UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 2017
or
| ¨ | Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission File Number 001-35853
BIOSTAGE, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 45-5210462 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| Incorporation or organization) | Identification No.) |
84 October Hill Road, Suite 11, Holliston, Massachusetts 01746
(Address of Principal Executive Offices, including zip code)
(774)233-7300
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.01 par value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES x NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | |
| Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company x | |
| Emerging growth company x |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act. YES ¨ NO x
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant, computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of June 30, 2017 was approximately $15,081,053. Shares of the registrant’s common stock held by each officer and director and each person known to the registrant to own 10% or more of the outstanding voting power of the registrant have been excluded in that such persons may be deemed affiliates. This determination of affiliate status is not a determination for other purposes.
At March 30, 2018, there were 2,859,419 shares of the registrant’s common stock issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Company’s definitive Proxy Statement in connection with the 2018 Annual Meeting of Stockholders (the “Proxy Statement”), to be filed within 120 days after the end of the Registrant’s fiscal year, are incorporated by reference into Part III of this Form 10-K. Except with respect to information specifically incorporated by reference in this Form 10-K, the Proxy Statement is not deemed to be filed as part hereof.
BIOSTAGE, INC.
TABLE OF CONTENTS
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2017
INDEX
| i |
This Annual Report on Form 10-K contains statements that are not statements of historical fact and are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934 (the “Exchange Act”), each as amended. The forward-looking statements are principally, but not exclusively, contained in “Item 1: Business” and “Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations.” These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Forward-looking statements include, but are not limited to, statements about management’s confidence or expectations and our plans, objectives, expectations and intentions that are not historical facts. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “seek,” “expect,” “plans,” “aim,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “intends,” “think,” “continue,” “potential,” “is likely,” “permit,” “objectives,” “optimistic,” “new,” “goal,” “target,” “strategy” and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We discuss many of these risks in detail under the heading “Item 1A. Risk Factors” beginning on page 18 of this Annual Report on Form 10-K. You should carefully review all of these factors, as well as other risks described in our public filings, and you should be aware that there may be other factors, including factors of which we are not currently aware, that could cause these differences. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this report. We may not update these forward-looking statements, even though our situation may change in the future, unless we have obligations under the federal securities laws to update and disclose material developments related to previously disclosed information. Biostage, Inc. is referred to herein as “we,” “our,” “us,” and “the Company.”
| 1 |
| Item 1. | Business. |
BUSINESS
We are a biotechnology company developing bioengineered organ implants based on our novel CellframeTM technology. Our Cellframe technology is comprised of a biocompatible scaffold seeded with the patient’s own stem cells. Our platform technology is being developed to treat life-threatening conditions of the esophagus, bronchus and trachea. By focusing on these underserved patients, we hope to dramatically improve the treatment paradigm for these patients. Our unique Cellframe technology combines the clinically proven principles of tissue engineering, cell biology and material science.
We believe that our Cellframe technology may provide surgeons a new paradigm to address life-threatening conditions of the esophagus, bronchus, and trachea due to cancer, infection, trauma or congenital abnormalities. Our novel technology harnesses the body’s response and modulates it toward the healing process to regenerate tissue and restore the continuity and integrity of the organ. We are pursuing the Cellspan TM esophageal implant as our first product candidates to address pediatric esophageal atresia and esophageal cancer, and we are also developing our technology’s applications to address conditions of the bronchus and trachea.
In collaboration with world-class institutions, such as Mayo Clinic and Connecticut Children’s Medical Center, we are advancing our technology toward filing an Investigational New Drug (IND) application with the U.S. Food and Drug Administration (FDA) for our Cellspan esophageal implant product candidates. We plan to conduct additional pre-clinical studies in 2018 for our pediatric esophageal atresia program as well as our adult esophageal cancer indication. In 2019, we plan to file an IND with the FDA for each of these esophageal indications.
Our Cellframe technology platform: how it works
Our Cellframe process begins with the collection of an adipose (fat) tissue biopsy from the patient followed by the use of standard tissue culture techniques to isolate and expand the patient’s own (autologous) mesenchymal (multipotent) stem cells, or MSC. The cells are seeded onto a proprietary biocompatible, synthetic scaffold, produced to mimic the dimensions of the organ to be regenerated, and incubated in a proprietary organ bioreactor. The scaffold is electrospun from polyurethane (PU) to form a non-woven, hollow tube. The specific microstructures of the Cellspan implants are designed to allow the cultured cells to attach to and cover the scaffold fibers.
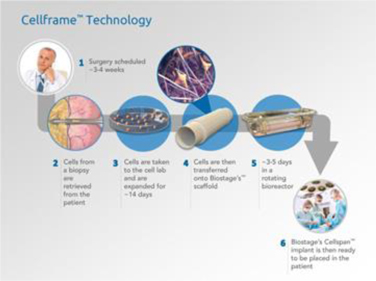
| 2 |
We have conducted large-animal studies to investigate the use of the Cellspan implants for the reconstitution of the continuity and integrity of tubular shape organs, such as the esophagus and the large airways, following a full circumferential resection of a clinically relevant segment, just as would occur in a clinical setting. We announced favorable preliminary preclinical results of large-animal studies for the esophagus, bronchus and trachea in November 2015. Based on the results of those studies, we chose the esophagus to be the initial focus for our organ regeneration technology.

Illustration of intersection of Cellspan esophageal implant and native
esophagus at time of implant and proposed mechanism of action
In May 2016, we reported an update of results from additional, confirmatory pre-clinical large-animal studies. We disclosed that the studies had demonstrated in a predictive large-animal model the ability of our Cellspan organ implant to successfully stimulate the regeneration of a section of esophagus that had been surgically removed. Cellspan esophageal implants, consisting of a proprietary biocompatible synthetic scaffold seeded with the recipient animal’s own stem cells, were surgically implanted in place of the esophagus section that had been removed. After the surgical full circumferential resection of a portion of the thoracic esophagus, the Cellspan implant stimulated the reconstitution of full esophageal structural integrity and continuity.

