
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington D. C. 20549
FORM
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the year ended | |
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the transition period from _____________ to _____________ |
Commission
file number
(Exact name of registrant as specified in its charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
(Address of principal executive offices)
(Issuer’s telephone number)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading symbol(s) | Name of each exchange on which registered | ||
| None | N/A | N/A |
Securities registered pursuant to Section 12(g) of the Act: Common stock, $0.001 par value
Indicate
by check mark whether the registrant is a well-known seasoned issuer as defined in Rule 405 of the Securities Act. ☐ Yes ☒
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ☐ Yes ☒
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days. ☒
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files). ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | |
| Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for completing with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by the check mark whether the registration has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined by Rule 12b-2 of the Exchange Act) ☐ Yes
As
of June 30, 2023, the aggregate market value of the issued and outstanding common stock held by non-affiliates of the registrant was
$
As of June 17, 2024, there were shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE - None.
CELL SOURCE, INC.
FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2023
INDEX
CAUTIONARY NOTE REGARDING FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements regarding, among other things, our anticipated financial and operating results. Forward-looking statements reflect our management’s current assumptions, beliefs, and expectations. Words such as “anticipate,” “believe,” “estimate,” “seek,” “expect,” “intend,” “could,” “plan,” and similar expressions are intended to identify forward-looking statements. While we believe that the expectations reflected in our forward-looking statements are reasonable, we can give no assurance that such expectations will prove correct. Forward-looking statements are subject to risks and uncertainties that could cause our actual results to differ materially from the future results, performance, or achievements expressed in or implied by any forward-looking statement we make. Some of the relevant risks and uncertainties that could cause our actual performance to differ materially from the forward-looking statements contained in this report are discussed below under the heading “Risk Factors” and elsewhere in this Annual Report on Form 10-K. We caution investors that these discussions of important risks and uncertainties are not exclusive, and our business may be subject to other risks and uncertainties which are not detailed there. Investors are cautioned not to place undue reliance on our forward-looking statements. We make forward-looking statements as of the date on which this Annual Report on Form 10-K is filed with the U.S. Securities and Exchange Commission (“SEC”), and we assume no obligation to update the forward-looking statements after the date hereof whether as a result of new information or events, changed circumstances, or otherwise, except as required by law.
| i |
Summary of Principal Risk Factors
Our business operations are subject to numerous risks, factors and uncertainties, including those outside our control that could cause our actual results to be harmed, including risks regarding the following:
Risks Related to our Business and Industry
| ● | We may not receive regulatory approvals for our product candidates or there may be a delay in obtaining such approvals. |
| ● | Clinical trials for our product candidates are expensive and time consuming and their outcome is uncertain. |
| ● | We may be required to suspend or discontinue clinical trials due to unexpected side effects or safety risks. |
| ● | Delays in our clinical trials could delay our ability to obtain regulatory approval and our ability to commercialize our products. |
| ● | The results of our clinical trial are uncertain and could substantially delay or prevent us from bringing products to market. |
| ● | Pre-clinical studies and Phase 1 or 2 clinical trials of our product candidates may not predict the results of subsequent human trials. |
| ● | Our clinical trials may fail to demonstrate evidence substantial evidence of the safety and efficacy of our product candidates. |
| ● | We are subject to various government regulations, may become subject to increased government regulation and may fail to comply with regulatory requirements. |
| ● | Regulatory approval of our product candidates may be withdrawn at any time. |
| ● | Even if approved, our products may not gain market acceptance. |
| ● | We do not own any patents and rely on the patents we license from Yeda Research and Development Company Limited. |
| ● | Our success will depend in part on our ability to obtain and maintain patent protection for the patents we license. |
| ● | Confidentiality agreements may not prevent unauthorized disclosure of trade secrets or proprietary information. |
| ● | We are dependent on collaborative partners and service providers. |
| ● | We will be unable to operate profitability if we are unable to keep up with rapid technological developments. |
| ● | Our ability to sell products will depend to a large extent upon reimbursement from insurance companies. |
| ● | We may expend our limited resources to pursue a particular product candidate or indications and fail to capitalize on more profitable products or indications. |
| ● | Clinical trial data that we publish may change as more patient data becomes available. |
| ● | We rely on key personnel. |
| ● | We may be subject to foreign exchange fluctuations. |
| ii |
| ● | We may be subject to potential product and clinical trials liability and liabilities related to working with hazardous materials. |
| ● | If we conduct clinical trials outside of the United States, the FDA may not accept data from such trials. |
| ● | The Russia-Ukraine and Israel-Hamas wars and other geopolitical and macroeconomic events have resulted in disruption of global credit and financial markets and may make necessary debt or equity financing more difficult, costly or dilutive. |
Risks Related to Our Capital Resources and Impairments
| ● | We have a limited operating history and a history of operating losses and expect to incur significant additional losses. |
| ● | We will need to secure additional financing. |
| ● | There is substantial doubt about our ability to continue as a going concern. |
| ● | We are an early stage company with an unproven business strategy. |
| ● | We are in default of payment obligations under promissory notes. |
Risks Related to Our Common Stock
| ● | We may issue additional shares of preferred stock in the future. |
| ● | There is not an active liquid trading market for our Common Stock. |
| ● | We may not be able to attract the attention of brokerage firms because we became a public company by means of a reverse acquisition. |
| ● | Voting power of our shareholders is highly concentrated in insiders. |
| ● | We do not intend to pay dividends on our Common Stock for the foreseeable future. |
| ● | Shareholders may be diluted by future issuances of Common Stock. |
| ● | As an issuer of “penny stock”, protection provided by federal securities laws relating to forward looking statements does not apply to us. |
| ● | Our ability to use net operating loss carryforwards may be limited. |
| ● | Our issuance of Common Stock upon exercise of warrants or options may depress the price of our Common Stock. |
Other General Risk Factors
| ● | Applicable regulatory requirements may make it difficult to retain or attract qualified directors and officers. |
| ● | Our stock price could be adversely affected if we fail to maintain effective internal controls over financial reporting. |
| ● | Failure to comply with the Sarbanes-Oxley Act of 2002 could harm our business and stock price. |
| ● | Our stock price may decline or be volatile, regardless of our operating results. |
| ● | Our quarterly operating results may fluctuate significantly or fall below expectations of investors or securities analysts. |
| ● | As a result of reduced disclosure requirements applicable to a “smaller reporting company,” the information we provide to investors may be different than information provided by other public companies. |
| ● | We could be subject to securities class action litigation. |
| iii |
PART I
ITEM 1. BUSINESS.
Overview
Our Business
We are a cell therapy company focused on immunotherapy. Since our inception, we have been involved with the development of proprietary immune system management technology (“Cell Source technology”) licensed from Yeda Research & Development Company Limited (“Yeda”), the commercial arm of the Weizmann Institute of Science (“Weizmann Institute”) in Israel. The current focus of our Research and Development efforts is at the University of Texas MD Anderson Cancer Center (“MD Anderson”) in Houston, Texas.
This technology addresses one of the most fundamental challenges within human immunology: how to tune the immune response such that it tolerates selected desirable foreign cells, but continues to attack all other (undesirable) targets. In simpler terms, a number of potentially life-saving treatments have limited effectiveness today because the patient’s immune system rejects them. For example, while HSCT – hematopoietic stem cell transplantation (e.g. bone marrow transplantation) has become a preferred therapeutic approach for treating blood cell cancer, most patients do not have a matched family donor. Although matched unrelated donors and cord blood can each provide an option for such patients, haploidentical stem cell transplants (sourced from partially mismatched family members) are rapidly gaining favor as a treatment of choice. This is still a risky and difficult procedure primarily due to potential conflicts between host and donor immune systems and as well as viral infections that often follow even successful HSCT while the compromised new immune system works to reconstitute itself by using the transplanted stem cells. Today, rejection is partially overcome using aggressive immune suppression treatments that leave the patient exposed to many dangers by compromising their immune system.
The unique advantage of Cell Source technology lies in the ability to induce sustained tolerance of transplanted cells (or organs) by the recipient’s immune system in a setting that requires only mild immune suppression, while avoiding the most common transplant related complications. The scientific term for inducing such tolerance in a transplantation setting is chimerism, where the recipient’s immune system tolerates the co-existence of the (genetically different) donor type and host (recipient) type cells. Attaining sustained chimerism is an important prerequisite to achieving the intrinsic GvL (graft versus leukemia) effect of HSCT and supporting the reconstitution of normal hematopoiesis (generation of blood cells, including those that protect healthy patients from cancer) in blood cancer patients. Preclinical data, as well as initial clinical data, show that Cell Source’s Veto Cell technology (currently in a trial in the US) can provide superior results in allogeneic (donor-derived) HSCT by allowing for haploidentical stem cell transplants under a mild conditioning regimen, while avoiding the most common post-transplant complications. Combining this with CAR (Chimeric Antigen Receptor) T cell therapy employing Veto Cells, as a VETO CAR-T treatment, we plan to treat patients in relapse as well as those in remission and use the cancer killing power of CAR-T to protect the patient while their immune system fully reconstitutes, thus providing an end-to-end solution for blood cancer treatment by potentially delivering a fundamentally safer and more effective allogeneic HSCT: prevention of relapse; avoidance of graft versus host disease (GvHD); prevention of viral infections; and enhanced persistence of GvL effect. This means that the majority of patients will be able to find a donor, and will have access to a potentially safer procedure with higher long term survival rates than what either donor-derived HSCT or autologous CAR-T each on their own currently provide.
The ability to induce permanent chimerism (and thus sustained tolerance) in patients – which allows the transplantation to overcome rejection without having to compromise the rest of the immune system – may open the door to effective treatment of a number of severe medical conditions, in addition to blood cancers, which are characterized by this need. These include:
| ● | The broader set of cancers, including solid tumors, that can potentially be treated effectively using genetically modified cells such as CAR-T cell therapy, but also face efficacy and economic constraints due to limited persistence based on immune system issues (i.e., the need to be able to safely and efficiently deliver allogeneic CAR-T therapy). Inducing sustained tolerance to CAR-T cells may bring reduced costs and increased efficacy by allowing for off-the-shelf (vs. patient-derived) treatments with more persistent cancer killing capability. |
| 4 |
| ● | Organ failure and transplantation. A variety of conditions can be treated by the transplantation of vital organs. However, transplantation is limited both by the insufficient supply of available donor organs and the need for lifelong, daily anti-reject treatments post-transplant. Haploidentical organ transplants, with sustained chimerism, have the potential to make life saving transplants accessible to the majority of patients, with the prospect of improved life quality and expectancy. |
| ● | Non-malignant hematological conditions (such as type one diabetes and sickle cell anemia) which could, in many cases, also be more effectively treated by stem cell transplantation if the procedure could be made safer and more accessible by inducing sustained tolerance in the stem cell transplant recipient. |
Corporate History
Cell Source, Inc. (the “Company”) is a Nevada corporation formed on June 6, 2012 under the name Ticket to See, Inc. (“TTSI”). Cell Source Ltd. (“Cell Source Israel”) was founded in 2011 in order to commercialize a suite of inventions that were the result of over ten (10) years of research at the Weizmann Institute. Pursuant to a Research and License Agreement by and between Cell Source Israel and Yeda, dated October 3, 2011, as amended in April, 2014, November, 2016, March, 2018, August 2019, December, 2019, November, 2020, and December, 2021 (the “Yeda License Agreement”), Yeda, the commercial arm of the Weizmann Institute, granted Cell Source Israel an exclusive, worldwide license to certain patents, discoveries, inventions, and other intellectual property generated (together with others) by Yair Reisner, Ph.D. (“Dr. Reisner”), former head of the Immunology Department at the Weizmann Institute.
Implications of being a Smaller Reporting Company
As a company with less than $100 million in revenue during our last fiscal year and a public float of less than $250 million, we qualify as a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. A “smaller reporting company” may take advantage of reduced reporting requirements and disclosure obligations that are otherwise applicable to public companies. These provisions include, but are not limited to:
| ● | being permitted to present only two years of audited financial statements and only two years of related Management’s Discussion & Analysis of Financial Condition and Results of Operations in this report on Form 10-K; |
| ● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act; and |
| ● | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements. |
We have elected to take advantage of certain of the reduced disclosure obligations and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our stockholders may be different than you might receive from other public reporting companies in which you hold equity interests. Decreased disclosures in our SEC filings due to our status as a “smaller reporting company” may make it harder for investors to analyze our results of operations and financial prospects.
| 5 |
Hematological Malignancies
Hematological malignancies (blood cancers) comprise a variety of lymphomas and leukemias. A very important treatment protocol for these malignancies involves the use of HSCT. Approximately 100,000 of these transplants are performed annually worldwide (table below). Our technology has immediate relevance for, at a minimum, the roughly 40,000 worldwide stem cell transplants that are allogeneic (using cells taken from another individual, not the patient). According to the Center for International Blood and Marrow Transplant Research (“CIMBTR”), there were 22,827 stem cell transplants performed in the US in 2021, of which 9,349 were allogeneic. The journal Bone Marrow Transplantation reports that 46,143 stem cell transplants were performed in Europe in 2022, of which 19,011 were allogeneic.
HSCT often has a curative effect when successful. However, it is very risky. HSCT typically involves destroying the patient’s native immune system with radiation or chemotherapy (myeloablation) before the transplantation, and then suppressing immune response (immunosuppression) with drugs to manage the conflicts between host and donor cells. The majority of patients are unable to find a matched family donor. Over 50% of all adult haploidentical transplant patients in the US suffering from AML (the most common indication for allogeneic HSCT) die within three years of transplantation. Among adult patients receiving haploidentical HSCT, of those who die in the first 100 days post-transplant, 38% die from either infections (associated with a compromised immune system), graft rejection or GvHD (Graft versus Host Disease).
Myeloablation and immunosuppression are dangerous and difficult to tolerate, especially in patients over age 50. Therefore, HSCT has been used mainly with younger patients.
Another very important treatment protocol for blood cancers is CAR-T cell therapy. This novel approach uses the patient’s own immune system cells to directly attack cancer cells. CAR-T cells are made by removing a specific set of cells from the blood, genetically modifying them in order to intensify the immune system’s natural response to cancer, and then re-injecting them into the patient. This form of cellular therapy has produced exceptional near term results in blood cancer patients and is currently being tested against a variety of different cancer types.
CAR T-cell therapy has been approved by the U.S. Food and Drug Administration as standard therapy for some patients with lymphoma (drug names Yescarta, Kymriah, Tecartus and Breyanzi), leukemia (drug name Kymriah, Tecartus) and multiple myeloma (drug names Abecma and Carvykti).
| 6 |
These approved treatments use the patient’s own cells in order to create CAR-T cells, which involves high cost (can reach close to $500,000 per infusion) and significant safety risks (e.g. high rate of relapse, significant incidence of Cytokine Release Syndrome (CRS)). While a number of companies are attempting to develop allogeneic or “off-the-shelf” CAR-T, they face several challenges including rejection by the host’s immune system and GvHD. The currently approved autologous CAR-T treatments, while showing high early response rates, have not shown long term survival results for blood cancer that exceed those of allogeneic HSCT.
This means that:
| a) | many blood cancer patients are not candidates for the primary treatment (HSCT) that represents a potential cure; | |
| b) | there is high mortality among those patients who are candidates for HSCT and do undergo the procedure; | |
| c) | CAR-T cell therapy, which is currently used in limited indications and has had relatively slow adoption, has yet to demonstrate long term survival that substantively exceeds that of HSCT. |
There is widespread awareness of the need for improved immune-system management technologies for HSCT – both to improve outcomes of transplantations for the traditional target set of patients and to expand the use of the procedure by making transplantation safe enough to become appropriate for a broader set of patients.
There is also a strong awareness for the need of an off-the-shelf approach to CAR-T that overcomes rejection, avoids GvHD, and has increased persistence so as to deliver longer-term efficacy.
We aspire to use VETO CAR-T, combining Veto Cell technology with allogeneic CAR-T cell therapy, to dramatically improve the outcomes of the allogeneic transplantations already being performed, and thereby to rapidly penetrate the current market. However, our target population greatly exceeds those patients who currently undergo HSCT or CAR-T, as the firm’s tolerizing technology could potentially make allogeneic transplantation and off-the-shelf CAR-T an option for a much larger proportion of the diseased population. The following table shows the incidence of the specific hematological malignancies on which we will focus:
Initial Malignancy Indications (note estimates for North America and EU only) | Incidence (Annual New Cases) | Annual HSCT | ||||||
| Lymphoma | 222,149 | 12,484 | ||||||
| Leukemia | 151,655 | 15848 | ||||||
| Multiple Myeloma | 80,570 | 20,992 | ||||||
| Total | 454,374 | 49,324 | ||||||
Source: National Cancer Institute, World Health Organization, Leukemia & Lymphoma Society, Lymphoma, European Cancer Information System, Coalition Europe, EMBT, HRSA, CIMBTR
For the purposes of this document, it is assumed that the immediate candidates for Cell Source-enabled HSCT will be the subset of cancer patients that today receive transplantations as part of their cancer treatment (rightmost column in table above). We believe that a portion of these patients will benefit from Veto Cell adjunct therapy, as such therapy aspires to improve the success and reduce the risk and mortality of a procedure that they are having anyway. With time, as Veto Cell treatment becomes more widespread and data is accumulated, we believe that the percentage of patients that will be referred for Veto Cell enabled HSCT will increase significantly.
It is also important to note that incidence of these diseases is increasing. The global market for blood cancer therapeutics was estimated at $48 billion in 2022 and is projected to increase in size to reach $98 billion by 2030 according to DataBridgeGlobal Blood Cancer Drug Market – Industry Trends and Forecast to 2030 October, 2023. The aging of the US population and the increased incidence of hematologic malignancies are expected to significantly increase the number of older patients who receive allogeneic HSCT.
| 7 |
HSCT Market Trends
There are four important market trends affecting the hematological malignancies market:
| 1) | As noted above, increasing incidence of these disorders, largely driven by the aging population. | |
| 2) | Improvement and proliferation of HSCT treatments. | |
| 3) | A “virtuous circle” of lowered death rate due to better transplantations leading to more aggressive focus on HSCT. | |
| 4) | The growing use of milder conditioning regimens, which makes the procedure more survivable for older patients (see table below). |
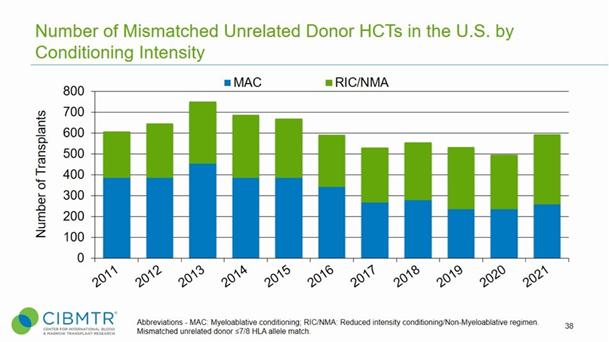
However, despite the above trends, the use of HSCT, especially allogeneic, remains limited because of the risks associated with the myeloablative treatments required to reduce the host immune response, viral infections and GvHD. This means that the “gold-standard” of treatment is largely unavailable to an age cohort that constitute a significant proportion of sufferers of these diseases.
The Company addresses this issue in a distinctive manner by significantly reducing the need for myeloablative treatment while avoiding the risk of GvHD, thereby improving the outlook for allogeneic transplantations and enabling their use in a much larger population set.
CAR-T cell therapy
One of the most promising new approaches to treating hematological malignancies is by using genetically modified T cells in treatments such as CAR-T and TCR. CAR-T cell therapy for blood cancers, which has already been approved by the FDA, has shown the ability to attain remissions in a significant proportion of those patients treated. That said, the number of patients treated has been relatively low, in part due to the significant costs associated with this treatment. Since the approved treatment products rely on autologous (patient derived) production of the CAR-T cells, the costs can run into the hundreds of thousands of dollars for a single treatment, with the cost of the infusion in some cases exceeding $480,000 in the US. The broader hope for CAR-T cell therapy is for an allogeneic or “off the shelf” version that is expected to significantly lower the treatment costs.
| 8 |
Cell Source has completed a preclinical proof-of-concept in collaboration with Professor Zelig Eshhar, the inventor of CAR-T cell technology, combining CAR-T and Veto Cell technology so as to allow for a successful allogeneic approach to CAR-T cell therapy.
Relevant Non-Malignant Diseases
While Hematological malignancies represent the Company’s initial focus, the Company’s selective immune response blocking technology may also be effective in treating certain non-malignant organ diseases as well as blood and immune system disorders. This would represent an additional growth opportunity for the Company.
The target non-malignant diseases are widespread. The Company’s first non-malignant disorder target is expected to be is support of organ transplantations (kidney, liver, etc.). Over 100,000 kidney and approximately 35,000 liver transplants are conducted worldwide each year. As with bone marrow transplantations, organ transplantations require substantial and ongoing immunosuppression to prevent rejection. This ongoing treatment is dangerous, quality-of-life and life expectancy reducing, and costly. The Company’s Veto Cell technology can potentially be used to selectively reduce immune response to the transplanted organ, thus broadening the donor pool and reducing or possibly eliminating the need for daily, life-long immunosuppression post transplantation.
A second target within non-malignant disorders are blood diseases such as sickle cell disease, aplastic anemia, beta thalassemia and scleroderma. Sickle cell anemia, for example, can be effectively treated by HSCT. However, because of HSCT’s riskiness, the procedure is currently used only in extreme cases. If successful in enabling safer HSCT, the Company can make this treatment available to a broader set of sickle cell anemia sufferers. Preclinical data have also shown the potential effectiveness of Veto Cells in preventing the development of Type 1 Diabetes.
Market Access and Channels
The market for stem cell transplantation is relatively concentrated. There are over 1,700 transplantation centers worldwide, of which some 700 are in North America and Western Europe.
A relatively small subset of these (often termed “Centers of Excellence”) tends to set the practice standards for the entire transplantation community. Therefore, as discussed in the “Strategy” section, the Company plans to focus its initial penetration strategy on a relatively small group of influential centers. There are over 150 centers in the US today that provide CAR-T cell therapy treatments.
Reimbursement issues for our therapies are expected to be relatively straightforward. Once clinical effectiveness and regulatory approval are established, the value-proposition for payors and providers is expected to be clear and compelling. Issues connected with immunosuppression and rejection constitute a major component of bone marrow transplantation costs, and significant improvement in this area is expected to bring substantive cost-savings for payors.
Sector Focus
We are in the overall arena of immunotherapy. The cancer immunotherapy market was estimated at approximately $125 billion for 2023 and is expected to grow at a compound annual growth rate of 8.3% from 2024 to 2030, according to Grandview Research and is projected to grow to over $210 billion by 2030, according to Market Digits.
Within the immunotherapy field, our initial focus is on allogeneic therapies (treatments using donor derived-as opposed to patient derived-cells), with a focus on haploidentical transplantations (transplantations that use cells from partially matched-as opposed to fully matched-donors and recipients). While potentially valuable, allogeneic therapies are relatively complex, risky, and expensive. A key driver of this complexity and associated costs is the conflict between host and donor immune systems, as discussed above.
Our technology, which in preclinical studies, as well as in a first-in-human proof of concept, has shown the ability to enable tolerance of donor cells without affecting other immune processes, is fundamentally enabling. We expect it to significantly increase the safety, reduce the overall treatment cost, and therefore broaden the scope of indications for such procedures.
| 9 |
The delivery method for Veto Cell treatments would take the form of a non-invasive cell suspension treatment administered intravenously. For HSCT treatments, Veto Cells are derived from stem cells taken from the same donor who is providing the stem cells for the transplantation itself. In the case of VETO CAR-T cell therapy, this will initially be combined with HSCT, but a more generic “off the shelf” modality offering is planned, which would eventually be marketed as a pre-packaged suspension of cells and medium, prepared and stored in advance.
Our Value Drivers
Our current positioning in the cancer immunotherapy value chain is typical of an early clinical stage company: developing, validating and attaining regulatory approvals for the various applications of our technology platforms. Going forward, once the products are commercialized, physician and patient interest in these treatments is expected to drive insurer reimbursement for patients – a key demand lever. The generic value chain for biotechnology development commences with an invention which is formulated, patented and successful in pre-clinical animal trials. We have already passed this stage with our Veto Cell technology platform, for which we have an exclusive license to use from Yeda, the owner of these patents. We are currently at the stage of human trials (testing both safety and efficacy). Finally, the offering earns regulatory approval and patient treatment, along with the ensuing revenues, can commence. This can be a particularly lengthy process in the United States and therefore some medical treatments are approved in Europe or Asia and generate revenues there prior to commencing U.S. sales. Recently expanded “fast track” regulation in the U.S. is aimed at getting critical treatments for life threatening conditions to patients more quickly.
Our successful preclinical validation of the Veto Cell treatment involved basic laboratory research including both in-vivo (live) animal trials and in-vitro (in a glass dish) human cell trials. This validated the protocol prior to commencing human clinical trials. Human clinical trials fine-tune the treatment protocol and confirm both safety and efficacy in treating patents. In parallel, the patents on the core technology go into the national phase in various countries and are emended with claims associated with exact treatment protocols, bolstering the protection afforded by already issued patents on the base technology.
In some cases, successful biotech companies have been able to capitalize on positive human clinical results (even prior to full approval for patient treatment) by either signing lucrative non-dilutive distribution option deals or by being partially or fully acquired by larger market participants. High profile CAR-T acquisitions have included KITE Pharmaceuticals, a CAR-T cell therapy company, which was acquired outright by Gilead Sciences in 2017 for $11.9 billion in cash, prior to having attained FDA approval and prior to commencing any product sales, and Juno Therapeutics, which was acquired by Celgene Corporation in 2018 for approximately $9 billion, also without having FDA approval for its CAR-T cell therapy technology. Multi-billion dollar oncology transactions have continued, including the 2023 acquisition by Pfizer of Seagen $43B, and Abbvie’s $10B acquisition of Immunogen in early 2024. There is no indication or assurance that we are currently under consideration for any option or acquisition deal.
We are currently conducting a human clinical trial for approval for the Veto Cell based treatments in the United States. We have had positive preclinical results for three of our cell therapy treatments. Yeda, the proprietary owners of the patents underlying our technologies from whom we license our patents, has been granted patents for its original Veto Cell. The revised versions of the Veto Cell are the subject of patent applications which have been granted in some jurisdictions and are pending in others. These newer patent applications both leverage the priority of the already granted patents and extend the protection period for more advanced versions. We are currently engaged in our first human clinical trial. If such trials are successful, they will demonstrate both safety (the patients survived and were not harmed) and initial indications of efficacy (there are signs of successful engraftment under a mild conditioning regimen, with a reduction in GvHD, and in the case of cancer patients prolonging the progression free period).
| 10 |
Science and Technology Overview
The patent portfolio that we license from Yeda includes a variety of cell therapy applications. The portfolio includes both granted and pending patents. The total relevant patent portfolio consists of 16 patent “families” (i.e. grouping of similar patent applications in different territorial jurisdictions) which currently include: 68 granted patents; 4 allowed/accepted patents; and a further 68 pending patents. The key terms of the agreement pursuant to which we license all of Yeda’s patents related to our technology is set forth in the section entitled “Intellectual Property” herein. The license period (per product, per country) is for the full life of the patents and expires at the later of the patent expiration date in that country or 15 years after the date that the FDA or local equivalent regulatory authority in each country approves that particular product for sale in that country. Provided that Cell Source either sponsors research or pays either a nominal license fee of $50,000 per year (total for use of all the products), or pays royalties on product sales on at least one product as per the license agreement, the license will remain in effect continuously and expire only with the expiration of the patent or 15 years after regulatory approval (later of the two) per product per country as described above.
Professor Yair Reisner, the inventor of Veto Cell technology, left the Weizmann Institute and relocated to MD Anderson in Houston, Texas. He was awarded a $6 million grant from the Cancer Research and Prevention Institute of Texas. This, coupled with research funding from the University itself, provided him with a total funding commitment of $10 million for five years. Professor Reisner is now the Head of Stem Cell Research at the Department of Stem Cell Transplantation & Cellular Therapy as well as the Reisner Laboratory at MD Anderson.
Cell Source is currently sponsoring ongoing research by Professor Reisner and members of his team, some of whom have also relocated from the Weizmann Institute to MD Anderson, for developing existing and new applications for Veto Cell technology and plans to license any new intellectual property developed there on an exclusive basis, as it does from Yeda. MD Anderson is the largest HSCT center in the United States, performing over 850 transplantations per year. MD Anderson is currently conducting a human clinical trial sponsored by Cell Source for its Anti-Viral Veto Cell. Professor Richard Champlin (who Chairs their Department of Stem Cell Transplantation and Cellular Therapy and is a longtime associate and collaborator of Professor Reisner) serves as Principal Investigator for this trial.
Although Yeda has applied for and been granted various patents related to our technology, a granted patent only provides Yeda, and the Company by virtue of its exclusive license, the right to use the underlying invention. However, in order for our cell therapy and cancer therapy to be legally sold and administered to patients, the FDA or similar regulatory agencies must approve its use. In other words, having a patent provides legal “freedom to operate” for a certain technology, and may provide the ability to prevent others from using the same technology without the patent holder’s permission. However, in order to legally manufacture and distribute products, a company must go through all of the typical approval steps delineated in the “Overview” section above.
The following sections provide an overview of each platform. Further information on the underlying science is available upon written request and the execution of an appropriate nondisclosure agreement.
