanding of the life sciences industry, including biotechnology and pharmaceuticals, as well as the legal cannabis industry. He holds MSc and MBA degrees from McGill University. His professional experience includes working at Merck & Co., and his capital markets career includes roles in biotechnology equity research and corporate finance. Mr. Lustig was the founder and CEO of Origin House, which was sold to Cresco Labs Inc. (CSE: CL; OTCQX: CRLBF) in 2020, where he currently serves as a director and as Head of Capital Markets. In addition to being a director of a number of public companies, he founded the Lustig Family Medical Cannabis Research & Care Fund of the Cedars Cancer Foundation that provides cannabis to palliative cancer patients. We believe that Mr. Lustig is qualified to serve as a member of our Board because of his industry-specific and capital markets experience.
Martin Schmieg, Director, rejoined the Company’s Board on November 24, 2020. Having served as a member of BriaCell’s Board from 2016 to March 2019, Mr. Schmieg is a “C” level executive with a diversified background in the global biotech, med-tech and pharmaceutical industries, with 40 years of business experience. He currently serves as Co-Founder and CEO of ClearIt, LLC, a private company based in Massachusetts. As a hands-on leader, Mr. Schmieg’s early career focused on accounting and financial management responsibilities, serving as Chief Financial Officer to privately held Cytometrics, Inc. and Advanced Bionics Corporation, and publicly traded Sirna Therapeutics, Inc. and Isolagen, Inc. We believe that Mr. Schmeig is qualified to serve as a member of our Board because of his long-term familiarity with the Company and his perspective and experience in relevant industries.
Rebecca Taub, MD, Director, has been a Director of the Company since her appointment on March 18, 2019. Dr. Taub currently serves as the President of Research and Development for Madrigal Pharmaceuticals, a clinical-stage biopharmaceutical company. She previously served as Vice President of Research and Development from July 2016 through her recent promotion to President of Research and Development on June 27, 2019. She has also served as Madrigal’s Chief Medical Officer since July 2016. Dr. Taub served as the CEO and a Director of Madrigal from September 2011 through Madrigal’s merger with Synta Pharmaceuticals Corp. in July 2016. Prior to joining Madrigal, Dr. Taub served as Senior Vice President, Research and Development of VIA Pharmaceuticals from 2008 to 2011 and as Vice President, Research, Metabolic Diseases at Hoffmann-LaRoche from 2004 to 2008. In those positions, Dr. Taub oversaw clinical development and drug discovery programs in cardiovascular and metabolic diseases, including the conduct of a series of Phase I and II proof of conduct clinical trials. Dr. Taub led drug discovery programs, including target identification, lead optimization and advancement of preclinical candidates into clinical development. From 2000 through 2003, Dr. Taub worked at Bristol-Myers Squibb Co. and DuPont Pharmaceutical Company, in a variety of positions, including Executive Director of CNS and metabolic diseases research. Before becoming a pharmaceutical executive, Dr. Taub was a tenured Professor of Genetics and Medicine at the University of Pennsylvania, and remains an adjunct professor. Dr. Taub is the author of more than 120 research articles. Before joining the faculty of the University of Pennsylvania, Dr. Taub served as an Assistant Professor at the Joslin Diabetes Center of Harvard Medical School, Harvard University and an associate investigator with the Howard Hughes Medical Institute. Dr. Taub received her M.D. from Yale University School of Medicine and her B.A. from Yale College. We believe that Dr. Taub is qualified to serve as a member of our Board due to her extensive experience as a pharmaceutical executive heading up major development programs in non-alcoholic steatohepatitis.
Jane Gross, Director, was appointed to the Company’s Board in November 2021. Dr. Gross is a highly experienced biotech executive with over 30 years in leading research and development teams from discovery through preclinical evaluation and clinical development of therapeutics for the treatment of cancer and autoimmune and inflammatory diseases. Dr. Gross currently serves as an Independent Director for aTyr Pharmaceuticals (Nasdaq: LIFE), a biotechnology company developing novel therapeutics for respiratory diseases and multiple cancer indications. Dr. Gross’s experience includes roles as Chief Scientific Officer and SVP, Research and Non-Clinical Development at Aptevo Therapeutics (Nasdaq: APVO), during which she led the discovery of novel antibody-based, bispecific protein therapeutics as immunotherapies to treat diseases like cancer. Previously, Dr. Gross served as VP, Applied Research and Non-Clinical Development at Emergent BioSolutions (NYSE: EBS), during which she successfully introduced a drug to patients from the design stage into the clinic stage. Formerly, as VP, Immunology Research at ZymoGenetics, Dr. Gross discovered and developed 30+ new product candidates, completed partnerships and out-licensing of assets, and helped position ZymoGenetics for a successful acquisition by Bristol Myers Squibb (NYSE: BMY) in 2010. Dr. Gross earned her Ph.D. in Immunology from the University of California, Berkeley and her Post-Doctoral Fellowship from the University of Washington in Immunology. We believe that Dr. Gross is qualified to serve as a member of our Board due to her extensive industry experience and academic background.
| 70 |
Family Relationships and Other Arrangements
There are no family relationships among our directors and executive officers. There are no arrangements or understandings between or among our executive officers and directors pursuant to which any director or executive officer was or is to be selected as a director or executive officer.
Composition of our Board6
Under our amended articles of incorporation, our Board consists of a minimum of three directors and up to that number which was last set by ordinary resolution of the shareholders. Our Board is currently comprised of seven directors, and under the Business Corporations Act (British Columbia) (“BCBCA”), as a reporting issuer, we must have no fewer than three directors. Under the BCBCA, a director may be removed with or without cause by a resolution passed by at least two-thirds of the votes cast by shareholders present in person or by proxy at a meeting and who are entitled to vote. The directors are appointed at the annual general meeting of shareholders and the term of office for each of the directors will expire at the time of our next annual shareholders meeting. Our amended articles of incorporation provide that, between annual general meetings of our shareholders, the directors may appoint one or more additional directors, but the number of additional directors may not at any time exceed one-third of the number of directors who held office at the expiration of the last meeting of our shareholders. Under the BCBCA, there is no minimum number of directors required to be resident Canadians as defined in the BCBCA.
Director Term Limits and Other Mechanisms of Board Renewal
Our Board has not adopted director term limits or other automatic mechanisms of Board renewal. Rather than adopting formal term limits, mandatory age-related retirement policies and other mechanisms of Board renewal, the nominating and corporate governance committee of our Board will develop a skills and competencies matrix for our Board as a whole and for individual directors. The nominating and corporate governance committee conducts a process for the assessment of our board of directors, each committee and each director regarding his or her effectiveness and contribution, and reports evaluation results to our Board on a regular basis.
Director Independence
Under the Nasdaq Rules, independent directors must comprise a majority of a listed company’s board of directors. For purposes of the Nasdaq Rules, an independent director means a person other than an executive officer or employee of the company who, in the opinion of the board of directors, has no relationship with the company that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. Under NI 58-101, a director is considered to be independent if he or she is independent within the meaning of Section 1.4 of National Instrument 52-110—Audit Committees. Section 1.4 of NI 52-110 generally provides that a director is independent if he or she has no direct or indirect relationship with the issuer which could, in the view of the issuer’s board of directors, be reasonably expected to interfere with the exercise of the director’s independent judgment.
Our Board has undertaken a review of the independence of each director. Based on information provided by each director concerning his or her background, employment and affiliations, our Board has determined that Dr. Gross, Dr. Taub, Mr. Embro-Pantalony, Mr. Schmieg, and Mr. Bondarenko, representing five of the seven members of our Board, are “independent” as that term is defined under the Nasdaq Rules. In making this determination, our Board considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our Board deemed relevant in determining their independence, including the beneficial ownership of our shares by each non-employee director. Dr. Williams is not independent by virtue of being the Company’s Chief Executive Officer. Mr. Lustig is not independent by virtue of being a significant securityholder of the Company.
Certain members of our Board are also members of the boards of other public companies. Our Board has not adopted a director interlock policy, but is kept informed of other public directorships held by its members.
| 71 |
Mandate of the Board of Directors
Our Board is responsible for supervising the management of our business and affairs, including providing guidance and strategic oversight to management. Our Board’s mandate includes, among other things, the following matters:
| ● | succession planning, including appointing, training and monitoring senior management; | |
| ● | developing the corporate goals and objectives that management is responsible for meeting and reviewing the performance of our senior officers against such corporate goals and objectives; | |
| ● | taking steps to satisfy itself as to the integrity of our executive officers and that our executive officers create a culture of integrity throughout the organization; | |
| ● | reviewing and approving our code of conduct and reviewing and monitoring compliance with the code of conduct and our enterprise risk management processes; | |
| ● | reviewing and approving management’s strategic and business plans and our financial objectives, plans and actions, including significant capital allocations and expenditures; and | |
| ● | reviewing and approving material transactions not in the ordinary course of business. |
Meetings of Independent Directors
Our Board holds regularly-scheduled quarterly meetings as well as ad hoc meetings from time to time. The independent members of our Board also meet, as required, without the non-independent directors and members of management after each regularly scheduled board meeting.
A director who has a material interest in a matter before our Board or any committee on which he or she serves is required to disclose such interest as soon as the director becomes aware of it. In situations where a director has a material interest in a matter to be considered by our Board or any committee on which he or she serves, such director may be required to absent himself or herself from the meeting while discussions and voting with respect to the matter are taking place. Directors are also required to comply with the relevant provisions of the BCBCA regarding conflicts of interest.
Position Descriptions
Our Board has adopted written terms of reference for the chairman which set out his or her key responsibilities, including duties relating to determining the frequency, dates and locations of meetings and setting Board meeting agendas, chairing Board and shareholder meetings and carrying out any other or special assignments or any functions as may be requested by our Board or management, as appropriate.
Our Board has also adopted written terms of reference for each of the committee chairs which set out each of the committee chair’s key responsibilities, including duties relating to determining the frequency, dates and locations of meetings and setting committee meeting agendas, chairing committee meetings, reporting to our Board and carrying out any other special assignments or any functions as may be requested by our Board.
In addition, our Board, in conjunction with our Chief Executive Officer, will develop and implement a written position description for the role of our Chief Executive Officer.
Orientation and Continuing Education
We have implemented an orientation program for new directors under which a new director meets separately with the chairman of our Board, members of the senior executive team and the secretary.