Illustration of esophageal reconstitution over Cellspan esophageal
implant following time of implant and proposed mechanism of action
Study animals were returned to a solid diet three weeks after the implantation surgery. The scaffold portions of the Cellspan implants, which are intended to be in place only temporarily, were retrieved approximately three weeks post-surgery via the animal’s mouth in a non-surgical endoscopic procedure. Within 2.5 to 3 months, a complete inner epithelium layer and other specialized esophagus tissue layers were regenerated. Two animals in the study were kept in life for almost two years to evaluate the long-term viability of the newly regenerated tubular conduit and were then sacrificed for histological data. Prior to their sacrifice, these animals demonstrated normal weight gain, appeared healthy and free of any significant side effects and received no specialized care.
| 3 |
Platform technology in life-threatening orphan indications
In November 2016, we were granted Orphan Drug Designation for our Cellspan esophageal implant by the FDA to restore the structure and function of the esophagus subsequent to esophageal damage due to cancer, injury or congenital abnormalities. Orphan drug designation provides a seven-year marketing exclusivity period against competition in the U.S. from the date of a product’s approval for marketing. This exclusivity would be in addition to any exclusivity we may obtain from our patents. Additionally, orphan designation provides certain incentives, including tax credits and a waiver of the Biologics License Application fee. We also plan to apply for orphan drug designation for our Cellspan esophageal implant in Europe. Orphan drug designation in Europe provides market exclusivity in Europe for ten years from the date of the product’s approval for marketing.
We are now advancing the development of our Cellframe technology, specifically a Cellspan esophageal implant, in large-animal studies with collaborators. In order to seek approval for the initiation of clinical trials for Biostage Cellspan esophageal implants in humans, GLP studies to support the safety of the Cellspan esophageal implant are required to submit an Investigational New Drug (IND) application with the FDA. As we believe that our recent studies provided sufficient confirmatory proof of concept data, we have initiated the Good Laboratory Practice (GLP) studies to demonstrate that our technology, personnel, systems and practices are sufficient for advancing into human clinical trials. We have conducted a number of IND-enabling GLP studies demonstrating safety and feasibility of the Cellspan implant. These studies were done primarily in pursuit of our adult esophageal cancer program. Additional GLP studies are necessary in support of the adult esophageal cancer program, which will also be supportive of an IND for the pediatric esophageal atresia program. We intend to perform those studies in 2018. We will also pursue additional non-GLP studies for the pediatric esophageal atresia program in 2018 to optimize that product candidate.
First-In-Human Use of Esophageal Implant Product Candidate
On August 7, 2017, we announced the use of our Cellspan Esophageal Implant product candidate in a patient at a major U.S. hospital via an FDA-approved single-use expanded access application. The patient was a 75-year old male with a life-threatening cancerous mass in his chest that spanned his heart, a lung and his esophagus. The surgery was performed in May 2017 to remove the tumor, repair the heart, part of one lung, and a section of the esophagus. The Cellspan Esophageal Implant was interpositioned into the gap in the esophagus created by the removal of the tumor. The patient’s surgeon informed us at that time that the surgery was a success and the patient was later discharged from the hospital. In February 2018 the surgeon informed us that the patient had died after living approximately eight months after surgery. The surgeon stated that the cause of death was a stroke, and that the stroke was unrelated to the esophageal implant. The surgeon also informed us that a preliminary autopsy had shown that the esophageal implant resulted in a regenerated esophageal tube in the patient, except for a very small (approximately 5mm) hole on the lateral wall that was right up against a synthetic graft inserted as part of the patient’s heart repair on the pericardium in that same surgery. The synthetic graft on the pericardium was not related to our esophageal implant product and may have acted as an irritant to esophageal regeneration where it contacted the esophageal implant. The surgeon also informed us that the esophageal regeneration in this patient was consistent with the regeneration previously observed in our large-animal studies. The surgeon informed us that following completion of the autopsy the hospital will pursue a publication and we expect to be in a position to release additional information on this landmark case at that time.
Our product candidates are currently in development and have not yet received regulatory approval for sale anywhere in the world.
Changing the surgical treatment of Esophageal Cancer
|
|
| |
| Illustration of esophageal cancer site | Illustration of potential human application of Cellspan esophageal implant at site of esophageal cancer (depicting implant prior to esophageal tissue reconstitution over implant) |


According to the World Health Organization’s International Agency for Research on Cancer, there are approximately 450,000 new cases of esophageal cancer worldwide each year. A portion of all patients diagnosed with esophageal cancer are treated via a surgical procedure known as an esophagectomy. The current standard of care for an esophagectomy requires a complex surgical procedure that involves moving the patient’s stomach or a portion of their colon into the chest to replace the portion of esophagus resected by the removal of the tumor. These current procedures have high rates of complications and can lead to a severely diminished quality of life and require costly ongoing care. Our Cellspan esophageal implants aim to provide a simpler surgical procedure, with reduced complications, that may result in a better quality of life after the operation and reduce the overall cost of these patients to the healthcare system.
| 4 |
Focus on Pediatric Esophageal Atresia: a congenital abnormality in need of a better solution
Each year, several thousand children worldwide are born with a congenital abnormality known as esophageal atresia, a condition where the baby is born with an esophagus that does not extend completely from the mouth to the stomach. When a long segment of the esophagus is lacking, the current standard of care is a series of surgical procedures where surgical sutures are applied to both ends of the esophagus in an attempt to stretch them and pull them together so they can be connected at a later date. This process can take weeks and the procedure is plagued by serious complications and may carry high rates of failure. Such approach also requires, in time, at least two separate surgical interventions. Other options include the use of the child’s stomach or intestine that would be pulled up into the chest to allow a connection to the mouth. We are working to develop a Cellspan esophageal implant solution to address the complications of esophageal atresia, that could potentially be life-changing or organ-sparing, or both.
Our Mission and Our Strategy
Our mission is to revolutionize regenerative medicine by bioengineering patient-specific Cellspan implants that use the patient’s own cells to stimulate organ regeneration and restore an organ’s structure and continuity. Our business strategy to accomplish this mission includes:
Targeting life-threatening medical conditions. We are focused on creating products to help physicians treat life-threatening conditions like esophageal cancer, central lung cancer and damage to the trachea caused by cancer, trauma or infection. We are also developing products for the treatment of congenital abnormalities of the esophagus and the airways. We are not targeting less severe conditions that have reasonable existing treatment options. Solutions for life-threatening medical conditions present a favorable therapeutic index, or risk/benefit relationship, by providing the opportunity of a significant medical benefit for patients who have poor or no treatment alternatives. We believe that product candidates targeting life-threatening medical conditions may be eligible for review and approval by regulatory authorities under established expedited review programs, which may result in savings of time in the regulatory approval process. Also, we believe that products targeting life-threatening medical conditions may be more likely to receive favorable reimbursement compared with treatments for less critical medical conditions.
