MEDIWOUND LTD.
INTRODUCTION
In this annual report, the terms “MediWound,” “we,”
“us,” “our” and “the company” refer to MediWound Ltd. and its subsidiaries.
This annual report includes other statistical, market and industry
data and forecasts which we obtained from publicly available information and independent industry publications and reports that we believe
to be reliable sources. These publicly available industry publications and reports generally state that they obtain their information
from sources that they believe to be reliable, but they do not guarantee the accuracy or completeness of the information. Although we
believe that these sources are reliable, we have not independently verified the information contained in such publications. Certain estimates
and forecasts involve uncertainties and risks and are subject to change based on various factors, including those discussed under the
headings “Special Note Regarding Forward-Looking Statements” and “ITEM 3.D. Risk Factors” in this annual report.
Throughout this annual report, we refer to various
trademarks, service marks and trade names that we use in our business. Solely for convenience, the trademarks, service marks and trade
names are referred to herein without the use of ® and ™ symbols. However, the omission of such symbols are not intended to
indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable
licensors to these trademarks, service marks and trade names. The “MediWound” design logo, “MediWound,” “NexoBrid,”
“EscharEx” and other trademarks or service marks of MediWound Ltd. appearing in this annual report are the property of MediWound
Ltd. We have several other trademarks, service marks and pending applications relating to our solutions. Other trademarks and service
marks appearing in this annual report are the property of their respective holders. Our use or display of other companies’ trademarks,
service marks or trade names is not intended to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
All historical share and per-share numbers for the year ended December
31, 2022 and any prior fiscal periods appearing in this annual report reflect a retroactive adjustment for our 1-for-7 reverse share split
effected on December 20, 2022.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
In addition to historical facts, this annual report on Form 20-F
contains forward-looking statements within the meaning of Section 27A of the U.S. Securities Act of 1933, as amended (the “Securities
Act”), Section 21E of the U.S. Securities Exchange Act of 1934, as amended (the “Exchange Act”) and the safe harbor
provisions of the U.S. Private Securities Litigation Reform Act of 1995. We make forward-looking statements in this annual report that
are subject to risks and uncertainties. These forward-looking statements include information about possible or assumed future results
of our business, financial condition, results of operations, liquidity, plans and objectives. In some cases, you can identify forward-looking
statements by terminology such as “believe,” “may,” “estimate,” “continue,” “anticipate,”
“intend,” “should,” “plan,” “expect,” “predict,” “potential,”
or the negative of these terms or other similar expressions. The statements we make regarding the following matters are forward-looking
by their nature:
|
|
•
|
our commercialization, marketing and manufacturing capabilities and strategy and the
ability of our marketing team to cover European regional burn centers and units;
|
|
|
•
|
the timing and conduct of our trials of NexoBrid, EscharEx and our other pipeline
product candidates, including statements regarding the timing, progress and results of current and future preclinical studies and clinical
trials, and our research and development programs;
|
|
|
•
|
the clinical utility, potential advantages and timing or likelihood of regulatory
filings and approvals of EscharEx and our other pipeline products;
|
|
|
•
|
our expectations regarding future growth, including our ability to develop new products;
|
|
|
•
|
our estimates regarding expenses, future revenues, capital requirements and our need
for additional financing;
|
i
|
|
•
|
anticipated funding under our contracts with the U.S. Biomedical Advanced Research
and Development Authority;
|
|
|
•
|
our ability to maintain adequate protection of our intellectual property;
|
|
|
•
|
our estimates regarding the market opportunity for NexoBrid, EscharEx and our other
pipeline products;
|
|
|
•
|
our expectation regarding the duration of our inventory of intermediate drug substances
and products;
|
|
|
•
|
the impact of our research and development expenses as we continue developing product
candidates; and
|
|
|
•
|
the impact of government laws and regulations. |
The preceding list is not intended to be an exhaustive
list of all our forward-looking statements. The forward-looking statements are based on our beliefs, assumptions and expectations of future
performance, taking into consideration the information currently available to us. These statements are only predictions based upon our
current expectations and projections about future events. These statements may be found in the sections of this annual report on Form
20-F entitled “ITEM 3.D. Risk Factors,” “ITEM 4. Information on the Company,” “ITEM 5. Operating and Financial
Review and Prospects,” “ITEM 10.E. Taxation-United States Federal Income Taxation-Passive Foreign Investment Company Considerations”
and elsewhere in this annual report, including the section entitled “ITEM 4.B. Business Overview” and “ITEM 4.B. Business
Overview-Our Focus,” which contain information obtained from independent industry sources. Actual results could differ materially
from those anticipated in these forward-looking statements due to various important factors, including all the risks discussed in “ITEM
3.D. Risk Factors” and information contained in other documents filed with or furnished to the Securities and Exchange Commission.
You should not rely upon forward-looking statements as predictions
of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee
that future results, levels of activity, performance and events and circumstances reflected in the forward-looking statements will be
achieved or will occur. Except as required by law, we undertake no obligation to publicly update any forward-looking statements for any
reason after the date of this annual report to conform these statements to actual results or to changes in our expectations.
ii
PART I
Item 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
Not applicable.
Item 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
|
|
B. |
Capitalization and Indebtedness |
Not applicable.
|
|
C. |
Reasons for the Offer and Use of Proceeds |
Not applicable.
Our business faces significant risks. You should
carefully consider all of the information set forth in this annual report and in our other filings with the United States Securities and
Exchange Commission (the “SEC”), including the following risk factors which we face and which are faced by our industry. Our
business, financial condition and results of operations could be materially and adversely affected by any of these risks. In that event,
the trading price of our ordinary shares would likely decline and you might lose all or part of your investment. This report also contains
forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking
statements, as a result of certain important factors including the risks described below and elsewhere in this report and our other SEC
filings. See “Special Note Regarding Forward-Looking Statements” on page i.
Risks Related to Development, Clinical Testing and Regulatory Approval
Product development is a lengthy and expensive
process, with an uncertain outcome.
We intend to develop and commercialize pipeline product candidates
based on our patented enzymatic technology platform for (i) marketing authorization of EscharEx in the U.S. and other jurisdictions, (ii)
NexoBrid in the U.S. for children and for all ages in other jurisdictions and (iii) our other pipeline products in variety of jurisdictions
across the world. However, before obtaining regulatory approval for the sale of our pipeline product candidates in any jurisdiction, we
must conduct, at our own expense, clinical studies to demonstrate that the products are safe and effective.
Preclinical and clinical testing is expensive, is difficult to
design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more of our clinical trials
can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, preclinical testing and the
clinical trial process. Even if preclinical or clinical trials are successful, we still may be unable to commercialize the product, as
success in preclinical trials, clinical trials or previous clinical trials does not ensure that later clinical trials will be successful.
A number of events could delay or prevent our ability to complete
necessary clinical trials for our pipeline product candidates, including:
|
|
•
|
regulators may not authorize us to conduct a clinical trial within a country or at
a prospective trial site or may require us to change the design of a study; |
|
|
•
|
delays may occur in reaching agreement on acceptable clinical trial terms with regulatory
authorities or prospective sites, or obtaining institutional review board or ethics committee approval or opinion; |
|
|
•
|
our preclinical tests or clinical trials may produce negative or inconclusive results,
and we may decide, or regulators may require us, to conduct additional trials or to abandon strategic projects; |
|
|
•
|
the number of patients required for our clinical trials may be larger than we anticipate,
enrollment in our clinical trials may be slower or more difficult than we expect, or patients may not participate in necessary follow-up
visits to obtain required data, any of which would result in significant delays in our clinical testing process;
|
|
|
•
|
our third-party contractors, such as a research institute, may fail to comply with
regulatory requirements or meet their contractual obligations to us. For example, due to a deviation associated with a third-party testing
lab used during the manufacturing process, our commercial partner in the U.S., Vericel, was unable to release NexoBrid in into the commercial
channel until an agreement with the FDA on this matter, resulting a delay of few month in the launch of NexoBrid in the U.S.;
|
|
|
•
|
we may be forced to suspend or terminate our clinical trials if the participants are
being exposed, or are thought to be exposed, to unacceptable health risks or if any participant experiences an unexpected serious adverse
event; |
|
|
•
|
regulators or institutional review boards may require that we hold, suspend or terminate
clinical research for various reasons, including noncompliance with regulatory requirements; |
|
|
•
|
undetected or concealed fraudulent activity by a clinical researcher, if discovered,
could preclude the submission of clinical data prepared by that researcher, lead to the suspension or substantive scientific review of
one or more of our marketing applications by regulatory agencies, and result in the recall of any approved product distributed pursuant
to data determined to be fraudulent;
|
|
|
•
|
the cost of our clinical trials may be greater than we anticipate; |
|
|
•
|
an audit of preclinical or clinical studies by regulatory authorities may reveal noncompliance
with applicable protocols or regulations, which could lead to disqualification of the results and the need to perform additional studies;
|
|
|
•
|
political unrest and wars, such as the war in Gaza and the conflict between Russia
and Ukraine, which could delay or disrupt business activity, and if such political unrest escalates or spills over to or otherwise impacts
additional regions, it could also heighten many of the other risk factors described in this Annual Report; |
|
|
•
|
delays may occur in obtaining our clinical materials; and |
|
|
•
|
epidemics or pandemics, such as the COVID-19 pandemic, which can affect the overall
healthcare infrastructure, including the ability to recruit patients, the ability to conduct studies at medical sites and the pace with
which governmental agencies, such as the FDA and foreign regulatory authorities, will review and approve regulatory submissions.
|
Moreover, we do not know whether preclinical tests or clinical
trials will begin or be completed as planned or will need to be restructured. Significant delays could also shorten the patent protection
period during which we may have the exclusive right to commercialize our pipeline product candidates or could allow our competitors to
bring products to the market before we do, impairing our ability to commercialize our pipeline product candidates.
Development and commercialization of EscharEx
and NexoBrid for pediatric use in the United States as well as our pipeline product candidates worldwide requires successful completion
of the regulatory approval process, which may suffer delays or fail.
In the United States, as well as other jurisdictions, we are required
to apply for and receive marketing authorization before we can market our products, as we have already received for NexoBrid in the United
States, the European Union (“EU”) and other international markets. This process can be time-consuming and complicated and
may result in unanticipated delays. To secure marketing authorization, an applicant generally is required to submit an application that
includes the data supporting preclinical and clinical safety and efficacy as well as detailed information on the manufacturing and control
of the product, proposed labeling and other information. Before marketing authorization is granted, regulatory authorities generally require
the inspection of the manufacturing facility or facilities and quality systems (including those of third parties) at which the product
candidate is manufactured and tested, to assess compliance with strictly enforced current good manufacturing practices (“cGMP”)
and similar foreign requirements such as Good Manufacturing Practices (“GMP”) in the EU, as well as potential audits of the
non-clinical and clinical trial sites that generated the data cited in the marketing authorization application to assess compliance with
requisite good clinical practices (“GCP”).
We cannot predict how long the applicable regulatory authority
or agency will take to grant marketing authorization or whether any such authorizations will ultimately be granted. Regulatory agencies,
including the FDA and the European Medicines Agency (the “EMA”), have substantial discretion in the approval process, and
the approval process and the requirements governing clinical trials vary from country to country. The policies of the FDA, the EMA or
other regulatory authorities may change or may not be explicit, and additional government regulations may be enacted that could prevent,
limit or delay regulatory approval of EscharEx, NexoBrid or our pipeline product candidates. For instance, the regulatory landscape related
to clinical trials in the EU recently evolved. The EU Clinical Trials Regulation (“CTR”) which was adopted in April 2014 and
repeals the EU Clinical Trials Directive, became applicable on January 31, 2022. While the EU Clinical Trials Directive required a separate
clinical trial application (“CTA”) to be submitted in each member state in which the clinical trial takes place, to both the
competent national health authority and an independent ethics committee, the CTR introduces a centralized process and only requires the
submission of a single application for multi-center trials. The CTR allows sponsors to make a single submission to both the competent
authority and an ethics committee in each member state, leading to a single decision per member state. The assessment procedure of the
CTA has been harmonized as well, including a joint assessment by all member states concerned, and a separate assessment by each member
state with respect to specific requirements related to its own territory, including ethics rules. Each member state’s decision is
communicated to the sponsor via the centralized EU portal. Once the CTA is approved, clinical study development may proceed. The CTR foresees
a three-year transition period. The extent to which ongoing and new clinical trials will be governed by the CTR varies. Clinical trials
for which an application was submitted (i) prior to January 31, 2022 under the EU Clinical Trials Directive, or (ii) between January 31,
2022 and January 31, 2023 and for which the sponsor has opted for the application of the EU Clinical Trials Directive remain governed
by said Directive until January 31, 2025. After this date, all clinical trials (including those which are ongoing) will become subject
to the provisions of the CTR. Compliance with the CTR requirements may impact our development plans. It is currently unclear to what extent
the United Kingdom (“UK”) will seek to align its regulations with the EU. The UK regulatory framework in relation to clinical
trials is derived from existing EU legislation (as implemented into UK law, through secondary legislation). On January 17, 2022, the UK
Medicines and Healthcare products Regulatory Agency (“MHRA”) launched an eight-week consultation on reframing the UK legislation
for clinical, which aims to streamline clinical trials approvals, enable innovation, enhance clinical trials transparency, enable greater
risk proportionality, and promote patient and public involvement in clinical trials. The MHRA published its consultation outcome on March
21, 2023 in which it confirmed that it would update the existing legislation. The resulting legislative changes will ultimately determine
the extent to which the UK regulations align with the CTR. A decision by the UK not to closely align its regulations with the new approach
that has been adopted in the EU may have an effect on the cost of conducting clinical trials in the UK as opposed to other countries.
Additionally, the EU pharmaceutical legislation is currently undergoing
a complete review process, in the context of the Pharmaceutical Strategy for Europe initiative, launched by the European Commission in
November 2020. The European Commission’s proposal for revision of several legislative instruments related to medicinal products
(potentially reducing the duration of regulatory data protection, revising the eligibility for expedited pathways, etc.) was published
on April 26, 2023. The proposed revisions, remain to be agreed and adopted by the European Parliament and European Council and the proposals
may, however, therefore be substantially revised before adoption, which is not anticipated before early 2026. The revisions may have a
significant impact on the biopharmaceutical industry in the long term. If we are slow or unable to adapt to changes in existing requirements
or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing authorization
that we may have obtained and we may not achieve or sustain profitability. We also cannot predict the likelihood, nature or extent of
government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad.
If such actions impose constraints on the FDA’s ability to engage in oversight and implementation activities in the normal course,
or if we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are
unable to maintain regulatory compliance, we may be subject to enforcement action and our business may be negatively impacted.
In addition, any regulatory approval that we will receive may also
contain requirements for potentially costly post-marketing testing, including Phase IV clinical trials, and surveillance to monitor the
safety and efficacy of the product candidate. For example, as part of the EMA regulatory approval process, we agreed to provide further
data from a post-marketing U.S. Phase III clinical trial of NexoBrid, which serves to address this post-marketing commitment to EMA. Once
a product is approved, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising,
promotion, import, export and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements
include submission of safety and other post-marketing information and reports, registration and continued compliance with cGMP and similar
foreign requirements and GCP for any clinical trials that we conduct post-approval. Although our manufacturing facility is cGMP-certified,
we may face difficulties in obtaining regulatory approval for the manufacturing and quality control process of our pipeline product candidates.
Any delays or failures in obtaining regulatory and marketing authorization
for EscharEx in the United States, or for NexoBrid or our pipeline product candidates worldwide, would adversely affect our business,
prospects, financial condition and results of operations.
Even though the FDA has approved NexoBrid for eschar removal in
adults with deep partial thickness and/or full thickness thermal burns, we will still face extensive and ongoing regulatory requirements
and obligations for EscharEx, NexoBrid for pediatric use, and for any product candidates for which we obtain approval.
Any regulatory approvals that we have received for NexoBrid and
may receive for NexoBrid, EscharEx or any of our product candidates will require the submission of reports to regulatory authorities and
surveillance to monitor the safety and efficacy of the product, may contain significant limitations related to use restrictions for specified
age groups, warnings, precautions or contraindications, and may include burdensome post-approval study or risk management requirements.
For example, the FDA-approved label for NexoBrid includes certain warnings and precautions regarding hypersensitivity reactions, pain
management, proteolytic injury to non-target tissue and coagulopathy.
In addition, the manufacturing processes, labeling, packaging,
distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for NexoBrid are and will remain
subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information
and reports, registration, as well as on-going compliance with cGMPs, and GCPs for any clinical trials that we conduct post-approval.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic, unannounced inspections
by the FDA and other regulatory authorities for compliance with cGMP regulations and standards. If we or a regulatory authority discover
previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facilities
where the product is manufactured, a regulatory authority may impose restrictions on that product, the manufacturing facility or us, including
requiring recall or withdrawal of the product from the market or suspension of manufacturing.
In addition, later discovery of previously unknown adverse events
or other problems with our products, manufacturers or manufacturing processes or failure to comply with regulatory requirements, may yield
various results, including:
•
restrictions on manufacturing such products;
•
restrictions on the labeling or marketing of products;
•
restrictions on product manufacturing, distribution or use;
•
requirements to conduct post-marketing studies or clinical trials;
• warning
letters or untitled letters;
• withdrawal
of the products from the market;
• refusal
to approve pending applications or supplements to approved applications that we submit;
•
recall of products;
• fines,
restitution or disgorgement of profits or revenues;
• suspension
or withdrawal of marketing authorizations;
•
refusal to permit the import or export of our products;
• product
seizure; or
•
injunctions or the imposition of civil or criminal penalties.
Further, the policies of the FDA and other regulatory authorities
may change, and additional government regulations may be enacted that could impose extensive and ongoing regulatory requirements and obligations
on any product candidate for which we obtain marketing authorization. We also cannot predict the likelihood, nature or extent of government
regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad.
Changes in funding or disruptions at FDA and
other government agencies caused by funding shortages or global health concerns could hinder their ability to hire and retain key leadership
and other personnel, or otherwise prevent new or modified products from being developed, approved or commercialized in a timely manner
or at all, or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely,
which could negatively impact our business.
The ability of FDA and foreign regulatory authorities to review
and approve new products can be affected by a variety of factors, including government budget and funding levels, statutory, regulatory,
and policy changes, FDA’s and foreign regulatory authorities’ ability to hire and retain key personnel and accept the payment
of user fees, and other events that may otherwise affect FDA’s and foreign regulatory authorities’ ability to perform routine
functions. Average review times at the FDA and foreign regulatory authorities have fluctuated in recent years as a result. In addition,
government funding of other government agencies that fund research and development activities is subject to the political process, which
is inherently fluid and unpredictable. Disruptions at FDA and other agencies such as the EMA, following its relocation to Amsterdam and
resulting staff changes, may also slow the time necessary for new drugs and biologics to be reviewed and/or approved by necessary regulatory
authorities, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December
22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as FDA, have had to furlough critical
FDA employees and stop critical activities.
Separately, in response to the COVID-19 pandemic,
the FDA postponed most inspections of domestic and foreign manufacturing facilities at various points. Even though the FDA has since resumed
standard inspection operations, the FDA has continued to monitor and implement changes to its inspectional activities to ensure the safety
of its employees and those of the firms it regulates as it adapts to the evolving COVID-19 pandemic, and any resurgence of the virus or
emergence of new variants may lead to further inspectional or administrative delays. Regulatory authorities outside the United States
have adopted similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs,
or if global health concerns continue to prevent FDA or other regulatory authorities from conducting their regular inspections,
reviews, or other regulatory activities, it could significantly impact the ability of FD or, other regulatory authorities to timely
review and process our regulatory submissions, which could have a material adverse effect on our business.
NexoBrid, EscharEx or our pipeline product
candidates may cause unanticipated and undesirable side effects or have other properties, which are currently unknown to us.
NexoBrid, EscharEx and all of our current pipeline product candidates
rely on our patented enzymatic platform technology, although their specific formulations or mode of applications may vary. Like most pharmaceutical
products, our approval labels for NexoBrid in the United States, Europe and other international markets list certain side effects. If
we or others identify previously unknown problems with NexoBrid, EscharEx or their underlying proteolytic enzymes, including adverse events
of unanticipated severity or frequency, problems with our manufacturers or manufacturing processes, or failure to comply with regulatory
requirements, the following consequences, among others, may result, including, without limitation:
|
|
•
|
restrictions on the marketing or manufacturing of the product, withdrawal of the product
from the market or voluntary or mandatory product recalls;
|
|
|
•
|
fines, warning letters or holds on clinical trials;
|
|
|
•
|
harm to our reputation, reduced demand for our products and loss of market acceptance;
|
|
|
•
|
refusal by the applicable regulatory authority to approve pending applications or
supplements to approved applications filed by us, or suspension or revocation of product license approvals;
|
|
|
•
|
product seizure or detention, or refusal to permit the import or export of products;
and
|
|
|
• |
injunctions or the imposition of civil or criminal penalties. |
Any of these events could prevent us from achieving or maintaining
market acceptance of NexoBrid, and future market acceptance of EscharEx, our pipeline product candidates or future product candidates,
which would adversely affect our business, prospects, financial condition and results of operations.
Regulatory approval for NexoBrid, EscharEx
and other pipeline product candidates is and may be limited to specific indications and conditions for which clinical safety and efficacy
have been demonstrated, and the prescription of off-label uses could adversely affect our business.
The marketing authorization for NexoBrid in the European Union
and other international markets is limited to the treatment of deep partial- and full-thickness burns in adults. In the United States,
the marketing authorization for NexoBrid is limited to eschar removal in adults with deep partial thickness and/or full thickness thermal
burns. Any additional regulatory approval of NexoBrid for severe burns and any regulatory approval we may receive for any of our pipeline
product candidates in the future, would be limited to those specific indications for which such pipeline product candidate had been deemed
safe and effective by the FDA, EMA, or another regulatory authority Additionally, labeling restrictions in the U.S. and EU limit the manner
in which a product may be used. For example, NexoBrid’s label in the U.S. and EU provides that it may only be used in specialized
burns centers or by burn specialists and that it is not to be applied to more than 30% and 15% of the patient’s total body surface
area, respectively. If physicians prescribe the medication for unapproved, or “off-label,” uses or in a manner that is inconsistent
with the manufacturer’s labeling, it could produce results such as reduced efficacy or other adverse effects, and the reputation
of our products in the marketplace may suffer. In addition, should any of our future products have a significant price difference and
if they are used interchangeably, off-label uses may cause a decline in our revenues or potential revenues. Furthermore, while physicians
may choose to prescribe treatments for uses that are not described in the product’s labeling and for uses that differ from those
approved by regulatory authorities, we cannot promote the products for any indications other than those that are specifically approved
by the European Commission, the FDA or other regulatory authorities. Regulatory authorities restrict communications by companies on the
subject of off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to enforcement
actions by those authorities. In the United States, “off-label promotion” by pharmaceutical companies has resulted in significant
litigation under the Federal False Claims Act, violations of which may result in substantial civil penalties and fines as well as exclusion
from government health care programs. More generally, failure to follow the rules and guidelines of regulatory agencies relating to promotion
and advertising, such as that promotional materials not be false or misleading, can result in refusal to approve a product, the suspension
or withdrawal of an approved product from the market, product recalls, fines, disgorgement of money, operating restrictions, injunctions
or criminal prosecution.
Risks Related to Manufacturing
We may not be able to expand our production
or processing capabilities or satisfy future demand.
Our global demand for NexoBrid surpasses the current manufacturing
capabilities. We are currently seeking to expand our manufacturing capabilities in order to increase our capacity to manufacture NexoBrid
and future product candidates and satisfy near term demand. The new GMP-compliant state-of-the-art manufacturing facility is projected
to be completed by mid-2024, with full-scale manufacturing currently expected to commence in 2025. However, we cannot guarantee that we
will be able to obtain the requisite approvals, including meeting regulatory and quality requirements, or if we do, that the facility
will satisfy additional growing demand. Conversely, there can be no assurance that even if we obtain a new facility, demand for our products
will increase proportionately to the increased production capability. Furthermore, we cannot assure that this or similar projects will
be implemented in a timely and cost-efficient manner, and that our current production will not be adversely affected by the operational
challenges of implementing the expansion project.
If our manufacturing facility in Yavne, Israel
was to suffer a serious accident, or if a force majeure event were to materially affect our ability to operate and produce NexoBrid, EscharEx
and our pipeline product candidates, all of our manufacturing capacity could be shut down for an extended period.
We currently rely on a single manufacturing facility
in Yavne, Israel, and we expect that all of our revenues in the near future will be derived from products manufactured at this facility.
If this facility were to suffer an accident or a force majeure event such as war, missile or terrorist attack, earthquake, major fire
or explosion, major equipment failure or power failure lasting beyond the capabilities of our backup generators or similar event, our
revenues would be materially adversely affected and any of our clinical trials could be materially delayed. In this situation, our manufacturing
capacity could be shut down for an extended period, we could experience a loss of raw materials, work in process or finished goods inventory
and our ability to operate our business would be harmed. In addition, in any such event, the reconstruction of our manufacturing facility
and storage facilities, and obtaining regulatory approval for the new facilities could be time-consuming. During this period, we would
be unable to manufacture NexoBrid or our pipeline product candidates. In addition, we currently have a limited inventory of NexoBrid that
we can supply to our customers if we are unable to further manufacture NexoBrid.
We are subject to a number
of other manufacturing risks, any of which could substantially increase our costs and limit the supply of NexoBrid, EscharEx and our pipeline
product candidates.
The process of manufacturing NexoBrid, EscharEx and our pipeline
product candidates is complex, highly regulated and subject to the risk of product loss due to contamination, equipment failure or improper
installation or operation of equipment, or vendor or operator error. Even minor deviations from normal manufacturing processes or quality
requirements for our products could result in reduced production yields, product defects and other supply disruptions. If microbial, viral
or other contaminations are discovered in NexoBrid, EscharEx or our pipeline product candidates or in the manufacturing facilities in
which NexoBrid, EscharEx or our pipeline product candidates are or will be made, such manufacturing facilities may need to be closed to
investigate and remedy the contamination.
We may experience any contaminations, major equipment failures,
or other similar manufacturing problems of such magnitude, any adverse developments affecting manufacturing operations for NexoBrid, EscharEx
or our pipeline product candidates which may result in additional shipment delays, inventory shortages, lot failures, withdrawals or recalls,
or other interruptions in the supply of NexoBrid, EscharEx or our pipeline product candidates. We may also have to take inventory write-offs
and incur other charges and expenses for our products that fail to meet specifications, undertake costly remediation efforts, or seek
costlier manufacturing alternatives.
Our ability to continue manufacturing and distributing
our products depends on our continued adherence to cGMP regulations.
The manufacturing processes for our products are governed by detailed
cGMP and similar foreign regulations, both for our marketed products in the EU and the U.S. and product candidates in clinical testing
in the U.S., EU and Israel. Failure by our manufacturing and quality operations unit to adhere to established regulations or to meet a
specification or procedure set forth in cGMP and similar foreign requirements could require that a product or material be rejected and
destroyed. Our adherence to cGMP and similar foreign regulations and the effectiveness of our quality control systems are periodically
assessed through inspections of our manufacturing facility by regulatory authorities. Such inspections could result in deficiency citations,
which would require us to take action to correct those deficiencies to the satisfaction of the applicable regulatory authorities. If critical
deficiencies are noted or if we are unable to prevent recurrences, we may have to recall products or suspend operations until appropriate
measures can be implemented. Since cGMP and similar foreign regulations reflect ever-evolving standards, we need to regularly update our
manufacturing processes and procedures to comply with cGMP and similar foreign regulations. These changes may cause us to incur additional
costs and may adversely impact our profitability. For example, more sensitive testing assays (if and when they become available, or due
to the discontinuation of the availability of the disposables currently used in production) may be required, or existing procedures or
processes may require revalidation, all of which may be costly and time-consuming and could delay or prevent the manufacturing of NexoBrid
or launch of a new product.
We depend on a sole supplier to obtain our
intermediate drug substance, bromelain SP, which is necessary for the production of our products.
We currently procure bromelain SP, a key substance starting material
in the manufacturing of NexoBrid, EscharEx and our pipeline product candidates, from a single supplier, Challenge Bioproducts Corporation
Ltd. (“CBC”). CBC’s manufacturing facilities are located in the Republic of China and it uses proprietary methods to
manufacture bromelain SP. Our supply agreement with CBC has no fixed expiration date and can be voluntarily terminated by us, with at
least six months’ advance written notice, or by CBC, with at least 24 months’ advance written notice. Although we have a contractual
right to procure this material from other suppliers, subject to payment of a one-time, non-material licensing fee to CBC, procuring this
material from any other source would require time and effort which may interrupt our supply of bromelain SP and may cause an interruption
of the supply of NexoBrid, EscharEx and our pipeline product candidates to the marketplace and for future clinical trials or other development
purposes. Regulatory authorities could require that we conduct additional studies in support of a new supplier, which could result in
significant additional costs or delays. Furthermore, there can be no assurance that we would be able to procure alternative supplies of
bromelain SP at all or at comparable quality or competitive prices or upon fair and reasonable contractual terms and conditions. Although
we believe that we currently store sufficient inventory of bromelain SP in our warehouse and CBC warehouse to continue full capacity operations
for non-EU markets for approximately two years, this inventory may prove insufficient, and any interruption or failure to source additional
bromelain SP from CBC or other third parties in a timely manner, or at all, would adversely affect our business, prospects, financial
condition and results of operations.
In addition, we rely on CBC to successfully scale-up its CBC’s
manufacturing facilities in order to meet future demand of our products pipelines. If CBC will not be able to obtain the requisite approvals,
including meeting regulatory and quality requirements or will not be able to scale-up its manufacturing facility in a timely manner, then
we may not be able to satisfy demand for our future products.
Our sole supplier of intermediate drug substance,
bromelain SP, is located in Taiwan, which exposes us to risks that harm our ability to manufacture NexoBrid, EscharEx and our pipeline
product candidates and substantially harm our business.
The manufacturing facilities of CBC, our sole supplier of bromelain
SP, a key substance as starting material in the manufacturing of NexoBrid, EscharEx and our pipeline product candidates, are located in
Taiwan. We believe one of the most significant risks associated with these
facilities being located in Taiwan is the risk that production may be interrupted or limited due to strains on the local infrastructure.
In addition, facilities located in Taiwan may be adversely affected by tensions, hostilities or trade disputes involving China, the United
States or other countries. There is considerable potential political instability in Taiwan related
to its disputes with China. Although we do not do business in North Korea, any future increase in
tensions between South Korea and North Korea, such as an outbreak or escalation of military hostilities,
or between Taiwan and China could materially adversely affect our operations in Asia or the global
economy, which in turn may seriously harm our business.
In addition, if CBC experiences any closures and labor shortages
as a result of rising tensions between the People’s Republic of China and Taiwan, we may face difficulty sourcing bromelain SP,
which could negatively affect our revenues.
Risks Related to Commercialization
Our revenue growth depends initially on our
ability to commercialize NexoBrid.
We currently have a marketing authorization in the United States,
the European Economic Area (“EEA”) (which consists of the 27 EU member states plus Norway, Liechtenstein, Switzerland and
Iceland), U.K., Israel, Russia, Ukraine, South Korea, Taiwan, United Arab Emirates Eurasian countries, Japan and India for a single product,
NexoBrid for eschar removal in adults with deep partial thickness and/or full thickness thermal burns, which we refer to as severe burns.
We are currently relying, for a significant portion of our revenues from sales of products, on sales of NexoBrid in the U.S., Europe and
in other international markets for the treatment of severe burns. We anticipate that, for at least the next several years, our ability
to generate revenues and become profitable will depend on the commercial success of NexoBrid in these markets, the procurement of the
Biomedical Advanced Research and Development Authority (“BARDA”) as well as successful launches in new markets such as the
U.S. and Japan.
The commercialization success of NexoBrid in
the U.S. is dependent on the actions of our partner Vericel.
On May 6, 2019, we entered into an exclusive license and supply agreements with Vericel
Corporation (”Vericel”) to commercialize NexoBrid in all countries of North America (the “Vericel License Agreement”).
In accordance with the Vericel License Agreement, Vericel paid us $17.5 million in cash as an upfront payment at the execution of the
Vericel License Agreement and an additional milestone payment of $7.5 million upon the achievement of BLA approval for NexoBrid. Vericel
is obligated to pay us up to $125 million, in the aggregate, upon attainment of certain sales milestones. Vericel is also obligated to
pay us tiered royalties on net sales of NexoBrid ranging from mid-high single-digit to mid-teen percentages, subject to certain customary
reductions, as well as a percentage of gross profits on committed purchases by BARDA and a royalty on additional sales to BARDA. The success
of our business depends largely on Vericel's success in commercializing NexoBrid. If Vericel does not succeed in launching and commercializing
NexoBrid in the U.S. or does not comply with the terms of our agreement, and as a result a dispute between us and Vericel arises, our
ability to generate revenues from NexoBrid will be substantially harmed.
We are dependent on our contract with BARDA
and/or MTEC/DOD to procure from us NexoBrid or to fund our development activities for NexoBrid for field use in the United States. If
these contracts will be suspend or terminate their procurement obligation of NexoBrid it will adversely impact our future revenues.
In September 2015, we were awarded the first BARDA Contract for
treatment of thermal burn injuries. This contract was amended several times over the years to extend its term until September 2024 (the
Company is pursuing an extension of the contract) and its aggregate amount which was awarded to $165 million as of the end of 2022. In
March 2023 BARDA expanded its awarded contract by providing supplemental funding of $10 million to support a $3 million replenishment
of expired product previously procured for emergency preparedness, the pediatric indication sBLA submission to the FDA, and enrollment
of an additional 50 patients in the expanded access treatment protocol (NEXT) (collectively the "First BARDA Contract").
The First BARDA Contract provided funding and technical support
for the pivotal U.S. Phase 3 clinical study (DETECT), the randomized, controlled pivotal clinical trial for use in the pediatric population
(CIDS), the marketing approval registration process for NexoBrid as well as its procurement and availability under the expanded access
treatment protocol (NEXT) in the U.S.
The total amount of the First BARDA Contract
is comprised of up to $110 million to support research and development activities and up to $65 million to procure NexoBrid for U.S. emergency
preparedness.
As of December 31, 2023, the Company has received approximately
$88 million in funding in the aggregate, under the First BARDA Contract, and an additional $16.5 million for procurement of NexoBrid for
U.S. emergency preparedness.
In Addition, In February, 2023, the Company was entered in to a
contract with the U.S. Department of Defense (DOD), through the Medical Technology Enterprise Consortium (MTEC), to develop NexoBrid as
a non-surgical solution for field-care burn treatment for the U.S. Army. The contract provided funding up to $2.7 million. During 2023,
the DOD through MTEC and directly through MTEC awarded the Company additional funding of $10.3 million to advance the development of a
new temperature stable formulation of NexoBrid.
However, BARDA and/or MTEC/DOD may terminate
the contract at any time, at its convenience, without any further funding obligations. There can be no assurances that BARDA and/or MTEC/DOD
will not terminate the contract. Changes in government budgets and agendas may result in a decreased and de-prioritized emphasis on supporting
the development of NexoBrid for field use. Any reduction or delay in MTEC/DOD funding may force us to suspend the program or seek alternative
funding, which may not be available on non-dilutive terms, terms favorable to us or at all. Further, we cannot provide any assurances
as to when or whether BARDA's to further fund $48.5 million (which includes $3 million allocated for replenishing of expired product,
which was a part of the supplemental funding in March 2023) for additional procurement of NexoBrid will be exercised.
The commercial success of NexoBrid, EscharEx
and our pipeline product candidates will depend upon their degree of market acceptance.
NexoBrid, EscharEx and our pipeline product candidates may not
gain market acceptance by physicians and their teams, healthcare payors, patients and others in the medical community. Although many physicians
in burn centers throughout Europe, the United States and other international markets have used NexoBrid for severe burns as part of our
clinical trials or since NexoBrid’s commercial launch in Europe, and other international markets, we cannot guarantee that use of
NexoBrid will be accepted in the market. We need to successfully integrate NexoBrid into the overall treatment of burns in burn centers.
If NexoBrid, EscharEx and our pipeline product candidates do not achieve an adequate level of acceptance, we may not generate revenue
and we may not achieve or sustain profitability. The degree of market acceptance of NexoBrid in U.S., Europe and in other international
countries where we receive marketing authorization, and of EscharEx and our pipeline product candidates, will depend on a number of factors,
some of which are beyond our control, including:
|
|
•
|
the willingness of physicians, burn care teams and hospital administrators to administer
our products and the acceptance of our products as part of the medical department routine;
|
|
|
•
|
the consent of hospitals to fund/purchase NexoBrid or obtain third-party coverage
or reimbursement for our products;
|
|
|
•
|
the ability to offer NexoBrid, EscharEx and our pipeline product candidates for sale
at an attractive value;
|
|
|
•
|
the efficacy and potential advantages of NexoBrid, EscharEx and our pipeline product
candidates relative to current standard of care;
|
|
|
•
|
the prevalence and severity of any side effects; and
|
|
|
•
|
the efficacy, potential advantages and timing of introduction to the market of alternative
treatments.
|
Failure to achieve market acceptance for NexoBrid, EscharEx or
any of our pipeline product candidates, if and when they are approved for commercial sale, will have a material adverse effect on our
business, financial condition and results of operations.
We may be unsuccessful in commercializing our
products due to unfavorable pricing regulations or third-party coverage and reimbursement policies.
We cannot predict the pricing and reimbursement of NexoBrid, EscharEx
or our pipeline product candidates. The regulations that govern marketing authorizations, pricing and reimbursement for new products vary
widely from country to country, among regions within some countries and among some hospitals. In some foreign jurisdictions, including
the EU, the pricing of prescription pharmaceuticals is subject to governmental control. In other countries, coverage negotiations must
occur at the regional or hospital level in order to be included in the hospital formulary. Pricing negotiations with governmental authorities
at the regional or hospital level can take considerable time after the receipt of marketing authorization for a product candidate. Additionally,
while we are executing a country-specific market access strategy, which includes pricing and/or reimbursement targets for NexoBrid in
most of Europe, we cannot guarantee that we will receive favorable hospital, regional or national funding or pricing and reimbursement.
As a result, even after obtaining regulatory approval for a product in a particular country, we may be subject to price regulations or
denied or limited by reimbursement or formulary inclusion, which may delay or limit our commercial launch of the product and negatively
impact the revenue we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability
to recoup our investment in NexoBrid, EscharEx or our pipeline product candidates, even after obtaining regulatory approval.
Additionally, we cannot be sure that coverage and reimbursement
will be available for NexoBrid, EscharEx or any pipeline product candidate that we commercialize in the future, and, if reimbursement
is available, whether the level of reimbursement will be adequate. Coverage and reimbursement may affect the demand for, the price of,
or the budget allocated for reimbursement for any product for which we obtain marketing authorization. Obtaining coverage and adequate
reimbursement for our products may be particularly difficult because of the higher prices often associated with products administered
under the supervision of a physician. If coverage and reimbursement are not available or are available only at limited levels, we may
not be able to successfully commercialize NexoBrid, EscharEx or any pipeline product candidate that we successfully develop. Eligibility
for reimbursement does not guarantee that any product will be paid for in all cases or at a rate that covers our costs. Interim payments
for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Payment rates may vary according
to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower cost products that
are already reimbursed and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory
discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently
restrict imports of products from countries where they may be sold at lower prices than in certain other countries, such as the United
States. In the United States, third-party payors often rely on the coverage policies and payment limitations imposed by Medicare and other
government payors, in setting their own coverage policies and reimbursement rates. Our inability to promptly obtain coverage and profitable
payment rates from hospital budget, government-funded and private payors for NexoBrid, EscharEx or any pipeline product candidate could
have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall
financial condition.
Recently enacted and future legislation in
the United States may increase the difficulty and cost for us to commercialize NexoBrid and seek marketing authorizations for and, if
approved, commercialize EscharEx and our pipeline product candidates in the United States and in foreign jurisdictions and affect the
prices at which our products may be sold.
The United States and several other jurisdictions are considering,
or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that may affect the
ability to sell NexoBrid, EscharEx or any of our pipeline product candidates profitably, if approved. We cannot predict the initiatives
that may be adopted in the future. The continuing efforts of hospitals, governments, insurance companies, managed care organizations and
other payors of healthcare services to contain or reduce costs of healthcare may adversely affect:
|
|
•
|
the market acceptance or demand for NexoBrid, EscharEx or any of our pipeline product
candidates, if approved; |
|
|
•
|
the ability to set a price that we believe is fair for NexoBrid, EscharEx or any of
our pipeline product candidates, if approved;
|
|
|
•
|
our ability to generate revenues and achieve or maintain profitability; |
|
|
•
|
the level of taxes that we are required to pay; and |
|
|
•
|
the availability of capital. |
Among policy makers and payors in the United States
and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare
costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these
efforts and has been significantly affected by major legislative initiatives. In March 2010, the Patient Protection and Affordable Care
Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act(the “ACA”) was
signed into law and intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies
against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees
on the health industry and impose additional health policy reforms.
Among the provisions of the ACA of importance to our potential
product candidates are the following:
|
|
•
|
an annual, nondeductible fee on any entity that manufactures or imports certain branded
prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare
programs;
|
|
|
•
|
an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid
Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively;
|
|
|
•
|
addressed a new methodology by which rebates owed by manufacturers under the Medicaid
Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
|
|
|
•
|
a new Medicare Part D coverage gap discount program, in which manufacturers must agree
to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap
period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D;
|
|
|
•
|
extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed
to individuals who are enrolled in Medicaid managed care organizations;
|
|
|
•
|
expansion of eligibility criteria for Medicaid programs by, among other things, allowing
states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals
with income at or below 133% of the Federal Poverty Level, thereby potentially increasing manufacturers’ Medicaid rebate liability;
|
|
|
•
|
expansion of the entities eligible for discounts under the Public Health Service pharmaceutical
pricing program;
|
|
|
•
|
a new requirement to annually report drug samples that manufacturers and distributors
provide to physicians; and
|
|
|
• |
a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative
clinical effectiveness research. |
Since its enactment, there have been judicial, executive and congressional
challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the
ACA brought by several states without specifically ruling on the constitutionality of the ACA. Thus, the ACA will remain in effect in
its current form. Further, prior to the U.S. Supreme Court ruling, President Biden issued an executive order that initiated a special
enrollment period for purposes of obtaining health insurance coverage through the ACA marketplace from February 15, 2021 through August
15, 2021. The executive order instructed certain governmental agencies to review and reconsider their existing policies and rules that
limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work
requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA.
In addition, other legislative changes have been proposed and adopted
since the Affordable Care Act was enacted. These changes included aggregate reductions to Medicare payments to providers, which went into
effect in April 2013 and, due to subsequent legislative amendments, will stay in effect through 2031, with the exception of a temporary
suspension from May 1, 2020 through March 31, 2022, unless additional Congressional action is taken. In January 2013, the American Taxpayer
Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, and increased
the statute of limitations period for the government to recover overpayments to providers from three to five years. These laws may result
in additional reductions in Medicare and other healthcare funding, which could negatively impact the market for NexoBrid, EscharEx and
our other product candidates, if approved, and, accordingly, our financial operations.
There has been heightened governmental scrutiny recently over the
manner in which drug manufacturers set prices for their marketed products, which have resulted in several Congressional inquiries and
proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and
manufacturer patient programs, and reform government program reimbursement methodologies for drug products. In March 2021, the American
Rescue Plan Act of 2021 was signed into law, which eliminates the statutory Medicaid drug rebate cap for single source and innovator multiple
source drugs, beginning January 1, 2024. The rebate was previously capped at 100% of a drug’s average manufacturer price, In August
2022, the Inflation Reduction Act of 2022, or IRA, was signed into law. Among other things, the IRA requires manufacturers of certain
drugs to engage in price negotiations with Medicare (beginning in 2026), with prices that can be negotiated subject to a cap; imposes
rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation (first due in 2023); and replaces
the Part D coverage gap discount program with a new discounting program (beginning in 2025). The IRA permits the Secretary of the Department
of Health and Human Services to implement many of these provisions through guidance, as opposed to regulation, for the initial years.
On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the drug price
negotiation program is currently subject to legal challenges. For that and other reasons, it is currently unclear how the IRA will be
effectuated, or the impact of the IRA on our business. At the state level, legislatures have increasingly passed legislation and implemented
regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts,
restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage
importation from other countries and bulk purchasing.
We expect that other possible healthcare reform measures may result
in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and additional
downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government
programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare
reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our drugs. Legislative and regulatory
proposals have been made to expand post-approval requirements and restrict sales and promotional activities for drugs. We cannot be sure
whether additional legislative changes will be enacted, or whether the FDA or comparable regulations, guidance or interpretations will
be changed, or what the impact of such changes on the marketing authorizations of our product candidates, if any, may be. In addition,
increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing authorization,
as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In the EU, similar developments may affect our ability to profitably
commercialize our product candidates, if approved. In addition to continuing pressure on prices and cost containment measures, legislative
developments at the EU or member state level may result in significant additional requirements or obstacles that may increase our operating
costs. The delivery of healthcare in the EU, including the establishment and operation of health services and the pricing and reimbursement
of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers
have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context.
In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement
of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to
develop and market products, this could prevent or delay marketing authorization of our product candidates, restrict or regulate post-approval
activities and affect our ability to commercialize our product candidates, if approved. In markets outside of the United States and EU,
reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific
products and therapies.
In December 2021, Regulation No 2021/2282 on Health Technology
Assessment (“HTA”) amending Directive 2011/24/EU, was adopted. This regulation which entered into force in January 2022 intends
to boost cooperation among EU member states in assessing health technologies, including new medicinal products, and providing the basis
for cooperation at the EU level for joint clinical assessments in these areas. The regulation foresees a three-year transitional period
and will permit EU member states to use common HTA tools, methodologies, and procedures across the EU, working together in four main areas,
including joint clinical assessment of the innovative health technologies with the most potential impact for patients, joint scientific
consultations whereby developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising
technologies early, and continuing voluntary cooperation in other areas. Individual EU member states will continue to be responsible for
assessing non-clinical (e.g., economic, social, ethical) aspects of health technology, and making decisions on pricing and reimbursement.
We face competition from the existing standard
of care, and we are furthermore subject to the risk that potential changes in medical practice and technology, or the development by our
competitors of products, treatments or procedures that are similar, more advanced, safer or more effective than ours, will render our
product candidates obsolete.
The medical, biotechnology and pharmaceutical industries are intensely
competitive and subject to significant technological and practice changes. We may face competition from many different sources with respect
to NexoBrid, EscharEx and our pipeline product candidates or any product candidates that we may seek to develop or commercialize in the
future. Possible competitors may be medical practitioners, pharmaceutical and wound care companies, academic and medical institutions,
governmental agencies and public and private research institutions, among others. Should any competitor’s product candidates receive
regulatory or marketing authorization prior to ours, they may establish a strong market position and be difficult to displace, or may
diminish the need for our products.
Our commercial opportunity could be reduced or eliminated if our
competitors develop and commercialize products, treatments or procedures that are safer, more effective, have fewer or less severe side
effects, are more convenient or are less expensive than any product that we may develop. In addition, we face competition from the current
standard of care for eschar removal in severe burns, which includes surgery, where eschar removal can occur by tangential excision, dermabrasion
or hydro jet, and non-surgical alternatives, such as topical medications applied to the eschar to facilitate the natural healing process.
In chronic and other hard-to-heal wounds, we expect to face competition from current standard of care for debridement via sharp debridement
or from the current non-surgical standard of care, either enzymatic debridement, primarily Smith & Nephew Plc’s SANTYL®,
a collagenase-based product indicated for debriding chronic dermal ulcers and severely burned areas, or autolytic debridement. Many of
our current or future competitors may have significantly greater financial resources and expertise in research and development, manufacturing,
preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we may have. Mergers
and acquisitions in the pharmaceutical and biotechnology industries or wound care markets may result in even more resources being concentrated
among a smaller number of our competitors. For example, Healthpoint Biotherapeutics, which marketed SANTYL, was acquired by Smith &
Nephew Plc in 2012. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative
arrangements with large and established companies. These companies compete with us in recruiting and retaining qualified scientific and
management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies
complementary to, or necessary for, our programs
Risks Related to Our Financial Position and Need for Additional
Capital
If we fail to manage our growth effectively, our business could
be disrupted.
Our future financial performance and ability to successfully commercialize
our products and to compete effectively will depend, in part, on our ability to manage any future growth effectively. We have made and
expect to continue to make significant investments to enable our future growth through, among other things, new product development, clinical
trials for new indications, expansion of our marketing and sales infrastructure and continued exploring for potential business development
opportunities. Our global demand for NexoBrid surpasses the current manufacturing capabilities. We are currently seeking to expand our
manufacturing capabilities in order to increase our capacity to manufacture NexoBrid and future product candidates and satisfy near term
demand., we initiated a facility scale-up in 2022 to meet the growing global demand for NexoBrid. The new GMP-compliant state-of-the-art
manufacturing facility is projected to be completed by mid-2024, with full-scale manufacturing currently expected to commence in 2025.
We expect the cost will be approximately $12.7 million. We must also be prepared to expand our workforce and train, motivate and manage
additional employees as the need for additional personnel arises. Even following expansion, our facilities, personnel, systems, procedures
and controls may not be adequate to support our future operations, or we may expand, but then fail to grow our sales of NexoBrid or our
pipeline product candidates sufficiently to support such operational growth. Any failure to manage future growth effectively could have
a material adverse effect on our business and results of operations.
We have a history of net losses. We expect
to continue to incur substantial and increasing net losses in the coming years, and we may never achieve or maintain profitability.
We have incurred significant net losses, including a net loss of
$6.7 million for the year ended December 31, 2023 and $19.6 million for the year ended December 31, 2022. As of December 31, 2023, we
had an accumulated deficit of $174.8 million. We expect to incur substantial net losses in the coming years. These losses and negative
cash flows have had, and will continue to have, an adverse effect on our shareholder's equity and working capital.
We expect to incur significant expenses and
increasing operating losses in the coming years.
We anticipate that our expenses and future capital requirements
may increase if and as we:
|
|
•
|
accelerate our clinical development activities, particularly with respect to our clinical
development of EscharEx for the debridement of chronic and other hard-to-heal wounds and our clinical trials for our other pipeline product
candidates;
|
|
|
•
|
further scale-up the manufacturing process for NexoBrid; |
|
|
•
|
seek regulatory and marketing authorizations for our products and any pipeline product
candidate that successfully completes clinical trials;
|
|
|
•
|
initiate additional preclinical, clinical or other studies for NexoBrid, EscharEx
and our pipeline product candidates, and seek to identify and validate new products;
|
|
|
•
|
commercialize NexoBrid and any pipeline product candidates for which we obtain marketing
authorization; |
|
|
•
|
acquire rights to other product candidates and technologies; |
| |
• |
change or add suppliers;
|
| |
• |
maintain, expand and protect our intellectual property portfolio;
|
| |
• |
attract and retain skilled personnel; and
|
| |
• |
experience any delays or encounter issues with any of the above. |
In
addition, we maintain cash, cash equivalents and bank deposits at financial institutions in Israel, Germany and the United States. Our
funds at these institutions exceed insured limits and some are not insured at all. Although we spread our cash, cash equivalents and bank
deposits among several financial institutions in order to reduce the risks associated with maintaining all of our balances at one financial
institution, in the event of failure of any financial institution where we maintain our cash and cash equivalents or bank deposits, there
can be no assurance that we would be able to access uninsured funds in such financial institution in a timely manner or at all. Any inability
to access or delay in accessing these funds could adversely affect our business and financial position.
