Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-Q
| x | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended March 31, 2015
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-36544
Sage Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 27-4486580 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
215 First Street
Cambridge, Massachusetts 02142
(Address of principal executive office) (Zip Code)
Registrant’s telephone number, including area code:
(617) 299-8380
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | x (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of May 1, 2015, there were 28,366,696 shares of the registrant’s Common Stock, $0.0001 par value per share, outstanding.
Table of Contents
FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q contains forward-looking statements that involve risks and uncertainties. We make such forward-looking statements pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 and other federal securities laws. All statements other than statements of historical facts contained in this Quarterly Report on Form 10-Q are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “may”, “will”, “should”, “expects”, “intends”, “plans”, “anticipates”, “believes”, “estimates”, “predicts”, “potential”, “continue” or the negative of these terms or other comparable terminology. These forward-looking statements include, but are not limited to, statements about:
| • | the accuracy of our estimates regarding expenses, future revenues and capital requirements; |
| • | our plans to develop and commercialize our product candidates, initially as treatments for status epilepticus, refractory status epilepticus, super-refractory status epilepticus, essential tremor and severe postpartum depression; |
| • | our ability to complete our ongoing clinical trials and to advance our product candidates into additional clinical trials, including pivotal clinical trials, and successfully complete such clinical trials; |
| • | regulatory developments in the United States and foreign countries; |
| • | the performance of our third-party manufacturers and contract research organizations; |
| • | our ability to obtain and maintain intellectual property protection for our proprietary assets; |
| • | the size of the potential markets for our product candidates and our ability to serve those markets; |
| • | the rate and degree of market acceptance of our product candidates for any indication once approved; |
| • | our ability to obtain additional financing; |
| • | the success of competing products that are or become available for the indications that we are pursuing; |
| • | the loss of key scientific or management personnel; and |
| • | other risks and uncertainties, including those listed under Part II, Item 1A. Risk Factors. |
Any forward-looking statements in this Quarterly Report on Form 10-Q reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Factors that may cause actual results to differ materially from current expectations include, among other things, those listed under Part II, Item 1A. Risk Factors and elsewhere in this Quarterly Report on Form 10-Q. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
2
Table of Contents
This Quarterly Report on Form 10-Q also contains estimates, projections and other information concerning our industry, our business, and the markets for certain diseases, including data regarding the estimated size of those markets, and the incidence and prevalence of certain medical conditions. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances reflected in this information. Unless otherwise expressly stated, we obtained this industry, business, market and other data from reports, research surveys, studies and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data and similar sources.
3
Table of Contents
Sage Therapeutics, Inc.
| Page | ||||||
| PART I – FINANCIAL INFORMATION | ||||||
| Item 1. |
Financial Statements (Unaudited) | 5 | ||||
| Consolidated Balance Sheets as of March 31, 2015 and December 31, 2014 | 5 | |||||
| Consolidated Statements of Operations and Comprehensive Loss for the three months ended March 31, 2015 and 2014 | 6 | |||||
| Consolidated Statements of Cash Flows for the three months ended March 31, 2015 and 2014 | 7 | |||||
| Notes to Consolidated Financial Statements | 8 | |||||
| Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 16 | ||||
| Item 3. |
Quantitative and Qualitative Disclosures About Market Risk | 26 | ||||
| Item 4. |
Controls and Procedures | 27 | ||||
| PART II – OTHER INFORMATION | ||||||
| Item 1. |
Legal Proceedings | 29 | ||||
| Item 1A. |
Risk Factors | 29 | ||||
| Item 2. |
Unregistered Sales of Equity Securities and Use of Proceeds | 53 | ||||
| Item 6. |
Exhibits | 53 | ||||
| 54 | ||||||
4
Table of Contents
PART I — FINANCIAL INFORMATION
| Item 1. | Financial Statements |
Sage Therapeutics, Inc. and Subsidiary
(in thousands, except share and per share data)
(Unaudited)
| March 31, 2015 |
December 31, 2014 |
|||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 113,162 | $ | 127,766 | ||||
| Prepaid expenses and other current assets |
1,367 | 1,056 | ||||||
|
|
|
|
|
|||||
| Total current assets |
114,529 | 128,822 | ||||||
| Property and equipment, net |
269 | 163 | ||||||
| Restricted cash |
39 | 39 | ||||||
| Deferred tax assets |
641 | 641 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 115,478 | $ | 129,665 | ||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 2,782 | $ | 2,429 | ||||
| Accrued expenses |
5,522 | 4,687 | ||||||
| Deferred tax liabilities |
641 | 641 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
8,945 | 7,757 | ||||||
| Other liabilities |
23 | 23 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
8,968 | 7,780 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Note 4) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.0001 par value; 5,000,000 shares authorized at March 31, 2015 and December 31, 2014, respectively; no shares issued or outstanding at March 31, 2015 and December 31, 2014, respectively |
— | — | ||||||
| Common stock, $0.0001 par value; 120,000,000 shares authorized at March 31, 2015 and December 31, 2014, respectively; 25,694,560 and 25,621,791 shares issued and outstanding at March 31, 2015 and December 31, 2014, respectively |
3 | 3 | ||||||
| Additional paid-in capital |
190,223 | 188,727 | ||||||
| Accumulated deficit |
(83,716 | ) | (66,845 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
106,510 | 121,885 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 115,478 | $ | 129,665 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these consolidated financial statements.
5
Table of Contents
Sage Therapeutics, Inc. and Subsidiary
Consolidated Statements of Operations and Comprehensive Loss
(in thousands, except share and per share data)
(Unaudited)
| Three Months Ended March 31, | ||||||||
| 2015 | 2014 | |||||||
| Operating expenses: |
||||||||
| Research and development |
$ | 12,900 | $ | 4,173 | ||||
| General and administrative |
3,997 | 1,617 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
16,897 | 5,790 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(16,897 | ) | (5,790 | ) | ||||
| Interest income (expense), net |
21 | — | ||||||
| Other income (expense), net |
5 | — | ||||||
|
|
|
|
|
|||||
| Net loss and comprehensive loss |
(16,871 | ) | (5,790 | ) | ||||
| Accretion of redeemable convertible preferred stock to redemption value |
— | (326 | ) | |||||
|
|
|
|
|
|||||
| Net loss attributable to common stockholders |
$ | (16,871 | ) | $ | (6,116 | ) | ||
|
|
|
|
|
|||||
| Net loss per share attributable to common stockholders—basic and diluted |
$ | (0.66 | ) | $ | (3.70 | ) | ||
|
|
|
|
|
|||||
| Weighted average number of common shares used in net loss per share attributable to common stockholders—basic and diluted |
25,655,883 | 1,652,726 | ||||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these consolidated financial statements.
6
Table of Contents
Sage Therapeutics, Inc. and Subsidiary
Consolidated Statements of Cash Flows
(in thousands)
(Unaudited)
| Three Months Ended March 31, | ||||||||
| 2015 | 2014 | |||||||
| Cash flows from operating activities |
||||||||
| Net loss |
$ | (16,871 | ) | $ | (5,790 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Stock-based compensation expense |
1,349 | 160 | ||||||
| Non-cash licensing and consulting fees |
424 | 127 | ||||||
| Depreciation and amortization |
22 | 11 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Prepaid expenses and other current assets |
(134 | ) | (52 | ) | ||||
| Accounts payable |
343 | (471 | ) | |||||
| Accrued expenses and other |
261 | 399 | ||||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(14,606 | ) | (5,616 | ) | ||||
|
|
|
|
|
|||||
| Cash flows from investing activities |
||||||||
| Purchase of property and equipment |
(110 | ) | (3 | ) | ||||
|
|
|
|
|
|||||
| Net cash used in investing activities |
(110 | ) | (3 | ) | ||||
|
|
|
|
|
|||||
| Cash flows from financing activities |
||||||||
| Proceeds from the issuance of Series B preferred stock, net of issuance costs |
— | 14,995 | ||||||
| Proceeds from the issuance of Series C preferred stock, net of issuance costs |
— | 37,981 | ||||||
| Proceeds from the issuance of common stock and restricted stock, net |
147 | 2 | ||||||
| Payment of offering costs |
(35 | ) | — | |||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
112 | 52,978 | ||||||
|
|
|
|
|
|||||
| Net increase (decrease) in cash and cash equivalents |
(14,604 | ) | 47,359 | |||||
| Cash and cash equivalents at beginning of period |
127,766 | 8,066 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at end of period |
$ | 113,162 | $ | 55,425 | ||||
|
|
|
|
|
|||||
| Supplemental disclosure of non-cash investing and financing activities |
||||||||
| Accretion of redeemable convertible preferred stock to redemption value |
$ | — | $ | 326 | ||||
| Deferred public offering costs included in accounts payable or accrued expenses |
$ | 151 | $ | 842 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
7
Table of Contents
SAGE THERAPEUTICS, INC. AND SUBSIDIARY
Notes to Consolidated Financial Statements
(Amounts in thousands, except share and per share data)
(Unaudited)
| 1. | Nature of the Business |
Sage Therapeutics, Inc. (“Sage” or the “Company”) is a biopharmaceutical company committed to developing and commercializing novel medicines to treat life-threatening, rare central nervous system (“CNS”) disorders, where there are inadequate or no approved existing therapies. The Company is targeting CNS indications where patient populations are easily identified, acute treatment is typically initiated in the hospital setting, clinical endpoints are well-defined, and development pathways are feasible. This focus allows the Company to make highly informed decisions when advancing its product candidates through the development process. The Company’s initial product candidates are aimed at treating different stages of status epilepticus, a life-threatening condition in which the brain is in a state of persistent seizure.
The Company was incorporated under the laws of the state of Delaware on April 16, 2010 and commenced operations on January 19, 2011 as Sterogen Biopharma, Inc. On September 13, 2011, the Company changed its name to Sage Therapeutics, Inc. under its Second Amended and Restated Certificate of Incorporation.
The Company is subject to risks and uncertainties common to early-stage companies in the biotech industry, including, but not limited to, development by competitors of new technological innovations, dependence on key personnel protection of proprietary technology, compliance with government regulations, and ability to secure additional capital to fund operations.
We have incurred losses and negative cash flows from operations since our inception. As of March 31, 2015, we had an accumulated deficit of $83.7 million. From our inception through March 31, 2014, we raised aggregate net proceeds of $90.6 million from the issuance of Series A, Series B and Series C redeemable convertible preferred stock. In July 2014, we raised net proceeds of $94.0 million from the sale of common stock in our initial public offering. In April 2015, we raised net proceeds of approximately $129.2 million from the sale of common stock in our underwritten public offering. We believe our cash balance of $113.2 million as of March 31, 2015 will be sufficient to fund our anticipated level of operations for at least the next 12 months.
| 2. | Summary of Significant Accounting Policies |
Basis of Presentation
The unaudited interim consolidated financial statements of the Company included herein have been prepared, pursuant to the rules and regulations of the Securities and Exchange Commission (the “SEC”). Certain information and footnote disclosures normally included in financial statements prepared in accordance with accounting principles generally accepted in the United States of America have been condensed or omitted from this report, as is permitted by such rules and regulations. Accordingly, these consolidated financial statements should be read in conjunction with the consolidated financial statements as of and for the year ended December 31, 2014.
The unaudited interim consolidated financial statements have been prepared on the same basis as the audited consolidated financial statements. In the opinion of the Company’s management, the accompanying unaudited interim consolidated financial statements contain all adjustments which are necessary to present fairly the Company’s financial position as of March 31, 2015, the results of its operations for the three months ended March 31, 2015 and 2014, and its cash flows for the three months ended March 31, 2015 and 2014. Such adjustments are of a normal and recurring nature. The results for the three months ended March 31, 2015 are not indicative of the results for the year ending December 31, 2015, or for any future period.
On July 23, 2014, the Company completed the sale of 5,750,000 shares of its common stock in its initial public offering (the “IPO”), at a price to the public of $18.00 per share, resulting in net proceeds to the Company of $94.0 million after deducting underwriting discounts and commissions and offering costs paid by the Company. The shares began trading on the Nasdaq Global Market on July 18, 2014.
In connection with preparing for the IPO, the Company’s board of directors and stockholders approved a 1-for-3.15 reverse stock split of the Company’s common stock effective July 2, 2014. All share and per share amounts in the unaudited consolidated financial statements contained herein and notes thereto have been retroactively adjusted, where necessary, to give effect to this reverse stock split. In connection with the closing of the IPO, all of the Company’s outstanding redeemable convertible preferred stock automatically converted into shares of common stock as of July 23, 2014, resulting in the issuance by the Company of an additional 18,007,575 shares of common stock. The significant increase in common stock outstanding in July 2014 will impact the year-over-year comparability of the Company’s net loss per share calculations over the next year.
On April 20, 2015, the Company completed the sale of 2,628,571 shares of common stock in its underwritten public offering of its common stock at a price to the public of $52.50 per share, resulting in net proceeds to the Company of approximately $129.2 million after deducting underwriting discounts and commissions and offering costs paid by the Company.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiary as disclosed in Note 2, Summary of Significant Accounting Policies, within the “Notes to Consolidated Financial Statements” accompanying its Annual Report on Form 10-K. Intercompany accounts and transactions have been eliminated.
8
Table of Contents
Recently Issued Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”), issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606), (“ASU 2014-09”). ASU 2014-09 outlines a new, single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. This new revenue recognition model provides a five-step analysis in determining when and how revenue is recognized. The new model will require revenue recognition to depict the transfer of promised goods or services to customers in an amount that reflects the consideration a company expects to receive in exchange for those goods or services. In April 2015, the FASB proposed a one year delay in the effective date of ASU 2014-09, which was originally effective for public entities for annual reporting periods beginning after December 15, 2016 and interim periods within those periods. Early adoption is not permitted. Companies may use either a full retrospective or a modified retrospective approach to adopt ASU 2014-09. The Company is currently assessing the method of adoption and the impact of this new accounting guidance will have on its consolidated financial statements and footnote disclosures.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements — Going Concern (Subtopic 205-40). The new guidance addresses management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. Management’s evaluation should be based on relevant conditions and events that are known and reasonably knowable at the date that the financial statements are issued. The standard will be effective for the first interim period within annual reporting periods beginning after December 15, 2016. Early adoption is permitted. The Company is evaluating the effect that this guidance will have on its consolidated financial statements.
In April 2015, the FASB issued guidance simplifying the presentation of debt issuance costs. To simplify presentation of debt issue costs, the amendments require that debt issue costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. The guidance becomes effective for the Company in its year ending December 31, 2016, and early adoption is permitted. The Company is currently assessing the impact that this standard will have on its consolidated financial statements.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Fair Value Measurements
Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three categories:
| Level 1 | - | Quoted market prices in active markets for identical assets or liabilities. At March 31, 2015 and December 31, 2014, the Company’s Level 1 assets consisted of money market funds totaling $113,162 and $127,766 respectively. | ||
| Level 2 | - | Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. At March 31, 2015 and December 31, 2014, the Company had no Level 2 assets or liabilities. | ||
| Level 3 | - | Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. At March 31, 2015 and December 31, 2014, the Company had no Level 3 assets or liabilities. | ||
The Company’s financial instruments generally consist of cash equivalents, accounts payable and accrued expenses. The carrying amounts for the applicable financial instruments reported in the balance sheets approximate their fair values at March 31, 2015 (unaudited) and December 31, 2014.
9
Table of Contents
Deferred Offering Costs
The Company capitalizes certain legal, accounting and other third-party fees that are directly associated with in-process equity financings as other assets until such financings are consummated. After consummation of the equity financing in July 2014, $2,285 of these costs were recorded in stockholders’ equity (deficit) as a reduction of additional paid-in capital generated as a result of the initial public offering. As of March 31, 2015 and December 31, 2014, the Company had recorded $186 and $10, respectively, of deferred offering costs, which are included in prepaid expenses and other current assets in the accompanying consolidated balance sheet. These costs were in contemplation of the Company’s public offering of common stock which closed in April 2015.
Segment Data
The company manages its operations as a single segment for the purposes of assessing performance and making operating decisions. The Company’s singular focus is on advancing medicines to treat central nervous system disorders, where there are inadequate or no approved existing therapies, including status epilepticus. All tangible assets are held within the U.S.
| 3. | Accrued Expenses |
Accrued expenses consist of the following:
| March 31, 2015 | December 31, 2014 | |||||||
| Employee-related expenses |
$ | 619 | $ | 1,279 | ||||
| Development costs |
4,290 | 2,788 | ||||||
| Professional services |
432 | 574 | ||||||
| Other accrued expenses |
181 | 46 | ||||||
|
|
|
|
|
|||||
| $ | 5,522 | $ | 4,687 | |||||
|
|
|
|
|
|||||
| 4. | Commitments and contingencies |
CyDex License Agreement
In October 2011, the Company entered into a research and development license with CyDex Pharmaceuticals, Inc. (“CyDex”) for the development of drug product using licensed technology for a period of one year. Under the terms of the license agreement, the Company paid an initial licensing fee of $200 and an additional fee of $100 for CyDex to perform research and development services to evaluate the licensed technology for formulation with the Company’s developmental product.