Developing products that have a relatively short time to market. Since the number of patients diagnosed with esophageal cancer in the U.S. each year is relatively small, we expect the number of patients that we would likely need to enroll in a clinical trial will also be relatively small. A small number of patients implies a relatively fast enrollment time and a less expensive clinical development program. Therefore, we expect to be able to conduct a clinical trial in a relatively short period of time compared to clinical trials in indications with larger patient populations. We intend to work closely with regulatory agencies and clinical experts to design and size the clinical studies appropriately based on the specific conditions our products are intended to treat.
Using our Cellframe technology as a platform to address multiple organs. We believe that pre-clinical data we have produced to date may suggest that our Cellframe technology is a novel and innovative approach to restoring organ function that may provide an ability to develop products that would address life-threatening conditions impacting organs like the esophagus, bronchus and trachea, and perhaps lower portions of the gastrointestinal (GI) tract. We believe that our Cellframe technology may allow physicians to treat certain life-threatening conditions in ways not currently possible, and in some combination, to save patients’ lives, avoid or reduce complications experienced in the current standard of care, and improve the patients’ quality of life, while at the same time reducing the overall cost of patient care to the healthcare system.
Supplying the finished organ implant to the surgeon. Our technology includes our proprietary organ bioreactor, as well as our proprietary biocompatible scaffold that is seeded with the patient’s own cells. We believe there is considerable value in supplying the final cell-seeded scaffold implant to the surgeon so that the hospital and surgeon may focus solely on performing the implantation.
Collaborating with leading medical and research institutions. We have and will continue to collaborate with leading medical and research institutions. We have a co-development initiative with Mayo Clinic for regenerative medicine organ implant products for the esophagus and airways based on our Cellframe technology. We are also collaborating with Connecticut Children’s Medical Center on a co-development project to translate our Cellframe technology for pediatric esophageal atresia from pre-clinical studies to clinical trials. We believe the use of our product candidates by leading surgeons and institutions will increase the likelihood that other surgeons and institutions will use our products.
| 5 |
Our Technology
Our Cellframe technology is comprised of our proprietary bioengineered organ scaffold seeded with the patient’s own stem cells in our proprietary organ bioreactor prior to implantation. We believe that our Cellframe technology combines a highly-engineered, biocompatible scaffold and a robust population of cells that, by tapping into the stem cell niche of the surrounding native tissue after implantation, may potentially enable a tubular organ to remodel or regenerate tissue to close the gap created by a surgical resection of a portion of that organ. This unique combination of technologies, developed through our extensive testing performed during the last few years, may potentially provide solutions to life-threatening conditions for patients with unmet medical needs.
We believe that our technology is unique, in that its mode of action appears to be different from other tissue engineering organ scaffold products developed previously, of which we are aware. Prior to our development of the Cellframe technology, our approach attempted to implant an organ scaffold that would be incorporated into the patient’s body by the surrounding native tissue growing into the scaffold. To our knowledge, all previous research and development efforts by other investigators were based on that same concept. Our Cellframe technology appears to work very differently. We believe that the unique combination of our highly-engineered organ scaffold with a population of the patient’s own mesenchymal stem cells enables an organ to develop new native tissue around our scaffold, but not into it, so the scaffold acts as a type of frame or staging for the new tissue. As a result, our scaffold is not incorporated into the body. Instead, it is retrieved from the body via an endoscopic procedure, not surgically, after sufficient tissue remodeling and regeneration has occurred to restore the organ’s integrity and function.
A Cellframe technology-based organ implant includes two key components: a biocompatible synthetic scaffold and the patient’s own stem cells.
Biocompatible Scaffold Component
Our proprietary biocompatible scaffold component of the Cellspan esophageal implant is constructed primarily of polyurethane (PU; a plastic polymer). This material was chosen based on extensive testing of various materials. The scaffold is made using a manufacturing process known as electrospinning. The combination of the electrospinning process, which provides control over the desired microstructure of the scaffold fabric, with the PU results in a scaffold that we believe has favorable biocompatibility characteristics.
The Patient’s Cells
Based on current pre-clinical development efforts, the cells we seed onto the scaffold are obtained from the patient’s adipose tissue (abdominal fat). This fat tissue is obtained from a standard biopsy before the implant surgery. Mesenchymal stem cells are extracted and isolated from the adipose tissue biopsy. The isolated cells are then expanded, or grown, for a short period prior to surgery in order to derive a sufficient cell population to be seeded on the scaffold. The cells are then seeded on the scaffold in our proprietary organ bioreactor and incubated there before the implant surgery.
We believe the Cellspan esophageal implant has the potential to provide a major advance over the current therapeutic options for treating esophageal cancer, damage from infection or trauma and congenital abnormalities. We believe our Cellframe technology has the potential to overcome the major challenges in restoring organ function for a damaged esophagus. With our Cellspan esophageal implant we are developing a surgical procedure that has the objective of reconstituting the continuity of the patient’s esophagus without having to relocate another organ in its place. In addition, by reducing or eliminating complications that occur in the current standard of care, we expect to reduce the costs of addressing and treating those additional complications. Because these substantial costs can be reduced or even eliminated with our technology, we believe our products, if successfully developed, can help save lives, improve the quality of life for patients and reduce overall healthcare costs.
| 6 |
Unmet Patient Needs and Cellspan Implant Solutions
Esophageal Cancer
There are approximately 456,000 new diagnoses of esophageal cancer globally each year, according to the World Health Organization’s International Agency for Research on Cancer. According to the American Cancer Society, there are approximately 17,000 new diagnoses of esophageal cancer in the U.S. each year, and there are more than 15,000 deaths from esophageal cancer each year. Esophageal cancer is very deadly - the five-year survival rate for people with esophageal cancer is 18% in the U.S. Approximately 5,000 esophagectomy surgeries occur in the U.S. annually to treat esophageal cancer, and approximately 10,000 esophagectomies occur in Europe annually. We believe that approximately one half of the world’s esophageal cancer cases occur in China, which would represent the largest patient population for our adult esophageal product candidate. We believe that our Cellspan esophageal implant, if approved, has the potential to provide a major advance over the current esophagectomy procedures for addressing esophageal cancer, which have high complication and morbidity rates.