We may need additional capital in the future, which may cause dilution
to our existing shareholders, restrict our operations or require us to relinquish rights to our pipeline product candidates or intellectual
property. If additional capital is not available, we may have to delay, reduce or cease operations.
We may seek additional funding in the future, which may consist
of equity offerings, collaborations, licensing arrangements or any other means to develop our pipeline product candidates, increase our
commercial manufacturing capabilities, operate our sales and marketing capabilities or other general corporate purposes.
Our prior registered equity offerings diluted then-existing shareholders,
and to the extent that we raise additional capital through, for example, the sale of equity or convertible debt securities under our shelf
registration statement, our existing shareholders’ ownership interest will be further diluted, and the terms may include liquidation
or other preferences that adversely affect our shareholders’ rights. The incurrence of indebtedness or the issuance of certain equity
securities could result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations
on our ability to incur additional debt or to issue additional equity, limitations on our ability to acquire or license intellectual property
rights and other operating restrictions that could adversely impact our ability to conduct our business. In addition, the issuance of
additional equity securities by us, or the possibility of such issuance, may cause the market price of our ordinary shares to decline.
Securing additional financing may also divert our management’s attention from our day-to-day activities, which may adversely affect
our ability to develop and commercialize NexoBrid, EscharEx and our pipeline product candidates.
Additional funding may not be available to us on acceptable terms,
or at all. In the event that we enter into collaborations or licensing arrangements in order to raise capital, we may be required to accept
unfavorable terms, including relinquishing or licensing to a third party on unfavorable terms our rights to product candidates or intellectual
property that we otherwise would seek to develop or commercialize ourselves or reserve for future potential arrangements when we might
be able to achieve more favorable terms.
If we are unable to raise additional capital when required or on
acceptable terms, we may be required to:
|
|
•
|
delay, scale back or discontinue the development, manufacturing scale-up or commercialization
of NexoBrid, EscharEx or our pipeline product candidates; |
|
|
•
|
seek additional corporate partners for NexoBrid, EscharEx or one or more of our pipeline
product candidates on terms that are less favorable than might otherwise be available; or |
|
|
•
|
relinquish or license to additional parties, on unfavorable terms, our rights to NexoBrid,
EscharEx or our pipeline product candidates that we otherwise would seek to develop or commercialize ourselves. |
|
|
•
|
any such consequence will have a material adverse effect on our business, operating
results and prospects and on our ability to develop our pipeline product candidates. |
We believe that our existing cash and cash equivalents, short-term
and restricted bank deposits will be sufficient to fund our operations and capital expenditure for at least twelve months from the date
of this report. Our estimates may prove to be wrong, and we could use our available capital resources sooner than we currently expect.
Further, changing circumstances, some of which may be beyond our control, could cause us to consume capital significantly faster than
we currently anticipate, and we may need to seek additional funds sooner than planned.
We make business decisions based on forecasts
of future sales of our products and pipeline product candidates that may be inaccurate.
Our market estimates are based on many assumptions, including,
but not limited to, reliance on external market research, our own internal research, population estimates, estimates of disease diagnostic
rates, treatment trends, and market estimates by third parties. Any of these assumptions can materially impact our forecasts and we cannot
be assured that the assumptions are accurate. If the market for any of our products or product candidates is less than this data would
suggest, the potential sales for the product or pipeline product candidates in question could be adversely affected, and our inventories
and net losses could increase.
Because of the numerous risks and uncertainties associated with
biopharmaceutical product development and commercialization, we are unable to accurately predict the timing or amount of future expenses
or when, or if, we will be able to achieve or maintain profitability. We have financed our operations primarily through the sale of equity
securities, licensing agreements and government grants. The size of our future net losses will depend, in part, on the rate of growth
or contraction of our expenses and the level and rate of growth, if any, of our revenues. If we are unable to successfully commercialize
NexoBrid, EscharEx or one or more of our pipeline product candidates or if revenue from NexoBrid, EscharEx or any pipeline product candidate
that receives marketing authorization is insufficient, we will not achieve profitability. Even if we do achieve profitability, we may
not be able to sustain or increase profitability.
Exchange rate fluctuations between the U.S.
dollar and the Israeli shekel, the Euro and other non-U.S. currencies may negatively affect our earnings.
The dollar is our functional and reporting currency. However, a
significant portion of our operating expenses are incurred in Israeli shekels and Euros. As a result, we are exposed to the risks that
the shekel may appreciate relative to the dollar, or, if the shekel instead devalues relative to the dollar, that the inflation rate in
Israel may exceed such rate of devaluation of the shekel, or that the timing of such devaluation may lag behind inflation in Israel. In
any such event, the dollar cost of our operations in Israel would increase and our dollar-denominated results of operations would be adversely
affected. We cannot predict any future trends in the rate of inflation in Israel or the rate of devaluation (if any) of the shekel against
the dollar. For example, the shekel depreciated relative to the dollar, on average, by 3.1% in 2023, by 13.2% in 2022 and appreciated
by 3.3% on average in 2021. If the dollar or Euro cost of our operations in Israel increases, our dollar- and Euro-measured results of
operations will be adversely affected. Our operations also could be adversely affected if we are unable to effectively hedge against currency
fluctuations in the future.
To the extent that we may receive revenues from sales in certain
countries, such as certain countries in the Asia Pacific region, where our sales are expected to be denominated in dollars, a strengthening
of the dollar in relation to other currencies could make our products less competitive in those foreign markets and collection of receivables
more difficult. For further information, see “ITEM 11. Quantitative and Qualitative Disclosures About Market Risk” elsewhere
in this annual report.
Risks Related to Healthcare Laws and Other Legal Compliance Matters
Certain of our business practices could become
subject to scrutiny by regulatory authorities, as well as to lawsuits brought by private citizens. Failure to comply with applicable law
or an adverse decision in lawsuits may result in adverse consequences to us.
The laws governing our conduct in the United States and in foreign
jurisdictions are enforceable by criminal, civil and administrative penalties. In the United States, violations of laws such as the Federal
Food, Drug and Cosmetic Act (the “FDCA”), the Public Health Service Act, the Federal False Claims Act, provisions of the U.S.
Social Security Act, including the “Anti-Kickback Statute,” or any regulations promulgated under their authority, may result
in significant administrative, civil and criminal sanctions, jail sentences, fines or exclusion from federal and state programs, as may
be determined by the U.S. Department of Justice, the Office of Inspector General of the U.S. Department of Health and Human Services (the
“OIG”), the Centers for Medicare & Medicaid Services, (“CMS“) other regulatory authorities and the courts.
There can be no assurance that our activities will not come under the scrutiny of regulators and other government authorities or that
our practices will not be found to violate applicable laws, rules and regulations or prompt lawsuits by private citizen “relators”
under federal or state false claims laws.
The federal Anti-Kickback Statute prohibits, among other things,
knowingly and willfully offering, paying, soliciting or receiving any remuneration (including any kickback, bribe or rebate), directly
or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase,
lease or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare
programs. The term “remuneration” has been broadly interpreted to include anything of value. Although there are a number of
statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are
drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations
may be subject to scrutiny if they do not qualify for an exception or safe harbor.
For example, even common business arrangements, such as discounted
terms and volume incentives for customers in a position to recommend or choose drugs and devices for patients, such as physicians and
hospitals, can result in substantial legal penalties, including, among other things, exclusion from Medicare and Medicaid programs if
not carefully structured to comply with applicable requirements. Also, certain business practices, such as payment of consulting fees
to healthcare providers, sponsorship of educational or research grants, charitable donations, interactions with healthcare providers and
financial support for continuing medical education programs, must be conducted within narrowly prescribed and controlled limits to avoid
any possibility of unlawfully inducing healthcare providers to prescribe or purchase particular products or rewarding past prescribing.
Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct
per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based
on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean
that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the Anti-Kickback
Statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to
violate it in order to have committed a violation. Violations of the federal Anti-Kickback Statute may result in significant civil monetary
penalties for each violation, plus up to three times the remuneration involved. Moreover, a claim including items or services resulting
from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False
Claims Act. Accordingly, civil penalties for such conduct can further be assessed under the federal False Claims Act. Violations can also
result in criminal penalties, including criminal fines and imprisonment. Similarly, violations can result in exclusion from participation
in government healthcare programs, including Medicare and Medicaid.
Significant enforcement activity has also taken place under federal
and state false claims act statutes. Violations of the federal False Claims Act can result in treble damages, and a penalty for each false
claim submitted for payment. Pharmaceutical, device and other healthcare companies have been prosecuted under these laws for, among other
things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product.
Companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of products for unapproved,
and thus non-covered, uses. The government may further prosecute conduct constituting a false claim under the criminal False Claims Act.
The criminal False Claims Act prohibits the making or presenting of a claim to the government knowing such claim to be false, fictitious,
or fraudulent and, unlike the civil False Claims Act, requires proof of intent to submit a false claim.
The federal False Claims Act, as well as certain state false claims
acts, also permits relators to file complaints in the name of the United States (and if applicable, particular states). These relators
may be entitled to receive up to 30% of total recoveries and have been active in pursuing cases against pharmaceutical companies. Where
practices have been found to involve improper incentives to use products, the submission of false claims, or other improper conduct, government
investigations and assessments of penalties against manufacturers have resulted in substantial damages and fines. In addition, to avoid
exclusion from participation in federal healthcare programs, many manufacturers have been required to enter into Corporate Integrity Agreements
that prescribe allowable corporate conduct and impose reporting and disclosure obligations by the manufacturer to the government. Failure
to satisfy requirements under the FDCA can also result in a variety of administrative, civil and criminal penalties, including injunctions
or consent decrees that prescribe allowable corporate conduct.
The federal Health Insurance Portability and Accountability Act
of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing,
or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully
embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and
knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent
statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the Anti-Kickback Statute, a
person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Additionally, there has been a recent trend of increased federal
and state regulation of payments and transfers of value provided to healthcare professionals and/or entities. The ACA, among other things,
imposed annual reporting requirements on certain manufacturers of drugs, devices, biologicals and medical supplies for payments and other
transfers of value provided by them, directly or indirectly, to physicians (defined to include doctors, dentists, optometrists, podiatrists
and chiropractors), certain non-physician practitioners (physician assistants, nurse practitioners, clinical nurse specialists, certified
registered nurse anesthetists, anesthesiologist assistants, and certified nurse midwives), and teaching hospitals, as well as ownership
and investment interests held by physicians and their family members. A manufacturer’s failure to submit timely, accurately and
completely the required information for all payments, transfers of value or ownership or investment interests may result in significant
civil monetary penalties.
In addition, we are subject to analogous state and foreign laws
and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving
healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state and foreign laws that
require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance
guidance promulgated by the governments or otherwise restrict payments that may be made to healthcare providers. For instance, payments
made to physicians in certain EU member states must be publicly disclosed. Moreover, agreements with physicians must often be subject
of prior notification and/or approval by the physician’s employer, their competent professional organization, and/or the competent
authorities of the individual EU member states, state and foreign laws that require drug manufacturers to report information related to
payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws
requiring the registration of pharmaceutical sales representatives.
Efforts to ensure that our business arrangements with third parties
will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities
will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable
fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other
governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, including,
without limitation, damages, fines, imprisonment, exclusion from participation in government healthcare programs, such as Medicare and
Medicaid, and the curtailment or restructuring of our operations, which could have a material adverse effect on our business. If any of
the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable
laws, it may be subject to criminal, civil or administrative sanctions, including exclusions from participation in government healthcare
programs, which could also materially affect our business.
As a public company with securities registered under the U.S. Securities
Exchange Act of 1934, as amended (the “Exchange Act”), we are subject to the U.S. Foreign Corrupt Practices Act (the “FCPA”).
The FCPA and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to
officials for the purpose of obtaining or retaining business. While we continue to maintain and enhance internal policies mandating compliance
with these anti-bribery laws, we may operate in parts of the world that have experienced governmental corruption to some degree and in
certain circumstances, strict compliance with anti-bribery laws may conflict with local customs and practices or may require us to interact
with doctors and hospitals, some of which may be state controlled, in a manner that is different than in the United States. Our internal
control policies and procedures may not be sufficient to effectively protect us against reckless or criminal acts committed by our employees
or agents. Violations of these laws, or allegations of such violations, could disrupt our business and result in a material adverse effect
on our financial condition, results of operations and cash flows.
Actual or perceived failures to comply with
applicable data protection, privacy and security laws, regulations, standards and other requirements could adversely affect our business,
results of operations, and financial condition.
The global data protection landscape is rapidly evolving, and we
are or may become subject to numerous state, federal and foreign laws, requirements and regulations governing the collection, use, disclosure,
retention, and security of personal information, such as information that we may collect in connection with clinical trials. Implementation
standards and enforcement practices are likely to remain uncertain for the foreseeable future, and we cannot yet determine the impact
future laws, regulations, standards, or perception of their requirements may have on our business. This evolution may create uncertainty
in our business, affect our ability to operate in certain jurisdictions or to collect, store, transfer use and share personal information,
necessitate the acceptance of more onerous obligations in our contracts, result in liability or impose additional costs on us. The cost
of compliance with these laws, regulations and standards is high and is likely to increase in the future. Any failure or perceived failure
by us to comply with federal, state or foreign laws or regulations, our internal policies and procedures or our contracts governing our
processing of personal information could result in negative publicity, government investigations and enforcement actions, claims by third
parties and damage to our reputation, any of which could have a material adverse effect on our operations, financial performance and business.
In the United States, HIPAA, as amended by the Health Information
Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, or collectively HIPAA, imposes, among other
things, certain standards relating to the privacy, security, transmission and breach reporting of individually identifiable health information.
While we do not believe that we are currently acting as a covered entity or business associate under HIPAA and thus are not directly regulated
under HIPAA, any person may be prosecuted under HIPAA’s criminal provisions either directly or under aiding-and-abetting or conspiracy
principles. Consequently, depending on the facts and circumstances, we could face substantial criminal penalties if we knowingly receive
individually identifiable health information from a HIPAA-covered healthcare provider or research institution that has not satisfied HIPAA’s
requirements for disclosure of individually identifiable health information. Certain states have also adopted comparable privacy and security
laws and regulations, some of which may be more stringent than HIPAA. Such laws and regulations will be subject to interpretation by various
courts and other governmental authorities, thus creating potentially complex compliance issues for us and our customers and strategic
partners. For example, the California Consumer Privacy Act (“CCPA”), which went into effect on January 1, 2020, among other
things, creates new data privacy obligations for covered companies and provides individual privacy rights to California residents, including
the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages
for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions,
including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact
our processing of personal information depending on the context. Further, the California Privacy Rights Act (“CPRA”) recently
passed in California. The CPRA generally went into effect on January 1, 2023, and imposes additional data protection obligations
on covered businesses, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk
data, and opt outs for certain uses of sensitive data. It also created a new California data protection agency authorized to issue substantive
regulations and could result in increased privacy and information security enforcement. Additional compliance investment and potential
business process changes may be required. Similar laws have passed in other states and are continuing to be proposed at the state and
federal level, reflecting a trend toward more stringent privacy legislation in the United States. The enactment of such laws could have
potentially conflicting requirements that would make compliance challenging.
Furthermore, the Federal Trade Commission (“FTC”) and
many state Attorneys General continue to enforce federal and state consumer protection laws against companies for online collection, use,
dissemination and security practices that appear to be unfair or deceptive. For example, according to the FTC, failing to take appropriate
steps to keep consumers’ personal information secure can constitute unfair acts or practices in or affecting commerce in violation
of Section 5(a) of the Federal Trade Commission Act. The FTC expects a company’s data security measures to be reasonable and appropriate
in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available
tools to improve security and reduce vulnerabilities.
In the event that we are subject to or affected by HIPAA, the CCPA,
the CPRA or other domestic privacy and data protection laws, any liability from failure to comply with the requirements of these laws
could adversely affect our financial condition.
We are subject to data privacy and security laws in the EU as well
as the EEA, including Regulation 2016/679, or the General Data Protection Regulation (“GDPR”) with respect to our collection,
control, processing, sharing, disclosure and other use of personal data of individuals located in the EEA or in the context of our activities
within the EEA. In addition, some of the personal data we process in respect of clinical trial participants is special category or sensitive
personal data under the GDPR, and subject to additional compliance obligations and to local law derogations. The GDPR went into effect
in May 2018, and companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory
enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global
revenues of the noncompliant company, whichever is greater. In addition to fines, a breach of the GDPR may result in regulatory investigations,
reputational damage, orders to cease/ change our data processing activities, enforcement notices, assessment notices (for a compulsory
audit) and/ or civil claims (including class actions). Among other requirements, the GDPR regulates transfers of personal data subject
to the GDPR to third countries that have not been found to provide adequate protection to such personal data, including the United States,
and the efficacy and longevity of current transfer mechanisms between the EEA and the United States remains uncertain. Case law from the
Court of Justice of the EU (“CJEU”) states that reliance on the standard contractual clauses, or SCCs - a standard form of
contract approved by the European Commission as an adequate personal data transfer mechanism - alone may not necessarily be sufficient
in all circumstances and that transfers must be assessed on a case-by-case basis. On October 7, 2022, President Biden signed an Executive
Order on “Enhancing Safeguards for United States Signals Intelligence Activities,” which introduced new redress mechanisms
and binding safeguards to address the concerns raised by the CJEU in relation to data transfers from the EEA to the United States and
which formed the basis of the new EU-US Data Privacy Framework (“DPF”), as released on December 13, 2022. The European Commission
adopted its Adequacy Decision in relation to the DPF on July 10, 2023, rendering the DPF effective as a GDPR transfer mechanism to U.S.
entities self-certified under the DPF. The DPF also introduced a new redress mechanism for EU citizens which addresses a key concern in
the previous CJEU judgments and may mean transfers under SCCs are less likely to be challenged in future. We currently rely on the
EU standard contractual clauses We expect the existing legal complexity and uncertainty regarding international personal data transfers
to continue. In particular, we expect the DPF Adequacy Decision to be challenged and international transfers to the United States and
to other jurisdictions more generally to continue to be subject to enhanced scrutiny by regulators.
As supervisory authorities issue further guidance on personal data
export mechanisms, including circumstances where the SCCs cannot be used, and/or start taking enforcement action, we could suffer additional
costs, complaints and/or regulatory investigations or fines, and/or if we are otherwise unable to transfer personal data between and among
countries and regions in which we operate, it could affect the manner in which we provide our services, the geographical location or segregation
of our relevant systems and operations, and could adversely affect our financial results.
Further, since January 2021, we may also be subject to the UK GDPR,
which, together with the UK Data Protection Act 2018, retains the GDPR in UK national law. The UK GDPR mirrors the fines under the GDPR,
meaning the potential of parallel fines of up to the greater of £17.5 million or 4% of global turnover. On October 12, 2023, the
UK Extension to the DPF came into effect (as approved by the UK Government), as a UK GDPR data transfer mechanism to U.S. entities self-certified
under the UK Extension to the DPF. The European Commission has adopted an adequacy decision in favor of the UK, enabling data transfers
from EU member states to the UK without additional safeguards. However, the UK adequacy decision will automatically expire in June 2025
unless the European Commission re-assesses and renews/extends that decision and remains under review by the Commission during this period.
In September 2021, the UK government launched a consultation on its proposals for wide-ranging reform of UK data protection laws following
Brexit. There is a risk that any material changes which are made to the UK data protection regime could result in the European Commission
reviewing the UK adequacy decision, and the UK losing its adequacy decision if the European Commission deems the UK to no longer provide
adequate protection for personal data. The relationship between the UK and the EU in relation to certain aspects of data protection law
remains uncertain, and it is unclear how UK data protection laws and regulations will develop in the medium to longer term.
Our business and operations may suffer in the
event of information technology system failures, cyberattacks or deficiencies in our cybersecurity.
We collect and maintain information in digital form that is necessary
to conduct our business, and we are increasingly dependent on information technology systems and infrastructure to operate our business.
In the ordinary course of our business, we collect, store and transmit large amounts of confidential information, including intellectual
property, proprietary business information and personal information of customers and our employees and contractors. It is critical that
we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We have implemented physical
and electronic security measures to protect our proprietary information, maintain standard operation procedures and conduct business continuity
and disaster recovery simulations, penetration testing and train our employees. However, we cannot provide assurances that our data protection
and security measures will be fully implemented or complied with, will not be breached or will provide adequate protection for our confidential
information or information technology systems.
Our information technology systems and those of our third-party
service providers, strategic partners and other contractors or consultants are vulnerable to attack and damage or interruption from computer
viruses and malware (e.g. ransomware), malicious code, natural disasters, terrorism, war, telecommunication and electrical failures, hacking,
cyberattacks, phishing attacks, worms/trojans and other social engineering schemes, employee theft or misuse, human error, fraud, denial
or degradation of service attacks, sophisticated nation-state and nation-state-supported actors or unauthorized access or use by persons
inside our organization, or persons with access to systems inside our organization. We have also outsourced elements of our information
technology infrastructure, and as a result a number of third-party vendors may or could have access to our confidential information. Data
breaches, cyber-attacks and other similar incidents are increasing in frequency, levels of persistence, sophistication and intensity and
are evolving in nature. Moreover, geopolitical tensions, particularly the Hamas-Israel and the Russia-Ukraine conflicts, have contributed
to a surge in cyber-attacks targeting Israeli companies and products globally, posing a threat to critical infrastructure.
There is a risk that third parties may obtain and improperly utilize
our proprietary information to our competitive disadvantage. Attacks upon information technology systems are increasing in their frequency,
levels of persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with
a wide range of motives and expertise. We may also face increased cybersecurity risks due to our reliance on internet technology and the
number of our employees continue to work remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities.
Furthermore, because the techniques used to obtain unauthorized access to, or to sabotage, systems change frequently and often are not
recognized until launched against a target, we may be unable to anticipate these techniques or implement adequate preventative measures.
We may also experience security breaches that may remain undetected for an extended period. Even if identified, we may be unable to adequately
investigate or remediate incidents or breaches due to attackers increasingly using tools and techniques that are designed to circumvent
controls, to avoid detection, and to remove or obfuscate forensic evidence. We may not be able to detect or prevent the unauthorized use
of our confidential information or take appropriate and timely steps to enforce our intellectual property rights.
We and certain of our service providers are from time to time subject
to cyberattacks and security incidents. While we do not believe that we have experienced any significant system failure, accident or security
breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of
our development programs and our business operations, whether due to a loss, corruption or unauthorized disclosure of our trade secrets,
personal information or other proprietary or sensitive information or other similar disruptions. It could also expose us to risks, including
an inability to provide our services and fulfill contractual demands, and could cause management distraction and the obligation to devote
significant financial and other resources to mitigate such problems. If a security breach or other incident were to result in the unauthorized
access to or unauthorized use, disclosure, release or other processing of personal information, it may be necessary to notify individuals,
governmental authorities, supervisory bodies, the media and other parties pursuant to privacy and security laws, and the costs associated
with the investigation, remediation and potential notification of the breach to counter-parties and data subjects could be material. Any
security compromise affecting us, our service providers, strategic partners, other contractors, consultants, or our industry, whether
real or perceived, could harm our reputation, erode confidence in the effectiveness of our security measures and lead to regulatory scrutiny.
To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or systems, or inappropriate
disclosure of confidential or proprietary or personal information, we could incur liability, including litigation exposure, penalties
and fines, we could become the subject of regulatory action or investigation, our competitive position could be harmed and the further
development and commercialization of our products and services could be delayed.
Laws and regulations affecting government contracts
make it more costly and difficult for us to successfully conduct our business.
We must comply with numerous laws and regulations relating to the
formation, administration and performance of government contracts, which can make it more difficult for us to retain our rights under
our BARDA contracts. These laws and regulations affect how we conduct business with government agencies. Among the most significant government
contracting regulations that affect our business are:
|
|
•
|
the Federal Acquisition Regulations (“FAR”) and agency-specific regulations
supplemental to the FAR, which comprehensively regulate the procurement, formation, administration and performance of government contracts;
|
| |
•
|
business ethics and public integrity obligations, which govern conflicts of interest
and the hiring of former government employees, restrict the granting of gratuities and funding of lobbying activities and include other
requirements such as the Anti-Kickback Statute and Foreign Corrupt Practices Act; |
|
|
•
|
export and import control laws and regulations; and
|
|
|
•
|
laws, regulations and executive orders restricting the use and dissemination of information
classified for national security purposes and the exportation of certain products and technical data.
|
Any material changes in applicable laws and regulations could
restrict our ability to maintain our BARDA contracts or obtain new contracts with the U.S. federal government.
We could be subject to product liability lawsuits,
which could result in costly and time-consuming litigation and significant liabilities.
The development of biopharmaceutical products involves an inherent
risk of product liability claims and associated adverse publicity. Our products may be found to be harmful or to contain harmful substances.
This exposes us to substantial risk of litigation and liability or may force us to discontinue production of certain products. Although
we have product liability insurance covering up to $10 million for claims in countries where NexoBrid is sold through our sales force
or through our distributors, the coverage may not insure us against all claims that may be asserted against us. Product liability insurance
is costly and often limited in scope. There can be no assurance that we will be able to obtain or maintain insurance on reasonable terms
or to otherwise protect ourselves against potential product liability claims that could impede or prevent commercialization of NexoBrid,
EscharEx or our pipeline product candidates. Furthermore, a product liability claim could damage our reputation, whether or not such claims
are covered by insurance or are with or without merit. A product liability claim against us or the withdrawal of a product from the market
could have a material adverse effect on our business or financial condition. Furthermore, product liability lawsuits, regardless of their
success, would likely be time-consuming and expensive to resolve and would divert management’s time and attention, which could seriously
harm our business.
We are subject to extensive environmental,
health and safety, and other laws and regulations.
Our business involves the controlled use of chemicals. The risk
of accidental contamination or injury from these materials cannot be eliminated. If an accident, spill or release of any such chemicals
or substances occurs, we could be held liable for resulting damages, including for investigation, remediation and monitoring of the contamination,
including natural resource damages, the costs of which could be substantial. We are also subject to numerous environmental, health and
workplace safety laws and regulations, including those governing laboratory procedures. Although we maintain workers’ compensation
insurance to cover the costs and expenses that may be incurred because of injuries to our employees resulting from the use of these materials,
this insurance may not provide adequate coverage against potential liabilities. Additional or more stringent laws and regulations affecting
our operations may be adopted in the future. We may incur substantial capital costs and operating expenses and may be required to obtain
consents to comply with any of these or certain other laws or regulations and the terms and conditions of any permits required pursuant
to such laws and regulations, including costs to install new or updated pollution control equipment, modify our operations or perform
other corrective actions at our respective facilities. In addition, fines and penalties may be imposed for noncompliance with environmental,
health and safety and other laws and regulations or for the failure to have, or comply with the terms and conditions of, required environmental
or other permits or consents.
The enactment of legislation implementing changes in tax legislation
or policies in different geographic jurisdictions could materially impact our business, financial condition and results of operations.
We conduct business globally and file income tax returns in multiple
jurisdictions. Our consolidated effective income tax rate could be materially adversely affected by several factors, including: changing
tax laws, regulations and treaties, or the interpretation thereof (such as the Inflation Reduction Act of 2022 signed into law in the
United States on August 16, 2022 which, among other changes, introduced a 15% corporate minimum tax on certain corporations and a 1% excise
tax on certain stock repurchases by United States corporations, which the U.S. Treasury indicated may also apply to certain stock redemptions
by a foreign corporation funded by certain United States affiliates); tax policy initiatives and reforms in effect and under consideration
(such as those related to the Organization for Economic Co-Operation and Development’s (“OECD”) Base Erosion and Profit
Shifting, or BEPS, project, the European Commission’s state aid investigations and other initiatives); the practices of tax authorities
in jurisdictions in which we operate; the resolution of issues arising from tax audits or examinations and any related interest or penalties.
Such changes may include (but are not limited to) the taxation of operating income, investment income, dividends received or (in the specific
context of withholding tax) dividends, royalties and interest paid.
We are unable to predict what tax reforms may be proposed or enacted
in the future or what effect such changes would have on our business, but such changes, to the extent they are brought into tax legislation,
regulations, policies or practices in jurisdictions in which we operate, could increase the estimated tax liability that we have expensed
to date and paid or accrued on our consolidated financial statements, and otherwise affect our future results of operations, cash flows
in a particular period and overall or effective tax rates in the future in countries where we have operations, reduce post-tax returns
to our shareholders and increase the complexity, burden and cost of tax compliance.
Risks Related to Our Intellectual Property Rights
Our success depends in part on our ability
to obtain and maintain protection for the intellectual property relating to, or incorporated into, our technology and products.
Our commercial success depends in part on our ability to obtain
and maintain patent protection and trade secret protection for our intellectual property and proprietary technologies, our products and
their uses, as well as our ability to operate without infringing upon the proprietary rights of others. We rely on a combination of patents,
trademark and trade secret laws, non-disclosure and confidentiality agreements, licenses, assignments of invention agreements and other
restrictions on disclosure and use to protect our intellectual property rights.
As of December 31, 2023, we had been granted a total of 88 patents
and have 19 pending patent applications. The family of patents that covers NexoBrid specifically includes 35 granted patents worldwide.
EscharEx is covered in 15 patents and 4 national phase applications. However, there can be no assurance that patent applications relating
to our products, processes or technologies will result in patents being issued, that any patents that have been issued will be adequate
to protect our intellectual property or that we will enjoy patent protection for any significant period of time. Additionally, any issued
patents may be challenged by third parties, and patents that we hold may be found by a judicial authority to be invalid or unenforceable.
Other parties may independently develop similar or competing technology or design around any patents that may be issued to or held by
us. Our current patents will expire or they may otherwise cease to provide meaningful competitive advantage, and we may be unable to adequately
develop new technologies and obtain future patent protection to preserve our competitive advantage or avoid adverse effects on our business.
Our patent protection may be limited, subjecting us to challenges
by competitors.
At present, we consider our patents relating to our enzymatic platform
technology, which underlies NexoBrid, EscharEx and our current pipeline product candidates, to be material to the operation of our business
as a whole. Our patents which cover NexoBrid claim specific mixtures of proteolytic enzymes, methods of producing such mixtures and methods
of treatment using such mixtures. Although the protection achieved is significant for NexoBrid, EscharEx and our pipeline product candidates,
when looking at our patents’ ability to block competition, the protection offered by our patents may be, to some extent, more limited
than the protection provided by patents which claim chemical structures that were previously unknown. If our patents covering NexoBrid
in various jurisdictions were subject to a successful challenge or if a competitor were able to successfully design around them, our business
and competitive advantage could be significantly affected.
In addition, the patent landscape in the biotechnology field is
highly uncertain and involves complex legal, factual and scientific questions, and changes in either patent laws or in the interpretation
of patent laws in the United States and other countries may diminish the value and strength of our intellectual property or narrow the
scope of our patent protection. In addition, we may fail to apply for or be unable to obtain patents necessary to protect our technology
or products or enforce our patents due to lack of information about the exact use of our process by third parties. Even if patents are
issued to us, they may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, which could limit our ability to
prevent competitors from using similar technology or marketing similar products, or limit the length of time our technologies and products
have patent protection. In addition, we are a party to license agreement with Mark Klein, that imposes various obligations upon us as
a licensee, including the obligation to make milestone and royalty payments contingent on the sales of NexoBrid. If we fail to comply
with these obligations, the licensor may terminate the license, in which event we might not be able to market any product that is covered
by the licensed intellectual property, including NexoBrid.
In order to preserve and enforce our patents and other intellectual
property rights, we may need to assert claims or file lawsuits against third parties. Such lawsuits could entail significant costs to
us and divert our management’s attention from developing and commercializing our products. Lawsuits may ultimately be unsuccessful
and may also subject us to counterclaims and cause our intellectual property rights to be challenged, narrowed, invalidated or held to
be unenforceable.
The timing of a patent application, grant,
and expiration may put us at a disadvantage compared to our competitors.
Our material patents also may not afford us protection against
competitors with similar technology. Because patent applications in the United States and many other jurisdictions are typically not published
until 18 months after their filing, if at all, and because publications of discoveries in scientific literature often lag behind actual
discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their
issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in
such patent applications. As a result, the patents we own and license may be invalidated in the future, and the patent applications we
own and license may not be granted. For example, if a third party has also filed a patent application covering an invention similar to
one covered in one of our patent applications, we may be required to participate in an adversarial proceeding known as an “interference
proceeding,” declared by the U.S. Patent and Trademark Office or its foreign counterparts, to determine priority of invention. The
costs of these proceedings could be substantial and our efforts in them could be unsuccessful, resulting in a loss of our anticipated
patent position. In addition, if a third party prevails in such a proceeding and obtains an issued patent, we may be prevented from practicing
technology or marketing products covered by that patent. Additionally, patents and patent applications owned by third parties may prevent
us from pursuing certain opportunities such as entering into specific markets or developing certain products. Finally, we may choose to
enter into markets where certain competitors have patents or patent protection over technology that may impede our ability to compete
effectively.
We may not be able to protect our intellectual
property rights in all jurisdictions.
Effective protection of our intellectual property rights may be
unavailable or limited in some countries, and even if available, we may fail to pursue or obtain necessary intellectual property protection
in such countries, because filing, prosecuting, maintaining and defending patents on product candidates in all countries throughout the
world would be prohibitively expensive. In addition, the legal systems of certain countries do not favor the aggressive enforcement of
patents and other intellectual property rights, and the laws of certain foreign countries do not protect our rights to the same extent
as the laws of the United States. As a result, our intellectual property may not provide us with sufficient rights to exclude others from
commercializing products similar or identical to ours. Competitors may use our technologies in jurisdictions where we have not obtained
patent protection to develop their own products, and we may be unable to prevent such competitors from importing such infringing products
into territories where we have patent protection but where enforcement is not as strong as in the United States or into jurisdictions
in which we do not have patent protection. These products may compete with our product candidates and our patents and other intellectual
property rights may not be effective or sufficient to prevent them from competing in those jurisdictions.
Our currently issued NexoBrid patents are nominally due to expire
at various dates between 2025 and 2029. However, because of the extensive time required for development, testing and regulatory review
of a potential product, and although such delays may entitle us to patent term extensions, it is possible that, before NexoBrid can be
commercialized in additional international jurisdictions and/or before any of our future products can be commercialized, any related patent
may expire or remain in force for only a short period following commercialization, thereby reducing any advantages of the patent. The
international PCT patent applications relating to EscharEx were filed on January 30, 2017. National phase applications corresponding to
these PCT applications were filed in several jurisdictions and the expiration date of the 15 patents that issued and those that will be
issued is January 30, 2037, absent patent-term adjustment and/or extensions. Our pending and future patent applications may not lead to
the issuance of patents or, if issued, the patents may not provide us with any competitive advantage. We also cannot guarantee that:
|
|
•
|
any of our present or future patents or patent claims or other intellectual property
rights will not lapse or be invalidated, circumvented, challenged or abandoned;
|
|
|
•
|
our intellectual property rights will provide competitive advantages or prevent competitors
from making or selling competing products;
|
|
|
•
|
our ability to assert our intellectual property rights against potential competitors
or to settle current or future disputes will not be limited by our agreements with third parties;
|
|
|
•
|
any of our pending or future patent applications will be issued or have the coverage
originally sought;
|
|
|
•
|
our intellectual property rights will be enforced in jurisdictions where competition
may be intense or where legal protection may be weak; or
|
|
|
• |
we will not lose the ability to assert our intellectual property rights against, or
to license our technology to, others and collect royalties or other payments. |
We may be unable to identify all past or future
unauthorized uses of our intellectual property.
Additionally, unauthorized use of our intellectual property may
have occurred or may occur in the future. Any failure to identify unauthorized use of, and otherwise adequately protect, our intellectual
property could adversely affect our business, including by reducing the demand for our products. Any reported adverse events involving
counterfeit products that purport to be our products could harm our reputation and the sale of our products. Moreover, if we are required
to commence litigation related to unauthorized use, whether as a plaintiff or defendant, such litigation would be time-consuming, force
us to incur significant costs and divert our attention and the efforts of our management and other employees, which could, in turn, result
in lower revenue and higher expenses.
In addition to patented technology, we rely
on our unpatented proprietary technology, trade secrets, processes and know-how.
We rely on proprietary information, such as trade secrets, know-how
and confidential information, to protect intellectual property that may not be patentable or that we believe is best protected by means
that do not require public disclosure. We generally seek to protect this proprietary information by entering into confidentiality agreements,
or consulting, services or employment agreements that contain non-disclosure and non-use provisions with our employees, consultants, contractors,
scientific advisors and third parties. However, we may fail to enter into the necessary agreements, and even if entered into, these agreements
may be breached or otherwise fail to prevent disclosure, third-party infringement or misappropriation of our proprietary information,
may be limited as to their term and may not provide an adequate remedy in the event of unauthorized disclosure or use of proprietary information.
We have limited control over the protection of trade secrets used by our suppliers and service providers and could lose future trade secret
protection if any unauthorized disclosure of such information occurs. In addition, our proprietary information may otherwise become known
or be independently developed by our competitors or other third parties. To the extent that our employees, consultants, contractors, scientific
advisors and other third parties use intellectual property owned by others in their work for us, disputes may arise as to the related
rights or resulting know-how and inventions. Costly and time-consuming litigation could be necessary to enforce and determine the scope
of our and relevant third parties’ proprietary rights and failure to obtain or maintain protection for our proprietary information
could adversely affect our competitive business position. In addition, if a third party is able to establish that we are using their proprietary
information without their permission, we may be required to obtain a license to such information or, if such a license is not available,
re-design our products to avoid any such unauthorized use or temporarily delay or permanently stop manufacturing or sales of the affected
products. Furthermore, laws regarding trade secret rights in certain markets where we operate may afford little or no protection to our
trade secrets.
Some of our employees were previously employed at universities
or other biotechnology or pharmaceutical companies, including potential competitors. While we take steps to prevent our employees from
using the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have
inadvertently or otherwise used or disclosed intellectual property, trade secrets or other proprietary information of any such employee’s
former employer. Litigation may be necessary to defend against these claims and, even if we are successful in defending ourselves, could
result in substantial costs to us or be distracting to our management. If we fail to defend any such claims successfully, in addition
to paying monetary damages, we may lose valuable intellectual property rights or personnel.
If we are unable to protect our trademarks
from infringement, our business prospects may be harmed.
We own trademarks that identify “MediWound,” “NexoBrid”
and “EscharEx,” among others, and have registered these trademarks in certain key markets. Although we take steps to monitor
the possible infringement or misuse of our trademarks, it is possible that third parties may infringe, dilute or otherwise violate our
trademark rights. Any unauthorized use of our trademarks could harm our reputation or commercial interests. In addition, our enforcement
against third-party infringers or violators may be unduly expensive and time-consuming, and the outcome may be an inadequate remedy.
We may be subject to claims that we infringe,
misappropriate or otherwise violate the intellectual property rights of third parties.
Our development, marketing or sale of NexoBrid, EscharEx or our
pipeline product candidates may infringe or be accused of infringing one or more claims of an issued patent to which we do not hold a
license or other rights. We may also be subject to claims that we are infringing, misappropriating or otherwise violating other intellectual
property rights, such as trademarks, copyrights or trade secrets. Third parties could therefore bring claims against us or our strategic
partners that would cause us to incur substantial expenses, including litigation costs or costs associated with settlement, and, if successful
against us, could cause us to pay substantial damages. Further, if such a claim were brought against us, we could be forced to temporarily
delay or permanently stop manufacturing or sales of NexoBrid, EscharEx or our pipeline product candidates that are the subject of the
suit.
If we are found to be infringing, misappropriating or otherwise
violating the patent or other intellectual property rights of a third party, or in order to avoid or settle claims, we may choose or be
required to seek a license from a third party and be required to pay license fees or royalties or both, which could be substantial. These
licenses may not be available on acceptable terms, or at all. Even if we were able to obtain a license, the rights may be nonexclusive,
which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing
a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened claims, we or our strategic
partners are unable to enter into licenses on acceptable terms.
There have been substantial litigation and other proceedings regarding
patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition, to the extent that we gain
greater visibility and market exposure as a public company in the United States, we face a greater risk of being involved in such litigation.
In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including interference,
opposition, re-examination and similar proceedings before the U.S. Patent and Trademark Office and its foreign counterparts, regarding
intellectual property rights with respect to NexoBrid, EscharEx or our pipeline product candidates. The cost to us of any patent litigation
or other proceeding, even if resolved in our favor, could be substantial. A negative outcome could result in liability for monetary damages,
including treble damages and attorneys’ fees if, for example, we are found to have willfully infringed a patent. A finding of infringement
could prevent us from developing, marketing or selling a product or force us to cease some or all of our business operations. Some of
our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially
greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could
have a material adverse effect on our ability to compete in the marketplace, and patent litigation and other proceedings may also absorb
significant management time.
Under applicable employment laws, we may not
be able to enforce covenants not to compete.
We generally enter into non-competition agreements with our employees.
These agreements prohibit our employees, if they cease working for us, from competing directly with us or working for our competitors
or clients for a limited period. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees
work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former employees or consultants
developed while working for us. For example, Israeli labor courts have required employers seeking to enforce non-compete undertakings
of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material
interests of the employer which have been recognized by the courts, such as the protection of a company’s trade secrets or other
intellectual property. If we cannot demonstrate that such interests will be harmed, we may be unable to prevent our competitors from benefiting
from the expertise of our former Israeli employees or consultants and our ability to remain competitive may be diminished. As to our U.S.
operations, on the U.S. federal level, there was movement in 2023 by federal agencies to make noncompete agreements unenforceable in general.
The Federal Trade Commission proposed a new rule to ban employers nationwide from using non-compete agreements with their employees and
independent contractors, and the General Counsel of the National Labor Relations Board issued a memo in March 2023 opining that many types
of non-compete and non-solicitation restrictions unlawfully interfere with employees’ protected rights under Section 7 of the National
Labor Relations Act. If any of these proposed new U.S. federal restrictions becomes effective, or if any state in which we have operations
continues to expand restrictions or bans the use of non-compete restrictions, that could adversely impact our ability to protect our investment
in our key employees in our U.S. locations, and harm our competitive position.
We may become subject to
claims for remuneration or royalties for assigned service invention rights by our employees, which could result in litigation and
adversely affect our business.
A significant portion of our intellectual property has been developed
for us by our employees in the course of their employment. Under the Israeli Patent Law, 5727-1967, or the Patent Law, inventions conceived
by an employee in the course and as a result of or arising from his or her employment with a company are regarded as “service inventions,”
which belong to the employer, absent a specific agreement between the employee and employer giving the employee proprietary rights. The
Patent Law also provides under Section 134 that if there is no agreement between an employer and an employee as to whether the employee
is entitled to consideration for service inventions, and to what extent and under which conditions, the Israeli Compensation and Royalties
Committee, or the Committee, a body constituted under the Patent Law, shall determine these issues. Section 135 of the Patent Law provides
criteria for assisting the Committee in making its decisions. According to case law handed down by the Committee, an employee’s
right to receive consideration for service inventions is a personal right and is entirely separate from the proprietary rights in such
invention. Therefore, this right must be explicitly waived by the employee. A decision handed down in May 2014 by the Committee clarifies
that the right to receive consideration under Section 134 can be waived and that such waiver can be made orally, in writing or by behavior
like any other contract. The Committee will examine, on a case by case basis, the general contractual framework between the parties, using
interpretation rules of the general Israeli contract laws. Further, the Committee has not yet determined one specific formula for calculating
this remuneration, nor the criteria or circumstances under which an employee’s waiver of his right to remuneration will be disregarded.
Similarly, it remains unclear whether waivers by employees in their employment agreements of the alleged right to receive consideration
for service inventions should be declared as void being a depriving provision in a standard contract. We generally enter into assignment-of-invention
agreements with our employees pursuant to which such individuals assign to us all rights to any inventions created in the scope of their
employment or engagement with us. Although our employees have agreed to assign to us service invention rights and have specifically waived
their right to receive any special remuneration for such service inventions beyond their regular salary and benefits, we may face claims
demanding remuneration in consideration for assigned inventions. As a consequence of such claims, we could be required to pay additional
remuneration or royalties to our current or former employees or be forced to litigate such claims, which could negatively affect our business.
Risks Related to an Investment in Our Ordinary Shares
The market price of our ordinary shares may
be subject to fluctuation and you could lose all or part of your investment.
Our ordinary shares were first offered publicly in our IPO in March
2014 at a price of $98.00 per share, and our ordinary shares have subsequently traded as high as $127.12 per share and as low as $7.10
per share through December 31, 2023. The market price of our ordinary shares on the Nasdaq Global Market may fluctuate as a result of
a number of factors, some of which are beyond our control, including, but not limited to:
|
|
• |
actual or anticipated variations in our and our competitors’ results of operations
and financial condition;
|
|
|
•
|
market acceptance of our products;
|
|
|
•
|
general economic and market conditions and other factors, including factors unrelated
to our operating performance;
|
|
|
•
|
the mix of products that we sell and related services that we provide;
|
|
|
•
|
changes in earnings estimates or recommendations by securities analysts, if our ordinary
shares continue to be covered by analysts;
|
|
|
•
|
publication of the results of preclinical or clinical trials for NexoBrid, EscharEx
or any of our pipeline product candidates;
|
| |
•
|
failure by us to achieve a publicly announced milestone; |
|
|
•
|
delays between our expenditures to develop and market new or enhanced products and
the generation of sales from those products;
|
|
|
•
|
development of technological innovations or new competitive products by others;
|
|
|
•
|
announcements of technological innovations or new products by us;
|
|
|
•
|
regulatory developments and the decisions of regulatory authorities as to the marketing
of our current products or the approval or rejection of new or modified products;
|
|
|
•
|
developments concerning intellectual property rights, including our involvement in
litigation;
|
|
|
•
|
changes in our expenditures to develop, acquire or license new products, technologies
or businesses;
|
|
|
•
|
changes in our expenditures to promote our products;
|
|
|
•
|
changes in the structure of healthcare payment systems;
|
|
|
•
|
our sale or proposed sale, or the sale by our significant shareholders, of our ordinary
shares or other securities in the future;
|
|
|
•
|
changes in key personnel;
|
|
|
•
|
success or failure of our research and development projects or those of our competitors;
and
|
|
|
• |
the trading volume of our ordinary shares. |
These factors and any corresponding price fluctuations may materially and adversely affect the market price of our ordinary shares and
result in substantial losses being incurred by our investors. In the past, following periods of market volatility, public company shareholders
have often instituted securities class action litigation. If we were involved in securities litigation, it could impose a substantial
cost upon us and divert the resources and attention of our management from our business.
Future sales of our ordinary shares could reduce the market price
of our ordinary shares.
If we or our existing shareholders, our directors or their affiliates
or certain of our executive officers, sell a substantial number of our ordinary shares in the public market, the market price of our ordinary
shares could decrease significantly. The perception in the public market that we or our shareholders might sell our ordinary shares could
also depress the market price of our ordinary shares and could impair our future ability to obtain capital, especially through an offering
of equity securities.
We have made significant offerings of our ordinary shares in the
past and may do so again in the future. For example, on April 23, 2019, the SEC declared effective our shelf registration statement on
Form F-3, which registered the resale of 1,605,732 shares that are subject to registration rights. All shares sold pursuant to an offering
covered by that registration statement (or a subsequent shelf registration that we may file to replace it after it expires) will be freely
transferable. See “ITEM 7.B. Related Party Transactions-Registration Rights Agreement.” Sales by us or our shareholders of
a substantial number of ordinary shares in the public market could cause the market price of our ordinary shares to decline or could impair
our ability to raise capital through a future sale of, or pay for acquisitions using, our equity securities.
In addition, as of March 15, 2024, 1,161,033
ordinary shares were subject to outstanding option and RSU awards granted to employees and office holders under our share incentive plans,
including 430,675 ordinary shares issuable under currently exercisable share options and RSUs. We have filed a registration statements
on Form S-8 registering the issuance of all such ordinary shares issuable under our share incentive plans. As of March 15, 2024, 680,108
shares remained available for future awards under our 2014 Equity Incentive Plan, which shares will be rolled over to a new share incentive
plan— the 2024 Share Incentive Plan— that we will adopt and bring for shareholder approval soon. Shares included in
such registration statements on Form S-8 may be freely sold in the public market upon issuance, except for shares held by affiliates,
who have certain restrictions on their ability to sell.
As a foreign private issuer, we are permitted
to, and actually do, follow certain home country corporate governance practices instead of otherwise applicable SEC and Nasdaq requirements.
As a foreign private issuer, we are permitted to, and do, follow
certain home country corporate governance practices instead of those otherwise required under the Nasdaq Stock Market listing rules for
domestic U.S. issuers. For instance, we follow home country practice in Israel with regard to the (i) quorum requirement for shareholder
meetings (ii) shareholder approval for certain transactions other than a public offering involving issuances of a 20% or more interest
in the company. See “ITEM 16G. Corporate Governance.” We may in the future elect to follow home country practices in Israel
with regard to other matters as well, such as the formation and composition of the nominating and corporate governance committee, separate
executive sessions of independent directors and the requirement to obtain shareholder approval for certain dilutive events (such as for
the establishment or amendment of certain equity-based compensation plans, issuances that will result in a change of control of the company,
and certain acquisitions of the stock or assets of another company). Following our home country governance practices as opposed to the
requirements that would otherwise apply to a U.S. company listed on the Nasdaq Global Market may provide less protection to you than what
is accorded to investors under the Nasdaq Stock Market listing rules applicable to domestic U.S. issuers. See “ITEM 16G. Corporate
Governance.”
As a foreign private issuer, we are not subject to the provisions
of Regulation FD or U.S. proxy rules and are exempt from filing certain Exchange Act reports. Loss of our foreign private issuer status
would be accompanied by a significant increase in compliance costs.
As a foreign private issuer, we are exempt from the rules and regulations
under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders
are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we
are not required under the Exchange Act to file annual and current reports and financial statements with the SEC as frequently or as promptly
as U.S. domestic companies whose securities are registered under the Exchange Act, and we are generally exempt from filing quarterly reports
with the SEC under the Exchange Act. Moreover, we are not required to comply with Regulation FD, which prohibits the selective disclosure
of material nonpublic information to, among others, broker-dealers and holders of a company’s securities under circumstances in
which it is reasonably foreseeable that the holder will trade in the company’s securities on the basis of the information. Even
though we intend to comply voluntarily with Regulation FD, these exemptions and leniencies will reduce the frequency and scope of information
and protections to which you are entitled as an investor.