The $200 payment was recorded as research and development expense as the acquired technology was in-process research and development, and the $100 payment was recorded to research and development expense in 2011 and 2012 as services were performed.
In December 2012, the Company exercised its option to enter into a commercial license and supply agreement for CyDex’s proprietary technology and paid $100 for the perpetual license, which was recorded as research and development expense.
In August 2013, the Company entered into a commercial license agreement as a result of which the December 2012 license was terminated and the December 2012 supply agreement was amended. Specifically, CyDex granted the Company an exclusive license to the CyDex technology for use in the fields of status epilepticus and traumatic brain injury. In exchange, the Company is required to pay upfront, milestone and royalty-based compensation. In addition, CyDex granted the Company a research license to Captisol for allopregnanolone for use in proof of concept studies. The August 2013 agreement will continue in effect unless and until terminated. In consideration for the amended license rights, the Company paid $300. The Company is obligated to make milestone payments based on achievement of clinical development and regulatory milestones of $900 and $3,750, respectively. Also under this agreement, the Company is required to pay royalties in the low single digits based on levels of net sales.
10
Table of Contents
Under the amended supply agreement with CyDex, the Company is required to purchase all of its supply of Captisol from CyDex and CyDex is required to supply the Company with Captisol, subject to certain limitations.
In April 2014, the Company amended its commercial license and supply agreements with CyDex to expand the fields of use to include the treatment, prevention or diagnosis of any disease or symptom in humans or animals. In consideration for the amended terms, the Company paid $200 upfront and is obligated to make milestone payments, once per field, based on the achievement of clinical development and regulatory milestones for the development of SAGE-547 in the fields of status epilepticus and traumatic brain injury of $750 and $3,750, respectively. For the development in two additional fields, the Company is obligated to make milestone payments, once per field, based on the achievement of clinical development and regulatory milestones of $1,250 and $8,500, respectively.
In March 2015, a clinical development milestone was met for one of the programs. Accordingly, the Company recorded research and development expense for the three months ended March 31, 2015 of $250.
In April 2015, an additional clinical development milestone was met for one of the programs. Accordingly, the Company will record research and development expense for the three months ended June 30, 2015 of $500.
University of California License Agreement
In October 2013, the Company entered into a non-exclusive license agreement with The Regents of the University of California whereby the Company was granted a non-exclusive license to certain clinical data and clinical material for use in the development and commercialization of biopharmaceutical products in the licensed field, including status epilepticus and post-partum depression. In May 2014, the license agreement was amended to add the treatment of essential tremor to the licensed field of use, materials and milestone fee provisions of the agreement.
The Company will be required to pay clinical development milestones of up to $100 and pay royalties of less than 1% on net sales for a period of fifteen years following the sale of the first commercial product.
The license will terminate on the earlier to occur of (i) 27 years after the effective date or (ii) 15 years after the last-derived product is first commercially sold.
In March 2015, a clinical development milestone was met. Accordingly, the Company recorded research and development expenses for the three months ended March 31, 2015 totaling $75.
Consulting Agreement
In January 2014, the Company entered into a consulting agreement with a nonemployee advisor whereby the Company is obligated to make cash payments of up to $2,000 and to issue up to 126,984 shares of common stock upon attainment of certain clinical development and regulatory milestones.
In January and March 2014, the first clinical development milestones for each of two programs included in the consulting agreement were met. Accordingly, the Company recorded research and development expense for the year ended December 31, 2014 of $177, comprised of $50 in cash and $127 related to the issuance of 15,872 shares of the Company’s common stock.
In March 2015, the second clinical development milestone for one of the programs included in the consulting agreement was met. Accordingly, the Company recorded research and development expense for the three months ended March 31, 2015 of $574, comprised of $150 in cash and $424 related to the issuance of 7,936 shares of the Company’s common stock.
In April 2015, the third clinical development milestone for one of the programs included in the consulting agreement was met. Accordingly, the Company will record research and development expense for the three months ended June 30, 2015 of $1,087, comprised of $300 in cash and $787 related to the issuance of 15,873 shares of the Company’s common stock.
11
Table of Contents
| 5. | Stock-Based Compensation |
2014 Stock Option Plan
On July 2, 2014, the Company’s stockholders approved the 2014 Stock Option and Incentive Plan (the “2014 Stock Option Plan”), which became effective upon the completion of the IPO. The 2014 Stock Option Plan provides for the grant of restricted stock awards, incentive stock options, non-statutory stock options, among others. The 2014 Stock Option Plan replaced the Company’s 2011 Stock Option and Grant Plan (the “2011 Stock Option Plan”). The Company will grant no further stock options or other awards under the 2011 Stock Option Plan. Any options or awards outstanding under the 2011 Stock Option Plan remained outstanding and effective. As of March 31, 2015, the total number of shares reserved under all equity plans is 4,240,053 and the Company had 1,459,354 shares available for future issuance under such plans.
The 2014 Stock Option Plan provides for an annual increase, to be added on the first day of each fiscal year, by up to 4% of the Company’s issued and outstanding shares of common stock on the immediately preceding December 31. On January 1, 2015, 773,779 shares of common stock, representing 3% of the Company’s issued and outstanding shares of common stock as of December 31, 2014, were added to the 2014 Stock Option Plan. Such shares are included in the equity plan totals specified in the paragraph above.
2014 Employee Stock Purchase Plan
On July 2, 2014, the Company’s stockholders approved the 2014 Employee Stock Purchase Plan. A total of 282,000 shares of common stock were initially authorized for issuance under this plan. The 2014 Employee Stock Purchase Plan became effective upon the completion of the IPO. As of March 31, 2015, no shares have been issued under this plan.
Stock-Based Compensation
Terms of restricted stock awards and stock option agreements, including vesting requirements, are determined by the Compensation Committee of the Board of Directors or the Board of Directors, subject to the provisions of the applicable stock option plan. Options and restricted stock awards granted by the Company generally vest based on the grantee’s continued service with the Company during a specified period following grant. Awards generally vest ratably over four years, with a 25% cliff vesting at the one year anniversary for new employee awards. During 2013, the Company also granted a pool of option awards which vest ratably over one year. During the three months ended March 31, 2015, the Company granted options to purchase 497,100 shares of common stock to employees, which contain performance-based vesting criteria. Performance-based vesting criteria for these options primarily relate to milestone events specific to the Company’s corporate goals, including but not limited to certain clinical and regulatory development milestones related to the Company’s product candidates. Recognition of stock-based compensation expense associated with these performance-based stock options commences when the performance condition is considered probable of achievement using management’s best estimates. As of March 31, 2015, no performance-based milestones had been achieved, achievement of the milestones was not deemed probable and no expense has been recognized related to these awards.
All awards are exercisable from the date of grant for a period of ten years.
12
Table of Contents
The stock-based compensation expense recognized during the three months ended March 31, 2015 and 2014 was as follows:
| Three Months Ended March 31, | ||||||||
| 2015 | 2014 | |||||||
| Stock compensation expense: |
||||||||
| Research and development |
$ | 523 | $ | 106 | ||||
| General and administrative |
826 | 54 | ||||||
|
|
|
|
|
|||||
| $ | 1,349 | $ | 160 | |||||
|
|
|
|
|
|||||
For stock option awards, the fair value of the options is estimated at the grant date using the Black-Scholes option-pricing model, taking into account the terms and conditions upon which options are granted. The fair value of the options is amortized on a straight-line basis over the requisite service period of the awards. The weighted average grant date exercise price per share relating to outstanding stock options granted under the Company’s stock option plans during the three months ended March 31, 2015 and 2014 was $38.28 and $4.64, respectively. The weighted average Black-Scholes value per share relating to outstanding stock options granted under the Company’s stock option plans during the three months ended March 31, 2015 was $28.76.
The fair value of each option granted to employees and directors during the three months ended March 31, 2015 and 2014 under the Company’s stock option plans has been calculated on the date of grant using the following weighted average assumptions:
Black-Scholes Assumptions:
| Three Months Ended March 31, 2015 |
Three Months Ended March 31, 2014 |
|||||||
| Expected dividend yield |
0 | % | 0 | % | ||||
| Expected volatility |
93 | % | 99.85 | % | ||||
| Risk free interest rate |
1.46 | % | 1.98 | % | ||||
| Expected term |
5.89 years | 5.98 years | ||||||
Expected dividend yield: The Company has not paid and does not anticipate paying any dividends in the foreseeable future.
Risk-free interest rate: The Company determined the risk-free interest rate by using a weighted average equivalent to the expected term based on the U.S. Treasury yield curve in effect as of the date of grant.
Expected volatility: As the Company has only been a public company since July 2014, there is not sufficient historical volatility for the expected term of the options. Therefore, the Company used an average historical share price volatility based on an analysis of reported data for a peer group of comparable companies.
Expected term (in years): Expected term represents the period that the Company’s share option grants are expected to be outstanding. As the Company has only been a public company since July 2014, there is not sufficient historical term data to calculate the expected term of the options. Therefore, the Company elected to utilize the “simplified” method to estimate the expected term of option grants issued to employees. Under this approach, the weighted average expected life is presumed to be the average of the vesting term and the contractual term of the option.
Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from estimates. The Company estimates forfeitures based on historical termination behavior. For the three months ended March 31, 2015 and 2014, a forfeiture rate of 10% was applied.
For options granted to nonemployees, the expected life of the option used is ten years, which is the contractual term of each such option. All other assumptions used to calculate the grant date fair value are generally consistent with the assumptions used for options granted to employees.
13
Table of Contents
The table below summarizes activity related to stock options:
| Shares | Weighted-Average Exercise Price |
Weighted-Average Remaining Life (in years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding as of December 31, 2014 |
1,996,615 | $ | 7.01 | 8.98 | $ | 59,362 | ||||||||||
| Granted |
904,475 | 38.28 | ||||||||||||||
| Exercised |
(39,594 | ) | 3.72 | 1,756 | ||||||||||||
| Forfeited |
(80,797 | ) | 26.01 | 1,595 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding as of March 31, 2015 |
2,780,699 | $ | 16.67 | 9.07 | $ | 93,152 | ||||||||||
|
|
|
|||||||||||||||
| Vested or expected to vest as of March 31, 2015 |
2,462,001 | $ | 15.89 | 9.04 | $ | 84,546 | ||||||||||
|
|
|
|||||||||||||||
| Exercisable as of March 31, 2015 |
458,113 | $ | 1.06 | 8.44 | $ | 22,370 | ||||||||||
|
|
|
|||||||||||||||
As of March 31, 2015, the Company had unrecognized stock-based compensation expense related to its unvested stock option awards of $17,223, which is expected to be recognized over the remaining weighted average vesting period of 3.01 years. The total fair value of shares vested for the three months ended March 31, 2015 and 2014 was $380 and 81, respectively. During the three months ended March 31, 2015 and 2014, stock option exercises resulted in proceeds of $147 and $2, respectively. The intrinsic value of stock options exercised during the three months ended March 31, 2015 and 2014 was $1,756 and $30, respectively.
Restricted Stock Awards
The Company granted restricted stock awards to certain officers, employees, directors, and consultants of the Company. During the three months ended March 31, 2015 and 2014, the Company recorded $82 and $22, respectively, of stock-based compensation expense related to its restricted stock.
The table below summarizes activity relating to restricted stock:
| Shares | Weighted Average Grant Date Fair Value Per Share |
|||||||
| Outstanding as of December 31, 2014 |
170,832 | $ | — | |||||
| Issued |
— | — | ||||||
| Vested |
(33,175 | ) | — | |||||
| Forfeited |
— | — | ||||||
| Repurchased |
— | — | ||||||
|
|
|
|||||||
| Outstanding as of March 31, 2015 |
137,657 | — | ||||||
|
|
|
|||||||
As of March 31, 2015 and 2014, the Company had unrecognized stock-based compensation expense related to its unvested restricted stock awards of $266 and $123, respectively, which is expected to be recognized over the remaining weighted average vesting period of 0.91 years and 1.9 years, respectively.
Unvested shares are subject to repurchase by the Company, at the issuance price, upon the employee’s termination at the Company’s sole discretion. No shares of restricted stock were repurchased in the three months ended March 31, 2015.
14
Table of Contents
| 6. | Net Loss Per Share |
Basic and diluted net loss per share attributable to common stockholders was calculated as follows for the three months ended March 31, 2015 and 2014:
| Three Months Ended March 31, | ||||||||
| 2015 | 2014 | |||||||
| Basic net loss per share attributable to common stockholders: |
||||||||
| Numerator: |
||||||||
| Net loss |
$ | (16,871 | ) | $ | (6,116 | ) | ||
|
|
|
|
|
|||||
| Denominator: |
||||||||
| Weighted average common shares outstanding—basic |
25,655,883 | 1,652,726 | ||||||
| Dilutive effect of common share equivalents resulting from common share options and preferred common shares (as converted) |
— | — | ||||||
|
|
|
|
|
|||||
| Weighted average common shares outstanding—diluted |
25,655,883 | 1,652,726 | ||||||
|
|
|
|
|
|||||
| Net loss per share attributable to common stockholders—basic and diluted |
$ | (0.66 | ) | $ | (3.70 | ) | ||
|
|
|
|
|
|||||
The following common stock equivalents outstanding as of March 31, 2015 and 2014 were excluded from the computation of diluted net loss per share attributable to common stockholders for the periods presented because including them would have been anti-dilutive:
| Three Months Ended March 31, | ||||||||
| 2015 | 2014 | |||||||
| Options to purchase common stock |
1,736,871 | 1,269,418 | ||||||
| Restricted stock |
137,184 | 268,960 | ||||||
| Redeemable convertible preferred stock (presented on a weighted average basis) |
— | 15,121,600 | ||||||
|
|
|
|
|
|||||
| 1,874,055 | 16,659,978 | |||||||
|
|
|
|
|
|||||
| 7. | Income Taxes |
The Company did not record a federal or state income tax benefit for the Company’s losses for the three months ended March 31, 2015 and 2014 due to the Company’s conclusion that a valuation allowance is required.
| 8. | Related Party Transactions |
Since inception, the Company has received consulting and management services from Third Rock Ventures LLC, which through its affiliates, owns 37.8% of the Company’s common stock at March 31, 2015. The Company incurred $8 and $99 for these services during the three months ended March 31, 2015 and 2014, respectively. At March 31, 2015, the Company owed Third Rock Ventures LLC $4, which is included in accounts payable. At December 31, 2014, the Company owed Third Rock Ventures LLC $5, which is included in accrued expenses.
15
Table of Contents
| Item 2. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited consolidated financial statements and related notes appearing elsewhere in this Quarterly Report on Form 10-Q (“Quarterly Report”) and the Annual Report on Form 10-K (“Annual Report”) and the audited financial information and the notes thereto.
Our actual results and timing of certain events may differ materially from the results discussed, projected, anticipated, or indicated in any forward-looking statements. We caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained in this Quarterly Report. In addition, even if our results of operations, financial condition and liquidity, and the development of the industry in which we operate are consistent with the forward-looking statements contained in this Quarterly Report, they may not be predictive of results or developments in future periods.
The following information and any forward-looking statements should be considered in light of factors discussed elsewhere in the Quarterly Report, including those risks identified under Part II, Item 1A. Risk Factors.
We caution readers not to place undue reliance on any forward-looking statements made by us, which speak only as of the date they are made. We disclaim any obligation, except as specifically required by law and the rules of the SEC, to publicly update or revise any such statements to reflect any change in our expectations or in events, conditions or circumstances on which any such statements may be based, or that may affect the likelihood that actual results will differ from those set forth in the forward-looking statements.
Overview
We are a biopharmaceutical company committed to developing and commercializing novel medicines to treat life-threatening, rare central nervous system, or CNS, disorders, where there are inadequate or no approved existing therapies. We are targeting CNS indications where patient populations are easily identified, acute treatment is typically initiated in the hospital setting, clinical endpoints are well-defined and development pathways are feasible.
