
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For
the quarterly period ended
OR
For the transition period from to
Commission
File No.
(Exact name of registrant as specified in its charter)
| No.
| ||
| (State of incorporation) | (IRS Employer Identification No.) |
(Address of principal executive offices, including zip code)
Registrant’s
telephone number, including area code:
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| The
|
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒ NO ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒ NO ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
| ☒ | Smaller reporting company | |||||
| Emerging growth company |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ☐
On November 1, 2021 there were shares of the registrant’s common stock, $0.001 par value per share, outstanding.
Celcuity Inc.
Table of Contents
As used in this report, the terms “we,” “us,” “our,” “Celcuity,” and the “Company” mean Celcuity Inc., unless the context indicates another meaning.
| 2 |
PART I. FINANCIAL INFORMATION
ITEM 1. Financial Statements
Celcuity Inc.
Condensed Balance Sheets
| September 30, 2021 | December 31, 2020 | |||||||
| (unaudited) | ||||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Deposits | ||||||||
| Deferred transaction costs | ||||||||
| Payroll tax receivable | ||||||||
| Prepaid assets | ||||||||
| Total current assets | ||||||||
| Property and equipment, net | ||||||||
| Operating lease right-of-use assets | ||||||||
| Total Assets | $ | $ | ||||||
| Liabilities and Stockholders’ Equity: | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Finance lease liabilities | ||||||||
| Operating lease liabilities | ||||||||
| Accrued expenses | ||||||||
| Total current liabilities | ||||||||
| Finance lease liabilities | ||||||||
| Operating lease liabilities | ||||||||
| Note payable, non-current | - | |||||||
| Total Liabilities | ||||||||
| Stockholders’ Equity: | ||||||||
| Preferred stock, $ par value: shares authorized; shares issued and outstanding as of September 30, 2021 and December 31, 2020 | - | - | ||||||
| Common stock, $ par value: shares authorized; and shares issued and outstanding as of September 30, 2021 and December 31, 2020, respectively | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ Equity | ||||||||
| Total Liabilities and Stockholders’ Equity | $ | $ | ||||||
See accompanying notes to the financial statements
| 3 |
Celcuity Inc.
Condensed Statements of Operations
(unaudited)
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||
| 2021 | 2020 | 2021 | 2020 | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | $ | $ | $ | ||||||||||||
| General and administrative | ||||||||||||||||
| Total operating expenses | ||||||||||||||||
| Loss from operations | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Other income (expense) | ||||||||||||||||
| Interest expense | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Interest income | ||||||||||||||||
| Loss on sale of fixed assets | ( | ) | ||||||||||||||
| Other income (expense), net | ( | ) | ( | ) | ||||||||||||
| Net loss before income taxes | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Income tax benefits | ||||||||||||||||
| Net loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | ( | ) | ||||
| Net loss per share, basic and diluted | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | ( | ) | ||||
| Weighted average common shares outstanding, basic and diluted | ||||||||||||||||
See accompanying notes to the financial statements
| 4 |
Celcuity Inc.
Condensed Statements of Changes in Stockholders’ Equity
Three Months and Nine Months Ended September 30, 2021
| Common Stock | Additional | Accumulated | ||||||||||||||||||
| Shares | Amount | Paid-In Capital | Deficit | Total | ||||||||||||||||
| Balance at December 31, 2020 | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Exercise of common stock warrants | ||||||||||||||||||||
| Exercise of common stock options, net of shares withheld for exercise price | ( | ) | ||||||||||||||||||
| Issuance of common stock upon closing of follow-on offering, net of underwriting discounts and offering costs | ||||||||||||||||||||
| Issuance of common stock in an at-the-market (“ATM”) offering | ||||||||||||||||||||
| Issuance costs associated with ATM offering | - | ( | ) | ( | ) | |||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance at March 31, 2021 (unaudited) | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | ||||||||||||||||||||
| Employee stock purchases | ||||||||||||||||||||
| Exercise of common stock options, net of shares withheld for exercise price | ||||||||||||||||||||
| Issuance of common stock warrants, note payable | - | |||||||||||||||||||
| Issuance of common stock, licensing agreement | ||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance at June 30, 2021 (unaudited) | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Exercise of common stock options, net of shares withheld for exercise price | ||||||||||||||||||||
| Issuance of common stock upon closing of follow-on offering, net of underwriting discounts and offering costs | ||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance at September 30, 2021 (unaudited) | $ | $ | $ | ( | ) | $ | ||||||||||||||
See accompanying notes to the financial statements
| 5 |
Celcuity Inc.
Condensed Statements of Changes in Stockholders’ Equity
Three Months and Nine Months Ended September 30, 2020
| Common Stock | Additional | Accumulated | ||||||||||||||||||
| Shares | Amount | Paid-In Capital | Deficit | Total | ||||||||||||||||
| Balance at December 31, 2019 | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance at March 31, 2020 (unaudited) | ( | ) | ||||||||||||||||||
| Stock-based compensation | ||||||||||||||||||||
| Employee stock purchases | ||||||||||||||||||||
| Issuance of common stock in an at-the-market (“ATM”) offering | ||||||||||||||||||||
| Issuance costs associated with ATM offering | - | ( | ) | ( | ) | |||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance at June 30, 2020 (unaudited) | ( | ) | ||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Issuance costs associated with ATM offering | - | ( | ) | ( | ) | |||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance at September 30, 2020 (unaudited) | $ | $ | $ | ( | ) | $ | ||||||||||||||
See accompanying notes to the financial statements
| 6 |
Celcuity Inc.
Condensed Statements of Cash Flows
(unaudited)
| Nine Months Ended September 30, | ||||||||
| 2021 | 2020 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used for operations: | ||||||||
| Depreciation | ||||||||
| Stock-based compensation | ||||||||
| Issuance of common stock, licensing agreement | ||||||||
| Amortization of debt issuance costs and discount | ||||||||
| PIK interest | ||||||||
| Loss on sale of fixed assets | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Payroll tax receivable | ( | ) | ||||||
| Prepaid assets and deposits | ( | ) | ||||||
| Accounts payable | ( | ) | ||||||
| Accrued expenses | ||||||||
| Non-cash operating lease, net | ( | ) | ( | ) | ||||
| Net cash used for operating activities | ( | ) | ( | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchases of property and equipment | ( | ) | ( | ) | ||||
| Proceeds from sale of property and equipment | ||||||||
| Net cash used for investing activities | ( | ) | ( | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from exercise of common stock warrants | ||||||||
| Proceeds from exercise of employee stock options | ||||||||
| Proceeds from employee stock purchases | ||||||||
| Proceeds from follow-on offering, net of underwriting discounts and offering costs | ||||||||
| Proceeds from note payable, net of debt issuance costs and discount of $ | ||||||||
| Gross proceeds from an ATM offering | ||||||||
| Payments for secondary registration statement costs | ( | ) | ( | ) | ||||
| Payments for finance leases | ( | ) | ( | ) | ||||
| Net cash provided by financing activities | ||||||||
| Net change in cash and cash equivalents | ( | ) | ||||||
| Cash and cash equivalents: | ||||||||
| Beginning of period | ||||||||
| End of period | $ | |||||||
| Supplemental disclosures of non-cash investing and financing activities: | ||||||||
| Property and equipment included in accounts payable | $ | $ | ||||||
| Issuance of common stock warrants and final fee recognized as discount to note payable | $ | $ | ||||||
See accompanying notes to the financial statements
| 7 |
CELCUITY INC.
NOTES TO CONDENSED FINANCIAL STATEMENTS (unaudited)
(For the Three and Nine Months Ended September 30, 2021 and 2020)
1. Organization
Nature of Business
Celcuity Inc., a Delaware corporation (the “Company”), is a clinical-stage biotechnology company seeking to extend the lives of cancer patients by pursuing an integrated companion diagnostic (CDx) and therapeutic (Rx) strategy. Our CELsignia companion diagnostic platform is uniquely able to analyze live patient tumor cells to identify new groups of cancer patients likely to benefit from targeted therapies. This enables a CELsignia CDx to support advancement of new indications for already approved targeted therapies. Our therapeutic efforts are focused on in-licensing and developing molecularly targeted therapies that address the same cancer driver our companion diagnostics can identify. By pursuing an integrated companion diagnostic and therapeutic strategy, we believe we are uniquely positioned to achieve our goal of helping cancer patients receive the therapeutic best suited to treat their cancer driver. The Company was co-founded in 2012 by Brian F. Sullivan and Dr. Lance G. Laing and is based in Minnesota. The Company has not generated any revenues to date.
Follow-on Offering
On
July 1, 2021, the Company completed a follow-on offering whereby it sold shares of common stock at a public offering price
of $ per share. The aggregate gross proceeds from the sale of shares in the follow-on offering was approximately $
On
February 26, 2021, the Company completed a follow-on offering whereby it sold shares of common stock (including shares
of common stock in connection with the full exercise of the underwriters’ option to purchase additional shares) at a public offering
price of $ per share. The aggregate gross proceeds from the sale of shares in the follow-on offering, including the sale of shares
pursuant to the full exercise of the underwriters’ option to purchase additional shares, was approximately $
2. Basis of Presentation, Summary of Significant Accounting Policies and Recent Accounting Pronouncements
Basis of Presentation
The accompanying unaudited financial statements include the accounts of the Company and have been prepared in accordance with Article 10 of Regulation S-X promulgated by the Securities and Exchange Commission (“SEC”). Accordingly, as permitted by Article 10, the unaudited financial statements do not include all of the information required by accounting principles generally accepted in the United States (“U.S. GAAP”). The balance sheet at December 31, 2020 was derived from the audited financial statements at that date and does not include all the disclosures required by U.S. GAAP. In the opinion of management, all adjustments which are of a normal recurring nature and necessary for a fair presentation have been reflected in the financial statements. These unaudited condensed financial statements should be read in conjunction with the audited financial statements as of and for the year ended December 31, 2020 and the related footnotes thereto included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2020. Operating results for the three and nine months ended September 30, 2021 are not necessarily indicative of the results to be expected during the remainder of the current year or for any future period.
Accounting Estimates
Management uses estimates and assumptions in preparing these unaudited condensed financial statements in accordance with U.S. GAAP. Those estimates and assumptions affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities, and the reported revenues and expenses. Actual results could differ from those estimates and the difference could be material. Significant items subject to such estimates and assumptions include the valuation of stock-based compensation and prepaid or accrued clinical trial costs.
Risks and Uncertainties
The Company is subject to risks common to companies in the development stage including, but not limited to, dependency on the clinical and commercial success of its diagnostic tests, ability to obtain regulatory approval of its diagnostic tests, the clinical and commercial success of its initial drug product, gedatolisib, the need for substantial additional financing to achieve its goals, uncertainty of broad adoption of its approved products, if any, by physicians and consumers, and significant competition.
