of focus, including allergy/inflammation the field of kinase inhibition for cancer and other diseases. Some of these competitive drugs and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. Specifically, there are a large number of companies developing or marketing treatments for cancer, including many major pharmaceutical and biotechnology companies.
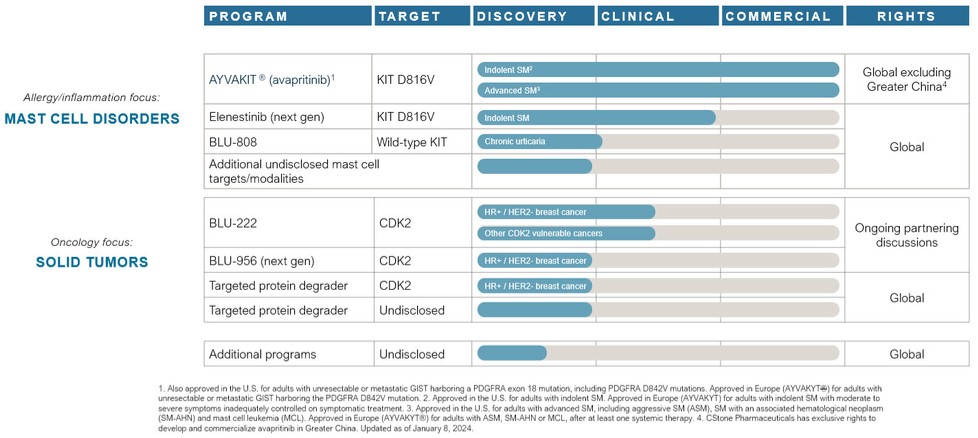
AYVAKIT/AYVAKYT and elenestinib (BLU-263) face competition for advanced SM from Novartis AG’s midostaurin and imatinib, and may face competition from drug candidates in development, including that being developed by Cogent Biosciences, Inc. and Hoth Therapeutics. Avapritinib and elenestinib may face non-advanced SM competition from drug candidates in development, including those being developed by AB Science S.A., Allakos Inc., Cogent Biosciences, Inc., Hoth Therapeutics, Invea Therapeutics Inc., and Theseus Pharmaceuticals Inc.
AYVAKIT/AYVAKYT may face competition from drug candidates in development for PDGFRA-driven GIST, including those being developed by AB Science S.A., ARIAD Pharmaceuticals, Inc., a wholly-owned subsidiary of Takeda Pharmaceutical Company Limited, AROG Pharmaceuticals, Inc., AstraZeneca plc, Cogent Biosciences, Inc., Deciphera Pharmaceuticals, LLC, Exelixis, Inc., Ningbo Tai Kang Medical Technology Co. Ltd. and Xencor, Inc.
We are developing BLU-222 for cancers vulnerable to CDK2 inhibition, including CCNE1-aberrant cancers, which, if approved, will face competition from indication-specific therapies such as AstraZeneca’s capivasertib, AstraZeneca and Merck’s olaparib, AstraZeneca and Daiichi Sankyo’s trastuzumab deruxtecan, Clovis Oncology’s rucaparib, Eisai’s lenvatinib, Genentech’s bevacizumab, GSK’s niraparib, GSK’s dostarlimab, Menarini Group & Stemline Therapeutics’ elacestrant, Merck’s pembrolizumab, and Novartis' alpelisib. In addition, BLU-222 may face competition from drug candidates in development, including those being developed by Acrivon Therapeutics, Allorion Therapeutics, Inc., Anrui Biomedical Technology, Arvinas, AstraZeneca plc, Aucentra Therapeutics, Avenzo Therapeutics, Bayer, BeiGene, BioTheryX, Inc., Cedilla Therapeutics, Inc., Cyclacel Pharmaceuticals Inc., Eli Lilly and Company, Ensem Therapeutics, Exelixis, Gilead Sciences, Inc., IMPACT Therapeutics, Inc., Incyclix Bio, LLC, Incyte Corporation, Monte Rosa Therapeutics, Inc., Pfizer Inc., Plexium, Inc., Regor Therapeutics Inc., Relay Therapeutics, Inc., Repare Therapeutics, Inc., Satya Pharma Innovations Pvt. Ltd., and Zentalis Pharmaceuticals, Inc.
We are developing BLU-808 for chronic urticaria and other allergy/inflammation disorders, which if approved, will face competition from omalizumab developed by Genentech and Novartis. In addition, BLU-808 may face competition from drug candidates in development for chronic urticaria, including those developed by Allakos Inc., Amgen Inc., AstraZeneca plc, Celldex Therapeutics, Inc., Escient Pharmaceuticals, Inc., Evommune, Inc, Incyte Corp., Jasper Therapeutics, Inc., Modulus Discovery Inc., Novartis AG, Regeneron Pharmaceuticals, Inc., Sanofi S.A., Third Harmonic Bio, Inc, Hangshou Highlightll Pharma, Leo Pharma A/S, Acelyrin Inc., Taiho Pharmaceutical Co., LTD, and Enanta Pharmaceuticals, Inc.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize drugs that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any drugs that we or our collaborators may develop. Our competitors also may obtain FDA or other regulatory approval for their drugs more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we or our collaborators are able to enter the market. The key competitive factors affecting the success of all of our drug candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.