For so long as we qualify as a foreign private issuer, we are not
required to comply with the proxy rules applicable to U.S. domestic companies, including the requirement applicable to emerging growth
companies to disclose the compensation of our Chief Executive Officer and other two most highly compensated executive officers on an individual,
rather than an aggregate, basis. Nevertheless, the regulations promulgated under the Israeli Companies Law, 5759-1999 (the “Israeli
Companies Law”) require us to disclose the annual compensation of our five most highly compensated officers on an individual, rather
than on an aggregate, basis. See “ITEM 6.B. Compensation.” Under the Companies Law regulations, this disclosure is required
to be included in the proxy statement for our annual meeting of shareholders each year, which we furnish to the SEC under cover of a Report
of Foreign Private Issuer on Form 6-K. Because of that disclosure requirement under Israeli law, we are also including such information
in this annual report, pursuant to the disclosure requirements of Form 20-F.
We would lose our foreign private issuer status if a majority of
our outstanding ordinary shares are held of record by U.S. shareholders and we fail to meet additional requirements necessary to avoid
loss of foreign private issuer status. Although we have elected to comply with certain U.S. regulatory provisions, our loss of foreign
private issuer status would make such provisions mandatory. The regulatory and compliance costs to us under U.S. securities laws as a
U.S. domestic issuer may be significantly higher. If we lose our foreign private issuer status, we will be required to file periodic reports
and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive than the forms available
to a foreign private issuer. We would also be required to follow U.S. proxy disclosure requirements, including the requirement to disclose
more detailed information about the compensation of our senior executive officers on an individual basis. We may also be required to modify
certain of our policies to comply with accepted governance practices associated with U.S. domestic issuers. Such conversion and modifications
will involve additional costs. In addition, we would lose our ability to rely upon exemptions from certain corporate governance requirements
on U.S. stock exchanges that are available to foreign private issuers.
We have never paid cash dividends on our share
capital, and we do not anticipate paying any cash dividends in the foreseeable future.
We have never declared or paid cash dividends on our share capital,
nor do we anticipate paying any cash dividends on our share capital in the foreseeable future. We currently intend to retain all available
funds and any future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our ordinary
shares will be an investor’s sole source of gain for the foreseeable future. In addition, Israeli law limits our ability to declare
and pay dividends, and may subject our dividends to Israeli withholding taxes. See “ITEM 8.A. Consolidated Statements and Other
Financial Information-Dividend Policy,” “ITEM 10.B. Articles of Association-Dividend and liquidation rights” and “ITEM
10.E. Taxation-Israeli Tax Considerations and Government Programs.”
If we are unable to satisfy the requirements
of Section 404 of the Sarbanes-Oxley Act, or if our internal control over financial reporting or our disclosure controls and procedures
are not effective, investors may lose confidence in the accuracy and the completeness of the reports we furnish or file with the SEC,
the reliability of our financial statements may be questioned and our share price may suffer.
We are required to comply with the internal control, evaluation
and certification requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”). Pursuant to Section
404(a) of the Sarbanes-Oxley Act, we are required to furnish a report by management on the effectiveness of our internal control over
financial reporting. Because we have become an accelerated filer (as would be the case if we were furthermore to qualify as or a large
accelerated filer), we are required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes Oxley Act.
To maintain the effectiveness of our disclosure controls and procedures
and our internal control over financial reporting, we expect that we will need to continue to enhance existing, and implement new, financial
reporting and management systems, procedures and controls to manage our business effectively and support our growth in the future. The
process of evaluating our internal control over financial reporting requires an investment of substantial time and resources, including
by our Chief Financial Officer and other members of our senior management. The determination and any remedial actions required could divert
internal resources and take a significant amount of time and effort to complete and could result in us incurring additional costs that
we did not anticipate, including the hiring of outside consultants.
Irrespective of compliance with Section 404, any failure of our
internal controls could have a material adverse effect on our stated results of operations and harm our reputation. As a result, we may
experience higher than anticipated operating expenses, as well as higher independent auditor fees during and after the implementation
of these changes. If we are unable to implement any of the required changes to our internal control over financial reporting effectively
or efficiently, it could adversely affect our operations, financial reporting or results of operations. Further, if our internal controls
over financial reporting are not effective, the reliability of our financial statements may be questioned and our share price may suffer.
Our U.S. shareholders may suffer adverse tax consequences if we
are characterized as a passive foreign investment company.
Generally, if for any taxable year 75% or more of our gross income
is passive income, or at least 50% of the average quarterly value of our assets (which may be determined in part by the market value of
our ordinary shares, which is subject to change) are held for the production of, or produce, passive income, we would be characterized
as a passive foreign investment company (“PFIC”) for U.S. federal income tax purposes. Based on our current estimates of our
gross income and gross assets and the nature of our business, we do not believe we were classified as a PFIC for the taxable year ended
December 31, 2023. There can be no assurance that we will not be considered a PFIC for the current or any future taxable year. PFIC status
is determined as of the end of the taxable year and depends on a number of factors, including the value of a corporation’s assets
and the amount and type of its gross income. Furthermore, the value of our gross assets is likely to be determined in large part by reference
to our market capitalization. As such, a decline in the value of our ordinary shares or an increase in the value of our passive assets
(including cash and short term investments), for example, may result in our becoming a PFIC. If we are characterized as a PFIC, our U.S.
shareholders may suffer adverse tax consequences, including having gains realized on the sale of our ordinary shares treated as ordinary
income, rather than as capital gain, the loss of the preferential rate that may be applicable to dividends received on our ordinary shares
by individuals who are U.S. Holders (as defined in “ITEM 10.E. Taxation-United States Federal Income Taxation”), and having
interest charges apply to distributions by us and the proceeds of share sales. Certain elections exist that may alleviate some of the
adverse consequences of PFIC status and would result in an alternative treatment (such as mark-to-market treatment) of our ordinary shares.
However, we do not intend to provide the information necessary for U.S. holders to make qualified electing fund elections if we are classified
as a PFIC. See “ITEM 10.E. Taxation-United States Federal Income Taxation-Passive Foreign Investment Company Considerations.”
If a U.S. person is treated as owning at least
10% of our ordinary shares, such holder may be subject to adverse U.S. federal income tax consequences.
If a U.S. person is treated as owning (directly, indirectly, or
constructively) at least 10% of the value or voting power of our ordinary shares, such person may be treated as a “U.S. shareholder”
with respect to each “controlled foreign corporation” in our group (if any). Since our group includes one or more U.S. subsidiaries,
certain of our non-U.S. subsidiaries will be treated as controlled foreign corporations (regardless of whether or not we are treated as
a controlled foreign corporation). A U.S. shareholder of a controlled foreign corporation may be required to report annually and include
in its U.S. taxable income its pro rata share of “Subpart F income,” “global intangible low-taxed income,” and
investments in U.S. property by controlled foreign corporations, regardless of whether the Company makes any distributions. An individual
that is a U.S. shareholder with respect to a controlled foreign corporation generally would not be allowed certain tax deductions or foreign
tax credits that would be allowed to a U.S. shareholder that is a U.S. corporation. Failure to comply with these reporting obligations
may subject a U.S. shareholder to significant monetary penalties and may prevent the statute of limitations with respect to such U.S.
shareholder’s U.S. federal income tax return for the year for which reporting was due from starting. We cannot provide any assurances
that we will assist holders of ordinary shares in determining whether any of our non-U.S. subsidiaries is treated as a controlled foreign
corporation or whether any holder of ordinary shares is treated as a U.S. shareholder with respect to any such controlled foreign corporation
or furnish to any U.S. shareholders information that may be necessary to comply with the aforementioned reporting and tax paying obligations.
The United States Internal Revenue Service has provided limited guidance on situations in which investors may rely on publicly available
information to comply with their reporting and taxpaying obligations with respect to foreign-controlled controlled foreign corporations.
A U.S. holder should consult its tax advisors regarding the potential application of these rules to an investment in the ordinary shares.
Risks Primarily Related to our Operations in Israel
Our headquarters, manufacturing and other significant
operations are located in Israel and, therefore, our operations may be disrupted and our results of operations or financial condition
may be adversely affected by Israel’s current war against the terrorist organization Hamas and other terrorist organizations and
by any adverse consequences of that war.
Our
headquarters, manufacturing and research and development facilities are located in Yavne, Israel. In addition, the majority of our key
employees and officers are residents of Israel.
In recent years, Israel has been engaged in sporadic armed conflicts
with Hamas, an Islamist terrorist group that controls the Gaza Strip, with Hezbollah, an Islamist terrorist group that controls large
portions of southern Lebanon, and with Iranian-backed military forces in Syria. In addition, Iran has threatened to attack Israel, may
be developing nuclear weapons and has targeted cyber-attacks against Israeli entities. On October 7, 2023, Hamas terrorists infiltrated
Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched
extensive rocket attacks on Israeli population within the State of Israel, mostly in southern and central Israel, including areas near
our headquarters, manufacturing and research and development facilities. These attacks resulted in extensive deaths, injuries and kidnapping
of civilians and soldiers. Following the attack, Israel’s security cabinet declared war against Hamas and a military campaign against
this terrorist organization, accompanied by the draft of several hundred thousand Israeli military reservists, commenced in parallel to
Hamas’ continued rocket and terror attacks. Following the attack by Hamas on Israel’s southern border, Hezbollah in Lebanon
also launched missile, rocket and shooting attacks against Israeli military sites, troops and Israeli towns in northern Israel. In response
to these attacks, the Israeli army has carried out a number of targeted strikes on sites belonging to Hezbollah in southern Lebanon, and
the two sides have been trading attacks on one another since that time. Furthermore, the Houthi movement, which controls parts of Yemen,
has been launching attacks on marine vessels traversing the Red Sea, which were thought to either be in route towards Israel or to be
partly owned by Israeli businessmen. It is possible that other terrorist organizations, including Palestinian military organizations in
the West Bank, as well as other hostile countries, such as Iran, will join the hostilities. It is currently not possible to predict the
duration or severity of the ongoing conflict. Up to the present time, the war has not had a material effect on our business, operations
and financial conditions. However, the ongoing conflict is still evolving and developing, and could still disrupt our business and operations.
As of December 31, 2023, we had 88 employees based in Israel. Certain
of our employees, were and may be in the future called perform military reserve duty in connection with the current war. Even in the absence
of an active military conflict, our employees could be called in the future to perform reserve military duty for up to 54 days (and in
the case of non-officer commanders or officers, up to 70 or 84 days, respectively) of each three-year period until they reach the age
of 40 (and in some cases, depending on their specific military profession, up to 45 or even 49 years of age). In certain emergency circumstances
(as was the case with the current war), these employees may be called to immediate and unlimited active duty. Our business and results
of operations could be disrupted by the absence of a significant number of employees related to military service, for a significant period
of time.
Our commercial insurance does not cover losses that may occur as
a result of an event associated with the current war, or any other terrorist attack that might cause us damages. The reinstatement value
of direct damages that are caused by terrorist attacks or acts of war that the Israeli government is currently committed to covering might
not be maintained or, if maintained, might not be sufficient to compensate us fully for damages incurred. Any losses or damages incurred
by us as a result of the current (or any other) armed conflict involving Israel could have a material adverse effect on our business and
results of operations
Boycotts and similar activities
promoted by certain Middle Eastern countries and other political groups against Israeli businesses may adversely impact our operations
and our ability to sell our products.
Several countries, principally in the Middle East, restrict doing
business with Israel and Israeli companies, and anti-Israel political activists have been encouraging additional countries to impose restrictions
on doing business with, and to boycott the products of, Israel and Israeli companies whether as a result of hostilities in the region
or otherwise. More countries may impose restrictions on doing business with Israel and Israeli companies if hostilities on Israel continue
or increase. Recently, Israel has signed bilateral peace agreements with several Middle Eastern (including Arab) countries, forging new
economic ties with them. Nevertheless, if the actions of boycott activists become more widespread and successful, that may adversely impact
our ability to sell our products. Israel’s ongoing war against the terrorist group Hamas may assist those activists seeking to harm
the economic interest of Israeli companies such as ours. For example, in light of the ongoing war, Moody’s decided to downgrade
Israel’s credit rating from A1 to A2, as well as Israel’s outlook rating from “stable” to “negative”.
This downgrade and similar economic measures could slow the flow of international investment to Israel, negatively impacting the business
environment in which we operate.
Provisions of Israeli law and our articles of association may delay,
prevent or otherwise impede a merger with, or an acquisition of, us, even when the terms of such a transaction are favorable to us and
our shareholders.
Israeli corporate law regulates mergers, requires tender offers
for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers or significant
shareholders and regulates other matters that may be relevant to such types of transactions. For example, a tender offer for all of a
company’s issued and outstanding shares can only be completed if the acquirer receives positive responses from the holders of at
least 95% of the issued share capital. Completion of the tender offer also requires approval of a majority of the offerees that do not
have a personal interest in the tender offer, unless, following consummation of the tender offer, the acquirer would hold at least 98%
of the company’s outstanding shares. Furthermore, the shareholders, including those who indicated their acceptance of the tender
offer, may, at any time within six months following the completion of the tender offer, petition an Israeli court to alter the consideration
for the acquisition, unless the acquirer stipulated in its tender offer that a shareholder that accepts the offer may not seek such appraisal
rights. See “ITEM 10.B. Articles of Association-Acquisitions Under Israeli law” for additional information.
Furthermore, Israeli tax considerations may make potential transactions
unappealing to us or to our shareholders whose country of residence does not have a tax treaty with Israel exempting such shareholders
from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect
to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of a
number of conditions, including, in some cases, a holding period of two years from the date of the transaction during which sales and
dispositions of shares of the participating companies are subject to certain restrictions. Moreover, with respect to certain share swap
transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no disposition of the shares
has occurred.
We have received Israeli government grants
for certain research and development activities. The terms of those grants require us to satisfy specified conditions and to pay penalties
in addition to repayment of the grants upon certain events.
Our research and development efforts have been
financed in part through grants from the Israeli Innovation Authority (“IIA”), formerly operating as the Israeli Office of
the Chief Scientist (the “OCS”). The total gross amount of grants actually received by us from the IIA, including accrued
interest (or such other interest rate that the IIA may set in the future) and net of royalties actually paid as of December 31, 2023,
totaled approximately $13.8 million and the amortized cost (using the interest method) of the liability as of that date totaled approximately
$7.8 million. As of December 31, 2023, we had accrued and paid net royalties to the IIA in an amount of $1.9 million. As of December 31,
2018 we determined that we will no longer be supported by the IIA. As a result, since 2020 we did not submit applications for IIA grants
and we do not plan to submit in 2024.
The IIA grants that we have received are repayable by payment of
royalties from the sale of products developed as part of the programs for which grants were received. Our obligation to pay these royalties
is contingent on our actual sale of such products and services. In the absence of such sales, no payment of such royalties is required.
Even following full repayment of any IIA grants, we must nevertheless continue to comply with the requirements of the Encouragement of
Research, Development and Technological Innovation in the Industry Law, 5744-1984 (formerly known as the Law for the Encouragement of
Industrial Research and Development, 5744-1984), and related regulations (collectively, the “Innovation Law”). When a company
develops know-how, technology or products using IIA grants, the terms of these grants and the Innovation Law restrict the transfer outside
of Israel of such know-how, and the manufacturing or manufacturing rights of such products, technologies or know-how, without the prior
approval of the IIA. Therefore, if aspects of our technologies are deemed to have been developed with IIA funding, the discretionary approval
of an IIA committee would be required for any transfer to third parties outside of Israel of know-how or manufacturing or manufacturing
rights related to those aspects of such technologies. We may not receive those approvals. Furthermore, the IIA may impose certain conditions
on any arrangement under which it permits us to transfer technology or development out of Israel.
The transfer of IIA-supported technology or know-how or manufacturing
or manufacturing rights related to aspects of such technologies outside of Israel may involve the payment of significant penalties and
other amounts, depending upon the value of the transferred technology or know-how, the amount of IIA support, the time of completion of
the IIA-supported research project and other factors. If our products are manufactured outside of Israel, assuming we receive prior approval
from the IIA for the foreign manufacturing, we may be required to pay increased royalties. The increase in royalties depends on the manufacturing
volume that is performed outside of Israel. These restrictions and requirements for payment may impair our ability to sell our technology
assets outside of Israel or to outsource or transfer development or manufacturing activities with respect to any product or technology
outside of Israel. Furthermore, the consideration available to our shareholders in a transaction involving the transfer outside of Israel
of technology or know-how developed with IIA funding (such as a merger or similar transaction) may be reduced by any amounts that we are
required to pay to the IIA.
It may be difficult to enforce a judgment of
a U.S. court against us, our officers and directors or the Israeli experts named in this annual report in Israel or the United States,
to assert U.S. securities laws claims in Israel or to serve process on our officers and directors and these experts.
We are incorporated in Israel. All of our executive officers and
three of our directors listed in this annual report reside outside of the United States, and most of our assets and most of the assets
of these persons are located outside of the United States. Therefore, a judgment obtained against us, or any of these persons, including
a judgment based on the civil liability provisions of the U.S. federal securities laws, may not be collectible in the United States and
may not be enforced by an Israeli court. It also may be difficult for you to effect service of process on these persons in the United
States or to assert U.S. securities law claims in original actions instituted in Israel. Israeli courts may refuse to hear a claim based
on an alleged violation of U.S. securities laws reasoning that Israel is not the most appropriate forum in which to bring such a claim.
In addition, even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the
claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proven as a fact by expert witnesses, which can
be a time-consuming and costly process. Certain matters of procedure will also be governed by Israeli law. There is little binding case
law in Israel that addresses the matters described above. As a result of the difficulty associated with enforcing a judgment against us
in Israel, you may not be able to collect any damages awarded by either a U.S. or foreign court.
Your rights and responsibilities as a shareholder
will be governed by Israeli law, which differs in some material respects from the rights and responsibilities of shareholders of U.S.
companies.
Since we are incorporated under Israeli law, the rights and responsibilities
of our shareholders are governed by our articles of association and Israeli law. These rights and responsibilities differ in some respects
from the rights and responsibilities of shareholders in U.S.-based corporations. In particular, a shareholder of an Israeli company has
a duty to act in good faith and in a customary manner in exercising its rights and performing its obligations towards the company and
other shareholders and to refrain from abusing its power in the company, including, among other things, in voting at the general meeting
of shareholders on certain matters, such as an amendment to the company’s articles of association, an increase of the company’s
authorized share capital, a merger of the company and approval of related party transactions that require shareholder approval. A shareholder
also has a general duty to refrain from discriminating against other shareholders. In addition, a controlling shareholder or a shareholder
who knows that it possesses the power to determine the outcome of a shareholders’ vote or to appoint or prevent the appointment
of an office holder in the company or has another power with respect to the company, has a duty to act in fairness towards the company.
However, Israeli law does not define the substance of this duty of fairness. See “ITEM 6.C. Board Practices.” Some of the
parameters and implications of the provisions that govern shareholder behavior have not been clearly determined. These provisions may
be interpreted to impose additional obligations and liabilities on our shareholders that are not typically imposed on shareholders of
U.S. corporations. Additionally, the quorum requirements for meetings of our shareholders are lower than is customary for domestic issuers.
As permitted under the Companies Law, pursuant to our articles of association, the quorum required for an ordinary meeting of shareholders
will consist of at least two shareholders present in person, by proxy or by other voting instrument in accordance with the Companies Law,
who hold at least 25% of our outstanding ordinary shares. For an adjourned meeting at which a quorum is not present, the meeting may generally
proceed irrespective of the number of shareholders present at the end of half an hour following the time fixed for the meeting.
General Risk Factors
If equity research analysts
do not continue to publish research or reports about our business or if they issue unfavorable commentary or downgrade our ordinary shares,
the price of our ordinary shares could decline.
The trading market for our ordinary shares relies in part on the
research and reports that equity research analysts publish about us and our business. We do not have control over these analysts and we
do not have commitments from them to write research reports about us. The price of our ordinary shares could decline if no research reports
are published about us or our business, or if one or more equity research analysts downgrades our ordinary shares or if those analysts
issue other unfavorable commentary or cease publishing reports about us or our business.
Our business could be adversely
impacted by climate change.
The intensifying effects of climate change present physical, liability,
and transition risks with both macro and micro implications for companies and financial markets. There is increasing concern that a gradual
increase in global average temperatures due to increased concentration of carbon dioxide and other greenhouse gases in the atmosphere
are causing significant changes in weather patterns around the globe and an increase in the frequency and severity of natural disasters
(such as floods, droughts, wildfires and severe storms). Such events could, among other things, disrupt our operations, including by damaging
or destroying our facilities or those of our suppliers, which may cause us to suffer losses and additional costs to maintain or resume
operations or as a result of supply chain-related delays or cancellations, which could have an adverse impact on our business and results
of operations. In addition, implementing changes to mitigate risks associated with such events may result in substantial short- and long-term
additional operational expenses, which may materially affect our profitability.
Expectations, regulations and scrutiny relating
to environmental, social and governance (ESG) matters may impose additional costs and expose us to new risks.
There is an increasing focus from certain investors, clients, regulators,
employees and other key stakeholders or third parties concerning environmental, social and governance (“ESG”) factors, including
those relating to climate change, supply chain matters and human capital management. Such increased scrutiny may result in increased costs,
increased risk of litigation or reputational damage relating to our ESG practices or performance, enhanced compliance or disclosure obligations,
or other adverse impacts on our business, financial condition or results of operations. In addition, if we communicate certain initiatives
and goals regarding ESG matters, we could fail, or be perceived to fail, in our achievement of such initiatives or goals, or we could
be criticized for the scope of such initiatives or goals. If we fail to satisfy the expectations of investors and other key stakeholders
or our initiatives are not executed as planned, our reputation and financial results could be materially and adversely affected. Additionally,
the emphasis on ESG matters has resulted and may result in the adoption of new laws and regulations, including new reporting requirements
in various jurisdictions. Our failure to comply with any applicable rules or regulations could lead to penalties and adversely impact
our reputation, customer attraction and retention, access to capital and employee retention. Such ESG matters may also impact our suppliers,
customers and business partners, which may augment or cause additional impacts on our business, financial condition or results of operations.
Item 4.
INFORMATION ON THE COMPANY
|
|
A.
|
History and Development of the Company
|
Our History
Our legal name is MediWound Ltd. and our commercial name is MediWound.
We are a company limited by shares organized under the laws of
the State of Israel. MediWound was founded in January 2000. We are registered with the Israeli Registrar of Companies. Our registration
number is 51-289494-0. Our principal executive offices are located at 42 Hayarkon Street, Yavne 8122745, Israel, and our telephone number
is +972 (77)-971-4100.
Our agent for service of process in the United States is Puglisi
& Associates, located at 850 Library Avenue, Suite 204, Newark, Delaware 19711, and its telephone number is +1 (302) 738-6680.
The SEC maintains an internet site that contains reports, proxy
and information statements, and other information regarding issuers that file electronically with the SEC at: http://www.sec.gov. Our
website address is www.MediWound.com. Information contained on, or that can be accessed through, our website does not constitute a part
of this annual report and is not incorporated by reference herein. We have included our website address in this annual report solely for
informational purposes.
Principal Capital Expenditures
For a description of our
principal capital expenditures and divestitures, see Item 5. “Operating and Financial Review and Prospects-Liquidity and Capital
Resources” and Note 15A(3) to our consolidated financial statements included elsewhere in this Annual Report.
We are a global leader in next-generation enzymatic therapeutics
focused on non-surgical tissue repair. We specialize in the development, production and commercialization of solutions that seek to improve
existing standards of care. We are committed to providing rapid and effective biologics that improve patient experiences and outcomes,
while reducing costs and unnecessary surgeries.
Our first drug, NexoBrid, is an FDA-approved orphan biologic for
eschar removal in severe burns that can replace surgical interventions and minimize associated costs and complications. NexoBrid is a
topically administered biological product that enzymatically removes nonviable burn tissue, or eschar, in patients with deep partial and/or
full-thickness thermal burns without harming viable tissue. NexoBrid is approved in over 40 countries, including in the United States,
European Union and Japan, where it has been designated as an orphan biologic drug.
Utilizing the same core biotherapeutic enzymatic
platform technology, we have developed a strong R&D pipeline including our lead drug under development, EscharEx. EscharEx, a phase
III-ready biologic, is a multimodal debridement therapy for the treatment of chronic wounds, with significant potential advantages over
the $360 million dominant product and an opportunity to expand the market. It is a concentrate of proteolytic enzymes enriched in bromelain
for topical, easy to use daily applications. In several Phase II trials, EscharEx was shown to be well-tolerated, and demonstrated positive
efficacy results in debridement and promotion of granulation tissue in various hard-to-heal wounds, with only a few daily applications.
EscharEx’s mechanism of action is mediated by proteolytic enzymes that cleave to and remove necrotic tissue preparing the wound
bed for healing. The Phase III study in patients with VLU, is expected to initiate in the second half of 2024.
MediWound’s pipeline also includes MW005, a topical therapeutic
for the treatment of basal cell carcinoma that has demonstrated positive results in Phase I/II study.
We manufacture NexoBrid and our product candidates in our cGMP
certified sterile manufacturing facility at our headquarters in Yavne, Israel.
Key Recent Developments
NexoBrid
In May 2023, we were awarded an additional $10 million from
BARDA. The supplemental funding was intended to support a $3 million replenishment of expired product previously procured for emergency
preparedness, the pediatric indication sBLA submission to the FDA, and enrollment of an additional 50 patients in the ongoing expanded
access treatment protocol (NEXT).
In September 2023, NexoBrid was launched in the U.S. by our partner,
Vericel, with more than 50 burn centers submitting packages to Pharmacy and Therapeutics (P&T) committees and more than 25 already
approved. Also, in the third quarter of 2023, NexoBrid was launched in Japan for treating adults and pediatric burn patients, and launched
in the fourth quarter in India.
In September 2023, the Supplemental BLA for pediatric indication
was accepted for review by the U.S. Food and Drug Administration (FDA), with a decision expected in the second half of 2024.In December
2023 we received European Commission approval for the removal of eschar in deep partial- and full-thickness thermal burns for all ages.
In November 2023, we expanded NexoBrid’s European market
presence by establishing a collaboration with PolyMedics Innovations (PMI) for the promotion of NexoBrid in Germany, Austria, Belgium,
the Netherlands, and Luxembourg.
During 2023, the DOD through MTEC and directly through MTEC awarded
the Company additional funding of $10.3 million to advance the development and production of a new, temperature-stable formulation of
NexoBrid, positioning it as the first-line non-surgical solution for treating severe burn injuries in pre-hospital settings.
EscharEx
During 2023 we aligned the U.S. Phase III clinical study protocol
with the European Medicine Agency (EMA) and the FDA, and are expected to submit a final protocol in the first half of 2024. 216 patients
will be treated globally across 40 sites with either EscharEx or a gel vehicle placebo, with an interim assessment to be performed once
67% of participants complete the study. Study initiation is expected in the second half of 2024.
During 2023 we established research collaborations with 3M, Mölnlycke
and MIMEDX to support the EscharEx Phase III clinical study.
In February 2024 we published a post-hoc
head-to-head comparative analysis of EscharEx vs SANTYL® using data from a Phase II randomized controlled study, which demonstrated
that EscharEx showed significant superiority over SANTYL in multiple clinical outcome measures: incidence of complete debridement; median
time to achieve complete debridement; incidence of achieving wound bed preparation (WBP); median time to achieve WBP; and time to wound
closure.
MW005
In July 2023, we announced positive results in
our U.S. Phase I/II study of MW005 for the treatment of basal cell carcinoma. Fifteen patients were treated with MW005 and completed the
study. Results showed MW005 was well-tolerated, with a high level of patient compliance. Based on clinical assessments, eleven out of
fifteen patients achieved complete clearance of their BCCs; the majority of these patients also had histologically confirmed complete
clearance.
Operations
In July 2023, we signed a turnkey scale-up agreement (the “Scale-up
Agreement”) to establish, commission, and validate a cutting-edge, sterile, and GMP-compliant manufacturing facility. Construction
of the new GMP-compliant state-of-the-art manufacturing facility is on track for mid-2024 completion, and is projected to achieve 6-fold
manufacturing capacity increase in 2025.
Alongside entering the Scale-Up agreement, we have also secured
a new lease agreement (the “Lease Agreement”) with the current property owner. This agreement allows us to continue the utilization
of our existing facilities and the planned manufacturing site. This property, located in Yavne, Israel, serves as the base for our administrative
headquarters, research and development laboratories, and manufacturing plant. The duration of the lease extends until 2035, with an option
for a further three-year extension until 2038.
Our Focus:
Burn
Care
NexoBrid, a concentrate of proteolytic enzymes enriched in bromelain,
is an easy to use, topically-applied product that removes eschar in four hours without harming the surrounding healthy tissues. Eschar
removal is a critical first step in the successful healing of severe burns. Under existing SOC, burn eschar may be removed either by employing
certain existing topical agents that have been found to be minimally effective or that take a significantly longer period of time to work,
or by resorting to non-selective surgery, which is traumatic and may result in loss of blood and viable tissue. NexoBrid’s rapid
and selective debridement alleviates the known risks associated with eschar, such as infection, eventual sepsis, wound deterioration and
consequential scarring, and it allows physicians to reach an informed decision on further treatment at an earlier stage by direct visual
assessment of the actual burn depth. Furthermore, NexoBrid minimizes the burden associated with invasive surgical procedures, reduces
the need for skin grafting and sacrifice of healthy tissue from donor sites on a patient’s body and generally results in a more
favorable overall long-term patient outcome. NexoBrid has been investigated in hundreds of patients across more than 22 countries and
four continents in ten completed Phase II, Phase III and post-marketing clinical studies. Over 14,000 burn patients have been treated
with NexoBrid in the market since 2013 and the safety and efficacy data reported from post-marketing data sources are consistent with
the data available from clinical trials and no new safety signals have been observed. There have been hundreds of presentations and several
award winning abstracts of NexoBrid in international and national scientific conferences, as well as about 120 peer-reviewed papers, resulting
in the support of burn specialists and key opinion leaders. Awareness of NexoBrid continues to grow through our marketing efforts in countries
where the drug is approved.
Burn Wounds
Burns are life threatening and debilitating traumatic injuries
causing considerable morbidity and mortality. A burn may result from thermal, electrical or chemical means that destroy the skin to varying
depths. According to Critical Care, an international clinical medical journal, burns are also among the most expensive traumatic injuries
because of long and costly hospitalization, rehabilitation and wound and scar treatment.
Most burn injuries involve part of or the entire thickness of the
skin and in some cases, the deeper subcutaneous fat tissue or underlying structures. The severity of the burn depends on three main factors:
|
|
•
|
The extent of the surface that the burn occupies is usually referred to as percent
of total body surface area (“TBSA”). A burn on an adult’s entire palm would generally amount to 1% TBSA, and the average
hospitalized patient has a burn covering approximately 9% TBSA. Burns covering more than 15-20% TBSA usually require hospitalization and
may result in dehydration, shock and increased risk of mortality.
|
|
|
•
|
The depth of the burn, referred to in terms of “degree” is generally classified into four categories:
|
|
|
○
|
Superficial or first-degree burns. Such burns
do not penetrate the basal membrane and usually heal naturally.
|
|
|
○
|
Dermal/partial thickness or second-degree burns.
Such burns are characterized by varying amounts of damaged dermis and can be further subdivided into superficial and deep partial-thickness
burns. Superficial partial-thickness burns may heal spontaneously after removal of the covering thin eschar. Conversely, deep partial-thickness
burns are often difficult for physicians to accurately diagnose before eschar removal and may progress and transform into full-thickness
burns if not debrided in a timely manner, depending on the magnitude of latent tissue death of the surrounding skin.
|
|
|
○
|
Full thickness or third-degree burns. Such
burns are characterized by death of the entire dermal tissue down to the subcutaneous fat and must be debrided and treated by autografting,
which is the process of harvesting skin from healthy donor sites on a patient’s body and transplanting it on the post-debridement,
clean wound bed.
|
|
|
○
|
Fourth-degree burns. Such burns, which are
rare, extend beyond the subcutaneous fat tissue into the underlying structures, such as muscle or bone, and also require debridement and
further substantial treatment.
|
|
|
•
|
Other factors include the age of the victim, the body part where the burn occurred
and any co-morbidities of the patient. For example, some patients may require hospitalization regardless of the TBSA or degree of the
burn, such as children, the elderly or victims with burns to the extremities, joints or head/neck area or with co-morbidities such as
smoke inhalation, diabetes or obesity.
|
When patients are hospitalized for a severe burn, the first step
in the treatment after patient stabilization and resuscitation is usually eschar removal. The eschar is the burned tissue in the wound,
which is deprived of blood and isolated from all natural systemic defense mechanisms. Eschar removal is an essential first step in the
treatment of patients with severe burns, allowing for:
|
|
•
|
the prevention of local infection, sepsis (a systemic inflammatory response caused
by severe infection) and additional damage to surrounding viable tissue; and
|
|
|
•
|
the initiation of the body’s healing process and scar prevention.
|
In addition to minimizing the possibility of additional complications,
once the eschar is removed, a physician may properly diagnose the true extent of the trauma by a direct visual assessment of the clean
wound bed. An informed treatment strategy can be decided upon only if the depth of the burn and extent of the tissue damage is known.
Diagnosis of burn depth is difficult, especially because the burn commonly changes its appearance during the first days after injury due
to burn progression. Burns that are initially difficult to classify due to the presence of eschar are referred to as “indeterminate”
burns. This ambiguity can delay the assessment of the burn depth and formulation of proper treatment. Unless the burns are life-threatening,
definitive treatment is postponed for several days post-injury until diagnosis is clearer, when burn progression by death of the surrounding
and underlying tissue has already occurred and ended. During this delay, local and systemic effects of post-burn inflammation and bacterial
contamination can occur. Therefore, earlier, selective eschar removal is essential to prevent eschar-related complications and to allow
the physician to reach an informed decision on further treatment.
Currently, there are two main treatment modalities for debridement:
|
|
○
|
Surgical debridement predominantly includes tangential excision, a procedure in which
a surgeon amputates the entire dead tissue mass, layer after layer, down to healthy, viable tissue. The excision is extended into healthy
intact tissue to make sure that no trace of the eschar remains, resulting in up to an estimated 30-50% of healthy tissue being excised
during this procedure. Other methods include dermabrasion, in which a mechanically powered, hand-held rotating abrading cylinder is used
to slowly scrape off tissue, and hydro surgery, in which a high-pressure flow of water abrades the tissue. These alternative methods have
attempted to limit the trauma associated with tangential excision, but entail spray of contaminated eschar or take a significantly longer
time to complete than tangential excision.
|
|
|
○
|
The benefits of surgical eschar removal are that it is usually fast and effective.
Disadvantages include the significant trauma of the procedure, associated blood loss, risk of surgery in delicate areas of the body such
as hands, added costs, and, most importantly, the loss of viable tissue that necessitates additional surgical procedures for harvesting
skin from healthy donor sites and autografting.
|
|
|
○
|
Due to the disadvantages of surgery in extensive burns some surgeons limit their debriding
surgery to only a part of the affected area in a single session (15-30% TBSA in most centers), thus delaying full debridement by days.
After several days, complications related to eschar contamination may begin and some of the benefits of the earlier debridement may not
be realized. On the other hand, when excising burns immediately, all suspected necrotic tissue will be excised, inevitably resulting in
over-excision, especially in “indeterminate” burns, as after surgical excision, the remaining skin often no longer has any
spontaneous healing potential and will heal only by autografting.
|
|
|
•
|
Non-surgical debridement |
|
|
○
|
Non-surgical debridement includes many different treatment options that do not require
direct surgical removal of the skin to remove eschar. With non-surgical debridement, the eschar is naturally, but slowly, removed by contaminant
microorganisms, tissue autolysis, or self-decomposition, and the inflammatory process that may lead to serious local and systemic complications.
In seeking to facilitate such natural processes, topical medication, anti-microbial agents, enzymes and biological/chemical applications
are often applied onto the eschar.
|
|
|
○
|
The benefits of this approach are that it is non-surgical, reduces trauma to the patient
and is easier to apply. Disadvantages include numerous dressing changes and mechanical scraping with limited debridement efficacy. This
prolongs the eschar removal process, which may lead to death of the tissue surrounding the initial burn wound, causing partial-thickness
wounds to transform into full-thickness wounds and forming granulation tissue that may develop into heavy scars.
|
As demonstrated in our clinical trials, NexoBrid combines the advantages
of surgical and non-surgical debridement modalities by providing rapid and effective eschar removal while not harming viable tissue. This
allows for earlier direct visual assessment of the burn wound in order to formulate proper treatment.
Market Opportunity
Severe burns require specialized care in hospitals or burn centers.
Approximately 160,000 patients with severe burns are hospitalized every year in the United States and Europe. The prevalence of patients
with severe burns is even higher in emerging economies. For example, approximately 400,000 patients are hospitalized every year with burns
in India according to a study conducted by IMS Health. The severe burn patients are predominantly treated by specialists in approximately
250 burn centers in Europe and the United States, as well as at burn units of large hospitals in Europe. We believe these patients can
benefit from NexoBrid’s effective and selective, non-surgical eschar removal.
In addition to our current marketing of NexoBrid in Europe and
the United States, we have signed local distribution agreements for distribution of NexoBrid in Europe, Asia-Pacific countries, members
of the Commonwealth of Independent States (“CIS”) and the Middle East and we plan to target additional markets in these territories
by leveraging our approved registration file for additional regional marketing authorizations.
In addition to the market opportunities for NexoBrid discussed
above, we believe that NexoBrid has the potential to play a critical role in the event of a burn mass casualty incident (“BMCI”),
which is generally defined as any incident in which emergency medical services resources, such as personnel and equipment, are overwhelmed
by the number and severity of casualties. A variety of public emergencies may give rise to a BMCI, such as terrorist attacks, natural
disasters, fires and explosions. One example of a BMCI is a mass burn casualty disaster, which is defined by the American Burn Association
as a catastrophic event in which the number of burn victims exceeds the capacity of the local burn center to provide optimal care. If
a significant number of burn victims arrive at a burn center following an event, some victims may go untreated until the bottleneck is
resolved. The use of non-surgical means that are capable of providing rapid eschar removal without harming healthy tissue, particularly
during public health emergencies, could potentially reduce the time, labor and resource burdens associated with the current standard-of-care,
thereby enabling the treatment of more patients. In the event of a mass burn casualty disaster, healthcare professionals can use NexoBrid
to begin treatment at the patient’s bedside without the need for a surgical team and facilities. NexoBrid has demonstrated in clinical
studies, with statistical significance, its ability to non-surgically and rapidly remove eschar in a single four-hour application. Once
the acute treatment has been completed, the wound can be covered with available means and further managed once the BMCI is under control
and the bottlenecks resolved. NexoBrid has been recognized by BARDA as a medical countermeasure for treatment of burns in the event of
a BMCI. Additionally, during the war in Israel, the entire non-U.S. NexoBrid inventory has been deployed to hospitals in Israel and the
military, to effectively treat those affected by the war.
BARDA Contracts
In In September 2015, we were awarded the First BARDA Contract
for treatment of thermal burn injuries. This contract was amended several times over the years to extend its term until September 2024
(the Company is pursuing an extension of the contract) and its total value, up to a total amount of $165 million as of the end of 2022.
In May 2023 BARDA expanded its awarded contract by providing supplemental funding of $10 million to support a $3 million replenishment
of expired product previously procured for emergency preparedness, the pediatric indication sBLA submission to the FDA, and enrollment
of an additional 50 patients in the expanded access treatment protocol (NEXT).
The First BARDA Contract provided funding and technical support
for the pivotal U.S. Phase 3 clinical study (DETECT), the randomized, controlled pivotal clinical trial for use in the pediatric population
(CIDS), the marketing approval registration process for NexoBrid as well as its procurement and availability under the expanded access
treatment protocol (NEXT) in the U.S.
The total amount of the First BARDA Contract is comprised of up to $110 million to support
research and development activities and up to $65 million to procure NexoBrid for U.S. emergency preparedness.
As of December 31, 2023, the Company has received approximately
$88 million in funding in the aggregate, under the First BARDA Contract, and an additional $16.5 million for procurement of NexoBrid for
U.S. emergency preparedness.
The first BARDA Contract may be terminated by BARDA at any time
at BARDA’s discretion.
In September 2018, we were awarded an additional, separate BARDA contract (the “Second
BARDA Contract”), to develop NexoBrid for the treatment of Sulfur Mustard injuries as part of BARDA’s preparedness for mass
casualty events. The Second BARDA Contract provides approximately $12 million of funding to support research and development activities
up to pivotal studies in animals under the U.S. FDA Animal Rule, and contains options for BARDA to provide additional funding of up to
$29 million for additional development activities, animal pivotal studies, and the BLA submission for licensure of NexoBrid for the treatment
of sulfur mustard injuries. The Second BARDA Contract was expired in 2023. As of December 31, 2023, the Company has received approximately
$4.4 million of funding from the Second BARDA contract.
DOD and MTEC contracts
In February, 2023, the Company was entered in to a contract with
the U.S. Department of Defense (DoD), through the Medical Technology Enterprise Consortium (MTEC), for development and production of a
new, temperature-stable formulation of NexoBrid, positioning it as the first-line non-surgical solution for treating severe burn injuries
in pre-hospital settings. The contract provides funding up to $2.7 million.
During 2023, the DOD through MTEC and directly through MTEC awarded
the Company additional funding of $10.3 million to advance this development of a new temperature stable formulation of NexoBrid.
The MTEC contract may be terminated by MTEC at any time at MTEC’s
discretion.
NexoBrid Clinical History
NexoBrid, our innovative biopharmaceutical product, has received
marketing authorizations from the U.S., European Commission and the Israeli, Argentinean, South Korean, Russian, Peruvian, Chilean, Taiwanese,
Ukrainian, other Eurasian states, United Arab Emirates, Japanese and Indian Ministries of Health for the removal of eschar in adults with
deep partial- and full-thickness thermal burns. The active ingredient of NexoBrid is a concentrate of proteolytic enzymes enriched in
bromelain extracted from pineapple stems. Proteolysis is a breakdown of proteins into smaller building blocks, polypeptides or amino acids.
Our research and development strategy is centered around our validated proteolytic enzyme platform technology, focused on next-generation
bio-active therapies for burn and wound care and biological medicinal products for tissue repair. For each indication, our research and
development team further develops and optimizes our enzymatic platform technology, creating unique and differentiated products meeting
separate needs based on the specific indication, which is the basis for NexoBrid, EscharEx and all other pipeline product candidates.
One vial of NexoBrid containing 2 grams of concentrate of proteolytic enzymes enriched in bromelain is sufficient for treating a burn
wound area of 1% total body surface area (“TBSA”). We developed NexoBrid to fulfill the previously unmet need for a non-surgical
effective and selective debriding agent that combines the efficacy and speed of surgery with the non-invasiveness of non-surgical methods.
NexoBrid enhances the ability of physicians to conduct an earlier direct visual assessment of the burn depth to reach an informed decision
on further treatment as well as to reduce the surgical burden and achieve a favorable long-term patient outcome.
NexoBrid has been investigated in hundreds of patients across 22
countries and four continents in ten completed Phase II and Phase III and post-marketing clinical studies. While we are marketing our
product for the removal of eschar in burn wounds under the name “NexoBrid,” in clinical trials the product has been referred
to as “Debridase” and “Debrase.”
The following table sets forth information regarding the completed
clinical trials of NexoBrid:
|
|
Trial 1
|
Trial 2
|
Trial 3
|
Trial 4
|
Trial 5
|
Trial 6
|
Trial 7
|
Trial 8
|
Trial 9
|
Trial 10 |
|
Study Type
|
Retrospective Phase II
Investigator initiated
|
Dose range Phase II
|
Prospective Phase II
IND/FDA
|
Phase II
IND/FDA
|
Phase III
EMA
|
Phase IIIb
EMA
|
Phase II
EMA
|
Post approval safety study
EMA
|
Phase III
IND/FDA
|
Phase III
IND/FDA |
|
Design
|
Data collected from files of patients treated with NexoBrid
|
Parallel, controlled, observer-
blind, randomized, single-center
|
Parallel, controlled, observer-
blind, three-arm, randomized,
multi-center
|
Parallel, controlled, open label, three-arm, randomized, single-center
|
Parallel, controlled, open label, two-arm, randomized, multi-center
|
Parallel, controlled, blinded, two-arm, multi-center
|
Open label,
single-arm,
multi-center
|
Observational retrospective data collection
|
Parallel, controlled, open label, three-arm, randomized, multi-center
|
multicenter, multinational, randomized, controlled, open-label
study in children |
|
Main Objectives
|
Safety and efficacy
|
Comparison of efficacy and safety
|
Safety and efficacy
|
Safety
|
Safety
Efficacy
|
Long-term scar assessment
Quality of life
|
Safety and pharmacokinetics
Efficacy
|
Effectiveness of the risk minimization activities
|
Safety
Efficacy
|
Safety
Efficacy
|
|
Wound Types
|
Deep partial/full thickness thermal burns
|
Deep partial /full thickness thermal burns
|
Deep partial /full thickness thermal burns
|
Deep partial /full thickness thermal burns
|
Deep partial/ full thickness thermal burns
|
Scar formation
|
Deep partial/full thickness thermal burns
|
Burns which were treated with NexoBrid in the market
|
Deep partial/ full thickness thermal burns
|
Deep partial/ full thickness thermal burns
|
|
Number of Patients
|
154
|
20
|
140
|
30
|
182
|
89
|
36
|
160
|
175
|
145 |
|
Study Length
|
1985-2000
|
2002-2005
|
2003-2004
|
2006-2007
|
2006-2009
|
2011
|
2009-2015
|
2017-2019
|
2015-2020
|
2015-2023 |
|
Location
|
Israel
|
Israel
|
International
|
United States
|
International
|
International
|
International
|
Europe
|
International
|
International |
Completed clinical trails
Pediatric investigational plan - CIDS study
The CIDS study was a Phase III, multicenter, multinational, randomized,
controlled, open-label study in children with thermal burns. The study objectives were to evaluate the efficacy and safety of treatment
with NexoBrid compared with SOC in hospitalized children with severe thermal burns of 1% to 30% TBSA. We expanded this study also to United
States burn centers, following approval of the study protocol by the FDA. The study was conducted in accordance with a study design endorsed
by the FDA and the EMA as part of the agreed Pediatric Investigational Plan (“PIP”) to support extension of the indication
to pediatric patients. The CIDS study included pediatric patients of all ages, from newborn to eighteen years of age, offering NexoBrid
to this important and sensitive group of patients. The primary endpoints evaluate early eschar removal, surgical burden, and cosmesis
and function with a 12-month follow-up. Secondary endpoints included reduction in the need for surgical excision for eschar removal (surgical
need), blood loss, reduction of the need for autograft in DPT wounds and non-inferiority in cosmesis and function at twenty-four months
follow-up from wound closure. Additional extended long term cosmesis and function assessment at more than 30 months from wound closure
was added to the protocol. Non-inferiority of the time to complete wound closure and other standard safety measurements were also compared
with the SOC control arm.
The study was expanded to include burn centers in the United States
following agreement with the FDA, under the same protocol with alignment to the U.S. Phase III study (DETECT) protocol for the adult population.
The non-inferiority of cosmesis and function at twelve months and twenty-four months from wound closure were defined as safety measurements.
In addition, reduction in surgical need was measured only by reduction in incidence of surgical excision for eschar removal.
The Phase III study met its three primary endpoints with
a high degree of statistical significance. NexoBrid demonstrated a significant reduction in the time to achieve complete eschar removal
and a significant reduction in the wound area requiring surgical excision (surgical need) while demonstrating non-inferiority to SOC in
quality of scars as measured by MVSS. The study also met certain secondary endpoints showing statistically significant reduction in the
incidence of surgical excision and a reduction in the need for autograft in deep partial thickness burns, as well as a favorable trend
in the reduction of blood loss during the eschar removal process. In addition, the study confirmed NexoBrid to be safe and well-tolerated
for all ages.
Based on these results, we received in December 2023 the European
Commission approval for the removal of eschar in deep partial- and full-thickness thermal burns for all ages.
Additionally, In September 2023, the Supplemental BLA for pediatric
indication was accepted for review by the U.S. Food and Drug Administration (FDA), with a decision expected in the second half of 2024.
Ongoing clinical trials
Expanded access treatment protocol (“NEXT”)
The NEXT protocol, which we initiated in October 2019, is an
open-label, single-arm treatment protocol which allows for the treatment of up to 250 burn patients with deep partial- and full-thickness
thermal burns up to 30% TBSA. In September 2020, the FDA agreed to allow the NEXT protocol to be expanded to include pediatric as well
as adult burn patients. The NEXT protocol is being funded by BARDA. See “BARDA Contracts” above. NEXT has been designed to
be consistent with current real-life burn treatment practices in the U.S. and up to 30 U.S. burn centers are anticipated to participate.
We received FDA concurrence that patients can be treated under the NEXT protocol in a burn MCI that is not a declared national emergency.
Following the commercial availability of NexoBrid in the U.S., NEXT is scheduled to be closed during the first half of 2024.
Wound
Care
EscharEx, a phase III-ready biologic, is a multimodal
debridement therapy for the treatment of chronic wounds, with significant potential advantages over the $360 million dominant product
and an opportunity to expand the market. It is a concentrate of proteolytic enzymes enriched in bromelain for topical, easy to use daily
applications. In several Phase II trials, EscharEx was shown to be well-tolerated, and demonstrated its positive efficacy results in debridement
and promotion of granulation tissue in various hard-to-heal wounds, with only a few daily applications. EscharEx’s mechanism of
action is mediated by proteolytic enzymes that may cleave to and remove the necrotic tissue, preparing wound bed for healing.
Chronic and Other Hard-to-Heal Wounds
The chronic and other hard-to-heal wound market consists of a broader
addressable population of more than 14 million patients in Europe and the United States alone suffering from chronic wounds such as VLUs,
DFUs, pressure ulcers and additional patients suffering from surgical/traumatic hard-to-heal wounds. Chronic and other hard-to-heal wounds
represent a $25 billion burden to the U.S. healthcare system. Chronic and hard-to-heal wounds are caused by impairment in the biochemical
and cellular healing processes due to local or systemic conditions and generally can take several weeks to heal, if not longer. Such wounds
can lead to significant morbidity, including pain, infection, impaired mobility, hospitalization, reduced productivity, amputation and
mortality. In each of the various wound types, the presence of the eschar and/or other devitalized tissue is a frequent cause for “chronification”
of wounds and the removal of this non-viable material is the key step to commence healing. The non-viable material needs to be removed
to prevent further deterioration of the wound that may result in additional adverse patient outcomes. If not effectively treated, these
wounds can lead to potentially severe complications including further infection, osteomyelitis, fasciitis, amputation and mortality. Most
advanced wound care therapies, including negative pressure wound therapy, such as V.A.C. Therapy, and skin substitutes such as Apligraf
and Dermagraft and human amniotic tissue products, are complementary to our lead product candidate, EscharEx, as these products require
a clean wound bed to effectively heal a wound. Four common chronic and other hard-to-heal wounds are:
|
|
•
|
Venous leg ulcers. VLUs develop as a result
of vascular insufficiency, or the inability for the vasculature of the leg to return blood back toward the heart properly. Based on our
comprehensive market research study on EscharEx that involved more than 200 healthcare professionals in the U.S. and Europe, which was
last updated in 2022, the VLU overall prevalence is approximately 3.3 million (1% of total U.S. population). Furthermore, the annual incidence
of VLUs in the U.S. alone, is approximately 960,000 (accounting for 45% recurrence), of which approximately 690,000 undergo debridement
in a given year. These ulcers usually form on the sides of the lower leg, above the ankle and below the calf, and are slow to heal and
often recur if preventative steps are not taken. The risk of VLUs can increase as a result of a blood clot forming in the deep veins of
the legs, obesity, smoking, lack of physical activity or work that requires many hours of standing.
|
|
|
•
|
Diabetic foot ulcers. Diabetes can lead to
a reduction in blood flow, which can cause patients to lose sensation in their feet and may prevent them from noticing injuries, sometimes
leading to the development of DFUs, which are open sores or ulcers on the feet that may take several weeks to heal, if ever. Based on
our comprehensive market research study conducted in 2015 on EscharEx that involved more than 200 healthcare professionals in the U.S.
and Europe and, which was updated in 2022, there were estimated 31 million diabetics in the United States in 2019 (9.4% of the U.S. population).