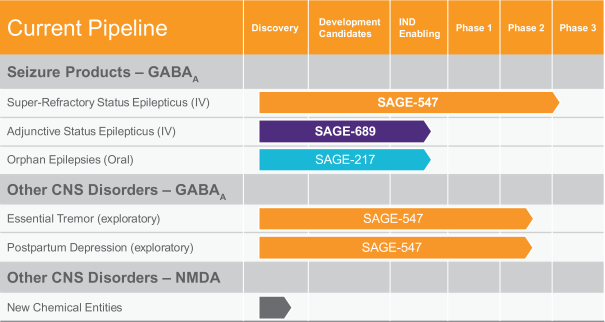
Our initial product candidates, which are summarized in the table below, are aimed at treating different stages of status epilepticus, or SE, a life-threatening condition in which the brain is in a state of persistent seizure, as well as other seizure and non-seizure disorders. The lead product candidate in our SE program, SAGE-547, is an intravenous, or IV, agent entering Phase 3 clinical development as an adjunctive therapy, a therapy combined with current therapeutic approaches, for the treatment of super-refractory SE, or SRSE. The current standard of care for SRSE is empiric, and there are no therapies at present that have been specifically approved for this indication. Over the course of 2014, the U.S. Food and Drug Administration, or FDA, granted us orphan drug designation and Fast Track designation for our investigational new drug application for SAGE-547 as a treatment for SRSE. On April 2, 2015, we announced that at a recent End-of-Phase 2 meeting with the FDA, general agreement was reached on the design and key elements for our planned Phase 3 clinical program for SAGE-547 for the treatment of SRSE and we expect to initiate the Phase 3 trial in mid-2015. If successful, we believe the results from this Phase 3 clinical trial, together with other clinical data obtained from the SAGE-547 development program, could form the basis of a New Drug Application, or NDA, submission for SAGE-547.
On May 14, 2015, we reported final results from our Phase 1/2 clinical trial of SAGE-547 in SRSE. SAGE-547 demonstrated robust activity with 77% of 22 evaluable patients meeting the key efficacy endpoint of being successfully weaned off their anesthetic agents while SAGE-547 was being administered. In addition, 77% of the total evaluable patients were successfully weaned off SAGE-547 without recurrence of SRSE in the 24 hour period following treatment. SAGE-547 also demonstrated favorable tolerability and a benefit-risk profile supporting development for this acutely ill patient population. Overall, 64% of patients experienced at least one serious adverse event, though none were drug-related as determined by the Safety Review Committee. Independent of treatment response, six patient deaths occurred within the study period, all driven by underlying medical conditions.
We continue to use SAGE-547 to explore additional potential uses of GABAA receptor modulators in clinical trials for essential tremor, a debilitating neurological disorder that causes involuntary, rhythmic shaking with no known cause with over 10 million people in the United States with essential tremor, and severe post-partum depression, or severe PPD, a distinct and readily identified form of major depressive disorder estimated to affect up to 20% of women following childbirth. If these exploratory trials are successful, we plan to use the data from them to help guide the design of second-generation GABAA receptor modulators for the chronic treatment of these diseases.
Our next-generation product candidates, SAGE-689 and SAGE-217, utilize similar mechanistic pathways as SAGE-547 and are designed to have pharmaceutical properties which optimize both their non-clinical profiles and potential clinical profiles for the treatment of different stages of SE, as well as other seizure and non-seizure disorders.
16
Table of Contents
17
Table of Contents
Since our inception in April 2010, we have devoted substantially all of our resources to organizing and staffing our company, business planning, raising capital, identifying and developing our product candidates, preparing to conduct and conducting non-clinical and clinical trials of our product candidates, providing general and administrative support for these operations and protecting our intellectual property. We have funded our operations to date through sales of our common stock and redeemable convertible preferred stock; the issuance of convertible notes and through proceeds from our initial public offering of common stock, or IPO, and our follow-on offering of common stock that was completed in April 2015.
We have not generated any revenue to date. We have incurred net losses in each year since our inception, and we have an accumulated deficit of $83.7 million as of March 31, 2015. Our net losses were $16.9 million, $36.1 million and $18.3 million for the three months ended March 31, 2015 and the years ended December 31, 2014 and 2013, respectively. These losses have resulted principally from costs incurred in connection with research and development activities and general and administrative costs associated with our operations. We expect to incur significant expenses and increasing operating losses for the foreseeable future.
We expect that our expenses will increase substantially in connection with our ongoing activities, as we:
| • | advance clinical development of SAGE-547, our lead product candidate in our SE program, including completing our planned Phase 3 clinical trial for SAGE-547 in SRSE, late stage non-clinical studies of SAGE-547 and initial preparations for a potential commercial launch; |
| • | advance our clinical trials to establish proof of principle in additional indications including severe PPD and essential tremor; |
| • | advance development of SAGE-689 as an adjunctive second-line therapy for the treatment of SE, including conducting a Phase 1 clinical trial; |
| • | advance development of SAGE-217 as an oral monotherapy for orphan epilepsies such as Dravet syndrome and Rett syndrome, including conducting a Phase 1 clinical trial; |
| • | continue our research and development efforts for other drug candidates in the treatment of CNS disorders including on our early-stage novel allosteric modulators for NMDA; |
| • | seek regulatory approvals for our product candidates; |
| • | add personnel, including personnel to support our product development and future commercialization; |
| • | add operational, financial and management information systems; |
| • | maintain, leverage and expand our intellectual property portfolio; and |
| • | operate as a public company. |
As a result, we will need additional financing to support our continuing operations. Until such time that we can generate significant revenue from product sales, if ever, we expect to finance our operations through a combination of public or private equity or debt financings or other sources, which may include collaborations with third parties. Arrangements with collaborators or others may require us to relinquish rights to certain of our technologies or product candidates. In addition, we may never successfully complete development of any of our product candidates, obtain adequate patent protection for our technology, obtain necessary regulatory approval for our product candidates or achieve commercial viability for any approved product candidates. Adequate additional financing may not be available to us on acceptable terms, or at all. Our inability to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue our business strategy. We will need to generate significant revenue to achieve profitability, and we may never do so.
We expect that our existing cash and cash equivalents as of March 31, 2015, will enable us to fund our operating expenses and capital expenditures requirements for at least the next 12 months. See “—Liquidity and Capital Resources.”
18
Table of Contents
Financial Operations Overview
Revenue
We have not generated any revenue from product sales since our inception and do not expect to generate any revenue from the sale of products in the near future. If our development efforts result in clinical success and regulatory approval or collaboration agreements with third parties for our product candidates, we may generate revenue from those product candidates.
Operating Expenses
Our operating expenses since inception have consisted of research and development activities and general and administrative costs.
Research and Development Expenses
Research and development expenses, which consist primarily of costs associated with our product research and development efforts, are expensed as incurred. Research and development expenses consist primarily of:
| • | personnel costs, including salaries, related benefits, stock-based compensation and related travel expenses for employees engaged in scientific research and development functions; |
| • | expenses incurred under agreements with contract research organizations, or CROs, and investigative sites that conduct our non-clinical studies and clinical trials; |
| • | expenses associated with manufacturing clinical trial materials and developing external manufacturing capabilities; |
| • | costs of outside consultants, including their fees, stock-based compensation and related travel expenses; |
| • | other expenses related to our non-clinical studies and expenses related to our regulatory activities; and |
| • | payments made under our third-party licensing agreements. |
Costs for certain development activities are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and our clinical sites.
We have been developing SAGE-547, SAGE-689 and SAGE-217 and focusing on other research and development programs related to exploratory efforts, target validation and lead optimization for our earlier-validated programs. Our direct research and development expenses are tracked on a program-by-program basis and consist primarily of external costs, such as fees paid to investigators, central laboratories, CROs and contract manufacturing organizations, or CMOs in connection with our non-clinical studies and clinical trials; third-party license fees related to our product candidates; fees paid to outside consultants who perform work on our programs; and costs related to manufacturing or purchasing clinical trial materials. We do not allocate employee-related costs and other indirect costs to specific research and development programs because these costs are deployed across multiple product programs under research and development and, as such, are separately classified as unallocated research and development expenses.
19
Table of Contents
The following table summarizes our research and development expenses by program:
| Three Months Ended March 31, |
||||||||||||
| Increase (Decrease) |
||||||||||||
| 2015 | 2014 | |||||||||||
| (in thousands) | ||||||||||||
| SAGE-547 |
$ | 6,578 | $ | 1,174 | $ | 5,404 | ||||||
| SAGE-689 |
1,069 | 860 | 209 | |||||||||
| SAGE-217 |
1,407 | 667 | 740 | |||||||||
| Other research and development programs |
1,563 | 273 | 1,290 | |||||||||
| Unallocated expenses |
2,283 | 1,199 | 1,084 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research and development expenses |
$ | 12,900 | $ | 4,173 | $ | 8,727 | ||||||
|
|
|
|
|
|
|
|||||||
Research and development activities are central to our business. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will continue to increase in the foreseeable future as we initiate clinical trials for certain product candidates and pursue later stages of clinical development of our product candidates.
We cannot determine with certainty the duration and completion costs of the current or future clinical trials of our product candidates or if, when, or to what extent we will generate revenue from the commercialization and sale of any of our product candidates. The duration, costs, and timing of clinical trials and development of our product candidates will depend on a variety of factors, including:
| • | the scope, rate of progress, and expense of our ongoing as well as any additional non-clinical studies, clinical trials and other research and development activities; |
| • | future clinical trial results; |
| • | uncertainties in clinical trial enrollment rate or design; |
| • | significant and changing government regulation; and |
| • | the timing and receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate, or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
General and Administrative Expenses
General and administrative expenses consist primarily of personnel costs, consisting of salaries, related benefits, stock-based compensation and related travel expenses of our executive, finance, business and corporate development and other administrative functions. General and administrative expenses also include expenses incurred under agreements with third parties relating to initial commercial evaluation and planning, facilities and other expenses, including rent, depreciation, maintenance of facilities, insurance and supplies; and professional fees for audit, tax and legal services, including legal expenses to pursue patent protection of our intellectual property.
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support the expected growth in our research and development activities and the potential commercialization of our product candidates. We also anticipate increased expenses associated with being a public company, including costs related to audit, legal, regulatory and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, director and officer insurance premiums, and investor relations costs. Additionally, if and when we believe that a regulatory approval of the first product candidate appears likely, we anticipate an increase in payroll and related expenses as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of our product candidates.
Interest Income (Expense), net and Other Income (Expense), net
Interest income (expense), net, and other income (expense), net, were insignificant for the three months ended March 31, 2015 and 2014.
20
Table of Contents
Results of Operations
Comparison of Three Months Ended March 31, 2015 and 2014
The following table summarizes our results of operations for the three months ended March 31, 2015 and 2014:
| Three Months Ended March 31, |
||||||||||||
| Increase (Decrease) |
||||||||||||
| 2015 | 2014 | |||||||||||
| (in thousands) | ||||||||||||
| Operating expenses: |
||||||||||||
| Research and development |
$ | 12,900 | $ | 4,173 | $ | 8,727 | ||||||
| General and administration |
3,997 | 1,617 | 2,380 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
16,897 | 5,790 | 11,107 | |||||||||
| Loss from operations |
(16,897 | ) | (5,790 | ) | (11,107 | ) | ||||||
| Interest income (expense), net |
21 | — | 21 | |||||||||
| Other income (expense), net |
5 | — | 5 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (16,871 | ) | $ | (5,790 | ) | $ | (11,081 | ) | |||
|
|
|
|
|
|
|
|||||||
Research and development expenses
| Three Months Ended March 31, |
||||||||||||
| Increase (Decrease) |
||||||||||||
| 2015 | 2014 | |||||||||||
| (in thousands) | ||||||||||||
| SAGE-547 |
$ | 6,578 | $ | 1,174 | $ | 5,404 | ||||||
| SAGE-689 |
1,069 | 860 | 209 | |||||||||
| SAGE-217 |
1,407 | 667 | 740 | |||||||||
| Other research and development programs |
1,563 | 273 | 1,290 | |||||||||
| Unallocated expenses |
2,283 | 1,199 | 1,084 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research and development expenses |
$ | 12,900 | $ | 4,173 | $ | 8,727 | ||||||
|
|
|
|
|
|
|
|||||||
Research and development expenses for the three months ended March 31, 2015 were $12.9 million compared to $4.2 million for the three months ended March 31, 2014. The increase of $8.7 million period over period was primarily due to the following:
| • | an increase of $5.4 million in expenses of our SAGE-547 program, due to the advancement of the program into clinical development, including Phase 1/2 clinical trial costs, CMC and toxicology. The costs in both periods include development milestones earned by consultants and licensors; |
| • | an increase of $0.2 million in expenses of our SAGE-689 program, with advancement of the lead optimization programs into IND-enabling non-clinical development activities (e.g., toxicology studies, process development, and drug substance manufacturing); |
| • | an increase of $0.7 million in expenses of our SAGE-217 program with advancement of the lead optimization program into IND-enabling non-clinical development (e.g., toxicology studies, process development, and drug substance manufacturing); |
| • | an increase of $1.3 million in expenses of our other research and development programs and discovery efforts for our next clinical candidates and back-up programs; and |
| • | an increase of $1.1 million in employee-related expenses, including an increase of $0.4 million of non-cash stock-based compensation expense and the effects of hiring additional full-time employees to support the growth in our research and development activities. |
General and administrative expenses
| Three Months Ended March 31, |
||||||||||||
| 2015 | 2014 | Increase (Decrease) |
||||||||||
| (in thousands) | ||||||||||||
| Personnel-related |
$ | 1,923 | $ | 627 | $ | 1,296 | ||||||
| Professional fees |
1,358 | 737 | 621 | |||||||||
| Facilities |
103 | 98 | 5 | |||||||||
| Other |
613 | 155 | 458 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total general and administrative expenses |
$ | 3,997 | $ | 1,617 | $ | 2,380 | ||||||
|
|
|
|
|
|
|
|||||||
21
Table of Contents
General and administrative expenses for the three months ended March 31, 2015 were $4.0 million, compared to $1.6 million for the three months ended March 31, 2014. The increase of $2.4 million in general and administrative expenses was primarily due to the $1.3 million increase in personnel-related costs due to the effects of hiring additional, full-time employees during 2014 to support operations, finance, human resources and early commercial planning activities, including an increase of $0.8 million in non-cash stock-based compensation expense. The increase of $0.6 million in professional fees were associated with being a public company, including costs related to audit, legal, regulatory and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, as well as investor relations costs.
Other income (expense), net
Interest income (expense), net, and other income (expense), net, were insignificant for the three months ended March 31, 2015 and 2014.
Liquidity and Capital Resources
Since our inception in April 2010, we have not generated any revenue and have incurred recurring net losses. As of March 31, 2015, we had an accumulated deficit of $83.7 million. From our inception through December 31, 2014, we have received net proceeds of $184.6 million from the sales of redeemable convertible preferred stock, the issuance of convertible notes and the proceeds from our IPO. On April 20, 2015, we completed the sale of 2,628,571 shares of common stock in our underwritten public offering of our common stock at a price to the public of $52.50 per share, resulting in gross proceeds of $138.0 million before deducting underwriting discounts and commissions and offering expenses payable by us.
As of March 31, 2015, our primary sources of liquidity were our cash and cash equivalents, which totaled $113.2 million. We invest our cash equivalents in highly liquid, interest-bearing investment-grade and government securities in order to preserve principal.
The following table summarizes the primary sources and uses of cash for the periods presented below:
| Three Months Ended March 31, |
||||||||
| 2015 | 2014 | |||||||
| (in thousands) | ||||||||
| Net cash provided by (used in): |
||||||||
| Operating activities |
$ | (14,606 | ) | $ | (5,616 | ) | ||
| Investing activities |
(110 | ) | (3 | ) | ||||
| Financing activities |
112 | 52,978 | ||||||
|
|
|
|
|
|||||
| Net increase (decrease) in cash and cash equivalents |
$ | (14,604 | ) | $ | 47,359 | |||
|
|
|
|
|
|||||
Operating activities
Operating activities used $14.6 million of cash in the three months ended March 31, 2015. The cash flow used in operating activities resulted primarily from our net loss of $16.9 million for the period and cash used for changes in our operating assets and liabilities of $0.5 million and by non-cash charges of $1.8 million. Our net loss was primarily attributable to research and development activities related to our lead programs in development and our general and administrative expenses, as we had no revenue in the period. Our non-cash charges during the three months ended March 31, 2015 primarily consisted of stock-based compensation expenses of $1.3 million and a non-cash licensing and consulting fee of $0.4 million. Net cash used in changes in our operating assets and liabilities consisted primarily of an increase in accounts payable of $0.3 million. Our prepaid expenses and other current assets, accounts payable and accrued expense balances were affected by the timing of vendor invoicing and payments.
