TABLE OF CONTENTS
|
|
|
Page |
|
|
|
3 |
|
A. |
Directors and Senior Management
|
3 |
|
B. |
Advisers |
3 |
|
C. |
Auditors |
3 |
|
|
|
3 |
|
|
|
3 |
|
A. |
[Reserved.] |
3 |
|
B. |
Capitalization and Indebtedness
|
3 |
|
C. |
Reasons for the Offer and Use of Proceeds
|
3 |
|
D. |
Risk factors |
3 |
|
|
|
25
|
|
A. |
History and Development of the Company
|
25 |
|
B. |
Business Overview |
25 |
|
C. |
Organizational Structure |
44 |
|
D. |
Property, Plants and Equipment |
44 |
|
|
|
44 |
|
|
|
44 |
|
A. |
Operating Results |
45 |
|
B. |
Liquidity and Capital Resources
|
48 |
|
C. |
Research and development, patents and
licenses, etc. |
50 |
|
D. |
Trend Information |
50 |
|
E. |
Critical Accounting Estimates |
50 |
|
|
|
51 |
|
A. |
Directors and senior management
|
51
|
|
B. |
Compensation of Directors and Executive
Officers |
53 |
|
C. |
Board Practices |
55 |
|
D. |
Employees |
61 |
|
E. |
Share Ownership |
63 |
|
F. |
Disclosure of a Registrant’s Action
to Recover Erroneously Awarded Compensation |
63 |
|
|
|
63 |
|
A. |
Major shareholders |
63 |
|
B. |
Related Party Transactions |
64 |
|
C. |
Interests of Experts and Counsel
|
64 |
|
|
|
65 |
|
A. |
Consolidated Statements and Other Financial
Information |
65 |
|
B. |
Significant Changes |
65 |
|
|
|
65 |
|
A. |
Offer and Listing Details |
65 |
|
B. |
Plan of Distribution |
65 |
|
C. |
Markets for Ordinary Shares |
65 |
|
D. |
Selling Shareholders |
65 |
|
E. |
Dilution |
65 |
|
F. |
Expenses of the Issue |
65 |
|
|
|
65 |
|
A. |
Share Capital |
65 |
|
B. |
Memorandum and Articles of Association
|
65 |
|
C. |
Material Contracts |
66 |
|
D. |
Exchange controls |
66 |
|
E. |
Taxation |
66 |
|
F. |
Dividends and paying agents |
74 |
|
G. |
Statement by experts |
74 |
|
H. |
Documents on display |
74 |
|
I. |
Subsidiary Information |
74 |
|
|
|
74 |
|
|
|
74 |
|
|
|
|
|
|
|
75 |
|
|
|
75 |
|
|
|
75
|
|
|
|
75 |
|
|
|
75 |
|
|
|
75 |
|
|
|
75 |
|
|
|
75 |
|
|
|
75 |
|
|
|
76 |
|
|
|
76 |
|
|
|
76 |
|
|
|
76 |
|
|
|
|
|
|
|
77 |
|
|
|
77 |
|
|
|
77 |
|
|
|
79 |
CERTAIN INFORMATION
Unless otherwise stated, all of our financial information
presented in this annual report has been prepared in accordance with generally accepted accounting principles in the United States of
America, or U.S. GAAP.
In this Annual Report on Form 20-F, or the Annual
Report, unless the context indicates otherwise, references to “NIS” are to the legal currency of Israel, “U.S. dollars,”
“$” or “dollars” are to United States dollars, and the terms “we,” “us,” “our company,”
“our,” and “Check-Cap” refer to Check-Cap Ltd. Unless otherwise indicated, U.S. dollar translation of NIS amounts
presented in this Annual Report are translated using the rate of $1.00 = NIS 3.519, the exchange rate published by the Bank of Israel
on December 30, 2022, and U.S. dollar translation of Euro amounts presented in this Annual Report are translated using the rate of $1.00
= Euro 0.938, based on the exchange rates published by the Bank of Israel on December 30, 2022.
We effected a 1-for-20 reverse share split of our
Ordinary Shares, effective as of November 23, 2022, in accordance with the approval of our shareholders at a meeting held on August 11,
2022. All share numbers in this Annual Report are reflected on a post-reverse stock split basis.
USE OF TRADE NAMES
AND TRADEMARKS
Throughout this Annual Report, we refer to various
trademarks, service marks and trade names that we use in our business. The “CHECK-CAP” and “C-Scan” trademarks
and design logos are the property of Check-Cap Ltd. Other trademarks and service marks appearing in this Annual Report are the property
of their respective holders. We do not intend our use or display of other companies’ tradenames, trademarks or service marks to
imply a relationship with, or endorsement or sponsorship of us by these other companies. Solely for convenience, trademarks and trade
names referred to in this Annual Report appear without the ® or TM symbols, but such references are not intended to indicate, in
any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these
trademarks and trade names.
FORWARD-LOOKING STATEMENTS
This Annual Report contains forward-looking statements,
about our expectations, beliefs or intentions regarding, among other things, our ongoing and planned product development and clinical
trials; the timing of, and our ability to make, regulatory filings and obtain and maintain regulatory approvals for our product candidates;
our intellectual property position; the degree of clinical utility of our products, particularly in specific patient populations; our
ability to develop commercial functions; expectations regarding product launch and revenue; our results of operations, cash needs; financial
condition, liquidity, prospects, growth and strategies; the industry in which we operate; and the trends that may affect the industry
or us. In addition, from time to time, we or our representatives have made or may make forward-looking statements, orally or in writing.
Forward-looking statements can be identified by the use of forward-looking words such as “believe,” “expect,”
“intend,” “plan,” “may,” “should” or “anticipate” or their negatives or other
variations of these words or other comparable words or by the fact that these statements do not relate strictly to historical or current
matters. These forward-looking statements may be included in, but are not limited to, various filings made by us with the U.S. Securities
and Exchange Commission, or the SEC, press releases or oral statements made by or with the approval of one of our authorized executive
officers. Forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made.
Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and
uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking
statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated
in forward-looking statements, including, but not limited to, the factors summarized below.
This Annual Report identifies important factors
which could cause our actual results to differ materially from those indicated by the forward-looking statements, particularly those set
forth under the heading “Risk Factors.” The risk factors included in this Annual Report are not necessarily all of the important
factors that could cause actual results to differ materially from those expressed in any of our forward-looking statements. Given these
uncertainties, readers are cautioned not to place undue reliance on such forward-looking statements. Factors that could cause our actual
results to differ materially from those expressed or implied in such forward-looking statements include, but are not limited to:
|
• |
our needs for additional capital to fund our operations and our inability to obtain additional capital
on acceptable terms, or at all; |
|
• |
the initiation, timing, progress and results of our clinical trials and other product development efforts;
|
|
• |
our reliance on one product; |
|
• |
the clinical development, commercialization and market acceptance of C-Scan; |
|
• |
our ability to receive de novo classification or premarket approval and other regulatory approvals for
C-Scan; |
|
• |
our ability to successfully complete clinical trials and obtain desired performance. |
|
• |
our reliance on single-source suppliers; |
|
• |
our ability to achieve an acceptable cost of goods; |
|
• |
our reliance on third parties, such as for purposes of our clinical trials and clinical development and
the manufacturing, marketing and distribution of C-Scan; |
|
• |
our ability to establish and maintain strategic partnerships and other corporate collaborations;
|
|
• |
our ability to achieve reimbursement and coverage from government and private third-party payors;
|
|
• |
the implementation of our business model and strategic plans for our business; |
|
• |
the scope of protection we are able to establish and maintain for intellectual property rights covering
C-Scan and our ability to operate our business without infringing the intellectual property rights of others; |
|
• |
competitive companies, technologies and our industry; |
|
• |
current or future unfavorable economic and market conditions
and adverse developments with respect to financial institutions and associated liquidity risk; and |
|
• |
those factors referred to in “Item 3. Key Information – D. Risk Factors,” “Item
4. Information on the Company,” and “Item 5. Operating and Financial Review and Prospects”, as well as in this Annual
Report generally. |
All forward-looking statements attributable to
us or persons acting on our behalf speak only as of the date of this Annual Report and are expressly qualified in their entirety by the
cautionary statements included in this Annual Report. We undertake no obligations to update or revise forward-looking statements to reflect
events or circumstances that arise after the date made or to reflect the occurrence of unanticipated events. In evaluating forward-looking
statements, you should consider these risks and uncertainties.
EXPLANATORY NOTE
Market data and certain industry data and forecasts
used throughout this Annual Report were obtained from internal company surveys, market research, consultant surveys commissioned by the
Company, publicly available information, reports of governmental agencies and industry publications and surveys. Industry surveys, publications,
consultant surveys commissioned by the Company and forecasts generally state that the information contained therein has been obtained
from sources believed to be reliable. However, this information may prove to be inaccurate because of the method by which some of the
data for the estimates is obtained or because this information cannot always be verified with complete certainty due to the limits on
the availability and reliability of raw data, the voluntary nature of the data gathering process and other limitations and uncertainties.
As a result, the market and industry data and forecasts included or incorporated by reference in this Annual Report, and estimates and
beliefs based on that data, may not be reliable. We have relied on certain data from third-party sources, including internal surveys,
industry forecasts and market research, which we believe to be reliable based on our management’s knowledge of the industry. However,
we have not ascertained the underlying economic assumptions relied upon therein. Forecasts are particularly likely to be inaccurate, especially
over long periods of time. In addition, we do not necessarily know what assumptions regarding general economic growth were used in preparing
the forecasts we cite. Statements as to our market position are based to the best of our knowledge on the most currently available data.
While we are not aware of any misstatements regarding the industry data presented in this Annual Report, our estimates involve risks and
uncertainties and are subject to change based on various factors, including those discussed under the heading “Risk Factors”
in this Annual Report.
PART
I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT
AND ADVISERS
A. Directors
and Senior Management
Not required.
B. Advisers
Not required.
C. Auditors
Not required.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not required.
ITEM 3. KEY INFORMATION
A. [Reserved.]
B.
Capitalization and Indebtedness
Not required.
C. Reasons
for the Offer and Use of Proceeds
Not required.
D. Risk
factors
In conducting our business,
we face many risks that may interfere with our business objectives. Some of these risks could materially and adversely affect our business,
financial condition and results of operations. In particular, we are subject to various risks resulting from changing economic, political,
industry, regulatory, business and financial conditions. The risks and uncertainties described below are not the only ones we face. Additional
risks and uncertainties not currently known to us or that we currently deem to be immaterial may also materially adversely affect our
business operations.
You should carefully consider
the following factors and other information in this Annual Report before you decide to invest in our ordinary shares. If any of the risks
referred to below occur, our business, financial condition and results of operations could suffer. In any such case, the trading price
of our ordinary shares could decline, and you may lose all or part of your investment.
Summary Risk Factors
Our business is subject to numerous risks and uncertainties,
including those highlighted in the section titled “Risk Factors” immediately following this summary. The principal factors
and uncertainties that make investing in our ordinary shares risky, include, among others:
Risks Related to Our Financial Position
|
• |
We have a history of losses, may incur future losses and may not achieve profitability; |
|
• |
We will require additional funding in order to complete the development, scale up
manufacturing and commercialization of C-Scan, which may cause dilution to our existing shareholders, restrict our operations or require
us to relinquish rights to C-Scan or intellectual property. If additional capital is not available, we may have to delay, reduce or cease
the development or commercialization of C-Scan; |
Risks Related to Our Business
|
• |
Although we received FDA approval of our
Investigational Device Exemption, or IDE (including our IDE amended application) for our U.S. pivotal study, the timing of the initiation
of the second part of the U.S. pivotal study and, if commenced, the outcome of the study is inherently uncertain, even if the study is
completed; |
|
• |
Clinical failure can occur at any stage of clinical development and we may not succeed in completing the
development of our product. Our clinical experience to date does not necessarily predict future results and may not have revealed certain
potential limitations of the technology and potential complications from C-Scan and may require further clinical validation. Any product
version we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval;
|
|
• |
We may face a number of challenges with respect to our commercialization efforts and may not succeed to
commercialize our product, obtain required regulatory approvals or manufacture commercial quantities of C-Scan at an acceptable cost to
enable us to generate significant revenues; |
|
• |
We expect to derive most of our revenues from sales of one product. Our inability to successfully commercialize
this product, or any subsequent decline in demand for this product, could severely harm our ability to generate revenues. |
|
• |
If healthcare professionals do not recommend our product to their patients, C-Scan may not achieve market
acceptance and we may not become profitable; |
|
• |
We expect to face competition from large, well-established manufacturers of traditional technologies for
CRC screening and detection of gastrointestinal disorders, as well as from new competitive technologies, especially within the field of
biomarkers; |
|
• |
We may rely on local distributors and/or strategic partners to market and distribute C-Scan in those countries
where we intend to market and distribute C-Scan; |
|
• |
We have limited manufacturing capabilities and if we are unable to scale up our manufacturing operations
to meet the necessary quantities for our upcoming clinical studies or anticipated market demand, our growth could be limited and our business,
financial condition and results of operations could be materially adversely affected; |
|
• |
Our reliance on sole or single source suppliers could harm our ability to conduct clinical trials and meet
demand for our product in a timely manner or within budget; |
|
• |
A health epidemic, pandemic or other outbreak, including the current COVID-19 pandemic, may materially
and adversely affect our business, financial condition and results of operations; |
|
• |
We may not be successful in establishing and maintaining strategic partnerships, which could adversely
affect our ability to develop and commercialize C-Scan; |
Risks Related to Regulations
|
• |
If we are unable to obtain, or experience significant delays in obtaining, FDA clearances or approvals,
or equivalent third country approvals for C-Scan or future products or product enhancements, our ability to commercially distribute and
market our products could suffer; |
|
• |
There is no guarantee that the FDA will grant a de novo classification request or PMA approval of C-Scan
and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business;
|
|
• |
If we or our future manufacturers or distributors do not obtain and maintain the necessary regulatory clearances
or approvals, or equivalent third country approvals in a specific country or region, we or our future distributors will not be able to
market and sell C-Scan or future products in that country or region; |
|
• |
The results of our current or future clinical trials may not support our product candidate requirements
or intended use claims or may result in the discovery of adverse side effects; |
|
• |
If we fail to obtain or maintain necessary regulatory clearances or CE Certificates for C-Scan or if there
are regulatory changes in our existing or future target markets, our ability to sell C-Scan and generate revenues could be harmed;
|
|
• |
Our failure to comply with radiation safety or radio frequency regulations in a specific country or region
could impair our ability to conduct our clinical trials, or commercially distribute and market C-Scan in that country or region;
|
|
• |
Our business is subject to complex environmental and health legislation in various jurisdictions that may
increase our costs and our risk of noncompliance; and |
|
• |
If we are unable to achieve reimbursement and coverage from government and private third-party payors for
procedures using C-Scan, or if reimbursement is insufficient to create an economic benefit for purchasing or using C-Scan when compared
to alternative procedures, demand for our products may not grow at the rate we expect. |
Risks Related to Our Intellectual Property
|
• |
If we are unable to protect our intellectual property rights, our competitive position could be harmed.
|
|
• |
Third-party claims of infringement or other claims against us could require us to redesign C-Scan, seek
licenses, or engage in future costly intellectual property litigation, which could negatively affect our future business and financial
performance. |
Risks Related to Our Operations in Israel
|
• |
Our principal offices, research and development facilities our manufacturing sites and some of our
suppliers are located in Israel and, therefore, our business, financial condition and results of operation may be adversely affected by
political, economic and military instability in Israel. |
|
• |
Pursuant to the terms of the Israeli government grants we received for research and development expenditures,
we are obligated to pay certain royalties on our revenues to the Israeli government. In addition, the terms of Israeli government grant
we received require us to satisfy specified conditions and to make additional payments in addition to repayment of the grants upon certain
events. |
Risks Related to Ownership of our Ordinary Shares
|
• |
Our ordinary shares could be delisted from the Nasdaq Capital Market. |
|
• |
Our stock price has and may be subject to fluctuation, and purchasers of our securities could incur substantial
losses. |
|
• |
Our business, operating results and growth rates may be adversely affected by current or future unfavorable
economic and market conditions and adverse developments with respect to financial institutions and associated liquidity risk. |
Risks Related to Our
Financial Position
We have a history of losses, may incur future losses and may not
achieve profitability.
We are a clinical and development-stage medical
diagnostics company with a limited operating history. We have incurred net losses in each fiscal year since we commenced operations in
2009. We incurred net losses of $19.1 million in 2022 and $17.2 million in 2021. As of December 31, 2022, our accumulated deficit was
$127.3 million. We expect our losses to continue for the foreseeable future as we continue our investment in research and development
and clinical trials to complete the development of our technology and to attain regulatory approvals, manufacturing scale up, begin the
commercialization efforts for C-Scan, increase our marketing and selling expenses, and incur additional costs as a result of being a public
company in the United States. Successful completion of our development program and, ultimately, the attainment of profitable operations
is dependent upon future events, including obtaining adequate financing to fulfill our development activities and our ability to manufacture
commercial quantities of C-Scan at an acceptable cost and generate significant revenues. The extent of our future operating losses and
the timing of becoming profitable are highly uncertain, and we may never achieve or sustain profitability.
We will require additional funding in order to complete the development,
scale up manufacturing and commercialization of C-Scan, which may cause dilution to our existing shareholders, restrict our operations
or require us to relinquish rights to C-Scan or intellectual property. If additional capital is not available, we may have to delay, reduce
or cease the development or commercialization of C- Scan.
Our operations have consumed substantial amounts
of cash. We expect that we will need to continue to raise and spend substantial amounts in order to complete the clinical development,
regulatory approval and commercialization of C-Scan. We intend to use our cash resources, including the proceeds of our March 2022 and
July 2021 registered direct offerings to finance these efforts. We recently announced the temporary postponement of the initiation
of the second part of the U.S. pivotal study that was expected in mid-2023 and that as a result, we adopted a plan of action, including
(among other things) a cost reduction plan in order to extend our cash runway to longer than initially planned, which we are in the process
of implementing. Based on such plan, we believe that we have sufficient capital to fund our ongoing operations and plans into the fourth
quarter of 2024. We will therefore need to raise additional funds to obtain the required capital prior to completing the development,
clinical trials and commercialization of C-Scan (see Item 5B “Operating and Financial Review and Prospects — Liquidity and
Capital Resources — Sources of Liquidity”). We may seek additional funding through equity offerings, debt financings, collaborations,
licensing arrangements or any other means to conduct clinical trials and develop, manufacture and market C-Scan or for our general corporate
purposes. Securing additional financing may divert our management’s attention from its day-to-day activities, which may adversely
affect our ability to develop and commercialize C-Scan. Additional financing may not be available to us on a timely basis on terms acceptable
to us, or at all. In addition, if we raise additional funds by issuing equity securities, you may experience significant dilution of your
ownership interest and the newly issued securities may have rights senior to those of the holders of our ordinary shares. In addition,
the issuance of additional equity securities by us, or the possibility of such issuance, may cause the market price of our ordinary shares
to decline. Alternatively, if we raise funds by obtaining loans from third parties, the terms of those financing arrangements may include
negative covenants, may require us to grant a lender a security interest in our assets or may include other restrictions on our business
that could impair our operational flexibility, and could also result in high interest expense. If we raise additional funds through collaborations,
licensing arrangements or other structured financing transactions, we may relinquish rights to certain of our technologies or products,
grant security interests in our assets or grant licenses to third parties on terms that are unfavorable to us. If adequate additional
financing on acceptable terms is not available, we may not be able to develop C-Scan at the rate or to the stage we desire, and we may
have to delay or abandon the commercialization of C-Scan. Any of these factors could materially adversely affect our business, financial
condition and results of operations.
Risks Related to Our
Business
Although we received FDA approval of our IDE (including our IDE
amended application) for our U.S. pivotal study, the timing of the initiation of the second part of the U.S. pivotal study and, if commenced,
the outcome of the study is inherently uncertain, even if the study is completed.
We received U.S. Food and Drug Administration,
or FDA, approval of our IDE and our IDE amended application for our planned U.S. pivotal study. While we initiated the first part of the
U.S. pivotal study in May 2022, following our internal assessment of the clinical data collected to date from our Israeli and U.S. calibration
studies, we have determined that the current efficacy results do not meet our goal in order to proceed to the powered portion of the U.S.
pivotal study and, therefore, the initiation of the second part of the U.S. pivotal study that was expected in mid-2023 is temporarily
postponed. We have adopted a plan of action that includes conducting additional clinical data analysis and approaching the FDA to make
amendments to the U.S. pivotal study protocol that are expected to be part of an IDE supplement submission to the FDA, and which are subject
to FDA approval. There can be no assurance that the FDA would approve such an IDE supplement or that we will be able to commence
or complete the powered potion of the U.S. pivotal study. Furthermore, in the event that the FDA or any other regulatory authority requires
us to complete additional studies or we are required to satisfy other FDA or other regulatory requests, the start of the second part of
the U.S. pivotal study may be further delayed. Even after we receive and incorporate guidance from the FDA, the FDA could disagree that
we have satisfied their requirements to commence our second part of the U.S. pivotal study or change their position on the acceptability
of our planned trial design or the clinical endpoints selected, which may require us to complete additional studies or clinical trials
or impose stricter approval conditions than we currently expect. As a result of the foregoing, the research and development, preclinical
studies and clinical testing is expensive and can take years to complete, and its outcome is inherently uncertain. Failure can occur at
any time during the development process. (see “Risk Factors – Risks Related to Our Business
– Clinical failure can occur at any stage of clinical development and we may not succeed in completing the development of our product.
Our clinical experience to date does not necessarily predict future results and may not have revealed certain potential limitations of
the technology and potential complications from C-Scan and may require further clinical validation. Any product version we advance through
clinical trials may not have favorable results in later clinical trials or receive regulatory approval.”).
Clinical failure can occur at any stage of
clinical development and we may not succeed in completing the development of our product. Our clinical experience to date does not necessarily
predict future results and may not have revealed certain potential limitations of the technology and potential complications from C-Scan
and may require further clinical validation. Any product version we advance through clinical trials may not have favorable results in
later clinical trials or receive regulatory approval.
Clinical failure can occur at any stage of clinical
development. To date, we have performed clinical studies with iterative versions of both scanning and non-scanning capsules, in conjunction
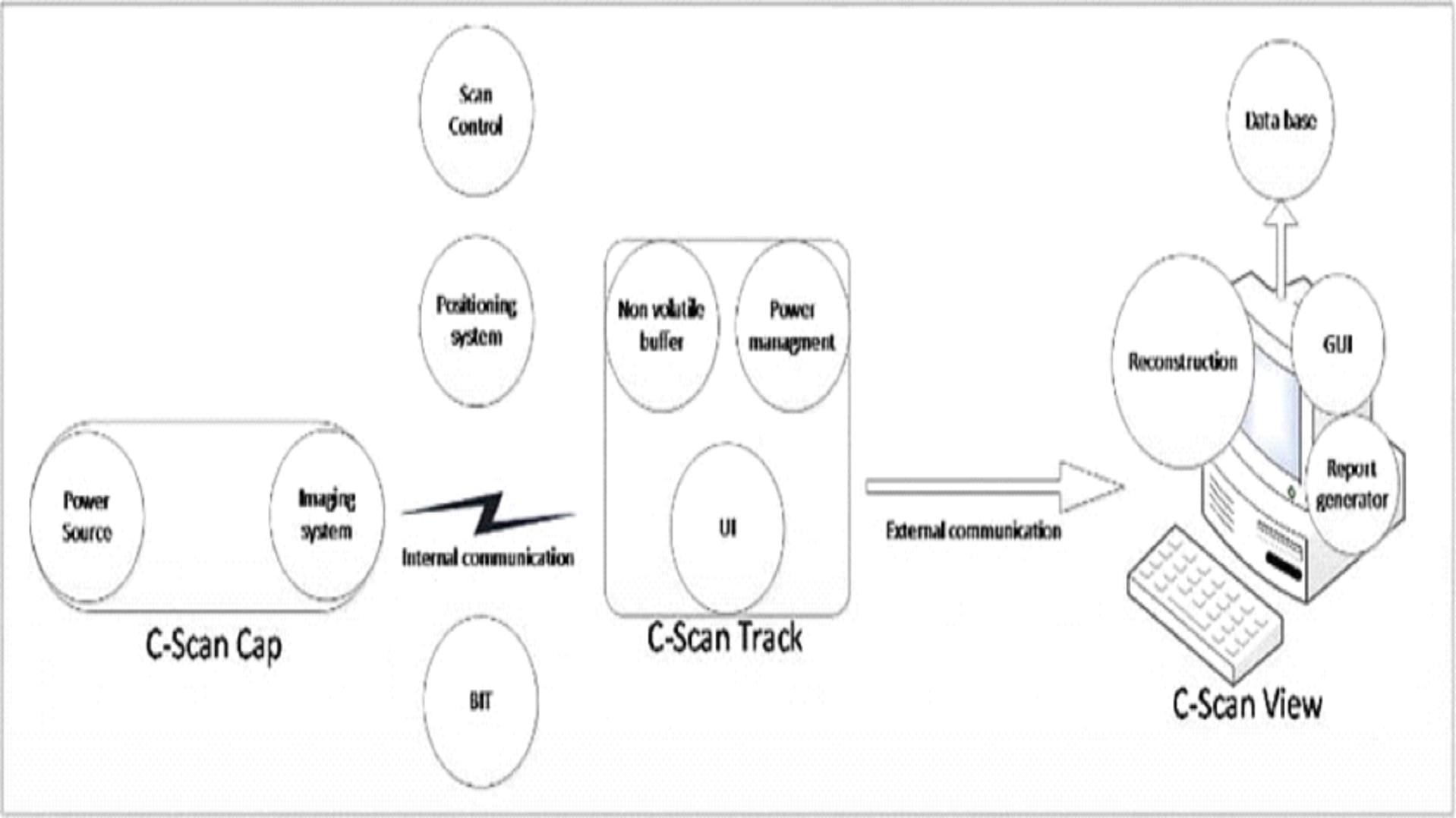
with iterative versions of the C-Scan Track , C-Scan View application and we are continuing to develop further iterative versions of our
C-Scan Cap and C-Scan Track. In addition, our clinical trials were conducted to date under differing protocols, while using specific inclusion
criteria and enriched and average risk patients’ enrolment. Therefore, we have a limited ability to identify potential problems
and/or inefficiencies concerning current and future versions of C-Scan in advance of its use in general and expanded groups of average
risk patients and we cannot assure you that its actual clinical performances will be satisfactory to support proposed indications and
regulatory approvals and clinical acceptance and adoption, or that its use will not result in unanticipated complications. For example,
in previous studies we have experienced technical failures of the C-Scan Cap and are continuously engaged in implementing a plan to ensure
improved reliability in later versions. To this end, we have been collecting additional clinical data in a study in Israel (initially
enrolling both average risk and high-risk patients and starting May 2022, have shifted to enrollment of only average risk patients), for
the purpose of further calibration of the C-Scan system, in parallel to conducting the first stage of the U.S. pivotal study that was
initiated in May 2022. As part of the approved amended IDE, we designed the U.S. pivotal study to include two stages: the first
stage, aimed to support our C-Scan calibration among the average risk population, which is intended to include up to 200 patients in the
U.S.; and the second stage, which is intended to include up to 800 subjects (400 in the US and 400 in Israel) and aims to demonstrate
C-Scan performance in a statistically significant manner. The initiation of the powered portion of the U.S. pivotal study was dependent
upon successful completion of the calibration portion of the U.S. pivotal study. Following our internal assessment of the clinical
data collected to date from our calibration studies, we have determined that the current efficacy results do not meet our goal in order
to proceed to the powered portion of the U.S. pivotal study. Therefore, the initiation of the powered portion of the U.S. pivotal
study that was expected in mid-2023 is temporarily postponed. We have adopted a plan of action that includes conducting additional
clinical data analysis and approaching the FDA to make amendments to the U.S. pivotal study protocol that are expected to be part of an
IDE supplement submission to the FDA, and which are subject to FDA approval. In addition, we plan to continue conducting our calibration
studies, albeit at a slower pace. However, there can be no assurance that we will be able to commence or complete the powered potion
of the U.S. pivotal study, that the FDA will approve our amendments to the U.S. pivotal study, or that the implementation of our plan
will be successful.
Furthermore, the results from laboratory, non-clinical
and completed clinical studies, as well as results from our ongoing clinical trials may not be indicative of final clinical results obtained
from our current C-Scan version or future versions of C-Scan on expanded average risk screening populations. In addition, the results
of our clinical trials are subject to human analyses and interpretation of the data accumulated, which could be affected by various errors
due to, among others, lack of sufficient clinical experience with C-Scan, assumptions used in the statistical analysis of results, interpretation
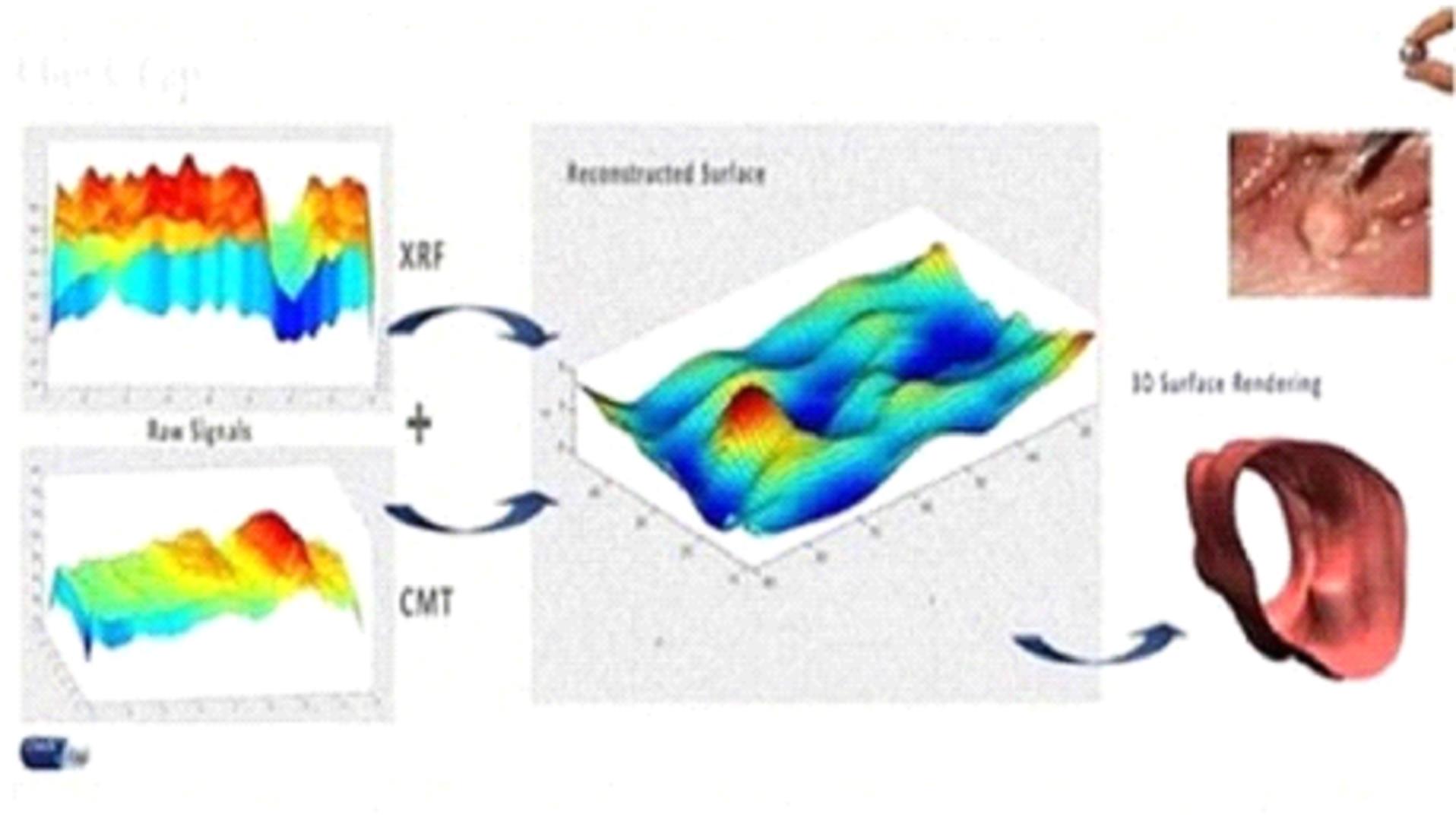
errors in the analysis of the clinical trials results, including the reconstructed images by C-Scan, or due to uncertainty in the actual
efficacy of C-Scan in its current clinical stage. Therefore, the safety and efficacy of C-Scan and the clinical results to date will require
further independent professional validation and require further clinical study. If C-Scan does not function as expected over time, we
may not be able to develop C-Scan at the rate or to the stage we desire, we could be subject to liability claims, our reputation may be
harmed, C-Scan may not achieve regulatory clearances, and C-Scan may not be widely adopted by healthcare providers and patients.
We may face a number of challenges with respect to our commercialization
efforts and may not succeed to commercialize our product, obtain required regulatory approvals or manufacture commercial quantities of
C-Scan at an acceptable cost to enable us to generate significant revenues.
Since commencing our operations, we have focused
on the research and development and limited clinical trials of C-Scan. Although we have received a CE-certificate from our notified body
(DEKRA - 0344) according to the EU Regulation 2017/745 on Medical Devices for the marketing and sale of C-Scan in the European Union,
valid until December 1, 2026, we have not commenced marketing and sales in the European Union and may require additional regulatory approval
in Member States of the European Union, for instance from the German Federal Office for Radiation Protection (Bundesamt für Strahlenschutz,
BfS) before we can commence marketing and sale of C-Scan such states. Furthermore, we may require additional regulatory certifications,
registrations and/or approvals from regulatory authorities in European jurisdictions outside the European Union, including with the UK
Medicines and Healthcare Products Regulatory Agency and the Swiss National Cooperative for the Disposal of Radioactive Waste (Nagra).
In Israel, we received approval from the Medical Devices and Accessories Division of the Israeli Ministry of Health, or AMAR, for the
marketing and sale of C-Scan in Israel, which is valid until December 31, 2024. We have not received approvals in other jurisdictions,
including the United States, and there can be no assurance that we will be able to receive regulatory approvals to commence marketing
and sales for C-Scan in the foreseeable future or ever. Our ability to generate revenues and achieve profitability depends on our ability
to successfully complete the development of our C-Scan product, demonstrate sufficient clinical evidence, obtain required regulatory approvals
and commercial licenses, manufacture commercial quantities of C-Scan at an acceptable cost to enable us to generate significant revenues.
The future success of our business cannot be determined
at this time, and we do not anticipate generating significant revenues from product sales for the foreseeable future. In addition, we
have no experience in commercializing C-Scan and may face a number of challenges with respect to our commercialization efforts, including,
among others, that:
|
• |
we may not have adequate financial or other resources to complete the development of our product, demonstrate
adequate clinical results, attain required regulatory approvals and licensures, obtain adequate manufacturing and capacity and begin the
commercialization efforts for C-Scan; |
|
• |
we may fail to obtain or maintain required regulatory approvals and licensures for C-Scan in our target
markets or may face adverse regulatory or legal actions relating to our system even if regulatory approval is obtained; |
|
• |
we may not demonstrate adequate clinical safety and clinical effectiveness results from our current or
future versions of C-Scan, to support regulatory body approval or market acceptance and adoption; |
|
• |
we may face ongoing limitations imposed by the Nuclear Regulatory Commission, or NRC, or other nuclear
regulatory commissions in jurisdictions in which we intend to commercialize C-Scan in relation to the disposal of our C-Scan Cap in the
sanitary system, such as requiring patients to retrieve our C-Scan Cap after use, which could impact engagement with clinical sites and
enrollment pace in our clinical studies and make C-Scan less attractive; |
|
• |
we may not be able to maintain an adequate supply chain due to reliance on single or sole suppliers for
critical components and scale up the manufacture of C-Scan to commercial quantities at an adequate quality or at an acceptable cost;
|
|
• |
we may not be able to establish adequate sales, marketing and distribution channels; |
|
• |
healthcare professionals and patients may not accept C-Scan; |
|
• |
we may not be aware of possible complications from the continued use of C-Scan because we have limited
clinical experience with respect to the actual use of C-Scan; |
|
• |
other technological breakthroughs in colorectal cancer, or CRC, screening, treatment and prevention may
reduce the demand for C-Scan; |
|
• |
changes in the market for CRC screening, new alliances between existing market participants and the entrance
of new market participants may interfere with our market penetration efforts; |
|
• |
government and private third-party payors may not agree to provide coding, coverage and payment adequate
to reimburse healthcare providers and patients for any or all of the purchase price in conjunction with clinical effectiveness of C-Scan,
which may adversely affect healthcare providers’ and patients’ willingness to purchase C-Scan; |
|
• |
uncertainty as to market demand may result in inefficient pricing of C-Scan; |
|
• |
we may not be able to adequately protect our intellectual property or may face third-party claims of intellectual
property infringement; and |
|
• |
we are dependent upon the results of ongoing clinical studies relating to C-Scan which may impact its perceived
value as against competing products. |
If we are unable to meet any one or more of these
challenges successfully, our ability to effectively commercialize C-Scan could be limited, which in turn could have a material adverse
effect on our business, financial condition and results of operations.
We expect to derive most of our revenues from
sales of one product. Our inability to successfully commercialize this product, or any subsequent decline in demand for this product,
could severely harm our ability to generate revenues.
We are currently dependent on the successful commercialization
of C-Scan to generate revenues. As a result, factors adversely affecting our ability to successfully commercialize, or the pricing of
or demand for, this product could have a material adverse effect on our financial condition and results of operations. If we are unable
to successfully commercialize or create market demand for C-Scan, we will have limited ability to generate revenues.
Furthermore, we may be vulnerable to fluctuations
in demand for C-Scan. Such fluctuations in demand may be due to many factors, many of which are beyond our control, including, among others:
|
• |
market acceptance of a new product, including healthcare professionals’ and patients’ preferences;
|
|
• |
market acceptance of the clinical safety and performance of C-Scan; |
|
• |
development of competitive or similarly cost-effective products by our competitors; |
|
• |
development delays of C-Scan, including delays in clinical trials or FDA clearance; |
|
• |
technological innovations in CRC screening, treatment and prevention; |
|
• |
adverse medical side effects suffered by patients using C-Scan, whether actually resulting from the use
of C-Scan or not; |
|
• |
changes in regulatory policies toward CRC screening or imaging technologies; |
|
• |
regulatory approval, changes in regulatory approval, clearance requirements and licensure for our product,
including by the NRC or other radiation safety authorities; |
|
• |
third-party claims of intellectual property infringement; |
|
• |
budget constraints and the availability of reimbursement or insurance coverage from third-party payors
for C-Scan; |
|
• |
increases in market acceptance of other technologies; |
|
• |
adverse responses from certain of our competitors to the offering of C-Scan; |
|
• |
licensure and perceived risk of manufacturing and using a product containing a radioactive source (including
associated agencies refusal to authorize capsule disposal), loss or theft; |
|
• |
licensure of medical centers (including clinical study sites) to use or offer product containing a radioactive
source; and |
|
• |
the shelf life of our C-Scan Cap. |
If healthcare professionals do not recommend our product to their
patients, C-Scan may not achieve market acceptance and we may not become profitable.
CRC screening candidates are generally referred
by their healthcare professional to approved tests and screening technology options. If healthcare professionals, including physicians,
do not recommend or prescribe our product to their patients, C-Scan may not achieve market acceptance and we may not become profitable.
In addition, physicians have historically been slow to change their medical diagnostic and treatment practices because of perceived liability
risks arising from the use of new products. Delayed adoption of C-Scan by healthcare professionals could lead to a delayed adoption by
patients and government and private third-party payors. Healthcare professionals may not recommend or prescribe C-Scan until certain conditions
have been satisfied including, among others:
|
• |
there is sufficient long-term clinical and health-economic evidence to convince them to alter their existing
screening methods and device recommendations; |
|
• |
there are recommendations from prominent physicians, educators and/or associations that C-Scan is safe
and effective; |
|
• |
we obtain favorable data from clinical and health-economic studies for C-Scan; |
|
• |
reimbursement or insurance coverage from government and private third-party payors is available;
|
|
• |
healthcare professionals obtain required approvals and licensures for the handling, storage, dispensing
and disposal of C-Scan; and |
|
• |
healthcare professionals become familiar with the complexities of C-Scan. |
We cannot predict when, if ever, healthcare professionals
and patients may adopt the use of C-Scan. Even if favorable data is obtained from clinical and health-economic studies for the regulatory
approval of C-Scan, there can be no assurance that prominent physicians would endorse it or that future clinical studies will continue
to produce favorable data regarding C- Scan. In addition, prolonged market exposure may also be a pre-requisite to reimbursement or insurance
coverage from government and private third-party payors. If C-Scan does not achieve an adequate level of acceptance by patients, healthcare
professionals and government and private third-party payors, we may not generate significant product revenues and we may not become profitable.
We expect to face competition from large, well-established manufacturers
of traditional technologies for CRC screening and detection of gastrointestinal disorders, as well as from new competitive technologies,
especially within the field of biomarkers.
Competition for C-Scan comes from traditional well-entrenched
manufacturers of tests and equipment for CRC screening, such as colonoscopy, sigmoidoscopy, CT Colonography, or CTC, optical capsule endoscopy,
fecal occult blood tests, or FOBTs, and fecal immunochemical tests, or FITs. The principal manufacturers of equipment for optical colonoscopy
and sigmoidoscopy include Olympus, Pentax, Hoya and Fuji Film. The principal manufacturers of equipment for CTC include General Electric
Healthcare Systems, Siemens Medical Solutions, Philips Healthcare. and Toshiba Corporation. The principal manufacturer of equipment for
optical capsule endoscopy includes Medtronic plc. All of these companies have substantially greater financial resources than we do, and
they have established reputations as well as worldwide distribution channels for providing medical instruments to physicians.
In addition, several companies have developed or
are developing non-invasive technologies based on stool, serum (from blood and other bodily fluids), or molecular diagnostics, or MDx,
tests that are used to indicate the presence of CRC and polyps in the colon. These companies include Exact Sciences, Polymedco, Epigenomics
AG, Gene News, EDP Biotech Corporation, Illumina, Inc., Quest Diagnostics, VolitionRx Nu.Q diagnostic, Freenome, Grail, Guardant, Genoscopy
and Universal DX. Exact Sciences commenced sales of its Cologuard ®, a non-invasive stool DNA screening test for colorectal cancer
in 2015, and is constantly seeking performance improvements through additional studies, alongside commercial expansion. During the past
years, there has been an extensive effort on the part of our competitors to utilize molecular biology and advanced computational techniques
to develop methods capable of identifying cell-free cancer biomarkers.
In December 2022, Guardant Health released results
from ECLIPSE (Evaluation of ctDNA LUNAR Assay In an Average Patient Screening Episode), an over 20,000 patient registrational study evaluating
the performance of its blood test for detecting colorectal cancer (CRC) in average-risk adults. The test demonstrated 83% sensitivity
in detecting individuals with CRC. Specificity was 90% in both individuals without advanced neoplasia and in those who had a negative
colonoscopy result. This test also demonstrated 13% sensitivity in detecting advanced adenomas. In this study, two configurations of a
multimodal blood-based screening test were evaluated independently - a cell-free DNA (cfDNA)-only test and a cfDNA test with protein biomarkers.
The announced results were derived from the cfDNA-only test, which outperformed the cfDNA test with protein biomarkers. Based on these
study results, Guardant Health plans to complete its premarket approval submission to the FDA in the first quarter of 2023.
In January 2023, Geneoscopy Inc., released results
from the CRC-PREVENT trial – a pivotal clinical trial evaluating the efficacy of its noninvasive, stool-based, at-home diagnostic
screening test to detect colorectal cancer (CRC) and advanced adenomas (AA) in average-risk individuals. The CRC-PREVENT trial included
8,289 individuals with diverse racial, ethnic, and socioeconomic backgrounds across more than 2,900 zip codes in all lower 48 states,
with colonoscopies performed in more than 3,800 endoscopy centers. Efficacy results from the study demonstrated 94% sensitivity for detecting
CRC, 45% sensitivity for detecting advanced adenoma (AA), and 88% specificity for no findings on a colonoscopy. The company submitted
a Premarket Approval (PMA) application to the FDA in January 2023.
These advancement efforts are assisted by large
capital investments, supported in turn by claims that certain products may promise the possibility of defeating certain cancers in their
early stages.
In addition, procedures for bowel cleansing that
are less onerous are constantly being developed, which could make our entry into the market more difficult. For instance, bowel cleansing
initiated by the ingestion of pills or food-substitute diet regimes rather than through drinking large amounts of distasteful liquids
may be viewed as an improvement to the cleansing process, but other screening methods may be even more palatable to patients.
If we are unable to convince patients and physicians
to adopt C-Scan over the current technologies marketed by our competitors or future new technologies that may be marketed by our existing
or potential competitors, our business and results of operations may suffer.
We may rely on local distributors and/or strategic partners to market
and distribute C-Scan in those countries where we intend to market and distribute C-Scan.
We may rely on local distributors and/or strategic
partners for the marketing and distribution of C-Scan. Our success in generating sales in countries or regions where we will engage local
distributors and/or strategic partners will depend in large part on the efforts of these third parties over whom we have limited control.
If we are unable to identify and engage with suitable local distributors and/or strategic partners in the countries where we intend to
market and distribute C-Scan, our business, financial condition and results of operations could be negatively affected.
We have limited manufacturing capabilities and if we are unable
to scale up our manufacturing operations to meet the necessary quantities for our upcoming clinical studies or anticipated market demand,
our growth could be limited and our business, financial condition and results of operations could be materially adversely affected.
We currently have limited resources, facilities
and experience in manufacturing sufficient quantities of C-Scan to meet the quantities we need for the demand we may expect from commercialization
efforts, if we are able to obtain regulatory clearance in the United States for C-Scan. We do not have experience in manufacturing C-Scan
in the commercial quantities that we expect to require to meet demand for C-Scan, nor can we be certain of our manufacturing costs. We
have in the past, and we may in the future, face certain technical challenges as we increase manufacturing capacity, including, among
others, equipment design and automation, material procurement and lower than expected yields and increased scrap costs, as well as challenges
related to maintaining quality control and assurance standards, manufacturing commercial quantities of C-Scan capsules at an acceptable
cost and logistics associated with the handling of radioactive materials and the shelf life of our C-Scan Cap, which have resulted in
and could result in delays in our clinical trial and commercialization plans and lost revenue. We may be unable to establish or maintain
reliable, high-volume manufacturing capacity. Even if we can establish and maintain this capacity, the cost of doing so may increase the
cost of C-Scan and reduce our ability to successfully compete. In addition, while we have established our own X-ray source production
line in Israel to support our clinical needs and potentially future sales in Israel and in Europe, subject to the successful progress
of our clinical studies and sufficient available capital, we plan to expand in the future our in-house X-ray production line to support
future commercial quantities demand and may not be successful in such efforts. Despite our continuing cost-cutting efforts, we may not
be able to achieve adequate cost that will be profitable or adequate quality at an acceptable cost. C-Scan includes several components
that are based on new technologies and are difficult to manufacture and some are being supplied to us by single source suppliers (see
“Risk Factors – Risks Related to Our Business – Our reliance on sole or single source
suppliers could harm our ability to conduct clinical trials and meet demand for our product in a timely manner or within budget.”).
Furthermore, we may encounter similar or unforeseen challenges initiating and later expanding production of any new products. If we are
unable to scale up our manufacturing capabilities to meet market demand, or to achieve adequate product cost, our growth could be limited
and our business, financial condition and results of operations could be materially adversely affected.
In addition, we received and may receive in the
future grants from the Government of the State of Israel through the IIA (formerly the OCS) for the financing of a portion of our research
and development expenditures and to support the funding of our transition from research and development to manufacturing, pursuant to
the Encouragement of Research, Development and Technological Innovation in the Industry Law 5744-1984 (formerly known as the Encouragement
of Industrial Research and Development Law 5744-1984), or the Innovation Law. The terms of the IIA grants subject us to certain restrictions
relating to (among other things) the transfer of the manufacturing of IIA-funded products outside Israel. See “Risk
Factors – Risks Related to Our Operations in Israel – Pursuant to the terms of the Israeli government grants we received for
research and development expenditures, we are obligated to pay certain royalties on our revenues to the Israeli government. In addition,
the terms of Israeli government grants we received require us to satisfy specified conditions and to make additional payments in addition
to repayment of the grants upon certain events.” Such restrictions may impair our ability to outsource or transfer development
or manufacturing activities with respect to any product or technology outside of Israel.
Our reliance on sole or single source suppliers could harm our ability
to conduct clinical trials and meet demand for our product in a timely manner or within budget.
We currently depend on sole or single source suppliers
for some of the components and materials necessary for the production of C-Scan. For example, we currently have a sole or single supplier
for the motor used to rotate the collimated X-ray source in C-Scan, for the customized X-ray detectors and batteries used in C-Scan, as
well as for the X-Ray enriched isotope substance. There is a limited number of manufacturers worldwide who are capable of manufacturing
the motor, the customized X-ray detectors and the batteries that we currently use in C-Scan. In addition, the application-specific integrated
circuit, or ASIC, residing in C-Scan is currently manufactured for us by a single semiconductor fabrication plant, or FAB. Furthermore,
we do not currently have written contracts with all our sole or single suppliers. While our current suppliers have been able to supply
the required quantities of such components to date, if the supply of these components is disrupted or terminated or if our current suppliers
are unable to supply required quantities of components now or in the commercialization period, we may not be able to find alternative
sources for these key components in a timely manner. Although we are planning to maintain strategic inventory of key components, the inventory
may not be sufficient to satisfy the demand for C-Scan if such supply is interrupted or may be subject to risk of loss due do catastrophic
events, such as a global political crisis or war, or a fire at our storage facility. In addition, to partially mitigate the risks of reliance
on sole and single source suppliers, we are seeking alternate manufacturers for some of our components which requires us to dedicate significant
resources and investment. There can be no assurance that we will be successful in seeking alternate suppliers or establish our own production
line. As a result of the foregoing, we may be unable to meet our clinical trials timelines and meet the demand for C-Scan, which could
harm our ability to generate revenues, lead to customer dissatisfaction and damage our reputation. If we are required to change the supplier
of any of these key components, there may be a significant delay in locating a suitable alternative manufacturer. In addition, we may
be required to verify that the new manufacturer maintains facilities and procedures that comply with FDA and other applicable quality
standards and with all applicable regulations and guidelines. As part of our mitigation plan, in December 2021, we signed an amendment
to the agreement, or the Amended Service Agreement, with the sole supplier of the X-ray source used in C-Scan and concurrently entered
into a Sub-lease Agreement, or the Sub-lease Agreement with the supplier, according to which lease approximately 70 square meters of laboratory
space at the supplier premises. According to the Amended Service Agreement, we independently produce the X-ray source using our own production
employees. The Amended Service Agreement extended the terms of the service agreement we had with the sole supplier, until the Sub-lease
Agreement entered into effect on September 21, 2022, after receipt of certain certificates including a business license from the Petach
Tikva municipality. Subject to the successful progress of our clinical studies and sufficient available capital, we plan to continue to
expand our in-house production line for our X-ray source in Israel, however we cannot assure you that we will commence or successfully
complete this effort and we may be required to find an alternative supplier for the X-ray source, which we may not be able to do in a
timely manner. The delays associated with the introduction of a new manufacturer for certain key components or new suppliers for certain
materials, could delay our ability to manufacture C-Scan in a timely manner or within budget. Furthermore, in the event that the manufacturer
of a key component of C-Scan ceases operations or otherwise ceases to do business with us, we may not have access to the information necessary
to enable an alternative supplier to manufacture the component. The occurrence of any of these events could harm our ability to meet demand
for C-Scan in a timely manner or within budget.
The use of any of our C-Scan Cap, C-Scan Track or C-Scan View could
result in product liability or similar claims that could be expensive, damage our reputation and harm our business.
Our business exposes us to an inherent risk of
potential product liability or similar claims related to the manufacturing, marketing and sale of medical devices. The medical device
industry has historically been litigious, and we face financial exposure to product liability or similar claims if the use of any of our
C-Scan Cap, C-Scan Track or C-Scan View were to cause or contribute to injury or death, including, without limitation, harm to the body
caused by the procedure or inaccurate diagnoses from the procedure that could affect treatment options. There is also the possibility
that defects in the design or manufacture of any of these products might necessitate a product recall. Although we plan to maintain product
liability insurance, the coverage limits of these policies may not be adequate to cover future claims. In the future, we may be unable
to maintain product liability insurance on acceptable terms or at reasonable costs and such insurance may not provide us with adequate
coverage against potential liabilities. A product liability claim, regardless of merit or ultimate outcome, or any product recall could
result in substantial costs to us, damage to our reputation, customer dissatisfaction and frustration, and a substantial diversion of
management attention. A successful claim brought against us in excess of, or outside of, our insurance coverage could have a material
adverse effect on our business, financial condition and results of operations.
C-Scan is a complex medical device that requires training for qualified
personal and care for data analysis.
C-Scan is a complex medical device that requires
training for qualified personal, including physicians, and care for data analysis. Although our distributors will be required to ensure
that C-Scan is prescribed only by trained clinicians, the potential for misuse of C-Scan still exists due to its complexity. Such misuse
could result in adverse medical consequences for patients that could damage our reputation, subject us to costly product liability litigation
and otherwise have a material adverse effect on our business, financial condition and results of operations.
We depend on third parties to manage our clinical studies and trials,
perform related data collection and analysis, and to enroll patients for our clinical trials, and, as a result, we may face costs and
delays that are beyond our control.
We rely on third parties, such as third-party clinical
research organizations, or CROs, clinical investigators, clinical research coordinators, physicians and clinical sites, to manage our
clinical trials and perform data collection and analysis, and to enroll patients for our clinical trials. Although we have and expect
to continue to have contractual arrangements with these third parties, we control only certain aspects of their activities. Nevertheless,
we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol and legal, regulatory
and scientific standards, and our reliance on such third parties does not relieve us of our regulatory responsibilities. If such third
parties fail to comply with applicable regulatory requirements, the clinical data generated in our clinical trials may be deemed unreliable
and regulatory authorities may require us to perform additional clinical trials before approving our marketing applications, which would
delay the regulatory approval process. Furthermore, we may not be able to control the amount and timing of resources that these parties
devote to our studies and trials or the quality of these resources. In addition, our engagement with clinical centers for conducting our
clinical trial in the U.S depends upon site-specific licensing and regulatory requirements associated with the X-ray technology within
our C-Scan capsules related to nuclear regulatory compliance.
If these third parties fail to properly manage
our studies and trials or enroll patients for our clinical trials, we will be unable to complete them at all or in a satisfactory or timely
manner, which could delay or prevent us from obtaining regulatory approvals for, or achieving market acceptance of, our product.
In addition, termination of relationships with
third parties may result in delays, inability to enter into arrangements with alternative third parties or do so on commercially reasonable
terms. Switching or adding additional clinical sites involves additional cost and requires management time and focus. In addition, there
is a natural transition period when a new clinical site commences work. As a result, delays occur, which can materially impact our ability
to meet our desired clinical development timelines.
We currently expect to sell our products mainly in the United States,
Europe, Israel and Japan and, if we are unable to manage our operations in these territories, our business, financial condition and results
of operations could be materially adversely affected.
Our headquarters and substantially all of our operations
and employees are presently located in Israel, but we currently expect to market our products mainly in the United States, Europe, Israel
and Japan. Accordingly, we are subject to risks associated with international operations, and our international sales and operations will
require significant management attention and financial resources. In addition, our international sales and operations will subject us
to risks inherent in international business activities, many of which are beyond our control and include, among others:
|
• |
foreign certification, registration and other regulatory requirements; |
|
• |
customs clearance and shipping delays; |
|
• |
import and export controls; |
|
• |
multiple and possibly overlapping tax structures; |
|
• |
difficulty forecasting the results of our international operations and managing our inventory due to our
reliance on third-party distributors; |
|
• |
differing laws and regulations, business and clinical practices, licensures, government and private third-party
payor reimbursement policies and patient preferences; |
|
• |
differing standards of intellectual property protection among countries; |
|
• |
difficulties in staffing and managing our international operations; |
|
• |
difficulties in penetrating markets in which our competitors’ products are more established and achieving
a competitive sale price for our product; |
|
• |
currency exchange rate fluctuations and foreign currency exchange controls and tax rates; and |
|
• |
political and economic instability, war or acts of terrorism or natural disasters, emergence of a pandemic,
or other widespread health emergencies (or concerns over the possibility of such an emergency, including for example, the COVID-19 pandemic).
|
If we are unable to manage our international operations effectively, our business,
financial condition and results of operations could be materially adversely affected.
We may not be successful in establishing and maintaining strategic
partnerships, which could adversely affect our ability to develop and commercialize C-Scan.
A part of our strategy is to evaluate and, as deemed
appropriate, enter into partnerships in the future when strategically attractive, including potentially with major medical device companies.
We face significant competition in seeking appropriate partners for C-Scan, and the negotiation process is time-consuming and complex.
In order for us to successfully partner C- Scan, potential partners must view C-Scan as economically valuable in markets they determine
to be attractive in light of the terms that we are seeking and other available products for licensing by other companies. Even if we are
successful in our efforts to establish strategic partnerships, the terms that we agree upon may not be favorable to us, and we may not
be able to maintain such strategic partnerships if, for example, development or approval of a product is delayed or sales of an approved
product are disappointing. Any delay in entering into strategic partnership agreements related to C-Scan could delay the development and
commercialization of our product candidates and reduce their competitiveness even if they reach the market.
In addition, our strategic partners may breach
any future agreement with us, and we may not be able to adequately protect our rights under these agreements. Furthermore, our strategic
partners will likely negotiate for certain rights to control decisions regarding the development and commercialization of C-Scan, if approved,
and may not conduct those activities in the same manner as we would do so.
If we fail to establish and maintain strategic
partnerships related to C-Scan, we will bear all the risks and costs related to the development and commercialization of C-Scan, and we
will need to seek additional significant financing, hire additional employees and otherwise develop expertise which we do not have and
for which we have not budgeted.
If we lose our key personnel or are unable to attract and retain
additional personnel, our business and ability to compete will be harmed.
Our success relies upon the continued service and
performance of the principal members of our management and research and development team, including Alex Ovadia and Yoav Kimchy. In order
to implement our business strategy, we will need to retain our key personnel with expertise in the areas of research and development,
clinical testing, government regulation, manufacturing, finance, marketing and sales. Our product development plans depend in part on
our ability to retain engineers with expertise in a variety of technical fields. Larger companies may have a competitive advantage in
hiring such engineers, due to the relatively high compensation they are able to offer. The loss of a number of these persons or our inability
to attract and retain qualified personnel could harm our business and our ability to compete. We do not maintain key man insurance for
Alex Ovadia or Yoav Kimchy.
Substantially all of our operations are currently conducted at our
facility near Haifa, Israel, and at our own X-ray source production line in Petach Tikva, Israel and any disruption at our facilities
could materially adversely affect our business, financial condition and results of operations.
Substantially all of our operations are currently
conducted at our facility near Haifa, Israel (other than certain production activities conducted by subcontractors) and at our own X-ray
source production line in Petach Tikvah, Israel. We take precautions to safeguard our facilities, including obtaining insurance coverage
and implementing health and safety protocols. However, a natural or other disaster, such as a fire, flood, earthquake, an armed conflict
involving Israel (as detailed further under “Risks Related to Our Operations in Israel”)
or a terrorist attack, could damage or destroy our facilities and our manufacturing equipment or inventory, cause substantial delays in
our operations and otherwise cause us to incur additional unanticipated expenses. In addition, the insurance we maintain against fires,
floods and other natural disasters and the war and terrorism insurance we maintain may not be adequate to cover our losses in any particular
case. Damage to our facilities or our other property or to any of our suppliers’ facilities and properties, whether located in Israel
or elsewhere, due to fire, a natural disaster or casualty event or an armed conflict or terrorist attack, could materially adversely affect
our business, financial condition and results of operations.
A security breach or disruption or failure in a computer or communications
systems could adversely affect us.
Despite the implementation of security measures,
our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer
viruses, unauthorized access, cyber-attacks, natural disasters, fire, terrorism, war, and telecommunication and electrical failures. If
such an event were to occur and interrupt our operations, it could result in a material disruption of our clinical program. For example,
the loss of clinical trial data from ongoing or planned clinical trials could result in delays in our regulatory approval efforts and
significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss
of or damage to our data or applications, loss of trade secrets or inappropriate disclosure of confidential or proprietary information,
including protected health information or personal data of clinical trial participants or employees or former employees, access to our
clinical data, or disruption of the manufacturing process, we could incur liability and the further development of C-Scan could be delayed.
We may also be vulnerable to cyber-attacks by hackers or other malfeasance. This type of breach of our cybersecurity may compromise our
confidential information and/or our financial information and adversely affect our business or result in legal proceedings. Further, these
cybersecurity breaches may inflict reputational harm upon us that may result in decreased market value and erode public trust.
We or the third parties upon whom we depend may be adversely affected
by natural disasters and/or health epidemics or pandemics, such as the COVID-19 pandemic, and our business continuity and disaster recovery
plans may not adequately protect us from a serious disaster.
Natural disasters could severely disrupt our operations
and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power
outage or other event occurred that prevented us from using all or a significant portion of our office, manufacturing and/or lab spaces,
that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, subcontractors,
suppliers, CROs, clinical sites, clinical investigators, clinical coordinators, third parties ongoing activities and schedules or that
otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our plans and business for a substantial
period of time.