Clinical Trial Costs
The Company records prepaid assets or accrued expenses for prepaid or estimated clinical trial costs conducted by third-party service providers, which includes the conduct of preclinical studies and clinical trials. These costs can be a significant component of the Company’s research and development expenses. The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with service agreements with its third-party service providers. The Company makes significant judgments and estimates in determining the accrued liabilities balance in each reporting period. As actual costs become known, the Company adjusts its prepaid assets or accrued expenses. The Company has not experienced any material differences between accrued costs and actual costs incurred. However, the status and timing of actual services performed, number of patients enrolled, and the rate of patient enrollments may vary from the Company’s estimates, resulting in an adjustment to expense in future periods. Changes in these estimates that result in material changes to the Company’s prepaid assets or accrued expenses could materially affect the Company’s results of operations.
| 8 |
Application of New or Revised Accounting Standards
Pursuant to the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), a company constituting an “emerging growth company” is, among other things, entitled to rely upon certain reduced reporting requirements. The Company is an emerging growth company but has irrevocably elected not to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards. As a result, the Company will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for public companies that are not emerging growth companies.
Recently Adopted Accounting Pronouncements
In August 2020, the FASB issued ASU No. 2020-06, Debt - Debt with Conversion and other Options (Subtopic 470-20) and Derivatives and Hedging - Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity (“ASU 2020-06”), which simplifies accounting for convertible instruments by removing major separation models required under current U.S. GAAP. ASU 2020-06 removes certain settlement conditions that are required for equity contracts to qualify for derivative scope exceptions and also simplifies the diluted earnings per share calculation in certain areas. The standard is effective for public business entities, excluding entities eligible to be smaller reporting companies as defined by the SEC, for fiscal years and interim periods within those fiscal years beginning after December 15, 2021. For all other entities, the standard will be effective for fiscal years beginning after December 15, 2023. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020, and adoption must be as of the beginning of the Company’s annual fiscal year. The Company’s early adoption of this accounting standard on April 8, 2021, in conjunction with the closing of a loan agreement, did not have an impact on the Company’s financial statements and related disclosures.
Basic and diluted net loss per common share is determined by dividing net loss attributable to common stockholders by the weighted-average common shares outstanding during the period. For all periods presented, the common shares underlying the options and warrants have been excluded from the calculation because their effect would be anti-dilutive. Therefore, the weighted-average shares outstanding used to calculate both basic and diluted loss per common share are the same.
For the three and nine months ended September 30, 2021 and 2020, potentially dilutive securities excluded from the computations of diluted weighted-average shares outstanding were options to purchase and shares of common stock, respectively, warrants to purchase and shares of common stock, respectively, and and shares of restricted common stock, respectively.
4. Commitments
Operating and Finance Leases
The
Company leases its corporate space in Minneapolis, Minnesota. In September 2017, the Company entered into a non-cancelable operating
lease agreement for building space. The new lease commenced, and the Company moved to the facility in May 2018, in conjunction with the
termination of its then existing lease. Rent expense is recorded on a straight-line basis over the lease term. In July 2020 the Company
signed an amendment to extend this lease through April 30, 2022. The lease amendment provides for monthly rent, real estate taxes and
operating expenses. As a result of the lease amendment, the Company recorded an incremental $
In May 2018, the Company entered into a non-cancelable finance lease agreement for office equipment with a five-year term. The underlying assets are included in furniture and equipment. The lease contains a bargain purchase option at the end of the lease.
When an implicit rate is not provided, the Company uses its incremental borrowing rate based on the information available at the lease commencement date in determining the present value of the lease payments.
| 9 |
Supplemental balance sheet information consisted of the following at September 30, 2021:
| Operating Lease | ||||
| Right-of-use assets | $ | |||
| Operating lease liability | $ | |||
| Less: short term portion | ( | ) | ||
| Long term portion | $ | |||
| Finance Lease | ||||
| Furniture and equipment | $ | |||
| Less: Accumulated depreciation | ( | ) | ||
| Net book value of property and equipment under finance lease | $ | |||
| Finance lease liability | $ | |||
| Less: short term portion | ( | ) | ||
| Long term portion | $ | |||
Maturity analysis under lease agreements consisted of the following as of September 30, 2021:
| Operating Leases | Finance Leases | |||||||
| 2021 | $ | $ | ||||||
| 2022 | ||||||||
| 2023 | ||||||||
| Total minimum lease payments | ||||||||
| Less: Present value discount | ( | ) | ( | ) | ||||
| Less: Amount representing services | ( | ) | ||||||
| Present value of net minimum lease payments | $ | $ | ||||||
Remaining Lease Term | Discount Rate | |||||||
| Weighted Average | ||||||||
| Operating lease | % | |||||||
| Finance lease | % |
Lease costs for the period ended September 30, 2021:
| Three-month Period | Nine-month Period | |||||||
| Operating lease cost | $ | $ | ||||||
| Finance lease cost: | ||||||||
| Amortization | ||||||||
| Interest | ||||||||
| Variable lease cost | ||||||||
| $ | $ |
Supplemental cash flow information related to leases for the period ended September 30, 2021:
| Three-month Period | Nine-month Period | |||||||
| Cash paid for amounts included in operating and finance leases: | ||||||||
| Operating cash outflow from operating leases | $ | $ | ||||||
| Operating cash outflow from finance leases | ||||||||
| Financing cash outflow from finance leases | ||||||||
| $ | $ | |||||||
| 10 |
Clinical Research Studies
The Company enters into contracts in the normal course of business for clinical trials, preclinical studies, manufacturing and other services and products for operating purposes. The Company currently has six Phase II clinical trial agreements in place to evaluate targeted therapies selected with one of our CELsignia tests. The Company also has a license agreement in place with Pfizer to research, develop, manufacture and commercialize gedatolisib. In conjunction with the license agreement, the Company is continuing a Phase I study – B2151009 related to gedatolisib. Timing of milestone payments are uncertain and the contracts generally provide for termination following a certain period after notice, therefore the Company believes that non-cancelable obligations under the agreements are not material.
5. Stockholders’ Equity
On
July 1, 2021, the Company completed a follow-on offering whereby it sold shares of common stock at a public offering price
of $ per share. The offering generated approximately $
On
February 26, 2021, the Company completed a follow-on offering whereby it sold shares of common stock (including shares
of common stock in connection with the full exercise of the underwriters’ option to purchase additional shares) at a public offering
price of $ per share. The aggregate gross proceeds from the sale of shares in the follow-on offering, including the sale of shares
pursuant to the full exercise of the underwriters’ option to purchase additional shares, was approximately $
On June 5, 2020, the Company entered into an At Market Issuance Sales Agreement (the “ATM Agreement”) with B. Riley FBR, Inc. (the “Agent”). Pursuant to the ATM Agreement, the Company was able to offer and sell from time to time, at its option, shares of common stock having an aggregate offering price of up to $, par value $ per share (the “Placement Shares”), through the Agent.
The Placement Shares were registered under the Securities Act of 1933, as amended, pursuant to the Registration Statement on Form S-3 (File No. 333-227466), which was originally filed with the SEC on September 21, 2018 and declared effective by the SEC on October 4, 2018, the base prospectus contained within the Registration Statement, and a prospectus supplement that was filed on June 5, 2020. Sales of the Company’s common stock, if any, under this prospectus supplement were able to be made by any method deemed to be an “at the market offering” as defined in Rule 415 promulgated under the Securities Act of 1933, as amended.
As of the nine months ended September 30, 2021, the Company sold shares of common stock pursuant to the ATM Agreement, at an average selling price of $ per share.
On February 23, 2021, in conjunction with the Company’s follow-on offering, the ATM Agreement was terminated.
| 2021 | 2020 | |||||||||||||||
| Shares | Weighted Average Exercise Price | Shares | Weighted Average Exercise Price | |||||||||||||
| Options outstanding at beginning of year | $ | $ | ||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ( | ) | ||||||||||||||
| Forfeited | ( | ) | ( | ) | ||||||||||||
| Balance at September 30 | $ | $ | ||||||||||||||
| Options exercisable at September 30: | $ | $ | ||||||||||||||
| Weighted Average Grant Date Fair Value for options granted during the period: | $ | $ | ||||||||||||||
| 11 |
| Options Outstanding | Options Exercisable | |||||||||||||||||||||
| Options Outstanding | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price | Aggregate Intrinsic Value | Options Exercisable | Weighted Average Exercise Price | Aggregate Intrinsic Value | ||||||||||||||||
| $ | $ | $ | ||||||||||||||||||||
The
Company recognized stock-based compensation expense for stock options of $ and $ for the three months ended September 30,
2021 and 2020, respectively and $ and $ for the nine months ended September 30, 2021 and 2020, respectively. In May
2020, the Company modified the exercise price on stock option awards to $
| 2021 | 2020 | |||||||
| Risk-free interest rate | % - | % - | ||||||
| Expected volatility | % - | % - | ||||||
| Expected life (years) | to | to | ||||||
| Expected dividend yield | ||||||||
The inputs for the Black-Scholes valuation model require management’s significant assumptions. Prior to the Company’s initial public offering, the price per share of common stock was determined by the Company’s board based on recent prices of common stock sold in private offerings. Subsequent to the initial public offering, the price per share of common stock is determined by using the closing market price on the Nasdaq Capital Market on the grant date. The risk-free interest rates are based on the rate for U.S. Treasury securities at the date of grant with maturity dates approximately equal to the expected life at the grant date. The expected life is based on the simplified method in accordance with the SEC Staff Accounting Bulletin Nos. 107 and 110. The expected volatility is estimated based on historical volatility information of peer companies that are publicly available in combination with the Company’s calculated volatility since being publicly traded.
All assumptions used to calculate the grant date fair value of non-employee options are generally consistent with the assumptions used for options granted to employees. In the event the Company terminates any of its consulting agreements, the unvested options issued in connection with the agreements would also be cancelled.
The
Company had
The
Company initially reserved a maximum of shares of common stock for issuance under the 2017 Amended and Restated Stock Incentive
Plan (the “2017 Plan”). The number of shares reserved for issuance was automatically increased by shares on January
1, 2020 and by shares on January 1, 2021 and will increase automatically on January 1 of each of 2022 through 2027 by the
Total unrecognized compensation cost related to stock options and restricted stock is estimated to be recognized as follows:
| 2021 | $ | |||
| 2022 | ||||
| 2023 | ||||
| 2024 | ||||
| 2025 | ||||
| Total estimated compensation cost to be recognized | $ |
| 12 |
The
Company recognized stock-based compensation expense related to its employee stock purchase plan of $and $for the three months ended September 30, 2021
and 2020, respectively and $and $for the nine months ended September 30, 2021
and 2020, respectively. The Company initially reserved a total of shares for issuance under the employee stock
purchase plan. The number of shares reserved for issuance was automatically increased by shares on January 1, 2020 and shares on January 1, 2021 and will increase automatically
on each subsequent January 1 by the
| Three Months Ended | Nine Months Ended | |||||||||||||||
| September 30, | September 30, | |||||||||||||||
| 2021 | 2020 | 2021 | 2020 | |||||||||||||
| Stock-based compensation expense in operating expenses: | ||||||||||||||||
| Research and development | $ | $ | $ | $ | ||||||||||||
| General and administrative | ||||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||||
7. Debt
On
April 8, 2021, the Company entered into a loan and security agreement (the “Loan Agreement”) with Innovatus Life Sciences
Lending Fund I, LP, a Delaware limited partnership (“Innovatus”) in its capacity as Collateral Agent and sole Lender. The
Lender agreed to loan up to $
The
Company is entitled to make interest-only payments for thirty-six months, or up to forty-eight months if certain conditions are met.