The annual incidence of DFUs in the United States alone is approximately 990,000 (accounting for 45% recurrence), of which approximately
820,000 undergo debridement in a given year.
|
|
|
•
|
Pressure ulcers. Pressure ulcers , also known
as pressure sores, or bed sores, are injuries to the skin or the tissue beneath the skin caused by pressure applied to the skin and subsequent
death of the tissue as a result of the reduced blood supply. These often occur in patients who are hospitalized or confined to a chair
or bed, and usually form over bony areas, where there is little cushion between the bone and the skin, such as heels, elbows, the sacral
area, and back of the head. Annually, 2.5 million pressure ulcers are treated in the United States in acute care facilities alone.
|
|
|
•
|
Surgical/traumatic wounds. Surgical wounds
form as a result of various types of surgical procedures such as investigative or corrective, minor or major, open (traditional) or minimal
access surgery, elective or emergency, and incisions (simple cuts) or excision (removal of tissue), among others. Traumatic wounds form
as a result of cuts, lacerations or puncture wounds, which have caused damage to the skin and underlying tissue. Severe traumatic wounds
may require surgical intervention to close the wound and stabilize the patient. Surgical/traumatic hard-to-heal wounds develop for various
reasons, such as local surgical complications, suboptimal closure techniques, presence of foreign materials, exposed bones or tendons
and infection. In the United States, millions receive post-surgical wound care annually.
|
Market Opportunity
Currently, surgery (sharp debridement) is generally considered
a first-line option. Sharp debridement is an effective method to debride a wound. However, this method requires surgically skilled physicians
performing surgery with patients under, anesthesia, which in elderly patients with various co-morbidities is accompanied with a higher
risk of local and systemic complications. Surgery may also involve hemorrhage which could be more difficult to control due to a high incidence
of use of anticoagulants in this population. Surgery on wounds may very easily become infected, with the infection propagating to surrounding
soft and boney tissues ending in life threatening major complication or amputation. Very often even minor, limited sharp debridement exposes
other sensitive tissue, such as tendons, deep vessels/nerves and bones that may become infected or may be severely damaged, necessitating
additional, more extensive debridement or even amputation. Due to these limitations, chronic wounds are treated by conservative methods,
with autolytic and enzymatic debridement being the most commonly-used non-sharp methods. This includes a collagenase-based enzymatic debriding
ointment, hydrogels and other topical dressings, which require numerous application sessions and a long time (6-8 weeks) to achieve a
clean wound bed, if they achieve this at all. Thus, there is an unmet medical need for a non-surgical rapid and effective debridement
agent for all care settings. Given the high demand for an effective non-surgical debridement technique and the clinical data generated
to date, EscharEx has the potential to expand the current use of enzymatic debridement across all sites of care and achieve substantial
market share. As documented in the Phase II study described below, EscharEx significantly improved the rate of complete debridement after
few once-daily applications, thus potentially facilitating wound debridement without the need for surgery. Based on our comprehensive
market research study on EscharEx that involved more than 200 healthcare professionals in the U.S. and Europe, which was last updated
in 2022, the EscharEx TAM for VLUs and DFUs is estimated at approximately $2 billion in the U.S. This market research and physician feedback
suggests potential market share for EscharEx at approximately 30%. Due to traditional drug pricing disparities between the U.S. and countries
within the EU, we expect the TAM for the market in the EU to be less than that of the U.S., but to be roughly similar on a unit volume
basis.
EscharEx Clinical History
EscharEx is a topical agent being developed for
debridement of chronic wounds, in order to fulfill an unmet need for a non-surgical rapid and effective debridement option. EscharEx is
based on the same active substance as NexoBrid but differs in other aspects, such as in formulation and presentation. Based on our current
pre-clinical studies, EscharEx demonstrated even higher potency in lower doses, which could further contribute to EscharEx’s potential
efficacy and tolerability. EscharEx has been designed in accordance with the current treatment workflow and reimbursement programs, providing
a non-surgical easy-to-use, potent product for daily application, which we believe will enhance patient compliance and improve quality
of care. Based on the feedback received from different stakeholders, we believe that EscharEx can address the unmet medical need for a
non-surgical rapid and effective product, particularly in the outpatient setting, where the majority of patients are treated, and has
a greater potential to achieve substantial market share.
EscharEx is more differentiated from NexoBrid, which further limits
the chances for competition between the two products.
Non-clinical safety studies performed with NexoBrid support EscharEx
development, and we have already completed successful bridging toxicology studies. In a pre-IND meeting the FDA stated that existing toxicology
data for EscharEx, including cross-referenced NexoBrid data, could be sufficient to support initiation of clinical studies in the product.
Following discussions with the regulatory authorities,
we aligned the Phase III study protocol with EMA and the FDA, and are on track to submit a final protocol in the first half of 2024. 216
patients will be treated globally across 40 sites with either EscharEx or a gel vehicle placebo, with an interim assessment to be performed
once 67% of participants complete the study. Study initiation is expected in the second half of 2024. Additionally, we established research
collaborations with 3M, Mölnlycke and MIMEDX to support the EscharEx Phase III clinical study.
Completed clinical trials
We completed a first Phase II feasibility study in Israel for chronic
and other hard-to-heal wounds. In January 2017 we completed and announced the final results of a second Phase II prospective study in
Israel and Europe. In November 2017, we announced the final results of a second cohort of the second Phase II study. Based on the completed
studies, we believe that our product candidate may be effective for debridement of chronic and other hard-to-heal wounds.
First Phase II feasibility study-Israel
This first Phase II feasibility study was conducted
in Israel to study the efficacy of our technology on chronic and other hard-to-heal wounds. The study assessed 24 patients at two sites.
The results showed that our technology demonstrated positive efficacy results in debriding various chronic and other hard-to-heal wound
etiologies, such as VLUs, DFUs, pressure sores and trauma on diseased skin.
Second Phase II study-Israel/E.U. - First Cohort
This second Phase II study was a prospective, controlled, assessor-blinded,
randomized, multi-center Phase II study in Israel and Europe. The study objectives were to evaluate the efficacy and safety of EscharEx
in comparison to the Gel Vehicle at a ratio of 2:1 for the treatment of a variety of chronic and other hard-to-heal wounds in three etiologies:
DFUs, VLUs and post-surgical or traumatic hard-to-heal wounds.
The primary endpoint assessed incidence of complete non-viable
tissue removal (debridement) at the end of the debridement period (within up to 10 daily applications) and the secondary endpoints assessed
various efficacy and safety endpoints, including wound bed preparation and wound healing.
In January 2017 we reported final results of the
first cohort of 73 patients. The average wound age in the EscharEx arm was more than double (72.8
weeks) that of the gel vehicle group (30.8 weeks). The average wound size was 33.6 cm2 in the EscharEx arm vs. 25.8 cm2 in the gel vehicle
group. Despite the larger wounds and that wounds treated with EscharEx were older than wounds treated with gel vehicle (72.8 vs. 30.8
weeks), the study met its primary endpoint. EscharEx demonstrated a statistically significantly higher incidence of complete debridement
at the end of the debridement period. Patients treated with EscharEx demonstrated a higher incidence of complete debridement (55% or 27/49)
compared with patients treated with the hydrogel6 vehicle (29% or 7/24) with p=0.047.
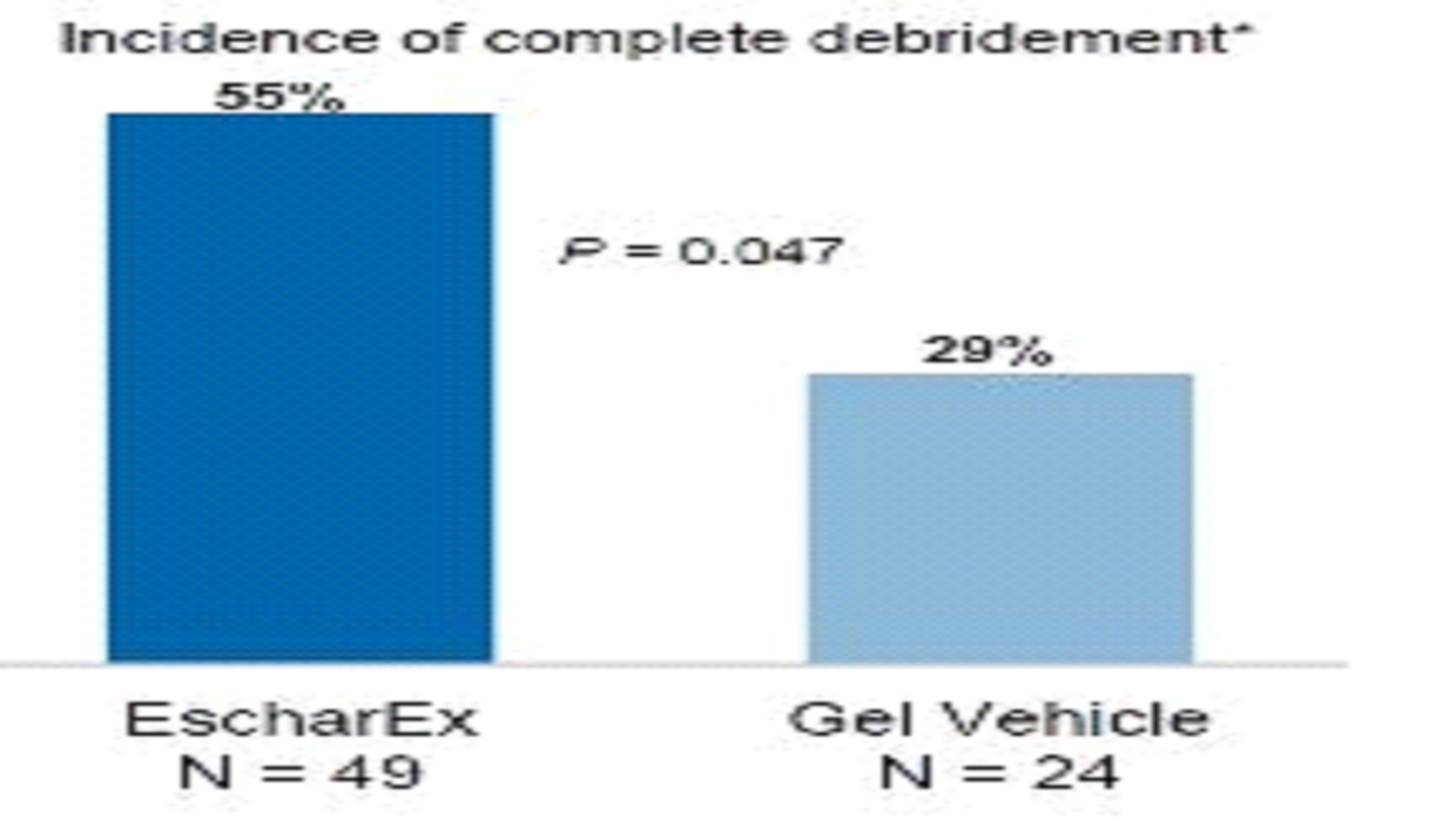
*w/i
10 daily applications
Predefined sub-group analyses showed that 50% of patients with
DFUs treated with EscharEx (8/16) achieved complete debridement at the end of the debridement period compared with 14.3% of patients with
DFUs treated with hydrogel vehicle (1/7). In addition, 62.5% of patients with VLUs treated with EscharEx (10/16) achieved complete debridement
at the end of the debridement period compared with 25% of patients with VLUs treated with hydrogel vehicle (2/8). Post hoc analysis showed
that 56.3% of patients with VLU or DFU in the EscharEx group had complete debridement at the end of the debridement period compared with
20.0% in hydrogel vehicle group (p=0.028).
The study included secondary endpoints that provide
further insight into number of efficacy and safety parameters. The secondary endpoint of time to complete debridement demonstrated a clear
trend (p=0.075) that strongly suggests that not only was there a difference in the incidence of debridement, as demonstrated by the primary
endpoint, but that debridement occurred earlier in the group treated by EscharEx. The advantage in time to complete debridement was corroborated
by the statistically significant post hoc result in the subgroup of patients with VLUs or DFUs that were treated with EscharEx (p=0.024).
Post hoc analysis showed that of patients who achieved complete
debridement in the EscharEx group, 93% (25/27) completed the debridement within 7 days (4-5 applications on average).
The overall patient demographics were comparable
across both arms. No deleterious effect on wound healing was observed and no material differences were found in reported adverse events.
The overall safety data were comparable between the arms.
Second Phase II study-Israel/E.U. - Second Cohort
After successfully completing the first cohort
of the study which included 73 patients recruited in 15 clinical sites, we initiated a second cohort of patients to evaluate safety and
tolerability over extended periods of application. In this second cohort, we recruited 38 patients from two etiologies, either VLUs or
DFUs, over extended periods of application (24-72 hours) with up to eight applications, randomizing the patients to two study arms EscharEx
or gel vehicle at a ratio of 2:1. The primary objective was to assess safety.
EscharEx met its primary safety endpoint in this cohort, and the
overall patient demographics and wound baseline characteristics were comparable across the arms in the second cohort. No related systemic
adverse events were reported and adverse events related to local application were mild to moderate, reversible and resolved during the
trial. Vital signs, pain scores, infection rates, laboratory parameters and blood loss were comparable between the two arms of the trial.
Overall, no material safety concerns were identified.
EscharEx U.S. Phase II Study in Venous Leg Ulcer (VLU) Patients
In December 2019, we initiated a U.S. Phase II adaptive design
clinical study of EscharEx for the treatment of VLUs. The study was a multicenter, prospective, randomized, placebo-controlled, adaptive
design study, evaluating the safety and efficacy of EscharEx in debridement of VLUs compared to gel vehicle (placebo control) and non-surgical
standard-of-care of either enzymatic or autolytic debridement (NSSOC). The study enrolled 120 patients, with 119 treated at approximately
20 clinical sites, primarily in the U.S. Study participants were treated with either EscharEx (n=46), gel vehicle control (n=43), or non-surgical
standard-of-care (n=30), with a three-month follow-up. The single primary endpoint was incidence of complete debridement (non-viable tissue
removal), clinically assessed, within up to 8 treatment applications during the assessment period (within 14 days), compared to gel vehicle
placebo control. Secondary and exploratory endpoints assessed time to achieve complete debridement, reduction of pain, reduction of wound
area, granulation tissue and wound quality of life, enabling evaluation of clinical benefits compared to both gel vehicle and NSSOC. Incidence
and time to achieve wound closure were assessed as safety measurements.
In May 2022 we announced our results from this study. The study met its primary endpoint
with a high degree of statistical significance, demonstrating that patients treated with EscharEx had a statistically significant higher
incidence of complete debridement during the 14-day measurement period within up to 8 applications compared to gel vehicle (EscharEx:
63% (29/46) vs. gel vehicle: 30% (13/43), p-value=0.004). EscharEx efficacy results remained statistically significant compared to gel
vehicle after adjusting for pre-specified covariates ascribed to patient baseline characteristics, wound size, wound age and region.

The study met key secondary and exploratory endpoints. Patients
treated with EscharEx had a statistically significant higher incidence of complete debridement, during the same 14-day measurement period,
compared to patients treated by non-surgical standard-of-care ("NSSOC") (EscharEx: 63% (29/46) vs. NSSOC: 13% (4/30)) and the time to
achieve complete debridement was significantly shorter. Estimated median time to complete debridement was 9 days for patients treated
with EscharEx and 59 days for patients treated with NSSOC (p-value=0.016). On average, complete debridement was achieved after 3.6 applications
of EscharEx compared to 12.8 applications with NSSOC. Patients treated with EscharEx demonstrated significantly higher incidence of greater
than 75% granulation tissue at the end of the treatment period compared to gel vehicle (p-value <0.0001). Favorable trends were observed
in wound area reduction and reduction of pain compared to gel vehicle.

In addition, the study showed that EscharEx was
well tolerated, and the overall safety results were comparable between the arms as assessed by the data safety monitoring board. Importantly,
there were no observed deleterious effects on wound closure and no material differences in reported adverse events. Estimated time to
complete wound closure was 64 days for patients treated with EscharEx compared to 78 days for patients treated with NSSOC.
Post-hoc
analyses from this study assessed the incidence and time to wound bed completely covered with granulation tissue. The incidence of achieving
complete debridement and complete cover of the wound bed with granulation tissue (i.e., wound bed preparation, WBP) during the daily treatment
period was 50.0% for EscharEx vs. 25% for the Gel Vehicle (p-value= 0.01) and 10% for NSSOC (p-value<
0.0001). The estimated median time to achieve WBP was 11 days for EscharEx vs. 85 days for the Gel
Vehicle (p-value= 0.002) and 63 days for the NSSOC (p-value= 0.0106).
Furthermore, it was shown that patients reaching WBP in the study are 4 times more likely to achieve wound closure (p=0004).
Post-hoc analyses from this study assessed the
incidence and time to complete debridement, complete granulation, and wound closure in patients treated with EscharEx (n=46) compared
to a sub-group of patients who were treated with SANTYL (n=8). Baseline characteristics (age, gender, wound age, wound size) were comparable
in both groups. The incidence of complete debridement during the daily treatment period (the first two weeks of the study) was 63.0% (95%
CI=47.5-76.8) for EscharEx vs. 0% for SANTYL; p=0.001. The estimated median time to achieve complete debridement during the study was
9 days (95% CI=5-15 days) for EscharEx vs. not achieved for SANTYL (95% CI=22-Not Applicable); p=0.023. The incidence of achieving complete
debridement and complete cover of the wound bed with granulation tissue (i.e., wound bed preparation, WBP) during the daily treatment
period was 50.0% (95% CI = 34.9%-65.1%) for EscharEx vs. 0% for SANTYL; p=0.015. The incidence of achieving WBP throughout the study was
78.3% (95% CI = 63.6-89.1) for EscharEx vs. 37.5% for SANTYL (95% CI=8.5-75.5); p=0.03. The estimated median time to achieve WBP was 11
days (95% CI =7-50 days) for EscharEx vs. not achieved for SANTYL (95% CI=22-Not Applicable); p=0.014. 15 of the 46 patients (32.6%) treated
with EscharEx completely closed their wounds during the study, compared to 2 out of 8 patients (25%) treated with SANTYL (NSS).
In those patients who achieved complete wound closure, the average time to wound closure was 48.4 days (SD=23.5) for EscharEx vs. 76.0
days (SD=2.8) for SANTYL; p=0.05. Patient reported applicational pain was comparable in both groups. The safety results and overall incidence
of adverse wound reactions were comparable between arms.
EscharEx Pharmacology Study
In May 2022, we announced positive results from our U.S. Phase
II pharmacology study of EscharEx for debridement of lower leg ulcers. The study was a prospective, open label, single-arm study, conducted
at three U.S. clinical sites. The study evaluated the clinical performance, safety, and pharmacology effect of EscharEx in the debridement
of lower leg ulcers (VLUs and DFUs). The study evaluated the safety and efficacy of debridement as measured by incidence of, and time
to complete debridement. In addition, the study evaluated the pharmacological effects of EscharEx as measured by the changes from baseline
to end of treatment period in (1) wound biofilm presence in wound biopsies, (2) bacterial burden measured by MolecuLight® fluorescence
images, and (3) biomarkers of wound healing and inflammation in wound fluid. Twelve patients with either VLUs or DFUs were enrolled in
the study. Patients were treated with up to eight daily applications of EscharEx and then continued follow-up for 2 weeks. Punch biopsies
and wound fluids were collected prior to the first, and after the last treatment. Biofilm presence was analyzed from wound biopsies. Wound
fluids were analyzed to evaluate biomarkers of wound healing and inflammation, i.e., MMPs, cytokines, chemokines, growth factors and HNE.
Fluorescent imaging was used during treatment to measure wound size and bacterial load. Fluorescent imaging was also utilized to identify
the highest fluorescence area to obtain the biopsy. EscharEx demonstrated safe and effective debridement with a few daily applications.
In addition, evaluation of wounds’ tissue samples (biopsies) and fluorescence images, indicated reduction of wound area, biofilm
and bacterial bioburden following the treatment with EscharEx.
Seventy percent of patients achieved complete debridement
during the course of treatment within up to 8 applications. On average, complete debridement was achieved after 3.9 applications of EscharEx.
Additionally, an average reduction of 35% in wound size was achieved by the end of the 2-week follow-up period. In all patients that were
positive for biofilm at baseline, the biofilm was reduced substantially to single individual microorganisms or completely removed by the
end of treatment. Seven patients had positive red fluorescence (indicative of bacteria) at baseline and average red fluorescence was reduced
from 1.69 cm2 pre-treatment to 0.60 cm2 post treatment. Biomarker analysis from wound fluid safety data showed that EscharEx was well-tolerated.
The development of EscharEx for the debridement of chronic and
other hard-to-heal wound indications is in Phase 2 studies, and there is no certainty that EscharEx will achieve all of the objectives
of the trials as required or that the FDA will allow at this stage to initiate further studies or that we will successfully complete the
development to obtain a marketing authorization for EscharEx. See “ITEM 3.D. Risk Factors-Development and commercialization of EscharEx
in the United States and our pipeline product candidates worldwide requires successful completion of the regulatory approval process,
and may suffer delays or fail.”
Non-Melanoma
Skin Cancer
MW005 is a topically applied biological product candidate for the
treatment of non-melanoma skin cancers, based on the same active substance of NexoBrid and EscharEx, a concentrate of proteolytic enzymes
enriched in bromelain. The clinical development plan of MW005 is supported by the results from several toxicological and other preclinical
studies, as well as vast clinical experience from NexoBrid and EscharEx, which share the same API. We launched a new clinical program
to evaluate our drug product candidate MW005 in patients with non-melanoma skin cancer.
In July 2023, we announced the final results of
this study. Fifteen patients were treated with MW005 and completed the study. Results showed MW005 was well-tolerated, with a high level
of patient compliance. Based on clinical assessments, eleven out of fifteen patients achieved complete clearance of their BCCs; the majority
of these patients also had histologically confirmed complete clearance.
Although we have conducted preclinical trials, the development
of MW005 for non-melanoma skin cancer indications is still in its preliminary phase and there is no certainty that it will achieve all
the aims of the trials as required and/or successfully complete the approval process for such indication. See “ITEM 3.D. Risk Factors-Development
and commercialization of EscharEx in the United States and our pipeline product candidates worldwide requires successful completion of
the regulatory approval process, and may suffer delays or fail.”
Research and Development
Our research and development strategy is centered around our validated
proteolytic enzyme platform technology, focused on next-generation protein-based therapies for burn and wound care, and for tissue repair,
which underlies NexoBrid and EscharEx, into additional product candidates for high-value indications. For more information regarding our
research and development expenses, see “ITEM 5.C. Research and Development, Patents and Licenses, etc.”
Pre-Clinical Clinical Studies
We conduct clinical studies and preclinical studies to support
the efficacy and safety of our products and their ingredients and to extend and validate their benefits for human health. Preclinical
studies allow us to substantiate the safety of our products and obtain preliminarily indications of their pharmacological and safety profile.
As of the date hereof, we have conducted more than 50 non-GLP and GLP preclinical studies. All pre-clinical safety and toxicology studies
were conducted according to the principles of Good Laboratory Practices (“GLP”), and thirteen clinical studies, according
to the principles of Good Clinical Practices (“GCP”), for NexoBrid, EscharEx and our pipeline product candidates. As a result,
we have developed significant experience in planning, designing, executing, analyzing and publishing clinical studies. Our research and
development team manages our clinical studies and coordinates the project planning, trial design, execution, outcome analyses and clinical
study report submission. During the design, execution and analyses of our studies, our research and development team consults with key
opinion leaders and top-tier consultants in the relevant field of research to optimize both design and execution, as well as to strengthen
the scientific, medical and regulatory compliance level of the investigational plan. Our clinical studies have been conducted in collaboration
with leading medical and research centers throughout the world.
Manufacturing, Supply and Production
We operate a manufacturing facility in Yavne, Israel.
This facility allows us to manufacture sterile biopharmaceutical products, such as NexoBrid. The facility is designed to meet current
cGMP requirements and similar foreign requirements, as certified by the U.S., EU member states competent authorities, the Israeli Ministry
of Health, South Korean ministry of health and Japanese ministry of health. Our facility is subject to audits for reassessment of cGMP
compliance and similar foreign requirements, which are performed periodically by regulatory authorities and was re-approved as cGMP-compliant
for an additional three year term as of the audit date, until 2025. Additionally, as part of the regulatory approval process for NexoBrid
our plant was inspected in 2022 by the FDA and the Pharmaceuticals and Medical Devices Agency (“PMDA”) of Japan to confirm
it meets all regulatory requirements. In addition, other regional applicable authorities may also need to inspect our plant to confirm
it meets all regulatory requirements in order to obtain marketing authorization in these jurisdictions. Applicable changes in our production
processes for NexoBrid must be approved by the EMA and similar authorities in other jurisdictions.
Our global demand for NexoBrid surpasses the current manufacturing
capabilities. We are currently seeking to expand our manufacturing capabilities in order to increase our capacity to manufacture NexoBrid
and future product candidates and satisfy near term demand. The new GMP-compliant state-of-the-art manufacturing facility is projected
to be completed by mid-2024, with full-scale manufacturing currently expected to commence in 2025. We expect the cost will be approximately
$12.7 million.
The starting material used by us in the manufacturing of NexoBrid
and our other product candidates is bromelain SP, which is derived from pineapple plant stems. We have entered into an agreement with
CBC, dated January 11, 2001, as amended on February 28, 2010, pursuant to which CBC uses proprietary methods to manufacture bromelain
SP and supplies us with this intermediate drug substance in bulk quantities. According to the terms of the agreement, CBC shall not, and
shall not permit related companies or a third party to, manufacture, use, supply or sell the raw materials for the use or production of
a product directly or indirectly competing with any of our products. Our supply agreement with CBC has no fixed expiration date and can
be voluntarily terminated by us, with at least six months’ advance written notice, or by CBC, with at least 24 months’ advance
written notice.
Upon obtaining bromelain SP from CBC, we further process it into
the drug substance and then into the drug product to finally create the powder form of NexoBrid. The necessary inactive ingredients contained
in NexoBrid, or the excipients, are readily available and generally sold to us by multiple suppliers. In addition to this powder, we manufacture
a sterile gel substance by combining water for injections produced by us at our facility and additional excipients.
Marketing, Sales and Distribution
We commercialize globally NexoBrid via multiple sales channels:
Europe
In Europe and Israel, we sell NexoBrid, primarily through our own
sales force consisting of a marketing team of specialized and knowledgeable sales representatives in Europe, focusing on leading burn
centers and Key Opinion Leaders (KOL) management. We have obtained national reimbursement for NexoBrid in Belgium, Italy, and Greece and
we continue to locally execute our market access strategy for most of Europe to obtain procurement by burn centers and hospitals as part
of their budget, or under local, regional or national reimbursement, depending on the specific process required in each country. We believe
that additional burn units in large hospitals as well as smaller hospitals will follow the treatment trends once established by the burn
centers. See “-Government Legislation and Regulation-Pharmaceutical Coverage, Pricing and Reimbursement.” Furthermore, we
are establishing additional distribution channels through local partners to extend outreach in EU (Sweden, Finland, France, Switzerland,
Greece, Malta, Bulgaria and Cyprus,), where NexoBrid is already approved for marketing as part of the European marketing authorization.
On November 2023, we expanded NexoBrid’s European market presence by establishing a collaboration with PolyMedics Innovations (PMI)
for the promotion of NexoBrid in Germany, Austria, Belgium, the Netherlands and Luxembourg. In addition to receiving marketing authorization
for NexoBrid in the European Union, key opinion leaders in the burn care field worldwide are already aware of NexoBrid’s efficiency
in removing eschar due to hundreds of scientific presentations and several award-winning abstracts at international and national conferences
and about 120 peer-reviewed papers.
North
America
Vericel License and Supply Agreements
On May 6, 2019, we entered into exclusive license and supply agreements with Vericel
to commercialize NexoBrid in all countries of North America (which we refer to as the “Territory”).
License Agreement.
We entered into the Vericel License Agreement pursuant to which
we granted Vericel an exclusive license, with the right to grant sublicenses, to develop and commercialize NexoBrid and any improvements
of NexoBrid (the “Licensed Product”) in the Territory.
Pursuant to the terms of the Vericel License Agreement, Vericel
will have exclusive control regarding the commercialization of Licensed Products in the Territory and must use commercially reasonable
efforts to commercialize Licensed Products within the Territory. We and Vericel have made customary representations and warranties and
have agreed to certain customary covenants, including confidentiality and indemnification.
Within 10 days of signing the Vericel License Agreement, Vericel
paid us an upfront fee of $17.5 million (the “Upfront Payment”) and upon the U.S. regulatory approval of the BLA for NexoBrid
Vericel paid us $7.5 million. Vericel is obligated to pay us up to $125 million, in the aggregate, upon attainment of certain sales milestones.
The first sales milestone of $7.5 million is triggered when annual net sales of the Licensed Products in the Territory exceed $75 million.
Vericel is also obligated to pay us tiered royalties on net sales of Licensed Products at rates ranging from mid-high single-digit to
mid-teen percentages, subject to certain customary reductions, as well as a percentage of gross profits on committed purchases by BARDA
and a royalty on additional sales to BARDA. The royalties will expire on a product-by-product and country-by-country basis upon the latest
to occur of (i) twelve years following the first commercial sale of such Licensed Product in such country, (ii) the earliest date on which
there are no valid claims of MediWound patent rights covering such Licensed Product in such country, and (iii) the expiration of the regulatory
exclusivity period for such Licensed Product in such country (the “Royalty Term”). Such royalties are subject to reduction
in the event that (a) Vericel must license additional third-party intellectual property in order to develop, manufacture or commercialize
a Licensed Product, or (b) biosimilar competition occurs with respect to the Licensed Product in any country within the Territory. After
the expiration of the applicable royalties for the Licensed Product in any country within the Territory, the license for such Licensed
Product in such country would become a fully paid-up, royalty-free, perpetual and irrevocable license.
The Vericel License Agreement expires on the date of expiration
of all royalty obligations due under the agreement unless earlier terminated in accordance with its terms. Either party may terminate
the agreement upon the failure of the other party to comply with its material obligations under the agreement if that failure is not remedied
within certain specified cure periods or in the event of a party’s insolvency. In addition, Vericel may terminate the agreement
upon a 150-day written notice to us.
Supply Agreement.
On May 6, 2019, concurrently with our entry into the License Agreement,
we entered into a supply agreement with Vericel (the “Supply Agreement”) pursuant to which we are obligated to supply Vericel
with NexoBrid for sale in the Territory on an exclusive basis for the first five years of the term of the Supply Agreement. The Supply
Agreement requires us to take steps to ensure that our manufacturing capacity meets Vericel’s demand for NexoBrid. In addition,
after the exclusivity period or upon supply failure, Vericel will be permitted to establish an additional or alternate source of supply.
Pursuant to the Supply Agreement, we will supply NexoBrid to Vericel
based on Vericel’s fixed orders on a unit price basis. After a specified period, the unit price, on an annual basis, may be increased
based on the United States Producer Price Index for Chemical Manufacturing published by the Bureau of Labor Statistics.
The Supply Agreement’s initial term is five
years (the “Initial Term”), with Vericel required to provide us with notice regarding whether it plans to extend the Initial
Term for an additional two years by the third anniversary of the Supply Agreement. In May 2022, Vericel notified us on its election to
extend the Initial Term for an additional 2 years until 2026. After the Initial Term and optional two-year extension, Vericel, at its
sole discretion, may choose to extend the Supply Agreement’s term for additional one-year periods for a potential total term of
fifteen years.
In September 2023, NexoBrid was launched in the U.S. by Vericel.
The Supply Agreement will automatically terminate upon the expiration
or termination of the License Agreement. Either party may terminate the Supply Agreement upon the failure of the other party to comply
with its material obligations under the Supply Agreement if such failure is not remedied within certain specified cure periods. After
the Initial Term, Vericel may terminate the Supply Agreement upon 12 months’ prior written notice to us, and we may terminate the
Supply Agreement upon 36 months prior written notice to Vericel.
BARDA
Pursuant to the First BARDA Contract, BARDA has completed the procurement
of NexoBrid valued at $16.5 million, for emergency stockpile as part of the HHS mission to build national preparedness for public health
medical emergencies. BARDA purchased inventory is being managed by MediWound under vendor managed inventory.
Under our exclusive license and supply agreements with Vericel,
we will receive a double-digit royalty on any additional future BARDA purchases of NexoBrid. Please see “Vericel License and Supply
Agreements” above.
Other
International Markets
In other international markets, we sell NexoBrid through local
distributors with which we have distribution agreements, focusing on Asia Pacific, EMEA and CEE. We have signed local distribution agreements
for distribution in Russia, Mexico, India, Bangladesh, Sri Lanka, Japan, Australia, New-Zealand, Singapore, Ukraine, Taiwan and United
Arab Emirates.
Our distributors in Russia, South Korea, Taiwan, Japan, India, United Arab Emirates
and Eurasian countries have obtained marketing authorization. Our additional distributors have filed or are in the process of filing for
market authorization in their respective territories and are expected to launch NexoBrid after receipt of local regulatory approval, which
may take a year or more to be granted, and, consequently, may occur in certain markets during 2023. We have launched NexoBrid in South
Korea, Russia, Taiwan Japan and India and expect additional launches following receipt of local marketing authorizations. We plan to enter
other international markets through collaboration with local distributors and leverage our approved registration file in Europe to obtain
regional marketing authorizations. For a breakdown of our consolidated revenues by geographic markets and by categories of operations
for the years ended December 31, 2022 and 2023, please see “Item 5.A Operating and Financial Review and Prospects-Operating Results.”
Intellectual Property
Our intellectual property and proprietary technology are important
to the development, manufacture and sale of NexoBrid, EscharEx and our future pipeline product candidates. We seek to protect our intellectual
property, core technologies and other know-how through a combination of patents, trademarks, trade secrets, non-disclosure and confidentiality
agreements, licenses, assignments of invention and other contractual arrangements with our employees, consultants, partners, suppliers,
customers and others. Additionally, we rely on our research and development program, clinical trials, know-how and marketing and distribution
programs to advance our products and product candidates. As of December 31, 2023, we had been granted a total of 88 patents and have 19
pending patent applications. The family of patents that covers NexoBrid specifically includes 35 granted patents worldwide. EscharEx is
covered by 15 patents and 4 national phase applications.
The main patents for our proteolytic enzyme technology which underlies
NexoBrid, EscharEx and our current pipeline product candidates have been issued in Europe, the United States and other international markets.
Our patents which cover NexoBrid claim specific mixtures of proteolytic enzymes, methods of producing such mixtures and methods of treatment
using such mixtures. Although the protection achieved is significant for NexoBrid, EscharEx and our pipeline product candidates, when
looking at our patents’ ability to block competition, the protection offered by our patents may be, to some extent, more limited
than the protection provided by patents which claim chemical structures which were previously unknown. Absent patent-term extensions,
the NexoBrid patents are nominally set to expire in 2025 and in 2029 in the United States. The NexoBrid patents issued in Europe and in
other foreign jurisdictions are nominally set to expire in 2025. The patents and the national phase applications relating to EscharEx,
if the national phase applications are granted, will expire on January 30, 2037, absent any patent-term adjustment and/or extensions.
While our policy is to obtain patents by application, license or
otherwise, to maintain trade secrets and to seek to operate without infringing on the intellectual property rights of third parties, technologies
related to our business have been rapidly developing in recent years. Additionally, patent applications that we may file or license from
third parties may not result in the issuance of patents, and our issued patents and any issued patents that we may receive in the future
may be challenged, invalidated or circumvented. For example, we cannot predict the extent of claims that may be granted or enforceable
in our patents nor can we be certain of the priority of inventions covered by pending third‑party patent applications filed in the
U.S. If third parties prepare and file patent applications that also claim technology or therapeutics to which we have rights, we may
have to participate in proceedings to determine priority of invention, which could result in substantial costs to us, even if the eventual
outcome is favorable to us. Moreover, because of the extensive time required for clinical development and regulatory review of a product
we may develop, it is possible that, before NexoBrid can be commercialized in additional jurisdictions and/or before any of our future
products can be commercialized, related patents will expire a short period following commercialization, thereby reducing the advantage
of such patent. Loss or invalidation of certain of our patents, or a finding of unenforceability or limited scope of certain of our intellectual
property rights, could have a material adverse effect on us. See “ITEM 3.D. Risk Factors - Our success depends in part on our ability
to obtain and maintain protection for the intellectual property relating to, or incorporated into, our technology and products.”
In addition to patent protection, we also rely on trade secrets,
including unpatented know‑how, technology innovation, drawings, technical specifications and other proprietary information in attempting
to develop and maintain our competitive position. We also rely on protection available under trademark laws, and we currently hold various
registered trademarks, including “MediWound,” “NexoBrid” and “EscharEx” in various jurisdictions,
including the United States, the European Union and Israel.
Klein License Agreement
In September 2000, we signed an exclusive license agreement, as
amended in June 2007, with Mark Klein, a third party, for use of certain patents and intellectual property (the “Klein License Agreement”).
Under the Klein License Agreement, we received an exclusive license to use the third party’s patents and intellectual property to
develop, manufacture, market and commercialize NexoBrid and its pipeline product candidates for the treatment of burns and other wounds.
The claims of such patents are directed to a process of preparing a mixture of escharase and proteolytic enzymes and cover the underlying
proteolytic mixture of escharase and proteolytic enzymes prepared by that specific process. Pursuant to the Klein License Agreement, we
are obligated to keep accounting records related to the sales of NexoBrid and its pipeline product candidates and pay royalties as discussed
below. The Klein License Agreement may be terminated by Mark Klein, subject to notice and dispute resolution provisions of the Klein License
Agreement, in the event of our breach, bankruptcy petition, insolvency or failure to achieve a development milestone within six months
of a target date. We have already achieved all development milestones under the Klein License Agreement.
As consideration under the Klein License Agreement, we paid an
aggregate amount of $1.0 million following the achievement of certain development milestones. In addition, we undertook to pay royalties
of 1.5‑2.5% from revenues, 10% of royalties received from sublicensing and 2% of lump‑sum payments received from sublicensing
up to $1 million and 4% above $1 million, in each case relating to products based on the licensed patents and intellectual property, for
a term of 10‑15 years, as applicable, from the date of the first commercial delivery in a major country. In addition, under the
Klein License Agreement, we agreed to pay a one‑time lump‑sum amount of $1.5 million upon reaching aggregate revenues of $100
million from the sale of such products.
Competition
The medical, biotechnology and pharmaceutical industries are intensely
competitive and subject to significant technological change and changes in practice. While we believe that our innovative technology,
knowledge, experience and scientific resources provide us with competitive advantages, we may face competition from many different sources
with respect to NexoBrid, EscharEx, and our existing pipeline product candidates or any product candidates that we may seek to develop
or commercialize in the future. Possible competitors may include medical practitioners, pharmaceutical and wound care companies, academic
and medical institutions, governmental agencies and public and private research institutions, among others. Any product that we successfully
develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
In addition, we face competition from the current SOC. The current
SOC for eschar removal in severe burns is surgery, where eschar removal can be performed by tangential excision, dermabrasion or hydro
jet, or non‑surgical alternatives, such as applying topical medications to the eschar to facilitate the natural healing process.
Consequently, we face competition from traditional surgical procedures and topical agents. However, based on our clinical trials, we believe
that NexoBrid has a sustainable competitive advantage over the current non‑surgical alternatives and is less invasive than surgery
in removing eschar in patients with burn wounds. See “-NexoBrid and Our Clinical History” for the results of our clinical
trials. Although we are in the clinical and preclinical phases for our pipeline product candidates for debridement of chronic and other
hard‑to‑heal wounds and treatment of low-risk basal cell carcinoma and connective tissue disorders and other indications,
respectively, if one of our pipeline product candidates receives approval in the future, we would compete with traditional surgery and
existing non‑surgical and other treatments. In chronic and other hard‑to‑heal wounds, we expect to face competition
from current standard of care for debridement by sharp debridement or from the current non-surgical standard of care, either enzymatic
debridement, primarily Smith & Nephew Plc’s SANTYL, a collagenase-based ointment, approved by the FDA for debriding chronic dermal ulcers.
SANTYL is currently the market-leading enzymatic debridement product, with more than $360 million in estimated annual sales in the United
States.
In addition to the currently available products, other products
may be introduced to debride chronic and other hard‑to‑heal wounds or treat superficial and nodular basal cell carcinoma and
connective tissue disorders during the time that we engage in necessary development. Accordingly, if one of our pipeline product candidates
is approved, our main challenge in the market would be to educate physicians seeking alternatives to surgery or less effective non-surgical
methods to use our product instead of already existing treatments. While we are still in the development stages, based on our studies,
we believe that our pipeline product candidates will be more effective than the current non‑surgical alternatives and less invasive
than surgery in removing non-viable material in chronic and other hard‑to‑heal wounds or in tumor resection and may be comparable
or perhaps better than currently available treatments for connective tissue disorders.
NexoBrid received orphan drug status in the United States on August
20, 2003 for debridement of deep partial‑ and full‑thickness burns in hospitalized patients. In the United States, a sponsor
that develops an orphan drug has marketing exclusivity for seven years post‑approval by the FDA. The exclusive marketing rights
in both regions are subject to certain exceptions, including the development of a clinically significant benefit over the prevalent SOC.
Once the market exclusivity for our orphan indication expires in a given jurisdiction, subject to other protections such as patents, we
could face competition from other companies that may attempt to develop other products for the same indication.
Government Legislation and Regulation
Our business is subject to extensive government regulation. Regulation
by governmental authorities in the United States, the European Union and other jurisdictions is a significant factor in the development,
manufacture and marketing of NexoBrid and in ongoing research and development activities.
European Union
The approval process of medicinal products in the European Union
generally involves satisfactorily completing each of the following:
|
|
•
|
laboratory tests, animal studies and formulation studies all performed in accordance
with the applicable EU GLP or GMP regulations;
|
|
|
•
|
submission to the relevant authorities of a CTA, which must be approved before human
clinical trials may begin;
|
|
|
•
|
performance of adequate and well‑controlled clinical trials to establish the
safety and efficacy of the product for each proposed indication;
|
|
|
•
|
submission to the relevant competent authorities of a marketing authorization application
(“MAA”), which includes the data supporting preclinical and clinical safety and efficacy as well as detailed information on
the manufacture and composition and control of the product development and proposed labeling as well as other information;
|
|
|
•
|
inspection by the relevant national authorities of the manufacturing facility or facilities
and quality systems (including those of third parties) at which the product is produced, to assess compliance with strictly enforced GMP;
|
|
|
•
|
potential audits of the non‑clinical and clinical trial sites that generated
the data in support of the MAA; and
|
|
|
•
|
review and approval by the relevant competent authority of the MAA before any commercial
marketing, sale or shipment of the product.
|
Quality/preclinical studies
In order to assess the potential safety and efficacy of a product,
tests include laboratory evaluations of product characterization, analytical tests and controls, as well as studies to evaluate toxicity
and pharmacological effects in animal studies. The conduct of the preclinical tests and formulation of the compounds for testing must
comply with the relevant E.U. regulations and requirements. The results of such tests, together with relevant manufacturing control information
and analytical data, are submitted as part of the CTA. Non-clinical studies are performed to demonstrate the health or environmental safety
of new biological substances. Non-clinical studies must be conducted in compliance with the principles of GLP as set forth in EU Directive
2004/10/EC. In particular, non-clinical studies, both in vitro and in vivo, must be planned, performed, monitored, recorded, reported
and archived in accordance with the GLP principles, which define a set of rules and criteria for a quality system for the organizational
process and the conditions for non-clinical studies. These GLP standards reflect the Organization for Economic Co-operation and Development
requirements.
Clinical trial approval
Clinical drug development is often described as consisting of four
temporal phases (Phase I‑IV). See, for example, the EMA’s note for guidance on general considerations for clinical trials
(CPMP/ICH/291/95).
|
|
•
|
Phase I (Most typical kind of study: Human Pharmacology);
|
|
|
•
|
Phase II (Most typical kind of study: Therapeutic Exploratory);
|
|
|
•
|
Phase III (Most typical kind of study: Therapeutic Confirmatory); and
|
|
|
•
|
Phase IV (Variety of Studies: Therapeutic Use).
|
Studies in Phase IV are all studies other than routine surveillance
performed after drug approval and are related to the approved indication.
The phase of development provides an inadequate basis for classification
of clinical trials because one type of trial may occur in several phases. The phase concept is a description, not a set of requirements.
The temporal phases do not imply a fixed order of studies since for some drugs in a development plan the typical sequence will not be
appropriate or necessary.
Clinical trials of medicinal products in the EU must be conducted
in accordance with EU and national regulations and the International Conference on Harmonization (“ICH”) guidelines on GCP
as well as the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. If
the sponsor of the clinical trial is not established within the EU, it must appoint an EU entity to act as its legal representative. The
sponsor must take out a clinical trial insurance policy, and in most EU member states, the sponsor is liable to provide ‘no fault’
compensation to any study subject injured in the clinical trial. The regulatory landscape related to clinical trials in the EU has been
subject to recent changes. The EU Clinical Trials Regulation (“CTR”) which was adopted in April 2014 and repeals the EU Clinical
Trials Directive, became applicable on January 31, 2022. Unlike directives, the CTR is directly applicable in all EU member states without
the need for member states to further implement it into national law. The CTR notably harmonizes the assessment and supervision processes
for clinical trials throughout the EU via a Clinical Trials Information System, which contains a centralized EU portal and database.
While the Clinical Trials Directive required a separate CTA to
be submitted in each member state, to both the competent national health authority and an independent ethics committee, much like the
FDA and IRB respectively, the CTR introduces a centralized process and only requires the submission of a single application to all member
states concerned. The CTR allows sponsors to make a single submission to both the competent authority and an ethics committee in each
member state, leading to a single decision per member state. The CTA must include, among other things, a copy of the trial protocol and
an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation.
The assessment procedure of the CTA has been harmonized as well, including a joint assessment by all member states concerned, and a separate
assessment by each member state with respect to specific requirements related to its own territory, including ethics rules. Each member
state’s decision is communicated to the sponsor via the centralized EU portal. Once the CTA is approved, clinical study development
may proceed.
The CTR foresees a three-year transition period. The extent to
which ongoing and new clinical trials will be governed by the CTR varies. Clinical trials for which an application was submitted (i) prior
to January 31, 2022 under the Clinical Trials Directive, or (ii) between January 31, 2022 and January 31, 2023 and for which the sponsor
has opted for the application of the Clinical Trials Directive remain governed by said Directive until January 31, 2025. After this date,
all clinical trials (including those which are ongoing) will become subject to the provisions of the CTR.
Medicines used in clinical trials must be manufactured in accordance
with GMP. Other national and EU-wide regulatory requirements may also apply.
Pediatric investigation plan (“PIP”)
We initiated a PIP study in November 2014.
On January 26, 2007, Regulation (EC) 1901/2006 came into force
with its primary purpose being the improvement of the health of children without subjecting children to unnecessary trials, or delaying
the authorization of medicinal products for use in adults. The regulation established the Pediatric Committee (“PDCO”), which
is responsible for coordinating the EMA’s activities regarding medicinal products for children. The PDCO’s main role is to
determine which studies the applicant needs to perform in the pediatric population as part of the PIP.
All applications for marketing authorization for new medicinal
products that were not authorized in the EU prior to January 26, 2007 must include the results of studies carried out in children of different
ages. The PDCO determines the requirements and procedures of such studies, describing them in a PIP. This requirement also applies when
a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The
PDCO can grant deferrals for some medicines, allowing a company to delay development of the medicine in children until there is enough
information to demonstrate its effectiveness and safety in adults. The PDCO can also grant waivers when development of a medicine in children
is not needed or is not appropriate, because the product is likely to be ineffective or unsafe in children, the disease or condition for
which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit
over existing treatments for pediatric patients. Before an MAA can be filed, or an existing marketing authorization can be amended, the
EMA confirms that the applicant complied with the studies’ requirements and measures listed in the PIP. Since the regulation became
effective, several incentives for the development of medicines for children become available in the European Union, including:
|
|
•
|
medicines that have been authorized for marketing in the EU with the results of PIP
studies included in the product information are eligible for an extension of their supplementary protection certificate extension (if
any is in effect at the time of approval) by six months. This is the case even when the studies’ results are negative;
|
|
|
•
|
for orphan medicines, such as NexoBrid, the incentive is an additional two years of
market exclusivity instead of one;
|
|
|
•
|
scientific advice and protocol assistance at the EMA are free of charge for questions
relating to the development of medicines for children; and
|
|
|
•
|
medicines developed specifically for children that are already authorized, but are
not protected by a patent or supplementary protection certificate, can apply for a pediatric use marketing authorization (“PUMA”).
If a PUMA is granted, the product will benefit from 10 years of market protection as an incentive.
|
In December 2023 we received European Commission approval
for the removal of eschar in deep partial- and full-thickness thermal burns for all ages.
Marketing authorization
Authorization to market a product in the EU member states proceeds
under one of four procedures: a centralized authorization procedure, a mutual recognition procedure, a decentralized procedure or a national
procedure. Marketing authorization may be granted only to an applicant established in the European Union. Through our wholly‑owned
German subsidiary, we received approval for NexoBrid pursuant to the centralized authorization procedure.