In early 2020, the World Health Organization declared
the rapidly spreading coronavirus disease (COVID-19) outbreak a pandemic. We experienced disruptions to our operations as a result of
the COVID-19 pandemic, including disruptions to the Company’s clinical studies. For example, during a period of few months
during the year ending December 31, 2022, there was a global shortage in Iodine due to COVID-19, which is critical patient intake used
in the C-Scan procedure. As a result, we faced delays in our clinical trials. The extent to which the COVID-19 pandemic shall impact
our operations in the future will depend on future developments, which are highly uncertain and cannot be predicted with confidence. In
particular, any future spread of COVID-19 globally or the reinstatement of restrictions or other actions that may be required to contain
COVID-19 or treat its impact could materially adversely impact our operations and workforce, including our research and clinical trials
and our ability to continue to raise capital, could affect the operations of key governmental agencies, such as the FDA, which may delay
our development plans, and could result in the inability of our suppliers to deliver components or raw materials on a timely basis or
at all, each of which in turn could have a material adverse impact on our business, financial condition and results of operation.
The disaster recovery and business continuity plans
we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result
of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business.
Risks Related to
Regulations
If we are unable to obtain, or experience significant delays in
obtaining, FDA clearances or approvals, or equivalent third country approvals for C-Scan or future products or product enhancements, our
ability to commercially distribute and market our products could suffer.
Our products are subject to rigorous regulation
by the FDA and numerous other federal, state and foreign governmental authorities and notified bodies. The process of obtaining regulatory
clearances or approvals, or equivalent third-country approvals to market a medical device can be costly and time consuming, and we may
not be able to obtain these clearances or approvals, or equivalent third-country approvals on a timely basis, if at all. In particular,
we expect to eventually generate a portion of our revenues from sales of C-Scan and future products in the United States, the European
Union, or third countries. Before a new medical device, or a new use of, or claim for, an existing product can be marketed in the United
States, it must first receive clearance under Section 510(k) of the Federal Food, Drug, and Cosmetic Act, or FDA approval of a premarket
approval application, or PMA, unless an exemption applies. The FDA will clear marketing of a low or moderate risk medical device through
the 510(k) process if sufficiently similar predicate devices have previously been cleared via this pathway. In the 510(k) clearance process,
the FDA must only determine that the proposed device is “substantially equivalent” to a device legally on the market, known
as a “predicate” device, with respect to intended use/indications for use, technological characteristics and principles of
operation in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence.
High risk devices deemed to pose the greatest risk,
such as life-sustaining, life-supporting, or implantable devices, or devices not deemed substantially equivalent to a previously cleared
device, require approval of a PMA. The PMA process is more costly, lengthy and more uncertain than the 510(k) clearance process. The PMA
pathway requires an applicant to demonstrate the safety and effectiveness of the device based, in part, on the data obtained in clinical
trials. A PMA application must be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial,
manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the device for its intended
use.
In instances where a device is novel and there
is no suitable predicate device, but that device is deemed to be of low or moderate risk, the FDA can reclassify the device to class I
or class II via de novo reclassification. This process involves the submission of a reclassification petition, and the FDA accepting that
“special controls” are adequate to ensure the product’s performance and safety. The FDA now allows “direct”
de novo reclassification petitions, a mechanism by which a sponsor can directly submit a detailed de novo reclassification petition as
the device’s initial submission without having to first receive a not substantially equivalent, or NSE, decision on a 510(k) submission.
These processes can be expensive and lengthy. FDA’s
510(k) clearance process may take 6 to 9 months, but it can last longer. Direct de novo reclassification typically may take at least 9
to 12 months from filing to clearance if no comments or disapproval from the FDA. The PMA pathway is much more costly and uncertain than
the 510(k) clearance process or de novo reclassification, and generally may take at least 12 to 18 months, or even longer, from the time
the application is filed with FDA to ultimate approval if no comments or disapproval from the FDA.
We are not aware of any legally marketed predicate
device upon which FDA could base a determination of substantial equivalence under a 510(k) clearance process. Our proposed strategy, therefore,
is to submit a direct de novo reclassification petition for C-Scan. To support this petition, our objective is to demonstrate that the
device poses a low or moderate risk to patients. We cannot assure you that FDA will not demand that we obtain PMA approval of C-Scan.
FDA can delay, limit or deny clearance or approval
of an application for many reasons, including, among others:
|
• |
we may not be able to demonstrate to FDA’s satisfaction that our products are safe and effective
for their intended use; |
|
• |
in the case of a 510(k) submission, we may not be able to demonstrate to FDA’s satisfaction that
our products are substantially equivalent to a legally marketed predicate; |
|
• |
the data from our non-clinical studies and clinical trials may be insufficient to support clearance or
approval; |
|
• |
in the case that PMA submission is required, that the manufacturing process or facilities we use may not
meet applicable requirements; and |
|
• |
changes in FDA’s 510(k) clearance, de novo reclassification, or PMA approval processes and policies,
or the adoption of new regulations may require additional data and delays in getting to market. |
We may not obtain the necessary regulatory clearances,
approvals or equivalent third country approvals to market C-Scan or future products in the United States or elsewhere. Any delay in, or
failure to receive or maintain, clearance, approval for C-Scan or other products under development could prevent us from generating revenue
from these products or achieving profitability.
There is no guarantee that the FDA will grant a de novo classification
request or PMA approval of C-Scan and failure to obtain necessary clearances or approvals for our future products would adversely affect
our ability to grow our business.
C-Scan and some of our future products may require
FDA clearance of a 510(k), de novo classification, or may require FDA approval of a PMA. The FDA may not approve or provide clearance
of C- Scan or our future products for the indications that are necessary or desirable for successful commercialization. Indeed, the FDA
may deny our requests for 510(k) clearance, de novo reclassification or PMA approval for C-Scan or any other future product, new intended
uses or modifications to these products once they are cleared or approved for marketing.
Our strategy is to submit a direct de novo classification
petition for C-Scan following completion of a U.S. pivotal study. A de novo reclassification generally applies when the 510(k) pathway
is not available due to the lack of a suitable predicate device and the FDA believes the device poses a low to moderate risk. De novo
petitions can either be submitted in lieu of a 510(k) notice, such as in our case, or after a 510(k) notice has been filed and found NSE
(Not Substantial Equivalent). If a 510(k) notice is found NSE, a de novo petition must be submitted within 30 days from the receipt of
the NSE determination.
To support our direct de novo petition, our objective
is to demonstrate that the device poses a low to moderate risk to patients. If the FDA determines that C-Scan is not a candidate for de
novo classification, it will require approval of the device for market through the PMA process. A PMA application must be supported by
extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data, to demonstrate
to the FDA’s satisfaction the safety and efficacy of the device for its intended use. By statute, the FDA has 180 days to review
the “accepted application,” although, generally, FDA can extend the review of the application and final review may take between
one and three years. During this review period, the FDA may request additional information or clarification of information already provided
or even request new data that may require us to conduct additional tests. Also during the review period, an advisory panel of experts
from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability
of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with quality
system regulations. The FDA’s review of a PMA could significantly delay our plans to get to market. There is also no guarantee that
the FDA would approve a PMA. Failure to receive clearance or approval for C-Scan or future products would have an adverse effect on our
ability to expand our business.
Even if the FDA ultimately accepts our de novo
reclassification petition, to support our petition, we still may be required to conduct additional clinical trials before a petition can
be filed.
Such studies or trials can be costly and the results
uncertain. If we are unable to successfully complete required clinical studies or trials, we will not receive FDA clearance or approval.
If we or our future manufacturers or distributors do not obtain
and maintain the necessary regulatory clearances or approvals, or equivalent third country approvals in a specific country or region,
we or our future distributors will not be able to market and sell C-Scan or future products in that country or region.
We intend to market C-Scan in a number of international
markets. To be able to market and sell C-Scan in a specific country or region, we and/or our distributors must comply with the regulations
of that country or region. While the regulations of some countries do not impose barriers to marketing and selling part or all of our
products or only require notification, others require that we and/or our distributors obtain the approval of specified regulatory authorities
such as the Nuclear Regulatory Commission (NRC) in the U.S., the Swiss National Cooperative for the Disposal of Radioactive Waste (Nagra)
and the German Federal Office for Radiation Protection (Bundesamt für Strahlenschutz, BfS), in addition to the CE mark that is required
for medical devices throughout the EU. These regulations, and the time required for regulatory review, vary from country to country. Obtaining
regulatory approvals is expensive and time-consuming, and we cannot be certain that we or our distributors will receive regulatory approvals
required for C-Scan or any future products in each country or region in which we plan to market such products. With regard to obtaining
the CE-certification, we have engaged with Dekra Certification as our Notified Body. If we modify C-Scan or any future products, we or
our distributors may need to apply for new regulatory approvals or our Notified Body may need to review the planned changes before we
are permitted to sell the respective products. We may not meet the quality and safety standards required to maintain the authorizations
that we or our distributors have received. If we or if permitted, our distributors are unable to maintain our authorizations or CE Certificates
in a particular country or region, we will no longer be able to sell C-Scan and/or any potential future products in that country or region,
and our ability to generate revenues will be materially and adversely affected.
If the indications for use or instructions for use for which the
iodinated oral contrast medium is approved are not sufficiently broad to support its use throughout the C-Scan procedure, the FDA or the
competent regulatory authorities in the European Union (EU) Member States and other foreign countries may consider that contrast agent
is being used off-label.
Ingestion of C-Scan requires the preparatory use
of iodinated oral contrast medium to provide a coating for colonic imaging. We cannot be sure that the indications for which iodinated
oral contrast medium are approved in the United States, or in other countries is sufficiently broad to cover such use. If the FDA or the
competent regulatory authorities in other countries consider that iodinated oral contrast medium is not approved for the purpose for which
it is used with the system, we may be considered to promote the off-label use of the iodinated oral contrast medium. Because the promotion
of off-label use of drugs or medicinal products is prohibited in the United States, and in other countries, we could face both related
issues with the FDA and/or the competent authorities of and/or other countries. In these circumstances, the FDA and/or the competent regulatory
authorities and/or other countries may require us to obtain appropriate regulatory approvals for the iodinated oral contrast medium prior
to marketing C-Scan with such substances. Under such circumstances, should we fail to obtain approval of the contrast agent for use with
C-Scan, in a timely fashion, or at all, this could delay or prevent regulatory clearance or approval of the C-Scan, and our business and
financial condition will be adversely affected.
If we are unable to successfully complete clinical trials with respect
to C-Scan, we may be unable to receive regulatory approvals or clearances for C-Scan and/or our ability to achieve market acceptance of
C-Scan will be harmed.
The development of the C-Scan system requires the
submission of data generated from clinical trials, which can be long, expensive and uncertain processes, subject to delays and failure
at any stage. The data obtained from the studies and trials may be inadequate to support regulatory clearances or approvals, or equivalent
third-country approval, or to allow market acceptance of the products being studied. C-Scan technology is currently undergoing clinical
development and clinical trials. To date, we have performed clinical studies with several versions of C-Scan including several versions
of our non-scanning capsules.
The development of sufficient and appropriate clinical
protocols to demonstrate safety, clinical performance and clinical effectiveness are required, and we may not adequately develop such
protocols to support clearance, approval, or equivalent third country approval. The clinical trials that were conducted using prior versions
of C-Scan, were conducted under differing protocols, used groups of patients different and/or smaller in size from those we intend to
study in future clinical trials with future versions of C-Scan, and the results from such clinical trials were subject to human interpretations
and based on statistical assumptions that could be affected by erroneous considerations. Further, the FDA, the competent regulatory authorities
of other countries, or our Notified Body in the EU may require us to submit data on a greater number of patients than we originally anticipated
and/or for a longer follow-up period or they may change the data collection requirements or data analysis applicable to our clinical trials.
The commencement or completion of any of our clinical
studies or trials may be delayed or halted, or be inadequate to support regulatory clearance, approval or product acceptance, or equivalent
third country approval, for numerous reasons, including, among others:
|
• |
patients do not enroll in the clinical trial at the rate we expect; |
|
• |
patients do not comply with trial protocols; |
|
• |
patient follow-up is not at the rate we expect; |
|
• |
patients withdraw from the study; |
|
• |
patients experience severe adverse side effects, including damage to the colon wall or related to excessive
radiation exposure as a result of capsule malfunction or break down or retention and may require to undergo a surgical procedure;
|
|
• |
patient death during a clinical trial, even though their death may be unrelated to our product; |
|
• |
FDA, institutional review boards, or IRBs, or other regulatory authorities do not approve a clinical trial
protocol or a clinical trial, or place a clinical trial on hold; |
|
• |
IRBs, Ethics Committees and third-party clinical investigators may delay or reject our trial protocol and
Informed Consent Form; |
|
• |
third-party clinical investigators decline to participate in a study or trial or do not perform a study
or trial on our anticipated schedule or consistent with the investigator agreements, study or trial protocol, good clinical practices,
quality of clinical data or other FDA or IRBs, Ethics Committees, or any other applicable requirements; |
|
• |
third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate
manner or consistent with the study or trial protocol or investigational or statistical plans; |
|
• |
regulatory inspections of our studies, trials or manufacturing facilities may require us to, among other
things, undertake corrective action or suspend or terminate our studies or clinical trials; |
|
• |
changes in governmental regulations or administrative actions; |
|
• |
we may not be able to develop C-Scan at the rate or to the stage we desire; |
|
• |
the interim or final results of the study or clinical trial are inconclusive or unfavorable as to safety
or efficacy; |
|
• |
a regulatory agency or our Notified Body concludes that our trial design is or was inadequate to demonstrate
safety and efficacy; |
|
• |
the capsule disposal was not authorized by regulatory agencies or patients failed to collect the capsule
following procedure completion; |
|
• |
the capsule was lost in the sewer system; |
|
• |
loss of clinical data; and |
|
• |
a health epidemic, pandemic or other outbreak, including the current COVID-19 pandemic. |
The results of non-clinical and clinical studies
do not necessarily predict future clinical trial results, and predecessor clinical trial results may not be repeated in subsequent clinical
trials. Additionally, the FDA and/or other third country regulatory entities may disagree with our interpretation of the data from our
non-clinical studies and clinical trials, or may find the clinical trial design, conduct or results inadequate to demonstrate safety or
efficacy, and may require us to pursue additional non-clinical studies or clinical trials, which could further delay the clearance, approval
of our products. The data we collect from our non-clinical testing, and other clinical trials may not be sufficient to support regulatory
clearance and approval.
If the third parties on which we rely to conduct our clinical trials
and clinical development do not perform as contractually required or expected, we may not be able to obtain regulatory clearance or approval,
or equivalent third country approval for, or commercialize, C-Scan or future products.
We do not have the ability to independently conduct
our clinical trials for C-Scan and we must rely on third parties, such as contract research organizations, medical institutions, clinical
investigators and contract laboratories to conduct such trials. If these third parties do not successfully carry out their contractual
duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy
of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other
reasons, our non-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not
be able to obtain regulatory clearance, approval for, or successfully commercialize, C-Scan or future products on a timely basis, if at
all, and our business, operating results and prospects may be adversely affected. Furthermore, our third-party clinical trial investigators
may be delayed in conducting our clinical trials for reasons outside of their control.
The results of our current or future clinical trials may not support
our product candidate requirements or intended use claims or may result in the discovery of adverse side effects.
Even if our current or future clinical trials are
completed as planned, we cannot be certain that their results will support our product requirements or intended use claims, which could
inhibit our marketing strategies, or that the FDA, foreign authorities or our Notified Body will agree with our conclusions regarding
them. Success in non-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we
cannot be sure that clinical trials will replicate the results of prior trials. The clinical trial process may fail to demonstrate that
C- Scan, or any future products, are safe and effective for the desired or proposed indicated uses, which could cause us to abandon a
product and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions
and, ultimately, our ability to commercialize C-Scan, or any future products, and generate revenues. It is also possible that patients
enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile.
If we fail to obtain or maintain necessary regulatory clearances
or CE Certificates for C-Scan, or if there are regulatory changes in our existing or future target markets, our ability to sell C-Scan
and generate revenues could be harmed.
C-Scan is a medical device that is subject to extensive
regulations that are in particular intended to assure its safety, effectiveness and compliance with applicable consumer laws. These laws
require medical devices to obtain approval or other market clearance before being placed on the market. If we fail to obtain or maintain
these regulatory approvals or clearances, our ability to sell C-Scan and generate revenues will be materially harmed.
These laws and regulations relate inter
alia to the design, development, testing, manufacturing, storage, labeling, packaging, content and language of the instructions
for use of the device, sale, promotion, distribution, importing and exporting, shipping, post-sale surveillance and recall from C-Scan’s
markets, and all countries in which we intend to sell C-Scan apply some form of regulations of this kind. Most notably, we must comply
with the EU law on medical devices, in particular, the European Medical Device Regulation (MDR) and EU Member States’ laws implementing
and complementing it, and other applicable EU and Member States’ law, and we are subject to extensive regulation in the United States
by the FDA and other federal, state and local authorities.
We are and will be subject to audits by our Notified
Body under the MDR. During this audit, the third-party assessor or Notified Body will examine the maintenance and implementation of our
quality control system, device post-marketing vigilance system and any changes or modifications made to our products.
On May 26, 2021, the MDR became formally applicable
and replaced the former regulatory framework for medical devices in the EU under the medical devices directives. The MDR strengthens the
medical devices rules in the EU. In particular, the MDR results in several medical devices being classified in higher risk classes and
therefore face elevated regulatory requirements. In addition, the MDR generally elevates regulatory requirements to medical devices. As
a result, it has become more difficult to market medical devices and costs incurred for clinical evaluation, conformity assessment and
post marketing surveillance have increased and will increase further; such regulatory changes may adversely affect our business, financial
condition and results of operations or restrict our operations.
Even if C-Scan or future products are cleared or approved by regulatory
authorities or after obtaining CE Certificates from our Notified Body, modifications to C-Scan or future products may require new regulatory
clearances or approvals, new CE Certificates, or may require us to recall or cease marketing it until the necessary clearances, approvals
or CE Certificates are obtained.
Once cleared, approved or marketed, modifications
to C-Scan or future products may require new regulatory approvals, clearances, including CE Certificates from our Notified Body, 510(k)
clearances or premarket approvals, or require us to recall or cease marketing the modified devices until these clearances or approvals
are obtained. Any modification to a 510(k)-cleared device that could significantly affect its safety or efficacy, or that would constitute
a major change in its intended use, requires a new 510(k) clearance or, possibly, a new de novo classification petition or PMA. The FDA
requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval,
supplement or clearance. A manufacturer may determine that a modification could not significantly affect safety or efficacy and does not
represent a major change in its intended use, so that no new 510(k) clearance is necessary. However, the FDA can review a manufacturer’s
decision and may disagree. The FDA may also on its own initiative determine that a new clearance or approval is required. Subject to compliance
with FDA guidance we may make modifications to C-Scan in the future that we believe do not or will not require additional clearances or
approvals. Further, our products could be subject to recall if the FDA determines, for any reason, that our products are not safe or effective.
Any recall or FDA requirement that we seek additional approvals or clearances could result in significant delays, fines, increased costs
associated with modification of a product, loss of revenue and potential operating restrictions imposed by the FDA.
If a manufacturer determines that a modification
to an FDA-cleared device could significantly affect its safety or efficacy, or would constitute a major change in its intended use, then
the manufacturer must file for a new 510(k) clearance or possibly a new de novo classification petition or premarket approval application.
Where we determine that modifications to our products require a new clearance or approval, we may not be able to obtain those additional
clearances or approvals for the modifications or additional indications in a timely manner, or at all.
Any modification to a PMA-approved device that
affects the device’s safety or effectiveness must either be approved in a PMA Supplement, or if the modification does not impact
the device’s safety or effectiveness, described in a 30-Day Notice or in the device’s Annual Report. The FDA may not approve
a modification described in a PMA Supplement, in which case the modified device cannot be marketed. The FDA can also disagree that a change
described in an Annual Report is appropriately described in either filing, and request that the company file a PMA Supplement and/or request
that the company cease marketing the modified device until the PMA Supplement is approved.
Similar rules also apply in foreign jurisdictions.
In the EU, we must inform the Notified Body that was involved in the conformity assessment of the medical devices we market or sell in
the EU of any planned substantial changes to our quality system with regard to C-Scan or other devices we may place on the market, if
such changes could affect compliance with the essential requirements laid down in the Annexes to the MDR (Essential Requirements), respectively,
or changes to the devices’ intended purpose. The Notified Body will then assess the changes and verify the products’ conformity
with the Essential Requirements. If the assessment is favorable, the Notified Body will issue a new CE Certificate or an addendum to the
existing CE Certificate attesting compliance with the Essential Requirements.
If the Notified Body or a relevant regulatory authority
disagrees with our assessments and repeals an authorization or an existing CE Certificate, we may be required to apply for a new CE Certificate
or other new regulatory clearances or approvals for modifications, and until such new CE-Certificate or other regulatory clearance or
change approval is obtained, we may be required to recall and/or to stop marketing the modified devices.
Obtaining clearances and approvals, or new or amended
CE Certificates for device modifications can be a time-consuming process, and delays in obtaining required future clearances, approvals,
or CE Certificates could adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn could harm
our future growth.
Even if C-Scan and future products are cleared
or approved by regulatory authorities or after obtaining CE Certificates from our Notified Body, if we or our suppliers fail to comply
with ongoing FDA or other foreign regulatory authority requirements, or if we experience unanticipated problems with our products, our
products could be subject to restrictions or withdrawal from the market.
The manufacturing processes, reporting requirements,
post-approval clinical data and promotional activities associated with any product for which we obtain clearance, approval or CE Certificates,
or equivalent third country approval will be subject to continuous regulatory review, oversight and periodic inspections by the FDA other
domestic and foreign regulatory authorities and our Notified Body. In particular, we and certain of our suppliers are required to comply
with the FDA’s Quality System Regulations, or QSR, as well as current good manufacturing practices, or cGMP. In the EU, we will
also be subject to the quality management system requirements laid down in the Annexes to the MDR. Such compliance can be facilitated
by, a certificate of compliance with the current version of the ISO 13485 (which, at this point, is ISO 13485:2016). Through compliance
with the ISO 13485:2016 standard, we will benefit from a presumption of conformity with the relevant quality management system requirements
laid down in the Annexes to the MDR. These regulations and standards govern inter alia the methods and documentation of the design, testing,
production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain clearance or approval,
CE Certificates, or equivalent third country approval. Regulatory authorities, such as the FDA, and our Notified Body enforce the QSR
and other regulations through periodic inspections. For example, the failure by us or one of our suppliers to comply with applicable statutes
and regulations falling within the competence of the FDA, or the failure to timely and adequately respond to any adverse inspectional
observations or product safety issues, could result in, among other things, any of the following enforcement actions:
|
• |
untitled letters, warning letters, fines, injunctions, corporate integrity agreements, consent decrees
and civil penalties; |
|
• |
unanticipated expenditures to address or defend such actions; |
|
• |
customer notifications for repair, replacement or refunds; |
|
• |
recall, detention or seizure of our products; |
|
• |
operating restrictions or partial suspension or total shutdown of production; |
|
• |
refusing or delaying our requests for 510(k) clearance, de novo classification or premarket approval of
new products or modified products; |
|
• |
operating restrictions; |
|
• |
withdrawing 510(k) clearances, de novo classification, or PMA approvals that have already been granted;
|
|
• |
suspension or withdrawal of our CE Certificates; |
|
• |
refusal to grant export approval for our products; or |
|
• |
civil or criminal prosecution. |
If any of these actions were to occur, our reputation
would be harmed, our product sales and profitability would suffer and we may not be able to generate revenue. Furthermore, our key suppliers
may not currently be or may not continue to be in compliance with all applicable regulatory requirements which could result in our failure
to produce our products on a timely basis and in the required quantities, if at all.
Even if regulatory clearance or approval of a product
is granted, or after obtaining CE Certificates, such clearance or approval, or CE Certificates may be subject to limitations on the intended
uses for which the product may be marketed and reduce our potential to successfully commercialize the product and generate revenue from
the product. If FDA or the competent regulatory authorities of foreign countries determines that our promotional materials, labeling,
training or other marketing or educational activities constitute the promotion of an unapproved use or the promotion of an intended purpose
not covered by our CE mark, they could request that we cease or modify our training or promotional materials or subject us to regulatory
enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider
our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties
under other statutory authorities, such as laws prohibiting false claims for reimbursement.
In addition, we may be required to conduct costly
post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with medical device reporting
requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown
problems with our products, including unanticipated adverse side effects or adverse side effects of unanticipated severity or frequency,
manufacturing problems, or failure to comply with regulatory requirements such as the Quality System Regulations, may result in changes
to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory
recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension or
withdrawal of regulatory approvals or CE Certificates, product seizures, injunctions or the imposition of civil or criminal penalties,
all of which would adversely affect our business, financial condition and operating results and prospects.
C-Scan may in the future be subject to product recalls that could
harm our reputation, business and financial results.
The FDA and similar foreign governmental authorities
have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture
or a public health/safety issue. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there
is a reasonable probability that the device would cause injury or death. In addition, foreign governmental bodies have the authority to
require the recall of our products in the event of material deficiencies or defects in design or manufacture. Manufacturers may, under
their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us
or one of our distributors could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies
and issues. Once marketed, recalls of any of our products, including C-Scan, would divert managerial and financial resources and have
an adverse effect on our business, financial condition and results of operations. FDA requires that certain classifications of recalls
be reported to the FDA within 10 working days after the recall is initiated. Companies are required to maintain certain records of recalls,
even if they are not reportable to FDA. We may initiate voluntary recalls involving our products in the future that we determine do not
require us to notify the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls.
A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take
enforcement action against us based on our failure to report the recalls when they were conducted.
If C-Scan or future products cause or contribute to a death or a
serious injury, or malfunction in such a way that causes or contributes to a death or serious injury, we will be subject to medical device
reporting regulations, which can result in corrective actions or enforcement actions from regulatory authorities.
Under FDA medical device reporting regulations,
medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death
or serious injury or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction
of our device (or any similar future product) were to recur. If we fail to investigate and report these events to FDA within the required
timeframes, or at all, the FDA could take enforcement action against us. Any such adverse event involving our products also could result
in future corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any
corrective action, whether voluntary or involuntary, including any legal action taken against us, will require us to devote sufficient
time and capital to the matter, distract management from operating our business, and may harm our reputation and financial results.
Under the EU MDR we will now have to comply with
the increased requirements of medical devices vigilance provisions. In particular, according to Art. 83 MDR, we are required to have a
post-market surveillance system in place in a manner that is proportionate to the risk class and appropriate for the type of device, as
an integral part of the quality management system. Depending on the devices’ risk class, either a post market surveillance report
or a periodic safety update report, or PSUR, must be created and provided to the competent authority upon request. The PSUR also has to
be provided to the Notified Body. Furthermore, requirements on vigilance and incident reporting are increased and market surveillance
competencies of the authorities are strengthened.
Our failure to comply with radiation safety or radio frequency regulations
in a specific country or region could impair our ability to conduct our clinical trials, or commercially distribute and market C-Scan
in that country or region.
C-Scan includes a small X-ray source and wireless
radio frequency transmitter and receiver, and is therefore subject to equipment authorization requirements in a number of countries and
regions. In the United States, the EU and Japan, authorities often require advance clearance of all radiation and radio frequency devices
before they can be sold or marketed in these jurisdictions, subject to limited exceptions. Competent authorities for such additional approval
requirements include the Swiss National Cooperative for the Disposal of Radioactive Waste (Nagra) and the German Federal Office for Radiation
Protection (Bundesamt für Strahlenschutz, BfS). Modifications to the approved C-Scan version design and specifications may require
new or further regulatory clearances or approvals before we are permitted to market and sell a modified C-Scan version. If we are unable
to obtain any required clearances or approvals from the authorities responsible for the radiation as well as the radio frequency regulations
in these and other jurisdictions, the sale or use of C-Scan could be prevented in these countries. Any such action could negatively affect
our business, financial condition and results of operations.
Our business is subject to complex environmental and health legislation
in various jurisdictions that may increase our costs and our risk of noncompliance.
Our research and development and manufacturing
processes, including through our service providers, involve the handling of potentially harmful radioactive and other hazardous materials.
Therefore, we and our service providers may be subject to various environmental, health and safety laws and regulations, including governing
the use, shipping, handling, storage and disposal of these materials, and we incur expenses related to compliance with these laws and
regulations. If we are found to have violated applicable environmental, health and safety laws, whether as a result of human error, equipment
failure or other causes, we could be held liable for damages, penalties and costs of remedial actions and could be subject to work stoppages
or delays, which could materially adversely affect our business, financial condition and results of operations. The risk of contamination
or injury from these materials cannot be eliminated. If an accident or release of any radioactive or other hazardous material occurs,
we could be held liable for resulting damages, including for investigation, remediation and monitoring of the contamination, including
the costs of which could be substantial. In addition, we may be required to pay damages or civil judgments in respect of third-party claims,
including those relating to personal injury (including exposure to radioactive materials) or contribution claims. In the future, we could
be subject to additional environmental requirements or existing environmental laws could become more stringent, which could lead to greater
compliance costs and increasing risks and penalties associated with violations. For example, changes to, or restrictions on, permitting
requirements or processes, hazardous or radioactive material storage or handling might require an unplanned capital investment or relocation.
If we or our service providers fail to comply with existing or new environmental laws or regulations, our business, financial condition
and results of operations could be materially adversely affected.
If we are unable to achieve reimbursement and coverage from government
and private third-party payors for procedures using C-Scan, or if reimbursement is insufficient to create an economic benefit for purchasing
or using C-Scan when compared to alternative procedures, demand for our products may not grow at the rate we expect.
The demand for C-Scan will depend significantly
on the eligibility of the procedures performed using C-Scan for reimbursement through government-sponsored healthcare payment systems
and private third-party payors. Reimbursement practices vary significantly from country to country and within some countries, by region,
and we must obtain reimbursement approvals on a country-by-country and/or region-by-region basis. In general, the process of obtaining
reimbursement and coverage approvals has been longer outside of the United States. We may not be able to obtain reimbursement approvals
in a timely manner or at all and existing reimbursement and coverage policies may be revised from time to time by government and private
third-party payors. If physicians, hospitals and other healthcare providers are unable to obtain sufficient coverage and reimbursement
from government and private third-party payors for procedures using C-Scan, if reimbursement is, or is perceived by our customers to be,
insufficient to create an economic incentive for purchasing or using C-Scan, or if such reimbursement does not adequately compensate physicians
and health care providers compared to the other procedures they offer, demand for our products may not grow at the rate we expect.
Federal and state privacy laws, and equivalent laws of third countries,
may increase our costs of operation and expose us to civil and criminal sanctions.
The Health Insurance Portability and Accountability
Act of 1996, as amended, and the regulations that have been issued under it, to which we refer collectively as HIPAA, and similar laws
outside the United States, contain substantial restrictions and requirements with respect to the use and disclosure of individuals’
protected health information. The HIPAA privacy rules prohibit “covered entities,” such as healthcare providers and health
plans, from using or disclosing an individual’s protected health information, unless the use or disclosure is authorized by the
individual or is specifically required or permitted under the privacy rules. Under the HIPAA security rules, covered entities must establish
administrative, physical and technical safeguards to protect the confidentiality, integrity and availability of electronic protected health
information maintained or transmitted by them or by others on their behalf. While we do not believe that we are a covered entity under
HIPAA, many of our customers may be covered entities subject to HIPAA. Such customers may require us to enter into business associate
agreements, which will obligate us to safeguard certain health information we obtain in the course of our relationship with them, restrict
the manner in which we use and disclose such information and impose liability on us for failure to meet our contractual obligations.
In addition, under The Health Information Technology
for Economic and Clinical Health Act of 2009, or HITECH, which was signed into law as part of the U.S. stimulus package in February 2009,
certain of HIPAA’s privacy and security requirements are now also directly applicable to “business associates” of covered
entities and subject them to direct governmental enforcement for failure to comply with these requirements. We may be deemed as a “business
associate” of some of our customers. As a result, we may be subject as a “business associate” to civil and criminal
penalties for failure to comply with applicable privacy and security rule requirements. Moreover, HITECH created a new requirement obligating
“business associates” to report any breach of unsecured, individually identifiable health information to their covered entity
customers and imposes penalties for failing to do so.
In addition to HIPAA, most U.S. states have enacted
patient confidentiality laws that protect against the disclosure of confidential medical information, and many U.S. states have adopted
or are considering adopting further legislation in this area, including privacy safeguards, security standards, and data security breach
notification requirements. These U.S. state laws, which may be even more stringent than the HIPAA requirements, are not preempted by the
federal requirements, and we are therefore required to comply with them to the extent they are applicable to our operations.
These and other possible changes to HIPAA or other
U.S. federal or state laws or regulations, or comparable laws and regulations in countries where we conduct business, could affect our
business and the costs of compliance could be significant. Failure by us to comply with any of the standards regarding patient privacy,
identity theft prevention and detection, and data security may subject us to penalties, including civil monetary penalties and in some
circumstances, criminal penalties. In addition, such failure may damage our reputation and adversely affect our ability to retain customers
and attract new customers.
The protection of personal data, particularly patient
data, is subject to strict laws and regulations in many countries. The collection and use of personal health data in the EU is governed
by the General Data Protection Regulation Reg. EU 2016/679, which became applicable on May 25, 2018, or the GDPR. The GDPR imposes a number
of requirements, including an obligation to seek the consent of individuals to whom the personal data relate, the information that must
be provided to the individuals, notification of data processing obligations to the competent national data protection authorities of individual
EU Member States and the security and confidentiality of the personal data. The GDPR also imposes strict rules on the transfer of personal
data out of the EU to the U.S. Failure to comply with the requirements of the GDPR and the related national data protection laws of the
EU Member States may result in fines and other administrative penalties and harm our business. We may incur extensive costs in ensuring
compliance with these laws and regulations, particularly if we are considered to be a data controller within the meaning of the GDPR.
The adoption of healthcare reform and deficit reduction measures
in the United States may adversely affect our business and financial results.
The Patient Protection and Affordable Care Act
of 2010, or the PPACA, which was modified on March 30, 2010 by the enactment of the Health Care and Education Reconciliation Act of 2010,
affects the way healthcare is financed by both governmental and private insurers, and significantly impacts the device industry. The PPACA
is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud
and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical
and medical device manufacturers, and impose additional health policy reforms. The PPACA imposes, among other things, an annual excise
tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States beginning in 2013, resulting
in an anticipated cost to the medical device industry of up to $20 billion over the next decade. We likely will be subject to the excise
tax with respect to C-Scan if it is approved for sale in the United States. The PPACA also limits the rate of growth in Medicare payments
to providers and authorizes certain voluntary demonstration projects beginning no later than 2013 around development of bundling payments
for acute, inpatient hospital services, physician services, and post-acute services for episodes of hospital care. In addition, the PPACA
provides for the establishment of an Independent Payment Advisory Board, or IPAB, that, beginning in 2014, could recommend changes in
Medicare payments to physicians and other providers that would take effect unless Congress passes an alternative measure to achieve the
same amount of savings. The IPAB has not yet been created. The PPACA also increases fraud and abuse penalties and expands the scope and
reach of the Federal Civil False Claims Act and government enforcement tools, which may adversely impact healthcare companies.
There have been judicial and congressional challenges
to the PPACA. The U.S. Supreme Court heard a constitutional challenge to the PPACA and in June 2012 held that the PPACA is constitutional.
However, states are allowed to opt out of the expansion of eligibility criteria for Medicaid under the PPACA and many states have chosen
to do so, causing many uninsured patients to remain without coverage. In addition to the PPACA, the effect of which cannot presently be
quantified given its recent enactment, various healthcare reform proposals have also emerged at the state level. If a law is enacted,
many if not all of the provisions of the PPACA may no longer apply to prescription drugs. While we are unable to predict what changes
may ultimately be enacted, to the extent that future changes affect how any future products are paid for and reimbursed by government
and private payers our business could be adversely impacted. On December 14, 2018, a federal district court in Texas ruled that the PPACA
is unconstitutional as a result of the Tax Cuts and Jobs Act, the federal income tax reform legislation previously passed by Congress
and signed by President Trump on December 22, 2017, that eliminated the individual mandate portion of the PPACA. The case, Texas, et al,
v. United States of America, et al., (N.D. Texas), is an outlier, but in 2019, the Fifth Circuit Court of Appeals subsequently upheld
the lower court decision which was then appealed to the United States Supreme Court. On June 17, 2021, the U.S. Supreme Court held that
the plaintiffs did not have standing to challenge the individual mandate because they have not shown a past or future injury. In November
2020, Joseph Biden was elected President and, in January 2021, the Democratic Party obtained control of the Senate. As a result of these
electoral developments, it is unlikely that continued legislative efforts will be pursued to repeal PPACA. Instead, it is possible that
legislation will be pursued to enhance or reform PPACA. We are not able to state with any certainty what will be the impact of this court
decision on our business pending further court action and possible appeals.
We cannot predict whether future healthcare initiatives
will be implemented at the federal or state level or the effect any future legislation or regulation will have on us. However, we anticipate
that the PPACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria
and an additional downward pressure on the price that we receive for any approved product, and could adversely affect our business. Any
reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors.
Insurers may also refuse to provide any coverage of uses of approved products for medical indications other than those for which the FDA
has granted market approvals, all of which may adversely affect our business, financial condition and results of operations, possibly
materially.
In addition, other legislative changes have been
proposed and adopted since the PPACA was enacted. In August 2011, President Obama signed into law the Budget Control Act of 2011, which,
among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions.
The Joint Select Committee did not achieve a targeted deficit reduction of an amount greater than $1.2 trillion for the years 2013 through
2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare
payments to healthcare providers of up to 2.0% per fiscal year, starting in 2013. In January 2013, President Obama signed into law the
American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several categories of healthcare providers
and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. If we
ever obtain regulatory approval and commercialization of C-Scan or any future product candidates, these laws may result in additional
reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our
financial operations. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and
promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether
the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of C-Scan
or any future product candidates may be.
Although we cannot predict the full effect on our
business of the implementation of existing legislation or the enactment of additional legislation pursuant to healthcare and other legislative
reform, we believe that legislation or regulations that would reduce reimbursement for, or restrict coverage of, C-Scan or any future
product candidates, could adversely affect how much or under what circumstances healthcare providers will prescribe or administer our
products. This could materially and adversely affect our business by reducing our ability to generate revenue, raise capital, obtain additional
collaborators and market C-Scan or any future product candidates. In addition, we believe the increasing emphasis on managed care in the
United States has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact any
future product sales.
In addition to healthcare reform, other deficit
reduction measures could affect reimbursement for our device and related procedures. For example, beginning April 1, 2013, Medicare payments
for all items and services have been reduced by 2% under the sequestration (i.e., automatic spending reductions) required by the Budget
Control Act of 2011, as amended by the American Taxpayer Relief Act of 2012. These cuts will remain in effect until 2024 unless Congress
enacts legislation to cancel or delay the cuts. These payment reductions, or similar efforts to reduce Medicare spending to control the
federal deficit, could adversely affect our business by reducing reimbursement to the providers who purchase and use our devices and perform
related procedures.
The implementation of the reporting and disclosure
obligations of the Physician Payment Sunshine Act’s provisions relating to healthcare reform could adversely affect our business.
A health care reform provision, generally referred to as the Physician Payment Sunshine Act or Open Payments Program, has imposed new
reporting and disclosure requirements for drug and device manufacturers with regard to payments or other transfers of value made to certain
practitioners (including physicians, dentists and teaching hospitals), and for such manufacturers and for group purchasing organizations,
with regard to certain ownership interests held by physicians in the reporting entity. On February 1, 2013, the Centers for Medicare and
Medicaid Services, or CMS, released the final rule to implement the Physician Payment Sunshine Act. As required under the Physician Payment
Sunshine Act, CMS will publish information from these reports on a publicly available website, including amounts transferred and physician,
dentist and teaching hospital identities.
The final rule implementing the Physician Payment
Sunshine Act is complex, ambiguous, and broad in scope. If we participate in federal healthcare programs, meaning our product is reimbursed
by a federal healthcare program such as Medicare, Medicaid, or Children’s Health Insurance Program, our product would be considered
a “covered device.” Within 180 days of becoming “covered,” we would be required to collect and report detailed
information regarding certain financial relationships we have with physicians and teaching hospitals. The Physician Payment Sunshine Act
preempts similar state reporting laws, although we may be required to report under certain of such state laws. Our compliance with the
new final rule imposes additional costs on us and requires additional resources including dedicated personnel with experience and expertise
in this area. Failure to comply may expose us to federal and/or state enforcement action and fines.
If we fail to comply with the U.S. federal
Anti-Kickback Statute and similar state and third-country laws, we could be subject to criminal and civil penalties and exclusion from
federally funded healthcare programs including the Medicare and Medicaid programs and equivalent third-country programs, which would have
a material adverse effect on our business and results of operations.
A provision of the Social Security Act, commonly
referred to as the federal Anti-Kickback Statute, prohibits the knowing and willful offer, payment, solicitation or receipt of any form
of remuneration, directly or indirectly, in cash or in kind, to induce or reward the referring, ordering, leasing, purchasing or arranging
for, or recommending the ordering, purchasing or leasing of, items or services payable, in whole or in part, by Medicare, Medicaid or
any other federal healthcare program. PPACA, among other things, clarified that a person or entity needs not to have actual knowledge
of the federal Anti-Kickback Statute or specific intent to violate it. Although there are a number of statutory exemptions and regulatory
safe harbors to the federal Anti- Kickback Statute protecting certain common business arrangements and activities from prosecution or
regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exemption or
safe harbor may be subject to scrutiny. The federal Anti-Kickback Statute is very broad in scope and many of its provisions have not been
uniformly or definitively interpreted by existing case law or regulations. In addition, most of the states have adopted laws similar to
the federal Anti-Kickback Statute, and some of these laws are even broader than the federal Anti-Kickback Statute in that their prohibitions
may apply to items or services reimbursed under Medicaid and other state programs or, in several states, apply regardless of the source
of payment. Violations of the federal Anti-Kickback Statute may result in substantial criminal, civil or administrative penalties, damages,
fines and exclusion from participation in federal healthcare programs.
All of our financial relationships with healthcare
providers, purchasers, and others who provide products or services to federal healthcare program beneficiaries are potentially governed
by the federal Anti-Kickback Statute and similar state laws. We believe our operations are in compliance with the federal Anti-Kickback
Statute and similar state laws. However, we cannot be certain that we will not be subject to investigations or litigation alleging violations
of these laws, which could be time-consuming and costly to us and could divert management’s attention from operating our business,
which in turn could have a material adverse effect on our business. In addition, if our arrangements were found to violate the federal
Anti- Kickback Statute or similar state laws, the consequences of such violations would likely have a material adverse effect on our business,
results of operations and financial condition.
There are other federal and state laws that may
affect our ability to operate, including the federal civil False Claims Act, which prohibits, among other things, individuals or entities
from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly making,
using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly
concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. PPACA
amended the Social Security Act to provide that the government may assert that a claim including items or services resulting from a violation
of the federal Anti- Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Moreover,
we may be subject to other federal false claim laws, including, among others, federal criminal healthcare fraud and false statement statutes
that extend to non-government healthcare benefit programs. Moreover, there are analogous state laws. Violations of these laws can result
in substantial criminal, civil or administrative penalties, damages, fines and exclusion from participation in federal healthcare programs.
Similar restrictions are imposed by the national
legislation of many third countries in which our medical devices will be marketed. Moreover, the provisions of the Foreign Corrupt Practices
Act of 1997 and other similar anti-bribery laws in other jurisdictions generally prohibit companies and their intermediaries from providing
money or anything of value to officials of foreign governments, foreign political parties, or international organizations with the intent
to obtain or retain business or seek a business advantage. Recently, there has been a substantial increase in anti-bribery law enforcement
activity by U.S. regulators, with more aggressive and frequent investigations and enforcement by both the U.S. Securities and Exchange
Commission and the Department of Justice. A determination that our operations or activities violated United States or foreign laws or
regulations could result in imposition of substantial fines, interruption of business, loss of supplier, vendor or other third-party relationships,
termination of necessary licenses and permits, and other legal or equitable sanctions. In addition, lawsuits brought by private litigants
may also follow as a consequence.
Our failure to comply with the necessary regulatory approval regarding
the use of radioactive materials could significantly impair our ability to develop, manufacture and/or sell C-Scan.
The manufacture of C-Scan requires the use and
storage of radioactive materials. In order to use such materials in the development and manufacture of C-Scan in Israel, we are required
to obtain a permit from the Israeli Commissioner for Environmental Radiation, or the Commissioner, pursuant to the Israeli Pharmaceutical
Regulations (Radioactive Elements and By-Products), 5740–1980. Should we fail to comply with the conditions of our currently existing
permit, the Commissioner would have authority to cancel our permit. Should the Commissioner determine that our activities or facilities,
or the activities or facilities adjacent to our premises, constitute a danger to the health and well-being of a person, the public or
the environment, the cancellation or suspension of our permit could be immediate and without prior notice. Furthermore, we cannot guarantee
the annual renewal of our permit and/or annual renewal subject to identical conditions, as the approval of an annual application and the
conditions thereof are at the discretion of the Commissioner. Similar requirements and regulations may apply to the manufacture of C-Scan
in other countries. Cancellation of or failure to renew our permit could have materially adverse consequences on our ability to manufacture
and sell our products and therefore on our ability to continue our business and operations.
Environmental, social and corporate governance
(ESG) issues, including those related to climate change and sustainability, may have an adverse effect on our business, financial condition
and results of operations and damage our reputation.
There is an increasing focus from certain investors,
customers, consumers, employees and other stakeholders concerning ESG matters. Additionally, public interest and legislative pressure
related to public companies’ ESG practices continue to grow. If our ESG practices fail to meet regulatory requirements or investor,
employee or other stakeholders’ evolving expectations and standards for responsible corporate citizenship in areas including environmental
stewardship, support for local communities, Board of Director and employee diversity, human capital management, employee health and safety
practices, product quality, supply chain management, corporate governance and transparency, our reputation and employee retention may
be negatively impacted, and our suppliers may be unwilling to continue to do business with us.
Investors and other stakeholders are increasingly
focusing on environmental issues, including climate change, energy and water use, plastic waste and other sustainability concerns. Concern
over climate change may result in new or increased legal and regulatory requirements to reduce or mitigate impacts to the environment.
Increased regulatory requirements may result in increased demands or requirements regarding components of our products and their environmental
impact on sustainability. Complying with these demands or requirements could cause us to incur additional manufacturing, operating or
product development costs.
In addition, new sustainability rules and regulations
have been adopted and may continue to be introduced in various states and other jurisdictions. For example, the SEC has published proposed
rules that would require companies to provide significantly expanded climate-related disclosures in their periodic reporting, which may
require us to incur significant additional costs to comply and impose increased oversight obligations on our management and board of directors.
If we do not adapt to or comply with new regulations,
or fail to meet evolving investor, industry or stakeholder expectations and concerns regarding ESG issues, investors may reconsider their
capital investment in our Company, which could have a material adverse effect on our business or financial condition.
Risks Related to Our
Intellectual Property
If we are unable to protect our intellectual property rights, our
competitive position could be harmed.
Our success and ability to compete depends in large
part upon our ability to protect our intellectual property. Although we have patents issued in Israel, Europe, United States, Japan, China,
India, Hong Kong, Canada, South Korea, Brazil and Australia, we continue to file and prosecute in many of the same countries and additional
countries. We face several risks and uncertainties in connection with our intellectual property rights, including, among others:
|
• |
pending and future patent applications may not result in the issuance of patents or, if issued, may not
be issued in a form that will be advantageous to us; |
|
• |
our issued patents may be challenged, invalidated or legally circumvented by third parties; |
|
• |
our patents may not be upheld as valid and enforceable or prevent the development of competitive products;
|
|
• |
the eligibility of certain inventions related to diagnostic medicine, more specifically diagnostic methods
and processes, for patent protection in the United States has been limited recently which may affect our ability to enforce our issued
patents in the United States or may make it difficult to obtain broad patent protection going forward in the United States; |
|
• |
the eligibility to protect methods for treating humans, which is available in the US, is generally not
available in other countries, for example in Europe; |
|
• |
for a variety of reasons, we may decide not to file for patent protection on various improvements or additional
features; and |
|
• |
intellectual property protection and/or enforcement may be unavailable or limited in some countries where
laws or law enforcement practices may not protect our proprietary rights to the same extent as the laws of the United States, the European
Union, Canada or Israel. |
Consequently, our competitors could develop, manufacture
and sell products that directly compete with our products, which could decrease our sales and diminish our ability to compete. In addition,
competitors could attempt to develop their own competitive technologies that fall outside of our intellectual property rights. If our
intellectual property does not adequately protect us from our competitors’ products and methods, our competitive position could
be materially adversely affected.
Because the medical device industry is litigious, we are susceptible
to intellectual property suits that could cause us to incur substantial costs or pay substantial damages or prohibit us from selling C-Scan.
There is a substantial amount of litigation over patent and other
intellectual property rights in the medical device industry. Whether a product infringes a patent involves complex legal and factual issues,
the determination of which is often uncertain. Searches typically performed to identify potentially infringed patents of third parties
are often not conclusive and because patent applications can take many years to issue, there may be applications now pending, which may
later result in issued patents which our current or future products may infringe. In addition, our competitors or other parties may assert
that C-Scan and the methods it employs may be covered by patents held by them. If C-Scan or any of its components infringes a valid patent,
we could be prevented from manufacturing or selling it unless we can obtain a license or redesign the product to avoid infringement. For
example, in March 2021, we entered into an exclusive license agreement with the University of Missouri pursuant to which we were granted
an exclusive license in the medical field to certain patents held by the University of Missouri that the University of Missouri claimed
included background intellectual property in C-Scan. While we do not currently expect to utilize such patents, should we sell C-Scan units
incorporating such patents, we shall pay the University royalties ranging from $0.30 to $5.00 per C-Scan unit depending on the number
of units sold, up to $15,000,000 in the aggregate. Third parties may currently have, or may eventually be issued, patents on which our
current or future products or technologies may infringe.
A license may not always be available or may require
us to pay substantial royalties. We also may not be successful in any attempt to redesign our product to avoid infringement. Infringement
and other intellectual property claims, with or without merit, can be expensive and time-consuming to litigate and could divert our management’s
attention from operating our business.
The steps we have taken to protect our intellectual property may
not be adequate, which could have a material adverse effect on our ability to compete in the market.
In addition to patents, we rely on confidentiality,
non-compete, non-disclosure and assignment of inventions provisions, as appropriate, with our employees, consultants, subcontractors,
suppliers and clinical investigators to protect and otherwise seek to control access to, and distribution of, our proprietary information.
These measures may not be adequate to protect our intellectual property from unauthorized disclosure, third-party infringement or misappropriation,
for the following reasons:
|
• |
the agreements may be breached, may not provide the scope of protection we believe they provide or may
be determined to be unenforceable, in part or in whole; |
|
• |
we may have inadequate remedies for any breach; |
|
• |
proprietary information could be disclosed to our competitors; or |
|
• |
others may independently develop substantially equivalent or superior proprietary information and techniques
or otherwise gain access to our trade secrets or disclose such technologies. |
If, for any of the above reasons, our intellectual
property is disclosed or misappropriated, it could harm our ability to protect our rights and could have a material adverse effect on
our business, financial condition and results of operations.
Furthermore, although our employees and consultants
have agreed to assign to us all rights to any intellectual property created in the scope of their employment or engagement with us and
most of our current employees and consultants, have agreed to waive their economic rights with respect to our intellectual property, we
cannot assure you that such claims will not be brought against us by current or former employees or consultants, despite their contractual
representations and obligations toward us, or by any of the medical and/or governmental institutions that employ or engage such consultants,
claiming alleged rights to our intellectual property or demanding remuneration in consideration for assigned intellectual property rights,
which could result in litigation and adversely affect our business, financial condition and results of operations.
Third-party claims of infringement or other claims against us could
require us to redesign C-Scan, seek licenses, or engage in future costly intellectual property litigation, which could negatively affect
our future business and financial performance.
Substantial litigation over intellectual property
rights exists in the medical device industry in general and in the medical imaging or screening sectors in particular. We expect that
we may be subject to third-party infringement claims as our profile grows, we generate any future revenues, the number of competitors
grows or the functionality of products and technology in different industry segments converges. For example, in March 2021, we entered
into an exclusive license agreement with the University of Missouri with respect to certain patents held by the University of Missouri
that the University of Missouri claimed included background intellectual property in C-Scan. In consideration for the grant of an exclusive
license to those patents in the medical field, we agreed to pay royalties ranging from $0.30 to $5.00 per C-Scan unit depending on the
number of units sold up to $15,000,000 in the aggregate. Third parties may currently have, or may eventually be issued, patents on which
our current or future products or technologies may infringe.
In addition, litigation in which we are accused
of infringement may cause negative publicity, adversely impact prospective customers, cause product shipment delays, prohibit us from
manufacturing, marketing or selling our current or future products, require us to develop non-infringing technology, make substantial
payments to third parties or enter into royalty or license agreements, which may not be available on acceptable terms, or at all. If a
successful claim of infringement were made against us and we could not develop non-infringing technology or license the infringed or similar
technology in a timely and cost-effective manner, our ability to generate significant revenues may be substantially harmed and we could
be exposed to significant liability. A court could enter orders that temporarily, preliminarily or permanently enjoin us, our suppliers,
distributors or our customers from making, using, selling, offering to sell or importing our current or future products, or could enter
an order mandating that we undertake certain remedial activities. Claims that we have misappropriated the confidential information or
trade secrets of third parties can have a similar negative impact on our reputation, business, financial condition or results of operations.
We may also become involved in litigation in connection
with our brand name rights. We do not know whether others will assert that our brand name infringes their trademark rights. In addition,
names we choose for our products may be claimed to infringe names held by others. If we have to change the names we use, we may experience
a loss in goodwill associated with our brand name, customer confusion and a loss of sales.
Third parties may challenge the validity of our issued patents or
challenge patent applications in administrative proceedings before various patent offices which, if successful, could negatively affect
our future business and financial performance.
Various patent offices, including in the United
States and Europe, provide administrative proceedings by which a third party can challenge the validity of an issued patent or challenge
an application that is being examined absent any threat of litigation. In some instances, including in the United States, the administrative
proceedings provide a more efficient and favorable forum to challenge our patents which may lead to more opportunities for competitors
to do so, particularly smaller competitors with limited resources. Moreover, the standards utilized in these administrative proceedings,
at least in the United States, provide certain legal advantages versus challenging the validity of a patent in a district court. If a
third party is successful in one of these administrative proceedings, the patent will no longer be enforceable in the corresponding jurisdiction.
With this loss in patent rights, we will not be able to prevent third parties from offering identical or similar competing products which
may result in lower profits and a less substantial market share.
We may need to initiate lawsuits to protect or enforce our patents
and other intellectual property rights, which could be expensive and, if we lose, could cause us to lose some of our intellectual property
rights, which would harm our ability to compete in the market.
We rely on patents to protect a portion of our
intellectual property and our competitive position. Patent law relating to the scope of claims in the technology fields in which we operate
is still evolving and, consequently, patent positions in the medical device industry are generally uncertain. In order to protect or enforce
our patent rights, we may initiate patent and related litigation against third parties, such as infringement suits or interference proceedings.
Any lawsuits that we initiate could be expensive, take significant time and divert our management’s attention from other business
concerns and the outcome of litigation to enforce our intellectual property rights in patents, copyrights, trade secrets or trademarks
is highly unpredictable. Litigation also puts our patents and other registered intellectual property at risk of being invalidated or interpreted
narrowly and our patent applications at risk of not being issued. In addition, we may provoke third parties to assert claims against us.
We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, including attorney fees, if any, may not
be commercially valuable. The occurrence of any of these events could have a material adverse effect on our business, financial condition
and results of operations.
We rely on trademark protection to distinguish our products from
the products of our competitors; however, if a third party is entitled to use our trademark, we could be forced to rebrand, which could
result in loss of brand recognition and our ability to distinguish our products may be impaired, which could adversely affect our business.
We rely on trademark protection to distinguish
our products from the products of our competitors. In jurisdictions where we have not registered our trademarks and logos and are using
them, and as permitted by applicable local law, we rely on common law trademark protection. Third parties may oppose our trademark applications,
or otherwise challenge our use of the trademarks, and may be able to use our trademarks in jurisdictions where they are not registered
or otherwise protected by law. If our trademarks are successfully challenged or if a third party is using confusingly similar or identical
trademarks in particular jurisdictions before we do, we may be prevented from using our brands and/or domain names and/or could be forced
to rebrand our products, which could result in loss of brand recognition, and could require us to devote additional resources to marketing
new brands. If others are able to use our trademarks, our ability to distinguish our products may be impaired, which could adversely affect
our business. Further, we cannot assure you that competitors will not infringe upon our trademarks, or that we will have adequate resources
to enforce our trademarks.
We may not be able to enforce covenants not to compete at all or,
we may be unable to enforce them for the duration contemplated in our employment contracts and may, therefore, be unable to prevent competitors
from benefiting from the expertise of some of our former employees involved in research and development activities.
We currently have non-compete agreements with substantially
all of our employees who are involved in research and development, all of whom are located in Israel. These agreements prohibit our employees,
if they cease working for us, from directly competing with us or working for our competitors for a limited period of time following termination
of employment. In many jurisdictions, courts are increasingly refusing to enforce restrictions on competition by former employees or have
interpreted them narrowly. For example, in Israel, where currently all of our employees reside, courts have required employers seeking
to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm
one of a limited number of material interests of the employer which have been recognized by the courts, such as the secrecy of a company’s
confidential commercial information or its intellectual property. If we cannot demonstrate that harm would be caused to us, an Israeli
court may refuse to enforce our non-compete restrictions or reduce the contemplated period of non- competition such that we may be unable
to prevent our competitors from benefiting from the expertise of our former employees.
Risks Related to
Our Operations in Israel
Our principal offices, research and development facilities, our
manufacturing sites and some of our suppliers are located in Israel and, therefore, our business, financial condition and results of operation
may be adversely affected by political, economic and military instability in Israel.
Our principal offices and research and development
facilities are located near Haifa, in the northern part of Israel and our own X-ray production line is located in Petach Tikva, Israel.
In addition, all of our employees and officers, and one of our directors, are residents of Israel. Accordingly, political, economic and
military conditions in Israel may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed
conflicts have taken place between Israel and its neighboring countries. In recent years, Israel has been engaged in sporadic armed conflicts
with Hamas, an Islamist terrorist group that controls the Gaza Strip, with Hezbollah, an Islamist terrorist group that controls large
portions of southern Lebanon, and with Iranian-backed military forces in Syria. In addition, Iran has threatened to attack Israel, may
be developing nuclear weapons and has targeted cyber attacks against Israeli entities. Some of these hostilities were accompanied by missiles
being fired from the Gaza Strip against civilian targets in various parts of Israel, and negatively affected business conditions in Israel.
Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect
our operations and results of operations.
Popular uprisings in various countries in the Middle
East and North Africa are affecting the political stability of those countries. Such instability may lead to deterioration in the political
and trade relationships that exist between the State of Israel and these countries. Furthermore, several countries, principally in the
Middle East, restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions on doing business
with Israel and Israeli companies if hostilities in the region continue or intensify. Such restrictions may seriously limit our ability
to sell our products to customers in those countries. Similarly, Israeli corporations are limited in conducting business with entities
from several countries. Parties with whom we may do business could decline to travel to Israel during periods of heightened unrest or
tension. In addition, the political and security situation in Israel may result in parties with whom we may have agreements involving
performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure
provisions in such agreements. In addition, any hostilities involving Israel could have a material adverse effect on our facilities including
our corporate office or on the facilities of our local suppliers, in which event all or a portion of our inventory may be damaged, and
our ability to deliver products to customers could be materially adversely affected.
Furthermore, the war and terrorism insurance we
maintain may not be adequate to cover our losses associated with armed conflicts and terrorist attacks. Although the Israeli government
in the past covered the reinstatement value of certain damages that were caused by terrorist attacks or acts of war, we cannot assure
you that this government coverage will be maintained, or if maintained, will be sufficient to compensate us fully for damages incurred.
Any losses or damages incurred by us could have a material adverse effect on our business.
In addition, our operations could also be disrupted
by the obligations of personnel to perform military service. As of December 31, 2022, we had 85 employees and independent contractors,
all of whom were based in Israel, except one in the U.S. Some of these Israeli employees and independent contractors may be called upon
to perform up to 54 days in each three year period (and in the case of military officers, up to 84 days in each three year period) of
military reserve duty until they reach the age of 40 (and in some cases, depending on their specific military profession up to 45 or even
49 years of age) and, in certain emergency circumstances, may be called to immediate and unlimited active duty. In response to increases
in terrorist activity, there have been periods of significant call-ups of military reservists and it is possible that there will be similar
large-scale military reserve duty call-ups in the future. Our operations could be disrupted by the absence of a significant number of
employees related to military service, which could materially adversely affect our business and results of operations.
Any hostilities involving Israel, terrorist activities
or political instability in the region or the interruption or curtailment of trade between Israel and its present trading partners, or
significant downturns in the economic or financial condition of Israel, could adversely affect our operations and product development,
cause our revenues to decrease and adversely affect our share price.
Furthermore, the Israeli government is currently
pursuing extensive changes to Israel’s judicial system. In response to the foregoing developments, individuals, organizations and
institutions, both within and outside of Israel, have voiced concerns that the proposed changes may negatively impact the business environment
in Israel including due to reluctance of foreign investors to invest or conduct business in Israel, as well as to increased currency fluctuations,
downgrades in credit rating, increased interest rates, increased volatility in securities markets, and other changes in macroeconomic
conditions. Such proposed changes may also adversely affect the labor market in Israel or lead to political instability or civil unrest.
To the extent that any of these negative developments do occur, they may have an adverse effect on our business, our results of operations
and our ability to raise additional funds.