Innovatus
also has the right, at its election, after June 1, 2021 and until the third anniversary of the Loan Agreement, to convert up to
| 13 |
In
connection with the Loan Agreement and the funding of the first tranche of the Term Loans, the Company incurred debt issuance costs of
approximately $
Long-term debt consisted of the following:
| September 30, 2021 | ||||
| Note payable | $ | |||
| Add: PIK interest (added to principal) | ||||
| Add: final fee | ||||
| Less: unamortized debt issuance costs | ( | ) | ||
| Less: unamortized debt discount | ( | ) | ||
| Total long-term debt | $ | |||
Future principal payments, including the final fee, are as follows:
| Years Ending December 31, | ||||
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| Total | $ | |||
8. License Agreement
On
April 8th, the Company entered into a license agreement with Pfizer to research, develop, manufacture and commercialize gedatolisib,
a potent, well-tolerated, reversible dual inhibitor that targets PI3K and mTOR, for the treatment, diagnosis and prevention of all diseases.
The Company paid Pfizer $
The
Company is also required to make milestone payments to Pfizer upon achievement of certain development and commercial milestone events,
up to an aggregate of $
The Company has the right to terminate the license agreement for convenience upon 90 days’ prior written notice. Pfizer may not terminate the agreement for convenience. Either the Company or Pfizer may terminate the license agreement if the other party is in material breach and such breach is not cured within the specified cure period. In addition, either the Company or Pfizer may terminate the license agreement in the event of specified insolvency events involving the other party.
| 14 |
ITEM 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our unaudited condensed financial statements and the related notes appearing under Item 1 of Part I of this Quarterly Report on Form 10-Q (this “Quarterly Report”). Some of the information contained in this discussion and analysis or set forth elsewhere in this Quarterly Report, including information with respect to our plans and strategy for our business and expected financial results, includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” discussed in our Annual Report on Form 10-K for the year ended December 31, 2020, in Exhibit 99.4 to our Current Report on Form 8-K, filed with the SEC on April 8, 2021 and elsewhere in this Quarterly Report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a clinical-stage biotechnology company seeking to extend the lives of cancer patients by pursing an integrated companion diagnostic (CDx) and therapeutic (Rx) strategy that leverages our CELsignia CDx platform. CELsignia is uniquely able to analyze live patient tumor cells to identify new groups of cancer patients likely to benefit from targeted therapies. This enables a CELsignia CDx to support advancement of new indications for already approved targeted therapies. Our therapeutic strategy aims to utilize CELsignia’s unique insights into tumor cell biology to identify, in-license, and develop potential first-in-class or best-in-class targeted therapies that treat the same cancer driver a CELsignia CDx can identify. We believe this integrated CDx and Rx strategy will maximize the impact our CELsignia platform has on the treatment landscape for cancer patients.
The first drug candidate we are developing internally is gedatolisib, a potent, well-tolerated, small molecule dual inhibitor, administered intravenously, that selectively targets all Class 1 isoforms of PI3K and mammalian target of rapamycin (mTOR). In April 2021, we obtained exclusive global development and commercialization rights to gedatolisib under a license agreement with Pfizer, Inc. Our interest in gedatolisib was prompted after we conducted a study of various PI3K targeted therapeutics while developing our CELsignia PI3K Activity test. Our CELsignia platform allows us to obtain proprietary insights about the relative effectiveness of PI3K targeted therapies. This study found that gedatolisib inhibited higher levels of PI3K-involved signaling activity than the other PI3K targeted therapeutics we evaluated and demonstrated superior drug synergy when combined with other targeted therapies. Gedatolisib’s initial clinical development program will focus on the treatment of patients with estrogen receptor positive (ER+), HER2-negative, advanced or metastatic breast cancer. Additional clinical development programs are expected to focus on other tumor types that involve a hormonal signaling pathway, such as endometrial, ovarian, or prostate cancer.
Supporting the development of a potential first-in-class targeted therapy for breast cancer, like gedatolisib, with our CELsignia platform is a natural extension of our strategy to use our CELsignia CDx to enable new indications for other companies’ targeted therapies. By combining companion diagnostics designed to enable proprietary new drug indications with targeted therapies that treat signaling dysregulation our CDx identifies, we believe we are uniquely positioned to improve the standard-of-care for many early and late-stage breast cancer patients. Our goal is to play a key role in the multiple treatment approaches required to treat breast cancer patients at various stages of their disease. With each program, we are:
| ● | Leveraging the proprietary insights CELsignia provides into live patient tumor cell function | |
| ● | Using a CELsignia CDx to identify new patients likely to respond to the paired targeted therapy | |
| ● | Developing a new targeted therapeutic option for breast cancer patients | |
| ● | Maximizing the probability of getting regulatory approval to market the targeted therapy indication |
Therapeutic (Rx) Product Development
Gedatolisib
Gedatolisib (PF-05212384) is a potent, reversible dual inhibitor that selectively targets PI3K and mTOR. Gedatolisib was originally developed by Wyeth and clinical development was continued by Pfizer after it acquired Wyeth. We exclusively licensed global rights to gedatolisib from Pfizer in April 2021. An on-going Phase 1b trial evaluating patients with ER+/HER2- metastatic breast cancer was initiated in 2016 and subsequently enrolled 138 patients. Patient enrollment for the four expansion arms of the trial is complete. Based on the favorable preliminary results reported to date from the Phase 1b trial, we intend to initiate, subject to feedback from the FDA, a Phase 2/3 clinical trial evaluating gedatolisib in combination with palbociclib and an endocrine therapy in patients with ER+/HER2- advanced or metastatic breast cancer in the first half of 2022.
Background
Breast cancer is the most prevalent cancer in women, accounting for 30% of all female cancers and 13% of cancer-related deaths in the United States. The National Cancer Institute estimated that approximately 270,000 new cases of breast cancer would be diagnosed in the United States in 2019, and approximately 42,000 breast cancer patients would die of the disease. Approximately 190,000, or 70%, of these new cases are for ER+/HER2- breast cancer.
| 15 |
Four different breast cancer subtypes are currently identified using molecular tests that determine the level of ER and HER2 expression. About 70% of breast cancers are ER+/HER2-, which is indicative of hormone dependency. Despite progress in treatment strategies, metastatic ER+/HER2- breast cancer (mBC) remains an incurable disease, with a median overall survival (OS) of three years and a five-year survival rate of 25%.
Four different classes of targeted therapies are currently used to treat ER+/HER2- tumors: endocrine-based therapies, CDK4/6 inhibitors, PI3K inhibitors and mTOR inhibitors. Each of the CDK4/6 inhibitors, PI3K inhibitors and mTOR inhibitors are generally used to respond to the related mechanisms of resistance to endocrine therapy, namely, activation of the CDK4/6, PI3K and mTOR pathways. These drugs generated revenues of over $8 billion globally in 2020.
As specifically relates to gedatolisib, activation of the PI3K/mTOR pathway has been implicated in a wide variety of human cancers, involving either activating mutations, or other unknown drivers of pathway amplification. These include cancers of the breast, prostate, endometrial, colon, rectum, and lung, among others.
PI3K constitutes a lipid kinase family involved in the regulation of diverse cellular processes, including cell proliferation, survival, cytoskeletal organization, and glucose transport. Class I PI3Ks are of particular therapeutic interest. They are heterodimers, comprising a catalytic (p110α, p110β, p110δ, or p110γ) and a regulatory (p85α, p55α, p50α, p85β, p55γ, or p101) subunit. Oncogenic PI3K signaling is activated by cell-surface receptors such as receptor tyrosine kinases, G-protein-coupled receptors, and also by well-known oncogenic proteins such as RAS.
Activities associated with PI3K involve complex essential cell regulatory mechanisms including feedforward and feedback signaling loops. Overactivation of the pathway is frequently present in human malignancies and plays a key role in cancer progression. Each of the four catalytic isoforms of class I PI3K preferentially mediate signal transduction and tumor cell survival based on the type of malignancy and the genetic or epigenetic alterations an individual patient harbors. For example, studies have demonstrated the p110α catalytic isoform is necessary for the growth of tumors driven by PIK3CA mutations and/or oncogenic RAS and receptor tyrosine kinases; the p110β catalytic isoform mediates tumorigenesis arising from the loss of the dephosphorylase activity of PTEN; and the p110δ catalytic isoform is highly expressed in leukocytes, making it a desirable target for inhibition in the treatment of hematologic malignancies. Due to the multiple subcellular locations, activities, and importance of the different PI3K complexes in regulating many types of cancer cell proliferation, control of PI3K activity is an important target in cancer therapy.
mTOR is as a critical effector in cell-signaling pathways commonly dysregulated in human cancers. The mTOR signaling pathway integrates both intracellular and extracellular signals and serves as a central regulator of cell metabolism, growth, proliferation, and survival. mTOR is a serine/threonine protein kinase, a downstream effector of PI3K, and regulated by hormones, growth factors, and nutrients, that is contained in two functionally distinct protein assemblies: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 belongs to a complex network of regulatory feedback loops, and once certain levels of activation are reached, is normally responsible for limiting the proliferative signals transmitted by upstream effectors such as PI3K/AKT activity. Equally complex mTORC2 regulates AKT phosphorylation, GSK3β, and control over glycolysis, and participates in organizing the cellular actin cytoskeleton. In addition, mTORC1 activation leads to the direct reduction of mTORC2 activity and mTOR can activate the functional domain of the ER, leading to ligand-independent hormone receptor activation. In cancer, dysfunctional signaling leads to various constitutive activities of mTOR complexes, making mTOR a good therapeutic target.
PI3K/mTOR as Resistance Mechanism to Endocrine and CDK4/6 Inhibitors
The upregulation of the PI3K/AKT/mTOR pathway promotes hormone dependent and independent ER transcriptional activity, which contributes to endocrine resistance, leading to tumor cell growth, survival, motility, and metabolism. It has also been demonstrated in vivo that PI3K and mTOR inhibition can restore sensitivity to endocrine therapy, providing a strong rationale for the combination of the two therapies.