Our licensed technology portfolio consists of 17 patent families, 78 granted patents and 59 pending patent applications. The following table lists the patents and patent applications that Yeda holds and which we have a license to use in each of the below-referenced countries:
| Anti Third Party Central Memory T Cells, Methods of Producing Same and Use of Same in Transplantation and Disease Treatment | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 9,738,872 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Europe | 2365823 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. | |||||
| China | ZL200980153053.4 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Israel | 212587 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. | |||||
| India | 285832 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2506311 | 29-Oct-2009 | 29-Oct-2029 | Granted | Yeda Research and Development Co. Ltd. | |||||
| 11 |
| Use of Anti Third Party Central Memory T Cells for Anti-Leukemia/Lymphoma Treatment | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 9,421,228 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Europe | 2613801 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Canada | 2,810,632 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| China | ZL201180053858.9 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Israel | 225102 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Japan | 5,977,238 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK1187528 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Republic of Korea | 10-1788826 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Singapore | 188473 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Mexico | 357746 | 08-Sep-2011 | 08-Sep-2031 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Anti Third Party Central Memory T Cells, Methods of Producing Same and Use of Same in Transplantation and Disease Treatment | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA (Divisional) |
11,324,777 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Continuation) |
2023-0024587-A1 | 06-Sep-2012 | 06-Sep-2032 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 2753351 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Canada | 2,848,121 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| China | ZL201280054739.X | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Israel | 231397 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2012305931 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| New Zealand | 622749 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Japan | 6,196,620 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK1200099 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Republic of Korea | 10-2073901 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Singapore | 11201400513P | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Brazil | BR 11 2014 005355 3 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Mexico | 351226 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| South Africa | 2014/01993 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| India | 375463 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2636503 | 06-Sep-2012 | 06-Sep-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| 12 |
| Use of Anti Third Party Central Memory T Cells | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| Europe | 3322424 | 14-Jul-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| China | CN 108025026 A | 14-Jul-2016 | 16-Jul-2036 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Methods Of Transplantation And Disease Treatment | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 10,933,124 | 14-Jul-2016 | 14-JuL-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Genetically Modified Anti-Third Party Central Memory T Cells and Use of Same in Immunotherapy | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 11,179,448 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Continuation) | 11,844,827 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Continuation) | 18/515,358 | 14-July-2016 | 16-Jul-2036 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 3322425 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Canada | 2,991,690 | 14-July-2016 | 16-Jul-2036 | Pending | Yeda Research and Development Co. Ltd. | |||||
| China | CN 108135938 A | 14-July-2016 | 16-Jul-2036 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Israel | 256916 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2016291825 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Japan | 7,057,748 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK1255063 | 14-July-2016 | 16-Jul-2036 | Granted | Yeda Research and Development Co. Ltd. | |||||
| 13 |
| Veto Cells Generated from Memory T-Cells | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 10,961,504 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Continuation) | 11,773,372 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Continuation) | 2023-0383254-A1 | 27-Jun-2017 | 27-Jun-2037 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 3475414 | 27-Jun-2017 | 27-Jun-2037 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Canada | 3,029,001 | 27-Jun-2017 | 27-Jun-2037 | Pending | Yeda Research and Development Co. Ltd. | |||||
| China | CN 109661463 A | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Israel | 263924 | 27-Jun-2017 | 27-Jun-2037 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2017289879 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Japan | 7334043 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | 40007502A | 27-Jun-2017 | 27-Jun-2037 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Singapore | 11201811563R | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Republic of Korea | 10-2444170 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Mexico | MX/a/2019/000022 | 27-Jun-2017 | 27-Jun-2037 | Pending | Yeda Research and Development Co. Ltd. | |||||
| India | 474458 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2779844 | 27-Jun-2017 | 27-Jun-2037 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Methods of Transplantation and Disease Treatment | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 10,751,368 | 18-Jan-2018 | 18-Jan-2038 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Genetically Modified Veto Cells and Use of Same in Immunotherapy | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 11,555,178 | 18-Jan-2018 | 18-Jan-2038 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Europe | 3571295 | 18-Jan-2018 | 18-Jan-2038 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Israel | 268126 | 18-Jan-2018 | 18-Jan-2038 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Anti-Viral Central Memory CD8+ Veto Cells in Haploidentical Stem Cell Transplantation | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| Canada | 3,149,379 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Japan | 2022-506965 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK40076176 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Brazil | BR 11 2022 001988 2 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Israel | 290337 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Mexico | MX/a/2022/001578 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| USA | 2023-0398214-A1 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2020326568 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| India | 202227011269 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 4009991 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| China | CN 114466925 A | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2022105687 | 06-Aug-2020 | 06-Aug-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| 14 |
| Veto CAR-T Cells | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 2023-0321235-A1 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 4196573 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Canada | 3,189,051 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| China | CN 116322717 A | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Israel | 300470 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2021323525 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Japan | 2023-509690 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK40096222 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Singapore | 11202300925X | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Mexico | MX/a/2023/001771 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| India | 202327016034 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2023105361 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Brazil | BR 11 2023 002424 2 | 11-Aug-2021 | 11-Aug-2041 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Conditioning Protocols for use with Anti-Viral Central Memory CD8+ Veto Cells in Haploidentical Stem Cell Transplantation | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| PCT | IL2023/051121 | 31-Oct-2023 | 31-Oct-2043 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Use of Veto Cells for the Treatment of Sickle Cell Disease | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA (Continuation) | 2022-0265726-A1 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 4055145 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Canada | 3,160,296 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| China | CN 114901801 A | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Israel | 292720 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Nigeria | NG/PT/PCT/2022/6020 | 05-Nov-2020 | 05-Nov-2040 | Granted | Yeda Research and Development Co. Ltd. | |||||
| South Africa | 2022/06164 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK40078965 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Mexico | MX/a/2022/005418 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Brazil | BR 11 2022 008700 4 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| 15 |
| Use of Veto Cells in Treatment of T Cell Mediated Autoimmune Diseases | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA (Continuation) | 2022-0265725-A1 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Europe | 4055146 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Canada | 3,160,301 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| China | CN 115175688 A | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Japan | 2022-525357 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Israel | 292722 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2020379319 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2022114785 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK40078964 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Mexico | MX/a/2022/005416 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Brazil | BR 11 2022 008638 5 | 05-Nov-2020 | 05-Nov-2040 | Pending | Yeda Research and Development Co. Ltd. | |||||
| A Combination Therapy for a Stable and Long Term Engraftment | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 10,369,172 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Europe | 2793914 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Israel | 233303 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2012355990 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| New Zealand | 627272 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | HK1202810 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Republic of Korea | 2109643 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Singapore (Divisional) | 10201801905W | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Mexico | 370404 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2657758 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| A Combination Therapy for a Stable and Long Term Engraftment Using Specific Protocols for T/B Cell Depletion | ||||||||||
| Country | Patent Number | Filed | Expires | Status | Assignee | |||||
| USA | 10,434,121 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| USA (Continuation) |
11,504,399 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Europe | 2797421 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Canada | 2,859,952 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Israel | 233302 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Australia | 2012355989 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Australia (Divisional) | 2016259415 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| New Zealand | 627549 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Japan | 6,313,219 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Hong Kong | 1202775 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Singapore | 11201403456U | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Brazil | BR 11 2014 015959 9 | 20-Dec-2012 | 20-Dec-2032 | Pending | Yeda Research and Development Co. Ltd. | |||||
| Mexico | 372502 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| South Africa | 2014/05298 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| Russian Federation | 2648354 | 20-Dec-2012 | 20-Dec-2032 | Granted | Yeda Research and Development Co. Ltd. | |||||
| 16 |
Veto Cell Technology Platform
Background
Our Veto Cell technology is a unique immunotherapy platform technology that enables the selective attenuation of the immune system. In other words, pre-clinical studies as well as initial human clinical trial results show that the treatment has the ability to reduce the immune response to selective “threats,” with low risk for adverse side effects.
What makes the Veto Cell approach distinctive is the degree to which it leverages the inherent specificity of the human immune system. The immune system defends the body by creating a specific stream of T-cell clones for each of millions of possible individual threats. A given T-cell will attack only its specific target, ignoring all other threats. Our technology enables the physician to selectively attenuate immune response, thus effectively “switching-off” an individual stream of T-cell clones without affecting other such streams of T-cell clones dispatched by the immune system to attack unwanted incursions.
The technology is based on the discovery that certain T-cells can acquire the property of attracting and proactively neutralizing immune attacks on them.
The technology has achieved distinctive results in animal live trial models. See, e.g., Eran Ophir et al. Murine anti-third party central-memory CD8+ promote hematopoietic chimerism under mild conditioning: lymph-node sequestration and deletion of anti-donor T cells, BLOOD, Feb. 14, 2013, at 1220; Towards off-the-shelf genetically modified T cells: prolonging functional engraftment in Mice by CD8 veto T cells, Leukemia 32, 2018; 1038-1040. Veto cells for safer nonmyeloablative haploidentical HSCT and CAR T cell therapy Seminars in Hematology 56, 2019; 173-182. It has also demonstrated both safety and efficacy thus far in human clinical trials.
The main objective of the trial is to achieve engraftment of the T-cell depleted transplant without GvHD by using Veto cells, and to define the optimal Veto cell dose. The initial version of the protocol has been very successful in achieving these primary endpoints in all of the first 10 patients. None of the patients experienced toxicity nor any other adverse outcome associated with the Veto cells. While a subsequently treated patient had initial engraftment, the patient developed secondary graft failure, associated with a viral infection which is known to cause this. Another patient with aplastic anemia, a nonmalignant disorder, achieved successful engraftment, but later developed autoimmune hemolytic anemia, and eventually died; this was not linked to the Veto cells.
Four patients who engrafted subsequently had reduced white blood cell counts, which responded to treatment. Importantly, none of these patients have rejected their transplant. These reduced white blood cell counts are antibody related issues, which are more common after T-cell depleted transplants and haploidentical transplants. Since this protocol uses a reduced intensity preparative regimen to reduce toxicity, there is limited depletion of B-cells. In other settings, the use of Rituximab prior to a T-cell deplete transplant has effectively addressed this issue, without adverse outcomes. The current protocol already includes Rituximab for patients with B-cell blood cell cancers, and we now plan to give Rituximab to all patients going forward. We have structured the balance of the trial to both determine the maximum Veto cell dose tolerance and also to ensure that we can avoid antibody related issues. If it continues to succeed in human clinical trials, we believe that it may have meaningful and potentially broad impact on the field of stem cell transplantation:
| 1) | Significantly improve outcomes of transplantations by reducing the host (transplant recipient) rejection rate of T-cell depleted stem cells (e.g. from bone marrow) – thus supporting successful engraftment of the transplanted cells, which is the treatment for the blood cancer itself. In order to improve the safety of this cancer treatment, Veto Cell technology has shown in both preclinical studies and initial clinical data that it can markedly reduce both the risk of GvHD and the need for using aggressive amounts of immunosuppression treatments. We have shown in preclinical studies, and are beginning to see in the clinic, the reduction of viral infections that typically threaten patients post transplantation. This safer means of deliver stem cell transplants would significantly reduce the HSCT mortality rate and therefore lead to broader use of this treatment. Furthermore, by adding CAR-T to the HSCT protocol, which we have already done successfully in preclinical studies, we can bridge between the initial transplantation and the conclusion of immune reconstitution, thus providing both short-term and ongoing protection against remission. This has the potential to significantly improve efficacy beyond that of the current outcomes of either CAR-T or HSCT on their own. |
| 17 |
| 2) | Substantively increase the number of transplantations by enabling successful engraftment under lower levels of immune suppression and therefore making the therapy accessible to older and sicker patients (who today may not survive ablation). | |
| 3) | Further increase the number of transplantations by making transplantation appropriate for other indications (for which today transplantation would be considered an inappropriately risky treatment). See, e.g. BMT 2021 Correction of Sickle Cell Disease by Allogeneic Hematopoietic Cell Transplantations with Anti-3rd Party Veto Cells, Bone Marrow Transplantation, March 3, 2021. |
In addition, our Veto Cell technology may possibly play a role in the treatment of a number of additional serious and currently poorly treated non-malignant diseases. Finally, based on preclinical studies using genetically modified cells, we believe that Veto Cells will be able to act as critical enabler for other cell therapies, most notably CAR-T cell therapy, which has recently shown strong initial indications of being effective in the near term in treating blood cancer.
Yeda has been granted two patents that extend the usage of Veto Cell technology as a critical enabler for other cell therapy treatments. One patent application highlights, based on preclinical data, the ability of Veto Cells to accompany other cell therapy treatments and help them overcome rejection and avoid GvHD in an allogeneic treatment setting. The other patent application involves a genetically modified Veto Cell that can have sustained survival in the patient’s body while avoiding rejection and GvHD. A more recent patent application was filed and has since been published which shows data that confirm combining Veto Cells with CAR-T cells, have the potential to make CAR-T cells, which to date have been effective primarily in an autologous (patient’s own cells) setting, succeed in an allogeneic setting. What follows is a description of the significance of these two new patent applications:
| - | Gene modified cell therapy is considered to be one of the most promising cancer treatment approaches in decades, with companies like Kite Pharma and JUNO Therapeutics having recently been acquired at multi-billion-dollar valuations after having successfully treated relatively small numbers of patients in clinical trials. | |
| - | While gene modified treatments such as CAR-T have shown remarkable results in cancer treatment trials, their published successes to date have been mostly limited to “autologous” blood cell cancer treatments using the patient’s own cells. There are concerns that this type of “personalized” treatment may not have favorable economics on a large-scale basis. | |
| - | The ideal, more lucrative commercial path for CAR-T and similar genetically engineered cell therapies is to become allogeneic or off the shelf product with drug-like distribution economics and to treat a broad spectrum of cancers including solid tumors. Allogene Therapeutics, an early-stage clinical company focused on allogeneic CAR-T, has in the past attained a valuation of $6 billion, whereas Legend Biotech valuation has recently reached a valuation of $10 billion and Fate Therapeutics valuation at one point reached the $10B level, underlining the importance of off-the-shelf CAR as a potential cancer treatment. Cell Source licenses Yeda’s patent applications for combining Veto Cells with genetically modified T cells and is currently developing a protocol to bring Veto and CAR-T combined cell therapy to the clinic. | |
| - | In the case of blood cancer treatment, we believe that a VETO CAR-T combined treatment will provide sustained protection for patients in relapse as well as a fundamentally superior approach for those in remission |
| 18 |
Cell Source previously completed a collaboration, through its licensing agreement with Yeda, with Professor Zelig Eshhar, the inventor of CAR-T cells. Professor Eshhar has served as both a scientist at the Weizmann Institute and on the Scientific Advisory Board of KITE Pharma. This collaboration confirmed the strength of combining Veto Cell technology with CAR-T cell therapy. Cell Source is now working towards the introduction of allogeneic VETO CAR-T HSCT combined cell treatment for lymphoma and leukemia and, eventually, off the shelf VETO CAR-T for these and other cancers, including solid tumors.
Furthermore, Yeda holds a granted patent, licensed to Cell Source, and has a pending patent based on more advanced data, for an Anti-Viral Veto Cell. Below is an explanation of the potential for this application:
| - | Other than primary disease (typically blood cell cancer) the leading causes of death in unrelated donor bone marrow transplants are rejection, GvHD, where the donor bone marrow rejects the host or recipient), and infections, which collectively are responsible for 38% of deaths after haploidentical donor transplants within the first 100 days post-transplant. | |
| - | It is well established that GvHD can be prevented by T cell depletion of the bone marrow transplant. However, this procedure is also associated with an increased rate of graft rejection. Preclinical studies and initial clinical results show that this problem can be overcome by adding Veto Cells to the bone marrow transplant, as well as allowing for a reduced intensity conditioning (RIC) regimen. However, viruses such as CMV and EBV remain a major threat to patients post-transplant. | |
| - | Cell Source has developed a next generation Veto Cell that not only facilitates mismatched transplants but also protects the transplant recipient against these common viruses. During the initial period after a stem cell transplantation the patient’s body undergoes an immune system reconstitution period. While the “new” immune system is building up, the patient is particularly vulnerable to viral infections, which develop in over 90% of bone marrow transplant recipients during the first 100 days post transplantation. Veto cells can fend off CMV and similar viral infection until such time as the patient’s own immune system reconstitutes to the point that it can fight off the infection on its own. | |
| - | Combining GvHD prevention by using T cell depleted transplants with anti-rejection action, under a mild conditioning regimen, as well as virus prevention, Veto Cell could potentially significantly increase survival rates post-transplant. Further adding the short-term cancer killing of CAR-T can combine to deliver even better long-term survival outcomes. | |
| - | Based on preclinical data, and as partially demonstrated in the case of a patient in the current trial, Veto Cells can also be used to facilitate organ transplants (e.g. kidney transplant combined with a bone marrow transplant) with partially mismatched donors and either reduce or eliminate the need for lifelong daily anti-rejection treatment currently given to even fully matched donor organ recipients. Among the data presented at the ASH annual meeting in late 2022, was the following: “One patient with a prior kidney transplant from the same donor had immunosuppressive therapy stopped without kidney rejection. This approach deserves further study in allogeneic HSCT for malignant and nonmalignant hematologic diseases, as well as enhancement of tolerance for organ transplantation.” |
Mechanism
Our Veto Cell is a CD8 central memory anti-3rd party T-cell that has five critical properties:
| 1) | It has an outer surface coating that triggers attack by specific host T-cells (and only those specific T-cells). | |
| 2) | It can annihilate an attacking T-cell without itself being damaged (specifically, it exposes or releases a death-signaling molecule when an attacking T-cell binds to it). | |
| 3) | It has been oriented to attack cells of a simulated third party (i.e., neither host nor donor), or a set of viral peptides, and thus exhibits markedly reduced risk of GvHD or graft rejection. |
| 19 |
| 4) | It is long-lived and endures in the body for extended periods. | |
| 5) | It migrates to the thymus and lymph nodes. |
The outcome is that when a large number of these cells are introduced into the body, they effectively eliminate the T-cell clones that the immune system dispatches to attack the desirable, transplanted stem cells.
Thus, for example, if a population of Veto Cells is derived from a donor, they will express the same peptide as do the donor’s cells. Therefore, the specific stream of host T-cells that would ordinarily attack the donor stem cells, are instead directed to “decoy” Veto Cells and disabled before they reach the transplantation.
Described in a Blood editorial as a “substantial advance in Cell Therapy,” a notable characteristic of our Veto Cell is that this mechanism is quite specific. Only those specific T-cell clones that were generated to attack cells from this specific donor are disabled. The rest of the immune system essentially remains intact. The conclusion of the ASH abstract for December 2022 states: “our data demonstrate reliable engraftment of haploidentical TCD (T Cell Depleted) HSCT combined with anti-viral CM (Central Memory) Veto CD8 T cells following a well-tolerated reduced intensity conditioning and show low rates of GVHD in the absence of immunosuppression (post transplantation).”
This is in marked contrast with conventional T cell depleted immunosuppression which degrades the entire immune system and is therefore associated with severe risk of infection and, in the case of stem cell transplantations, high mortality. It is also fundamentally superior to current T cell replete approaches, which, while attaining engraftment, are characterized by a marked incidence of GvHD, which can be debilitating, chronic, and sometimes even fatal.
This effect is long-lived. Firstly, the Veto Cells themselves are long-lived memory cells. Secondly, when infused with stem cells the latter migrate to the thymus where, over time, they create a new “identity” in the host and initiate chimerism where the host and donor cells peacefully co-exist. This chimerism has the effect of “educating” new T-cells being generated by the thymus to tolerate donor cells and this tolerance can become permanent. Furthermore, by inducing permanent tolerance to donor cells, Veto Cells may be able to enable both acceptance (i.e. mitigate both host rejection and GvH (graft) rejection) and thus persistence (i.e. extended survival resulting in enhanced efficacy) of important cell therapy treatments such as CAR-T cells, TCRs and NK cells in treating both blood cell and solid tumor cancers. Beyond this, Veto Cells are also effective not only in neutralizing host anti-donor rejecting cells, but also in the prevention of viruses such as EBV and CMV that are a common cause of post-transplantation morbidity and mortality.
Target Indications
Our Veto Cell technology, an intravenously administered cell suspension, if successful, could initially be used in stem cell (e.g. bone marrow) transplantations and other treatments associated with malignant disorders (i.e., cancers). Veto Cell technology may also be applied to selected non-malignant conditions. The following sections provide a brief overview of the use of the Veto Cell technology in both of these scenarios.
i. Stem Cell Transplantation
In order to describe the effect of Veto Cells in transplantation, it is helpful to first briefly review the state of the art:
In a conventional stem cell transplant, the recipient first receives myeloablative conditioning – powerful chemotherapy and/or radiation therapy intended to destroy his/her own bone marrow cells. This has a threefold purpose:
| 1) | It destroys the host T-cells so they will not attack (reject) the donor bone marrow cells. | |
| 2) | It makes space in the host bone marrow for the new donor cells. | |
| 3) | It destroys diseased host blood cells so that they do not proliferate and cause relapse following the procedure. |
| 20 |
In practice however, there are three major problems:
| ● | Host rejection – the myeloablative conditioning does not destroy all of the host T-cells. Those that remain may aggressively attack the donor bone marrow cells before they can engraft. | |
| ● | GvHD – the transplanted cells include donor T-cells which recognize the host’s body as foreign and attack it. | |
| ● | Infections are a common complication from HSCT and result in 28% of early patients deaths in haploidentical transplants of adult patients in the US. |
Rejection, GvHD and viral infections are all potentially life-threatening complications in and of themselves and also lead to the use of dangerous and costly immunosuppression medications.
ii. Veto Cell in Transplantation
The Veto Cell technology addresses not only rejection but also GvHD and infections. In a transplantation scenario, a population of donor Veto Cells is created to “escort” the stem cells when they are transplanted. This population is created by identifying donor cells with Veto Cell properties, exposing them to simulated 3rd party cells (e.g., selecting only those that react to a third person and therefore by definition will not react to either host or donor) or to viral peptides, and expanding their population in the lab.
The Veto Cells are then introduced into the host following the transplantation of the stem cells. The host mounts its normal immune response to the donor cells by generating a population of T-cell clones that will bind to any cells expressing markers from this specific donor. In a conventional transplantation, these T-cells would bind to and destroy donor stem-cells thus causing rejection of the transplant.
However, when the transplantation treatment involves a large number of Veto Cells, this rejection mechanism is “ambushed.” Since the Veto Cells express the same donor markers as the stem cells, the host T-cell clones will attempt to bind to the donor-derived Veto Cells as noted above, which act as decoys by attracting and then counterattacking and killing the clones before they ever reach the stem cell transplantation. These same Veto Cells concurrently attack viruses such as CMV and EBV which are a common source of infections that threaten HSCT patients. Based on additional preclinical data, in June of 2016 Yeda filed a U.S. provisional patent application, which has since been granted, and in 2019 a further patent application based on additional data, also licensed by Cell Source, which show the ability of Veto Cells to be directed against these types of viruses that typically cause infections in bone marrow transplant patients. This additional functionality, when combined with attacking host anti-donor rejecting cells, may even further enhance survival rates for patients.
iii. VETO CAR-T combined therapy
HSCT are well known to be an effective treatment for hematological malignancies. Making these treatments safer and more accessible by reducing the need for harmful immune suppression, avoiding GvHD and fending off common post-transplantation viruses are expected to facilitate, through successful Veto Cell treatments, a broader and more successful use of HSCT for not only the most severe cases, but also for older or weaker patients who are not capable of tolerating high intensity conditioning (high levels of radiation and chemotherapy). This is expected to significantly increase the number of patients who can receive successful cancer treatments that require allogeneic HSCT.
CAR-T cell therapy has shown strong cancer killing efficacy in the near term, mainly in an autologous setting. Longer term efficacy to date has been significantly lower, and to date there has been little success in establishing a successful approach to allogeneic CAR-T therapy. Having worked with Zelig Eshhar, the inventor of CAR-T therapy, to combine the CAR-T cell and the Veto Cell in to single cell which both directly attacks cancer cells and facilitates T cell depleted HSCT under RIC, Cell Source is now working to combine Veto Cell powered HSCT with CAR-T cell therapy for blood cell cancers into a single treatment, thus providing a comprehensive end-to-end solution which addresses both short-term cancer killing (via CAR-T) and long term relapse prevention (through a more safely delivered reconstituted immune system).
| 21 |
iv. Enabling Third Party Cell Therapies
Based on preclinical studies using genetically modified cells, in July of 2015 Yeda filed two U.S. provisional patent applications, both of which have since been granted, which are also licensed exclusively by Cell Source on a worldwide basis. These patent applications show the ability of Veto Cells to enhance the performance of cell therapy treatments involving genetically modified receptors. When combined with CAR-T or TCR cell therapy for example, these would potentially greatly enhance the ability of these treatments to be used in an allogeneic or “off the shelf” setting, and also increase their efficacy by avoiding both rejection and GvHD, thus increasing their persistence (survival in the patient’s body). A new patent, based on the Cell Source collaboration with Dr. Zelig Eshhar, the inventor of CAR-T technology, showing the effectiveness of Veto Cells combined with CAR-T cell therapy, was filed by Yeda, under license to Cell Source, in 2020.
This combined VETO CAR-T or similar treatment (e.g. combining Veto with NK cells, or other types of cancer treatments) is expected to result in broadly applicable effective treatments for both blood cell cancers and, eventually, a variety of solid tumor cancers as well.
v. In Non-Malignant Diseases
There are two major categories of non-malignant disorders that the Veto Cell technology aspires to address: organ transplantation and non-malignant hematological disorders.
In the case of organ transplantations and congenital non-malignant hematological disorders, the goal of the Veto Cells is to enable transplantation (stem cell or organ) by reducing host/donor immune system conflicts. This could potentially allow for mismatched (partial vs. full identity match between donor and host) kidney transplants, for example, and also obviate the need for lifelong daily anti-rejection medication which is the current standard of care. Such an outcome could improve quality of life, reduce cost of care and significantly increase life expectancy for a broader audience of prospective transplant recipients.
In the case of congenital non-malignant diseases such as sickle cell disease and aplastic anemia, the body’s bone marrow produces “flawed” cells. An effective treatment is HSCT which replaces the flawed host bone marrow with healthy donor cells. These cells then produce healthy blood cells, basically curing the anemia. As noted elsewhere however, today HSCT is a risky procedure because of the graft/host immune conflicts. It is therefore used infrequently to treat sickle cell disease. The Veto Cell tolerizing technology would increase the target population for this treatment by significantly reducing these conflicts and by extension the procedure’s risk. Yeda has filed patent applications, licensed to Cell Source, based on preclinical data that show Veto Cells’ effectiveness in reversing Sickle Cell Disease and their use in the treatment of T-cell mediated auto immune diseases such as preventing the development of Type 1 Diabetes.
Development Status
The Veto Cell platform has been extensively tested by in vitro studies (on both human and mouse disease) and confirmed in animal trials. The results appear to be consistently effective.
1. Inducing chimerism:
The following images show some example data from the Veto Cell animal studies. Skin of black mice has been grafted onto the backs of white mice. The data show that T-cells from host and donor mice are fully coexisting in the treatment group using the Veto Cells (“chimerism”).
| 22 |

2. Successful bone marrow transplantation under reduced levels of immune suppression:
The anti-rejection effect in the data below shows mice with lymphoma treated with Veto Cell therapy.
The control group mice (left side) all die by day 27. By contrast, the Veto Cell treatment group (right side) show far better results.
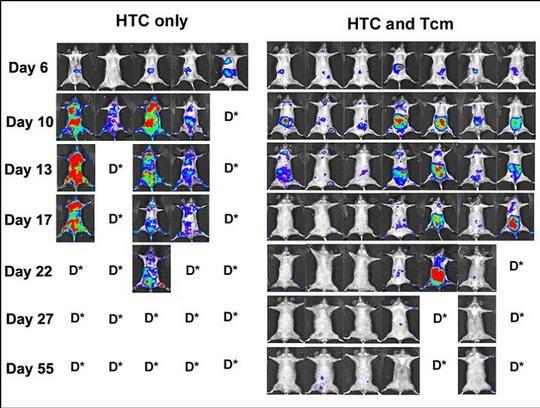
| 23 |
3. Effective cancer killing while avoiding rejection in allogeneic CAR-T wen combined with Veto Cells:
The cancer killing in the data below shows mice with leukemia treated with CAR=T Veto Cells:
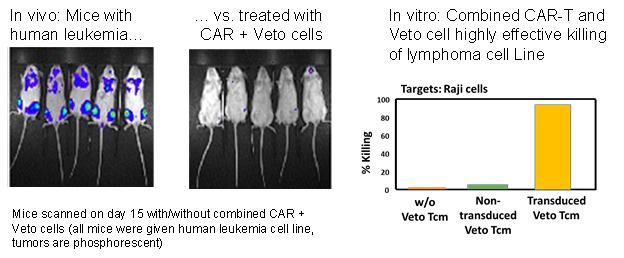
The control group mice (left side) all have extensive tumors whereas the Veto Cell treatment group (right side) show far better results. Right side exhibit shows lymphoma killing any Veto transduced cells in vitro.
Administration
We envision that Veto Cell therapy will be administered in an in-patient setting, typically as part of the existing procedures involved with stem cell transplantations. Blood will be taken from the donor. The blood will be sent to a regional Company center where the Veto Cells will be developed and expanded – a process that lasts up to two weeks. The Veto Cells will then be sent to the transplantation center where they will be infused to the patient intravenously along with the transplantation.
Patent Status
Cell Source’s CD8 TcM (central memory T-cells) Veto Cell, are protected by granted patents in the US, Mexico, Europe, China, Japan, Hong Kong, Korea, Singapore, Israel, India and the Russian Federation as well as Canada, Australia, New Zealand and South Africa. More recent patent applications, including those for the Genetically Modified Veto Cell and the Anti-Viral Veto Cell have been granted in the US and are in the national phase in a broad set of jurisdictions.
Development Roadmap
The Veto Cell platform roadmap comprises two main programs as outlined in the table below. The specific clinical trials planned for each are detailed in the Clinical Trials section of this document.
| Offering | Objective | Major Activities | Estimated timing | |||
| VETO CAR-T (with and without HSCT) | Validate and introduce new commercial treatment to deliver safer and more successful haploidentical HSCT combined with CAR-T cell therapy | 1. Establish initial safety and efficacy for Anti-viral Veto Cell, then augment existing trial protocol with CAR-T | ●2025 | |||
| 2. Commence multi-center registration study | ●2026-2027 | |||||
| 3. Introduce approved product to high profile US HSCT centers | ●2028-2029 | |||||
| Veto Cell Organ Transplantation | Validate efficacy of Veto Cells in attaining engraftment and reducing need for ongoing post-transplant anti-rejection treatment for haploidentical kidney transplants | 1. Finalize treatment protocol and commence Phase 1/2 study |
●2024-2025
| |||
| 2. Show sustained tolerance post-transplant without need for daily anti-rejection therapy | ●2026-2027 |
| 24 |
Products and Services
Currently, we do not have any products, and there is no assurance that we will be able to develop any products.
The following products are currently planned:
| 1. | VETO CAR-T HSCT cell therapy for donor mismatched allogeneic stem cell transplantations for treatment of blood cancer. |
This is our flagship (as an initial platform for increasing transplantation success) and is focused on haploidentical allogeneic stem cell transplantations. Treatment will comprise infusion of VETO CAR-T cells derived from the donor and processed in a Company (or subcontracted) facility that will be accessible to the transplantation center at the time of transplantation.
| 2. | Veto Cell based haploidentical organ transplantation initially for kidney and then also for liver transplants. |
This therapy will involve a partially mismatched donor organ transplant followed by an Anti-Viral Veto powered HSCT using stem cells derived from the same donor.
| 3. | Off the shelf VETO CAR-T cell therapy. |
This treatment would be use Veto Cells to increase persistence and hence efficacy of CAR-T cell therapy, without the use of HSCT, for blood cancers and eventually solid tumors as well.
| 4. | Veto Cell tolerance therapy for non-malignant disorders. |
This is the application of Veto Cell technology to treatment of non-malignant (i.e., non-cancerous) diseases, as discussed in the Technology section. Target indications for Veto Cell therapy for nonmalignant disorders are likely to be: tolerizing therapy for allogeneic transplantations for sickle cell anemia, aplastic anemia, etc. (by using stem cell transplantations as referenced in no. 2 above) and tolerizing therapy for a broader range of congenital immune system related disorders, possibly including preventing the development of Type 1 Diabetes in Diabetes prone or early onset Diabetes patients.
Our Overall Development Status and Future Development Program
Prior to commercializing its products, the Company must conduct human clinical trials and obtain FDA approval and/or approvals from comparable foreign regulatory authorities.
| 25 |
Generally speaking, as a preclinical biotechnology firm, Cell Source needs to go through several necessary steps in order to commercialize its products and commence revenue generation. These steps are per product, but can run in parallel for multiple products, which are each in different stages of the development “pipeline”, so that, for example, when a certain product is already in a human clinical trial, another product may still be in preclinical development and a third may be awaiting regulatory approval to commence human trials. These can also take place in parallel, and varied stages, for the same product in different geographic jurisdictions. The typical steps per product (and range of time frame for each) are:
| 1) | Complete development of human treatment protocol (2-5 years) | |
| 2) | Apply for and receive approval to commence human trials (9-18 months) | |
| 3) | Recruit patients (1-6 months) | |
| 4) | Conduct Phase 1 trials showing safety of product (1-2 years) | |
| 5) | Apply for and receive approval to conduct trials showing product efficacy (6-12 months) | |
| 6) | Data collecting and analysis (6-12 months) | |
| 7) | Conduct Phase 2 efficacy trials (2-3 years) | |
| 8) | Data collecting and analysis (6-12 months) | |
| 9) | Apply for and receive approval to conduct trials showing efficacy in larger numbers of patients (6-12 months) | |
| 10) | Conduct Phase 3 efficacy trials with larger numbers of patients (2-4 years) | |
| 11) | Data collecting and analysis (6-12 months) | |
| 12) | Apply for and receive approval for production scale manufacturing facilities (6-12 months) | |
| 13) | Contract third party or establish own production facilities (6-30 months) | |
| 14) | Contract third party or establish own distribution platform (6-18 months) | |
| 15) | Commence manufacturing and distribution (6-12 months) |
Notably, steps 12-15 can be conducted in parallel with some of the steps above. In the case of Cell Source and other firms that treat terminal patients with either rare diseases or those for which there is currently no effective treatment, or where preclinical studies indicate a reasonable expectation to increase life expectancy and survival rates by a substantive margin, several of these steps can be combined and or shortened, subject to regulatory discretion. For example, Phase 1 and 2 (safety and efficacy) can be combined in a single concurrent step; approvals for subsequent steps can be accelerated; in some countries patients can already be treated commercially after the end of Phase 2, foregoing the requirement for Phase 3 data prior to commencing commercial treatments.