Pursuant to the terms of the Israeli government grants we received
for research and development expenditures, we are obligated to pay certain royalties on our revenues to the Israeli government. In addition,
the terms of Israeli government grant we received require us to satisfy specified conditions and to make additional payments in addition
to repayment of the grants upon certain events.
We have received grants from the Government of
the State of Israel through the IIA (formerly the OCS) for the financing of a portion of (i) our research and development expenditures
and (ii) a portion of the development of our manufacturing line, pursuant to the Innovation Law and related regulations and guidelines.
As of December 31, 2022, we had received funding from the IIA for research and development of C-Scan in the aggregate amount of approximately
$5.6 million. As of December 31, 2022, we had not paid any royalties to the IIA and had a contingent obligation to the IIA with respect
to such funding (including interest at the rate of 12-month LIBOR) in the amount of approximately $6.2 million. In addition, in January
2021, we received an IIA grant approval to support the funding of our transition from research and development to manufacturing. The final
IIA grant amounted to $620,000 (NIS 2.18 million) (along with a co-investment by us of the same amount), which we are not required to
repay to the IIA, subject to the terms and conditions set forth in the grant approval, of which we received approximately $222,000 (NIS
784,000) in February 2023, $82,000 (NIS 264,000) in January 2022 and $349,000 (NIS 1,134,000) in 2021. We may apply for additional IIA
grants in the future; however, there is no assurance that such applications will be approved in the amount requested or at all. Furthermore,
the funds available for IIA grants out of the annual budget of the State of Israel have been reduced in the past and may be further reduced
in the future. We cannot predict whether we will be entitled to any future grants, or the amounts of any such grants.
Under the terms of the Innovation Law as currently
in effect, products developed with IIA funding are required to be manufactured in Israel, unless the IIA approved grant program includes
a pre-determined portion of manufacturing that may be performed outside Israel (as certain of our IIA approved grants included). The approval
of the IIA is required for the transferring of manufacturing outside Israel in excess of such pre-determined portion (however, only a
notice to the IIA, as opposed to approval, is required for the transfer outside Israel of up to 10% of the cumulative manufacturing in
excess of such pre-approved portion). If manufacturing of IIA-funded products is transferred outside Israel (following IIA approval) in
excess of the pre-determined percentage included in the grant approval, then the royalty repayment rate will be increased by 1% with respect
to the additional approved percentage to be manufactured outside Israel and the royalty repayment for the entire approved program may
be increased to up to three times the amount of the grants received, depending on the percentage manufactured outside Israel (plus accrued
interest). We may explore from time to time whether certain other components of C-Scan can be assembled outside of Israel. For example,
we may in the future explore whether it would be possible to assemble the capsule without the X-ray source in Israel, and have the X-ray
source subsequently manufactured and assembled into C-Scan at a certified radioisotope production facility or at a distribution center
outside Israel.
Over the years, we received approval of grant applications
that included a certain predetermined percentage of manufacturing to be performed outside of Israel of the X-ray source but additional
examination of these approvals and consequent manufacturing is required to determine liabilities to the IIA, if any. IIA prior approval
is also required for the transfer of IIA-funded know-how to a third party outside of Israel (including by way of license), which we may
not receive (and any such approval would typically be subject to payment of a redemption fee, calculated according to a formula under
the Innovation Law, which may be in the amount of up to six times the amount of the grants received (less paid royalties, if any, and
depreciation, but no less than the total grants received), plus accrued interest. Even following the full repayment of any IIA grants,
we must nevertheless continue to comply with the requirements of the Innovation Law and related regulations and guidelines. The foregoing
restrictions and requirements for payment may impair our ability to sell our technology assets outside of Israel or to outsource or transfer
development or manufacturing activities with respect to any product or technology outside of Israel. Furthermore, the consideration available
to our shareholders in a transaction involving the transfer outside of Israel of technology or know-how developed with IIA funding (such
as a merger or similar transaction) may be reduced by any amounts that we are required to pay to the IIA.
If we fail to comply with any of the conditions
and restrictions imposed by the Innovation Law and related regulations and guidelines, or by the specific terms under which we received
the grants, we may be required to refund any grants previously received together with interest and penalties, and, in certain circumstances,
may be subject to criminal charges.
Your rights and responsibilities as a shareholder are governed by
Israeli law, which differ in some material respects from the rights and responsibilities of shareholders of U.S. companies.
The rights and responsibilities of the holders
of our ordinary shares are governed by our amended articles of association and by Israeli law. These rights and responsibilities differ
in some material respects from the rights and responsibilities of shareholders in U.S. based corporations. In particular, a shareholder
of an Israeli company has a duty to act in good faith and in a customary manner in exercising its rights and performing its obligations
towards the company and other shareholders, and to refrain from abusing its power in the company, including, among other things, in voting
at a general meeting of shareholders on matters such as amendments to a company’s articles of association, increases in a company’s
authorized share capital, mergers and acquisitions and certain related party transactions requiring shareholder approval under Israeli
law. In addition, a controlling shareholder of an Israeli company or a shareholder who is aware that it possesses the power to determine
the outcome of a shareholder vote or to appoint or prevent the appointment of a director or executive officer in the company or has other
powers towards the company, has a duty of fairness toward the company. There is limited case law available to assist us in understanding
the nature of this duty or the implications of these provisions. These provisions may be interpreted to impose additional obligations
and liabilities on holders of our ordinary shares that are not typically imposed on shareholders of U.S. corporations.
It may be difficult to enforce a judgment of a U.S. court against
us, certain of our officers and directors or the Israeli experts named in this Annual Report in Israel or the United States, to assert
U.S. securities laws claims in Israel or to serve process on certain of our officers and directors and these experts.
We are incorporated in Israel. A majority of our
executive officers and directors are not residents of the United States, and a substantial portion of our assets are located outside the
United States. Therefore, a judgment obtained against us, or any of these persons, including a judgment based on the civil liability provisions
of the U.S. federal securities laws, may not be collectible in the United States and may not be enforced by an Israeli court. It also
may be difficult for you to effect service of process on these persons in the United States or to assert U.S. securities law claims in
original actions instituted in Israel. Israeli courts may refuse to hear a claim based on an alleged violation of U.S. securities laws
on the grounds that Israel is not the most appropriate forum in which to bring such a claim. In addition, even if an Israeli court agrees
to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable,
the content of applicable U.S. law must be proven as a fact by expert witnesses, which can be a time consuming and costly process. Certain
matters of procedure will also be governed by Israeli law. There is little binding case law in Israel that addresses the matters described
above. As a result of the difficulty associated with enforcing a judgment against us in Israel, you may not be able to collect any damages
awarded by either a U.S. or foreign court.
Provisions of Israeli law and our amended articles of association
may delay, prevent or otherwise impede a merger with, or an acquisition of, us, even when the terms of such a transaction are favorable
to us and our shareholders.
Israeli corporate law regulates mergers, requires
tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors,
officers or significant shareholders and regulates other matters that may be relevant to such types of transactions. For example, a full
tender offer for all of a public company’s issued and outstanding shares can only be completed if the acquirer receives positive
responses from the holders of at least 95% of the issued share capital and the approval of a majority of the offerees that do not have
a personal interest in the tender offer, unless at least 98% of the company’s outstanding shares are tendered. Furthermore, the
shareholders, including those who indicated their acceptance of the tender offer (unless the acquirer stipulated in its tender offer that
a shareholder that accepts the offer may not seek appraisal rights), may, at any time within six months following the completion of the
tender offer, petition an Israeli court for an appraisal right, to alter the consideration for the acquisition. In addition, a statutory
merger may not be consummated unless at least 50 days have passed from the date on which a proposal for approval of the merger was filed
by each party with the Israeli Registrar of Companies and at least 30 days have passed from the date on which the merger was approved
by the shareholders of each party.
We may become subject to claims for payment of compensation for
assigned service inventions by our current or former employees, which could result in litigation and adversely affect our business.
Under the Israeli Patents Law, 5727-1967, or the
Patents Law, inventions conceived by an employee during the scope of his or her employment are regarded as “service inventions”
and are owned by the employer, absent a specific agreement between the employee and employer giving the employee service invention rights.
Section 134 of the Patents Law provides that if no agreement between an employer and an employee exists that prescribes whether, to what
extent, and on what conditions the employee is entitled to remuneration for his or her service inventions, then such matters may, upon
application by the employee, be decided by a government-appointed compensation and royalties committee established under the Patents Law,
or the Committee. Although our employees have agreed to assign to us all rights to any intellectual property created in the scope of their
employment and most of our current employees, including all those involved in the development of our intellectual property, have agreed
to waive their economic rights with respect to service inventions, we cannot assure you that claims will not be brought against us by
current or former employees demanding remuneration in consideration for assigned service inventions. If any such claims were filed, we
could potentially be required to pay remuneration to our current or former employees for such assigned service inventions, or be forced
to litigate such claims, which could negatively affect our business.
Risks Related to
Ownership of our Ordinary Shares
We incur and will continue to incur significant costs as a result
of operating as a public company in the United States, and our management is required to devote substantial time to compliance initiatives.
As a public company whose securities are traded
in the United States, we incur and will continue to incur significant legal, accounting and other expenses. The Sarbanes-Oxley Act of
2002, as well as rules and regulations implemented by the U.S. Securities and Exchange Commission and the Nasdaq Stock Market, impose
various requirements on public companies, including requiring the establishment and maintenance of effective disclosure and financial
controls. If we lose our status as a “non-accelerated filer”, we will be required to include an attestation report on
internal control over financial reporting issued by our independent registered public accounting firm. Our management and other personnel
devote a substantial amount of time to these compliance initiatives. Changes in the laws, rules and regulations affecting public companies
would result in increased costs to us as we respond to their requirements. These rules and regulations could make it more difficult or
more expensive for us to obtain certain types of insurance, including directors’ and officers’ liability insurance, and we
may be required to accept reduced policy limits and coverage or incur substantial costs to obtain or maintain the same or similar coverage.
The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board
of directors, our board committees or as executive officers. We cannot predict or estimate the amount or timing of additional costs we
may incur in order to comply with such requirements.
If we fail to maintain effective internal control over financial
reporting, the price of our ordinary shares may be adversely affected.
Our internal control over financial reporting may
have weaknesses and conditions that could require correction or remediation, the disclosure of which may have an adverse impact on the
price of our ordinary shares. We are required to establish and maintain appropriate internal control over financial reporting. Failure
to establish those controls, or any failure of those controls once established, could adversely affect our public disclosures regarding
our business, prospects, financial condition or results of operations. In addition, management’s assessment of internal control
over financial reporting may identify weaknesses and conditions that need to be addressed in our internal control over financial reporting
or other matters that may raise concerns for investors. In addition, as a “non-accelerated filer,” we are exempt from the
provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation
report on the effectiveness of internal control over financial reporting. Decreased disclosures in our SEC filings due to our status as
a “non-accelerated filer” may make it harder for investors to analyze our results of operations and financial prospects and
may make our ordinary shares a less attractive investment.. Any actual or perceived weaknesses and conditions that need to be addressed
in our internal control over financial reporting or disclosure of management’s assessment of our internal control over financial
reporting may have an adverse impact on the price of our ordinary shares.
Our ordinary shares could be delisted from the Nasdaq Capital Market.
Nasdaq has established certain standards for the
continued listing of a security on the Nasdaq Capital Market. The standards for continued listing include, inter
alia, that the minimum bid price for the listed securities be at least $1.00 per share. Under these rules, a security is considered
deficient if it fails to achieve at least a $1.00 closing bid price for a continuous period of 30 business days.
On December 23, 2021, we received a letter from
the Staff, indicating that we are not in compliance with the minimum bid price requirement for continued listing set forth in Listing
Rule 5550(a)(2), which requires listed securities to maintain a minimum bid price of $1.00 per share, or the Minimum Bid Price Requirement.
We had 180 calendar days (i.e., until June 21, 2022) to regain compliance with the Minimum Bid Price Requirement pursuant to Nasdaq Listing
Rules. On June 22, 2022, we received a letter from the Staff notifying us that Nasdaq granted us a 180-day extension, until December
19, 2022 to regain compliance with the Minimum Bid Price Requirement. On November 23, 2022, we effected a 1-for-20 reverse share
split of our ordinary shares, in accordance with the approval of our shareholders at a meeting held on August 11, 2022. On December
9, 2022, following the increase of the share price in Nasdaq from November 25, 2022 to December 8, 2022, we were notified by Nasdaq that
we regained compliance with the minimum $1.00 bid price rule.
If we are delisted from Nasdaq, our ordinary shares
may be eligible for trading on an over-the-counter market in the United States. In the event that we are not able to obtain a listing
on another U.S. stock exchange or quotation service for our ordinary shares, it may be extremely difficult or impossible for shareholders
to sell their ordinary shares in the United States. Moreover, if we are delisted from Nasdaq, but obtain a substitute listing for our
ordinary shares in the United States, it will likely be on a market with less liquidity, and therefore, experience potentially more price
volatility than experienced on Nasdaq. Shareholders may not be able to sell their ordinary shares on any such substitute U.S. market in
the quantities, at the times, or at the prices that could potentially be available on a more liquid trading market. As a result of these
factors, if our ordinary shares are delisted from Nasdaq, the price of our ordinary shares is likely to decline. A delisting of our ordinary
shares from Nasdaq could also adversely affect our ability to obtain financing for our operations and/or result in a loss of confidence
by investors, or employees.
We are a foreign private issuer and, as a result, we are not subject
to U.S. proxy rules and are subject to the Securities Exchange Act of 1934 reporting obligations that, to some extent, are more lenient
and less frequent than those applicable to a U.S. issuer.
We report under the Securities Exchange Act of
1934, as amended, or the Exchange Act as a foreign private issuer. Because we qualify as a foreign private issuer under the Exchange Act,
we are exempt from certain provisions of the Exchange Act that are applicable to U.S. public companies, including (i) the sections of
the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange
Act; (ii) the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and
liability for insiders who profit from trades made in a short period of time; and (iii) the rules under the Exchange Act requiring the
filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports
on Form 8-K, upon the occurrence of specified significant events. We intend to furnish quarterly reports to the SEC on Form 6-K for so
long as we are subject to the reporting requirements of Section 13(g) or 15(d) of the Exchange Act, although the information we furnish
may not be the same as the information that is required in quarterly reports on Form 10-Q for U.S. domestic issuers. In addition, while
U.S. domestic issuers that are not large accelerated filers or accelerated filers are required to file their annual reports on Form 10-K
within 90 days after the end of each fiscal year, foreign private issuers are not required to file their annual report on Form 20-F until
120 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation FD (Fair Disclosure), aimed at
preventing issuers from making selective disclosures of material information. Although we intend to make interim reports available to
our shareholders in a timely manner, you may not have the same protections afforded to shareholders of companies that are not foreign
private issuers.
As a foreign private issuer, we are permitted, to follow, and follow
certain home country corporate governance practices instead of otherwise applicable Nasdaq requirements, which may result in less protection
than is accorded to investors under rules applicable to domestic U.S. issuers.
As a foreign private issuer, we are permitted to
follow certain home country corporate governance practices instead of those otherwise required under the Listing Rules of the Nasdaq Stock
Market for domestic U.S. issuers. For instance, we follow home country practice in Israel with regard to, among other things, director
nomination procedures, the approval of compensation of officers and quorum requirements at general meetings of our shareholders. In addition,
we follow our home country law instead of the Listing Rules of the Nasdaq Stock Market that require us to obtain shareholder approval
for certain dilutive events, such as the establishment or amendment of certain equity based compensation plans, an issuance that will
result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or greater
interest in the company, and certain acquisitions of the stock or assets of another company. Following our home country governance practices
as opposed to the requirements that would otherwise apply to a United States company listed on Nasdaq may provide less protection to you
than what is accorded to investors under the Listing Rules of the Nasdaq Stock Market applicable to domestic U.S. issuers.
If we lose our status as a foreign private issuer under the SEC’s
rules, our compliance costs will increase.
We would lose our foreign private issuer status
if more than 50 percent of our outstanding voting securities are directly or indirectly held of record by residents of the United States
and if a majority of our directors or executive officers are U.S. citizens or residents and we fail to meet additional requirements necessary
to avoid loss of foreign private issuer status. Although we have elected to comply with certain U.S. regulatory provisions, our loss of
foreign private issuer status would make such provisions mandatory. The regulatory and compliance costs for us under U.S. securities laws
as a U.S. domestic issuer may be significantly higher. If we are not a foreign private issuer, we will be required to file periodic reports
and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive than the forms available
to a foreign private issuer. We would also be required to follow U.S. proxy disclosure requirements, including the requirement to disclose
more detailed information about the compensation of our senior executive officers on an individual basis. We may also be required to modify
certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion and modifications
will involve additional costs. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements
on U.S. stock exchanges that are available to foreign private issuers.
Exchange rate fluctuations between the U.S. dollar and the NIS and
the Euro and inflation may negatively affect our earnings and we may not be able to hedge our currency exchange risks successfully.
The dollar is our functional and reporting currency.
However, a significant portion of our operating expenses, including personnel and facilities related expenses, are incurred in NIS. As
a result, we are exposed to the risks that the NIS may appreciate relative to the U.S. dollar, or, if the NIS instead depreciates relative
to the U.S. dollar, that the inflation rate in Israel may exceed such rate of depreciation of the NIS, or that the timing of such depreciation
may lag behind inflation in Israel. In any such event, the dollar cost of our operations in Israel would increase and our dollar- denominated
results of operations would be adversely affected. In year 2022, the NIS depreciated by 13.15% relative to the U.S. dollar compared to
an appreciation of 3.266% in year 2021 and the Israeli rate of inflation increased by 5.3% and 2.8% during years 2022 and 2021, respectively,
and decreased by 0.7% in year 2020, and therefore has not had a material adverse effect on our financial condition during these years.
In addition, we may incur operating expenses denominated in Euros, and therefore, our operating results may also be subject to fluctuations
due to changes in the U.S. dollar/Euro exchange rate. We cannot predict any future trends in the rate of inflation in Israel or the rate
of devaluation (if any) of the NIS, the Euro and other foreign currencies against the U.S. dollar. Although we engage in currency hedging
arrangements from time to time, these measures may not adequately protect us from fluctuations in the exchange rates of the NIS, the Euro
and other foreign currencies in relation to the U.S. dollar (and/or from inflation of such foreign currencies), and involve costs and
risk of their own.
We have never declared or paid a dividend and currently do not intend
to pay cash dividends in the foreseeable future. Any return on investment may be limited to the value of our securities.
We have never declared and do not anticipate paying
cash dividends on our ordinary shares in the foreseeable future. Our board of directors has discretion to declare and pay dividends on
our ordinary shares and will make any determination to do so based on a number of factors, such as our operating results, financial condition,
current and anticipated cash needs and other business and economic factors that our board of directors may deem relevant. In addition,
we are only permitted to pay dividends out of “profits” (as defined by the Israeli Companies Law, 1999, or the Israeli Companies
Law), provided that there is no reasonable concern that the dividend distribution will prevent us from meeting our existing and foreseeable
obligations, as they become due. If we do not pay dividends, our ordinary shares may be less valuable because a return on your investment
will only occur if the trading price of our securities appreciates. Further, you should not rely on an investment in us if you require
dividend income from your investments.
If securities or industry analysts do not publish research or reports
about us or our business or publish unfavorable research about us or our business, the price of our securities and their trading volume
could decline.
The trading market for our securities will depend
in part on the research and reports that securities or industry analysts publish about us or our business. We currently have limited research
coverage by securities and industry analysts. If one or more of the analysts who covers us downgrades our securities, the price of our
securities would likely decline. We do not have control over these analysts and we do not have commitments from them to continue to write
research reports about us or our business. The price of our ordinary shares could decline if one or more equity research analysts downgrade
our ordinary shares or if those analysts issue other unfavorable commentary or cease publishing reports about us or our business.
Our stock price has and may be subject to fluctuation, and purchasers
of our securities could incur substantial losses.
Our stock price has been subject to considerable
fluctuation since our initial public offering in February 2015, with the closing price per share having varied from a low of $1.93 to
a high of $1,411.22, and may be subject to fluctuation in the future. The stock market in general has experienced extreme volatility that
has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able
to sell their securities at or above the purchase price. The market price for our ordinary shares on the Nasdaq Capital Market may fluctuate
as a result of a number of factors, some of which are beyond our control, including, among others:
|
• |
we may not be able to develop C-Scan at the rate or to the stage we desire; |
|
• |
inability to obtain the approvals necessary to commence further clinical trials; |
|
• |
unsatisfactory results of clinical trials; |
|
• |
announcements of regulatory approval or the failure to obtain it, or specific label indications or patient
populations for its use, or changes or delays in the regulatory review process; |
|
• |
any intellectual property infringement actions in which we may become involved; |
|
• |
announcements concerning our competitors or the medical device industry in general; |
|
• |
achievement of expected product sales and profitability or our failure to meet expectations; |
|
• |
our commencement of, or involvement in, litigation; |
|
• |
any major changes in our board of directors or management; |
|
• |
legislation in the United States relating to the sale or pricing of medical device; |
|
• |
future substantial sales of our ordinary shares; |
|
• |
changes in earnings estimates or recommendations by securities analysts, if our ordinary shares are covered
by analysts; |
|
• |
the trading volume of our ordinary shares; or |
|
• |
natural disasters and political and economic instability, including wars, terrorism, political unrest,
results of certain elections and votes, emergence of a pandemic, or other widespread health emergencies (or concerns over the possibility
of such an emergency, including for example, the COVID-19 pandemic), boycotts, adoption or expansion of government trade restrictions,
and other business restrictions. |
In addition, the stock market in general, and Nasdaq
Stock Market in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate
to the operating performance of small companies. Broad market and industry factors may negatively affect the market price of our ordinary
shares, regardless of our actual operating performance. Further, a systemic decline in the financial markets and related factors beyond
our control may cause our share price to decline rapidly and unexpectedly.
The trading market for our ordinary shares is not always active,
liquid and orderly, which may inhibit the ability of our shareholders to sell ordinary shares.
Since our initial public offering in February 2015,
the trading market for our ordinary shares has not always been active, liquid or orderly. The lack of an active market at times may impair
your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market
may also reduce the fair market value of your shares. An inactive market may also impair our ability to raise capital by selling shares.
In December 2020, we amended our articles of association to increase
our authorized share capital. There are certain risks associated with this increase.
In December 2020, our shareholders approved an
increase to our authorized share capital and to amend our Articles of Association accordingly. As a result, our authorized and registered
share capital is NIS 864,000,000 divided into 18,000,000 ordinary shares, nominal (par) value NIS 48 each. The increase of our authorized
share capital is designed to enable us to have sufficient authorized share capital that would allow us to meet our future business needs
as they arise. These needs could include, among other things, the sale of shares in public and private offerings to raise additional capital,
the purchase of property or assets, the use of shares for various equity compensation and other employee benefit plans and arrangements,
the declaration of share splits, and other bona fide corporate purposes. The possible future issuance of equity securities consisting
of ordinary shares or securities convertible into ordinary shares could affect our current shareholders in a number of ways, including
the following: (i) diluting the voting power of the current holders of ordinary shares; (ii) diluting the market price of the ordinary
shares, to the extent that the new ordinary shares are issued and sold at prices below current trading prices of the existing ordinary
shares, or if the issuance consists of equity securities convertible into ordinary shares, to the extent that the securities provide for
the conversion into ordinary shares at prices that could be below current trading prices of the ordinary shares; and (iii) diluting the
book value per share of the outstanding ordinary shares. Furthermore, the authorized shares could, in theory, also be used to resist or
frustrate a third-party transaction that is favored by a majority of the independent shareholders (for example, by permitting issuances
that would dilute the share ownership of a person seeking to effect a change in the composition of our board of directors or management
or contemplating a tender offer or other transaction for the combination of our company with another company).
We have broad discretion in how we use the net proceeds from our
financings, and we may not use these proceeds effectively.
Our management has broad discretion as to the application of
the net proceeds of our financings. Our shareholders may not agree with the manner in which our management chooses to allocate and spend
the net proceeds. Moreover, our management may use the net proceeds for corporate purposes that may not increase our market value.
Our business, operating results and growth rates may be adversely
affected by current or future unfavorable economic and market conditions and adverse developments with respect to financial institutions
and associated liquidity risk.
Our business depends on the economic health of the global economies.
If the conditions in the global economies remain uncertain or continue to be volatile, or if they deteriorate, including as a result of
the impact of military conflict, such as the war between Russia and Ukraine, terrorism or other geopolitical events, our business, operating
results and financial condition may be materially adversely affected. Economic weakness, inflation and increases in interest rates, limited
availability of credit, liquidity shortages and constrained capital spending could negatively affect our financial condition, results
of operations or cash flows. In addition, the military conflict may affect the ability to purchase materials sourced in Russia.
For example, one of our suppliers sources the enriched X-ray isotope substance used in C-Scan from Russia and while we have sufficient
amounts of such material for our U.S. pivotal study, we could experience disruptions in our supply chain in the future if sanctions and
export control restrictions on Russian entities and individuals are still imposed, and we may not be able to find alternative sources
for such key material in a timely manner.
In addition, increases in inflation raise our costs
for commodities, labor, materials and services and other costs required to operate our business, and failure to secure these on reasonable
terms may adversely impact our financial condition. Additionally, increases in inflation, along with the uncertainties surrounding COVID-19,
geopolitical developments and global supply chain disruptions, have caused, and may in the future cause, global economic uncertainty and
uncertainty about the interest rate environment, which may make it more difficult, costly or dilutive for us to secure additional financing.
A failure to adequately respond to these risks could have a material adverse impact on our financial condition, results of operations
or cash flows.
More recently, the closures of SVB and Signature Bank and their
placement into receivership with the FDIC created bank-specific and broader financial institution liquidity risk and concerns. Although
the Department of the Treasury, the Federal Reserve and the FDIC jointly released a statement that depositors at SVB and Signature Bank
would have access to their funds, even those in excess of the standard FDIC insurance limits, under a systemic risk exception, future
adverse developments with respect to specific financial institutions or the broader financial services industry may lead to market-wide
liquidity shortages, impair the ability of companies to access near-term working capital needs, and create additional market and economic
uncertainty. There can be no assurance that future credit and financial market instability and a deterioration in confidence in economic
conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, liquidity shortages,
volatile business environment or continued unpredictable and unstable market conditions. If the current equity and credit markets deteriorate,
or if adverse developments are experienced by financial institutions, it may cause short-term liquidity risk and also make any necessary
debt or equity financing more difficult, more costly, more onerous with respect to financial and operating covenants and more dilutive.
Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our strategy,
our financial condition, results of operations or cash flows and stock price and could require us to alter our plans. In addition, there
is a risk that one or more of our service providers, financial institutions, manufacturers and suppliers and other partners may be adversely
affected by the foregoing risks, which could directly affect our ability to attain our operating goals on schedule and on budget.
Risks Related to
Taxation
There is a risk that we could be treated as a domestic (U.S.) corporation
for U.S. federal income tax purposes by reason of the transactions related to our acquisition of all of the business operations and substantially
all of the assets of Check-Cap LLC on May 31, 2009, or the Reorganization.
Section 7874(b) of the Internal Revenue Code of
1986, as amended, or the Code, generally provides that a foreign corporation (i.e., a corporation created or organized under the laws
of a jurisdiction outside of the United States) would be treated as a domestic (U.S.) corporation for U.S. federal income tax purposes
if, pursuant to a plan or a series of related transactions, (1) the foreign corporation acquires, directly or indirectly, substantially
all of the assets of a domestic corporation (or substantially all of the properties constituting a trade or business of a domestic partnership),
(2) after the acquisition, the former shareholders of the acquired corporation by reason of holding shares of the acquired corporation
(or, in the case of an acquisition with respect to a domestic partnership, the former partners of the domestic partnership by reason of
holding a capital or profits interest in the domestic partnership) own at least 80% of the stock (by vote or value) of the acquiring corporation,
and (3) after the acquisition, the expanded affiliated group that includes the acquiring corporation does not have substantial business
activities in the foreign country in which, or under the laws of which, the acquiring corporation is created or organized when compared
to the total business activities of such expanded affiliated group. On the basis of analysis of the relevant facts and circumstances and
the relevant law (including the temporary regulations under Section 7874 applicable at the time of the Reorganization), it was determined
that the third condition described in the preceding sentence was not met with respect to the Reorganization and, therefore, that the inversion
tax rules of Section 7874(b) would not apply to treat us as a domestic corporation for U.S. federal income tax purposes. However, since
this determination was made on the basis of all of the relevant facts and circumstances, and it is not clear which facts and circumstances
the Internal Revenue Service, or the IRS, may consider more important than others, this conclusion is not free from doubt.
If Section 7874(b) were to apply to the Reorganization
(and we were to be treated as a domestic corporation for U.S. federal income tax purposes), then, among other things, (i) we would be
subject to U.S. federal income tax on our worldwide taxable income (if and when we have taxable income); (ii) certain payments (e.g.,
interest and dividends) that we make (or have made) to our foreign investors may be (or may have been) subject to U.S. withholding taxes;
(iii) we may be subject to significant penalties for the failure to file certain tax returns and reports, including reports with respect
to our foreign bank accounts; and (iv) the U.S. unitholders of Check-Cap LLC would not have been subject to U.S. federal income tax on
royalties that are deemed to be paid to them under Section 367(d) of the Code as a result of the Reorganization. As discussed under Item
5B “Operating and Financial Review and Prospects – Liquidity and Capital Resources – Application of Critical Accounting
Policies and Estimates – Royalties provision – Reimbursement liability to Check-Cap LLC unitholders,” as part of the
Reorganization, we committed to reimburse the unitholders of Check-Cap LLC for any tax burdens that may be imposed on them due to the
Reorganization, including royalties that are deemed to be paid to the U.S. unitholders under Section 367(d) of the Code.
Prospective investors are urged to consult their
own advisors on these issues. The balance of this discussion, including the discussion under Item 10E “Additional Information –
Taxation – U.S. Federal Income Taxation,” assumes that we will be and have been treated as a foreign corporation for U.S.
federal income tax purposes.
We may be eligible for tax benefits from government programs, which
require us to meet certain conditions, including regarding the location of our property, plant and equipment and manufacturing in Israel.
We can provide no assurance that we would continue to be eligible for such benefits and/or that any such benefits will not be terminated
in the future.
Our manufacturing facilities in Israel may qualify
as a “Benefited Enterprise” under the Israeli Law for Encouragement of Capital Investments, 5719-1959, which would entitle
us to receive certain tax benefits. In order to be eligible for such benefits, we would be required to meet certain conditions, including
the making of a minimum capital investment in our productive assets and the carrying on of a required portion of our manufacturing in
Israel. The amount of the benefit will be determined in accordance with various conditions, including the location of our property, plant
and equipment and the location of certain of our sub-contractors. If we cease to meet the required conditions for eligibility, the tax
benefits could be cancelled and we could be required to pay increased taxes or to refund the amounts of the benefits received with interest
and penalties. We can provide no assurance as to the amount of future capital investment in our productive assets, our future manufacturing
location and the future location of our property, plant and equipment and certain of our sub-contractors, and therefore, we cannot provide
assurance that we will be eligible for such tax benefits or assurance as to the amount of such tax benefits. Even if we continue to meet
the relevant requirements, the tax benefits that Benefited Enterprises receive may not be continued in the future at their current levels
or at all. If these tax benefits were reduced or eliminated, the amount of taxes that we would be required to pay would likely increase,
as all of our operations would consequently be subject to corporate tax at the standard rate, which could adversely affect our results
of operations. See Item 10E “Additional Information—Taxation—Israeli Tax Considerations and Government Programs—Law
for the Encouragement of Capital Investments, 5719-1959” for additional information concerning these tax benefits.
There is a risk that we may be classified as a passive foreign investment
company, or PFIC, which could result in adverse U.S. federal income tax consequences to U.S. investors.
In general, we will be treated as a passive foreign
investment company, or a PFIC, for any taxable year in which either (1) at least 75% of our gross income (including our pro rata share
of the gross income of our 25% or more- owned corporate subsidiaries) is passive income or (2) at least 50% of the average value of our
assets (including our pro rata share of the assets of our 25% or more-owned corporate subsidiaries) is attributable to assets that produce,
or are held for the production of, passive income. Passive income generally includes dividends, interest, rents, royalties, and gains
from the disposition of passive assets. If we are determined to be a PFIC for any taxable year (or portion thereof) that is included in
the holding period of a U.S. Holder (as defined in Item 10E “Additional Information—Taxation—U.S. Federal Income Taxation”)
of our securities, the U.S. Holder may be subject to increased U.S. federal income tax liability upon a sale or other disposition of our
securities or the receipt of certain excess distributions from us and may be subject to additional reporting requirements. We believe
that we were a PFIC for the taxable year ended December 31, 2022 and may be a PFIC for the taxable year ending December 31, 2023. Our
actual PFIC status for our current taxable year or any subsequent taxable year is uncertain and will not be determinable until after the
end of such taxable year. Accordingly, there can be no assurance with respect to our status as a PFIC for our taxable year ending December
31, 2023 or any subsequent taxable year.
U.S. investors are urged to consult their own tax
advisors regarding the possible application of the PFIC rules. For more information, see Item 10E “Additional Information—Taxation—U.S.
Federal Income Taxation—U.S. Holders—Passive Foreign Investment Company Rules.”
ITEM 4. INFORMATION ON OUR COMPANY
A. History
and Development of the Company Our History
Our legal and commercial name is Check-Cap Ltd.
We were formed as a company in Israel on April 5, 2009. On May 31, 2009, we acquired all of the business operations and substantially
all of the assets of Check-Cap LLC, a Delaware limited liability company formed in December 2004. On May 15, 2015, we formed our wholly-owned
subsidiary Check-Cap US, Inc., a Delaware corporation.
On February 24, 2015, we successfully completed
an initial public offering in the United States and the listing of our securities on the Nasdaq Capital Market.
We are subject to the provisions of the Israeli
Companies Law. Our principal executive offices are located at Check-Cap Building, 29 Abba Hushi Avenue, P.O. Box 1271, Isfiya, 3009000,
Israel. Our telephone number is +972-4-8303400 and our website is located at www.check-cap.com (the
information contained therein or linked thereto shall not be considered incorporated by reference in this Annual Report). Our U.S. agent
is Puglisi & Associates, located at 850 Library Avenue, Suite 204, Newark, Delaware 19711.
We use our website (http://www.check-cap.com)
as a channel of distribution of company information. The information we post through this channel may be deemed material. Accordingly,
investors should monitor our website, in addition to following our press releases, SEC filings and public conference calls and webcasts.
The contents of our website are not, however, a part of this Annual Report.
Principal Capital Expenditures
For a discussion of our capital expenditures, see
Item 5 “Operating and Financial Review and Prospects—Liquidity and Capital Resources.”
B. Business
Overview Our Company
We are a clinical stage medical diagnostics company
aiming to redefine CRC screening through the introduction of C-Scan®, the first and only patient-friendly preparation-free test designed
to detect polyps before they may transform into cancer to enable early intervention and cancer prevention.
The disruptive capsule-based screening technology
aims to increase screening adherence worldwide and enable millions of people to stay healthy. The system utilizes ultra-low-dose X- rays
to scan the inner lining of the colon for precancerous polyps, and other structural abnormalities. CRC is the third most commonly diagnosed
cancer, with more than 1.9 million new cases identified every year globally. Nearly 935,000 deaths occur annually worldwide as a result
of CRC. While approximately 0.5% of the average-risk screening population presents with cancerous polyps in the colon and rectum at any
given time, approximately 25% of the same population presents with benign polyps that could potentially turn into cancer over time. It
can take up to 10 years before a pre-cancerous polyp develops into invasive cancer. As such, there is a crucial detection window for the
prevention of colorectal cancer, through the detection of these benign polyps. While routine screening is recommended by The American
Cancer Society for healthy people aged 45 years and older, screening adherence remains low. Currently, colonoscopy is the gold standard
for the detection of colorectal polyps, but about 1 in 3 adults among the targeted screening population avoids having a colonoscopy in
the U.S., and adherence in other regions of the world such as Europe and Asia is even lower, due to the invasiveness of the procedure
and bowel preparation. Most patient-friendly CRC screening tests currently available, or poised to enter the market, such as fecal or
liquid biopsy tests, are primarily designed to detect cancer and demonstrate low sensitivity in detecting pre-cancerous polyps. As such,
they do not necessarily provide patients with the time window to pre-empt the disease. C-Scan is non-invasive and requires no bowel cleansing
or sedation, allowing the patients to continue their daily routine with no interruption as the capsule is propelled through the gastrointestinal
tract by natural motility. C-Scan is comprised of three main components: (1) C-Scan Cap, an ingestible X-ray scanning capsule; (2) C-Scan
Track, three miniaturized patches worn on the patient’s back for integrated positioning, control and data recording; and (3) C-Scan
View, a proprietary software to process and represent 2D and 3D maps of the inner surface of the colon. We believe that this solution
has the potential to become an alternative for both physicians and patients and to increase the number of people completing CRC screening.
Our C-Scan Cap is swallowed and propelled by natural
motility through the gastrointestinal tract and excreted naturally with no need for retrieval for data collection (currently, during clinical
trials in the U.S., the C-Scan capsule is not intended to be disposed of in the sanitary sewer system, and is therefore collected by the
patient, and returned to the medical facility administering the device for further decay and disposal). Unlike other existing CRC screening
methods, this process should not disrupt a patient’s normal activities or require fasting. Our C-Scan Cap employs ultra-low-dose
X-rays, which allow the C-Scan system to scan the interior lining of the colon even when surrounded by intestinal content. As such, we
believe that patients using C-Scan will not be required to undergo any prior bowel cleansing.
Our C-Scan Cap is being designed to transmit position,
motility and the data it collects to the C-Scan Track that will be attached to the patient’s back. The external data recorder is
being designed to enable the download of the data to our C-Scan View application to allow physicians to analyze the data collected by
our C-Scan Cap. The C-Scan Track is being designed to provide the physician with localization data aligned with a reconstructed image.
We intend for physicians to be able to review the colon’s inner structural information.
Colonic polyps are tissue growths that occur on
the lining of the colon. Polyps in the colon are common, and certain types of polyps may become cancerous over time. In the event that
polyps are identified by C-Scan, the patient may be advised to undergo a subsequent traditional colonoscopy procedure to examine, remove
and biopsy the polyps. For those patients who require a subsequent colonoscopy, concerns regarding pain, discomfort and embarrassment
may still remain. We do not, however, believe that these concerns will make the use of C-Scan any less attractive to physicians and patients.
Although patients who are initially screened utilizing a traditional colonoscopy could avoid the need for a second colonoscopy if polyps
are discovered, we believe that C-Scan will still be attractive to physicians and patients who object to or cannot undergo a colonoscopy
as a large number of these patients if screened will not require a subsequent colonoscopy and those found with suspicious findings, will
likely agree to undergo colonoscopy.
We initiated our first clinical studies in 2010,
consisting of two single-center feasibility studies with non-scanning (no X-ray source) capsules for the purposes of measuring gastrointestinal
tract activity, colon contractions and associated capsule motility, and shortening capsule transit time.
In 2013, we initiated a multi-center prospective
clinical feasibility study, designed to allow for the recruitment of 100 subjects, to establish clinical proof of concept, safety and
functionality of C-Scan in patients eligible for CRC screening. Analysis conducted on the first 66 capsules swallowed by participants
showed that 65 of 66 capsules swallowed were naturally eliminated, without major or minor side effects, after 62±40.7 hours. The
average calculated radiation exposure was 0.06 ± 0.04 mSv (similar to a single chest radiograph). Both pedunculated and sessile polyps
were detected in several patients and validated later by colonoscopy.
In September 2017, we completed a multi-center
study of C-Scan in support of CE Mark submission. The objective of the study was to assess safety and the clinical performance of C- Scan
in detecting patients with polyps. The three-center trial enrolled 66 patients, with a mean age of 59 years. Following capsule ingestion,
subjects swallowed small doses of contrast agent and fiber supplements with each meal throughout capsule passage. Average capsule transit
time was 52±32 hours, and the average total X-ray dose was 0.05 mSv (CT colonography effective dose is ~ 6.0 mSv). No bowel preparation,
sedation, or change in diet was required. Both confirmatory colonoscopy, performed by an independent investigator, and C-Scan review,
performed by a central review group, were blinded to results. The study demonstrated a 44% sensitivity in the 45 subjects included in
the analysis for polyps, with specificity (ability to correctly identify lack of polyps) of 89%. Sensitivity strongly correlated (R-squared
= 0.98) to the percentage of the colon scanned. Sensitivity was 78% (p<0.05) and 100% (p<0.05) for subjects where greater than 50%
and 70% of the colon was scanned, respectively. Specificity was consistent for all subjects.
In the fourth quarter of 2017, we initiated an
interim clinical study for the purpose of introducing an advanced version of C-Scan, Version 3, which incorporated the then latest algorithms
and system optimization and tailored scanning of the colon to the patient’s natural colonic movements to maximize the amount of
the colon that is tracked and imaged. In March 2018, we announced results from the interim study. Evaluable results of 21 patients showed
average colon imaging coverage of 64%, a 40% improvement over 46% average colon imaging coverage in the multi-center study. Sensitivity
was 78% (p<0.05) for subjects with greater than 50% colon imaging coverage and 100% (p<0.05) for subjects with greater than 70%
colon imaging coverage. Specificity was consistent at around 89%.
Following our certification to ISO 13485:2016 by
our Notified Body, and completion of our multi-center clinical study and achievement of compliance with the requirements of the Medical
Devices Directive, in September 2017, we submitted a request for CE marking for the marketing and sale of C-Scan in the European Union.
We received the CE Mark certification from our notified body (DEKRA - 0344) on January 9, 2018 and a renewal certificate according to
MDR (EU Regulation 2017/745) was granted on January 12, 2022, which is valid until December 1, 2026. In September 2018, we received approval
from AMAR for the marketing and sale of C-Scan in Israel. Following the MDR approval we received from our notified body according to the
EU Regulation 2017/745, the AMAR approval was renewed and is valid until December 31, 2024.
During the first quarter of 2018, we initiated
a multi-center, open label, home monitoring, prospective study designed to determine the performance characteristics of C-Scan system
Version 3, for detecting pre-cancerous polyps compared with the fecal immunochemical test (FIT), in each case using colonoscopy as the
reference method, for the purpose of collecting additional evidence of clinical effectiveness and clinical utility to support market adoption.
The study included 90 evaluable patients who either had known polyps or were considered to be of average risk. Each patient ingested a
C-Scan capsule and also underwent a FIT and a comparative colonoscopy performed by independent gastroenterologists, who were blinded to
the corresponding test’s results. The C-Scan clinical evaluation was obtained using the evaluable patient population implementing
a gender-based motility analysis and the results of both C-Scan and FIT were compared to colonoscopy. The primary efficacy endpoint of
the study was sensitivity (ability to correctly identify patients with polyps) and specificity (ability to correctly identify patients
with lack of polyps) of the C-Scan system compared to FIT in detecting subjects with polyps ≥10 mm. In July 2019, we announced final
results from our post-CE approval study. The results demonstrate that C-Scan achieved a sensitivity of 76% (p=0.0005) in patients with
polyps ≥10 mm, while FIT achieved a sensitivity of 29% (p=0.005) in patients with polyps ≥10 mm. C- Scan achieved a specificity
of 82% in all patients, while FIT achieved a specificity of 96% in all patients. In addition, C-Scan detected all 4 patients (100%) with
polyps ≥40 mm, while the FIT detected only 1 of the 4 patients (25%) with polyps ≥40mm. Overall, C-Scan achieved a sensitivity
of 66% (p=0.01) in all patients, including patients with polyps <10mm, while FIT achieved a sensitivity of 23% (p<0.0001) in all
patients, including patients with polyps <10mm. In total, 142 patients enrolled in the study and after factoring in technical and physiological
dropouts and protocol violations, the number of evaluable patients was 90. No serious adverse events were reported, and the adverse events
were mild in severity.
We conducted pre-submission meetings with the FDA
during the period of December 2016 and February 2017 for the purpose of receiving feedback on the regulatory pathway for our system in
the United States. We also sought feedback on a proposed protocol for a feasibility or pilot study, the primary purposes of which is to
establish the safety of the C-Scan system and evaluate user compliance and satisfaction. In December 2018, we received from the FDA conditional
approval of our IDE application to initiate a U.S. pilot study of C-Scan and received final approval from the FDA in February 2019.
In April 2019, we initiated the U.S. pilot study
of C-Scan. The U.S. pilot study (NCT03735407 ) was a prospective, multi-center, open label, single arm study was designed to evaluate
the safety, usability and subject compliance of the C-Scan system. The study included 28 evaluable patients, more than two thirds of whom
were considered to be of average risk for colorectal cancer. Each patient ingested a C-Scan capsule and also underwent a fecal immunochemical
test (FIT) as well as a comparative colonoscopy, which was performed by an independent gastroenterologist who was blinded to the corresponding
test results. The study was performed at two sites, the NYU Grossman School of Medicine and Mayo Clinic, Rochester. The primary endpoint
of the study was to evaluate the incidence of device or procedure related serious adverse events. Secondary endpoints included patient
compliance, subject satisfaction and device and procedure related performance. Due to sample size, the study was not designed to be powered
for statistical significance. In December 2019, we announced the results of the pilot study. No device or procedure related serious adverse
events (SAEs) were reported and all device or procedure related adverse events were mild in severity. In total, 45 patients enrolled in
the study, of which 40 patients underwent the study procedure. 39 patients complied with the procedure and completed a questionnaire following
the procedure and reported higher satisfaction with the C-Scan System procedure compared to colonoscopy. A total of 28 patients were evaluable
after factoring in technical and physiological dropouts and protocol violations. Analysis of the evaluable patient results revealed agreement
between C-Scan and colonoscopy in detection of polyps was consistent with data from the post-CE approval study.
In preparation for our U.S. pivotal study, we have
continued to optimize C-Scan’s functionality and patient experience by additional clinical data collection in a study in Israel,
using advanced C-Scan versions, which incorporate mainly advanced algorithms, improved detection capability and reduced energy consumption.
During the current study in Israel, we initially enrolled both average risk and high-risk patients (e.g., “enriched population”)
and in May 2022, shifted to enrollment of only average risk patients, for the purpose of further calibration of the C-Scan system, in
parallel to conducting the first stage of the U.S. pivotal study that was initiated in May 2022, as described below.
In November 2020, we finalized our proposed U.S
pivotal study design and submitted our IDE application to the FDA and in March 2021, our IDE application was approved by the FDA.
In January 2022, we submitted to the FDA an amended
IDE application to amend the U.S. pivotal study design to add a first part to the study that is designed to enable further calibration
of the C-Scan system and enhancement of C-Scan algorithms, specifically for the average risk U.S. population and in February 2022, our
IDE amended application was approved by the FDA. Following this amended IDE, the U.S. pivotal study consists of two parts. The first part
is designed to enable further calibration of the C-Scan system for the average risk U.S. population, and is intended to include up to
200 patients in the U.S. The second part is a statistically powered, randomized study which will compare the performance of C-Scan to
traditional colonoscopy, and is intended to include up to 800 patients. The goal of the pivotal study in the United States is to
(i) demonstrate device safety as evidenced by a lack of device related serious adverse events; and (ii) provide efficacy data concerning
C- Scan’s performance.
In May 2022, we initiated the first part of the
U.S. pivotal study. In December 2022, we submitted an IDE supplement to the FDA to include enrollment of patients at age 45-75 instead
of 50-75, which was approved by the FDA in January 2023. To date, we have enrolled more than 300 average risk patients as
part of our Israeli study and only 17 average risk patients in the first part of our U.S. pivotal study, mainly as a result of slower
than expected U.S. site recruitment pace, due to licensing requirements with local states associated with the X-ray technology within
our C-Scan capsule. The initiation of the powered potion of the U.S. pivotal study was dependent upon successful completion of the calibration
portion of the U.S. pivotal study. Following our internal assessment of the clinical data collected to date from our calibration
studies, we have determined that the current efficacy results do not meet our goal in order to proceed to the powered portion of the U.S.
pivotal study. As a result, we have adopted a plan of action that includes conducting additional clinical data analysis and approaching
the FDA to make amendments to the U.S. pivotal study protocol that are expected to be part of an IDE supplement submission to the FDA,
and which are subject to FDA approval. In addition, we plan to continue conducting our calibration studies, albeit at a slower pace, to
collect additional clinical data and we also implementing a cost reduction plan, in order to extend our cash runway. The initiation of
the powered portion of the U.S. pivotal study that was expected in mid-2023 is therefore temporarily postponed.
Following and subject to the successful completion
of our U.S. pivotal study and available capital, our current strategy is to submit a direct de novo reclassification petition for FDA
approval for the marketing of C-Scan in the United States. Direct de novo reclassification typically takes at least 9 to 12 months from
filing to clearance if there are no comments or disapproval from the FDA. If the FDA determines that C-Scan is not a candidate for de
novo reclassification, it will require approval of the device for market through the PMA process. The PMA pathway is much more costly
and uncertain than the 510(k) clearance process or de novo reclassification, and generally takes at least 12 to 18 months, or even longer,
from the time the application is filed with FDA to ultimate approval if there are no comments or disapproval from the FDA.
Since our formation, we have not generated any
revenue. We do not anticipate generating any significant revenue for the foreseeable future and we do not yet have any specific launch
dates for our product. We incurred net losses of $19.1 in 2022, $17.2 million in 2021, and $13.8 million in 2020. As of December 31, 2022,
we had an accumulated deficit of $127.3 million and a total shareholders’ equity of $41.3 million.
Our Solution
CRC screening can reduce the incidence of and mortality
from the disease by enabling detection of precancerous polyps in the colon at an earlier, more treatable stage. CRC is one of the few
cancers that can be prevented through screening because pre-cancerous polyps, from which colon cancers often develop, can be identified
and removed. Today, there is a range of options for CRC screening in the average-risk population, with current technology falling into
two general categories: (i) structural exams that enable physicians to visualize the colon for abnormalities, such as optical colonoscopy
(which is currently regarded as the “gold standard” for CRC screening), sigmoidoscopy, CTC and optical capsules. All such
exams require aggressive bowel preparation and are invasive exams; and (ii) stool and serum based tests, such as FOBTs, FITs, stool DNA,
and blood tests, which test for blood in stool and irregularities in blood and DNA. Notwithstanding the many CRC screening alternatives
and despite the fact that the tests are encouraged by clinicians and insurers and the proven clinical value of screening for CRC, a large
portion of the population is still reluctant to perform CRC screening.
C-Scan is designed to enable early detection of
precancerous polyps while providing a patient friendly solution without requiring any colon cleansing. The purpose of C-Scan is to detect
colorectal polyps in subjects who, based on demographics and medical history factors, are considered average risk for CRC. If polyps are
detected, the patient is elevated to high risk for CRC, which studies have shown increases the adherence of patients to undergo a colonoscopy
procedure. If a positive result is obtained through the C-Scan, the patient should be referred to colonoscopy.
Although C-Scan utilizes X-Ray technology (exposing
the patient to approx. 0.05 mSv), we believe that the potential risks associated with such radiation exposure are low compared to the
potential risks associated with other procedures such as perforation, bleeding or sedation related effects (optical colonoscopy and sigmoidoscopy)
and dehydration and damage to kidneys. Unlike FOBTs, FITs and stool DNA tests, our capsule-based imaging modality generates structural
information on the colon, which could assist in the detection of pre-cancerous polyps. We therefore do not believe that the ultra-low-dose
radiation in our capsule will make C-Scan less attractive to physicians and patients than other less-effective products that do not employ
any radiation.
The Radiation Safety Division of the Soreq Nuclear
Research Center found, as set forth in its report of November 2010 that was prepared at our request and based on the information provided
by us and the relevant methods and principles known at such time, or the Report, that the radiation dose to the patient in the proposed
screening procedure utilizing the scanning device developed by us at that time in routine operation and normal conditions is low relative
to the radiation dose involved in conventional imaging procedures using X-rays (such as fluoroscopy and CT) and is also low when compared
to the radiation dose involved in established screening procedures such as mammography, all as more fully described in the Report In addition,
Michael S. Gossman, M.S., DABR, FAAPM, FACR, the Chief Medical Physicist in Radiation Oncology and Radiation Safety Officer, Regulation
Directive Medical Physics, concluded, as set forth in his report dated November 14, 2018 that was prepared at our request and submitted
to the FDA in connection with the IDE application for the U.S. pilot study, following analysis of both normal capsule transit and extreme
conditions, that C-Scan is radiation dose safe for medical diagnostics and exposes patients to very low dose radiation during normal operation
and that even in extreme and unlikely scenarios, patient exposure to radiation is unlikely to cause severe harm to patients undergoing
the C-Scan procedure.
We believe that gastroenterologists will adopt
our technology and recommend the use of our capsule. This may increase the number of people undergoing CRC screening and may encourage
more people with polyps to undergo a polypectomy – a therapeutic procedure during which polyps are removed.
Our goal is to become a leading supplier of CRC
screening technology and to establish our technology as a leading CRC screening method. Key elements of our strategy include:
|
• |
seeking to obtain regulatory approvals for the sale of C-Scan in the United States; |
|
• |
enter into partnerships in the future when strategically attractive, including potentially with major medical
device companies; |
|
• |
obtaining government and private third-party reimbursement for our technology; |
|
• |
improving and enhancing our existing technology portfolio and developing new technologies; and |
|
• |
successfully marketing our product to establish a large customer base, primarily in the U.S. |
Our Technology
Our technology is based on an ingestible capsule
(C-Scan Cap), which is swallowed by the patient and propelled by natural motility through the gastrointestinal tract. Our capsule transmits
information to a receiving device (C-Scan Track) worn on the patient’s body that stores the information for off-line analysis. Our
C-Scan Cap consists of an X-ray source and several X- ray detectors. The X-ray source is contained in a rotating radiation shield, enabling
the generation of 360-degree angular scans. The collection of successive angular scans is intended to enable the virtual reconstruction
of a portion of the colon’s inner surface. During movement of our capsule longitudinally through the colon, successive images of
portions of the colon enable the three- dimensional reconstruction of the colon. C-Scan is also intended to enable structural identification
of polyps, and masses, which protrude inward into the colon, through the detection of irregularities in the topography of the colon’s
inner surface.
C-Scan is intended to be prescribed to patients
by physicians. Prior to capsule ingestion, patients will swallow 15ml of iodinated oral contrast medium, three times a day, combined with
oral fiber, and continue to do so in order to enhance the contrast of the colon surface. The capsule is propelled by natural motility
through the gastrointestinal tract. During transit, information is transmitted to the C-Scan Track, which stores the information for off-line
analysis. After our C-Scan Cap is expelled from a patient’s body, the C-Scan Track data will be downloaded into our workstation
(C-Scan View) and uploaded to a cloud-based server, through which an expert analyst will perform pre-analysis to be followed by a physician’s
final interpretation that includes a findings report and a determination of whether the patient is considered average or elevated risk.
Currently, our internal analysts are expected to perform the complete interpretation of the data and prepare a report accordingly. We
anticipate that we will begin involving physicians in this process upon the initiation of the statistically powered portion of the U.S
pivotal study. Our proprietary software is being designed to process the data and produce a two and three-dimensional visualization of
the colon’s inner surface.
C-Scan consists of the following three main subsystems
that together enable the generation of high-resolution 3D imaging of the colon’s inner surface, further described below: (i) an
ultra- low-dose X-ray based colon scanning capsule (C-Scan Cap); (ii) C-Scan Track; and (iii) a client\server PC-based application (C-Scan-View).
The C-Scan system enables a patient-friendly, preparation
free and painless evaluation of colorectal abnormalities. Using the C-Scan system, suspicious findings that may be colorectal polyps can
be identified, thus assisting the physician in deciding to elevate the patient to high-risk category and refer the patient to a colonoscopy
procedure. Studies have shown that the adherence of an elevated-risk patient to undergo a colonoscopy procedure is higher than that of
an average risk patient.
The C-Scan system provides information for the
physician, enabling him to make a “YES / NO” decision for suspected presence of polyps in the colon, based on the premise
that the presence of polyps in the colon is associated with the potential development of CRC. The C-Scan system is intended to be used
as a preliminary tool to assist in the detecting of subjects who are at elevated risk for polyps and thus may increase the adherence of
those subjects to undergo colonoscopy.
The C-Scan system is designed to evaluate the presence
of polyps, without the need for fasting and prior bowel cleansing, through identifying suspects in the human colon supported by physiological
data, such as measured transit time of the capsule through the GI tract.
The C-Scan system presented in Figure 1 includes
three main units, C-Scan Cap, C-Scan Track and C-Scan View. The C-Scan procedure steps are illustrated in Figure 2. The C-Scan Track is
attached to the patient’s back, after which, the patient ingests the C-Scan capsule. The capsule starts moving through the patient’s
gastrointestinal system until it reaches the colon where it starts scanning the colon walls and transmitting the acquired data to the
C-Scan Track. Following the natural excretion of the capsule, the patient returns the C-Scan Track to enable data loading and analysis
using C-Scan View. The analysis report includes an indication of either “NO- Average Risk” in cases where no suspicious findings
were identified or “YES- Elevated Risk” in cases where suspicious findings were identified. An operational block diagram is
presented below in Figure 4.
Figure 1: Illustration of C-Scan system units

Figure 2: Illustration of C-Scan system
procedure
C-Scan Cap
C-Scan Cap is an X-ray scanning capsule, which
enables detection of suspected polyps. C-Scan Cap is ingested by the patient and propelled by peristalsis, natural motility, it passes
through the gastrointestinal tract and is excreted naturally, with either no need for retrieval or required retrieval of the capsule,
depending on the local agency guidance ( currently, during clinical trials in the U.S., the C-Scan capsule is not intended to be disposed
of in the sanitary sewer system, and is therefore collected by the patient, and returned to the medical facility administering the device
for further decay and disposal.
C-Scan Cap is designed to measure, collect and transmit structural information, and
is comprised of the following components:
|
• |
X-ray Source – Including radioactive material sealed in a cylindrical housing. |
|
• |
Collimator – Radiation shield around the source, which absorbs most of the radiation. Several radial
holes enable emission of radiation in defined directions. |
|
• |
X-ray Sensor – Comprised of several solid state X-ray detectors for measuring the scattered radiation
intensity. |
|
• |
Tilt Sensor – Indication of capsule motion (3D acceleration). |
|
• |
Rotation Motor – For rotating the collimator and X-ray Source. |
|
• |
Compass sensor – Indication of true north (reference coordinate system). |
|
• |
Pressure sensor – indicating the hydrostatic pressure inside the colon. |
|
• |
Source Concealment Mechanism – Conceals the source inside the radiation shield. |
|
• |
R-T – Radio frequency transceiver device to communicate with the receiver. |
|
• |
Batteries – Electrical power supply for the capsule. |
|
• |
Memory – Data storage. The capsule should be able to store up to an hour of measured data.
|
|
• |
C-Scan Track Coil – Transmits a continuous electromagnetic field utilized by an external localization
system to track 3D position. |
Image for illustration purpose only
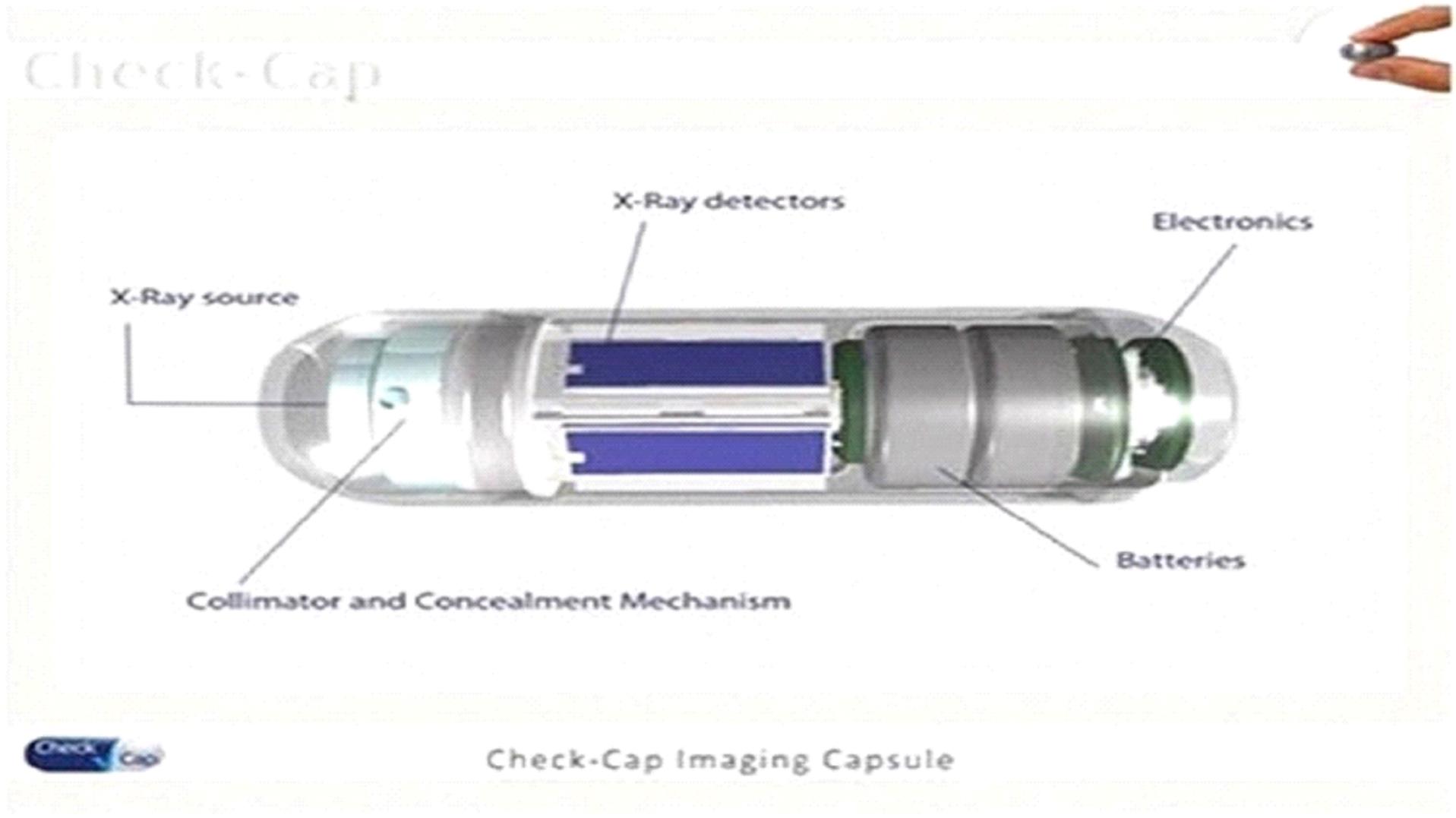
C-Scan Track
C-Scan Track is a small, disposable system attached
to the patient’s back via biocompatible adhesive skin patches. C-Scan Track communicates with the C-Scan Cap and enables data download
for analysis purposes. Both the C-Scan Track and C-Scan Capsule are equipped with an electromagnetic capability allowing for capsule position
and orientation estimations through the C-Scan Track. A dedicated scan control algorithm (SCA) identifies the C-Scan Cap’s movements
in the colon and, accordingly, commands the C-Scan Cap when to perform a scan. C-Scan Track also measures the transit time from capsule
ingestion to excretion through radio frequency communication to a non-volatile memory device, and enable data retrieval, to an external
processor.