During the three months ended March 31, 2014, operating activities used $5.6 million of cash, primarily resulting from our net loss of $5.8 million, partially offset by non-cash charges of $0.3 million and cash used by changes in our operating assets and liabilities of $0.1 million. Our net loss was primarily attributed to research and development activities related to our lead programs in development and our general and administrative expenses, as we had no revenue in the period. Our net non-cash charges during the three months ended March 31, 2014 primarily consisted of stock-based compensation expenses of $0.2 million and a non-cash licensing and consulting fee of $0.1 million. Net cash used in changes in our operating assets and liabilities consisted primarily of increases in accounts payable and accrued expenses totaling $0.1 million, offset by a $0.1 million increase in prepaid expenses and other current assets. Our prepaid expenses and other current assets, accounts payable and accrued expense balances were affected by the timing of vendor invoicing and payments.
22
Table of Contents
Investing activities
During the three months ended March 31, 2015, we used $0.1 million of cash for purchases of property and equipment.
During the three months ended March 31, 2014, we had no significant purchases of property and equipment.
Financing activities
During the three months ended March 31, 2015 and 2014, net cash provided by financing activities was $0.1 million and $53.0 million, respectively. Net cash provided by financing activities in the three months ended March 31, 2015 consisted mainly of $0.1 million of proceeds from the issuance of common stock from exercises of options. Net cash provided by financing activities in the three months ended March 31, 2014 consisted of $53.0 million from the issuance of Series B and Series C redeemable convertible preferred stock.
Operating Capital Requirements
To date, we have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize one of our current or future product candidates. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for,
23
Table of Contents
our product candidates and begin to commercialize any approved products. We expect to incur additional costs associated with operating as a public company. In addition, subject to obtaining regulatory approval of any of our product candidates, we expect to incur significant commercialization expenses for product sales, marketing and manufacturing. Accordingly, we anticipate that we will need substantial additional funding in connection with our continuing operations.
Based on our current operating plan, we expect that our existing cash and cash equivalents as of
March 31, 2015, including the net proceeds from our IPO which closed on July 23, 2014 and the sale of common stock in our underwritten public offering of common stock that closed on April 20, 2015, will enable us to fund our operating
expenses and capital expenditure requirements through mid-2017. In that time, we expect that our expenses will increase substantially as we continue clinical development of SAGE-547, including completing our Phase 3 clinical trial, fund IND-enabling
activities and Phase 1 clinical development for
SAGE-689, fund IND-enabling activities and Phase 1 clinical development for SAGE-217, fund new and ongoing research and development activities and working capital, and fund other general corporate
purposes. We have based our estimates on assumptions that could change, and we may use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and
commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures necessary to complete the development and commercialization of our product candidates.
Our future capital requirements will depend on many factors, including:
| • | the costs, timing, and outcome of regulatory reviews and approvals; |
| • | the ability of our product candidates to progress through clinical development successfully; |
| • | the initiation, progress, timings, costs, and results of non-clinical studies and clinical trials for our other programs and potential product candidates; |
| • | the number and characteristics of the product candidate we pursue; |
| • | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
| • | the extent to which we acquire or in-license other products and technologies; and |
| • | our ability to establish any future collaboration arrangements on favorable terms, if at all. |
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends and may require the issuance of warrants, which could potentially dilute your ownership interest. If we raise additional funds through collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or research programs or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves.
24
Table of Contents
Contractual Obligations and Commitments
The following table summarizes our contractual obligations at March 31, 2015 and the effect such obligations are expected to have on our liquidity and cash flow in future periods:
| Payments Due by Period | ||||||||||||||||||||
| Total | Less Than 1 year |
1-3 Years | 3-5 Years | More Than 5 years |
||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Operating lease commitments (1) |
$ | 751 | $ | 365 | $ | 386 | $ | — | $ | — | ||||||||||
| Milestones under a licensing agreement (2) |
500 | 500 | — | — | — | |||||||||||||||
| Milestones under a consulting agreement (4) |
300 | 300 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total (1)(2)(3)(4) |
$ | 1,551 | $ | 1,165 | $ | 386 | $ | — | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (1) | We lease office space in Cambridge, Massachusetts under operating lease agreements that initially expire on February 28, 2017 and July 31, 2017. The minimum lease payments in the table do not include related common area maintenance charges or real estate taxes, which costs are variable. |
| (2) | We have acquired exclusive and non-exclusive rights to use, research, develop and offer for sale certain products and patents under three separate licensing agreements, including amendments entered into in April and May 2014, with Washington University, CyDex Pharmaceuticals, Inc. and The Regents of the University of California. The licensing rights obligate us to make payments to the licensors for license fees, milestones, license maintenance fees and royalties. We are obligated to make future remaining milestone payments under these agreements of up to $29.5 million upon achieving certain pre-commercialization milestones, such as clinical trials and regulatory approvals. |
In April 2015, a clinical development milestone was met for one of the programs. Accordingly, we will record research and development expense for the three months ended June 30, 2015 of $500. This amount is shown in the table.
Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain milestones. These contingent milestones may not be achieved. We have not included any of these amounts in the table as we cannot estimate or predict when, or if, these amounts will become due.
In addition, under the licensing agreements, we will owe single-digit royalties on sales of commercial products, if any, developed using the licensed technologies. Under two of these license agreements, we are obligated to pay to the licensors a percentage of fees received if and when we sublicense the technologies. As of March 31, 2015, we had not developed a commercial product using the licensed technologies and we had not entered into any sublicense agreements for the technologies. We have not included any of these amounts in the table as we cannot estimate or predict when, or if, these amounts will become due.
| (3) | We enter into contracts in the normal course of business with CROs for clinical trials, non-clinical research studies and testing, manufacturing and other services and products for operating purposes. These contracts generally provide for termination upon notice, and therefore we believe that our non-cancelable obligations under these agreements are not material. |
| (4) | Under a January 2014 consulting agreement, we are obligated to make remaining milestone payments of up to $1.8 million and to issue up to 103,176 shares of our common stock to a nonemployee consultant upon achieving certain clinical trial milestones and regulatory approval milestones. |
In April 2015, the third clinical development milestone for one of the programs included in the consulting agreement was met. Accordingly, we will record research and development expense for the three months ended June 30, 2015 of $1,087, comprised of $300 in cash and $787 related to the issuance of 15,873 shares of our common stock. The cash portion is shown in the table.
Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain milestones. These contingent milestones may not be achieved. We have not included any of these amounts in the table as we cannot estimate or predict when, or if, these amounts will become due.
25
Table of Contents
Off-Balance Sheet Arrangements
We do not currently have, nor did we have during the periods presented, any off-balance sheet arrangements as defined by SEC rules.
Application of Critical Accounting Policies
We have prepared our consolidated financial statements in accordance with U.S. generally accepted accounting principles. Our preparation of these consolidated financial statements requires us to make estimates, assumptions, and judgments that affect the reported amounts of assets, liabilities, expenses, and related disclosures at the date of the consolidated financial statements, as well as revenue and expenses recorded during the reporting periods. We evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results could therefore differ materially from these estimates under different assumptions or conditions.
There have been no material changes to our critical accounting policies from those described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in our Annual Report on Form 10-K filed by us with the SEC on March 6, 2015.
Recently Issued Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”), issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606), (“ASU 2014-09”). ASU 2014-09 outlines a new, single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. This new revenue recognition model provides a five-step analysis in determining when and how revenue is recognized. The new model will require revenue recognition to depict the transfer of promised goods or services to customers in an amount that reflects the consideration a company expects to receive in exchange for those goods or services. In April 2015, the FASB proposed a one year delay in the effective date of ASU 2014-09, which was originally effective for public entities for annual reporting periods beginning after December 15, 2016 and interim periods within those periods. Early adoption is not permitted. Companies may use either a full retrospective or a modified retrospective approach to adopt ASU 2014-09. We are currently assessing the method of adoption and the impact of adopting this new accounting guidance will have on our consolidated financial statements and footnote disclosures.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements — Going Concern (Subtopic 205-40). The new guidance addresses management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. Management’s evaluation should be based on relevant conditions and events that are known and reasonably knowable at the date that the financial statements are issued. The standard will be effective for the first interim period within annual reporting periods beginning after December 15, 2016. Early adoption is permitted. We are evaluating the effect that this guidance will have on our consolidated financial statements.
In April 2015, the FASB issued guidance simplifying the presentation of debt issuance costs. To simplify presentation of debt issue costs, the amendments require that debt issue costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. The guidance becomes effective for us in the year ending December 31, 2016, and early adoption is permitted. We are currently assessing the impact that this standard will have on our consolidated financial statements.
| Item 3. | Quantitative and Qualitative Disclosure about Market Risk |
We had cash and cash equivalents of approximately $113.2 million at March 31, 2015. The primary objectives of our investment activities are to preserve principal, provide liquidity and maximize income without significantly increasing risk. Our primary exposure to market risk relates to fluctuations in interest rates, which are affected by changes in the general level of U.S. interest rates. Given the short-term nature of our cash and cash equivalents, we believe that a sudden change in market interest rates would not be expected to have a material impact on our financial condition and/or results of operation. We do not have any foreign currency or other derivatives financial instruments.
We do not believe that our cash and cash equivalents have significant risk of default or illiquidity. While we believe our cash and cash equivalents do not contain excessive risk, we cannot provide absolute assurance that in the future our investments will not be subject to adverse changes in market value. In addition, we maintain significant amounts of cash and cash equivalents at one or more financial institutions that are in excess of federally insured limits.
26
Table of Contents
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation had a material effect on our results of operations during the year ended March 31, 2015.
| Item 4. | Controls and Procedures |
Management’s Evaluation of our Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) or 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act) that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is (1) recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and (2) accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
As of March 31, 2015, our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this report. In designing and evaluating our disclosure controls and procedures, our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our principal executive officer and principal financial officer have concluded based upon the evaluation described above that, as of March 31, 2015, our disclosure controls and procedures were effective at the reasonable assurance level.
27
Table of Contents
We continue to review and document our disclosure controls and procedures, including our internal controls and procedures for financial reporting, and may from time to time make changes aimed at enhancing their effectiveness and to ensure that our systems evolve with our business.
Changes in Internal Control Over Financial Reporting
During the three months ended March 31, 2015, there have been no changes in our internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15(d)-15(f) promulgated under the Securities Exchange Act of 1934, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
28
Table of Contents
PART II — OTHER INFORMATION
As of the date of this filing, we were not party to any legal matters or claims. In the future, we may become party to legal matters and claims arising in the ordinary course of business, the resolution of which we do not anticipate would have a material adverse impact on our financial position, results of operations or cash flows.
| Item 1A. | Risk Factors |
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as the other information in this Quarterly Report on Form 10-Q and in our other public filings before making an investment decision. Our business, prospects, financial condition, or operating results could be harmed by any of these risks, as well as other risks not currently known to us or that we currently consider immaterial. If any such risks or uncertainties actually occur, our business, financial condition or operating results could differ materially from the plans, projections and other forward-looking statements included in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this report and in our other public filings. The trading price of our common stock could decline due to any of these risks, and as a result, you may lose all or part of your investment.
Risks Related to Product Development, Regulatory Approval and Commercialization
We depend heavily on the success of the product candidates within our seizure program, of which SAGE-547 is entering Phase 3 clinical development and SAGE-689 and SAGE-217 are in non-clinical development. We cannot be certain that we will be able to obtain regulatory approval for, or successfully commercialize, any of our product candidates.
We currently have no drug products for sale and may never be able to develop marketable drug products. Our business depends heavily on the successful non-clinical and clinical development, regulatory approval and commercialization of the product candidates in our lead program in status epilepticus, or SE, of which only one product candidate, SAGE-547, is entering Phase 3 clinical development for the treatment of super-refractory SE, or SRSE, and our other product candidates, SAGE-689 and SAGE-217, are in non-clinical development. SAGE-547 will require substantial additional clinical development, testing and regulatory approval before we are permitted to commence its commercialization. The non-clinical studies and clinical trials of our product candidates are, and the manufacturing and marketing of our product candidates will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and, if approved, market any product candidate. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through non-clinical studies and clinical trials that the product candidate is safe and effective for use in each target indication. Drug development is a long, expensive and uncertain process, and delay or failure can occur at any stage of any of our clinical trials. This process can take many years and may include post-marketing studies and surveillance, which will require the expenditure of substantial resources. Of the large number of drugs in development in the United States, only a small percentage will successfully complete the U.S. Food and Drug Administration, or FDA, regulatory approval process and will be commercialized. Accordingly, even if we are able to obtain the requisite financing to continue to fund our development and non-clinical studies and clinical trials, we cannot assure you that any of our product candidates will be successfully developed or commercialized.
Both SAGE-689 and SAGE-217 are in non-clinical development and have yet to begin the clinical development process. We plan to file Investigational New Drug Applications, or INDs, for both SAGE-689 and SAGE-217 late in 2015 and to begin a Phase 1 clinical trial for each of SAGE-689 and SAGE-217 thereafter.
We are not permitted to market our product candidates in the United States until we receive approval of a New Drug Application, or an NDA, from the FDA, or in any foreign countries until we receive the requisite approval from such countries.
Obtaining approval of an NDA is a complex, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of any of our product candidates for many reasons, including, among others:
| • | we may not be able to demonstrate that our product candidates are safe and effective in treating SE, refractory SE, or RSE, SRSE, essential tremor, or severe postpartum depression or severe PPD, as applicable, to the satisfaction of the FDA; |
| • | the results of our non-clinical studies and clinical trials may not meet the level of statistical or clinical significance required by the FDA for marketing approval; |
| • | the FDA may disagree with the number, design, size, conduct, implementation or differing drug formulation used in of our non-clinical studies and clinical trials; |
| • | the FDA may require that we conduct additional non-clinical studies and clinical trials; |
| • | the FDA or the applicable foreign regulatory agency may not approve the formulation, labeling or specifications of any of our product candidates; |
| • | the contract research organizations, or CROs, that we retain to conduct our non-clinical studies and clinical trials may take actions outside of our control that materially adversely impact our non-clinical studies and clinical trials; |
| • | the FDA may find the data from non-clinical studies and clinical trials insufficient to demonstrate that our product candidates’ clinical and other benefits outweigh their safety risks; |
29
Table of Contents
| • | the FDA may disagree with our interpretation of data from our non-clinical studies and clinical trials; |
| • | the FDA may not accept data generated at our non-clinical studies and clinical trial sites; |
| • | if our NDA, if and when submitted, is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional non-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
| • | the FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval or post-approval; |
| • | the FDA or the applicable foreign regulatory agency may determine that the manufacturing processes or facilities of third-party contract manufacturers with which we contract do not conform to applicable requirements, including current Good Manufacturing Practices, or cGMPs; or |
| • | the FDA or applicable foreign regulatory agency may change its approval policies or adopt new regulations. |
Any of these factors, many of which are beyond our control, could jeopardize our ability to obtain regulatory approval for and successfully market our product candidates. Any such setback in our pursuit of regulatory approval would have a material adverse effect on our business and prospects.
We cannot be certain that our planned Phase 3 clinical trial of SAGE-547 will be sufficient to support the submission of an NDA for this product candidate, and in any event we must obtain additional clinical and non-clinical data before an NDA may be submitted.
In general, the FDA requires two pivotal trials to support approval of an NDA, but in certain circumstances, will approve an NDA based on only one pivotal trial. If successful, we believe the results from our planned Phase 3 clinical trial of SAGE-547, together with other safety and efficacy data from the SAGE-547 development program, could form the basis of an NDA submission for SAGE-547. However, depending upon the outcome of the current program, the FDA may require that we conduct additional pivotal trials before we can submit an NDA for SAGE-547. To allow dosing in patients below the age of two we would need to either conduct additional clinical trial(s) or amend the protocol for our planned Phase 3 clinical trial.
Furthermore, we will need to complete several other clinical studies prior to submitting an NDA to the FDA, including an absorption, metabolism, and excretion pharmacokinetics study in healthy volunteers, studies to test the effect of SAGE-547 on exposure to phenytoin and in patients with severe renal impairment and patients with hepatic impairment, as well as a study to test the abuse potential of SAGE-547. If the result of these additional clinical studies are not positive or yield unanticipated results, it may delay or prevent the submission or approval of an NDA for SAGE-547.