The nominating and corporate governance committee will be responsible for coordinating orientation and continuing director development programs relating to the committee’s mandate. The chairman of our Board will be responsible for overseeing director continuing education designed to maintain or enhance the skills and abilities of our directors and to ensure that their knowledge and understanding of our business remains current.
| 72 |
Code of Conduct
Our board of directors has adopted a Code of Ethics that applies to all of our directors, officers and employees. We have made the Code of Ethics available on our website https://briacell.com/corporate/corporate-governance/. We intend to disclose future amendments to, or waivers of, our Code of Ethics, as and to the extent required by SEC regulations, at the same location on our website identified above or in public filings.
Monitoring Compliance with the Code of Conduct
Our nominating and corporate governance committee will be responsible for reviewing and evaluating the code of conduct at least annually and will recommend any necessary or appropriate changes to our Board for consideration. The nominating and corporate governance committee will assist our Board with the monitoring of compliance with the code of conduct, and will be responsible for considering any waivers therefrom (other than waivers applicable to members of the nominating and corporate governance committee, which shall be considered by the audit committee, or waivers applicable to our directors or executive officers, which shall be subject to review by our Board as a whole).
Requirement for Directors and Officers to Disclose Interest in a Contract or Transaction
In accordance with the BCBCA, each director and officer must disclose the nature and extent of any interest that he or she has in a material contract or material transaction whether made or proposed with us, if the director or officer is a party to the contract or transaction, is a director or an officer or an individual acting in a similar capacity of a party to the contract or transaction, or has a material interest in a party to the contract or transaction. Subject to certain limited exceptions under the BCBCA, no director may vote on a resolution to approve a material contract or material transaction which is subject to such disclosure requirement.
As of the date hereof, except as otherwise disclosed in this Annual Report on Form 10-K, to the knowledge of the Board or the management of the Company, there are no material interests, whether direct or indirect, of any informed person of the Company, any proposed director of the Company, or any associate or affiliate of any informed person or proposed director, in any transaction since the commencement of the Company’s most recently completed financial year or in any proposed transaction which has materially affected or would materially affect the Company of any of its subsidiaries.
Benefits upon Termination of Employment
The service contracts with our directors do not provide for any benefits upon termination of employment, other than a “tail” directors and officers insurance policy.
Complaint Reporting
In order to foster a climate of openness and honesty in which any concern or complaint pertaining to a suspected violation of the law, our code of conduct or any of our policies, or any unethical or questionable act or behavior, our code of conduct will require that our employees promptly report the violation or suspected violation. In order to ensure that violations or suspected violations can be reported without fear of retaliation, harassment or an adverse employment consequence, we will adopt a whistleblowing policy which will contain procedures that are aimed to facilitate confidential, anonymous submissions of complaints by our directors, officers, employees and others.
Committees of the Board
We currently have an audit committee, a compensation committee and a nominating and corporate governance committee, with each committee having a written charter.
| 73 |
Audit Committee
Our Audit Committee is currently comprised of Vaughn C. Embro-Pantalony, Martin Schmieg and Jane A. Gross, and chaired by Mr. Embro-Pantalony. Our Board has determined that each of Mr. Schmieg and Mr. Embro-Pantalony is financially literate and meets the independence requirements for directors, including the heightened independence standards for members of the audit committee under Rule 10A-3 under the Exchange Act and NI 52-110. Our Board has determined that Mr. Embro-Pantalony is “financially sophisticated” within the meaning of the Nasdaq Rules, “financially literate” within the meaning of NI 52-110, and a “financial expert” as defined by Rule 10A-3 under the Exchange Act.
We have adopted an Audit Committee Charter setting forth the purpose, composition, authority and responsibility of the audit committee. The primary function of the audit committee is to assist the Board in fulfilling its financial oversight responsibilities by reviewing the financial reports and other financial information provided by the company to regulatory authorities and the Company’s shareholders, the Company’s systems of internal controls regarding finance and accounting and the Company auditing, accounting and financial reporting processes. Consistent with this function, the Committee will encourage continuous improvement of, and should foster adherence to, Company’s policies, procedures and practices at all levels. The Committee’s primary duties and responsibilities are to:
| ● | Serve as an independent and objective party to monitor the Company’s financial reporting and internal control system and review Company’s financial statements; | |
| ● | Review and appraise the performance of the Company’s external auditors; and | |
| ● | Provide an open avenue of communication among the Company’s auditors, financial and senior management and the Board. |
The Audit Committee meets at least annually, or more frequently as circumstances dictate. As part of its job to foster open communication, the Audit Committee meets at least annually with the external auditors.
To fulfill its responsibilities and duties, the Audit Committee:
| ● | Reviews and updates the Audit Committee’s charter annually; | |
| ● | Reviews the Company’s financial statements, Management Discussion & Analysis and any annual and interim earnings, press releases before the Company publicly discloses this information and any reports or other financial information (including quarterly financial statements), which are submitted to any governmental body, or to the public, including any certification, report, opinion, or review rendered by the external auditors; | |
| ● | Reviews annually, the performance of the external auditors who shall be ultimately accountable to the Board and the Committee as representatives of the shareholders of the Company; | |
| ● | Obtains annually, a formal written statement of external auditors setting forth all relationships between the external auditors and the Company, consistent with Independence Standards Board Standard I; | |
| ● | Reviews and discusses with the external auditors any disclosed relationships or services that may impact the objectivity and independence of the external auditors; | |
| ● | Takes, or recommends that the full Board takes, appropriate action to oversee the independence of the external auditors; | |
| ● | Recommends to the Board the selection and, where applicable, the replacement of the external auditors nominated annually for shareholder approval; | |
| ● | Reviews and approves the Company’s hiring policies regarding partners, employees and former partners and employees of the present and former external auditors of the Company; | |
| ● | Reviews and pre-approves all audit and audit-related services and the fees and other compensation related thereto; | |
| ● | In consultation with the external auditors, reviews with management the integrity of the Company’s financial reporting process, both internal and external; |
| 74 |
| ● | Considers the external auditors’ judgments about the quality and appropriateness of the Company’s accounting principles as applied in its financial reporting; | |
| ● | Considers and approves, if appropriate, changes to the Company’s auditing and accounting principles and practices as suggested by the external auditors and management; | |
| ● | Reviews significant judgments made by management in the preparation of the financial statements and the view of the external auditors as to appropriateness of such judgments; | |
| ● | Following completion of the annual audit, reviews separately with management and the external auditors any significant difficulties encountered during the course of the audit, including any restrictions on the scope of work or access to required information; | |
| ● | Reviews any significant disagreement among management and the external auditors in connection with the preparation of the financial statements; | |
| ● | Reviews with the external auditors and management the extent to which changes and improvements in financial or accounting practices have been implemented; | |
| ● | Reviews any complaints or concerns about any questionable accounting, internal accounting controls or auditing matters; | |
| ● | Reviews certification process; and | |
| ● | Reviews any related-party transactions. |
Principal Accountant’s Fees
External Audit Service Fees
The following table sets forth the aggregate fees paid to the Company’s external auditors, Chartered Professional Accountants, by the Company during the financial years ended July 31, 2022 and 2021:
| Year ended July 31, 2022 | Year ended July 31, 2021 | |||||||
| Audit Fees | $ | 232,884 | $ | 211,000 | ||||
| Audit-Related Fees | - | 21,950 | ||||||
| Tax Fees | 11,900 | 11,900 | ||||||
| All Other Fees | 17,134 | 17,139 | ||||||
| Total: | $ | 261,918 | $ | 261,989 | ||||
Compensation Committee7
Our compensation committee is comprised of Mr. Embro-Pantalony and Mr. Schmieg and is chaired by Mr. Schmieg. The Compensation Committee is appointed by the Board to assist in promoting a culture of integrity throughout the Company, to assist the Board in setting director and senior executive compensation, and to develop and submit to the Board recommendations with respect to other employee benefits as the Compensation Committee sees fit. In the performance of its duties, the Compensation Committee is guided by the following principles:
| ● | offering competitive compensation to attract, retain and motivate highly qualified executives in order for the Company to meet its goals; and |
| ● | acting in the interests of the Company and the shareholders by being fiscally responsible. |
The Board relies on the knowledge and experience of the members of the Compensation Committee to set appropriate levels of compensation for senior officers. Neither the Company nor the Compensation Committee currently has, or has had at any time since incorporation, any contractual arrangement with any executive compensation consultant who has a role in determining or recommending the amount or form of senior officer compensation.
| 75 |
When determining compensation payable, the Compensation Committee considers both external and internal data. External data includes general market conditions and well as information regarding compensation paid to directors, CEOs and CFOs of companies of similar size and at a similar stage of development in the industry. Internal data includes annual reviews of the performance of the directors, CEO and CFO in light of the Company’s corporate objectives and considers other factors that may have impacted the Company’s success in achieving its objectives.
Nominating and Corporate Governance Committee
The Nominating and Corporate Governance Committee is appointed by the Board to assist in fulfilling its corporate governance responsibilities under applicable laws. The Nominating and Corporate Governance Committee is responsible for, among other things, developing the Company’s approach to governance issues and establishing sound corporate governance practices that are in the interests of shareholders and that contribute to effective and efficient decision-making.
Our Nominating and Corporate Governance Committee is currently comprised of Mr. Embro-Pantalony and Dr. Taub and is chaired by Mr. Embro-Pantalony.
Exculpation, Insurance and Indemnification of Directors and Officers
Under the BCBCA, a company may indemnify: (i) a current or former director or officer of that company; (ii) a current or former director or officer of another corporation if, at the time such individual held such office, the corporation was an affiliate of the company, or if such individual held such office at the company’s request; or (iii) an individual who, at the request of the company, held, or holds, an equivalent position in another entity (an “indemnifiable person”) against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by him or her in respect of any civil, criminal, administrative or other legal proceeding or investigative action (whether current, threatened, pending or completed) in which he or she is involved because of that person’s position as an indemnifiable person, unless: (i) the individual did not act honestly and in good faith with a view to the best interests of such company or the other entity, as the case may be; or (ii) in the case of a proceeding other than a civil proceeding, the individual did not have reasonable grounds for believing that the individual’s conduct was lawful. A company cannot indemnify an indemnifiable person if it is prohibited from doing so under its articles or by applicable law. A company may pay, as they are incurred in advance of the final disposition of an eligible proceeding, the expenses actually and reasonably incurred by an indemnifiable person in respect of that proceeding only if the indemnifiable person has provided an undertaking that, if it is ultimately determined that the payment of expenses was prohibited, the indemnifiable person will repay any amounts advanced. Subject to the aforementioned prohibitions on indemnification, a company must, after the final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by an indemnifiable person in respect of such eligible proceeding if such indemnifiable person has not been reimbursed for such expenses, and was wholly successful, on the merits or otherwise, in the outcome of such eligible proceeding or was substantially successful on the merits in the outcome of such eligible proceeding. On application from an indemnifiable person, a court may make any order the court considers appropriate in respect of an eligible proceeding, including the indemnification of penalties imposed or expenses incurred in any such proceedings and the enforcement of an indemnification agreement. As permitted by the BCBCA, under Article 21.1, we are required to indemnify our directors and former directors (and such individual’s respective heirs and legal representatives) and we will indemnify any such person to the extent permitted by the BCBCA.