The specific detailed next steps the company must take to get the treatments or products to market include the following:
In the case of the Megadose Drug Combination, the Hematology and Bone Marrow Transplantation Unit of the University of Parma in Italy on May 14, 2014 requested and on October 23, 2014 obtained approval from the Italian Medicine Association (the Italian equivalent of the U.S. FDA) to conduct human clinical trials using the “Megadose + Drug Combination.” While we are not mentioned in the application nor in the approval, we may indirectly benefit from the outcome of the trial, if successful, although we are not the sponsor of this trial. There are no written or verbal agreements between the hospital and Cell Source regarding the use of the technology. That said, Cell Source is aware and in favor of the hospital’s plans to use the technology and would of course find a positive initial outcome encouraging. Since the treatment is being done on compassionate grounds as a non-commercial clinical trial, there is no legal requirement for the hospital to obtain approval to use the treatment protocol. The hospital successfully treated the first cancer patient using the Megadose Drug Combination technology that Cell Source exclusively licenses from Yeda. The patient who was suffering from late-stage multiple myeloma, was released from hospital within a month of being treated and has since been cancer free for over seven years, with no GvHD, as initially reported in Blood Advances, vol. 1 no. 24 2166-2175 which was published online October 27, 2017.
While Cell Source was not a sponsor of the trial, the results provide a positive initial indication with respect to the technology. The patient received a bone marrow transplantation from a haploidentical or “mismatched” donor under a RIC regimen (i.e., a relatively low level of immune suppression treatment). There was successful initial engraftment of the transplantation in the absence of GvHD.
| 26 |
In November 2018, we executed a sponsored research agreement with MD Anderson, which was amended in December 2020 and in October 2021. The Company engaged MD Anderson to perform research services in the amount of approximately $1,500,000 from January 1, 2019 to December 31, 2021 (approximately $500,000 each year for three years). The agreement was amended in December 2020 in order to increase the research budget for the year ending December 31, 2021 from approximately $500,000 to $800,000. In November 2022, the agreement was amended to extend the agreement by one year to November 27, 2023 and define the budget during such one-year period to be approximately $1,300,000.
In February 2019, we executed a second agreement with MD Anderson for the production of Veto Cells and the conducting of a Phase ½ FDA trial for the Anti-Rejection, Anti-Viral Veto Cell. The treatment protocol was submitted to the FDA by MD Anderson in February of 2019. Cell Source has conducted Veto Cell production development in cooperation with the Medical Center at the Julius Maximilian University of Würzburg in Germany.
For the Anti-Viral Veto Cell product candidate, MD Anderson is currently conducting a Phase 1/2 human clinical trial, sponsored by Cell Source. The trial has successfully completed the four three treatment cohorts, with 12 patients each receiving a haploidentical HSCT under reduced intensity conditioning with Veto Cells. This first in human dose optimization trial has thus far shown that the initial dose is in fact the optimal dose, as all nine patients had successful stem cell engraftment after 42 days, in the absence of severe GvHD. Cell Source then continued the trial with a higher dose level. Cell Source anticipates that the US Phase 1/2 trial, once augmented with CAR-T cancer killing capability in the anticipated VETO CAR-T cell HSTC combined therapy, will last through 2025 or 2026. This would be followed by completion of a Phase 2 trial and Phase 3 trial, which could each last another 2-3 years. While under a fast track FDA program such as RMAT initial marketing approval could potentially be attained after a Phase 2 registration study as early as in 2027 or 2028, full approval, if successful, may not be attained until 2028 or later. Cell Source has concluded an initial proof-of-concept collaboration with Professor Zelig Eshhar, the inventor of CAR-T cell therapy, with respect to combining CAR-T cell therapy with Veto Cells. This is expected to lead to the augmentation of the existing Veto Cell treatment protocol at MD Anderson to include VETO CAR-T cells in 2024 or 2025, which may lead to fast track approval by 2027 or 2028 but may last until 2028 or 2029.
As referenced above, it is possible that Cell Source treatments could qualify for any or all of Fast Track, Breakthrough Therapy, Accelerated Approval, RMAT or Priority Review designation under the FDA, which would hasten their approval if successful. The estimated costs for each step of development, in terms of clinical trials, are delineated below:
Cell Source estimates the cost of clinical trials alone to be at least $5 million over the coming two years and overall company financing requirements of at least $50 million in order to reach commercialization for the Veto Cell products. This would mean that Cell Source will need to secure one or more significant capital infusions in order to reach the point that meaningful revenues could be generated.
The following table summarizes the development plan through 2028:

| 27 |
Competition
In the area of allogeneic HSCT and related GvHD and virus management, our competitors include: the so-called “Baltimore” protocol, which employs a T-cell replete approach under RIC; companies who which have been focused on reducing GvHD in a T-cell depleted setting, with high intensity conditioning (e.g. Bristol Myers Squibb, Merck (OncoImmune), Takeda); companies working to treat GvHD after it occurs (e.g. Abbvie, Incyte and Kadmon (now part of Sanofi)); and finally cell therapy companies developing anti-viral treatments (e.g. Atara, Allovir). In the area of CAR-T cell therapy, our competitors include allogeneic CAR-T companies (e.g. Allogene, Legend, Fate) and autologous CAR-T focused players (e.g. Novartis, Gilead (KITE), BMS/Celgene (JUNO)).
Haploidentical HSCT is gaining popularity in the US, outflanking UCB (umbilical cord blood) and growing more quickly than MUD (matched unrelated donor) based transplants. In the US, the majority of haploidentical HSCT are performed under RIC, mostly using T- cell replete transplants with post-transplant cyclophosphamide treatment. While this “Baltimore” RIC approach has gained popularity (mainly due to safety reasons) – as a T-cell replete approach it carries the risk of marked GvHD. Although some T-cell depleted approaches have shown reductions in GvHD, they face significant safety issues due to their aggressive use of immune suppression. Similarly, while currently approved CAR-T therapy for blood cancer has shown compelling short-term efficacy, the initial longer term data have shown a marked drop off in overall survival rates. Cell Source’s distinctive combination of a T-cell depleted HSCT with RIC, complemented by anti-viral activity and, eventually, enhanced by CAR-T short-term cancer killing, aims to provide the “best of both worlds” with safer, more effective HSTC leading to a reconstituted immune system, supported by “bridging” CAR-T remission induction and relapse prevention during the immune reconstitution period.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Earlier stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. While our commercial opportunities may be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than our own products, we believe that if our human trials show efficacy at the same levels of our animal trials, we would have the potential to develop at least a niche market share. Also, a number of large US cancer centers such as Johns Hopkins in Baltimore, Fred Hutchinson in Seattle, City of Hope in Duarte, CA and Dana Farber in Boston are conducting clinical trials and providing treatments on a compassionate care basis that can be funded on a not for profit basis and provide competition to Cell Source.
We expect that our ability to compete effectively will depend upon our capacity to:
| ● | successfully complete adequate and well-controlled clinical trials that demonstrate statistically significant safety and efficacy and to obtain all requisite regulatory approvals in a timely and cost-effective manner; | |
| ● | effectively use patents and possibly exclusive partnership agreements with important third-party treatment providers and collaboration partners to maintain a stable competitive stance for our Technology; | |
| ● | attract and retain appropriate clinical and commercial personnel and service providers; and | |
| ● | establish adequate distribution relationships for our products. |
Failure in efficiently developing and executing these capabilities may have an adverse effect on our business, financial condition or results of operations.
Strategy Overview
Our strategy is based on two underlying drivers: (a) that both preclinical and clinical data show Veto Cell technology to be consistently effective and have significant advantages over competitors; and (b) that the lead indications (the most common blood cancers) are relatively common, have high mortality and have limited treatment options today.
| 28 |
Based on the foregoing drivers, we have developed a business plan with the objective of obtaining regulatory approvals and subsequently launching product sales with a focus on the United States, Europe and Asia.
Key Strategy Elements
We are pursuing a staged entry strategy. The first several years will be narrowly focused, both in terms of market segments (blood cancers, kidney disease) and products (VETO CAR-T and VETO Organ Transplants).
Subsequently, we plan to broaden the segmentation strategy to include, stand-alone cancer treatments without HSCT (e.g. for solid tumors) and additional HSCT indications (e.g. selected genetic non-malignant diseases).
Our strategy can be summarized as follows:
| Strategy Element | Introductory period (years 1 -3 post FDA approval) |
Years 4+ | ||
| Market Segments | ● Lymphoma and leukemia ● Multiple myeloma ● Kidney disease |
● Same as before plus solid tumor cancer targets, liver failure, sickle cell disease, beta thalassemia, diabetes and other non-malignant hematological disorders; | ||
| Product Rollout | ● VETO CAR-T (with or without HSCT) for B-cell malignancies ● Veto Cell Kidney transplants |
● VETO CAR-T for solid tumors ● Veto Cell liver transplants ● Veto HSCT for non-malignant disorders | ||
| Customer/ Geographic Focus | ● United States ● Western Europe ● China |
● Major markets worldwide | ||
| Channels/Go to Market | ● Direct relationships with leading transplantation centers ● Partnerships with global pharma players |
● Out-licensing to, or outright acquisition by, global pharma players | ||
| Pricing | ● Consistent with other cell therapy offerings currently associated with transplantations and immuno-oncology |
● Potentially higher volume, lower cost for “off the shelf” Offerings | ||
| Operations | ● Three production centers: - US - Western Europe - Far East ● Initial capacity leased from or situated adjacent to major transplantation center. |
● Regional production centers owned outright or JV with Partners |
Segment Selection
Within the general market for immune therapies, we have selected target market segments (i.e., medical conditions) for initial focus based on two (2) key criteria:
| 1) | Severity of unmet medical need: degree of severity of the indication and the effectiveness of existing treatments. These criteria help determine the proper regulatory pathway. | |
| 2) | Technology relevance: relative value of the ability to manage immune response to the treatment of a given indication. |
| 29 |
We will initially focus on indications that score highly with respect to both criteria (e.g., blood cancers, kidney failure). These conditions may qualify for Fast Track status with the FDA, and, due to the cost and relative efficacy of current treatment alternatives, could potentially support profitable price points for effective new treatments.
Product Rollout
Cell Source plans to seek approval initially in the US and Europe and, in parallel but with a delayed start, in China and possibly Japan. A successful first-in-human Phase 1/2 trial in the US, which could be concluded by 2025 or 2026, would serve as a strong foundation for trials in other countries. Limited sales on a “compassionate grounds” basis may, depending on qualification for Breakthrough Therapy or other Accelerated Approval designation, commence as early as 2027 or 2028. Full approval by the FDA in the U.S. can take as long as 8 years, or until 2030.
Future products may include VETO CAR-T HSCT combined cell therapy for allogeneic stem cell transplantations as well as Veto Cell based organ transplantation. Following the initial market penetration and establishment of solid market positioning, we plan to broaden the product offering to address a wider variety of indications which may include custom Veto Cell developments for specific collaborations with other cell therapy treatments. For example, we believe that one area in which we could broaden our product offerings is to utilize our Veto Cell technology, if successful in humans, to address the rejection problems being faced by companies developing NK, TCR and blood cancer treatment, as an enabler for these treatments to help them overcome some of the rejection and persistence related performance issues their technology currently seems to be facing. If our Veto Cell technology proves to be successful in humans, we plan to continue to explore such potential applications in the future.
Customer/Geographic Focus
Assuming positive clinical trials, we will initially focus our sales efforts of Veto Cell anti-rejection therapy on centers dealing with late-stage B-cell malignancies. High profile, high volume HSCT facilities can be targeted to market this treatment.
Current plans are to introduce the products first in North America and Western Europe, and, perhaps concurrently, in China. Focusing on key transplantation facilities in target geographic markets will allow us to both refine the administration of our products and bolster our reputation in both these and a broader set of geographic markets.
After the introductory period, we plan to expand activities in these initial markets while simultaneously broadening geographic coverage. In Stage 2, we plan to initiate active marketing efforts in the remaining North American and Western European countries, Japan, Australia, and possibly Russia and India.
Marketing Strategy
The initial target market is concentrated and networked. It comprises the approximately 100 leading transplantation centers in the target geographies. As discussed in the “Market Access” and “Channels” section, these centers are well connected to each other and tend to quickly share innovations and best practices.
The planned penetration strategy is to introduce Veto Cell into the best-known and most influential centers in North America and Western Europe, and benefit from the exposure and industry leadership provided by these centers.
This initial penetration strategy includes incorporating some of these centers into clinical trials so as to expose and involve their medical leadership.
In the longer term, we plan to drive use and awareness within and across the broader oncology community in order to encourage oncologists to refer their patients to centers that already use our products and therapies and to encourage pull-influence on additional centers to adopt our products and therapies.
| 30 |
The broader provider community will be addressed both through a presence in leading peer-reviewed publications and by attending conferences where research and best clinical practices are shared, seminars are conducted, and networking opportunities are provided for the physicians. Furthermore, a dedicated sales force will approach leading bone marrow transplant physicians in the United States, as well as other key points of contact at the leading HSCT centers in the US as referenced above.
Operating Strategy
Veto Cell doses are to be prepared by Cell Source facilities or qualified production partners. This is to both protect trade secrets and directly control quality during the initial stages.
The graphic below outlines the general operating model in each geographic market.
Patient care facilities send frozen cells to a Cell Source processing center. Most likely, the first processing center will likely consist of production capacity leased from or service providers situated adjacent to a large transplantation center, such as MD Anderson in Texas. Such a transplantation center has appropriate equipment and infrastructure, along with available production capacity, and will also represent an immediate market for our offerings for use in their own procedures. The Cell Source processing center processes the cells and sends the treated cells and appropriate protocols back to the caregiver for infusion at time of transplantation.
In the introductory post regulatory approval phase, we plan on establishing one center in the U.S., one in Western Europe (most likely Germany), and one in the Far East. Specific locations and timing are to be determined. Initially, we plan to outsource production capacity from existing facilities operated by Contract Manufacturing Organizations (CMO) adjacent to large hospitals, or, where capacity is available, contract directly with major cancer treatment centers who have accredited GMP facilities and experienced cell production staff for Veto Cell production. Subsequently, sales from these centers can justify and fund stand-alone facilities.
The general goal of the initial centers is to support the FDA process, provide full coverage for the North American and European markets, and provide access to the Chinese market. Following the introductory period in each respective market, we may elect to migrate the production facilities from leased space in transplantation center laboratories or contract services with specialized CMOs to company-owned stand-alone facilities.
| 31 |
In general, we assume a capital cost per stand-alone production facility of at least $10 million. This estimate is based, in part, on the projected high costs of GMP “clean rooms,” each of which can cost $1 million or more to set up. We will need to obtain financing in order to fund the setup of such facilities. There can be no assurance that financing will be available in amounts or on terms acceptable to us, if at all.
Clinical Trials Overview
We will initially focus our clinical trials on stem cell transplantation for patients suffering from blood cancers (lymphoma, leukemia, myeloma), for which our Veto Cell technology constitutes a potential breakthrough. These indications have unmet needs as evidenced by the valuations of leading CAR-T players who thus far have chiefly presented data treating these diseases.
We commenced our first Phase ½ clinical trial in late 2019. This trial combines traditional Phase 1 safety with Phase 2 efficacy inasmuch as it is a safety trial conducted on sick patients, so as to both establish safety and show initial indications of efficacy concurrently. The goal is to demonstrate safety and initial efficacy in several indications. Management has structured the trials such that an additional goal of showing initial markers pointing to successful engraftment, in the absence of GvHD, while preventing viral infections, already within Phase 1/2.
The chart below provides an overview of the current trials plan, which can of course vary based on both finalization of human protocols and timing or regulatory approvals:
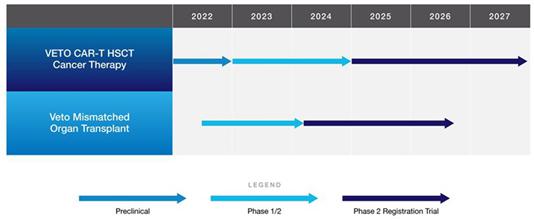
Trial Plans
Trials are planned for the US and Europe. The current initial US trial plans to treat 24 patients. We plan to focus on haploidentical (donor mismatched) stem cell bone transplantation under reduced intensity conditioning (reduced levels of immune suppression treatment) for B-cell malignancies and non-malignant indications (e.g. sickle cell disease). We are currently conducting a preclinical trial for VETO CAR-T cell therapy. Once we complete a proof of concept, we plan to augment the current clinical trial protocol for blood cell cancer to include VETO CAR-T cells, and to develop an Off-the-shelf VETO CAR-T treatment, without a stem cell transplant, for patients in relapse. In the future, we plan to conduct clinical trials for solid tumor patients as well. Also, once we have shown safety and efficacy for Veto Cell based stem cell transplants, we plan to combine these with haploidentical kidney transplants in patient trials.
Regulatory Issues Overview
We are currently seeking regulatory approval from the U.S. FDA, and also plan to apply to the European Medicines Agency (“EMA”) in Europe and to approach similar agencies elsewhere for approvals to both produce and sell our products.
We commenced a 24 patient human clinical trials for Anti-Viral Veto Cells, our lead product candidate, in the US in late 2019.
| 32 |
Regulatory Process and Expectations
We have developed and will continue to develop our clinical trial protocols with the support of highly experienced medical practitioners who have vast experience in working with their local regulators. MD Anderson, for example, as the largest stem cell transplantation center and leading cancer treatment facility in the US, has a thoroughgoing internal protocol approval process which serves to refine every aspect of each patient protocol, in great detail, in anticipation of any potential issues that the FDA would typically wish to see addressed.
The clinical trials outlined in the previous section are designed to lead to regulatory approval for Veto Cell-based therapy in treating blood cancers and stem cell transplantation applications and, thereafter, solid organ transplantations and, eventually, solid tumor cancers.
Interim Revenue Opportunities
While our focus is to conclude Phase 3 approval for cancer treatments, the Company is also exploring complementary shorter-term opportunities for generating revenue before additional FDA approvals are received, namely:
| 1) | Treating patients after the end of Phase 2 (based on US Fast Track approvals and/or European Marketing Authorization Approvals) with either partial or full insurance reimbursement available); and | |
| 2) | Potential upfront and milestone driven licensing revenues from collaborations with third parties. |
Intellectual Property
Pursuant to the Yeda License Agreement, Yeda granted the Company an exclusive worldwide license to certain patents, discoveries, inventions and other intellectual property generated (together with others) by Professor Yair Reisner at the Immunology Department at the Weizmann Institute. Under the Yeda License Agreement, the Company grants Yeda a 4% royalty on sales of patented products. Cell Source is required to pay Yeda a $50,000 annual license fee until such time as payment of royalties commences. The Yeda License Agreement also requires the Company to proceed with the development of the technologies on a timely basis.
The license period (per product, per country) is for the full life of the patents and expires at the later of the patent expiration date in that country or 15 years after the date that the FDA or local equivalent regulatory authority in each country approves that particular product for sale in that country. As long as Cell Source sponsors research or pays either a nominal license fee of $50,000 per year (total for use of all the products) or royalties on product sales on at least one product as per the license agreement, the license will remain in effect continuously and expire only with the expiration of the patent or 15 years after regulatory approval (later of the two) per product per country as described above. Cell Source voluntarily sponsors Research at the Weizmann Institute for the sake of developing its products and treatments from initial invention through to finalization of human treatment protocols.
The agreement with Yeda, as amended most recently on December 2, 2021, includes certain development milestones. If the Company fails to achieve any one of the milestones set forth in the Yeda License Agreement (as per the current amended version) which are listed below, then Yeda will be entitled to (i) modify the related license such that it will become non-exclusive or (ii) terminate the Yeda License Agreement upon thirty (30) days written notice:
| a. | by January 1, 2025, to have commenced Phase 2 clinical trials with respect to a Product; provided that in the interim the company continues to substantively sponsor research and clinical trials; | |
| b. | by January 1, 2028, to have either commenced Phase 3 clinical trials or to have received FDA or EMA marketing approval in a respect of a Product (“Marketing Approval”); | |
| c. | within 12 (twelve) months from the date of Marketing Approval, to have made a First Commercial Sale of a Product; or | |
| d. | in case commercial sale of any Product having commenced, there shall be a period of 12 (twelve) months or more during which no sales of any Product shall take place by the Company or its Sublicensees (except as a result of force majeure or other factors beyond the control of the Company). |
| 33 |
Additionally, the Yeda License Agreement also provides that:
| ○ | Title. All right, title and interest in and to the Licensed Information and the Patents (as those terms are defined in the Yeda License Agreement) and all right, title and interest in and to any drawings, plans, diagrams, specifications, other documents, models, or any other physical matter in any way containing, representing or embodying any of the foregoing, vest and shall vest in Yeda and subject to the license granted in the Yeda License Agreement.
| |
| ○ | Patents. Both Yeda and the Company shall consult with one another on the filing of patent applications for any portion of Licensed Information and/or corresponding to patent application existing at the time the Yeda License Agreement was executed. Yeda shall retain outside patent counsel that will be approved by Cell Source, to prepare, file and prosecute patent applications. All applications will be filed in Yeda’s name. | |
| ○ | Patents; Patent Infringements. Where the Company determines that a third party is infringing one or more of the Patents or is sued, in prosecuting or defending such litigation, the Company must pay any expenses or costs or other liabilities incurred in connection with such litigation (including attorney’s fees, costs and other sums awarded to the counterparty in such action). The Company agreed to indemnify Yeda against any such expenses or costs or other liabilities. | |
| ○ | License. With regard to the expiration of Patents, a Product is deemed to be covered by a Patent so long as such Product is protected by “Orphan Drug” status (or the like). The Company has an exclusive worldwide license under the Licensed Information and the Patents for the development, manufacture and sales of the Products. License remains in force in each country with respect to each Product until the later of (i) the expiration of the last Patent in such country covering such Product or (ii) the expiration of a 15-year period commencing the day FDA New Drug Approval is received for a Product in such country. |
The Company may grant a Sublicense only with Yeda’s prior written consent, which shall not be withheld unreasonably provided that:
| i. | the proposed Sublicense is for monetary consideration only; | |
| ii. | the proposed Sublicense is to be granted in a bona fide arm’s length commercial transaction; | |
| iii. | a copy of the agreement granting the Sublicense and all amendments thereof shall be made available to Yeda, 14 days before their execution and Cell Source shall submit to Yeda copies of all such Sublicenses and all amendments thereof promptly upon execution thereof; and | |
| iv. | the proposed Sublicense is made by written agreement, the provisions of which are consistent with the terms of the License and contain, inter alia, the following terms and conditions, including: the Sublicense shall expire automatically on the termination of the License for any reason. |
However, Yeda’s prior written consent is not needed if the sublicense is limited to China, and the Company grants it to a Chinese affiliated entity of the Company.
| ○ | Termination. The Yeda License Agreement terminates on the later of: (i) the expiration of the last of the Patents or (ii) the expiry of a continuous period of 20 years during which there shall not have been a First commercial sale of any product in any country. Yeda may terminate by written notice, effective immediately, if the Company challenges the validity of any of the Patents. If a challenge is unsuccessful, then in addition to Yeda’s right to termination, the Company shall pay to Yeda liquidated damages in the amount of $8,000,000. Either the Company or Yeda may terminate the Yeda License Agreement and the License by serving a written notice upon (i) occurrence of a material breach or (ii) the granting of a winding-up order. Additionally, Yeda may terminate for failure to reimburse Yeda for patent application and/or prosecution expenses. |
| 34 |
Our technology portfolio includes a patented platform termed “Veto Cell” (more formally described as “Anti 3rd party central memory T cell”), which is an immune tolerance biotechnology that enables the selective blocking of immune responses.
For a list of all the patents and pending patents that Yeda holds and which we have a license to use, please refer to the table in the section entitled “Science and Technology Overview” above.
Patents & Proprietary Rights
Our success will depend in part on our ability to protect our existing product candidates and the products we acquire or license by obtaining and maintaining a strong proprietary position. To develop and maintain our position, we intend to continue relying upon patent protection, orphan drug status, Hatch-Waxman exclusivity, trade secrets, know-how, continuing technological innovations and licensing opportunities. We intend to seek patent protection whenever available for any products or product candidates and related technology we acquire in the future.
We may also seek orphan drug status whenever it is available. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and Canada, and 10 years in the EU. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for a different clinical indication.
It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information made known to the individual during the course of the individual’s relationship with us is to be kept confidential and may not be disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual shall be our exclusive property.
Government Regulation and Product Approval
We submitted our first IND application to the FDA, which was done on our behalf by MD Anderson, in February 2019. Cell Source itself has not had any contact with any regulator anywhere regarding treatment approvals or clinical trials associated with regulatory approvals. We are aware that a hospital in Italy has independently requested and received approval to conduct a trial with a treatment protocol the patents for which we license from Yeda, which today forms part of the broader protocol that we plan to use in the US and European clinical trials, but we are not mentioned in the application nor in the approval. However, we may indirectly benefit from the outcome of the trial, if successful, although we are not the sponsor of this trial. There are no written or verbal agreements between the hospital and Cell Source regarding the use of the technology. That said, Cell Source is aware and in favor of the hospital’s plans to use the technology and would find a positive initial outcome encouraging. Since the treatment is being done on compassionate grounds as a non-commercial clinical trial, there is no legal requirement for the hospital to obtain approval to use the treatment protocol.
Cell Source commenced a human clinical trial, conducted on its behalf by MD Anderson, in 2019 to show initial safety, and possibly efficacy, results in the US.
Regulation by governmental authorities in the U.S. and other countries is a significant factor, affecting the cost and time of our research and product development activities, and will be a significant factor in the manufacture and marketing of any approved products. All of our products require regulatory approval by governmental agencies prior to commercialization. In particular, our products are subject to rigorous pre-clinical and clinical testing and other approval requirements by the FDA and similar regulatory authorities in other countries. Various statutes and regulations also govern or influence the manufacturing, safety, reporting, labeling, transport and storage, record keeping and marketing of our products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, the necessary regulatory approvals could harm our business.
| 35 |
The regulatory requirements relating to the testing, manufacturing and marketing of our products may change from time to time and this may impact our ability to conduct clinical trials and the ability of independent investigators to conduct their own research with support from us.
The clinical development, manufacturing and marketing of our products are subject to regulation by various authorities in the U.S., the EU and other countries, including, in the U.S., the FDA, in Canada, Health Canada, and, in the EU, the EMA. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act in the U.S. and numerous directives, regulations, local laws and guidelines in Canada and the EU and elsewhere govern the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. Product development and approval within these regulatory frameworks takes a number of years and involves the expenditure of substantial resources.
Regulatory approval will be required in all the major markets in which we seek to develop our products. At a minimum, approval requires the generation and evaluation of data relating to the quality, safety, and efficacy of an investigational product for its proposed use. The specific types of data required and the regulations relating to this data will differ depending on the territory, the treatment candidate involved, the proposed indication and the stage of development.
In general, new cell compositions are tested in animals until adequate evidence of safety is established to support the proposed clinical study protocol designs. Clinical trials for new products are typically conducted in three sequential phases that may overlap. In Phase 1, the initial introduction of the pharmaceutical into either healthy human volunteers or patients with the disease (typically 20 to 50 subjects), the emphasis is on testing for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase 2 involves studies in a limited patient population (typically 50 to 200 patients) to determine the initial efficacy of the pharmaceutical for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a treatment protocol shows preliminary evidence of some efficacy and is found to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to more fully evaluate clinical outcomes in a larger patient population in adequate and well-controlled studies designed to yield statistically sufficient clinical data to demonstrate efficacy and safety.
In the U.S., specific pre-clinical data, manufacturing and chemical data, as described above, need to be submitted to the FDA as part of an IND application, which, unless the FDA objects, will become effective thirty (30) days following receipt by the FDA. Phase 1 studies in human volunteers may commence only after the application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the EU. Currently, in each member state of the EU, following successful completion of Phase 1 studies, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase 2 studies. In many places in Europe, a two-tiered approval system mandates approval at the regional level prior to applying for national approval. Regional approval cycle times, including multiple iterations where questions are answered and the specific details of the protocol may be fine-tuned, can last several months prior to applying to the national (federal government level) regulator. The national regulatory authorities in the EU typically have between one and three months in which to raise any objections to the proposed study, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase 1 studies, further submissions to regulatory authorities are necessary in relation to Phase 2 and 3 studies to update the existing IND. Authorities may require additional data before allowing the studies to commence and could demand that the studies be discontinued at any time if there are significant safety issues. In addition to the regulatory review, a study involving human subjects has to be approved by an independent body. The exact composition and responsibilities of this body will differ from country to country. In the U.S., for example, each study will be conducted under the auspices of an independent institutional review board at each institution at which the study is conducted. This board considers among other things, the design of the study, ethical factors, the privacy of protected health information as defined under the Health Insurance Portability and Accountability Act, the safety of the human subjects and the possible liability risk for the institution. Equivalent rules to protect subjects’ rights and welfare apply in each member state of the EU, where one or more independent ethics committees, which typically operate similarly to an institutional review board, will review the ethics of conducting the proposed research. These ethical review committees typically exist at the regional level, where approval is required prior to applying for national approval. Other regulatory authorities around the rest of the world have slightly differing requirements involving both the execution of clinical trials and the import/export of pharmaceutical products. It is our responsibility to ensure we conduct our business in accordance with the regulations of each relevant territory.
| 36 |
By leveraging existing pre-clinical and clinical data, we are seeking to build upon an existing pre-clinical safety and efficacy database to accelerate our research. In addition, our focus on an end-stage population which has limited current treatment options may result in relatively shorter approval cycle times. Approval by the FDA in this category generally has been based on objective response rates and duration of responses rather than demonstration of survival benefit. As a result, trials of drugs to treat end-stage refractory cancer indications have historically involved fewer patients and generally have been faster to complete than trials of drugs for other indications. We are aware that the FDA and other similar agencies are regularly reviewing the use of objective endpoints for commercial approval and that policy changes may impact the size of trials required for approval, timelines and expenditures significantly. The trend over the past few years has been to shorten approval cycles for terminal patients in the U.S. by employing a “fast track” approach.
In order to gain marketing approval, we must submit a dossier to the relevant authority for review, which is known in the U.S. as an NDA and in the EU as a marketing authorization application, or MAA. The format is usually specific and laid out by each authority, although in general it will include information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product as well as the non-clinical and clinical data. Once the submitted NDA is accepted for filing by the FDA, it undertakes the review process that takes ten (10) months, unless an expedited priority review is granted which takes six (6) months to complete. Approval can take several months to several years, if multiple ten (10) month review cycles are needed before final approval is obtained, if at all.
The approval process can be affected by a number of factors. The NDA may be approvable requiring additional pre-clinical, manufacturing data or clinical trials which may be requested at the end of the ten (10) month NDA review cycle, thereby delaying marketing approval until the additional data are submitted and may involve substantial unbudgeted costs. The regulatory authorities usually will conduct an inspection of relevant manufacturing facilities, and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. Further inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor for adverse effects or other additional studies as deemed appropriate. After approval for the initial indication, further clinical studies are usually necessary to gain approval for any additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs in the indications on which we are focusing our efforts. These include accelerated approval under Subpart H of the agency’s NDA approval regulations, fast track drug development procedures and priority review. At this time, we have not determined whether any of these approval procedures will apply to any of our current treatment candidates.
The US, EU and other jurisdictions may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which, in the US, is generally a disease or condition that affects no more than 200,000 individuals. In the EU, orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than fifty (50) in 100,000 persons in the EU; without incentive it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and ten (10) years in the EU. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. Orphan drug designation must be requested before submitting an NDA or MAA. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process; however, this designation provides an exemption from marketing authorization (NDA) fees.
| 37 |
We are also subject to numerous environmental and safety laws and regulations, including those governing the use and disposal of hazardous materials. The cost of compliance with and any violation of these regulations could have a material adverse effect on our business and results of operations. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. Compliance with laws and regulations relating to the protection of the environment has not had a material effect on our capital expenditures or our competitive position. However, we are not able to predict the extent of government regulation, and the cost and effect thereof on our competitive position, which might result from any legislative or administrative action pertaining to environmental or safety matters.
In various countries, animal rights activism has led to either formal or informal boycotting of certain types of animal trials. This may have an adverse impact on our business as we rely on animal experiments as precursors to human trials.
Employees
Other than our Chief Executive Officer, we currently do not have any full-time employees, but retain the services of independent contractors/consultants on a contract-employment basis. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel. We anticipate that in the near future, other key personnel will enter into employment agreements with the Company on customary terms.
ITEM 1A. RISK FACTORS.
An investment in the Company’s Common Stock involves a high degree of risk. You should carefully consider the risks described below as well as other information provided to you in this Annual Report on Form 10-K, including information in the section of this document entitled “Information Regarding Forward Looking Statements.” The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our Common Stock could decline, and you may lose all or part of your investment.