The current standard of care for the esophagectomy requires either (A) a gastric pull-up, where the stomach is cut and sutured into a tubular shape, then pulled up through the diaphragm to replace a portion of the esophagus resected by the removal of the cancerous tumor; or (B) a colon interposition, where a portion of the colon is resected and used to replace the portion of the esophagus resected by the removal of the cancerous tumor. Esophagectomies have 90-day mortality rates of up to 19%. Serious complications, such as leakage at the anastomoses, which can lead to infections and sepsis, and pulmonary complications, such as impaired pulmonary function or pneumonia, occur in up to 30% of esophagectomy cases. Other complications from esophagectomies, such as a narrowing of the esophagus post-surgery, gastroesophageal reflux and dumping syndrome (repetitive nausea, dizziness and vomiting) can also pose significant quality of life issues for patients.
We believe that the Cellspan esophageal implant has the potential to provide physicians a new, simpler procedure to restore organ function while significantly reducing complication and morbidity rates compared with the current standard of care, and without creating significant quality of life issues for patients.
Esophageal Atresia
Esophageal Atresia (EA) is a rare congenital abnormality in which a baby is born without part of the esophagus. About 1 in 4,000 babies in the U.S. is born with EA. In some cases, the two sections can be connected surgically. However, in cases where the gap is too great for a simple surgical reconnection, the current standard of care is a gastric pull-up, a colon interposition, or a procedure known as the Foker process. In the Foker process, traction devices are surgically attached to the two ends of the esophagus. Traction is then applied, usually for several weeks during which time the baby remains in an Intensive Care Unit, to stimulate the ends of the esophagus to grow and narrow the gap. If the Foker process is successful in narrowing the gap sufficiently, a second surgery is necessary to connect the two ends of the esophagus. In addition to the Foker process being complex, it is also a very expensive procedure, because the baby will normally be several months in hospital for the process.
We believe that a pediatric Cellspan esophageal implant may provide pediatric surgeons with a better procedure to treat EA that would result in a connected esophagus with higher success rates, lower complications and lower overall costs to the healthcare system.
Central Lung Cancer
Lung cancer is the most common form of cancer and the most common cause of death from cancer worldwide. There are more than 450,000 new lung cancer diagnoses annually in the U.S. and Europe. In approximately 25% of all lung cancer cases, the cancerous tumor resides only in a bronchus and not in the lobes of the lungs, and is known as central lung cancer. Approximately 33,000 central lung cancer cases diagnosed in the U.S. and Europe are Stage I and II and are considered eligible for surgical resection, often with adjuvant chemotherapy and radiation. Approximately 5,000 of those patients are treated via pneumonectomy, a surgical procedure involving the resection of the cancer tumor, the whole bronchus below the tumor and the entire lung to which it is connected. It is a complex surgery and, due to the removal of a lung, results in a 50% reduction in the patient’s respiratory capacity. The procedure has reported rates of post-surgical (in hospital) mortality of 8% to 15%. Complication rates associated with pneumonectomy are reported as high as 50%, and include post-operative pneumonia, supraventricular arrhythmias and anastomotic leakage, placing patients at significant mortality risk post-discharge.
We believe that a Cellspan bronchial implant, once developed and approved for marketing, has the potential to provide physicians a treatment alternative superior to the sleeve pneumonectomy to address central lung cancer, a simpler procedure to restore organ function of the bronchus without sacrificing one of the patient’s lungs, resulting in fewer post-surgery complications, improved mortality rates and improved quality of life for the patient.
Life-threatening conditions of the Trachea
There are approximately 8,000 patients per year in the U.S. and Europe who suffer from a condition of the trachea that put the patient at high risk of death. These conditions can be due to tracheal trauma, tracheal stenosis or trachea cancer. There are approximately 40,000 tracheal trauma patients diagnosed each year in the U.S. Of those, approximately 1,000 are severe enough to need surgical resection procedures. Tracheal stenosis is a rare complication from tracheostomies, but may have a devastating impact on respiratory function for patients. Approximately 2,000 patients are diagnosed with stenosis from tracheostomy in the U.S. each year. Trachea cancer is a very rare but extremely deadly cancer. Trachea cancer patients in the U.S. have a median survival of 10 months from diagnosis and a 5-year survival of only 27%. There were approximately 200 cases of primary trachea cancer diagnosed in the U.S. in 2013. Based on these facts, we estimate that there are approximately 8,000 patients in the U.S. and Europe with conditions of the trachea that put them at high risk of death, but for whom there is currently no clinically effective tracheal implant or replacement method currently available.
We believe that a Cellspan tracheal implant may potentially provide physicians a treatment to re-establish the structural integrity and function of a damaged or diseased trachea to address life-threatening conditions due tracheal trauma, stenosis or cancer.
| 7 |
Our History
We were incorporated under the laws of the State of Delaware on May 3, 2012 by Harvard Bioscience, Inc. (“Harvard Bioscience”) to provide a means for separating its regenerative medicine business from its other businesses. Harvard Bioscience decided to separate its regenerative medicine business into our company, a separate corporate entity (the “Separation”), and it spun off its interest in our business to its stockholders in November 2013. Since the Separation we have been a separately-traded public company and Harvard Bioscience has not been a stockholder of our common stock or controlled our operations. Following the Separation, we continued to innovate our bioreactors based on our physiology expertise, we developed our materials science capabilities and we investigated and developed a synthetic tracheal scaffold. By that time, we had built and staffed cell biology laboratories at our Holliston facility, to give ourselves the ability to perform and control our scientific investigation and developments internally. At that point, we began the second phase of our company’s development.
In mid-2014, we increased the pace of our scientifically-based internal analysis and development of our first-generation tracheal implant product, the HART-Trachea. From large-animal studies conducted thereafter we found that the product elicited an unfavorable inflammatory response after implantation, which required additional development and testing. These requirements extended our expectations regarding our regulatory milestones and we announced the additional testing and extended milestone expectations in January 2015. During 2015 we isolated and tested all major variables of the organ scaffold and the cell source and protocols, examining the effects of alternatives against the then-existing product approach. Through extensive in vitro preclinical studies, and small-animal and large-animal studies, we made dramatic improvements, and discovered that the mechanism of action of this new approach was very different from our hypothesis regarding that of the first-generation product. We call this new implant approach our Cellframe technology. Our Cellframe technology uses a different scaffold material and microstructure, a different source and concentration of the patient’s cells and several other changes from our earlier trachea initiative. We believe that our Cellframe technology, although built on learnings from our earlier-generation product initiative, represents a new technology platform resulting from our rigorous science and development. We see the development of our Cellframe technology platform as the beginning of a new, third phase in our company’s progression.
We discontinued development of our earlier initiative in 2014; that first-generation product approach was significantly different from our new Cellframe technology and Cellspan product candidates currently in development. We have focused our development efforts on our Cellframe technology and Cellspan product candidates, which we have and will continue to develop internally, and with our collaborators, via a rigorous scientific development process.