The C-Scan Track is comprised of the following
components:
|
• |
Sticker Housings – Biocompatible and water-resistant stickers and housing integrating all functional
components, attached to the patient’s back, enabling approximately five days of continuous operation. |
|
• |
Recorder – Consists of receiver electronics embedded software and nonvolatile memory. |
|
• |
Antennas – Radio frequency antennas are embedded into the sticker housings and used to communicate
with the capsule. |
|
• |
Activation/Deactivation Circuit – Used to activate/deactivate the C-Scan Track through a specialized
protocol. |
|
• |
UI Indicators – Provides user with vocal and vibration indication as required. |
|
• |
PCB – Electronics’ printed circuit boards. |
|
• |
Microcontroller – Runs embedded software, logic that manages the C-Scan Track and SCA. |
|
• |
RF Transceivers – Several transceivers used to communicate with the capsule. |
|
• |
TILT/Compass Sensors – To determine the patient’s body movements. |
|
• |
Batteries – Electrical power supply for the C-Scan Track. |
|
• |
Memory – Non-volatile data storage to store data acquired by the system. |
Image for illustration purpose only
C-Scan View
The C-Scan View software is a client/server-based
application that enables procedure data download from the C-Scan Track, data analysis and report generation. The C-Scan View’s main
functions are:
|
• |
Load and display procedure information and data; |
|
• |
Image review, enabling the user to view structural information of the colon wall; |
|
• |
Produce procedure report; and |
|
• |
Store procedure results on server. |
The data from the C-Scan Track is loaded and processed
to create a reconstruction model of the colon wall, which is displayed in the C-Scan View as structural information of the colon wall,
as well as Whole-Gut-Transit-Time (WGTT) data. The data is analyzed by a team that includes expert analysts, who review the structural
information to identify suspicious findings that are protruding or bulging on the colon wall. The analysts consider capsule’s WGTT
as part of the analysis process. The gastroenterology physician reviews the analysis in the C-Scan View and generates the final report
as to whether the patient is assessed to be at average or at elevated risk, determined through a combined analysis consisting of both
structural analysis and transit time analyses.
Figure 4: Conceptual block-diagram of
the C-Scan system
C-Scan System Non-Clinical and Clinical History
We have developed and validated our capsule-based
imaging modality for providing structural information on colonic polypoid lesions and masses for CRC screening. Below is a summary of
the validation tests carried out by us in the laboratory, in phantoms, animals and humans, which were designed to evaluate this new imaging
modality’s performance and potential clinical value.
Non-Clinical Testing
Imaging Performance Testing
The C-Scan Cap transmits data as it transits the
colon. This data consists of imaged slices perpendicular to the capsule’s longitudinal axis; slices are then reconstructed by the
C-Scan View to produce 2D and 3D images of the inner surface of the colon. Following are performance measurements of the capsule imaging.
|
• |
Modulation Transfer Function, or MTF. The capsule was moved along
a longitudinal-edge phantom setup in 3mm steps. The figure below shows a typical raw signal after filtering for peak detection. The same
test was carried out using an angular-edge phantom setup, which demonstrated similar results to those shown below. These tests do not
take into account noise characteristics. |
Image for illustration purpose only
For each position of the capsule in the phantom,
the mean signal intensity (peak) was measured, the result of which is shown in the right figure below. Resulting line spread function,
or LSF, which is the differential of the curve in the left figure below.
Image for illustration purpose only
The graphs above demonstrate that the existing
design of C-Scan can detect objects of approximately 2-3mm when noise is not taken into account.
|
• |
Resolution Limit: Estimation of the Smallest Visible Object Size. In
order to estimate the size of the smallest visible object, both spatial resolution and noise characteristics must be taken into account.
The graph below presents the estimated MTF of C-Scan. Noise analysis indicates MTF 1/3 for minimum visibility, which demonstrates that
the smallest visible object that can be detected with the existing design of C-Scan (in the conditions used, which included a colon diameter
of 30mm) is of approximately 5-6 mm (see graph below). |
Image
for illustration purpose only

Image Reconstruction
Two main characteristics of C-Scan contribute to the image reconstruction performance:
|
• |
The number of photons hitting the detector per time frame. |
|
• |
The angular spread of the photon beam coming out of the capsule collimator. |
Based on the laboratory tests performed with a
previous version of C-Scan, polyps of 6 mm and larger should be visible and 10 mm polyps and larger are expected to be detected at higher
sensitivity. To further enhance the visibility of 6 mm – 9 mm polyps, a new design of the collimator was successfully incorporated
and tested in our advanced C-Scan Cap that is expected to enable 1.5 times the number of photons to be detected by the detectors, allowing
our recently enhanced algorithm to improve the imaging performance.
Animal Testing and Tissue Equivalent Phantom Image Reconstruction
The physics of our imaging modality was tested
in the laboratory on phantoms with tissue equivalent material and in animals to ensure that laboratory conditions mimic real life clinical
scenarios.
Following the initial proof of concept, we performed
a series of studies in order to evaluate the feasibility and preliminary safety of our technology. All studies were performed in pigs
ranging from 60 to 90 kg. Pigs, which are commonly used in gastrointestinal studies, were selected as the animal model for preliminary
evaluation of C-Scan based on the resemblance of the porcine colon size and morphology to the human colon. However, there are marked differences
between the colon of pigs and that of humans. The pig colon is much longer and has a larger diameter, in addition to other anatomical
differences. In the pig model, the pressure waves of peristalsis are believed to be more frequent and shorter than in humans. As a result,
we believe that colon content movement is substantially slower and more frequent in pigs than in humans. In these studies, we did not
intend to collect statistically significant data; hence, the tests were repeated a limited number of times until adequate data was collected.
The first test was performed to demonstrate imaging
proof-of-concept using a wired C-Scan. This technology included all the basic features intended to be included in the clinical C- Scan,
but on a larger scale due to the use of off-the-shelf components. The subsequent studies used versions of the C-Scan that integrated most
of the imaging components, software and electronics of C-Scan that we used with humans. Since off-the-shelf components were used, the
animal capsules were larger and heavier than the version of C-Scan that are used clinically.
Raw data from an animal colon showing a decrease
in X-ray florescence, or XRF, photon signals and an increase in Compton backscattering, or CMT, signals corresponding to the position
of a polyp that was detected when our C-Scan Cap passed over the polyp is shown in the image below. These two signals are combined in
order to form a three dimensional image below.
Image for illustration
purpose only
The animal studies conducted to date demonstrate
that our technology provides sufficient resolution, in these studies, for the detection of 10 mm polyps which is the size of clinically
significant polyps. The animal studies also demonstrated that 5 mm polyps can be detected, though with lower resolution than 10 mm polyps
in the first animal capsule. Animal health was maintained throughout the studies. No adverse effects related to passage of our capsule
were noted.
The capsules evaluated in the animal studies were
significantly larger than the capsules that we are using with humans. The differences in anatomy, physiology, and capsules may have several
effects on the data compared to use in the human population. Motility of the capsules through the digestive C-Scan was slow due to the
specific shape of the porcine gastrointestinal tract. In addition, because of the size of the capsule, it was retained in the stomach
for many hours and even days. Accordingly, the animal model required that normal ingestion be replaced by direct insertion of the capsule
into the small bowel. In order to simplify the development and animal testing, we used Tungsten radiation source with long half-life (120
days).
Following the success of the animal testing, a
series of in-vitro tests were conducted to simulate different clinical scenarios in the laboratory using a miniaturized human capsule.
Polyps were created and reconstruction of the laboratory phantoms with a human capsule was generated to assess the ability to detect polyps
as the capsule advances in the colon. The in-vitro tests demonstrated the imaging capabilities of our imaging technology. Below is the
reconstruction of a laboratory phantom image.
Image for illustration
purpose only
Polyp Detection Analysis
Laboratory tests were carried out to estimate the
capsule’s ability to detect polyps in phantoms and demonstrate sensitivity and specificity of such detection. Below is an example
of the reconstruction of a scan composed of three slices: XRF, CMT and a fused (combined) image.
Image for illustration
purpose only
Receiver Operating Characteristics
Standard receiver operating characteristics, or
ROC, curves were generated from phantom data with 8 mm polyp in a 30 mm barrel phantom with 3% iodine concentration mimicking the colon
contents. CMT, XRF and fused (combined) data were analyzed based on 2D slices that were generated and standard deviation indicator. There
were a few cases where the noise in the phantoms was high enough to generate polyp false positive condition separately for each data type,
especially in CMT. However, fusion of CMT and XRF data contributed to noise reduction and enabled to demonstrate 100% true positive and
0% false positive.
Image for illustration
purpose only
Clinical Trials
We initiated our first clinical study at University
Hospital, Hamburg, Germany in 2010. The purpose of this study was to monitor and record the colon contractions and the associated motility
of the capsule in the colon. This study was conducted with a passive capsule that contained no X-ray source or detectors. It included
several electronic components of C-Scan and had similar dimensions to the current capsule. 63 healthy volunteers were enrolled and no
adverse events were reported.
We completed a limited, single-center, feasibility
study at Rambam Medical Center, Haifa, Israel to assess the motility of a non-scanning capsule in healthy subjects. The objective of the
study was optimizing the daily routine of the subjects in order to shorten the transit time of our capsule. 15 subjects participated and
swallowed a capsule with the same weight and dimensions as our current C-Scan capsule. No adverse events were reported and all capsules
were retrieved. A structured daily routine determined the timing of the following: capsule ingestion, the subjects’ daily meals,
the contrast agent ingestion and one evening dose of 10 mg of Bisacodyl, and all subjects continued their regular active lifestyles (such
as work and exercise). The average transit time of the capsule in the 15 subjects was approximately 38 ± 19 hours, which is comparable
to the average transit time of our capsule in subjects participating in the multi-center feasibility study, in which the participants
do not ingest a daily dose of Bisacodyl, and participants are released to their homes and continue their regular lifestyles during the
study.
A 10 subject clinical proof-of-concept study, conducted
at Tel Aviv Sourasky Medical Center in Israel and using a prior version of C-Scan, did not identify any material safety or feasibility
issues. The study demonstrated the applicability of C-Scan to the human colon, generating images of the colon without any prior bowel
preparation. All subjects ingested the capsule easily with smooth passage within the designated transit time, on average, within 48-72
hours. There were no reported device-related adverse events. Mild effects on bowel movements were noted, which were determined to be related
to the contrast agent and passed within one to two days after the capsule excretion. Estimated total radiation exposure was calculated
using standard established factors for calculating effective radiation exposure, such as the duration of the capsule inside the body,
and was based on the activity of the radiation source inside the C-Scan Cap and radiation energy, both of which were measured for each
case study. The average calculated exposure for the entire procedure in the 10-case study, from ingestion of the capsule to excretion,
was 0.03 mSv (STD 0.007 mSv). This level of radiation exposure is similar to a single chest X-ray (approximately 0.06mSv) and two orders
of magnitude less than a CTC.
The 10-subject study constituted the initial phase
of a multi-center, prospective clinical feasibility study to establish the safety, functionality and preliminary efficacy of C-Scan in
patients eligible for CRC screening, by comparing results from the clinical feasibility study with those from non-invasive, low-sensitivity
FITs, as well as from optical colonoscopies. The feasibility study has been designed to allow for the recruitment of 100 subjects. The
study was conducted at multiple centers in Israel. The clinical feasibility study was used to evaluate the image resolution generated
by the capsule in a human colon without cathartic preparation, assess polyp imaging in various shapes and in different segments of the
colon, and evaluated the safety of the device in terms of total and segmental transit time and analyze the effects of the presence of
polyps and variable colon dimensions on these parameters. During the feasibility study we collected data regarding the overall imaging
of the colon’s internal surfaces during the passage of the capsule to support the development of a correlation map of polyps identified
through our imaging system with polyps imaged by optical colonoscopy and CTC. Additionally, the feasibility study allowed for the measurement
of total radiation exposure and the distribution of contrast material within the colon.
Analysis conducted on the first 66 capsules swallowed
by participants enrolled in the multi-center, prospective clinical feasibility study showed that 65 of 66 capsules swallowed were naturally
eliminated, without major or minor side effects, after 62±40.7 hours. The average calculated radiation exposure was 0.06 ± 0.04
mSv (similar to a single chest radiograph). Image reconstructions allowed 2D/3D views of the colonic wall and lumen with the typical contour
of different segments (hepatic flexure, triangular shape of the transverse colon). Both pedunculated and sessile polyps were detected
in several patients and validated later by colonoscopy.
In September 2017, we completed a multi-center
study of C-Scan in support of CE Mark submission. The objective of the study was to assess safety and the clinical performance of C- Scan
in detecting patients with polyps. The three-center trial enrolled 66 patients, with a mean age of 59 years. Following capsule ingestion,
subjects swallowed small doses of contrast agent and fiber supplements with each meal throughout capsule passage. Average capsule transit
time was 52±32 hours, and the average total X-ray dose was 0.05 mSv (CT colonography effective dose is ~ 6.0 mSv). No bowel preparation,
sedation, or change in diet was required. Both confirmatory colonoscopy, performed by an independent investigator, and C-Scan review,
performed by a central review group, were blinded to results. The study demonstrated a 44% sensitivity in the 45 subjects included in
the analysis for polyps, with specificity (ability to correctly identify lack of polyps) of 89%. Sensitivity strongly correlated (R-squared
= 0.98) to the percentage of the colon scanned. Sensitivity was 78% (p<0.05) and 100% (p<0.05) for subjects where greater than 50%
and 70% of the colon was scanned, respectively. Specificity was consistent for all subjects.
In the fourth quarter of 2017, we initiated an
interim clinical study for the purpose of introducing an advanced C-Scan version, Version 3, which incorporated the then latest algorithms
and system optimization and tailored scanning of the colon to the patient’s natural colonic movements to maximize the amount of
the colon that is tracked and imaged. In March 2018, we announced results from the interim study. Evaluable results of 21 patients showed
average colon imaging coverage of 64%, a 40% improvement over 46% average colon imaging coverage in the multi-center study. Sensitivity
was 78% (p<0.05) for subjects with greater than 50% colon imaging coverage and 100% (p<0.05) for subjects with greater than 70%
colon imaging coverage. Specificity was consistent at around 89%.
During the first quarter of 2018, we initiated
a multi-center, open label, home monitoring, prospective study designed to determine the performance characteristics of C-Scan Version
3, for detecting pre-cancerous polyps compared with the fecal immunochemical test (FIT), in each case using colonoscopy as the reference
method, for the purpose of collecting additional evidence of clinical effectiveness and clinical utility to support market adoption. The
study included 90 evaluable patients who either had known polyps or were considered to be of average risk. Each patient ingested a C-Scan
capsule and also underwent a FIT and a comparative colonoscopy performed by independent gastroenterologists, who were blinded to the corresponding
test’s results. The C-Scan clinical evaluation was obtained using the evaluable patient population implementing a gender-based motility
analysis and the results of both C-Scan and FIT were compared to colonoscopy. The primary efficacy endpoint of the study was sensitivity
(ability to correctly identify patients with polyps) and specificity (ability to correctly identify patients with lack of polyps) of the
C-Scan system compared to FIT in detecting subjects with polyps ≥10 mm. In July 2019, we announced final results from our post-CE
approval study. The results demonstrate that C-Scan achieved a sensitivity of 76% (p=0.0005) in patients with polyps ≥10 mm, while
FIT achieved a sensitivity of 29% (p=0.005) in patients with polyps ≥10 mm. C-Scan achieved a specificity of 82% in all patients,
while FIT achieved a specificity of 96% in all patients. In addition, C-Scan detected all 4 patients (100%) with polyps ≥40 mm,
while the FIT detected only 1 of the 4 patients (25%) with polyps ≥40mm. Overall, C-Scan achieved a sensitivity of 66% (p=0.01)
in all patients, including patients with polyps <10mm, while FIT achieved a sensitivity of 23% (p<0.0001) in all patients, including
patients with polyps <10mm. In total, 142 patients enrolled in the study and after factoring in technical and physiological dropouts
and protocol violations, the number of evaluable patients was 90. No serious adverse events were reported, and the adverse events were
mild in severity.
In December 2018, we received from the FDA conditional
approval of our IDE application to initiate a U.S. pilot study of the C-Scan Version 3 and received final approval from the FDA in February
2019. In April 2019, we initiated the U.S. pilot study of C-Scan. The U.S. pilot study (NCT03735407) was a prospective, multi-center,
open label, single arm study was designed to evaluate the safety, usability and subject compliance of the C-Scan. The study included 28
evaluable patients, more than two thirds of whom were considered to be of average risk for colorectal cancer. Each patient ingested a
C-Scan capsule and also underwent a fecal immunochemical test (FIT) as well as a comparative colonoscopy, which was performed by an independent
gastroenterologist who was blinded to the corresponding test results. The study was performed at two sites, the NYU Grossman School of
Medicine and Mayo Clinic, Rochester. The primary endpoint of the study was to evaluate the incidence of device or procedure related serious
adverse events. Secondary endpoints included patient compliance, subject satisfaction and device and procedure related performance. Due
to sample size, the study was not designed to be powered for statistical significance. In December 2019, we announced the results of the
study. No device or procedure related serious adverse events (SAEs) were reported and all device or procedure related adverse events were
mild in severity. In total, 45 patients enrolled in the study, of which 40 patients underwent the study procedure. 39 patients complied
with the procedure and completed a questionnaire following the procedure and reported higher satisfaction with C-Scan procedure compared
to colonoscopy. A total of 28 patients were evaluable after factoring in technical and physiological dropouts and protocol violations.
Analysis of the evaluable patient results revealed agreement between C-Scan and colonoscopy in detection of polyps was consistent with
data from the post-CE approval study.
In preparation of the U.S. pivotal study, we continued
to optimize the functionality of our advanced version of C-Scan, which incorporates mainly advanced algorithms, improved detection capability
and reduced energy consumption, through additional clinical data collection in a study in Israel (initially on average risk and high-risk
patients and in May 2022, shifted to enrollment of only average risk patients), for the purpose of further calibration of the C-Scan system,
which we have continued in parallel to conducting the first stage of the U.S. pivotal study that was initiated in May 2022.
In November 2020, we finalized our proposed U.S.
pivotal study design and submitted our IDE application to the FDA and in March 2021, our IDE application was approved by the FDA. In January
2022, we submitted a supplement to the FDA to amend the U.S. pivotal study design to add a first part to the study that is designed to
enable further device calibration of the C-Scan system and enhancement of C-Scan algorithms, specifically for the average risk U.S. population.
In February 2022, we received the FDA’s approval for our IDE supplement. Following this amended IDE, the U.S. pivotal study consists
of two parts. The first part is designed to enable further calibration of the C-Scan system for the average risk U.S. population, and
is intended to include up to 200 patients in the U.S. The second part is a statistically powered, randomized study which will compare
the performance of C-Scan to traditional colonoscopy, and is intended to include up to 800 patients. The goal of the pivotal study
in the United States is to (i) demonstrate device safety as evidenced by a lack of device related serious adverse events; and (ii) provide
efficacy data concerning C- Scan’s performance.
In May 2022, we initiated the first part of the
U.S. pivotal study, which focuses on device calibration and enhancement of C-Scan algorithm among average risk population, initially at
Mayo Clinic Rochester, Minnesota and New York University (NYU) School of Medicine. In addition, we recently added a new site as well as
expanded the NYU site.
In January 2023, we obtained approval from the
FDA for our protocol amendment to lower the patient age enrollment criteria to 45-75 years (from 50-75 years previously) in the U.S. pivotal
study. Our NYU site has already obtained approval of this amendment from its Institutional Review Board (IRB) and begun to implement
it to recruit younger patients as part of the first part of our U.S. pivotal study.
To date, we have enrolled more than 300 average
risk patients as part of our Israeli study and only 17 average risk patients in the first part of our U.S. pivotal study, mainly as a
result of slower than expected U.S. site recruitment pace, due to licensing requirements with local states associated with the X-ray technology
within our C-Scan capsule.
The initiation of the powered potion of the U.S.
pivotal study was dependent upon successful completion of the calibration portion of the U.S. pivotal study. Following our internal
assessment of the clinical data collected to date from our calibration studies, we have determined that the current efficacy results do
not meet our goal in order to proceed to the powered portion of the U.S. pivotal study. As a result, we adopted a plan of action
that includes conducting additional clinical data analysis and approaching the FDA to make amendments to the U.S. pivotal study protocol
that are expected to be part of an IDE supplement submission to the FDA, and which are subject to FDA approval. In addition, we plan to
continue conducting our calibration studies, albeit at a slower pace, to collect additional clinical data and we are also implementing
a cost reduction plan, in order to extend our cash runway. The initiation of the powered portion of the U.S. pivotal study that was expected
in mid-2023 is therefore temporarily postponed.
Research and Development
Our research and development strategy is centered
on developing C-Scan. Our research and development team consists of 76 individuals employed (including independent contractors) on a full-time
basis and 3 individuals on a part-time basis as of March 15, 2023, including our research and development employees, clinical team, quality
and assurance team and manufacturing team. All our research and development employees are located at our facility in Isfiya, Israel, except
for 5 full time employees who are located at our facility in Petach Tikva, Israel and one independent contractor who is located in the
U.S.
We have received grants from the Government of
the State of Israel through the IIA (formerly the OCS) for the financing of a portion of our research and development expenditures and
a portion of our expenditures relating to our transition to manufacturing pursuant to the Innovation Law and related regulations and guidelines.
As of December 31, 2022, we had received funding from the IIA for research and development of our C-Scan in the aggregate amount of approximately
$5.6 million. As of December 31, 2022, we had not paid any royalties to the IIA and had a contingent liability to the IIA with respect
to such funding (including interest at the rate of 12-month LIBOR) in the amount of approximately $6.2 million. In addition, in January
2021, we received an IIA grant approval to support the funding of our transition from research and development to manufacturing. The final
IIA grant amounted to approximately $620,000 (NIS 2.18 million) (along with a co-investment by us of the same amount), which we are not
required to repay to the IIA, subject to the terms and conditions set forth in the grant approval, of which we received approximately
$222,000 (NIS 784,000) in February 2023, $82,000 (NIS 264,000) in January 2022 and $349,000 (NIS 1,134,000) in 2021. We may apply for
additional IIA grants in the future; however, there is no assurance that such applications will be approved in the amount requested or
at all. We cannot predict whether we will be entitled to any future grants, or the amounts of any such grants. See “Risk
Factors – Risks Related to Our Operations in Israel – Pursuant to the terms of the Israeli government grants we received for
research and development expenditures, we are obligated to pay certain royalties on our revenues to the Israeli government. In addition,
the terms of Israeli government grants we received require us to satisfy specified conditions and to make additional payments in addition
to repayment of the grants upon certain events.”
We incurred approximately $14.3 million, $12.3 million and $10.0 million in research
and development expenses, net (after deducting participation by government grants) for the years ended December 31, 2022, 2021, and 2020,
respectively. For additional information, see “Management’s Discussion and Analysis of Financial Condition and Results of
Operations— Financial Operations Overview—Research and Development Expenses, Net.”
Intellectual Property
An important part of our competitive strategy is
to seek, when appropriate, protection for our products and proprietary technology through a combination of U.S. and foreign patents, trademarks,
trade secrets, non-disclosure and confidentiality, assignment of invention and other contractual arrangements with our employees, consultants
and suppliers. These measures, however, may not be adequate to protect our technology from unauthorized disclosure, third-party infringement
or misappropriation as these parties may breach these agreements, and we may not have adequate remedies for any such breach. We intend
to prosecute and defend our proprietary technology. The primary test for patent protection eligibility includes patentability, novelty,
non- obviousness, and utility.
We submit applications under the Patent Cooperation
Treaty, or PCT, which is an international patent law treaty that provides a unified procedure for filing a single initial patent application
to seek patent protection for an invention simultaneously in each of the member states. Although a PCT application is not itself examined
and cannot issue as a patent, it allows the applicant to seek protection in any of the member states through national-phase applications.
As of December 31, 2022, we had 56 granted patents
(not including separate validations in Europe) in main world markets, covering various aspects of our technology. In addition, we have
19 additional pending patent applications in the pipeline. We have submitted patent applications covering our technology in the United
States, member states of the European Patent Organization, Australia, Brazil, Canada, China, Hong Kong, India, Israel, Japan and South
Korea. We have received patent grants for our core patent by the United States Patent and Trademark Office as well as from the European
Patent Office, Australia, Brazil, China, Hong Kong, Israel, India, Canada, South Korea and Japan. We also filed patent applications describing
the use of our technology in several other medical applications.
Our registered U.S. Patent Number 7,787,926 discloses
an ingestible capsule with a radiation source and radiation detectors that, when used in conjunction with a radio opaque contrast agent,
is adapted to detect clinically relevant findings in the colon. Utilizing X-ray fluorescence and Compton back scatterings, the capsule
is able to measure the distance between the capsule and the colon wall and to distinguish between gas, intestinal contents, and clinically
significant findings in the gastrointestinal tract. If the appropriate maintenance, renewal, annuity or other governmental fees are paid,
the non-extended patent term for this patent will expire on August 28, 2026.
A PCT patent application (PCT/IL2008/000163), granted
in Europe, Israel, United States, Australia, India, China, Canada and Hong Kong, discloses additional features such as a rotating collimator
and improved scanning mechanisms, the capability to determine tissue density to differentiate between different types of polyps, as well
as the capability to determine capsule movement in the colon. If the appropriate maintenance, renewal, annuity or other governmental fees
are paid, the non-extended patent term for all national phases will expire on February 6, 2028, other than the U.S. patent that will expire
on April 4, 2030.
Another PCT application (PCT/IL2011/000462), granted
in Europe, Israel, United States, Canada, Australia, South Korea and Brazil, discloses a number of alternate fail safe concealment mechanisms
that can be utilized in the capsule to ensure that the X-ray source is blocked when the capsule is not scanning and is open when it is
scanning, allowing the capsule to image the colon. The fail-safe feature ensures that in the event of power failure, the radiation source
is blocked and X-rays do not escape. If the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended
patent term for all national phases will expire on June 9, 2031, other than the U.S. patent that will expire on July 5, 2032.
In another PCT patent application (PCT/IL2008/000765),
which was granted in the United States, Europe, Israel and Japan, we disclose an imaging catheter that utilizes X-ray fluorescence, Compton
back scattering and electron back scattering. The imaging catheter is designed for use in cardiac applications as well as intra-operative
imaging applications such as imaging inside blood vessels where optical imaging cannot be performed because of obscuring circumstances.
If the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non- extended patent term for all national phases
will expire on June 4, 2028, other than the U.S. patent that will expire on July 28, 2028.
In addition, during the year 2022 we were granted
two new patents:
"Position Estimation of Imaging Capsule in Gastrointestinal
Tract" granted in Israel, covering C-Scan's proprietary tracking technology, which enables real time tracking of the capsule and its activation
when it moves throughout the colon. This functionality allows for optimal scanning along the gastrointestinal (GI) tract while maintaining
low energy consumption during the procedure. The patent also covers the capsule positioning data recording utilized by the C-Scan analysis
suite, which enables gastroenterologists to make a clinical decision and generate a report with their diagnosis and recommendations.
Corresponding patents were granted in Japan, China, Europe and the United States.
"System and method for polyp detection through
capsule dynamics" granted in Japan. This patent was submitted following analysis of hundreds of patients that showed that certain
capsule dynamic properties correlate with increased likelihood for the presence of polyps. Corresponding patents were filed in Europe and
the United States.
While our policy is to obtain patents by application,
to maintain trade secrets and to seek to operate without infringing on the intellectual property rights of third parties, technologies
related to our business have been rapidly developing in recent years. Additionally, patent applications that we may file may not result
in the issuance of patents, and our issued patents and any issued patents that we may receive in the future may be challenged, invalidated
or circumvented. For example, we cannot predict the extent of claims that may be allowed or enforced in our patents nor be certain of
the priority of inventions covered by pending third-party patent applications. If third parties prepare and file patent applications that
also claim technology or therapeutics to which we have rights, we may have to partake in proceedings to determine priority of invention,
which could result in substantial costs to us, even if the eventual outcome is favorable to us. Moreover, because of the extensive time
required for clinical development and regulatory review of a product we may develop, it is possible that, before C-Scan can be commercialized,
related patents will have expired or will expire a short period following commercialization, thereby reducing the advantage of such patent.
Loss or invalidation of certain of our patents, or a finding of unenforceability or limited scope of certain of our intellectual property,
could have a material adverse effect on us. See “Item 3D “Key Information—Risk Factors—Risks Related to Our Intellectual
Property.”
In addition to patent protection, we rely on trade
secrets, including unpatented know-how, technology innovation, drawings, technical specifications and other proprietary. We also rely
on protection available under trademark laws, and hold the following registered trademarks in the United States and Hong Kong: “CHECK
CAP”, CHECK CAP (logo), “C-Scan” and C-Scan (logo) and the following registered trademarks in the European Union: CHECK
CAP (logo), “C-Scan” and C-Scan (logo), and the following trademark applications are pending in China: “CHECK CAP”,
CHECK CAP (logo), “C-Scan” and C-Scan (logo).
In March 2021, we entered into an exclusive license
agreement with the University of Missouri with respect to certain patents held by the University of Missouri that the University of Missouri
claimed included background intellectual property in C-Scan. In consideration for the grant of an exclusive license to those patents in
the medical field, we agreed to pay royalties ranging from $0.30 to $5.00 per C-Scan unit depending on the number of units sold up to
$15,000,000 in the aggregate.
Competition
Competition for C-Scan comes from traditional well-entrenched
manufacturers of tests and equipment for CRC screening, such as colonoscopy, sigmoidoscopy, CTC, optical capsule endoscopy, FOBTs and
FITs. The principal manufacturers of equipment for optical colonoscopy and sigmoidoscopy include Olympus, Pentax, Hoya and Fuji Film.
The principal manufacturers of equipment for CTC include General Electric Healthcare Systems, Siemens Medical Solutions, Philips Healthcare
and Toshiba Corporation. The principal manufacturer of equipment for optical capsule endoscopy includes Medtronic plc. All of these companies
have substantially greater financial resources than we do, and they have established reputations as well as worldwide distribution channels
for providing medical instruments to physicians.
In addition, several companies have developed or
are developing non-invasive technologies based on stool, serum (from blood and other bodily fluids), or molecular diagnostics (MDx), tests
that are used to indicate the presence of CRC and polyps in the colon. These companies include Exact Sciences, Polymedco, Epigenomics
AG, Gene News, EDP Biotech Corporation, Illumina, Inc., Quest Diagnostics, VolitionRx Nu.Q diagnostic, Freenome , Grail, , Guardant, Genoscopy
and Universal DX. Exact Sciences commenced sales of its Cologuard ®, a non-invasive stool DNA screening test for colorectal cancer
in 2015, and is constantly seeking performance improvements through additional studies, alongside commercial expansion. During the past
years, there has been an extensive effort on the part of our competitors to utilize molecular biology and advanced computational techniques
to develop methods capable of identifying cell-free cancer biomarkers.
In December 2022, Guardant Health released results
from ECLIPSE (Evaluation of ctDNA LUNAR Assay In an Average Patient Screening Episode), an over 20,000 patient registrational study evaluating
the performance of its blood test for detecting colorectal cancer (CRC) in average-risk adults. The test demonstrated 83% sensitivity
in detecting individuals with CRC. Specificity was 90% in both individuals without advanced neoplasia and in those who had a negative
colonoscopy result. This test also demonstrated 13% sensitivity in detecting advanced adenomas. In this study, two configurations of a
multimodal blood-based screening test were evaluated independently - a cell-free DNA (cfDNA)-only test and a cfDNA test with protein biomarkers.
The announced results were derived from the cfDNA-only test, which outperformed the cfDNA test with protein biomarkers. Based on these
study results, Guardant Health plans to complete its premarket approval submission to the FDA in the first quarter of 2023.
In January 2023, Geneoscopy Inc., released results
from the CRC-PREVENT trial – a pivotal clinical trial evaluating the efficacy of its noninvasive, stool-based, at-home diagnostic
screening test to detect colorectal cancer (CRC) and advanced adenomas (AA) in average-risk individuals. The CRC-PREVENT trial included
8,289 individuals with diverse racial, ethnic, and socioeconomic backgrounds across more than 2,900 zip codes in all lower 48 states,
with colonoscopies performed in more than 3,800 endoscopy centers. Efficacy results from the study demonstrated 94% sensitivity for detecting
CRC, 45% sensitivity for detecting advanced adenoma (AA), and 88% specificity for no findings on a colonoscopy. The company submitted
a Premarket Approval (PMA) application to the FDA in January 2023.
These advancement efforts are assisted by large
capital investments, supported in turn by claims that certain products may promise the possibility of defeating certain cancers in their
early stages.
Procedures for bowel cleansing that are less onerous
are constantly being developed, which could make our entry into the market more difficult. For instance, bowel cleansing initiated by
the ingestion of pills or food-substitute diet regimes rather than through drinking large amounts of distasteful liquids may be viewed
as an improvement to the cleansing process, but other screening methods may be even more palatable to patients.
Sales and Marketing
Our goal is to establish our position as a leading
player in the CRC screening market. Although we do not currently generate revenues, we expect to generate revenues through sales of C-Scan
following demonstration of acceptable clinical safety and effectiveness and obtaining required regulatory approvals and licensures.
Because we are still in the clinical and development
stage, we are subject to certain challenges, including, among others, that:
|
• |
our technology has been tested on a limited basis and therefore we cannot assure the product’s clinical
value; |
|
• |
we need to raise an amount of capital sufficient to complete the development of our technology, obtain
the requisite regulatory approvals and commercialize our current and future products; |
|
• |
following the receipt of CE Mark of conformity to protection standards for sale of the C-Scan system in
the European Union, we may need to obtain additional regulatory approvals in certain local jurisdictions in the European Union before
we can commence marketing and sale of C-Scan and will need to obtain the requisite regulatory approvals in the United States, Japan and
other markets where we plan to focus our commercialization efforts; |
|
• |
we need to obtain reimbursement coverage from third-party payors for procedures using C-Scan; |
|
• |
we need to scale-up our manufacturing capabilities of C-Scan in commercial quantities at an adequate quality
and at an acceptable cost; and |
|
• |
we need to establish and expand our user base while competing against other sellers of capsule endoscopy
systems as well as other current and future CRC screening technologies and methods. |
Our ability to operate our business and achieve
our goals and strategies is subject to numerous risks as described more fully in “Risk Factors.”
Our primary corporate objective is obtaining the
U.S. clearance/approval for C-Scan. However, based on the MDR approval that we received from our notified body according to the EU Regulation,
available capital and market assessment, we plan to evaluate opportunities in Europe and Israel, including possible engagement with strategic
partners and/or local distributors who are active in the gastroenterology field.
In Europe, we currently expect to offer C-Scan
as an imaging and screening tool for the general population. In the United States, we may choose to first obtain regulatory clearance/approval
for C- Scan in a screening sub-population, and after we have conducted more extensive clinical studies supported by compelling evidence
in the United States, we would anticipate applying to the FDA for the use of C-Scan as a primary screening tool.
Subject to the successful completion of our clinical
trials and the receipt of our initial FDA clearance/approval, we expect to launch the product in the U.S. market, where we will consider
to either set up our own sales force or align with a strategic partner. Initially, we anticipate selling C-Scan to the private sector.
In parallel to our efforts to obtain FDA approval, we plan to build and execute our strategy for reimbursement, primarily for Medicare
and private insurers
Subject to local regulatory approval, we also may
at a later stage market C-Scan in key markets in Asia.
Manufacturing and Suppliers
Our manufacturing operations are conducted at our
facility located in Isfiya, Israel and in our own X-ray production line in Petach Tikva. In Isfiya, we lease approximately 1,550 square
meters at this facility under a lease agreement expiring on December 31, 2023, and we have an option to extend the agreement for an additional
period of three years. We have the right to terminate the Isfiya lease agreement at any time, upon at least 60 days prior written notice.
In December 2021, we signed a Sub-lease Agreement with one of our suppliers, according to which we lease approximately 70 square meters
of laboratory production space at the supplier’s premises. The Sub-lease Agreement entered into effect on September 21, 2022, after
receipt of certain certificates, including a business license from the Petach Tikva municipality. The Sub-lease Agreement expires on January
31, 2025, and we have an option to extend the lease period for an additional two years. We have the right to terminate the agreement at
any time, upon at least 90 days prior written notice. The lessor has the right to terminate the Sub-lease Agreement, upon at least 12
months prior written notice. We currently have sufficient space to manufacture C-Scan for the contemplated clinical studies, but have
limited resources and facilities in commercially manufacturing large quantities of C-Scan and C-Scan View to meet the demand we expect
for our future commercialization efforts. We have faced and expect to face certain technical challenges as we increase manufacturing capacity,
including, among others, logistics associated with the handling of radioactive materials, equipment design and automation, material procurement,
lower than expected yields and increased scrap costs, as well as challenges related to maintaining quality control and assurance standards,
while maintaining commercially efficient pricing for C-Scan. Our production objective is to establish a scalable capacity in order to
meet such expanded demand for C-Scan and market expansion. If we are unable to scale up our manufacturing capabilities to meet our clinical
trials needs and future market demand, our growth could be limited and our business, financial condition and results of operations could
be materially adversely affected.
In July 2016, we entered into an agreement with
GE Healthcare to develop and demonstrate proof of principle of the process for high-volume manufacturing for the production of the X-
ray source and its assembly into our C-Scan capsule. The agreement was amended in November 2017 in order to further our manufacturing
collaboration with GE Healthcare. The agreement involves GE’s final assembly, packaging and shipping of C-Scan capsules. We supplied
GE Healthcare with supporting calibration and final assembly methodology and equipment and in August 2019, we announced the completion
of manufacturing line transfer implementation and qualification for C-Scan operated by GE Healthcare, primarily to enable the availability
of C-Scan for U.S. clinical trials. At present, we intend to ship systems for our U.S. clinical studies directly from our local site in
Israel.
In January 2021, we received an IIA grant approval
to support the funding of our transition from research and development to manufacturing. The final IIA grant amounted to $620,000 (NIS
2.18 million) including for the purpose of the establishment of our own X-Ray source manufacturing line in Israel. We do not currently
have any sales. We are planning to develop a scale-up plan to meet our expected commercial supply needs. During 2022, we expanded our
manufacturing capacity to support the expected larger clinical volume and subsequent higher volumes expected in the early commercialization
stage. We are continuously exploring whether certain components of C-Scan can be assembled outside of Israel. In addition, we are exploring
possible alternative sources for the key capsule components. All of the facilities in which manufacturing and assembly of our products
will be conducted (including those of sub-contractors) will need to comply with applicable regulations and standards for medical devices.
We currently depend on sole or single source suppliers
for some of the components necessary for the production of C-Scan. For example, we currently have a sole supplier for the motor used to
rotate the collimated X-ray source in C-Scan, for the customized X-ray detectors and batteries used in C-Scan. There is a limited number
of manufacturers worldwide who are capable of manufacturing the motor, the customized X-ray detectors and the batteries that we currently
use in C-Scan. In addition, the ASIC residing in C-Scan is currently manufactured for us by a single FAB. However, there are many alternative
FABs worldwide and the design of our current ASIC could be adapted in the event it became necessary to use an alternative FAB. Furthermore,
we do not currently have written contracts with all our sole or single suppliers. While our current suppliers have been able to supply
the required quantities of such components to date, if the supply of these components is disrupted or terminated or if our current suppliers
are unable to supply required quantities of components now or in the commercialization period, we may not be able to find alternative
sources for these key components in a timely manner. Although we are planning to maintain strategic inventory of key components, the inventory
may not be sufficient to satisfy the demand for C-Scan if such supply is interrupted, or subject to risk of loss due to catastrophic events
such as a global political crisis or war, or a fire at our storage facility. In addition, to partially mitigate the risks of reliance
on sole and single source suppliers, we are seeking alternate manufacturers for some of our components which requires us to dedicate significant
resources and investment. There can be no assurance that we will be successful in seeking alternate suppliers or establish our own production
line. As a result of the foregoing, we may be unable to meet our clinical trials timelines and to meet the demand for C-Scan, which could
harm our ability to generate revenues, lead to customer dissatisfaction and damage our reputation. If we are required to change the supplier
of any of these key components, there may be a significant delay in locating a suitable alternative manufacturer. In addition, we may
be required to verify that the new manufacturer maintains facilities and procedures that comply with FDA and other applicable quality
standards and with all applicable regulations and guidelines. As part of our mitigation plan, in December 2021, we signed the Amended
Service Agreement with the sole supplier of the X-ray source used in C-Scan, and we concurrently entered into the Sub-lease Agreement
with the supplier, according to which we agreed to lease approximately 70 square meters of laboratory production space at the supplier
premises. According to the Amended Service Agreement, we independently produce the X-ray source using our own production employees. The
Amended Service Agreement extended the terms of the service agreement we had with the sole supplier until the Sub-lease Agreement entered
into effect on September 21, 2022, after receipt of certain certificates, including a business license from the Petach Tikva municipality.
Upon successful progress of our clinical studies and available sufficient capital, we plan to continue to expand our own production line
for our X-ray source in Israel, however, we cannot assure you that we will commence this effort or succeed in expanding our in house production
line and we may be required to find an alternative source for the X-ray source, which we may not be able to do in a timely manner. The
delays associated with the introduction of a new manufacturer for certain key components or new suppliers for certain materials, could
delay our ability to manufacture C-Scan in a timely manner or within budget. Furthermore, in the event that the manufacturer of a key
component of C-Scan ceases operations or otherwise ceases to do business with us, we may not have access to the information necessary
to enable an alternative supplier to manufacture the component. The occurrence of any of these events could harm our ability to meet demand
for C-Scan in a timely manner or within budget.
Environmental Health and Safety Matters
We are subject to environmental health and safety
laws and regulations in Israel, governing, among other things, the use of radioactive materials, including the Israeli Radioactive Elements
and their Products, Regulation, 1980, the Hazardous Substances Law, 5753-1993 and regulations thereunder, including the Licensing of Business
Regulations (Disposal of Hazardous Substances), 5750-1990, the Israeli Work Safety Regulations (Occupational Safety and Health of Ionizing
Radiation Practitioners) 1992-5753 and Women Employment Regulations (Work with Ionizing Radiation), 1979-5739. Our current research and
development activities, including our production activities, require (and our currently contemplated commercial activities will require)
permits from various governmental authorities including, Israel’s Ministry of Environmental Protection, Israel’s Ministry
of Health, local municipal authorities and the NRC and Agreement State regulators. Failure to obtain or maintain any such permits or to
comply with their conditions could have a material adverse effect on our business, financial condition and results of operations. The
Ministry of Environmental Protection and the Ministry of Health conduct from time to time periodic inspections in order to review and
ensure our compliance with the various regulations.
These laws, regulations and permits could potentially
require expenditure by us for compliance or remediation. If we fail to comply with such laws, regulations or permits, we may be subject
to fines and other civil, administrative or criminal sanctions, including the revocation of permits and licenses necessary to continue
our business activities and could be subject to work stoppages or delays. In addition, we may be required to pay damages or civil judgments
in respect of third-party claims, including those relating to personal injury (including exposure to radioactive materials) or contribution
claims. Some environmental, health and safety laws apply strict, joint and several liability. We may be identified as a responsible party
under such laws. Such developments could have a material adverse effect on our business, financial condition and results of operations.
In addition, laws and regulations relating to environmental,
health and safety matters are often subject to change. In the event of any changes or new laws or regulations, we could be subject to
new compliance measures or to liabilities for activities which were previously permitted.
U.S. Government Regulation
C-Scan is a medical device subject to extensive
regulation by FDA and other U.S. federal and state regulatory bodies. To ensure that medical products distributed in the United States
are safe and effective for their intended use, FDA has promulgated regulations that govern, among other things, the following activities
that we or our partners perform and will continue to perform:
|
• |
product design and development; |
|
• |
validation and verifications; |
|
• |
product manufacturing (good manufacturing practices); |
|
• |
product storage, shipping and handling; |
|
• |
premarket clearance or approval; |
|
• |
advertising and promotion; |
|
• |
product marketing, sales and distribution; |
|
• |
post-market surveillance reporting death or serious injuries and medical device reporting. |
FDA’s Premarket Clearance and Approval Requirements
Unless an exemption applies, before we can commercially
distribute medical devices in the United States, we must obtain, depending on the type of device, either prior premarket notification,
or 510(k) clearance, de novo authorization or approval of a Premarket Approval Application, or PMA from the FDA. The FDA classifies medical
devices into one of three classes:
|
• |
Class I devices, which are subject to only general controls (e.g.,
labeling, medical devices reporting, and prohibitions against adulteration and misbranding) and, in some cases, to the 510(k) premarket
clearance requirements; |
|
• |
Class II devices, generally requiring 510(k) premarket clearance before they may be commercially marketed
in the United States; and |
|
• |
Class III devices, consisting of devices deemed by the FDA to pose the greatest risk, such as life-sustaining,
life-supporting or implantable devices, or devices deemed not substantially equivalent to a predicate device, generally requiring the
submission of a PMA approval supported by clinical trial data. |
510(k) Clearance Pathway
To obtain 510(k) clearance, we must submit a premarket
notification, or 510(k) notice, demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device
or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of premarket
approval applications. By regulation, a premarket notification must be submitted to the FDA at least 90 days before we intend to market
a device, and we must receive 510(k) clearance from the FDA before we actually market the device. The Medical Device User Fee Amendments,
or the MDUFA, performance goal for a traditional 510(k) clearance is 90 days. As a practical matter, however, the 510(k) clearance pathway
may take approximately between 6 to 9 months, but it can take significantly longer. FDA may require additional information, including
clinical data, to make a determination regarding substantial equivalence.
There are three types of 510(k)s: traditional,
special, and abbreviated. Special 510(k)s are typically for devices that have certain technological, design or labelling changes that
require a new 510(k) but where the method(s) to evaluate the changes are well established and the results of change evaluation can be
sufficiently reviewed in a summary or risk analysis format. Abbreviated 510(k)s are for devices that conform to special controls for the
device type or to a recognized standard. The special and abbreviated 510(k)s are intended to streamline review, and the FDA intends to
process special 510(k)s within 30 days of receipt.
After a device receives 510(k) clearance, any modification
that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will
require a new 510(k) clearance or, depending on the modification, require premarket approval. The FDA requires each manufacturer to determine
whether the proposed change requires submission of a 510(k), or a premarket approval, but the FDA can review any such decision and can
disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA can require
the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or premarket approval is obtained. If the
FDA requires us to seek 510(k) clearance or premarket approval for any modifications to a previously cleared product, we may be required
to cease marketing or recall the modified device until we obtain this clearance or approval. Also, in these circumstances, we may be subject
to significant regulatory fines or penalties.
De Novo Reclassification
Medical device types that the FDA has not previously
classified as Class I, II or III are automatically classified into Class III regardless of the level of risk they pose. To market low
to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, a manufacturer
may request a de novo down-classification. This procedure allows a manufacturer whose novel device is automatically classified into Class
III to request classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk,
rather than requiring the submission and approval of a PMA application. A medical device may be eligible for de novo classification if
the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially
equivalent or a manufacturer may request de novo classification directly without first submitting a 510(k) premarket notification to the
FDA and receiving a not substantially equivalent determination. The FDA is required to classify the device within 120 calendar days following
receipt of the de novo application, although in practice, the FDA’s review may take significantly longer. During the pendency of
the FDA’s review, the FDA may issue an additional information letter, which places the de novo request on hold and stops the review
clock pending receipt of the additional information requested. In the event the de novo requestor does not provide the requested information
within 180 calendar days, the FDA will consider the de novo request to be withdrawn. If the manufacturer seeks reclassification into Class
II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety
and effectiveness of the medical device. In addition, the FDA may reject the de novo request for classification if it identifies a legally
marketed predicate device that would be appropriate for a 510(k) or determines that the device is not low to moderate risk or that general
controls would be inadequate to control the risks and special controls cannot be developed. In the event the FDA determines the data and
information submitted demonstrate that general controls or general and special controls are adequate to provide reasonable assurance of
safety and effectiveness, the FDA will grant the de novo request for classification. When the FDA grants a de novo request for classification,
the device is granted marketing authorization and further can serve as a predicate for future devices of that type, through a 510(k) premarket
notification.
Our current strategy is to submit a direct de novo
reclassification petition for C-Scan. To support a direct de novo reclassification petition, our objective is to demonstrate that the
device poses a low to moderate risk to patients. If the FDA determines that C-Scan is not a candidate for de novo reclassification, it
will require approval of the device for market through the PMA process.
Alternatively, if we seek 510(k) clearance and
our device is found not substantially equivalent, or NSE, a de novo petition must be filed within 30 days from the receipt of the NSE
determination. The request should include a description of the device, labeling for the device, reasons for the recommended classification
and information to support the recommendation. The de novo process following an NSE determination has a 60-day review period, although
it is typical for the review to take far longer. If the FDA classifies the device into class II, the company will then receive an approval
order to market the device. This device type can then be used as a predicate device for future 510(k) submissions. However, if the FDA
subsequently determines that the device will remain in the class III category, then the device may not be marketed until the company has
obtained an approved PMA.
Premarket Approval Pathway
A premarket approval application must be submitted
if the device cannot be cleared through the 510(k) process or is found ineligible for de novo reclassification. PMAs are generally required
for Class III devices that support or sustain human life, are of substantial importance in preventing impairment of human health, or which
present a potential, unreasonable risk of illness or injury. The premarket approval application process is generally more costly and time
consuming than the 510(k) process. A premarket approval application must be supported by extensive data including, but not limited to,
technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA’s satisfaction the safety and effectiveness
of the device for its intended use.
After a PMA application is submitted, the FDA has
45 days to determine whether the application is sufficiently complete to permit a substantive review and thus whether the FDA will file
the application for review. After a premarket approval application is sufficiently complete, the FDA will accept the application and begin
an in-depth review of the submitted information. By statute, the FDA has 180 days to review the “accepted application,” although,
generally, review of the application may take at least 12 to 18 months, but it may take significantly longer. During this review period,
the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory
panel of experts from outside FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to
the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance
with quality system regulations.
Upon completion of the PMA application review,
the FDA may: (i) approve the PMA which authorizes commercial marketing with specific prescribing information for one or more indications,
which can be more limited than those originally sought; (ii) issue an approvable letter which indicates the FDA’s belief that the
PMA application is approvable and states what additional information the FDA requires, or the post-approval commitments that must be agreed
to prior to approval; (iii) issue a not approvable letter which outlines steps required for approval, but which are typically more onerous
than those in an approvable letter, and may require additional clinical trials that are often expensive and time consuming and can delay
approval for months or even years; or
(iv) deny the application. If the FDA issues an approvable or
not approvable letter, the applicant has 180 days to respond, after which the FDA’s review clock is reset.
New premarket approval applications or premarket
approval application supplements are required for modifications that affect the safety or effectiveness of the device, including, for
example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. Premarket
approval supplements often require the submission of the same type of information as a premarket approval application, except that the
supplement is limited to information needed to support any changes from the device covered by the original premarket approval application
and may not require as extensive clinical data or the convening of an advisory panel.
Breakthrough Device Program
The FDA has implemented a Breakthrough Device Program
that is intended to help patients receive more timely access to breakthrough medical technologies that have the potential to provide more
effective treatment or diagnosis for life-threatening or irreversibly debilitating diseases or conditions. A device also must meet one
of the following criteria: (i) it represents breakthrough technology; (ii) there is no approved or cleared alternative; (iii) it offers
significant advantages over existing cleared or approved devices; or (iv) availability of the device is in the best interest of patients.
Under the program, we are eligible to receive priority review and interactive communications from the FDA regarding device development
and clinical trial protocols, all the way through to commercialization decisions.
In February 2021, the FDA granted us a Breakthrough
Device designation for C-Scan for the indication as stated in the FDA’s Breakthrough Device designation approval, as follows: “C-
Scan is intended to identify adult subjects 50 years or older, within the average risk population, who are at elevated risk for polyps
larger than or equal to 1 cm. It is intended for adults who are poor candidates for colonoscopy. Polyps have been shown to be associated
with the development of colorectal cancer. C-Scan is not intended to replace colonoscopy. A positive C-Scan result should be followed
by colonoscopy.”
Clinical Trials
Clinical trials are almost always required to support
a premarket approval application or de novo reclassification petition and are sometimes required for a 510(k) premarket notification.
If the device presents a “significant risk,” as defined by the FDA, to human health, the FDA requires the device sponsor to
file an IDE application with the FDA and obtain IDE approval prior to commencing the human clinical trials. During the trial, the sponsor
must comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting, and recordkeeping. The investigators
must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational
devices, and comply with all reporting and recordkeeping requirements. Clinical trials for significant risk devices may not begin until
the IDE application is approved by the FDA and the appropriate institutional review boards, or IRBs, at the clinical trial sites. An IRB
is an appropriately constituted group that has been formally designated to review and monitor medical research involving subjects and
which has the authority to approve, require modifications in, or disapprove research to protect the rights, safety and welfare of human
research subjects. A nonsignificant risk device does not require FDA approval of an IDE; however, the clinical trial must still be conducted
in compliance with various requirements of FDA’s IDE regulations and be approved by an IRB at the clinical trials sites. We, the
FDA, or the IRB at each site at which a clinical trial is being performed may withdraw approval of a clinical trial at any time for various
reasons, including a belief that the risks to study subjects outweigh the benefits or a failure to comply with FDA or IRB requirements.
Even if a trial is completed, the results of clinical testing may not demonstrate the safety and effectiveness of the device, may be equivocal
or may otherwise not be sufficient to obtain approval or clearance of the product.
Similarly, in Europe the clinical study must be
approved by the local ethics committee and in some cases, including studies of high-risk devices, by the Ministry of Health in the applicable
country.
Sponsors of clinical trials of devices are required
to register with clinicaltrials.gov, a public database of clinical trial information. Information related to the device, patient population,
phase of investigation, study sites and investigators, and other aspects of the clinical trial is made public as part of the registration.
Pervasive and Continuing FDA Regulation
After a device is cleared or approved for marketing, numerous and pervasive regulatory
requirements continue to apply. These include:
|
• |
establishment registration and device listing with the FDA; |
|
• |
QSR requirements, which require manufacturers, including third-party manufacturers, to follow stringent design, testing, control,
documentation and other quality assurance procedures during all aspects of the design and manufacturing process; |
|
• |
labeling regulations and FDA prohibitions against the promotion of investigational products, or the promotion of ‘‘off-label’’
uses of cleared or approved products; |
|
• |
requirements related to promotional activities; |
|
• |
clearance or approval of product modifications to 510(k)-cleared devices that could significantly affect safety or effectiveness
or that would constitute a major change in intended use of one of our cleared devices, or approval of certain modifications to PMA-approved
devices; |
|
• |
medical device reporting regulations, which require that a manufacturer report to the FDA if a device it markets may have caused
or contributed to a death or serious injury, or has malfunctioned and the device or a similar device that it markets would be likely to
cause or contribute to a death or serious injury, if the malfunction were to recur; |
|
• |
correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections and product
recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present
a risk to health; |
|
• |
the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product that is in
violation of governing laws and regulations; and |
|
• |
post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health
or to provide additional safety and effectiveness data for the device. |
Quality Systems Regulation Requirements
Our manufacturing processes are required to comply
with the applicable portions of the QSR, which cover the methods and the facilities and controls for the design, manufacture, testing,
production, processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices
intended for human use. The QSR requires that each manufacturer establish a quality systems program by which the manufacturer monitors
the manufacturing process and maintains records that show compliance with FDA regulations and the manufacturer’s written specifications
and procedures relating to the devices. The QSR also requires, among other things, maintenance of records and certain documentation, a
device master file, device history file, and complaint files. QSR compliance is necessary to receive and maintain FDA clearance or approval
to market new and existing products. As a manufacturer, we are subject to periodic scheduled or unscheduled audits or inspections by the
FDA. Our failure to maintain compliance with the QSR requirements could result in the shut-down of, or restrictions on, our manufacturing
operations and the recall or seizure of our products, which would have a material adverse effect on our business. The discovery of previously
unknown problems with any of our products, including unanticipated adverse events or adverse events of increasing severity or frequency,
whether resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of medicine,
could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
The FDA has broad regulatory compliance and enforcement
powers. As manufacturers, we and our third-party manufacturers are subject to announced and unannounced inspections by the FDA to determine
our compliance with quality system regulation and other regulations. We have not yet been inspected by the FDA. If the FDA determines
that we failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may
result in any of the following sanctions:
|
• |
FDA untitled letters, FDA Form 483s, FDA warning letters, it has come to our attention letters, fines, injunctions, consent decrees
and civil penalties; |
|
• |
unanticipated expenditures to address or defend such actions; |
|
• |
customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
|
• |
recall, detention or seizure of our products; |
|
• |
operating restrictions, partial suspension or total shutdown of production; |
|
• |
refusing or delaying our requests for regulatory approvals or clearances of new products or modified products; |
|
• |
withdrawing of 510(k) clearances or PMA approvals that have already been granted; |
|
• |
refusal to grant export approval for our products; or |
|
• |
civil or criminal prosecution. |
The FDA can also publish Safety Communications
or Letters to Health Care Providers when the agency becomes aware of new issues involving a specific product or, or more broadly, a product
family. These communications are posted on the FDA’s website and describe the FDA’s analysis of a current issue and provide
specific regulatory approaches and clinical recommendations for patient management.
Regulatory Pathway for C-Scan
We have established a clinical and regulatory
strategy with our advisors and have conducted a pre submission meeting during the period of December 2016 and February 2017, with the
FDA (an informal interaction to facilitate a clearer understanding of FDA’s expectations). During this process, we received the
FDA’s feedback on our submission and our questions. We also sought feedback on a proposed protocol for a feasibility or pilot study,
the primary purposes of which was to establish the safety of the C-Scan system and evaluate user compliance and satisfaction. In December
2018, we received from the FDA conditional approval of our IDE application to initiate a U.S. pilot study of the C-Scan system and received
final approval from the FDA in February 2019. In April 2019, we initiated the U.S. pilot study of C-Scan that was completed by December
2019.
In November 2020, we finalized our proposed
U.S. pivotal study design and submitted our IDE application to the FDA and in March 2021, our IDE application was approved by the FDA.
In January 2022, we submitted to the FDA
an amended IDE application to amend the U.S. pivotal study design to add a first part to the study that is designed to enable further
calibration of the C-Scan system, specifically for the average risk U.S. population and in February 2022, our IDE amended application
was approved by the FDA. Following this amended IDE, the U.S. pivotal study consists of two parts. The first part is designed to enable
further calibration of the C-Scan system for the average risk U.S. population, and is intended to include up to 200 patients in the United
States. The second part is a statistically powered, randomized study which will compare the performance of C-Scan to traditional colonoscopy,
and is intended to include up to 800 patients. The goal of the pivotal study in the United States is to (i) demonstrate device safety
as evidenced by a lack of device related serious adverse events; and (ii) provide efficacy data concerning C- Scan’s performance.
In May 2022, we announced we had initiated
the first part of the U.S. pivotal study, among the average risk U.S. population. In December 2022, we submitted an IDE supplement to
the FDA to include enrollment of patients at age 45-75 instead of 50-75, which was approved by the FDA in January 2023.
As part of the U.S. pivotal study, we have
continued to optimize the functionality and patient experience of our advanced version of C-Scan, which incorporates mainly advanced algorithms,
improved detection capability and reduced energy consumption, through additional clinical data collection in a study in Israel (initially
on average risk and high-risk patients and starting May 2022, shifted to enrollment of only average risk patients) for the purpose of
further calibration of the C-Scan system, in parallel to conducting the first part of the U.S. pivotal study that was initiated in May
2022.
The initiation of the powered potion of
the U.S. pivotal study was dependent upon successful completion of the calibration portion of the U.S. pivotal study. Following
our internal assessment of the clinical data collected to date from our calibration studies, we have determined that the current efficacy
results do not meet our goal in order to proceed to the powered portion of the U.S. pivotal study. As a result, we have adopted
a plan of action that includes conducting additional clinical data analysis and approaching the FDA to make amendments to the U.S. pivotal
study protocol that are expected to be part of an IDE supplement submission to the FDA, and which are subject to FDA approval. In addition,
we plan to continue conducting our calibration studies, albeit at a slower pace, to collect additional clinical data and we also implementing
a cost reduction plan, in order to extend our cash runway. The initiation of the powered portion of the U.S. pivotal study that was expected
in mid-2023 is therefore temporarily postponed.