The BCBCA provides certain protections under Part 5 – Management, Division 5 - Indemnification of Directors and Officers and Payment of Expenses, to our current and former directors and officers, as well as other eligible parties defined in Section 159 of the BCBCA (the “Eligible Parties”, each an “Eligible Party”). The Company will indemnify the Eligible Parties, to the fullest extent permitted by law and subject to certain limitations listed in Section 163 of the BCBCA, against any proceeding in which an Eligible Party or any of the heirs and personal or other legal representatives of the Eligible Party, by reason of the Eligible Party being or having been a director or officer of, or holding or having held a position equivalent to that of a director or officer of, the Company or an associated corporation (a) is or may be joined as a party, or (b) is or may be liable for or in respect of a judgment, penalty or fine in, or expenses related tom, the proceeding.
| 76 |
We maintain insurance policies relating to certain liabilities that our directors and officers may incur in such capacity.
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation Table
The following table presents the compensation awarded to, earned by or paid to each of our named executive officers for the years ended July 31, 2022 and July 31, 2021.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock Awards ($)(1) | Option Awards ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||
| William V. Williams, MD, FRCP | 2022 | 560,992 | 150,000 | - | 100,152 | - | 811,144 | |||||||||||||||||||||
| President and Chief Executive Officer | 2021 | 217,995 | - | - | 612,324 | - | 830,319 | |||||||||||||||||||||
| Gadi Levin, CA, MBA | 2022 | 202,091 | 45,000 | - | 9,240 | - | 256,331 | |||||||||||||||||||||
| Chief Financial Officer and Corporate Secretary | 2021 | 114,278 | - | - | 229,621 | - | 343,899 | |||||||||||||||||||||
| Giuseppe Del Priore, MD, MPH(2) | 2022 | 199,665 | - | - | 215,881 | - | 415,546 | |||||||||||||||||||||
| Chief Medical Officer | 2021 | - | - | - | - | |||||||||||||||||||||||
| Miguel A. Lopez-Lago, PhD(3) | 2022 | 211,616 | 35,000 | - | 22,456 | - | 269,072 | |||||||||||||||||||||
| Chief Scientific Officer | 2021 | 40,909 | - | - | - | 40,909 | ||||||||||||||||||||||
| (1) | This column represents the grant date fair value of the award in accordance with stock-based compensation rules under Accounting Standards Codification Topic 718. For a more detailed discussion of the valuation model and assumptions used to calculate the fair value of each option award, refer to Note 2 of the financial statements included in this annual report. | |
| (2) | Giuseppe Del Priore was appointed as the Chief Medical Officer on February 16, 2022 |
| (3) | Miguel A. Lopez-Lago was appointed as the Chief Scientific Officer on May 26, 2022 |
Outstanding Equity Awards at Fiscal Year-End
The following table provides information regarding option awards held by each of our named executive officers that were outstanding as of July 31, 2022.
| Option Awards | Stock Awards | |||||||||||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | Number of shares or units of stock that have not vested (#) | Market value of shares or units of stock that have not vested ($) | ||||||||||||||||||
| William V. Williams, MD, FRCP | 200,000 | - | 4.24 | 03/29/26 | - | - | ||||||||||||||||||
| 22,300 | - | 8.47 | 01/13/27 | 5,575 | 33,384 | |||||||||||||||||||
| Gadi Levin, CA, MBA | 75,000 | - | 4.24 | 03/29/26 | - | - | ||||||||||||||||||
| 20,000 | - | 4.71 | 05/20/27 | 17,500 | 55,441 | |||||||||||||||||||
| Giuseppe Del Priore, MD, MPH | 150,000 | - | 7.51 | 02/16/27 | 112,500 | 647,642 | ||||||||||||||||||
| Miguel A. Lopez-Lago, PhD | 15,000 | - | 8.47 | 01/13/27 | 9,375 | 56,139 | ||||||||||||||||||
| 77 |
Non-Employee Director Compensation
The following table presents the total compensation for each person who served as a non-employee member of our Board and received compensation for such service during the fiscal year ended July 31, 2022. Other than as set forth in the table and described more fully below, we did not pay any compensation, make any equity awards or non-equity awards to, or pay any other compensation to any of the non-employee members of our Board in 2022.
| Name | Fees Earned or Paid in Cash | Stock Awards ($) | Option Awards ($) | All Other Compensation | Total ($) | |||||||||||||||
| Jamieson Bondarenko, CFA, CMT | 171,279 | - | 1,122,784 | - | 1,294,063 | |||||||||||||||
| Vaughn C. Embro-Pantalony, MBA, FCPA, FCMA, CDIR, ACC | 82,675 | - | 224,557 | - | 307,232 | |||||||||||||||
| Marc Lustig, MSC, MBA | 59,663 | - | 224,557 | - | 284,220 | |||||||||||||||
| Martin E. Schmieg | 72,500 | - | 224.557 | - | 297,057 | |||||||||||||||
| Rebecca Taub, MD | 60,000 | - | 44,911 | - | 104,911 | |||||||||||||||
| Jane A. Gross, PhD | 30,000 | - | 283,747 | - | 313,747 | |||||||||||||||
Employment Agreements
Dr. Williams V. Williams
On August 31, 2021, we entered into a compensation package with Dr. Williams, our Chief Executive Officer (the “2021 Compensation Package”). Pursuant to the 2021 Compensation Package, Mr. Williams receives $550,000 annually and may earn an equity incentive bonus compensation, which may include a direct stock award of up to $125,000 based upon a performance review as of December 31, 2021 (the “Performance Review”). In addition, the 2021 Compensation Package provides for an option award to purchase up to $250,000 in common shares of the Company, in connection with the Performance Review, which vests over a four year period and provides for an aggregate cash, stock and option award of up to $950,000.
On June 21, 2022, we entered into a compensation package with Dr. Williams (the “2022 Compensation Package”). Pursuant to the 2022 Compensation Package, Mr. Williams receives $650,000 annually and an annual bonus of $150,000. In addition, the 2022 Compensation Package provides for a performance stock option award of $250,000 and a total cash, bonus and option award of up to $1,050,000.
Giuseppe Del Priore
On February 14, 2022, we entered into an employment agreement with Giuseppe Del Priore, our Chief Medical Officer (the “Del Priore Employment Agreement”). The Del Priore Employment Agreement provides for a full-time position, $350,000 annual salary and standard employee benefit plan participation. In addition, Mr. Del Priore was granted an option to purchase 150,000 of the Company’s common shares. The Del Priore Employment Agreement provides that Mr. Del Priore is eligible for an annual bonus in either cash or options to purchase common shares of the Company based on the successful completion of certain corporate milestones selected by our Chief Executive Officer and reviewed in the sole discretion of our Board or a compensation committee.
Gadi Levin
On March 2, 2022, we entered into an executive employment agreement with Gadi Levin, our Chief Financial Officer (the “Levin Employment Agreement”), effective January 1, 2022. The Levin Employment Agreement provides for a part-time position (60%), $200,000 annual salary (“Base Salary”) and standard employee benefit plan participation. Our Board approved a annual discretionary bonus of 30% of Mr. Levin’s yearly salary and $100,000 in stock options, which vest over a four year period per calendar year. In addition, Mr. Levin was granted 20,000 options in accordance with the terms of the Company’s stock option plan. During August 2022, Mr. Levin’s annual salary was increased to 250,000, retroactively to January 1, 2022
| 78 |
Miguel Lopez-Lago
On May 26, 2022, we entered into an employment agreement with Miguel Lopez-Lago, our Chief Scientific Officer (the “Lopez-Lago Employment Agreement”). The Lopez-Lago Employment Agreement provides for $210,000 annually for Mr. Lopez-Lago’s duties as our Chief Scientist Officer.
Equity Compensation Plan Information
The following table summarizes the total number of outstanding awards and shares available for other future issuances of options under all of our equity compensation plans as of July 31, 2022. All of the outstanding awards listed below were granted under our stock option plan.
| Plan Category | Number of Shares to be Issued Upon Exercise of Outstanding Options, Warrants and Rights | Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Shares Remaining Available for Future Issuance Under the Equity Compensation Plan (Excluding Shares in First Column) | |||||||||
| Equity compensation plans approved by shareholders | 9,674,638 | $ | 5.83 | 61,501 | ||||||||
| Equity compensation plans not approved by shareholders | - | - | - | |||||||||
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding the beneficial ownership of our common shares as of October 27, 2022 by:
| ● | each of our named executive officers; | |
| ● | each of our directors; | |
| ● | all of our current directors and executive officers as a group; and | |
| ● | each shareholder known by us to own beneficially more than 5% of our common shares. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Common shares that may be acquired by an individual or group within 60 days of October 27, 2022, pursuant to the exercise of options or warrants, vesting of common shares or conversion of preferred stock or convertible debt, are deemed to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table. Percentage of ownership is based on 15,518,018 common shares issued and outstanding as of October 27, 2022.
| 79 |
Except as indicated in footnotes to this table, we believe that the shareholders named in this table have sole voting and investment power with respect to all common shares shown to be beneficially owned by them, based on information provided to us by such shareholders. Unless otherwise indicated, the address for each director and executive officer listed is: c/o BriaCell Therapeutics Corp., Suite 300 – 235 15th Street, West Vancouver, BC V7T 2X1.