Clinical Trials
Our Esophageal Cellspan Implant has been designated by the FDA as a combination product biologic. We believe that this is a favorable designation as it allows for orphan designation and a more participatory path to approval. To date, we have conducted numerous pre-clinical studies in our esophageal implant programs and continue to see consistent regeneration. We are analyzing these data and our human experience to better ascertain what specific pre-clinical studies are still needed prior to conducting a phase I human clinical trial. We are pursuing two indications in parallel. The two complementary indications we are pursuing are esophageal cancer and pediatric esophageal atresia. In order to market our product candidates, we will need to successfully complete clinical trials. The initial indication for which we intend to seek FDA approval will be to restore the function of the esophagus subsequent to esophageal damage or stenosis due to cancer, injury or infection.
Because esophageal cancer affects only approximately 17,000 patients per year in the U.S. we anticipate that our clinical trials will involve relatively few patients. Therefore, once commenced, we expect to be able to conduct a clinical trial in a relatively short period of time compared to clinical trials in indications with larger patient populations. We intend to work closely with regulatory agencies and clinical experts to design and size the clinical studies appropriately based on the specific conditions our products are intended to treat. We also intend to request expedited review from the FDA for the Cellspan esophageal implant product. Receipt of expedited review would reduce the overall time through the regulatory approval process. These expedited requests are submitted during the IND process.
We believe that we have excellent pre-clinical and clinical support of the pediatric atresia program through our collaboration with Connecticut Children’s Medical Center and our primary investigator Dr. Christine Finck. Essentially, we liken the pediatric atresia market to a rare disease market. Accordingly, the clinical trial population should reflect the ultra-orphan nature of the disease state.
We intend to pursue regulatory approval for the Cellspan esophageal implant in the U.S., initially. Following clinical trials in other foreign markets, we expect to pursue regulatory approval for the Cellspan esophageal implant in those foreign markets, as well. We believe that approximately one half of the world’s esophageal cancer cases occur in China, which would represent the largest potential patient population for our adult esophageal implant product candidate, and we are consequently beginning to prepare to address that market.
Research and Development
Our primary research and development activities are focused in three areas: materials science, cell biology and engineering. In materials science, we focus on designing and testing biocompatible organ scaffolds, testing the structural integrity and the cellularization capacities of the scaffolds. In cell biology, we focus on developing and testing isolation and expansion protocols, cell characterization and fate studies, investigating the effects of various cell types and concentrations, evaluating the biocompatibility of scaffolds, experimenting with different cell seeding methodologies, and developing protocols for implantation experiments. Our engineering group supports the materials science and cell biology groups across an array of their activities, i.e. designing, engineering and making our proprietary organ bioreactors. All three of our R&D groups combine to plan and execute the in vitro studies. A fundamental part of our R&D effort in developing the Cellframe technology has been dedicated to the discovery and development of small and large animal model studies. The large-animal model employs the use of Yucatan mini-pigs. Our Cellspan scaffolds were implanted in the cervical portion as well as the thoracic portion of the esophagus and the airways in studies to date.
Following the failure to receive the funding with respect to a securities purchase agreement in August 2017, and in an effort to conserve cash, we completed a reduction in headcount of 20 persons during October and November 2017, of whom 18 were directly involved in research and development activities. As a result, we had one research and development employee as of December 31, 2017. Following our raising additional capital in December 2017, January 2018 and February 2018, we re-hired four of our former research and development employees into key scientific and engineering positions, and retained two others as consultants, in January 2018. We believe that our new staffing level after those hires, combined with our consultant and co-development collaborator resources, is sufficient to pursue both of our esophageal programs.
In addition to our in-house engineering and scientific development team, we collaborate with leaders in the field of regenerative medicine who are performing the fundamental research and surgeries in this field to develop and test new products that will advance and improve the procedures being performed. We will work with our collaborators to further enhance our products to make them more efficient and easier to use by surgeons. In the U.S., our principal collaborations have been with Mayo Clinic and Connecticut Children’s Medical Center. Collaboration typically involves us developing new technologies specifically to address issues these researchers and clinicians face, and then working together to translate our technology from pre-clinical studies to clinical trials. In certain instances, we have entered into agreements that govern the ownership of the technologies developed in connection with these collaborations.
| 8 |
We incurred approximately $7.7 million and $7.6 million of research and development expenses in 2017 and 2016, respectively. As we have not yet applied for or received regulatory approval to market any clinical products and sales of our research bioreactor products have not been significant in relation to our operating costs, no significant amount of these research and development costs have been passed on to our customers.
Manufacturing
Biostage has developed a comprehensive manufacturing process for our product candidates, including: cell biology, scaffold production, cell isolation and expansion, seeding of cells on the scaffold, incubation and expansion processes in the bioreactor and product transportation. We currently perform certain manufacturing steps in-house and subcontract certain processes and activities, primarily those related to cell expansion, seeding and incubation, to experienced partners.
For our scaffolds we use a process called electrospinning to create the fabric part of the scaffold. Electrospinning is a well-known fabrication process. It is useful for cell culture applications as it can create extremely thin fibers (much thinner than a human hair) that can make a fabric with pores approximately the same size as a cell. The electrospinning process parameters can be tuned to create a structure that is very similar to the natural structure of the collagen fibers in human extracellular matrix. Our process and end product have been developed over many years and involve many trade secrets and proprietary know-how. Our Cellspan scaffolds are made from polyurethane, an inert polymer that is not bioresorbable. However, we also perform studies on the use of scaffolds made from bioresorbable materials. While we do not manufacture the cells, as they will come from the patient’s adipose tissue, for regulatory purposes we are responsible for the quality control of the cells and the seeding of the cells onto the scaffold in the bioreactor. For this we have, in collaboration with our partners, developed standard operating procedures for the seeding of cells on the scaffold. For U.S. clinical trials we anticipate that the seeding will be performed in an automated version of our bioreactor at a pre-qualified third-party contract manufacturer using current Good Manufacturing Procedures (cGMP) using our proprietary protocol and under the supervision of our staff.
For our scaffolds, our primary materials are medical-grade plastic resins and solvents used to liquefy the resins in our manufacturing process. These materials are readily available from a variety of suppliers and do not currently represent a large proportion of our total costs. For our bioreactors, we perform final assembly and testing of components that we buy from third parties like machine shops, parts distributors, molding facilities and printed circuit board manufacturers. These operations are performed primarily at our Holliston, MA headquarters.