Following and subject to the successful
completion of our U.S. pivotal study and available capital, our current strategy is to submit a direct de novo reclassification petition
for our system. Although the FDA could require us to submit a PMA, we believe that the device could be considered for evaluation under
the FDA’s de novo reclassification provisions, which allow a novel device to be reclassified into class I or class II. To support
this, our objective is to demonstrate that the device poses a low to moderate risk to patients.
We believe that important potential benefits
of C-Scan for CRC screening are the elimination of the need for bowel preparation, the elimination of the need for conscious sedation,
the minimally invasive, painless nature of the examination, and the ability to pursue normal daily activities immediately following the
procedure. Furthermore, the C-Scan system is being designed to generate information from segments of the colon (e.g.,
cecum and ascending colon) that are difficult to access by conventional optical colonoscopy (i.e.,
incomplete colonoscopies) without the risks and discomforts of operative examination or other invasive methods. We believe these benefits
will be attractive to a large number of patients from the target populations that so far refrained from any screening tests. Thus, we
anticipate that our capsule will increase the public compliance to screening recommendation.
If FDA determines that C-Scan is not a candidate
for de novo reclassification, it will require approval of the device for market through the PMA process. Because of the technological
characteristics of this device, the non-clinical tests (including lab and animals) and clinical data required may not be significantly
different between de novo and PMA regulatory processes. We believe that under both scenarios, we are required to conduct a multi-center
clinical study to establish the safety and efficacy and to demonstrate sensitivity and specificity of C-Scan in several hundreds of patients.
In February 2021, the FDA granted us a
Breakthrough Device designation for C-Scan for the indication as stated in the FDA’s Breakthrough Device designation approval, as
follows: “C- Scan is intended to identify adult subjects 50 years or older, within the average risk population, who are at elevated
risk for polyps larger than or equal to 1 cm. It is intended for adults who are poor candidates for colonoscopy. Polyps have been shown
to be associated with the development of colorectal cancer. C-Scan is not intended to replace colonoscopy. A positive C-Scan result should
be followed by colonoscopy.”
The FDA’s Breakthrough Device Program
aims to provide patients and providers with expedited access to innovative medical devices that offer more effective diagnosis by expediting
their development, assessment and review while preserving the statutory standards for premarket approval. In addition, the Centers for
Medicare & Medicaid Services (CMS) has created expedited and special reimbursement pathways and programs based on FDA’s Breakthrough
Device designation, for which we may be eligible.
FCC Clearance and Regulation
Because C-Scan includes a wireless radio
frequency transmitter and receiver, it is subject to equipment authorization requirements in the United States. The U.S. Federal Communications
Commission, or FCC, requires authorization of radio frequency devices before they can be sold or marketed in the United States, subject
to limited exceptions. The authorization requirements are intended to confirm that the proposed products comply with FCC radio frequency
emission, power level standards, and other technical requirements and will not cause interference. Our system is using the same frequency
band as other approved capsules. The current design of C-Scan complies with the FCC’s technical requirements and received FCC authorization.
We expect that our future advanced design will also be comply with the FCC’s technical requirements, so it is expected that it will
be authorized by the FCC as well.
Third-Party Coverage and Reimbursement
Coverage of and reimbursement for C-Scan
and related procedures, after approval, will be subject to the requirements of various third-party payors, including government-sponsored
healthcare payment systems and private third-party payors. Coverage policies and reimbursement methodologies vary significantly from program-to-program
and may be subject to federal and state regulations. For example, the Social Security Act requires all items and services to be “reasonable
and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member” in order
to be covered by Medicare. While Medicare might not provide separate reimbursement for a device, it, may include payment for the device
in the payment to the facility or physician for the related procedure. Third-party payors’ coverage and reimbursement policies,
including their interpretations of whether an item or service is “reasonable and necessary” or experimental and/or investigational,
and their payment methodologies, are subject to change pursuant to legislation, regulation, or, in the case of private payors, negotiations
with industry and providers.
Fraud and Abuse Laws
In the United States, the healthcare industry
is subject to extensive federal, state, and local regulation. Both federal and state governmental agencies subject the healthcare industry
to intense regulatory scrutiny, including heightened civil and criminal enforcement efforts. These laws constrain the sales, marketing
and other promotional activities of manufacturers of medical devices, by limiting the kinds of financial arrangements (including sales
programs) we may have with hospitals, physicians and other potential purchasers of the medical devices. The laws and regulations that
may affect our ability to operate include, but are not limited to:
|
• |
The federal Anti-Kickback Statute, which prohibits, among other things, knowingly
or willingly offering, paying, soliciting or receiving remuneration, directly or indirectly, in cash or in kind, to induce or reward the
purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any health care items or service for which
payment may be made, in whole or in part, by federal healthcare programs such as Medicare and Medicaid. This statute has been interpreted
to apply to arrangements between medical device manufacturers on one hand and prescribers, purchasers and formulary managers on the other.
Further, PPACA, among other things, clarified that a person or entity needs not to have actual knowledge of the federal Anti-Kickback
Statute or specific intent to violate it. Although there are a number of statutory exemptions and regulatory safe harbors to the federal
Anti-Kickback Statute protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the exemptions
and safe harbors are drawn narrowly, and practices that do not fit squarely within an exemption or safe harbor may be subject to scrutiny;
|
|
• |
The federal civil False Claims Act, which prohibits, among other things, individuals
or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly
making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly
concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. In addition,
PPACA amended the Social Security Act to provide that the government may assert that a claim including items or services resulting from
a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims
Act. Many medical device manufacturers and other healthcare companies have been investigated and have reached substantial financial settlements
with the federal government under the civil False Claims Act for a variety of alleged improper marketing activities, including providing
free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees,
grants, free travel, and other benefits to physicians to induce them to use the company’s products. In addition, in recent years
the government has pursued civil False Claims Act cases against a number of manufacturers for causing false claims to be submitted as
a result of the marketing of their products for unapproved, and thus non-reimbursable, uses. Device manufacturers also are subject to
other federal false claim laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to
non-government health benefit programs; |
|
• |
Analogous state laws and regulations, such as state anti-kickback and false claims
laws, may apply to items or services reimbursed under Medicaid and other state programs or, in several states, apply regardless of the
payor. Several states now require medical device manufacturers to report expenses relating to the marketing and promotion or require them
to implement compliance programs or marketing codes. For example, California, Connecticut and Nevada mandate the implementation of corporate
compliance programs, while Massachusetts and Vermont impose more detailed restrictions on device manufacturers’ marketing practices
and tracking and reporting of gifts, compensation and other remuneration to healthcare providers; |
|
• |
The federal Foreign Corrupt Practices Act of 1997 and other similar anti-bribery laws
in other jurisdictions generally prohibit companies and their intermediaries from providing money or anything of value to officials of
foreign governments, foreign political parties, or international organizations with the intent to obtain or retain business or seek a
business advantage. Recently, there has been a substantial increase in anti-bribery law enforcement activity by U.S. regulators, with
more frequent and aggressive investigations and enforcement proceedings by both the Department of Justice and the U.S. Securities and
Exchange Commission. Violations of these laws can result in the imposition of substantial fines, interruptions of business, loss of supplier,
vendor or other third-party relationships, termination of necessary licenses and permits, and other legal or equitable sanctions. Other
internal or government investigations or legal or regulatory proceedings, including lawsuits brought by private litigants, may also follow
as a consequence; and |
|
• |
The federal Physician Payment Sunshine Act, being implemented as the Open Payments
Program, requires manufacturers of “covered products” (drugs, devices, biologics, or medical supplies for which payment is
available under Medicare, Medicaid, or the Children’s Health Insurance Program) to track and publicly report payments and other
transfers of value that they provide to U.S. physicians and teaching hospitals, as well as any ownership interests that U.S. physicians
hold in applicable manufacturer. Applicable manufacturers must submit a report to the Centers for Medicare & Medicaid Services, or
CMS, by the 90th day of each calendar year disclosing payments and transfers of value made in the preceding calendar year. |
Violations of any of the laws described above or any other governmental
regulations that apply to us, may cause us to be subject to significant civil, criminal and administrative penalties, damages, fines,
exclusion from government-funded healthcare programs, like Medicare and Medicaid, and the curtailment or restructuring of our operations.
Privacy Laws
HIPAA/HITECH and related U.S. federal and
state laws protect the confidentiality of certain patient health information, including patient records, and restrict the use and disclosure
of that protected information. In particular, the U.S. Department of Health and Human Services promulgated patient privacy rules under
HIPAA.
These privacy rules protect medical records
and other personal health information by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting
of their protected health information and limiting most use and disclosures of health information to the minimum degree reasonably necessary
to accomplish the intended purpose. Because we intend to sell products, once approved, to persons and entities subject to HIPAA and are
exposed to personally-identifiable health information in the course of our operations, we also may be subject to certain elements of HIPAA,
particularly as a business associate to covered entities, as well as similar state laws. HIPAA imposes civil and criminal penalties for
violations of its provisions, which could be substantial. State privacy laws have their own penalty provisions which may be applicable.
NRC Regulatory Matters
As a manufacturer of a medical device containing
a sealed, radioactive source, we are subject to regulation by various governmental authorities that oversee the possession, use, and disposal
of radioactive material. These regulations govern, among other things, the design, development, testing, manufacturing, packaging, labeling,
distribution, import/export, sale, and marketing and disposal of our product.
The C-Scan capsule contains within the
device a radioactive sealed source, which contains a small amount of radioactive material. This material aids in the imaging and scanning
capabilities of the device as it passes through the patient. The C-Scan capsule is ingested by the patient, and passes through the patient’s
gastrointestinal tract before exiting the human body.
Depending on where we manufacture and distribute
the device, various countries nuclear regulatory laws and regulations may apply. In Israel, where we manufacture the device, Israel nuclear
regulatory laws apply to the radioactive material in the device. We adhere to the Israeli regulations and guidance for the design, development,
testing, manufacturing and packaging, as well as the applicable nuclear regulations in the manufacturing phase.
In the United States, the C-Scan capsule is undergoing clinical
trials. The U.S. Nuclear Regulatory Commission, or the NRC, and state nuclear regulatory agencies oversee the use of radioactive materials
in the United States. The NRC regulates the use of radioactive materials in medical devices under several parts of its regulations, including
10 C.F.R. Part 20, “Standards for Protection Against Radiation,” 10 C.F.R. Part 30, “Rules of General Applicability
to Domestic Licensing of Byproduct Material,” and 10 C.F.R. Part 35, “Medical Use of Byproduct Material.” NRC Agreement
States implement similar regulations under a nuclear regulatory framework developed by the state pursuant to authority delegated by the
NRC.
In accordance with applicable NRC regulations,
and relevant NRC Agreement State requirements, we comply with the radioactive materials safety and handling requirements applicable for
our development of our C-Scan technology applicable to clinical trials. Regarding disposal of the device, the NRC places conditions and
limitations on the disposal of radioactive material in the sanitary sewer. In 2022, after extensive engagement with the NRC, we submitted
a petition for rulemaking to the NRC, requesting that the agency amend the NRC regulations at 10 CFR 20.2003(b), which pertains to disposal
of radioactive material into the sanitary sewer system, to include the disposal of the C-Scan capsule. Under the NRC’s current
interpretation of the 10 CFR 20.2003(b) regulations, disposal of a device like the C-Scan capsule into the sanitary sewer system is not
currently permitted. The NRC’s review of the rulemaking request remains ongoing. Therefore, during the U.S. pivotal study, the C-Scan
capsule is not intended to be disposed of in the sanitary sewer system, and is instead intended to be collected by the patient and returned
to the medical facility administering the device for further storage, decay and disposal. For the collection and disposal phase of the
U.S. pivotal study, we have established, through a third party, a decay and disposal process of the C-Scan capsule that is in accordance
with the NRC and Agreement State requirements.
In the U.S, engagement with various sites
to participate in the U.S. pivotal study has been progressing slower than expected, in part due to compliance with, and the need for,
the site-specific licensing and compliance with radioactive materials regulatory requirements associated with the X-ray technology contained
within our C-Scan capsule.
The future commercial sale of C-Scan will
also be subject to extensive nuclear regulations in various jurisdictions around the world where the C-Scan capsule will be used, and
we intend to comply with such regulations as they become applicable. We expect the commercial manufacture and distribution of C-Scan will
also require us, or a company affiliated with our company, to obtain a number of licenses and certifications related to nuclear regulatory
compliance. We also expect that in commercialization, clinical centers/hospitals will need to obtain a number of licenses and certifications
related to nuclear regulatory compliance.
Regulation in Europe, Israel, Japan and Other Countries
The European Union has adopted numerous
directives and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices.
Each European Union member state has implemented legislation applying these directives and standards at the national level. Other countries,
such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the European Union with respect to medical devices.
Devices that comply with the requirements of the laws of the relevant member state applying the applicable European Union directive are
entitled to bear CE conformity marking and, accordingly, can be commercially distributed throughout the member states of the European
Union and other countries that comply with or mirror these directives. The method of assessing conformity varies depending on the type
and class of the product, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a
“Notified Body,” an independent and neutral institution appointed to conduct conformity assessment. This third-party assessment
consists of an audit of the manufacturer’s quality system and clinical information, as well as technical review of the manufacturer’s
product. An assessment by a Notified Body in one country within the European Union is required in order for a manufacturer to commercially
distribute the product throughout the European Union. In addition, compliance with ISO 13845:2016 on quality systems issued by the International
Organization for Standards, among other standards, establishes the presumption of conformity with the essential requirements for a CE
marking. In addition, many countries apply requirements in their reimbursement, pricing or health care systems that affect companies’
ability to market products.
On January 9, 2018, we obtained the CE
mark of conformity for C-Scan. On December 21, 2021, the European MDR issued a renewal of our CE mark according to the EU Regulation 2017/745
on Medical Devices for the marketing and sale of C-Scan in the European Union, valid until December 1, 2026.
C-Scan and our operations are also subject
to regulation by AMAR, which is responsible for the registration of medical devices in Israel, issuance of import licenses and monitoring
of marketing of medical equipment. In September 2018, we received approval from AMAR for the marketing and sale of C-Scan in Israel. Following
the approval we received from our notified body according to the EU Regulation 2017/745, the AMAR approval was renewed and is valid until
December 31, 2024.
In Japan, manufacturing and marketing medical
devices are regulated by the Pharmaceutical Affairs Law, or PAL. In accordance with the PAL, manufacturers must obtain a license for manufacturing
medical devices from the Ministry of Health, Labour and Welfare, or MHLW. A license is required for each manufacturing plant specified
by an MHLW Ministerial Ordinance.
A licensed manufacturer is responsible
only for manufacturing medical devices. In regard to the marketing of medical devices, the PAL specifies that a Marketing Authorization
Holder, or MAH, licensed by MHLW is responsible for putting medical devices into marketplace. Licenses for marketing medical devices are
divided into the following 3 types, which correspond to the classifications below:
|
• |
No. 1 type license for marketing – Specially controlled medical devices (Class III, IV) |
|
• |
No. 2 type license for marketing – Controlled medical devices (Class II) |
|
• |
No. 3 type license for marketing – General medical devices (Class I) |
Generally, the process for obtaining marketing
clearance for medical devices in Japan ranges from twelve months (for products with only very minor modifications from previously cleared
product versions) to a few years in the case of a completely new device.
In order for us to market our products
in countries other than the United States, Israel, the EU and Japan (which were described above), we must obtain regulatory approvals
and comply with extensive safety and quality regulations in these countries. These regulations, including the requirements for approvals
or clearance and the time required for regulatory review varies from country to country. It is customary that once a product has been
cleared for sales in the US and is CE marked in the EU, many other countries will follow. Failure to obtain regulatory approval or clearance
in any foreign country in which we plan to market our product may harm our ability to generate revenue and harm our business.
United Kingdom
The withdrawal of the United Kingdom (UK)
from the EU took effect on January 1, 2021, and there are 27 member states remaining in the EU. As of January 1, 2021, the UK is a “third
country” with regard to the EU (subject to the terms of the EU UK Trade Agreement) and EU law ceased to apply directly in the UK.
However, the UK has retained the EU regulatory regime with certain modifications as standalone UK legislation. Therefore, the UK regulatory
regime is currently similar to EU regulations, but under new legislation, the Medicines and Medical Devices Act, the UK may adopt changed
regulations that may diverge from the EU legislative regime for medicines and medical devices and their research, development and commercialization.
For a two-year period starting January 1, 2021, the UK has adopted transitional provisions, which inter alia apply to the importation
of medical devices into the UK and the acceptance of CE-certificates until the end of June 2023, subject to certain registrations with
the UK Medicines and Healthcare Products Regulatory Agency. From July 1, 2023, certification under the new UK medical device legislation
will be required to place products on the market in the UK. Medical devices, will need to be registered with the MHRA before they are
placed on the Great Britain market. Medical device manufacturer based outside the UK that wish to place a device on the Great Britain
market, will need to appoint a single UK Responsible Person, who will act on its behalf to carry out specified tasks, such as registration.
Third-Party Reimbursement
Reimbursement in the United States
In the United States, healthcare providers
that purchase and/or prescribe medical devices generally rely on third-party payors, such as Medicare, Medicaid, or private health insurance
plans, to reimburse all or a portion of the cost of the devices, as well as any related healthcare services utilizing the device.
Coverage is not guaranteed simply because
a product has received FDA clearance or approval or has a CPT or HCPCS code that describes the product or service. For example, Medicare’s
general definition of a medically necessary service is one that is reasonable and necessary for the diagnosis or treatment of an illness
or injury, or that improves the functioning of a malformed body member.
Colorectal cancer (CRC) is the fourth most
common cancer and the second leading cause of cancer deaths in the United States, and is expected to cause about 52,550 deaths during
2023. It is an important issue for the Medicare and the commercial population. Approximately 4.1 percent of men and women will be diagnosed
with colorectal cancer at some point during their lifetime, based on 2016–2018 data. In 2018, there were an estimated 1,365,135
people living with colorectal cancer in the United States. In 2022, the National Institutes of Health (NIH) National Cancer Institute
(NCI) estimated that there will be over 151,000 new cases of colon and rectal cancer in the United States with a median age at diagnosis
of 66 years. Colorectal cancer is most frequently diagnosed among people aged 6 – 74 years (NIH). The American Cancer Society’s
estimates for the number of colorectal cancer cases in the United States for 2023 forecast over 153,000 new cases of colon and rectal
cancer. From 2013 to 2019, while incidence rates dropped by about 1% each year, there is a rising incidence among younger adults since
at least the mid-1990s. From 2012 through 2016, it increased every year by 2% in people younger than 50 and 1% in people 50 to 64. While
the death rate from colorectal cancer has been dropping among both men and women for several decades, deaths from colorectal cancer among
people younger than 55 have increased 1% per year from 2008 to 2017.
Among all races, African Americans have the highest death rate. Based on data from
SEER 17 2012-2018, the five-year relative survival rate was 65.1%. The earlier colorectal cancer is caught, the better chance a person
has of surviving five years after being diagnosed. For colorectal cancer, 37.2% are diagnosed at the local stage. The 5-year relative
survival for localized colorectal cancer is 90.9%.
COVID-19 has caused an unprecedented disruption
in cancer prevention efforts, including CRC screening. In general, colorectal cancer screening decreased by 28% to as much as 100% in
different countries and at different times after the onset of the COVID-19 pandemic. During this period, completion of colonoscopies requiring
examination showed a decrease of 65.7%, surveillance colonoscopy showed a decrease of 44.6 to 79%, prescription colonoscopy decreased
60 to 81%, and referrals to colonoscopy showed a 43% decline. In a simulation model by Issaka, et al, COVID-19–related reductions
in care utilization resulted in an estimated 1,176,942 to 2,014,164 fewer colorectal cancer screenings, 8,346 to 12,894 fewer colorectal
cancer diagnoses, and 6113 to 9301 fewer early-stage colorectal cancer diagnoses between 2020 and 2023. In an extended period of limited
CRC screenings due to the pandemic, up to 43% of eligible adults could remain unscreened.
Since the 1990s, Medicare
has covered a number of CRC screening tests from non-invasive fecal based tests to colonoscopy, providing a range of choices for patients
to choose the most individually suitable modalities.
The cost-effectiveness of a device (or the
service in which a device is used) may be a key factor in obtaining third-party payer reimbursement for such device or service, but is
not commonly a factor for Medicare. Medicare covers certain colorectal cancer (CRC) screening tests and procedures for Medicare beneficiaries
that meet certain eligibility criteria, as authorized by Sections 1861(s)(2)(R) and 1861(pp) of the Social Security Act and regulations
at 42 CFR 410.37. However, Medicare coverage of CRC screening tests as defined under Section 1861(pp)(1)(D) states “Such other tests
or procedures, and modifications to tests and procedures under this subsection, with such frequency and payment
limits, as the Secretary determines appropriate, in consultation with appropriate organizations.” (emphasis added).
A key factor affecting the cost-effectiveness
of colorectal cancer screening is patient adherence. Adherence is strongly affected by patients’ willingness to use the device (or
service incorporating a device) as a screening tool for CRC.
Several models have been designed
to demonstrate the cost effectiveness of optical colonoscopy, CT colonography (CTC), fecal testing and capsule endoscopy. Today, several
technologies - FOBT / FIT, Flexible Sigmoidoscopy, Optical Colonoscopy, Barium Enema, and multi-target stool DNA – are covered for
Medicare beneficiaries meeting certain eligibility criteria for screening for colorectal cancer, while other technologies including CTC,
capsule endoscopy, and liquid biopsy are not currently covered by Medicare for CRC screening.
In 2009, the Centers for Medicare and Medicaid
Services (CMS) issued a decision memorandum concluding that the evidence was inadequate to support Medicare coverage of CT colonography
(CTC) for CRC screening. Their main argument for the decision was based on available evidence that screening with CTC would not necessarily
result in cost savings at least at current screening compliance rates. CTC was not seen as a tool which could potentially increase patients’
adherence. This procedure involves bowel preparation, as well as insufflations of the colon and the exposure of patients to very significant
amount of radiation. Extracolonic incidental findings on CT colonography are common, with the percentage of participants with extracolonic
findings ranging from 58% to 66%. Further, as CTC potentially detects lesions outside of the colon, CMS has no ability to pay for the
evaluation of incidental findings in a patient without signs or symptoms unless specifically directed by Congress to do so.
There are benefits of screening for advanced
adenomas. The adenoma detection rate (based on colonoscopies) was inversely associated with the risks of interval colorectal cancer, advanced-stage
interval cancer, and fatal interval cancer.
An important European study (C. Hassan et
al, “Cost-Effectiveness of Capsule Endoscopy in Screening for Colorectal Cancer,” Endoscopy; 2008 40: 414-421) assessed the
potential cost effectiveness of screening with optical capsule endoscopy and compared its cost-effectiveness with optical colonoscopy.
Cost-effectiveness of screening was measured in terms of cost per life-year saved through prevention or down staging of CRC. The conclusion
was that the cost effectiveness of capsule endoscopy in CRC screening will depend mainly on its ability to improve compliance in the general
population.
Third-party payors in the United States
began issuing coverage policies for capsule endoscopy in early 2002, subsequent to FDA approval and availability of the technology. Initially,
reimbursement policies provided coverage for capsule endoscopy of the small bowel only for the diagnosis of obscure gastrointestinal bleeding.
Subsequently, reimbursement coverage has been expanded to include other diagnoses and as of December 31, 2022, approximately 300 million
people in the United States have coverage of this technology for a number of capsule endoscopy of the small bowel indications including
obscure gastrointestinal bleeding, suspected Crohn’s disease, suspected small bowel tumors, celiac disease and other small bowel
pathologies.
Currently, there is no Medicare, Medicaid,
or commercial insurance reimbursement for capsule endoscopy of the colon in the United States for CRC screening, despite the availability
of CPT code 91113 Gastrointestinal tract imaging, intraluminal (e.g., capsule endoscopy), colon, with interpretation and report, which
became effective January 1, 2022.
In February 2020, CMS received a formal
request for a national coverage determination from Epigenomics to consider coverage of Epi proColon, one example of a blood-based biomarker
screening test for colorectal cancer. Over the last several years, blood-based biomarker tests have emerged as another potential non-invasive
option for the early detection of colorectal cancer. A blood-based biomarker (biological marker in the patient’s blood) is a measurable
DNA, RNA or protein component that indicates disease, in this case cancer. Blood-based cancer biomarkers include but are not limited to
specific gene mutations, methylation of genes, and antigens. The blood-based biomarker that is measured in a person’s blood can
be an indicator of a process, such as disease risk or progression, like progression to colorectal cancer, thought to be correlated with
a long term outcome, such as mortality. It is typically easier to measure a biomarker than the true outcome of interest, such as mortality
from colorectal cancer. However, the biomarker might not be a good surrogate for and not well correlated with the clinical outcome of
interest, such as cancer survival or mortality, or it might not identify a patient early enough to alter the clinical course of disease.
One example test, Epi proColon, functions as a blood-based colorectal cancer screening test by identifying the circulating mSEPT9 (Septin9)
gene in cell-free DNA isolated from plasma. CRC tumors have an increased likelihood of exhibiting aberrant methylation at the promoter
region of the SEPT9 gene DNA.
In the past CRC screening NCDs, CMS has
discussed appropriate test performance criteria such as point sensitivity and specificity compared to colonoscopy. The determination of
appropriateness is similar to the consideration of what is appropriate for the device to be, “at least as beneficial as an existing
and available medically appropriate alternative.” Through the evaluation of evidence from published studies, the clinical data for
the device must reach the threshold for being appropriate. The direct evidence suggests that there is clinical utility in using FOBT and
FIT for early detection of colorectal cancer (CRC) because these tests reduce mortality of the disease. Conversely, there is no direct
evidence on outcomes such as mortality for blood-based biomarker tests used in screening for colorectal cancer. By comparing the test
performance characteristics, defined as sensitivity, specificity, and positive predictive value, of new screening tests to gFOBT or FIT,
CMS can assess whether blood-based tests might translate into similar reductions in disease mortality for Medicare beneficiaries. Thus,
if the sensitivity and specificity of the new screening tests are as good as or better than the sensitivity and specificity of gFOBT or
FIT, then there is indirect evidence suggesting that the new blood-based biomarker tests may reduce colorectal cancer mortality.
In January 2021, CMS updated National Coverage
Determination 210.3, stating that, effective for dates of service on or after January 19, 2021, a blood-based biomarker test is covered
as an appropriate colorectal cancer screening test once every 3 years for Medicare beneficiaries (i) when performed in a Clinical Laboratory
Improvement Act (CLIA)-certified laboratory, (ii) when ordered by a treating physician and (iii) when all of the following requirements
are met: The patient is: age 50-85 years; and, asymptomatic (no signs or symptoms of colorectal disease including but not limited to lower
gastrointestinal pain, blood in stool, positive guaiac fecal occult blood test or fecal immunochemical test); and, at average risk of
developing colorectal cancer (no personal history of adenomatous polyps, colorectal cancer, or inflammatory bowel disease, including Crohn’s
Disease and ulcerative colitis; no family history of colorectal cancers or adenomatous polyps, familial adenomatous polyposis, or hereditary
nonpolyposis colorectal cancer). The blood-based biomarker screening test must have all of the following: FDA market authorization with
an indication for colorectal cancer screening; and, proven test performance characteristics for a blood-based screening test with both
sensitivity greater than or equal to 74% and specificity greater than or equal to 90% in the detection of colorectal cancer compared to
the recognized standard (accepted as colonoscopy at this time), as minimal threshold levels, based on the pivotal studies included in
the FDA labeling.
CMS concluded that the published evidence
does not show that the available blood-based biomarker CRC screening test improves health outcomes for Medicare beneficiaries. The indirect
evidence (comparing a blood-based screening test to another test that has been shown to improve mortality such as screening fecal immunochemical
test) does not show that blood- based screening test is equivalent in the detection of large adenomas or early stage cancer. The test
performance of the Epi proColon® test does not meet sensitivity and specificity levels established by prior evidence at which the
benefits of using the screening test outweigh harms to Medicare patients.
In July 2022, a group of colorectal cancer
survivors, patient advocates, and life-science professionals met to discuss opportunities for stakeholders to improve access to treatment
and screening in the US. Joined by White House Cancer Moonshot Coordinators, the group centered discussion on three initiatives:
|
• |
CRC education and awareness as part of First Lady Jill Biden’s cancer screening messaging efforts, |
|
• |
Comprehensive CRC screening solutions supporting current screening tools, enabling the workgroup to reach underserved communities,
|
|
• |
Building on previous successes of the CDC’s Colorectal Cancer Control Program (CRCCP). |
In December 2022, Guardant Health released
results from ECLIPSE (Evaluation of ctDNA LUNAR Assay In an Average Patient Screening Episode), an over 20,000 patient registrational
study evaluating the performance of its blood test for detecting colorectal cancer (CRC) in average-risk adults. The test demonstrated
83% sensitivity in detecting individuals with CRC. Specificity was 90% in both individuals without advanced neoplasia and in those who
had a negative colonoscopy result. This test also demonstrated 13% sensitivity in detecting advanced adenomas. In this study, two configurations
of a multimodal blood-based screening test were evaluated independently - a cell-free DNA (cfDNA)-only test and a cfDNA test with protein
biomarkers. The announced results were derived from the cfDNA-only test, which outperformed the cfDNA test with protein biomarkers. Based
on these study results, Guardant Health plans to complete its premarket approval submission to the FDA in the first quarter of 2023.
In January 2023, Geneoscopy Inc., released
results from the CRC-PREVENT trial – a pivotal clinical trial evaluating the efficacy of its noninvasive, stool-based, at-home diagnostic
screening test to detect colorectal cancer (CRC) and advanced adenomas (AA) in average-risk individuals. The CRC-PREVENT trial included
8,289 individuals with diverse racial, ethnic, and socioeconomic backgrounds across more than 2,900 zip codes in all lower 48 states,
with colonoscopies performed in more than 3,800 endoscopy centers. Efficacy results from the study demonstrated 94% sensitivity for detecting
CRC, 45% sensitivity for detecting advanced adenoma (AA), and 88% specificity for no findings on a colonoscopy. The company submitted
a Premarket Approval (PMA) application to the FDA in January 2023.
There can be no assurance that coverage
of the C-Scan by Medicare and/or commercial payors will be obtained in the near future or at all. Third-party payors may deny coverage
of the C-Scan if they determine that a procedure was not reasonable or necessary as determined by the payor, was experimental, does not
improve health outcomes, is not equivalent in the detection of large adenomas or early stage cancers, and/or was used for an unapproved
indication (among other factors).
Coverage Outside the United States
In countries outside the United States,
coverage for CRC screening may be obtained from various sources, including governmental authorities, private health insurance plans, and
labor unions. In some countries, private insurance systems may also offer payments for some therapies. Although not as prevalent as in
the United States, health maintenance organizations are emerging in certain European countries. Coverage systems in international markets
vary significantly by country and, within some countries, by region. Coverage approvals must be obtained on a country-by- country or region-by-region
basis.
C. Organizational
Structure
On May 15, 2015, we formed our wholly-owned subsidiary Check-Cap US, Inc., a Delaware
corporation.
D. Property,
Plants and Equipment
Our principal executive offices and operations
are conducted at a facility located in Isfiya, Israel since June 1, 2009. We also have an X-ray production line in Petach Tikva, Israel.
In Isfiya, we lease approximately 1,550 square meters at this facility under a lease agreement expiring on December 31, 2023, and we have
an option to extend the agreement for an additional period of three years. We have the right to terminate the Isfiya lease agreement at
any time, upon at least 60 days prior written notice. We currently plan to extend the Isfiya lease agreement by the additional three years.
In December 2021, we signed the Sub-lease Agreement with one of our suppliers, according to which we lease approximately 70 square meters
of laboratory production space at the supplier’s premises. The Sub-lease Agreement entered into in effect on September 21, 2022,
after receipt of certain certificates, including a business license from Petach Tikva municipality. The Sub-lease Agreement expires on
January 31, 2025, and we have an option to extend the lease period for an additional two years. We have the right to terminate the Sub-lease
Agreement at any time, upon at least 90 days prior written notice. The lessor has the right to terminate the Sub-lease Agreement, upon
at least 12 months prior written notice.
We believe that our current facilities
are adequate to meet our current needs for the clinical phase of our development. See Item 4B “Information on Our Company –
Business Overview – Manufacturing and Suppliers.”
ITEM 4A. Unresolved Staff Comments
None.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
You should read the
following discussion and analysis of our financial condition and results of operations in conjunction with our consolidated financial
statements and related notes included in this Annual Report beginning on page F-1. The following discussion and analysis contain forward-looking
statements that involve risks and uncertainties. Our actual results and the timing of selected events could differ materially from those
anticipated in these forward-looking statements as a result of various factors, including those set forth under Item 3D “Key Information
- Risk factors” and elsewhere in this Annual Report.
Overview
We are a clinical stage medical diagnostics
company aiming to redefine CRC screening through the introduction of C-Scan®, the first and only patient-friendly preparation-free
test designed to detect polyps before they may transform into cancer to enable early intervention and cancer prevention. The disruptive
capsule-based screening technology aims to increase screening adherence worldwide and enable millions of people to stay healthy. The system
utilizes ultra-low-dose X-rays to scan the inner lining of the colon for precancerous polyps, and other structural abnormalities. CRC
is the third most commonly diagnosed cancer, with more than 1.9 million new cases identified every year globally. Nearly 935,000 deaths
occur annually worldwide as a result of CRC. While approximately 0.5% of the average-risk screening population presents with cancerous
polyps in the colon and rectum at any given time, approximately 25% of the same population presents with benign polyps that could potentially
turn into cancer over time. It can take up to 10 years before a pre-cancerous polyp develops into invasive cancer. As such, there is a
crucial detection window for the prevention of colorectal cancer, through the detection of these benign polyps. While routine screening
is recommended by The American Cancer Society for healthy people aged 45 years and older, screening adherence remains low. Currently,
colonoscopy is the gold standard for the detection of colorectal polyps, but about 1 in 3 adults among the targeted screening population
avoids having a colonoscopy in the U.S., and adherence in other regions of the world such as Europe and Asia is even lower, due to the
invasiveness of the procedure and bowel preparation. Most patient-friendly CRC screening tests currently available, or poised to enter
the market, such as fecal or liquid biopsy tests, are primarily designed to detect cancer and demonstrate low sensitivity in detecting
pre-cancerous polyps. As such, they do not necessarily provide patients with the time window to pre-empt the disease. C-Scan is non- invasive
and requires no preparation or sedation, allowing the patients to continue their daily routine with no interruption as the capsule is
propelled through the gastrointestinal tract by natural motility. C-Scan is comprised of three main components: (1) C-Scan Cap, an ingestible
X-ray scanning capsule; (2) C-Scan Track, three miniaturized patches worn on the patient’s back for integrated positioning, control
and data recording; and (3) C-Scan View, a proprietary software to process and represent 2D and 3D maps of the inner surface of the colon.
We believe that this solution has the potential to become an alternative for both physicians and patients and to increase the number of
people completing CRC screening.
Check-Cap LLC was formed in 2004 as a Delaware
limited liability company to develop a novel and superior solution for colon cancer screening. In 2009, all of the business operations
and substantially all of the assets of Check-Cap LLC were transferred to Check Cap Ltd., a newly-incorporated Israeli company. Our headquarters
are located near Haifa, in the northern part of Israel. Our management team includes an experienced group of executives in the business,
research, clinical and regulatory fields. As of March 15, 2022, our research and development team consists of 78 experienced professionals
(including employees and independent contractors) in a variety of fields such as clinics, physics, algorithms, electronics, software,
mechanical engineering, verification and validation, and manufacturing.
C-Scan is being designed to create a reconstructed
three-dimensional representation of the interior surface of the colon and to enable detection of clinically significant polyps with a
high degree of sensitivity. Colonic polyps are tissue growths that occur on the lining of the colon. Polyps in the colon are extremely
common and certain types of polyps can become cancerous over time.
For the past several years, we have focused
on the research and development of our imaging technology. For details regarding our clinical trials, see Item 4B “Business Overview—C-
Scan System Non-Clinical and Clinical History.”
We have generated significant losses to
date, and we expect to continue to generate losses for at least the next several years as we continue our investment in research and development
and clinical trials in order to complete the development of C-Scan and to attain regulatory approvals, manufacturing scale up, begin the
commercialization efforts for our products, increase our marketing and selling expenses, and incur additional costs as a result of being
a public company in the United States. The extent of our future operating losses and the timing of becoming profitable are highly uncertain,
and we may never achieve or sustain profitability. As of December 31, 2022, we had accumulated losses of approximately $127.3 million.
Our financing activities are described below under “Liquidity and Capital Resources.”
Since our inception, we have devoted substantially
all of our efforts to research and development, clinical trials, recruiting management and technical staff, acquiring assets and raising
capital. We are still in the development and clinical stage and have not yet generated revenues. We have incurred losses of $19.1 million
and $17.2 million for the years ended December 31, 2022 and 2021, respectively. As of December 31, 2022, our accumulated deficit was approximately
$127.3 million. The extent of our future operating losses and the timing of becoming profitable are uncertain.
We have funded our operations to date primarily
through equity financings, sales of our ordinary shares and warrants, the exercise of warrants and other funding transactions and through
grants from the IIA.
Additional funding will be required to
complete our research and development and clinical trials, to attain regulatory approvals, to establish manufacturing infrastructure and
to begin the commercialization efforts of C-Scan. We have not yet commercialized our product. Even if we commercialize our product, we
may not become profitable in the foreseeable future. Our ability to achieve profitability depends on a number of factors, including our
ability to obtain regulatory approval for our product, successfully reduce the manufacturing cost of our C-Scan product, successfully
complete any post-approval regulatory obligations and successfully commercialize our product alone or in partnership.
To meet our capital needs, we are considering
multiple alternatives, including, but not limited to, additional equity financings and other funding transactions. While we have been
successful in raising financing in the past, there can be no assurance that we will be able to do so in the future on a timely basis on
terms acceptable to us, or at all. Uncertain market conditions and approval by regulatory bodies and adverse results from clinical trials
may (among other reasons) adversely impact our ability to raise capital in the future.
We recently announced the temporary postponement
of the initiation of the second part of the U.S. pivotal study that was expected in mid-2023 and that as a result, we adopted a plan of
action, including (among other things) a cost reduction plan in order to extend our cash runway to longer than initially planned, which
we are in the process of implementing. Based on such plan, we believe that current cash on hand will be sufficient to fund our ongoing
operations and plans into the fourth quarter of 2024. Management expects that we will continue to generate losses from the development,
manufacturing and infrastructure costs, clinical development and regulatory activities of C-Scan, which will result in negative cash flow
from operating activity. The actual amount of cash that we will need to operate is subject to many factors, including, but not limited
to, the timing, conduct of clinical trials for C-Scan and regulatory path along with cost to commercialize our product.
For a more detailed description of our business and plans, see Item 4B “Information
on Our Company – Business Overview.”
A. Operating Results
Financial Operations Overview
Revenue
We have not generated any revenue since our inception. To date,
we have funded our operations primarily through equity financings, as well as from grants that we received from the IIA. If our product
development efforts result in clinical success, regulatory approval and the successful commercialization, we expect to generate revenue
from sales of C-Scan.
Operating Cost and Expenses
Our operating costs and expenses are classified
into two categories: research and development expenses and general and administrative expenses. For each category, the largest component
is personnel costs, which consists of salaries, employee benefits and share-based compensation. Operating costs and expenses also include
allocated overhead costs for depreciation of equipment. Operating costs and expenses are generally recognized as incurred. We recently
announced the temporary postponement of the initiation of the second part of the U.S. pivotal study that was expected in mid-2023 and
that as a result, we adopted a plan of action including (among other things) a cost reduction plan in order to extend our cash runway
to longer than initially planned, which we are in the process of implementing. However, we expect personnel costs and operating costs
to continue to increase in the future as we continue the development of C-Scan, including the pivotal study and scale up of our manufacturability.
Research and Development Expenses, Net
Research and development activities are
central to our business model. We intend to increase our research and development operations in order to complete the development of C-Scan,
to conduct our clinical trials, including the pivotal study in the United States, and to attain regulatory approvals.
Since 2018, we have spent approximately $54.7 million
on research and development expenses as of December 31, 2022, of which $0.95 million was funded by government grants. Our total research
and development expenses, net of participations in the years ended December 31, 2022, 2021, and 2020 were approximately $14.3, $12.3 million,
and $10.0 million, respectively. All research and development expenses are expensed as incurred. We expect research and development expenses
to slightly increase in 2023 and may further increase according to the progress of our clinical program.
Research and development expenses consist primarily
of costs incurred for our development activities, including:
|
• |
employee-related expenses for research and development staff (including research and development, clinical,
operations, production and Q&A staff), including salaries, benefits and related expenses, share-based compensation and travel expenses;
|
|
• |
payments associated with clinical activities including payments made to third-party CROs, investigative
sites, patients, materials and consultants; |
|
• |
payments associated with the development activities of our advanced C-Scan system and non-clinical activities,
including payments made to third-party subcontractors, providers and consultants; |
|
• |
manufacturing development costs and manufacturing scale up costs; |
|
• |
costs associated with regulatory operations; |
|
• |
facilities, depreciation and other expenses, which include direct and allocated expenses for rent and maintenance
of facilities; and |
|
• |
costs associated with obtaining and maintaining patents. |
Our research and development expenses, net, are
net of grants for the research and development for C-Scan and for the transition from research and development to manufacturing that we
have received from the Government of Israel through the IIA. Under the terms of the Innovation Law as currently in effect, in exchange
for the research and development grants we received, we are required to pay the IIA royalties from our revenues up to an aggregate of
100% (which may be increased under certain circumstances) of the U.S. dollar-linked value of the grant, plus interest at the rate of 12-month
LIBOR. Pursuant to regulations under the Innovation Law, the rate of royalty repayment is 3% or 4% of revenues (which may be increased
under certain circumstances) from sales of products and services based on know-how funded by the IIA research and development grants we
have received. As of December 31, 2022, we had received funding from the IIA for the research and development of C-Scan in the aggregate
amount of approximately $5.6 million. As of December 31, 2022, we had not paid any royalties to the IIA and had a contingent obligation
to the IIA in respect of such research and development grants (including interest at the rate of 12-month LIBOR) in the amount of approximately
$6.2 million. In addition, in January 2021, we received an IIA grant approval to support the funding of our transition from research and
development to manufacturing. The final IIA grant amounted to $620,000 (NIS 2.18 million) (along with a co- investment by us of
the same amount), which we are not required to repay to the IIA, subject to the terms and conditions set forth in the grant approval,
of which we received approximately $222,000 (NIS 784,000) in February 2023, $82,000 (NIS 264,000) in January 2022 and $349,000 (NIS 1,134,000)
in 2021 . For additional information regarding the IIA grants, see Item 10E “Additional Information - Taxation - Israeli Tax Considerations
and Government Programs - The Encouragement of Research, Development and Technological Innovation in the Industry Law 5744-1984 (formerly
the Encouragement of Industrial Research and Development Law, 5744-1984).” There can be no assurance that we will continue to receive
grants from the IIA in amounts sufficient for our operations, if at all. See also “Item 3D “Key Information - Risk Factors—
Risks Related to Our Operations in Israel—Pursuant to the terms of the Israeli government grants
we received for research and development expenditures, we are obligated to pay certain royalties on our revenues to the Israeli government.
In addition, the terms of the Israeli government grants we received require us to satisfy specified conditions and to make additional
payments in addition to repayment of the grants upon certain events.”
General and Administrative Expenses
Our general and administrative expenses consist
primarily of salaries and other related costs, including share-based compensation expense, for persons serving as our
directors and executives, finance, legal, human resources and administrative personnel, professional service fees, directors’ and
officers’ liability insurance and other general corporate expenses, such as communication, office and travel expenses. Based on
our current plans, we expect that our general and administrative expenses will slightly decrease in 2023; however, we expect general and
administrative expenses to increase in the future, in part due to costs associated with being a public company in the United States, including
compliance under the Sarbanes-Oxley Act of 2002 and rules promulgated by the U.S. Securities and Exchange Commission. These public company-related
expense increases will likely include costs of additional legal fees, accounting and audit fees, directors’ and officers’
liability insurance premiums and costs related to investor relations.
Finance Income, net
Our finance income, net in 2022, 2021, and 2020
consists primarily of interest earned on our cash equivalents and short-term bank deposits and changes in provision for royalties.
Foreign currency transactions are translated into
U.S. dollars using the exchange rates prevailing at the dates of the transactions or valuation where items are re-measured. Foreign exchange
gains and losses resulting from the settlement of such transactions and from the translation of year-end exchange rates of monetary assets
and liabilities denominated in foreign currencies are recognized to “finance income, net” in the consolidated statement of
operations.
Taxes on Income
The standard corporate tax rate in Israel is 23%
for the 2018 tax year and thereafter.
We do not generate taxable income in Israel, as
we have historically incurred operating losses resulting in carryforward tax losses totaling approximately $105 million as of December
31, 2022. We anticipate that we will be able to carry forward these tax losses indefinitely to future tax years. However, a tax loss that
can be utilized in a certain tax year cannot be carried forward to future tax years. Accordingly, we do not expect to pay taxes in Israel
until we have taxable income after the full utilization of our carry forward tax losses.
Under the Law for the Encouragement of Capital
Investments, 5719-1959 and other Israeli legislation, we may be entitled to certain additional tax benefits, including reduced tax rates,
accelerated depreciation and amortization rates for tax purposes on certain assets, deduction of public offering expenses in three equal
annual installments and amortization of other intangible property rights for tax purposes. See Item 10E “Additional Information
— Taxation— Israeli Tax Considerations and Government Programs” for additional information concerning these tax benefits.
Results of Operations
For convenience purposes, the numbers set forth
in the management’s discussion and analysis below are, where applicable, rounded up and presented in millions, whereas the numbers
in the tables below are presented in thousands. As result, the percentages set forth in the year-over-year comparisons below are based
on numbers that have (where applicable) been rounded up to millions, which may slightly differ than the percentages that would result
from the corresponding numbers set forth in the table that are presented in thousands.
Year Ended December 31, 2022 Compared to Year Ended December 31, 2021
| |
|
|
|
| |
|
|
|
|
|
|
| |
|
(US$ in thousands, except per share data) |
|
|
Research and development expenses, net |
|
$ |
14,271 |
|
|
$ |
12,349 |
|
|
General and administrative expenses |
|
|
5,763 |
|
|
|
4,972 |
|
|
Operating loss |
|
|
20,034 |
|
|
|
17,321 |
|
|
Finance income, net |
|
|
|
|
|
|
|
|
|
Net loss |
|
|
|
|
|
|
|
|
Research and Development
Expenses, net. Research and development expenses, net for the year ended December 31, 2022 were approximately $14.3 million,
as compared with approximately $12.3 million for the year ended December 31, 2021 an increase of $1.9 million or 15.6%. The increase in
research and development expenses, net between 2022 and 2021 was primarily due to (i) an increase of approximately $0.8 million in salary
and related expense, mainly as a result of an expansion in head count that was offset in part by a reduction in bonuses expense in 2022
and by exchange rate fluctuations, (ii) an increase of approximately $0.6 million in subcontractors and consultants including regulatory
consultants, (iii) an increase of approximately $0.2 million in share-based compensation, (iv) an increase of approximately $0.8 million
in other research and development expenses, including clinical study expenses, and (iv) a $0.2 million decrease in the participation of
the IIA in research and development expenses. The increase in research and development expenses, net between 2022 and 2021, was offset
in part by a $0.8 million decrease in material expenses.
| |
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
Salaries and related expenses |
|
$ |
8,257 |
|
|
$ |
7,428 |
|
|
$ |
829 |
|
|
Share-based compensation |
|
|
493 |
|
|
|
329 |
|
|
|
164 |
|
|
Materials |
|
|
2,258 |
|
|
|
3,020 |
|
|
|
(762 |
) |
|
Subcontractors and consultants (*) |
|
|
1,375 |
|
|
|
820 |
|
|
|
555 |
|
|
Depreciation |
|
|
289 |
|
|
|
169 |
|
|
|
120 |
|
|
Cost for registration of patents |
|
|
140 |
|
|
|
153 |
|
|
|
(13 |
) |
|
Other research and development expenses(*) |
|
|
1,681 |
|
|
|
861 |
|
|
|
820 |
|
| |
|
|
14,493 |
|
|
|
12,780 |
|
|
|
1,713 |
|
|
Less participation of the IIA (formerly the OCS) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Total research and development expenses, net |
|
|
|
|
|
|
|
|
|
|
|
|
(*) Reclassified
General and Administrative Expenses.
Our general and administrative expenses for the year ended December 31, 2022 were approximately $5.8 million, as compared to approximately
$5.0 million for the year ended December 31, 2021, an increase of $0.8 million, or 15.9%. The increase in general and administrative
expenses is primarily due to (i) a $0.4 million increase in professional services expenses, (ii) a $0.2 million increase in share-based
compensation expenses, and (iii) a $0.4 million increase in other general expenses, mainly associated with directors’ and officers’
liability insurance, offset in part by a $0.3 million decrease in salaries and related expenses, mainly due to a reduction in bonuses
expense in 2022.
| |
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
Salaries and related expenses |
|
$ |
1,784 |
|
|
$ |
2,042 |
|
|
$ |
(258 |
) |
|
Share-based compensation |
|
|
386 |
|
|
|
162 |
|
|
|
224 |
|
|
Professional services |
|
|
1,092 |
|
|
|
710 |
|
|
|
382 |
|
|
Office rent and maintenance |
|
|
246 |
|
|
|
172 |
|
|
|
74 |
|
|
Depreciation |
|
|
116 |
|
|
|
36 |
|
|
|
80 |
|
|
Other general and administrative expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
Total general and administrative expenses |
|
|
|
|
|
|
|
|
|
|
|
|
Operating Loss.
Our operating loss for the year ended December 31, 2022 was approximately $20.0 million, as compared with approximately $17.3 million
for the year ended December 31, 2021, an increase of approximately $2.7 million, or 15.7%. The increase was as a result of the $1.9 million
increase in research and development expenses, net for the year ended December 31, 2022 and the $0.8 million increase in general and administrative
expenses for the year ended December 31, 2022, as compared to the year ended December 31, 2021.
Finance Income,
net. Our finance income, net for the year ended December 31, 2022 was approximately $926,000, as compared to approximately
$119,000 for the year ended December 31, 2021, an increase of approximately $807,000. The change in our finance income, net is primarily
due to an increase of $946,000 of interest income on short-term deposits recorded for the year ended December 31, 2022, as compared to
the year ended December 31, 2021, offset in part by a $173,000 decrease in fair value of derivatives recorded for the year ended December
31, 2022, as compared to the year ended December 31, 2021.
Net loss.
Our net loss for the year ended December 31, 2022 was approximately $19.1 million, as compared to approximately $17.2 million for the
year ended December 31, 2021, an increase of $1.9 million.
Year Ended December 31, 2021 Compared to Year Ended December 31, 2020
| |
|
|
|
| |
|
2021 |
|
|
|
|
| |
|
(US$ in thousands, except per share data) |
|
|
Research and development expenses, net |
|
$ |
12,349 |
|
|
$ |
10,008 |
|
|
General and administrative expenses |
|
|
4,972 |
|
|
|
3,924 |
|
|
Operating loss |
|
|
17,321 |
|
|
|
13,932 |
|
|
Finance income, net |
|
|
119 |
|
|
|
86 |
|
|
Net loss |
|
$ |
17,202 |
|
|
$ |
13,846 |
|
Research and Development
Expenses, net. Research and development expenses, net for the year ended December 31, 2021 were approximately $12.3 million,
as compared with approximately $10.0 million for the year ended December 31, 2020, an increase of $2.3 million or 23.4%. The increase
in research and development, net expenses between 2021 and 2020 was primarily due to (i) an increase of approximately $1.3 million in
salary and related expense, mainly as a result of an expansion in head count and currency exchange rate fluctuation, (ii) a $1.2 million
increase in material expenses, (iii) an increase of approximately $0.16 million in share-based compensation, and (iv) an increase of approximately
$0.4 million in other research and development expenses, including clinical expenses. The increase in research and development expenses,
net between 2021 and 2020, was offset in part by a $0.3 million decrease in subcontractors and consultants and a $0.43 million grant received
from the IIA, which amount was recorded as a deduction from research and development expenses in 2021.
|
|
2021 |
|
|
2020 |
|
|
Change |
|
| |
|
(US$ in thousands) |
|
|
Salaries and related expenses |
|
$ |
7,428 |
|
|
$ |
6,173 |
|
|
$ |
1,255 |
|
|
Share-based compensation |
|
|
329 |
|
|
|
165 |
|
|
|
164 |
|
|
Materials |
|
|
3,020 |
|
|
|
1,792 |
|
|
|
1,228 |
|
|
Subcontractors and consultants(*) |
|
|
820 |
|
|
|
1,103 |
|
|
|
(283 |
) |
|
Depreciation |
|
|
169 |
|
|
|
123 |
|
|
|
46 |
|
|
Cost for registration of patents |
|
|
153 |
|
|
|
164 |
|
|
|
(11 |
) |
|
Other research and development expenses(*) |
|
|
861 |
|
|
|
488 |
|
|
|
373 |
|
| |
|
|
12,780 |
|
|
|
10,008 |
|
|
|
2,772 |
|
|
Less participation of the IIA (formerly the OCS) |
|
|
(431 |
) |
|
|
- |
|
|
|
(431 |
) |
|
Total research and development expenses, net |
|
$ |
12,349 |
|
|
$ |
10,008 |
|
|
$ |
2,341 |
|
(*) Reclassified
General and Administrative
Expenses. Our general and administrative expenses for the year ended December 31, 2021 were approximately $5.0 million, as compared
to approximately $3.9 million for the year ended December 31, 2020, an increase of $1.0 million, or 27%. The increase in general
and administrative expenses is primarily due to a $0.3 million increase in salaries and related expenses, a $0.1 million increase in professional
services expenses and a $0.6 million increase in other general expenses, mainly associated with directors’ and officers’ liability
insurance, offset in part by a $0.1 million decrease in share-based compensation expenses.
|
|
2021 |
|
|
2020 |
|
|
Change |
|
| |
|
(US$ in thousands) |
|
|
Salaries and related expenses |
|
$ |
2,042 |
|
|
$ |
1,698 |
|
|
$ |
344 |
|
|
Share-based compensation |
|
|
162 |
|
|
|
243 |
|
|
|
(81 |
) |
|
Professional services |
|
|
710 |
|
|
|
574 |
|
|
|
136 |
|
|
Office rent and maintenance |
|
|
172 |
|
|
|
174 |
|
|
|
(2 |
) |
|
Depreciation |
|
|
36 |
|
|
|
25 |
|
|
|
11 |
|
|
Other general and administrative expenses |
|
|
1,850 |
|
|
|
1,210 |
|
|
|
640 |
|
|
Total general and administrative expenses |
|
$ |
4,972 |
|
|
$ |
3,924 |
|
|
$ |
1,048 |
|
Operating Loss. Our operating loss
for the year ended December 31, 2021 was approximately $17.3 million, as compared with approximately $13.9 million for the year ended
December 31, 2020, an increase of approximately $3.4 million, or 24.3%. The increase was as a result of the $2.3 million increase
in research and development expenses, net for the year ended December 31, 2021 and the $1.0 million increase in general and administrative
expenses for the year ended December 31, 2021, as compared to the year ended December 31, 2020.
Finance Income,
net. Our finance income, net for the year ended December 31, 2021 was approximately $119,000, as compared to approximately
$86,000 for the year ended December 31, 2020, an increase of approximately $33,000. The change in our finance income, net is primarily
due to an increase of $31,000 of interest income on short-term deposits recorded for the year ended December 31, 2021, as compared to
the year ended December 31, 2020.
Net loss.
Our net loss for the year ended December 31, 2021 was approximately $17.2 million, as compared to approximately $13.8 million for the
year ended December 31, 2020, an increase of $3.4 million.
B. Liquidity
and Capital Resources
Sources of Liquidity
To date, we have funded our operations primarily
through equity financings consummated prior to our initial public offering, our initial public offering, private placements, registered
direct and underwritten public offerings, our warrant exercise transaction and grants that we received from the IIA (formerly the OCS)
and the BIRD Foundation. As of December 31, 2022, we had approximately $42.1 million cash and cash equivalents, including restricted cash,
and had invested most of our available cash in short term bank deposits.
Effective November 23, 2022, our Board of Directors
effected a reverse share split of 1-for-20 (i.e., 20 ordinary shares were combined into one ordinary share). Share and per share amounts,
prior to November 23, 2022, have been adjusted to reflect the reverse share split.
During the first quarter of 2021, certain warrant
holders exercised warrants to purchase 1,210,235 ordinary shares, at exercise prices ranging from $15-$16 per share, for total gross proceeds
of approximately $19.2 million.
On July 2, 2021, we consummated a registered direct
offering of 1,296,297 ordinary shares and warrants to purchase up to 1,296,297 ordinary shares, or the July 2021 Offering. Each ordinary
share was sold together with one warrant to purchase one ordinary share at a combined purchase price of $27 per share and accompanying
warrant, for aggregate gross proceeds of approximately $35,000,000, excluding any proceeds that may be received upon exercise of the warrants.
Each warrant will be immediately exercisable and will expire on January 2, 2024. The exercise price of each warrant is $30 per ordinary
share, subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if at the time of exercise thereof,
there is no effective registration statement registering the issuance or resale of ordinary shares underlying the warrants. The ordinary
shares and the warrants are separable and were issued separately, but purchased together in this offering. We agreed to pay the placement
agent an aggregate fee equal to 7.0% of the gross proceeds of the offering, a management fee of 1.0% of the gross proceeds of the offering,
a non-accountable expenses allowance of $25,000, fees and expenses up to $100,000, and a clearing fee of $15,950. In addition, issued
to the placement agent or its designees warrants to purchase up to a total of 5.0% of the aggregate number of ordinary shares sold in
the transaction, or warrants to purchase up to 64,815 ordinary shares, substantially on the same terms as the warrants issued to the investors
in the private placement, except that the placement agent warrants will have an exercise price of $33.75 per share (which represents 125%
of the investors’ purchase price per share and accompanying warrant). Upon any exercise of the warrants for cash, we agreed to pay
the placement agent a total cash fee equal to 7.0% of the aggregate gross proceeds from the exercise of the warrants, a management fee
equal to 1.0% of the aggregate gross proceeds from the exercise of the warrants, and warrants to purchase up to 5.0% of the number of
ordinary shares issued upon the cash exercise of the warrants.
On March 3, 2022, we consummated a registered direct
offering of 1,000,000 ordinary shares and warrants to purchase up to 750,000 ordinary shares, or the March 2022 Offering. Each ordinary
share was sold together with one warrant to purchase 0.75 ordinary share at a combined purchase price of $10 per share and accompanying
warrant, for aggregate gross proceeds of $10,000,000, excluding any proceeds that may be received upon exercise of the warrants. Each
warrant will be immediately exercisable and will expire on March 3, 2027. The exercise price of each warrant is $13 per ordinary share,
subject to adjustment as set forth therein. The warrants may be exercised on a cashless basis if at the time of exercise thereof, there
is no effective registration statement registering the issuance or resale of ordinary shares underlying the warrants. The ordinary shares
and the warrants are separable and were issued separately, but purchased together in this offering. We agreed to pay the placement agent
an aggregate fee equal to 7.0% of the gross proceeds of the offering, a management fee of 1.0% of the gross proceeds of the offering,
a non-accountable expenses a of $70,000, and a clearing fee of $15,950. In addition, we agreed to issue to the placement agent or its
designees warrants to purchase up to a total of 5.0% of the aggregate number of ordinary shares sold in the transaction, or warrants to
purchase up to 50,000 ordinary shares, substantially on the same terms as the warrants issued to the investors in the private placement,
except that the placement agent warrants will have an exercise price of $12.5 per share (which represents 125% of the investors’
purchase price per share and accompanying warrant) and expiration date of March 1, 2027. Upon any exercise of the warrants for cash, we
agreed to pay the placement agent a total cash fee equal to 7.0% of the aggregate gross proceeds from the exercise of the warrants, a
management fee equal to 1.0% of the aggregate gross proceeds from the exercise of the warrants, and warrants to purchase up to 5.0% of
the number of ordinary shares issued upon the cash exercise of the warrants. Simultaneously with this offering, we entered into a warrant
amendment agreement, or the Warrant Amendment Agreement, with the investors in the March 2022 Offering. Pursuant to the Warrant Amendment
Agreement, certain warrants to purchase up to an aggregate of 926,297 ordinary shares of the Company that were issued to the investors
in the July 2021 Offering were amended to have a reduced exercise price of $13 per share and the term of exercise was extended to January
2, 2025.
Since our inception through December 31, 2022,
we had received funding from the IIA for research and development of C-Scan in the aggregate amount of approximately $5.6 million, of
which $31,000 was received in the year ended December 31, 2020. We did not receive IIA funding for research and development for the years
ended December 31, 2022 and 2021. As of December 31, 2022, we had not paid any royalties to the IIA and had a contingent obligation to
the IIA with respect to such funding (including interest at the rate of 12-month LIBOR) in the amount of approximately $6.2 million. In
addition, in January 2021, we received an IIA grant approval to support the funding of our transition from research and development to
manufacturing. The final IIA grant amounted to $620,000 (NIS 2.18 million) (along with a co-investment by us of the same amount), which
we are not required to repay to the IIA, subject to the terms and conditions set forth in the grant approval, of which we received approximately
$222,000 (NIS 784,000) in February 2023, $82,000 (NIS 264,000) in January 2022 and $349,000 (NIS 1,134,000) in 2021.