Risks related to our Business and our Industry
We may not receive regulatory approvals for our product candidates or there may be a delay in obtaining such approvals.
Our products and our ongoing development activities are subject to regulation by regulatory authorities in the countries in which we or our collaborators and distributors wish to test, manufacture or market our products. For instance, the FDA will regulate the product in the U.S. and equivalent authorities, such as the EMA, will regulate in Europe. Regulatory approval by these authorities will be subject to the evaluation of data relating to the quality, efficacy and safety of the product for its proposed use, and there can be no assurance that the regulatory authorities will find our data sufficient to support product approval.
The time required to obtain regulatory approval varies between countries. In the U.S., for products without “Fast Track” status, it can take up to eighteen (18) months after submission of an application for product approval to receive the FDA’s decision. Even with Fast Track status, FDA review and decision can take up to twelve (12) months.
Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements as well as case load at the regulatory agency at the time.
| 38 |
Clinical trials for our product candidates are expensive and time consuming, and their outcome is uncertain.
The process of obtaining and maintaining regulatory approvals for new therapeutic products is expensive, lengthy and uncertain. Costs and timing of clinical trials may vary significantly over the life of a project owing to any or all of the following non-exclusive reasons:
| ● | the duration of the clinical trial; | |
| ● | the number of sites included in the trials; | |
| ● | the countries in which the trial is conducted; | |
| ● | the length of time required and ability to enroll eligible patients; | |
| ● | the number of patients that participate in the trials; | |
| ● | the number of doses that patients receive; | |
| ● | the drop-out or discontinuation rates of patients; | |
| ● | per patient trial costs; | |
| ● | third party contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; | |
| ● | our final product candidates having different properties in humans than in laboratory testing; | |
| ● | the need to suspend or terminate our clinical trials; | |
| ● | insufficient or inadequate supply of quality of necessary materials to conduct our trials; | |
| ● | potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies; | |
| ● | problems engaging institutional review boards (“IRB”) to oversee trials or in obtaining and maintaining IRB approval of studies; | |
| ● | the duration of patient follow-up; | |
| ● | the efficacy and safety profile of a product candidate; | |
| ● | the costs and timing of obtaining regulatory approvals; and | |
| ● | the costs involved in enforcing or defending patent claims or other intellectual property rights. |
Late-stage clinical trials are especially expensive, typically requiring tens of millions of dollars, and take years to reach their outcomes. Such outcomes often fail to reproduce the results of earlier trials. It is often necessary to conduct multiple late-stage trials, including multiple Phase 3 trials, in order to obtain sufficient results to support product approval, which further increases the expense. Sometimes trials are further complicated by changes in requirements while the trials are underway (for example, when the standard of care changes for the disease that is being studied in the trial). Accordingly, any of our current or future product candidates could take a significantly longer time to gain regulatory approval than we expect, or may never gain approval, either of which could delay or stop the commercialization of our product candidates.
| 39 |
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidates.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects as a result of participating in our clinical trials.
Delays in our clinical trials could result in us not achieving anticipated developmental milestones when expected, increased costs and delay our ability to obtain regulatory approval and commercialize our product candidates.
Delays in our ability to commence or enroll patients for our clinical trials could result in us not meeting anticipated clinical milestones and could materially impact our product development costs and delay regulatory approval of our product candidates. We do not know whether planned clinical trials will be commenced or completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including:
| ● | delays in the development of manufacturing capabilities for our product candidates to enable their consistent production at clinical trial scale; | |
| ● | delays in the commencement of clinical trials as a result of clinical trial holds or the need to obtain additional information to complete an Investigational New Drug Application (IND); | |
| ● | delays in obtaining regulatory approval to commence new trials; | |
| ● | adverse safety events experienced during our clinical trials; | |
| ● | insufficient efficacy during trials leading to withdrawal of product candidate; | |
| ● | delays in obtaining clinical materials; | |
| ● | slower than expected patient recruitment for participation in clinical trials; and | |
| ● | delays in reaching agreement on acceptable clinical trial agreement terms with prospective sites or obtaining institutional review board approval. |
If we do not successfully commence or complete our clinical trials on schedule, the price of our common stock may decline.
| 40 |
Our product candidates must undergo rigorous clinical testing, the results of which are uncertain and could substantially delay or prevent us from bringing them to market.
Before we can obtain regulatory approval for a product candidate, we must undertake extensive clinical testing in humans to demonstrate safety and efficacy to the satisfaction of the FDA or other regulatory agencies. Clinical trials of new drug candidates sufficient to obtain regulatory marketing approval are expensive and take years to complete.
We cannot be certain of successfully completing clinical testing within the time frame we have planned, or at all. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our product candidates, including the following:
| ● | our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing or to abandon programs; | |
| ● | the results obtained in earlier stage clinical testing may not be indicative of results in future clinical trials; | |
| ● | clinical trial results may not meet the level of statistical significance required by the FDA or other regulatory agencies; | |
| ● | enrollment in our clinical trials for our product candidates may be slower than we anticipate, resulting in significant delays and additional expense; | |
| ● | we, or regulators, may suspend or terminate our clinical trials if the participating patients are being exposed to unacceptable health risks; and | |
| ● | the effects of our product candidates on patients may not be the desired effects or may include undesirable side effects or other characteristics that may delay or preclude regulatory approval or limit their commercial use, if approved. |
Completion of clinical trials depends, among other things, on our ability to enroll a sufficient number of patients, which is a function of many factors, including:
| ● | the therapeutic endpoints chosen for evaluation; | |
| ● | the eligibility criteria defined in the protocol; | |
| ● | the perceived benefit of the investigational drug under study; | |
| ● | the size of the patient population required for analysis of the clinical trial’s therapeutic endpoints; | |
| ● | our ability to recruit clinical trial investigators and sites with the appropriate competencies and experience; | |
| ● | our ability to obtain and maintain patient consents; and | |
| ● | competition for patients by clinical trial programs for other treatments. |
We may experience difficulties in enrolling patients in our clinical trials, which could increase the costs or affect the timing or outcome of these clinical trials. This is particularly true with respect to diseases with relatively small patient populations.
Preclinical studies and Phase 1 or 2 clinical trials of our product candidates may not predict the results of subsequent human clinical trials.
Preclinical studies, including studies of our product candidates in animal models, may not accurately predict the result of human clinical trials of those product candidates. In particular, promising animal studies suggesting the efficacy of our products may not predict the ability of these products to treat humans. Our technology may be found not to be efficacious when studied in human clinical trials.
| 41 |
To satisfy FDA or foreign regulatory approval standards for the commercial sale of our product candidates, we must demonstrate in adequate and controlled clinical trials that our product candidates are safe and effective. Success in early clinical trials, including Phase 2 trials, does not ensure that later clinical trials will be successful. Our initial results from Phase ½ clinical trials also may not be confirmed by later analysis or subsequent larger clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials.
Our clinical trials may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates or any future product candidates, which would prevent or delay or limit the scope of regulatory approval and commercialization.
To obtain the requisite regulatory approvals to market and sell any of our product candidates and any other future product candidates, we must demonstrate through clinical trials that our product candidates are safe and effective for use in each targeted indication. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical development process. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization. We may be unable to establish clinical endpoints that applicable regulatory authorities would consider clinically meaningful, and a clinical trial can fail at any stage of testing.
Further, the process of obtaining regulatory approval is expensive, often takes many years following the commencement of clinical trials, and can vary substantially based upon the type, complexity, and novelty of the product candidates involved, as well as the target indications, patient population, and regulatory agency. Prior to obtaining approval to commercialize our product candidates and any future product candidates in the United States or abroad, we or our potential future collaborators must demonstrate with substantial evidence from adequate and well-controlled clinical trials, and to the satisfaction of the FDA or comparable foreign regulatory authorities, that such product candidates are safe and effective for their intended uses.
Clinical trials that we conduct may not demonstrate the efficacy and safety necessary to obtain regulatory approval to market our product candidates. In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the clinical trial protocols, and the rate of dropout among clinical trial participants. If the results of our ongoing or future clinical trials are inconclusive with respect to the efficacy of our product candidates, if we do not meet the clinical endpoints with statistical and clinically meaningful significance, or if there are safety concerns associated with our product candidates, we may be delayed in obtaining marketing approval, if at all. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications.
Even if the trials are successfully completed, clinical data are often susceptible to varying interpretations and analyses, and we cannot guarantee that the FDA or comparable foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. We cannot guarantee that the FDA or comparable foreign regulatory authorities will view our product candidates as having efficacy even if positive results are observed in clinical trials. The FDA or comparable foreign regulatory authorities may not agree with our manufacturing strategy or find comparability between our clinical trial product candidates and proposed commercial product candidates even if positive results are observed in clinical trials, which may result in regulatory delays or a need to perform additional clinical studies. Moreover, results acceptable to support approval in one jurisdiction may be deemed inadequate by another regulatory authority to support regulatory approval in that other jurisdiction. To the extent that the results of the trials are not satisfactory to the FDA or comparable foreign regulatory authorities for support of a marketing application, approval of our product candidates and any future product candidates may be significantly delayed, or we may be required to expend significant additional resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates. Even if regulatory approval is secured for a product candidate, the terms of such approval may limit the scope and use of the specific product candidate, which may also limit its commercial potential.
| 42 |
We are subject to various government regulations.
The manufacture and sale of human therapeutic and diagnostic products in the U.S., Canada and foreign jurisdictions are governed by a variety of statutes and regulations. These laws require approval of manufacturing facilities, controlled research and testing of products and government review and approval of a submission containing manufacturing, preclinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current Good Manufacturing Practice (or cGMP) during production and storage, and control of marketing activities, including advertising and labeling.
The products we are currently developing will require significant development, preclinical and clinical testing and investment of substantial funds prior to their commercialization. The process of obtaining required approvals can be costly and time-consuming, and there can be no assurance that future products will be successfully developed and will prove to be safe and effective in clinical trials or receive applicable regulatory approvals. Markets other than the U.S. and Canada have similar restrictions. Potential investors and shareholders should be aware of the risks, problems, delays, expenses and difficulties which we may encounter in view of the extensive regulatory environment which controls our business.
We may become subject to increased government regulation.
Increased government regulation could: (i) reduce any future revenues; (ii) increase our operating expenses; and (iii) expose us to significant liabilities. We cannot be sure what effect any future material noncompliance by us with any future laws and regulations or any material changes in current laws and regulations could have on our business, operating results and financial condition.
We may fail to comply with regulatory requirements.
Our success will be dependent upon our ability, and our collaborative partners’ abilities, to maintain compliance with regulatory requirements, including cGMP, and safety reporting obligations. The failure to comply with applicable regulatory requirements can result in, among other things, fines, injunctions, civil penalties, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products, operating and production restrictions and criminal prosecutions.
Regulatory approval of our product candidates may be withdrawn at any time.
After regulatory approval has been obtained for medicinal products, the product and the manufacturer are subject to continual review, including the review of adverse experiences and clinical results that are reported after our products are made available to patients, and there can be no assurance that such approval will not be withdrawn or restricted. Regulators may also subject approvals to restrictions or conditions or impose post-approval obligations on the holders of these approvals, and the regulatory status of such products may be jeopardized if such obligations are not fulfilled. If post-approval studies are required, such studies may involve significant time and expense.
The manufacturer and manufacturing facilities we use to make any of our products will also be subject to periodic review and inspection by the FDA or EMA, as applicable. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the product or manufacturer or facility, including withdrawal of the product from the market. We will continue to be subject to the FDA or EMA requirements, as applicable, governing the labeling, packaging, storage, advertising, promotion, recordkeeping, and submission of safety and other post-market information for all of our product candidates, even those that the FDA or EMA, as applicable, had approved. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
| 43 |
Even if approved, our products may not gain market acceptance, in which case we may not be able to generate product revenues, which will materially adversely affect our business, financial condition, and results of operations.
Even if the FDA or any comparable foreign regulatory authority approves the marketing of any product candidates that we develop, physicians, healthcare providers, patients, or the medical community may not accept or use them. Additionally, the product candidates that we are developing are based on our proprietary platforms, which are new technologies. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenues or any profits from operations. The degree of market acceptance of any of our product candidates will depend on a variety of factors, including:
| ● | the timing of market introduction; | |
| ● | the terms of any approvals and the countries in which approvals are obtained; | |
| ● | the number and clinical profile of competing products; | |
| ● | our ability to provide acceptable evidence of safety and efficacy; | |
| ● | the prevalence and severity of any side effects; | |
| ● | relative convenience and ease of administration; | |
| ● | cost-effectiveness; | |
| ● | patient diagnostics and screening infrastructure in each market; | |
| ● | marketing and distribution support; | |
| ● | adverse publicity about our product candidates; | |
| ● | availability of coverage, adequate reimbursement and sufficient payment from health maintenance organizations and other insurers, both public and private, for our product candidates, or the procedures utilizing our product candidates, if approved; | |
| ● | the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors and government authorities; and | |
| ● | other potential advantages over alternative treatment methods. |
In addition, although we are not utilizing replication competent vectors, adverse publicity due to the ethical and social controversies surrounding the therapeutic use of such technology, and reported side effects from any clinical trials using these technologies or the failure of such trials to demonstrate that these therapies are safe and effective may limit market acceptance of our product candidates. If our product candidates are approved but fail to achieve market acceptance among physicians, patients, hospitals, cancer treatment centers or others in the medical community, we will not be able to generate significant revenue.
If our product candidates fail to gain market acceptance, this will have a material adverse impact on our ability to generate revenues to provide a satisfactory, or any, return on our investments. Even if some products achieve market acceptance, the market may prove not to be large enough to allow us to generate significant revenues.
We do not own any patents and rely on the patents we license from Yeda Research and Development Limited.
We do not currently own any patents and only have an exclusive worldwide license to certain intellectual property owned by Yeda pursuant to a license agreement between us and Yeda. Under the license agreement with Yeda, Yeda retains ownership of the licensed patents. If we were to default under the license agreement, then our rights to Yeda’s intellectual property would be extinguished and we would lose all rights to operate the license. In such an event, we would effectively cease to operate unless we re-obtained licensing with Yeda.
| 44 |
We are dependent on protecting our proprietary rights.
Our success and competitive position and future overall revenues will depend in part on our ability to obtain and maintain patent protection over the patents that we have an exclusive license to use for our product candidates, methods, process and other technologies to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. Although our patents and related technologies are owned by Yeda, under our exclusive license agreement, we are required to pay all patent related expenses for applications, renewals, etc., as well as any and all legal or other costs associated with the defending and protecting such proprietary rights. However, we cannot predict:
| ● | the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent the patents that we license; | |
| ● | whether or not others will obtain patents claiming aspects similar to those covered by the patents that we license; or | |
| ● | whether we will need to initiate litigation or administrative proceedings, which may be costly whether we win or lose. |
For a complete list of the patents that we license from Yeda, please see the section entitled “Science and Technology Overview” of this Annual Report on Form 10-K.
We may be required to obtain licenses from third parties to avoid infringing patents or other proprietary rights. No assurance can be given that any licenses required under any such patents or proprietary rights would be made available, if at all, on terms we find acceptable. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
A number of pharmaceutical, biopharmaceutical and biotechnology companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to or affect our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. Such conflict could limit the scope of the patents, if any, that we may be able to obtain. Such conflict may also result in the denial of our patent applications. In addition, if patents that cover our activities are issued to other companies, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited. In addition, we could incur substantial costs in defending ourselves in suits brought against us on patents that our products might infringe or in filing suits against others to have such patents declared invalid.
Patent applications in the U.S. are maintained in secrecy and not published if either: i) the application is a provisional application or, ii) the application is filed and we request no publication and certify that the invention disclosed “has not and will not” be the subject of a published foreign application. Otherwise, U.S. applications or foreign counterparts, if any, publish 18 months after the priority application has been filed. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we or any licensor were the first creator of inventions covered by pending patent applications or that we or such licensor was the first to file patent applications for such inventions. Moreover, we might have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost to us, even if the eventual outcome were favorable to us. There can be no assurance that our patents, if issued, would be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents.
Much of our know-how and technology may not be patentable. To protect our rights, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance, however, that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, our business may be adversely affected by competitors who independently develop competing technologies, especially if we obtain no, or only narrow, patent protection.
| 45 |
Confidentiality agreements with employees and third parties may not prevent unauthorized disclosure of trade secrets and other proprietary information.
In addition to the protection afforded by patents, we seek to rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or that we elect not to patent, processes for which patents are difficult to enforce, and any other elements of our product candidates, technology and product discovery and development processes that involve proprietary know-how, information, or technology that is not covered by patents. Any disclosure, either intentional or unintentional, by our employees, the employees of third parties with whom we share our facilities or third-party consultants and vendors that we engage to perform research, clinical trials or manufacturing activities, or misappropriation by third parties (such as through a cybersecurity breach) of our trade secrets or proprietary information could enable competitors to duplicate or surpass our technological achievements, thus eroding our competitive position in our market. Because we expect to rely on third parties in the development and manufacture of our product candidates, we must, at times, share trade secrets with them. Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
We are dependent on our collaborative partners and service providers the loss of which would hurt our business.
Our strategy is to enter into various arrangements with corporate and academic collaborators, licensors, licensees, service providers and others for the research, development, clinical testing and commercialization of our products. We intend to or have entered into agreements with academic, medical and commercial organizations to research, develop and test our products. In addition, we intend to enter into corporate partnerships to commercialize the Company’s core products. There can be no assurance that such collaborations can be established on favorable terms, if at all.
Should any collaborative partner or service provider fail to appropriately research, develop, test or successfully commercialize any product to which the Company has rights, our business may be adversely affected. Failure of a collaborative partner or service provider to successfully conduct or complete their activities or to remain a viable collaborative partner or commercial enterprise for any particular program could delay or halt the development or commercialization of any products arising out of such program. While management believes that collaborative partners and service providers will have sufficient economic motivation to continue their activities, there can be no assurance that any of these collaborations or provisions of required services will be continued or result in successfully commercialized products.
Notably, we maintain an exclusive worldwide license to certain intellectual property owned by Yeda pursuant to the Yeda License Agreement, as further discussed in the “Intellectual Property” section hereinafter. If we should default under the License Agreement, then our rights to Yeda’s intellectual property would extinguish, and we would lose all rights to operate the licenses. In such event, we would effectively cease to operate unless we re-obtained licensing with Yeda.
In addition, there can be no assurance that the collaborative research or commercialization partners will not pursue alternative technologies or develop alternative products either on their own or in collaboration with others, including our competitors, as a means for developing treatments for the diseases or conditions targeted by our programs.
If we are unable to keep up with rapid technological changes in our field or compete effectively, we will be unable to operate profitably.
We are engaged in a rapidly changing field. Other products and therapies that will compete directly with the products that we are seeking to develop and market currently exist or are being developed. Competition from fully integrated pharmaceutical companies and more established biotechnology companies is intense and is expected to increase. Most of these companies have significantly greater financial resources and expertise in discovery and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and marketing than us. Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and established biopharmaceutical or biotechnology companies. Many of these competitors have significant products that have been approved or are in development and operate large, well-funded discovery and development programs. Academic institutions, governmental agencies and other public and private research organizations also conduct research, seek patent protection and establish collaborative arrangements for therapeutic products and clinical development and marketing. These companies and institutions compete with us in recruiting and retaining highly qualified scientific and management personnel. In addition to the above factors, we will face competition based on product efficacy and safety, the timing and scope of regulatory approvals, availability of supply, marketing and sales capability, reimbursement coverage, price and patent position. There is no assurance that our competitors will not develop more effective or more affordable products, or achieve earlier patent protection or product commercialization, than our own.
| 46 |
Other companies may succeed in developing products earlier than ourselves, obtaining Health Canada, European Medicines Agency (the “EMA”) and FDA approvals for such products more rapidly than we will, or in developing products that are more effective than products we propose to develop. While we will seek to expand our technological capabilities in order to remain competitive, there can be no assurance that research and development by others will not render our technology or products obsolete or non-competitive or result in treatments or cures superior to any therapy we develop, or that any therapy we develop will be preferred to any existing or newly developed technologies.
Our ability and our collaborators’ ability to sell therapeutic products will depend to a large extent upon reimbursement from health care insurance companies.
Our success may depend, in part, on the extent to which reimbursement for the costs of therapeutic products and related treatments will be available from third-party payers such as government health administration authorities, private health insurers, managed care programs, and other organizations. Over the past decade, the cost of health care has risen significantly, and there have been numerous proposals by legislators, regulators and third-party health care payers to curb these costs. Some of these proposals have involved limitations on the amount of reimbursement for certain products. Similar federal or state health care legislation may be adopted in the future and any products that we or our collaborators seek to commercialize may not be considered cost-effective. Adequate third-party levels that are sufficient for realization of an appropriate return on investment in product development.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs, therapeutic platforms, and product candidates that we identify for specific indications. Additionally, we have contractual commitments under our collaboration agreements to use commercially reasonable efforts to develop certain programs and, thus, do not have unilateral discretion to vary from such agreed to efforts. In addition, we have contractual commitments to conduct certain development plans, and thus may not have discretion to modify such development plans, including clinical trial designs, without agreement from our collaboration partners. As a result, we may forego or delay pursuit of opportunities with other therapeutic platforms or product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs, therapeutic platforms, and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing, or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights.
Interim, topline, or preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data becomes available or as we make changes to our manufacturing processes and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose interim, topline, or preliminary data from our preclinical studies and clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations, and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. Further, modifications or improvements to our manufacturing processes for a therapy may result in changes to the characteristics or behavior of the product candidate that could cause our product candidates to perform differently and affect the results of our ongoing clinical trials. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available.
| 47 |
From time to time, we may also disclose preliminary or interim data from our preclinical studies and clinical trials. Preliminary or interim data from clinical trials are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects. Additionally, disclosure of preliminary or interim data by us or by our competitors could result in volatility in the price of our common stock.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions, or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate, and our company in general. If the interim, topline, or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, any of our potential product candidates may be harmed, which could harm our business, operating results, prospects, or financial condition.
We rely on key personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to grow effectively.
We are dependent on our Chief Executive Officer, Itamar Shimrat, our Executive Chairman, Dennis Brown, and on scientific and drug development consultants, the loss of services of one or more of whom could materially adversely affect us.
Other than our Chief Executive Officer, we currently do not have full-time employees, but we retain the services of independent contractors/consultants on a contract-employment basis. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel.
We may be subject to foreign exchange fluctuation.
We maintain our accounts in both U.S. dollars and Israeli Shekels. A portion of our expenditures are in foreign currencies, most notably in Israeli Shekels, and therefore we are subject to foreign currency fluctuations, which may, from time to time, impact our financial position and results. We may enter into hedging arrangements under specific circumstances, typically through the use of forward or futures currency contracts, to minimize the impact of increases in the value of the Israeli Shekel. In order to minimize our exposure to foreign exchange fluctuations we may hold sufficient Israeli Shekels to cover our expected Israeli Shekel expenditures.
We may be exposed to potential product and clinical trials liability.
Our business exposes us to potential product liability risks, which are inherent in the testing, manufacturing, marketing and sale of therapeutic products. Human therapeutic products involve an inherent risk of product liability claims and associated adverse publicity. While we will continue to take precautions we deem appropriate, there can be no assurance that we will be able to avoid significant product liability exposure. We do not currently maintain liability insurance coverage as such insurance is expensive and difficult to obtain. As we move forward with our own clinical trials, we plan to obtain liability insurance coverage in the jurisdictions applicable to such clinical trials. However, when we seek such insurance, it may not be available on acceptable terms, if at all. The inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims could prevent or inhibit our ability to conduct clinical trials in certain jurisdiction or the commercialization of our current or potential products. A product liability claim brought against us in a clinical trial or a product withdrawal could have a material adverse effect upon us and our financial condition. Should the insurance coverage be insufficient in amount or scope to address multiple and diverse claims, liabilities not covered by insurance could represent a significant financial liability for Cell Source. Since Yeda does not conduct human trials, there is no need for Cell Source to have insurance for trials there. As Cell Source continues to contract facilities at hospitals to conduct human trials on its behalf, it will ensure that full and proper insurance coverage will be in place with respect to such clinical facilities. Cell Source plans to insure its direct participation in clinical trials, above and beyond whatever insurance coverage is already held by the institutions and facilities providing services with respect to such clinical trials, as may be required.
| 48 |
We may become subject to liabilities related to risks inherent in working with hazardous materials.
Our discovery and development processes involve the controlled use of hazardous and radioactive materials. We are subject to federal, state, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by such laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our resources. We are not specifically insured with respect to this liability. Although we believe that we are in compliance in all material respects with applicable environmental laws and regulations and currently do not expect to make material capital expenditures for environmental control facilities in the near-term, there can be no assurance that we will not be required to incur significant costs to comply with environmental laws and regulations in the future, or that our operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
We may in the future conduct clinical trials for our products or product candidates outside the United States and the FDA may not accept data from such trials.
We may in the future choose to conduct one or more of our clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such study data by the FDA is subject to certain conditions. For example, the study must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The study population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical studies conducted outside of the United States must be representative of the population for whom we intend to label the product in the United States. In addition, such studies would be subject to the applicable local laws and FDA acceptance of the data would be dependent upon its determination that the studies also complied with all applicable U.S. laws and regulations. There can be no assurance the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept any such data, it would likely result in the need for additional trials, which would be costly and time consuming and delay aspects of our business plan.
Potential disruptions to our preclinical development efforts include, but are not limited to:
| ● | delays or disruptions in preclinical experiments and IND-enabling studies due to restrictions of on-site staff, limited or no access to animal facilities, and unforeseen circumstances at contract research organizations (CROs) and vendors; | |
| ● | limitations on employee or other resources that would otherwise be focused on the conduct of our preclinical work and any clinical trials we subsequently commence, including because of sickness of employees or their families, the desire of employees to avoid travel or contact with large groups of people, an increased reliance on working from home, school closures, or mass transit disruptions; | |
| ● | delays in necessary interactions with regulators, ethics committees, and other important agencies and contractors due to limitations in employee resources or forced furlough of government or contractor personnel; and | |
| ● | limitations in maintaining our corporate culture that facilitates the transfer of institutional knowledge within our organization and fosters innovation, teamwork, and a focus on execution. | |
| ● | interruption of key clinical trial activities, such as clinical trial site data monitoring and efficacy, safety and translational data collection, processing and analyses, due to limitations on travel imposed or recommended by federal, state, or local governments, employers and others or interruption of clinical trial subject visits, which may impact the collection and integrity of subject data and clinical study endpoints; |
| 49 |
| ● | delays or difficulties in initiating or expanding clinical trials, including delays or difficulties with clinical site initiation and recruiting clinical site investigators and clinical site staff; | |
| ● | delays or difficulties in enrolling and retaining patients in our clinical trials; | |
| ● | interruption of, or delays in receiving, supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns, or stoppages and disruptions in materials and reagents; | |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; | |
| ● | interruption or delays in the operations of the FDA and comparable foreign regulatory agencies; | |
| ● | delays in receiving approval from local regulatory authorities to initiate our planned clinical trials; | |
| ● | limitations on employee resources that would otherwise be focused on the conduct of our preclinical studies and clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; | |
| ● | interruption of, or delays in receiving, supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; | |
| ● | refusal of the FDA or comparable regulatory authorities to accept data from clinical trials in affected geographies; and | |
Future developments in these and other areas present material uncertainty and risk with respect to our clinical trials, business, financial condition, and results of operations.
Risks Related to Our Capital Resources and Impairments
We have a limited operating history and a history of operating losses and expect to incur significant additional operating losses.
Our planned principal operations are the development and commercialization of new cell therapy products focused on treatment of blood cancers, certain non-malignant disorders and organ transplantations. We are currently conducting research and development activities in order to facilitate the continued transition of the patented technology we license from the laboratory to clinical trials. We have a limited operating history. Therefore, there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We have generated net losses since we began operations, including net losses of $5,321,212 and $5,167,748 for the years ended December 31, 2023 and 2022, respectively. We expect to incur substantial additional net expenses over the next several years as our research, development, and commercial activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the preclinical and clinical development of our product candidates; obtaining necessary regulatory approvals from the U.S. Food and Drug Administration (the “FDA”) and international regulatory agencies; successful manufacturing, sales, and marketing arrangements; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
| 50 |
We will need to secure additional financing.
We anticipate that we will incur operating losses for the foreseeable future. As of December 31, 2023, we had cash in the amount of $22,203. Based on our current resources, we will not be able to continue to operate without additional immediate funding. If we are not successful in securing additional financing, we may be required to delay significantly, reduce the scope of or eliminate one or more of our research or development programs, downsize our general and administrative infrastructure, or seek alternative measures to avoid insolvency, including arrangements with collaborative partners or others that may require us to relinquish rights to certain of our technologies, product candidates or products. Our future capital requirements will depend on many factors, including:
| ● | the scope, timing, progress, costs, and results of discovery, preclinical development, and clinical trials for our current or future product candidates; | |
| ● | the number of clinical trials required for regulatory approval of our current or future product candidates; | |
| ● | the costs, timing, and outcome of regulatory review of any of our current or future product candidates; | |
| ● | the cost of manufacturing clinical and commercial supplies of our current or future product candidates; | |
| ● | the costs and timing of future commercialization activities, including manufacturing, marketing, sales, and distribution, for any of our product candidates for which we receive marketing approval; | |
| ● | the costs and timing of preparing, filing, and prosecuting patent applications, maintaining and enforcing our intellectual property rights, and defending any intellectual property-related claims, including any claims by third parties that we are infringing upon their intellectual property rights; | |
| ● | our ability to maintain existing, and establish new, strategic collaborations, licensing, or other arrangements and the financial terms of any such agreements, including the timing and amount of any future milestone, royalty, or other payments due under any such agreement; | |
| ● | the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval; | |
| ● | expenses to attract, hire, and retain skilled personnel; | |
| ● | the costs of operating as a public company; | |
| ● | our ability to establish a commercially viable pricing structure and obtain approval for coverage and adequate reimbursement from third-party and government payers; | |
| ● | the effect of competing technological and market developments; and | |
| ● | the extent to which we acquire or invest in businesses, products, and technologies. |
Our ability to raise additional funds will depend on financial, economic, political and market conditions and other factors, over which we may have no or limited control. Market volatility resulting other factors could also adversely impact our ability to access capital as and when needed. Additional funds may not be available when we need them, on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we could be required to:
| ● | delay, limit, reduce, or terminate preclinical studies, clinical trials, or other research and development activities, or eliminate one or more of our development programs altogether; and | |
| ● | delay, limit, reduce, or terminate our efforts to access manufacturing capacity, establish sales and marketing capabilities or other activities that may be necessary to commercialize our product candidates, or reduce our flexibility in developing or maintaining our sales and marketing strategy. |
| 51 |
The Russia-Ukraine and Israel-Hamas wars have disrupted global markets and my adversely impact our ability to obtain financing.
On February 24, 2022, Russian military forces invaded Ukraine, and the length, impact, and outcome of the ongoing war in Ukraine is highly unpredictable. On October 7, 2023, Hamas terrorists infiltrated Israel’s border with the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas has also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel. These attacks have resulted in extensive deaths, injuries and kidnapping. Following the attack, Israel’s security cabinet declared war against Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. The intensity and duration of Israel’s current war against Hamas is similarly difficult to predict. As a result of the Russia-Ukraine and Israel-Hamas wars and other geopolitical and macroeconomic events, the global credit and financial markets have experienced volatility and disruptions, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, and uncertainty about economic stability. If the equity and credit markets deteriorate, it may make any necessary debt or equity financing more difficult, more costly or more dilutive.
There is substantial doubt about our ability to continue as a going concern.
As of December 31, 2023, we had a working capital deficit and accumulated deficit of $15,611,543 and $41,667,388, respectively. During the year ended December 31, 2023, we incurred a net loss of $5,321,212. We have historically incurred operating losses and may continue to incur operating losses for the foreseeable future. We believe that these conditions raise substantial doubt about our ability to continue as a going concern for at least one year from the date these financial statements are issued. This may hinder our future ability to obtain financing or may force us to obtain financing on less favorable terms than would otherwise be available. We have not generated revenues to-date. Our primary source of operating funds since inception has been equity and debt financings. Our plans include continued efforts to raise additional capital through debt and equity financings. There is no assurance that these funds will be sufficient to enable us to fully complete our development activities or attain profitable operations. If we are unable to obtain such additional financing on a timely basis or, notwithstanding any request we may make, if our debt holders do not agree to convert their notes into equity or extend the maturity dates of their notes, we may have to curtail our development, marketing and promotional activities, which would have a material adverse effect on our business, financial condition and results of operations, and ultimately we could be forced to discontinue our operations and liquidate. There can be no assurance that we will be able to continue as a going concern.