Sales and Marketing
We expect that most surgeries using the Cellspan esophageal implant product will be performed at a relatively small number of major hospitals in the U.S., Canada and European countries that will establish themselves as specialized centers of excellence. We believe that a relatively small number of centers of excellence in each country would be able to treat a very large percentage of that country’s patients annually, given the expected number of patients to be treated each year. So, we expect our markets to be served by a concentrated number of treatment centers. Further, our three Cellspan product candidates are for the esophagus, the bronchi and the trachea, three organs all treated by thoracic surgeons. Therefore, all three products, once approved, would be marketed primarily to physicians practicing in a single surgical specialty, so we expect that the total number of physicians using our products will be a much smaller population than if our products were to be used by physicians in multiple areas of surgical specialties. Due to our expectation of a population of physicians in one surgical specialty being the primary users of our products in a concentrated number of centers of excellence in each national market, we expect to be able to support our markets with a fairly small field sales force.
We expect to price the product commensurate with the medical value created for the patient and the costs avoided with the use of our product. We further expect to be paid by the hospital that buys the product from us. Finally, we expect that the hospital would seek reimbursement from payers for the entire transplant procedure, including the use of our products.
Harvard Bioscience is the exclusive distributor for the research versions of our organ bioreactors. Harvard Bioscience can only sell those products to the research markets in accordance with the terms of our distribution agreement. We retain all rights to manufacture and sell all our products for clinical use.
| 9 |
Intellectual Property and Related Agreements
We actively seek to protect our products and proprietary information by means of U.S. and foreign patents, trademarks and contractual arrangements. Our success will depend in part on our ability to obtain and enforce patents on our products, processes and technologies to preserve our trade secrets and other proprietary information and to avoid infringing on the patents or proprietary rights of others.
We anticipate that we will sell products in various markets in the United States and various jurisdictions under brand name, logo and product design trademarks and service marks and that these marks will attain material importance in the future.
We also own select U.S. Patents as well as certain patents in Germany. These patents cover aspects of device and processes currently under development by our company. Patents for various processes and devices extend for varying periods according to the date of patent filing or grant and the legal term of patents in the country or countries in which the patent was obtained. The actual protection afforded by a patent can vary from country to country and depends on factors such as the type of patent, scope of protection and available legal remedies.
In addition to issued patents, we have several pending patent applications in the U.S. and key target jurisdictions. We believe that one or more of these pending patent applications may be of importance to material position depending upon factors such as the relevant patent jurisdiction, type of patent granted, and scope of patent claims ultimately allowed in a given jurisdiction. Depending upon factors such as the type of grant and the date on which the patent application was filed, we anticipate that the term of certain pending patents may extend to 2036.
We also rely on unpatented proprietary technologies in the development and commercialization of our products. We also depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as those of our advisors, consultants and other contractors. To help protect our proprietary know-how that may not be patentable, and our inventions for which patents may be difficult to enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require employees, consultants and advisors to enter into agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions that arise from their activities for us. Additionally, these confidentiality agreements require that our employees, consultants and advisors do not bring to us, or use without proper authorization, any third party’s proprietary technology.
| 10 |
Exclusive License Agreement and Sponsored Research Agreement - InBreath Bioreactor
We entered into a sponsored research agreement with Sara Mantero, Maria Adelaide Asnaghi, and the Department of Bioengineering of the Politecnico Di Milano, or PDM. Under the terms of this agreement, PDM was required to use its facilities and best efforts to conduct a research program relating to the development of bioreactors, clinical applications, and automated seeding processes. We were required to provide engineering support to PDM with respect to bioreactor designs. Intellectual property developed by PDM or its employees, including Dr. Mantero or Ms. Asnaghi, under this sponsored research agreement will be owned by Dr. Mantero or Ms. Asnaghi and covered by our exclusive license agreement described above. In addition, we have an option to an exclusive license for intellectual property relating to new technology that may not be covered by the exclusive license agreement. We will own any inventions and discoveries that we solely develop in connection with the research program and any inventions and discoveries that are jointly developed in connection with the research program will be owned jointly by the parties. We terminated the sponsored research agreement in 2017.
Sublicense Agreement with Harvard Bioscience
We have entered into a sublicense agreement with Harvard Bioscience pursuant to which Harvard Bioscience has granted us a perpetual, worldwide, royalty-free, exclusive, except as to Harvard Bioscience and its subsidiaries, license to use the mark “Harvard Apparatus” in the name Harvard Apparatus Regenerative Technology. The mark “Harvard Apparatus” is used under a license agreement between Harvard Bioscience and Harvard University, and we have agreed to be bound by such license agreement in accordance with our sublicense agreement. We currently have no affiliation with Harvard University.
Separation Agreements with Harvard Bioscience
On November 1, 2013, to effect the Separation, Harvard Bioscience distributed all of the shares of our common stock to the Harvard Bioscience stockholders (the “Distribution”). Prior to the Distribution Harvard Bioscience contributed the assets of its regenerative medicine business, and approximately $15 million in cash, to our company to fund our operations following the Distribution.
In connection with the Separation and immediately prior to the Distribution, we entered into a Separation and Distribution Agreement, Intellectual Property Matters Agreement, Product Distribution Agreement, Tax Sharing Agreement, Transition Services Agreement, and Sublicense Agreement with Harvard Bioscience to effect the Separation and Distribution and provide a framework for our relationship with Harvard Bioscience after the Separation. These agreements govern the current relationships among us and Harvard Bioscience and provided for the allocation among us and Harvard Bioscience of Harvard Bioscience’s assets, liabilities and obligations (including employee benefits and tax-related assets and liabilities) attributable to periods prior to the Separation.