We are a clinical and development-stage medical
diagnostics company and have not yet generated revenues. We have incurred net losses in each fiscal year since we commenced operations
in 2009. We incurred net losses of $19.1 million, $17.2 million, and $13.8 million in 2022, 2021, and 2020, respectively. As of December
31, 2022, our accumulated deficit was $127.3 million. The extent of our future operating losses and the timing of becoming profitable
are uncertain.
We recently adopted a cost reduction plan in order
to extend our cash runway to longer than initially planned, which we are in the process of implementing. Based on such plan, we
believe that current cash on hand will be sufficient to fund our ongoing operations and plans into the fourth quarter of 2024. We expect
that we will continue to generate losses from our research and development and clinical trials, regulatory activities, manufacturing infrastructure
and commercialization efforts of C-Scan, which will result in a negative cash flow from operating activity. The actual amount of cash
that we will need to operate is subject to many factors, including, but not limited to, the initiation, timing, progress and results of
our clinical trials and other product development efforts along with cost to commercialize our product.
We anticipate that we will need to raise substantial
additional financing in the future to fund our operations. In order to meet these additional cash requirements, we may incur debt, license
certain intellectual property, and seek to sell additional equity or convertible securities that may result in dilution to our shareholders.
If we raise additional funds through the issuance of equity or convertible securities, these securities could have rights or preferences
senior to those of our ordinary shares and could contain covenants that restrict our operations. There can be no assurance that we will
be able to obtain additional equity or debt financing on terms acceptable to us, if at all. Our future capital requirements will depend
on many factors, including:
|
• |
our ability to obtain funding from third parties, including any future collaborative partners; |
|
• |
the scope, rate of progress and cost of our clinical trials and other research and development programs;
|
|
• |
the time and costs required to gain regulatory approvals; |
|
• |
the terms and timing of any collaborative, licensing and other arrangements that we may establish;
|
|
• |
the costs of filing, prosecuting, defending and enforcing patents, patent applications, patent claims,
trademarks and other intellectual property rights; |
|
• |
any product liability or other lawsuits related to our product; |
|
• |
the expenses needed to attract and retain skilled personnel; |
|
• |
the costs and timing of future commercialization activities, including product manufacturing, marketing,
sales, and distribution; |
|
• |
the revenue, if any, received from commercial sales of our product; |
|
• |
the general and administrative expenses related to being a public company; |
|
• |
the effect of competition and market developments |
|
• |
future clinical trial results; |
|
• |
the COVID – 19 pandemic; and |
|
• |
the political and security situation in Israel. |
Historical Cash Flows
The following table summarizes our statement of
cash flows for the years ended December 31, 2022, 2021, and 2020.
| |
|
|
|
| |
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
Net cash used in operating activities |
|
$ |
(18,707 |
) |
|
$ |
(16,264 |
) |
|
$ |
(13,113 |
) |
|
Net cash used in investing activities |
|
$ |
(12,508 |
) |
|
$ |
(16,006 |
) |
|
$ |
(10,451 |
) |
|
Net cash provided by financing activities |
|
$ |
8,850 |
|
|
$ |
51,024 |
|
|
$ |
23,582 |
|
Operating Activities
Net cash used in operating activities for the year
ended December 31, 2022 was $18.7 million, as compared to $16.3 million for the year ended December 31, 2021. The increase in net cash
used in operating activities in 2022 was attributable primarily to the increase in operating loss and due to a decrease of accruals on
account of employee compensation and a decrease of trade accounts payable, accruals and other current liabilities, offset in part by a
decrease in prepaid and other current assets and non-current assets, as compared to 2021. Net cash used in operating activities for the
year ended December 31, 2021 was $16.3 million, as compared to $13.1 million for the year ended December 31, 2020. The increase in net
cash used in operating activities in 2021 was attributable primarily to the increase in operating loss and increase in prepaid and other
current assets and non-current assets offset in part by an increase in trade accounts payable, accruals and other current liabilities,
as compared to 2020
Investing Activities
Net cash used in investing activities for the year
ended December 31, 2022 was $12.5 million, comprised primarily of investment in short-term bank deposits in the amount of $12.0 million
and purchase of property and equipment in the amount of $0.5 million. Net cash used in investing activities for the year ended December
31, 2021 was $16.0 million, comprised primarily of investment in short-term bank deposits in the amount of $15 million and purchase of
property and equipment in the amount of $1.0 million. Net cash used in investing activities for the year ended December 31, 2020 was $10.5
million, comprised primarily of investment in short-term bank and other deposits in the amount of $10.1 million and purchase of property
and equipment in the amount of $0.4 million.
Financing Activities
Net cash provided by financing activities for the
year ended December 31, 2022 was $8.9 million, comprised of net proceeds from the issuance of ordinary shares in the March 2022 registered
direct offering. Net cash provided by financing activities for the year ended December 31, 2021 was $51.0 million, comprised primarily
of net proceeds of $31.8 million from the issuance of ordinary shares in the July 2021 registered direct offering and $19.2 from exercise
of warrants during the first quarter of 2021. Net cash provided by financing activities for the year ended December 31, 2020 was $23.6
million, comprised primarily of net proceeds of $4.73 million from the issuance of ordinary shares in the February 2020 private placement,
net proceeds of $10.2 million from the April 2020 and May 2020 registered direct offerings and net proceeds of $8.7 million from the July
2020 warrant exercise transaction.
We expect that our sources of cash for the year
ended December 31, 2023 will include cash held in our bank accounts and may include proceeds from issuance of ordinary shares as a result
of financing transactions or from the exercise of warrants.
Capital and Operating Expenditures
We expect to incur
losses from operations for the foreseeable future.
Our expected future expenditures related to product, clinical and regulatory clearances
include the following:
|
• |
completion of the clinical development of C-Scan; |
|
• |
conducting clinical trials in the United States and other territories for purposes of regulatory approval
and post-marketing validation; |
|
• |
development of advanced version and future generations of C-Scan and future products; |
|
• |
FDA and additional regulatory filing activities in countries we intend to commercialize our system;
|
|
• |
Manufacturing scale up costs; and |
We recently adopted a cost reduction plan in order
to extend our cash runway to longer than initially planned, which we are in the process of implementing. Accordingly, based on the
cost reduction plan, we believe that our cash and cash equivalents and short-term deposits will be sufficient to fund our operations and
plans, including the U.S. pivotal study, into the fourth quarter of 2024.
Management’s plans include additional fund
raising in the future in order to secure sufficient cash resources to finance our research and development and clinical trials, regulatory
activities, manufacturing infrastructure and future commercialization efforts for C-Scan.
To meet our capital needs, we are considering multiple
alternatives, including, but not limited to, additional equity financings and other funding transactions. While we have been successful
in raising financing in the past, there can be no assurance that we will be able to do so in the future on a timely basis on terms acceptable
to us, or at all. Uncertain market conditions and approval by regulatory bodies and adverse results from clinical trials may (among other
reasons) adversely impact our ability to raise capital in the future.
In the event we are unable to successfully raise
additional capital, we will not have sufficient cash flows and liquidity to finance our business operations as currently contemplated.
Accordingly, in such circumstances we would be compelled to immediately reduce general and administrative expenses and delay research
and development projects and clinical trials, until we are able to obtain sufficient financing. If such sufficient financing is not received
timely, we would then need to pursue a plan to license or sell our assets, seek to be acquired by another entity, cease operations and/or
seek bankruptcy protection.
See “Item 3D “Key Information –
Risk Factors—Risks Related to Our Financial Position.”
We have operating lease obligations consisting
of payments pursuant to a lease agreement for office facilities in effect as of December 31, 2022, as well as lease agreements for vehicles,
which generally run for a period of three years. See Note 5 to our audited consolidated financial statements presented elsewhere in this
Annual Report.
Royalties provision
Provision for royalties to an ASIC designer
In December 2007, we entered into an agreement
for the development of an application specific integrated circuit, or ASIC, component to be used as an amplifier for the capture of signals
at low frequencies from X-ray detectors contained in our product. The ASIC developer is entitled to receive royalties from us in the amount
of €0.5 (approximately $0.53) for every ASIC component that we will sell, up to €200,000 (approximately $213,299). The net
present value of the royalty liability to the ASIC designer is dependent upon our management estimates and assumption as to future product
shipments and interest rates used to calculate the present value of the cash payments required to repay the royalties to the ASIC designer.
In calculating the present value of future royalty payments to the ASIC designer, we used a discount factor of 17.6%, commensurate with
our risk at the date of initial recognition of the liability. Any updates in the expected product shipments and the liability will be
recorded to profit and loss each period. As of December 31, 2022, it was probable that we will be required to pay the above mentioned
royalties, and accordingly, a liability for this reimbursement has been accounted for in our financial statements in the amount of $94,000.
As more information is gathered to assist our management
in making forecasts, the liability will be updated.
C. Research
and development, patents and licenses, etc.
For a description of our research and development
programs and the amounts that we have incurred over the last three years pursuant to those programs, see Item 4B “Information on
Our Company—Business Overview—Research and Development.”
D. Trend
Information
Our results of operations and financial condition
may be affected by various trends and factors discussed in Item 3D “Key Information—Risk factors,” Item 4 “Information
on Our Company” and elsewhere in this Item 5 “Operating and Financial Review and Prospects.”
E. Critical
Accounting Estimates
Our consolidated financial statements are prepared
in accordance with U.S. GAAP. The preparation of our financial statements requires us to make estimates, judgments and assumptions that
can affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial
statements and the reported amounts of expenses during the reporting periods. We base our estimates, judgments and assumptions on historical
experience and other factors that we believe to be reasonable under the circumstances. Materially different results can occur as circumstances
change and additional information becomes known. See Note 2 to our audited consolidated financial statements presented elsewhere in this
Annual Report for a description of the significant accounting policies that we used to prepare our consolidated financial statements.
We believe the following critical policies reflect
the more significant judgments and estimates used in preparation of the Consolidated Financial Statements.
Share-based compensation
We recognize expense for our share-based compensation
based on the fair value of the awards granted. We estimate the fair value of equity-based payment awards on the date of grant. The value
of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in our consolidated
statement of operations.
We recognize compensation expenses for the value of our awards granted based on the graded-vesting method over the requisite service period
for each separately vesting portion of the award.
We recognize compensation cost for awards with
performance conditions if and when we conclude that it is probable that the performance conditions will be achieved. We reassess at each
reporting period the probability of vesting for awards with performance conditions and adjust compensation cost based on our probability
assessment.
We use the Black-Scholes-Merton option-pricing
model for our share-based awards. The option-pricing model requires a number of assumptions, of which the most significant are the fair
market value of the underlying ordinary shares, expected share price volatility and the expected option term. In the year ended December
31, 2022, 2021 and 2020, expected volatility was calculated based upon actual historical stock price movements over the most recent periods
ending on the grant date, equal to the expected term of the options.
The expected option term represents the period
of time that options granted are expected to be outstanding. The expected option term is determined based on the simplified method in
accordance with Staff Accounting Bulletin No. 110, as adequate historical experience is not available to provide a reasonable estimate.
The risk-free interest rate is based on the yield
from U.S. treasury bonds with an equivalent term. We have historically not paid dividends and have no foreseeable plans to pay dividends.
Equity
In the years ended December 31, 2022 and December
31, 2021 we completed certain financing transactions in which we issued ordinary shares and warrants. The warrants are immediately exercisable
at a fixed exercise price per ordinary share, subject to certain adjustments and will expire on the expiring date as determined in the
applicable warrant. The warrants may be exercised on a cashless basis if at the time of exercise thereof, there is no effective registration
statement registering the ordinary shares underlying the warrants. We analyzed the terms of the warrants and concluded that the terms
of the warrants did not include features that would preclude equity classification. Therefore, we classified the warrants as equity.
Recent Accounting Pronouncements
See Note 2 to our audited consolidated financial
statements presented elsewhere in this Annual Report for a description of the recent accounting policies.
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A. Directors
and senior management
The following table sets forth information for
our executive officers and directors as of March 30, 2023. Unless otherwise stated, the address for our directors and executive officers
is c/o Check-Cap Ltd., 29 Abba Hushi Avenue, P.O. Box 1271, Isfiya, 3009000, Israel.
|
Name |
Age |
Position(s)
|
|
Alex Ovadia |
61 |
Chief Executive Officer |
|
Mira Rosenzweig |
51 |
Chief Financial Officer |
|
Yoav Kimchy |
62 |
Chief Technology Officer |
|
Boaz Shpigelman |
51 |
Vice President, Research and Development |
|
Joshua (Shuki) Belkar |
54 |
Vice President, Operations |
|
Hanit Brenner-Lavie |
51 |
Vice President, Clinical Affairs |
|
Israel Hershko |
57 |
Vice President, Quality Assurance and Regulatory Affairs |
|
Steven Hanley (1)(2) |
55 |
Chairman of the Board of Directors |
|
Clara Ezed (3)(4) |
53 |
Director |
|
Mary Jo Gorman (1)(2)(3)(4) |
63 |
Director |
|
XiangQian (XQ) Lin |
41 |
Director |
|
Yuval Yanai (1)(2)(3)(4) |
70 |
Director |
| (1) |
Member of our Nominating Committee. |
| (2) |
Member of our Financing Committee. |
| (3) |
Member of our Compensation Committee. |
| (4) |
Member of our Audit Committee. |
Executive Officers and Directors
Alex
Ovadia has served as our Chief Executive Officer since February 26, 2018. Mr. Ovadia has served as a member of the Board of Directors
of our U.S. subsidiary, Check-Cap US, Inc., since March 2018. Prior to that, Mr. Ovadia served as our Chief Operating Officer and Israeli
Site Manager since July 1, 2015, in addition to serving as our Vice President of Research and Development since January 2013. Mr. Ovadia
has more than 25 years of experience leading global operations, in addition to worldwide management of complex projects. From 2001 to
2012, Mr. Ovadia served in various positions as global director and senior manager for CT research and development at Philips Healthcare
– CT Systems. Mr. Ovadia was also appointed as a member of Global CT R&D staff and as a member of management of Philips Medical
System Technologies Ltd. Prior to that, from 1990 to 2001, Mr. Ovadia served as project/systems engineering manager for large scale military
projects, mainly aircraft upgrades for U.S. and European governments performed by Elbit Systems Ltd. Mr. Ovadia holds a BSc degree in
electrical engineering from the Technion-Israel Institute of Technology.
Mira
Rosenzweig has served as our Chief Financial Officer since April 2019. Ms. Rosenzweig has 20 years of financial management experience.
Prior to joining Check-Cap, Ms. Rosenzweig served as Chief Financial Officer of Entera Bio Ltd. (Nasdaq: ENTX) since May 2014. Prior to
Entera Bio, from May 2013 to April 2014, Ms. Rosenzweig served as Chief Financial Officer of privately-held Paskal Technologies and, before
that, from September 2008 to November 2011, as Vice President and Chief Financial Officer of Camtek Ltd. (Nasdaq: CAMT). Prior to that,
Ms. Rosenzweig served in various finance roles including at Elron Electronic Industries Ltd. (TASE: ELRN). Ms. Rosenzweig began her career
as a senior accountant at Luboshitz Kasierer, which is now part of Ernst & Young. Ms. Rosenzweig holds a Bachelor of Arts in Accounting
and Economics from the University of Haifa, Israel, and is a Certified Public Accountant (Israel).
Yoav
Kimchy has served as our Chief Technology Officer since our inception in April 2009 and served as a member of our board of directors
from 2009 and until December 1, 2015. Dr. Kimchy founded Check-Cap LLC in December 2004, serving as its president and chief executive
officer until March 2008, and as its president and chief technical officer since March 2008. Dr. Kimchy has also served as a member of
the board of directors of Check-Cap Ltd. (Delaware), the manager of Check-Cap LLC, since December 2004. Between 2005 and 2012, Dr. Kimchy
also served as our vice president of research and development. Between 2000 and 2003, Dr. Kimchy served as the vice president of research
and development of V-Target Ltd. (Israel), a medical device company developing gamma imaging applications. Prior to that, from 1998 to
2000, Dr. Kimchy served as the Director of Cardiovascular Research at Impulse Dynamics Ltd. (Israel), a medical device company developing
a unique therapeutic pulsing technology for chronic heart failure. From 1994 to 1998, Dr. Kimchy served as a systems engineer and algorithm
specialist for an Israeli government contract project with the Israeli Navy. Dr. Kimchy also served as a lieutenant in the Israeli Navy.
Dr. Kimchy holds a B.Sc. degree in physics and mathematics from the Hebrew University of Jerusalem, an M.Sc. degree in biomedical engineering
from Tel Aviv University and a PhD from the Technion-Israel Institute of Technology.
Boaz
Shpigelman has served as our Vice President, Research and Development since March 2018. From 2012 to 2018, Mr. Shpigelman served
as our director of development. From 2007 to 2012, Mr. Shpigelman served as R&D Manager at Orsan Medical Technologies Ltd., which
develops technologies for brain monitoring, with particular attention to intracranial pressure and its influence on cerebral perfusion.
From 2003 to 2007, Mr. Shpigelman served as Vice R&D Manager at GI-View, a medical company that developed a single use auto maneuvering
colonoscopy. From 1993 to 2003, Mr. Shpigelman held various positions at Scitex/Creo, a company that developed devices for the graphic
art field, initially as a hardware/software developer and subsequently as a director in the input division. Mr. Shpigelman holds a BSc
degree in Electrical Engineering from the Technion-Israel Institute of Technology.
Joshua
(Shuki) Belkar has served as Vice President of Operation since June 2019. Mr. Belkar has more than 20 years of experience managing
operations at global medical device companies, including managing large volume manufacturing of capsule-based products. Prior to joining
Check-Cap, Mr. Belkar served as vice president of operations and global services at Mazor Robotics Ltd., which was recently acquired by
Medtronic Plc. Prior to Mazor Robotics, Mr. Belkar served in various executive roles at Medtronic, including director of operations, vice
president of manufacturing and operations and director of production. Before joining Medtronic Plc., Mr. Belkar served as manager
of production at Elscint Medical Systems Ltd. Mr. Belkar holds a BD degree in Social Sciences from the Open University, Israel, and an
MA degree in logistic management and an executive MBA degree, both from Bar-Ilan University, Israel.
Hanit
Brenner-Lavie, Ph.D. has served as Vice President of Clinical Affairs since September 2021. Dr. Brenner-Lavie brings more than
15 years of experience in global clinical and regulatory affairs, including experience within the medical devices industry. Most recently,
from 2017 to August 2021, Dr. Brenner-Lavie served as the Vice President of Clinical Affairs at Alma Lasers, a global company developing
medical technology for the surgical and medical markets, where she led the successful introduction of innovative medical devices utilizing
laser, ultrasound and radio frequency (RF) technologies to address various medical indications. Previously, from 2012 to 2017, Dr. Brenner-Lavie
held the position of Clinical Director at Lumenis Ltd., a leader in lasers and light-based technological solutions for medical and aesthetic
treatments, and Clinical Research Manager at Syneron Ltd. (today Candela Medical), a global leader in aesthetic medical devices. Dr. Brenner-Lavie
holds a Ph.D. in Medical Sciences from the Technion – Israel Institute of Technology and an M.Sc. degree in Medical Sciences from
the Hebrew University of Jerusalem.
Israel
Hershko has served as Vice President of Quality Assurance and Regulatory Affairs since August 2019. Mr. Hershko has more than 30
years of experience in quality assurance and regulatory affairs in the medical device industry. Mr. Hershko has led the transfer of several
new products into global markets from the regulatory and quality aspects. Prior to joining Check- Cap, Mr. Hershko held executive positions
as director of quality assurance and regulatory affairs in several medical device companies, including Medical Compression Systems Ltd.,
Q Core Medical Ltd., Syneron and GE Healthcare. Mr. Hershko holds a BA degree in Business from Derby University.
Steven
Hanley has served as a member of our Board of Directors since February 2015 and as the Chairman of our Board of Directors since
September 2017, and has served as a member of our Nominating Committee since October 2015 and as a member of our Financing Committee since
June 2016. Mr. Hanley served as a member of our Audit Committee from March 2015 until December 2017 and as a member of our Compensation
Committee from March 2015 until March 2019. Mr. Hanley is currently the Co-Founder, board member and Chief Executive Officer of MediBeacon
Inc., an optical diagnostic company based in St. Louis, Missouri formed upon acquiring assets and intellectual property from Covidien
in 2012. Mr. Hanley is an experienced global business leader who has managed highly complex pharmaceutical and medical device operations
with annual global revenue exceeding US$1 billion. As the President of Covidien plc’s Imaging Solutions business unit, Mr. Hanley
led a multifunctional organization that included sales, marketing, logistics, manufacturing, as well as research and development. Internationally,
Mr. Hanley’s track record includes numerous new drug and device product introductions and sales force expansion in Eastern Europe,
China and Latin America. Mr. Hanley is experienced working in different cultures and successfully navigating dynamic regulatory environments.
Over his nearly 18 years with the Covidien family of companies, Mr. Hanley developed a large network of business leaders and clinicians
to help determine market needs, commercial potential and product positioning. As a sales leader, Mr. Hanley called on radiologists, nuclear
medicine physicians, cardiologists, as well as surgeons in specialties including general, orthopedic, and OB/GYN. Mr. Hanley is Principal
and Founder of Neem LLC, which was founded in 2009 to focus on startup and entrepreneurial medical device and other life science companies
with whom the firm works to bridge the gap between breakthrough technology and commercialization. Mr. Hanley is the Chairman of the Board
of Managers for Daya CNS LLC, based in St Louis Missouri. In addition, Mr. Hanley is currently on the Advisory Board for Kogent Surgical
LLC, based in St Louis Missouri. Mr. Hanley provided consultancy services to us on behalf of Neem LLC from November 2009 until December
31, 2014 and served as a Scientific Advisor to our company from June 2011 until his election to our Board of Directors in February 2015.
Mr. Hanley holds B.A. and M.A. degrees in business administration from Marquette University.
Clara
Ezed has served as a member of our Board of Directors since June 2017, and has served as a member of our Audit Committee since
December 2017 and as a member of our Compensation Committee since March 2019. Since 2022, Ms. Ezed has served as a director at AXA Investment
Managers UK Ltd., a global investment management firm. Ms. Ezed serves as an Executive Board Director Business Development of DLRC Ltd.,
an EU based life-sciences consulting firm, since May 2021. Ms. Ezed has served as Head of EU Operations, EU General Counsel and VP European
Regulatory Affairs of La Jolla Pharmaceutical, a San Diego based biopharmaceutical company, from December 2017 to December 2020. Prior
to that, Ms. Ezed served as VP, Legal & Regulatory Affairs of Emas Pharma Ltd., a global contract research organization, from October
2013 to November 2017. From September 2009 to October 2013, Ms. Ezed served as VP, Regulatory Affairs & Drug Safety of European Medical
Advisory Services Ltd., a global contract research organization. Since 2011, Ms. Ezed has served as a director of a privately listed company,
Sebaroyale Ltd, focused on the development and commercialization of a range of cosmeceuticals. Ms. Ezed, a lawyer and pharmacist with
22 years’ experience working in the pharmaceutical industry, has managed a large and varied portfolio working in regulatory and
medical affairs, quality assurance, clinical operations and pharmacovigilance. Ms. Ezed holds a BSc. in Pharmacy from the University of
Strathclyde, an MBA degree from Middlesex University, a Postgraduate Diploma in Pharmacovigilance from the University of Hertfordshire,
a Postgraduate Diploma in Law from Nottingham University and a Postgraduate Diploma in Legal Professional Services from City Law School,
London. Ms. Ezed is a member of the Bar of England and Wales and a member of the General Pharmaceutical Council.
Dr.
Mary Jo Gorman has served as a member of our Board of Directors and as a member of our Audit Committee and Compensation Committee
since May 2015, and has served as a member of our Financing Committee since June 2016. Dr. Gorman currently serves on the board of directors
of Flosonics Medical, a privately-held medical company based in Canada, and serves on its compensation committee. Dr. Gorman also serves
on the not-for-profit boards of Barnes Jewish Hospital, Goldfarb School of Nursing and Stowe State University Foundation. From 2019 to
2021, Dr. Gorman served as the chief executive officer of Healthy Bytes, a venture backed healthcare services company, located in St.
Louis, Missouri. Dr. Gorman also serves as Managing Director at Prosper Capital, an early stage investment fund focused on women-led businesses.
In 2006, Dr. Gorman founded Advanced ICU Care, the largest telemedicine ICU services provider in the United States, in which she also
served as Chairman and Chief Executive Officer from 2006 and 2014. From December 2016 to 2018, Dr. Gorman served as interim CEO and as
a member of the audit committee of TripleCare, a U.S. provider of telemedicine-based healthcare services to skilled nursing facilities,
subsequently sold to Curavi Health. Dr. Gorman served on the board of directors of Curavi Health from 2019 to July 2020. From 1999 to
2006, Dr. Gorman served at IPC-The Hospitalist Company (Nasdaq:IPCM), a leading national physician group practice, as Chief Medical Officer
(2003-2006), Vice President of Medical Affairs (2001-2003) and Regional Medical Director (1999-2001). From 1996 to 1998, Dr. Gorman served
as President of Inpatient Care Group, a hospitalist group in St. Louis, Missouri, which she had founded to provide hospitalist services
to primary care physicians and hospitals and which was subsequently sold to IPC. From 1991 to 2008, Dr. Gorman served as President of
Critical Care Services, Inc., a privately held corporation which she had founded and which was later sold to Advanced ICU Care. Dr. Gorman
was awarded with the following awards: 2015 Distinguished Alumni Award, Southern Illinois University School of Medicine; 2013 Distinguished
Alumni Award, Olin School of Business, Washington University; 2011 EY Entrepreneurial Winning WomenTM Class of 2011; 2009 Top 25 Influential
Women in Healthcare by Modern HealthCare Magazine. Dr. Gorman holds a B.A. degree in Chemistry and Biology (Cum Laude) from St. Louis
University, an M.B.A degree from Olin School of Business, Washington University and an M.D. from Southern Illinois University School of
Medicine.
XiangQian
(XQ) Lin has served as a member of our Board of Directors since February 2015. Mr. Lin has served as the President and Chief Executive
Officer of the Esco Group since 2011, and is a life sciences entrepreneur and investor with a demonstrated track record across the United
States, Asia and Europe. In 2018, Mr. Lin served as the founder and managing partner of EVX Ventures, a venture capital firm which builds
and invests in biotech companies across Asia. Mr. Lin has co-founded multiple companies including Carmine Therapeutics and PairX Bio.
Mr. Lin holds a BSc degree in Economics and Finance from the Wharton School of Business, University of Pennsylvania.
Yuval
Yanai has served as a member of our Board of Directors since March 2015 and has served as the Chairman of our Audit Committee and
Compensation Committee since March 2015, as the Chairman of our Nominating Committee since October 2015 and as the Chairman of our Financing
Committee since June 2016. Mr. Yanai served as Senior Vice President and Chief Financial Officer of Given Imaging Ltd. from September
2005 through March 2014. From October 2000 through August 2005, Mr. Yanai served as Senior Vice President and Chief Financial Officer
of Koor Industries Ltd. Prior to that, from April 1998 to September 2000, Mr. Yanai served as Vice President and Chief Financial Officer
of NICE Systems Ltd., an Israeli global provider of Insight from Interactions, and, from 1991 to April 1998, he served as the Vice President,
Finance and Chief Financial Officer of Elscint Ltd., a former Israeli company engaged in the developing and manufacturing of medical imaging
devices. Mr. Yanai joined Elscint in 1985 and served as Corporate Controller and Corporate Treasurer through 1991. Mr. Yanai also serves
as an external director (within the meaning of the Israeli Companies Law) of S&P Global Maalot. Mr. Yanai also serves as an external
director (within the meaning of the Israeli Companies Law) of Clal Biotechnology Industries and serves as the Chairman of its audit, financial
reporting and compensation committees. Mr. Yanai also serves as a member of the board of directors of PulseNmore Ltd. and as a member
of its financial reporting and audit committees and as Chairman of its investment committee. Mr. Yanai also serves as a member of the
board of directors of Nobio Ltd. and BRH Medical Ltd., CellSound Aesthetics Ltd., Notal Vision, Inc. and as an acting CFO (part time)
of Art Medical Ltd. Previously, Mr. Yanai served as the Chairman of The Israeli Fund for UNICEF (pro bono) and as an external director
(within the meaning of the Israeli Companies Law) of Hadassah Medical Organization and as the Chairman of its finance and compensation
committees and as a member of its tenders and donation committee. Mr. Yanai also previously served as an external director (within the
meaning of the Israeli Companies Law) of Mazor Robotics Ltd. and Medical Compression Systems Ltd. and as a director of Medigus Ltd. and
Alcobra Ltd. Mr. Yanai also served as a director of Macrocure Ltd., Citycon Oj, Starplast Industries Ltd., Adama Ltd. (formerly Makteshim-Agan
Industries Ltd.), ECI Telecom Ltd., Equity One, Inc., BVR Systems Ltd., Tadiran Communication Ltd., The Elisra Group, Telrad Networks
Ltd. and Medical Compression Systems (D.B.N) Ltd. Mr. Yanai holds a B.Sc. degree in Accounting and Economics from Tel-Aviv University.
Arrangements Concerning Election of Directors; Family Relationships
We are not a party to, and are not aware of, any
voting agreements among our shareholders. In addition, there are no family relationships among our executive officers and directors.
Scientific Advisors
Certain of our officers and employees, including
our chief executive officer, our chief technology officer and our clinical director, consult from time to time, on an individual, as needed
basis with the individuals listed below with respect to matters of scientific relevance. We refer to these individuals as our Scientific
Advisors. The amount of consulting services provided by our Scientific Advisors ranges from several hours a month to several hours a year.
The names and biographies of the individuals who act as our Scientific Advisors are set forth below:
Prof.
Nadir Arber is a full Professor of Medicine and Gastroenterology. Prof. Arber is a holder of the Yechiel and Helen Lieber chair
for Cancer Research at Tel Aviv University, Sackler School of Medicine. Prof. Arber serves as the Director of the Integrated Cancer Prevention
Center (ICPC) at Tel Aviv Sourasky Medical Center in Tel Aviv. He chaired the grants committee of the Sackler School of Medicine at Tel
Aviv University. Prof. Arber is the Head of Cancer Research Center, Head of Djerassi Oncology Center. Prof. Arber, a noted expert in the
field of early detection and prevention, has been serving as the principal investigator (PI) of several international, multicenter clinical
trials in the field of early detection, prevention and therapy of gastrointestinal malignancies using NSAIDs and in particular, CRC. Prof.
Arber has published more than 330 publications. Prof. Arber received a MD from the Hadassah School of Medicine of the Hebrew University
of Jerusalem in 1987, an MSc degree from Sackler School of Medicine, Tel Aviv University in 1991 and a MHA degree from Rekanaty School
of Management, Tel Aviv University in 1991. Prof. Arber has served as a Scientific Advisor to the Company since January 2012. We have
entered into a consultation service agreement with Prof. Arber who is a leading investigator in our multicenter clinical study which is
held at Tel Aviv Sourasky Medical Center and other sites.
B. Compensation
of Directors and Executive Officers
The aggregate compensation paid and share-based
compensation and other payments expensed by us to our directors and executive officers with respect to the year ended December 31, 2022
was $3.1 million. This amount includes approximately $0.34 million set aside or accrued to provide pension, severance, retirement or similar
benefits or expenses, but does not include business travel, professional and business association dues and expenses reimbursed to office
holders, and other benefits commonly reimbursed or paid by companies in our industry. As of December 31, 2022, (i) options to purchase
1,456 ordinary shares granted to our directors and executive officers were outstanding under our 2006 Unit Option Plan, with a weighted
average exercise price of approximately $1,185.2 per share, (ii) options to purchase 68,213 ordinary shares granted to our directors and
executive officers were outstanding under our 2015 Equity Incentive Plan and the United States Sub-Plan to our 2015 Equity Incentive Plan,
with a weighted average exercise price of approximately $20.07 per share; and (iii) 6,180 restricted share units awarded to our directors
and executive officers were outstanding under our 2015 Equity Incentive Plan and the United States Sub-Plan to our 2015 Equity Incentive
Plan.
At our 2022 annual general meeting held on December
29, 2022, our shareholders approved, following the approval of our Compensation Committee and Board of Directors, the payment to each
of our directors of the following fees: (i) an annual fee of $25,000 for service on the Board of Directors and $2,500 for service on the
Audit Committee; and (ii) a per meeting fee of $850 for each meeting of the Board of Directors or any committee thereof attended in person
or via telephone or any resolution approved by written consent. In addition, our shareholders approved an annual fee of (i) $10,000 for
service as Chairman of the Board of Directors (other than an Active Chairman, who may be entitled to an increased fee in accordance with
our Compensation Policy) and (ii) $5,000 for service as Chairman of the Audit Committee. Our directors also benefit from directors’
and officers’ indemnification and exculpation agreements as well as from our directors’ and officers’ liability insurance
policy. The directors are also entitled to reimbursement of expenses (including travel, stay and lodging), subject to the Israeli Companies
Law and the regulations promulgated thereunder, and in accordance with our company practices and our Compensation Policy for Executive
Officers and Directors, or the Compensation Policy. The foregoing fees are the same annual and per meeting fees that have been paid to
our directors since our 2017 annual general meeting.
The directors are also entitled to reimbursement
of expenses (including travel, stay and lodging), subject to the Israeli Companies Law and the regulations promulgated thereunder, and
in accordance with our company practices and our Compensation Policy for Executive Officers and Directors, or the Compensation Policy.
We do not have any written agreements with any
current director providing for benefits upon the termination of such director’s relationship with us.
To our knowledge, there are no agreements and arrangements
between any director and any third party relating to compensation or other payment in connection with their candidacy or service on our
Board of Directors.
The table below sets forth the compensation paid
to our five most highly compensated office holders (within the meaning of the Israeli Companies Law), in each case during or with respect
to the year ended December 31, 2022. We refer to the five individuals for whom disclosure is provided herein as our “Covered Executives.”
For purposes of the table and the summary below,
“compensation” includes salary cost, consultancy fees, bonuses, equity-based compensation, retirement or termination payments,
benefits and perquisites such as car, phone and social benefits, to the extent applicable, and any undertaking to provide such compensation.
All amounts reported in the table are in terms of cost to us, as recognized in our financial statements for the year ended December 31,
2022, including compensation paid to such Covered Executive following the end of the year in respect of services provided during the year.
Each of the Covered Executives was covered by our directors’ and officers’ liability insurance policy and was entitled to
indemnification and exculpation in accordance with indemnification and exculpation agreements, our articles of association and applicable
law.
The exchange rate that we used to calculate the
“Salary Cost” as presented in the following table, was NIS 3.38 to US $1.00, and is provided herein for convenience (such
exchange rate is based on the average of the last-day exchange rate of each of the 12 months during 2022, as published by the Bank of
Israel).
| |
|
|
|
|
|
|
|
Share-Based Compensation (3) |
|
|
|
|
|
Name and Principal Position |
|
US$ |
|
|
Alex Ovadia – Chief Executive Officer |
|
$ |
405,742 |
|
|
$ |
3,603 |
|
|
$ |
309,320 |
|
|
$ |
718,665 |
|
|
Yoav Kimchy – Chief Technology Officer |
|
$ |
369,727 |
|
|
$ |
(7,074 |
) |
|
$ |
26,425 |
|
|
$ |
389,078 |
|
|
Mira Rosenzweig- Chief Financial Officer |
|
$ |
285,710 |
|
|
$ |
2,275 |
|
|
$ |
40,669 |
|
|
$ |
328,654 |
|
|
Boaz Shpigelman -Vice President, Research and Development |
|
$ |
285,883 |
|
|
$ |
(1,260 |
) |
|
$ |
33,055 |
|
|
$ |
317,678 |
|
|
Joshua (Shuki) Belkar- Vice President, Operation |
|
$ |
268,171 |
|
|
$ |
1,395 |
|
|
$ |
38,430 |
|
|
$ |
307,996 |
|
________________________
| (1) |
“Salary Cost” includes the Covered Executive’s gross salary plus payment of social benefits made by us on behalf
of such Covered Executive. Such benefits may include, to the extent applicable to the Covered Executive, payments, contributions and/or
allocations for savings funds, education funds, pension, severance, risk insurances, payments for social security and tax gross-up payments,
vacation, a leased car and associated expenses, medical insurances and benefits, convalescence or recreation pay and other benefits and
perquisites consistent with our policies. |
| (2) |
Represents an adjustment to the amount of the estimated 2021 annual bonus that was recorded in the year ended December 31, 2021 to
the actual 2021 annual bonus that was approved for payment in 2022. |
| (3) |
Represents the share-based compensation expenses recorded in our consolidated financial statements for the year ended December 31,
2022 based on the fair value of the grant date of the equity awards, in accordance with accounting guidance for equity-based compensation.
|
The following table sets forth information with respect to the options and RSUs granted
to the Covered Executives for the year ended December 31, 2022.
|
|
|
|
|
|
|
|
|
Number of Shares Underlying Award |
|
|
|
|
|
Benefit recognized in 2022 (in US$) |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Mira Rosenzweig(1) |
|
March 21, 2022 |
|
Options |
|
$10.184 |
|
1,225 |
|
March 21, 2032 |
|
$8,596 |
|
$3,948 |
| |
|
March 21, 2022 |
|
RSU |
|
|
|
525 |
|
March 21, 2032 |
|
$4,341 |
|
$1,994 |
|
Yoav Kimchy(1) |
|
March 21, 2022 |
|
Options |
|
$10.184 |
|
700 |
|
March 21, 2032 |
|
$4,912 |
|
$2,256 |
| |
|
March 21, 2022 |
|
RSU |
|
|
|
300 |
|
March 21, 2032 |
|
$2,480 |
|
$1,140 |
|
Boaz Shpigelman(1) |
|
March 21, 2022 |
|
Options |
|
$10.184 |
|
1,050 |
|
March 21, 2032 |
|
$7,368 |
|
$3,384 |
| |
|
March 21, 2022 |
|
RSU |
|
|
|
450 |
|
March 21, 2032 |
|
$3,721 |
|
$1,709 |
|
Joshua (Shuki) Belkar(1) |
|
March 21, 2022 |
|
Options |
|
$10.184 |
|
1,225 |
|
March 21, 2032 |
|
$8,596 |
|
$3,948 |
| |
|
March 21, 2022 |
|
RSU |
|
|
|
525 |
|
March 21, 2032 |
|
$4,341 |
|
$1,994 |
________________________
|
(1) |
All options and RSUs granted to the Covered Executives in fiscal year 2022 vest over a four-year period commencing on their date
of grant, such that 25% of the award shall vest on the first anniversary of the date of grant and 2.0833% will vest at the end of each
month thereafter. Alex Ovadia, our chief executive officer, was not granted any equity-based compensation in 2022. |
Employment Agreements with Covered Executives
We have entered into written employment agreements
with each of our executive officers. These agreements contain standard provisions for a company in our industry regarding non- competition,
confidentiality of information and assignment of inventions. The enforceability of covenants not to compete in Israel and the United States
is subject to limitations. For example, Israeli courts have recently required employers seeking to enforce non-compete undertakings of
a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material
interests of the employer which have been recognized by the courts, such as the secrecy of a company’s confidential commercial information
or its intellectual property.
We entered into an Employment Agreement dated as
of December 27, 2012, as amended, with Alex Ovadia, or Mr. Ovadia’s Employment Agreement, pursuant to which Mr. Ovadia served as
our Vice President of Research and Development, Israeli Site Manager and Chief Operations Officer until February 26, 2018. Effective as
of February 26, 2018, Mr. Ovadia was appointed as our Chief Executive Officer and Mr. Ovadia’s Employment Agreement was amended
accordingly, effective as of such date. The terms of Mr. Ovadia’s engagement in his capacity as our Chief Executive Officer were
approved by our shareholders at the extraordinary general meeting of shareholders held on April 2, 2018. Mr. Ovadia’s Employment
Agreement, as currently in effect, may be terminated by either us or Mr. Ovadia upon 90 days’ prior notice to the other party. Pursuant
to Mr. Ovadia’s Employment Agreement, we also have the right to immediately terminate Mr. Ovadia’s employment for “Cause”
(as defined in Mr. Ovadia’s Employment Agreement). Pursuant to Mr. Ovadia’s Employment Agreement, Mr. Ovadia was entitled
to an annual salary of NIS 924,000 ($273,372 per annum, paid in NIS based on an exchange rate of $1= NIS 3.38). Mr. Ovadia’s salary
was subsequently increased in May 2019 to a monthly salary of NIS 80,808 (approximately $23,908 based on an exchange rate of $1= NIS 3.38)
and effective as of December 1, 2019, to a monthly base salary NIS 84,500 (approximately $25,000 based on an exchange rate of $1= NIS
3.38), in each case by our Compensation Committee in reliance on an exemption from shareholder approval available under Israel law. Mr.
Ovadia is also entitled to a leased car and associated expenses up to an aggregate monthly cost of NIS 12,000 (approximately $3,550 based
on an exchange rate of $1= NIS 3.38). In addition, prior to fiscal year 2022, Mr. Ovadia’s employment terms provided that he may
be entitled to an annual bonus of up to 30% of his annual salary subject to receipt of requisite approvals and commencing as of fiscal
year 2022, Mr. Ovadia may be entitled to an annual bonus of up 50%, as approved by the shareholders at the annual general meeting held
on December 9, 2021. Payment of the annual bonus shall be subject to the achievement of certain milestones determined by the Compensation
Committee and Board of Directors. Mr. Ovadia’s Employment Agreement also contains confidentiality, intellectual property, and non-competition
and non-solicitation provisions.
We entered into an Employment Agreement
effective as of April 28, 2019 with Mira Rosenzweig, or Ms. Rosenzweig’s Employment Agreement, pursuant to which Ms. Rosenzweig
serves as our Chief Financial Officer. Ms. Rosenzweig’s Employment Agreement may be terminated by either us or Ms. Rosenzweig upon
90 days’ prior notice to the other party. Pursuant to Ms. Rosenzweig’s Employment Agreement, we also have the right to immediately
terminate Ms. Rosenzweig’s employment for “cause” (as described in Ms. Rosenzweig’s Employment Agreement). Pursuant
to Ms. Rosenzweig’s Employment Agreement, Ms. Rosenzweig is entitled to a salary of NIS 720,000 ($213,018 based on an exchange rate
of $1= NIS 3.38) per annum. In addition, prior to fiscal year 2022, Ms. Rosenzweig may be entitled to an annual bonus of up to 25% of
her annual salary and commencing as of fiscal year 2022, Ms. Rosenzweig may be entitled to an annual bonus of up to 30% of her annual
salary, subject to receipt of customary approvals. Payment of the annual bonus shall be subject to the achievement of certain milestones
determined by the Compensation Committee and Board of Directors. Ms. Rosenzweig’s Employment Agreement also contains confidentiality,
intellectual property, and non-competition and non-solicitation provisions.
We entered into an Employment Agreement
dated as of September 17, 2014, as amended from time to time, with Yoav Kimchy, or Mr. Kimchy’s Employment Agreement, pursuant to
which Mr. Kimchy serves our Chief Technology Officer. Mr. Kimchy’s Employment Agreement may be terminated by either us or Mr. Kimchy
upon 120 business days’ prior notice to the other party. Pursuant to Mr. Kimchy’s Employment Agreement, we also have the right
to immediately terminate Mr. Kimchy’s employment for “Cause” (as defined in Mr. Kimchy’s Employment Agreement)
or if Mr. Kimchy is unable to perform his duties for a period of two consecutive months by reason of illness or accident. Pursuant to
Mr. Kimchy’s Employment Agreement, Mr. Kimchy is entitled to a salary of NIS 1,003,800 ($296,982 based on an exchange rate of $1=
NIS 3.38) per annum. In addition, Mr. Kimchy may be entitled to an annual bonus of up to 30% of his annual salary, subject to receipt
of requisite approvals. Payment of the annual bonus shall be subject to the achievement of certain milestones determined by the Compensation
Committee and Board of Directors. Mr. Kimchy’s Agreement also contains confidentiality, intellectual property, and non-competition
and non-solicitation provisions.
We entered into an Employment Agreement
dated as of March 30, 2018, with Boaz Shpigelman, as amended from time to time, or Mr. Shpigelman’s Employment Agreement, pursuant
to which Mr. Shpigelman serves as our Vice President, Research and Development. Mr. Shpigelman’s Employment Agreement may be terminated
by either us or Mr. Shpigelman upon 90 days’ prior notice to the other party. Pursuant to Mr. Shpigelman’s Employment Agreement,
we also have the right to immediately terminate Mr. Shpigelman’s employment for “Cause” (as defined in Mr. Shpigelman’s
Employment Agreement). Pursuant to Mr. Shpigelman’s Employment Agreement, Mr. Shpigelman is entitled to a salary of NIS 711,000
($210,355 based on an exchange rate of $1= NIS 3.38) per annum. In addition, Mr. Shpigelman may be entitled to an annual bonus of up to
25% of his annual salary, subject to receipt of customary approvals. Payment of the annual bonus shall be subject to the achievement of
certain milestones determined by the Compensation Committee and Board of Directors. Mr. Shpigelman’s Employment Agreement also contains
confidentiality, intellectual property, and non-competition and non-solicitation provisions.
We entered into an Employment Agreement
effective as of June 16, 2019 with Joshua (Shuki) Belkar, or Mr. Belkar’s Employment Agreement, pursuant to which Mr. Belkar serves
as our Vice President, Operation. Mr. Belkar’s Employment Agreement may be terminated by either us or Mr. Belkar upon 90 days’
prior notice to the other party. Pursuant to Mr. Belkar’s Employment Agreement, we also have the right to immediately terminate
Mr. Belkar’s employment for “cause” (as described in Mr. Belkar’s Employment Agreement). Pursuant to Mr. Belkar’s
Employment Agreement, Mr. Belkar is entitled to a salary of NIS 672,000 ($198,817 based on an exchange rate of $1= NIS 3.38) per annum.
In addition, Mr. Belkar may be entitled to an annual bonus of up to 25% of his annual salary, subject to receipt of customary approvals.
Payment of the annual bonus shall be subject to the achievement of certain milestones determined by the Compensation Committee and Board
of Directors. Mr. Belkar’s Employment Agreement also contains confidentiality, intellectual property, and non-competition and non-solicitation
provisions.
Our agreements with our executive officers
do not provide for benefits upon the termination of their respective employment with us, other than payment of salary and benefits during
the required notice period for termination of these agreements, which varies under these individual agreements.
C. Board
Practices
Board of Directors
Under the Israeli Companies Law, the management
of our business, including strategy and policies, is vested in our board of directors. Our board of directors may exercise all powers
and may take all actions that are not specifically granted to our shareholders or to management. Our executive officers are responsible
for our day-to-day management and have individual responsibilities established by our board of directors. Our chief executive officer
is appointed by, and serves at the discretion of, our board of directors, subject to the employment agreement that we have entered into
with him. All other executive officers are appointed by our chief executive officer, and are subject to the terms of any applicable employment
agreements that we may enter into with them.
Under our amended articles of association, our
board of directors must consist of at least four and not more than eleven directors, including the external directors (if external directors
serve on the board of directors).
Our Board of Directors currently consists of five
members, four of whom satisfy the independence requirements of the Nasdaq Listing Rules, such that we comply with the Nasdaq Listing Rule
that requires that a majority of our board of directors be comprised of independent directors, within the meaning of Nasdaq Listing Rules.
Our directors are elected by the general meeting
of our shareholders by the vote of a majority of the ordinary shares present, in person or by proxy, and voting at that meeting. Each
director will hold office until the first annual general meeting of shareholders following his or her appointment, unless the tenure of
such director expires earlier pursuant to the Israeli Companies Law or unless he or she is removed from office as described below. In
addition, our amended articles of association allow our board of directors to appoint directors (other than the external directors) to
fill vacancies on our board of directors, for a term of office equal to the remaining period of the term of office of the director(s)
whose office(s) have been vacated.
Our Board of Directors elected to rely on the exemption
available to foreign private issuers under the Nasdaq Listing Rules and follow Israeli law and practice with regard to the process of
nominating directors, in accordance with which the board of directors (or a committee thereof) is authorized to recommend to the shareholders
director nominees for election (other than directors elected by the board of directors to fill a vacancy). Our Board of Directors established
a Nominating Committee, whose role is to select and recommend to the Board of Directors for selection, director nominees, while considering
the appropriate size and composition of the Board of Directors, the requirements of applicable law regarding service as a member of our
Board of Directors and the criteria for the selection of new members of the Board of Directors, and determined that our Nominating Committee
need not be composed solely of independent directors (within the meaning of Nasdaq Listing Rules). However, our Nominating Committee
currently consists solely of independent directors.
Under the Israeli Companies Law and our amended
articles of association, nominations for directors may also be added to the agenda of a future general meeting of shareholders, at the
request of any one or more shareholders holding at least 1% of our outstanding voting power. Any director nominated by a shareholder is
required to certify to us, as required by all director nominees, that he or she meets all the requirements of the Israeli Companies Law
for election as a director of a public company, and possesses the necessary qualifications and is able to dedicate sufficient time, to
fulfill his or her duties as a director of our company, taking into consideration our company’s size and special needs.
Under the Israeli Companies Law, our board of directors
must determine the minimum number of directors who are required to have accounting and financial expertise (as defined in regulations
promulgated under the Israeli Companies Law). In determining the number of directors required to have such expertise, our board of directors
must consider, among other things, the type and size of the company and the scope and complexity of its operations. Our board of directors
has determined that the minimum number of directors of our company who are required to have accounting and financial expertise is one.
External Directors
Under the Israeli Companies Law, companies incorporated
under the laws of the State of Israel that are “public companies,” must appoint at least two external directors who meet the
qualification requirements in the Companies Law.
However, pursuant to regulations promulgated under
the Israeli Companies Law, Israeli companies whose shares are listed on certain stock exchanges outside of Israel (including the Nasdaq
Capital Market) with no controlling shareholder (within the meaning of the Israeli Companies Law), such as ourselves, that satisfy the
requirements of the laws in the foreign jurisdiction where the company’s shares are listed, as they apply to companies incorporated
in such jurisdiction, with respect to the appointment of independent directors and the composition of the audit committee and compensation
committee, may elect to exempt themselves from the requirements of Israeli law with respect to (i) the requirement to appoint outside
directors and that one outside director serve on each committee of the board of directors authorized to exercise any of the powers of
the board of directors; (ii) certain limitations on the employment or service of an outside director or his or her spouse, children or
other relatives, following the cessation of the service as an outside director, by or for the company, its controlling shareholder or
an entity controlled by the controlling shareholder; (iii) the composition, meetings and quorum of the audit committee; and (iv) the composition
and meetings of the compensation committee. If a company has elected to avail itself from the requirement to appoint external directors
and at the time a director is appointed all members of the board of directors are of the same gender, a director of the other gender must
be appointed.
Following analysis of our qualification to rely
on the exemption, our Board of Directors determined to adopt the exemption, subject to and effective as of the approval by our shareholders
of a certain amendment to our articles of association, which was obtained at our 2017 annual general meeting held on June 22, 2017. If
in the future we were to have a controlling shareholder, we would again be required to comply with the requirements relating to external
directors and the composition of the audit committee and compensation committee under Israeli law.
Audit Committee
Our audit committee currently consists of Yuval
Yanai, May Jo Gorman and Clara Ezed. Yuval Yanai serves as the Chairman of the audit committee.
Composition of the Audit Committee
In accordance with regulations promulgated under
the Companies Law described above, we elected to “opt out” from the Israeli Companies Law requirement to appoint external
directors and related rules concerning the composition of the audit committee and compensation committee. Under such exemption, among
other things, the composition of our audit committee must comply with the requirements of SEC and Nasdaq rules.
Under the Nasdaq corporate governance rules, we
are required to maintain an audit committee consisting of at least three independent directors, within the meaning of the Exchange Act
and Nasdaq Listing Rules, each of whom must be able to read and understand fundamental financial statements, including the company’s
balance sheet, income statement and cash flow statement (and one of whom has past employment experience in finance or accounting, requisite
professional certification in accounting or other comparable experience or background that leads to financial sophistication) and none
of whom has participated in the preparation of our or any of our subsidiary’s financial statements at any time during the prior
three years.
All members of our audit committee meet the requirements
for financial literacy under the applicable rules and regulations of the U.S. Securities and Exchange Commission and the Nasdaq Listing
Rules. Our board of directors has determined that Mr. Yanai is an audit committee financial expert as defined by the U.S. Securities and
Exchange Commission rules and has the requisite financial sophistication required by the Nasdaq Listing Rules.
Each of the members of the audit committee qualifies
as an “independent director” within the meaning of Nasdaq Listing Rules and is “independent” as such term is defined
in Rule 10A- 3(b)(1) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which is different from the general Nasdaq
test for independence of board and committee members.
Audit Committee Role
Our board of directors adopted an audit committee
charter, which became effective upon the listing of our securities on the Nasdaq Capital Market, as subsequently amended, which sets forth
the responsibilities of the audit committee consistent with the rules of the U.S. Securities and Exchange Commission and the Nasdaq Listing
Rules, as well as the requirements for audit committees under the Israeli Companies Law, including the following:
|
• |
oversight of our independent registered public accounting firm and recommending the engagement, compensation
or termination of engagement of our independent registered public accounting firm to the board of directors or shareholders for their
approval, as applicable, in accordance with the requirements of the Israeli Companies Law; |
|
• |
recommending the engagement or termination of the person filling the office of our internal auditor; and
|
|
• |
recommending the terms of audit and non-audit services provided by the independent registered public accounting
firm for pre-approval by our board of directors or shareholders for their approval, as applicable, in accordance with the requirements
of the Israeli Companies Law. |
Our audit committee provides assistance to our
board of directors in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting,
internal control and legal compliance functions by pre-approving the services performed by our independent accountants and reviewing their
reports regarding our accounting practices and systems of internal control over financial reporting. Our audit committee also oversees
the audit efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants
are independent of management.
Under the Israeli Companies Law, our audit committee
is responsible for:
|
• |
determining whether there are deficiencies in the business management practices of our company, including
in consultation with our internal auditor or the independent auditor, and making recommendations to the board of directors to improve
such practices; |
|
• |
determining whether to approve certain related party transactions (including transactions in which an office
holder has a personal interest) and whether such transaction is extraordinary or material under Israeli Companies Law (see “—
Approval of Related Party Transactions under Israeli Law”); |
|
• |
determining whether a competitive process must be implemented for the approval of certain transactions
with controlling shareholders or its relative or in which a controlling shareholder has a personal interest (whether or not the transaction
is an extraordinary transaction), under the supervision of the audit committee or other party determined by the audit committee and in
accordance with standards to be determined by the audit committee, or whether a different process determined by the audit committee should
be implemented for the approval of such transactions; |
|
• |
determining the process for the approval of certain transactions with controlling shareholders or in which
a controlling shareholder has a personal interest that the audit committee has determined are not extraordinary transactions but are not
immaterial transactions; |
|
• |
where the board of directors approves the working plan of the internal auditor, to examine such working
plan before its submission to the board of directors and proposing amendments thereto; |
|
• |
examining our internal controls and internal auditor’s performance, including whether the internal
auditor has sufficient resources and tools to dispose of its responsibilities; |
|
• |
examining the scope of our auditor’s work and compensation and submitting a recommendation with respect
thereto to our board of directors or shareholders, depending on which of them is considering the compensation of our auditor; and
|
|
• |
establishing procedures for the handling of employees’ complaints as to the management of our business
and the protection to be provided to such employees. |
Compensation Committee and Compensation Policy
Our compensation committee currently consists Yuval
Yanai, Mary Jo Gorman and Clara Ezed. Yuval Yanai serves as the Chairman of the compensation committee.
Composition of the Compensation Committee
In accordance with regulations promulgated under
the Companies Law described above, we elected to “opt out” from the Israeli Companies Law requirement to appoint external
directors and related rules concerning the composition of the audit committee and compensation committee.
Under the Nasdaq Listing Rules, we are required
to maintain a compensation committee consisting of at least two directors, each of whom is an independent director within the meaning
of the Nasdaq Listing Rules. Our compensation committee currently complies with the provisions of Nasdaq Listing Rules relating to composition
requirements.
Compensation Committee Role
Our board of directors adopted a compensation committee
charter, which became effective upon the listing of our shares on the Nasdaq Capital Market, as subsequently amended, which sets forth
the responsibilities of the compensation committee consistent with the Nasdaq Listing Rules and the requirements for compensation committees
under the Israeli Companies Law, including the following:
|
• |
recommending to the board of directors for its approval (i) a compensation policy; (ii) whether a compensation
policy should continue in effect, if the then-current policy has a term of greater than three years (approval of either a new compensation
policy or the continuation of an existing compensation policy must in any case occur every three years); and (iii) periodic updates to
the compensation policy. See “— Compensation Committee and Compensation Policy.” In addition, the compensation committee
is required to periodically examine the implementation of the compensation policy; |
|
• |
the approval of the terms of employment and service of office holders (including determining whether the
compensation terms of a candidate for chief executive officer of the company need not be brought to approval of the shareholders); and
|
|
• |
reviewing and approving grants of options and other incentive awards to persons other than office holders
to the extent such authority is delegated by our board of directors, subject to the limitations on such delegation as provided in the
Israeli Companies Law. |
Compensation Policy
Under the Israeli Companies Law, the duties of
the compensation committee include the recommendation to the company’s board of directors of a policy regarding the terms of engagement
of office holders, as such term is defined in the Israeli Companies Law, to which we refer to as a compensation policy, and any extensions
and updates thereto. The compensation policy must be approved at least once every three years, first, by our board of directors, upon
recommendation of our compensation committee, and second, by the shareholders by the Special Approval for Compensation (as defined below
under “— Approval of Related Party Transactions under Israeli Law — Disclosure of Personal Interests of an Office Holder
and Approval of Certain Transactions”).
Our current Compensation Policy for Executive Officers
and Directors, which was approved by our shareholders at the 2020 annual general meeting held on December 10, 2020, serves as the basis
for decisions concerning the financial terms of employment or engagement of our office holders, including exculpation, insurance, indemnification
and any benefit, monetary payment or obligation of payment in respect of employment or engagement, including and any severance payment
or benefit.
The Compensation Policy must be determined and
later reevaluated according to certain factors, including: (i) the advancement of a company’s objectives, business plan and its
long- term strategy; (ii) the creation of appropriate incentives for executives, while considering (among other things) the company’s
risk management policy; (iii) the size and the nature of the company’s operations; and (iv) with respect to variable compensation,
the contribution of the office holder towards the achievement of the company’s long-term goals and the maximization of its profits,
all with a long-term objective and in accordance with the position of the office holder.
In accordance with the Israeli Companies law, our
Compensation Policy refers to the following factors:
|
• |
the knowledge, skills, expertise, professional experience and accomplishments of the relevant office holder;
|
|
• |
the office holder’s roles and responsibilities and prior compensation agreements with him or her;
|
|
• |
with respect to variable compensation – the possibility of reducing variable compensation at the
discretion of the board of directors, and the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation;
and |
|
• |
with respect to severance compensation, the period of employment or service of the office holder, the terms
of his or her compensation during such period, the company’s performance during such period, the person’s contribution towards
the company’s achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving
the company. |
In addition, in according with the Israeli Companies
Law, our Compensation Policy includes the following principles:
|
• |
the link between variable compensation (e.g., bonuses) and long-term performance and measurable criteria
(i.e., variable compensation must be determined based on long-term performance and measurable criteria). Only “non-material”
portion of variable compensation may be determined based on criteria that is not measurable, taking into account office holders’
contribution to the company; |
|
• |
the ratio of variable to fixed compensation, and the ceiling for the value of variable compensation, which
is determined at the time of payment, except that the ceiling for equity-based compensation is determined at the time of grant;
|
|
• |
the conditions under which an office holder would be required to repay compensation paid to him or her
if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company’s
financial statements; |
|
• |
the minimum holding or vesting period for variable, equity-based compensation, while taking into account
long-term objectives; and |
|
• |
maximum limits for severance compensation. |
Nominating Committee
Our Board of Directors has established a Nominating
Committee, whose role is to select and recommend to the Board of Directors for selection, director nominees, while considering the appropriate
size and composition of the Board of Directors, the requirements of applicable law regarding service as a member of our Board of Directors
and the criteria for the selection of new members of the Board of Directors. While our Board of Directors elected to rely on the exemption
available to foreign private issuers under the Nasdaq Listing Rules with respect to the process of nominating directors, and determined
that our Nominating Committee need not be composed solely of independent directors (within the meaning of Nasdaq Listing Rules), our Nominating
Committee currently consists solely of independent directors. The Nominating Committee is currently comprised of the following directors:
Yuval Yanai, Steven Hanley and Mary-Jo Gorman. Yuval Yanai serves as the Chairman of the Nominating Committee.
Financing Committee
Our Board of Directors has established a financing
committee, which currently consists of Steven Hanley, Yuval Yanai and Mary Jo Gorman. Our financing committee is responsible for the review,
approval and oversight of any potential financings, including the negotiations of any potential financings, and to provide a recommendation
to our Board of Directors concerning the approval of any potential financings.
Internal Auditor
Under the Israeli Companies Law, the board of directors
of an Israeli public company must appoint an internal auditor recommended by the audit committee. An internal auditor may not be:
|
• |
a person (or a relative of a person) who holds more than 5% of the company’s outstanding shares or
voting rights; |
|
• |
a person (or a relative of a person) who has the power to appoint a director or the general manager of
the company; |
|
• |
an office holder, within the meaning of the Israeli Companies Law (including a director and the general
manager) of the company (or a relative thereof); or |
|
• |
a member of the company’s independent accounting firm, or anyone on his or her behalf. |
The role of the internal auditor is to examine,
among other things, our compliance with applicable law and orderly business procedures. The audit committee is required to oversee the
activities and to assess the performance of the internal auditor as well as to approve the internal auditor’s work plan. We have
appointed Ms. Dana Gottesman Erlich of BDO as our internal auditor.