We are an early-stage company with an unproven business strategy and may never achieve commercialization of our candidate products or profitability.
We are at an early stage of development and commercialization of our technologies and product candidates. We have not yet begun to market any products and, accordingly, have not begun to generate revenues from the commercialization of our products. Our products will require significant additional clinical testing and investment prior to commercialization. A commitment of substantial resources by ourselves and, potentially, our partners to conduct time-consuming research and clinical trials will be required if we are to complete the development of our product candidates. There can be no assurance that any of our product candidates will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs or be successfully marketed. Most of our product candidates are not expected to be commercially available for several years, if at all.
We are in default of payment obligations under certain promissory notes.
As of December 31, 2023 and through the date of this filing, notes payable with principal amounts totaling $1,876,093 and $1,906,093, respectively, were past due. Although only one holder of a note with the principal amount of $250,000 has elected to pursue remedies against us, no assurance can be given that the other holders will not do so in the future. The institution of collection actions could have a material adverse effect on our business and could force us to seek relief through insolvency or other proceedings.
| 52 |
Risks Related to Our Common Stock
There may be additional issuances of shares of preferred stock in the future.
Our Articles of Incorporation permit us to issue up to 10,000,000 shares of preferred stock and our board of directors has authorized 1,350,000 shares of Series A Convertible Preferred, 2,000,000 shares of Series B Convertible Preferred, and 1,000,000 shares of Series C Convertible Preferred Stock, for issuance. Our board of directors could authorize the issuance of additional series of preferred stock in the future and such preferred stock could grant holders preferred rights on parity with the Series A Preferred and Series C Preferred as to dividend payments and liquidation preference. The issuances of other series of preferred stock could have the effect of reducing the amounts available to the holders Series A Preferred and Series C Preferred in the event of our liquidation, winding-up or dissolution. It may also reduce cash dividend payments on the Series A Preferred if we do not have sufficient funds to pay dividends on all Series A Preferred outstanding and outstanding parity preferred stock.
Our articles of incorporation allow for our board to create a new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our Common Stock.
Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. Our Board of Directors have the authority to issue up to 10,000,000 shares of our preferred stock, the terms of which may be determined by the Board without further stockholder approval. As a result, our Board of Directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of Common Stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our Common Stock. In addition, our Board of Directors could authorize the issuance of a series of preferred stock that has greater voting power than our Common Stock or that is convertible into our Common Stock, which could decrease the relative voting power of our Common Stock or result in dilution to our existing stockholders.
There is not an active liquid trading market for the Company’s Common Stock.
The Company is required to report under the Exchange Act and its Common Stock is eligible for quotation on the OTC Markets Expert Market. Quotations in Expert Market securities are restricted from public viewing. In addition, there is no regular active trading market in the Company’s Common Stock, and we cannot give an assurance that an active trading market will develop. If an active market for the Company’s Common Stock develops,
| ● | variations in our quarterly operating results; | |
| ● | announcements that our revenue or income are below analysts’ expectations; | |
| ● | general economic slowdowns; | |
| ● | sales of large blocks of the Company’s Common Stock; and | |
| ● | announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments. |
Our Common Stock is subject to the “penny stock” rules of the Securities and Exchange Commission, which may make it more difficult for stockholders to sell our Common Stock.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require that a broker or dealer approve a person’s account for transactions in penny stocks, and the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased.
| 53 |
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must obtain financial information and investment experience objectives of the person, and make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth the basis on which the broker or dealer made the suitability determination, and that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of the Company’s Common Stock if and when such shares are eligible for sale and may cause a decline in the market value of its stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stock.
We may not be able to attract the attention of brokerage firms because we became a public company by means of a reverse acquisition.
Because we became public through a “reverse acquisition,” securities analysts of brokerage firms may not provide coverage of us since there is little incentive to brokerage firms to recommend the purchase of our Common Stock. No assurance can be given that brokerage firms will want to conduct any secondary offerings on behalf of the Company in the future.
Voting power of our shareholders is highly concentrated by insiders.
Our officers, directors and affiliates currently own or have rights to acquires shares of common stock and preferred stock representing approximately 28% of the voting power of our outstanding securities. Such concentrated control of the Company may adversely affect the value of our shares of common stock. If you acquire shares of our common stock, you may have no effective voice in our management. Sales by our insiders or affiliates, along with any other market transactions, could affect the value of our ordinary shares.
We do not intend to pay dividends to holders of Common Stock for the foreseeable future.
We have paid no dividends on our Common Stock to date and it is not anticipated that any dividends will be paid to holders of our Common Stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, it is currently anticipated that any earnings will be retained to finance our future expansion and for the implementation of our business plan. As an investor, you should take note of the fact that a lack of a dividend can further affect the market value of our stock and could significantly affect the value of any investment in our Company.
Other General Factors
Applicable regulatory requirements, including those contained in and issued under the Sarbanes-Oxley Act of 2002, may make it difficult for the Company to retain or attract qualified officers and directors, which could adversely affect the management of its business and its ability to obtain or retain listing of its Common Stock.
The Company may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by the stock exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers.
| 54 |
Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. The Company may have difficulty attracting and retaining directors with the requisite qualifications. If the Company is unable to attract and retain qualified officers and directors, the management of its business and its ability to obtain or retain listing of our shares of Common Stock on any stock exchange (assuming the Company elects to seek and are successful in obtaining such listing) could be adversely affected.
If we fail to maintain effective internal controls over financial reporting, the price of our common stock may be adversely affected.
In the past, our management identified weaknesses in our internal controls and although our management believes such weaknesses have been remediated, our internal control over financial reporting may still or could in the future have weaknesses and conditions that could require correction or remediation, the disclosure of which may have an adverse impact on the price of our common stock. We are required to establish and maintain appropriate internal controls over financial reporting. Failure to establish those controls, or any failure of those controls once established, could adversely affect our public disclosures regarding our business, prospects, financial condition or results of operations. In addition, management’s assessment of internal controls over financial reporting may identify weaknesses and conditions that need to be addressed in our internal controls over financial reporting or other matters that may raise concerns for investors. Any actual or perceived weaknesses and conditions that need to be addressed in our internal control over financial reporting or disclosure of management’s assessment of our internal controls over financial reporting may have an adverse impact on the price of our common stock.
We are required to comply with certain provisions of Section 404 of the Sarbanes-Oxley Act of 2002 and if we fail to comply in a timely manner, our business could be harmed and our stock price could decline.
Rules adopted by the SEC pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 require an annual assessment of internal controls over financial reporting, and for certain issuers an attestation of this assessment by the issuer’s independent registered public accounting firm. The standards that must be met for management to assess the internal controls over financial reporting as effective are evolving and complex, and require significant documentation, testing, and possible remediation to meet the detailed standards. We expect to incur significant expenses and to devote resources to Section 404 compliance on an ongoing basis. It is difficult for us to predict how long it will take or how costly it will be to complete the assessment of the effectiveness of our internal control over financial reporting for each year and to remediate any deficiencies in our internal control over financial reporting. As a result, we may not be able to complete the assessment and remediation process on a timely basis. In addition, although attestation requirements by our independent registered public accounting firm are not presently applicable to us, we could become subject to these requirements in the future and we may encounter problems or delays in completing the implementation of any resulting changes to internal controls over financial reporting. In the event that our principal executive and financial officer determines that our internal control over financial reporting is not effective as defined under Section 404, we cannot predict how regulators will react or how the market prices of our shares will be affected; however, we believe that there is a risk that investor confidence and share value may be negatively affected.
As an issuer of “penny stock,” the protection provided by the federal securities laws relating to forward looking statements does not apply to us.
Although federal securities laws provide a safe harbor for forward-looking statements made by a public company that files reports under the federal securities laws, this safe harbor is not available to issuers of penny stocks. As a result, we will not have the benefit of this safe harbor protection in the event of any legal action based upon a claim that the material provided by us contained a material misstatement of fact or was misleading in any material respect because of our failure to include any statements necessary to make the statements not misleading. Such an action could hurt our financial condition.
| 55 |
Our issuance of Common Stock upon exercise of warrants or options may depress the price of our Common Stock.
As of December 31, 2023, we had 39,830,802 shares of Common Stock issued and outstanding and outstanding warrants to purchase 15,438,607 shares of Common Stock. The issuance of shares of Common Stock upon exercise of outstanding warrants or options could result in substantial dilution to our stockholders, which may have a negative effect on the price of our Common Stock.
Our stock price may be volatile or may decline regardless of our operating performance, resulting in substantial losses for investors.
The market price of our common stock may be highly volatile and may fluctuate substantially as a result of a variety of factors, some of which are related in complex ways. The market price of our common stock may fluctuate significantly in response to numerous factors, many of which are beyond our control, including the factors listed below and other factors describe in this “Risk Factors” section:
| ● | the commencement, enrollment, or results of current and future preclinical studies and clinical trials and trials we may conduct, or changes in the development status of our product candidates; | |
| ● | any delay in our regulatory filings for our product candidates and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such filings, including, without limitation, the issuance by the FDA of a “refusal to file” letter or a request for additional information; | |
| ● | adverse results or delays in clinical trials; | |
| ● | our decision to initiate a preclinical study or clinical trial, not to initiate a preclinical study or clinical trial or to terminate an existing preclinical study or clinical trial; | |
| ● | adverse actions taken by regulatory agencies with respect to our preclinical studies or clinical trials, manufacturing supply chain or sales and marketing activities, including failure to receive regulatory approval of our product candidates; | |
| ● | changes in laws or regulations, including, but not limited to, preclinical study or clinical trial requirements for approvals; | |
| ● | any adverse changes to our relationship with manufacturers or suppliers; | |
| ● | manufacturing, supply or distribution shortages; | |
| ● | our failure to commercialize our product candidates; | |
| ● | changes in the structure of healthcare payment systems; | |
| ● | additions or departures of key scientific or management personnel; | |
| ● | unanticipated serious safety concerns related to the use of our product candidates; | |
| ● | disputes or other developments relating to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies; | |
| ● | variations in our results of operations; | |
| ● | our cash position; | |
| ● | our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public; |
| 56 |
| ● | publication of research reports about us or our industry, or in vivo and ex vivo cell engineering products in particular, or positive or negative recommendations or withdrawal of research coverage by securities analysts; | |
| ● | announcements made by us or our competitors of new product and service offerings, acquisitions, strategic relationships, joint ventures, or capital commitments; | |
| ● | our inability to establish collaborations, if needed; | |
| ● | our ability to effectively manage our growth; | |
| ● | the size and growth of our initial target markets; | |
| ● | changes in the market valuations of similar companies; | |
| ● | press reports, whether or not true, about our business; | |
| ● | sales or perceived potential sales of our common stock by us or our stockholders in the future; | |
| ● | overall fluctuations in the equity markets; | |
| ● | ineffectiveness of our internal controls; | |
| ● | changes in accounting practices or principles; | |
| ● | changes or developments in the global regulatory environment; | |
| ● | litigation involving us, our industry or both, or investigations by regulators into our operations or those of our competitors; | |
| ● | general political and economic conditions; and | |
| ● | other events or factors, many of which are beyond our control. |
In addition, the stock market in general and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. If the market price of our common stock after this offering does not exceed the initial public offering price, you may not realize any return on, and may lose some or all of, your investment.
Our quarterly operating results may fluctuate significantly or may fall below the expectations of investors or securities analysts, each of which may cause our stock price to fluctuate or decline.
We expect our operating results to be subject to quarterly fluctuations. Our net loss and other operating results will be affected by numerous factors, including:
| ● | timing and variations in the level of expense related to the current or future development of our programs; | |
| ● | timing and status of enrollment for our clinical trials; | |
| ● | results of clinical trials, or the addition or termination of clinical trials or funding support by us or potential future partners; | |
| ● | our execution of any collaboration, licensing or similar arrangements, and the timing of payments we may make or receive under potential future arrangements or the termination or modification of any such potential future arrangements; |
| 57 |
| ● | any intellectual property infringement, misappropriation or violation lawsuit or opposition, interference or cancellation proceeding in which we may become involved; | |
| ● | additions and departures of key personnel; | |
| ● | strategic decisions by us or our competitors, such as acquisitions, divestitures, spin-offs, joint ventures, strategic investments or changes in business strategy; | |
| ● | if any product candidate we may develop receive regulatory approval, the timing and terms of such approval and market acceptance and demand for such product candidates; | |
| ● | the timing and cost to establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval and intend to commercialize on our own or jointly with current or future collaborators; | |
| ● | regulatory developments affecting current or future product candidates or those of our competitors; | |
| ● | the amount of expense or gain associated with the change in value of the success payments and contingent consideration; and | |
| ● | changes in general market and economic conditions. |
If our quarterly operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially. We believe that quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
If we take advantage of specified reduced disclosure requirements applicable to a “smaller reporting company”, the information that we provide to stockholders may be different than they might receive from other public companies.
As a company with less than $100 million in revenue during our last fiscal year and a public float of less than $250 million, we qualify as a “smaller reporting company”. As a smaller reporting company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include:
| ● | Only two years of audited financial statements in addition to any required unaudited interim financial statements with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; | |
| ● | Reduced disclosure about our executive compensation arrangements; | |
| ● | Exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting. |
As a result of our status as a “smaller reporting company,” the information that we provide stockholders may be different than you might get from other public companies in which you hold stock.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which would harm our business, operating results, or financial condition. Additionally, the dramatic increase in the cost of directors’ and officers’ liability insurance may cause us to opt for lower overall policy limits or to forgo insurance that we may otherwise rely on to cover significant defense costs, settlements, and damages awarded to plaintiffs.
| 58 |
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Under the Tax Cuts and Jobs Act of 2017, as modified by the Coronavirus Aid, Relief, and Economic Stability Act, or CARES Act, our federal net operating losses, or NOLs, generated in tax years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal NOLs in tax years beginning after December 31, 2020, is limited to 80% of taxable income. It is uncertain if and to what extent various states will conform to the Tax Cuts and Jobs Act of 2017, or the CARES Act. In addition. Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change,” generally defined as a greater than 50 percentage point change (by value) in its equity ownership by certain stockholders over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research and development tax credits) to offset its post-change income or taxes may be limited. Shifts in our stock ownership (some of which are outside our control). As a result, our ability to use our pre-change NOLs and tax credits to offset post-change taxable income, if any, could be subject to limitations. Similar provisions of state tax law may also apply. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
ITEM 1B. UNRESOLVED STAFF COMMENTS.
Not applicable.
ITEM 1C. CYBERSECURITY.
Due to the size of the Company and minimal activity level, record keeping requirements are also minimal. All records are tightly maintained in one secure location with no utilization of servers or cloud-based services. We do not engage any assessors, consultants, auditors or other third parties in connection with our cybersecurity risk management at this time.
As of the year ending December 31, 2023, the Company has had no cybersecurity incidents or been at risk from cybersecurity threats that have materially affected or were reasonably likely to materially affect our business strategy, results of operations or financial condition.
ITEM 2. PROPERTIES.
Our corporate headquarters is located at 57 West 57th Street, New York, NY 10019 under an annual lease. The telephone number at such address is (646) 416-7896. We believe that our facilities are adequate and suitable for our current operations. To the extent that other office space is required, we believe that such space is readily available.
ITEM 3. LEGAL PROCEEDINGS.
Except as described below, we are not involved in any pending legal proceeding or litigations and, to the best of our knowledge, no governmental authority is contemplating any proceeding to which we are a party or to which any of our properties is subject, which would reasonably be likely to have a material adverse effect on us.
In January 2019, the holder of a promissory note in the principal amount of $250,000 due on March 16, 2016 instituted a collection action in the Supreme Court of the State of New York, County of New York. On June 12, 2019, the plaintiff served a motion for summary judgment through the Secretary of State which was heard on July 12, 2019 and granted. The Company contended that it was not given sufficient notice under the applicable statute and did not have an opportunity to oppose the motion. Judgment was entered in October 2019 in the amount of $267,680. The Company brought a motion to vacate based on the jurisdictional defect of the motion in not providing the required amount of time, but that motion was denied in February 2021 without properly addressing the jurisdictional issues raised by the Company. The Company appealed the denial and then filed a motion to Renew and Reargue the motion to vacate based on the Court’s failure to address critical issues. That motion was also denied on April 15, 2021 without addressing the Company’s arguments. The Company appealed the second denial as well and pursued both appeals in a consolidated manner so as to resolve all issues together. Each of the appeals was denied. While the Company’s motions were pending, the plaintiff commenced steps to collect judgment. During the year ended December 31, 2021, $103,088 of a deposit made with the court by a third party on behalf of the Company was released to an officer of the court and has been accounted for as partial note repayment, with an additional $146,912 due under the note repaid by a release of the remaining deposit to an officer of the court and garnishment of Company funds during the year ended December 31, 2022, which was also accounted for as a note repayment. In August 2023, a supplemental judgment of $38,838 was entered against the Company. Inasmuch as there were no further opportunities to appeal, the Company was required to pay the remaining amount due, which was estimated to be approximately $113,000 and recorded as a liability as of December 31, 2023. As of May 31, 2024 after taking into account accrued and unpaid interest, approximately $117,000 was owed to the plaintiff and the plaintiff was seeking, among other things, additional monetary sanctions. In June 2024, the Company resolved this matter by making a final payment of $135,000 and the plaintiff agreed to cease the pursuit of additional sanctions against the Company and file a satisfaction of judgment.
In August 2022, a holder of 360,000 shares of the Company’s common stock filed a complaint against the Company, its President and legal counsel in the United States District Court, Southern District of New York, claiming unspecified damages for an alleged wrongful refusal to authorize the Company’s transfer agent to remove restrictive legends from the shares held by the shareholder. The complaints against the Company’s legal counsel and President were dismissed by the Court. Effective December 29, 2023, the parties reached a settlement agreement whereby the Company, in exchange for the dismissal with prejudice of the claims made by the plaintiff against the Company, agreed to (i) cause the Company’s transfer agent to remove the restrictive legend on the shares held by the plaintiff and (ii) issue the following securities to the plaintiff: 180,000 shares of its common stock (which was issued subsequent to December 31, 2023), a warrant to purchase 180,000 shares of its common stock at an exercise price of $0.75 per share and a convertible promissory note in the principal amount of $50,000.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable.
| 59 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market for Common Equity and Related Stockholder Matters
Our common stock is currently quoted under the symbol “CLCS” on the OTC Markets “Expert Market” due to our failure to timely file our reports with the Securities and Exchange Commission. Quotations in Expert Market Securities are restricted from public viewing. We plan to seek relisting of our common stock on the OTCQB following the filing of this report.
As of June 17, 2024, there were 213 holders of record of our common stock.
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain future earnings, if any, to finance the expansion of our business. As a result, we do not anticipate paying any cash dividends in the foreseeable future.
Warrants
As of December 31, 2023, we had outstanding warrants to purchase an aggregate of 15,438,607 shares of common stock with a weighted average exercise price of $1.09 per share.
Securities Authorized for Issuance Under Equity Compensation Plans
Our Board of Directors adopted the 2019 Equity Incentive Plan (the “Plan”) in August 2019. A total of 7,900,000 shares of common stock were initially reserved for issuance under the Plan and the number of reserved shares increases on the first day of each year in an amount equal to the lesser of 3% of the number of shares of common stock outstanding on the last day of the preceding year or the amount determined by our Board of Directors. As of December 31, 2023, a total of 4,982,190 shares of common stock were reserved for issuance under the Plan. The Plan permits the Board of Directors to issue stock options, stock appreciation rights, restricted stock, restricted stock units, performance and other awards to employees, consultants and directors of the Company. As of the date of filing, the Company’s shareholders have not approved the Plan. The number of stock options outstanding under the Plan, the weighted average exercise price of the outstanding options, and the number of securities remaining available for issuance as of December 31, 2023 were as follows:
EQUITY COMPENSATION PLAN TABLE
| Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted-average exercise price of outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
| Equity compensation plans approved by security holders | — | — | — | |||||||||
| Equity compensation plans not approved by security holders | 6,932,004 | $ | 0.83 | 4,982,190 | ||||||||
| Total | 6,932,004 | $ | 0.83 | 4,982,190 | ||||||||
| 60 |
Sales of Unregistered Securities
In November 2023 we issued a warrant to purchase 25,000 shares of our common stock at an exercise price of $1.25 per share toa holder of a convertible promissory note in connection with the amendment and extension of the maturity date of the note. We relied upon the exemption provided by Section 4(a)(2) of the Securities Act in connection with this transaction.
During the three months ended December 31, 2023, we issued 352,960 shares of common stock to accredited investors upon the conversion of 35,296 shares of the Company’s Series C Convertible Preferred Stock. We relied upon the exemption provided by Section 3(a)(9) of the Securities Act in connection with this transaction.
During the three months ended December 31, 2023, $305,000 of principal outstanding under convertible notes automatically converted into 40,667 shares of our Series C Convertible Preferred Stock and we elected to issue 23,801 shares of common stock in lieu of the payment of $19,055 of cash interest due under such notes. We relied upon the exemption provided by Section 4(a)(2) of the Securities Act in connection with these transactions.
During the three months ended December 31, 2023, we issued 106,668 units to accredited investors at a price of $7.50 per unit. Each unit consisted of one share of Series B Preferred Stock and a warrant to purchase 11.25 shares of common stock at an exercise price of $0.75 per share. We relied upon the exemption provided by Section 4(a)(2) of the Securities Act in connection with these transactions.
In December 2023, we issued 180,00 shares of our common stock, a warrant to purchase 180,000 shares of our common stock at an exercise price of $1.25 per share and a convertible promissory note in the principal amount of $50,000 in connection with the settlement of a litigation matter. The note is convertible into our common stock at an exercise price equal to 80% of the average volume weighted average price of the common stock during the ten trading days prior to the conversion. We relied upon the exemption provided by Section 4(a)(2) of the Securities Act in connection with this transaction.
In December 2023, we issued an aggregate of 808,420 shares of our common stock as payment-in-kind dividends to holders of our Series A Convertible Preferred Stock and holders of our Series C Convertible Preferred Stock. We relied upon the exception provided by Section 4(a)(2) of the Securities Act in connection with these transactions.
Issuer Purchases of Equity Securities
None.
ITEM 6. [RESERVED]
| 61 |
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
This Management Discussion and Analysis (“MD&A”) contains “forward-looking statements,” which represent our projections, estimates, expectations or beliefs concerning among other things, financial items that relate to management’s future plans or objectives or to our future economic and financial performance. In some cases, you can identify these statements by terminology such as “may,” “should,” “plans,” “believe,” “will,” “anticipate,” “estimate,” “expect,” “project” or “intend,” including their opposites or similar phrases or expressions.
You should be aware that these statements are projections or estimates as to future events and are subject to a number of factors that may tend to influence the accuracy of the statements. These forward-looking statements should not be regarded as a representation by the Company or any other person that the events or plans of the Company will be achieved. You should not unduly rely on these forward-looking statements, which speak only as of the date of this MD&A. Except as may be required under applicable securities laws, we undertake no obligation to publicly revise any forward-looking statement to reflect circumstances or events after the date of this MD&A or to reflect the occurrence of unanticipated events. You should, however, review the factors and risks we describe under “Risk Factors” in this Annual Report on Form 10-K. Actual results may differ materially from any forward-looking statement.
Overview
We are a cell therapy company focused on immunotherapy. Since our inception, we have been involved with the development of proprietary immune system management technology licensed from Yeda, the commercial arm of the Weizmann Institute. We have recently shifted the focus of our research and development efforts to MD Anderson.
This technology addresses one of the most fundamental challenges within human immunology: how to tune the immune response such that it tolerates selected desirable foreign cells, but continues to attack all other (undesirable) targets. In simpler terms, a number of potentially life-saving treatments have limited effectiveness today because the patient’s immune system rejects them. For example, while HSCT - hematopoietic stem cell transplantation (e.g. bone marrow transplantation) has become a preferred therapeutic approach for treating blood cell cancer, most patients do not have a matched family donor. Although matched unrelated donors and cord blood can each provide an option for such patients, haploidentical stem cell transplants (sourced from partially mismatched family members) are rapidly gaining favor as a treatment of choice. This is still a risky and difficult procedure primarily because of potential conflicts between host and donor immune systems and also due to viral infections that often follow even successful HSCT while the compromised new immune system works to reconstitute itself by using the transplanted stem cells. Today, rejection is partially overcome using aggressive immune suppression treatments that leave the patient exposed to many dangers by compromising their immune system.
The unique advantage of Cell Source technology lies in the ability to induce sustained tolerance of transplanted cells (or organs) by the recipient’s immune system in a setting that requires only mild immune suppression, while avoiding the most common post-transplant complications. The scientific term for inducing such tolerance in a transplantation setting is chimerism, where the recipient’s immune system tolerates the co-existence of the (genetically different) donor type and host (recipient) type cells. Attaining sustained chimerism is an important prerequisite to achieving the intrinsic GvL (graft versus leukemia) effect of HSCT and supporting the reconstitution of normal hematopoiesis (generation of blood cells, including those that protect healthy patients from cancer) in blood cancer patients. Preclinical data and initial clinical data show that Cell Source’s Veto Cell technology (currently in a clinical trial in the US) can provide superior results in allogeneic (donor-derived) HSCT by allowing for haploidentical stem cell transplants under a mild conditioning regimen, while avoiding the most common post-transplant complications. Combining this with CAR (Chimeric Antigen Receptor) T cell therapy as a unified VETO CAR-T treatment, we plan to treat patients in relapse as well as those in remission and use the cancer killing power of CAR-T to protect the patient while their immune system fully reconstitutes, thus providing an end-to-end solution for blood cancer treatment by potentially delivering a fundamentally safer and more effective allogeneic HSCT: prevention of relapse; avoidance GvHD; prevention of viral infections; and enhanced persistence of GvL effect. This means that the majority of patients will be able to find a donor, and will have access to a potentially safer procedure with higher long term survival rates than what either donor-derived HSCT or autologous CAR-T each on their own currently provide.
| 62 |
The ability to induce permanent chimerism (and thus sustained tolerance) in patients – which allows the transplantation to overcome rejection without having to compromise the rest of the immune system - may open the door to effective treatment of a number of severe medical conditions, in addition to blood cancers, which are characterized by this need. These include:
| ● | The broader set of cancers, including solid tumors, that can potentially be treated effectively using genetically modified cells such as CAR-T cell therapy, but also face efficacy and economic constraints due to limited persistence based on immune system issues (i.e., the need to be able to safely and efficiently deliver allogeneic CAR-T therapy). Inducing sustained tolerance to CAR-T cells may bring reduced and cost and increased efficacy by allowing for off-the-shelf (vs. patient-derived) treatments with more persistent cancer killing capability. | |
| ● | Organ failure and transplantation. A variety of conditions can be treated by the transplantation of vital organs. However, transplantation is limited both by the insufficient supply of available donor organs and the need for lifelong, daily anti-reject treatments post-transplant. Haploidentical organ transplants, with sustained chimerism, have the potential to make life saving transplants accessible to the majority of patients, with the prospect of improved life quality and expectancy. | |
| ● | Non-malignant hematological conditions (such as type one diabetes and sickle cell anemia) which could, in many cases, also be more effectively treated by stem cell transplantation if the procedure could be made safer and more accessible by inducing sustained tolerance in the stem cell transplant recipient. |
Human Capital Resources
Other than our Chief Executive Officer, we currently do not have any full-time employees, but retain the services of independent contractors/consultants on a contract-employment basis.
Recent Developments
Preclinical Results and Clinical Results
After two years of intensive collaboration with Professor Zelig Eshhar, the inventor of CAR-T cell therapy, preclinical data confirmed that Veto Cells can markedly extend persistence of genetically modified T cells from the same donor and that genetically modified Veto Cells can effectively inhibit tumors expressing an antigen recognized by the transgenic T cell receptor. Furthermore, human Veto Cells transfected with CAR exhibit anti-tumor activity in-vitro without losing their veto activity. These preclinical results have formed the basis of our current development of a clinical protocol for allogeneic VETO CAR-T HSCT combined therapy for blood cancer treatment. Cell Source plans to submit this protocol for approval by the end of 2024. The Phase 1/2 clinical trial at the University of Texas MD Anderson Cancer Center, using Cell Source’s Anti-viral Veto Cells, has successfully completed the first three treatment cohorts, with 12 patients each receiving a haploidentical HSCT under reduced intensity conditioning with Veto Cells. This first in human dose optimization trial has thus far shown that the initial dose is in fact the optimal dose, as all nine patients had successful stem cell engraftment after 42 days, in the absence of severe GvHD. Cell Source has continued the trial as it proceeds with the next cohorts of patients, using the higher dose level, in order to complete the dose finding process.
Private Placement of Series B Convertible Preferred Stock
Beginning in October 2023, the Company entered into subscription agreements with certain accredited investors in a private placement offering. Each unit, which is sold at a price of $7.50 per unit, consists of one (1) share of Series B Convertible Preferred Stock and a five-year warrant to purchase a certain number of shares of common stock at an exercise price of $0.75 per share. For every $100,000 of units acquired, the investor will receive warrants to purchase an aggregate of 150,000 shares of common stock.
From October 2023 through the date of filing, the Company sold 206,799 units for gross proceeds of $1,551,000 and issued warrants to purchase 2,326,500 shares of the Company’s common stock.
| 63 |
Risks and Uncertainties
On October 7, 2023, a conflict arose between Israel and Hamas militants on Israel’s southern border from the Gaza Strip. The intensity and duration of Israel’s current war against Hamas is difficult to predict, and as are such war’s economic implications on the Company’s business and operations. To the extent that any of these negative developments do occur, they may have an adverse effect on the Company’s business, results of operations and its ability to raise additional funds. As of December 31, 2023, the Company considered the impact of the war on its business and operational assumptions and estimates and determined there were no material adverse impacts on the Company’s consolidated results of operations and financial position as of December 31, 2023.
Consolidated Results of Operations
Year Ended December 31, 2023 Compared with the Year Ended December 31, 2022
Research and Development
Research and development expense was $1,577,995 and $1,996,173 for the years ended December 31, 2023 and 2022, respectively, a decrease of $418,178, or 21%. This decrease is mainly attributable to the achievement of five patient enrollment milestones achieved in 2022 for $527,525 under the sponsored research agreement with MD Andersen, whereas one $105,505 enrollment milestone was achieved in the 2023 period.
General and Administrative
General and administrative expense was $2,604,751 and $2,179,160 for the years ended December 31, 2023 and 2022, respectively, an increase of $425,591, or 20%. General and administrative expenses are primarily comprised of external consulting and professional fees, payroll and stock-based compensation expenses. The increase was primarily attributable to an increase of $219,000 in consulting expenses recorded in 2023 due to increased financial advisory services during the 2023 period, and an increase in stock-based compensation of $351,259 mainly as a result of an aggregate of 1,150,000 shares of the Company’s common stock issued in consideration for consulting services, partially offset by a decrease in legal fees of $41,458.
Loss on Legal Settlement
During the year ended December 31, 2023, we recognized a $142,600 loss on legal settlement. The loss on legal settlement is attributable to the issuance of a $50,000 convertible note payable, a five-year warrant to purchase 180,000 shares of the Company’s common stock at an exercise price of $0.75 per share, and the issuance of 180,000 shares of the Company’s common stock in satisfaction of unspecified damages for an alleged wrongful refusal to authorize the Company’s transfer agent to remove restrictive legends from the shares held by the shareholder.
Interest Expense
Interest expense for the years ended December 31, 2023 and 2022 was $668,117 and $707,115, respectively, a decrease of $38,998, or 6%.
Interest Expense - Amortization of Debt Discount
Amortization of debt discount was $380,569 and $285,300 for the years ended December 31, 2023 and 2022, respectively, an increase of $95,269, or 33%. The increase is primarily due to greater amount of notes outstanding and related debt discount amortization.
| 64 |
Change in Fair Value of Derivative Liability
During the year ended December 31, 2023, we recognized a gain on the change in fair value of derivative liability of $10,900. The change in fair value of derivative liability is attributable to the decrease in fair value of the Company’s common stock during 2023.