Government Regulation
Any product that we may develop based on our Cellframe technology, and any other clinical products that we may develop, will be subject to considerable regulation by governments. We were in the past informed by the FDA that our previous-generation tracheal product candidate would be regulated under the Biologics License Application, or BLA, pathway in the U.S. and we were informed by the European Medicines Agency (EMA) that the previous generation tracheal product would be regulated under the Advanced Therapy Medicinal Products(ATMP), pathway in the EU. On October 18, 2016, we also received written confirmation from FDA’s Center for Biologics Evaluation and Research(CBER), that FDA intends to regulate our Cellspan esophageal implant as a combination product under the primary jurisdiction of CBER. We further understand that CBER may choose to consult or collaborate with the FDA’s Center for Devices and Radiological Health (CDRH), with respect to the characteristics of the synthetic scaffold component of our product based on CBER’s determination of need for such assistance. Although our Cellframe technology differs in design and performance from the first-generation product candidate, we expect that Cellframe-based products will be regulated by the FDA and EMA under the same pathways as the first-generation tracheal product candidate. This expectation is based on the fact that the Cellframe technology is centered on the delivery of the patient’s own cells seeded on an implanted synthetic scaffold in order to restore organ function and our belief that the cells provide the primary mode of action. Of course, it is possible that some of our current and future products may use alternative regulatory pathways.
| 11 |
Regulatory Strategy
Domestic Regulation of Our Products and Business
The testing, manufacturing, and potential labeling, advertising, promotion, distribution, import and marketing of our products are subject to extensive regulation by governmental authorities in the U.S. and in other countries. In the U.S., the FDA, under the Public Health Service Act, the Federal Food, Drug and Cosmetic Act, and its implementing regulations, regulates biologics and medical device products.
The labeling, advertising, promotion, marketing and distribution of biopharmaceuticals, or biologics and medical devices also must be in compliance with the FDA and U.S. Federal Trade Commission (FTC), requirements which include, among others, standards and regulations for off-label promotion, industry sponsored scientific and educational activities, promotional activities involving the internet, and direct-to-consumer advertising. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures, injunctions and criminal prosecution. Further, we are required to meet regulatory requirements in countries outside the U.S., which can change rapidly with relatively short notice.
| 12 |
We have been informed by the FDA that our Cellspan esophageal implant product candidates are combination biologic/device products. Biological products must satisfy the requirements of the Public Health Services Act and the Food, Drug and Cosmetics Act and their implementing regulations. In order for a biologic product to be legally marketed in the U.S., the product must have a BLA approved by the FDA.
The BLA Approval Process
The steps for obtaining FDA approval of a BLA to market a biopharmaceutical, or biologic product in the U.S. include:
| • | completion of pre-clinical laboratory tests, animal studies and formulation studies under the FDA’s GLP regulations; |
| • | submission to the FDA of an IND application, for human clinical testing, which must become effective before human clinical trials may begin and which must include Institutional Review Board (IRB), approval at each clinical site before the trials may be initiated; |
| • | performance of adequate and well-controlled clinical trials in accordance with Good Clinical Practices (GCP), to establish the safety and efficacy of the product for each indication; |
| • | submission to the FDA of a BLA, which contains detailed information about the chemistry, manufacturing and controls for the product, extensive pre-clinical information, reports of the outcomes of the clinical trials, and proposed labeling and packaging for the product; |
| • | the FDA’s acceptance of the BLA for filing; |
| • | satisfactory review of the contents of the BLA by the FDA, including the satisfactory resolution of any questions raised during the review or by the advisory committee, if applicable; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP regulations, to assure that the facilities, methods and controls are adequate to ensure the product’s identity, strength, quality and purity; and |
| • | FDA approval of the BLA. |
Based on preliminary discussions with the FDA, we expect the clinical trials for our esophageal implant product candidates to be conducted in two sequential phases:
| · | A Phase 1, or Pilot Trial, where our product would be tested on a small number, perhaps five or six, of patients to demonstrate the product’s safety. If successful, that study would be followed by, |
| · | A Phase II Registration, or Pivotal Trial to test the product’s efficacy. We believe that the nature of our esophageal products and the sizes of their targeted patient populations would lead to a small number of patients in this trial, relative to most biotechnology clinical trials. |
| 13 |
Clinical testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the phases of clinical trials that are conducted under an IND and may, at its discretion, reevaluate, alter, suspend, or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA or the sponsor may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request that additional pre-clinical studies or clinical trials be conducted as a condition to product approval.
Companies also may seek Fast Track or Breakthrough Therapy designation for their products. Fast Track or Breakthrough Therapy products are those that are intended for the treatment of a serious or life-threatening condition and that demonstrate the potential to address unmet medical needs for such a condition. If awarded, the Fast Track or Breakthrough Therapy designation applies to the product only for the indication for which the designation was received.
If the FDA determines after review of preliminary clinical data submitted by the sponsor that a Fast Track or Breakthrough Therapy product may be effective, it may begin review of portions of a BLA before the sponsor submits the complete BLA (rolling review), thereby accelerating the date on which review of a portion of the BLA can begin. There can be no assurance that any of our products will be granted Fast Track or Breakthrough Therapy designation. And even if they are designated as Fast Track or Breakthrough Therapy products, we cannot assure you that our products will be reviewed or approved more expeditiously for their Fast Track or Breakthrough Therapy indications than would otherwise have been the case or will be approved promptly, or at all. Furthermore, the FDA can revoke Fast Track or Breakthrough Therapy designation at any time.
| 14 |
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive Accelerated Approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a product receiving Accelerated Approval perform adequate and well-controlled post-approval clinical trials to verify and further define the product’s clinical benefit and safety profile. There can be no assurance that any of our products will receive Accelerated Approval. Even if Accelerated Approval is granted, the FDA may withdraw such approval if the sponsor fails to conduct the required post-approval clinical trials, or if the post-approval clinical trials fail to confirm the early benefits seen during the accelerated approval process.
Priority Review Voucher
Fast Track or Breakthrough Therapy designation and Accelerated Approval should be distinguished from Priority Review designation although products awarded Fast Track or Breakthrough Therapy designation may also be eligible for Priority Review designation.
Products regulated by the CBER may receive Priority Review designation if they provide significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious or life-threatening disease. The agency has agreed to the performance goal of reviewing products awarded Priority Review designation within six months, whereas products under standard review receive a ten-month target. The review process, however, can be significantly extended by FDA requests for additional information or clarification regarding information already provided in the submission. Priority Review designation is requested at the time the BLA is submitted, and the FDA makes a decision as part of the agency’s review of the application for filing.