The centralized procedure provides for the grant of a single marketing
authorization, issued by the European Commission based on the opinion of the EMA’s Committee for Medicinal Products for Human Use
(“CHMP”), that is valid throughout the EU and the EEA countries, and including Norway, Iceland and Lichtenstein. The centralized
procedure is compulsory for medicines produced by certain biotechnological processes, products designated as orphan medicinal products,
advanced therapy medicinal products (“ATMPs”) and products with a new active substance indicated for the treatment of certain
diseases, and is optional for products which constitute a significant therapeutic, scientific, or technical innovation or for which a
centralized process is in the interest of patients. Products that have received orphan designation in the EU, such as NexoBrid, will qualify
for this centralized procedure, under which each product’s MAA is submitted to the EMA. Under the centralized procedure in the European
Union, the maximum time frame for the evaluation of an MAA by the EMA is 210 days (excluding clock stops, when additional written or oral
information is to be provided by the applicant in response to questions asked by the Committee of Medicinal Products for Human Use). In
general, if the centralized procedure is not followed, there are three alternative procedures where applications are filed with one or
more members state medicines regulators, each of which will grant a national marketing authorization:
|
|
•
|
Mutual recognition procedure. If an authorization
has been granted by one-member state, or the Reference Member State, an application may be made for mutual recognition in one or more
other member states, or the Concerned Member State(s).
|
|
|
•
|
Decentralized procedure. The decentralized
procedure may be used to obtain a marketing authorization in several European member states when the applicant does not yet have a marketing
authorization in any country.
|
|
|
•
|
National procedure. Applicants following the
national procedure will be granted a marketing authorization that is valid only in a single member state. Furthermore, this marketing
authorization is not based on recognition of another marketing authorization for the same product awarded by an assessment authority of
another member state. If marketing authorization in only one-member state is preferred, an application can be filed with the national
competent authority of a member state. The national procedure can also serve as the first phase of a mutual recognition procedure.
|
It is not always possible for applicants to follow the national
procedure. In the case of medicinal products in the category for which the centralized authorization procedure is compulsory, that procedure
must be followed. In addition, the national procedure is not available in the case of medicinal product dossiers where the same applicant
has already obtained marketing authorization in one of the other European Union member state or has already submitted an application for
marketing authorization in another member state and the application is under consideration. In the latter case, applicants must follow
a mutual recognition procedure.
After a drug has been authorized and launched, it is a condition
of maintaining the marketing authorization that all aspects relating to its quality, safety and efficacy must be kept under review. Sanctions
may be imposed for failure to adhere to the conditions of the marketing authorization. In extreme cases, the authorization may be revoked,
resulting in withdrawal of the product from sale.
Period of authorization and renewals
Under the above-described procedures, in order to grant the marketing
authorization, the EMA or the competent authorities of the EU member states make an assessment of the risk benefit balance of the product
on the basis of scientific criteria concerning its quality, safety and efficacy. Marketing authorization is valid for an initial five‑year
period and may be renewed thereafter on the basis of a re‑evaluation of the risk‑benefit balance by the EMA or by the competent
authority of the authorizing member state. To this end, the marketing authorization holder shall provide the EMA or other applicable competent
authority a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the
marketing authorization was granted, at least six months before the end of the initial five‑year period. Once renewed, the marketing
authorization is valid for an unlimited period, unless the EMA or other applicable competent authority decides, on justified grounds relating
to pharmacovigilance, to proceed with one additional five‑year renewal. Any authorization which is not followed by the actual placing
of the drug on the E.U. market (in case of centralized procedure) or on the market of the authorizing member state within three years
after authorization shall cease to be valid.
Regulatory data protection
Without prejudice to the law on the protection of industrial and
commercial property, some marketing authorizations benefit from an “8+2(+1)” year period of regulatory protection. During
the first eight years from the grant of the innovator company’s marketing authorization, data exclusivity applies. If granted, the
data exclusivity period prevents generic or biosimilar applicants from relying on the pre-clinical and clinical trial data contained in
the dossier of the reference product when applying for a generic or biosimilar MA in the EU during a period of eight years from the date
on which the reference product was first authorized in the EU. After the eight years have expired, a generic company can make use of the
preclinical and clinical trial data of the originator in their regulatory applications but still cannot market their product until the
end of the 10 year market exclusivity period. An additional one year of market exclusivity can be obtained if, during the first eight
years of those 10 years, the marketing authorization holder obtains an approval for one or more new therapeutic indications which, during
the scientific evaluation prior to their approval, are determined to bring a significant clinical benefit in comparison with existing
therapies. However, there is no guarantee that a product will be considered by the EU’s regulatory authorities to be a new chemical
or biological entity, and products may not qualify for data exclusivity. Under the current rules, a third party may reference the preclinical
and clinical data of the reference product beginning eight years after first approval, but the third party may market a generic version
only after 10 (or 11) years have lapsed.
There is a special regime for biosimilars, or biological medicinal products that are
similar to a reference medicinal product but that do not meet the definition of a generic medicinal product, for example, because of differences
in raw materials or manufacturing processes. For such products, the results of appropriate preclinical or clinical trials must be provided,
and guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product.
There are no such guidelines for complex biological products, such as gene or cell therapy medicinal products, and so it is unlikely that
biosimilars of those products will currently be approved in the EU. However, guidance from the EMA states that they will be considered
in the future in light of the scientific knowledge and regulatory experience gained at the time.
Additional data protection can be applied for when an applicant
has complied with all requirements as set forth in an approved PIP.
Post-Approval Requirements
Similar to the United States, both marketing authorization holders
and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the European Commission and/or the
competent regulatory authorities of the member states. The holder of a marketing authorization must establish and maintain a pharmacovigilance
system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations
include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports (“PSURs”).
All new MAA must include a risk management plan (“RMP”)
describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated
with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such
risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs,
or the conduct of additional clinical trials or post-authorization safety studies.
Failure to comply with the aforementioned EU and member state laws
may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to authorize the conduct of
clinical trials, or to grant marketing authorization, product withdrawals and recalls, product seizures, suspension, withdrawal or variation
of the marketing authorization, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions,
injunctions, suspension of licenses, fines and criminal penalties. These penalties could include delays or refusal to authorize the conduct
of clinical trials, or to grant the marketing authorization, product withdrawals and recalls, product seizures, suspension, withdrawal
or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing or clinical trials,
operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
The aforementioned EU rules are generally applicable in the EEA.
Brexit
The UK left the EU on January 31, 2020, following which existing
EU medicinal product legislation continued to apply in the United Kingdom during the transition period under the terms of the EU-UK Withdrawal
Agreement. The transition period, which ended on December 31, 2020, maintained access to the EU single market and to the global trade
deals negotiated by the EU on behalf of its members. The transition period provided time for the UK and EU to negotiate a framework for
partnership for the future, which was then crystallized in the Trade and Cooperation Agreement (“TCA”) and became effective
on January 1, 2021. The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP inspections
of manufacturing facilities for medicinal products and GMP documents issued but does not foresee wholesale mutual recognition of UK and
EU pharmaceutical regulations.
EU laws which have been transposed into UK law through secondary
legislation continue to be applicable as “retained EU law”. However, under the Retained EU Law (Revocation and Reform) Bill
2022, which is currently before the UK parliament, any retained EU law not expressly preserved and “assimilated” into domestic
law or extended by ministerial regulations (to no later than 23 June 2026) will automatically expire and be revoked by December 31, 2023.
In addition, new legislation such as the EU CTR or in relation to orphan medicines will not be applicable. The UK government has passed
a new Medicines and Medical Devices Act 2021, which introduces delegated powers in favor of the Secretary of State or an ‘appropriate
authority’ to amend or supplement existing regulations in the area of medicinal products and medical devices. This allows new rules
to be introduced in the future by way of secondary legislation, which aims to allow flexibility in addressing regulatory gaps and future
changes in the fields of human medicines, clinical trials and medical devices. As of January 1, 2021, the Medicines and Healthcare Products
Regulatory Agency (“MHRA”) is the UK’s standalone medicines and medical devices regulator. As a result of the Northern
Ireland protocol, different rules will apply in Northern Ireland than in England, Wales, and Scotland, together, Great Britain (“GB”);
broadly, Northern Ireland will continue to follow the EU regulatory regime, but its national competent authority will remain the MHRA.
The MHRA has published a guidance on how various aspects of the UK regulatory regime for medicines will operate in GB and in Northern
Ireland following the expiry of the Brexit transition period on December 31, 2020. The guidance includes clinical trials, importing, exporting,
and pharmacovigilance and is relevant to any business involved in the research, development, or commercialization of medicines in the
UK. The new guidance was given effect via the Human Medicines Regulations (Amendment etc.) (EU Exit) Regulations 2019 (the “Exit
Regulations”).
The MHRA has introduced changes to national licensing procedures,
including procedures to prioritize access to new medicines that will benefit patients, including a 150-day assessment and a rolling review
procedure. All existing EU MAs for centrally authorized products were automatically converted or grandfathered into UK MAs, effective
in GB (only), free of charge on January 1, 2021, unless the MA holder chose to opt-out. After Brexit, companies established in the UK
cannot use the centralized procedure and instead must follow one of the UK national authorization procedures or one of the remaining post-Brexit
international cooperation procedures to obtain an MA to commercialize products in the UK. For a period of three years from January 1,
2021, the MHRA may rely on a decision taken by the an Commission on the approval of a new (centralized procedure) MA when determining
an application for a GB authorization; or use the MHRA’s decentralized or mutual recognition procedures which enable MAs approved
in EU member states (or Iceland, Liechtenstein, Norway) to be granted in GB.
There will be no pre-MA orphan designation. Instead, the MHRA will
review applications for orphan designation in parallel to the corresponding MA application. The criteria are essentially the same, but
have been tailored for the market, i.e., the prevalence of the condition in GB, rather than the EU, must not be more than five in 10,000.
Should an orphan designation be granted, the period or market exclusivity will be set from the date of first approval of the product in
GB.
Data Privacy and Security Laws
Numerous state, federal and foreign laws, regulations, and standards
govern the collection, use, access to, confidentiality and security of health-related and other personal information, and could apply
now or in the future to our operations or the operations of our partners. In the United States, numerous federal and state laws and regulations,
including data breach notification laws, health information privacy and security laws and consumer protection laws and regulations govern
the collection, use, disclosure, and protection of health-related and other personal information. In addition, certain laws govern the
privacy and security of personal data, including health-related data in the EU/EEA and in other foreign jurisdictions. Privacy and security
laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can
result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
Manufacturing
The manufacturing of authorized drugs, for which a separate manufacturer’s
license is mandatory, must be conducted in strict compliance with the EMA’s cGMP requirements and comparable requirements of other
regulatory bodies, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure
their safety and proper identification. The EMA monitors compliance with its GMP requirements through mandatory registration of facilities
and inspections of those facilities. The EMA may have a coordinating role for these inspections while the responsibility for carrying
them out rests with the competent authority of the member state under whose responsibility the manufacturer falls. Failure to comply with
these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and could subject the applicant
to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product,
injunctive action or possible civil and criminal penalties. In January 2013, the EU and Israel signed the Protocol on Conformity Assessment
and Acceptance of Industrial Products (the “ACAA”), which covers medicinal products. The ACAA provides for mutual recognition
of the conclusions of inspections of compliance of manufacturers and importers with the principles and guidelines of EU GMP and equivalent
Israeli cGMP. Certification of the conformity of each batch to its specifications by either the importer or the manufacturer established
in Israel or in the EU shall be recognized by the other party without re‑control at import from one party to the other.
Marketing and promotion
The marketing and promotion of authorized medicinal products, including
industry‑sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public,
are strictly regulated in the European Union, notably under Directive 2001/83 and subject to laws concerning promotion of medicinal products,
interactions with physicians, misleading and comparative advertising and unfair commercial practices. The applicable legislation aims
to ensure that information provided by holders of marketing authorizations regarding their products is truthful, balanced and accurately
reflects the safety and efficacy claims authorized by the EMA or by the applicable national authority of the authorizing member state.
All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and
therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the EU.
Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are
governed by regulations in each member state and can differ from one country to another. Failure to comply with these requirements can
result in adverse publicity, warning letters, mandated corrective advertising and potential civil and criminal penalties.
United States
Review and approval of biologics
In addition to E.U. regulations, NexoBrid is marketed as a biologic
product in the United States for eschar removal in adults with deep partial thickness and/or full thickness thermal burns and is therefore
subject to various U.S. regulations. In the United States, the FDA regulates biologics under the Federal, Food, Drug and Cosmetic Act
(“FDCA”), the Public Health Service Act, and their respective implementation regulations. Biologics require the submission
of a BLA and licensure by the FDA prior to being marketed in the United States. The process of obtaining regulatory approvals and the
subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial
time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process,
approval process or after approval may subject an applicant to a variety of administrative or judicial sanctions as well as enforcement
actions brought by the FDA, the U.S. Department of Justice or other governmental entities. Possible sanctions may include the FDA’s
refusal to approve pending BLAs or supplements, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters,
product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government
contracts, restitution, disgorgement and civil or criminal penalties.
The process required by the FDA prior to marketing and distributing
a biologic in the United States generally involves the following:
|
|
•
|
completion of laboratory tests, animal studies and formulation studies in compliance
with the FDA’s GLP and GMP regulations, as applicable;
|
|
|
•
|
submission to the FDA of an investigational new drug application (“IND”),
which must become effective before clinical trials may begin;
|
|
|
•
|
approval by an independent institutional review board (“IRB”) at each
clinical site before each trial may be initiated;
|
|
|
•
|
performance of adequate and well‑controlled clinical trials in accordance with
GCP to establish the safety and efficacy of the product for each indication;
|
|
|
•
|
preparation and submission to the FDA of a BLA;
|
|
|
•
|
satisfactory completion of an FDA advisory committee review, if applicable;
|
|
|
•
|
satisfactory completion of one or more FDA inspections of the manufacturing facility
or facilities at which the product, or components thereof, are produced to assess compliance with cGMP requirements, and to assure that
the facilities, methods and controls are adequate to preserve the product’s safety, purity and potency, and of selected clinical
investigation sites to assess compliance with GCP; and
|
|
|
•
|
payment of user fees and FDA review and approval of the BLA to permit commercial marketing
of the product for particular indications for use in the United States.
|
Preclinical studies
Preclinical studies include laboratory evaluation of product chemistry,
toxicity and formulation, as well as animal studies to assess the potential safety and efficacy of the product candidate. Preclinical
safety tests must be conducted in compliance with FDA regulations regarding good laboratory practices. The results of the preclinical
tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND which must become effective
before clinical trials may commence. Some preclinical testing may continue even after the IND is submitted.
Clinical trials in support of a BLA
Clinical trials involve the administration of an investigational
product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include, among other
things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical
trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the study, the parameters
to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent
protocol amendments must be submitted to the FDA as part of the IND. An IND automatically becomes effective 30 days after receipt by the
FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical
hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin.
In addition, an IRB representing each institution participating
in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must
conduct continuing review and reapprove the study at least annually. The IRB must review and approve, among other things, the study protocol
and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. Information
about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination
on their website, ClinicalTrials.gov.
For purposes of BLA approval, clinical trials are typically conducted
in three sequential phases, which may overlap or be combined. In the United States, the three phases are generally described as follows:
|
|
Phase I:
|
The investigational product is initially
introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption,
metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness and to determine optimal dosage.
|
|
|
Phase II:
|
The investigational product is administered to a limited patient population to identify
possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to
determine dosage tolerance and optimal dosage.
|
|
|
Phase III:
|
The investigational product is administered to an expanded patient population, generally
at geographically dispersed clinical trial sites, in well‑controlled clinical trials to generate enough data to statistically evaluate
the efficacy and safety of the product for approval, to establish the overall risk‑benefit profile of the product, and to provide
adequate information for the labeling of the product. |
In some cases, the FDA may require, or companies may voluntarily
pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase IV studies
may be made a condition to approval of the BLA.
Progress reports detailing the results of the clinical trials must
be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase I, Phase II and Phase III clinical
trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate
a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health
risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted
in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Submission of a BLA to the FDA
The results of the preclinical studies and clinical trials, together
with other detailed information, including information on the manufacture, control and composition of the product, are submitted to the
FDA as part of a BLA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee
Act (PDUFA), as amended, applicants are required to pay user fees to the FDA for reviewing a BLA. These user fees, as well as the annual
program fees required for approved products, can be substantial. Each BLA submitted to the FDA for approval is typically reviewed for
administrative completeness and reviewability within 60 days following submission of the application. If found complete, the FDA will
“file” the BLA, which triggers a full review of the application. The FDA may refuse to file any BLA that it deems incomplete
or not properly reviewable at the time of submission. The FDA’s established goals are to review and act on standard applications
within ten months after it accepts the application for filing, or, if the application qualifies for priority review, six months after
the FDA accepts the application for filing. In both standard and priority reviews, the review process is often significantly extended
by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product
is safe, pure and potent and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the
product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application
review questions.
Before approving a BLA, the FDA generally inspects the facilities
at which the product is manufactured or facilities that are significantly involved in the product development and distribution process,
and will not approve the product unless cGMP compliance is satisfactory. Additionally, before approving a BLA, the FDA will typically
inspect one or more clinical sites to assure compliance with GCP. If the FDA determines that the application, manufacturing process or
manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing
or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application
does not satisfy the regulatory criteria for approval. After the FDA evaluates a BLA and conducts inspections of manufacturing facilities
where the investigational product will be produced, the FDA may issue an approval letter or a Complete Response letter. An approval letter
authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response letter
will describe all of the deficiencies that the FDA has identified in the BLA, except that where the FDA determines that the data supporting
the application are inadequate to support approval, the FDA may issue the Complete Response letter without first conducting required inspections,
testing submitted product lots, and/or reviewing proposed labeling. In issuing the Complete Response letter, the FDA may recommend actions
that the applicant might take to place the BLA in condition for approval, including requests for additional information or clarification.
The FDA may deny approval of a BLA if applicable statutory or regulatory
criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any
application may include many delays or may never be granted. If a product is approved, the approval will impose limitations on the indicated
uses for which the product may be marketed, will require that warning statements be included in the product labeling, may impose additional
warnings to be specifically highlighted in the labeling (e.g., a Black Box Warning), which can significantly affect promotion and sales
of the product, may require that additional studies be conducted following approval as a condition of the approval and may impose restrictions
and conditions on product distribution, prescribing or dispensing. For example, the FDA may approve the BLA with a Risk Evaluation and
Mitigation Strategy, or REMS, to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known
or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their
safe use. A REMS program may be required to include various elements, such as a medication guide or patient package insert, a communication
plan to educate healthcare providers of the drug’s risks, or other elements to assure safe use, such as limitations on who may prescribe
or dispense the drug, dispensing only under certain circumstances, special monitoring and the use of patient registries. Once a product
is approved, marketing the product for other indicated uses or making certain manufacturing or other changes requires FDA review and approval
of a supplemental BLA or a new BLA, which may require additional clinical data. In addition, further post‑marketing testing and
surveillance to monitor the safety or efficacy of a product may be required. Also, product approvals may be withdrawn if compliance with
regulatory standards is not maintained or if safety or manufacturing problems occur following initial marketing. In addition, new government
requirements may be established that could delay or prevent regulatory approval of our product candidates under development.
Post‑approval requirements
Any biologic products for which we receive FDA approvals are subject
to pervasive continuing regulation by the FDA. Certain requirements include, among other things, record‑keeping requirements, reporting
adverse experiences with the product, providing the FDA with updated safety and efficacy information annually or more frequently for specific
events, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying
with FDA promotion and advertising requirements. These promotion and advertising requirements include, among others, standards for direct‑to‑consumer
advertising, prohibitions against promoting drugs for uses or in patient populations that are not described in the drug’s approved
labeling, known as “off‑label use,” and other promotional activities, such as those considered to be false or misleading.
Failure to comply with FDA requirements can have negative consequences, including the immediate discontinuation of noncomplying materials,
adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal
penalties. Such enforcement may also lead to scrutiny and enforcement by other government and regulatory bodies. Although physicians may
prescribe legally available drugs for off‑label uses, manufacturers may not encourage, market or promote such off‑label uses.
As a result, “off‑label promotion” has formed the basis for litigation under the Federal False Claims Act, violations
of which are subject to significant civil fines and penalties.
The manufacturing of NexoBrid, EscharEx and our pipeline product
candidates are and will be required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations.
NexoBrid is manufactured at our production plant in Yavne, Israel, which is cGMP certified. The FDA’s cGMP regulations require,
among other things, quality control and quality assurance, as well as the corresponding maintenance of comprehensive records and documentation.
Biologic manufacturers and other entities involved in the manufacture and distribution of approved drugs and biologics are also required
to register their establishments and list any products they make with the FDA and to comply with related requirements in certain states.
These entities are further subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP
and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control
to maintain cGMP compliance. In addition, a BLA holder must comply with post‑marketing requirements, such as reporting of certain
adverse events. Such reports can present liability exposure, as well as increase regulatory scrutiny that could lead to additional inspections,
labeling restrictions or other corrective action to minimize further patient risk. Discovery of problems with a product after approval
may result in serious and extensive restrictions on the product, manufacturer or holder of an approved BLA, as well as lead to potential
market disruptions. These restrictions may include recalls, fines, warning letters, or untitled letters, clinical holds on clinical studies,
refusal of the FDA to approve pending applicants or supplements to approved applications, product seizure or detention, or refusal to
permit the import or export of products, suspension or revocation of a product license approval until the FDA is assured that quality
standards can be met, and continuing oversight of manufacturing by the FDA under a “consent decree,” which frequently includes
the imposition of costs and continuing inspections over a period of many years, as well as possible withdrawal of the product from the
market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented. Other types of
changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review
and approval. The FDA also may impose a number of post‑approval requirements as a condition of approval of a BLA. For example, the
FDA may require post‑marketing testing, or Phase IV testing, as well as REMS and/or surveillance to monitor the effects of an approved
product or place other conditions on an approval that could otherwise restrict the distribution or use of NexoBrid.
Orphan designation and exclusivity
On August 20, 2003, NexoBrid received orphan drug designation in
the United States. Under the Orphan Drug Act, the FDA may designate a drug product as an “orphan drug” if it is intended to
treat a rare disease or condition, meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which
there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment
of the disease or condition will be recovered from sales of the product. A company must request orphan product designation before submitting
a BLA. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan drug designation
does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product with orphan status receives the first FDA approval
for the disease or condition for which it has such designation, the product will be entitled to orphan product exclusivity. Orphan product
exclusivity means that FDA may not approve any other applications for the same product for the same disease or condition for seven years,
except in certain limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Competitors
may receive approval of different products for the disease or condition for which the orphan product has exclusivity and may obtain approval
for the same product but for a different indication. If a drug or drug product designated as an orphan product ultimately receives marketing
authorization for a disease or condition broader than that designated in its orphan product application, it may not be entitled to exclusivity.
In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation
was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients
with the rare disease or condition.
Expedited Development and Review Programs
The FDA offers a number of expedited development and review programs
for qualifying product candidates. The fast track program is intended to expedite or facilitate the process for reviewing new products
that meet certain criteria. Specifically, new products are eligible for fast track designation if they are intended to treat a serious
or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast
track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of
a fast track product has opportunities for frequent interactions with the review team during product development and, once a BLA is submitted,
the product may be eligible for priority review. A fast track product may also be eligible for rolling review, where the FDA may consider
for review sections of the BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for
the submission of the sections of the BLA, the FDA agrees to accept sections of the BLA and determines that the schedule is acceptable,
and the sponsor pays any required user fees upon submission of the first section of the BLA.
A product intended to treat a serious or life-threatening disease
or condition may also be eligible for breakthrough therapy designation to expedite its development and review. A product can receive breakthrough
therapy designation if preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing
therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
The designation includes all of the fast track program features, as well as more intensive FDA interaction and guidance beginning as early
as Phase I and an organizational commitment to expedite the development and review of the product, including involvement of senior managers.
Any marketing application for a biologic submitted to the FDA for approval, including a product with a fast track designation and/or breakthrough
therapy designation, may be eligible for other types of FDA programs intended to expedite the FDA review and approval process, such as
priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide a significant improvement
in the treatment, diagnosis or prevention of a serious disease or condition compared to marketed products. For products containing new
molecular entities, priority review designation means the FDA’s goal is to take action on the marketing application within six months
of the 60-day filing date, compared with ten months under standard review.
Additionally, products studied for their safety and effectiveness
in treating serious or life-threatening diseases or conditions may receive accelerated approval upon a determination that the product
has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured
earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality
or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative
treatments. As a condition of accelerated approval, the FDA will generally require the sponsor to perform adequate and well-controlled
post-marketing clinical studies to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical
benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which
could adversely impact the timing of the commercial launch of the product.
In 2017, FDA established a new regenerative medicine advanced therapy,
or RMAT, designation as part of its implementation of the 21st Century Cures Act, which was signed into law in December 2016. To qualify
for RMAT designation, the product candidate must meet the following criteria: (1) it qualifies as a RMAT, which is defined as a cell therapy,
therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with
limited exceptions; (2) it is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (3)
preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such a disease or condition.
Like fast track and breakthrough therapy designation, RMAT designation provides potential benefits that include more frequent meetings
with FDA to discuss the development plan for the product candidate and eligibility for rolling review and priority review. Products granted
RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely
to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites, including through expansion to
additional sites. Once approved, when appropriate, the FDA can permit fulfillment of post-approval requirements under accelerated approval
through the submission of clinical evidence, clinical studies, patient registries, or other sources of real world evidence such as electronic
health records; through the collection of larger confirmatory datasets; or through post-approval monitoring of all patients treated with
the therapy prior to approval.
Fast track designation, breakthrough therapy designation, priority
review, accelerated approval, and RMAT designation do not change the standards for approval but may expedite the development or approval
process.
Pediatric studies and exclusivity
Under the Pediatric Research Equity Act of 2003, a BLA or supplement
thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in
all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product
is safe and effective. Sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline
of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver
requests, and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then
review the information submitted, consult with each other and agree upon a final plan. The FDA or the applicant may request an amendment
to the plan at any time. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some
or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in the FDASIA.
Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
Separately, in the event the FDA issues a Written Request for pediatric
data relating to a product, a BLA sponsor who submits such data may be entitled to pediatric exclusivity. Pediatric exclusivity is another
type of non‑patent marketing exclusivity in the United States which, if granted, provides for the attachment of an additional six
months of marketing protection to the term of any existing exclusivity, including other non‑patent and orphan exclusivity. This
six‑month exclusivity may be granted if a BLA sponsor submits pediatric data that fairly respond to the Written Request from the
FDA for such data. The data do not need to show that the product is effective in the pediatric population studied; rather, if the clinical
trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric
studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity
or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the
regulatory period during which the FDA cannot accept or approve another application.
Patent term restoration and extension
A patent claiming a new drug product may be eligible for a limited
patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch‑Waxman Act”),
which permits a patent restoration of up to five years for the patent term lost during product development and the FDA regulatory review.
The restoration period granted is typically one‑half the time between the effective date of an IND and the submission date of a
BLA, plus the time between the submission date of a BLA and the ultimate approval date. Patent term restoration cannot be used to extend
the remaining term of a patent past a total of fourteen years from the product’s approval date. Only one patent applicable to an
approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of
the patent in question. A patent that covers multiple drugs for which approval is sought can only be extended in connection with one of
the approvals. The U.S. Patent and Trademark Office reviews and approves the application for any patent term extension or restoration
in consultation with the FDA.
Biosimilars and reference product exclusivity
The Patient Protection and Affordable Care Act, as amended by the
Health Care and Education Reconciliation Act (collectively, the “ACA”), which was signed into law in 2010, includes a subtitle
called the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), which created an abbreviated approval pathway
for biological products that are biosimilar to or interchangeable with an FDA-approved reference biological product.
Biosimilarity, which requires that there be no clinically meaningful
differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical
studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product
and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient
and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or
switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive
use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products,
as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval
pathway that are still being worked out by the FDA.
Under the BPCIA, an application for a biosimilar product may not
be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the
approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was
first licensed. During this 12‑year period of exclusivity, another company may still market a competing version of the reference
product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate
and well‑controlled clinical trials to demonstrate the safety, purity and potency of their product.
The BPCIA also created certain exclusivity periods for biosimilars
approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA
will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law. The BPCIA is complex and continues to be
interpreted and implemented by the FDA. In addition, recent government proposals have sought to reduce the 12‑year reference product
exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject
of recent litigation. As a result, the ultimate impact, implementation, and meaning of the BPCIA remains subject to significant uncertainty.
Review and Approval of Drug Products Outside
the European Union and the United States
In addition to the above regulations, we must obtain approval of
a product by the comparable regulatory authorities of foreign countries outside of the European Union and the United States before we
can commence clinical trials or marketing of NexoBrid in those countries. The approval process varies from country to country and the
time may be longer or shorter than that required for FDA or EU approval. In addition, the requirements governing the conduct of clinical
trials, product licensing, pricing and reimbursement vary greatly from country to country. In all cases, clinical trials are conducted
in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration
of Helsinki.
If we fail to comply with applicable regulatory requirements, we
may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products,
operating restrictions and criminal prosecution.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement
status of any products for which we obtain regulatory approval. In the United States, EU and other markets, sales of any products for
which we receive regulatory approval for commercial sale will depend to a large extent on the availability of reimbursement from third‑party
payors. Third‑party payors include governments, government health administrative authorities, managed care providers, private health
insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate
from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third‑party payors may
limit coverage to specific drug products on an approved list, or formulary, which might not include all of the drug products approved
for a particular indication by the FDA, European Commission or National Ministries of Health. Third‑party payors are increasingly
challenging the price and examining the medical necessity and cost‑effectiveness of medical products and services, in addition to
their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and
cost‑effectiveness of NexoBrid, in addition to the costs required to obtain the FDA or other Ministry of Health approvals. Additionally,
NexoBrid may not be considered medically necessary or cost‑effective. A payor’s decision to provide coverage for a drug product
does not guarantee that an adequate reimbursement rate will be approved. Adequate third‑party reimbursement may not be available
to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In the United States, the ACA substantially changed the way healthcare
is financed by both governmental and private insurers and significantly impacted the pharmaceutical industry. The ACA contains a number
of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse provisions,
which will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment
for performance initiatives and improvements to the physician quality reporting system and feedback program. Additionally, the ACA:
|
|
•
|
increases the minimum level of Medicaid rebates payable by manufacturers of brand‑name
drugs from 15.1% to 23.1%;
|
|
|
•
|
requires collection of rebates for drugs paid by Medicaid managed care organizations;
and
|
|
|
•
|
imposes a non‑deductible annual fee on pharmaceutical manufacturers or importers
who sell certain “branded prescription drugs” to specified federal government programs.
|
Since its enactment, there have been judicial, executive and congressional
challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the
ACA brought by several states without specifically ruling on the constitutionality of the ACA. Thus, the ACA will remain in effect in
its current form. Further, prior to the U.S. Supreme Court ruling, President Biden issued an executive order that initiated a special
enrollment period for purposes of obtaining health insurance coverage through the ACA marketplace from February 15, 2021 through August
15, 2021. The executive order instructed certain governmental agencies to review and reconsider their existing policies and rules that
limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work
requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA.
There has been heightened governmental scrutiny recently over the
manner in which drug manufacturers set prices for their marketed products, which have resulted in several Congressional inquiries and
proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and
manufacturer patient programs, and reform government program reimbursement methodologies for drug products. . In March 2021, the American
Rescue Plan Act of 2021 was signed into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s
average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. In August 2022, the Inflation
Reduction Act of 2022, or IRA, was signed into law. Among other things, the IRA requires manufacturers of certain drugs to engage in price
negotiations with Medicare (beginning in 2026), with prices that can be negotiated subject to a cap; imposes rebates under Medicare Part
B and Medicare Part D to penalize price increases that outpace inflation (first due in 2023); and replaces the Part D coverage gap discount
program with a new discounting program (beginning in 2025). The IRA permits the Secretary of the Department of Health and Human Services
to implement many of these provisions through guidance, as opposed to regulation, for the initial years. For that and other reasons, it
is currently unclear how the IRA will be effectuated, or the impact of the IRA on our business. We expect that additional U.S. federal
healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. federal government will pay
for healthcare products and services, which could result in reduced demand for our products or additional pricing pressures. At the state
level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological
product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing
cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
In the EU, pricing and reimbursement schemes vary widely from country to country and often within regions or provinces of countries. Some
countries may limit the annual budget of coverage or request that the company participate in the cost above certain use levels or for
treatments perceived as unsuccessful and impose monitoring processes on the use of the product. Some countries and hospitals may require
inclusion into the hospital formulary for payment from the hospital budget. Some countries and hospitals may require the completion of
additional studies that compare the cost‑effectiveness of a particular drug candidate to currently available therapies. For example,
the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems
provide reimbursement and to control the prices of medicinal products for human use. This Health Technology Assessment (“HTA process”)
which is currently governed by the national laws of the individual EU member states, is the procedure according to which the assessment
of the public health impact, therapeutic impact and the economic and societal impact of use of a given medicinal product in the national
healthcare systems of the individual country is conducted. The outcome of HTA regarding specific medicinal products will often influence
the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States.
On December 15, 2021, the Health Technology Regulation (“HTA Regulation”) was adopted. The HTA Regulation is intended to boost
cooperation among EU member states in assessing health technologies, including new medicinal products, and providing the basis for cooperation
at EU level for joint clinical assessments in these areas. When it enters into application in 2025, the HTA Regulation will be intended
to harmonize the clinical benefit assessment of HTA across the EU.
Further, EU member states may approve a specific price for a drug
product or may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the
market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward
pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers
are being erected to the entry of new products. In addition, in some countries, cross‑border imports from low‑priced markets
exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations
for drug products may not allow favorable reimbursement and pricing arrangements. Historically, products launched in the EU do not follow
price structures of the United States and generally prices tend to be significantly lower.
Healthcare Law and Regulation
Healthcare providers, physicians and third‑party payors play
a primary role in the recommendation and prescription of drug products that are granted marketing authorization. Arrangements with healthcare
providers, third‑party payors and other customers are subject to broadly applicable fraud and abuse and other healthcare laws and
regulations. Such restrictions under applicable federal, state and foreign healthcare laws and regulations, include the following:
|
|
•
|
the federal healthcare Anti‑Kickback Statute prohibits, among other things,
persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in
kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service for
which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. A person or entity does
not need to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation;
|
|
|
•
|
the federal False Claims Act imposes civil penalties, and provides for civil whistleblower
or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims
for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal
government. In addition, the government may assert that a claim including items or services resulting from a violation of the federal
Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act;
|
|
|
•
|
HIPAA, imposes criminal and civil liability for executing a scheme to defraud any
healthcare benefit program or making false statements relating to healthcare matters;
|
|
|
•
|
the federal false statements statute prohibits knowingly and willfully falsifying,
concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare
benefits, items or services;
|
|
|
•
|
the federal physician payment transparency requirements under the Affordable Care
Act require certain manufacturers of drugs, devices and medical supplies to report to Centers for Medicare & Medicaid Services information
related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors),
certain other non-physician practitioners such as physician assistants and nurse practitioners, and teaching hospitals and physician ownership
and investment interests;
|
|
|
•
|
analogous state and foreign laws and regulations, such as state anti‑kickback
and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non‑governmental
third‑party payors, including private insurers.
|
Violations of any of these laws or any other governmental laws
and regulations that may apply include, without limitation, significant civil, criminal and administrative penalties, damages, fines,
imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, disgorgement, contractual
damages, reputational harm, diminished profits and the curtailment or restructuring of our operations.
Some state laws require pharmaceutical companies to comply with
the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government
in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or
marketing expenditures. Additionally, certain state and local laws require the registration of pharmaceutical sales representatives.
Environmental, Health and Safety Matters
We are subject to extensive environmental, health and safety laws
and regulations in a number of jurisdictions, primarily Israel, governing, among other things: the use, storage, registration, handling,
emission and disposal of chemicals, waste materials and sewage; chemicals, air, water and ground contamination; air emissions and the
cleanup of contaminated sites, including any contamination that results from spills due to our failure to properly dispose of chemicals,
waste materials and sewage. Our operations at our Yavne manufacturing facility use chemicals and produce waste materials and sewage. Our
activities require permits from various governmental authorities including, local municipal authorities, the Ministry of Environmental
Protection and the Ministry of Health. The Ministry of Environmental Protection and the Ministry of Health, local authorities and the
municipal water and sewage company conduct periodic inspections in order to review and ensure our compliance with the various regulations.
These laws, regulations and permits could potentially require the
expenditure by us of significant amounts for compliance or remediation. If we fail to comply with such laws, regulations or permits, we
may be subject to fines and other civil, administrative or criminal sanctions, including the revocation of permits and licenses necessary
to continue our business activities. In addition, we may be required to pay damages or civil judgments in respect of third‑party
claims, including those relating to personal injury (including exposure to hazardous substances we use, store, handle, transport, manufacture
or dispose of), property damage or contribution claims. Some environmental, health and safety laws allow for strict, joint and several
liability for remediation costs, regardless of comparative fault. We may be identified as a responsible party under such laws. Such developments
could have a material adverse effect on our business, financial condition and results of operations.
In addition, laws and regulations relating to environmental, health
and safety matters are often subject to change. In the event of any changes or new laws or regulations, we could be subject to new compliance
measures or to penalties for activities which were previously permitted. For instance, new Israeli regulations were promulgated in 2012
relating to the discharge of industrial sewage into the sewer system. These regulations establish new and potentially significant fines
for discharging forbidden or irregular sewage into the sewage system.
Properties
Our principal executive offices are located at 42 Hayarkon Street,
Yavne 8122745, Israel. We have leased these facilities from a third party. The lease agreement will expire on 2035, with an option for
a further three year extension until 2038. The facilities consist of approximately 32,300 square feet of space, and the yearly lease fee
is approximately $648,000. These facilities house our administrative headquarters, our research and development laboratories and our manufacturing
plant.
During 2023 we initiated the expansion of the Company’s manufacturing
facility to address the increasing global demand for NexoBrid. This expansion project is scheduled for completion by the end of 2024 and
is projected to achieve 6-fold manufacturing capacity increase in 2025.
In July 2023, we entered into a turnkey scale-up agreement with
Biopharmax Group Ltd. To bolster our manufacturing infrastructure and support our long-term growth trajectory. The objective of this agreement
is to establish, commission, and validate a cutting-edge, sterile, and GMP-compliant manufacturing facility. The venture aims to increase
our production capacity significantly, projected to expand to six times the current capacity, aligning with our strategic plan to meet
the escalating global demand for NexoBrid. The new facility, equipped with fully operational clean rooms, will be exclusively designed
for NexoBrid production. It will comply with stringent regulations from the GMP, FDA, EMA, Israeli Ministry of Health, and relevant Israeli
regulatory bodies. An estimated $12.7 million will be invested in the project, set for completion by mid-2024, with full-scale manufacturing
expected to commence in 2025.
We have furthermore leased additional office space in Yavne, Israel,
consisting of approximately 4,000 square feet, for a term of 2 years, commencing in January 1, 2024, with an option for a further three
year extension until 2028. The yearly lease fee is approximately $120,000. For the initial two years of the lease term, the majority of
our lease payments are being covered by the DOD.
C.
Organizational Structure
The legal name of our company is MediWound Ltd. and we are organized
under the laws of the State of Israel. Our corporate structure consists of MediWound Ltd., our Israeli parent company, (i) MediWound Germany
GmbH, our active wholly‑owned subsidiary, which was incorporated on April 16, 2013 under the laws of the Federal Republic of Germany
(ii) MediWound US, Inc., which was incorporated on December 8, 2020 under the laws of the State of Delaware and (iii) MediWound UK Limited,
our inactive wholly‑owned subsidiary, which was incorporated on July 26, 2004 under the laws of England.
D.
Property, Plants and Equipment
See “ITEM 4.B. Business Overview-Properties”, “ITEM
4.B. Business Overview-Manufacturing, Supply and Production” and “ITEM 4.B. Business Overview-Environmental, Health and Safety
Matters”.
Item 4A. UNRESOLVED STAFF
COMMENTS
None.
Item 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
A. Operating
Results
The information contained in this section should
be read in conjunction with our consolidated financial statements for the year ended December 31, 2023 and related notes, and the information
contained elsewhere in this annual report. Our financial statements have been prepared in accordance with IFRS, as issued by the IASB.
Company Overview
We are a biopharmaceutical company that develops, manufactures,
and commercializes novel, cost effective, bio‑therapeutic, non-surgical solutions for tissue repair and regeneration. Our strategy
leverages our breakthrough enzymatic technology platform into diversified portfolio of biotherapeutics across multiple indications to
pioneer solutions for unmet medical needs. Our current portfolio is focused on next-generation protein-based therapies for burn care,
wound care and tissue repair.
Our first innovative biopharmaceutical product, NexoBrid®,
has received marketing authorization from the FDA and marketing authorization from the European Medicines Agency (the “EMA”)
and other international markets for removal of dead or damaged tissue, known as eschar, in adults with deep partial-thickness and full-thickness
thermal burns, also referred to as severe burns. NexoBrid, a concentrate of proteolytic enzymes enriched in bromelain, represents a new
paradigm in burn care management, and our clinical trials have demonstrated, with statistical significance, its ability to non‑surgically
and rapidly remove the eschar, without harming viable tissues, earlier relative to existing standard of care.
We commercialize NexoBrid globally through multiple sales channels.
We sell NexoBrid to burn centers in the European Union, United Kingdom and Israel, primarily through our direct sales force, focusing
on key burn centers and KOLs. In the United States, we entered into exclusive license and supply agreements with Vericel Corporation (Nasdaq:
VCEL) to commercialize NexoBrid in North America. We have established local distribution channels in multiple international markets, focusing
on Asia Pacific, EMEA, CEE and LATAM, which local distributors are also responsible for obtaining local marketing authorization within
the relevant territories.
We have been awarded two contracts with the U.S. Biomedical Advanced
Research and Development Authority ("BARDA"), for the advancement of the development and manufacturing, as well as the procurement of
NexoBrid which has initiated on January 2020, as a medical countermeasure as part of BARDA preparedness for mass casualty events.
EscharEx, our next-generation enzymatic therapy under development,
is a topical biological drug candidate for the debridement of chronic and other hard-to-heal wounds. Designed for the outpatient setting,
EscharEx is an easy-to-use concentrate of proteolytic enzymes enriched in bromelain; having the same API as NexoBrid. In several Phase
II trials, EscharEx was shown to be well-tolerated and demonstrated safety and efficacy in the debridement of various chronic and other
hard-to-heal wounds with only few daily applications. EscharEx’s mechanism of action is mediated by the proteolytic enzymes that
cleave and remove the necrotic tissue and prepare the wound bed for healing. On May 12, 2022, we announced positive results from our U.S.
Phase II clinical study of EscharEx for the debridement of VLUs. The study met its primary endpoint with a high degree of statistical
significance, demonstrating that patients treated with EscharEx had a statistically significant higher incidence of complete debridement
during the 14-day measurement period within up to 8 applications, compared to gel vehicle (EscharEx: 63% (29/46) vs. gel vehicle: 30%
(13/43), p-value=0.004). EscharEx efficacy superiority remained statistically significant after adjusting for pre-specified covariates
ascribed to patient baseline characteristics, wound size, wound age and regions.
The study met key secondary and exploratory endpoints. Patients
treated with EscharEx had a statistically significant higher incidence of complete debridement, during the same 14-day measurement period,
compared to patients treated by non-surgical standard-of-care ("NSSOC") (EscharEx: 63% (29/46) vs. NSSOC: 13% (4/30)) and the time to
achieve complete debridement was significantly shorter. Estimated median time to complete debridement was 9 days for patients treated
with EscharEx and 59 days for patients treated with NSSOC (p-value=0.016). On average, complete debridement was achieved after 3.6 applications
of EscharEx compared to 12.8 applications with NSSOC. Patients treated with EscharEx demonstrated significantly higher incidence of at
least 75% granulation tissue at the end of the treatment period compared to gel vehicle (p-value <0.0001). Favorable trends were observed
in wound area reduction and reduction of pain compared to gel vehicle.
In addition, the study showed that EscharEx was safe and well tolerated,
and the overall safety was comparable between the arms as assessed by the data safety monitoring board. Importantly, there were no observed
deleterious effects on wound closure and no material differences in reported adverse events. Estimated time to complete wound closure
was 64 days for patients treated with EscharEx compared to 78 days for patients treated with NSSOC.
EscharEx was also evaluated in a U.S. Phase II pharmacology study.
The study was prospective, open label, single-arm and conducted at three U.S. clinical sites. On July 7, 2022, we announced positive results
from this study. 70% of patients achieved complete debridement during the course of treatment within up to 8 applications. On average,
complete debridement was achieved after 3.9 applications of EscharEx. Additionally, an average reduction of 35% in wound size was achieved
by the end of the 2-week follow-up period. In all patients that were positive for biofilm at baseline, the biofilm was reduced substantially
to single individual microorganisms or completely removed by the end of treatment. Seven patients had positive red fluorescence (indicative
of bacteria) at baseline and average red fluorescence was reduced from 1.69 cm2 pre-treatment to 0.60 cm2 post treatment. Biomarker analysis
from wound fluid safety data showed that EscharEx is safe and well-tolerated.
Our third innovative product candidate, MW005, is a topically applied
biological drug candidate for the treatment of non-melanoma skin cancers, based on the same API as NexoBrid and EscharEx (a concentrate
of proteolytic enzymes enriched in bromelain). In July 2021, we initiated a phase I/II study of MW005 for the treatment of low-risk BCC.
On July 11, 2022, we announced positive initial data from this study. In the first cohort, eleven patients with either superficial or
nodular BCC were treated. Patients enrolled into the study received seven topical applications of MW005, once every other day. At the
end of eight weeks post treatment period, all patients undergo complete excision, and the specimen is subject to an independent histological
clearance examination.
In July 2023, we again announced positive results in our U.S. Phase I/II study of MW005
for the treatment of basal cell carcinoma. Fifteen patients were treated with MW005 and completed the study. Results showed MW005 to be
safe and well-tolerated, with a high level of patient compliance. Based on clinical assessments, eleven out of fifteen patients achieved
complete clearance of their BCCs; the majority of these patients also had histologically confirmed complete clearance.
Based on the data generated to date, MW005 is safe, well-tolerated
and an effective treatment for BCC with a majority of patients who completed the study demonstrating a complete histological clearance
of target lesions.
We manufacture NexoBrid and our product candidates in our cGMP
certified sterile manufacturing facility at our headquarters in Yavne, Israel. As of December 31, 2023, we had cash and cash equivalents
and short term and restricted bank deposits of $41.7 million. Our revenues were $18.7 million and $26.5 in 2023 and 2022, respectively.
Our net operating loss was $15.3 million and $8.3 in 2023 and 2022, respectively. We had an accumulated deficit of $174.8 million as of
December 31, 2023. We expect to incur significant expenses and operating losses in the coming years, as research and development activities
are central to our operations, which will offset by cash inflows from NexoBrid.
We expect to continue to invest in our research and development
efforts, including in respect of our NexoBrid ongoing clinical trials which are fully funded by BARDA, as well as the clinical development
and trials of EscharEx, MW005 and our other pipeline product candidates. In addition, we expect to continue to advance NexoBrid as a standard
of care, and expand its commercial reach in international markets, including for potential use as a medical countermeasure during mass
casualty events.
Key Components of Statements of Operations
Revenues
Sources of revenues. We
derive revenues from sales of NexoBrid to burn centers and hospitals burn units in USA, Europe and Israel as well as to local distributors
in other countries in accordance with distribution agreements we have in place, which also include revenues from licenses. We generate
revenues from BARDA procurement of NexoBrid for emergency stockpile pursuant to BARDA contract.
We generate revenues from development services provided to BARDA,
and to DOD/MTEC.
Our ability to generate additional, more significant revenues will
depend on the successful commercialization of NexoBrid.
Cost of Revenues
Our total cost of revenues includes expenses for the manufacturing
of NexoBrid, including: the cost of raw materials; employee‑related expenses, including salaries, equity based‑compensation
and other benefits and related expenses, lease payments, utility payments, depreciation, changes in inventory of finished products, royalties
and other manufacturing expenses. These expenses are partially reduced by an allotment of manufacturing costs associated with research
and development activities to research and development expenses.
Cost of revenues includes costs associated with the research and
development services provided to BARDA and MTEC, including salaries and related expenses, clinical trials, sub‑contractors and external
advisors.
We expect that our cost of revenues from sale of products will
continue to increase as we expand the sale of NexoBrid throughout the European Union, the United States and other international markets.
Operating Expenses
Research and Development Expenses
Research and development activities are central to our business
model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages
of clinical development, primarily due to the increased size and duration of later‑stage clinical trials. We expect research and
development costs to increase significantly for the foreseeable future as EscharEx progresses in its clinical program in the U.S. and
our other pipeline product candidates' progress in clinical trials. However, we do not believe that it is possible at this time to accurately
project total program‑specific expenses to reach commercialization. There are numerous factors associated with the successful development
of any of our product candidates, including future trial design and various regulatory requirements, many of which cannot be determined
with accuracy at this time based on our stage of development. Additionally, future commercial and regulatory factors beyond our control
will affect our clinical development programs and plans. Our actual spending could differ as our plans change and we invest in other drugs
or potentially reduce our anticipated funding on research for existing products. Research and development expenses consist primarily of
compensation for employees engaged in research and development activities, including salaries, equity‑based compensation, benefits
and related expenses, clinical trials, contract research organization sub‑contractors, development materials, external advisors
and the allotted cost of our manufacturing facility for research and development purposes.
Selling and Marketing Expenses
Selling and marketing expenses consist primarily of compensation
expenses for personnel engaged in sales and marketing, including salaries, equity based‑compensation and benefits and related expenses,
as well as promotion, marketing, market access, medical, and sales and distribution activities. These expenses are primarily comprised
of costs related to our subsidiary in Germany, which is focused on marketing NexoBrid in E.U., and costs related to maintain marketing
authorization.
General and Administrative Expenses
General and administrative expenses consist principally of compensation
for employees in executive and administrative functions, including salaries, equity‑based compensation, benefits and other related
expenses, professional consulting services, including legal and audit fees, as well as costs of office and overhead. We expect general
and administrative expenses to remain stable.
Financial Income/Financial Expense
Financial income includes interest income, revaluation of financial
instruments and exchange rate differences. Financial expense consists primarily of revaluation of financial instruments, financial expenses
in respect of deferred revenue, revaluation of lease liabilities and exchange rate differences. The market interest due on government
grants received from the IIA is also considered a financial expense, and is recognized beginning on the date we receive the grant until
the date on which the grant is expected to be repaid as part of the revaluation to fair value of liabilities in respect of government
grants.
Taxes on Income
The standard corporate tax rate in Israel is 23%.
We do not generate taxable income in Israel, as
we have historically incurred operating losses resulting in carry forward tax losses and other temporarily differences from R&D expenses
totaling approximately $172 million as of December 31, 2023. We anticipate that we will be able to carry forward these tax losses indefinitely
to future tax years. Accordingly, we do not expect to pay taxes in Israel until we have taxable income after the full utilization of our
carry forward tax losses.
Under the Law for the Encouragement of Capital Investments, 5719‑1959
(the “Investment Law”), we have been granted “Beneficiary Enterprise” status, which provides certain benefits,
including tax exemptions and reduced corporate tax rates. Income not eligible for Beneficiary Enterprise benefits is taxed at the regular
corporate tax rate. The benefit entitlement period starts from the first year that the Beneficiary Enterprise first earns taxable income,
and is limited to 12 years from the year in which the company requested to have tax benefits apply.