While we believe we and the FDA are in general agreement on the design and key elements of our planned Phase 3 clinical trial for SAGE-547, before beginning the trial, the FDA must review the final protocol for the trial, along with additional information supporting the proposed trial design. Concurrent with starting the Phase 3 clinical trial, the FDA will review certain updated chemistry, manufacturing and controls, or CMC, information, that we are required to submit. We also plan to share with the FDA the results of our long-term toxicity studies in two animal species, the first segment of which we submitted to the FDA in the second quarter of 2014. Additional long-term toxicity studies, required for an NDA submission, are ongoing. If the FDA does not approve the protocol for the planned trial in the form we submit it, or if the FDA is not satisfied with the additional CMC information we plan to provide, the start or continuation of the planned Phase 3 trial may be delayed or the design of the trial may change. The FDA may require that we conduct additional toxicity studies and other non-clinical studies before submitting an NDA for SAGE-547.
A Fast Track designation by the FDA may not actually lead to a faster development or regulatory review or approval process.
We have received Fast Track designation for our investigational new drug application, or IND, for SAGE-547 for the treatment of SRSE, and in the future we may seek Fast Track designation for other product candidates as well. If a product is intended for the treatment of a serious or life-threatening condition and the product demonstrates the potential to address unmet medical needs for this condition, the sponsor may apply for the FDA Fast Track designation. Fast Track designation does not necessarily lead to a faster development pathway or regulatory review process and does increase the likelihood of regulatory approval. The FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from our clinical development programs.
The number of patients suffering from SE, RSE and SRSE is small or has not been established with precision. If the actual number of patients with SE, RSE and SRSE is smaller than we anticipate, we may encounter difficulties in enrolling patients in our clinical trials, thereby delaying or preventing development of our product candidates, and if any of our product candidates are approved, we believe our revenue and ability to achieve profitability would be materially adversely affected.
There is no precise method of establishing actual number of patients with SE, RSE or SRSE in any geography over any time period. Moreover, SE, RSE and SRSE are acute episode conditions. If we are not able to identify patients at the time of SE, RSE or SRSE onset, we will have difficulty completing our clinical trials. We estimate that the annual incidence of SE, RSE and SRSE in the United States is up to 150,000, 35,000 and 25,000 patients, respectively. If the actual number of patients with SE, RSE or SRSE is lower than we believe, we may experience
30
Table of Contents
difficulty in enrolling patients in our clinical trials, thereby delaying development of our product candidates. Further, if any of our product candidates are approved, the markets for our product candidates for these indications would be smaller than we anticipate, which could limit our ability to achieve profitability.
Favorable results from the emergency-use cases of SAGE-547 do not ensure that clinical trials will be successful and the results in any future emergency-use cases may not be positive and could adversely impact our clinical development plans.
SAGE-547 has been administered to a small number of patients as part of emergency-use cases, which permitted the administration of SAGE-547 outside of clinical trials. No assurance can be given that positive results observed to date in these emergency-use cases are attributable to SAGE-547, as they were not carried out in the controlled environment of a clinical trial. Further, no assurance can be provided that administration of SAGE-547 to other patients in any future emergency-use cases or otherwise will have positive results. Emergency use is a term that is used to refer to the use of an investigational drug outside of a clinical trial to treat a patient with a serious or immediately life-threatening disease or condition and who has no comparable or satisfactory alternative treatment options. Regulators often allow emergency use on a case-by-case basis for an individual patient or for defined groups of patients with similar treatment needs. In the event there are negative results in future emergency-use cases, it could adversely affect or delay our clinical development of SAGE-547.
If serious adverse events or other undesirable side effects are identified during the use of SAGE-547 in emergency-use cases, investigator sponsored trials or exploratory clinical trials of SAGE-547, it may adversely affect our development of SAGE-547 for SRSE.
In addition to use in emergency cases as described above, SAGE-547 is currently being tested in an investigator sponsored clinical trial for the treatment of traumatic brain injury, or TBI, by one of our collaborators and may be subjected to testing for other indications in additional investigator sponsored trials. Currently, we are also testing SAGE-547 in a proof of concept trial in patients with essential tremor and a proof of concept trial in patients with severe PPD. If serious adverse events or other undesirable side effects, or unexpected characteristics of SAGE-547 are observed in emergency-use cases or in investigator sponsored clinical trials of SAGE-547 or our exploratory clinical trials, it may adversely affect or delay our clinical development of SAGE-547, or we may need to abandon its development for SRSE entirely, and the occurrence of these events would have a material adverse effect on our business.
Positive results from early non-clinical studies and clinical trials of our product candidates are not necessarily predictive of the results of later non-clinical studies and clinical trials of our product candidates. If we cannot replicate the positive results from our earlier non-clinical studies and clinical trials of our product candidates in our later non-clinical studies and clinical trials, we may be unable to successfully develop, obtain regulatory approval for and commercialize our product candidates.
Positive results from our non-clinical studies of our product candidates, and any positive results we may obtain from our early clinical trials of our product candidates, may not necessarily be predictive of the results from required later non-clinical studies and clinical trials. Similarly, even if we are able to complete our planned non-clinical studies or clinical trials of our product candidates according to our current development timeline, the positive results from our non-clinical studies and clinical trials of our product candidates may not be replicated in subsequent non-clinical studies or clinical trial results. For example, although 12 of the first 17 patients treated with SAGE-547 and evaluable for efficacy in our Phase 1/2 clinical trial met the key efficacy endpoint and none of the 20 patients enrolled in the study have yet experienced any severe adverse events related to SAGE-547, future patients enrolled and treated with SAGE-547 in our Phase 1/2 clinical trial or later-stage clinical trials may not have the same outcome. Also, our later-stage clinical trials will differ in important ways from our ongoing Phase 1/2 clinical trial of SAGE-547, which could cause the outcome of these later-stage trials to differ from our earlier stage clinical trials. For example, our planned Phase 3 clinical trial of SAGE-547 will be a placebo-controlled trial, while our Phase 1/2 clinical trial was open-label, and an intent-to-treat statistical analysis, which is a more rigorous statistical analysis, will be employed in evaluating the data in our planned Phase 3 clinical trial. In addition, the formulation of SAGE-547 we intend to use in our planned Phase 3 trial is somewhat different than the formulation used in the Phase 1/2 trial. We do not believe the change will negatively affect trial results, but we cannot be sure. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, non-clinical findings made while clinical trials were underway or safety or efficacy observations made in non-clinical studies and clinical trials, including previously unreported adverse events. Moreover, non-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in non-clinical studies and clinical trials nonetheless failed to obtain FDA approval. We have not completed any clinical trials for our product candidates yet, and if we fail to produce positive results in our planned non-clinical studies or clinical trials of any of our product candidates, the development timeline and regulatory approval and commercialization prospects for our product candidates, and, correspondingly, our business and financial prospects, would be materially adversely affected.
Failures or delays in the commencement or completion of our planned clinical trials of our product candidates could result in increased costs to us and could delay, prevent or limit our ability to generate revenue and continue our business.
We have an ongoing a Phase 1/2 clinical trial of SAGE-547 as a treatment for SRSE and ongoing proof of concept studies of SAGE-547 for patients with essential tremor and severe PPD. We will need to complete at least one additional trial prior to the submission of an NDA for SAGE-547 as a treatment for SRSE. Successful completion of our clinical trials is a prerequisite to submitting an NDA to the FDA and, consequently, the ultimate approval and commercial marketing of SAGE-547 for SRSE and our other product candidates. We do not know whether any of our clinical trials will begin or be completed on schedule, if at all, as the commencement and completion of clinical trials can be delayed or prevented for a number of reasons, including, among others:
| • | the FDA may deny permission to proceed with our planned clinical trials or any other clinical trials we may initiate, or may place a clinical trial on hold; |
31
Table of Contents
| • | delays in filing or receiving approvals of additional INDs that may be required; |
| • | negative results from our ongoing non-clinical studies; |
| • | delays in reaching or failing to reach agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| • | inadequate quantity or quality of a product candidate or other materials necessary to conduct clinical trials, for example delays in the manufacturing of sufficient supply of finished drug product; |
| • | difficulties obtaining Institutional Review Board, or IRB, approval to conduct a clinical trial at a prospective site or sites; |
| • | challenges in recruiting and enrolling patients to participate in clinical trials, including the small size of the patient population, acute nature of SRSE, the proximity of patients to trial sites, eligibility criteria for the clinical trial, the nature of the clinical trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications |
| • | severe or unexpected drug-related side effects experienced by patients in a clinical trial; |
| • | delays in validating any endpoints utilized in a clinical trial; |
| • | our inability to satisfy the CMC requirements of the FDA or file amendments to our IND as requested by the FDA prior to the initiation of a clinical trial; |
| • | the FDA may disagree with our clinical trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for our clinical trials; |
| • | reports from non-clinical or clinical testing of other CNS therapies that raise safety or efficacy concerns; and |
| • | difficulties retaining patients who have enrolled in a clinical trial but may be prone to withdraw due to rigors of the clinical trials, lack of efficacy, side effects, personal issues or loss of interest. |
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the IRBs at the sites where the IRBs are overseeing a clinical trial, a data and safety monitoring board, or DSMB, overseeing the clinical trial at issue or other regulatory authorities due to a number of factors, including, among others:
| • | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| • | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities that reveals deficiencies or violations that require us to undertake corrective action, including the imposition of a clinical hold; |
| • | unforeseen safety issues, including any that could be identified in our ongoing non-clinical carcinogenicity studies, adverse side effects or lack of effectiveness; |
| • | changes in government regulations or administrative actions; |
| • | problems with clinical supply materials; and |
| • | lack of adequate funding to continue clinical trials. |
Changes in regulatory requirements, FDA guidance or unanticipated events during our non-clinical studies and clinical trials of our product candidates may occur, which may result in changes to non-clinical studies and clinical trial protocols or additional non-clinical studies and clinical trial requirements, which could result in increased costs to us and could delay our development timeline.
Changes in regulatory requirements, FDA guidance or
unanticipated events during our non-clinical studies and clinical trials may force us to amend non-clinical studies and clinical trial protocols or the FDA may impose additional non-clinical studies and clinical trial requirements. Amendments or
changes to our clinical trial protocols would require resubmission to the FDA and IRBs for review and approval, which may adversely impact the cost, timing or successful completion of clinical trials. Similarly, amendments to our non-clinical
studies may adversely impact the cost, timing, or successful completion of those non-clinical studies. For example, we intend to seek a waiver from the need to perform a study of SAGE-547 on certain cardiac measures. If the FDA does not grant the
waiver, we will be required to conduct such a study, the results of which could delay the filing of an NDA for
SAGE-547. If we experience delays completing, or if we terminate, any of our non-clinical studies or clinical trials, or if we are
required to conduct additional non-clinical studies or clinical trials, the commercial prospects for our product candidates may be harmed and our ability to generate product revenue will be delayed.
We rely, and expect that we will continue to rely, on third parties to conduct any clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
32
Table of Contents
We do not have the ability to independently conduct clinical trials. We rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, to conduct clinical trials on our product candidates. We enter into agreements with third-party CROs to provide monitors for and to manage data for our ongoing clinical trials. We rely heavily on these parties for execution of clinical trials for our product candidates and control only certain aspects of their activities. As a result, we have less direct control over the conduct, timing and completion of these clinical trials and the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties may:
| • | have staffing difficulties; |
| • | fail to comply with contractual obligations; |
| • | experience regulatory compliance issues; |
| • | undergo changes in priorities or become financially distressed; or |
| • | form relationships with other entities, some of which may be our competitors. |
These factors may materially adversely affect the willingness or ability of third parties to conduct our clinical trials and may subject us to unexpected cost increases that are beyond our control. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific requirements and standards, and our reliance on CROs does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with regulations and guidelines, including current Good Clinical Practices, or cGCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial patients are adequately informed of the potential risks of participating in clinical trials. These regulations are enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for any products in clinical development. The FDA enforces cGCP regulations through periodic inspections of clinical trial sponsors, principal investigators and trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with cGCPs. In addition, our clinical trials must be conducted with product candidates produced under cGMPs regulations and will require a large number of test patients. Our failure or the failure of our CROs to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
Although we do design our clinical trials for our product candidates, CROs conduct all of the clinical trials. As a result, many important aspects of our drug development programs are outside of our direct control. In addition, the CROs may not perform all of their obligations under arrangements with us or in compliance with regulatory requirements, but we remain responsible and are subject to enforcement action that may include civil penalties up to and including criminal prosecution for any violations of FDA laws and regulations during the conduct of our clinical trials. If the CROs do not perform clinical trials in a satisfactory manner, breach their obligations to us or fail to comply with regulatory requirements, the development and commercialization of our product candidates may be delayed or our development program materially and irreversibly harmed. We cannot control the amount and timing of resources these CROs devote to our program or our clinical products. If we are unable to rely on clinical data collected by our CROs, we could be required to repeat, extend the duration of, or increase the size of our clinical trials and this could significantly delay commercialization and require significantly greater expenditures.
If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any clinical trials such CROs are associated with may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, we believe that our financial results and the commercial prospects for our product candidates in the subject indication would be harmed, our costs could increase and our ability to generate revenue could be delayed.
We rely completely on third-party suppliers to manufacture our clinical drug supplies for our product candidates, and we intend to rely on third parties to produce non-clinical, clinical and commercial supplies of any future product candidate.
We do not currently have, nor do we plan to acquire, the infrastructure or capability to internally manufacture our clinical drug supply of our product candidates, or any future product candidates, for use in the conduct of our non-clinical studies and clinical trials, and we lack the internal resources and the capability to manufacture any product candidates on a clinical or commercial scale. For example, SAGE-547 used in the emergency-use cases was manufactured at an academic site, the active pharmaceutical ingredient for SAGE-547 for our Phase 1/2 clinical trial was manufactured at an academic site and SAGE-547 as formulated for our Phase 1/2 clinical trial was manufactured at a third-party manufacturer’s site. The facilities used by our contract manufacturers to manufacture the active pharmaceutical ingredient and final drug product must complete a pre-approval inspection by the FDA and other comparable foreign regulatory agencies to assess compliance with applicable requirements, including cGMPs, after we submit our NDA or relevant foreign regulatory submission to the applicable regulatory agency.
We do not control the manufacturing process of, and are completely dependent on, our contract manufacturers to comply with cGMPs for manufacture of both active drug substances and finished drug products. If our contract manufacturers cannot successfully manufacture
33
Table of Contents
material that conforms to our specifications and the strict regulatory requirements of the FDA or applicable foreign regulatory agencies, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. In addition, we have no direct control over our contract manufacturers’ ability to maintain adequate quality control, quality assurance and qualified personnel. Furthermore, all of our third-party contract manufacturers are engaged with other companies to supply and/or manufacture materials or products for such companies, which exposes our third-party contract manufacturers to regulatory risks for the production of such materials and products. As a result, failure to satisfy the regulatory requirements for the production of those materials and products may affect the regulatory clearance of our contract manufacturers’ facilities generally. If the FDA or an applicable foreign regulatory agency determines now or in the future that these facilities for the manufacture of our product candidates are noncompliant, we may need to find alternative manufacturing facilities, which would adversely impact our ability to develop, obtain regulatory approval for or market our product candidates. Our reliance on contract manufacturers also exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may appropriate our trade secrets or other proprietary information.
We do not have long-term supply agreements in place with our contract manufacturers, and each batch of our product candidates is individually contracted under a quality and supply agreement. If we engage new contract manufacturers, such contractors must complete an inspection by the FDA and other applicable foreign regulatory agencies. We plan to continue to rely upon contract manufacturers and, potentially, collaboration partners to manufacture commercial quantities of our product candidates, if approved. Our current scale of manufacturing is adequate to support all of our needs for non-clinical studies and clinical trial supplies.
Even if we receive marketing approval for our product candidates in the United States, we may never receive regulatory approval to market our product candidates outside of the United States.
We have not yet selected any markets outside of the United States where we intend to seek regulatory approval to market our product candidates. In order to market any product outside of the United States, however, we must establish and comply with the numerous and varying safety, efficacy and other regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product candidate testing and additional administrative review periods. The time required to obtain approvals in other countries might differ from that required to obtain FDA approval. The marketing approval processes in other countries may implicate all of the risks detailed above regarding FDA approval in the United States as well as other risks. In particular, in many countries outside of the United States, products must receive pricing and reimbursement approval before the product can be commercialized. Obtaining this approval can result in substantial delays in bringing products to market in such countries. Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory process in others. Failure to obtain marketing approval in other countries or any delay or other setback in obtaining such approval would impair our ability to market our product candidates in such foreign markets. Any such impairment would reduce the size of our potential market, which could have a material adverse impact on our business, results of operations and prospects.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to generate any revenue.