| Number of Shares | Percentage of Common Shares | |||||||
| Name of Beneficial Owner | Beneficially Owned | Beneficially Owned | ||||||
| Directors and Named Executive Officers | ||||||||
| Jamieson Bondarenko, CFA, CMT(1) | 557,356 | 3.59 | % | |||||
| William V. Williams, MD, FRCP(2) | 406,358 | 2.62 | % | |||||
| Gadi Levin, CA, MBA(3) | 82,848 | * | ||||||
| Giuseppe Del Priore, MD, MPH(4) | 38,750 | * | ||||||
| Miguel A. Lopez-Lago, PhD(5) | 6,875 | * | ||||||
| Vaughn C. Embro-Pantalony, MBA, FCPA, FCMA, CDIR, ACC(6) | 72,024 | * | ||||||
| Marc Lustig, MSC, MBA | 1,660,000 | 10.70 | % | |||||
| Martin E. Schmieg(7) | 63,075 | * | ||||||
| Rebecca Taub, MD(8) | 17,500 | * | ||||||
| Jane A. Gross, PhD(9) | 47,500 | * | ||||||
| All current named executive officers and directors as a group (10 persons) | 2,952,285 | 19.02 | % | |||||
| 5% or Greater Shareholders | ||||||||
| Marc Lustig, MSC, MBA | 1,660,000 | 10.70 | % | |||||
| * | Represents beneficial ownership of less than 1%. |
Notes :
| 1. | Includes 150,000 options with an exercise price of $4.35, expiring on March 29, 2026, 187,500 options with an exercise price of $8.47, expiring on January 13, 2027 and 100,000 warrants to purchase common shares with an exercise price of $5.3125, expiring on February 26, 2026. | |
| 2. | Includes 200,000 options with an exercise price of $4.35, expiring on March 29, 2026, 16,725 options with an exercise price of $8.47, expiring on January 13, 2027, 12,725 options with an exercise price of C$8.38, expiring on August 2, 2027 and 27,272 warrants to purchase common shares with an exercise price of $5.3125, expiring on February 26, 2026. | |
| 3. | Includes 75,000 options with an exercise price of US$4.24, expiring on March 29, 2026, 2,500 options with an exercise price of US$4.71, expiring on May 20, 2027 and 2,538 options with an exercise price of C$8.38, expiring on August 2, 2027. | |
| 4. | Includes 37,500 options with an exercise price of US$7.51, expiring on February 16, 2027 and 1,250 options with an exercise price of C$8.38, expiring on August 2, 2027. | |
| 5. | 5,625 options with an exercise price of $8.47, expiring on January 13, 2027 and 1,250 options with an exercise price of C$8.38, expiring on August 2, 2027. | |
| 6. | Includes 25,000 options with an exercise price of US$4.24, expiring on March 29, 2026 and 37,500 options with an exercise price of $8.47, expiring on January 13, 2027. | |
| 7 | Includes 25,000 options with an exercise price of US$4.24, expiring on March 29, 2026 and 37,500 options with an exercise price of $8.47, expiring on January 13, 2027. | |
| 8. | Includes 10,000 options with an exercise price of US$4.24, expiring on March 29, 2026 and 7,500 options with an exercise price of $8.47, expiring on January 13, 2027. | |
| 9. | Includes 10,000 options with an exercise price of US$7.74, expiring on November 1, 2026 and 37,500 options with an exercise price of $8.47, expiring on January 13, 2027. |
Section 16(A) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our officers and directors, and persons who own more than 10% of a registered class of our equity securities, to file reports of ownership and changes in ownership with the SEC. Officers, directors and greater than 10% shareholders are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file.
Based on a review of the copies of such forms received, we believe that during the fiscal year ending July 31, 2022, all filing requirements applicable to our officers, directors and greater than 10% beneficial owners were complied with.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
There have been no transactions since August 1, 2020 to which we have been a party, including transactions in which the amount involved in the transaction exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end for the last two completed fiscal years, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described elsewhere in this Annual Report on Form 10-K. We are not a party to a current related party transaction, and no transaction is currently proposed, in which the amount of the transaction exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end for the last two completed fiscal years and in which a related person had or will have a direct or indirect material interest.
| 80 |
Director Independence
Our board of directors undertook a review of the independence of our directors and considered whether any director has a relationship with us that could compromise that director’s ability to exercise independent judgment in carrying out that director’s responsibilities. Our board of directors has affirmatively determined that Dr. Gross, Dr. Taub. Mr. Bondarenko, Mr. Empro-Pantalony, Mr. Lustig, and Mr. Schmieg are each an “independent director,” as defined under the Nasdaq rules.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Audit Fees
The aggregate fees billed to us by MNP LLP, our independent registered public accounting firm, for the indicated services for each of the last two fiscal years were as follows:
| 2022 | 2021 | |||||||
| Audit fees (1) | $ | 232,884 | $ | 211,000 | ||||
| Audit-related fees (2) | $ | - | $ | 21,950 | ||||
| Tax fees | $ | 11,900 | $ | 11,900 | ||||
| All other fees | $ | 17,134 | $ | 17,139 | ||||
| (1) | Audit fees consist of fees for professional services performed by MNP LLP for the audit and review of our financial statements. |
| (2) | Audit related fees consist of fees for preparation and filing of our registration statements, including issuance of comfort letters. |
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Auditors
Consistent with SEC policies and guidelines regarding audit independence, the Audit Committee is responsible for the pre-approval of all audit and permissible non-audit services provided by our independent registered public accounting firm on a case-by-case basis. Our Audit Committee has established a policy regarding approval of all audit and permissible non-audit services provided by our principal accountants. Our Audit Committee pre-approves these services by category and service. Our Audit Committee has pre-approved all of the services provided by our independent registered public accounting firm.
| 81 |
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
|
Exhibit Number |
Description of Exhibit | |
| (a)(1) Financial Statements | ||
| The financial statements required by this item are submitted in a separate section beginning on page F-1 of this Annual Report on Form 10-K. |
(b) Exhibits
| 82 |
| + | Indicates a management contract or compensatory plan or arrangement. |
ITEM 16. FORM 10-K SUMMARY
None.
| 83 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| BRIACELL THERAPEUTICS CORP. | |
| /s/ William V. Williams | |
| October 27, 2022 | Chief Executive Officer (Principal Executive Officer and Principal Accounting and Financial Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| SIGNATURE | TITLE | DATE | ||
| /s/ William V. Williams | Chief Executive Officer, President and Director | October 27, 2022 | ||
| William V. Williams | (Principal Executive Officer) | |||
| /s/ Gadi Levin | Chief Financial Officer and Corporate Secretary (Principal Accounting and Financial Officer) | October 27, 2022 | ||
| Gadi Levin | ||||
| /s/ Jamieson Bondarenko | Chairman of the Board of Directors | October 27, 2022 | ||
| Jamieson Bondarenko |
| /s/ Vaughn C. Embro-Pantalony | Director | October 27, 2022 | ||
| Vaughn C. Embro-Pantalony |
| /s/ Marc Lustig | Director | October 27, 2022 | ||
| Marc Lustig | ||||
|
/s/ Martin E. Schmieg |
Director | October 27, 2022 | ||
| Martin E. Schmieg |
| /s/ Rebecca Taub | Director | October 27, 2022 | ||
| Rebecca Taub |
| /s/ Jane A. Gross | Director | October 27, 2022 | ||
| Jane A. Gross |
| 84 |
Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
Expressed in United States Dollars
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of BriaCell Therapeutics Corp.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of BriaCell Therapeutics Corp. (the Company) as of July 31, 2022 and 2021, and the related consolidated statements of operations and comprehensive loss, changes in shareholders’ equity, and cash flows for each of the years in the two-year period ended July 31, 2022, and the related notes (collectively referred to as the consolidated financial statements).
In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of July 31, 2022 and 2021, and the results of its consolidated operations and its consolidated cash flows for each of the years in the two-year period ended July 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
| F-2 |
Critical Audit Matter description
The Company changed its accounting framework from the International Financial Reporting Standards (IFRS) to generally accepted accounting principles in the United States (US GAAP). The change in accounting framework caused a change in accounting treatment of certain warrant instruments from being classified as equity to liability. The transition treatment of these warrants requires the Company to perform detail accounting and complex accounting analysis and multiple complex calculations to account for the warrant liabilities. Thus, we identified the change in the accounting treatment of the warrant instruments as a critical audit matter.
How the Critical Audit Matter was Addressed in the Audit
The primary procedures MNP performed to address this critical audit matter included the following, among other procedures:
| ● | We obtained a transition memo from management to understand the implication of the changes from IFRS to US GAAP. We assessed the accounting treatment change for reasonability. | |
| ● | We obtained management’s recalculation of balances and adjustments due to the change in the accounting treatment and recalculated the balances and adjustments as at July 31, 2022 and July 31, 2021 to determine if the amounts were reasonably calculated and presented. |

Chartered Professional Accountants
Licensed Public Accountants
We have served as the Company’s auditor since 2015.