In addition, the PI3K/AKT/mTOR pathway, like other mitogenic pathways, can also promote the activities of cyclin D and CDK4/6 to drive proliferative cell cycling. Internal preclinical studies conducted by Pfizer provided evidence in cell-line xenograft models that the combination of PI3K and CDK4/6 inhibitors may overcome both intrinsic and adaptive resistance to endocrine therapy, leading to tumor regressions. In an MCF7 xenograft model (ER+/HER2-/PIK3CA mutant) the combination of gedatolisib with palbociclib and fulvestrant led to durable tumor regressions. Importantly, tumors regressed to minimal volumes within 20 days of triplet therapy, and continued to remain dormant, without further therapy, for up to 90 days.
| 16 |
Advantages of Gedatolisib over other PI3K and mTOR inhibitors
The important role the PI3K/AKT/mTOR pathway plays in cancer has led to significant investment in the development of many different PI3K and mTOR inhibitors for solid tumors. However, developing efficacious and well-tolerated therapies that target this pathway has been challenging. This reflects the inherent adaptability and complexity of the PI3K pathway, where numerous feedforward and feedback loops, crosstalk with other pathways, and compensatory pathways enable resistance to PI3K inhibition. Another major hurdle for the development of PI3K pathway inhibitors has been the inability to achieve optimal drug-target blockade in tumors while avoiding undue toxicities in patients. These challenges may explain why PI3K and mTOR inhibitors have not yielded the outstanding clinical activity many researchers expected.
We believe there is significant potential for gedatolisib to address previously treated breast cancer tumors and has the potential to be used in other tumor types where the PI3K/AKT/mTOR pathway is either: i) driving tumorigenesis directly; ii) cooperating with other dysregulated signaling pathways; or iii) a mechanism of resistance to other drug therapies.
As a result, we believe gedatolisib’s unique mechanism of action and intravenous formulation offer distinct advantages over currently approved and investigational therapies that target PI3K or mTOR alone or together.
| ● | Overcomes drug resistance that can occur with isoform-specific PI3K inhibitors. | |
| Gedatolisib is a pan-class I isoform PI3K inhibitor with low nanomolar potency for the p110α, p110β, p110γ, and p110δ isoforms. Each isoform is known to preferentially affect different signal transduction events that involve tumor cell survival, depending upon the aberrations associated with the linked pathway. A pan-PI3K inhibitor can thus treat tumors harboring abnormalities that signal through different PI3K isoforms, which would potentially induce anti-tumor activity in a broader population of patients than an isoform-specific PI3K inhibitor. In addition, it has been reported that inhibition of one PI3K isoform may be offset by the increased activity of the other isoforms through different adaptive mechanisms. Inhibiting all four PI3K isoforms, as gedatolisib does, can thus prevent the confounding effect of isoform interaction that may occur with isoform-specific PI3K inhibitors. | ||
| ● | Overcomes paradoxical activation of PI3K induced by mTOR inhibition. | |
| As a potent inhibitor of mTOR, in addition to PI3K, gedatolisib, inhibits the PI3K/AKT/mTOR pathway both upstream and downstream of AKT. Furthermore, it has been demonstrated that the PI3K pathway is activated following selective mTOR inhibition by relief of normal feedback regulatory mechanisms, thus providing a compelling rationale for simultaneous inhibition of PI3K and mTOR. | ||
| ● | Better tolerated by patients than oral PI3K and mTOR drugs. | |
| Gedatolisib is administered intravenously (IV) once weekly or on a four-week cycle of three weeks-on, one week-off, in contrast to the orally administered pan-PI3K or dual PI3K/mTOR inhibitors that are no longer being clinically developed. Oral pan-PI3K or PI3K/mTOR inhibitors have repeatably been found to induce significant side effects that were not well tolerated by patients. This typically leads to a high proportion of patients requiring dose reductions or treatment discontinuation. The challenging toxicity profile of these drug candidates ultimately played a significant role in the decisions to halt their development, despite showing promising efficacy. By contrast, gedatolisib stabilizes at lower concentration levels in plasma compared to orally administered PI3K inhibitors, resulting in less toxicity, while maintaining concentrations sufficient to inhibit PI3K/AKT/mTOR signaling. | ||
| Isoform-specific PI3K inhibitors administered orally were developed to reduce toxicities in patients. While the range of toxicities associated with isoform-specific inhibitors is narrower than oral pan-PI3K or PI3K/mTOR inhibitors, administering them orally on a continuous basis still leads to challenging toxicities. The experience with an FDA approved oral p110-α specific inhibitor, Piqray, illustrates the challenge. In its Phase 3 pivotal trial Piqray was found to induce a Grade 3 or 4 adverse event (AE) related to hyperglycemia in 39% of patients evaluated. In addition, 26% of patients discontinued treatment. By contrast, in the 103-patient dose expansion portion of the Phase 1b clinical trial with gedatolisib, only 7% of patients experienced Grade 3 or 4 hyperglycemia and less than 10% discontinued treatment. |
| 17 |
Clinical Experience with Gedatolisib
As of September 30, 2021, 492 patients with solid tumors have received gedatolisib in eight clinical trials sponsored by Pfizer. Of the 492 patients, 129 were treated with gedatolisib as a single agent in three clinical trials. The remaining 363 patients received gedatolisib in combination with other anti-cancer agents in five clinical trials. Additional patients received gedatolisib in combination with other anti-cancer agents in nine investigator sponsored clinical trials.
Phase 1 First-in-Human Study
Pfizer conducted a Phase 1, open-label, dose-escalation first-in human study of single-agent gedatolisib in patients with advanced solid tumors. The primary objective of Part 1 of the study was to determine the safety, tolerability, and maximum tolerated dose (MTD) of single-agent gedatolisib administered once weekly as an intravenous (IV) infusion. Seventy-seven patients with advanced solid tumors received doses of gedatolisib and the MTD was determined to be 154 mg IV once weekly (n = 42). Subsequent analysis determined that the recommended Phase 2 dose could be increased to 180 mg IV once weekly.
At the MTD, the majority of patients enrolled in the MTD group experienced only grade 1 treatment-related adverse events (AEs). Grade 3 treatment-related adverse events were noted in 23.8% of patients, and the most frequently reported included mucosal inflammation and stomatitis (7.1%), increased alternative lengthening of telomeres (ALT) (7.1%), and increased aspartate aminotransferase (AST) (4.8%). No treatment-related AEs of grade 4 or 5 severity were reported at any dose level.
Phase 1b ER+/HER2- mBC Clinical Trial Results (preliminary)
In 2016, Pfizer initiated a Phase 1b trial dose-finding trial with an expansion portion for safety and efficacy to evaluate gedatolisib when added to either the standard doses of palbociclib plus letrozole or palbociclib plus fulvestrant in patients with ER+/HER2- metastatic breast cancer. PI3K mutation status was not used as an eligibility criterion. Patient enrollment for the trial is complete.
The illustration below depicts how the combination of gedatolisib, palbociclib, and fulvestrant is intended to simultaneously block interdependent ER, PI3K, mTOR & CDK signaling pathways in ER+ breast cancer to address ER and CDKi resistance mechanisms.

A total of 138 patients with ER+/HER2- metastatic breast cancer were dosed in the clinical trial.
| ● | 35 patients were enrolled in two dose escalation arms to evaluate the safety and tolerability and determine the MTD of gedatolisib when used in combination with the standard doses of palbociclib and endocrine therapies. The MTD was determined to be 180 mg administered intravenously once weekly. | |
| ● | 103 patients were enrolled in one of four expansion arms (A, B, C, D) to determine if the triplet combination of gedatolisib plus palbociclib and letrozole or gedatolisib plus palbociclib and fulvestrant produced a superior objective response (OR), compared to historical control data of the doublet combination (palbociclib plus endocrine therapy). All patients received gedatolisib in combination with standard doses of palbociclib and endocrine therapy (either letrozole or fulvestrant). In Arms A, B, and C, patients received an intravenous dose of 180 mg of gedatolisib once weekly. In Arm D, patients received an intravenous dose of 180 mg of gedatolisib on a four-week cycle of three weeks-on, one week-off. Objective response was determined using Response Evaluation Criteria in Solid Tumors v1.0, or RECIST v1.0. |
| 18 |
| ○ | Arm A: mBC with progression and no prior endocrine-based systemic therapy or a CDK4/6 inhibitor in the metastatic setting. First-line endocrine-based therapy for metastatic disease (CDK4/6 treatment naive). | |
| ○ | Arm B: mBC with progression during one or two prior endocrine-based systemic therapy in the metastatic setting, with no prior therapy with any CDK inhibitor. Second- or third-line endocrine-based therapy for metastatic disease. | |
| ○ | Arm C: mBC with progression during one or two prior endocrine-based systemic therapies in the metastatic setting and following prior therapy with a CDK inhibitor. Second- or third-line endocrine-based therapy for metastatic disease. | |
| ○ | Arm D: mBC having progressed on a CDK inhibitor in combination with endocrine therapy as the most recent regimen for metastatic disease. Second- or third-line endocrine-based therapy for metastatic disease. |
A preliminary analysis for the 103 patients enrolled in the expansion portion of the Phase 1b clinical trial, as of the database cutoff date of January 11, 2021, showed:
| ● | Efficacy analysis for all arms in aggregate: |
| ○ | 60% objective response rate (ORR): 53 of the 88 evaluable patients had either a confirmed or unconfirmed partial response, or PR (48 confirmed, 5 unconfirmed). |
| ○ | 75% clinical benefit rate (CBR): 66 of the 88 evaluable patients had either a confirmed PR or had stable disease for 24 weeks. |
| ● | Best responses, as measured by RECIST v1.0, are shown in the following chart. The dotted line represents the cutoff for PR (defined as a 30% reduction from baseline). |
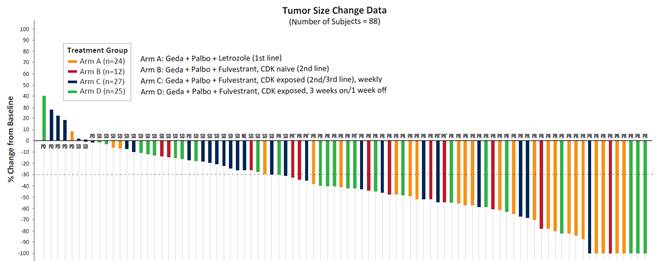
| ● | Preliminary safety analysis: |
| ○ | For all arms in aggregate, all patients experienced at least one Grade 1 or Grade 2 treatment-emergent adverse event. The most commonly reported adverse events regardless of grade and occurring in at least 30% of patients included stomatitis (81%), neutropenia (80%), nausea (75%), fatigue (68%), dysgeusia (46%), vomiting (45%), anemia (40%), diarrhea (34%), decreased appetite (32%), leukopenia (32%). |
| ○ | For all arms in aggregate, the Grade 3 and 4 treatment-emergent adverse events occurring in at least 20% of patients were neutropenia (67%), stomatitis (27%) and rash (20%). Neutropenia is a known class effect of CDK4/6 inhibitors. Stomatitis was reversible in most patients with a steroidal mouth rinse. All grades of treatment-related adverse events related to hyperglycemia was reported in 22% of patients; Grade 3 or 4 hyperglycemia was reported in 7% of patients. Gedatolisib was discontinued in 10% of patients. |
| ○ | For the patients in Arm D, who received the recommended phase two dose, Grade 3 and 4 treatment-emergent adverse events occurring in at least 20% of patients were neutropenia (67%) stomatitis and (22%). All grades of treatment-related adverse events related to hyperglycemia was reported in 22% of patients; Grade 3 or 4 hyperglycemia was reported in 7% of patients. |
| 19 |
| ○ | Gedatolisib was discontinued in 7% of patients. |
| ○ | As of the cutoff date, 22 patients were continuing to receive gedatolisib in combination with the other study drugs, 17 of whom have been on study treatment for more than two years. |
| ● | Preliminary best overall response data for each arm is presented in the table below: |
| Arm (evaluable patients) | A (N=24) | B (N=12) | C (N=27) | D (N=25) | ||||||||||||
| Patients | 1L: CDKi-naïve | 2L+: CDKi-naïve | 2L/3L: CDKi-pretreated | 2L/3L: Immediately prior CDKi | ||||||||||||
| Overall Response Rate (evaluable patients) | 83 | % | 75 | %1 | 33 | %2 | 60 | %3 | ||||||||
| Clinical Benefit Rate (evaluable patients) | 92 | % | 92 | % | 48 | % | 76 | % | ||||||||
1. Arm A: 20 of the 24 evaluable patients had a confirmed PR.
2. Arm B: 9 of the 12 evaluable patients had either a confirmed PR or unconfirmed PR (7 confirmed PR, 2 unconfirmed PR).
3. Arm C: 9 of the 27 evaluable patients had either a confirmed PR or unconfirmed PR (7 confirmed PR, 2 unconfirmed PR).
4. Arm D: 15 of the 25 evaluable patients had either a confirmed PR or unconfirmed PR (14 confirmed PR, 1 unconfirmed PR).