We do not currently have an infrastructure for the sales, marketing and distribution of pharmaceutical products. In order to market our product candidates, if approved by the FDA or any other regulatory body, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, or if we are unable to do so on commercially reasonable terms, our business, results of operations, financial condition and prospects will be materially adversely affected.
Even if we receive marketing approval for our product candidates, our product candidates may not achieve broad market acceptance, which would limit the revenue that we generate from their sales.
The commercial success of our product candidates, if approved by the FDA or other applicable regulatory authorities, will depend upon the awareness and acceptance of our product candidates among the medical community, including physicians, patients and healthcare payors. Market acceptance of our product candidates, if approved, will depend on a number of factors, including, among others:
| • | the efficacy of our product candidates as demonstrated in clinical trials, and, if required by any applicable regulatory authority in connection with the approval for the applicable indications, to provide patients with incremental health benefits, as compared with other available CNS therapies; |
| • | limitations or warnings contained in the labeling approved for our product candidates by the FDA or other applicable regulatory authorities; |
| • | the clinical indications for which our product candidates are approved; |
| • | availability of alternative treatments already approved or expected to be commercially launched in the near future; |
| • | the potential and perceived advantages of our product candidates over current treatment options or alternative treatments, including future alternative treatments; |
| • | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| • | the strength of marketing and distribution support and timing of market introduction of competitive products; |
34
Table of Contents
| • | publicity concerning our products or competing products and treatments; |
| • | pricing and cost effectiveness; |
| • | the effectiveness of our sales and marketing strategies; |
| • | our ability to increase awareness of our product candidates through marketing efforts; |
| • | our ability to obtain sufficient third-party coverage or reimbursement; or |
| • | the willingness of patients to pay out-of-pocket in the absence of third-party coverage. |
If our product candidates are approved but do not achieve an adequate level of acceptance by patients, physicians and payors, we may not generate sufficient revenue from our product candidates to become or remain profitable. Before granting reimbursement approval, healthcare payors may require us to demonstrate that our product candidates, in addition to treating these target indications, also provide incremental health benefits to patients. Our efforts to educate the medical community and third-party payors about the benefits of our product candidates may require significant resources and may never be successful.
Our product candidates may cause undesirable side effects that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt non-clinical studies and clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other regulatory authorities.
Further, clinical trials by their nature utilize a sample of the potential patient population. With a limited number of patients and limited duration of exposure, rare and severe side effects of our product candidates may only be uncovered with a significantly larger number of patients exposed to the product candidate. If our product candidates receive marketing approval and we or others identify undesirable side effects caused by such product candidates (or any other similar products) after such approval, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw or limit their approval of such product candidates; |
| • | regulatory authorities may require the addition of labeling statements, such as a “boxed” warning or a contraindication; |
| • | we may be required to change the way such product candidates are distributed or administered, conduct additional clinical trials or change the labeling of the product candidates; |
| • | we may be subject to regulatory investigations and government enforcement actions; |
| • | we may decide to remove such product candidates from the marketplace; |
| • | we could be sued and held liable for injury caused to individuals exposed to or taking our product candidates; and |
| • | our reputation may suffer. |
We believe that any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidates and could substantially increase the costs of commercializing our product candidates and significantly impact our ability to successfully commercialize our product candidates and generate revenues.
Even if we receive marketing approval for our product candidates, we may still face future development and regulatory difficulties.
Even if we receive marketing approval for our product candidates, regulatory authorities may still impose significant restrictions on our product candidates, indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. For example, prior to product launch, the U.S. Drug Enforcement Agency, or DEA, needs to determine the controlled substance schedule of SAGE-547, taking into account the recommendation of the FDA. Our product candidates will also be subject to ongoing FDA requirements governing the labeling, packaging, storage and promotion of the product and record keeping and submission of safety and other post-market information. The FDA has significant post-marketing authority, including, for example, the authority to require labeling changes based on new safety information and to require post-marketing studies or clinical trials to evaluate serious safety risks related to the use of a drug. The FDA also has the authority to require, as part of an NDA or post-approval, the submission of a REMS. Any REMS required by the FDA may lead to increased costs to assure compliance with new post-approval regulatory requirements and potential requirements or restrictions on the sale of approved products, all of which could lead to lower sales volume and revenue.
Manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMPs and other regulations. If we or a regulatory agency discover problems with our product candidates, such as adverse events of unanticipated severity or frequency, or problems with the facility where our product candidates are manufactured, a regulatory agency may impose restrictions on our product candidates, the manufacturer or us, including requiring withdrawal of our product candidates from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may, among other things:
| • | issue warning letters or untitled letters; |
35
Table of Contents
| • | seek an injunction or impose civil or criminal penalties or monetary fines; |
| • | suspend or withdraw marketing approval; |
| • | suspend any ongoing clinical trials; |
| • | refuse to approve pending applications or supplements to applications submitted by us; |
| • | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| • | seize or detain products, refuse to permit the import or export of products, or require that we initiate a product recall. |
Competing therapies could emerge adversely affecting our opportunity to generate revenue from the sale of our product candidates.
The biopharmaceuticals industry is highly competitive. There are many public and private biopharmaceutical companies, universities, governmental agencies and other research organizations actively engaged in the research and development of products that may be similar to our product candidates or address similar markets. It is probable that the number of companies seeking to develop products and therapies similar to our products will increase.
Currently, there are no therapies specifically approved for RSE or SRSE. However, many products approved for other indications, general anesthetics and anti-seizure drugs, are used off-label for various stages of SE therapy. Additionally, though not indicated, acupuncture, hypothermia, and electroconvulsive therapy are sometimes used prior to withdrawal of care for patients with SRSE.
In the field of neuroactive steroids focused on modulation of GABAA or NMDA receptors, our principal competitor is Marinus Pharmaceuticals, Inc., which is developing a reformulated form of Ganaxolone, a known GABAA positive allosteric modulator neuroactive steroid, for potential treatment of drug-resistant partial complex seizures and fragile X syndrome.
Many of our potential competitors, alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of treatments and the commercialization of those treatments. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
We may seek to establish collaborations, and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. For some of our product candidates, we may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of those product candidates.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such collaboration could be more attractive than the one with us for our product candidate. The terms of any collaborator or other arrangements that we may establish may not be favorable to us.
We may also be restricted under existing collaboration agreements from entering into future agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of the product candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
36
Table of Contents
In addition, any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Collaborations with pharmaceutical or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration would adversely affect us financially and could harm our business reputation.
We may not be successful in our efforts to identify or discover additional product candidates or we may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
The success of our business depends primarily upon our ability to identify, develop and commercialize products based on our proprietary chemistry platform. Although some of our product candidates are in non-clinical and clinical development, our research programs may fail to identify other potential product candidates for clinical development for a number of reasons. Our research methodology may be unsuccessful in identifying potential product candidates or our potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval.
Because we have limited financial and management resources, we focus on a limited number of research programs and product candidates and are currently focused on our SE program. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial drugs or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable drugs. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through future collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which would have a material adverse effect on our business and could potentially cause us to cease operations. Research programs to identify new product candidates require substantial technical, financial and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
We are subject to healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Although we do not currently have any products on the market, once we begin commercializing our products, we may be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal government and the states and foreign governments in which we conduct our business. Healthcare providers, physicians and others will play a primary role in the recommendation and prescription of our product candidates, if approved. Our future arrangements with third-party payors will expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our product candidates, if we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
| • | The federal anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid. |
| • | The federal False Claims Act imposes criminal and civil penalties, including those from civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government. |
| • | The federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information. |
| • | The federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services. |
| • | The federal transparency requirements, sometimes referred to as the “Sunshine Act,” under the Patient Protection and Affordable Care Act, require manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests. |
37
Table of Contents
| • | Analogous state laws and regulations, such as state anti-kickback and false claims laws and transparency laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures and drug pricing. |
Ensuring that our future business arrangements with third parties comply with applicable healthcare laws and regulations could be costly. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations, including anticipated activities to be conducted by our sales team, were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines and exclusion from government funded healthcare programs, such as Medicare and Medicaid, any of which could substantially disrupt our operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products, such as SAGE-547, SAGE-689, and SAGE-217, if approved. In particular, a product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. For example, if we receive marketing approval for SAGE-547 as a treatment for SRSE, physicians may nevertheless prescribe SAGE-547 to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
SAGE-547 will, and our other product candidates may, contain controlled substances, the manufacture, use, sale, importation, exportation, prescribing and distribution of which are subject to regulation by the DEA.
Before we can commercialize SAGE-547, and potentially our other product candidates, the DEA will need to determine the controlled substance schedule, taking into account the recommendation of the FDA. This may be a lengthy process that could delay our marketing of a product candidate and could potentially diminish any regulatory exclusivity periods for which we may be eligible. SAGE-547 will, and our other product candidates may, if approved, be regulated as “controlled substances” as defined in the Controlled Substances Act of 1970, or CSA, and the implementing regulations of the DEA, which establish registration, security, recordkeeping, reporting, storage, distribution, importation, exportation, inventory, quota and other requirements administered by the DEA. These requirements are applicable to us, to our third-party manufacturers and to distributors, prescribers and dispensers of our product candidates. The DEA regulates the handling of controlled substances through a closed chain of distribution. This control extends to the equipment and raw materials used in their manufacture and packaging, in order to prevent loss and diversion into illicit channels of commerce. A number of states and foreign countries also independently regulate these drugs as controlled substances.
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use, and may not be marketed or sold in the United States. A pharmaceutical product may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest risk of abuse and Schedule V substances the lowest relative risk of abuse among such substances.
We expect that SAGE-547 will, and our other product candidates may, be listed by the DEA as Schedule IV controlled substances under the CSA. Consequently, the manufacturing, shipping, storing, selling and using of the products will be subject to a high degree of regulation. Also, distribution, prescribing and dispensing of these drugs are highly regulated.
Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule.
Because of their restrictive nature, these laws and regulations could limit commercialization of our product candidates containing controlled substances. Failure to comply with these laws and regulations could also result in withdrawal of our DEA registrations, disruption in manufacturing and distribution activities, consent decrees, criminal and civil penalties and state actions, among other consequences.
38
Table of Contents
Even if approved, reimbursement policies could limit our ability to sell our product candidates.
Market acceptance and sales of our product candidates will depend on reimbursement policies and may be affected by healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels for those medications. Cost containment is a primary concern in the U.S. healthcare industry and elsewhere. Government authorities and these third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that reimbursement will be available for our product candidates and, if reimbursement is available, the level of such reimbursement. Reimbursement may impact the demand for, or the price of, our product candidates. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates.
In some foreign countries, particularly in Canada and European countries, the pricing of prescription pharmaceuticals is subject to strict governmental control. In these countries, pricing negotiations with governmental authorities can take six months or longer after the receipt of regulatory approval and product launch. To obtain favorable reimbursement for the indications sought or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidates with other available therapies. If reimbursement for our product candidates is unavailable in any country in which we seek reimbursement, if it is limited in scope or amount, if it is conditioned upon our completion of additional clinical trials, or if pricing is set at unsatisfactory levels, our operating results could be materially adversely affected.
Even though we have obtained orphan drug designation for SAGE-547 as a treatment for SE, there may be limits to the regulatory exclusivity afforded by such designation.
Even though we have obtained orphan drug designation for SAGE-547 for treatment of SE from the FDA, there are limitations to exclusivity afforded by such designation. In the United States, the company that first obtains FDA approval for a designated orphan drug for the specified rare disease or condition receives orphan drug marketing exclusivity for that drug for a period of seven years. This orphan drug exclusivity prevents the FDA from approving another application, including a full NDA to market the same drug for the same orphan indication, except in very limited circumstances, including when the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. For purposes of small molecule drugs, the FDA defines “same drug” as a drug that contains the same active moiety and is intended for the same use as the drug in question. To obtain orphan drug exclusivity for a drug that shares the same active moiety as an already approved drug, it must be demonstrated to the FDA that the drug is safer or more effective than the approved orphan designated drug, or that it makes a major contribution to patient care. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition or if another drug with the same active moiety is determined to be safer, more effective, or represents a major contribution to patient care.
Our future growth may depend, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability may depend, in part, on our ability to commercialize our product candidates in foreign markets for which we may rely on collaboration with third parties. If we commercialize our product candidates in foreign markets, we would be subject to additional risks and uncertainties, including:
| • | our customers’ ability to obtain reimbursement for our product candidates in foreign markets; |
| • | our inability to directly control commercial activities because we are relying on third parties; |
| • | the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements; |
| • | different medical practices and customs in foreign countries affecting acceptance in the marketplace; |
| • | import or export licensing requirements; |
| • | longer accounts receivable collection times; |
| • | longer lead times for shipping; |
| • | language barriers for technical training; |
| • | reduced protection of intellectual property rights in some foreign countries; |
| • | the existence of additional potentially relevant third party intellectual property rights; |
| • | foreign currency exchange rate fluctuations; and |
| • | the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute. |
Foreign sales of our product candidates could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
39
Table of Contents
Risks Related to Our Intellectual Property Rights
If we are unable to adequately protect our proprietary technology, or obtain and maintain issued patents that are sufficient to protect our product candidates, others could compete against us more directly, which would have a material adverse impact on our business, results of operations, financial condition and prospects.
We strive to protect and enhance the proprietary technologies that we believe are important to our business, including seeking patents intended to cover our products and compositions, their methods of use and any other inventions that are important to the development of our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, should they issue, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain the proprietary position of our product candidates. Our owned and licensed patent applications relate to SAGE-547, GABAA receptor modulators, including genus and species claims to SAGE-689 and NMDA receptor modulators.
We currently have no issued patents covering any of our lead product candidates, SAGE-547, SAGE-689, or SAGE-217. We cannot provide any assurances that any of our pending patent applications will mature into issued patents and, if they do, that such patents will include, claims with a scope sufficient to protect our product candidates or otherwise provide any competitive advantage. For example, the patent applications that may provide coverage for SAGE-547, only cover particular formulations and particular methods of using such formulations to treat seizure conditions, such as SE. As a result, if a patent issues from such patent applications, it would not prevent third-party competitors from creating, making and marketing alternative formulations, that fall outside the scope of our patent claims or practicing alternative methods. There can be no assurance that any such alternative formulations will not be equally effective as our formulation of SAGE-547. Moreover, other parties have developed technologies that may be related or competitive to our approach, and may have filed or may file patent applications and may have received or may receive patents that may overlap or conflict with our patent applications, either by claiming the same methods or formulations or by claiming subject matter that could dominate our patent position. Such third-party patent positions may limit or even eliminate our ability to obtain patent protection for certain inventions.
The patent positions of biotechnology and pharmaceutical companies, including our patent position, involve complex legal and factual questions, and, therefore, the issuance, scope, validity and enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated, or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, ex parte reexamination, or inter partes review proceedings, supplemental examination and challenges in district court. Patents may be subjected to opposition, post-grant review, or comparable proceedings lodged in various foreign, both national and regional, patent offices. These proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such proceedings may be costly. Thus, any patents, should they issue, that we may own or exclusively license may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to develop, market or otherwise commercialize our product candidates.
Furthermore, though a patent, if it were to issue, is presumed valid and enforceable, its issuance is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products. Even if a patent issues and is held to be valid and enforceable, competitors may be able to design around our patents, such as using pre-existing or newly developed technology. Other parties may develop and obtain patent protection for more effective technologies, designs or methods. We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, vendors, former employees and current employees. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries. If these developments were to occur, they could have a material adverse effect on our sales.
Our ability to enforce our patent rights depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
In addition, proceedings to enforce or defend our patents, if and when issued, could put our patents at risk of being invalidated, held unenforceable, or interpreted narrowly. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of our patents are invalid or otherwise unenforceable. If any of our patents, if and when issued, covering our product candidates are invalidated or found unenforceable, our financial position and results of operations would be materially and adversely impacted. In addition, if a court found that valid, enforceable patents held by third parties covered our product candidates, our financial position and results of operations would also be materially and adversely impacted.
40
Table of Contents
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
| • | any of our pending patent applications, if issued, will include claims having a scope sufficient to protect our product candidates or any other products or product candidates; |
| • | any of our pending patent applications will issue as patents at all; |
| • | we will be able to successfully commercialize our product candidates, if approved, before our relevant patents expire; |
| • | we were the first to make the inventions covered by each of our patents and pending patent applications; |
| • | we were the first to file patent applications for these inventions; |
| • | others will not develop similar or alternative technologies that do not infringe our patents; |
| • | others will not use pre-existing technology to effectively compete against us; |
| • | any of our patents, if issued, will be found to ultimately be valid and enforceable; |
| • | any patents issued to us will provide a basis for an exclusive market for our commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties; |
| • | we will develop additional proprietary technologies or product candidates that are separately patentable; or |
| • | that our commercial activities or products will not infringe upon the patents or proprietary rights of others. |
We rely upon unpatented trade secrets, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our employees and our collaborators and consultants. It is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees and consultants who are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could otherwise become known or be independently discovered by our competitors.