October 27, 2022

| F-3 |
BriaCell Therapeutics Corp
Consolidated Balance Sheets
As at July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data)
| July 31, 2022 | July 31, 2021 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Amounts receivable | ||||||||
| Prepaid expenses | ||||||||
| Total current assets | ||||||||
| NON-CURRENT ASSETS: | ||||||||
| Investments | ||||||||
| Intangible assets, net | ||||||||
| Total non-current assets | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES AND SHAREHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Trade payables | $ | $ | ||||||
| Accrued expenses and other payables | ||||||||
| Total current liabilities | ||||||||
| NON-CURRENT LIABILITIES: | ||||||||
| Warrant liability | ||||||||
| Government loans | ||||||||
| Total non-current liabilities | $ | $ | ||||||
| CONTINGENT LIABILITIES AND COMMITMENTS | ||||||||
| SHAREHOLDERS’ EQUITY: | ||||||||
| Share Capital of par value – Authorized: at July 31, 2022 and 2021; Issued and outstanding: and shares at July 31, 2022 and 2021, respectively | ||||||||
| Additional paid in capital | ||||||||
| Warrant reserve | ||||||||
| Accumulated other comprehensive loss | ( | ) | ( | ) | ||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total shareholders’ equity | ||||||||
| Total liabilities and shareholders’ equity | $ | $ | ||||||
These consolidated financial statements were approved and authorized for issue on behalf of the Board of Directors on October 27, 2022 by:
| On behalf of the Board: | ||
| “Jamieson Bondarenko” | “William Williams” | |
| Director | Director |
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
BriaCell Therapeutics Corp
Consolidated Statements of Operations and Comprehensive Loss
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data)
| Year Ended | ||||||||
| July 31, 2022 | July 31, 2021 | |||||||
| Research and development expenses | $ | $ | ||||||
| General and administrative expenses | ||||||||
| Total operating loss | ( | ) | ( | ) | ||||
| Financial expenses, net | ( | ) | ( | ) | ||||
| Loss and comprehensive loss | $ | ( | ) | $ | ( | ) | ||
| Net loss per share attributable to ordinary shareholders, basic and diluted | $ | ( | ) | $ | ( | ) | ||
| Weighted average number of shares used in computing net loss per share attributable to ordinary shareholders, basic and diluted | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
BriaCell Therapeutics Corp
Consolidated Statements of Changes in Shareholders’ Equity
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars , except share and per share data)
| Share capital | ADDITIONAL | ACCUMULATED OTHER | TOTAL SHAREHOLDERS’ | |||||||||||||||||||||
| Number | Amount | PAID IN CAPITAL | COMPREHENSIVE INCOME (LOSS) | ACCUMULATED DEFICIT | EQUITY (DEFICIT) | |||||||||||||||||||
| Balance, July 31, 2020 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||
| Issuance of warrants on convertible debt | - | |||||||||||||||||||||||
| Conversion feature | - | |||||||||||||||||||||||
| Issuance of shares for debt | ||||||||||||||||||||||||
| Issuance of shares in public offering | ||||||||||||||||||||||||
| Issuance of shares in private placement, net of issuance costs | ||||||||||||||||||||||||
| Reclassification of warrant liability | - | |||||||||||||||||||||||
| Exercise of warrants | ( | ) | ||||||||||||||||||||||
| Expiration of warrants | - | ( | ) | |||||||||||||||||||||
| Expiration and forfeiture of options | - | ( | ) | |||||||||||||||||||||
| Issuance of options | - | |||||||||||||||||||||||
| Loss for the year | - | ( | ) | ( | ) | |||||||||||||||||||
| Balance, July 31, 2021 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| Exercise of Broker Warrants | ||||||||||||||||||||||||
| Exercise of Private Placement Warrants | ||||||||||||||||||||||||
| Exercise of Public Offering Warrants | ||||||||||||||||||||||||
| Shares issuance costs | - | ( | ) | ( | ) | |||||||||||||||||||
| Issuance of options | - | |||||||||||||||||||||||
| Shares repurchased and canceled | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| Expiration of options | - | ( | ) | |||||||||||||||||||||
| Loss for the year | - | ( | ) | ( | ) | |||||||||||||||||||
| Balance, July 31, 2022 | ( | ) | ( | ) | ||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
BriaCell Therapeutics Corp
Consolidated Statements of Cash Flows
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data)
| Year Ended | ||||||||
| July 31, 2022 | July 31, 2021 | |||||||
| Cash flow from operating activities: | ||||||||
| Loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | ||||||||
| Share-based compensation | ||||||||
| Interest expense | ||||||||
| Gain from government grant | ( | ) | ||||||
| Expensed share issue costs in public offering | ||||||||
| Loan forgiveness | ( | ) | ||||||
| Loss on extinguishment of settlement of debt | ||||||||
| Change in fair value of warrants | ||||||||
| Changes in assets and liabilities: | ||||||||
| Decrease (increase) in amounts receivable | ( | ) | ||||||
| Decrease in prepaid expenses | ( | ) | ( | ) | ||||
| Increase (decrease) in accounts payable | ( | ) | ||||||
| Increase (decrease) in accrued expenses and other payables | ( | ) | ||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| Cash flow from financing activities: | ||||||||
| Proceeds from public offering, net | ||||||||
| Proceeds from private placement, net | ||||||||
| Proceeds from exercise of warrants | ||||||||
| Share and warrant buyback program | ( | ) | ||||||
| Repayment government grant | ( | ) | ||||||
| Repayment of unsecured convertible loan | ( | ) | ||||||
| Proceeds from issuance of unsecured convertible loan | ||||||||
| Share issuance costs | ( | ) | ||||||
| Repayment of short-term loans | ( | ) | ||||||
| Net cash provided by (used in) financing activities | ( | ) | ||||||
| Increase (decrease) in cash and cash equivalents | ( | ) | ||||||
| Cash and cash equivalents at beginning of year | ||||||||
| Cash and cash equivalents at end of year | $ | $ | ||||||
| Significant non-cash transactions: | ||||||||
| Shares issued for settlement of debt | $ | $ | ||||||
| Forgiveness of government grant | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-7 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 1: GENERAL
| a. | BriaCell Therapeutics Corp. (“BriaCell” or the “Company”) was incorporated under the Business Corporations Act (British Columbia) on July 26, 2006 and is listed on the Toronto Stock Exchange (“TSX”) under the symbol “BCT” and the Company also trades on the Nasdaq Capital Market (“NASDAQ”) under the symbols “BCTX” and “BCTXW”. | |
| b. | BriaCell is an immuno-oncology biotechnology company. BriaCell owns the US patent to Bria-IMT™, a whole-cell cancer vaccine (US Patent No.7674456) (the “Patent”). The Company is currently advancing its immunotherapy program, Bria-IMT™, to complete a 24-subject Phase I/IIa clinical trial and by research activities in the context of BriaDx™, a companion diagnostic test to identify patients likely benefitting from Bria-IMT™. | |
| c. | The
Company continues to devote substantially all of its efforts toward research and development activities. In the course of such activities,
the Company has sustained operating losses and expects such losses to continue in the foreseeable future. The Company’s accumulated
deficit as of July 31, 2022 was $ | |
| d. | The Company has a wholly-owned U.S. subsidiary, BriaCell Therapeutics Corp. (“BTC”), which was incorporated in April 3, 2014, under the laws of the state of Delaware. BTC has a wholly-owned subsidiary, Sapientia Pharmaceuticals, Inc. (“Sapientia” and together with BTC the “Subsidiaries”), which was incorporated in September 20, 2012, under the laws of the state of Delaware. The Company has one operating segment and reporting unit. | |
| e. | Since January 2020, the Coronavirus outbreak has dramatically expanded into a worldwide pandemic creating macro-economic uncertainty and disruption in the business and financial markets. Many countries around the world, including Canada and the United States have been taking measures designated to limit the continued spread of the Coronavirus, including the closure of workplaces, restricting travel, prohibiting assembling, closing international borders and quarantining populated areas. Such measures present concerns that may dramatically affect the Company’s ability to conduct its business effectively. | |
| The Company may face difficulties recruiting or retaining patients in our ongoing and planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to our clinical trial sites because of the outbreak of COVID-19. In the event that clinical trial sites are slowed down or closed to enrolment in our trials, this could have a material adverse impact on our clinical trial plans and timelines. The Company is continuing to assess its business plans and the impact COVID-19 is having on the Company’s clinical trial timelines and the Company’s ability to recruit candidates for clinical trials. The extent to which COVID-19 and global efforts to contain its spread will impact our operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scope of the outbreak and the actions taken to contain or treat the coronavirus outbreak. The Company currently believes that the execution of our clinical trials and research programs are delayed by at least one quarter due to COVID-19. |
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES
a. Basis of presentation of the financial statements:
The Company’s consolidated financial statements have been prepared in accordance with the United States generally accepted accounting principles (U.S. GAAP) as set forth in the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification (ASC).
Prior to July 2022, the Company prepared its financial statements in accordance with International Financial Reporting Standards (IFRS), as issued by the International Accounting Standards Board (IASB), as permitted in the United States based on the Company’s qualification as a “foreign private issuer” under the rules and regulations of the U.S Securities and Exchange Commission (the “SEC”). On August 1, 2022, the Company no longer qualified as a “foreign private issuer” as such term is defined in Rule 405 under the Securities Act of 1933 and therefore, as a domestic filer, prepared its consolidated financial statements in accordance with U.S. GAAP.
| F-8 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES (Cont.)
b. Use of estimates:
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates, judgments and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. The Company’s management believes that the estimates, judgment and assumptions used are reasonable based upon information available at the time they are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities at the dates of the consolidated financial statements, and the reported amount of expenses during the reporting periods. Actual results could differ from those estimates.
Going Concern
Preparation of the consolidated financial statement on a going concern basis, which contemplates the realization of assets and payments of liabilities in the ordinary course of business. Should the Company be unable to continue as a going concern, it may be unable to realize the carrying value of its assets, including its intangible assets and to meet its liabilities as they become due
Warrants and options
The Company uses the Black-Scholes option-pricing model to estimate fair value of options and the warrant liability at each reporting date. The key assumptions used in the model are the expected future volatility in the price of the Company’s shares and the expected life of the warrants.
Income Taxes
Provisions for taxes are made using the best estimate of the amount expected to be paid based on a qualitative assessment of all relevant factors. The Company reviews the adequacy of these provisions at the end of the reporting period. However, it is possible that at some future date an additional liability could result from audits by taxing authorities. Where the final outcome of these tax-related matters is different from the amounts that were initially recorded, such differences will affect the tax provisions in the period in which such determination is made.
c. Principal of consolidation:
The consolidated financial statements include the accounts of the Company and its subsidiaries. All intercompany balances and transactions have been eliminated upon consolidation.
d. Consolidated financial statements in U.S dollars:
The functional currency is the currency that best reflects the economic environment in which the Company and its subsidiary operates and conducts their transactions. The Company’s management believes that the functional currency of the Company and its subsidiaries is the U.S. dollar.
Accordingly, monetary accounts maintained in currencies other than the U.S. dollar are remeasured into U.S. dollars at each reporting period end in accordance with ASC No. 830 “Foreign Currency Matters.” All transaction gains and losses of the remeasured monetary balance sheet items are reflected in the statements of operations as financing income or expenses as appropriate.
The Company changed its functional currency from the Canadian dollar (C$) to the United States dollar (US$) as of May 1, 2021. The change in presentation currency is a voluntary change which is accounted for retrospectively.
| F-9 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES (Cont.)
e. Cash and cash equivalents:
Cash equivalents are short-term highly liquid deposits that are readily convertible to cash with original maturities of three months or less, at the date acquired.
f. Property and equipment, net:
Property
and equipment with individual values of over $
| % | |
| Computers and peripheral equipment |
g. Intangible assets, net:
Separately acquired intangible assets are measured on initial recognition at cost including directly attributable costs. Intangible assets acquired in a business combination are measured at fair value at the acquisition date. Expenditures relating to internally generated intangible assets, excluding capitalized development costs, are recognized in profit or loss when incurred.