Approval of Related Party Transactions under Israeli Law
Fiduciary Duties of Directors and Executive Officers
The Israeli Companies Law codifies the fiduciary
duties that office holders owe to a company. Each person listed in the table under “Management—Executive Officers and Directors”
is an office holder under the Israeli Companies Law.
An office holder’s fiduciary duties consist
of a duty of care and a duty of loyalty. The duty of care requires an office holder to act with the level of care with which a reasonable
office holder in the same position would have acted under the same circumstances. The duty of care includes a duty to use reasonable means
to obtain:
|
• |
information on the advisability of a given action brought for his or her approval or performed by virtue
of his or her position; and |
|
• |
all other important information pertaining to any such action. |
The duty of loyalty requires an office holder to act in good faith and in the best
interests of the company, and includes, among other things, the duty to:
|
• |
refrain from any conflict of interest between the performance of his or her duties to the company and his
or her other duties or personal affairs; |
|
• |
refrain from any activity that is competitive with the company; |
|
• |
refrain from exploiting any business opportunity of the company to receive a personal gain for himself
or herself or others; and |
|
• |
disclose to the company any information or documents relating to the company’s affairs which the
office holder received as a result of his or her position as an office holder. |
We may approve an act specified above which would
otherwise constitute a breach of the office holder’s duty of loyalty, provided that the office holder acted in good faith, the act
or its approval does not harm the company and the office holder discloses his or her personal interest a sufficient amount of time before
the date for discussion of approval of such act.
Disclosure of Personal Interests of an Office Holder and Approval of Certain Transactions
Disclosure of Personal Interests of an Office Holder
The Israeli Companies Law requires that an office
holder promptly disclose to the company any “personal interest” that he or she may be aware of and all related material information
or documents concerning any existing or proposed transaction with the company. An interested office holder’s disclosure must be
made promptly and in any event no later than the first meeting of the board of directors at which the transaction is considered. A personal
interest includes an interest of any person in an act or transaction of a company, including a personal interest of such person’s
relative or of a corporate entity in which such person or a relative of such person holds 5% or more of the outstanding shares or voting
rights, is a director or general manager or in which he or she has the right to appoint at least one director or the general manager,
but excluding a personal interest arising from one’s ownership of shares in the company. A personal interest includes the personal
interest of a person for whom the office holder holds a voting proxy or the personal interest of the office holder with respect to his
or her vote on behalf of a person for whom he or she holds a proxy even if such shareholder has no personal interest in the matter. An
office holder is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative
in a transaction that is not considered an extraordinary transaction. Under the Israeli Companies Law, an extraordinary transaction is
defined as any of the following: a transaction other than in the ordinary course of business; a transaction that is not on market terms;
or a transaction that may have a material impact on a company’s profitability, assets or liabilities.
Generally, a person who has a personal interest
in a matter which is considered at a meeting of the board of directors or the audit committee shall not be present at such a meeting or
vote on that matter unless, with respect to an office holder, the chairman of the audit committee or board of directors (as applicable)
determines that the office holder should be present during the discussions in order to present the transaction that is subject to approval
(provided that the office holder may not vote on the matter). If a majority of the members of the audit committee or the board of directors
(as applicable) has a personal interest in the approval of a transaction, then all directors may participate in discussions of the audit
committee or the board of directors (as applicable) on such transaction and the voting on approval thereof. If a majority of the members
of the board of directors has a personal interest in the approval of a transaction, shareholder approval is also required for such transaction.
Approval of Transactions with Officer Holders
If it is determined that an office holder has a
personal interest in a transaction that is not an extraordinary transaction, approval by the board of directors is required for the transaction,
unless the company’s articles of association provide for a different method of approval. Further, so long as an office holder has
disclosed his or her personal interest in a transaction, the board of directors may approve an act by the office holder that would otherwise
be deemed a breach of his or her duty of loyalty, provided that the transaction is in the company’s best interest and the office
holder acted in good faith. An extraordinary transaction in which an office holder has a personal interest requires approval first by
the company’s audit committee and subsequently by the board of directors.
Compensation of Officers Other than the Chief Executive Officer
The compensation of an office holder (other than
the chief executive officer) who is not a director generally requires approval first by the company’s compensation committee, then
by the company’s board of directors, according to the company’s compensation policy. In special circumstances the compensation
committee and board of directors may approve a compensation arrangement that is inconsistent with the company’s compensation policy,
provided that they have considered the same considerations and matters required for the approval of a compensation policy in accordance
with the Israeli Companies Law and such arrangement must be approved by a majority vote of the shares present and voting at a shareholders
meeting on the matter, provided that either: (i) such majority includes at least a majority of the shares held by all shareholders who
are not controlling shareholders and shareholders who do not have a personal interest in such compensation arrangement present and voting
on the matter, excluding abstentions; or (ii) the total number of shares of non-controlling shareholders and shareholders who do not have
a personal interest in the matter and who vote against the matter does not exceed 2% of the company’s aggregate voting rights. We
refer to this as the Special Approval for Compensation. However, if the shareholders of the company do not approve a compensation arrangement
with an executive officer that is inconsistent with the company’s compensation policy, the compensation committee and board of directors
may, in special circumstances, override the shareholders’ decision if each of the compensation committee and the board of directors
discuss the arrangement again, analyze the shareholders’ objection and provide detailed reasons for their decision.
An amendment to an existing arrangement with an
office holder (other than the chief executive officer) who is not a director requires only the approval of the compensation committee,
if the compensation committee determines that the amendment is not material in comparison to the existing arrangement. However, according
to regulations promulgated under the Israeli Companies Law, an amendment to an existing arrangement with an office holder (who is not
a director) who is subordinate to the chief executive officer shall not require the approval of the compensation committee, if (i) the
amendment is approved by the chief executive officer and the company’s compensation policy determines that a non-material amendment
to the terms of service of an office holder (other than the chief executive officer) may be approved by the chief executive officer and
(ii) the engagement terms are consistent with the company’s compensation policy.
Compensation of Chief Executive Officer
The compensation of a public company’s chief
executive officer generally requires the approval of first, the company’s compensation committee; second, the company’s board
of directors and third (except for a number of exceptions), the company’s shareholders by the Special Approval for Compensation.
However, if the shareholders of the company do not approve a compensation arrangement with a chief executive officer, the compensation
committee and board of directors may, in special circumstances, override the shareholders’ decision if each of the compensation
committee and the board of directors discuss the arrangement again, analyze the shareholders’ objection and provide detailed reasons
for their decision. However, an amendment to an existing arrangement with a chief executive officer who is not a director requires only
the approval of the compensation committee, if the compensation committee determines that the amendment is not material in comparison
to the existing arrangement.
According to regulations promulgated under the
Israeli Companies Law, the renewal or extension of an existing arrangement with a chief executive officer shall not require shareholder
approval if (i) the renewal or extension is not beneficial to the chief executive officer as compared to the prior arrangement or there
is no substantial change in the terms and other relevant circumstances; and (ii) the engagement terms are consistent with the company’s
compensation policy and the prior arrangement was approved by the shareholders by the Special Approval for Compensation.
Compensation of Directors
Arrangements regarding the compensation of a director
require the approval of the compensation committee, board of directors and (except for a number of exceptions) shareholders by ordinary
majority, in that order. The approval of the compensation committee and board of directors must be in accordance with the compensation
policy. In special circumstances, the compensation committee and board of directors may approve a compensation arrangement that is inconsistent
with the company’s compensation policy, provided that they have considered the same considerations and matters required for the
approval of a compensation policy in accordance with the Israeli Companies Law and that shareholder approval was obtained by the Special
Approval for Compensation.
With respect to compensation of an officer (including
chief executive officer) or director who is also a controlling shareholder, see “— Disclosure of Personal Interests of Controlling
Shareholders and Approval of Certain Transactions.”
Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain
Transactions
Pursuant to Israeli law, the disclosure requirements
regarding personal interests that apply to directors and executive officers also apply to a controlling shareholder of a public company.
In the context of a transaction involving a shareholder of the company, a controlling shareholder also includes a shareholder who holds
25% or more of the voting rights in the company if no other shareholder holds more than 50% of the voting rights in the company. For this
purpose, the holdings of all shareholders who have a personal interest in the same transaction will be aggregated. Extraordinary transactions
with a controlling shareholder or in which a controlling shareholder has a personal interest, including a private placement in which a
controlling shareholder has a personal interest, and the terms of engagement with a controlling shareholder or a relative thereof, directly
or indirectly (including through a corporation controlled by a controlling shareholder), for the provision of services to the company
and his or her terms of employment or service as an office holder or employment as other than an office holder, require the approval of
each of (i) the audit committee or the compensation committee with respect to the terms of service or employment by the company as an
office holder, an employee or service provider; (ii) the board of directors; and (iii) the shareholders, in that order. The shareholder
approval requires one of the following, which we refer to as a Special Majority:
|
• |
a majority of the shares held by all shareholders who do not have a personal interest in the transaction
and who are present and voting on the matter approves the transaction, excluding abstentions; or |
|
• |
the shares voted against the transaction by shareholders who have no personal interest in the transaction
and who are present and voting at the meeting do not exceed 2% of the voting rights in the company. |
Each shareholder voting on the approval of an extraordinary
transaction with a controlling shareholder must inform the company prior to voting whether or not he or she has a personal interest in
the approval of the transaction, otherwise, the shareholder is not eligible to vote on the proposal and his or her vote will not be counted
for purposes of the proposal.
To the extent that any such transaction with a
controlling shareholder is for a period of more than three years, approval is required once every three years, unless, with respect to
any such extraordinary transactions, the audit committee determines that the duration of the transaction is reasonable given the related
circumstances.
The compensation committee and board approval for
arrangements regarding the terms of service or employment of a controlling shareholder must be in accordance with the company’s
compensation policy. In special circumstances the compensation committee and board of directors may approve a compensation arrangement
that is inconsistent with the company’s compensation policy, provided that they have considered the same considerations and matters
required for the approval of a compensation policy in accordance with the Israeli Companies Law and that shareholder approval was obtained
by the Special Majority.
Pursuant to regulations promulgated under the Israeli
Companies Law, certain transactions with a controlling shareholder or his or her relative, or with directors, relating to terms of service
or employment that would otherwise require approval of a company’s shareholders may be exempt from shareholder approval upon certain
determinations of the audit committee and board of directors. In addition, disclosure of a personal interest in a private placement of
a public company (including disclosure of any material fact or document) is required by (i) a shareholder holding 5% or more of the company’s
issued and outstanding capital or its voting rights whose holdings will increase as result of the private placement and a shareholder
who will hold 5% or more of the company’s issued and outstanding capital or its voting rights as a result of the private placement,
if 20% or more of the company’s outstanding share capital prior to the private placement is issued in the private placement and
the payment for which is not only in cash or listed securities or the transaction is not on market terms; and (ii) a person or entity
that will become a controlling shareholder as a result of the private placement.
Shareholder Duties
Pursuant to the Israeli Companies Law, a shareholder
has a duty to act in good faith and in a customary manner toward the company and other shareholders and to refrain from abusing his or
her power in the company, including, among other things, in voting at a meeting of shareholder with respect to the following matters:
|
• |
an amendment to the company’s articles of association; |
|
• |
an increase of the company’s authorized share capital; |
|
• |
the approval of related party transactions and acts of office holders that require shareholder approval.
|
In addition, a shareholder has a general duty to
refrain from discriminating against other shareholders.
Certain shareholders have a duty of fairness toward
the company. These shareholders include any controlling shareholder, any shareholder who knows that he or she has the power to determine
the outcome of a shareholder vote and any shareholder who has the power to appoint or to prevent the appointment of an office holder of
the company or other power towards the company. The Israeli Companies Law does not define the substance of the duty of fairness, except
to state that the remedies generally available upon a breach of contract will also apply in the event of a breach of the duty to act with
fairness.
Exculpation, Insurance and Indemnification of Directors and Officers
Under the Israeli Companies Law, a company may
not exculpate an office holder from liability for a breach of the duty of loyalty. An Israeli company may exculpate an office holder in
advance from liability to the company, in whole or in part, for damages caused to the company as a result of a breach of duty of care,
but only if a provision authorizing such exculpation is included in its articles of association. Our amended articles of association include
such a provision, to the fullest extent permitted by law. The company may not exculpate in advance a director from liability arising out
of a prohibited dividend or other distribution to shareholders.
Under the Israeli Companies Law and the Israeli
Securities Law, 5728-1968, or the Israeli Securities Law, a company may indemnify an office holder in respect of the following liabilities
and expenses incurred for acts performed by him or her as an office holder, either pursuant to an undertaking made in advance of any such
event or following an event, provided its articles of association include a provision authorizing such indemnification:
|
• |
a financial liability imposed on him or her in favor of another person pursuant to a judgment, including
a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an office holder with respect to
such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the board of directors,
can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount or according to criteria
determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail the abovementioned foreseen
events and amount or criteria; |
|
• |
reasonable litigation expenses, including attorneys’ fees, incurred by the office holder (1) as a
result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation or proceeding,
provided that (i) no indictment was filed against such office holder as a result of such investigation or proceeding; and (ii) no financial
liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or proceeding or,
if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal intent; and
(2) in connection with a monetary sanction; |
|
• |
reasonable litigation expenses, including attorneys’ fees, incurred by the office holder or imposed
by a court in proceedings instituted against him or her by the company, on its behalf, or by a third party, or in connection with criminal
proceedings in which the office holder was acquitted, or as a result of a conviction for an offense that does not require proof of criminal
intent; and |
|
• |
expenses, including reasonable litigation expenses and legal fees, incurred by an office holder in relation
to an administrative proceeding instituted against such office holder, or certain compensation payments made to an injured party imposed
on an office holder by an administrative proceeding, pursuant to certain provisions of the Israeli Securities Law. |
Under the Israeli Companies Law and the Israeli
Securities Law, a company may insure an office holder against the following liabilities incurred for acts performed by him or her as an
office holder if and to the extent provided in the company’s articles of association:
|
• |
a breach of the duty of loyalty to the company, provided that the office holder acted in good faith and
had a reasonable basis to believe that the act would not harm the company; |
|
• |
a breach of the duty of care to the company or to a third party, to the extent such a breach arises out
of the negligent conduct of the office holder; |
|
• |
a financial liability imposed on the office holder in favor of a third party; and |
|
• |
expenses, including reasonable litigation expenses and legal fees, incurred by an office holder in relation
to an administrative proceeding instituted against such office holder or certain compensation payments to an injured party imposed on
an office holder by an administrative proceeding, pursuant to certain provisions of the Securities Law. |
Under the Israeli Companies Law, a company may
not indemnify, exculpate or enter into an insurance contract for office holder liability, for any of the following:
|
• |
a breach of the duty of loyalty, except for indemnification and insurance for a breach of the duty of loyalty
to the company to the extent that the office holder acted in good faith and had a reasonable basis to believe that the act would not prejudice
the company; |
|
• |
a breach of the duty of care committed intentionally or recklessly, excluding a breach arising out of the
negligent conduct of the office holder; |
|
• |
an act or omission committed with intent to derive illegal personal benefit; or |
|
• |
a fine, monetary sanction or forfeit levied against the office holder. |
Under the Israeli Companies Law, exculpation, indemnification
and insurance of office holders in a public company must be approved by the compensation committee and the board of directors and, with
respect to the chief executive officer and a director or (under certain circumstances), also by the shareholders. See “— Approval
of Related Party Transactions under Israeli Law.” However, the insurance of office holders shall not require shareholder approval
and may be approved only by the compensation committee, if the engagement terms are determined in the company’s compensation policy
and that policy was approved by the shareholders by the Special Approval for Compensation, provided that the policy is on market terms
and is not likely to materially impact the company’s profitability, assets or obligations.
Our amended articles of association permit us to
exculpate, indemnify and insure our office holders to the fullest extent permitted under the Israeli Companies Law and the Israeli Securities
Law. We have obtained directors’ and officers’ liability insurance for the benefit of our office holders and intend to continue
to maintain such coverage and pay all premiums thereunder to the fullest extent permitted by the Israeli Companies Law.
We have entered into indemnification and exculpation
agreements with each of our current officers and directors exculpating them from a breach of their duty of care to us to the fullest extent
permitted by the Israeli Companies Law and undertaking to indemnify them to the fullest extent permitted by the Israeli Companies Law
and the Israeli Securities Law, to the extent that these liabilities are not covered by insurance. This indemnification is limited to
events determined as foreseeable by our board of directors based on our activities, as set forth in the indemnification agreements. Under
such indemnification agreements, the maximum aggregate amount of indemnification that we may pay to any and all of our currently serving
or future officers and directors together may not exceed the higher of $5 million and 25% of our shareholders equity according to our
most recent financial statements at the time of payment. In the opinion of the SEC, however, indemnification of directors and office holders
for liabilities arising under the Securities Act of 1933, as amended, is against public policy and therefore unenforceable.
D. Employees
As of December 31, 2022, we had 85 employees and
independent contractors, all of whom were based in Israel other than one based in the United States, and of whom 10 persons were in administrative,
legal, accounting, information technology and human resources and 75 persons were in research and development. Under Israeli law, we and
our employees are subject to protective labor provisions, including the length of the workday, minimum wages for employees, annual leave,
sick pay, determination of severance pay and advance notice of termination of employment, as well as procedures for hiring and dismissing
employees and equal opportunity and anti-discrimination laws. While none of our employees are party to any collective bargaining agreements,
orders issued by the Israeli Ministry of Economy and Industry may make certain industry-wide collective bargaining agreements applicable
to us. These agreements affect matters such as the length of the workday and week, recuperation pay, travel expenses and pension rights.
We have never experienced labor-related work stoppages and believe that our relationships with our employees are a significant part of
our operations and that we maintain a good and positive relationship with our employees.
Israeli law generally requires the payment of severance
compensation by employers upon the retirement, death or dismissal of an employee. We fund our ongoing severance obligations by making
monthly payments to insurance policies. All of our current employees have agreed that upon termination of their employment, they will
be entitled to receive only the amounts accrued in the insurance policies with respect to severance pay. Furthermore, Israeli employees
and employers are required to pay predetermined sums to the National Insurance Institute, which is similar to the U.S. Social Security
Administration. These amounts also include payments for national health insurance.
In addition, we have various consulting arrangements
with experts including in regulatory, research and clinical matters, including with physicians.
E.
Share Ownership
Share Ownership of Executive Officers and Directors
For information concerning the beneficial ownership
of our ordinary shares by our executive officers and directors, see the table in Item 7A. “Major Shareholders and Related Party
Transactions—Major shareholders.” As of December 31, 2022, (i) options to purchase 1,665 ordinary shares granted to our directors
and executive officers were outstanding under our 2006 Unit Option Plan, with a weighted average exercise price of approximately $1,185.6
(ii) options to purchase 130,449 ordinary shares granted to our directors and executive officers were outstanding under our 2015 Equity
Incentive Plan and the United States Sub-Plan to our 2015 Equity Incentive Plan, with a weighted average exercise price of approximately
$24.24 per share; and (iii) 8,153 restricted share units awarded to our directors and executive officers were outstanding under our 2015
Equity Incentive Plan and the United States Sub-Plan to our 2015 Equity Incentive Plan.
Option and Incentive Plans
In connection with the transfer of all of the business
operations and substantially all of the assets of Check-Cap LLC to us in 2009, we assumed the Check-Cap LLC 2006 Unit Option Plan, or
the 2006 Plan. On June 23, 2015, our board of directors approved and adopted the Check-Cap Ltd. 2015 Equity Incentive Plan, or the 2015
Israeli Plan, and the Check-Ltd. 2015 United States Sub-Plan to Check-Cap Ltd. 2015 Equity Incentive Plan, or the 2015 U.S. Sub-Plan.
The 2015 Israeli Plan and 2015 U.S. Sub-Plan are referred together as the 2015 Plan. As of such date, we ceased to grant options under
the 2006 Plan and all equity-based awards made after such date shall be made under the 2015 Plan. Our shareholders approved the 2015 Plan
at an extraordinary general meeting of shareholders held on August 13, 2015.
The primary provisions of the 2006 Plan and 2015
Plan are described below.
2006 Plan
General.
The 2006 Plan provided for the grant of options to purchase our ordinary shares to our employees, consultants and service providers. For
the purpose of the 2006 Plan: (i) an “employee” means any person, including officers, directors or affiliates who are employed
by us or by our affiliates; (ii) a “consultant” means any person who is engaged by us to render consulting or advisory services
to us or to any of our entities provided that such services are provided in good faith and are (a) not in connection with the offer or
sale of our securities in a capital raising transaction and (b) not directly or indirectly promoting or maintaining a market for our securities;
(iii) a “service provider” means an employee, director, supplier or officer holder as defined in the Israeli Companies Law;
and (iv) an “affiliate” means any entity which is directly or indirectly our parent or subsidiary.
Administration
of the 2006 Plan. Our board of directors has had the authority to administer the 2006 Plan and to grant options under the 2006
Plan, including, the authority to determine the persons to whom options would be granted, the number of shares subject to each option,
the time or times at which the options would be granted, restrictions on the transferability of the options, and the schedule and conditions
on which such options may be exercised.
Awards under the
2006 Plan. The 2006 Plan provided for the grant of options pursuant to Sections 102 and 3(i) of the Israeli Income Tax Ordinance
[New Version], 5721-1961, which we refer to as the Tax Ordinance. The 2006 Plan provides that Section 102 options may be granted only
to employees who are Israeli residents and who do not own interests possessing more than 10% of the total combined voting power of all
classes of our equity or the equity of our affiliates immediately before such option is granted. Options granted to optionees who are
Israeli residents that are not intended to qualify as Section 102 Options are granted under Section 3(i) of the Tax Ordinance, which does
not provide for similar tax benefits, and are referred to as Section 3(i) options. The 2006 Plan was submitted for the approval of the
Israeli Tax Authority, which we refer to as the ITA, as required by applicable law. Options granted to employees under the 2006 Plan were
granted under Section 102(b)(2) of the Tax Ordinance, which permits the issuance to a trustee under the “capital gains track.”
In order to comply with the terms of the capital gains track, all options granted under a specific plan and subject to the provisions
of Section 102 of the Tax Ordinance, as well as the shares issued upon exercise of such options and other shares received subsequently
following any realization of rights with respect to such options, such as share dividends and share splits, must be registered in the
name of a trustee selected by the board of directors and held in trust for the benefit of the relevant employee, director or officer for
a period of two years from the date of the grant. However, under this track, we are not allowed to deduct an expense with respect to the
issuance of the options or shares.
Exercise Price;
Vesting. The exercise price of an option granted under the 2006 Plan has been determined by the board of directors or a committee
appointed by it. The first option grant to an employee generally vested over a period of three years and nine months commencing on the
date of grant, such that 8.33% vest on the first anniversary of the date of grant and an additional 8.33% vest on each subsequent three-month
period thereafter, for 33 months. Additional options granted to an employee generally vest over a period of three years commencing on
the three months anniversary of the date of grant, such that 8.33% is fully vested on the date of grant and an additional 8.33% vest on
each subsequent three-month period thereafter, for 33 months.
Options Term;
Termination of Employment or Service. Options granted under the 2006 Plan generally expire within ten years of the grant date or
upon the earlier termination of employment of, or services provided by, the optionee, as applicable, subject to the extended period of
exercisability upon termination of employment or services, as applicable. Upon termination of the employment of or services rendered by
an optionee, as applicable (other than for cause, disability or death), generally vested options may be exercised within three months
after the date of such termination or within such shorter time period (not to be less than 30 days) or such longer time period (not to
exceed five years) as our board of directors or a committee appointed by it shall determine, but in any event no later than the expiration
date of the options. If the employment or services of the optionee are terminated because of death or disability (or if the optionee dies
within three months after termination of employment or services other than for cause), the optionee’s options may be exercised by
the optionee or the optionee’s legal representative or authorized assignee to the extent exercisable on the date of such termination
or within 12 months thereafter or as otherwise determined by the board of directors or a committee appointed by it. If the employment
or services of the optionee are terminated for cause, all outstanding options will, to the extent not previously exercised, be of no force
and effect as of the date of termination, unless otherwise determined by the board of directors or a committee appointed by it.
M&A Transaction.
The 2006 Plan provides that in the event of a merger or consolidation of our company in which our company is not the surviving entity,
an acquisition of all or substantially all of the outstanding capital of our company or the sale of all or substantially all of our assets,
the optionee shall be provided the opportunity to (i) exercise his or her options in connection with the transaction and to receive in
the transaction such consideration as the holder of ordinary shares shall receive in the transaction; or (ii) retain his or her options
or receive a substitute option from the surviving company, if any.
As of December 31, 2022, we had granted options
to purchase an aggregate 8,825 ordinary shares under the 2006 Plan, of which options to purchase an aggregate 1,292 ordinary shares had
been exercised into our ordinary shares, options to purchase an aggregate 5,781 ordinary shares had been forfeited without having been
exercised and options to purchase an aggregate 1,752 ordinary shares were outstanding with a weighted average exercise price of approximately
$1,273.9 per share. As of December 31, 2022, no shares are available for future awards under the 2006 Plan.
2015 Equity Incentive Plan
Awards under the
2015 Plan. Awards under the 2015 Plan may be options granted pursuant to Section 102 of the Tax Ordinance, or Section 102 Options,
or Section 3(i) of the Tax Ordinance, or Section 3(i) Options, “incentive stock options”, or ISOs, within the meaning of Section
422 of the U.S. Internal Revenue Code of 1986, as amended, and options not intended to qualify as ISOs, or Non-statutory Stock Options,
stock appreciation rights, or SARs, restricted stock awards, or RSAs, and restricted stock units, or RSUs, or any combination of the foregoing.
Unless the Administrator (as defined below) determines
otherwise and subject to applicable law, Section 102 Options may be granted only to Israeli employees, executives and directors (excluding
controlling shareholders) and Section 3(i) Options may be granted to consultants, controlling shareholders and non-Israeli employees,
executives and directors, in each case of our company or any subsidiary. The Section 102 Options may be granted either pursuant to Section
102(c) of the Tax Ordinance, which are not required to be held in trust by a trustee, or pursuant to Section 102(b), which are required
to be held in trust for a specified period to qualify for certain tax benefits. Options granted pursuant to Section 102(b) shall be designated
to qualify for capital gain tax treatment in accordance with Section 102(b)(2) of the Tax Ordinance or ordinary income tax in accordance
with Section 102(b)(1) of the Tax Ordinance and thereafter, only such type of Section 102(b) options shall be granted until the Administrator
has determined otherwise in accordance with applicable law (which may not be prior to one year after the first grant of such type of Section
102(b) options).
Unless the Administrator determines otherwise and
subject to applicable law, ISOs may be granted only to non-Israeli employees and Non-statutory Stock Options may be granted to non- Israeli
employees and consultants, in each case of our company or any subsidiary. To the extent that the aggregate fair market value of the ordinary
shares with respect to which ISOs are exercisable for the first time by a participant during any calendar year under all company plans
exceeds $100,000, then unless the Administrator determines otherwise at any time and subject to applicable law, such options shall be
treated as Non-statutory Stock Options.
Administration
of the 2015 Plan. The Plan 2015 will be administered by the Board of Directors or, subject to applicable law, a committee appointed
by the Board of Directors, or Committee and the Administrator. Subject to the provisions of the 2015 Plan and, in the case of a Committee,
the specific duties delegated by the Board of Directors, and subject to the approval of any relevant authorities and compliance with all
applicable laws, the Administrator shall have the full power and authority at its sole discretion, from time to time and at any time,
among other things:
|
• |
To determine whether and to what extent awards are to be granted to participants under the 2015 Plan and
to select the eligible recipients of awards under the 2015 Plan; |
|
• |
To approve forms of agreement for use under the 2015 Plan; |
|
• |
To determine the terms and conditions of any award under the 2015 Plan, including the exercise price, the
time or times and the extent to which the awards may be exercised (which may be based on performance criteria), any vesting acceleration
or waiver of forfeiture restrictions, and any restriction or limitation regarding any award or the ordinary shares relating thereto, based
in each case on such factors as the Administrator, at its sole discretion, shall determine; |
|
• |
To determine the fair market value of the shares covered by each award; |
|
• |
To make an election as to the type of Section 102 Option; |
|
• |
To prescribe, amend and rescind rules and regulations relating to the 2015 Plan, including rules and regulations
relating to sub-plans established for the purpose of qualifying for preferred tax treatment under foreign tax laws; |
|
• |
To authorize conversion or substitution under the 2015 Plan of any or all awards and to cancel or suspend
awards, as necessary, provided the material interests of the participants are not harmed; |
|
• |
To construe and interpret the terms of the 2015 Plan and awards granted pursuant to the 2015 Plan; and
|
|
• |
To alter, revise or otherwise adjust the terms of the 2015 Plan and the award agreement, as may be required
pursuant to any applicable laws of local or foreign jurisdictions. |
Term of Awards.
The term of each option shall be stated in the award agreement but in no event may it be more than ten years from the date of grant.
Unless the Administrator determines otherwise and subject to applicable law, no ISO may be granted under the 2015 Plan to a grantee who
possess more than 10% of the total combined voting power of our company or any of our affiliate (a “10% Shareholder”) unless
the option terminates on a date that is not later than the day preceding the fifth anniversary of the date of grant. Unless otherwise
specified in the applicable award agreement, the term of a SAR will be ten years.
Exercise Price.
The exercise price of any award under the 2015 Plan shall be determined by the Administrator, subject to applicable law. Unless
the Administrator determines otherwise and subject to applicable law, in the case of ISOs and Non-Statutory Stock Options, the exercise
price per share shall be no less than the fair market value per ordinary share on the date of grant and in the case of an ISO granted
to a 10% Shareholder, no less than 110% of the fair market value per ordinary share on the date of grant.
Non-Transferability
of Awards. Unless the Administrator determines otherwise and subject to applicable law: (i) options and SARs may not be sold, pledged,
assigned, hypothecated, transferred, or disposed of in any manner other than by will or by the laws of descent or distribution and may
be exercised, during the lifetime of the participant, only by the participant. For as long as options or shares purchased upon the exercise
of options are held by a trustee on behalf of the participant, all rights of the participant with respect to such options and shares shall
be personal, and may not be transferred, assigned, pledged or mortgaged, other than by will or laws of descent and distribution; (ii)
RSUs may not be sold, pledged, transferred, assigned or encumbered; and (iii) RSAs may not be sold, transferred, pledged, assigned or
otherwise disposed of during the restricted period, provided that the Administrator may provide for the lapse of such restrictions in
installments and may accelerate or waive such restrictions in whole or in part.
Termination
of Employment or Service.
Options and SARs.
If a participant ceases to be an employee or consultant, in the absence of specified period in the award agreement and unless the Administrator
determines otherwise, such participant may exercise his/her options (to the extent vested on the date of such termination) or SARs within
three months following such termination (but in no event later than the expiration date of the option or SAR). If a participant retires,
he/she may continue to enjoy such rights with respect to awards under the 2015 Plan, on such terms and conditions as the Administrator
may determine. If the participant’s employment or service is terminated as a result if his/her death or permanent disability, the
participant (or, if the participant died, the participant’s estate or any person who acquired the right to exercise the option by
bequest or inheritance), may exercise his/her options (to the extent vested on the date of such termination) and/or SARs within such additional
period of time following such termination as specified in the award agreement (which may not be less than six months), or in the absence
of a specified period in the award agreement, until 12 months following such termination or any longer period determined by the Administrator,
but in no event later than the expiration date of the option or SAR.
RSAs. If
a participant’s service of employment is terminated prior to the restricted period, subject to the terms of the award agreement
or as otherwise determined by the Administrator, the participant’s restricted stock and any associated dividends that remain subject
to forfeiture will then be forfeited automatically.
RSUs. If
a participant’s service of employment is terminated prior to the RSU vesting, subject to the terms of the award agreement or as
otherwise determined by the Administrator, the participant’s RSUs that remain subject to forfeiture will then be forfeited automatically.
M&A Transaction.
In the event of a Transaction (as defined in the 2015 Plan, which generally includes (among other things) a sale of all or substantially
all of our assets or shares, a merger, consolidation or amalgamation with or into another company or a scheme of arrangement for effecting
any of the foregoing or such other transaction determined as such by the Administrator, all subject to the conditions and limitations
in the 2015 Plan), unless otherwise determined by the Administrator, in its sole discretion, any award granted under the 2015 shall be
assumed or substituted by us or the successor company, under terms determined by the Administrator or the terms of the 2015 Plan applied
by the successor company to such assumed or substituted awards, all in accordance with the terms of the 2015 Plan. Regardless of whether
or not the awards are assumed or substituted, the Administrator may, at its sole discretion, among other things, provide for the exercise
of any exercisable and vested awards, the cancellation of unexercised and unvested awards, the acceleration of unvested awards, or the
cancellation of awards for payment in cash, shares or other property of our company or the acquiring company or other party to the Transaction,
under such terms as the Administrator may determine, all in accordance with the terms of the 2015 Plan.
Term and Termination
of the 2015 Plan. The 2015 Plan became effective upon its adoption by our Board of Directors and shall continue in effect for a
term of ten years. Our Board of Directors may at any time amend, alter, suspend or terminate the 2015 Plan. No amendment, alteration,
suspension or termination of the 2015 Plan shall impair the rights of any participant, unless mutually agreed otherwise in writing between
the participant and the Administrator.
As of December 31, 2022, we had granted options
to purchase an aggregate 221,541 ordinary shares under the 2015 Plan, of which options to purchase an aggregate 350 ordinary shares had
been exercised into ordinary shares, an aggregate 31,051 ordinary shares had been forfeited without having been exercised and options
to purchase an aggregate 190,140 ordinary shares were outstanding as of such date, with a weighted average exercise price of approximately
$24.8 per share. In addition, as of December 31, 2022, we had issued 35,152 RSUs under the 2015 Plan, of which 5,988 RSUs had been forfeited,
9,461 RSUs had vested, and 19,703 RSUs were outstanding as of such date. As of December 31, 2022, no ordinary shares are available for
future awards under the 2015 Plan. In January 2023, our Board of Directors resolved to increase the number of ordinary shares reserved
for issuance under the 2015 Plan by 30,000 shares.
F. Disclosure
of a registrant’s action to recover erroneously awarded compensation.
Not applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. Major
shareholders
The following table sets forth information with
respect to the beneficial ownership of our ordinary shares as of March 15, 2023 by (i) each person or entity known to us to beneficially
own more than 5% of our outstanding ordinary shares; (ii) each of our current executive officers and directors individually; and (iii)
all of our current executive officers and directors as a group.
The percentage of beneficial ownership of our ordinary
shares is based on 5,846,989 ordinary shares, NIS 48.00 par value per share outstanding as of March 15, 2023. Beneficial ownership is
determined in accordance with the rules of the SEC and generally includes voting power or investment power with respect to securities.
All ordinary shares subject to options and warrants currently exercisable or exercisable into ordinary shares within 60 days of March
15, 2023, and underlying RSUs that shall vest within 60 days of March 15, 2023, are deemed to be outstanding and beneficially owned by
the shareholder holding such options, warrants or RSUs for the purpose of computing the number of shares beneficially owned by such shareholder.
Such shares are also deemed outstanding for purposes of computing the percentage ownership of the person holding the option, warrant or
RSU. They are not, however, deemed to be outstanding and beneficially owned for the purpose of computing the percentage ownership of any
other shareholder.
This table is based upon information supplied by
officers and directors and is believed to be accurate. Except as indicated in footnotes to this table, we believe that the shareholders
named in this table have sole voting and investment power with respect to all shares shown to be beneficially owned by them, based on
information provided to us by such shareholders. Unless otherwise noted below, each shareholder’s address is: c/o 29 Abba Hushi
Avenue, P.O. Box 1271, Isfiya, 3009000, Mount Carmel, Israel.
| |
|
Ordinary Shares Beneficially Owned |
|
| |
|
|
|
|
|
|
|
Directors and Executive Officers |
|
|
|
|
|
|
|
Alex Ovadia (1) |
|
|
19,271 |
|
|
|
* |
|
|
Yoav Kimchy (2) |
|
|
7,440 |
|
|
|
* |
|
|
Boaz Shpigelman (3) |
|
|
4,823 |
|
|
|
* |
|
|
Joshua (Shuki) Belkar (4) |
|
|
3,370 |
|
|
|
* |
|
|
Hanit Brenner-Lavie (5) |
|
|
790 |
|
|
|
* |
|
|
Israel Hershko (6) |
|
|
3,279 |
|
|
|
* |
|
|
Mira Rosenzweig (7) |
|
|
3,660 |
|
|
|
* |
|
|
Steven Hanley (8) |
|
|
1,253 |
|
|
|
* |
|
|
Clara Ezed (9) |
|
|
1,207 |
|
|
|
* |
|
|
Mary Jo Gorman (10) |
|
|
1,247 |
|
|
|
* |
|
|
XiangQian (XQ) Lin (11) |
|
|
2,150 |
|
|
|
* |
|
|
Yuval Yanai (12) |
|
|
1,254 |
|
|
|
* |
|
|
All director and executive officers as a group (12 persons)(13)
|
|
|
49,743 |
|
|
|
0.84 |
% |
* Less than 1% of the Company’s ordinary shares.
| (1) |
Includes (i) 752 outstanding ordinary shares, and (ii) 18,519 ordinary shares subject to options currently exercisable or options
and/or RSUs that will become exercisable or vested within 60 days of the date of this table |
| (2) |
Includes (i) 2,289 ordinary shares directly held by Yoav Kimchy, (ii) 3,730 ordinary shares subject to options and RSUs held by Yoav
Kimchy currently exercisable or options and/or RSUs held by Yoav Kimchy that will become exercisable or vested within 60 days of the date
of this table, (iii) 1,334 ordinary shares directly held by Sigalit Kimchy, the wife of Yoav Kimchy, and (iv) 87 ordinary shares subject
to options and RSUs held by Sigalit Kimchy currently exercisable or options and/or RSUs that will become exercisable or vested within
60 days of the date of this table. Yoav Kimchy and Sigalit Kimchy have joint beneficial ownership over the shares beneficially held by
them. |
| (3) |
Includes (i) 914 outstanding ordinary shares, and (ii) 3,909 ordinary shares subject to options
currently exercisable or options and/or RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (4) |
Includes (i) 385 ordinary shares, and (ii) 2,985 ordinary shares subject to options currently exercisable or options
and/or RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (5) |
Includes 790 ordinary shares subject to options currently exercisable or options and/or RSUs that will become exercisable or vested
within 60 days of the date of this table. |
| (6) |
Includes (i) 565 outstanding ordinary shares, and (ii) 2,714 ordinary shares subject to options currently exercisable or options
and/or RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (7) |
Includes (i) 415 outstanding ordinary shares, and (ii) 3,245 ordinary shares subject to options currently exercisable or options
and/or RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (8) |
Includes (i) 258 outstanding ordinary shares, and (ii) 995 ordinary shares subject to options currently exercisable or options and/or
RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (9) |
Includes (i) 212 outstanding ordinary shares, and (ii) 995 ordinary shares subject to options currently exercisable or options and/or
RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (10) |
Includes (i) 252 outstanding ordinary shares, and (ii) 995 ordinary shares subject to options currently exercisable or options and/or
RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (11) |
Includes (i) 953 outstanding ordinary shares, and (ii) 1,197 ordinary shares subject to options currently exercisable or options
and/or RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (12) |
Includes (i) 259 outstanding ordinary shares, and (ii) 995 ordinary shares subject to options currently exercisable or options and/or
RSUs that will become exercisable or vested within 60 days of the date of this table. |
| (13) |
Includes (i) 8,588 outstanding ordinary shares, and (ii) 41,155 ordinary shares subject to options currently exercisable or options
and/or RSUs that will become exercisable or vested within 60 days of the date of this table. |
As of March 15, 2023, based on information provided
to us by our transfer agent in the United States and other information reasonably available to us, we had 51 holders of record of our
ordinary shares in the United States. Such holders of record held, as of that date, 99.87 % of our outstanding ordinary shares. The number
of record holders is not representative of the number of beneficial holders of our ordinary shares, as 99.81 % of our outstanding ordinary
shares are recorded in the name of Cede & Co. as nominee for the Depository Trust Company, in whose name all shares held in “street
name” are held in the United States.
None of our shareholders have voting rights different
from the voting rights of other shareholders. To the best of our knowledge, we are not owned or controlled, directly or indirectly, by
another corporation or by any government. We are not aware of any arrangement that may, at a subsequent date, result in a change of control
of our company.
To our knowledge, other than as disclosed in the
table above, our other filings with the SEC and this Annual Report, there has been no significant change in the percentage ownership held
by any major shareholder since January 1, 2020.
B. Related
Party Transactions
Other than the executive and director compensation,
executive officer employment agreements, indemnification and exculpation arrangements and directors’ and officers’ liability
insurance policy discussed in “Management,” and the transactions described below, since January 1, 2020, we have not been
or are not a party to any related party transactions. The descriptions provided below are summaries of the terms of such agreements, do
not purport to be complete and are qualified in their entirety by the complete agreements.
Pontifax Warrants
On October 14, 2014, we issued warrants to purchase
an aggregate of 924 of our ordinary shares at an exercise price of NIS 48 per share, or the Pontifax Warrants, to the Pontifax Funds in
consideration of their commitment to provide to us, for no consideration, the following services, if and to the extent requested by us:
(i) business development services, in such scope and substance as shall be agreed between us and the Pontifax; and (ii) a representative
designated by Pontifax to serve as the chairman of our board of directors. The Pontifax Funds subsequently agreed that the exercise price
of fifty-percent of their warrants will increase to equal the price at which our ordinary shares are sold to the public in the initial
public offering of our securities, or if units are sold in the initial public offering of our securities, the exercise price per share
will increase to be equal to the effective price per share of the ordinary shares underlying the units sold to the public in the offering.
The Pontifax Funds also agreed that such portion of their warrants would vest and become exercisable only upon the consummation of the
initial public offering of our securities prior to their expiration date. The remaining warrants with an NIS 48 exercise price would vest
on a quarterly basis in eight equal installments during a period of 24 months from issuance. In addition, the Pontifax Funds agreed to
reduce the term of their respective warrants such that these warrants will now expire after eight years (instead of ten years) following
their issuance, i.e., on October 14, 2022. Upon the closing of our initial public offering any unvested portion of the warrants became
fully vested and exercisable. In September 2018, Pontifax exercised 462 of the Pontifax Warrants, with an exercise price of 48 per share,
into 376 ordinary shares on a cashless basis. On October 14, 2022, the remaining Pontifax Warrants to purchase 462 ordinary shares, with
an exercise price of $1,214.4 per share, expired.
Transactions with Check-Cap LLC and the Members and Manager of
Check-Cap LLC
On May 31, 2009, we entered into an asset transfer
agreement with Check-Cap LLC pursuant to which Check-Cap LLC transferred all of its business operations and substantially all of its assets
to us. Our shareholders’ holdings on the date of the asset transfer transaction reflected their interests as members of Check-Cap
LLC. In the framework of the asset transfer agreement and under the Shareholders Agreement, we undertook to use commercially reasonable
efforts to procure that distributions or advance funds are made to our shareholders holding (at the date of the transaction) ordinary
shares, Series A preferred shares and/or Series B preferred shares (i.e., the shareholders who are also members of Check-Cap LLC), as
would be necessary to eliminate the tax impact on such shareholders of the Reorganization and the transfer of all of the business operations
and substantially all of the assets from Check-Cap LLC to us. Notwithstanding the foregoing, we will not advance payments to such shareholders
to address the fact that they will no longer receive a “pass through” of losses generated by us as they previously received
while owning units of Check-Cap LLC. These advances, if and to the extent made, will be deducted from any distributions such shareholders
are entitled to receive from us. Since we do not expect to be profitable in year 2023, the liability based on the discounted outflows
is $0. As a result, as of December 31, 2022, the balance of the reimbursement liability totaled $0 ($0 as of December 31, 2021 and $14,000
as of December 31, 2020).
In connection with the asset transfer agreement
entered into in May 2009, we assumed the former obligation of Check-Cap LLC to distribute any proceeds it collects on the $1 million key
man life insurance policy with respect to Yoav Kimchy, the Company’s chief technology officer and a former director, to the former
holders of the Series A preferred units in an amount equal to their respective capital contributed to Check-Cap LLC, less any amounts
previously distributed to them, plus any accrued and unpaid dividends due to them as of the date of distribution. On November 16, 2016,
we cancelled the key man life insurance policy with respect to Yoav Kimchy.
C. Interests
of Experts and Counsel
Not applicable.
ITEM 8. FINANCIAL INFORMATION
A. Consolidated
Statements and Other Financial Information.
Consolidated Financial Statements
See Item 18 “Financial Statements.”
Legal Proceedings
From time to time, we may be involved in litigation
that arises through the normal course of business. As of the date of this filing, we are not a party to any material litigation nor are
we aware of any such threatened or pending litigation.
There are no material proceedings in which any
of our directors, officers or affiliates or any registered or beneficial shareholder of more than 5% of our ordinary shares, or any associate
of any of the foregoing is an adverse party or has a material interest adverse to our interest.
Dividend Policy
We have never declared or paid dividends on our
ordinary shares and currently do not intend to pay cash dividends on our ordinary shares in the foreseeable future. We currently intend
to retain all of our future earnings, if any, to finance the growth and development of our business. See Item 3D “Key Information
– Risk factors - Risks Related to the Company.”
Our ability to distribute dividends also may be
limited by future contractual obligations and by Israeli law. The Israeli Companies Law restricts our ability to declare dividends. Unless
otherwise approved by a court, we can distribute dividends only from “profits” (as defined by the Israeli Companies Law),
and only if there is no reasonable concern that the dividend distribution will prevent us from meeting our existing and foreseeable obligations
as they become due. Subject to the foregoing, payment of future dividends, if any, will be at the discretion of our board of directors
and will depend on various factors, such as our financial condition, operating results, current and anticipated cash needs and other business
and economic factors that our board of directors may deem relevant. See Exhibit 2.1 “Description of Securities—Dividend and
Liquidation Rights.” The payment of dividends may be subject to Israeli withholding taxes. See Item 10E. “Additional Information—Taxation—Israeli
Tax Considerations and Government Programs—Taxation of Our Shareholders—Taxation of Non-Israeli Shareholders on Receipt of
Dividends.” Furthermore, if we pay a dividend out of income attributed to our Benefited Enterprise that was generated during the
tax exemption period, we may be subject to tax on the grossed- up amount of such distributed income at the corporate tax rate which would
have been applied to our Benefited Enterprise’s income had we not enjoyed the exemption. See Item 10E. “Additional Information—Taxation
– Israeli Tax Considerations and Government Programs — Law for the Encouragement of Capital Investments, 5719-1959 —
Tax Benefits Subsequent to the 2005 Amendment.”
B. Significant
Changes
Except as disclosed elsewhere in this Annual Report,
there have been no other significant changes since December 31, 2022, until the date of the filing of this Annual Report.
ITEM 9. THE OFFER AND LISTING
A. Offer
and Listing Details
Our units were listed on the Nasdaq Capital Market
on February 19, 2015 under the symbol “CHEKU.” Prior to that date, there was no public trading market for our securities.
Our initial public offering was priced at $1,440 per unit on February 20, 2015. Each unit consisted of one ordinary share and one-half
of a Series A Warrant to purchase one ordinary share. Each unit was issued with one and one-half non-transferrable Long Term Incentive
Warrants. On March 18, 2015, the units separated and ceased to exist. Since such date, our ordinary shares and Series A Warrants were
listed on the Nasdaq Capital Market under the symbols “CHEK” and “CHEKW”, respectively. The Series A Warrants
expired on February 24, 2020 and ceased to be listed on the Nasdaq Capital Market since February 20, 2020. The Long Term Incentive Warrants
expired on February 24, 2022. Since May 8, 2018, our Series C Warrants have also been listed on the Nasdaq Capital Market under the symbol
“CHEKZ”. On April 4, 2018, we effected a 1-for-12 reverse share split of our ordinary shares. On November 23, 2022, we effected
a 1-for-20 reverse share split of our ordinary shares.
B. Plan
of Distribution
Not applicable.
C. Markets
for Ordinary Shares
See “—Offer and Listing Details”
above.
D. Selling
Shareholders
Not applicable.
E. Dilution
Not applicable.
F. Expenses
of the Issue
Not applicable.
ITEM 10. ADDITIONAL INFORMATION
A. Share
Capital
Not applicable.
B. Memorandum
and Articles of Association
A copy of our amended and restated articles of
association is incorporated by reference attached as Exhibit 1.1 to this Annual Report. Other than as disclosed below, the information
called for by this Item is set forth in Exhibit 2.1 to this Annual Report and is incorporated by reference into this Annual Report.
Registration Number and Purposes of the Company
Our registration number with the Israeli Registrar
of Companies is 51-425981-1. Our purpose as set forth in our amended articles of association is to engage in any lawful activity.
Shareholder Meetings
Under Israeli law, we are required to hold an annual
general meeting of our shareholders once every calendar year that must be held no later than 15 months after the date of the previous
annual general meeting. All meetings other than the annual general meeting of shareholders are referred to in our amended articles of
association as special general meetings. Our board of directors may call special general meetings whenever it sees fit, at such time and
place, within or outside of Israel, as it may determine. In addition, the Israeli Companies Law provides that our board of directors is
required to convene a special general meeting upon the written request of (i) any two of our directors or one-quarter of the serving members
of our board of directors; or (ii) one or more shareholders holding, in the aggregate, either (a) 5% or more of our outstanding shares
and 1% of our outstanding voting power or (b) 5% or more of our outstanding voting power.
Furthermore, the Israeli Companies Law requires
that resolutions regarding the following matters be approved by our shareholders at a general meeting:
|
• |
approval of certain related party transactions; |
|
• |
increases or reductions of our authorized share capital; |
|
• |
the exercise of our board of director’s powers by a general meeting, if our board of directors is
unable to exercise its powers and the exercise of any of its powers is essential for our proper management. |
Subject to the provisions of the Israeli Companies
Law and regulations promulgated thereunder, shareholders entitled to participate and vote at general meetings are the shareholders of
record on a date to be decided by the board of directors, which, as a company listed on an exchange outside Israel, may be between four
and 40 days prior to the date of the meeting.
The Israeli Companies Law requires that a notice
of any annual general meeting or special general meeting be provided to shareholders at least 21 days prior to the meeting and if the
agenda of the meeting includes, among other things, the appointment or removal of directors, the approval of transactions with office
holders or interested or related parties, an approval of a merger or the approval of the compensation policy, notice must be provided
at least 35 days prior to the meeting.
Under the Israeli Companies Law, our shareholders
are not permitted to take action via written consent in lieu of a meeting.
Borrowing powers
Pursuant to the Israeli Companies Law and our amended
articles of association, our board of directors may exercise all powers and take all actions that are not required under law or under
our amended articles of association to be exercised or taken by our shareholders, including the power to borrow money for company purposes.
For details regarding the approvals required under
the Israeli Companies Law for the approval of director compensation, see “Item 6C “Directors, Senior Management and Employees
— Board Practices -Approval of Related Party Transactions under Israeli Law — Disclosure of Personal Interests of an Office
Holder and Approval of Certain Transactions—Compensation of Directors.”
C. Material
Contracts
We have not entered into any material contracts
other than in the ordinary course of business and other than those described in Item 4. “Information on Our Company,” Item
7B “Major Shareholders and Related Party Transactions - Related Party Transactions” or elsewhere in this Annual Report.
D. Exchange
controls
There are currently no Israeli currency control
restrictions on remittances of dividends on our ordinary shares, proceeds from the sale of the shares or interest or other payments to
non- residents of Israel, except for shareholders who are subjects of countries that are, or have been, in a state of war with Israel.
E. Taxation
The following description is not intended to constitute
a complete analysis of all tax consequences relating to our ordinary shares, Series C Warrants and Series D Warrants (sometimes referred
to collectively or individually as our “securities”). You should consult your own tax advisor concerning the tax consequences
of your particular situation, as well as any tax consequences that may arise under the laws of any state, local, non-U.S. or other taxing
jurisdiction.
Israeli Tax Considerations and Government Programs
The following is a brief summary of the material
Israeli tax laws applicable to us and certain Israeli Government programs that benefit us. This section also contains a discussion of
material Israeli tax consequences concerning the ownership and disposition of our securities purchased by investors who are non-Israeli
resident shareholders. This summary does not discuss all the aspects of Israeli tax law that may be relevant to a particular investor
in light of his or her personal investment circumstances or to some types of investors subject to special treatment under Israeli law.
Examples of such investors include residents of Israel or traders in securities who are subject to special tax regimes not covered in
this discussion. Because parts of this discussion are based on new tax legislation that has not yet been subject to judicial or administrative
interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion.
The discussion below is subject to change, including due to amendments under Israeli law or changes to the applicable judicial or administrative
interpretations of Israeli law, which change could affect the tax consequences described below.
General Corporate Tax Structure in Israel
Israeli resident companies are generally subject
to corporate tax, currently at the rate of 23% (effective as of January 1, 2018) of a company’s taxable income. However, the effective
tax rate payable by a company that derives income from an Approved Enterprise, a Benefited Enterprise a Preferred Enterprise or a Preferred
Technological Enterprise (as discussed below) may be considerably less. Capital gains derived by an Israeli resident company are subject
to tax at the prevailing corporate tax rate.
Law for the Encouragement of Industry (Taxes), 5729-1969
The Law for the Encouragement of Industry (Taxes),
5729-1969, generally referred to as the Industry Encouragement Law, provides several tax benefits for “Industrial Companies.”
The Industry Encouragement Law defines an “Industrial Company” as a company resident in Israel, of which 90% or more of its
income in any tax year, other than income from defense loans, is derived from an “Industrial Enterprise” owned by it and located
in Israel or in the “Area” (as such term is defined under Section 3A of the Israeli Income Tax Ordinance (New Version), 5721-1961,
referred to as the Ordinance). An “Industrial Enterprise” is defined as an enterprise whose principal activity in a given
tax year is industrial production.
The following corporate tax benefits, among others,
are available to Industrial Companies:
|
• |
amortization over an eight-year period, beginning from the year in which such rights were first used, of
the cost of purchased know-how and patents and rights to use a patent and know-how which are used for the development or advancement of
the Industrial Enterprise; |
|
• |
under limited conditions, a Parent Company (as such term defined in the Industry Encouragement Law) may
elect to file consolidated tax returns with related Israeli Industrial Companies; and |
|
• |
expenses related to a public offering are deductible in equal amounts over three years beginning from the
year of the offering. |
Eligibility for the benefits under the Industry
Encouragement Law is not subject to receipt of prior approval from any governmental authority. We believe that we may qualify as an “Industrial
Company” within the meaning of the Industry Encouragement Law; however, there can be no assurance that we will qualify as an Industrial
Company or that the benefits described above will be available in the future.
Law for the Encouragement of Capital Investments, 5719-1959
The Law for the Encouragement of Capital Investments,
5719-1959, generally referred to as the Investment Law, was originally enacted in order to provide certain incentives for capital investments
in production facilities (or other eligible assets).
The Investment Law has been amended several times
in recent years, primarily: Amendment 60, effective as of April 1, 2005, referred to as the 2005 Amendment; Amendment 68, effective as
of January 1, 2011, referred to as the 2011 Amendment; Amendment 71, effective as of January 1, 2014, referred to as the 2014 Amendment;
Amendment 73, effective as of January 1, 2017, referred to as the 2017 Amendment; and Amendment 74, effective as of August 15, 2021, referred
to as the 2021 Amendment. Pursuant to the foregoing amendments, generally tax benefits that were granted in accordance with the provisions
of the Investment Law prior to each such amendment remain in force; however, any benefits granted subsequent to the respective amendment
are subject to the provisions of the Investment Law as amended.
Tax Benefits Prior to the 2005 Amendment
Prior to the 2005 Amendment, a capital investment
in eligible production facilities (or other eligible assets) could, upon application to the Investment Center of the Israeli Ministry
of Economy (formerly named the Ministry of Industry, Trade and Labor), be designated as an “Approved Enterprise” and accordingly,
entitled to certain tax benefits under the Investment Law. Each certificate of approval for an Approved Enterprise relates to a specific
investment program in the Approved Enterprise, delineated both by the financial scope of the investment and by the physical characteristics
of the facility or the asset. We do not have any Approved Enterprises.
Tax Benefits Subsequent to the 2005 Amendment
Pursuant to the 2005 Amendment, a company whose
facilities meet certain criteria set forth in the 2005 Amendment may claim certain tax benefits offered by the Investment Law (as further
described below) directly in its tax returns, without the need to obtain prior approval. In order to receive the tax benefits, a company
must make an investment which meets all of the conditions, including exceeding a minimum entitling investment amount, set forth in the
Investment Law. Such investment allows a company to receive “Benefited Enterprise” status, and may be made over a period of
no more than three years ending at the end of the year in which the company chose to have the tax benefits apply to its Benefited Enterprise,
referred to as the “Year of Election.”
The extent of the tax benefits available under
the 2005 Amendment to qualifying income of a Benefited Enterprise depends on, among other things, the geographic location in Israel of
the Benefited Enterprise. The location will also determine the period for which tax benefits are available. Under the “Exemption
Track” the tax benefits include an exemption from corporate tax on undistributed income generated by the Benefited Enterprise for
a period of two to ten years, depending on the geographic location of the Benefited Enterprise in Israel, and a reduced corporate tax
rate of 10% to 25% for the remainder of the benefits period, depending on the level of foreign investment in the company in each year.
The benefits period is for a duration of seven or ten years, depending on the location of the Benefited Enterprise, from the later of
the first year in which the company generated taxable income from its Benefited Enterprise and the Year of Election, but in any event
not more than 12 or 14 years from the Year of Election, depending on the location of the Benefited Enterprise. A company qualifying for
tax benefits under the Exemption Track which pays a dividend (as well as a deemed dividend, such as investments in foreign resident subsidiaries,
granting loans to related parties, repurchases of shares, acquisitions of securities/shares, capital reductions and additional events
which reflect the transfer of funds out of the Benefitted Enterprise activity) out of income derived during the tax exemption period by
its Benefited Enterprise will be subject to corporate tax in respect of the amount of the dividend (grossed-up to reflect the pre-tax
income that it would have had to earn in order to distribute the dividend) at the corporate tax rate which would have otherwise been applicable.
Dividends paid out of income attributed to a Benefited Enterprise are generally subject to withholding tax at source at the rate of 20%
or such lower rate as may be provided in an applicable tax treaty. With respect to such dividends, the recently enacted 2021 Amendment
introduced a new dividend distribution ordering rule as well as a Temporary Order to incentivize the distribution thereof (see discussion
below under “Tax Benefits under the 2021 Amendment”).
Applying reduced tax rates in accordance with a certain tax treaty is subject to the receipt in advance of a valid certificate
from the Israel Tax Authority allowing for a reduced tax rate.
The benefits available to a Benefited Enterprise
are subject to the fulfillment of conditions stipulated in the Investment Law and its regulations. If a company does not meet these conditions,
it may be required to refund the amount of tax benefits, as adjusted by the Israeli consumer price index, plus interest, or other monetary
penalties.
We currently have one Benefited Enterprise program
under the Investment Law, which, we believe, entitles us to certain tax benefits with respect to income to be derived from our Benefited
Enterprise. We chose 2010 as the Year of Election. We believe that we are located in the Zone A specified development zone and therefore,
believe we are entitled to a 10-year benefit period, during which taxable income from our Benefited Enterprise program (once generated)
will be tax exempt, commencing with the year we will first earn taxable income relating to such enterprise, subject to a 14-year limitation
from the Year of Election, and therefore, the tax benefit period will in any event end in 2023.
Tax Benefits under the 2011 Amendment and the 2014
Amendment
The 2011 Amendment canceled the availability of
the benefits granted to companies under the Investment Law prior to 2011 and, instead, introduced new benefits for income generated by
a “Preferred Company” through its “Preferred Enterprise” or “Special Preferred Enterprise” (as such
terms are defined in the Investment Law) as of January 1, 2011. The definition of a Preferred Company includes, among other limitations
detailed under the Investment Law, a company incorporated in Israel that is not wholly-owned by a governmental entity, and that, among
other things, owns a Preferred Enterprise or Special Preferred Enterprise and is controlled and managed from Israel. Under the 2014 Amendment,
effective as of January 1, 2014, a Preferred Company is entitled to a reduced corporate tax rate of 16% with respect to its income derived
by its Preferred Enterprise in 2014 and thereafter, unless the Preferred Enterprise is located in a specified development zone (referred
to as “Zone A”), in which case the rate will be 9% for the tax years 2014-2016 and, pursuant to the 2017 Amendment (as discussed
below), 7.5% from 2017 and thereafter. We believe our facilities are located in the Zone A specified development zone.
Dividends paid out of income attributed to a Preferred
Enterprise are generally subject to withholding tax at source at the rate of 20% or such lower rate as may be provided in an applicable
tax treaty. However, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if such dividends
are subsequently distributed to individuals or a non-Israeli company, withholding tax at a rate of 20% or such lower rate as may be provided
in an applicable tax treaty will apply).
The 2011 Amendment also provided transitional provisions
to address companies already enjoying existing tax benefits under the Investment Law. These transitional provisions provide, among other
things, that unless an irrevocable request is made to apply the provisions of the Investment Law as amended in 2011 with respect to income
to be derived as of January 1, 2011, a Benefited Enterprise can elect to continue to benefit from the benefits provided to it before the
2011 Amendment came into effect, provided that certain conditions are met.
We are currently examining whether we are eligible
for the tax benefits under the 2011 Amendment and the possible effect, if any, of the provisions of the 2011 Amendment on our financial
statements.