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our product candidates, if approved.
Our success will depend in part on our ability to operate without infringing the intellectual property and proprietary rights of third parties. We cannot assure you that our business, products and methods do not or will not infringe the patents or other intellectual property rights of third parties.
The pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our product candidates or the use of our technologies infringes patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. As we continue to develop and, if approved, commercialize our current product candidates and future product candidates, competitors may claim that our technology infringes their intellectual property rights as part of business strategies designed to impede our successful commercialization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, third parties may have currently pending patent applications which may later result in issued patents that our product candidates may infringe, or which such third parties claim are infringed by our technologies. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on us. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.
Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. Any claim relating to intellectual property infringement that is successfully asserted against us may require us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing another party’s patents, for past use of the asserted intellectual property and royalties and other consideration going forward if we are forced to take a license. In addition, if any such claim were successfully asserted against us and we could not obtain such a license, we may be forced to stop or delay developing, manufacturing, selling or otherwise commercializing our product candidates.
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time-consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to do one or more of the following:
| • | cease developing, selling or otherwise commercializing our product candidates; |
| • | pay substantial damages for past use of the asserted intellectual property; |
41
Table of Contents
| • | obtain a license from the holder of the asserted intellectual property, which license may not be available on reasonable terms, if at all; and |
| • | in the case of trademark claims, redesign, or rename, some or all of our product candidates to avoid infringing the intellectual property rights of third parties, which may not be possible and, even if possible, could be costly and time-consuming. |
Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors. These agreements generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. For example, even if we have a consulting agreement in place with an academic advisor pursuant to which such academic advisor is required to assign any inventions developed in connection with providing services to us, such academic advisor may not have the right to assign such inventions to us, as it may conflict with his or her obligations to assign all such intellectual property to his or her employing institution.
Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. Patent and Trademark Office, or U.S. PTO, and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Even if the patent applications we own or license are issued, competitors may infringe these patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid, is unenforceable and/or is not infringed, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court.
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent, if and when issued, covering one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or
42
Table of Contents
amendment of our patents in such a way that they no longer cover our product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such a loss of patent protection would have a material adverse impact on our business.
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States, assuming that rights are obtained in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. The statutory deadlines for pursuing patent protection in individual foreign jurisdictions are based on the priority date of each of our patent applications. For the patent families related to SAGE-547, SAGE-689 and SAGE-217, and many of the other patent families that we own or license, the relevant statutory deadlines have not yet expired. For each of the patent families that we believe provide coverage for our lead product candidates, we will need to decide whether and where to pursue protection outside the United States.
Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to biotechnology. For example, an April 2014 report from the Office of the United States Trade Representative identified a number of countries, including India and China, where challenges to the procurement and enforcement of patent rights have been reported. Several countries, including India and China, have been listed in the report every year since 1989. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Furthermore, proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We are dependent on licensed intellectual property. If we were to lose our rights to licensed intellectual property, we may not be able to continue developing or commercializing our product candidates, if approved. If we breach any of the agreements under which we license the use, development and commercialization rights to our product candidates or technology from third parties or, in certain cases, we fail to meet certain development deadlines, we could lose license rights that are important to our business.
We are a party to a number of license agreements under which we are granted rights to intellectual property that are important to our business and we expect that we may need to enter into additional license agreements in the future. Our existing license agreements impose, and we expect that future license agreements will impose on us, various development, regulatory and/or commercial diligence obligations, payment of milestones and/or royalties and other obligations. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, the licensor may have the right to terminate the license, in which event we would not be able to market products covered by the license. Our business could suffer, for example, if any current or future licenses terminate, if the licensors fail to abide by the terms of the license, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms.
As we have done previously, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we cannot provide any assurances that third-party patents do not exist that might be enforced against our current product candidates or future products in the absence of such a license. We may fail to obtain any of these licenses on commercially reasonable terms, if at all. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are
43
Table of Contents
unable to do so, we may be unable to develop or commercialize the affected product candidates, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation.
Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise between us and our licensors regarding intellectual property subject to a license agreement, including:
| • | the scope of rights granted under the license agreement and other interpretation-related issues; |
| • | whether and the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
| • | our right to sublicense patent and other rights to third parties under collaborative development relationships; |
| • | our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our product candidates, and what activities satisfy those diligence obligations; and |
| • | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners. |
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We have entered into several licenses to support our various programs. We completed an exclusive license agreement with Washington University, or WU, under certain patent families that comprise a variety of small molecule allosteric modulators of GABAA receptors and for which we have the worldwide right to develop and commercialize. A patent family that discloses and claims SAGE-689 is licensed to us under this agreement. We are obligated to pay WU certain clinical/regulatory milestones and single-digit royalties on products developed from this technology. Termination of our license agreement with WU would have a material adverse impact on our ability to develop and commercialize SAGE-689.
We have also entered into an exclusive license agreement with CyDex Pharmaceuticals, Inc., or CyDex, a wholly owned subsidiary of Ligand Pharmaceuticals, Inc., to use its Captisol technology to develop SAGE-547 for the field of use, which includes all fields for the treatment, prevention or diagnosis of any disease or symptom in humans or animals. We are obligated to pay CyDex certain clinical/regulatory milestones and single-digit royalties on SAGE-547. In addition, we entered into a supply agreement with CyDex, pursuant to which they supply us with Captisol to formulate SAGE-547. Absent an alternative agreement by the parties, our rights under our exclusive license agreement terminate in the event that the supply agreement terminates. Currently, our SAGE-547 product candidate in clinical development is formulated in Captisol. Termination of our license agreement with CyDex would have a material adverse impact on our ability to develop and commercialize SAGE-547 in its current formulation.
We also entered into a non-exclusive license with The Regents of the University of California, or the Regents. Pursuant to this agreement the Regents granted us a non-exclusive, non-transferable license under all personal property rights of the Regents covering the tangible personal property in an IND application package owned by the Regents, or the Data, and a specified quantity of cGMP grade allopregnanolone, or the Material, to (i) use the Data for reference or incorporation in an IND for use of the Material as a treatment of SE, essential tremor and/or postpartum depression and (ii) use the Material or modifications of the Material to develop a pharmaceutical formulation for clinical trials for SE, essential tremor and/or postpartum depression. This agreement requires us to pay milestone payments in connection with the first derived product, which would include SAGE-547, that meets the relevant milestones and we must also pay single-digit royalties for each derived product for a period of 15 years following the first commercial sale of such derived product. Termination of our license agreement with the Regents would have a material adverse impact on our ability to develop and commercialize derived products, which would include SAGE-547.
We may enter into additional license(s) to third-party intellectual property that are necessary or useful to our business. Our current licenses and any future licenses that we may enter into impose various royalty payment, milestone, and other obligations on us. For example, as is the case for the Washington University license, the licensor may retain control over patent prosecution and maintenance under a license agreement, in which case, we may not be able to adequately influence patent prosecution or prevent inadvertent lapses of coverage due to failure to pay maintenance fees. If we fail to comply with any of our obligations under a current or future license agreement, our licensor(s) may allege that we have breached our license agreement and may accordingly seek to terminate our license with them. In addition, future licensor(s) may decide to terminate our license at will. Termination of any of our current or future licenses could result in our loss of the right to use the licensed intellectual property, which could materially adversely affect our ability to develop and commercialize a product candidate or product, if approved, as well as harm our competitive business position and our business prospects.
In addition, if our licensors fail to abide by the terms of the license, if the licensors fail to prevent infringement by third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms our business could suffer.
Some intellectual property which we have licensed may have been discovered through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements, and a preference for U.S. industry. Compliance with such regulations may limit our exclusive rights, subject us to expenditure of resources with respect to reporting requirements, and limit our ability to contract with non-U.S. manufacturers.
44
Table of Contents
Some of the intellectual property rights we have licensed may have been generated through the use of U.S. government funding and may therefore be subject to certain federal regulations. For example, some of the intellectual property rights licensed to us under the license agreements with WU and the Regents may have been generated using U.S. government funds. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future product candidates pursuant to the Bayh-Dole Act of 1980, or Bayh-Dole Act. These U.S. government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third party if it determines that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). The U.S. government also has the right to take title to these inventions if we fail, or the applicable licensor fails, to disclose the invention to the government and fail to file an application to register the intellectual property within specified time limits. In addition, the U.S. government may acquire title to these inventions in any country in which a patent application is not filed within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us, or the applicable licensor, to expend substantial resources. In addition, the U.S. government requires that any products embodying the subject invention or produced through the use of the subject invention be manufactured substantially in the United States. The manufacturing preference requirement can be waived if the owner of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. manufacturers may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property.
We currently do not plan to apply for additional U.S. government funding, but if we do, and we discover compounds or drug candidates as a result of such funding, intellectual property rights to such discoveries may be subject to the applicable provisions of the Bayh-Dole Act.
If we do not obtain additional protection under the Hatch-Waxman Amendments and similar foreign legislation by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of our product candidates, one or more of the U.S. patents we own or license may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. For example, we may not be granted an extension if the active ingredient of SAGE-547, allopregnanolone, is used in another drug company’s product candidate and that product candidate is the first to obtain FDA approval. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our ability to generate revenues could be materially adversely affected.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological and legal complexity, and is therefore costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation: the Leahy-Smith America Invents Act, referred to as the America Invents Act. The America Invents Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. It is not yet clear what, if any, impact the America Invents Act will have on the operation of our business. However, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that may issue from our patent applications, all of which could have a material adverse effect on our business and financial condition.
In addition, recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. The full impact of these decisions is not yet known. For example, on March 20, 2012 in Mayo Collaborative Services, DBA Mayo Medical Laboratories, et al. v. Prometheus Laboratories, Inc., the Court held that several claims drawn to measuring drug metabolite levels from patient samples and correlating them to drug doses were not patentable subject matter. The decision appears to impact diagnostics patents that merely apply a law of nature via a series of routine steps and it has created uncertainty around the ability to obtain patent protection for certain inventions. Additionally, on June 13, 2013 in Association for Molecular Pathology v. Myriad Genetics, Inc., the Court held that claims to isolated genomic DNA are not patentable, but claims to complementary DNA molecules are patent eligible because they are not a natural product. The effect of the decision on patents for other isolated natural products is uncertain. In Alice Corporation Pty. Ltd. v. CLS Bank International, et al., a case involving patent claims directed to a method for mitigating settlement risk, the Court held that the patent eligibility of claims directed to abstract ideas, products of nature, and laws of nature should be determined using the same framework set forth in Prometheus. The U.S. PTO recently issued a set of guidelines setting forth procedures for determining subject matter eligibility of claims directed to abstract ideas, products of nature, and laws of nature in line with the Prometheus, Myriad, and Alice decisions. The guidance does not limit the application of Myriad to DNA but, rather, applies the decision to other natural products.
45
Table of Contents
In addition to increasing uncertainty with regard to our ability to obtain future patents, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on these and other decisions by the U.S. Congress, the federal courts and the U.S. PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce any patents that may issue in the future.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Our employees have been previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We also engage advisors and consultants who are concurrently employed at universities or who perform services for other entities.
Although we are not aware of any claims currently pending against us, we may be subject to claims that we or our employees, advisors or consultants have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third party. We have and may in the future also be subject to claims that an employee, advisor or consultant performed work for us that conflicts with that person’s obligations to a third party, such as an employer, and thus, that the third party has an ownership interest in the intellectual property arising out of work performed for us. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying monetary claims, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our product candidates, which would materially adversely affect our commercial development efforts.
Numerous factors may limit any potential competitive advantage provided by our intellectual property rights.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, provide a barrier to entry against our competitors or potential competitors, or permit us to maintain our competitive advantage. Moreover, if a third party has intellectual property rights that cover the practice of our technology, we may not be able to fully exercise or extract value from our intellectual property rights. The following examples are illustrative:
| • | others may be able to develop and/or practice technology that is similar to our technology or aspects of our technology but that is not covered by the claims of patents, should such patents issue from our patent applications; |
| • | we might not have been the first to make the inventions covered by a pending patent application that we own; |
| • | we might not have been the first to file patent applications covering an invention; |
| • | others may independently develop similar or alternative technologies without infringing our intellectual property rights; |
| • | pending patent applications that we own or license may not lead to issued patents; |
| • | patents, if issued, that we own or license may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors; |
| • | third parties may compete with us in jurisdictions where we do not pursue and obtain patent protection; |
| • | we may not be able to obtain and/or maintain necessary or useful licenses on reasonable terms or at all; |
| • | third parties may assert an ownership interest in our intellectual property and, if successful, such disputes may preclude us from exercising exclusive rights over that intellectual property; |
| • | we may not develop or in-license additional proprietary technologies that are patentable; and |
| • | the patents of others may have an adverse effect on our business. |
Should any of these events occur, they could significantly harm our business and results of operations.
General Company-Related Risks
As our product candidates reach alter stage clinical development, we will need to develop and expand our company, and we may encounter difficulties in managing this development and expansion, which could disrupt our operations.
As of the date of this filing, we had 29 full-time employees and no part-time employees, and as our product candidates reach later stage clinical development, we expect to increase our number of employees and the scope of our operations. To manage our anticipated development and expansion, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Also, our management may need to divert a disproportionate amount of its
46
Table of Contents
attention away from its day-to-day activities and devote a substantial amount of time to managing these development activities. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. This may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The physical expansion of our operations may lead to significant costs and may divert financial resources from other projects, such as the development of our product candidates. If our management is unable to effectively manage our expected development and expansion, our expenses may increase more than expected, our ability to generate or increase our revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our product candidates, if approved, and compete effectively will depend, in part, on our ability to effectively manage the future development and expansion of our company.
Our future success depends on our ability to retain our President and Chief Executive Officer and to attract, retain and motivate qualified personnel.
We are highly dependent on Dr. Jeffrey M. Jonas, our President and Chief Executive Officer. We have entered into an employment agreement with Dr. Jonas, but he may terminate his employment with us at any time. Although we do not have any reason to believe that we will lose the services of Dr. Jonas in the foreseeable future, the loss of his services might impede the achievement of our research, development and commercialization objectives. We also do not have any key-man life insurance on Dr. Jonas. We rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us and may not be subject to our standard non-compete agreements. Recruiting and retaining qualified scientific personnel and sales and marketing personnel will also be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific personnel from universities and research institutions. Failure to succeed in clinical trials may make it more challenging to recruit and retain qualified scientific personnel.
Our employees may engage in misconduct or other improper activities, including violating applicable regulatory standards and requirements or engaging in insider trading, which could significantly harm our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with the regulations of the FDA and applicable non-U.S. regulators, provide accurate information to the FDA and applicable non-U.S. regulators, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of, including trading on, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may be ineffective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We face potential product liability exposure, and, if claims are brought against us, we may incur substantial liability.
The use of our product candidates in clinical trials and the sale of our product candidates, if approved, exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with our product candidates. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, including as a result of interactions with alcohol or other drugs, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we become subject to product liability claims and cannot successfully defend ourselves against them, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:
| • | withdrawal of patients from our clinical trials; |
| • | substantial monetary awards to patients or other claimants; |
| • | decreased demand for our product candidates or any future product candidates following marketing approval, if obtained; |
| • | damage to our reputation and exposure to adverse publicity; |
| • | increased FDA warnings on product labels; |
| • | litigation costs; |
| • | distraction of management’s attention from our primary business; |
| • | loss of revenue; and |
| • | the inability to successfully commercialize our product candidates or any future product candidates, if approved. |
47
Table of Contents
We maintain product liability insurance coverage for our clinical trials with a $10 million annual aggregate coverage limit. Nevertheless, our insurance coverage may be insufficient to reimburse us for any expenses or losses we may suffer. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses, including if insurance coverage becomes increasingly expensive. If and when we obtain marketing approval for our product candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may not be able to obtain this product liability insurance on commercially reasonable terms. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial, particularly in light of the size of our business and financial resources. A product liability claim or series of claims brought against us could cause our stock price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, business and prospects could be materially adversely affected.
We will incur increased costs as a result of operating as a public company, and our management team will be required to devote substantial time to new compliance initiatives.
Now that we are a public company, and particularly after we are no longer considered an “emerging growth company,” we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002 and rules subsequently implemented by the Securities and Exchange Commission and The NASDAQ Stock Market have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we will be required to furnish a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our consolidated financial statements.