Intangible assets with finite useful lives are amortized over their useful lives and reviewed for impairment annually and whenever there is an indication that the asset may be impaired. The evaluation is performed at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities. Recoverability of these group of assets is measured by a comparison of the carrying amounts to the future undiscounted cash flows the group of assets is expected to generate. If such review indicates that the carrying amount of intangible assets is not recoverable, the carrying amount of such assets is reduced to fair value.
The amortization period and the amortization method for an intangible asset are reviewed at least at each year end.
Intangible assets with indefinite useful lives are not systematically amortized and are tested for impairment annually, or whenever there is an indication that the intangible asset may be impaired. The useful life of these assets is reviewed annually to determine whether their indefinite life assessment continues to be supportable. If the events and circumstances do not continue to support the assessment, the change in the useful life assessment from indefinite to finite life is accounted for prospectively as a change in accounting estimate and on that date the asset is tested for impairment. Commencing from that date, the asset is amortized systematically over its useful life.
The useful lives of intangible assets are as follows:
| Patents | |
| Useful life | |
| Amortization method | |
| In-house development or purchase |
For
the years ended July 31, 2022 and 2021,
| F-10 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES (Cont.)
h. Impairment of long-lived assets:
The Company’s long-lived assets to be held or used, including intangible assets that are subject to amortization, are reviewed for impairment in accordance with ASC 360 “Property, Plants and Equipment”, whenever events or changes in circumstances indicate that the carrying amount of an asset (or asset group) may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset (or asset group) to the future undiscounted cash flows expected to be generated by the assets (or asset group). If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amount of the assets exceeds their fair value.
For
the years ended July 31, 2022 and 2021,
i. Research and Development expenses:
Research and development expenses are recognized in the consolidated statements of operations when incurred. Research and development expenses consist of intellectual property, development and production expenditures.
j. Fair value of financial instruments:
The accounting guidance for fair value provides a framework for measuring fair value, clarifies the definition of fair value, and expands disclosures regarding fair value measurements. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the reporting date. The accounting guidance establishes a three-tiered hierarchy, which prioritizes the inputs used in the valuation methodologies in measuring fair value as follows:
| Level 1 | — | Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. | |
| Level 2 | — | Observable inputs that are based on inputs not quoted on active markets but corroborated by market data. | |
| Level 3 | — | Unobservable inputs are used when little or no market data are available. |
The carrying amounts of cash and cash equivalents, amounts receivables, trade payable and accrued expenses and other payables approximate their fair value due to the short-term maturity of such instruments.
| F-11 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES (Cont.)
k. Leases:
The Company accounts for leases according to ASC 842, “Leases”. The Company determines if an arrangement is a lease and the classification of that lease at inception based on: (1) whether the contract involves the use of a distinct identified asset, (2) whether the Company obtains the right to substantially all the economic benefits from the use of the asset throughout the period, and (3) whether the Company has a right to direct the use of the asset. An ROU asset represents the right to use an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease agreement. An ROU asset is measured based on the discounted present value of the remaining lease payments, plus any initial direct costs incurred and prepaid lease payments, excluding lease incentives. The lease liability is measured at lease commencement date based on the discounted present value of the remaining lease payments. The implicit rate within the operating leases is generally not determinable, therefore the Company uses the Incremental Borrowing Rate (“IBR”) based on the information available at commencement date in determining the present value of lease payments. The Company’s IBR is estimated to approximate the interest rate for collateralized borrowing with similar terms and payments and in economic environments where the leased asset is located. An option to extend the lease is considered in connection with determining the ROU asset and lease liability when it is reasonably certain that the Company will exercise that option. An option to terminate is considered unless it is reasonably certain that the Company will not exercise the option.
The Company elected the practical expedient for lease agreements with a term of twelve months or less and does not recognize right-of-use (“ROU”) assets and lease liabilities in respect of those agreements. The Company also elected the practical expedient to not separate lease and non-lease components for its leases.
The Company accounts for share-based compensation in accordance with ASC No. 718, “Compensation – Stock Compensation”, which requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the award is recognized as an expense over the requisite service periods, which is the vesting period of the respective award, on a straight-line basis when the only condition to vesting is continued service.
The Company has selected the Black-Scholes option-pricing model as the most appropriate fair value method for its option awards. The Company recognizes forfeitures of equity-based awards as they occur.
| F-12 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES (Cont.)
m. Income Taxes:
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes”, which prescribes the use of the liability method whereby deferred tax asset and liability account balances are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, to reduce deferred tax assets to their estimated realizable value, if needed.
ASC
740 offers a two-step approach for recognizing and measuring a liability for uncertain tax positions. The first step is to evaluate the
tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more
likely than not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of
any related appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50%
likely to be realized upon ultimate settlement. As of July 31, 2022, and 2021
The Company’s basic net loss per share is calculated by dividing net loss attributable to ordinary shareholders by the weighted-average number of shares of ordinary shares outstanding for the period, without consideration of potentially dilutive securities. The diluted net loss per share is calculated by giving effect to all potentially dilutive securities outstanding for the period using the treasury share method or the if-converted method based on the nature of such securities. Diluted net loss per share is the same as basic net loss per share in periods when the effects of potentially dilutive ordinary shares are anti-dilutive.
| F-13 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 2: SIGNIFICANT ACCOUNTING POLICIES (Cont.)
o. Recently issued and adopted accounting standards:
As an “emerging growth company,” the Jumpstart Our Business Startups Act (“JOBS Act”) allows the Company to delay adoption of new or revised accounting pronouncements applicable to public companies until such pronouncements are made applicable to private companies. The Company has elected to use this extended transition period under the JOBS Act. The adoption dates discussed below reflects this election.
| 1. | In June 2016, the FASB issued ASU No. 2016-13 (Topic 326), Financial Instruments—Credit Losses: Measurement of Credit Losses on Financial Instruments, which replaces the existing incurred loss impairment model with an expected credit loss model and requires a financial asset measured at amortized cost to be presented at the net amount expected to be collected. The guidance will be effective for the Company for fiscal years beginning after December 15, 2022. Early adoption is permitted. Effective August 1, 2021, the Company early adopted ASU 2016-13. Adoption of the new standard did not have a material impact on the financial statements. |
| 2. | In August 2020, the FASB issued ASU 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity (“ASU 2020-06”). The final guidance issued by the FASB for convertible instruments eliminates two of the three models in ASC 470-20 that require separate accounting for embedded conversion features. Separate accounting is still required in certain cases. Additionally, among other changes, the guidance eliminates some of the conditions for equity classification in ASC 815-40-25 for contracts in an entity’s own equity. The guidance also requires entities to use the if-converted method for all convertible instruments in the diluted earnings per share calculation and include the effect of share settlement for instruments that may be settled in cash or shares, except for certain liability-classified share-based payment awards. ASU 2020-06 is effective for the company for fiscal years beginning after December 15, 2023, and interim periods within those fiscal years. Early adoption is permitted for fiscal years beginning after December 15, 2020. Effective August 1, 2021, the Company early adopted ASU 2020-06. Adoption of the new standard did not have a material impact on the financial statements. |
| 3. | In November 2021, the FASB issued ASU No. 2021-10, Government Assistance (Topic 832): Disclosure by Business Entities about Government Assistance (ASU 2021-10), which improves the transparency of government assistance received by most business entities by requiring the disclosure of: (1) the types of government assistance received; (2) the accounting for such assistance; and (3) the effect of the assistance on a business entity’s financial statements. This guidance is effective for financial statements issued for annual periods beginning after 15 December 2021. Early adoption is permitted. Adoption of the new standard did not have a material impact on the financial statements. |
| F-14 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 3: INTANGIBLE ASSETS. NET
Acquired intangible assets with finite lives consisted of the following as of July 31, 2022 and 2021:
| July 31, | ||||||||
| 2022 | 2021 | |||||||
| Patents | $ | $ | ||||||
| Gross intangible assets | ||||||||
| Less – accumulated amortization | ( | ) | ( | ) | ||||
| Intangible assets, net | $ | $ | ||||||
The attributable intellectual property relates to Sapientia’s various patents, which the Company is amortizing over 20 years, consistent with its accounting policy.
Amortization
expenses for the years ended July 31, 2022 and 2021, were $
The estimated future amortization expense of intangible assets as of July 31, 2022 is as follows:
| 2023 | $ | |||
| 2024 | ||||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 2028 and thereafter | ||||
| $ |
| F-15 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 4: LOANS
(a) Short-terms loans
During
the year ended July 31, 2021, the Company received seven unsecured loans from directors and an officer in the total amount of $
During March 2021, all the above mentioned short-term loans and accrued interest were repaid.
Total
interest expense in respect to all short-term loans for year ended July 31, 2022 and 2021 is and $
(b) Government grants
On
April 24, 2020, the Company received a $
On
December 13, 2021, the Company repaid the CEBA loan in the amounts of $
For
the year ended July 31, 2022 and 2021, the Company recorded an interest expense of nil and $
On
May 1, 2020 the Company received $
For
the year ended July 31, 2022 and 2021, the Company recorded an interest expense of and $
| F-16 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 4: LOANS (Cont.)
(c) 2020 Convertible Loan
On
November 16, 2020 (“Closing Date”), the Company closed a brokered private placement of an unsecured convertible debenture
unit of the Company (the “Debenture Unit”) to a single subscriber, purchased at a price of $
The
Debenture Unit was comprised of (A) $
The
Debenture was convertible, at the option of the holder thereof, from the period beginning on May 16, 2021, until the repayment of the
In
consideration for the services rendered by ThinkEquity, a division of Fordham Financial Management, Inc. (the “Broker”),
the Broker received a cash commission of $
The Company has determined that the Debenture Unit contained the following freestanding financial instruments: The term loan and the warrants. The warrants and the Broker Warrants are not indexed to the Company’s own stock and were classified as liabilities, initially measured at fair value, and subsequently measured at fair value through earnings.
The Company has determined that the term loan contained embedded derivatives required to be bifurcated from the host debt instrument pursuant to ASC 815-15. In addition, since the conversion feature was not bifurcated, the Company concluded that the term loan also includes a beneficial conversion feature which was accounted for as an equity component.