| ● | Preliminary progression free survival (PFS) data for each arm is presented in the table below: |
| Arm (enrolled patients) | A (N=31) | B (N=13) | C (N=32) | D (N=27) | ||||||||||
| Median PFS (months) (95% CI) | >29 (Not Yet Reached) | 11.9 (3.7, NR) | 5.1 (3.4, 7.5) | 13.2 (9.0, 16.7) |
In light of the preliminary results reported to date from the Phase 1b trial, we intend to initiate, subject to feedback from the FDA, a Phase 2/3 clinical trial evaluating gedatolisib in combination with palbociclib and an endocrine therapy in patients with ER+/HER2- advanced or metastatic breast cancer in the first half of 2022.
We expect to use the CELsignia PI3K Activity Test to help support development of gedatolisib for breast cancer indications. Our internal studies demonstrate how measurement of PI3K-involved signaling may provide a sensitive and specific method of identifying patients most likely to benefit from PI3K inhibitors. We believe CELsignia tests uniquely enable us to pursue indications simultaneously for unselected patient populations and CELsignia selected patient sub-groups. This approach can greatly reduce the risk of pursing an indication for a large, but unselected patient population, as we plan to do for the initial gedatolisib indication. By combining the capabilities of CELsignia PI3K Activity Test with a potent pan-PI3K/mTOR inhibitor like gedatolisib, we believe we are uniquely suited to maximize the probability of obtaining regulatory approval to market gedatolisib.
Phase 2 Pilot Clinical Trial for HER2+/PIK3CA+ Patients
The Korean Cancer Study Group sponsored a Phase 2 pilot clinical trial to evaluate gedatolisib combined with a trastuzumab biosimilar (Herzuma®), in patients with HER2+/PIK3CA+ metastatic breast cancers whose disease had progressed after treatment with three or more prior HER2 targeted therapy regimens. The clinical trial commenced in December 2019 and interim efficacy data from the first 16 patients enrolled was presented at the San Antonio Breast Cancer Symposium in December 2020. Patients received a trastuzumab biosimilar (8 mg/kg IV for 1st cycle loading dose, and then 6 mg/kg IV every 3 weeks) plus gedatolisib (180 mg, weekly IV). The primary endpoint was objective response, a reduction of at least 30% in tumor volume by RECIST v1.1.
| 20 |
As of a data cutoff date of October 30, 2020, nine of 16 patients achieved a partial response, an ORR of 56%, and four patients had stable disease. Thirteen of 16 patients thus received either a partial response or stable disease, resulting in a clinical benefit rate of 81%. Best responses are shown in the following chart. The dotted lines represent the cutoff for progressive disease (>20% tumor growth) and for partial response (>30% tumor regression).
Best Response
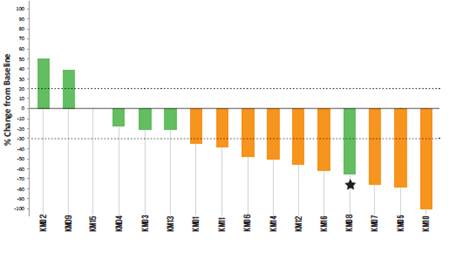
* Patient whose target lesion decreased by 63% but a new leptomeningeal seeding occurred.
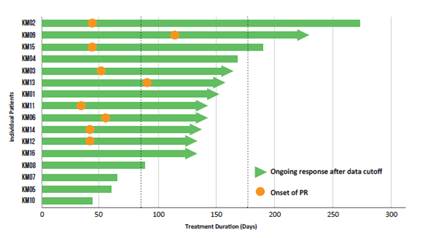
The duration of treatment for the 16 patients evaluated is shown in the chart below. As of the October 30, 2020 data cutoff, 16 patients (80%) remained on therapy. Four patients discontinued treatment, one due to disease progression, one due to an adverse event of Grade 1 diarrhea, one participant decision, and one patient being unable to undergo the required MRI imaging due to a titanium rod implant from non-treatment related worsening of scoliosis. At the time of data cut-off, the median time on treatment for these 20 patients was 10.1 cycles (approximately 10 months) and all 10 patients who had achieved an objective response remained on therapy assessment. At the time of the analysis, nine patients had a continuing response. The dashed lines show the response at 3 months and 6 months.
Duration of Treatment
CELsignia Development and CDx Programs
Our proprietary CELsignia diagnostic platform is the only commercially ready technology we are aware of that uses a patient’s living tumor cells to identify the specific abnormal cellular process driving a patient’s cancer and the targeted therapy that best treats it. This enables us to identify patients whose tumors may respond to a targeted therapy, even though they lack a previously associated molecular mutation. By identifying cancer patients whose tumors lack an associated genetic mutation but have abnormal cellular activity a matching targeted therapeutic is designed to inhibit, CELsignia CDx can expand the markets for a number of already approved targeted therapies. Our current CDx identifies breast and ovarian cancer patients whose tumors have cancer drivers potentially responsive to treatment with human epidermal growth factor receptor 2-negative (HER2), mesenchymal-epithelial transition factor (c-MET), or phosphatidylinositol 3-kinases (PI3K) targeted therapeutics. While U.S. Food and Drug Administration (“FDA”) approval or clearance is not currently required for CELsignia tests offered as a stand-alone laboratory developed test, if we are partnered with a drug company to launch a CELsignia test as a companion diagnostic for a new drug indication, we would be required to obtain premarket approval, or PMA, in conjunction with the pharmaceutical company seeking a new drug approval for the matching therapy.
| 21 |
Our CELsignia platform provides an important advantage over traditional molecular diagnostics. Current molecular diagnostics analyze fragmented cells to obtain a snapshot of the genetic mutations present in a patient’s tumor. Using cell fragments prevents molecular diagnostics from analyzing the dynamic cellular activities, known as cell signaling, that regulate cell proliferation or survival. Cancer can develop when critical cell signaling, regulating physiologic activity such as cell proliferation, becomes abnormal or dysregulated. Since genetic mutations are often only weakly correlated to the dysregulated cell signaling activity driving a patient’s cancer, a molecular diagnostic is prone to providing an incomplete diagnosis. CELsignia tests overcome this limitation by measuring dynamic cell signaling activity in a cancer patient’s living tumor cells. When a CELsignia test detects abnormal signaling activity, a more accurate diagnosis of the patient’s cancer driver is obtained.
We are supporting the advancement of new potential indications for seven different targeted therapies, controlled by other pharmaceutical companies, that would rely on a CELsignia CDx to select patients. Six Phase 2 trials are underway to evaluate the efficacy and safety of these therapies in CELsignia selected patients. These patients are not currently eligible to receive these drugs and are not identifiable with a molecular test.
Our first analytically validated and commercially ready test using our CELsignia platform, the CELsignia HER2 Pathway Activity Test for breast cancer, diagnoses two new sub-types of HER2-negative breast cancer that traditional molecular diagnostics cannot detect. Our internal studies show that approximately 15-20% of HER2-negative breast cancer patients have abnormal HER2 signaling activity similar to levels found in HER2-positive breast cancer cells. As a result, these HER2-negative patients have undiagnosed HER2-driven breast cancer and would be likely to respond to the same anti-HER2 targeted therapies only HER2-positive patients receive today. We have three interventional clinical trials underway to evaluate the efficacy of HER2 targeted therapies in breast cancer patients selected with our CELsignia HER2 Pathway Activity Test.
Our second CELsignia test for breast cancer evaluates independent c-Met signaling activity and its involvement with HER family signaling in HER2-negative breast cancer tumor cells. Our internal studies show that approximately 20%-25% of HER2-negative breast cancer patients have abnormal c-Met signaling activity that is co-activated with abnormal HER family signaling. These studies suggest that this sub-group of HER2-negative breast cancer patients may best respond to treatment with a combination of HER family and c-Met inhibitors. We have two interventional clinical trials underway to evaluate the efficacy of HER2 and c-Met targeted therapies, in previously treated metastatic HER2-negative breast cancer patients selected with our CELsignia Multi-Pathway Activity Test, or CELsignia MP Test.
Our third CELsignia test for breast cancer evaluates PI3K signaling in HER2-negative breast cancer tumor cells. Our internal studies demonstrate how measurement of PI3K-involved signaling may provide a more sensitive and specific method of identifying patients most likely to benefit from PI3K inhibitors than current genetic tests that measure PI3K mutations. We intend to combine these three tests to expand the CELsignia MP Test. With this next generation CELsignia test, we plan to provide an analysis of EGFR/HER1, HER2, HER3, c-MET, and PI3K-node involved signaling activity for each patient tumor specimen received.
We completed development of our first CELsignia test for ovarian cancer in 2020. This test identifies a new sub-group of ovarian cancer patients with tumors that have abnormal c-Met and HER2 signaling activity. These findings suggest that a significant sub-group of ovarian cancer patients may respond to treatment with a combination of ErbB and c-Met inhibitors. Nearly 14,000 women a year die from ovarian cancer, a disease that has less than a 50% five-year survival rate and a limited range of targeted therapy options. There is thus a significant unmet need for additional therapeutic options for ovarian cancer patients. As a companion diagnostic, our CELsignia test for ovarian cancer will be intended to help pharmaceutical companies obtain new drug indications and expand treatment options for this challenging tumor type. We are currently in discussions with pharmaceutical companies about collaborating on future clinical trials.