Comparison of Period to Period Results of Operations
We are providing within this section a supplemental discussion
that compares our historical statement of operations data in accordance with IFRS, as issued by the IASB. The below table and the below
discussion provides data for each of the years ended December 31, 2023 and 2022. The below discussion of our results of operations omits
a comparison of our results for the years ended December 31, 2022 and 2021. In order to view that discussion, please see “Item 5.
Operating and Financial Review and Prospects-A. Operating Results- Comparison of Period to
Period Results of Operations- Year Ended December 31, 2022 Compared to
Year Ended December 31, 2021” in our Annual Report on Form 20-F for the year ended December 31, 2022, which we filed with the SEC
on March 16, 2023.
| |
|
Years Ended December 31, |
|
| |
|
2023 |
|
|
2022 |
|
| |
|
(in thousands) |
|
|
Condensed statements of operations data: |
|
|
|
|
|
|
|
Revenues
|
|
$ |
18,686 |
|
|
$ |
26,496 |
|
|
Cost of revenues
|
|
|
15,108 |
|
|
|
13,331 |
|
|
Gross profit
|
|
|
3,578 |
|
|
|
13,165 |
|
| |
|
|
|
|
|
|
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
7,467 |
|
|
|
10,181 |
|
|
Selling and marketing
|
|
|
4,844 |
|
|
|
3,725 |
|
|
General and administrative
|
|
|
6,768 |
|
|
|
6,920 |
|
|
Other (income) expenses
|
|
|
(211 |
) |
|
|
684 |
|
|
Operating loss
|
|
|
(15,290 |
) |
|
|
(8,345 |
) |
| |
|
|
|
|
|
|
|
|
|
Financial income (expenses), net
|
|
|
8,759 |
|
|
|
(11,176 |
) |
|
Loss before taxes on income
|
|
|
(6,531 |
) |
|
|
(19,521 |
) |
| |
|
|
|
|
|
|
|
|
|
Taxes on income
|
|
|
(185 |
) |
|
|
(78 |
) |
| |
|
|
|
|
|
|
|
|
|
Net loss
|
|
$ |
(6,716 |
) |
|
$ |
(19,599 |
) |
Year Ended December 31, 2023 Compared to Year Ended December 31,
2022
Revenues
| |
|
Years Ended December 31, |
|
| |
|
2023 |
|
|
2022 |
|
| |
|
(in thousands) |
|
|
Revenues from sale of products |
|
$ |
6,261 |
|
|
$ |
5,347 |
|
| |
|
|
|
|
|
|
|
|
|
Revenues from development services |
|
|
12,265 |
|
|
|
12,943 |
|
| |
|
|
|
|
|
|
|
|
|
Revenues from license agreements and royalties
|
|
|
160 |
|
|
|
8,206 |
|
| |
|
|
|
|
|
|
|
|
| |
|
|
18,686 |
|
|
|
26,496 |
|
We generated total revenues of approximately $18.7
million for the year ended December 31, 2023 compared to approximately $26.5 million for the year ended December 31, 2022. The decrease
is primarily attributed to the BLA approval millstone payment from Vericel that took place in 2022.
Revenues from sale of products
Revenues from sales of products in 2023 were $6.3 million, a 17%
increase compared to $5.3 million in 2022 mainly attributed to new customers.
Revenues from development services
Revenues from development services decreased by 5% from $12.9 million in 2022 to $12.3
million in 2023. The decrease mainly driven from BARDA.
Revenues from license agreements and royalties
In 2023, revenues from license agreements and royalties were $0.2
million compared to $8.2 million in 2022, the decrease mainly driven by the BLA approval milestone of $7.5 million from Vericel in 2022.
Our revenues, as reported in our consolidated financial statements,
are based on the location of the customers, as shown in the below table:
| |
|
Years Ended December 31, |
|
| |
|
2023 |
|
|
2022 |
|
| |
|
(in thousands) |
|
|
International (excluding U.S.) |
|
$ |
5,608 |
|
|
$ |
4,624 |
|
|
U.S. |
|
|
13,078 |
|
|
|
21,872 |
|
| |
|
|
18,686 |
|
|
|
26,496 |
|
BARDA contributed 56% and 51% of the Company’s total revenues
in 2023 and 2022 respectively. Vericel contributed 4% and 28% of the Company’s total revenues in 2023 and 2022 respectively. DoD/MTEC
contributed 10% and 2.8% of the Company’s total revenues in 2023 and 2022 respectively.
Costs and Expenses
Cost of revenues
| |
|
Years Ended December 31, |
|
| |
|
2023 |
|
|
2022 |
|
| |
|
(in thousands) |
|
|
Cost of revenues from sales of products
|
|
$ |
4,927 |
|
|
$ |
3,184 |
|
|
Cost of revenues from development services
|
|
|
10,177 |
|
|
|
9,829 |
|
|
Cost of revenues from license agreements
|
|
|
4 |
|
|
|
318 |
|
| |
|
|
15,108 |
|
|
|
13,331 |
|
Cost of revenues as a percentage of total revenues increased from 50% for 2022 to 81%
for 2023.
Cost of revenues from sales of products as a percentage of revenues from sales of products
increased to approximately 79% for the year ended December 31, 2023, from approximately 60% in the year ended December 31, 2022. The increase
of cost of revenues from sales of product is primarily driven by revenue mix changes.
Cost of revenues from development services as a percentage of revenues
from development services was approximately 83% in the year ended December 31, 2023, compared to approximately 76% in the year ended December
31, 2022, mainly attributed to BARDA.
Cost of revenues from license agreements and royalties as a percentage
of revenues from license agreements was 3% in the year ended December 31, 2023 compared to approximately 4% in the year ended December
31,2022.
Research and development expenses,
Research and development expenses decreased by 27% from approximately
$10.2 million in the year ended December 31, 2022 to approximately $7.5 million in the year ended December 31, 2023, attributed mainly
to the completion of EscharEx Phase II study.
Selling and marketing expenses
Selling and marketing expenses increased by 30% in 2023 compared
to 2022, from approximately $3.7 million in the year ended December 31, 2022 to approximately $4.8 million in the year ended December
31, 2023, mainly related to consulting services, and various marketing expenses.
General and administrative expenses
General and administrative expenses decreased by 2% in 2023 compared
to 2022 from approximately $6.9 million in the year ended December 31, 2022 to approximately $6.8 million in the year ended December 31,
2023.
Other (income) expenses
Other one-time incomes for the year ended December 31, 2023
were $(0.2) million associated with non-cash income resulted with termination of sub-lease agreement with CBI. Other one-time expenses
for the year ended December 31, 2022 were $0.7 million associated with management replacement and Vericel milestone payment fee.
|
Financial income, net |
|
Years Ended December 31, |
|
| |
|
2023 |
|
|
2022 |
|
| |
|
(in thousands) |
|
|
Financial income |
|
$ |
10,651 |
|
|
$ |
461 |
|
|
Financial expenses |
|
|
(1,892 |
) |
|
|
(11,637 |
) |
| |
|
|
8,759 |
|
|
|
(11,176 |
) |
Financial income
Financial income increased from approximately $0.5 million in the
year ended December 31, 2022 to approximately $10.7 million in the year ended December 31, 2023. The increase was primarily driven by
an $8.3 million adjustment resulting from the revaluation of warrants and by $2.3 million of interest income from short term deposits
in 2023.
Financial expense
Financial expenses decreased from approximately $11.6 million in
the year ended December 31, 2022 to approximately $1.9 million in the year ended December 31, 2023. The decrease in financial expenses
was primarily driven by $9 million adjustment resulting from the revaluation of warrants in 2022.
B.
Liquidity and Capital Resources
Our primary uses of cash are to fund working capital requirements,
manufacturing costs, research and development activities related to EscharEx and other products candidates, capital expenditure requirements,
as well as sales and marketing activities associated with the commercialization of NexoBrid in Europe.
On March 7, 2022, the Company completed a public offering in total
of 744,048 new ordinary shares which were issued in consideration to offering price of $13.44 per share. The net proceeds were $8,653,
after deducting commissions and other offering expenses. In addition, on March 22, 2022 the underwriters exercised their options to purchase
an additional 89,012 ordinary shares at the same public offering price. The net consideration to the Company, less underwriting discounts
and commissions was at additional of $1,021.
On September 26, 2022, the Company completed a registered direct
(the “RD”) offering in an aggregate amount of $13,257 represent a combine purchase price of $12.25 for issuance of 1,082,223
ordinary shares and 1,082,223 warrants that become exercisable on November 28, 2022, at an exercise price of $13.475 per ordinary share
which will expire in four years, The net proceeds from this offering in the amount of $11,698 have been received on September 28, 2022.
The issuance expenses related to the non-current financial liability were recorded through profit and lost and the issuance expenses related
to the issuance of shares recorded as a deduction from the proceed in equity.
On October 6, 2022 we entered the PIPE Securities Purchase Agreement
with the several purchasers listed on the signature pages thereto (the “PIPE Purchasers”), in connection with the offer and
sale of 1,407,583 pre-funded warrants to purchase up to 1,407,583 Ordinary Shares (the “Pre-Funded Warrants”) and 1,407,583
ordinary warrants to purchase up to 1,407,583 Ordinary Shares (the “PIPE Ordinary Warrants,” and together with the Pre-Funded
Warrants, the “PIPE Warrants”) (the “PIPE Offering”). The combined purchase price for one Pre-Funded Warrant and
associated PIPE Ordinary Warrant was $12.243. The Pre-Funded Warrants have an exercise price of $0.007 per Ordinary Share and the PIPE
Ordinary Warrants have an exercise price of $13.475 per Ordinary Share and each become exercisable on November 28, 2022. The PIPE Offering
closed on October 6, 2022. The gross proceeds from the PIPE Offering were approximately $17.23 million. As of December 31, 2022, all Pre-Funded
Warrants have been exercised and none of the PIPE Ordinary Warrants have been exercised.
H.C. Wainwright & Co., LLC (“Wainwright”) acted
as the exclusive placement agent for the RD Offering and the PIPE Offering (together, the “2022 Offerings”). Upon closing
of the Offerings, we issued Wainwright (or its designees) the warrants to purchase up to 124,491 ordinary shares (the “Wainwright
Warrants”). The warrants have substantially the same terms as the RD Warrants and the Series A Warrants, except that the Wainwright
Warrants have an exercise price equal to $15.3125 per share (which represents 125% of the offering price per Ordinary Share in the Offerings)
and will expire four (4) years after November 28, 2022, but no more than five (5) years following the commencement of the sales pursuant
to the RD Offering.
On February 3, 2023, we entered into a securities purchase
agreement (the “2023 Securities Purchase Agreement”) with the purchasers listed on the signature pages thereto (the “2023
Purchasers”), in connection with the offer and sale of 1,964,286 ordinary shares (the “2023 Offering”). The purchase
price per ordinary share was $14.00. The 2023 Offering closed on February 7, 2023. The gross proceeds from the Offering were approximately
$27.5 million.
The Company believes that its existing cash and cash equivalents, short-term and restricted
bank deposits of $41.7 million as of December 31, 2023, will be sufficient to fund its operations and capital expenditure for at least
twelve months from the date of issuance of these consolidated financial statements.
Our future capital requirements will depend on many factors, including
our revenue growth, timing of milestone payments, the timing and extent of our spending on research and development efforts, and international
expansion. We may also seek to invest in or acquire complementary businesses or technologies. To the extent that existing cash and cash
from operations are insufficient to fund our future activities, we may need to raise additional funding through debt and equity financing.
Additional funds may not be available on favorable terms or at all
The accompanying consolidated financial statements have been prepared
on a basis which assumes that the Company will continue as a going concern. From inception to December 31, 2023, the Company has incurred
cash outflows from operations, losses from operations, and has an accumulated deficit of $174.8 million.
Cash Flows
The following table summarizes our consolidated
statement of cash flows for the periods presented. The below discussion beneath the table omits a description of our cash flows for the
year ended December 31, 2023. In order to view that discussion, please see “Item 5. Operating and Financial Review and Prospects
Liquidity and Capital Resources” in our Annual Report on Form 20-F for the year ended December 31, 2022, which we filed with the
SEC on March 16, 2023:
| |
|
Year Ended December 31, |
|
| |
|
2023 |
|
|
2022 |
|
| |
|
|
|
|
|
|
|
Net cash provided by (used in): |
|
|
|
|
|
|
|
Operating activities
|
|
$ |
(10,465 |
) |
|
$ |
(11,885 |
) |
|
Investing activities
|
|
|
(34,321 |
) |
|
|
(481 |
) |
|
Financing activities
|
|
|
22,917 |
|
|
|
35,764 |
|
Net cash used in operating activities
Net cash used in all periods resulted primarily
from our net loss adjusted for non-cash charges and measurements and changes in components of working capital. Adjustments for non-cash
items include depreciation and amortization, equity-based compensation, revaluation of contingent liabilities warrants, and lease liability,
and changes in assets and liabilities items.
Net cash used in operating activities decreased to approximately
$10.5 million in the year ended December 31, 2023 compared to net cash used in operating activities of approximately $11.9 million in
the year ended December 31, 2022, primarily as a result of our operational net loss, partially offset by various non-cash items such as
depreciation, shared based compensation, financial income, net and revaluation of contingent considerations and warrants.
Net cash used in investing activities
Net cash used in investing activities primarily derives from investment
in short term banks deposits and from purchases of property and equipment mainly related to scaling up our production facility, offset
by interest received from short term bank deposit. Net cash used in investing activities was $34.3 million in the year ended December
31, 2023, compared to $0.5 million provided during the year ended December 31, 2022, primarily result of investment in short- term bank
deposits and investments in property and equipment.
Net cash provided by financing activities
Net cash provided by financing activities was $22.9
million in the year ended December 31, 2023 compared to $35.8 million in the year ended December 31, 2022. The decrease in net cash provided
by financing activities was primarily a result of the proceeds from the company’s 2022 Offerings, net of issuance expenses.
Israeli Corporate-Level Tax Considerations and Government Programs
The following is a brief summary of the material Israeli tax laws
applicable to us, and certain Israeli Government programs that benefit us and therefore impact our results of operations and financial
condition. To the extent that the discussion is based on new tax legislation that has not yet been subject to judicial or administrative
interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion.
The discussion below is subject to change, including due to amendments under Israeli law or changes to the applicable judicial or administrative
interpretations of Israeli law, which change could affect the tax consequences described below.
General Corporate Tax Structure in Israel
Generally, Israeli companies are subject to a corporate tax on
their taxable income. Effective January 1, 2018 and thereafter, the corporate tax rate is 23%. However, the effective tax rate payable
by a company that derives income from an Approved Enterprise, a Beneficiary Enterprise, a Preferred Enterprise or Technology Enterprise
(as discussed below) may be considerably less. Capital gains derived by an Israeli company are generally subject to the prevailing regular
corporate tax rate.
Law for the Encouragement of Industry (Taxes), 5729-1969
The Law for the Encouragement of Industry (Taxes), 5729-1969 (the
“Industry Encouragement Law”), provides several tax benefits for “Industrial Companies.”
The Industry Encouragement Law defines an “Industrial Company”
as an Israeli resident-company which was incorporated in Israel, of which 90% or more of its income in any tax year, other than income
from certain government loans, is derived from an “Industrial Enterprise” owned by it and located in Israel or in the “Area”,
in accordance with the definition under section 3A of the Israeli Income Tax Ordinance (New Version) 1961. An “Industrial Enterprise”
is defined as an enterprise whose principal activity in a given tax year is industrial production.
The following tax benefits, among others, are available to Industrial
Companies:
|
|
•
|
amortization of the cost of purchased a patent, rights to use a patent, and know-how,
which are used for the development or advancement of the Industrial Enterprise, over an eight-year period, commencing on the year in which
such rights were first exercised;
|
|
|
•
|
under limited conditions, an election to file consolidated tax returns with related
Israeli Industrial Companies controlled by it; and
|
|
|
•
|
expenses related to a public offering are deductible in equal amounts over a three
years period commencing on the year of the offering.
|
Eligibility for benefits under the Industry Encouragement Law is
not contingent upon approval of any governmental authority.
We believe that we currently qualify as an Industrial Company within
the meaning of the Industry Encouragement Law. However, there can be no assurance that we will continue to qualify as an Industrial Company
or that the benefits described above will be available in the future.
Law for the Encouragement of Capital Investments, 5719-1959
The Investment Law provides certain incentives for capital investments
in production facilities (or other eligible assets).
The Investment Law was significantly amended several times during
recent years, with the three most significant changes effective as of April 1, 2005 (the “2005 Amendment”), as of January
1, 2011 (the “2011 Amendment”), and as of January 1, 2017 (the “2017 Amendment”). Pursuant to the 2005 Amendment,
tax benefits granted in accordance with the provisions of the Investment Law prior to its revision by the 2005 Amendment remain in force
but any benefits granted subsequently are subject to the provisions of the amended Investment Law. Similarly, the 2011 Amendment introduced
new benefits to replace those granted in accordance with the provisions of the Investment Law in effect prior to the 2011 Amendment. However,
companies entitled to benefits under the Investment Law as in effect prior to January 1, 2011 were entitled to choose to continue to enjoy
such benefits, provided that certain conditions are met, or elect instead, irrevocably, to forego such benefits and have the benefits
of the 2011 Amendment apply. The 2017 Amendment introduces new benefits for Technological Enterprises, alongside the existing tax benefits.
Prior to 2011, we did not utilize any of the benefits for which we were eligible under the Investment Law.
The following is a summary of the Investment Law subsequent to
its amendments as well as the relevant changes contained in the new legislation.
Tax Benefits Subsequent to the 2005 Amendment
The 2005 Amendment applies to new investment programs and investment
programs commencing after 2004, but does not apply to investment programs approved prior to April 1, 2005 (“Approved Enterprise”).
The 2005 Amendment provides that terms and benefits included in any certificate of approval that was granted before the 2005 Amendment
became effective (April 1, 2005) will remain subject to the provisions of the Investment Law as in effect on the date of such approval.
Pursuant to the 2005 Amendment, the Israeli Authority for Investments and Development of the Israeli Ministry of Economy (the “Investment
Center”) will continue to grant Approved Enterprise status to qualifying investments. The 2005 Amendment, however, limits the scope
of enterprises that may be approved by the Investment Center by setting criteria for the approval of a facility as an Approved Enterprise.
The 2005 Amendment provides that Approved Enterprise status will
only be necessary for receiving cash grants. As a result, it is no longer necessary for a company to obtain the advance approval of the
Investment Center in order to receive the tax benefits previously available under the alternative benefits track. Rather, a company may
claim the tax benefits offered by the Investment Law directly in its tax returns, provided that its facilities meet the criteria for tax
benefits set forth in the 2005 Amendment. Companies or programs under the new provisions receiving these tax benefits are referred to
as Beneficiary Enterprises. Companies that have a Beneficiary Enterprise, are entitled to approach the Israel Tax Authority for a pre‑ruling
regarding their eligibility for tax benefits under the Investment Law, as amended. Tax benefits are available under the 2005 Amendment
to production facilities (or other eligible facilities), which are generally required to derive more than 25% of their business income
from export to specific markets with a population of at least 14 million in 2012 (such export criteria will further increase in the future
by 1.4% per annum). In order to receive the tax benefits, the 2005 Amendment states that a company must make an investment which meets
certain conditions, including exceeding a minimum investment amount specified in the Investment Law. Such investment allows a company
to receive “Beneficiary Enterprise” status, and may be made over a period of no more than three years ending in the year in
which the company chose to have the tax benefits apply to its Beneficiary Enterprise. Where the company requests to apply the tax benefits
to an expansion of existing facilities, only the expansion will be considered to be a Beneficiary Enterprise and the company’s effective
tax rate will be the weighted average of the applicable rates. In this case, the minimum investment required in order to qualify as a
Beneficiary Enterprise is required to exceed a certain percentage of the value of the company’s production assets before the expansion.
The extent of the tax benefits available under the 2005 Amendment
to qualifying income of a Beneficiary Enterprise depends on, among other things, the geographic location in Israel of the Beneficiary
Enterprise. The location will also determine the period for which tax benefits are available. Such tax benefits include an exemption from
corporate tax on undistributed income for a period of between two to ten years, depending on the geographic location of the Beneficiary
Enterprise in Israel, and a reduced corporate tax rate of between 10% to 25% for the remainder of the benefits period, depending on the
level of foreign investment in the company in each year. A company qualifying for tax benefits under the 2005 Amendment which pays a dividend
out of income attributed to its Beneficiary Enterprise during the tax exemption period will be subject to corporate tax in respect of
the amount of the dividend distributed (grossed‑up to reflect the pre‑tax income that it would have had to earn in order to
distribute the dividend) at the corporate tax rate that would have otherwise been applicable. Dividends paid to Israeli shareholders out
of income attributed to a Beneficiary Enterprise (or out of dividends received from a company whose income is attributed to a Beneficiary
Enterprise) are generally subject to withholding tax at source at the rate of 15% (in the case of non-Israeli shareholders - subject to
the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 15% or such lower rate as may be provided
in an applicable tax treaty, applicable to dividends and distributions out of income attributed to a Beneficiary Enterprise). The reduced
rate of 15% is limited to dividends and distributions out of income attributed to a Beneficiary Enterprise during the benefits period
and actually paid at any time up to 12 years thereafter, except with respect to a qualified Foreign Investment Company (as such term is
defined in the Investment Law), in which case the 12‑year limit does not apply.
The benefits available to a Beneficiary Enterprise are subject to the fulfillment of
conditions stipulated in the Investment Law and its regulations. If a company does not meet these conditions, it would be required to
refund the amount of tax benefits, as adjusted by the Israeli consumer price index, and interest, or other monetary penalties.
We currently have Beneficiary Enterprise programs under the Investment
Law, which we believe will entitle us to certain tax benefits. The majority of any taxable income from our Beneficiary Enterprise programs
(once generated) would be tax exempt for a period of ten years commencing in the year in which we will first earn taxable income relating
to such enterprises, subject to the 12-year limitation from the year the company chose to have its tax benefits apply.
Tax Benefits Under the 2011 Amendment
The 2011 Amendment canceled the availability of the tax benefits
granted under the Investment Law prior to 2011 and, instead, introduced new tax benefits for income generated by a “Preferred Company”
through its “Preferred Enterprise” (as such terms are defined in the Investment Law) as of January 1, 2011. The definition
of a Preferred Company includes a company incorporated in Israel that is not fully owned by a governmental entity, and that has, among
other things, Preferred Enterprise status and is controlled and managed from Israel. The tax benefits under the 2011 Amendment for a Preferred
Company meeting the criteria of the law include, among others, a reduced corporate tax rate of 15% for preferred income attributed to
a Preferred Enterprise in 2011 and 2012, unless the Preferred Enterprise was located in a specified development zone, in which case the
rate was 10%. Under the 2011 Amendment, such corporate tax rate was reduced in 2013 from 15% and 10%, respectively, to 12.5% and 7%, respectively,
and then increased to 16% and 9%, respectively, in 2014 and thereafter until 2016. Pursuant to the 2017 Amendment, in 2017 and thereafter,
the corporate tax rate for Preferred Enterprise which is located in a specified development zone was decreased to 7.5%, while the reduced
corporate tax rate for other development zones remains 16%. Income attributed to a Preferred Company from a “Special Preferred Enterprise”
(as such term is defined in the Investment Law) would be entitled, during a benefits period of 10 years, to reduced tax rates of 8%, or
5% if the Special Preferred Enterprise is located in a certain development zone. As of January 1, 2017, the definition of “Special
Preferred Enterprise” includes less stringent conditions. Dividends paid to Israeli shareholders out of preferred income attributed
to a Preferred Enterprise or to a Special Preferred Enterprise are generally subject to withholding tax at source at the rate of 20% (in
the case of non-Israeli shareholders - subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax
rate, 20%, or such lower rate as may be provided in an applicable tax treaty(. However, if such dividends are paid to an Israeli company,
no tax is required to be withheld (although, if such dividends are subsequently distributed to individuals or a non‑Israeli company,
the aforesaid will apply).
The 2011 Amendment also provided transitional provisions to address
companies already enjoying existing tax benefits under the Investment Law. These transitional provisions provide, among other things,
that: unless an irrevocable request is made to apply the provisions of the Investment Law as amended in 2011 with respect to income to
be derived as of January 1, 2011, a Beneficiary Enterprise can elect to continue to benefit from the benefits provided to it before the
2011 Amendment came into effect, provided that certain conditions are met.
We have examined the possible effect, if any, of these provisions
of the 2011 Amendment on our financial statements and have decided, at this time, not to opt to apply the new benefits under the 2011
Amendment. There can be no assurance that we will comply with the conditions required to remain eligible for benefits under the Investment
Law in the future or that we will be entitled to any additional benefits thereunder.
New Tax benefits under the 2017 Amendment that
became effective on January 1, 2017.
The 2017 Amendment was enacted as part of the Economic Efficiency
Law that was published on December 29, 2016, and is effective as of January 1, 2017. The 2017 Amendment provides new tax benefits for
two types of “Technology Enterprises,” as described below, and is in addition to the other existing tax beneficial programs
under the Investment Law.
The 2017 Amendment provides that a technology company satisfying
certain conditions will qualify as a “Preferred Technology Enterprise” and will thereby enjoy a reduced corporate tax rate
of 12% on income that qualifies as “Preferred Technology Income,” as defined in the Investment Law. The tax rate is further
reduced to 7.5% for a Preferred Technology Enterprise located in development zone A. In addition, a Preferred Technology Company will
enjoy a reduced corporate tax rate of 12% on capital gain derived from the sale of certain “Benefitted Intangible Assets”
(as defined in the Investment Law) to a related foreign company if the Benefitted Intangible Assets were acquired from a foreign company
on or after January 1, 2017 for at least NIS 200 million, and the sale receives prior approval from the Israeli Innovation Authority (the
“IIA”).
The 2017 Amendment further provides that a technology company satisfying
certain conditions will qualify as a “Special Preferred Technology Enterprise” and will thereby enjoy a reduced corporate
tax rate of 6% on “Preferred Technology Income” regardless of the company’s geographic location within Israel. In addition,
a Special Preferred Technology Enterprise will enjoy a reduced corporate tax rate of 6% on capital gain derived from the sale of certain
“Benefitted Intangible Assets” to a related foreign company if the Benefitted Intangible Assets were either developed by Special
Preferred Technology Enterprise or acquired from a foreign company on or after January 1, 2017, and the sale received prior approval from
IIA. A Special Preferred Technology Enterprise that acquires Benefitted Intangible Assets from a foreign company for more than NIS 500
million will be eligible for these benefits for at least ten years, subject to certain approvals as specified in the Investment Law. Dividends
distributed by a Preferred Technology Enterprise or a Special Preferred Technology Enterprise, to Israeli shareholders paid out of Preferred
Technology Income, are generally subject to withholding tax at source at the rate of 20% (in the case of non-Israeli shareholders - subject
to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 20%, or such lower rate as may be provided
in an applicable tax treaty. However, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if
such dividends are subsequently distributed to individuals or a non-Israeli company, the aforesaid will apply). If such dividends are
distributed to a foreign company that holds solely or together with other foreign companies 90% or more in the Israeli company and other
conditions are met, the withholding tax rate will be 4% (or a lower under the tax treaty, if applicable, subject to the receipt in advance
of a valid certificate from the Israeli Tax Authority allowing for a reduced tax rate).
C.
Research and Development, Patents and Licenses, etc.
Our research and development strategy is centered on developing
our patented proteolytic enzyme technology, which underlies NexoBrid and EscharEx, into additional products for high value indications.
Our research and development team is located at our facilities in Yavne, Israel, and consists of 26 employees as of December 31, 2022
and is supported by highly experienced consultants in various research and development disciplines.
We have received government grants (subject to our obligation to
pay royalties) as part of the NexoBrid and EscharEx research and development programs approved by the IIA. The total gross amount of grants
actually received by us from the IIA, including accrued interest and net of royalties actually paid, totaled approximately $13.8 million
as of December 31, 2023 and the amortized cost (using the interest method) of the liability totaled approximately $7.8 million and $7.6
million as of December 31, 2023 and 2022, respectively. Because the repayment of IIA grants is in the form of future royalties, the balance
of the commitments to the IIA is presented as an amortized liability on our balance sheet. As of December 31, 2022, we had accrued and
paid royalties to the IIA totaling $2 million.
We received funds from BARDA in accordance with the terms of our BARDA contracts. As
of December 31, 2023 we had accrued $93 million of BARDA’s participation in NexoBrid’s research and development programs.
For a description of our research and development policies for
the last three years, see “ITEM 4.B. Business Overview-Research and Development.”
D.
Trend Information
We continue to closely monitor macro-economic conditions, including
the headwinds caused by supply chain problems, inflation, increased interest rates and other trends that have been adversely impacting
economic activity on a global scale. We have been assessing, on an ongoing basis, the implications of those global conditions for our
operations, supply chain, liquidity, cash flow and product orders, and will act in an effort to mitigate adverse consequences as needed.
To the extent inflation increases our costs and expenses, we could consider price increases to offset those cost pressures.
Specific developments that may potentially impact our operating
performance in an adverse manner include:
|
|
• |
further actions taken by central banks in Europe and the U.S. to increase interest
rates as a means to slow down inflation, which may worsen credit/financing conditions for our customers who purchase our products;
|
|
|
• |
potential contraction of economic activities and recessionary conditions that could
arise as a result of interest rate increases and a decrease in consumer demand; and
|
|
|
• |
the continued depreciated value of the Euro relative to the U.S. dollar, which may
have an adverse impact on the U.S.- denominated value of our European-derived revenues for purposes of our financial statements.
|
We cannot provide any assurances as to the extent of our resilience
to the adverse impact of these specific developments in future periods. See also “ITEM 3.D. - Risk Factors -“We depend on
a sole supplier to obtain our intermediate drug substance, bromelain SP, which is necessary for the production of our products.”
Other than the foregoing and as disclosed elsewhere in this annual
report, we are not aware of any trends, uncertainties, demands, commitments or events for the period from January 1, 2022 to the present
time that are reasonably likely to have a material adverse effect on our net revenue, income, profitability, liquidity or capital resources,
or that would cause the disclosed financial information to be not necessarily indicative of future operating results or financial condition.
E. Critical
Accounting Estimates
Our consolidated financial statements are prepared in conformity
with IFRS, as issued by the IASB. The preparation of these historical financial statements in conformity with IFRS requires management
to make estimates, assumptions and judgments in certain circumstances that affect the reported amounts of assets, liabilities and contingencies
as of the date of the financial statements and the reported amounts of revenue and expenses during the reporting periods. We evaluate
our assumptions and estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that
we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values
of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different
assumptions or conditions. Our critical accounting estimates are described in Note 2 to our consolidated financial statements included
elsewhere in this annual report.
Item 6. DIRECTORS,
SENIOR MANAGEMENT AND EMPLOYEES
A. Directors
and Senior Management
The following table sets forth the name, age and position of each
of our executive officers and directors as of March 15, 2024:
|
Name
|
|
Age
|
|
Position
|
|
Executive Officers
|
|
|
|
|
|
Ofer Gonen
|
|
51
|
|
Chief Executive Officer
|
|
Shmulik Hess
|
|
51
|
|
Chief Operating Officer & Chief Commercial Officer
|
|
Ety Klinger |
|
62
|
|
Chief Research and Development Officer
|
|
Hani Luxenburg
|
|
51
|
|
Chief Financial Officer
|
|
Yaron Meyer |
|
45
|
|
Executive Vice President, General Counsel and Corporate Secretary
|
|
Robert J. Snyder
|
|
74 |
|
Chief Medical Officer |
| |
|
|
|
|
|
Directors
|
|
|
|
|
|
Nachum (Homi) Shamir (1)(2)(3)(5)
|
|
69
|
|
Chairman of the Board of Directors
|
|
Vickie R. Driver (1)(4)(5)
|
|
70
|
|
Director
|
|
David Fox (2)(3)(5) |
|
66
|
|
Director |
|
Shmuel (Milky) Rubinstein (4)(5)
|
|
84
|
|
Director
|
|
Stephen T. Willis (1)(2)(4)(5) |
|
67 |
|
Director |
|
|
|
|
(1)
|
Member of our audit committee.
|
|
(2)
|
Member of our compensation committee.
|
|
(3)
|
Member of our nominating and governance committee.
|
|
(4)
|
Member of our research and development committee. |
|
(5)
|
Independent director under the listing rules of the Nasdaq Stock Market.
|
Executive Officers
Ofer Gonen has served as
our Chief Executive Officer since July 2022 and on our board of directors since September 2003. Mr. Gonen is also the Chief Executive
Officer of Cactus Acquisition Corp. 1 (Nasdaq: CCTS) and served as the Chief Executive Officer of Call Biotechnology Industries (TASE:
CBI) from February 2017 until June 2022. Mr. Gonen has more than 20 years of experience in managing life science investments and business
collaborations in both the US and Israel. Mr. Gonen serves as a board member of several private and publicly-traded portfolio companies
of CBI, including Gamida Cell (Nasdaq: GMDA) and Cactus, as well as a managing partner at the Anatomy Medical Fund. Before joining CBI,
Mr. Gonen was the General Manager of Biomedical Investments Ltd., a partner at Arte Venture Group, as well as a technology consultant
to various Israeli venture capital funds. Mr. Gonen gained extensive experience in R&D and management of defense-oriented projects
at the prestigious “Talpiot” program of the Israeli Defense Forces. He holds a B.Sc. in Physics, Mathematics and Chemistry
from the Hebrew University of Jerusalem, and an M.A. in Economics and Finance from Tel Aviv University, with distinction.
Shmulik Hess has served
as our Chief Operating Officer since December 2023. Dr. Hess has over two decades of extensive expertise in drug development and commercial
operations in healthcare. Prior to joining MediWound, he served as Chief Executive Officer at Tabby Therapeutics, Enlivex Therapeutics
(Nasdaq: ENLV), and Valin Technologies. Formerly, Dr. Hess served as a global operations executive at SciGen Ltd (acquired by VBI Vaccines).
Dr. Hess is the inventor of multiple patents and author of numerous publications in peer reviewed scientific journals. He received his
Ph.D. in Pharmaceutical Science from the Hebrew University, Israel and was a research fellow at Harvard-MIT Health Sciences and Technology
(HST).
Ety Klinger has served
as our Chief Research and Development Officer since May 2014. Prior to joining MediWound, Dr. Klinger was Vice President of Research and
Development at Proteologics Ltd since July 2011, where she was responsible for discovery projects in the ubiquitin system, conducted in
collaboration with GlaxoSmithKline plc and Teva. Prior to this, Dr. Klinger served for 17 years in numerous leadership positions at Teva’s
global innovative R&D division and served as Teva’s Board representative at various biotechnology companies. Dr. Klinger was
a key member of the Copaxone® development team. As
a project leader she led the chemistry, manufacture and control, preclinical, clinical and post‑marketing R&D activities of
various innovative treatments for multiple sclerosis (MS), autoimmune and neurological diseases. From 2006 to 2011, as a Senior Director
at Teva, Dr. Klinger was a member of Teva’s global innovative R&D management team. From 2006 to 2008, she served as the Head
of MS and Autoimmune Diseases at Teva, and led the Life Cycle Management (LCM) of innovative R&D. Dr. Klinger holds a B.Sc. in Biology
from the Hebrew University in Jerusalem, a M.S. and a Ph.D. in Biochemistry from Tel‑Aviv University and an MBA degree from Tel
Aviv University and Northwestern University.
Hani Luxenburg has served
as our Chief Financial Officer since May 2023. Ms. Luxenberg has over two decades of progressive leadership experience managing financial
and accounting operations. Prior joining MediWound, Ms. Luxenburg served as Chief Financial Officer at BIRD Aerosystems. Prior to that,
she held senior finance roles at AstraZeneca, Alvarion Technologies Ltd., and Ernst & Young. Ms. Luxenburg is a certified public accountant
and holds a Bachelor of Arts in Economics and Accounting from the University of Haifa, along with a Bachelor of Law from IDC Herzliya.
In addition, she is a member of the Israel Bar Association.
Yaron Meyer has served
as our Executive Vice President since March 2019 and as our General Counsel and Corporate Secretary since December 2013. From April 2008
to November 2013, he served as the Corporate Secretary of CBI. From November 2010 to November 2013, he served as the General Counsel and
Corporate Secretary of D‑Pharm Ltd. From April 2008 to May 2010, he served as a legal counsel of Clal Industries Ltd. From May 2005
to April 2008, he worked as an associate at Shibolet & Co. Advocates. Mr. Meyer holds an LL.B. degree from Haifa University, Israel.
Dr. Robert Snyder has served
as our Chief Medical Officer since January 2023. Dr. Robert J. Snyder (DPM, MSc, MBA, CWSP, FFPM RCPS) is Dean, Professor, Director of
Clinical Research and Fellowship Director in Wound Care and Research at Barry University School of Podiatric Medicine. He is certified
in foot and ankle surgery by the American Board of Podiatric Surgery and is also a board-certified wound specialist. Dr. Snyder is past-president
of the Association for the Advancement of Wound Care and past-president of the American Board of Wound Management. Dr. Snyder has completed
an MBA in Health Management from The George Washington University and the Global Clinical Scholars Research Training Program at Harvard
Medical School. Dr. Snyder is a key opinion leader and sought-after speaker, lecturing extensively throughout the United States and abroad.
He has published several book chapters and over 165 papers in peer reviewed and trade journals on wound care, and was the recipient of
the Dr. Robert Warriner Memorial Award for excellence in wound management. Dr. Snyder serves as the Associate Editor for JAPMA and on
the editorial advisory boards of Ostomy Wound Management, Wounds and as a periodic reviewer for the Lancet and NEJM. He has been a Principal
Investigator on more than 65 randomized controlled trials for innovative wound healing modalities and products.
Directors
Nachum (Homi) Shamir has
served as Chairman of our board of directors since August 2022. Mr. Shamir most recently the Chairman, and Chief Executive Officer of
Luminex Corporation from 2014 through its sale to DiaSorin S.p.A.(“DiaSorin”) in 2021. Mr. Shamir continued to serve as President
of Luminex after its sale to DiaSorin pursuant to a transition agreement with DiaSorin until June 2022. Additionally, Mr. Shamir has served
as President and Chief Executive Officer of Given Imaging from 2006 through its sale to Covidien (now Medtronic) in 2014. Mr. Shamir currently
serves on the Board of Directors of IsoPlexis Corporation (Nasdaq: ISO) and Strata Skin Sciences (Nasdaq: SSKN); and as Chairman of the
Board of Cactus Acquisition Corp. (Nasdaq: CCTS). Mr. Shamir holds a Bachelor of Science degree from the Hebrew University of Jerusalem
and a Masters of Public Administration from Harvard University.
Vickie R. Driver has served
as a member of our Board since May 2017. She is board certified in foot surgery by the American Board of Podiatric Surgery and is a Fellow
at the American College of Foot and Ankle Surgeons, licensed in Virginia, Massachusetts, and Rhode Island. Dr Driver serves as Chair,
Board of Directors for the Wound Care Collaborative Community, an important collaboration with the FDA, CMS and the NIH and has received
a prestigious honor of receiving The Robert A. Warriner III, MD Memorial Award. She is System Wide Medical Director of the Wound Care
and Hyperbaric Centers at INOVA Healthcare in the DC Metropolitan area and is Professor, University of Virginia, School of Medicine. She
is also Fellow, Royal College of Physicians and Surgeons-Glasgow, PM and Inaugural Fellow, Assoc for the Advancement of Wound Care, FAAWC.
She currently serves as Honorary Visiting Professor at Cardiff University (UK) in the Department of Medicine and Professor at Barry University
(USA). She proudly serves as a member of the Wound Healing Society (WHS) Board of Directors and as member Board of Directors for the Critical
Limb Ischemia (CLI) Global Society. She has completed her tenure as president for the Advancement of Wound Care Association (AAWC) and
served for 9 years on the Board of Directors. Dr. Driver is a former Professor of Surgery in the Department of Orthopedics at Brown University
(Clinical) and Associate Professor of Surgery at Boston University. She has also chaired the Wound Care Experts and U.S. Food and Drug
Administration (“FDA”) Clinical Endpoints Project [WEF-CEP]. The project was successful in developing the research to expand
the wound healing clinical endpoints considered by FDA. She and her team proposed a combined effort to develop the Wound-care Experts/FDA-Clinical
Endpoints Project [WEF-CEP] to strategically identify clinically meaningful, evidence-based, and patient-centered wound care endpoints
that are relevant for clinical research and trials. The goal was to collaboratively work with the FDA to expand the list of acceptable
primary endpoints, recognizing that new and innovative treatments, devices, and drugs may not have complete healing as the focus. She
has served as a senior investigator for more than 70 important multi‑center randomized clinical trials, as well as developed and
supervised multiple research fellowship training programs. She has served and chaired multiple committees for large national and international
pivotal clinical trials, has authored over 150 publications and abstracts and is former Director, Translational Medicine at Novartis Institute
for BioMedical Research. Dr. Driver is credited with the development and directorship of multiple major multidisciplinary Limb Preservation-
Wound Healing Centers of Excellence, including Military/VA, Hospital and University based programs. Dr. Driver served on the Inaugural
Educational Committee at the American College of Wound Healing and Tissue Repair at University of Illinois School of Medicine and was
Scientific Director, Colorado Prevention Center, Wound Care Laboratory at the University of Colorado. Dr. Driver has held several leadership,
teaching, research and clinical positions at Academic Medical Centers, Veterans Administration Medical Centers, and Military Medical Centers.
Dr. Driver received a Doctorate of Podiatric Medicine and Surgery from the California College of Podiatric Medicine and Surgery and a
master’s degree in medical education from Samuel Merritt University.
Mr. David Fox has served
as a member of our board of directors since April 2020. Mr. Fox has served as a member of our board of directors since April 2020.
Mr. Fox was most recently a partner at Kirkland & Ellis LLP and served as a member of its Global Executive Management Committee until
2019. Prior to joining Kirkland, Mr. Fox was a partner at Skadden, Arps, Slate, Meagher & Flom LLP, where he was a member of
its governing committee. Mr. Fox is a member of the executive committee and board of directors of the Park Avenue Armory, which
enables artists to create and audiences to experience epic, adventurous work while also offering arts education programs at no cost to
public school students, and is chairman of the advisory board of New Alternatives for Children, an organization that provides support
to families caring for medically fragile children. Mr. Fox is the principal of David Fox & Co. LLC an advisory business and
CEO of Bald Productions LLC, a movie and television development and production company. He is also an advisor to Longacre Square
Partners, a communications and special situations advisory firm and to Nardello & Co, a global investigations firm. In addition,
Mr. Fox serves on the executive committee and the board of governors, and is an honorary fellow, of the Hebrew University, Jerusalem.
He holds an LL.B. degree from Jerusalem University, Israel.
Shmuel "Milky" Rubinstein has
served as a member of our board since August 2023. Mr. Rubinstein brings a distinguished record of leadership in the pharmaceutical and
biotechnology sectors to our board. Currently serving as Chairman of Trima Pharma, Milky's expertise extends across various prominent
board roles, including Strata Skin Sciences (SSKN), Medison Biotech, and Keystone Dental. Notably, he held the esteemed position of CEO
at Taro Pharmaceuticals (TARO), overseeing its successful acquisition by SUN Pharma. Milky's extensive board engagements also encompass
Kamada (KMDA), Exalenz (Acquired by VIVO), and Clal Biotechnology Industries (CBI). With a proven track record, Milky Rubinstein's insights
are poised to contribute significantly to our company's strategic direction and growth.
Stephen T. Wills
has served as a member of our Board since May 2017, as Chairman of our Board since October 2017 and as Executive Chairman of our board
since May 2019. Mr. Wills serves as Chief Financial Officer (since 1997) and Chief Operating Officer (since 2011) of Palatin Technologies,
Inc. (NYSE: PTN), a biopharmaceutical company developing targeted, receptor‑specific peptide therapeutics for the treatment of diseases
with significant unmet medical need and commercial potential. He also serves as Chief Financial Officer of Cactus Acquisition Corp. 1
Limited (Nasdaq: CCTS), a special purpose acquisition company, since 2021 until February 2024, when majority
ownership was acquired by a new Sponsor. Mr. Wills serves on the boards of Gamida Cell Ltd. (Nasdaq: GMDA), a leading cellular
and immune therapeutics company since March 2019 (audit chairperson and compensation and finance committee member) and of Amryt Pharma,
a biopharmaceutical company focused on developing and delivering treatments to help improve lives of patients with rare and orphan diseases
since September 2019 (chairman of audit committee and member of the compensation and finance committee) until
April 2023, when Amryt was acquired by Chiesi Farmaceutici. Mr. Wills also serves on the board of trustees and executive committee
of The Hun School of Princeton, a college preparatory day and boarding school since 2013, and its chairman since June 2018
until his retirement in June 2023. Mr. Wills served on the board of directors of Caliper Corporation, a psychological assessment
and talent development company since March 2016 and as chairman from December 2016 until December 2019, when Caliper was acquired by PSI.
Mr. Wills served as executive chairman and interim principal executive officer of Derma Sciences Inc. a provider of advanced wound care
product from December 2015 to February 2017, when Derma Sciences was acquired by Integra Lifesciences (Nasdaq: IART). Previously, Mr.
Wills served on the Board of Derma Sciences as the lead director and chairman of the audit committee from June 2000 to December 2015.
Mr. Wills served as the Chief Financial Officer of Derma Sciences from 1997 to 2000. Mr. Wills served as the president and Chief Operating
Officer of Wills, Owens & Baker, P.C., a public accounting firm from 1991 to 2000. Mr. Wills, a certified public accountant, earned
his Bachelor of Science in accounting from West Chester University, and a Master of Science in taxation from Temple University.
There are no family relationships among any of our executive officers or directors.
There are no arrangements or understandings with major shareholders, customers, suppliers
or others, pursuant to which any person referred to above was selected as a director or member of senior management.
B.
Compensation
Compensation of Directors and Executive Officers
The table below reflects the compensation granted to our five most
highly compensated officers during or with respect to the year ended December 31, 2023. All amounts reported in the table reflect the
cost to the company, as recognized in our financial statements for the year ended December 31, 2023.
|
Name and Position
|
|
Salary &
Social
Benefits(1) |
|
|
Bonus |
|
|
Share-Based
Payment(2)
|
|
|
Other Compensation
(3) |
|
|
Total |
|
| |
|
(thousand U.S. dollars)(4) |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ofer Gonen, Chief Executive Officer
|
|
|
480 |
|
|
|
152 |
|
|
|
484 |
|
|
|
17 |
|
|
|
1,133 |
|
|
Ety Klinger, Chief Research
& Development Officer |
|
|
297 |
|
|
|
58 |
|
|
|
102 |
|
|
|
22 |
|
|
|
479 |
|
|
Yaron Meyer, Executive Vice President, General Counsel
& Corporate Secretary |
|
|
250 |
|
|
|
44 |
|
|
|
80 |
|
|
|
5 |
|
|
|
379 |
|
|
Hani Luxenburg, Chief Financial Officer (5)
|
|
|
166 |
|
|
|
43 |
|
|
|
9 |
|
|
|
26 |
|
|
|
244 |
|
|
Boaz Gur-Lavie, former Chief
Financial Officer (6) |
|
|
140 |
|
|
|
22 |
|
|
|
46 |
|
|
|
16 |
|
|
|
224 |
|
|
(1)
|
Represents the officer’s gross salary plus payment of mandatory social benefits
made by the company on behalf of such officer. Such benefits may include, to the extent applicable to the executive, payments, contributions
and/or allocations for savings funds (e.g., Managers’ Life Insurance Policy), education funds (referred to in Hebrew as “keren
hishtalmut”), pension, severance, risk insurances (e.g., life or work disability insurance) and payments for social security.
|
|
(2) |
Represents the equity‑based compensation expenses recorded in the company’s
consolidated financial statements for the year ended December 31, 2023 based on the options’ grant date fair value in accordance
with accounting guidance for equity‑based compensation. |
|
(3) |
Represents the other benefits to such officer, which includes either or both of (i)
car expenses, including lease costs, gas and maintenance, provided to the officers, (ii) vacation benefits and (iii) severance payment.
|
|
(4) |
Converted (i) from NIS into U.S. dollars at the rate of NIS 3.69 = U.S$1, based on
the average representative rate of exchange between the NIS and the U.S. dollar in the year ended December 31, 2023 as reported by the
Bank of Israel in the year ended December 31, 2023. |
|
(5) |
Ms. Luxenburg Joined the Company in May 2023. |
|
(6) |
Mr. Gur Lavie worked in the Company until July 2023. |
The aggregate compensation paid and equity‑based
compensation and other payments expensed by us and our subsidiaries to our directors and executive officers with respect to the year ended
December 31, 2023 was $3.8 million. As of December 31, 2023, options to purchase 467,146 ordinary shares, exercisable at a weighted average
exercise price of $15.3 per share, and restricted share units (“RSUs”) that may be settled for 44,067 ordinary shares, in
each case granted to our directors and executive officers, were outstanding under our equity incentive plans. We do not have any written
agreements with any director for benefits upon the termination of such director’s relationship with our company or its subsidiaries.
Employment Agreements with Executive Officers
We have entered into written employment agreements with all of
our executive officers, which include standard provisions for a company in our industry regarding non‑competition/solicitation,
confidentiality of information and assignment of inventions. Except for Ofer Gonen, our Chief Executive Officer, our executive officers
will not receive benefits upon the termination of their respective employment with us, other than payment of salary and benefits (and
limited accrual of vacation days) during the required notice period for termination of their employment, which varies for each individual.
Upon termination of his employment, Mr. Gonen is entitled to a one‑time termination payment of six months of salary.
Directors’ Service Contracts
Other than with respect to our directors that are also executive
officers, there are no arrangements or understandings between us, on the one hand, and any of our directors, on the other hand, providing
for benefits upon termination of their service as directors of our company.
Share Incentive Plans
2014 Equity Incentive Plan
In March 2014, we adopted and obtained shareholder approval for
our 2014 Equity Incentive Plan, which was amended as of December 18, 2018 (the “2014 Plan”). The 2014 Plan provides for the
grant of options, restricted shares, RSUs and other share‑based awards to our and our subsidiaries’ and affiliates’
directors, employees, officers, consultants and advisors, among others and to any other person whose services are considered valuable
to us or them, to continue as service providers, to increase their efforts on our behalf or behalf of a subsidiary or affiliate and to
promote the success of our business. Following the approval of the 2014 Plan by the Israeli tax authorities, we are only granting options
or other equity incentive awards under the 2014 Plan, which shares will be rolled over to a new share incentive plan— the 2024 Share
Incentive Plan— that we will adopted and bring for shareholder approval soon.
The 2014 Plan is administered by our board of directors or by a
committee designated by the board of directors, which determine, subject to Israeli law, the grantees of awards and the terms of the grant,
including exercise prices, vesting schedules, acceleration of vesting and the other matters necessary in the administration of the 2014
Plan. The 2014 Plan enables us to issue awards under various tax regimes, including, without limitation, pursuant to Sections 102 and
3(i) of the Ordinance, as discussed under “-2003 Share Incentive Plan” above, and under Section 422 of the U.S. Internal Revenue
Code of 1986, as amended (the “Code”).