As such, the proceeds were allocated to the warrants, bifurcated embedded derivatives and the equity component. The residual proceeds were allocated to the liability component. The cash commission and Broker Warrants were expensed in the statement of operations and comprehensive loss.
The
Debenture’s net proceeds were $
On
March 1, 2021, the Debenture was repaid and the Company recorded a charge in the consolidated statements of operations and comprehensive
loss of $
NOTE 5: ACCRUED EXPENSES AND OTHER PAYABLES
Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Clinical activities | $ | |||||||
| Professional services | ||||||||
| Other | ||||||||
| $ | $ | |||||||
| F-17 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 6: CONTINGENT LIABILITIES AND COMMITMENTS
a. Legal proceedings:
On May 19, 2021, Alpha Capital Anstalt (“Alpha”) filed a lawsuit in the New York State Supreme Court, Commercial Division, New York County against BriaCell Therapeutics Corp. (“BriaCell”), alleging that BriaCell breached a loan contract when it refused to reprice and extend the term of warrants purported held by Alpha in spring 2021, seeking monetary and injunctive relief for delivery of those amended warrants. Counterclaiming and defending against Alpha’s complaint, BriaCell alleges that Alpha’s loan to BriaCell is unenforceable both because the loan is criminally usurious under New York law and because Alpha acted as an unregistered securities dealer in violation of American securities law. BriaCell also has alleged that Canadian securities law, regulation, and rules prohibited it from amending the warrants to comply with Alpha’s spring 2021 demands. On May 11, 2022, Alpha moved to dismiss BriaCell’s operative Amended Counterclaim. The parties have fully briefed that motion, and the Court has calendared oral argument on that motion for February 7, 2023. Expert discovery is ongoing and may affect the value of the parties’ respective claims and damages.
The Company disagrees with Alpha’s claims, is defending these claims, and has filed a counter claim. At this time, whilst it is impossible to provide any guarantee as to the outcome of the lawsuit, it is the Company’s assessment, based on advice from the Company’s legal counsel at this time, and based on the information known by the Company, that it’s more likely than not that BriaCell will not have to pay Alpha in the litigation.
b. Lease
In
July 2021, the Company ended its lease agreement in Berkeley, California. During the same time, the Company started a month-to-month
lease arrangement for office and lab space in New York, New York in the amount of approximately $
NOTE 7: FAIR VALUE MEASUREMENTS
The following table presents information about our financial instruments that are measured at fair value on a recurring basis as of July 31, 2022 and 2021:
| Fair Value Measurements at July 31, | ||||||||||||||||||||||||
| July 31, 2022 | July 31, 2021 | |||||||||||||||||||||||
| Level 1 | Level 2 | Total | Level 1 | Level 2 | Total | |||||||||||||||||||
| Financial Assets: | ||||||||||||||||||||||||
| Cash and cash equivalents | $ | |||||||||||||||||||||||
| Total assets measured at fair value | $ | |||||||||||||||||||||||
| Financial liabilities: | ||||||||||||||||||||||||
| Warrants liability | ||||||||||||||||||||||||
| Total liabilities measured at fair value | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
We classify our cash equivalents and the liability in respect of publicly traded warrants within Level 1 because we use quoted market prices in active markets.
The fair value of the warrant liability for non-public warrants is measured using inputs other than quoted prices included in Level 1 that are observable for the liability either directly or indirectly, and thus are classified as Level 2 financial instruments.
| F-18 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 8: SHAREHOLDERS’ EQUITY
a. Authorized share capital
The
authorized share capital consists of an number of common shares with
b. Issued share capital
During the years ended July 31, 2021 and 2022, the Company issued shares as follows:
| i) | On
August 18, 2020, the Company issued Shares to Sichenzia Ross Ference LLP or certain members or employees of Sichenzia Ross
Ference LLP as compensation for legal services. The shares were valued at $ per share and the Company recorded a loss on the
extinguishment of debt of $ | |
| ii) | On
February 26, 2021, the Company completed an underwritten public offering in the United States. The aggregate gross proceeds to the
Company from the offering were approximately $ |
In
addition, the Company issued the underwriter warrants (“Public Offering Broker Warrants”). Each Public Offering Broker
Warrant entitles the holder to purchase one Share at an exercise price per Public Offering Broker Warrant that is equal to $
The
Company granted the underwriter a 45-day option to purchase up to additional Shares and/or Pre-Funded Warrants and/or
In addition, the Company issued the underwriter Public Offering Broker Warrants.
| F-19 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 8: SHAREHOLDERS’ EQUITY (Cont.)
b. Issued share capital (continued)
During the year ended July 31, 2021, the Company accounted for the Public
Offering as follows: The
Pre-funded Warrants were recorded in equity. The Public Offering Warrants and the Over-allotment Warrants were recorded as a liability
with fair value of $
The
fair value of the Over-allotment Warrants at the issuance date was $
At
July 31, 2022 the fair value of the Public Offering Warrants and Public Offering Broker Warrants were $
As
a result, for the year ended July 31, 2022, the Company recorded a loss on the revaluation of the total warrant liability of $
February 26, 2021 (Issuance date) | April 12, 2021 (Issuance date) | July 31, 2022 | July 31, 2021 | |||||||||||||
| Share price | $ | $ | $ | $ | ||||||||||||
| Exercise price | $ | $ | $- | $- | ||||||||||||
| Expected life (years) | - | - | ||||||||||||||
| Volatility | % | % | % | % | ||||||||||||
| Dividend yield | % | % | % | % | ||||||||||||
| Risk free rate | % | % | % | % | ||||||||||||
| F-20 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 8: SHAREHOLDERS’ EQUITY (Cont.)
b. Issued share capital (continued)
| iii) | On
June 3, 2021, the Company entered into securities purchase agreements (each a “Purchase Agreement”) with certain institutional
and accredited investors (the “Investors”) pursuant to which the Company issued (i) Shares, (ii) pre-funded
warrants to purchase up to an aggregate of |
In
connection with the Private Placement, the Company agreed to: 1) pay the placement agent a cash commission equal to
The
Private Placement Pre-funded Warrants were recorded in equity. The fair value of the Private Placement Warrants was $
$
On
June 25, 2021, and June 26, 2021,
| iv) | The following table presents the summary of the changes in the fair value of the warrants: |
| Warrants liability | ||||
| Balance as of July 31, 2020 | $ | |||
| Convertible Debt Warrants | ||||
| Issuance of Public Offering Warrants | ||||
| Issuance of Public Offering Broker Warrants | ||||
| Issuance of Private Placement Warrants | ||||
| Exercise of Warrants | ( | ) | ||
| Reclassification of warrant liability to warrant reserve following change in functional currency | ||||
| Change in fair value | ||||
| Balance as of July 31, 2021 | $ | |||
| Issuance of warrants | ||||
| Warrant buyback program | ( | ) | ||
| Exercise of warrants | ( | ) | ||
| Change in fair value | ||||
| Balance as of July 31, 2022 | $ | |||
| v) | During
the year ended July 31, 2022, | |
| vi) | During
the year ended July 31, 2022, |
| F-21 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 8: SHAREHOLDERS’ EQUITY (Cont.)
c. Share buyback program
On September 9, 2021 the Company approved a repurchase program whereby the Company may purchase through the facilities of the TSX or NASDAQ (i) up to common shares (the “Common Shares”) and (ii) up to publicly traded BCTXW warrants (the “Listed Warrants”) in total, representing % of the Common Shares and % of the Listed Warrants comprising the “public float” as of September 8, 2021, over the next 12 months (the “Buyback”). Independent Trading Group (ITG) Inc. will act as the Company’s advisor and dealer manager in respect of the Buyback. The Company received final regulatory approval on September 22, 2021. As of July 31, 2022, the Company repurchased a total of shares with a value of $ (net of commissions) and publicly traded warrants for $ (net of commissions) with a fair value of $. All of the warrants and shares repurchased have been cancelled.
d. Share Purchase Warrants
| Number of warrants outstanding | Weighted average exercise price | |||||||
| Balance, July 31, 2020 | $ | |||||||
| Granted from the issuance of a convertible note | ||||||||
| Granted in the Public Offering | ||||||||
| Granted in the Over-allotment Option | ||||||||
| Granted in the Private Placement | ||||||||
| Expired | ( | ) | ( | ) | ||||
| Exercised | ( | ) | ( | ) | ||||
| Balance, July 31, 2021 | $ | |||||||
| Expired | ( | ) | ( | ) | ||||
| Exercised | ( | ) | ( | ) | ||||
| Repurchased and cancelled | ( | ) | ( | ) | ||||
| Balance, July 31, 2022 | $ | |||||||
| F-22 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 8: SHAREHOLDERS’ EQUITY (Cont.)
d. Share Purchase Warrants (continued)
| Number of Warrants outstanding as of July 31, 2022 | Exercise Price | Number of Warrants Exercisable as of July 31, 2022 | Expiry Date | |||||||||
| $ | ||||||||||||
| $ | – | |||||||||||
| $ | ||||||||||||
e) Compensation Warrants
A summary of changes in compensation warrants for the years ended July 31, 2022 and 2021 is presented below:
| Number of warrants outstanding | Weighted average exercise price | |||||||
| Balance, July 31, 2020 | ||||||||
| Granted from the issuance of a convertible note | ||||||||
| Granted in the Public Offering | ||||||||
| Granted in the Over Allotment | ||||||||
| Granted in the Private Placement | ||||||||
| Expired | ( | ) | ( | ) | ||||
| Exercised | ||||||||
| Balance, July 31, 2021 | $ | |||||||
| Exercised | ( | ) | ( | ) | ||||
| Balance, July 31, 2022 | $ | |||||||
| F-23 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 8: SHAREHOLDERS’ EQUITY (Cont.)
e) Compensation Warrants (continued)
As of July 31, 2022, compensation warrants outstanding were as follows:
| Number of Warrants as of July 31, 2022 | Exercise Price | Exercisable As of July 31, 2022 | Expiry Date | |||||||||
| $ | ||||||||||||
| $ | ||||||||||||
| $ | ||||||||||||
The
Company has adopted a stock option plan (the “Stock Option Plan”) under which it is authorized to grant options to officers,
directors, employees and consultants enabling them to acquire up to
The Company estimates the fair value of stock options granted using the Black-Scholes option-pricing model. The option-pricing model requires a number of assumptions, of which the most significant are the expected stock price volatility and the expected option term.