Gain on Extinguishment of Note Payable
During the year ended December 31, 2023, we recognized $41,920 of gain on extinguishment of note payable. The gain on extinguishment of note payable is attributable to the exchange of a promissory note in the principal amount of $100,000 for 176,000 shares of common stock.
Liquidity and Going Concern
We measure our liquidity in a number of ways, including the following:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash | $ | 22,203 | $ | 222,665 | ||||
| Working capital deficiency | $ | (15,611,543 | ) | $ | (12,633,895 | ) | ||
During the year ended December 31, 2023, we had not generated any revenues, had a net loss of $5,321,212 and had used cash in operations of $2,347,891. As of December 31, 2023, we had a working capital deficiency of $15,611,543 and an accumulated deficit of $41,667,388. As of December 31, 2023 and through the date of this filing, notes payable with principal amounts totaling $1,876,093 and $1,906,093, respectively, were past due and are classified as current liabilities on the consolidated balance sheet as of December 31, 2023. We will continue to incur net operating losses to fund operations. These conditions raise substantial doubt about our ability to continue as a going concern for at least one year from the date these financial statements are issued.
We are currently funding our operations on a month-to-month basis. Our ability to continue our operations is dependent on the execution of management’s plans, which include the raising of capital through the debt and/or equity markets, until such time that funds provided by operations are sufficient to fund working capital requirements. Subsequent to December 31, 2023 and as more fully described in Note 13, Subsequent Events, the Company received aggregate proceeds of $646,672 from the issuance of notes payable and convertible notes payable- related parties and $751,000 from the sale of Series B Preferred Stock. We may need to incur additional liabilities with certain related parties to sustain our existence. If we were not to continue as a going concern, we would likely not be able to realize our assets at values comparable to the carrying value or the fair value estimates reflected in the balances set out in the preparation of our financial statements.
There can be no assurances that we will be successful in generating additional cash from equity or debt financings or other sources to be used for operations. Should we not be successful in obtaining the necessary financing to fund our operations, we would need to curtail certain or all operational activities and/or contemplate the sale of our assets, if necessary.
During the years ended December 31, 2023 and 2022, our sources and uses of cash were as follows:
Net Cash Used in Operating Activities
We experienced negative cash flows from operating activities for the years ended December 31, 2023 and 2022 in the amounts of $2,347,891 and $3,138,686, respectively. The net cash used in operating activities for the year ended December 31, 2023 was primarily due to cash used to fund a net loss of $5,321,212 adjusted for net non-cash expenses in the aggregate amount of $754,834, partially offset by $2,218,487 of net cash provided by changes in the levels of operating assets and liabilities. The net cash used in operating activities for the year ended December 31, 2022 was primarily due to cash used to fund a net loss of $5,167,748, adjusted for net non-cash expenses in the aggregate amount of $465,542, partially offset by $1,563,520 of net cash provided by changes in the levels of operating assets and liabilities.
| 65 |
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the years ended December 31, 2023 and 2022 was $2,147,429 and $3,268,256, respectively. The net cash provided by financing activities during the year ended December 31, 2023 was attributable to $1,596,978 of proceeds from the issuance of convertible notes payable, $799,918 of proceeds received from Convertible Series B Preferred stock subscriptions, and $30,000 of proceeds from the issuance of notes payable, offset by $279,467 of repayments of financing liability. The net cash provided by financing activities during the year ended December 31, 2022 was attributable to $3,275,000 of proceeds from the issuance of convertible notes payable and $168,094 of proceeds from the issuance of notes payable, offset by the repayments of notes payable in the amount of $146,912, and $27,926 of repayment of financing liability.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Recently Issued Accounting Pronouncements
For recently issued accounting announcements, see “Recent Accounting Standards” in Note 3, Summary of Significant Accounting Policies in the notes to our consolidated financial statements included in this Annual Report on Form 10-K.
Critical Accounting Estimates
The preparation of financial statements and related disclosures are in conformity with U.S. GAAP. These accounting principles require us to make estimates and judgments that can affect the reported amounts of assets and liabilities as of the date of the financial statements, as well as the reported amounts of revenue and expense during the periods presented. We believe that the estimates and judgments upon which we rely are reasonable based upon information available to us at the time that we make these estimates and judgments. To the extent that there are material differences between these estimates and actual results, our financial results will be affected. The accounting policies that reflect our more significant estimates and judgments and which we believe are the most critical to aid in fully understanding and evaluating our reported financial results are described below.
We consider an accounting estimate to be critical if: (i) the accounting estimate requires us to make assumptions about matters that were highly uncertain at the time the accounting estimate was made, and (ii) changes in the estimate that are reasonably likely to occur from period to period or use of different estimates that we reasonably could have used in the current period, would have a material impact on our financial condition or results of operations.
Management has identified certain critical accounting estimates which are outlined below. In addition, there are other items within our financial statements that require estimation but are not deemed critical, as defined above. Changes in estimates used in these and other items could have a material impact on our financial statements.
Valuation of the Company’s Common Stock Price
Since the Company’s common stock historically was not actively traded on a public market, the fair value of the Company’s restricted equity instruments is estimated by management based on observations of the sales prices of both restricted and freely tradable common stock, or instruments convertible into common stock. During the year ended December 31, 2023, the Company obtained a third-party valuation of its common stock as of December 31, 2023 and July 1, 2023 which was considered in management’s estimation of fair value during the year ended December 31, 2023. The third-party valuation was performed in accordance with regulation of Section 409A of the Internal Revenue Code (“IRC”) as well as FASB ASC Topic 718.
| 66 |
The independent appraisal utilized the market approach, specifically the Backsolve method. The Backsolve method utilizes the economics from a direct transaction in the Company’s securities, specifically, issuances of convertible notes and Series B Convertible Preferred Stock during 2023, in determining fair value. When applying the Backsolve method, the Company evaluated the below valuation inputs:
| 1) | Total equity value of the Company. | |
| 2) | Application the Black-Scholes option pricing method (“OPM”) to the various classes of convertible securities outstanding as of each of the valuation dates outlined above. | |
| 3) | Application of a probability-weighted average present value of various classes of convertible securities. | |
| 4) | The total value of each share class was divided by the security’s respective fully diluted shares outstanding, in order to calculate the per share value for each security on a marketable basis. |
Under the OPM, it was determined the Company’s common stock had a fair value of $0.26 and $0.34 per share as of December 31, 2023 and July 1, 2023, respectively, which included a discount for lack of marketability of 25%. Furthermore, the independent appraisal determined the Company’s expected volatility was 65% and 80% as of December 31, 2023 and July 1, 2023 respectively, by evaluating historical and implied volatilities of guideline companies.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
Our consolidated financial statements are presented following the signature page to this report.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
None.
| 67 |
ITEM 9A. CONTROLS AND PROCEDURES.
Disclosure Controls and Procedures
Disclosure controls are procedures that are designed with the objective of ensuring that information required to be disclosed in our reports filed under the Exchange Act, such as this Annual Report, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls are also designed with the objective of ensuring that such information is accumulated and communicated to our management, including the Principal Executive and Financial Officer, as appropriate to allow timely decisions regarding required disclosure. Internal controls are procedures which are designed with the objective of providing reasonable assurance that (1) our transactions are properly authorized, recorded and reported; and (2) our assets are safeguarded against unauthorized or improper use, to permit the preparation of our consolidated financial statements in conformity with United States generally accepted accounting principles.
In connection with the preparation of this Annual Report, management, with the participation of our Principal Executive and Financial Officer, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e) and 15d-15(e)). Based upon that evaluation, our Principal Executive and Financial Officer concluded that, as of December 31, 2023, our disclosure controls and procedures were effective.
Internal Control over Financial Reporting
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Exchange Act. Internal control over financial reporting is a process designed by, or under the supervision of, our Principal Executive and Financial Officer, and effected by the Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP including those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and the disposition of our assets, (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP and that receipts and expenditures are being made only in accordance with authorizations of our management and Board of Directors, and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements.
Under the supervision and with the participation of our management, including our principal executive and financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2023, based on the Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) (2013 Framework). Based on this evaluation under the 2013 Framework, our principal executive and financial officer concluded that our internal control over financial reporting was effective as of December 31, 2023.
Changes in Internal Controls
There were no changes in our internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act) during the quarter ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations of the Effectiveness of Control
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations of any control system, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected.
Attestation Report of Registered Public Accounting Firm
This Annual Report does not contain an attestation report of our independent registered public accounting firm regarding internal control over financial reporting since the rules for smaller reporting companies provide for this exemption.
ITEM 9B. OTHER INFORMATION.
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS.
None.
| 68 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
Below are the names and certain information regarding the Company’s executive officers and directors:
| Name | Age | Title(s) | ||
| Dennis Brown | 75 | Director (Chairman) | ||
| Itamar Shimrat | 64 | Chief Executive Officer, Chief Financial Officer | ||
| Darlene Soave | 80 | Director | ||
| George Verstraete | 70 | Director |
Dr. Dennis M. Brown, PhD, was elected Director of the Company on June 30, 2014 and as Chairman of the Board on May 18, 2015. Dr. Brown became the Chair of our Audit Committee in September 2015. Dr. Brown is a founder and Chief Scientific Officer of Kintara Therapeutics, Inc. (Nasdaq: KTRA). Dr. Brown has more than thirty years of drug discovery and development experience. Since 2000 to the present, Dr. Brown has served as Chairman of Mountain View Pharmaceutical’s Board of Directors and is the President of Valent. Dr. Brown has focused over the past 5 years on the development of Kintara (formerly under the name DelMar Pharmaceuticals), (serving as its Chief Scientific Officer since January 25, 2013 and Director since February 11, 2013. His extensive technical expertise, successful track record as an inventor, executive and director in the field of medical technology position him as an authoritative voice on the scientific, intellectual property, finance and commercialization and well as general management issues for Cell Source both now and in the future. In 1999 he founded ChemGenex Therapeutics, which merged with a publicly traded Australian company in 2004 to become ChemGenex Pharmaceuticals (ASX: CXS/NASDAQ: CXSP), of which he served as President and a Director until 2009. He was previously a co-founder of Matrix Pharmaceutical, Inc., where he served as Vice President (VP) of Scientific Affairs from 1985-1995 and as VP, Discovery Research, from 1995-1999. He also previously served as an Assistant Professor of Radiology at Harvard University Medical School and as a Research Associate in Radiology at Stanford University Medical School. He received his B.A. in Biology and Chemistry (1971), M.S. in Cell Biology (1975) and Ph.D. in Radiation and Cancer Biology (1979), all from New York University. Dr. Brown is an inventor of about 34 issued U.S. patents and applications, many with foreign counterparts. Dr. Brown’s scientific knowledge and experience qualifies him to serve on our Board of Directors.
Itamar Shimrat, CEO and CFO and Director, is a Canadian businessman and a founding member of Cell Source Israel. Since Cell Source Israel’s inception, Mr. Shimrat served as a Chief Financial Officer and he served as a Director from Cell Source Israel’s inception until May 2022. In October 2013, he was appointed Chief Executive Officer. From March 2009 through September 2011, Mr. Shimrat served as Chief Financial Officer and Director of Rainbow Energy Ltd. From September 2011 through October 2013, Mr. Shimrat served as Chief Financial Officer and Director of Cell Source Ltd. From August 2012 to present, Mr. Shimrat served as Director of Step Up - Olim Madrega Inc. From October 2013 to March 2022, Mr. Shimrat served as Chief Executive Officer and Director of Cell Source Ltd. Previously, Mr. Shimrat served as an Executive Vice President at First International Bank of Israel from March 2005 until April, 2008. Prior to 2008, he served as a senior manager at McKinsey & Company’s Tel Aviv office after having being elected Partner at Mitchell Madison Group and consulting for Bain & Co. Mr. Shimrat led major profit improvement programs for leading corporations ranging from American Express and Barclays to El Al Airlines. He has been a Director of two private companies: Rainbow Energy Ltd., a company in the renewable energy industry, and Step Up - Olim Madrega Ltd., a company in the wheelchair industry, and also was on the Allocations Committee of Matan, a leading Israeli philanthropic organization. He holds an MBA with Distinction from the Ivey Business School of the University of Western Ontario in Canada. Itamar brings to Cell Source significant knowledge and experience in the area of corporate finance.
Darlene Soave was appointed to the Company’s Board of Directors effective March 25, 2021. Ms. Soave, a native of Detroit, was a Co- founder and Director of Soave Enterprises, a diversified management and investment company. For over 40 years, she played key roles in its business successes, which were primarily achieved through investing in well-run companies, providing them with the tools and resources necessary to further enhance their businesses, and gauging real-world results through the use of proprietary performance metrics. From the start, a defining characteristic of Soave Enterprises was its nurturing a highly entrepreneurial culture. The company rapidly grew from humble beginnings into one of the industry’s largest and most respected environmental services and waste management groups. Over the following decades, Soave Enterprises went on to flourish in a broad array of industries and endeavors. These include: Coastal Florida luxury condominiums; Chicago-area Budweiser distributorships; Metal recycling operations in the Midwest; Mercedes-Benz retail operations in Kansas City; and a Washington, D.C.-area master-planned residential community. Today, its diversified portfolio generates annual revenues exceeding $1.8 billion. Through the DG Group Inc., Ms. Soave’s current holdings encompass a vertically integrated portfolio of over 500 income-producing residential properties in addition to various other commercial assets. DG Group identifies, grows, and sustains its portfolio businesses by leveraging the combined strengths of their management teams and financial resources. DG Group’s holdings also include an array of companies that are committed to health-enhancing and potentially life-saving technologies. A hallmark of DG Group is its singular approach to combining state-of-the-art technology with quality, hands-on leadership. It was Ms. Soave’s lifelong advocacy and personal embodiment of ‘anti-aging’ that captured her initial interest and ultimately led to her investment and involvement in Cell Source. Ms. Soave has served on the Board of Directors of Soave Enterprises, The Detroit Historical Society, The Detroit Symphony Orchestra, Barrett House (for abused women and children), Friends of Fleck, and the Restoration Board of the Detroit Opera House. Ms. Soave’s extensive business experience qualifies her to serve on our Board of Directors.
| 69 |
George Verstraete was appointed to the Company’s Board of Directors in March 2022. Mr. Verstraete is an entrepreneur who has owned and/or managed various enterprises since 1980. An expert in real estate development and property management, Mr. Verstraete currently serves as President of DGR Management. He also serves as a Director of Southern Desert Operations LLC.
The Company’s directors are elected at the annual meeting of shareholders to hold office until the annual meeting of shareholders for the ensuing year or until their successors have been duly elected and qualified. Officers are elected annually by the Board of Directors and serve at the discretion of the Board.
Board Leadership Structure and Role in Risk Oversight
Due to the small size and early stage of the Company, we have not adopted a formal policy on whether the Chairman and Chief Executive Officer positions should be separate or combined. Dr. Brown serves as the Chairman whereas Mr. Shimrat serves as the Chief Executive Officer.
Our Board of Directors (“Board”) is primarily responsible for overseeing our risk management processes on behalf of the Company. The Board receives and reviews periodic reports from management, auditors, legal counsel, and others, as considered appropriate regarding our Company’s assessment of risks. In addition, the Board focuses on the most significant risks facing our Company and our Company’s general risk management strategy, and also ensures that risks undertaken by our Company are consistent with the Board’s appetite for risk. While the Board oversees our company’s risk management, management is responsible for day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks facing our company and that our board leadership structure supports this approach.
Involvement in Certain Legal Proceedings
Our directors and executive officers have not been involved in any of the following events during the past ten years:
| 1. | any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; |
| 2. | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| 3. | being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities; |
| 4. | being found by a court of competent jurisdiction in a civil action, the SEC or the Commodity Futures Trading Commission to have violated a Federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| 5. | being subject of, or a party to, any Federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of any Federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| 6. | being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
| 70 |
Code of Ethics
We have not adopted a Code of Business Conduct and Ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions because of the small number of persons involved in the management of the Company.
Board Meetings and Attendance
During the year ended December 31, 2023, the Company’s Board of Directors held no meetings and acted by written consent on 21 occasions.
Nominating Committee
We have not adopted any procedures by which security holders may recommend nominees to our Board of Directors.
Audit Committee
The Audit Committee of the Board of Directors operates under a charter that has been approved by the Board of Directors. The Audit Committee of the Board of Directors is responsible for overseeing our accounting and financial reporting processes and the audits of our financial statements. From April 2019 until July 2022 the members of the Audit Committee were Dennis Brown (Chair), Ben Friedman and Itamar Shimrat. Mr. Friedman resigned his position as a member of the Company’s Board of Directors in July 2022 and has since been replaced by George Verstraete.
The Board of Directors determined that Mr. Shimrat, also a member of the Audit Committee, is an “audit committee financial expert,” as that is defined in Item 407(d)(5) of Regulation S-K. The Board of Directors has determined that Dr. Brown is an “independent director” and an “audit committee financial expert” based upon said definitions. The Audit Committee held 4 meetings during the fiscal year ended December 31, 2023.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors and executive officers and persons who own more than 10% of the issued and outstanding shares of our common stock to file reports of initial ownership of common stock and other equity securities and subsequent changes in that ownership with the SEC. Officers, directors and greater than ten percent stockholders are required by SEC regulation to furnish us with copies of all Section 16(a) forms they file. To our knowledge, during the fiscal year ended December 31, 2023, all Section 16(a) filing requirements applicable to our officers, directors and greater than 10% beneficial owners were complied with, except that Forms 4 required to be filed by (i) each of Dennis Brown, Darlene Soave and Phyllis Friedman Investment ULC to report the issuance of shares of common stock as payment of in-kind dividends on the shares of Series A Preferred Stock that they own, (ii) Darlene Soave to report the purchase of Series B Preferred Stock and (iii) Darlene Soave and George Verstraete to repot the receipt of warrants have not yet been filed.
| 71 |
ITEM 11. EXECUTIVE COMPENSATION.
The following table sets forth all compensation earned in respect of the Company’s principal executive officer (“PEO”) for 2023 and 2022:
| Name and Principal Position | Year | Salary | Bonus | Stock Awards | Option Awards | All Other Compensation | Total | |||||||||||||||||||||
| Itamar Shimrat | 2023 | $ | 204,290 | $ | - | $ | - | $ | - | $ | - | $ | 204,290 | |||||||||||||||
| 2022 | $ | 208,088 | $ | - | $ | - | $ | - | $ | - | $ | 208,088 | ||||||||||||||||
Director Compensation
The following table sets forth certain information concerning the compensation of our non-employee directors for the fiscal year ended December 31, 2023:
| Change in | |||||||||||||||||||||||||||
| Present Value | |||||||||||||||||||||||||||
| Fees Earned | and Nonqualified | ||||||||||||||||||||||||||
| or | Option | Deferred | |||||||||||||||||||||||||
| Paid In | Stock | Awards | Compensation | All Other | |||||||||||||||||||||||
| Year | Salary | Awards | (1) | Earnings | Compensation | Total | |||||||||||||||||||||
| Dennis Brown | 2023 | $ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||||
| Darlene Soave | 2023 | $ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||||
| George Verstraete | 2023 | $ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||||
Outstanding Equity Awards at Fiscal Year End
The following table presents the outstanding equity awards held as of December 31, 2023 by our sole executive officer.
OUTSTANDING EQUITY AWARDS AT FISCAL YEAR-END
| Option Awards | Stock Awards | |||||||||||||||||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) | Option Exercise Price ($) | Option Expiration Date | Number of Shares or Units of Stock That Have Not Vested (#) | Market Value of Shares or Units of Stock That Have Not Vested ($) | Equity Incentive Plan Awards: Number of Unexercised Shares, Units or Other Rights That Have Not Vested (#) | Equity Incentive Plan Awards: Market or Payout Value of Unearned Shares, Units or Other Rights That Have Not Vested ($) | |||||||||||||||||||||
| Itamar Shimrat | 1,350,000 | — | — | $ | 1.00 | (1) | 3/7/2026 | — | — | — | — | |||||||||||||||||||
| (1) | The exercise price of this option is equal to the Fair Market Value of the Shares on the date of grant (as defined in the Company’s 2019 Equity Incentive Plan). |
Risk Management
The Company does not believe risks arising from its compensation policies and practices for its employees are reasonably likely to have a material adverse effect on the Company.
Equity Grant Committee
In August 2020, the Board of Directors created an Equity Grant Committee and authorized it to make equity awards to employees, consultants and other service providers. Mr. Brown and Mr. Shimrat are the current members of the Equity Grant Committee. The Equity Grant Committee acted by written consent on 7 occasions during the year ended December 31, 2023.
Compensation Committee Interlocks and Insider Participation
Currently, the Board of Directors does not have a standing compensation committee, or a committee performing similar functions, except for the Equity Grant Committee. During the fiscal year ended 2023, the entire Board of Directors deliberated with respect to executive compensation.
| 72 |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The following tables set forth certain information, as of the date set forth below, with respect to the beneficial ownership of the outstanding Common Stock, Series A Preferred Stock, Series B Preferred Stock and Series C Preferred Stock by (i) any holder of more than five (5%) percent of the applicable class; (ii) each of the Company’s executive officers and directors; and (iii) the Company’s directors and executive officers as a group. Except as otherwise indicated, each of the stockholders listed below has sole voting and investment power over the shares beneficially owned.
Shares of Common Stock Beneficially Owned(1)
As of June 17, 2024
| Name and Address of Beneficial Owner(2): | Common Stock | Percent (3) | Percent of Total Voting Power(4) | |||||||||
| Itamar Shimrat, Chief Executive Officer, Chief Financial Officer and Director | 1,925,004 | (5) | 4.54 | % | 3.04 | % | ||||||
| Dennis Brown, Director (Executive Chairman) | 646,599 | (6) | 1.56 | % | 1.04 | % | ||||||
| Darlene Soave, Director | 7,846,455 | (7) | 16.26 | % | 22.60 | % | ||||||
| George Verstraete, Director | 2,375,000 | (8) | 5.47 | % | 3.69 | % | ||||||
| All directors and executive officers as a group (4 persons) | 12,793,063 | 24.37 | % | 27.57 | % | |||||||
| Yair Reisner 1515 Holcombe Boulevard Houston, Texas 77030 | 3,782,004 | (9) | 8.44 | % | 5.74 | % | ||||||
| YEDA Research & Development Co., Ltd. P.O. Box 905 Rehovot, 76100, Israel | 3,155,348 | 7.69 | % | 5.09 | % | |||||||
| Phyllis Friedman Investment ULC Toronto, Ontario, Canada | 5,936,836 | (10) | 14.06 | % | 9.48 | % | ||||||
| (1) | Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock subject to options or warrants currently exercisable or convertible, or exercisable or convertible within 60 days of June 17, 2024 are deemed outstanding for computing the percentage of the person holding such option or warrant but are not deemed outstanding for computing the percentage of any other person. |
| (2) | Except as otherwise indicated, the address of each beneficial owner is c/o Cell Source, Inc., 57 West 57th Street, Suite 400, New York, New York 10019. |
| (3) | Based on 41,019,348 shares of common stock issued and outstanding as of June 17, 2024. |
| (4) | Includes voting power attributable to ownership of common stock, Series A Preferred Stock, Series B Preferred Stock and Series C Preferred Stock. |
| (5) | Includes 1,350,000 shares of common stock issuable upon the exercise of options to purchase common stock at an exercise price of $1.00 per share. |
| (6) | Includes 450,000 shares of common stock issuable upon the exercise of options to purchase common stock at an exercise price of $0.75 per share and 66,670 shares of common stock issuable upon conversion of 6,667 shares of Series A Preferred Stock. |
| (7) | Includes 1,333,340 shares of common stock issuable upon conversion of 133,334 shares of Series A Preferred Stock, 466,670 shares of common stock issuable upon the conversion of 46,667 shares of Series B Preferred Stock 4,265,000 shares of common stock issuable upon the exercise of warrants having an exercise price of $0.75 per share held by a trust of which Ms. Soave serves as trustee, 788,028 shares of common issuable upon the exercise of warrants having an exercise price of $1.25 per share held by a trust of which Ms. Soave serves as trustee and 375,000 shares of common stock issuable upon the exercise of options to purchase common stock at an exercise price of $1.00 per share. |
| (8) | Includes 375,000 shares of common stock upon the exercise of options to purchase common stock at an exercise price of $0.75 per share and 2,000,000 shares of common stock issuable upon the exercise of warrants having an exercise price of $1.00 per share. |
| (9) | Includes 3,782,004 shares of common stock issuable upon the exercise of options to purchase common stock at an exercise price of $0.75 per share. |
| (10) | Includes 435,530 shares of common stock issuable upon conversion of 43,553 shares of Series A Preferred Stock, 300,000 shares of common stock issuable upon the conversion of 30,000 shares of Series B Preferred Stock, 300,000 shares of common stock issuable upon the exercise of options to purchase common stock at an exercise price of $0.75 per share and 337,500 shares of common stock issuable upon the exercise of warrants to purchase common stock at an exercise price of $0.75 per share. |
| 73 |
Shares of Preferred Stock Beneficially Owned(1)
As of June 17, 2024
| Name and Address of Beneficial Owner(2): | Series A Preferred Stock | Percent(3) |
Series B Preferred Stock | Percent(4) | Series C Preferred Stock |
Percent(5) | Percent of Total Voting Power(6) | |||||||||||||||||||||
| Itamar Shimrat, Chief Executive Officer, Chief Financial Officer and Director | – | – | – | – | – | – | 3.04 | % | ||||||||||||||||||||
| Dennis Brown, Director (Executive Chairman) | 6,667 | * | – | – | – | – | 1.04 | % | ||||||||||||||||||||
| Darlene Soave, Director | 133,334 | 9.93 | % | 1,006,227 | (7) | 81.82 | % | – | – | 22.60 | % | |||||||||||||||||
| George Verstraete, Director | – | – | – | – | – | – | 3.69 | % | ||||||||||||||||||||
| All directors and executive officers as a group (4 persons) | 140,001 | 10.43 | % | 1,006,227 | (7) | 81.82 | % | - | - | 27.57 | % | |||||||||||||||||
| Hua Tuo Online (Hong Kong) Limited Suite 5207, 52/F Central Plaza 18 Harbour Road Wanchai, Hong Kong | 133,334 | 9.93 | % | – | – | – | – | 2.34 | % | |||||||||||||||||||
| Acuity Investments LLC 124 South Gay Street Knoxville, Tennessee 37902 | 130,402 | (7) | 9.72 | % | – | – | – | – | 3.31 | % | ||||||||||||||||||
| IGEA Ventures 1057 Commerce Avenue Union, New Jersey 07083 | 108,667 | 8.10 | % | – | – | – | – | 2.81 | % | |||||||||||||||||||
| Ingram Tynes 820 Shades Creek Parkway, Suite 2300 Birmingham, Alabama 35209 | 23,330 | 1.74 | % | – | – | 40,000 | 7.26 | % | 1.42 | % | ||||||||||||||||||
| Timothy Dieschbourg 1101 Bette Lane Glenview Illinois 60025 | – | – | 36,800 | 17.80 | % | – | – | 1.25 | % | |||||||||||||||||||
| Cipayo Ltd 80 Main Street Road Town Tortola VG 1110, VI | – | – | 20,000 | 9.67 | % | – | – | * | ||||||||||||||||||||
| Michael J. La Morgese 3410 Baron Road Pompano Beach Florida 33062 | – | – | 13,333 | 6.44 | % | 17,000 | 3.09 | % | 1.21 | % | ||||||||||||||||||
| Next Generation Trust Company as Custodian FBO Tom Karsen IRA 71 Livingston Ave. Suite 204 Roseland New Jersey 07068 | - | - | 13,333 | 6.44 | % | - | - | 0.46 | % | |||||||||||||||||||
| Susan McCracken 4459 Decatur Drive Ferndale, Washington 98248 | – | – | – | – | 33,333 | 6.05 | % | * | ||||||||||||||||||||
| Mario Dell’Area 217 Park Drive Eastchester, New York 10709 | 76,500 | 5.7 | % | – | – | – | – | 1.79 | % | |||||||||||||||||||
*Less than 1%
| (1) | Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock subject to options or warrants currently exercisable or convertible, or exercisable or convertible within 60 days of June 17, 2024 are deemed outstanding for computing the percentage of the person holding such option or warrant but are not deemed outstanding for computing the percentage of any other person. |
| (2) | Except as otherwise indicated, the address of each beneficial owner is c/o Cell Source, Inc., 57 West 57th Street, Suite 400, New York, New York 10019. |
| (3) | Based on 1,342,195 shares of Series A Preferred Stock issued and outstanding as of June 17, 2024. |
| (4) | Based on 206,799 shares of Series B Preferred Stock issued and outstanding as of June 17, 2024. |
| (5) | Based on 550,815 shares of Series C Preferred Stock issued and outstanding as of June 17, 2024. |
| (6) | Includes voting power attributable to ownership of common stock, Series A Preferred Stock, Series B Preferred Stock and Series C Preferred Stock. |
| (7) | Includes 959,960 shares of Series B Preferred Stock issuable upon conversion of an outstanding convertible note held by a trust of which Ms. Soave serves as trustee. |
| (8) | Includes 99,468 shares of Series A Preferred Stock owned by Acuity Capital, LLC. |
| 74 |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Certain Relationships and Related Transactions
The Company maintains an exclusive worldwide license to certain intellectual property of Yeda, the commercial arm of the Weizmann Institute, which currently owns 3,155,348 shares of Company common stock. Dr. Reisner, who heads the Reisner Laboratory at MD Anderson, owns options to purchase 3,782,004 shares of Company common stock. See the section entitled “Intellectual Property” in this Annual Report on Form 10-K.
In March 2018, the Yeda License Agreement was amended to reduce the Company’s funding obligation for the period from October 2017 through September 2018 to $500,000 and $100,000 for the period from October 2018 through June 2019. In addition, the License Agreement was amended to provide that the Company would fund an additional $100,000 of research during 2018 and the Company’s obligation to fund the original research was reduced by $50,000. The Company funded the additional $100,000 of additional research in April 2018 and $50,000 was credited against the amount that would have otherwise been funded by the Company for the period from July 2018 through September 2018. After giving effect to these amendments and this credit, the Company was required to fund $100,000 for the three-month period ended June 30, 2018, $50,000 for the three month period ended December 2018, and was required to fund $25,000 for the three month period ended March 2019 and $25,000 for the three month period ended June 2019. In connection with Amendments No. 5, 6 and 7 to the License Agreement, the latest of which was dated November 15, 2020, the research conducted (“Further Research”) under the Agreement was extended to cover the period from July 1, 2019 to March 31, 2020 and the associated research budget for this period was a total of $235,000. In addition, these amendments and an amendment effective December 2, 2021 amended the milestones and related completion dates. If the Company fails to achieve any of the milestones by the dates set forth in the agreement, Yeda is entitled to terminate the license upon written notice to the Company. To date, the Company has been deemed to have met all of the milestones and the next milestone in the agreement is January 1, 2025. Either Yeda or the Company may terminate the agreement and the license after the commitment of a material breach by the other party and in certain other instances as detailed in the agreement.
For the years ended December 31, 2023 and 2022, the Company recorded expenses in operations of approximately $58,000 related to its Agreement with Yeda.
In May 2017, the Company received a loan of $180,000 from an entity owned by Ben Friedman and a loan of $45,000 from an entity owned by David Zolty. Each of Mr. Friedman and Mr. Zolty was a director of the Company at the time of the loans but resigned their positions as directors in July 2022. The loans, which are non-interest bearing and became due on May 18, 2018, remained outstanding as of December 31, 2023. As of December 31, 2023, the Company had accrued an obligation to issue warrants to purchase 49,500 and 198,000 shares, respectively, at an exercise price of $0.75 per share to the entity owned by Mr. Friedman and Mr. Zolty as a result of the Company’s failure to repay these notes by the maturity date.
In December 2018, the Company received a non-interest-bearing short-term advance in the amount of $100,000 from David Zolty, who was a director at the time of the loan. Because the short-term advance was not repaid by the Company on or before January 15, 2019, the Company was required under the terms of the advance to (i) issue to Mr. Zolty warrants to purchase 100,000 shares of common stock on such date and (ii) to further issue warrants to purchase 25,000 shares of common stock for each month that the advance remains outstanding after such date. As of December 31, 2023, the entire amount of the advance was outstanding. As of December 31, 2023, the Company was required to issue warrants to purchase an aggregate of 1,575,000 shares at an exercise price of $0.75 per share to Mr. Zolty in connection with this advance.