In addition to our CELsignia tests for HER2-negative breast cancer and ovarian cancer, we expect to develop CELsignia tests to diagnose eight new potential cancer sub-types we have discovered in lung, ovarian, kidney, and bladder cancers. Approved or investigational drugs are currently available to treat these new potential cancer sub-types. We expect to launch these additional tests on a staggered basis over the next few years while continuing our research to identify additional new cancer sub-types.
| 22 |
Our overall commercialization strategy is to develop diagnostics that expand the patient population eligible for targeted therapies. In furtherance of this strategy, we will seek collaborations with pharmaceutical companies to field clinical trials to advance the clinical development of their targeted therapies with the eventual goal of obtaining FDA approval of a new drug indication. Collaborations are expected to involve initially Phase I or Phase II interventional clinical trials to evaluate the efficacy of our collaboration partners’ targeted therapies on patients selected with one of our CELsignia tests. These trials would not be intended to separately evaluate the CELsignia tests, whether as standalone tests or companion diagnostics. While FDA approval or clearance is not currently required for CELsignia tests offered as a stand-alone laboratory developed test, if we are partnered with a drug company to launch a CELsignia test as a companion diagnostic for a new drug indication, we would be required to obtain premarket approval, or PMA, in conjunction with the pharmaceutical company seeking a new drug approval for the matching therapy.
We are currently collaborating on six Phase II clinical trials to evaluate the efficacy of our collaboration partners’ targeted therapies in patients selected with one of our CELsignia tests. The goal of these trials is to support the development of six potential new drug indications to treat patient groups found responsive by our CELsignia test to their approved targeted therapies. These clinical trials include:
| ● | FACT-1 Clinical Trial to Evaluate Efficacy of Genentech’s HER2 Targeted Therapies. We are collaborating with NSABP Foundation, Inc. (“NSABP”) and Genentech, Inc. (“Genentech”) to evaluate the efficacy and safety of Genentech’s drugs, Herceptin (trastuzumab) and Perjeta (pertuzumab), and chemotherapy in breast cancer patients selected with our CELsignia test. Based on NSABP’s updated estimates of patient enrollment rates to reflect the impact of COVID-19 and the impact in the third quarter of hospitalizations associated with the SARS-CoV-2 Delta variant, interim results are expected to be available in the second half of 2022 and final results approximately nine months later. The goal is to demonstrate that patients who have an abnormal HER2 signaling pathway, as identified by our CELsignia test, respond to treatment with a matching targeted therapy. |
| ● | FACT-2 Clinical Trial to Evaluate Efficacy of Puma’s HER2 Targeted Therapy. We are collaborating with Puma Biotechnology, Inc. (“Puma”) and West Cancer Center to conduct a Phase II single-arm interventional trial to evaluate the efficacy and safety of Puma’s drug, Nerlynx (neratinib), and chemotherapy in breast cancer patients selected with our CELsignia test. Based on West Cancer Center’s updated estimates of patient enrollment rates to reflect the impact of COVID-19 and the impact in the third quarter of hospitalizations associated with the SARS-CoV-2 Delta variant, interim results are expected to be available in the second half of 2022 and final results approximately nine months later. The goal of the trial is to demonstrate that triple-negative breast cancer patients who have a hyperactive HER2 signaling tumor, as identified by the CELsignia test, respond to treatment with Nerlynx, a matching HER2 therapy. |
| ● | FACT-3 Clinical Trial to Evaluate Efficacy of Pfizer’s pan-HER and c-Met Targeted Therapies. In January 2021, we announced a clinical trial collaboration with Sarah Cannon Research Institute and Pfizer to conduct a Phase II clinical trial. This open-label Phase II trial will evaluate the efficacy and safety of two Pfizer targeted therapies, Vizimpro (dacomitinib), a pan-HER inhibitor, and Xalkori (crizotinib), a c-Met inhibitor, in previously treated metastatic HER2-negative breast cancer patients selected with our CELsignia MP Test. Based on the Sarah Cannon Research Institute’s estimates of patient enrollment rates, interim results are expected 12-15 months after the protocol is activated and final results 12-15 months later. The goal of the trial is to demonstrate that previously treated HER2-negative metastatic breast cancer patients who have hyperactive HER2 and c-Met signaling tumors, as identified by the CELsignia test, respond to treatment with Vizimpro in combination with Xalkori. |
| ● | FACT-4 Clinical Trial to Evaluate Efficacy of Puma’s HER2 Targeted Therapy. In December 2020, we announced a clinical trial collaboration with Massachusetts General Hospital and Puma, a biopharmaceutical company, to conduct a Phase II clinical trial. This open-label Phase II trial will evaluate the efficacy and safety of Puma’s drug, Nerlynx (neratinib), and Faslodex (fulvestrant), an AstraZeneca drug, in previously treated metastatic HR-positive (HR+), HER2-negative breast cancer patients selected with our CELsignia HER2 Pathway Activity Test. Based on Massachusetts General Hospital’s estimates of patient enrollment rates, we expect to obtain interim results 12-15 months after the protocol is activated and final results 12 to 15 months later. The goal of the trial is to demonstrate that previously treated HR+, HER2-negative metastatic breast cancer patients who have hyperactive HER2 signaling tumors, as identified by the CELsignia test, respond to treatment with Nerlynx in combination with Faslodex, a hormonal therapy that targets the estrogen receptor. |
| 23 |
| ● | FACT-5 Clinical Trial to Evaluate Efficacy of Puma’s pan-HER Inhibitor and chemotherapy. In October 2021, we announced a clinical trial collaboration with University of Rochester Wilmot Cancer Center and Puma, to conduct a Phase II clinical trial. This open-label Phase II trial will evaluate the efficacy and safety of Puma’s drug, Nerlynx (neratinib), and the chemotherapy capecitabine, in previously treated metastatic HER2-negative breast cancer patients with brain metastases selected with our CELsignia HER2 Pathway Activity Test. Based on University of Rochester Wilmot Cancer Center estimates of patient enrollment rates, we expect to obtain interim results 12-15 months after the protocol is activated and the final results 12 to 15 months later. We expect enrollment to begin by mid-2022. The goal of the trial is to demonstrate that previously treated HER2-negative metastatic breast cancer patients with brain metastases who have hyperactive HER2 signaling tumors, as identified by the CELsignia test, respond to treatment with Nerlynx in combination with capecitabine. |
| ● | FACT-6 Clinical Trial to Evaluate Efficacy of Novartis’s c-Met Inhibitor and Puma’s pan-HER Inhibitor. In March 2021, we announced a clinical trial collaboration with MD Anderson, Novartis AG, and Puma, to conduct a Phase I/II clinical trial. This open-label Phase I/II trial will evaluate the efficacy and safety of Novartis’ c-Met inhibitor, Tabrecta (capmatinib), and Puma’s pan-HER inhibitor, Nerlynx (neratinib), in previously treated metastatic HER2-negative breast cancer patients selected with our CELsignia MP Test. Based on MD Anderson’s estimates of patient enrollment rates, we expect to obtain interim results 12-15 months after the protocol is activated and final results 12-15 months later. The goal of the trial is to demonstrate that previously treated HER2-negative metastatic breast cancer patients who have hyperactive HER2 and c-Met signaling tumors, as identified by the CELsignia test, respond to treatment with Tabrecta in combination with Nerlynx. |
Impact of COVID-19 on our Business
Health and Safety
To help protect the health and safety of our employees, suppliers and collaborators, we took proactive, aggressive action from the earliest signs of the outbreak. We enacted rigorous safety measures in our laboratory and administrative offices, including implementing social distancing protocols, allowing working from home for those employees that do not need to be physically present in a lab to perform their work, suspending travel, implementing temperature checks at the entrances to our facilities, extensively and frequently disinfecting our workspaces and providing masks to those employees who must be physically present. We expect to continue with these measures until the COVID-19 pandemic is better contained and we may take further actions as government authorities require or recommend or as we determine to be in the best interests of our employees, suppliers, and collaborators.
Clinical Trials and Collaborations
As a result of the COVID-19 pandemic, governmental authorities implemented numerous and constantly evolving measures to try to contain the virus, such as travel bans and restrictions, limits on gatherings, quarantines, shelter-in-place orders, and business shutdowns. While many of these measures have been relaxed in recent months, new variants of the coronavirus, such as the delta variant, appear likely to cause new or former protective measures to be reconsidered. As we continue to advance our clinical trial collaborations, we are in close contact with our current clinical sponsors, and principal investigators, as well as prospective pharmaceutical company and clinical collaborators, to assess the impact of COVID-19 on our trial enrollment timelines and collaboration discussions. Based on delays in the enrollment of patients in our ongoing clinical trials due to the increased hospitalizations associated with the SARS-CoV-2 Delta variant, we now expect interim results from the FACT-1 and FACT-2 trials to be delayed until the second half of 2022 and final results approximately nine months later. As the impact of COVID-19 on our industry becomes clearer, we may need to reassess the timing of our anticipated clinical milestones. Prospective clinical trial collaborations with pharmaceutical companies and sponsors may also be delayed but the impact on the timing of finalizing agreements is not yet known.
Research and Development
While our facility currently remains operational, the evolving measures to try to contain the virus have impacted and may further impact our workforce and operations, as well as those of our vendors and suppliers. Our laboratory remains operational as of this date, but, in response to the COVID-19 pandemic, we have implemented protective policies that reduce the number of research and development staff operating in our laboratory at any one time. However, in light of the focus of healthcare providers and hospitals on fighting the virus, several of the clinical sites that provide us tumor tissue for research have halted this service, reducing the number of new tumor tissue specimens we would typically expect to receive. These various constraints may slow or diminish our research and development activities, particularly if the pandemic continues to experience rising infections from the delta variant of the coronavirus or otherwise. In addition, cancer research-related industry meetings, such as the American Association for Cancer Research (AACR), were previously delayed for several months and could be again in the future as a result of the virus. So far, our submissions to present research results at these meetings have been accepted, but the release of the results have been postponed in conjunction with the delayed meeting schedules. We cannot be certain as to whether these types of delays will continue and what the impact will be on our ability to present research results in the future.
| 24 |
Results of Operations
We have not generated any revenue from sales to date, and we continue to incur significant research and development and other expenses related to our ongoing operations. As a result, we are not and have never been profitable and have incurred losses in each period since our inception in 2012. For the three months ended September 30, 2021 and 2020, we reported a net loss of approximately $6.0 million and $2.5 million, respectively, and for the nine months ended September 30, 2021 and 2020, we reported a net loss of approximately $22.9 million and $6.9 million, respectively. As of September 30, 2021, we had an accumulated deficit of approximately $49.2 million. As of September 30, 2021, we had cash and cash equivalents of approximately $90.4 million.
Components of Operating Results
Revenue
To date, we have not generated any revenue. Initially, our ability to generate revenue will depend primarily upon our ability to obtain partnership agreements with pharmaceutical companies to provide companion diagnostics for such pharmaceutical partners’ existing or investigational targeted therapies. We expect these partnerships to generate significant revenue from the sale of tests to identify patients eligible for clinical trials, from milestone payments, and, potentially, from royalties on the incremental drug revenues our tests enable. Once a new drug indication is received that requires use of our companion diagnostic to identify eligible patients, we expect to generate revenues from sales of tests to treating physicians. With the execution of the Pfizer license agreement in April 2021, whereby we acquired exclusive world-wide licensing rights to develop and commercialize gedatolisib, we expect to conduct clinical trials to support potential regulatory approval to market gedatolisib. If we obtain regulatory approvals to market gedatolisib, we expect to generate revenue from sales of the drug for the treatment of breast cancer patients.