Tax Benefits under the 2017 Amendment
The 2017 Amendment was enacted as part of the Economic
Efficiency Law that was published on December 29, 2016, and became effective as of January 1, 2017. The 2017 Amendment provides new tax
benefits for two types of “Technological Enterprises”, as described below, and is in addition to the other existing tax beneficial
programs under the Investment Law.
The 2017 Amendment provides two new tax incentive
tracks – the “Preferred Technological Enterprise” track and “Special Preferred Technological Enterprise”
track (as such terms are defined in the Investment Law). The benefits available to a Preferred Technological Enterprise or Special Preferred
Technological Enterprise are subject to the fulfillment of conditions stipulated in the Investment Law.
A Preferred Technological Enterprise is entitled
(among other things) to a reduced corporate tax rate of 12% with respect to its income which qualifies as “Preferred Technology
Income”, as defined in the Investment Law, unless the Preferred Technological Enterprise is located in a specified development zone
(referred to as “Zone A”), in which case the rate will be 7.5%. A Special Preferred Technological Enterprises is entitled
to a reduced corporate tax rate of 6% with respect to its Preferred Technology Income, regardless of the company’s enterprise geographic
location within Israel.
We are currently examining whether we are eligible
for the tax benefits under the 2017 Amendment and the possible effect, if any, of the provisions of the 2017 Amendment on our financial
statements.
Tax Benefits under the 2021 Amendment
The 2021 Amendment introduced a new dividend distribution
ordering rule to cause the distribution of earnings that were tax-exempt under the historical Approved or Beneficial Enterprise regimes
(Trapped Earnings), to be on a pro-rata basis from any dividend distribution, which is applicable to distributions starting from August
15, 2021 onwards. Accordingly, the corporate income tax claw-back will apply to any dividend distribution, as long as the company has
Trapped Earnings.
The termination or substantial reduction of any of the benefits
available under the Investment Law could materially increase our future tax liabilities.
The Encouragement of Research, Development and Technological Innovation
in the Industry Law 5744-1984 (formerly known as the Encouragement of Industrial Research and Development Law, 5744-1984)
Under the Encouragement of Research, Development
and Technological Innovation in the Industry Law 5744-1984 (formerly known as the Encouragement of Industrial Research and Development
Law, 5744-1984), referred to as the Innovation Law, research and development programs that meet specified criteria and are approved by
the IIA are eligible for grants. As of December 31, 2022, we had received funding from the IIA for the financing of a portion of our research
and development expenditures for C-Scan in the aggregate amount of approximately $5.6 million. As of December 31, 2022, we had not paid
any royalties to the IIA and had a contingent liability to the IIA with respect to such funding (including interest at the rate of 12-month
LIBOR) in the amount of approximately $6.2 million. In addition, in January 2021, we received an IIA grant approval to support the funding
of our transition from research and development to manufacturing. The final IIA grant amounted to $620,000 (NIS 2.18 million) (along with
a co-investment by us of the same amount), subject to the terms and conditions set forth in the grant approval, which we are not required
to repay to the IIA, of which we received approximately $222,000 (NIS 784,000) in February 2023, $82,000 (NIS 264,000) in January 2022
and $349,000 (NIS 1,134,000) in 2021.
Under the Innovation Law as currently in effect,
the research and development grants are typically up to 50% of the project’s approved expenditures. The grantee is required to pay
royalties to the State of Israel from the sale of products (and associated services) developed using IIA research and development funding.
Regulations under the Innovation Law, as currently in effect, generally provide for the payment of royalties of 3% or 4% (and at an increased
rate under certain circumstances) on sales of products and services based on technology and know-how developed using such IIA research
and development grants, until 100% (which may be increased under certain circumstances) of the grant, linked to the U.S. dollar and bearing
interest at the LIBOR rate, is repaid.
The terms of IIA grants that we have received (the
research and development grants and the transition to manufacturing grants) require that products developed with IIA funded be manufactured
in Israel, unless the IIA approved grant program includes a pre-determined portion of manufacturing that may be performed outside Israel
(as certain of our IIA approved grants included). The approval of the IIA is required for the transferring of manufacturing outside Israel
in excess of such pre-determined portion (however, only a notice to the IIA, as opposed to approval, is required for the transfer outside
Israel of up to 10% of the cumulative manufacturing in excess of such pre-approved portion). If manufacturing of IIA-funded products is
transferred outside Israel (following IIA approval) in excess of the pre-determined percentage included in the grant approval, then the
royalty repayment rate is increased by 1% with respect to the additional approved percentage to be manufactured outside Israel and the
royalty repayment for the entire approved program may be increased to up to three times the amount of the grants received, depending on
the percentage manufactured outside Israel (plus accrued interest). We may explore from time to time whether certain other components
of C-Scan can be assembled outside of Israel. For example, we may in the future explore whether it would be possible to assemble the capsule
without the X-ray source in Israel, and have the X-ray source subsequently manufactured and assembled into C-Scan at a certified radioisotope
production facility or at a distribution center outside Israel.
Over the years, we received approval of grant applications
(research and development grants and the transition to manufacturing grant) that included a certain predetermined percentage of manufacturing
of the X-ray source to be performed outside of Israel but additional examination of these approvals and consequent manufacturing is required
to determine liabilities to the IIA, if any.
IIA prior approval is also required for the transfer
of IIA-funded know-how to a third party outside of Israel (including by way of license), which we may not receive (and any such approval
would be subject to payment of a redemption fee, calculated according to a formula under the Innovation Law, which may be in the amount
of up to six times the amount of the grants received, (less paid royalties, if any, and depreciation, but no less than the total grants
received), plus accrued interest). Even following the full repayment of any IIA grants, we must nevertheless continue to comply with the
requirements of the Innovation Law. If we fail to comply with any of the conditions and restrictions imposed by the Innovation Law and
regulations and guidelines thereunder, or by the specific terms under which we received the grants, we may be required to refund any grants
previously received together with interest and penalties, and, in certain circumstances, may be subject to criminal charges.
Taxation of our Shareholders
Capital Gains
Taxes Applicable to Non-Israeli Resident Shareholders. A non-Israeli resident who derives capital gains from the sale of securities
of an Israeli resident company will be exempt from Israeli tax so long as (i) the capital gains are not attributed to a permanent establishment
that the non-resident maintains in Israel, (ii) the securities were not received from a relative or in a tax free reorganization transaction
and (iii) the securities are not traded on the Tel Aviv Stock Exchange on the date of sale (provided, however, that other exemptions may
apply if a an Israeli resident company is listed for trading on the Tel Aviv Stock Exchange). However, non-Israeli corporations will not
be entitled to the foregoing exemption if Israeli residents: (i) have a controlling interest of 25% or more in such non-Israeli corporation;
or (ii) are the beneficiaries of, or are entitled to, 25% or more of the revenues or profits of such non-Israeli corporation, whether
directly or indirectly.
Additionally, a sale of securities by a non-Israeli
resident may be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, under the Convention
Between the Government of the United States of America and the Government of the State of Israel with respect to Taxes on Income, as amended,
generally referred to as the United States-Israel Tax Treaty, the sale, exchange or other disposition of shares by a shareholder who (i)
is a U.S. resident (for purposes of the treaty); (ii) holds the shares as a capital asset; and (iii) is entitled to claim the benefits
afforded to such person by the treaty, is generally exempt from Israeli capital gains tax. Such exemption will not apply if, among other
things: (i) the capital gain arising from such sale, exchange or other disposition is treated as industrial or commercial profits attributed
to a permanent establishment in Israel, subject to certain conditions; (ii) the shareholder holds, directly or indirectly, shares representing
10% or more of the voting capital of the corporation during any part of the 12-month period preceding the disposition, subject to certain
conditions; (iii) the capital gain arising from such sale, exchange or disposition is treated as royalties; or (iv) such U.S. resident
is an individual and was present in Israel for 183 days or more during the relevant taxable year. In such case, the sale, exchange or
disposition of our securities would be subject to Israeli tax, to the extent applicable; however, under the United States-Israel Tax Treaty,
the taxpayer would be permitted to claim a credit for such taxes against the U.S. federal income tax imposed with respect to such sale,
exchange or disposition, subject to the limitations under U.S. law applicable to foreign tax credits. The United States-Israel Tax Treaty
does not relate to U.S. state or local taxes. The United States-Israel Tax Treaty is currently under review and subject to change.
In some instances where our shareholders may be
liable for Israeli tax on the sale of their securities, the payment of the consideration may be subject to the withholding of Israeli
tax at source. Shareholders may be required to demonstrate that they are exempt from tax on their capital gains and to obtain an exemption
from withholding tax certificate from the Israel Tax Authority in order to avoid withholding at source at the time of sale.
Taxation of Non-Israeli
Shareholders on Receipt of Dividends. Non-Israeli residents are generally subject to Israeli withholding tax on the receipt of
dividends paid on our ordinary shares at the rate of 25%, unless relief is provided in a treaty between Israel and the shareholder’s
country of residence (subject to the receipt of a valid certificate from the Israeli Tax Authority allowing for a reduced tax rate). With
respect to a person who is a “substantial shareholder” at the time of receiving the dividend or at any time during the preceding
12 months, the applicable withholding tax rate is 30%, unless such “substantial shareholder” holds such shares through a nominee
company, in which case the rate is 25%. A “substantial shareholder” is generally a person who alone or together with such
person’s relative (as such term is defined in the Ordinance) or another person who collaborates with such person on a permanent
basis, holds, directly or indirectly, at least 10% of any of the “means of control” of the corporation. “Means of control”
generally include the right to vote, receive profits, nominate a director or an executive officer, receive assets upon liquidation, or
order someone who holds any of the aforesaid rights how to act, regardless of the source of such right.
However, a distribution of dividends to non-Israeli
residents is subject to withholding tax at source at a rate of 15% if the dividend is distributed from income attributed to an Approved
Enterprise or a Benefited Enterprise and 20% if the dividend is distributed from income attributed to a Preferred Enterprise or a Preferred
Technological Enterprise, unless a reduced tax rate is provided under an applicable tax treaty. The corporate income tax claw-back may
apply upon any dividend distribution, as long as the company has Trapped Earnings (see discussion above under “Tax
Benefits under the 2021 Amendment”). We cannot assure you that in the event we declare a dividend we will designate the income
out of which the dividend is paid in a manner that will reduce shareholders’ tax liability.
Under the United States-Israel Tax Treaty, the
maximum rate of tax withheld at source in Israel on dividends paid to a holder of our ordinary shares who is a U.S. resident (for purposes
of the United States-Israel Tax Treaty) is 25%. With respect to dividends paid to a U.S. corporation that held 10% or more of the capital
of the paying corporation throughout the tax year in which the dividend is distributed and the preceding tax year and provided that not
more than 25% of the gross income of the paying corporation for such prior taxable year (if any) consists of certain interest or dividends,
the maximum rate of tax withheld at source is 12.5%; provided, however, that if the paying corporation is an Approved Enterprise, the
applicable withholding tax rate under such circumstances is reduced to 15% (rather than 12.5%). We believe that the reference in the United
States-Israel Tax Treaty to an Approved Enterprise under the Investment Law is deemed to include also a Benefitted Enterprise Preferred
Enterprise and Preferred Technological Enterprise under the Investment Law. The United States-Israel Tax Treaty is currently under review
and subject to change.
U.S. residents who are subject to Israeli withholding
tax on a dividend may be entitled to a credit or deduction for U.S. federal income tax purposes in the amount of the taxes withheld, subject
to detailed rules contained in U.S. tax legislation.
Exercise or Lapse
of Series C Warrant or Series D Warrant. A holder of a Series C Warrant or Series D Warrant generally will not recognize gain or
loss upon the exercise of a Series C Warrant or Series D Warrant for cash. An ordinary share acquired pursuant to the exercise of a Series
C Warrant or Series D Warrant for cash generally will have a tax basis equal to the holder’s tax basis in the Series C Warrant or
Series D Warrant (as the case may be), increased by the amount paid to exercise the Series C Warrant or Series D Warrant. The holding
period of such ordinary share generally would begin on the day after the date of exercise of the Series C Warrant or Series D Warrant.
If a Series C Warrant or Series D Warrant is allowed to lapse unexercised, the holder generally will recognize a capital loss equal to
such holder’s tax basis in the Series C Warrant or Series D Warrant (as the case may be).
It is possible that a cashless exercise would be
treated as a taxable exchange in which gain or loss is recognized. In such event, a holder could be deemed to have surrendered a number
of Series C Warrants or Series D Warrants with a fair market value equal to the exercise price for the number of Series C Warrants or
Series D deemed exercised. For this purpose, the number of Series C Warrants or Series D Warrants deemed exercised would be equal to the
number of Series C Warrants or Series D Warrants, as applicable, that would entitle the holder to receive upon exercise the number of
ordinary shares issued pursuant to the cashless exercise of the Series C Warrants or Series D Warrants (as the case maybe). In this situation,
the holder would recognize capital gain or loss in an amount equal to the difference between the fair market value of the Series C Warrants
or Series D Warrants deemed surrendered to pay the exercise price and the holder’s tax basis in the Series C Warrants or Series
D Warrants deemed surrendered.
Adjustments with
Respect to Warrants. The terms of the Series C Warrant and Series D Warrant provide for an adjustment to the number of ordinary
shares for which the warrant may be exercised or adjustment to the exercise price of the warrant in certain events. An adjustment of the
exercise price or an adjustment that has the effect of preventing dilution generally is not taxable. However, the holders of the Series
C Warrants and Series D Warrants may be treated as receiving a constructive distribution from us if, for example, the adjustment increases
the warrant holders’ proportionate interest in our assets or earnings and profits (e.g., through a decrease in the exercise price
of the Series C Warrants or Series D Warrants) as a result of a distribution of cash to the holders of our ordinary shares, which is taxable
to the holders of such ordinary shares as described under “—Taxation of our Shareholders” above. Such constructive distribution
would be subject to tax as described under that section in the same manner as if the holders of the Series C Warrants or Series D Warrants
received a cash distribution from us equal to the fair market value of such increased interest. Holders of Series C Warrants and Series
D Warrants are urged to consult their own tax advisors on these issues.
Surtax
Subject to the provisions of an applicable tax
treaty, individuals who are subject to tax in Israel are also subject to an additional tax at a rate of 3% on annual income (including,
but not limited to, dividends, interest and capital gain) exceeding NIS 698,280 for 2023, which amount is linked to the annual change
in the Israeli consumer price index.
Estate and Gift Tax
Israeli law presently does not impose estate or
gift taxes.
EACH HOLDER OF OUR SECURITIES IS URGED TO CONSULT
ITS OWN TAX ADVISOR WITH RESPECT TO THE PARTICULAR TAX CONSEQUENCES TO SUCH HOLDER OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF OUR
SECURITIES, INCLUDING THE APPLICABILITY AND EFFECT OF ANY ISRAELI TAX LAWS AND ANY APPLICABLE TAX TREATIES.
If a United States person is treated as owning at least 10% of our
shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a United States person is treated as owning
(directly, indirectly or constructively) at least 10% of the value or voting power of our shares, such person may be treated as a “United
States shareholder” with respect to each “controlled foreign corporation” in our group (if any). A United States shareholder
of a controlled foreign corporation may be required to annually report and include in its U.S. taxable income its pro rata share of “Subpart
F income,” “global intangible low-taxed income” and investments in U.S. property by controlled foreign corporations,
whether or not we make any distributions, and may be subject to tax reporting obligations. An individual that is a United States shareholder
with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign tax credits that would
be allowed to a United States shareholder that is a U.S. corporation. A failure to comply with these reporting obligations may subject
you to significant monetary penalties and may prevent the statute of limitations with respect to your U.S. federal income tax return for
the year for which reporting was due from starting. We cannot provide any assurances that we will assist any shareholder in determining
whether such shareholder is treated as a United States shareholder with respect to any “controlled foreign corporation” in
our group (if any) or furnish to any United States shareholders information that may be necessary to comply with the aforementioned reporting
and tax paying obligations. A United States investor should consult its tax advisors regarding the potential application of these rules
to its investment in the shares.
U.S. Federal Income Taxation
The following are certain material U.S. federal
income tax consequences of the acquisition, ownership and disposition of our securities.
The discussion below of the U.S. federal income
tax consequences to “U.S. Holders” will apply to a beneficial owner of our securities that is for U.S. federal income tax
purposes:
|
• |
an individual citizen or resident of the United States; |
|
• |
a corporation (or other entity treated as a corporation) that is created or organized (or treated as created
or organized) in or under the laws of the United States, any state thereof or the District of Columbia; |
|
• |
an estate whose income is includible in gross income for U.S. federal income tax purposes regardless of
its source; or |
|
• |
a trust if (i) a U.S. court can exercise primary supervision over the trust’s administration and
one or more U.S. persons are authorized to control all substantial decisions of the trust; or (ii) it has a valid election in effect under
applicable U.S. Treasury regulations to be treated as a U.S. person. |
A beneficial owner of our securities that is described
above is referred to herein as a “U.S. Holder.” If a beneficial owner of our securities is not described as a U.S. Holder
and is not an entity treated as a partnership or other pass-through entity for U.S. federal income tax purposes, such owner will be considered
a “Non-U.S. Holder.” Certain material U.S. federal income tax consequences of the acquisition, ownership and disposition of
our securities applicable specifically to Non-U.S. Holders are described below under the heading “Non-U.S. Holders.”
This discussion is based on the Code, its legislative
history, Treasury regulations promulgated thereunder, the United States-Israel Tax Treaty, or the Treaty, published rulings and court
decisions, all as currently in effect. These authorities are subject to change or differing interpretations, possibly on a retroactive
basis.
This discussion does not address all aspects of
U.S. federal income taxation that may be relevant to any particular holder based on such holder’s individual circumstances. In particular,
this discussion considers only holders that own and hold our securities as capital assets within the meaning of Section 1221 of the Code
(generally, property held for investment), and does not address the potential application of the alternative minimum tax or the U.S. federal
income tax consequences to holders that are subject to special rules, including, but not limited to:
|
• |
financial institutions or financial services entities; |
|
• |
persons that are subject to the mark-to-market accounting rules under Section 475 of the Code; |
|
• |
governments or agencies or instrumentalities thereof; |
|
• |
regulated investment companies; |
|
• |
real estate investment trusts; |
|
• |
certain expatriates or former long-term residents of the United States; |
|
• |
persons that actually or constructively own 5% or more of our shares (by vote or value); |
|
• |
persons that acquired our securities pursuant to an exercise of employee options, in connection with employee
incentive plans or otherwise as compensation; |
|
• |
persons that hold our securities as part of a straddle, constructive sale, hedging, conversion or other
integrated transaction; |
|
• |
persons whose functional currency is not the U.S. dollar; |
|
• |
passive foreign investment companies; or |
|
• |
controlled foreign corporations. |
This discussion does not address any aspect of
U.S. federal non-income tax laws, such as gift or estate tax laws, or state, local or non-U.S. tax laws or, except as discussed herein,
any tax reporting obligations applicable to a holder of our securities. Additionally, this discussion does not consider the tax treatment
of partnerships or other pass-through entities or persons who hold our securities through such entities. If a partnership (or other entity
classified as a partnership for U.S. federal income tax purposes) is the beneficial owner of our securities, the U.S. federal income tax
treatment of a partner (or person or entity treated as a partner) in the partnership generally will depend on the status of the partner
and the activities of the partnership. This discussion also assumes that any distribution made (or deemed made) to a holder in respect
of our securities and any consideration received (or deemed received) by a holder in connection with the sale or other disposition of
our securities will be in U.S. dollars. In addition, as described in “Risk Factors – Risks Related to Taxation”, there
is a risk that we could be treated as a domestic (U.S.) corporation for U.S. federal income tax purposes by reason of the Reorganization;
this discussion also assumes that we will be and have been treated as a foreign corporation for U.S. federal income tax purposes. Moreover,
this discussion assumes that a holder owns a sufficient number of Series C Warrants and/or Series D Warrants, such that the holder will
not have a fractional warrant upon the exercise of a Series C Warrant and/or, Series D Warrant, as the case may be.
We have not sought, and will not seek, a ruling
from the IRS or an opinion of counsel as to any U.S. federal income tax consequence described herein. The IRS may disagree with the description
herein, and its determination may be upheld by a court. Moreover, there can be no assurance that future legislation, regulations, administrative
rulings or court decisions will not adversely affect the accuracy of the statements in this discussion.
EACH HOLDER OF OUR SECURITIES IS URGED TO CONSULT
ITS OWN TAX ADVISOR WITH RESPECT TO THE PARTICULAR TAX CONSEQUENCES TO SUCH HOLDER OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF OUR
SECURITIES, INCLUDING THE APPLICABILITY AND EFFECT OF ANY STATE, LOCAL, AND NON-U.S. TAX LAWS, AS WELL AS U.S. FEDERAL TAX LAWS AND ANY
APPLICABLE TAX TREATIES.
U.S. Holders
Taxation of Cash Distributions
As noted above, we currently do not intend to pay
cash dividends on our ordinary shares in the foreseeable future. Subject to the passive foreign investment company, or PFIC, rules discussed
below, a U.S. Holder generally will be required to include in gross income as ordinary income the amount of any cash dividend paid in
respect of our ordinary shares. A cash distribution on our ordinary shares generally will be treated as a dividend for U.S. federal income
tax purposes to the extent the distribution is paid out of our current or accumulated earnings and profits (as determined for U.S. federal
income tax purposes). Such dividend generally will not be eligible for the dividends-received deduction generally allowed to U.S. corporations
in respect of dividends received from other U.S. corporations. The portion of such cash distribution, if any, in excess of such earnings
and profits will be applied against and reduce (but not below zero) the U.S. Holder’s adjusted tax basis in the ordinary shares.
Any remaining excess generally will be treated as gain from the sale or other taxable disposition of such ordinary shares. We do not intend
to calculate our earnings and profits under U.S. federal income tax principles. Therefore, a U.S. Holder should expect that a distribution
will be reported as a dividend even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain
under the rules described above.
With respect to non-corporate U.S. Holders, any
such cash dividends may be subject to U.S. federal income tax at the lower applicable regular long term capital gains tax rate (see “—
Taxation on the Disposition of Ordinary Shares or Series C Warrants or Series D Warrants” below) provided that (a) our ordinary
shares are readily tradable on an established securities market in the United States or we are eligible for the benefits of the Treaty,
(b) we are not a PFIC, as discussed below, for either the taxable year in which the dividend was paid or the preceding taxable year, and
(c) certain holding period requirements are met. Therefore, if our ordinary shares are not readily tradable on an established securities
market, and we are not eligible for the benefits of the Treaty, then cash dividends paid by us to non-corporate U.S. Holders will not
be subject to U.S. federal income tax at the lower regular long term capital gains tax rate. Under published IRS authority, shares are
considered for purposes of clause (a) above to be readily tradable on an established securities market in the United States only if they
are listed on certain exchanges, which presently include the Nasdaq Capital Market. Although our ordinary shares are currently listed
and traded on the Nasdaq Capital Market, U.S. Holders nevertheless should consult their own tax advisors regarding the availability of
the lower rate for any cash dividends paid with respect to our ordinary shares.
Dividends paid to a U.S. Holder with respect to
our ordinary shares generally will be foreign source income, which may be relevant in calculating such U.S. Holder’s foreign tax
credit limitations. Subject to certain conditions and limitations, non-refundable Israeli tax withheld on dividends may be deducted from
such U.S. Holder’s taxable income or credited against such U.S. Holder’s U.S. federal income tax liability. The election to
deduct, rather than credit, foreign taxes, is made on a year-by-year basis and applies to all foreign taxes paid by a U.S. Holder or withheld
from a U.S. Holder that year. The limitation on foreign taxes eligible for credit is calculated separately with respect to specific classes
of income. For this purpose, dividends that we distribute generally should constitute “passive category income,” or, in the
case of certain U.S. Holders, “general category income.” A foreign tax credit for foreign taxes imposed on distributions may
be denied if a U.S. Holder does not satisfy certain minimum holding period requirements. As a result of recent changes to the U.S. foreign
tax credit rules, a withholding tax generally will need to satisfy certain additional requirements in order to be considered a creditable
tax for a U.S. Holder. We have not determined whether these requirements have been met and, accordingly, no assurance can be given
that any withholding tax on dividends paid by us will be creditable. The rules relating to the determination of the foreign tax credit
are complex, and U.S. Holders should consult their tax advisors to determine whether and to what extent they will be entitled to this
credit.
Adjustments with Respect to Warrants
The terms of each Series C Warrant and Series D
Warrant provide for an adjustment to the number of ordinary shares for which the warrant may be exercised or to the exercise price of
the warrant in certain events. An adjustment that has the effect of preventing dilution generally is not taxable. However, the U.S. Holders
of the Series C Warrants and Series D Warrants would be treated as receiving a constructive distribution from us if, for example, the
adjustment increases the warrant holders’ proportionate interest in our assets or earnings and profits (e.g., through a decrease
in the exercise price of the Series C Warrants or Series D Warrants) as a result of a distribution of cash to the holders of our ordinary
shares, which is taxable to the U.S. Holders of such ordinary shares as described under “— Taxation of Cash Distributions,”
above. Such constructive distribution would be subject to tax as described under that section in the same manner as if the U.S. Holders
of the Series C Warrants and Series D Warrants received a cash distribution from us equal to the fair market value of such increased interest.
U.S. Holders of Series C Warrants and Series D Warrants are urged to consult their own tax advisors on these issues.
Taxation on the Disposition of Ordinary Shares or Series C Warrants or Series D Warrants
Upon a sale or other taxable disposition of our
ordinary shares or the Series C Warrants or Series D Warrants, and subject to the PFIC rules discussed below, a U.S. Holder generally
will recognize capital gain or loss in an amount equal to the difference between the amount realized and the U.S. Holder’s adjusted
tax basis in the securities.
The regular U.S. federal income tax rate on capital
gains recognized by U.S. Holders generally is the same as the regular U.S. federal income tax rate on ordinary income, except that long
term capital gains recognized by non-corporate U.S. Holders generally are subject to U.S. federal income tax at a maximum regular rate
of 20%. Capital gain or loss will constitute long term capital gain or loss if the U.S. Holder’s holding period for the securities
exceeds one year. The deductibility of capital losses is subject to various limitations. Any such gain or loss that a U.S. Holder recognizes
generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes.
As discussed in “Israel Tax Consideration
and Government Programs − Taxation of our Shareholders” above. an Israeli capital gains tax may apply to any gains from
the disposition of our ordinary shares or the Series C Warrants or Series D Warrants by a U.S. Holder. If such Israeli tax applies to
any such gain, a U.S. Holder generally will not be entitled to claim a foreign tax credit for such tax unless the U.S. holder is eligible
for benefits under the Treaty, if such holder is considered a resident of the United States for purposes of the Treaty, and otherwise
meets the requirements for claiming benefits under the Treaty. In lieu of claiming a credit, a U.S. Holder may be able to elect to deduct
the Israeli taxes in computing taxable income, subject to applicable limitations. An election to deduct foreign taxes instead of
claiming foreign tax credits applies to all foreign taxes paid or accrued in the relevant taxable year. U.S. Holders should consult their
own tax advisors regarding the deduction or credit for any such Israeli tax and their eligibility for the benefits of the Treaty.
Additional Taxes
U.S. Holders that are individuals, estates or trusts
and whose income exceeds certain thresholds generally may be subject to a 3.8% Medicare contribution tax on unearned income, including,
without limitation, dividends on, and gains from the sale or other taxable disposition of, our ordinary shares or the Series C Warrants
or Series D Warrants, subject to certain limitations and exceptions. U.S. Holders should consult their own tax advisors regarding the
effect, if any, of such tax on their ownership and disposition of our ordinary shares or the Series C Warrants or Series D Warrants.
Exercise or Lapse of a Series C Warrant or Series D Warrant
Subject to the PFIC rules discussed below, a U.S.
Holder generally will not recognize gain or loss upon the exercise of a Series C Warrant or Series D Warrant for cash. An ordinary share
acquired pursuant to the exercise of a Series C Warrant or Series D Warrant for cash generally will have a tax basis equal to the U.S.
Holder’s tax basis in the Series C Warrant or Series D Warrant, as applicable, increased by the amount paid to exercise the Series
C Warrant or Series D Warrant. The holding period of such ordinary share generally would begin on the day after the date of exercise of
the Series C Warrant or Series D Warrant. If a Series C Warrant or Series D Warrant is allowed to lapse unexercised, a U.S. Holder generally
will recognize a capital loss equal to such holder’s tax basis in the Series C Warrant or Series D Warrant.
The tax consequences of a cashless exercise of
Series C Warrants and Series D Warrants are not clear under current tax law. A cashless exercise may be tax-free, either because it is
not a realization event (i.e., not a transaction in which gain or loss is realized) or because the transaction is treated as a recapitalization
for U.S. federal income tax purposes. In either tax-free situation, a U.S. Holder’s tax basis in the ordinary shares received would
equal the U.S. Holder’s basis in the Series C Warrants or Series D Warrants surrendered. If the cashless exercise were treated as
not being a realization event, the U.S. Holder’s holding period in the ordinary shares could be treated as commencing on the date
following the date of exercise of the Series C Warrants or Series D Warrants. If the cashless exercise were treated as a recapitalization,
the holding period of the ordinary shares received would include the holding period of the Series C Warrants or Series D Warrants, as
applicable.
It is also possible that a cashless exercise could
be treated as a taxable exchange in which gain or loss is recognized. In such event, a U.S. Holder could be deemed to have surrendered
a number of Series C Warrants or Series D Warrants, as applicable, with a fair market value equal to the exercise price for the number
of Series C Warrants or Series D Warrants deemed exercised. For this purpose, the number of Series C Warrants or Series D Warrants deemed
exercised would be equal to the amount needed to receive on exercise the number of ordinary shares issued pursuant to the cashless exercise
of the Series C Warrants or Series D Warrants. In this situation, the U.S. Holder would recognize capital gain or loss in an amount equal
to the difference between the fair market value of the Series C Warrants or Series D Warrants deemed surrendered to pay the exercise price
and the U.S. Holder’s tax basis in such Series C Warrants or Series D Warrants deemed surrendered. Such gain or loss would be long-term
or short-term depending on the U.S. Holder’s holding period in the Series C Warrants or Series D Warrants, as applicable. In this
case, a U.S. Holder’s tax basis in the ordinary shares received would equal the sum of the fair market value of the Series C Warrants
or Series D Warrants deemed surrendered to pay the exercise price and the
U.S. Holder’s tax basis in the Series C Warrants
or Series D Warrants deemed exercised, and a U.S. Holder’s holding period for the ordinary shares should commence on the date following
the date of exercise of the Series C Warrants or Series D Warrants. There also may be alternative characterizations of any such taxable
exchange that would result in similar tax consequences, except that a U.S. Holder’s gain or loss would be short-term.
Due to the absence of authority on the U.S. federal
income tax treatment of a cashless exercise of Series C Warrants and Series D Warrants, it is unclear which, if any, of the alternative
tax consequences and holding periods described above would be adopted by the IRS or a court of law. Accordingly, U.S. Holders should consult
their tax advisors regarding the tax consequences of a cashless exercise of Series C Warrants and Series D Warrants.
Passive Foreign Investment Company Rules
A foreign (i.e., non-U.S.) corporation will be
a PFIC if either (a) at least 75% of its gross income in a taxable year of the foreign corporation, including its pro rata share of the
gross income of any corporation in which it is considered to own at least 25% of the corporation by value, is passive income, or (b) at
least 50% of the average value of its assets in a taxable year of the foreign corporation, including its pro rata share of the assets
of any corporation in which it is considered to own at least 25% of the corporation by value, are held for the production of, or produce,
passive income. Passive income generally includes dividends, interest, rents and royalties (other than certain rents or royalties derived
from the active conduct of a trade or business), and gains from the disposition of passive assets.
We believe that we were a PFIC for the taxable
year ended December 31, 2022 and may be a PFIC for the taxable year ending December 31, 2023. Our PFIC status for our current taxable
year or any subsequent taxable year is uncertain and will not be determinable until after the end of such taxable year. Accordingly, there
can be no assurance with respect to our status as a PFIC for our taxable year ending December 31, 2023 or any subsequent taxable year.
If we are determined to be a PFIC for any taxable
year (or portion thereof) that is included in the holding period of a U.S. Holder of our ordinary shares or Series C Warrants or Series
D Warrants, and, in the case of our ordinary shares, the U.S. Holder did not make a timely qualified electing fund or a QEF, election
for our first taxable year as a PFIC in which the U.S. Holder held (or was deemed to hold) the ordinary shares, a purging election, a
QEF election along with a purging election, or a mark-to-market election, each as described below, such holder generally will be subject
to special rules for regular U.S. federal income tax purposes with respect to:
|
• |
any gain recognized by the U.S. Holder on the sale or other disposition of its ordinary shares or Series
C Warrants or Series D Warrants; and |
|
• |
any “excess distribution” made to the U.S. Holder (generally, any distributions to such U.S.
Holder during a taxable year of the U.S. Holder that are greater than 125% of the average annual distributions received by such U.S. Holder
in respect of the ordinary shares during the three preceding taxable years of such U.S. Holder or, if shorter, such U.S. Holder’s
holding period for the ordinary shares). |
Under these rules,
|
• |
the U.S. Holder’s gain or excess distribution will be allocated ratably over the U.S. Holder’s
holding period for the ordinary shares or Series C Warrants or Series D Warrants; |
|
• |
the amount allocated to the U.S. Holder’s taxable year in which the U.S. Holder recognized the gain
or received the excess distribution or to the period in the U.S. Holder’s holding period before the first day of our first taxable
year in which we qualified as a PFIC will be taxed as ordinary income; |
|
• |
the amount allocated to other taxable years (or portions thereof) of the U.S. Holder and included in its
holding period will be taxed at the highest ordinary tax rate in effect for that year and applicable to the U.S. Holder; and |
|
• |
the interest charge generally applicable to underpayments of tax will be imposed in respect of the tax
attributable to each such other taxable year of the U.S. Holder. |
Although a determination as to our PFIC status
is made annually, an initial determination that we are a PFIC generally will apply for subsequent years to a U.S. Holder that held (or
was deemed to hold) our ordinary shares or Series C Warrants or Series D Warrants while we were a PFIC, whether or not we meet the test
for PFIC status in those subsequent years. If we are determined to be a PFIC in any taxable year, and then cease to meet the test for
PFIC status in a subsequent taxable year, a U.S. Holder may be able to make a purging election to eliminate this continuing PFIC status
with respect to its ordinary shares in certain circumstances. A purging election generally creates a deemed sale of such ordinary shares
at their fair market value on the last day of our tax year during which we qualified as a PFIC (or, in the case of a purging election
made in connection with a QEF election, the first day of our taxable year in which qualify as a QEF with respect to such U.S. Holder).
Any gain recognized by the purging election generally will be treated as an excess distribution subject to the special tax and interest
charge rules described above. As a result of the purging election, the U.S. Holder generally will increase the adjusted basis in its ordinary
shares by the amount of gain recognized and will also have a new holding period in its ordinary shares for purposes of the PFIC rules.
In general, if we are determined to be a PFIC,
a U.S. Holder may avoid the PFIC tax consequences described above with respect to the ordinary shares by making a timely QEF election
(or a QEF election along with a purging election). Pursuant to the QEF election, a U.S. Holder generally will be required to include in
income its pro rata share of our net capital gains (as long term capital gain) and other earnings and profits (as ordinary income), on
a current basis, in each case whether or not distributed, in the taxable year of the U.S. Holder in which or with which our taxable year
ends if we are treated as a PFIC for that taxable year. However, a U.S. Holder may make a QEF election only if we agree to provide certain
tax information to such holder annually. At this time, we do not intend to provide U.S. Holders with such information as may be required
to make a QEF election effective. In any event, a QEF election may not be made with respect to warrant.
Alternatively, if a U.S. Holder, at the close of
its taxable year, owns ordinary shares in a PFIC that are treated as marketable stock, the U.S. Holder may make a mark-to-market election
with respect to such ordinary shares for such taxable year. If the U.S. Holder makes a valid mark-to-market election for the first taxable
year of the U.S. Holder in which the U.S. Holder holds (or is deemed to hold) the ordinary shares and for which we are determined to be
a PFIC, such holder generally will not be subject to the PFIC rules described above with respect to its ordinary shares as long as such
shares continue to be treated as marketable stock. Instead, in general, the U.S. Holder will include as ordinary income for each year
that we are treated as a PFIC the excess, if any, of the fair market value of its ordinary shares at the end of its taxable year over
the adjusted tax basis in its ordinary shares. The U.S. Holder also will be allowed to take an ordinary loss in respect of the excess,
if any, of the adjusted tax basis of its ordinary shares over the fair market value of its ordinary shares at the end of its taxable year
(but only to the extent of the net amount of previously included income as a result of the mark-to-market election). The U.S. Holder’s
adjusted tax basis in its ordinary shares will be adjusted to reflect any such income or loss amounts, and any further gain recognized
on a sale or other taxable disposition of the ordinary shares in a taxable year in which we are treated as a PFIC generally will be treated
as ordinary income. Special tax rules may also apply if a U.S. Holder makes a mark-to-market election for a taxable year after the first
taxable year in which the U.S. Holder holds (or is deemed to hold) our ordinary shares and for which we are determined to be a PFIC. Currently,
a mark-to-market election may not be made with respect to warrants.
The mark-to-market election is available only for
stock that is regularly traded on a national securities exchange that is registered with the U.S. Securities and Exchange Commission,
including the Nasdaq Capital Market, or on a foreign exchange or market that is regulated or supervised by a governmental authority of
the country in which the exchange or market is located and which (A) meets certain requirements, that are enforced by law, relating to
trading volume, listing, financial disclosure, surveillance and other requirements that are designed to (i) prevent fraudulent and manipulative
acts and practices, (ii) remove impediments to and perfect the mechanism of a free and open, fair and orderly market and (iii) protect
investors and (B) has rules that effectively promote the active trading of listed stock. Although our ordinary shares are currently listed
and traded on the Nasdaq Capital Market, U.S. Holders nevertheless should consult their own tax advisors regarding the availability and
tax consequences of a mark-to-market election with respect to our ordinary shares under their particular circumstances.
If we are a PFIC and, at any time, have a foreign
subsidiary that is classified as a PFIC, a U.S. Holder of our ordinary shares generally should be deemed to own a portion of the shares
of such lower-tier PFIC, and generally could incur liability for the deferred tax and interest charge described above if we receive a
distribution from, or dispose of all or part of our interest in, or the
U.S. Holder were otherwise deemed to have disposed
of an interest in, the lower-tier PFIC. A mark-to-market election generally would not be available with respect to such a lower-tier PFIC.
U.S. Holders are urged to consult their own tax advisors regarding the tax issues raised by lower-tier PFICs.
A U.S. Holder that owns (or is deemed to own) ordinary
shares in a PFIC during any taxable year of the U.S. Holder generally is required to file an IRS Form 8621 (whether or not a mark-to-market
election is or has been made) with such U.S. Holder’s U.S. federal income tax return and provide such other information as may be
required by the U.S. Treasury Department. Failure to file IRS Form 8621 for each applicable taxable year may result in substantial penalties
and the statute of limitations on the assessment and collection of U.S. federal income taxes of such U.S. Holder for the related taxable
year may not close until three years after the date on which the required information is filed.
The rules dealing with PFICs and purging and mark-to-market
elections are very complex and are affected by various factors in addition to those described above. Accordingly, U.S. Holders of our
securities should consult their own tax advisors concerning the application of the PFIC rules to our securities under their particular
circumstances.
Non-U.S. Holders
Cash dividends paid or deemed paid to a Non-U.S.
Holder with respect to our ordinary shares generally will not be subject to U.S. federal income tax unless such dividends are effectively
connected with the Non-U.S. Holder’s conduct of a trade or business within the United States (and, if required by an applicable
income tax treaty, are attributable to a permanent establishment or fixed base that such holder maintains or maintained in the United
States).
In addition, a Non-U.S. Holder generally will not
be subject to U.S. federal income tax on any gain attributable to a sale or other taxable disposition of our securities unless such gain
is effectively connected with its conduct of a trade or business in the United States (and, if required by an applicable income tax treaty,
is attributable to a permanent establishment or fixed base that such holder maintains or maintained in the United States) or the Non-U.S.
Holder is an individual who is present in the United States for 183 days or more in the taxable year of such sale or other disposition
and certain other conditions are met (in which case, such gain from U.S. sources generally is subject to U.S. federal income tax at a
30% rate or a lower applicable tax treaty rate).
Dividends and gains that are effectively connected
with the Non-U.S. Holder’s conduct of a trade or business in the United States (and, if required by an applicable income tax treaty,
are attributable to a permanent establishment or fixed base that such holder maintains or maintained in the United States) generally will
be subject to regular U.S. federal income tax at the same regular U.S. federal income tax rates as applicable to a comparable U.S. Holder
and, in the case of a Non-U.S. Holder that is a corporation for U.S. federal income tax purposes, may also be subject to an additional
branch profits tax at a 30% rate or a lower applicable tax treaty rate.
Backup Withholding and Information Reporting
In general, information reporting for U.S. federal
income tax purposes should apply to distributions made on our securities within the United States to a U.S. Holder (other than an exempt
recipient) and to the proceeds from sales and other dispositions of our securities by a U.S. Holder (other than an exempt recipient) to
or through a U.S. office of a broker. Payments made (and sales and other dispositions effected at an office) outside the United States
will be subject to information reporting in limited circumstances. In addition, certain information concerning a U.S. Holder’s adjusted
tax basis in its securities and adjustments to that tax basis and whether any gain or loss with respect to such securities is long term
or short term also may be required to be reported to the IRS, and certain holders may be required to file an IRS Form 8938 (Statement
of Specified Foreign Financial Assets) to report their interest in our securities.
Moreover, backup withholding of U.S. federal income
tax, currently at a rate of 24%, generally will apply to dividends paid on our securities to a U.S. Holder (other than an exempt recipient)
and the proceeds from sales and other dispositions of our securities by a U.S. Holder (other than an exempt recipient), in each case who:
|
• |
fails to provide an accurate taxpayer identification number; |
|
• |
is notified by the IRS that backup withholding is required; or |
|
• |
in certain circumstances, fails to comply with applicable certification requirements. |
A Non-U.S. Holder generally may eliminate the requirement
for information reporting and backup withholding by providing certification of its foreign status, under penalties of perjury, on a duly
executed applicable IRS Form W-8 or by otherwise establishing an exemption.
Backup withholding is not an additional tax. Rather,
the amount of any backup withholding will be allowed as a credit against a U.S. Holder’s or a Non-U.S. Holder’s U.S. federal
income tax liability and may entitle such holder to a refund, provided that certain required information is timely furnished to the IRS.
Holders are urged to consult their own tax advisors
regarding information reporting, the application of backup withholding, and the availability of and procedures for obtaining an exemption
from backup withholding in their particular circumstances.
THE DISCUSSION ABOVE
IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PROSPECTIVE INVESTOR. EACH PROSPECTIVE INVESTOR
IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES RELATING TO THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR ORDINARY
SHARES, SERIES C WARRANTS OR SERIES D WARRANTS, IN EACH CASE IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES, INCLUDING THE CONSEQUENCES
OF ANY PROPOSED CHANGE IN APPLICABLE LAWS.
F. Dividends
and paying agents
Not applicable.
G. Statement
by experts
Not applicable.
H. Documents
on display
We are subject to the informational requirements
of the Exchange Act. Accordingly, we are required to file reports and other information with the SEC, including annual reports on Form
20-F and reports on Form 6-K. As a foreign private issuer, we are exempt under the Exchange Act from, among other things, the rules prescribing
the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and
short swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange
Act to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are
registered under the Exchange Act. The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements
and other information regarding registrants like us that file electronically with the SEC. You can also inspect this Annual Report on
such website.
A copy of each document (or a translation thereof
to the extent not in English) concerning our company that is referred to in this Annual Report is available for public view (subject to
confidential treatment of certain agreements pursuant to applicable law) at our principal executive offices at Check-Cap Building, 29
Abba Hushi Avenue, P.O. Box 1271, Isfiya, 3009000, Israel.
I. Subsidiary
Information
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURE
ABOUT MARKET RISK
Foreign Currency Exchange Rate Risk
Some of our assets and liabilities are affected
by fluctuations in the exchange rate between the U.S. dollar and the NIS and between the U.S. dollar and the Euro. For example, salaries
and related expenses for Israeli employees are paid in NIS and some of our suppliers are located in Europe and require payment in Euros.
During 2022, we entered into zero-cost collars
or minimal cost collars contracts to hedge against the risk of changes in exchange rates on future cash flow from payments denominated
in NIS. As of December 31, 2022, we had outstanding foreign exchange collars in the notional amount of approximately $5.3 million with
a fair value liability net of $56.
As of December 31, 2022, our total assets and liabilities
(including leases) linked to the NIS amounted to approximately $1.3 million and $3.3 million, respectively. A 10% depreciation of the
dollar in relation to the NIS would cause an exchange rate loss of approximately $180,000.
As of December 31, 2022, we had no assets linked
to the Euro and our total liabilities linked to the Euro amounted to approximately $117,000. A 10% depreciation of the dollar in relation
to the Euro would cause an exchange rate gain of approximately $11,000.
During the year ended December 31, 2022, the exchange
rate between the U.S. dollar and the NIS increased by 13.15 % and the exchange rate between the U.S. dollar and the Euro increased by
6.12 %. During the year ended December 31, 2021, the exchange rate between the U.S. dollar and the NIS decreased by 3.27% and the exchange
rate between the U.S. dollar and the Euro increased by 8.39%. During the year ended December 31, 2020, the exchange rate between the U.S.
dollar and the NIS decreased by 6.97% and the exchange rate between the U.S. dollar and the Euro decreased by 8.53%.
Interest Rate Risk
Our obligation to pay royalties to the IIA is linked
to LIBOR and we are therefore exposed to changes in LIBOR.
The primary objective of our investment activities
is to preserve principal while maximizing the interest income we receive from our investments, without increasing risk. Currently, we
do not have any outstanding borrowings.
In addition, we invest and intend to invest our
cash balances, including certain of the net proceeds from our financings pending their ultimate use, primarily in bank deposits, and fixed-income
securities issued by corporations and securities issued by the U.S. and Israeli governments. We are exposed to market risks resulting
from changes in interest rates relating primarily to our financial investments in cash and deposits. We do not use derivative financial
instruments to limit exposure to interest rate risk. Our interest income increased in 2022 mainly due to the increase of the interest
rates as a result of changes in the financial markets especially in the U.S and the increase in our cash amounts. Our interest rates
may decline in the future as a result of changes in the financial markets. The effect of a hypothetical 10% change in interest rates would
have impacted our interest income by approximately $100,000 for the year ended December 31, 2022. The impact would have been
immaterial for the years ended December 31, 2021 and 2020.
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN
EQUITY SECURITIES
Not applicable.
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
Not applicable.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS
OF SECURITY HOLDERS AND USE OF PROCEEDS
Not applicable.
ITEM 15. CONTROLS AND PROCEDURES
(a) Disclosure
Controls and Procedures. Our management, including our Chief Executive Officer and Chief
Financial Officer have evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e)
and 15d-15(e) of the Exchange Act) as of the end of the period covered by this Annual Report, or the Evaluation Date. Based on such evaluation,
the Chief Executive Officer and the Chief Financial Officer have concluded that, as of the Evaluation Date, the Company’s disclosure
controls and procedures are effective.
(b) Management’s
Annual Report on Internal Control Over Financial Reporting. Our management is responsible
for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f)
of the Exchange Act. Our management, including our Chief Executive Officer and our Chief Financial Officer, conducted an evaluation of
the effectiveness of our internal control over financial reporting based on the framework and criteria established in Internal Control—Integrated
Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) as of the Evaluation Date. Based
on that evaluation, our management has concluded that our internal control over financial reporting was effective as of the Evaluation
Date. As all internal control systems, no matter how well designed, have inherent limitations, our internal control over financial reporting
may not prevent or detect misstatements.
(c) Attestation
Report of Registered Public Accounting Firm. Not applicable (we are exempt from this requirement due to our status under
the Exchange Act as a non-accelerated filer as of the current time).
(d) Changes
in Internal Control Over Financial Reporting. Based on the evaluation conducted by our Chief Executive Officer and our Chief Financial
Officer pursuant to Rules 13a-15(d) and 15d-15(d) under the Exchange Act, our management has concluded that there was no change in our
internal control over financial reporting that occurred during the year ended December 31, 2022 that materially affected, or is reasonably
likely to materially affect, our internal control over financial reporting.
ITEM 16. [RESERVED]
ITEM 16A. Audit Committee Financial Expert.
Our Board of Directors
has determined that Mr. Yuval Yanai, a member of our audit committee is an audit committee financial expert as defined by rules of the
U.S. Securities and Exchange Commission and is an independent director under Nasdaq Listing Rules.
ITEM 16B. Code of Ethics.
We have adopted a Code of Business Conduct and
Ethics that applies to all of our directors and employees, including our chief executive officer, chief financial officer, controller
or principal accounting officer, or other persons performing similar functions, which complies with the “code of ethics” contemplated
by Item 16B of Form 20-F promulgated by the U.S. Securities and Exchange Commission. A copy of our Code of Business Conduct and Ethics
is available on our website at http://ir.check-cap.com/corporate-governance.
Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report and is not incorporated
by reference herein. If we make any amendment to the Code of Business Conduct and Ethics or grant any waivers, including any implicit
waiver, from a provision of the code of ethics, we will disclose the nature of such amendment or waiver on our website to the extent required
by the rules and regulations of the U.S. Securities and Exchange Commission. Under Item 16B of the U.S. Securities and Exchange Commission’s
Form 20-F, if a waiver or amendment of the Code of Business Conduct and Ethics applies to our principal executive officer, principal financial
officer, principal accounting officer or controller and relates to standards promoting any of the values described in Item 16B(b) of Form
20-F, we will disclose such waiver or amendment on our website in accordance with the requirements of Instruction 4 to such Item 16B.
ITEM 16C. Principal Accountant Fees and Services.
The following table represents aggregate fees billed
to us for professional services rendered for fiscal years ended December 31, 2022 and 2021 by Deloitte Brightman Almagor Zohar & Co.,
a Firm in the Deloitte Global Network, an independent registered accounting firm.
| |
|
|
|
|
|
|
|
Audit Fees (1)
|
|
$ |
110,000 |
|
|
$ |
140,000 |
|
|
Tax Fees (2)
|
|
$ |
15,264 |
|
|
$ |
5,060 |
|
|
Other Fees (3)
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
| (1) |
The audit fees for the years ended December 31, 2022 and 2021 were for professional services rendered for the audits of our financial
statements, consents and in connection with certain of our filings with the U.S. Securities and Exchange Commission. |
| (2) |
Tax fees for the year ended December 31, 2022, are for services rendered by our auditors in connection with a tax audit and tax study.
Tax fees for the year ended December 31, 2021 are for services rendered by our auditors in connection with a tax audit. |
| (3) |
Other fees for the year ended December 31, 2022 and 2021, are for services rendered by our auditors in connection with a benchmark
study. |
Audit Committee Pre-approval Policies and Procedures
Our audit committee has adopted a pre-approval
policy for the engagement of our independent registered public accounting firm to perform certain audit and non-audit services. Pursuant
to this policy, which is designed to assure that such engagements do not impair the independence of our auditors, the audit committee
pre-approves annually a catalog of specific audit and non-audit services in the categories of audit services, audit-related services and
tax services that may be performed by our independent registered public accounting firm. If a type of service, that is to be provided
by our auditors, has not received such general pre-approval, it will require specific pre-approval by our audit committee. The policy
prohibits retention of the independent registered public accounting firm to perform the prohibited non-audit functions defined in applicable
SEC rules.
ITEM 16D. Exemptions from the Listing Standards
for Audit Committees.
Not applicable.
ITEM 16E. Purchases of Equity Securities by the
Issuer and Affiliated Purchasers.
Not applicable.
ITEM 16F. Change in Registrant’s Certifying
Accountant.
Not applicable.
ITEM 16G. Corporate governance.
Under the Israeli Companies Law, companies incorporated
under the laws of the State of Israel whose shares are publicly traded, including companies with shares listed on the Nasdaq Capital Market,
are considered public companies under Israeli law and are required to comply with various corporate governance requirements relating to
such matters as external directors, the audit committee, the compensation committee and an internal auditor. These requirements are in
addition to the corporate governance requirements imposed by the Listing Rules of the Nasdaq Stock Market and other applicable provisions
of U.S. securities laws to which we became subject (as a foreign private issuer) upon the closing of our initial public offering and the
listing of our securities on the Nasdaq Capital Market. Under the Listing Rules of the Nasdaq Stock Market, a foreign private issuer,
such as us, may generally follow its home country rules of corporate governance in lieu of the comparable requirements of the Listing
Rules of the Nasdaq Stock Market, except for certain matters including (among others) the composition and responsibilities of the audit
committee and the independence of its members within the meaning of the rules and regulations of the U.S. Securities and Exchange Commission.
We currently rely on this “home country practice exemption” solely with respect to the following items:
|
• |
Nomination of our directors. Israeli law and
our amended articles of association do not require director nominations to be made by a nominating committee of our board of directors
consisting solely of independent directors, as required under the Listing Rules of the Nasdaq Stock Market. Our Board of Directors elected
to rely on the exemption available to foreign private issuers under the Nasdaq Listing Rules and follow Israeli law and practice with
regard to the process of nominating directors, in accordance with which directors are recommended by the board of directors for election
by the shareholders (other than directors elected by the board of directors to fill a vacancy). Our Board of Directors established a Nominating
Committee, whose role is to select and recommend to the Board of Directors for selection, director nominees, while considering the appropriate
size and composition of the Board of Directors, the requirements of applicable law regarding service as a member of our Board of Directors
and the criteria for the selection of new members of the Board of Directors, and determined that our Nominating Committee need not be
composed solely of independent directors (within the meaning of Nasdaq Listing Rules). However, our Nominating Committee currently
consists solely of independent directors (within the meaning of Nasdaq Listing Rules). |
|
• |
Compensation of officers. We follow Israeli
law and practice with respect to the approval of officer compensation. While our compensation committee currently complies with the provisions
of the Nasdaq Listing Rules relating to composition requirements and Israeli law generally requires that the compensation of the chief
executive officer and all other executive officers be approved, or recommended to the board for approval, by the compensation committee
(and in certain instances, shareholder approval is required), Israeli law includes relief from compensation committee approval in certain
instances. For details regarding the approvals required under the Israeli Companies Law and regulation promulgated thereunder for the
approval of compensation of the chief executive officer, all other executive officers and directors, see Item 6C “Directors, Senior
Management and Employees— Board Practices — Approval of Related Party Transactions under Israeli Law — Disclosure of
Personal Interests of an Office Holder and Approval of Certain Transactions”. |
|
• |
Shareholder approval. We will seek shareholder
approval for all corporate actions requiring such approval under the requirements of the Israeli Companies Law, rather than seeking approval
for corporate actions in accordance with Nasdaq Listing Rule 5635. In particular, under the Nasdaq Listing Rule, shareholder approval
is generally required for: (i) an acquisition of shares/assets of another company that involves the issuance of 20% or more of the acquirer’s
shares or voting rights or if a director, officer or 5% shareholder has greater than a 5% interest (or such persons collectively have
a 10% or greater interest) in the target company or the assets to be acquired or the consideration to be received and the present or potential
issuance of ordinary shares, or securities convertible into or exercisable for ordinary shares, could result in an increase in outstanding
common shares or voting power of 5% or more; (ii) the issuance of shares leading to a change of control; (iii) adoption/amendment of a
stock option or purchase plan or other equity compensation arrangements, pursuant to which stock may be acquired by officers, directors,
employees or consultants (with certain limited exception); and (iv) issuances of 20% or more of the shares or voting rights (including
securities convertible into, or exercisable for, equity) of a listed company via a private placement (and/or via sales by directors/officers/5%
shareholders) if such equity is issued (or sold) at below the greater of the book or market value of shares. We will seek shareholder
approval for all actions requiring such under the Israeli Companies Law. Under the Israeli Companies Law, the adoption of, and material
changes to, equity-based compensation plans generally require the approval of the board of directors. For details regarding the approvals
required under the Israeli Companies Law for the approval of compensation of the chief executive officer, all other executive officers
and directors, see “Item 6C “Directors, Senior Management and Employees — Board Practices -Approval of Related Party
Transactions under Israeli Law — Disclosure of Personal Interests of an Office Holder and Approval of Certain Transactions.”
For details regarding the approvals required under the Israeli Companies Law for the approval of transactions with and compensation of
controlling shareholders, see “Item 6C “Directors, Senior Management and Employees — Board Practices - Approval of Related
Party Transactions under Israeli Law — Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions.”
For details regarding the approvals required under the Israeli Companies Law for certain acquisitions of our shares and mergers, see Exhibit
2.1. “Description of Securities — Acquisitions under Israeli Law.” |
|
• |
Quorum requirement. Under our amended and
restated articles of association and as permitted under the Israeli Companies Law, a quorum for any meeting of shareholders shall be the
presence of at least two shareholders present in person, by proxy or by a written ballot, who hold at least 25% of the voting power of
our shares (or if a higher percentage is required by law, such higher percentage) instead of 33 1/3% of the issued share capital required
under the Nasdaq Listing Rules. If the meeting was adjourned for lack of a quorum, at the adjourned meeting, at least two shareholders
present in person or by proxy shall constitute a quorum, unless the meeting of shareholders was convened at the demand of shareholders,
in which case, the quorum shall be the presence of one or more shareholders holding at least 5% of our issued share capital and at least
one percent of the voting power of our shares, or one or more shareholders with at least 5% of the voting power of our shares. |
Except as stated above, we currently intend to
comply with the rules generally applicable to U.S. domestic companies listed on Nasdaq. We may in the future decide to use the foreign
private issuer exemption with respect to some or all of the other Nasdaq corporate governance rules. Following our home country governance
practices, as opposed to the requirements that would otherwise apply to a company listed on Nasdaq, may provide less protection than is
accorded to investors under Nasdaq listing requirements applicable to domestic issuers. For more information, see “Item 3D “Key
Information - Risk Factors Risks Related to the Company”- As a foreign private issuer, we are permitted, and intend, to follow certain
home country corporate governance practices instead of otherwise applicable Nasdaq requirements, which may result in less protection than
is accorded to investors under rules applicable to domestic U.S. issuers.”
ITEM 16H. Mine Safety Disclosure.
Not applicable.
ITEM 16I. Disclosure Regarding Foreign Jurisdictions
that Prevent Inspections.
Not applicable.
PART III
ITEM 17. FINANCIAL STATEMENTS
Not applicable.
ITEM 18. FINANCIAL STATEMENTS
Our Financial Statements beginning on pages F-1 through F- 37 are hereby incorporated
herein by reference. These Financial Statements are filed as part of this Annual Report.
ITEM 19. EXHIBITS
Exhibit Index
101.SCH Inline XBRL Taxonomy Extension
Schema Document
101.CAL Inline
XBRL Taxonomy Extension Calculation Linkbase Document
101.DEF Inline
XBRL Taxonomy Extension Definition Linkbase
101.LAB Inline
XBRL Taxonomy Extension Label Linkbase
101.PRE Inline
XBRL Taxonomy Extension Presentation Linkbase
|
(1) |
Incorporated by reference to the Registration Statement on Form F-1 of the Registrant (File No. 333-201250). |
|
(2) |
Incorporated by reference to Annex B of Exhibit
99.3 to the Form 6-K filed by the Registrant with the Form 6-K filed by the Registrant with the Securities Exchange Commission on July
6, 2015. |
|
(3) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on August 12, 2016.
|
|
(4) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on June 24, 2015.
|
|
(5) |
Incorporated by reference to the Registration Statement on Form F-1/A by the Registrant with the Securities and Exchange Commission
on April 25, 2018. |
|
(6) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on May 4, 2018.
|
|
(7) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on February 6, 2019.
|
|
(8) |
Incorporated by reference to the Annual Report on Form 20-F filed by the Registrant with the SEC on March 6, 2020. |
|
(9) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on April 22, 2020.
|
|
(10) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on May 4, 2020.
|
|
(11) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on May 12, 2020.
|
|
(12) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on July 24, 2020.
|
|
(13) |
Incorporated by reference to the Registration Statement on Form F-1 by the Registrant with the SEC on May 20, 2020. |
|
(14) |
Incorporated by reference to the Annual Report on Form 20-F filed by the Registrant with the Securities and Exchange Commission on
March 15, 2016. |
|
(15) |
Incorporated by reference to the Annex A of Exhibit
99.1 to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on October 11, 2020. |
|
(16) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on July 2, 2021.
|
|
(17) |
Incorporated by reference to the Form 6-K filed by the Registrant with the Securities and Exchange Commission on March 3, 2022.
|
|
(18) |
Incorporated by reference to the Annual Report on Form 20-F filed by the Registrant with the SEC on March 18, 2021. |
|
(19) |
Incorporated by reference to the Annual Report on Form 20-F filed by the Registrant with the SEC on April
6, 2022. |
|
(20) |
Incorporated by reference to the Registration Statement
on Form F-1/A by the Registrant with the Securities and Exchange Commission on February 17, 2015. |
|
(21) |
Incorporated by reference to the Form 6-K filed by
the Registrant with the Securities and Exchange Commission on November 22, 2017. |
|
(22) |
Incorporated by reference to the Annual Report on
Form 20-F filed by the Registrant with the SEC on March 15, 2016. |
SIGNATURES
The Registrant hereby certifies that it meets all of the requirements for filing on
Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
|
Date: March 31, 2023
|
CHECK-CAP LTD.
By: /s/
Alex Ovadia
Name: Alex Ovadia
Title: Chief Executive Officer (Principal Executive
Officer)
By: /s/
Mira Rosenzweig
Name: Mira Rosenzweig
Title: Chief Financial Officer (Principal Financial
Officer and Principal Accounting Officer) |
79