Options granted under the 2014 Plan to U.S. residents may qualify
as “incentive stock options” within the meaning of Section 422 of the Code, or may be non‑qualified. The exercise price
for “incentive stock options” must not be less than the fair market value on the date on which an option is granted, or 110%
of the fair market value if the option holder holds more than 10% of our share capital.
We currently intend to grant awards under the 204 Plan under the
capital gains track of Section 102(b)(2) of the Ordinance only to our employees, directors and officers who are not controlling shareholders
and are considered Israeli residents.
Awards under the 2014 Plan may be granted until ten years from
the date on which the 2014 Plan was approved by our board of directors.
Options granted under the 2014 Plan generally vest over three or
four years commencing on the date of grant, such that 33% or 25%, respectively, vests annually on the anniversary of the date of grant.
Options, other than certain incentive share options, that are not exercised within ten years from the grant date expire, unless otherwise
determined by our board of directors or its designated committee, as applicable. Share options that qualify as “incentive stock
options” and are granted to a person holding more than 10% of our voting power will expire within five years from the date of the
grant. In the event of the death of a grantee while employed by or performing service for us or a subsidiary or within three months thereafter,
or the termination of a grantee’s employment or services for reasons of disability, the grantee, or in the case of death, his or
her legal successor, may exercise options that have vested prior to termination within a period of one year from the date of disability
or death. If we terminate a grantee’s employment or service for cause, all of the grantee’s vested and unvested options will
expire on the date of termination. If a grantee’s employment or service is terminated for any other reason, the grantee may exercise
his or her vested options within three months of the date of termination. Any expired or unvested options return to the pool for reissuance.
In the event of a merger or consolidation of our company or a sale of all, or substantially all, of our shares or assets or other transaction
having a similar effect on us, then without the consent of the option holder, our board of directors or its designated committee, as applicable,
may but is not required to (i) cause any outstanding award to be assumed or an equivalent award to be substituted by such successor corporation,
or (ii) in case the successor corporation refuses to assume or substitute the award (a) provide the grantee with the option to exercise
the award as to all or part of the shares or (b) cancel the options against payment in cash in an amount determined by the board of directors
or the committee as fair in the circumstances. Notwithstanding the foregoing, our board of directors or its designated committee may upon
such event amend or terminate the terms of any award, including conferring the right to purchase any other security or asset that the
board of directors shall deem, in good faith, appropriate. Our board of directors or its designated committee may, in its discretion,
approve that any awards granted under the 2014 Plan shall be subject to additional conditions in the case of a merger or a consolidation.
Restricted share awards are ordinary shares that are awarded to
a participant subject to the satisfaction of the terms and conditions established by the board of directors or a committee designated
by the board of directors. Until such time as the applicable restrictions lapse, restricted shares are subject to forfeiture and may not
be sold, assigned, pledged or otherwise disposed of by the participant who holds those shares. Generally, if a grantee’s employment
or service is terminated for any reason prior to the expiration of the time when the restrictions lapse, shares that are still restricted
will be forfeited.
Prospective 2024 Share Incentive Plan
Because the 2014 Plan expires in March 2024, we expect to adopt
a new share incentive plan—the 2024 Share Incentive Plan (the “2024 Plan”) - in the near future and bring it to our
shareholders for approval. Outstanding grants under the 2014 Plan will remain subject to the 2014 Plan even after the expiration of that
plan, but any ordinary shares available under the 2014 Plan as of the adoption of the 2024 Plan, or that subsequently become available
under the 2014 Plan due to the expiration, cancellation, forfeiture or other surrender of outstanding grants under the 2014 Plan, will
become available for new grants under the 2024 Plan. We will provide a description of the new 2024 Plan in the proxy statement for the
general meeting of shareholders at which such plan will be presented for shareholder approval, and in subsequent documents filed with
or furnished to, the SEC.
The following table provides information regarding the outstanding
options to purchase our ordinary shares, and RSUs held by each of our directors and executive officers who beneficially owns greater than
1% of our ordinary shares (after including shares underlying options or RSUs) as of March 15, 2024:
|
Name |
|
Number of
Options |
|
Number of
RSUs |
|
Grant Date |
|
Exercise
Price - $ |
|
Vested
Options/RSU's |
|
Expiration Date |
|
Ofer Gonen |
|
85,700 |
|
|
|
07/06/2022 |
|
14.42 |
|
37,500 |
|
06/06/2032 |
| |
|
|
|
20,089 |
|
07/06/2022 |
|
0 |
|
15,625 |
|
06/06/2032 |
| |
|
86,000 |
|
|
|
31/05/2023 |
|
11.89 |
|
21,500 |
|
30/05/2033 |
C.
Board Practices
Board of Directors
Authorities
Under the Israeli Companies Law, the management of our company
is vested in our board of directors. Our board of directors may exercise all powers and may take all actions that are not specifically
granted to our shareholders or to management. Our executive officers are responsible for our day‑to‑day management and have
individual responsibilities established by our board of directors. Our Chief Executive Officer is appointed by, and serves at the discretion
of, our board of directors, subject to the employment agreement that we have entered into with him. All other executive officers are also
appointed by our board of directors, and are subject to the terms of any applicable employment agreements that we may enter into with
them.
Composition
Under our articles of association, our board of directors must
consist of at least five and not more than nine directors, including, to the extent then required to appoint them, at least two external
directors, who may be required to be appointed under the Israeli Companies Law. At any time the minimum number of directors (other than
the external directors) shall not fall below three. Other than external directors, for whom special election requirements apply under
the Israeli Companies Law when they are required to be elected, as detailed below, the Israeli Companies Law and our articles of association
provide that directors are elected annually at the general meeting of our shareholders by a vote of the holders of a majority of the voting
power represented present and voting, in person or by proxy, at that meeting. We have only one class of directors.
In accordance with the exemption available to foreign private issuers
under Nasdaq rules, we are not required to comply with the requirements of the Nasdaq rules with regard to having a majority of independent
directors on our board of directors, as long as we follow Israeli law and practice, in accordance with which our board of directors includes
at least two external directors. However, because we have elected under the Israeli Companies Law regulations to opt-out from compliance
with Israeli law requirements related to appointment of external directors and audit and compensation committee composition (as described
under “External Directors” below), we are not permitted to also exempt ourselves from the Nasdaq majority independent directors
requirement. Our board of directors has determined that six of our eight current directors are independent under the Nasdaq Stock Market
listing rules such that we comply with the Nasdaq majority independence rule.
Under the Israeli Companies Law, our board of directors must determine
the minimum number of directors who are required to have accounting and financial expertise. In determining the number of directors required
to have such expertise, our board of directors must consider, among other things, the type and size of the company and the scope and complexity
of its operations. Our board of directors has determined that the minimum number of directors of our company who are required to have
accounting and financial expertise is one.
Nominations and Election
In accordance with the Nasdaq listing rules, we have appointed
a committee of the board of directors that is authorized to recommend to our board, for nomination for election by our shareholders,
director nominees. Please see “Committees of the Board of Directors— Nominating and Governance Committee” below for
more information.
Under the Israeli Companies Law and our articles of association,
nominees for directors may also be proposed by any shareholder holding at least 1% of our outstanding voting power. However, any such
shareholder may propose a nominee only if a written notice of such shareholder’s intent to propose a nominee has been given to our
Secretary (or, if we have no such Secretary, our Chief Executive Officer). Pursuant to our Articles of Association, any such notice must
include certain information, including, among other things, a description of all arrangements between the nominating shareholder and the
proposed director nominee(s) and any other person pursuant to which the nomination(s) are to be made by the nominating shareholder, the
consent of the proposed director nominee(s) to serve as our director(s) if elected and a declaration signed by the nominee(s) declaring
that there is no limitation under the Israeli Companies Law preventing their election, and that all of the information that is required
under the Israeli Companies Law to be provided to us in connection with such election has been provided. Under the Israeli Companies Law
regulations, any such shareholder nomination must be delivered to our registered Israeli office within seven days after we publish notice
of our upcoming annual general meeting of shareholders (or within 14 days after we publish a preliminary notification of an upcoming annual
general meeting).
In addition, our articles of association allow our board of directors
to appoint directors to fill vacancies on our board of directors for a term of office equal to the remaining period of the term of office
of the director(s) whose office(s) have been vacated.
We are not a party to, and are not aware of, any voting agreements
among our shareholders. In addition, there are no family relationships among our executive officers and directors.
External Directors
Under the Israeli Companies Law, the boards of directors of companies,
whose shares are publicly traded, including companies with shares traded in the United States, are generally required to include at least
two members who qualify as external directors.
Under regulations promulgated under the Israeli Companies Law,
Israeli public companies whose shares are traded on certain U.S. stock exchanges, such as the Nasdaq Global Market, and that lack a controlling
shareholder (as defined below) are exempt from the requirement to appoint external directors. Any such company is also exempt from the
Israeli Companies Law requirements related to the composition of the audit and compensation committees of the Board. Eligibility for these
exemptions is conditioned on compliance with U.S. stock exchange listing rules related to majority Board independence and the composition
of the audit and compensation committees of the Board, as applicable to all listed domestic U.S. companies. Eligibility is furthermore
conditioned on our election of a female or male director at any time when we hold elections of directors and the Board is then composed
of solely male or solely female members.
On December 5, 2022, in light of our Board’s determination
that Clal Biotechnology Industries Ltd. was no longer a “controlling shareholder” of our company under the Israeli Companies
Law definition (provided further below), the Board elected, pursuant to the Israeli Companies Law regulations, to exempt our company from
compliance with the (i) requirement to appoint external directors, and (ii) required composition of the audit committee and compensation
committees of the Board under the Israeli Companies Law. At the time that it made that election, our Board affirmatively determined that
we meet the conditions for exemption from the external director requirement, including that a majority of the members of our Board, along
with each of the members of the audit and compensation committees of the Board, are independent under the Nasdaq Listing Rules. Our Board
has confirmed subsequently in an ongoing manner that we continue to fulfill those conditions for exemption from the Israeli Companies
Law requirements related to (i) the appointment of external directors, and (ii) the composition of the audit committee and compensation
committees of the Board.
Our election to exempt our company from compliance with the
external director and related requirements can be reversed at any time by our Board, in which case we would need to hold a shareholder
meeting to once again to appoint external directors, whose election would be for a three-year term. The election of each external director
would require a majority vote of the shares present and voting at a shareholders meeting, provided that either:
• the majority voted in favor of election includes a majority
of the shares held by non-controlling shareholders who do not have a personal interest in the election of the external director (other
than a personal interest not deriving from a relationship with a controlling shareholder) that are voted at the meeting, excluding abstentions,
which we refer to as a disinterested majority; or
• the total number of shares held by non-controlling,
disinterested shareholders (as described in the previous bullet-point) voted against the election of the director does not exceed two
percent (2%) of the aggregate voting rights in the company.
The term “controlling shareholder” is defined in
the Israeli Companies Law as a shareholder with the ability to direct the activities of the company, other than by virtue of being an
office holder. A shareholder is presumed to be a controlling shareholder if the shareholder holds 50% or more of the voting rights in
a company or has the right to appoint the majority of the directors of the company or its general manager (i.e., its CEO).
For further information concerning the Israeli Companies Law provisions
related to external directors, please see “Item 6. Directors, Senior Management and Employees- C. Board Practices- Board of Directors-
External Directors” in our annual report on Form 20-F for the year ended December 31, 2022, which we filed with the SEC on March
16, 2023.
Leadership Structure of the Board
In accordance with the Israeli Companies Law and our articles of
association, our board of directors is required to appoint one of its members to serve as chairman of the board of directors. Our board
of directors has appointed Homi Shamir to serve as chairman of the board of directors.
Committees of the Board of Directors
Audit Committee
Israeli Companies Law composition requirements
Under the Israeli Companies Law, we are required to have an audit
committee comprised of at least three directors. To the extent we are then required to appoint external directors, this committee must
include all of the external directors, one of whom must serve as chairman of the committee. There are additional requirements as to the
composition of the audit committee under the Israeli Companies Law. However, when we elected to exempt our company from the external director
requirement, we concurrently elected to exempt our company from all of such requirements (which exemption is conditioned on our fulfillment
of all Nasdaq listing requirements related to the composition of the audit committee).
Nasdaq listing rules composition requirements
Under the Nasdaq Stock Market listing rules, we are required to
maintain an audit committee consisting of at least three independent directors, each of whom is financially literate and one of whom has
accounting or related financial management expertise.
Our audit committee consists of Stephen T. Wills
(chairperson), Vickie R. Driver and Nachum Shamir, each of whom is an independent director
in accordance with Rule 10A‑3(b)(1) under the Exchange Act and satisfies the independent director requirements under the Nasdaq
Stock Market listing rules. All members of our audit committee meet the requirements for financial literacy under the applicable listing
rules of the Nasdaq Stock Market. Our board of directors has determined that Stephen T Wills is an “audit committee financial expert,”
as defined in the SEC regulations, and possesses accounting and financial expertise, as defined under the Israeli Companies Law.
Audit committee role
Our audit committee provides assistance to our board of directors
in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting, internal control
and legal compliance functions by pre‑approving the services performed by our independent accountants and reviewing their reports
regarding our accounting practices and systems of internal control over financial reporting. Our audit committee also oversees the audit
efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants are independent
of management.
Our board of directors has adopted an audit committee charter that
sets forth the responsibilities of the audit committee consistent with the rules and regulations of the SEC and the Nasdaq Stock Market
listing rules, as well as the requirements for such committee under the Israeli Companies Law, including the following:
|
|
• |
overseeing our independent registered public accounting firm and recommending the
engagement, compensation or termination of engagement of our independent registered public accounting firm to the board of directors in
accordance with Israeli law; |
|
|
• |
recommending the engagement or termination of the person filling the office of our
internal auditor; |
|
|
• |
recommending the terms of audit and non‑audit services provided by the independent
registered public accounting firm for pre‑approval by our board of directors; |
| |
• |
pre-approving all audit, audit-related and all permitted non-audit services, and related
fees and terms, to be provided to the Company by the independent auditor under applicable law and regulations; |
| |
• |
Establishing policies for hiring employees or former employees of the independent
auditor in accordance with applicable law and regulations; |
| |
• |
reviewing periodically with management, the internal auditor and the independent auditor,
the adequacy and effectiveness of the Company’s system of internal control over financial reporting; |
| |
• |
evaluating whether management is effectively communicating to employees and other
persons retained by MediWound the importance of internal accounting and financial control effectiveness; |
| |
• |
reviewing with management and the independent auditor the annual and quarterly financial
statements of MediWound prior to filing (or submission, as the case may be) with the SEC; |
| |
• |
discussing with management, and review prior to submission, any responses to SEC comments
regarding the Company’s financial statements or financial reporting; |
| |
• |
discussing with management and MediWound’s independent auditors generally the
types of financial information (including earnings guidance) to be disclosed in earnings press releases and earnings calls, as well as
to analysts and rating agencies; |
| |
• |
reviewing and discussing with management and MediWound independent auditors MediWound’s
earnings press releases, including the use of any pro forma, adjusted or other non-GAAP financial information, before their release to
the public; |
| |
• |
reviewing with the MediWound’s general counsel and/or external counsel legal
and regulatory matters that could have a material impact on the financial statements;
|
| |
• |
establishing procedures for (i) the receipt, retention, and treatment of complaints
received by MediWound regarding accounting, internal accounting controls or auditing matters; and (ii) the confidential, anonymous submission
by employees of MediWound of concerns regarding questionable accounting or auditing matters, and review any complaints or concerns received
pursuant to such procedures; |
| |
• |
reviewing with management and the independent auditor risks of material misstatements
due to fraud, and the process and controls implemented by the Company to manage the risks; |
| |
• |
recommending to the Board the retention and termination of the internal auditor, and
the internal auditor’s engagement fees and terms, in accordance with the Companies Law, approve the internal auditor’s work
plan, and review and discuss the internal auditor’s work on a quarterly basis; |
| |
• |
reviewing and monitoring, as appropriate, (i) litigation or other legal matters that
could have a significant impact on MediWound’s financial results; and (ii) significant findings of any examination by regulatory
authorities or agencies, in the areas of securities, accounting or tax; |
| |
• |
receiving reports of suspected business irregularities and legal compliance issues
through periodic and, when appropriate, immediate reporting by members of MediWound’s management, legal counsel, the independent
or internal auditor or pursuant to any “whistleblower policy” adopted by the Committee. |
| |
• |
establishing procedures for handling complaints by MediWound’s employees with
respect to deficiencies in its business operations, including the protection to be granted to such complaining employees; |
| |
• |
overseeing MediWound’s policies and procedures regarding compliance with applicable
financial and accounting related standards, rules and regulations; |
| |
• |
reviewing and considering the approval of related party transactions, including transactions
between MediWound and a controlling shareholder (as defined under the Israeli Companies Law) or a transaction with another person in which
a controlling shareholder has a personal interest, and transactions involving an office holder of MediWound (as defined in the Israeli
Companies Law) that may present a conflict of interest between the duties of such office holder to the Company and his or her personal
interests, in each case in accordance with Nasdaq listing rules, the Israeli Companies Law or as referred by the Board; |
| |
• |
confirming that MediWound’s independent auditors are informed of the committee’s
understanding of the Company’s related party transactions that are significant to the Company; and reviewing and discussing with
the Company’s independent auditors the auditors’ evaluation of the Company’s identification of, accounting for, and
disclosure of, its related party transactions, including any significant matters arising from the audit regarding MediWound’s related
party transactions; |
| |
• |
discussing MediWound’s policies with respect to risk assessment and risk management,
and reviewing contingent liabilities and risks that may be material to MediWound and relevant major legislative and regulatory developments
that could materially impact MediWound’s contingent liabilities and risks; |
| |
•
|
reviewing periodically the Company’s major financial risk exposures and the
Company’s policies for managing such risks; |
| |
• |
conducting or authorizing investigations into any matters within the committee’s
scope of responsibilities; and |
| |
• |
reviewing and approving any material change or waiver in MediWound’s Code of
Conduct regarding directors or executive officers, and disclosures made in the Company’s annual report. |
Our audit committee may not approve any actions requiring its approval
(see “-Approval of Related Party Transactions Under Israeli Law”), unless at the time of the approval a majority of the committee’s
members are present, which majority consists of unaffiliated directors including at least one external director, to the extent we then
have external directors serving on the Board).
Compensation Committee
Israeli Companies Law composition requirements
Under the Israeli Companies Law, the board of directors of a public
company must appoint a compensation committee. If a company is required to appoint external directors, the committee must consist of at
least three members, including all of the external directors, one of whom must serve as chairman of the committee. There are additional
requirements as to the composition of the compensation committee under the Companies Law. However, when we elected to exempt our company
from the external director requirement, we concurrently elected to exempt our company from all of such requirements (including the three-member
minimum). Our exemption under the Companies Law is conditioned on our fulfillment of all Nasdaq listing requirements related to the composition
of the compensation committee.
Israeli Companies Law committee duties
The duties of the compensation committee include the recommendation
to the company’s board of directors of a policy regarding the terms of engagement of office holders, which we refer to as a compensation
policy. That policy must be adopted by the company’s board of directors, after considering the recommendations of the compensation
committee, and must be approved by the company’s shareholders, which approval requires what we refer to as a Special Majority Approval
for Compensation. A Special Majority Approval for Compensation requires shareholder approval by a majority vote of the shares present
and voting at a meeting of shareholders called for such purpose, provided that either (a) such majority includes at least a majority of
the shares held by all shareholders who are not controlling shareholders and do not have a conflict of interest (referred to under the
Israeli Companies Law as a “personal interest”) in such compensation arrangement or (b) the total number of shares of non-controlling
shareholders and shareholders who do not have a personal interest in the compensation arrangement and who vote against the arrangement
does not exceed 2% of the company’s aggregate voting rights.
Compensation policy requirements
We have adopted a compensation policy, most recently at the extraordinary
general meeting of shareholders held on November 28, 2022, which policy serves as the basis for decisions concerning the financial terms
of employment or engagement of office holders, including exculpation, insurance, indemnification or any monetary payment or obligation
of payment or other benefit in respect of employment or engagement. Under the Israeli Companies Law, the compensation policy must relate
to certain factors, including advancement of the company’s objectives, the company’s business plan and its long-term strategy,
and creation of appropriate incentives for office holders. It must also consider, among other things, the company’s risk management,
size and the nature of its operations. The compensation policy must furthermore consider the following additional factors:
|
|
•
|
the knowledge, skills, expertise and accomplishments of the relevant office holder;
|
|
|
•
|
the office holder’s roles and responsibilities and prior compensation agreements
with him or her;
|
|
|
•
|
the relationship between the terms offered and the average compensation of the other
employees of the company, including those employed through manpower companies;
|
|
|
•
|
the impact of disparities in salary upon work relationships in the company;
|
|
|
•
|
the possibility of reducing variable compensation at the discretion of the board of
directors;
|
|
|
•
|
the possibility of setting a limit on the exercise value of non-cash variable equity-based
compensation; and
|
|
|
•
|
as to severance compensation, the period of service of the office holder, the terms
of his or her compensation during such service period, the company’s performance during that period of service, the person’s
contribution towards the company’s achievement of its goals and the maximization of its profits, and the circumstances under which
the person is leaving the company.
|
The compensation policy
must also include the following principles:
|
|
•
|
the link between variable compensation and long-term performance, which variable compensation
shall, other than office holder who report to the CEO, be primarily based on measurable criteria;
|
|
|
•
|
the relationship between variable and fixed compensation, and the ceiling for the
value of variable compensation;
|
|
|
•
|
the conditions under which an office holder would be required to repay compensation
paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated
in the company’s financial statements;
|
|
|
•
|
the minimum holding or vesting period for variable, equity-based compensation; and
|
|
|
•
|
maximum limits for severance compensation.
|
The
compensation committee is responsible for (a) recommending the compensation policy to the company’s board of directors for its approval
(and subsequent approval by its shareholders) and (b) duties related to the compensation policy and to the compensation of a company’s
office holders as well as functions previously fulfilled by a company’s audit committee with respect to matters related to approval
of the terms of engagement of office holders, including:
|
|
•
|
recommending whether a compensation policy should continue in effect, if the then-current
policy has a term of greater than three years (approval of either a new compensation policy or the continuation of an existing compensation
policy must in any case occur every three years, other than following a company’s initial public offering, in which case such approval
must occur within 5 years of the initial public offering);
|
|
|
•
|
recommending to the board of directors periodic updates to the compensation policy
and assessing implementation of the compensation policy;
|
|
|
•
|
approving compensation terms of executive officers, directors and employees that require
approval of the compensation committee;
|
|
|
•
|
determining whether the compensation terms of a chief executive officer nominee, which
were determined pursuant to the compensation policy, will be exempt from approval of the shareholders because such approval would harm
the ability to engage with such nominee; and
|
|
|
•
|
determining, subject to the approval of the board and under special circumstances,
whether to override a determination of the company’s shareholders regarding certain compensation related issues.
|
A copy of our current compensation policy serves as an exhibit to this annual report
on Form 20-F.
Nasdaq listing rules composition
requirements
Under Nasdaq corporate governance rules, we are required to maintain
a wholly-independent compensation committee consisting of at least two independent directors or, if we choose to follow requirements under
Israeli law, we must disclose that fact in this annual report. Each of the members of the compensation committee is required to be independent
under the Nasdaq rules relating to compensation committee members and Rule 10C‑1(b)(1) under the Exchange Act, which are different
than the general test for independence of board members.
Our compensation committee consists of Nachum Shamir (chairperson),
David Fox and Stephen T. Wills, each of whom is an independent director under the Nasdaq Stock Market listing rules and each of whom satisfies
the above-described additional requirements for compensation committee members under the Nasdaq rules and Exchange Act.
Compensation committee charter and role
Our board of directors has adopted a compensation committee charter
setting forth the responsibilities of the compensation committee, which include:
|
|
•
|
the responsibilities set forth in the compensation policy;
|
|
|
•
|
reviewing and approving the granting of options and other incentive awards to the
extent such authority is delegated by our board of directors; and
|
|
|
• |
reviewing, evaluating and making recommendations regarding the compensation and benefits
for our non-employee directors. |
Nominating and Governance Committee
Under Nasdaq corporate governance rules, director nominees must either
be selected, or recommended for the Board's selection, either by independent directors constituting a majority of the Board's independent
directors in a vote in which only independent directors participate, or a nominations committee comprised solely of independent directors.
If we choose to exempt ourselves from that requirement in accordance with our home country practices, we must disclose that fact in this
annual report. Each of the members of the nominating committee is required to be independent under the Nasdaq rules.
Our Board has established a nominating and governance committee, whose members consist of Nachum Shamir
and David Fox, each of whom has been determined by our Board to meet that Nasdaq independence requirement, with Mr. Fox serving as chair.
Our board of directors has adopted a nominating and governance committee charter setting forth the responsibilities of the committee,
which include:
|
|
•
|
overseeing and assisting our board in reviewing and recommending nominees for election of directors;
|
|
|
•
|
assessing the performance of the members of our board; and
|
|
|
•
|
establishing and maintaining effective corporate governance policies and practices, including, but not
limited to, developing and recommending to our board a set of corporate governance guidelines applicable to our business.
|
Research and Development Committee
Our Board has established a research and development committee,
which is composed of Vickie R Driver, Stephen T. Wills and Sharon Malka, with Dr. Driver serving as chairperson. The primary functions
of the research and development committee include:
|
|
•
|
overseeing the Company's scientific, technical, research and development strategy,
and the implementation thereof; and
|
|
|
•
|
advising our board of directors and management regarding program prioritization, clinical
development strategy, regulatory strategy and interactions, and related matters. |
Board Diversity Matrix (As of March 1, 2024)
|
Country of Principal Executive Offices
|
Israel |
|
Foreign Private Issuer
|
Yes |
|
Disclosure Prohibited under Home Country Law
|
No |
|
Total Number of Directors
|
5 |
| |
|
|
|
|
|
|
|
|
Part I: Gender Identity
|
|
|
|
|
|
|
|
|
Directors |
1 |
|
4 |
|
0 |
|
0 |
|
Part II: Demographic Background
|
|
|
|
|
|
|
|
|
Underrepresented Individual in Home Country
Jurisdiction |
* |
|
|
|
|
|
|
|
LGBTQ+
|
* |
|
|
|
|
|
|
|
Did Not Disclose Demographic Background
|
* |
|
|
|
|
|
|
(*) There are no directors who fall into any demographic background category who have agreed to disclose
that information publicly.
Internal Auditor
Under the Israeli Companies Law, the board of directors of an Israeli
public company must appoint an internal auditor recommended by the audit committee. An internal auditor may not be:
|
|
•
|
a person (or a relative of a person) who holds 5% or more of the company’s outstanding
shares or voting rights;
|
|
|
•
|
a person (or a relative of a person) who has the power to appoint a director or the
general manager of the company (i.e., the chief executive officer);
|
|
|
•
|
an office holder (including a director) of the company (or a relative thereof); or
|
|
|
•
|
a member of the company’s independent accounting firm, or anyone on its behalf.
|
The role of the internal auditor is to examine, among other things,
our compliance with applicable law and orderly business procedures.
The audit committee is required to oversee the activities and to
assess the performance of the internal auditor as well as to review the internal auditor’s work plan. Our internal auditor is Mr.
Yisrael Gewirtz.
Fiduciary Duties of Directors and Executive Officers
The Israeli Companies Law codifies the fiduciary duties that office
holders owe to a company. Each person listed in the table under “Executive Officers and Directors” is an office holder under
the Israeli Companies Law.
An office holder’s fiduciary duties consist of a duty of
care and a duty of loyalty. The duty of care requires an office holder to act with the level of care with which a reasonable office holder
in the same position would have acted under the same circumstances. The duty of loyalty requires that an office holder act in good faith
and in the best interests of the company.
The duty of care includes a duty to use reasonable means to obtain:
|
|
•
|
information on the advisability of a given action brought for his or her approval
or performed by virtue of his or her position; and
|
|
|
•
|
all other important information pertaining to any such action.
|
The duty of loyalty includes a duty to:
|
|
•
|
refrain from any conflict of interest between the performance of his or her duties
to the company and his or her other duties or personal affairs;
|
|
|
•
|
refrain from any activity that is competitive with the business of the company;
|
|
|
•
|
refrain from exploiting any business opportunity of the company to receive a personal
gain for himself or herself or others; and
|
|
|
• |
disclose to the company any information or documents relating to the company’s
affairs which the office holder received as a result of his or her position as an office holder. |
Disclosure of personal interests of an office holder and approval of certain transactions
The Israeli Companies Law requires that an office holder promptly
disclose to the board of directors any personal interest that he or she may be aware of and all related material information or documents
concerning any existing or proposed transaction with the company. An interested office holder’s disclosure must be made promptly
and in any event no later than the first meeting of the board of directors at which the transaction is considered. A personal interest
includes an interest of any person in an act or transaction of a company, including a personal interest of such person’s relative
or of a corporate body in which such person or a relative of such person is a 5% or greater shareholder, director or general manager or
in which he or she has the right to appoint at least one director or the general manager, but excluding a personal interest stemming from
one’s ownership of shares in the company.
A personal interest furthermore includes the personal interest
of a person for whom the office holder holds a voting proxy or the personal interest of the office holder with respect to his or her vote
on behalf of a person for whom he or she holds a proxy even if such shareholder has no personal interest in the matter. An office holder
is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction
that is not considered an extraordinary transaction. Under the Israeli Companies Law, an extraordinary transaction is defined as any of
the following:
|
|
•
|
a transaction other than in the ordinary course of business;
|
|
|
•
|
a transaction that is not on market terms; or
|
|
|
•
|
a transaction that may have a material impact on a company’s profitability,
assets or liabilities.
|
If it is determined that an office holder has a personal interest
in a transaction which is not an extraordinary transaction, approval by the board of directors is required for the transaction, unless
the company’s articles of association provide for a different method of approval. Further, so long as an office holder has disclosed
his or her personal interest in a transaction, the board of directors may approve an action by the office holder that would otherwise
be deemed a breach of his or her duty of loyalty. However, a company may not approve a transaction or action that is not in the best interest
of the company or that is not performed by the office holder in good faith. An extraordinary transaction in which an office holder has
a personal interest requires approval first by the company’s audit committee and subsequently by the board of directors. The compensation
of, or an undertaking to indemnify or insure, an office holder who is not a director requires approval first by the company’s compensation
committee, then by the company’s board of directors. If such compensation arrangement or an undertaking to indemnify or insure is
inconsistent with the company’s stated compensation policy, or if the office holder is the chief executive officer (apart from a
number of specific exceptions), then such arrangement is further subject to a Special Majority Approval for Compensation. Arrangements
regarding the compensation, indemnification or insurance of a director require the approval of the compensation committee, board of directors
and shareholders by ordinary majority, in that order, and under certain circumstances, a Special Majority Approval for Compensation. Generally,
a person who has a personal interest in a matter which is considered at a meeting of the board of directors or the audit committee may
not be present at such a meeting or vote on that matter unless the chairman of the relevant committee or board of directors (as applicable)
determines that he or she should be present in order to present the transaction that is subject to approval. If a majority of the members
of the audit committee or the board of directors (as applicable) has a personal interest in the approval of a transaction, then all directors
may participate in discussions of the audit committee or the board of directors (as applicable) on such transaction and the voting on
approval thereof, but shareholder approval is also required for such transaction.
Disclosure of personal interests of controlling shareholders and
approval of certain transactions
Pursuant to Israeli law, the disclosure requirements regarding
personal interests that apply to directors and executive officers also apply to a controlling shareholder of a public company. In the
context of a transaction involving a shareholder of the company, a controlling shareholder also includes a shareholder who holds 25% or
more of the voting rights in the company if no other shareholder holds more than 50% of the voting rights in the company. For this purpose,
the holdings of all shareholders who have a personal interest in the same transaction will be aggregated. The approval of the audit committee
or the compensation committee, the board of directors and the shareholders of the company, in that order, is required for (a) extraordinary
transactions with a controlling shareholder or in which a controlling shareholder has a personal interest, (b) the engagement with a controlling
shareholder or his or her relative, directly or indirectly, including through a company under the control of the controlling shareholder,
for the provision of services to the company, (c) the terms of engagement and compensation of a controlling shareholder or his or her
relative who is an office holder or (d) the employment of a controlling shareholder or his or her relative by the company, other than
as an office holder. In addition, the shareholder approval requires one of the following, which we refer to as a Special Majority:
|
|
•
|
at least a majority of the shares held by all shareholders who do not have a personal
interest in the transaction and who are present and voting at the meeting approves the transaction, excluding abstentions; or
|
|
|
•
|
the shares voted against the transaction by shareholders who have no personal interest
in the transaction and who are present and voting at the meeting do not exceed 2% of the voting rights in the company.
|
To the extent that any such transaction with a controlling shareholder
is for a period extending beyond three years, approval is required once every three years, unless, with respect to certain transactions,
the audit committee determines that the duration of the transaction is reasonable given the circumstances related thereto. Arrangements
regarding the compensation, indemnification or insurance of a controlling shareholder in his or her capacity as an office holder require
the approval of the compensation committee, board of directors and shareholders by a Special Majority, in that order, and the terms thereof
may not be inconsistent with the company’s stated compensation policy.
Pursuant to regulations promulgated under the Israeli Companies
Law, certain transactions with a controlling shareholder or his or her relative, or with directors, that would otherwise require approval
of a company’s shareholders may be exempt from shareholder approval upon certain determinations of the audit committee and board
of directors.
Shareholder duties
Pursuant to the Israeli Companies Law, a shareholder has a duty
to act in good faith and in a customary manner toward the company and other shareholders and to refrain from abusing his or her power
in the company, including, among other things, in voting at a general meeting and at shareholder class meetings with respect to the following
matters:
|
|
•
|
an amendment to the company’s articles of association;
|
|
|
•
|
an increase of the company’s authorized share capital;
|
|
|
•
|
a merger; or
|
|
|
•
|
the approval of related party transactions and acts of office holders that require
shareholder approval.
|
A shareholder also has a general duty to refrain from discriminating
against other shareholders. In addition, certain shareholders have a duty of fairness toward the company. These shareholders include any
controlling shareholder, any shareholder who knows that he or she has the power to determine the outcome of a shareholder vote and any
shareholder who has the power to appoint or to prevent the appointment of an office holder of the company or other power towards the company.
The Israeli Companies Law does not define the substance of the duty of fairness, except to state that the remedies generally available
upon a breach of contract will also apply in the event of a breach of the duty to act with fairness.
Exculpation, Insurance and Indemnification of Directors and Officers
Under the Israeli Companies Law, a company may not exculpate an
office holder from liability for a breach of the duty of loyalty. An Israeli company may exculpate an office holder in advance from liability
to the company, in whole or in part, for damages caused to the company as a result of a breach of duty of care but only if a provision
authorizing such exculpation is included in its articles of association. Our articles of association include such a provision. A company
may not exculpate in advance a director from liability arising out of a prohibited dividend or distribution to shareholders.
Under the Israeli Companies Law, a company may indemnify an office
holder in respect of the following liabilities and expenses incurred for acts performed by him or her as an office holder, either pursuant
to an undertaking made in advance of an event or following an event, provided its articles of association include a provision authorizing
such indemnification:
|
|
•
|
financial liability imposed on him or her in favor of another person pursuant to a
judgment, including a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an office holder
with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the
board of directors, can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount
or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail
the abovementioned foreseen events and amount or criteria;
|
|
|
•
|
reasonable litigation expenses, including attorneys’ fees, incurred by the office
holder (1) as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation
or proceeding, provided that (i) no indictment was filed against such office holder as a result of such investigation or proceeding, and
(ii) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation
or proceeding or, if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal
intent; and (2) in connection with a monetary sanction; and
|
|
|
•
|
reasonable litigation expenses, including attorneys’ fees, incurred by the office
holder or imposed by a court in proceedings instituted against him or her by the company, on its behalf, or by a third party, or in connection
with criminal proceedings in which the office holder was acquitted, or as a result of a conviction for an offense that does not require
proof of criminal intent.
|
Under the Israeli Companies Law, a company may insure an office
holder against the following liabilities incurred for acts performed by him or her as an office holder, if and to the extent provided
in the company’s articles of association:
|
|
•
|
a breach of the duty of loyalty to the company, provided that the office holder acted
in good faith and had a reasonable basis to believe that the act would not harm the company;
|
|
|
•
|
a breach of duty of care to the company or to a third party, to the extent such a
breach arises out of the negligent conduct of the office holder; and
|
|
|
•
|
a financial liability imposed on the office holder in favor of a third party.
|
Under the Israeli Companies
Law, a company may not indemnify, exculpate or insure an office holder against any of the following:
|
|
•
|
a breach of the duty of loyalty, except for indemnification and insurance for a breach
of the duty of loyalty to the company to the extent that the office holder acted in good faith and had a reasonable basis to believe that
the act would not harm the company;
|
|
|
•
|
a breach of duty of care committed intentionally or recklessly, excluding a breach
arising out of the negligent conduct of the office holder;
|
|
|
•
|
an act or omission committed with intent to derive illegal personal benefit; or
|
|
|
•
|
a fine or forfeit levied against the office holder.
|
Under the Israeli Companies
Law, exculpation, indemnification and insurance of office holders in a public company must be approved by the compensation committee and
the board of directors and, with respect to certain office holders or under certain circumstances, also by the shareholders. See “-Approval
of Related Party Transactions Under Israeli Law.”
Our articles of association permit us to exculpate, indemnify and
insure our office holders to the fullest extent permitted or to be permitted by the Israeli Companies Law. We have obtained directors’
and officers’ liability insurance for the benefit of our office holders and intend to continue to maintain such coverage and pay
all premiums thereunder to the fullest extent permitted by the Israeli Companies Law. In addition, we have entered into agreements with
each of our directors and executive officers exculpating them from liability to us for damages caused to us as a result of a breach of
duty of care and undertaking to indemnify them, in each case, to the fullest extent permitted by our articles of association and Israeli
Law.
The maximum indemnification amount set forth in those agreements
is limited to an amount equal to the greater of (i) 25% of our total shareholders’ equity based on our most recently financial statements
of the time of the actual payment of the indemnification; (ii) $50 million; (iii) 40% of our total market cap (which shall mean the average
closing price of the Company’s ordinary shares over the 30 trading days prior to the actual payment of indemnification multiplied
by the total number of issued and outstanding shares of the Company as of the date of actual payment); and (iv) in connection with or
arising out of a public offering of our securities, the aggregate amount of proceeds from the sale by us and/or any shareholder of ours
securities in such offering. The maximum amount set forth in those agreements is in addition to amounts actually paid, if any, under insurance
policies and/or by a third-party pursuant to an indemnification arrangement.
As of December 31,
2023, we had 100 employees, 88 of whom were based in Israel and 12 based throughout Europe and employed by our German subsidiary. The
distribution of our employees according to main areas of activity is as follows: 14 employees in the administrative department, 26 employees
in the research and development department, 48 employees in the manufacturing department and 12 employees in the sales and marketing department.
As of December 31, 2023, we did not employ a significant number of temporary employees.
Israeli labor laws govern the length of the workday and workweek,
minimum wages for employees, procedures for hiring and dismissing employees, determination of severance pay, annual leave, sick days,
advance notice of termination, payments to the National Insurance Institute and other conditions of employment, and include equal opportunity
and anti-discrimination laws. While none of our employees is party to any collective bargaining agreements, certain provisions of the
collective bargaining agreements between the Histadrut (General Federation of Labor in Israel) and the Coordination Bureau of Economic
Organizations (including the Industrialists’ Associations) are applicable to our employees in Israel by order of the Israeli Ministry
of the Economy. These provisions primarily concern pension fund benefits for all employees, insurance for work-related accidents, recuperation
pay and travel expenses. We generally provide our employees with benefits and working conditions beyond the required minimums.
We have never experienced any employment-related work stoppages
and believe our relationships with our employees are good.
For information regarding the share ownership
of our directors and executive officers, see “ITEM 6.B. Compensation-2014 Equity Incentive Plan” and “ITEM 7.A. Major
Shareholders.”
F. Disclosure of Any Action to Recover
Erroneously Awarded Compensation
None.
Item 7. MAJOR
SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
The following table sets forth
information with respect to the beneficial ownership of our shares as of March 15, 2024 by:
|
|
•
|
each person or entity known by us to own beneficially more than 5% of our outstanding shares;
|
|
|
•
|
each of our directors and executive officers individually; and
|
|
|
• |
all of our executive officers and directors as a group. |
The beneficial ownership of ordinary shares
is determined in accordance with the rules of the SEC and generally includes any ordinary shares over which a person exercises sole or
shared voting or investment power. The percentage of shares beneficially owned is based on 9,491,718 ordinary shares issued and outstanding
as of March 15, 2024. Ordinary shares that are issuable under stock options or RSUs that are currently exercisable or exercisable within
60 days of March 15, 2024 are deemed to be outstanding and to be beneficially owned by the person holding the stock option for the purpose
of computing the number of shares and percentage ownership of that person. Those shares are not deemed outstanding, however, for the purpose
of computing the percentage ownership of any other person. All of our shareholders, including the shareholders listed below, have the
same voting rights attached to their ordinary shares. See “ITEM 10.B. Articles of Association.” None of our principal shareholders
nor our directors or executive officers possesses different or special voting rights with respect to their ordinary shares. Unless otherwise
noted below, each shareholder’s address is c/o MediWound Ltd., 42 Hayarkon Street, Yavne 8122745, Israel.
A description of any material relationship that our principal shareholders
have had with us or any of our predecessors or affiliates within the past three years is included under “ITEM 7.B. Related Party
Transactions.”
|
Name of Beneficial Owner
|
|
Number of
Shares
Beneficially
Held |
|
|
Percentage of
Class |
|
|
Directors
and Executive Officers |
|
|
|
|
|
|
|
Nachum (Homi) Shamir
|
|
|
* |
|
|
|
* |
|
|
Ofer Gonen
|
|
|
98,730 |
|
|
|
1.1 |
% |
|
Vickie R. Driver |
|
|
* |
|
|
|
* |
|
|
David Fox
|
|
|
* |
|
|
|
* |
|
|
Shmuel (Milky) Rubinstein
|
|
|
* |
|
|
|
* |
|
|
Stephen T. Wills
|
|
|
* |
|
|
|
* |
|
|
Shmulik Hess
|
|
|
* |
|
|
|
* |
|
|
Ety Klinger
|
|
|
* |
|
|
|
* |
|
|
Hany Luxenburg
|
|
|
* |
|
|
|
* |
|
|
Yaron Meyer
|
|
|
* |
|
|
|
* |
|
|
Robert Snyder
|
|
|
* |
|
|
|
* |
|
|
All executive officers and directors as a group (11 persons)(1)
|
|
|
|
|
|
|
3.8 |
% |
| |
|
|
|
|
|
|
|
|
|
Principal
Shareholders (who are not Directors or Executive Officers) |
|
|
|
|
|
|
|
|
|
Clal Biotechnology Industries Ltd. and
affiliates (2)
|
|
|
1,481,522 |
|
|
|
16.0 |
% |
|
Israel Biotech Fund II, L.P. and affiliates
(3)
|
|
|
959,652 |
|
|
|
9.9 |
% |
|
Deep Insight Limited Partnership and affiliates
(4)
|
|
|
959.652 |
|
|
|
9.9 |
% |
|
Point72 Associates, LLC and affiliates
(5)
|
|
|
821,500 |
|
|
|
8.9 |
% |
|
Rosalind Advisors, Inc. and affiliates
(6)
|
|
|
643,255 |
|
|
|
6.8 |
% |
| (1) |
Shares beneficially owned consist of 79,068 ordinary shares held directly or indirectly by such executive officers and directors
and 267,058 ordinary shares issuable upon exercise of outstanding options that are currently exercisable or exercisable within 60 days
of March 15, 2024. |
| (2) |
Based solely on Schedule 13D/A filed on September 1, 2023, Clal Biotechnology
Industries Ltd. (“CBI”) owns directly 308,811 ordinary shares, and may be deemed to share voting and investment power over
the 1,172,710 ordinary shares owned directly by Clal Life Sciences L.P. (“CLS”), the general partner of which, Clal Application
Center Ltd., is wholly owned by CBI. Each of Access Industries Holdings LLC (“AIH”), Access Industries, LLC (“Access
LLC”), Access Industries Management, LLC (“AIM”), Clal Industries Ltd. (“Clal Industries”) and Mr. Blavatnik
may be deemed to share voting and investment power over the ordinary shares owned directly by CBI and CLS because (i) Len Blavatnik controls
AIM, AIH, Access LLC and AI International GP Limited (the general partner of AI SMS, as defined below), (ii) AIM controls Access LLC and
AIH, (iii) Access LLC controls a majority of the outstanding voting interests in AIH, (iv) AIH owns a majority of the equity of AI SMS
L.P. (“AI SMS”), (v) AI SMS controls AI Diversified Holdings Ltd. (“Holdings Limited”), (vi) Holdings Limited
owns AI Diversified Parent S.à r.l., which owns AI Diversified Holdings S.à r.l., which owns Access AI Ltd (“Access AI”),
(vii) Access AI wholly owns Clal Industries, (viii) Clal Industries is the controlling shareholder of CBI, and (ix) CBI is the sole shareholder
of Clal Application Center Ltd. The Reporting Persons, other than CBI and CLS, and each of their affiliated entities and the officers,
partners, members and managers thereof, disclaims beneficial ownership of these securities. The address of Clal Industries Ltd. is the
Triangular Tower, 3 Azrieli Center, Tel Aviv 67023, Israel and the address of Access Industries Holdings LLC is c/o Access Industries
Inc., 40 West 57th Street, New York, New York 10019, United States. |
| (3) |
Based solely on Schedule 13G/A filed on January 8, 2024, the 959,652 ordinary shares include 408,397 ordinary shares that are issuable
upon the exercise of warrants held directly by Israel Biotech Fund II, L.P. (“IBF II”). Israel Biotech Fund GP Partners II,
L.P. (“IBF GP”) is the sole general partner of IBF II, and I.B.F Management Ltd. (“IBF Management”) is the sole
general partner of IBF GP. IBF GP and IBF Management may be deemed to share voting and dispositive power with respect to the ordinary
shares that are beneficially owned by IBF II. The address IBF Management is HaOgen Tower, 4 Oppenheimer St., Rehovot 7670104, Israel and
the address of the other reporting persons is 75 Fort Street, Clifton House, PO Box, 1350, KY1-1108, Grand Cayman. |
| (4) |
Based solely on Schedule 13G/A filed on January 8, 2024, the 959,652 ordinary shares include 408,397 ordinary shares that are issuable
upon the exercise of warrants held directly by Deep Insight Limited Partnership (“Deep Insight”). Deep Insight Fund GP Limited
Partnership (“Deep Insight GP LP”) is the sole general partner of Deep Insight, Deep Insight GP Ltd. (“Deep Insight
GP Company”) is the sole general partner of Deep Insight GP LP, Deep Insight Management Ltd. (“Deep Insight Management”)
is the management company of Deep Insight GP LP and each of Barak Ben-Eliezer and Dr. Eyal Kishon hold 50% of the outstanding shares of
Deep Insight GP Company and Deep Insight Management. Deep Insight GP LP, Deep Insight GP Company, Deep Insight Management, Barak Ben-Eliezer
and Dr. Eyal Kishon may be deemed to share voting and dispositive power with respect to the Ordinary Shares that are beneficially owned
by Deep Insight. Barak Ben-Eliezer and Dr. Eyal Kishon disclaim beneficial ownership of the Ordinary Shares reported by Deep Insight herein.
The address of each of the reporting persons is 2 Rachel Imeinu St., Modiin, Israel 7177190. |
| (5) |
Based solely on Schedule 13G filed on January 8, 2024, Point72 Associates, LLC (“Point72 Associates”) owns 821,500 ordinary
shares. Pursuant to an investment management agreement, Point72 Asset Management, L.P. (“Point72 Asset Management”) maintains
investment and voting power with respect to the securities held by Point72 Associates. Point72 Capital Advisors Inc. is the general partner
of Point72 Asset Management. Steven Cohen controls each of Point72 Asset Management and Point72 Capital Advisors Inc. The address of the
reporting persons is 72 Cummings Point Road, Stamford, CT 06902. |
| (6) |
Based solely on Schedule 13G/A filed on February 14, 2024, Rosalind Master Fund L.P. (“RMF”) is the record owner of 479,990
shares of ordinary shares and 163,265 shares of ordinary shares issuable upon exercise of warrants. Rosalind Advisors, Inc. is the investment
advisor to RMF and may be deemed to be the beneficial owner of shares held by RMF. Steven Salamon is the portfolio manager of Rosalind
Advisors, Inc. and may be deemed to be the beneficial owner of shares held by RMF. Gilad Aharon is the portfolio manager and member of
the Advisor which advises RMF. Notwithstanding the foregoing, Rosalind Advisors, Inc. and Mr. Salamon disclaim beneficial ownership of
the shares. The address of RMF is P.O. Box 309 Ugland House, Grand Cayman KY1-1104, Cayman Islands, and the address of the rest of the
reporting persons is 15 Wellesley Street West, Suite 326, Toronto, Ontario M4Y 0G7 Canada. |
Changes in Ownership of Major Shareholders
To our knowledge, other than as disclosed in the
table above, our other filings with the SEC and this Annual Report, there has been no significant change in the percentage ownership held
by any major shareholder since January 1, 2021, except as follows: as reported in their filings with the SEC, during 2023, CBI and its
affiliates have decreased their holdings from approximately 34% to 16.1% as a result of a dilution from the 2023 Offering t as described
in Item 5. “Operating and Financial Review and Prospects-Liquidity and Capital Resources.
Voting Rights
The major shareholders listed above do not have voting rights with
respect to their ordinary shares that are different from the voting rights of other holders of our ordinary shares.
Change in Control Arrangements
We are not aware of any arrangement that may at a subsequent date,
result in a change of control of the Company.
Registered Holders
As of March 15, 2024, we had one holder of record
of our ordinary shares in the United States, which is Cede & Co., the nominee of The Depository Trust Company. This shareholder held
in the aggregate 82.8% of the 9,256,368 ordinary shares issued and outstanding as of March 15, 2024. The number of record holders in the
United States is not representative of the number of beneficial holders nor is it representative of where such beneficial holders are
resident since many of these ordinary shares were held by brokers or other nominees.
|
|
B. |
Related Party Transactions |
Information Rights Agreement
We have entered into an information rights agreement with CBI,
which provides CBI with certain information rights relating to our financial information of the company and certain other information
necessary for CBI to meet Israeli Securities Law requirements. CBI is not required to reimburse us for expenses we incur in providing
such information.