The Company has agreed to issue warrants to purchase 134,000 shares of common stock at an exercise price of $0.75 per share to Mr. Zolty in consideration of Mr. Zolty making a $134,000 payment to Yeda on the Company’s behalf in 2016. As of December 31, 2023, the Company had not issued the warrants to Mr. Zolty.
| 75 |
Darlene Soave was appointed to the Company’s Board of Directors effective March 25, 2021. On October 28, 2019, the Company issued a convertible note payable to Ms. Soave that has a principal amount of $6,000,000. The convertible note, as amended in October 2022, bears interest at 10% per annum and matures on October 28, 2024. Ms. Soave has the right, at her option, to convert the note into shares of the Company’s Series B Convertible Preferred Stock at a price of $7.50 per share at any time after the designation and sale of the Series B Convertible Preferred Stock. Interest accruing under the note will be payable upon the maturity of the note and may be paid at the Company’s option in either cash or shares of the Company’s common stock (calculated based upon $0.75 per share for purposes of calculating the number of shares of common stock to be issued). For each $500,000 advanced under the note, Ms. Soave will be issued a five-year warrant to purchase 400,000 shares of the Company’s common stock at an exercise price of $1.25 per share. During the years ended December 31, 2023 and 2022, the Company received no additional advances. As of December 31, 2023, $3,500,000 of principal under the convertible note was outstanding and the Company had issued warrants to purchase 2,800,000 shares of common stock at an exercise price of $1.25 per share to Ms. Soave. Ms. Soave has assigned the note to a trust for which she serves as trustee.
George Verstraete was appointed to the Company’s Board of Directors on March 10, 2022. The Company and Mr. Verstraete entered into a promissory note agreement dated March 10, 2022, whereby Mr. Verstraete, at his discretion, can loan up to $6,000,000 to the Company. Mr. Verstraete has agreed to loan an aggregate of $2,500,000 to the Company under the note. The note bears interest at a rate of 10% per annum and will mature twelve months from the date of issuance. Mr. Verstraete has the right, at his option, to convert the note into shares of the Company’s Series B Convertible Preferred Stock at a conversion price of $7.50 per share at any time after the creation and sale of the Series B Convertible Preferred Stock. Interest accruing under the note will be payable upon the maturity of the note and may be paid at the Company’s option in either cash or shares of the Company’s common stock (calculated based upon $0.75 per share for purposes of calculating the number of shares of common stock to be issued). For each $500,000 advanced under the note, Mr. Verstraete will be issued a warrant to purchase 400,000 shares of the Company’s common stock at an exercise price of $1.25 per share. Each warrant will have a five-year term. As of December 31, 2022, the Company had received $2,500,000 under the note. In connection with these advances, the Company issued five-year immediately vested warrants to purchase an aggregate of 2,000,000 shares of common stock at an exercise price $1.25 per share. The warrants had an issuance date relative fair value of $233,400 which will be amortized over the term of the note. In February 2023, the Company entered into an agreement whereby $413,018 of payments made by Mr. Verstraete to third parties on behalf of the Company in June 2022 were characterized as convertible notes payable - related parties. Additional advances of $250,000, $100,000, $150,000 and $105,000 were made in April 2023, May 2023, July 2023, and September 2023, respectively, which increased the outstanding principal balance to $3,518,018. Mr. Verstraete has assigned the note to a trust for which Darlene Soave serves as trustee. Effective May 17, 2023, the Company issued warrants to purchase 906,914 shares of common stock at an exercise price of $1.25 per share to the trust in consideration of the advances made under the note in 2023.
Each of Ms. Soave and Mr. Verstraete was issued options to purchase 375,000 shares of the Company’s common stock at an exercise price of $1.00 per share in September 2022 in connection with their service as a director of the Company. The options have terms of five (5) years.
In November 2023, a trust for which Darlene Soave serves as trustee purchased 20,000 shares of the Company’s Series B Convertible Preferred Stock and warrants to purchase 225,000 shares of common stock for a purchase price of $150,000.
In November 2023, Phyllis Friedman Investment ULC purchased 10,000 shares of the Company’s Series B Convertible Preferred Stock and warrants to purchase 112,500 shares of common stock for a purchase price of $75.000.
Director Independence
None of our directors are independent, as that term is defined under the Nasdaq Marketplace Rules.
| 76 |
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES.
The following is a summary of fees for professional services rendered by our independent registered public accounting firm for the years ended December 31, 2023 and 2022:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Audit Fees | $ | 189,000 | $ | 169,435 | ||||
| Tax Fees | - | 15,090 | ||||||
| Total | $ | 189,000 | $ | 184,525 | ||||
Audit fees represent fees for professional services performed for the audit of our annual financial statements and the review of our quarterly financial statements, as well as services that are normally provided in connection with statutory and regulatory filings or engagements.
Tax fees were for tax compliance services for the years ended December 31, 2023 and 2022.
The Audit Committee is responsible for the appointment, compensation and oversight of the work of the independent registered public accountants and approves in advance any services to be performed by the independent registered public accountants, whether audit-related or not. The Audit Committee reviews each proposed engagement to determine whether the provision of services is compatible with maintaining the independence of the independent registered public accountants. The fees shown above were pre-approved either by our Board or our Audit Committee.
| 77 |
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
Financial Statements
See Index to Financial Statements immediately following the signature page of this Annual Report.
Financial Statement Schedules
All financial statement schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
Exhibits
The following exhibits are included as part of this Annual Report:
| 78 |
| 79 |
| (1) | Incorporated by reference to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on July 1, 2014. | |
| (2) | Incorporated by reference to the Company’s Registration Statement on Form S-1 filed with the Securities and Exchange Commission on June 6, 2012. | |
| (3) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 26, 2014. | |
| (4) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 6, 2014. | |
| (5) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 19, 2014. | |
| (6) | Incorporated by reference to the Company’s Registration Statement on Form S-1 filed with the Securities and Exchange Commission on August 8, 2014. | |
| (7) | Incorporated by reference to the Company’s Registration Statement Form S-1/A filed with the Securities and Exchange Commission on September 23, 2014. | |
| (8) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 30, 2014. | |
| (9) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on December 2, 2014. | |
| (10) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on April 1, 2015. | |
| (11) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 3, 2015. |
| 80 |
| (12) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on June 10, 2015. | |
| (13) | Incorporated by reference to the Company’s Form 8-K filed with the Securities and Exchange Commission on July 28, 2015. | |
| (14) | Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 13, 2015. | |
| (15) | Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on April 14, 2016. | |
| (16) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on May 13, 2016. | |
| (17) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 15, 2016. | |
| (18) | Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on July 25, 2018. | |
| (19) | Incorporated by reference to the Company’s Form 10-K/A filed with the Securities and Exchange Commission on June 19, 2019. | |
| (20) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on May 20, 2019. | |
| (21) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 14, 2019. | |
| (22) | Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on March 30, 2020. | |
| (23) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on May 15, 2020. | |
| (24) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on August 14, 2020. | |
| (25) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on November 13, 2020. | |
| (26) | Incorporated by reference to the Company’s Form 10-K filed with the Securities and Exchange Commission on April 15, 2021. | |
| (27) | Incorporated by reference to the Company’s Form 10-Q filed by the Company with the Securities and Exchange Commission on August 12, 2021. | |
| (28) | Incorporated by reference to the Company’s Form 10-Q filed with the Securities and Exchange Commission on November 15, 2021. | |
| (29) | Incorporated by reference to the Company’s Form 10-K filed by the Company with the Securities and Exchange Commission on April 15, 2022. | |
| (30) | Incorporated by reference to the Company’s Form 10-K filed by the Company with the Securities and Exchange Commission on August 8, 2021. | |
| (31) | Incorporated by reference to the Company’s Form 10-Q filed by the Company with the Securities and Exchange Commission on November 9, 2021 | |
| (32) | Incorporated by reference to the Company’s Form 10-K filed by the Company with the Securities and Exchange Commission on August 8, 2023. | |
| * | Filed Herewith | |
| ** | Certain information has been excluded from this exhibit because (i) it is not material and (ii) would be competitively harmful if publicly disclosed. |
ITEM 16. FORM 10-K SUMMARY.
Not applicable.
| 81 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| CELL SOURCE, INC. | ||
| Dated: June 24, 2024 | By: | /s/ Itamar Shimrat |
| Name: | Itamar Shimrat | |
| Title: | Chief Executive Officer and | |
| Chief Financial Officer | ||
| (Principal Executive, Financial and Accounting Officer) | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| SIGNATURE | TITLE | DATE | |||
| By: | /s/ Dennis Brown | Chairman | June 24, 2024 | ||
| Dennis Brown | |||||
| By: | /s/ Itamar Shimrat | Chief Executive Officer and Chief Financial Officer | June 24, 2024 | ||
| Itamar Shimrat | (Principal Executive, Financial and Accounting Officer) | ||||
| By: | /s/ Darlene Soave | Director | June 24, 2024 | ||
| Darlene Soave | |||||
| By: | /s/ George Verstraete | Director | June 24, 2024 | ||
| George Verstraete | |||||
| 82 |
CELL SOURCE, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of
Cell Source, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Cell Source, Inc. and Subsidiary (the “Company”) as of December 31, 2023 and 2022, the related consolidated statements of operations, changes in stockholders’ deficiency and cash flows for each of the two years in the period ended December 31, 2023, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 2, the Company has a significant working capital deficiency, has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the below critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing separate opinions on the critical audit matter or on the accounts or disclosures to which it relates.
Valuation of Common Stock Price
As discussed in Note 3 to the consolidated financial statements, the Company obtained third-party valuations of its common stock price as of December 31, 2023 and July 1, 2023, which was used in management’s estimation of fair value of its equity instruments during the year ended December 31, 2023. The estimates used by management are considered highly complex and subjective. The independent valuations utilized the market approach, specifically the Backsolve method. The Backsolve method utilizes the economics from a direct transaction in the Company’s securities in determining fair value. The Backsolve method utilizes the Black-Scholes option pricing method (“OPM”) which allocated a probability-weighted present value to the Company’s convertible securities with the following steps being applied:
| a) | Establishment of total enterprise or equity value; | |
| b) | Analysis of equity rights for each class of security; | |
| c) | Selection of appropriate model for valuation purposes; | |
| d) | Determination of key valuation inputs; and | |
| e) | Computation of the fair value of the subject security. |
Under the OPM, it was determined the Company’s common stock fair value was $0.26 and $0.34 per share as of December 31, 2023 and July 1, 2023, respectively, which included a discount for lack of marketability of 25%. Furthermore, the independent valuations determined the Company’s expected volatility was 65% and 80% as of December 31, 2023 and July 1, 2023, respectively, by evaluating historical and implied volatilities of guideline companies.
We identified the valuation of the Company’s common stock price as a critical audit matter. The Company’s common stock price is highly subjective and requires a higher degree of auditor judgment as the Company’s common stock price was determined by using highly subjective estimates made by management. Further, specialized valuation skills were needed to assess the Company’s process and value the common stock price using the OPM.
The primary procedures we performed to address this critical audit matter included the following:
| ● | We identified and considered the relevance, reliability and sufficiency of the sources of data used by the Company in developing the assumptions used to determine the common stock price. | |
| ● | We obtained an understanding of the factors considered and assumptions made by management and the Company’s valuation specialist in developing the estimate of the common stock price, the sources of data relevant to these factors and assumptions and the procedures used to obtain the data and the methods used to calculate the estimate. | |
| ● | We obtained an understanding of the factors considered and assumptions made by management and the Company’s valuation specialist in developing the estimate of the common stock price, the sources of data relevant to these factors and assumptions and the procedures used to obtain the data and the methods used to calculate the estimate. |
/s/ Marcum llp
We have served as the Company’s auditor since 2014.
June 24, 2024
| F-2 |
CELL SOURCE, INC.
CONSOLIDATED BALANCE SHEETS
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash | $ | $ | ||||||
| Prepaid expenses | ||||||||
| Other current assets | ||||||||
| Total Assets | $ | $ | ||||||
| Liabilities and Stockholders’ Deficiency | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued expenses | ||||||||
| Accrued expenses - related parties | ||||||||
| Accrued interest | ||||||||
| Accrued interest - related parties | ||||||||
| Accrued compensation | ||||||||
| Notes payable, net of debt discount of $ and $ as of December 31, 2023 and 2022, respectively | ||||||||
| Notes payable - related parties | ||||||||
| Convertible notes payable, net of debt discount of $ and $ as of December 31, 2023 and 2022, respectively | ||||||||
| Convertible notes payable - related parties, net of debt discount of $ | ||||||||
| Derivative liabilities | ||||||||
| Financing liability | ||||||||
| Advances payable | ||||||||
| Advances payable - related party | ||||||||
| Accrued dividend payable | ||||||||
| Total Liabilities | ||||||||
| Commitments and contingencies (Note 11) | ||||||||
| Stockholders’ Deficiency: | ||||||||
| Preferred stock, $ par value, shares authorized | ||||||||
| Series A Convertible Preferred Stock, shares designated, shares issued and
outstanding as of December 31, 2023 and 2022; liquidation preference of $ | ||||||||
| Series B Convertible Preferred Stock, shares designated, and shares issued and
outstanding as of December 31, 2023 and 2022, respectively; liquidation preference of $ | ||||||||
| Series C Convertible Preferred Stock, shares designated, and shares issued
and outstanding as of December 31, 2023 and 2022, respectively; liquidation preference of $ | ||||||||
| Common stock, $ par value, shares authorized; and shares issued and outstanding as of December 31, 2023 and 2022, respectively | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total Stockholders’ Deficiency | ( | ) | ( | ) | ||||
| Total Liabilities and Stockholders’ Deficiency | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
CELL SOURCE, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Operating Expenses: | ||||||||
| Research and development | $ | $ | ||||||
| Research and development - related party | ||||||||
| Loss on legal settlement | ||||||||
| General and administrative | ||||||||
| Total Operating Expenses | ||||||||
| Loss From Operations | ( | ) | ( | ) | ||||
| Other (Expense) Income: | ||||||||
| Interest expense | ( | ) | ( | ) | ||||
| Interest expense - related parties | ( | ) | ( | ) | ||||
| Interest expense - amortization of debt discount | ( | ) | ( | ) | ||||
| Interest expense - amortization of debt discount - related party | ( | ) | ( | ) | ||||
| Change in fair value of derivative liability | ||||||||
| Gain on extinguishment of note payable | ||||||||
| Total Other Expense | ( | ) | ( | ) | ||||
| Net Loss | ( | ) | ( | ) | ||||
| Dividend attributable to Series A, Series B, and Series C preferred stockholders | ( | ) | ( | ) | ||||
| Net Loss Applicable to Common Stockholders | $ | ( | ) | $ | ( | ) | ||
| Net Loss Per Common Share - Basic and Diluted | $ | ) | $ | ) | ||||
| Weighted Average Common Shares Outstanding - | ||||||||
| Basic and Diluted | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
CELL SOURCE, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ DEFICIENCY
FOR THE YEARS ENDED DECEMBER 31, 2023 AND 2022
| Convertible Preferred | Convertible Preferred | Convertible Preferred | Additional | Total | ||||||||||||||||||||||||||||||||||||||||
| Stock - Series A | Stock - Series B | Stock - Series C | Common Stock | Paid-In | Accumulated | Stockholders’ | ||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | Deficiency | ||||||||||||||||||||||||||||||||||
| Balance, January 1, 2022 | $ | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||||||||||||||||||||
| Conversion of convertible notes payable and accrued interest into Series C Convertible Preferred Stock and common stock | - | - | ||||||||||||||||||||||||||||||||||||||||||
| Series A and C Convertible Preferred Stock dividends: | ||||||||||||||||||||||||||||||||||||||||||||
| Accrual of earned dividends | - | - | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| Payment of dividends in kind | - | - | - | |||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants in connection with issuance of convertible notes payable | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Warrants issued in satisfaction of accrued interest | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation: | ||||||||||||||||||||||||||||||||||||||||||||
| Warrants | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Options | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| Balance, December 31, 2022 | $ | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||||||||||||||||||||
| Issuance of Series B Convertible Preferred Stock and warrants for cash, net | - | - | - | |||||||||||||||||||||||||||||||||||||||||
| Conversion of convertible notes payable and accrued interest into Series C Convertible Preferred Stock and common stock | - | - | ||||||||||||||||||||||||||||||||||||||||||
| Series A, B, and C Convertible Preferred Stock dividends: | ||||||||||||||||||||||||||||||||||||||||||||
| Accrual of earned dividends | - | - | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| Payment of dividends in kind | - | - | - | |||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants in connection with issuance of convertible notes payable | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants in connection with issuance of notes payable | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock in connection with extinguishment of note payable | - | - | - | |||||||||||||||||||||||||||||||||||||||||
| Conversion of Series C Convertible Preferred Stock into common stock | - | - | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| Warrants issued in satisfaction of accrued interest | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants in connection with modification of convertible note payable | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation: | ||||||||||||||||||||||||||||||||||||||||||||
| Warrants | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||
| Common stock | - | - | - | |||||||||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| Balance, December 31, 2023 | $ | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
CELL SOURCE, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| For Years Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash Flows From Operating Activities: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Gain on extinguishment of note payable | ( | ) | ||||||
| Change in fair value of derivative liability | ( | ) | ||||||
| Loss on legal settlement | ||||||||
| Interest expense - amortization of debt discount | ||||||||
| Interest expense - amortization of debt discount - related party | ||||||||
| Non-cash interest expense - warrants | ( | ) | ||||||
| Stock-based compensation: | ||||||||
| Options | ||||||||
| Warrants | ||||||||
| Common stock | ( | ) | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses | ||||||||
| Other current assets | ( | ) | ||||||
| Accounts payable | ||||||||
| Accrued expenses | ||||||||
| Accrued expenses - related parties | ||||||||
| Accrued interest | ||||||||
| Accrued interest - related parties | ||||||||
| Accrued compensation | ||||||||
| Net Cash Used In Operating Activities | ( | ) | ( | ) | ||||
| Cash Flows From Financing Activities: | ||||||||
| Proceeds from issuance of convertible notes payable | ||||||||
| Proceeds from issuance of convertible notes payable - related party | ||||||||
| Proceeds from issuance of notes payable | ||||||||
| Repayment of notes payable | ( | ) | ||||||
| Proceeds from issuance of Series B Convertible Preferred Stock and warrants, net | ||||||||
| Repayment of financing liability | ( | ) | ( | ) | ||||
| Net Cash Provided By Financing Activities | ||||||||
| Net (Decrease) Increase In Cash | ( | ) | ||||||
| Cash - Beginning of Year | ||||||||
| Cash - End of Year | $ | $ | ||||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||
| Cash paid for: | ||||||||
| Interest | $ | $ | ||||||
| Income taxes | $ | $ | ||||||
| Non-cash investing and financing activities: | ||||||||
| Accrual of earned preferred stock dividends | $ | ( | ) | $ | ( | ) | ||
| Common stock issued in connection with payment of Series A and C Convertible Preferred Stock dividends in-kind | $ | $ | ||||||
| Financing of Directors and Officer’s insurance | $ | $ | ||||||
| Conversion of Series C Convertible Preferred Stock into common stock | $ | $ | ||||||
| Conversion of accrued expenses into note principal | $ | $ | ||||||
| Accrual of warrant obligations in connection with issuance of notes payable | $ | $ | ||||||
| Warrants issued in satisfaction of accrued interest | $ | ( | ) | $ | ||||
| Issuance of warrants in connection with the issuance of notes payable and convertible notes payable | $ | $ | ||||||
| Issuance of warrants in satisfaction of accrued interest | $ | $ | ||||||
| Conversion of convertible notes payable and accrued interest into Series C Preferred Stock and common stock | $ | $ | ||||||
| Issuance of embedded derivative liabilities in connection with issuance of note payable | $ | $ | ||||||
| Issuance of common stock in connection with extinguishment of note payable | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
CELL SOURCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 – Business Organization, Nature of Operations and Risks and Uncertainties
Organization and Operations
Cell Source, Inc. (“Cell Source”, “CSI” or the “Company”) is a Nevada corporation formed on June 6, 2012 that is the parent company of Cell Source Limited (“CSL”), a wholly owned subsidiary which was founded in Israel in 2011 in order to commercialize a suite of inventions relating to certain cancer treatments. The Company is a biotechnology company focused on developing cell therapy treatments based on the management of immune tolerance. The Company’s lead prospective product is its patented Veto Cell immune system management technology, which is an immune tolerance biotechnology that enables the selective blocking of immune responses. CSL’s Veto Cell immune system management technology is based on technologies patented, owned, and licensed to CSL by Yeda Research and Development Company Limited, an Israeli corporation (“Yeda”) (see Note 11, Commitments and Contingencies). The Company’s target indications include: lymphoma, leukemia and multiple myeloma through the facilitation of safer and more accessible stem cell (e.g. bone marrow) transplantation acceptance, treatment of end stage kidney disease and other non-malignant organ diseases through improved organ transplantation (broadened donor pool, reduced dependence on post-transplant anti-rejection therapy), and ultimately treating a variety of cancers and non-malignant diseases.
Risks and Uncertainties
On October 7, 2023, a conflict arose between Israel and Hamas militants on Israel’s southern border from the Gaza Strip. The intensity and duration of Israel’s current war against Hamas is difficult to predict, and as are such war’s economic implications on the Company’s business and operations. To the extent that any of these negative developments do occur, they may have an adverse effect on the Company’s business, results of operations and its ability to raise additional funds. As of December 31, 2023, the Company considered the impact of the war on its business and operational assumptions and estimates and determined there were no material adverse impacts on the Company’s consolidated results of operations and financial position as of and for the year ended December 31, 2023.
Note 2 – Going Concern and Management’s Plans
During
the years ended December 31, 2023 and 2022, the Company had not generated any revenues, had recurring net losses of $
The
Company is currently funding its operations on a month-to-month basis. While there can be no assurance that it will be successful, the
Company is in active negotiations to raise additional capital. The Company’s primary sources of operating funds since inception
have been equity and debt financings. Management’s plans include continued efforts to raise additional capital through debt and
equity financings. There is no assurance that these funds will be sufficient to enable the Company to fully complete its development
activities or attain profitable operations. If the Company is unable to obtain such additional financing on a timely basis or, notwithstanding
any request the Company may make, if the Company’s debt holders do not agree to convert their notes into equity or extend the maturity
dates of their notes, the Company may have to curtail its development, marketing and promotional activities, which would have a material
adverse effect on the Company’s business, financial condition and results of operations, and ultimately the Company could be forced
to discontinue its operations and liquidate. Subsequent to December 31, 2023 and as more fully described in Note 13, Subsequent Events, the Company received aggregate
proceeds of $
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”), which contemplate continuation of the Company as a going concern and the realization of assets and satisfaction of liabilities in the normal course of business. The carrying amounts of assets and liabilities presented in the financial statements do not necessarily purport to represent realizable or settlement values. The consolidated financial statements do not include any adjustment that might result from the outcome of this uncertainty.
| F-7 |
Note 3 – Summary of Significant Accounting Policies
Principles of Consolidation
The Company’s financial statements are consolidated and include the accounts of CSI and CSL. All significant intercompany transactions have been eliminated in the consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates, judgments and assumptions that affect the amounts reported in the financial statements and the amounts disclosed in the related notes to the financial statements. Management bases its estimates and judgements on historical experience and on various other assumptions that it believes are reasonable under the circumstances. The amounts of assets and liabilities reported in the Company’s balance sheets and the amounts of expenses reported for each of the periods presented are affected by estimates and assumptions, which are used for, but not limited to, fair value of the Company’s common stock price using the Backsolve method. Certain of the Company’s estimates could be affected by external conditions, including those unique to the Company and general economic conditions. It is reasonably possible that these external factors could have an effect on the Company’s estimates and could cause actual results to differ from those estimates. Estimates and assumptions are periodically reviewed and the effects of any material revisions are reflected in the consolidated financial statements in the period that they are determined to be necessary. See the Valuation of Common Stock Price section of this note for additional detail of the use of estimates in estimating the fair value of the Company’s common stock.
Cash and Cash Equivalents
The
Company considers all highly-liquid instruments with an original maturity of three months or less when purchased to be cash equivalents.
As of December 31, 2023 and 2022, the Company did
Convertible Instruments
The Company evaluates its convertible instruments to determine if those contracts or embedded components of those contracts qualify as derivative financial instruments to be separately accounted for in accordance with Topic 815 of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”). The accounting treatment of derivative financial instruments requires that the Company record embedded conversion options and any related freestanding instruments at their fair values as of the inception date of the agreement and at fair value as of each subsequent balance sheet date. Any change in fair value is recorded as non-operating, non-cash income or expense for each reporting period at each balance sheet date. The Company reassesses the classification of its derivative instruments at each balance sheet date. If the classification changes as a result of events during the period, the contract is reclassified as of the date of the event that caused the reclassification. Embedded conversion options and any related freestanding instruments are recorded as a discount to the host instrument and are amortized as interest expense over the term of the related debt instrument.
The Company primarily uses the Black-Scholes option pricing model to estimate the fair value of its warrants and embedded conversion options. The Black-Scholes option pricing model includes subjective input assumptions that can materially affect the fair value estimates.
| F-8 |
Preferred Stock
The Company applies the accounting standards for distinguishing liabilities from equity when determining the classification and measurement of its preferred stock. Preferred shares subject to mandatory redemption are classified as liability instruments and are measured at fair value. Conditionally redeemable preferred shares (including preferred shares that feature redemption rights that are either within the control of the holder or subject to redemption upon the occurrence of uncertain events not solely within the Company’s control) are classified as temporary equity. At all other times, preferred shares are classified as stockholders’ equity.
Fair Value of Financial Instruments
The Company measures the fair value of financial assets and liabilities based on ASC 820 “Fair Value Measurements and Disclosures” (“ASC 820”), which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
ASC 820 defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. ASC 820 also establishes a fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. ASC 820 describes three levels of inputs that may be used to measure fair value:
Level 1 — quoted prices in active markets for identical assets or liabilities;
Level 2 — quoted prices for similar assets and liabilities in active markets or inputs that are observable; and
Level 3 — inputs that are unobservable (for example, cash flow modeling inputs based on assumptions).
The carrying amounts of the Company’s financial instruments, such as cash, other current assets, accounts payable, accrued expenses and other current liabilities approximate fair values due to the short-term nature of these instruments. The carrying amounts of Company’s credit obligations approximate fair value because the effective yields on these obligations, which include contractual interest rates, are comparable to rates of returns for instruments of similar credit risk.
Income Taxes
CSI is the parent of CSL, a wholly owned Israeli subsidiary. The Company is subject to federal and New York state and city income taxes in the United States and federal income taxes in the State of Israel.
The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of items that have been included or excluded in the consolidated financial statements or tax returns. Deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (“temporary differences”) at enacted tax rates in effect for the years in which such temporary differences are expected to reverse.
The Company utilizes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return.
The
Company’s policy is to classify assessments, if any, for tax-related interest as interest expense and penalties as general and
administrative expenses in the consolidated statements of operations. Tax related interest and penalties of $
| F-9 |
Research and Development
Research and development expenses are recognized to operations as they are incurred and consist of fees paid to academic institutions (for sponsored research), consultants, hospitals for clinical trials and related clinical manufacturing costs, as well as license fees to the owners of the licensed intellectual property and milestone payments based on the number of patients treated in clinical trials. The Company records prepaid expenses on its consolidated balance sheets for the payment of research and development expenses in advance of services being provided. As of December 31, 2023 and 2022, the Company did not have any research and development expenses that were prepaid or capitalized.
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award. The fair value of the award is measured on the grant date and is then recognized over the period the services are required to be provided in exchange for the award, usually the vesting period. Upon the exercise of an option or warrant, the Company issues new shares of common stock out of its authorized shares.
Because the Company’s common stock historically was not actively traded on a public market, the fair value of the Company’s restricted equity instruments is estimated by management based on observations of the sales prices of both restricted and freely tradable common stock, or instruments convertible into common stock. The Company obtained a third-party valuation of its common stock as of December 31, 2023, July 1, 2023 and December 31, 2022, which was considered in management’s estimation of fair value during the years ended December 31, 2023 and 2022. The third-party valuation was performed in accordance with regulation of Section 409A of the Internal Revenue Code (“IRC”) as well as FASB ASC Topic 718. The estimates used by management are considered highly complex and subjective. The Company anticipates that once its shares become more actively traded, the use of such estimates will no longer be necessary to determine the fair value of its common stock.
The independent appraisal utilized the market approach, specifically the Backsolve method. The Backsolve method utilizes the economics from a direct transaction in the Company’s securities in determining fair value. The Backsolve method utilizes the Black-Scholes option pricing method (“OPM”) which allocated a probability-weighted present value to the Company’s convertible securities. The following steps were applied under the OPM:
| ● | Establishment of total enterprise or equity value; | |
| ● | Analysis of equity rights for each class of security; | |
| ● | Selection of appropriate model for valuation purposes; | |
| ● | Determination of key valuation inputs; and | |
| ● | Computation of the fair value of the subject security. |
Under
the OPM, it was determined the Company’s common stock had a fair value of $, $ and $ per share as of December 31, 2023,
July 1, 2023 and December 31, 2022, respectively, which included a discount for lack of marketability of
Foreign Currency Translation
The Company’s functional and reporting currency is the United States Dollar. The functional currency of the Company’s operating subsidiary is their local currency (The New Israeli Shekel). Assets and liabilities are translated based on the exchange rates at the balance sheet date, while revenue and expense accounts are translated at the actual exchange rates in the effect of the date of the transaction during the year. Equity accounts are translated at historical exchange rates. The resulting translation gain and loss adjustments are accumulated as a component of other comprehensive income. Foreign currency gains and losses resulting from transactions denominated in foreign currencies, including intercompany transactions, are included in results of operations. The translation gains and losses for the years ended December 31, 2023 and 2022 are immaterial.
| F-10 |
The Company computes basic net loss per share by dividing net loss by the weighted average number of common shares outstanding for the period and excludes the effects of any potentially dilutive securities. Diluted earnings per share includes the dilution that would occur upon the exercise or conversion of all dilutive securities into common stock using the “treasury stock” and/or “if converted” methods, as applicable.
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Options | ||||||||
| Warrants | ||||||||
| Convertible notes [1] [2] | ||||||||
| Convertible preferred stock | ||||||||
| Total | ||||||||
| [1] | Convertible
notes are assumed to be converted at the rate of $
|
| [2] | As of December 31, 2022, excludes shares of common stock underlying convertible notes that are expected to become convertible into shares of Series B Convertible Preferred Stock since such stock had not been designated by the Company as of December 31, 2022. |
Comprehensive Income (Loss)
The
Company reports comprehensive income (loss) and its components in its consolidated financial statements. Comprehensive income (loss)
consists of net loss and foreign currency translation adjustments affecting stockholders’ deficiency that, under U.S. GAAP, are
excluded from net loss. The differences between net loss as reported and comprehensive income (loss) are immaterial. As of December 31,
2023, the exchange rate between the New Israeli Shekel and the U.S. Dollar was
Sequencing Policy
As a result of the issuance of a convertible note payable on December 29, 2023 that is convertible into the Company’s common stock at a variable conversion price with no floor (see Note 8, Notes Payable for details), the Company adopted a sequencing policy under ASC 815-40-35, whereby, in the event that reclassification of contracts from equity to assets or liabilities is necessary pursuant to ASC 815 due to the Company’s inability to demonstrate it has sufficient authorized shares as a result of certain securities with a potentially indeterminable number of shares, shares will be allocated on the basis of the earliest issuance date of potentially dilutive instruments, with the earliest grants receiving the first allocation of shares. Pursuant to ASC 815, issuance of securities to the Company’s employees or directors are not subject to the sequencing policy.
| F-11 |
Subsequent Events
The Company evaluates events that have occurred after the balance sheet date but through the date these consolidated financial statements are issued. Based upon that evaluation, the Company did not identify any recognized or non-recognized subsequent events that would have required adjustment or disclosure in the consolidated financial statements except as disclosed in Note 13, Subsequent Events.