As we continue to grow, we will need to hire additional qualified accounting and financial personnel with appropriate public company experience.
As we continue to grow our organization, we will need to establish and maintain effective disclosure and financial controls and make changes in our corporate governance practices. We will need to hire additional accounting and financial personnel with appropriate public company experience and technical accounting knowledge, and it may be difficult to recruit and maintain such personnel. Even if we are able to hire appropriate personnel, our existing operating expenses and operations will be impacted by the direct costs of their employment and the indirect consequences related to the diversion of management resources from product development efforts.
Our ability to use our net operating loss carryforwards and certain tax credit carryforwards may be subject to limitation.
As of December 31, 2014, we had federal and state net operating loss carryforwards of $55.8 million and $55.4 million, respectively, which begin to expire in 2031. As of December 31, 2014, we also had federal and state research and development tax credit carryforwards of $0.7 million and $0.3 million, respectively, which begin to expire in 2031 and 2027, respectively. As of December 31, 2014, we had federal orphan drug tax credit carryforwards of $3.6 million, which begin to expire in 2034. Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, changes in our ownership may limit the amount of our net operating loss carryforwards and tax credit carryforwards that could be utilized annually to offset our future taxable income, if any. This limitation would generally apply in the event of a cumulative change in ownership of our company of more than 50% within a three-year period. Any such limitation may significantly reduce our ability to utilize our net operating loss carryforwards and research and development tax credit carryforwards before they expire. The completion of our recent public offering and our initial public offering, or IPO, together with private placements and other transactions that have occurred since our inception, may trigger such an ownership change pursuant to Section 382. Any such limitation, whether as the result of our IPO, recent follow-on offering, prior private placements, sales of our common stock by our existing stockholders or additional sales of our common stock by us, could have a material adverse effect on our results of operations in future years. We have not completed a study to assess whether an ownership change for purposes of Section 382 has occurred, or whether there have been multiple ownership changes since our inception, due to the significant costs and complexities associated with such study.
48
Table of Contents
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. A severe or prolonged economic downturn could result in a variety of risks to our business, including, weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
We or the third parties upon whom we depend may be adversely affected by natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business.
Our internal computer systems, or those of our third-party CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product candidates’ development programs.
Despite the implementation of security measures, our internal computer systems and those of our third-party CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of clinical trial data for our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology or product candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the further development of our product candidates could be delayed.
We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction.
Risks Related to Our Financial Position and Need for Capital
We are a biopharmaceutical company with a limited operating history and have not generated any revenue from product sales. We have incurred significant operating losses since our inception and anticipate that we will incur continued losses for the foreseeable future.
We are a biopharmaceutical company with a limited operating history on which to base your investment decision. Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We were incorporated in April 2010. Our operations to date have been limited primarily to organizing and staffing our company, raising capital and conducting research and development activities for our product candidates. We have never generated any revenue from product sales. We have not obtained regulatory approvals for any of our product candidates.
We have funded our operations to date through proceeds from sales of redeemable convertible preferred stock and, to a lesser extent, the issuance of convertible notes. On July 23, 2014, we completed the sale of 5,750,000 shares of our common stock in our IPO, at a price to the public of $18.00 per share, resulting in net proceeds of $94.0 million after deducting underwriting discounts and commissions and offering expenses paid by us. On April 20, 2015, we completed the sale of 2,628,571 shares of our common stock in a public offering, at a price to the public of $52.50 per share, resulting in net proceeds of $138.0 million after deducting underwriting discounts and commissions and offering expenses paid by us. From our inception through March 31, 2015, we had received net proceeds of $184.6 million from such transactions. As of March 31, 2015, our cash and cash equivalents were $113.2 million. We have incurred significant net losses in each year since our inception, including net losses of $16.9 million for the three months ended March 31, 2015 and $36.1 million and $18.3 million for the years ended December 31, 2014 and 2013, respectively. Substantially all of our operating losses have resulted from costs incurred in connection with our research and development programs and from general and administrative costs associated with our operations. We expect to incur increasing levels of operating losses over the next several years and for the foreseeable future. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ deficit and working capital. We expect our research and development expenses to significantly increase in connection with our clinical trials of our product candidates. In addition, if we obtain marketing approval for our product candidates, we will incur significant sales, marketing and outsourced-manufacturing expenses. As a public company, we will incur additional
49
Table of Contents
costs associated with operating as a public company. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if at all. Even if we do become profitable, we may not be able to sustain or increase our profitability on a quarterly or annual basis.
Our ability to become profitable depends upon our ability to generate revenue.
To date, we have not generated any revenue from our lead product candidates, SAGE-547, SAGE-689 and SAGE-217, and we do not know when, or if, we will generate any revenue. We do not expect to generate significant revenue unless and until we obtain
marketing approval of, and begin to sell,
SAGE-547, SAGE-689 or SAGE-217. Our ability to generate revenue depends on a number of factors, including, but not limited to, our ability to:
| • | initiate and successfully complete clinical trials that meet their clinical endpoints; |
| • | initiate and successfully complete all safety studies required to obtain U.S. and foreign marketing approval for our product candidates; |
| • | commercialize our product candidates, if approved, by developing a sales force or entering into collaborations with third parties; and |
| • | achieve market acceptance of our product candidates in the medical community and with third-party payors. |
Absent our entering into a collaboration or partnership agreement, we expect to incur significant sales and marketing costs as we prepare to commercialize our product candidates. Even if we initiate and successfully complete pivotal clinical trials of our product candidates, and our product candidates are approved for commercial sale, and despite expending these costs, our product candidates may not be a commercially successful drug. We may not achieve profitability soon after generating product sales, if ever. If we are unable to generate product revenue, we will not become profitable and may be unable to continue operations without continued funding.
We will need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product development efforts or other operations.
We are currently advancing our product candidates through non-clinical and clinical development. Developing small molecule products is expensive, and we expect our research and development expenses to increase substantially in connection with our ongoing activities, particularly as we advance our product candidate in clinical trials. Depending on the status of regulatory approval or, if approved, commercialization of our product candidates, as well as the progress we make in selling our product candidates, we may require additional capital to fund operating needs thereafter. We may also need to raise additional funds sooner if we choose to pursue additional indications and/or geographies for our product candidates or otherwise expand more rapidly than we presently anticipate.
As of March 31, 2015, our cash and cash equivalents were $113.2 million. We expect that our existing cash and cash equivalents will be sufficient to fund our current operations for at least the next 12 months. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. In any event, we will require additional capital to obtain regulatory approval for, and to commercialize, our product candidates. Raising funds in the current economic environment may present additional challenges. Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all of our stockholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidate or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any product candidate or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights.
We may seek additional capital through a combination of private and public equity offerings, debt financings, collaborations and strategic and licensing arrangements. To the extent that we raise additional capital through the sale of common stock or securities convertible or exchangeable into common stock, your ownership interest in our company will be diluted. In addition, the terms of any such securities may
50
Table of Contents
include liquidation or other preferences that materially adversely affect your rights as a stockholder. Debt financing, if available, would increase our fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic partnerships and licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, our intellectual property, future revenue streams or grant licenses on terms that are not favorable to us.
Risks Related to Our Common Stock
Market volatility may affect our stock price and the value of your investment.
Our IPO was completed on July 23, 2014, in which we sold shares of our common stock at a price of $18.00 per share. There has been a public market for our common stock for only a short period of time. Although our common stock is listed on the NASDAQ Global Market, an active public market for our common stock may not emerge or be sustained.
The market price for our common stock, similar to other biopharmaceutical companies, is likely to be volatile. The market price of our common stock may fluctuate significantly in response to a number of factors, most of which we cannot control, including, among others:
| • | plans for, progress of or results from non-clinical studies and clinical trials of our product candidates; |
| • | the failure of the FDA to approve our product candidates; |
| • | announcements of new products, technologies, commercial relationships, acquisitions or other events by us or our competitors; |
| • | the success or failure of other CNS therapies; |
| • | regulatory or legal developments in the United States and other countries; |
| • | failure of our product candidates, if approved, to achieve commercial success; |
| • | fluctuations in stock market prices and trading volumes of similar companies; |
| • | general market conditions and overall fluctuations in U.S. equity markets; |
| • | variations in our quarterly operating results; |
| • | changes in our financial guidance or securities analysts’ estimates of our financial performance; |
| • | changes in accounting principles; |
| • | our ability to raise additional capital and the terms on which we can raise it; |
| • | sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders; |
| • | additions or departures of key personnel; |
| • | discussion of us or our stock price by the press and by online investor communities; and |
| • | other risks and uncertainties described in these risk factors. |
We have a significant stockholder, which will limit your ability to influence corporate matters and may give rise to conflicts of interest.
A fund affiliated with Third Rock Ventures, or TRV, is our largest stockholder. As of March 31, 2015, TRV beneficially owned approximately 37.8% of our common stock. Accordingly, TRV exerts and will continue to exert significant influence over us and any action requiring the approval of the holders of our common stock, including the election of directors and amendments to our organizational documents, such as increases in our authorized shares of common stock and approval of significant corporate transactions. Furthermore, the interests of TRV may not always coincide with your interests or the interests of other stockholders and TRV may act in a manner that advances its best interests and not necessarily those of other stockholders, including seeking a premium value for its common stock, which might affect the prevailing market price for our common stock.
Our executive officers, directors, principal stockholders and their affiliates will continue to exercise significant control over our company, which will limit your ability to influence corporate matters and could delay or prevent a change in corporate control.
As of March 31, 2015, existing holdings of our executive officers, directors, principal stockholders and their affiliates, including investment funds affiliated with ARCH Venture Fund VII, L.P., or ARCH, TRV, and entities affiliated with Fidelity Investment, or Fidelity, represent beneficial ownership, in the aggregate, of approximately 65.2% of our outstanding common stock. As a result, these stockholders, if they act together, will be able to influence our management and affairs and control the outcome of matters submitted to our stockholders for approval, including the election of directors and any sale, merger, consolidation, or sale of all or substantially all of our assets. These stockholders acquired their shares of common stock for substantially less than the price of the shares of common stock being acquired in the recently completed IPO, and these stockholders may have interests, with respect to their common stock, that are different from those of investors in the recently completed IPO and the concentration of voting power among these stockholders may have an adverse effect on the price of our common stock. In addition, this concentration of ownership might adversely affect the market price of our common stock by:
| • | delaying, deferring or preventing a change of control of us; |
51
Table of Contents
| • | impeding a merger, consolidation, takeover or other business combination involving us; or |
| • | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
Future sales of our common stock may cause our stock price to decline.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur could significantly reduce the market price of our common stock and impair our ability to raise adequate capital through the sale of additional equity securities.
We have broad discretion in how we use the proceeds from our IPO and recent public offering and may not use these proceeds effectively, which could affect our results of operations and cause our stock price to decline.
We will have considerable discretion in the application of the net proceeds from our recently completed public offering and our IPO. We may use the net proceeds for purposes that do not yield a significant return or any return at all for our stockholders. In addition, pending their use, we may invest the net proceeds from the follow-on offering or from the IPO in a manner that does not produce income or that loses value.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, even one that may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws may delay or prevent an acquisition of us or a change in our management. These provisions include a classified board of directors, a prohibition on actions by written consent of our stockholders and the ability of our board of directors to issue preferred stock without stockholder approval. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which limits the ability of stockholders owning in excess of 15% of our outstanding voting stock to merge or combine with us. Although we believe these provisions collectively provide for an opportunity to obtain greater value for stockholders by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer rejected by our board were considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
We are an “emerging growth company,” and as a result of the reduced disclosure and governance requirements applicable to emerging growth companies, our common stock may be less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we have taken advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. Since we have chosen not to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, our auditors are not required to attest to the effectiveness of our internal control over financial reporting. As a result, investors may become less comfortable with the effectiveness of our internal controls and the risk that material weaknesses or other deficiencies in our internal controls go undetected may increase. Since we have chosen to provide reduced disclosures in our periodic reports and proxy statements while we are an emerging growth company, investors have access to less information and analysis about our executive compensation, which may make it difficult for investors to evaluate our executive compensation practices. We cannot predict if investors will find our common stock less attractive because we have chosen to rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We will remain an “emerging growth company” until the earlier of (a) the last day of the fiscal year following the fifth anniversary of the completion of our IPO, (b) the last day of the fiscal year in which we have total annual gross revenue of at least $1.0 billion, (c) the date on which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million as of the prior June 30th , and (d) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We do not intend to pay dividends on our common stock and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We have never declared or paid any cash dividend on our common stock and do not currently intend to do so in the foreseeable future. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends in the foreseeable future. Therefore, the success of an investment in shares of our common stock will depend upon any future appreciation in their value. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which you purchased them.
52
Table of Contents
If equity research analysts stop publishing research or reports about our business or if they issue unfavorable commentary or downgrade our common stock, the price of our common stock could decline.
The trading market for our common stock relies in part on the research and reports that equity research analysts publish about us and our business. We do not control these analysts. The price of our common stock could decline if one or more equity research analysts downgrade our common stock or if analysts issue other unfavorable commentary or cease publishing reports about us or our business.
| Item 2. | Unregistered Sales of Equity Securities and Use of Proceeds |
Unregistered Sales of Equity Securities
None.
Use of Proceeds from Initial Public Offering of Common Stock
On July 23, 2014, we closed the sale of 5,750,000 shares of common stock to the public (inclusive of 750,000 shares of common stock sold by us pursuant to the full exercise of an overallotment option granted to the underwriters) at a price of $18.00 per share, before underwriting discounts. The offer and sale of the shares in our initial public offering was registered under the Securities Act pursuant to registration statements on Form S-1 (File No. 333-196849), which was filed with the SEC on June 17, 2014 and amended subsequently and declared effective by the SEC on July 17, 2014. Following the sale of the shares in connection with the closing of our initial public offering, the offering terminated. The offering did not terminate before all the securities registered in the registration statements were sold. JPMorgan Securities Co. and Goldman Sachs & Co. acted as joint book-running managers of the offering, and Canaccord Genuity Inc. and Leerink Partners acted as co-managers of the offering.
We raised approximately $94.0 million in net proceeds after deducting underwriting discounts and commissions and offering expenses paid by us. None of these expenses consisted of direct or indirect payments made by us to directors, officers or persons owning 10% or more of our common stock or to their associates, or to our affiliates. There has been no material change in the planned use of proceeds from our initial public offering as described in our final prospectus filed with the SEC on July 18, 2014 pursuant to Rule 424(b)(4). We invested the funds received in cash equivalents and other short-term investments in accordance with our investment policy.
The exhibits filed as part of this Quarterly Report on Form 10-Q are set forth on the Exhibit Index, which is incorporated herein by reference.
53
Table of Contents
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| SAGE THERAPEUTICS, INC. | ||||||
| May 15, 2015 | By: | /s/ Jeffrey M. Jonas | ||||
| Jeffrey M. Jonas, M.D. | ||||||
| Chief Executive Officer, President and Director (Principal Executive Officer) | ||||||
| May 15, 2015 | By: | /s/ Kimi Iguchi | ||||
| Kimi Iguchi | ||||||
| Chief Financial Officer (Principal Financial and Accounting Officer) | ||||||
54
Table of Contents
EXHIBIT INDEX
| Incorporated by Reference to: | ||||||||||
| Exhibit |
Description |
Form or |
Exhibit |
Filing |
SEC File | |||||
| 31.1* |
Certification of Principal Executive Officer pursuant to Exchange Act rules 13a-14 or 15d-14. | |||||||||
| 31.2* |
Certification of Principal Financial Officer pursuant to Exchange Act rules 13a-14 or 15d-14. | |||||||||
| 32.1+ |
Certification of Principal Executive Officer and Principal Financial Officer pursuant to Exchange Act rules 13a-14(b) or 15d-14(b) and 18 U.S.C. Section 1350. | |||||||||
| 101.INS* |
XBRL Instance Document. | |||||||||
| 101.SCH* |
XBRL Taxonomy Extension Schema Document. | |||||||||
| 101.CAL* |
XBRL Taxonomy Extension Calculation Document. | |||||||||
| 101.DEF* |
XBRL Taxonomy Extension Definition Linkbase Document. | |||||||||
| 101.LAB* |
XBRL Taxonomy Extension Labels Linkbase Document. | |||||||||
| 101.PRE* |
XBRL Taxonomy Extension Presentation Link Document. | |||||||||
| * | Filed herewith. |
| + | The certification furnished in Exhibit 32.1 hereto is deemed to be furnished with this Quarterly Report on Form 10-Q and will not be deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, except to the extent that the Registrant specifically incorporates it by reference. |
55