Research and Development
Since our inception, we have primarily focused on research and development of our CELsignia platform, development and validation of our CELsignia tests, and research related to the discovery of new cancer sub-types. Beginning in April 2021, we are also focusing on development of gedatolisib, a PI3K/mTOR targeted therapy. Research and development expenses primarily include:
| ● | employee-related expenses related to our research and development activities, including salaries, benefits, recruiting, travel and stock-based compensation expenses; | |
| ● | laboratory supplies; | |
| ● | consulting fees paid to third parties; | |
| ● | clinical trial costs; | |
| ● | facilities expenses; and | |
| ● | legal costs associated with patent applications. |
Internal and external research and development costs are expensed as they are incurred. As we initiate clinical trials to evaluate efficacy of targeted therapies in cancer patients selected with one of our CELsignia tests and to develop gedatolisib, the proportion of research and development expenses allocated to external spending will grow at a faster rate than expenses allocated to internal expenses.
General and Administrative
General and administrative expenses consist primarily of salaries, benefits and stock-based compensation related to our executive, finance and support functions. Other general and administrative expenses include professional fees for auditing, tax, and legal services associated with being a public company, director and officer insurance, investor relations and travel expenses for our general and administrative personnel.
Sales and Marketing
Sales and marketing expenses consist primarily of professional and consulting fees related to these functions. To date, we have incurred immaterial sales and marketing expenses as we continue to focus primarily on the development of our CELsignia platform and corresponding CELsignia tests. We would expect to begin to incur increased sales and marketing expenses in anticipation of the commercialization of our first CELsignia tests and the commercialization of our first drug, gedatolisib. These increased expenses are expected to include payroll-related costs as we add employees in the commercial departments, costs related to the initiation and operation of our sales and distribution network and marketing related costs.
Interest Expense
Interest expense is primarily due to a loan agreement and finance lease obligations.
Interest Income
Interest income consists of interest income earned on our cash and cash equivalent balances.
| 25 |
Results of Operations
Three Months Ended September 30, | Increase (Decrease) | |||||||||||||||
| 2021 | 2020 | $ | Percent Change | |||||||||||||
| Statements of Operations Data: | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 4,960,515 | $ | 1,962,610 | $ | 2,997,905 | 153 | % | ||||||||
| General and administrative | 639,271 | 517,465 | 121,806 | 24 | ||||||||||||
| Total operating expenses | 5,599,786 | 2,480,075 | 3,119,711 | 126 | ||||||||||||
| Loss from operations | (5,599,786 | ) | (2,480,075 | ) | (3,119,711 | ) | 126 | |||||||||
| Other income (expense) | ||||||||||||||||
| Interest expense | (433,072 | ) | (28 | ) | (433,044 | ) | n/a | |||||||||
| Interest income | 5,612 | 5,805 | (193 | ) | (3 | ) | ||||||||||
| Loss on sale of fixed assets | - | - | - | n/a | ||||||||||||
| Other income (expense), net | (427,460 | ) | 5,777 | (433,237 | ) | n/a | ||||||||||
| Net loss before income taxes | (6,027,246 | ) | (2,474,298 | ) | (3,552,948 | ) | 144 | |||||||||
| Income tax benefits | - | - | - | - | ||||||||||||
| Net loss | $ | (6,027,246 | ) | $ | (2,474,298 | ) | $ | (3,552,948 | ) | 144 | % | |||||
Nine Months Ended September 30, | Increase (Decrease) | |||||||||||||||
| 2021 | 2020 | $ | Percent Change | |||||||||||||
| Statements of Operations Data: | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 20,266,965 | $ | 5,576,250 | $ | 14,690,715 | 263 | % | ||||||||
| General and administrative | 1,768,058 | 1,428,578 | 339,480 | 24 | ||||||||||||
| Total operating expenses | 22,035,023 | 7,004,828 | 15,030,195 | 215 | ||||||||||||
| Loss from operations | (22,035,023 | ) | (7,004,828 | ) | (15,030,195 | ) | 215 | |||||||||
| Other income (expense) | ||||||||||||||||
| Interest expense | (824,283 | ) | (93 | ) | (824,190 | ) | n/a | |||||||||
| Interest income | 7,803 | 81,639 | (73,836 | ) | (90 | ) | ||||||||||
| Gain on sale of fixed assets | (263 | ) | - | (263 | ) | n/a | ||||||||||
| Other income (expense), net | (816,743 | ) | 81,546 | (898,289 | ) | n/a | ||||||||||
| Net loss before income taxes | (22,851,766 | ) | (6,923,282 | ) | (15,928,484 | ) | 230 | |||||||||
| Income tax benefits | - | - | - | - | ||||||||||||
| Net loss | $ | (22,851,766 | ) | $ | (6,923,282 | ) | $ | (15,928,484 | ) | 230 | % | |||||
| 26 |
Research and Development
Our research and development expenses for the three months ended September 30, 2021 were approximately $5.0 million, representing an increase of approximately $3.0 million, or 153%, compared to the same period in 2020. The increase of $3.0 million primarily resulted from expenses related to the development of gedatolisib. Employee related expenses, including consulting fees, accounted for $1.1 million of the increase. The increase of $1.1 million also included an increase of $0.2 million in non-cash stock-based compensation. The remaining increase of $1.9 million is related to clinical trials, patent legal fees and costs associated with the transfer of the gedatolisib-related activities from Pfizer to Celcuity.
Our research and development expenses for the nine months ended September 30, 2021 were approximately $20.3 million, representing an increase of approximately $14.7 million, or 263%, compared to the same period in 2020. The increase primarily resulted from a $10.0 million upfront license fee related to the execution of the Pfizer license agreement, which included $5.0 million of non-cash expense for the issuance of common stock. The remaining $4.7 million increase primarily resulted from expenses related to the development of gedatolisib. Employee related expenses, including consulting fees, accounted for a $1.8 million increase. The increase of $1.8 million also included an increase of $0.2 million in non-cash stock-based compensation. The remaining increase of $2.9 million is related to clinical trials, patent legal fees and costs associated with the transfer of the gedatolisib-related activities from Pfizer to Celcuity.
Conducting a significant amount of research and development is central to our business model. We plan to increase our research and development expenses for the foreseeable future as we seek to develop gedatolisib, discover new cancer sub-types, and develop and validate additional CELsignia tests to diagnose such sub-types. We also expect to incur increased expenses to support companion diagnostic business development activities with pharmaceutical companies as we develop additional CELsignia tests and initiate a clinical trial for gedatolisib.
General and Administrative
Our general and administrative expenses for the three months ended September 30, 2021 were approximately $0.6 million, representing an increase of approximately $0.1 million, or 24%, compared to the same period in 2020. The increase primarily resulted from a $0.1 million increase in non-cash stock-based compensation.
Our general and administrative expenses for the nine months ended September 30, 2021 were approximately $1.8 million, representing an increase of approximately $0.3 million, or 24%, compared to the same period in 2020. The increase primarily resulted from a $0.1 million increase in non-cash stock-based compensation. In addition, other general and administrative expenses increased $0.2 million due to professional fees associated with being a public company and director and officer insurance.
We anticipate that our general and administrative expenses will increase in future periods, reflecting both increased costs in connection with the potential future commercialization of CELsignia tests and gedatolisib, an expanding infrastructure, and increased professional fees associated with being a public company.
Interest Expense
Interest expense for the three months ended September 30, 2021 was $0.4 million and represents an increase of $0.4 million compared to the same periods in 2020. For the nine months ended September 30, 2021, interest expense was $0.8 million and represents an increase of $0.8 million compared to the same period in 2020. The increase is due to the loan agreement that was executed in April 2021 and includes $0.2 million and $0.4 million of non-cash interest expense for the three- and nine-month periods ended September 30, 2021, respectively.
Interest Income
Interest income for the three months ended September 30, 2021 was flat compared to the same period in 2020. Interest income for the nine months ended September 30, 2021 represents a decrease of approximately $0.1 million compared to the same period in 2020. This decrease was primarily the result of lower market interest rates.
Liquidity and Capital Resources
Since our inception, we have incurred losses and cumulative negative cash flows from operations. Through September 30, 2021, we have raised capital of approximately $13.7 million and $7.5 million through private placements of common equity and unsecured convertible notes, respectively. On September 22, 2017, we closed on the initial public offering of our common stock, which generated approximately $23.3 million of additional cash after taking into account underwriting discounts and commissions and offering expenses. On June 5, 2020, we entered into an At Market Issuance Sales Agreement with B. Riley, FBR, Inc (the “ATM Agreement”). The ATM Agreement allowed us to sell shares of common stock up to an aggregate offering price of $10.0 million. Through September 30, 2021, we generated approximately $0.1 million of additional cash through sales pursuant to the ATM Agreement, after taking into account commissions and offering expenses. On February 26, 2021, we completed a follow-on offering of our common stock, which generated approximately $25.8 million of additional cash after taking into account underwriting discounts and offering expenses. In conjunction with the follow-on offering, the ATM Agreement was terminated. On April 8, 2021, we entered into a loan agreement with Innovatus Life Sciences Lending Fund I, LP (“Innovatus”), whereby Innovatus agreed to loan up to $25 million in three tranches consisting of (i) a $15.0 million non-contingent term A loan that was funded on April 8, 2021, (ii) a $5 million term B loan to be funded upon our request no later than March 31, 2022, and (iii) a $5 million term C loan to be funded upon our request no later than March 31, 2023. Funding of the term B and C loan is subject to our ability to achieve certain milestones. On July 1, 2021, we completed a follow-on offering of our common stock, which generated approximately $52.8 million of additional cash after taking into account underwriting discounts of approximately $3.4 million and offering expenses of approximately $0.1 million.
| 27 |
Cash from these capital raising activities has been our primary source of funds for our operations since inception. As of September 30, 2021, our cash and cash equivalents were approximately $90.4 million, and we had an accumulated deficit of approximately $49.2 million.
We expect that our research and development and general and administrative expenses will increase as we continue to develop gedatolisib, our CELsignia platform and additional CELsignia tests, conduct research related to the discovery of new cancer sub-types, conduct clinical trials, and pursue other business development activities. We would also expect to incur sales and marketing expenses as we commercialize our CELsignia tests and gedatolisib. We expect to use cash on hand to fund our research and development expenses, capital expenditures, working capital, sales and marketing expenses, and general corporate expenses, as well as for the increased costs associated with being a public company.
Based on our current business plan, we believe that our current cash on hand will provide sufficient cash to finance operations and pay obligations when due for at least the next twelve months.
We may seek to raise additional capital to expand our business, pursue strategic investments, and take advantage of financing or other opportunities that we believe to be in the best interests of the Company and our stockholders. Additional capital may be raised through the sale of common or preferred equity or convertible debt securities, entry into debt facilities or other third-party funding arrangements. The sale of equity and convertible debt securities may result in dilution to our stockholders and those securities may have rights senior to those of our common shares. Agreements entered into in connection with such capital raising activities could contain covenants that would restrict our operations or require us to relinquish certain rights. Additional capital may not be available on reasonable terms, or at all.