Recent Accounting Standards
In June 2022, the FASB issued ASU 2022-03, Fair Value Measurement (Topic 820), “Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions” which clarifies that contractual restrictions on equity security sales are not considered part of the security unit of account and, therefore, are not considered in measuring fair value. In addition, the restrictions cannot be recognized and measured as separate units of account. Disclosures on such restrictions are also required. The amendments are effective for fiscal years beginning after December 15, 2023, including interim periods within those fiscal years, and are required to be applied prospectively, with any adjustments from the adoption recognized in earnings and disclosed. The adoption of ASU 2022-03 on January 1, 2024 did not have a material impact on the Company’s financial position, results of operations, and cash flows.
In July 2023, the FASB issued ASU 2023-03 to amend various SEC paragraphs in the Accounting Standards Codification to primarily reflect the issuance of SEC Staff Accounting Bulletin No. 120. ASU No. 2023-03, “Presentation of Financial Statements (Topic 205), Income Statement—Reporting Comprehensive Income (Topic 220), Distinguishing Liabilities from Equity (Topic 480), Equity (Topic 505), and Compensation—Stock Compensation (Topic 718): Amendments to SEC Paragraphs Pursuant to SEC Staff Accounting Bulletin No. 120, SEC Staff Announcement at the March 24, 2022 EITF Meeting, and Staff Accounting Bulletin Topic 6.B, Accounting Series Release 280—General Revision of Regulation S-X: Income or Loss Applicable to Common Stock.” ASU 2023-03 amends the ASC for SEC updates pursuant to SEC Staff Accounting Bulletin No. 120; SEC Staff Announcement at the March 24, 2022 Emerging Issues Task Force (“EITF”) Meeting; and Staff Accounting Bulletin Topic 6.B, Accounting Series Release 280 - General Revision of Regulation S-X: Income or Loss Applicable to Common Stock. These updates were immediately effective and did not have a significant impact on the Company’s consolidated financial statements.
In October 2023, the FASB issued ASU 2023-06, “Disclosure Improvements: Codification Amendments in Response to the SEC’s Disclosure Update and Simplification Initiative,” to amend certain disclosure and presentation requirements for a variety of topics within the ASC. These amendments align the requirements in the ASC to the removal of certain disclosure requirements set out in Regulation S-X and Regulation S-K, announced by the SEC. The effective date for each amended topic in the ASC is either the date on which the SEC’s removal of the related disclosure requirement from Regulation S-X or Regulation S-K becomes effective, or on June 30, 2027, if the SEC has not removed the requirements by that date. Early adoption is prohibited. The Company is currently evaluating the impact of this standard but does not expect it to have a material impact on its consolidated financial statements.
In November 2023, the FASB issued ASU 2023-07, Improvements to Reportable Segments Disclosures (Topic 280), which updates reportable segment disclosure requirements, primarily through enhanced disclosures about significant segment expenses on both an annual and interim basis. The guidance becomes effective for fiscal years beginning after December 15, 2023 and interim periods within fiscal years beginning after December 15, 2024, with early adoption permitted. Since this new ASU addresses only disclosures, the Company does not expect the adoption of this ASU to have any material effects on its financial condition, results of operations or cash flows. The Company is currently evaluating any new disclosures that may be required upon adoption of ASU 2023-07.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures. The amendments in this update address investor requests for more transparency about income tax information through improvements to income tax disclosures primarily related to the rate reconciliation and income taxes paid information. This update also includes certain other amendments to improve the effectiveness of income tax disclosures. The amendments in ASU 2023-09 are effective for fiscal years beginning after December 15, 2024 with early adoption permitted. The Company is currently evaluating the impact of this standard but does not expect it to have a material impact on its consolidated financial statements.
| F-12 |
Note 4 – Fair Value
The following table provides a summary of the changes in fair value, including net transfers in and/or out, of all of the Company’s instruments recorded at fair value, which were all Level 3 liabilities measured at fair value on a recurring basis using unobservable inputs during the years ended December 31, 2023 and 2022:
| Accrued | Accrued | Derivative | ||||||||||||||
| Interest | Compensation | Liability | Total | |||||||||||||
| Balance - January 1, 2022 | $ | $ | $ | $ | ||||||||||||
| Accrual of warrant obligation | ||||||||||||||||
| Issuance of warrants | ( | ) | ( | ) | ||||||||||||
| Change in fair value | ( | ) | ||||||||||||||
| Balance - December 31, 2022 | $ | $ | $ | $ | ||||||||||||
| Accrual of warrant obligation | ||||||||||||||||
| Accrual of common stock obligation | ||||||||||||||||
| Issuance of warrants | ( | ) | ( | ) | ||||||||||||
| Issuance of warrants and conversion option | ||||||||||||||||
| Change in fair value | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Balance - December 31, 2023 | $ | $ | $ | $ | ||||||||||||
See Note 6, Accrued Compensation, Note 8, Notes Payable, Note 10, Stockholders’ Deficiency, and Note 11, Commitments and Contingencies for additional details.
Financial assets are considered Level 3 when their fair values are determined using pricing models, discounted cash flow methodologies or similar techniques and at least one significant model assumption or input is unobservable. The Company’s Level 3 liabilities shown in the above table consist of warrants with “down-round protection”, as the Company is unable to determine if it will have sufficient authorized common stock to settle such arrangements, warrants deemed to be derivative liabilities according to the Company’s sequencing policy in accordance with ASC 815-40-35-12, the embedded conversion options within its convertible notes payable and an accrued obligation to issue warrants and common stock.
In applying the Black-Scholes option pricing model utilized in the valuation of Level 3 liabilities, the Company used the following approximate assumptions:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Risk-free interest rate | % - | % | % - | % | ||||
| Expected term (years) | - | - | ||||||
| Expected volatility | %- | % | % - | % | ||||
| Expected dividends | % | % | ||||||
The expected term used is the contractual life of the instrument being valued. Since the Company’s stock has not been publicly traded for a sufficiently long period of time or with significant volume, the Company is utilizing an expected volatility based on a review of the historical volatilities, over a period of time, equivalent to the expected life of the instrument being valued, of similarly positioned public companies within its industry. The risk-free interest rate was determined from the implied yields from U.S. Treasury zero-coupon bonds with a remaining term consistent with the expected term of the instrument being valued.
As
of December 31, 2023 and 2022, the Company had an obligation to issue and shares of common stock, respectively, to certain
service providers and as a legal settlement to a certain holder of the Company’s common stock that had a fair value of $
| F-13 |
Note 5 – Accrued Expenses
Accrued expenses consisted of the following:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Accrued other professional fees | $ | $ | ||||||
| Accrued legal fees | ||||||||
| Accrued research and development | ||||||||
| Other accrued expenses | ||||||||
| Accrued director compensation | ||||||||
| Accrued third-party payments | ||||||||
| Total accrued expenses | $ | $ | ||||||
See Note 11, Commitments and Contingencies for details on accrued expenses - related parties.
Note 6 – Accrued Compensation
Accrued compensation consisted of the following:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Pension insurance | $ | $ | ||||||
| Severance | ||||||||
| Witholding tax | ||||||||
| Accrued payroll | ||||||||
| Vacation | ||||||||
| Stock-based compensation - common stock | ||||||||
| Social security | ||||||||
| Stock-based compensation - warrants | ||||||||
| Total accrued compensation | $ | $ | ||||||
Note 7 – Advances Payable
Advances payable and advances payable – related party represent cash received from lenders for working capital purposes. See Note 12, Related Party Transactions.
During
the years ended December 31, 2023 and 2022, the Company did
| F-14 |
Note 8 – Notes Payable
The Company has a variety of outstanding debt instruments consisting of: a) notes payable, b) notes payable to related parties, c) convertible notes payable, and d) convertible notes payable to related parties. The notes within each of those groups are described in the sections below.
As
of December 31, 2023 and through the date of this filing, notes payable with principal amounts totaling $
During
the years ended December 31, 2023 and 2022, the Company recorded interest expense of $
a) Notes payable consist of the following:
| December 31, | ||||||||||||||||
| Issuance Date | Interest Rate | Maturity Date | 2023 | 2022 | ||||||||||||
| March 26, 2015 | % | Past Due | $ | $ | ||||||||||||
| May 15, 2015 | % | Past Due | ||||||||||||||
| May 10, 2016 | % | Past Due | ||||||||||||||
| July 20, 2016 - October 13, 2016 | % | Past Due | ||||||||||||||
| i) | February 26, 2018 | % | Past Due | |||||||||||||
| ii) | June 16, 2022 | % | Past Due | |||||||||||||
| iii) | December 18, 2023 | % | ||||||||||||||
| Total principal outstanding | $ | $ | ||||||||||||||
| Less: unamortized debt discount | ( | ) | ||||||||||||||
| Notes payable, net | $ | $ | ||||||||||||||
Details regarding certain of these notes are as follows (which numbering corresponds to the above table):
| i) |
During
the year ended December 31, 2022, the Company paid $ |
| ii) | |
| iii) |
| F-15 |
b) Notes payable due to related parties consist of the following:
| December 31, | ||||||||||||||
| Issuance Date | Interest Rate | Maturity Date | 2023 | 2022 | ||||||||||
| November 26, 2014 | % | Past Due | $ | $ | ||||||||||
| July 20, 2015 | % | Past Due | ||||||||||||
| Total principal outstanding | $ | $ | ||||||||||||
| Less: unamortized debt discount | ||||||||||||||
| Notes payable - related parties, net | $ | $ | ||||||||||||
c) Convertible notes payable consist of the following:
| December 31, | ||||||||||||||||||||||
| Issuance Date | Interest Rate | Maturity Date | Conversion Price | Convertible Into | 2023 | 2022 | ||||||||||||||||
| July 24, 2015 | % | Past Due | $ | Common Stock | $ | $ | ||||||||||||||||
| October 7, 2015 | % | Past Due | $ | Common Stock | ||||||||||||||||||
| May 18, 2017 | % | Past Due | $ | Series A Convertible Preferred Stock | ||||||||||||||||||
| i) | August 5, 2022 - December 29, 2022 | % | $ | Common Stock & Series B Convertible Preferred Stock | ||||||||||||||||||
| ii) | May 8, 2023 - June 27, 2023 | % | Past Due | $ | Common Stock | |||||||||||||||||
| ii) | July 31, 2023 - August 22, 2023 | % | $ | Common Stock & Series C Convertible Preferred Stock | ||||||||||||||||||
| iii) | December 29, 2023 | % | $ | Common Stock | ||||||||||||||||||
| Total principal outstanding | $ | $ | ||||||||||||||||||||
| Less: unamortized debt discount | ( | ) | ( | ) | ||||||||||||||||||
| Convertible notes payable, net | $ | $ | ||||||||||||||||||||
Details regarding certain of these notes are as follows (which numbering corresponds to the above table):
| i) |
During
the year ended December 31, 2022, the Company issued convertible note payable in the aggregate principal amount of $
On
November 29, 2023, the Company and a certain investor agreed to extend the maturity date of a certain convertible note payable in
the principal amount of $
In connection with the note
amendment, the Company agreed to issue the investor
During
the years ended December 31, 2023 and 2022, an aggregate principal amount of $ |
| F-16 |
| ii) |
During
the year ended December 31, 2023, the Company issued convertible notes payable in the aggregate principal amount of $ |
| iii) |
During
the year ended December 31, 2022, an aggregate of $
During
the year ended December 31, 2023, $
| F-17 |
d) Convertible notes payable due to related parties consist of the following:
| December 31, | ||||||||||||||||||||||
| Issuance Date | Interest Rate | Maturity Date | Conversion Price | Convertible Into | 2023 | 2022 | ||||||||||||||||
| i) | May 18, 2017 | % | Past Due | $ | Series B Convertible Preferred Stock | $ | $ | |||||||||||||||
| ii) | October 28, 2019 | $ | Common Stock & Series B Convertible Preferred Stock | |||||||||||||||||||
| iii) | March 10, 2022 | % | $ | Common Stock & Series B Convertible Preferred Stock | ||||||||||||||||||
| Total principal outstanding | $ | $ | ||||||||||||||||||||
| Less unamortized debt discount | ( | ) | ||||||||||||||||||||
| Convertible notes payable - related parties, net | $ | $ | ||||||||||||||||||||
| i) | |
| ii) |
As
of December 31, 2023 and 2022, there was an aggregate principal amount of $ |
| iii) |
| F-18 |
| In
February 2023, $ | |
| The
Company received additional advances of $ | |
Mr. Verstraete has assigned the Verstraete Note to a trust for which Darlene Soave, a director of the Company, serves as trustee. On March 10, 2023, the Company and the trust agreed to extend the maturity date of the Verstraete Note to September 10, 2023. On November 8, 2023, the Company and the trust agreed to extend the maturity date of the Verstraete Note to March 10, 2024.
Subsequent to December 31, 2023, the Company and the trust agreed to extend the maturity date of the promissory note to September 10, 2024. See Note 13, Subsequent Events, for additional details regarding the note extension. | |
| The Series B Convertible Preferred Stock had been designated by the Board on September 21, 2023 and the Company sold shares of its Series B Convertible Preferred Stock. The Company adopted ASU 2020-06 effective January 1, 2022 which eliminated the need to assess whether a beneficial conversion feature needed to be recognized upon either (a) the future issuance of new convertible notes; or (b) the resolution of any contingent beneficial conversion features. As a result, the Company did not evaluate for a beneficial conversion feature underlying the Series B Convertible Notes and Series B Convertible Preferred Stock. |
Note 9 – Income Taxes
The loss before income taxes consists of the following US and Israeli components:
| For The Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| United States | $ | ( | ) | $ | ( | ) | ||
| Israel | ( | ) | ( | ) | ||||
| Loss before income taxes | $ | ( | ) | $ | ( | ) | ||
The provision for income taxes consists of the following expenses (benefits):
| For The Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Deferred | ||||||||
| Federal | $ | ( | ) | $ | ( | ) | ||
| Foreign | ( | ) | ||||||
| U.S. State and local | ( | ) | ( | ) | ||||
| ( | ) | ( | ) | |||||
| Change in valuation allowance | ||||||||
| Provision for income taxes | $ | $ | ||||||
| F-19 |
The provision for income taxes differs from the United States federal statutory rate as follows:
| For The Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Expected federal statutory rate | ( | )% | ( | )% | ||||
| State and local taxes, net of federal tax benefit | ( | )% | ( | )% | ||||
| Statutory rate differential - domestic vs. foreign | ( | )% | ( | )% | ||||
| Permanent differences | % | % | ||||||
| Prior period true-up and other | % | % | ||||||
| Change in valuation allowance | % | % | ||||||
| Income tax provision (benefit) | % | % | ||||||
Deferred tax assets consist of the following:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Net operating loss carryforwards | $ | $ | ||||||
| Stock-based compensation | ||||||||
| Accrued expenses | ||||||||
| Deferred research and development expenses | ||||||||
| Deferred tax assets | ||||||||
| Valuation allowance | ( | ) | ( | ) | ||||
| Deferred tax assets, net | $ | $ | ||||||
As
of December 31, 2023 and 2022, the Company had approximately $
The
Company has assessed the likelihood that deferred tax assets will be realized in accordance with the provisions of ASC 740 Income
Taxes (“ASC 740”). ASC 740 requires that such a review considers all available positive and negative evidence, including
the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies. ASC 740 requires that
a valuation allowance be established when it is “more likely than not” that all, or a portion of, deferred tax assets will
not be realized. After the performance of such reviews as of December 31, 2023 and 2022, management believes that uncertainty exists
with respect to future realization of its deferred tax assets and has, therefore, established a full valuation allowance as of those
dates. Thus, the Company increased the valuation allowance by approximately $
Management
has evaluated and concluded that there were
| F-20 |
The Company’s 2021 domestic federal tax returns remain subject to audit, while the Company’s 2023 and 2022 domestic federal tax returns have not been filed yet. The Company’s Israeli federal tax returns remain subject to audit, beginning with the 2014 tax returns. No tax audits were commenced or were in process during the years ended December 31, 2023 and 2022.
Note 10 – Stockholders’ Deficiency
Authorized Capital
Effective July 26, 2023, the Company amended the certificates of designation which established the Series A Convertible Preferred Stock and Series C Convertible Preferred Stock to increase the number of shares designated from to shares for the Series A Convertible Preferred Stock and from to shares for the Series C Convertible Preferred Stock.
On September 21, 2023, the Company’s Board of Directors approved the designation of shares of the authorized shares of preferred stock as Series B Convertible Preferred Stock, par value $ per share.
As
of December 31, 2023, the Company was authorized to issue shares of common stock, par value of $ per share, and
shares of preferred stock, par value of $ per share. The holders of the Company’s common stock are entitled
to
Common Stock
During the year ended December 31, 2023, certain investors converted an aggregate of shares of Series C Convertible Preferred Stock into an aggregate of shares of the Company’s common stock.
During the year ended December 31, 2023, the Company issued an aggregate of immediately-vested shares of the Company’s common stock to consultants with a grant date fair value of $ which was immediately recognized in the consolidated statement of operations.
During the years ended December 31, 2023 and 2022, the Company issued and shares of common stock, respectively, in connection with the conversion of convertible notes payable and accrued interest. See Note 8, Notes Payable – Convertible Notes Payable for additional details.
See Note 8, Notes Payable for details associated with the issuance of shares of common stock with an issuance date fair value of $ in connection with the extinguishment of a note payable during the year ended December 31, 2023.
See elsewhere in Note 10, Stockholders’ Deficiency for details of issuances of common stock in connection with accrued dividends.
Series A Convertible Preferred Stock
The Series A Convertible Preferred Stock has a stated value of $ per share. The Series A Convertible Preferred Stock contains the following terms:
Conversion.
Each share of Series A Convertible Preferred Stock is convertible into shares of common stock (subject to adjustment as provided
in the related certificate of designation of preferences, rights and limitations) at the option of the holder at any time. The number
of shares of common stock which are issuable upon conversion of the Series A Convertible Preferred Stock shall be equal to the number
of shares of Series A Convertible Preferred Stock to be converted, multiplied by the stated value of $ per share, divided by the
conversion price in effect at the time of conversion, initially at $
Mandatory Conversion. Series A Preferred Stock will automatically convert into common stock at the earlier of (a) any of the Company’s treatment candidates receiving Food and Drug Administration or European Medicines Agency approval; or (b) five years from the final closing of the offering.
| F-21 |
Liquidation Preference. Upon any liquidation, dissolution or winding-up of the Company, the holders of Series A Preferred Stock will be entitled to be paid for each share of Series A Preferred Stock held thereby, but only to the extent the assets of the Company are legally available for distribution to its stockholders, in an amount equal to the stated value per share plus any accrued but unpaid dividends, before any distribution or payment may be made to the holders of any junior securities.
Voting
Rights.
Dividends.
During
the year ended December 31, 2023, the Company accrued additional preferred dividends of $
During
the year ended December 31, 2022, the Company accrued additional preferred dividends of $
Series B Convertible Preferred Stock
On October 30, 2023, the Company filed the Certificate of Designation with the Office of the Secretary of State for the State of Nevada, which established the Series B Convertible Preferred Stock. The Series B Convertible Preferred Stock has a stated value of $ per share. The Series B Convertible Preferred Stock contains the following terms:
Conversion.
Each share of Series B Convertible Preferred Stock is convertible into shares of common stock (subject to adjustment as provided
in the related certificate of designation of preferences, rights and limitations) at the option of the holder at any time. The number
of shares of common stock which are issuable upon conversion of the Series B Convertible Preferred Stock shall be equal to the number
of shares of Series B Convertible Preferred Stock to be converted, multiplied by the stated value of $ per share, divided by the
conversion price in effect at the time of conversion, initially at $
Mandatory Conversion. On the earlier of (i) October 30, 2027 or (ii) any of the Company’s treatment candidates receiving approval from the U.S. or European agencies, all of the outstanding shares of Series B Convertible Preferred Stock will automatically convert to common stock.
Liquidation Preference. In the event of the liquidation, dissolution or winding-up of the Company, the holders of Series B Preferred Stock will be entitled to be paid for each share of Series B Preferred Stock held thereby, but only to the extent the assets of the Company are legally available for distribution to its stockholders, in an amount equal to the stated value per share plus any accrued but unpaid dividends. The Series B Convertible Preferred Stock will rank senior to common stock and any other class of capital stock which does not expressly rank senior to or pari passu with the Series B Preferred Stock and will rank pari passu with the Series A and Series C Convertible Preferred Stock.
Voting
Rights.
| F-22 |
Dividends.
The Company determined that the Series B Convertible Preferred Stock was perpetual preferred stock.
Beginning
in October 2023, the Company entered into subscription agreements with certain accredited investors in a private placement offering.
Each unit, which is sold at a price of $ per unit, consists of one (1) share of Series B Convertible Preferred Stock and a five-year
warrant to purchase a certain number of shares of common stock at an exercise price of $
During
the year ended December 31, 2023, the Company sold an aggregate of units to certain investors for net proceeds of $
During
the year ended December 31, 2023, the Company accrued preferred dividends of $
Series C Convertible Preferred Stock
The Series C Convertible Preferred Stock has a stated value of $ per share. The Series C Convertible Preferred Stock contains the following terms:
Conversion.
Each share of Series C Preferred Stock is convertible into shares of common stock (subject to adjustment as provided in the related
certificate of designation of preferences, rights and limitations) at the option of the holder at any time. The number of shares of common
stock which are issuable upon conversion of the Series C Preferred Stock shall be equal to the number of shares of Series C Preferred
Stock to be converted, multiplied by the stated value of $ per share, divided by the conversion price in effect at the time of conversion,
initially at $
Mandatory Conversion. On the earlier of (i) July 27, 2025 or (ii) any of the Company’s treatment candidates receiving approval from the U.S. or European agencies, all of the outstanding shares of Series C Preferred Stock will automatically convert to common stock.
Liquidation Preference. In the event of the liquidation, dissolution or winding-up of the Company, the holders of Series B Preferred Stock will be entitled to be paid for each share of Series C Preferred Stock held thereby, but only to the extent the assets of the Company are legally available for distribution to its stockholders, in an amount equal to the stated value per share plus any accrued but unpaid dividends. The Series C Preferred Stock will rank senior to common stock and any other class of capital stock which does not expressly rank senior to or pari passu with the Series C Preferred Stock and will rank pari passu with the Series A and Series B Preferred Stock.
Voting
Rights.
Dividends.
| F-23 |
During the year ended December 31, 2023, shares of Series C Preferred Stock were converted into shares of common stock at the shareholder’s election.
During
the years ended December 31, 2023 and 2022, the Company accrued additional preferred dividends of $
During the years ended December 31, 2023 and 2022, the Company issued and shares of Series C Convertible Preferred Stock, respectively, in connection with conversions of notes payable into Series C Convertible Preferred Stock. See Note 8, Notes Payable – Convertible Notes Payable for additional details.
Equity Incentive Plan
On August 13, 2019, the Company’s Board of Directors approved the adoption of the Company’s 2019 Equity Incentive Plan (the “Plan”). A total of shares of common stock were initially reserved for issuance under the Plan and the number of reserved shares increases on the first day of each year in an amount equal to the lesser of % of the number of shares of common stock outstanding on the last day of the preceding year or the amount determined by our Board of Directors. The Plan permits the Board of Directors to issue stock options, stock appreciation rights, restricted stock, restricted stock units, performance and other awards to employees, consultants and directors of the Company. As of December 31, 2023, a total of shares of common stock were reserved for issuance under the Plan. Of this amount, as of December 31, 2023, a total of shares were available for future issuance under the Plan. As of the date of filing, the Company’s shareholders have not approved the Plan.
Stock-Based Compensation
During
the year ended December 31, 2023, the Company recognized stock-based compensation expense of $ (consisting of $ expense
related to warrants (of which, $ has been included within stockholder’s deficiency and $(
During the year ended December 31, 2023, $ of stock-based compensation expense was included within general and administrative expenses and $ was included within research and development on the consolidated statement of operations. During the year ended December 31, 2022, $ of stock-based compensation expense was included within general and administrative expenses and $ was included within research and development on the consolidated statement of operations. As of December 31, 2023, there was $ of unrecognized stock-based compensation expense which will be recognized over a weighted average remaining amortization period of years.
| F-24 |
Stock Warrants
On
May 25, 2023, the Company issued immediately vested -year warrants to an investor to purchase an aggregate amount of
On
August 9, 2023, the Company issued immediately vested four-year warrants to a former director of the Company to purchase an aggregate
amount of
On
December 15, 2023, the Company entered into an advisory agreement for a term of December 15, 2023 to March 15, 2024. As compensation
for services performed, the Company issued a -year warrant which vests one-third each month to the advisor to purchase an aggregate
amount of
The
Company entered into an advisory agreement for a term of December 15, 2023 to May 15, 2024. As compensation for services performed, the
Company issued a -year warrant to purchase an aggregate amount of
On
December 29, 2023, the Company issued an immediately vested five-year warrant to purchase
During
the years ended December 31, 2023 and 2022, the Company issued warrants to purchase an aggregate of
See Note 4, Fair Value and Note 11, Commitments and Contingencies for additional details.
| F-25 |
A summary of the warrant activity during the year ended December 31, 2023 is presented below:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | |||||||||||||||
| Number of | Exercise | Life | Intrinsic | |||||||||||||
| Warrants | Price | In Years | Value | |||||||||||||
| Outstanding, January 1, 2023 | $ | |||||||||||||||
| Issued | ||||||||||||||||
| Expired/Canceled | ( | ) | ||||||||||||||
| Outstanding, December 31, 2023 | $ | $ | ||||||||||||||
| Exercisable, December 31, 2023 | $ | $ | ||||||||||||||
| Warrants Outstanding | Warrants Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Outstanding | Average | Exercisable | ||||||||||||
| Exercise | Number of | Remaining Life | Number of | |||||||||||
| Price | Warrants | In Years | Warrants | |||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
Stock Options
On
September 13, 2022, the Company granted an aggregate of
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Risk-free interest rate | N/A | % | ||||||
| Expected term (years) | N/A | |||||||
| Expected volatility | N/A | % | ||||||
| Expected dividends | N/A | % | ||||||
The expected term used is the contractual life of the instrument being valued. Since the Company’s stock has not been publicly traded for a sufficiently long period of time or with significant volume, the Company is utilizing an expected volatility based on a review of the historical volatilities, over a period of time, equivalent to the expected life of the instrument being valued, of similarly positioned public companies within its industry. The risk-free interest rate was determined from the implied yields from U.S. Treasury zero-coupon bonds with a remaining term consistent with the expected term of the instrument being valued.
| F-26 |
There were no stock options granted during the year ended December 31, 2023. The weighted average estimated grant date fair value of the stock options granted during the year ended December 31, 2022 was approximately $ per share, respectively.
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | |||||||||||||||
| Number of | Exercise | Life | Intrinsic | |||||||||||||
| Options | Price | In Years | Value | |||||||||||||
| Outstanding, January 1, 2023 | $ | |||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Forfeited/cancelled | ||||||||||||||||
| Outstanding, December 31, 2023 | $ | $ | ||||||||||||||
| Exercisable, December 31, 2023 | $ | $ | ||||||||||||||
| Options Outstanding | Options Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Outstanding | Average | Exercisable | ||||||||||||
| Exercise | Number of | Remaining Life | Number of | |||||||||||
| Price | Option | In Years | Options | |||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
Note 11 – Commitments and Contingencies
Yeda Research and License Agreement
On
October 3, 2011, the Company entered into a Research and License Agreement (the “Agreement”) with Yeda Research and Development
Company Limited (“Yeda”) for Veto Cell technology and an exclusive option to negotiate an additional license for organ regeneration
technology. Yeda is the technology transfer and commercial arm of the Weizmann Institute, for research conducted at the Weizmann Institute
for an invention comprising methods of bone marrow transplantation and cell therapy utilizing Veto Cells. As Yeda is a founder and a
significant shareholder of the Company, it is a related party. If the Company fails to achieve any of the milestones by the dates set
forth in the Agreement, Yeda is entitled to terminate the license upon written notice to the Company. In December, 2021, the Company
and Yeda amended the agreement to extend the next milestone to January 1, 2025. To date, the Company has been deemed to have met all
of the milestones. Either Yeda or the Company may terminate the agreement and the license after the commitment of a material breach by
the other party and in certain other instances as detailed in the agreement. The Company paid Yeda a total of $
During
the years ended December 31, 2023 and 2022, the Company recorded research and development expenses of $
| F-27 |
MD Anderson Sponsored Research Agreements
In
October 2021, the Sponsored Research Agreement with The University of Texas M.D. Anderson Cancer Center (“MD Anderson”) dated
November 2018 was amended to extend the agreement by one year to November 27, 2022 and define the budget during such one-year period
to be approximately $
The
Company recognized $
Litigation
Certain conditions may exist as of the date the consolidated financial statements are issued, which may result in a loss to the Company, but which will only be resolved when one or more future events occur or fail to occur. The Company assesses such contingent liabilities, and such assessment inherently involves an exercise of judgment. In assessing loss contingencies related to legal proceedings that are pending against the Company, or unasserted claims that may result in such proceedings, the Company evaluates the perceived merits of any legal proceedings or unasserted claims, as well as the perceived merits of the amount of relief sought or expected to be sought therein.
If the assessment of a contingency indicates that it is probable that a material loss has been incurred and the amount of the liability can be estimated, then the estimated liability would be accrued in the Company’s consolidated financial statements. If the assessment indicates that a potential material loss contingency is not probable, but is reasonably possible, or is probable but cannot be estimated, then the nature of the contingent liability and an estimate of the range of possible losses, if determinable and material, would be disclosed.
In January 2019, the holder of a promissory note in the principal amount of $
| F-28 |
In
August 2022, a holder of
shares of the Company’s common stock filed a complaint against the Company, its President and legal counsel in the United
States District Court, Southern District of New York, claiming unspecified damages for an alleged wrongful refusal to authorize the
Company’s transfer agent to remove restrictive legends from the shares held by the shareholder. The complaints against the
Company’s legal counsel and President were dismissed by the Court. Effective December 29, 2023, the parties reached a
settlement agreement whereby the Company, in exchange for the dismissal with prejudice of the claims made by the plaintiff against
the Company, agreed to (i) cause the Company’s transfer agent to remove the restrictive legend on the shares held by the
plaintiff and (ii) issue the following securities to the plaintiff:
shares of its common stock (which was issued subsequent to December 31, 2023), a warrant to purchase
See Note 8, Notes Payable for details associated with the issuance of the convertible note payable in connection with the settlement.
Loss contingencies considered remote are generally not disclosed, unless they involve guarantees, in which case the guarantees would be disclosed. There can be no assurance that such matters will not materially and adversely affect the Company’s business, financial position, and results of operations or cash flows. Aside from the matters discussed above, there are no other known contingencies through the date of this filing.
Note 12 – Related Party Transactions
See Note 8, Notes Payable for details associated with the issuance of convertible notes payable, the Soave Note and the Verstraete Note, to directors of the Company.
As
of December 31, 2023 and 2022, the Company was required to issue warrants to purchase an aggregate of
Note 13 – Subsequent Events
The Company has evaluated events that have occurred after the balance sheet and through the date the financial statements were issued. Based upon the evaluation, the Company did not identify any recognized or non-recognized subsequent events that would have required adjustment or disclosure in the financial statements, except as disclosed below.
Subsequent
to December 31, 2023, the Company entered into an advisory agreement with a certain advisor to perform independent advisory services
in connection with business operations from January 5, 2024 to June 4, 2024. In consideration of services to be performed, the Company
shall issue to the advisor
Subsequent to December 31, 2023, the Company issued shares of the Company’s common stock to a certain investor on January 1, 2024 as payment for convertible note payable late fees incurred.
Subsequent to December 31, 2023, the Company issued shares of the Company’s common stock to certain consultants as compensation for services performed.
Subsequent to December 31, 2023, the Company issued shares of the Company’s common stock to a certain holder of the Company’s common stock in connection with a legal settlement, which is discussed in Note 11, Commitments and Contingencies – Litigation.
Subsequent
to December 31, 2023, the Company received gross proceeds of $
Subsequent
to December 31, 2023, a convertible note with $
Subsequent
to December 31, 2023, the Company received proceeds of $
Subsequent
to December 31, 2023, the Company received additional advances of $
Subsequent to December 31, 2023, the Company entered into note amendment agreements with a trust controlled by Ms. Soave to extend the maturity dates of the Soave Note and the Verstraete Note to October 28, 2024 and September 10, 2024, respectively.
| F-29 |