Expected volatility was calculated based upon the Company’s historical share price and historical volatilities of similar entities in the related sector index. The expected term of the options granted is derived from output of the option valuation model and represents the period of time that options granted are expected to be outstanding. The risk-free interest rate is based on the yield from U.S. treasury bonds with an equivalent term. The Company has historically not paid dividends and has no foreseeable plans to pay dividends.
Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Dividend yield | % | % | ||||||
| Expected volatility of the share prices | % | %- | % | |||||
| Risk-free interest rate | %- | % | %- | % | ||||
| Expected term (in years) | ||||||||
| F-24 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 9: SHARE - BASED COMPENSATION (Cont.)
Number of options | Weighted average exercise price | Weighted average remaining contractual term (in years) | Aggregate intrinsic value | |||||||||||||
| Balance as of July 31, 2021 | $ | $ | ||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Forfeited | ( | ) | ||||||||||||||
| Expired | ( | ) | ||||||||||||||
| Balance as of July 31, 2022 | ||||||||||||||||
| Exercisable as of July 31, 2022 | $ | $ | ||||||||||||||
The weighted-average grant date per-share fair value of stock options granted during 2022 and 2021 was $ and $, respectively. As of July 31, 2022, there are $ of total unrecognized costs related to share-based compensation that is expected to be recognized over a period of up to years.
| Exercise price | Options outstanding as of July 31, 2022 | Weighted average remaining contractual term (years) | Options exercisable as of July 31, 2022 | Weighted average remaining contractual term (years) | Expiry Date | |||||||||||||||
| $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||
Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Research and development expenses | $ | $ | ||||||
| General and administrative expenses | ||||||||
| Total share-based compensation | $ | $ | ||||||
| F-25 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 10: TAXES ON INCOME
a. Components of income taxes excluding cumulative effects of changes in accounting principles, other comprehensive income, and equity in net results of affiliated companies accounted for after-tax for the years ended December 31 were as follows:
b. The Company recorded loss before taxes on income for the period indicated as follows:
Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Domestic | $ | ( | ) | $ | ( | ) | ||
| Foreign | ( | ) | ( | ) | ||||
| $ | ( | ) | $ | ( | ) | |||
c. The reconciliation of the combined Canadian federal and provincial statutory income tax rate of 27% (2021 - 27%) to the effective tax rate is as follows:
| July 31, 2022 | July 31, 2021 | |||||||
| Net loss before recovery of income taxes | $ | ( | ) | $ | ( | ) | ||
| Expected income tax (recovery) expense | ( | ) | ( | ) | ||||
| Tax rate changes and effect of taxes of subsidiaries at foreign rates | ||||||||
| Share-based compensation and other non-deductible expenses | ||||||||
| Foreign exchange loss | ||||||||
| Share issuance cost booked directly to equity | ( | ) | ( | ) | ||||
| Valuation allowance | ||||||||
| Income tax (recovery) | $ | $ | ||||||
d.
The Company had
e. Significant components of the Company’s deferred tax assets are as follows:
| July 31, 2022 | July 31, 2021 | |||||||
| Deferred Tax Assets: | ||||||||
| Property, plant and equipment | $ | |||||||
| Marketable Securities | ||||||||
| Warrant liability | ||||||||
| Share issuance costs | ||||||||
| Operating tax losses carried forward | ||||||||
| Operating tax losses carried forward- USA | ||||||||
| Total deferred tax assets | ||||||||
| Valuation allowance | ( | ) | ( | ) | ||||
| Net deferred tax assets | $ | $ | ||||||
| Deferred Tax Liability: | ||||||||
| Intellectual Property | $ | ( | ) | $ | ( | ) | ||
| Convertible Debentures | ( | ) | ||||||
| Total net deferred tax liabilities | ( | ) | ( | ) | ||||
| Valuation allowance | ||||||||
| Net deferred tax assets (liabilities) | $ | $ |
f.
The Company has net deferred tax assets relating primarily to net operating loss (“NOL”) carryforwards, warrant liability and share issuance costs. Subject to certain limitations, the Company may use these deferred tax assets
to offset taxable income in future periods. Due to the Company’s history of losses and uncertainty regarding future earnings, a
full valuation allowance has been recorded against the Company’s deferred tax assets, as it is more likely than not that such assets
will not be realized. The net change in the total valuation allowance for the year ended July 31, 2022, was $
At
July 31, 2022, the Company had US federal NOL carryforwards of approximately $
Utilization of the NOL carryforwards and credits may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code (“IRC”) Sections 382 and 383, and similar state provisions. The Company has not completed an IRC 382/383 analysis regarding the limitation of NOL and credit carryforwards. If a change in ownership were to have occurred, the annual limitation may result in the expiration of NOL carryforwards and credits before utilization. If eliminated, the related asset would be removed from the deferred tax asset schedule with a corresponding reduction in the valuation allowance.
The Company has adopted the provisions of ASC 740-10, which clarifies the accounting for uncertain tax positions. ASC 740-10 requires that the Company recognize the impact of a tax position in its financial statements if the position is more likely than not to be sustained upon examination based on the technical merits of the position. For the year ended July 31, 2022, the Company had no material unrecognized tax benefits, and based on the information currently available, no significant changes in unrecognized tax benefits are expected in the next 12 months.
The Company’s policy is to recognize interest and penalties related to uncertain tax positions as income tax expense. The Company has no accruals for interest or penalties on its accompanying consolidated balance sheets as of July 31, 2022, and 2021, and has not recognized interest or penalties in the consolidated statements of operations for the years ended July 31, 2022, and 2021.
The Company is subject to taxation in the United States, New York, California, and Canada. The Company is subject to tax examination by tax authorities in those jurisdictions for periods after 2015. However, to the extent allowed by law, the taxing authorities may have the right to examine the periods where NOLs and credits were generated and carried forward, and make adjustments to the amount of the NOL and credit carryforwards. The Company is not currently under examination by federal or state jurisdictions.
| F-26 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 11: RELATED PARTY TRANSACTIONS AND BALANCES
Parties are considered to be related if one party has the ability, directly or indirectly, to control the other party or exercise significant influence over the other party in making operating and financial decisions. This would include the Company’s senior management, who are considered to be key management personnel by the Company. Parties are also related if they are subject to common control or significant influence. Related parties may be individuals or corporate entities. A transaction is considered to be a related party transaction when there is a transfer of resources or obligations between related parties.
a. The following related party salaries and directors’ fees are included in the consolidated statements of operations and comprehensive loss:
Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Directors (*) | $ | $ | ||||||
| Officers (**) | ||||||||
| $ | $ | |||||||
| (*) |
| (**) |
b. The following related party balances are included in the consolidated balance sheets:
| As of July 31, | ||||||||
| 2022 | 2021 | |||||||
| Directors (*) | $ | $ | ||||||
| Officers (**) | ||||||||
| $ | $ | |||||||
| F-27 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 12: FINANCIAL EXPENSE, NET
Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Interest income | $ | $ | ||||||
| Interest expense | ( | ) | ( | ) | ||||
| Change in fair value of warrant liability | ( | ) | ( | ) | ||||
| Gain on government grant | ||||||||
| Foreign exchange loss | ( | ) | ( | ) | ||||
| Loss on extinguishment of debt | ( | ) | ||||||
| Financial expenses, net | $ | ( | ) | $ | ( | ) | ||
Basic net loss per ordinary share is computed by dividing net loss for each reporting period by the weighted-average number of ordinary shares outstanding during each period. Diluted net loss per ordinary share is computed by dividing net loss for each reporting period by the weighted average number of ordinary shares outstanding during the period, plus dilutive potential ordinary shares considered outstanding during the period, in accordance with ASC No. 260-10 “Earnings Per Share”. The Company experienced a loss in the year ended July 31, 2022 and 2021; hence all potentially dilutive ordinary shares were excluded due to their anti-dilutive effect.
| Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Numerator: | ||||||||
| Net loss available to shareholders of ordinary shares | ( | ) | ( | ) | ||||
| Denominator: | ||||||||
| Shares used in computing net loss per ordinary shares, basic and diluted | ||||||||
| Year ended July 31, | ||||||||
| 2022 | 2021 | |||||||
| Employee stock options and warrants excluded from the computation of diluted per share amounts as their effect would be antidilutive | ||||||||
| F-28 |
BriaCell Therapeutics Corp
Notes to the Consolidated Financial Statements
For the Years Ended July 31, 2022 and 2021
(Expressed in US Dollars, except share and per share data and unless otherwise indicated)
NOTE 14: LONG-LIVED ASSETS BY GEOGRAPHIC LOCATION
| As of July 31, | ||||||||
| 2022 | 2021 | |||||||
| United States | $ | $ | ||||||
| Total long-lived assets* | $ | $ | ||||||
| (*) |
NOTE 15: SUBSEQUENT EVENTS
The Company evaluated the possibility of subsequent events existing in the Company’s consolidated financial statements through October 27, 2022, the date that the consolidated financial statements were available for issuance. The Company is not aware of any subsequent events which would require recognition or disclosure in the consolidated financial statements, except as noted below.
a) Approval of Omnibus Incentive Plan
On August 2, 2022, the Company approved an omnibus equity incentive plan (“Omnibus Plan), which will permit the Company to grant incentive stock options, preferred share units, restricted share units (“RSU’s”), and deferred share units (collectively, the “Awards”) for the benefit of any employee, officer, director, or consultant of the Company or any subsidiary of the Company. The maximum number of Shares available for issuance under the Omnibus Plan shall not exceed 15% of the issued and outstanding Shares, from time to time, less the number of Shares reserved for issuance under all other security-based compensation arrangements of the Company, including the existing Stock Option Plan. The Omnibus Plan remains subject to approval by the shareholders of the Company (the “Shareholders”) and final approval of the Toronto Stock Exchange (“Exchange”) and will replace the Company’s existing Stock Option Plan upon receipt of such approvals (“Approvals”).
The Company may make grants under the Omnibus Plan, however, the grants cannot be settled until the Approvals have been received.
b) Option and RSU grants
On August 2, 2022, the Company granted options, under the Stock Option Plan, to directors, officers and employees with an exercise price of CAD$. The options vest quarterly in advance over a -year period and expire on . of the options were issued to officers of the Company.
The Company also granted, under the Omnibus Plan, RSU’s to the CEO. The RSU’s vested immediately.
| F-29 |