2021 Registration Rights Agreement
We are party to an amended and restated registration rights agreement,
dated April 6, 2021, with certain of our shareholders (the “2021 Registration Rights Agreement”). The 2021 Registration Rights
Agreement, which was approved by our shareholders at our 2021 annual general meeting of shareholders, replaced the registration rights
agreement, dated March 3, 2014 (the “Original Registration Rights Agreement”), that we had entered into in connection with
our initial public offering with certain of our pre-IPO shareholders, which expired by its own terms on its seven-year anniversary. The
ordinary shares held by most of our pre-IPO shareholders who were party to the Original Registration Rights Agreement were no longer entitled
to registration rights under that agreement as of the time that it expired, given their ability to freely sell their shares in the open
market under Rule 144 of the Securities Act. However, each of CBI and Professor Lior Rosenberg, and their affiliated entities that hold
ordinary shares (consisting of Clal Life Sciences LP and L.R. Research & Development Ltd., respectively) remained entitled to registration
rights as of the time of the expiration of the Original Registration Rights Agreement, and we therefore entered into the Registration
Rights Agreement with them as a means of extending those rights. The 2021 Registration Rights Agreement provides to the holders of our
ordinary shares that are party to the agreement the right to demand that we file a registration statement or request that their ordinary
shares be covered by a registration statement that we are otherwise filing. In May 2022, we filed, and the SEC declared effective, on
June 3, 2022, a shelf registration statement on Form F-3 that registered the resale of the 1,819,780 shares that were then entitled to
registration rights under the 2021 Registration Rights Agreement. That registration statement remains in effect as of the date of this
Annual Report. The registration rights under the 2021 Registration Rights Agreement are described in more detail in Exhibit 2.1 to this
Annual Report, which is incorporated by reference in ITEM 10.B. Articles of Association.
Founders’ and Shareholders’ Agreement
In January 2001,
we entered into a founders’ and shareholders’ agreement (the “Founders Agreement”), with CBI, Prof. Lior Rosenberg,
and LR, a private company which is wholly-owned by Prof. Rosenberg. The Founders Agreement was amended in 2006. Pursuant to the Founders
Agreement, in exchange for the issuance of ordinary shares and certain rights thereunder and the payment of certain fixed amounts, Prof.
Rosenberg granted to us a perpetual, exclusive, non-revocable, royalty-free, sub-licensable, worldwide license for intellectual property
relating to debridement using products based on our proteolytic enzyme technology. As of the date hereof, all of the payments under the
Founders Agreement were paid by us to Prof. Rosenberg in accordance with the Founders Agreement. The Founders Agreement also provided
for anti-dilution, pre-emptive rights, a right of first refusal on the sale of our ordinary shares and bring-along rights, all of which
were subsequently terminated. In September 2022 we entered into an additional license agreement with LR for intellectual property rights
related to the development of a synthetic hyaluronic acid polyurethane dressing for debrided and non-debrided burns. Under this license
LR received an upfront payment of $150,000.
Agreements with Directors and Officers
Employment Agreements
We have entered into employment agreements with each of our executive
officers, which include standard provisions for a company in our industry regarding non-competition/solicitation, confidentiality of information
and assignment of inventions. However, the enforceability of the non-competition provisions may be limited under applicable law. Our executive
officers, other than our CEO, will not receive benefits upon the termination of their respective employment with us, other than payment
of salary and benefits (and limited accrual of vacation days) during the required notice period for termination of their employment, which
varies for each individual. Our CEO, is entitled upon termination of employment (other than a termination by the Company for Cause, as
defined below), to receive a single lump-sum payment equal 6 times his salary as of the last day of employment, less deductions and withholdings
under applicable law, subject him signing a separation agreement and release of known and unknown claims in a customary form provided
by the Company.
Options
Since our inception, we have granted options to purchase our ordinary
shares to our directors and executive officers. Such option agreements may contain acceleration provisions upon certain merger, acquisition
or change of control transactions. We describe our option plans under “ITEM 6.B. Compensation-2003 Israeli Share Option Plan”
and “ITEM 6.B. Compensation-2014 Equity Incentive Plan.” Upon the consummation of a merger or acquisition transaction, an
executive officer’s options will be assumed or substituted by the surviving company, if applicable, or, in the compensation committee’s
sole discretion, will vest immediately or be amended, modified or terminated. Our compensation committee approved accelerated vesting
in the case of a merger or an acquisition transaction for certain of our directors and executive officers with respect to the option agreements
dated December 23, 2015, June 22, 2017, January 16, 2018, December 31, 2018, May 2, 2019, April 23, 2020, March 4, 2021 and February 15,
2023, April 3, 2023, August 15, 2023, December 1, 2023 and February 26, 2024.
RSUs
Under the 2014 Plan, we have granted RSUs to our executive officers
and our chairman of the board. The RSU agreements generally provide for vesting of RSUs over a four-year period of continuous employment
or service, with 25% of the RSUs vesting at the lapse of one year following the vesting commencement date, and the remaining 75% of the
RSUs vesting in three equal installments, at the lapse of each of the following three years. Absent a specific acceleration provision,
if a grantee’s service is terminated for any reason, all RSUs that have not vested will immediately terminate. RSUs that have vested
but have not been settled yet for underlying ordinary shares may generally be settled within the three months following the termination
of the service of the grantee, other than in the case of termination due to death or disability (in which case the grantee or his/her
estate will have one year to settle the vested RSUs for underlying ordinary shares) or termination for cause (in which case all unsettled
RSUs will immediately terminate). Upon the consummation of a merger or acquisition transaction, an executive officer’s or the chairman’s
RSUs will be assumed or substituted by the surviving company, if applicable, or, in the compensation committee’s sole discretion,
will vest immediately or be amended, modified or terminated. The RSUs that we grant may contain acceleration provisions upon certain merger,
acquisition or change of control transactions, if approved by our board of directors with respect to a specific grant. The RSUs are generally
subject to the further terms of the 2014 Plan, which we describe under “ITEM 6.B. Compensation-2014 Equity Incentive Plan.”
Exculpation, indemnification and insurance
Our articles of association permit us to exculpate, indemnify and
insure each of our directors and office holders to the fullest extent permitted by the Israeli Companies Law. Additionally, we have entered
into indemnification agreements with each of our directors and executive officers, undertaking to indemnify them to the fullest extent
permitted by Israeli law, including with respect to liabilities resulting from a public offering of our shares, to the extent that these
liabilities are not covered by insurance. We have also obtained Directors and Officers insurance for each of our executive officers and
directors. See “ITEM 6.C. Board Practices-Exculpation, Insurance and Indemnification of Directors and Officers.”
|
|
C. |
Interests of Experts and Counsel |
None.
|
Item 8. |
FINANCIAL INFORMATION |
|
|
A. |
Consolidated Statements and Other Financial Information |
Consolidated Financial Statements
See Item 18. “Financial Statements”.
Legal and Arbitration Proceedings
From time to time, we may be party to litigation or subject to
claims incident to the ordinary course of business.
Settlement of Litigation Involving Our Company
and Teva
Under a previously reported settlement agreement, as amended, we
were obligated to pay Teva $10.2 million, of which twelve quarterly equal installments were paid from January 1, 2021 until December 31,
2023. In addition, commencing on January 1, 2021, we agreed to pay Teva an aggregate annual amount of $1 million in four quarterly equal
installments, unless we do not recognize any revenues generated from the sale or license of NexoBrid in any such quarter, up to an aggregate
amount equal to $7.2 million regardless of the number of quarters required for purposes of the payment of such aggregate amount. We also
agreed to indemnify Teva and its controlled affiliates from and against claims relating to a certain milestone related to PolyHeal under
an agreement associated with our collaboration agreements with Teva, for up to an amount of $10.2 million, if a notice of such claim had
been received by us prior to December 31, 2023, however, no notice of a claim was received by such date and therefore the indemnification
rights under this settlement agreement expired.
Dividend Policy
We have never declared or paid cash dividends to our shareholders
and we do not intend to pay cash dividends in the foreseeable future. We intend to reinvest any earnings in developing and expanding our
business. Any future determination relating to our dividend policy will be at the discretion of our board of directors and will depend
on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements,
business prospects, our strategic goals and plans to expand our business, applicable law and other factors that our board of directors
may deem relevant.
No significant changes have occurred since December 31, 2022, except
as otherwise disclosed in this annual report.
|
Item 9. |
THE OFFER AND LISTING |
Our ordinary shares trade on the Nasdaq Global Market under the
symbol “MDWD”.
Not applicable.
See “-Listing Details” above.
Not applicable.
Not applicable.
Not applicable.
|
Item 10. ADDITIONAL INFORMATION |
Not applicable.
|
|
B. |
Articles of Association |
A copy of our amended and restated articles of association is attached
as Exhibit 1.1 to this Annual Report. Other than as disclosed below, the information called for by this Item is set forth in Exhibit 2.1
to this Annual Report on Form 20-F, which information is incorporated by reference into this Item 10.B. of this annual report.
"Except described below or otherwise described in this annual report
in Item 4.A “History and Development of the Company”, Item 4.B “Business Overview”, “Item 5.B 'Operating
and Financial Review and Prospects—Liquidity and Capital Resources”, Item 6.C “Board Practices” and Item 7.B “Related
Party Transactions”, we are not currently, nor have we been for the two years immediately preceding the date of this Annual Report,
party to any material contract other than contracts entered into in the ordinary course of business."
For a description of the registration rights that are subject to
our 2021 Registration Rights Agreement, see “Item 7.B. Related Party Transactions – 2021 Registration Rights Agreement.”
For a description of our exclusive license and supply agreements
with Vericel, see “Item 4.B. Business Overview- Marketing, Sales and Distribution- Vericel
License and Supply Agreements.”
For a description of our license agreement with Mark Klein, see
“Item 4.B. Business Overview-Intellectual Property-Klein License Agreement.”
We have entered into an agreement with Challenge Bioproducts Corporation
Ltd. (“CBC”), a corporation organized and existing under the laws of the Republic of China, dated January 11, 2001, as amended
on February 28, 2010, pursuant to which CBC uses proprietary methods to manufacture bromelain SP and supplies us with this intermediate
drug substance in bulk quantities. According to the terms of the agreement, CBC shall not, and shall not permit related companies or a
third party to, manufacture, use, supply or sell the raw materials for the use or production of a product directly or indirectly competing
with any of our products. Our supply agreement with CBC has no fixed expiration date and can be voluntarily terminated by us, with at
least six months’ advance written notice, or by CBC, with at least 24 months’ advance written notice.
For a description of the main lease agreement, see “Item
4. Properties.”
For a description of the turnkey scale-up agreement with Biopharmax
Group Ltd., see “Item 4. Properties.”
2022 Registration Rights Agreement
In connection with the PIPE Offering (as described in “ITEM
5. Operating and Financial Review and Prospects— B. Liquidity and Capital Resources”), in October 2022, we entered into a
registration rights agreement with the several investors named in the PIPE Securities Purchase Agreement (the “2022 Registration
Rights Agreement”). Pursuant to the 2022 Registration Rights Agreement, within 45 calendar days of the date of the closing of the
PIPE, we were required to file a registration statement to register for resale of (i) Pre-Funded Warrant Shares, (ii) the PIPE Ordinary
Warrant Shares and (iii) the RD Ordinary Share Warrant (together, the “Registrable Securities”). Pursuant to the 2022 Registration
Rights Agreement, we agreed to cause such registration statement to be declared effective under the Securities Act of 1933, as amended
(the “Securities Act”), as promptly as possible after the filing thereof, but in any event no later 75 days, or in the event
of a full review by the SEC, 110 days, after the closing date of the PIPE. We further agreed to use our best efforts to keep such registration
statement continuously effective under the Securities Act until the date that all Registrable Securities covered by such registration
statement have been sold or otherwise may be sold pursuant to Rule 144 under the Securities Act. On November 10, 202, we filed a registration
statement on Form F-1 pursuant to our obligations under the 2022 Registration Rights Agreement, which became effective on November 25,
2022.
In 1998, Israeli currency control regulations were liberalized
significantly, so that Israeli residents generally may freely deal in foreign currency and foreign assets, and non-residents may freely
deal in Israeli currency and Israeli assets. There are currently no Israeli currency control restrictions on remittances of dividends
on the ordinary shares or the proceeds from the sale of the shares provided that all taxes were paid or withheld; however, legislation
remains in effect pursuant to which currency controls can be imposed by administrative action at any time.
Non-residents of Israel may freely hold and trade our securities.
Neither our articles of association nor the laws of the State of Israel restrict in any way the ownership or voting of ordinary shares
by non-residents, except that such restrictions may exist with respect to citizens of countries which are in a state of war with Israel.
Israeli residents are allowed to purchase our ordinary shares.
The following description is not intended to constitute a complete
analysis of all tax consequences relating to the acquisition, ownership and disposition of our ordinary shares. You should consult your
own tax advisor concerning the tax consequences of your particular situation, as well as any tax consequences that may arise under the
laws of any state, local, foreign or other taxing jurisdiction.
Israeli Tax Considerations for Our Shareholders
Capital gains taxes applicable to non‑Israeli resident shareholders
A non‑Israeli resident (whether an individual or a corporation)
who derives capital gains from the sale of shares in an Israeli resident company that were purchased after the company was listed for
trading on the Tel Aviv Stock Exchange or on a recognized stock exchange outside of Israel, will generally be exempt from Israeli capital
gain tax so long as the shares were not held through a permanent establishment that the non‑resident maintains in Israel (and with
respect to shares listed on a recognized stock exchange outside of Israel, so long as the particular capital gain is otherwise subject
to the Israeli Income Tax Law (Inflationary Adjustments) 5745‑1985. These provisions dealing with capital gain are not applicable
to a person whose gains from selling or otherwise disposing of the shares are deemed to be business income. However, non‑Israeli
corporations will not be entitled to the foregoing exemption if Israeli residents (i) have a controlling interest of more than 25% in
such non‑Israeli corporation or (ii) are the beneficiaries of, or are entitled to, 25% or more of the revenues or profits of such
non‑Israeli corporation, whether directly or indirectly.
If not exempt, a non-Israeli resident shareholder would generally
be subject to tax on capital gain at the ordinary corporate tax rate (23% in 2023), if generated by a company, or at the rate of 25%,
if generated by an individual, or 30%, if generated by an individual who is a “substantial shareholder” (as defined under
the Tax Ordinance), at the time of sale or at any time during the preceding 12-month period (or if the shareholder claims a deduction
for interest and linkage differences expenses in connection with the purchase and holding of such shares). A “substantial shareholder”
is generally a person who alone or together with such person’s relative or another person who collaborates with such person on a
permanent basis, holds, directly or indirectly, at least 10% of any of the “means of control” of the corporation. “Means
of control” generally include, among others, the right to vote, receive profits, nominate a director or an executive officer, receive
assets upon liquidation, or order someone who holds any of the aforesaid rights how to act, regardless of the source of such right. Individual
and corporate shareholders dealing in securities in Israel are taxed at the tax rates applicable to business income (a corporate tax rate
for a corporation (23% in 2023) and a marginal tax rate of up to 47% for an individual in 2023 (excluding excess tax as discussed below))
unless contrary provisions in a relevant tax treaty apply.
Additionally, a sale of shares by a non‑Israeli resident
may also be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, under the Convention
Between the Government of the United States of America and the Government of the State of Israel with respect to Taxes on Income, as amended
(the “United States‑Israel Tax Treaty”), the sale, exchange or other disposition of shares by a shareholder who is a
United States resident (for purposes of the United States‑Israel Tax Treaty) holding the shares as a capital asset and is entitled
to claim the benefits afforded to such a resident by the United States‑Israel Tax Treaty (a “Treaty U.S. Resident”)
is generally exempt from Israeli capital gains tax unless: (i) the capital gain arising from such sale, exchange or disposition is attributed
to real estate located in Israel; (ii) the capital gain arising from such sale, exchange or disposition is attributed to royalties; (iii)
the capital gain arising from the such sale, exchange or disposition can be attributable to a permanent establishment of the shareholder
maintained in Israel, under certain terms; (iv) such Treaty U.S. Resident holds, directly or indirectly, shares representing 10% or more
of the voting capital of a company during any part of the 12‑month period preceding such sale, exchange or disposition, subject
to certain conditions; or (v) such Treaty U.S. Resident is an individual and was present in Israel for a period or periods aggregating
to 183 days or more during the relevant taxable year. In each case, the sale, exchange or disposition of our ordinary shares would be
subject to such Israeli tax, to the extent applicable; However, under the United States‑Israel Tax Treaty, such Treaty U.S. Resident
would be permitted to claim a credit for such taxes against the U.S. federal income tax imposed with respect to such sale, exchange or
disposition, subject to the limitations in U.S. laws applicable to foreign tax credits. The United States-Israel Tax Treaty does not provide
such credit against any U.S. state or local taxes. In some instances where our shareholders may be liable for Israeli tax on the sale
of their ordinary shares, the payment of the consideration may be subject to the withholding of Israeli tax at source. Shareholders may
be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of
sale. Specifically, in transactions involving a sale of all of the shares of an Israeli resident company, in the form of a merger or otherwise,
the Israel Tax Authority may require from shareholders who are not liable for Israeli tax to sign declarations in forms specified by this
authority or obtain a specific exemption from the Israel Tax Authority to confirm their status as non‑Israeli resident, and, in
the absence of such declarations or exemptions, may require the purchaser of the shares to withhold taxes at source.
Taxation of non‑Israeli shareholders on receipt of dividends
Non‑Israeli residents (whether individuals or corporations)
are generally subject to Israeli income tax on the receipt of dividends paid on our ordinary shares at the rate of 25% unless a relief
is provided in a treaty between Israel and a shareholder's country of residence (provided that a valid certificate from the Israeli Tax
Authority allowing for a reduced withholding tax rate is obtained in advance). With respect to a person who is a “substantial shareholder”
at the time of receiving the dividend or on any time during the preceding 12 months, the applicable tax rate is 30%. Such dividends are
generally subject to Israeli withholding tax at a rate of 25% so long as the shares are registered with a nominee company (whether or
not the recipient is a substantial shareholder), unless relief is provided in a treaty between Israel and the shareholder’s country
of residence and provided that a valid certificate from the Israel Tax Authority allowing for a reduced withholding tax rate is obtained
in advance. However, a distribution of dividends to non‑Israeli residents is generally subject to withholding tax at source at a
rate of 15% if the dividend is distributed from income attributed to a Beneficiary Enterprise, or such a reduced tax rate as may be provided
under an applicable tax treaty (provided that a valid certificate from the Israel Tax Authority allowing for a reduced withholding tax
rate or such lower tax rate as may be provided in an applicable tax treaty is obtained in advance). For example, under the United States‑Israel
Tax Treaty, the maximum rate of tax withheld at source in Israel on dividends paid to a holder of our ordinary shares who is a Treaty
U.S. Resident is 25%. However, generally, the maximum rate of withholding tax on dividends, not generated by an Approved Enterprise or
Beneficiary Enterprise, that are paid to a U.S. corporation holding 10% or more of the outstanding voting capital throughout the tax year
in which the dividend is distributed as well as during the previous tax year, is 12.5%, provided that not more than 25% of the gross income
for such preceding year consists of certain types of dividends and interest. Notwithstanding the foregoing, dividends distributed from
income attributed to an Approved Enterprise or Beneficiary Enterprise are not entitled to such reduction under the tax treaty but are
subject to a withholding tax rate of 15% for such a U.S. corporation, provided that the condition related to our gross income for the
previous year (as set forth in the previous sentence) is met. The aforementioned rates under the United States-Israel Tax Treaty would
not apply if the dividend income is derived through a permanent establishment of the U.S. resident in Israel. If the dividend is attributable
partly to income derived from an Approved Enterprise, Beneficiary Enterprise or Preferred Enterprise, and partly to other sources of income,
the withholding rate will be a blended rate reflecting the relative portions of the two types of income. We cannot assure you that we
will designate the profits that we may distribute in a way that will reduce shareholders’ tax liability.
A non‑Israeli resident who receives dividends from which
tax was withheld, is generally exempt from the obligation to file tax returns in Israel with respect to such income, provided that (i)
such income was not derived from a business conducted in Israel by the taxpayer, (ii) the taxpayer has no other taxable sources of income
in Israel with respect to which a tax return is required to be filed and (iii) the tax payer is not obligated to pay the excess tax (as
further explained below).
Excess Tax
Individuals who are subject to tax in Israel (whether any such
individual is an Israeli resident or non-Israeli resident) are also subject to an additional tax at a rate of 3% on annual income exceeding
a certain level, which amount is linked to the annual change in the Israeli consumer price index, including but not limited to, dividends,
interest and capital gain. In 2023, the additional tax was at a rate of 3% on annual income exceeding NIS 698,280.
United States Federal Income Taxation
The following is a description of the material U.S. federal income
tax consequences of the ownership and disposition of our ordinary shares by a U.S. Holder that holds the ordinary shares as capital assets.
This description does not address tax considerations applicable to holders that may be subject to special tax rules, including, without
limitation:
|
|
•
|
banks, financial institutions or insurance companies;
|
|
|
•
|
real estate investment trusts, regulated investment companies or grantor trusts;
|
|
|
•
|
dealers or traders in securities, commodities or currencies;
|
|
|
•
|
tax‑exempt entities or organizations, including an “individual retirement
account” or “Roth IRA” as defined in Section 408 or 408A of the Code, respectively;
|
|
|
•
|
certain former citizens or long‑term residents of the United States;
|
|
|
•
|
persons that received our shares as compensation for the performance of services;
|
|
|
•
|
persons that holds our shares as part of a “hedging,” “integrated”
or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes;
|
|
|
•
|
partnerships (including entities classified as partnerships for U.S. federal income
tax purposes) or other pass‑through entities, or holders that will hold our shares through such an entity;
|
|
|
•
|
S corporations;
|
|
|
•
|
holders that acquired ordinary shares as a result of holding or owning our preferred
shares;
|
|
|
•
|
U.S. Holders (as defined below) whose “functional currency” is not the
U.S. dollar;
|
|
|
•
|
persons that are residents of ordinarily resident in or have a permanent establishment
in a jurisdiction outside the United States; or
|
|
|
•
|
holders that own directly, indirectly or through attribution 10.0% or more of the
voting power or value of our shares.
|
Moreover, this description does not address the U.S. federal estate,
gift or alternative minimum tax consequences, Medicare consequences, or any state, local or foreign tax consequences, of the ownership
and disposition of our ordinary shares.
This summary is based on the Internal Revenue Code of 1986, as
amended (the “Code”), administrative pronouncements, judicial decisions and final, temporary and proposed Treasury regulations,
all as currently in effect and available. These authorities are subject to change or differing interpretation, possibly with retroactive
effect. U.S. Holders should consult their tax advisors concerning the U.S. federal, state, local and foreign tax consequences of owning
and disposing of our ordinary shares in their particular circumstances. For purposes of this summary, a “U.S. Holder” is a
beneficial owner of our ordinary shares who is, for U.S. federal income tax purposes:
|
|
•
|
an individual who is a citizen or individual resident of the United States;
|
|
|
•
|
a corporation, or other entity taxable as a corporation for U.S. federal income tax
purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia;
|
|
|
•
|
an estate, the income of which is subject to U.S. federal income taxation regardless
of its source; or
|
|
|
•
|
a trust that (1) is subject to the primary supervision of a U.S. Court and one or
more U.S. persons that have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under
applicable Treasury regulations to be treated as a U.S. person.
|
If a partnership (or other entity treated as a partnership for
U.S. federal income tax purposes) holds our ordinary shares, the tax treatment of a partner in such partnership generally will depend
upon the status of the partner and upon the activities of the partnership. Investors who are partners in a partnership should consult
their tax advisors as to the particular U.S. federal income tax consequences of owning and disposing of our ordinary shares in their particular
circumstances.
A “Non‑U.S. Holder” is a beneficial owner of
our ordinary shares that is neither a U.S. Holder nor a partnership for U.S. federal income tax purposes.
Unless otherwise indicated, this discussion assumes that the company
is not, and will not become, a “passive foreign investment company,” or a PFIC, for U.S. federal income tax purposes. See
“ITEM 10.E. Taxation-United States Federal Income Taxation-Passive Foreign Investment Company Considerations” below. Further,
this summary does not address the U.S. federal estate and gift, state, local or non‑U.S. tax consequences to U.S. Holders of owning
and disposing of our ordinary shares. Investors should consult their own tax advisors regarding the U.S. federal, state and local, as
well as non‑U.S. income and other tax consequences of owning and disposing of our ordinary shares in their particular circumstances.
Distributions
If you are a U.S. Holder, the gross amount of any
distribution made to you with respect to our ordinary shares before reduction for any Israeli taxes withheld therefrom, other than certain
distributions, if any, of our ordinary shares distributed pro rata to all our shareholders, generally will be includible in your income
as dividend income to the extent such distribution is paid out of our current or accumulated earnings and profits as determined under
U.S. federal income tax principles. We do not expect to maintain calculations of our earnings and profits under U.S. federal income tax
principles. Therefore, if you are a U.S. Holder you should expect that the entire amount of any distribution generally will be taxable
as dividend income to you. Non‑corporate U.S. Holders may qualify for the lower rates of taxation with respect to dividends on ordinary
shares applicable to long‑term capital gains (i.e., gains from the sale of capital assets held for more than one year), provided
that certain conditions are met, including certain holding period requirements and the absence of certain risk reduction transactions.
However, such dividends will not be eligible for the dividends received deduction generally allowed to corporate U.S. Holders. If you
are a U.S. Holder, dividends paid to you with respect to our ordinary shares will generally be treated as foreign source income, which
may be relevant in calculating your foreign tax credit limitation. Subject to certain conditions and limitations, Israeli tax withheld
on dividends may be deducted from your taxable income or credited against your U.S. federal income tax liability. The limitation on foreign
taxes eligible for credit is calculated separately with respect to specific classes of income. For this purpose, dividends that we distribute
generally should constitute “passive category income.” A foreign tax credit for foreign taxes imposed on distributions may
be denied if you do not satisfy certain minimum holding period requirements. Applicable U.S. Treasury Regulations have imposed additional
requirements that must be met for a foreign tax to be creditable, depending on the nature of such foreign
tax, although temporary relief from the application of certain aspects of these Regulations has been provided until new guidance or regulations
are issued. The rules relating to the determination of the foreign tax credit are complex, and you should consult your tax advisor
to determine whether and to what extent you will be entitled to this credit.
Subject to the discussion below under “-Backup Withholding
Tax and Information Reporting Requirements,” if you are a Non‑U.S. Holder, you generally will not be subject to U.S. federal
income (or withholding) tax on dividends received by you on your ordinary shares, unless you conduct a trade or business in the United
States and such income is effectively connected with that trade or business (or, if required by an applicable income tax treaty, the dividends
are attributable to a permanent establishment or fixed base that such holder maintains in the United States).
Sale, Exchange or Other Taxable Disposition
of Ordinary Shares
If you are a U.S. Holder, you generally will recognize
gain or loss on the sale, exchange or other taxable disposition of our ordinary shares equal to the difference between the amount realized
on such sale, exchange or other taxable disposition and your adjusted tax basis in our ordinary shares, and such gain or loss will be
capital gain or loss. The initial tax basis in an ordinary share generally will be equal to the cost of such ordinary share. Except with
respect to foreign currency gain or loss, if you are a non‑corporate U.S. Holder, capital gain from the sale, exchange or other
taxable disposition of ordinary shares is generally eligible for a preferential rate of taxation applicable to capital gains, if your
holding period for such ordinary shares exceeds one year (i.e., such gain is long‑term capital gain). The deductibility of capital
losses for U.S. federal income tax purposes is subject to limitations under the Code. Any such gain or loss that a U.S. Holder recognizes
generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes. Because a U.S. Holder may use foreign
tax credits against only the portion of United States federal income tax liability that is attributed to foreign source income in the
same category, a U.S. Holder’s ability to utilize a foreign tax credit with respect to the foreign tax imposed on any such sale
or other taxable disposition, if any, may be significantly limited. Applicable
U.S. Treasury Regulations further restrict the availability of any such credit. However, a recent notice from the IRS indicates that the
U.S. Department of the Treasury and the IRS are considering proposing amendments to such Regulations and allows, subject to certain conditions,
taxpayers to defer the application of many aspects of such Regulations for taxable years beginning on or after December 28, 2021 and ending
before the date that a notice or other guidance withdrawing or modifying the temporary relief is issued (or any later date specified in
such notice or other guidance).
Subject to the discussion below under “-Backup Withholding
Tax and Information Reporting Requirements,” if you are a Non‑U.S. Holder, you generally will not be subject to U.S. federal
income or withholding tax on any gain realized on the sale or exchange of such ordinary shares unless:
|
|
•
|
such gain is effectively connected with your conduct of a trade or business in the
United States (or, if required by an applicable income tax treaty, the gain is attributable to a permanent establishment or fixed base
that such holder maintains in the United States); or
|
|
|
•
|
you are an individual and have been present in the United States for 183 days or more
in the taxable year of such sale or exchange and certain other conditions are met.
|
Passive Foreign Investment Company Considerations
If we were to be classified as a “passive foreign investment
company,” or “PFIC,” in any taxable year, a U.S. Holder would be subject to special rules generally intended to reduce
or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. Holder could derive from investing in a non‑U.S.
company that does not distribute all of its earnings on a current basis.
A non‑U.S. corporation will be classified as a PFIC for federal
income tax purposes in any taxable year in which, after applying certain look‑through rules with respect to the income and assets
of subsidiaries, either:
|
|
•
|
at least 75% of its gross income is “passive income”; or
|
|
|
•
|
at least 50% of the average quarterly value of its total gross assets (which may be
determined in part by the market value of our ordinary shares, which is subject to change) is attributable to assets that produce “passive
income” or are held for the production of passive income.
|
Passive income for this purpose generally includes dividends, interest,
royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which
produce passive income, and includes amounts derived by reason of the temporary investment of funds raised in offerings of our ordinary
shares. If a non‑U.S. corporation owns at least 25% by value of the stock of another corporation, the non‑U.S. corporation
is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation and as receiving directly
its proportionate share of the other corporation’s income. If we are classified as a PFIC in any year with respect to which a U.S.
Holder owns our ordinary shares, we will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding years during
which the U.S. Holder owns our ordinary shares unless we cease to be a PFIC and the U.S. holder has made a “deemed sale” election
under the PFIC rules.
Based on our current estimates of our gross income
and the estimated fair market value of our gross assets and the nature of our business, we do not believe we were classified as a PFIC
for the taxable year ending December 31, 2023. However, we must determine our PFIC status annually based on tests which are factual in
nature, and our status in future years will depend on our income, assets and activities in those years. Further, because the value of
our gross assets is likely to be determined in large part by reference to our market capitalization, a decline in the value of our ordinary
shares or an increase in the value of our passive assets (including cash and short term investments) may result in our becoming a PFIC.
There can be no assurance that we will not be considered a PFIC for any taxable year. If we were a PFIC and you are a U.S. Holder, then
unless you make one of the elections described below, a special tax regime will apply to both (a) any “excess distribution”
by us to you (generally, your ratable portion of distributions in any year which are greater than 125% of the average annual distribution
received by you in the shorter of the three preceding years or your holding period for our ordinary shares) and (b) any gain realized
on the sale or other disposition of the ordinary shares. Under this regime, any excess distribution and realized gain will be treated
as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period,
(b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for
such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to
tax at the U.S. Holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed
below) and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable
in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to long‑term
capital gains discussed above under “Distributions.” Certain elections may be available that would result in an alternative
treatment (such as mark‑to‑market treatment) of our ordinary shares.
If a U.S. Holder makes a valid mark‑to‑market election
for the first tax year in which such U.S. Holder holds (or is deemed to hold) ordinary shares in a corporation and for which such corporation
is determined to be a PFIC, the U.S. Holder generally will recognize as ordinary income any excess of the fair market value of the ordinary
shares at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of
the adjusted tax basis of the ordinary shares over their fair market value at the end of the taxable year (but only to the extent of the
net amount of income previously included as a result of the mark‑to‑market election). If a U.S. Holder makes the election,
the U.S. Holder’s tax basis in the ordinary shares will be adjusted to reflect these income or loss amounts. Any gain recognized
on the sale or other disposition of ordinary shares in a year when we are a PFIC will be treated as ordinary income and any loss will
be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark‑to‑market
election). The mark‑to‑market election is available only if we are a PFIC and our ordinary shares are “regularly traded”
on a “qualified exchange.” Our ordinary shares will be treated as “regularly traded” in any calendar year in which
more than a de minimis quantity of the ordinary shares, are traded on a qualified exchange on at least 15 days during each calendar quarter.
Nasdaq is a qualified exchange for this purpose and, consequently, if the ordinary shares are regularly traded, the mark‑to‑market
election will be available to a U.S. Holder. If we are a PFIC, the general tax treatment for U.S. Holders described in this section would
apply to indirect distributions and gains deemed to be realized by U.S. Holders in respect of any entity in which we hold equity that
is also a PFIC (a "lower tier PFIC"). Because a mark‑to‑market election generally would not be available with respect to any
lower‑tier PFICs, a U.S. Holder may continue to be subject to the PFIC rules with respect to such holder’s indirect interest
in any investments held by us that are treated as an equity interest in such lower-tiers PFICs.
We do not intend to provide the information necessary for U.S.
Holders to make qualified electing fund elections if we are classified as a PFIC. U.S. Holders should consult their tax advisors to determine
whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular
circumstances.
If a U.S. Holder owns ordinary shares during any year in which
we are a PFIC, the U.S. Holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign
Investment Company or Qualified Electing Fund) or successor form with respect to the company, generally with the U.S. Holder’s federal
income tax return for that year. If the company was a PFIC for a given taxable year, then you should consult your tax advisor concerning
your annual filing requirements.
U.S. Holders should consult their tax advisors regarding whether
we are a PFIC and the potential application of the PFIC rules.
Backup Withholding Tax and Information Reporting
Requirements
U.S. backup withholding tax and information reporting requirements
may apply to certain payments to certain holders of stock. Information reporting generally will apply to distributions on, and to proceeds
from the sale, exchange or redemption of, our ordinary shares made within the United States, or by a United States payor or United States
middleman, to a holder of our ordinary shares, other than an exempt recipient (including a payee that is not a United States person that
provides an appropriate certification and certain other persons). Payments made (and sales or other dispositions effected at an office)
outside the U.S. will be subject to information reporting in limited circumstances. A payor will be required to withhold backup withholding
tax from any payments of dividends on, or the proceeds from the sale or redemption of, ordinary shares within the United States, or by
a United States payor or United States middleman, to a holder, other than an exempt recipient, if such holder fails to furnish its correct
taxpayer identification number or otherwise fails to comply with, or establish an exemption from, such backup withholding tax requirements,
or to report dividends required to be shown on the holder’s U.S. federal income tax returns. Back up withholding is not an additional
tax. Any amounts withheld under the backup withholding rules will be allowed as a credit against the beneficial owner’s U.S. federal
income tax liability, if any, and any excess amounts withheld under the backup withholding rules may be refunded, provided that the required
information is timely furnished to the IRS.
Foreign Asset Reporting
Certain U.S. Holders who are individuals and certain entities may
be required to report information relating to an interest in our ordinary shares, subject to certain exceptions (including an exception
for shares held in accounts maintained by certain financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial
Assets) with their federal income tax return. U.S. Holders are urged to consult their tax advisors regarding their information reporting
obligations, if any, with respect to their ownership and disposition of our ordinary shares.
F.
Dividends and Paying Agents
Not applicable.
G.
Statement by Experts
Not applicable.
H.
Documents on Display
We are required to make certain filings with the SEC. The SEC maintains
an internet website that contains reports, proxy statements and other information about issuers, like us, that file electronically with
the SEC. The address of that site is www.sec.gov.
We also make available on our website, free of charge, our annual
reports on Form 20-F and the text of our reports on Form 6-K, including any amendments to these reports, as well as certain other SEC
filings, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. Our website address is www.mediwound.com.
The information contained on our website is not incorporated by reference in this document.
I.
Subsidiary Information
Not applicable.
J.
Annual Report to Security Holders
Not Applicable.
Item 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES
ABOUT MARKET RISK
We are exposed to a variety of risks, including foreign currency
exchange fluctuations, changes in interest rates and inflation. We regularly assess currency, interest rate and inflation risks to minimize
any adverse effects on our business as a result of those factors.
Foreign Currency Risk
The U.S. dollar is our functional and reporting currency. A significant
portion of our operating expenses are denominated in Israeli shekels, accounting for approximately 45%, 45% and 44% of our operating expenses
in the years ended December 31, 2023, 2022 and 2021, respectively. We also have expenses in other non‑dollar currencies, in particular
the Euro, and for the next few years, we expect that a substantial portion of our revenues will be denominated in U.S dollar. A devaluation
of the shekel in relation to the U.S. dollar has the effect of reducing the U.S. dollar amount of our expenses or payables that are payable
in shekels, unless those expenses or payables are linked to the U.S. dollar. Conversely, any increase in the value of the shekel in relation
to the U.S. dollar has the effect of increasing the U.S. dollar value of our unlinked shekel expenses, which would have a negative impact
on our profit margins.
Because exchange rates between the U.S. dollar and both the shekel
and the Euro (as well as between the U.S. dollar and other currencies) fluctuate continuously, such fluctuations have an impact on our
results and period‑to‑period comparisons of our results. The effects of foreign currency re‑measurements are reported
in our consolidated financial statements of operations. The following table presents information about the changes in the exchange rates
of the shekel against the U.S. dollar and changes in the exchange rates of the Euro against the U.S. dollar:
|
|
|
Appreciation (Devaluation) of |
|
|
Period |
|
Shekel against the U.S. dollar
(%)
|
|
|
Euro
against the U.S. dollar
(%)
|
|
|
|
|
|
|
|
|
|
|
2021 |
|
|
3.3
|
|
|
|
7.7
|
|
|
2022 |
|
|
(13.2 |
) |
|
|
5.8
|
|
|
2023 |
|
|
(3.1 |
) |
|
|
(3.7 |
) |
A 10% increase (decrease) in the value of the NIS and Euro against
the U.S. dollar would have increased (decreased) our net profit by (loss) approximately $1.54 million for the year ended December 31,
2023.
As we are marketing and selling NexoBrid in Europe and conducting
clinical trials of outside the United States, we will continue to monitor exposure to currency fluctuations. We do not currently engage
in currency hedging activities in order to reduce this currency exposure, but we may begin to do so in the future. Instruments that may
be used to hedge future risks may include foreign currency forward and swap contracts. These instruments may be used to selectively manage
risks, but there can be no assurance that we will be fully protected against material foreign currency fluctuations.
Other Market Risks
We do not believe that we have material exposure to interest rate
risk due to the fact that we have no long‑term debt.
We do not believe that we have any material exposure to inflationary
risks. We do not believe that the rate of inflation in Israel has had a material impact on our business to date. However, our costs in
Israel will increase if inflation in Israel exceeds the devaluation of the shekel against the U.S. dollar (to the extent that it devalues
at all) or if the timing of such devaluation lags behind inflation in Israel.
Item 12. DESCRIPTION OF
SECURITIES OTHER THAN EQUITY SECURITIES
Not applicable.
PART II
Item 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
Item 14. MATERIAL
MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
None.
Item 15. CONTROLS AND PROCEDURES
(a) Disclosure Controls and Procedures
Our management, including our Chief Executive Officer and Chief
Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a‑15(e)
and 15d‑15(e) under the Exchange Act) as of December 31, 2023. Based on such evaluation, our Chief Executive Officer and Chief Financial
Officer have concluded that, as of December 31, 2023, our disclosure controls and procedures were effective.
(b) Management Annual Report on Internal Control over Financial
Reporting
Our management, under the supervision of our Chief Executive Officer
and Chief Financial Officer, is responsible for establishing and maintaining adequate internal control over financial reporting as defined
in Rules 13a‑15(f) and 15d‑15(f) under the Exchange Act.
Our management, including our Chief Executive Officer and Chief
Financial Officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this
assessment, our management used the criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring
Organizations of the Treadway Commission (COSO). Our management has concluded, based on its assessment, that our internal control over
financial reporting was effective as of December 31, 2023.
(c) Attestation report of the registered public accounting firm
Somekh Chaikin, a member firm of KPMG International, an independent
registered public accounting firm, to which we refer as KPMG Israel, which audited the financial statements included in this annual report
containing the disclosure required by this Item 15 has issued an attestation report regarding the effectiveness of our internal control
over financial reporting. That report is included in “Item 18. Financial Statements” on page F-[1] of this annual report,
which attestation report is incorporated by reference in this Item 15(c).
(d) Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting
(as such term is defined in Rules 13a‑15(f) and 15d‑15(f) under the Exchange Act) that occurred during the period covered
by this annual report that have materially affected, or that are reasonably likely to materially affect, our internal control over financial
reporting.
Item 16. [Reserved]
Item 16A. AUDIT COMMITTEE FINANCIAL EXPERT
Our board of directors has determined that Stephen Wills qualifies
as an “audit committee financial expert,” as defined under the U.S. federal securities laws and has the requisite financial
experience defined by the Nasdaq Marketplace Rules. In addition, Stephen Wills is independent as such term is defined in Rule 10A‑3(b)(1)
under the Exchange Act and under the listing standards of the Nasdaq Global Market.
Item 16B. CODE OF ETHICS
We have adopted a code of business conduct and ethics applicable
to our executive officers, directors and all other employees. A copy of the code is delivered to every employee of MediWound Ltd. and
its subsidiaries and is available to our investors and others on our website http://ir.mediwound.com/ or by contacting our investor relations
department. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report and
is not incorporated by reference herein. Any waivers of this code for executive officers or directors will be disclosed through the filing
of a Form 6‑K or on our website. We granted no waivers under our code of ethics in 2023.
Item 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Principal Accountant Fees and Services
The cost for the following professional services rendered by Somekh
Chaikin, a member firm of KPMG International Haifa, Israel, Auditor firm ID: 1057, our independent registered public accounting firm for
the year ended December 31, 2023 and 2022:
|
|
|
2023 |
|
|
2022 |
|
|
Audit Fees |
|
$ |
375,000 |
|
|
$ |
270,000 |
|
|
Tax Fees |
|
|
23,395
|
|
|
|
28,549
|
|
|
Total |
|
$ |
398,395 |
|
|
$ |
298,549 |
|
“Audit fees”
are the aggregate fees paid for the audit of our annual financial statements for the years 2023 and 2022. This category also includes
services that generally the independent accountant provides, such as consents and assistance with and review of documents filed with the
SEC.
“Tax fees” include
fees for professional services rendered by our independent registered public accounting firm for tax compliance, transfer pricing and
tax advice on actual or contemplated transactions.
The Audit Committee pre‑approves all audit and non‑audit
services provided by the independent registered public accounting firm.
Item 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
Item 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED
PURCHASERS
Not applicable.
Item 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
Not applicable.
Item 16G. CORPORATE GOVERNANCE
As a foreign private issuer, we are permitted to comply with Israeli
corporate governance practices instead of the Nasdaq Stock Market requirements, provided that we disclose those Nasdaq Stock Market requirements
with which we do not comply and the equivalent Israeli requirement that we follow instead. We currently rely on this “foreign private
issuer exemption” with respect to the following requirements:
|
|
•
|
Quorum. As permitted under the Israeli Companies
Law pursuant to our articles of association, the quorum required for an ordinary meeting of shareholders will consist of at least two
shareholders present in person, by proxy or by other voting instrument in accordance with the Israeli Companies Law, who hold at least
25% of the voting power of our shares (and in an adjourned meeting, with some exceptions, at least two shareholders), instead of 33 1/3%
of the issued share capital required under the Nasdaq Stock Market listing rules.
|
|
|
•
|
Shareholder approval. We do not intend to
follow Nasdaq Stock Market rules which require shareholder approval in order to enter into any transaction, other than a public offering,
involving the sale, issuance or potential issuance by the Company of ordinary shares (or securities convertible into or exercisable for
ordinary shares) equal to 20% or more of the outstanding share capital of the Company or 20% or more of the voting power outstanding before
the issuance for less than the greater of book or market value of the ordinary shares. We will follow Israeli law with respect to any
requirement to obtain shareholder approval in connection with any private placements of equity securities. |
Item 16H. MINE SAFETY DISCLOSURE
Not applicable.
Item 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT
INSPECTIONS.
Not applicable.
Item 16J. DISCLOSURE REGARDING INSIDER TRADING POLICIES.
The disclosure under this item is not yet required in this annual
report.
Item 16K. CYBERSECURITY
Cybersecurity Risk Management and Strategy
We have adopted a risk-based
approach to protecting our information technology systems and confidential information through the adoption of certain technical and administrative
safeguards, including certain information technology policies intended to protect the confidentiality, integrity, and availability of
our critical systems and information. Key elements of our cybersecurity risk management program include:
|
• |
technical controls deployed to protect more critical systems and data,
such as multi-factor authentication, firewalls, network segregation, and secure file transfer protocols; |
|
• |
disaster recovery and business continuity plan; |
|
• |
monitoring of our information technology systems for information security
risks; |
|
• |
based on their level of risk, cybersecurity assessments of third-party
software / platforms; and |
|
• |
regular cybersecurity awareness training of our employees, designed
to help identify any new threats and to address security incidents. |
We have not identified risks from known cybersecurity
threats, including as a result of any prior cybersecurity incidents, that have materially affected or are reasonably likely to materially
affect us, including our operations, business strategy, results of operations, or financial condition. See “Risk Factors—Risks
Related to Healthcare Laws and Other Legal Compliance Matters—Our business and operations may suffer in the event of information
technology system failures, cyberattacks or deficiencies in our cybersecurity.”
Cybersecurity Governance
Our Board considers cybersecurity risk as part
of its overall risk oversight function and oversees management’s implementation of our cybersecurity risk management program.
The Committee receives periodic reports from management
on our cybersecurity risks. In addition, management updates the Committee, as necessary, regarding any material cybersecurity incidents,
as well as any incidents with lesser impact potential.
The full Board also receives briefings from management
on our cyber risk management program. Board members receive presentations on cybersecurity topics from our Chief Operations Officer
(CISO), internal security staff or external experts as part of the Board’s continuing education on topics that impact public companies.
The Committee reports to the full Board regarding
its activities, including those related to cybersecurity. The full Board also receives periodic briefings from management on our cyber
risk management program, as well as from internal security staff or external experts engaged from time to time as part of the Board’s
continuing education on topics that impact public companies.
Our management team, including Chief Operations
Officer, is responsible for assessing and managing our material risks from cybersecurity threats. The team has primary responsibility
for our overall cybersecurity risk management program and supervises both our internal cybersecurity personnel and any retained external
cybersecurity consultants which are engaged as required. Our management team’s combined experience includes over 10 years of organizational
risk management experience.
Our management team supervises efforts to prevent,
detect, mitigate, and remediate cybersecurity risks and incidents through various means, which may include briefings from internal security
personnel; threat intelligence and other information obtained from governmental, public or private sources, including external consultants
engaged by us; and alerts and reports produced by security tools deployed in the IT environment.
PART III
Item 17. FINANCIAL STATEMENTS
Not applicable.
Item 18. FINANCIAL STATEMENTS
See pages F‑1 through F-47 of this annual report.
Item 19. EXHIBITS
|
101.INS |
Inline XBRL Instance Document |
|
|
|
|
101.SCH |
Inline XBRL Taxonomy Extension Schema Document |
|
|
|
|
101.CAL |
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
101.DEF |
Inline XBRL Taxonomy Definition Linkbase Document |
|
|
|
|
101.LAB |
Inline XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
101.PRE |
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
104 |
Cover Page Interactive Data File (the cover page iXBRL tags are embedded within the
Inline XBRL document) |
129
|
†
|
Portions of this exhibit have been omitted pursuant to Instruction 4(a) to Exhibits
to Form 20-F because they are both (i) not material and (ii) the type that the Registrant treats as private or confidential.
|
|
* |
Indicates management contract or compensatory plan or arrangement. |
|
(1)
|
Previously filed with the SEC on March 3, 2014 pursuant to the Registrant’s
registration statement on Form F‑1 (File No. 333‑193856) and incorporated by reference herein. |
|
(2)
|
Previously furnished to the SEC on May 5, 2021 as Appendix B to the Registrant’s
proxy statement for its 2021 annual general meeting of shareholders held on June 15, 2021, attached as Exhibit 99.1 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. |
|
(3)
|
Previously filed with the SEC on February 10, 2014 pursuant to the Registrant’s
registration statement on Form F‑1 (File No. 333‑193856) and incorporated by reference herein. |
|
(4)
|
Previously filed with the SEC on February 25, 2020 pursuant to the Registrant’s
annual report on Form 20-F for the year ended December 31, 2019 (File No. 001-36349) and incorporated by reference herein.
|
|
(5)
|
Previously furnished to the SEC on October 21, 2022 as Appendix A to the Registrant’s
proxy statement for its extraordinary general meeting of shareholders held on November 28, 2022, attached as Exhibit 99.1 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
|
|
(6)
|
Previously filed with the SEC on February 25, 2021 pursuant to the Registrant’s
annual report on Form 20-F for the year ended December 31, 2020 (File No. 001-36349) and incorporated by reference herein. |
|
(7)
|
Previously filed with the SEC on January 25, 2016 as Exhibit 4.14 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2015 (File No. 001‑36349) and incorporated by reference herein.
|
|
(8)
|
Previously filed with the SEC on February 21, 2017 as Exhibit 4.15 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2016 (File No. 001‑36349) and incorporated by reference herein.
|
|
(9)
|
Previously filed with the SEC on March 19, 2018 as Exhibit 4.16 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2017 (File No. 001‑36349) and incorporated by reference herein.
|
|
(10)
|
Previously filed with the SEC on March 25, 2019 as Exhibit 4.17 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2018 (File No. 001‑36349) and incorporated by reference herein.
|
|
(11)
|
Previously filed with the SEC on March 19, 2018 as Exhibit 4.17 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2017 (File No. 001‑36349) and incorporated by reference herein.
|
|
(12)
|
Previously filed with the SEC on March 25, 2019 as Exhibit 4.20 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2018 (File No. 001‑36349) and incorporated by reference herein.
|
|
(13)
|
Previously filed with the SEC on March 25, 2019 as Exhibit 4.21 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2018 (File No. 001‑36349) and incorporated by reference herein.
|
|
(14)
|
Previously filed with the SEC by Vericel Corporation on August 6, 2019 as Exhibit
10.9 to its quarterly report on Form 10-Q for the quarter ended June 30, 2019 (File No. 001‑35280) and incorporated by reference
herein. |
|
(15)
|
Previously filed with the SEC by Vericel Corporation on August 6, 2019 as Exhibit
10.10 to its quarterly report on Form 10-Q for the quarter ended June 30, 2019 (File No. 001‑35280) and incorporated by reference
herein. |
|
(16)
|
Previously furnished to the SEC on June 9, 2022 as Appendix A to the Registrant’s
proxy statement for its 2022 annual general meeting of shareholders held on July 19, 2022, attached as Exhibit 99.1 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
|
|
(17)
|
Previously filed with the SEC on March 17, 2022 as Exhibit 4.11.7 to the Registrant’s
annual report on Form 20‑F for the year ended December 31, 2021 (File No. 001‑36349) and incorporated by reference herein.
|
|
(18)
|
Previously furnished to the SEC on September 26, 2022 as Exhibit 4.1 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. |
|
(19)
|
Previously furnished to the SEC on September 26, 2022 as Exhibit 4.2 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein. |
|
(20)
|
Previously furnished to the SEC on September 26, 2022 as Exhibit 4.4 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
|
|
(21)
|
Previously furnished to the SEC on September 26, 2022 as Exhibit 10.3 to the Registrant’s
report of foreign private issuer on Form 6‑K (File No. 001‑36349) and incorporated by reference herein.
|
130