Cash Flows
| Nine Months Ended | ||||||||
| September 30, | ||||||||
| 2021 | 2020 | |||||||
| (unaudited) | ||||||||
| Net cash provided by (used in): | ||||||||
| Operating activities | $ | (14,187,938 | ) | $ | (5,038,438 | ) | ||
| Investing activities | (71,459 | ) | (75,494 | ) | ||||
| Financing activities | 92,988,298 | 75,317 | ||||||
| Net increase (decrease) in cash and cash equivalents | $ | 78,728,901 | $ | (5,038,615 | ) | |||
Operating Activities
Net cash used in operating activities was approximately $14.2 million for the nine months ended September 30, 2021 and consisted primarily of a net loss of approximately $22.9 million, offset by non-cash expense items of approximately $7.3 million and working capital changes of $1.4 million. Non-cash expense items of approximately $7.3 million primarily consisted of $5.0 million for issuance of common stock related to a license agreement, $1.7 million of stock-based compensation expense, non-cash interest expense of $0.4 million and depreciation expense of $0.2 million. The approximately $1.4 million of working capital changes was primarily due to an increase in accounts payable. The net cash used in operating activities was approximately $5.0 million for the nine months ended September 30, 2020 and consisted primarily of a net loss of approximately $6.9 million, adjusted for working capital changes of approximately $0.3 million and non-cash expense items of approximately $1.6 million. Non-cash expense items of approximately $1.6 million primarily consisted of $1.3 million of stock-based compensation expense and depreciation expense of $0.3 million.
Investing Activities
Net cash used in investing activities for the nine months ended September 30, 2021 and 2020 were flat at approximately $0.1 million consisting of purchases of property and equipment.
| 28 |
Financing Activities
Net cash provided by financing activities for the nine months ended September 30, 2021 was approximately $93.0 million. The $93.0 million primarily consisted of net proceeds from the sale of shares of our common stock through two follow-on offerings totaling $78.5 million and $14.4 million from net proceeds related to the closing of a loan agreement. The remaining $0.1 million was the result of proceeds from the exercise of common stock warrants and employee stock options and proceeds from employee stock purchases. The net cash provided by financing activities for the nine months ended September 30, 2020 was approximately $0.1 million and primarily reflects net proceeds from the sale of shares of our common stock through the ATM Agreement and employee stock purchases.
Recent Accounting Pronouncements
From time-to-time new accounting pronouncements are issued by the Financial Accounting Standards Board or other standard setting bodies and adopted by us as of the specified effective date. Unless otherwise discussed in Note 2 to our unaudited condensed financial statements included in Item 1 of Part I of this Quarterly Report, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
Critical Accounting Policies and Use of Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our unaudited condensed financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses during the reporting periods. These items are monitored and analyzed by us for changes in facts and circumstances, and material changes in these estimates could occur in the future. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Changes in estimates are reflected in reported results for the period in which they become known. Actual results may differ materially from these estimates.
Our significant accounting policies are more fully described in Note 2 to our unaudited condensed financial statements included in Item 1 of Part I of this Quarterly Report.
Private Securities Litigation Reform Act
The Private Securities Litigation Reform Act of 1995 provides a “safe harbor” for forward-looking statements. Such forward-looking information is included in this Quarterly Report and in other materials filed or to be filed by us with the SEC (as well as information included in oral statements or other written statements made or to be made by us). Forward-looking statements include all statements based on future expectations. This Quarterly Report contains forward-looking statements that involve risks and uncertainties including, but not limited to, (i) our clinical trial plans and the estimated costs for such trials, including the timing of launching a Phase 2/3 clinical trial for gedatolisib; (ii) our expectations with respect to costs and timelines to develop, validate and launch CELsignia tests and to continue to develop gedatolisib; (iii) our beliefs related to the perceived advantages of our CELsignia tests compared to traditional molecular or other diagnostic tests; (iv) the expected benefits of gedatolisib; (v) our expectations regarding the timeline of patient enrollment and results from clinical trials, including the existing clinical trial for gedatolisib; (vi) the future payments that may be owned to Pfizer under the license agreement; (vii) our expectations regarding partnering with pharmaceutical companies and other third parties; (viii) our expectations regarding revenue from sales of CELsignia tests and revenue from milestone or other payment sources; (ix) our plans with respect to research and development and related expenses for the foreseeable future; (x) our expectations regarding business development activities, including companion diagnostic related activities with pharmaceutical companies, expanding our sales and marketing functions and the costs associated with such activities; (xi) our expectations with respect to the CELsignia tests and the analytical capabilities and potential impact of such tests; (xii) our beliefs regarding the ability of our cash on hand to fund our research and development expenses, capital expenditures, working capital, sales and marketing expenses, and general corporate expenses, as well as the increased costs associated with being a public company; and (xiii) our expectations regarding the impact that the COVID-19 pandemic and related economic effects will have on our business and results of operations.
In some cases, you can identify forward-looking statements by the following words: “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “ongoing,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would,” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. Forward-looking statements are only predictions and are not guarantees of performance. These statements are based on our management’s beliefs and assumptions, which in turn are based on their interpretation of currently available information.
| 29 |
These statements involve known and unknown risks, uncertainties and other factors that may cause our results or our industry’s actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Certain risks, uncertainties and other factors include, but are not limited to, our limited operating history; the unknown impact of the COVID-19 pandemic on our business; our initial success being heavily dependent on the success of our CELsignia HER2 Pathway Activity Test; our inability to develop and commercialize gedatolisib; our inability to determine whether our CELsignia tests are currently commercially viable; challenges we may face in developing and maintaining relationships with pharmaceutical company partners; the complexity and timeline for development of CELsignia tests and gedatolisib; the uncertainty and costs associated with clinical trials; the uncertainty regarding market acceptance by physicians, patients, third-party payors and others in the medical community, and with the size of market opportunities available to us; the pricing of molecular and other diagnostic products and services that compete with us; uncertainty with insurance coverage and reimbursement for our CELsignia tests; difficulties we may face in managing growth, such as hiring and retaining a qualified sales force and attracting and retaining key personnel; changes in government regulations; and obtaining and maintaining intellectual property protection for our technology and time and expense associated with defending third-party claims of intellectual property infringement, investigations or litigation threatened or initiated against us. These and additional risks, uncertainties and other factors are described more fully in our Annual Report on Form 10-K for the year ended December 31, 2020 and in Exhibit 99.4 to our Current Report on Form 8-K, filed with the SEC on April 8, 2021 and elsewhere in this Quarterly Report. Copies of filings made with the SEC are available through the SEC’s electronic data gathering analysis and retrieval system (EDGAR) at www.sec.gov.
You should read the cautionary statements made in this Quarterly Report as being applicable to all related forward-looking statements wherever they appear in this Quarterly Report. We cannot assure you that the forward-looking statements in this Quarterly Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. You should read this Quarterly Report completely. Other than as required by law, we undertake no obligation to update these forward-looking statements, even though our situation may change in the future.
ITEM 3. Quantitative and Qualitative Disclosures about Market Risk
As a smaller reporting company, we are not required to provide disclosure pursuant to this item.
ITEM 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our Chief Executive Officer and Chief Financial Officer, referred to collectively herein as the Certifying Officers, are responsible for establishing and maintaining our disclosure controls and procedures. The Certifying Officers have reviewed and evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”)) as of September 30, 2021. Based on that review and evaluation, the Certifying Officers have concluded that, as of the end of the period covered by this Quarterly Report, our disclosure controls and procedures, as designed and implemented, are effective and provide reasonable assurance that information required to be disclosed by us in the periodic and current reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the periods specified by the SEC’s rules and forms.
Changes in Internal Control Over Financial Reporting.
There were no changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the three months ended September 30, 2021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 30 |
PART II. — OTHER INFORMATION
ITEM 1. Legal Proceedings
From time to time we may be involved in disputes or litigation relating to claims arising out of our operations. We are not currently a party to any legal proceedings that could reasonably be expected to have a material adverse effect on our business, financial condition and results of operations.
ITEM 1A. Risk Factors
As a smaller reporting company, we are not required to provide disclosure pursuant to this item. However, in addition to other information set forth in this Quarterly Report, including the important information in the section entitled “Private Securities Litigation Reform Act,” you should carefully consider the “Risk Factors” discussed in our Annual Report on Form 10-K for the year ended December 31, 2020 and in Exhibit 99.4 to our Current Report on Form 8-K, filed with the SEC on April 8, 2021 for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in this Quarterly Report. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial might materially adversely affect our actual business, financial condition and/or operating results.
ITEM 2. Unregistered Sales of Equity Securities and Use of Proceeds
Use of Proceeds from Initial Public Offering of Common Stock
On September 22, 2017, we completed our initial public offering of 2,760,000 shares of our common stock at a price to the public of $9.50 per share. The total number of shares of common stock sold in the offering includes the exercise of an overallotment we granted to Craig-Hallum Capital Group LLC, the sole managing underwriter of the offering, to purchase 360,000 shares of common stock. The shares of common stock were registered for sale pursuant to Registration Statements on Form S-1 (Registration Nos. 333-220128 and 333-220527), filed with the SEC and declared effective on September 19, 2017 (the “Effective Date”). The aggregate offering price for the registered shares of common stock was approximately $26.2 million. The offering commenced on September 20, 2017 and did not terminate before all of the shares of common stock that were registered were sold.
The aggregate offering price for the shares of common stock sold in the offering was approximately $26.2 million. We received net proceeds of approximately $23.3 million from the offering, after deducting underwriting discounts and commissions of approximately $1.8 million and offering expenses of approximately $1.1 million. No payments for the foregoing expenses were made by us to any of our officers, directors or persons owning ten percent or more of our common stock, or to the associates of any of the foregoing, or to its affiliates, other than payments in the ordinary course of business to our officers for salaries and bonuses.
There has been no material change in the planned use of proceeds as described in our Prospectus filed with the SEC on September 20, 2017. From the Effective Date through September 30, 2021, we have used approximately $21.5 million in furtherance of our planned use of proceeds, which includes funding additional research and development for discovery of new cancer sub-types and development and validation of new CELsignia tests; clinical trials to support clinical claims; development of operational processes and capital expenditures; and working capital and other general corporate purposes.
Recent Unregistered Sales of Equity Securities
None
ITEM 3. Defaults Upon Senior Securities
None.
ITEM 4. Mine Safety Disclosures
Not applicable.
ITEM 5. Other Information
None.
| 31 |
ITEM 6. Exhibits
EXHIBIT INDEX
* Filed herewith.
** Furnished herewith.
+ Certain portions have been omitted from this exhibit.
| 32 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned thereunto duly authorized.
| Dated: November 9, 2021 | CELCUITY INC. | |
| By | /s/ Brian F. Sullivan | |
| Brian F. Sullivan | ||
| Chairman and Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| By | /s/ Vicky Hahne | |
| Vicky Hahne | ||
| Chief Financial Officer | ||
| (Principal Financial and Accounting Officer) |
| 33 |