Separately, but somewhat related, is a product’s ability to qualify its sponsor to receive a Priority Review Voucher, or PRV. For a product aimed at prevention or treatment of a “rare pediatric disease” as defined by Food, Drug and Cosmetics Act at 21 USC 360ff, and that also meets certain other qualifying attributes, the product’s sponsor may qualify, apply for and receive a PRV, from the FDA. A PRV entitles its holder to Priority Review for a drug application, and the PRV is transferable. Some companies who have received PRV’s have sold their PRV’s to other companies who have then used the PRV to receive Priority Review for a drug application with the FDA. Recent transfers of PRV’s from one company to another have occurred at prices in the $125 – 150 million range. We intend to apply for rare pediatric disease designation for our pediatric esophageal implant product candidate as a first step in pursuit of a PRV. A PRV is earned only upon marketing approval of the product. There is no certainty that our pediatric esophageal product will achieve marketing approval from the FDA, or that if it does, that FDA would award us a PRV. Further, if received, there is no certainty that the value of a PRV at that future date will compare favorably with the values reflected in recent transfers of PRV’s.
| 15 |
Orphan Drug Designations
The Orphan Drug Act provides incentives to manufacturers to develop and market drugs and biologics for rare diseases and conditions affecting fewer than 200,000 persons in the U.S. at the time of application for orphan drug designation.. In September 2014 the FDA granted orphan designation to our HART-Trachea product in the U.S. In November 2016, we were granted Orphan Drug Designation for our Cellspan esophageal implant by the FDA to restore the structure and function of the esophagus subsequent to esophageal damage due to cancer, injury or congenital abnormalities. The first developer to receive FDA marketing approval for an orphan biologic is entitled to a seven-year exclusive marketing period in the U.S. for that product. The marketing exclusivity prevents FDA approval of another application for the same product for the same indication for a period of seven years. Orphan status also entitles the product’s sponsor to certain other benefits, such as a waiver of the BLA user fee, which is currently a $2 million value. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
International
We plan to seek required regulatory approvals and comply with extensive regulations governing product safety, quality, manufacturing and reimbursement processes in order to market our products in other major foreign markets. The regulation of our products in the Asian and European markets, and in other foreign markets varies significantly from one jurisdiction to another. The classification of the particular products and related approval or CE marking procedures can involve additional product testing and additional administrative review periods. The time required to obtain these foreign approvals or to CE mark our products may be longer or shorter than that required in the U.S., and requirements for approval may differ from the FDA requirements. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
Legislation similar to the Orphan Drug Act has been enacted in other jurisdictions, including the EU. The orphan legislation in the EU is available for therapies addressing conditions that affect five or fewer out of 10,000 persons. The marketing exclusivity period is for ten years, although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Employees
At December 31, 2017, we had 5 employees working in our business, of whom 4 were full-time and one was part-time. At that date, all of our employees were based in the U.S. None of our employees are unionized. In general, we consider our relations with our employees to be good.
Competition
We are not aware of any companies whose products are directly competitive with our cell-seeded biocompatible synthetic scaffold system. However, in our key markets we may in the future compete with multiple pharmaceutical, biotechnology, and medical device, including, among others, Aldagen, Asterias Biotherapeutics, Athersys, BioTime, Caladrius Biosciences, Celgene, Cytori Therapeutics, E. I. du Pont de Nemours and Company, InVivo Therapeutics, Mesoblast, Miramatrix Medical, Nanofiber Solutions, Neuralstem, Organovo, Osiris Therapeutics, Pluristem, Smiths Medical, Tissue Genesis, Inc., Tissue Growth Technologies, United Therapeutics, Vericel Corporation and W.L. Gore and Associates. In addition, there are many academic and clinical centers that are developing regenerative technologies that may one day become competitors with us.
Many of our potential competitors have substantially greater financial, technological, research and development, marketing, and personnel resources than we do. We cannot forecast if or when these or other companies may develop competitive products.
| 16 |
We expect that other products will compete with products and potential products based on efficacy, safety, cost, and intellectual property positions. While we believe that these will be the primary competitive factors, other factors include, in certain instances, obtaining marketing exclusivity under the Orphan Drug Act, availability of supply, manufacturing, marketing and sales expertise and capability, and reimbursement coverage.
Executive Officers of the Registrant
The following table shows information about our executive officers as of December 31, 2017.
| Name | Age | Position(s) | ||
| James McGorry | 61 | Chief Executive Officer and Member of the Board of Directors | ||
| Thomas McNaughton | 57 | Chief Financial Officer |
James McGorry - Chief Executive Officer and Director
Mr. McGorry has served as our President and Chief Executive Officer (CEO) since July 6, 2015. He has served as a Member of our Board of Directors since February 2013. Mr. McGorry has more than 30 years of experience as a life science business leader in biologics, personalized medicine and medical devices, including multiple product launches. Prior to becoming President and CEO at Biostage, Mr. McGorry most recently served as Executive Vice President and General Manager, Translational Oncology Solutions for Champions Oncology and previously was Executive Vice President of Commercial Operations at Accellent. During a 12-year tenure at Genzyme, he held leadership positions across several therapeutic areas, including Bio Surgery, Cardiac Surgery, Oncology and Transplant. Mr. McGorry also was President of Clineffect Systems, an electronic medical records company. He began his life sciences career with Baxter Healthcare Corporation, where he spent 11 years in positions of increasing responsibility. Mr. McGorry also served as an officer in the United States Army for six years, including commanding a special operations Green Beret SCUBA detachment. Mr. McGorry has an MBA with a concentration in healthcare from Duke University, Fuqua School of Business, and a B.S. in engineering from the United States Military Academy at West Point where he was the president of his class. We believe Mr. McGorry’s qualifications to sit on our Board of Directors include his extensive executive leadership positions at several biotechnology and healthcare companies over the past 25 years.
Thomas McNaughton - Chief Financial Officer
Mr. McNaughton has served as our Chief Financial Officer since May 3, 2012. Mr. McNaughton joined Harvard Bioscience as its Chief Financial Officer in September 2008, and served in that role until the spin-off of our company from Harvard Bioscience on November 1, 2013. During 2008 and prior to joining Harvard Bioscience, Mr. McNaughton was a consultant providing services primarily to an angel-investing group and a silicon manufacturing start-up. From 2005 to 2007, he served as Vice President of Finance and Chief Financial Officer for Tivoli Audio, LLC, a venture capital-backed global manufacturer of premium audio systems. From 1990 to 2005, Mr. McNaughton served in various managerial positions in the areas of financial reporting, treasury, investor relations, and acquisitions within Cabot Corporation, a global manufacturer of fine particulate products, and served from 2002 to 2005 as Finance Director, Chief Financial Officer of Cabot Supermetals, a $350 million Cabot division that provided high purity tantalum and niobium products to the electronics and semiconductor industries. Mr. McNaughton practiced from 1982 to 1990 as a Certified Public Accountant in the audit services group of Deloitte & Touche, LLP. He holds a B.S. in accounting and finance with distinction from Babson College.
Available Information and Website
Our website address is www.biostage.com. Our Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and exhibits and amendments to those reports filed or furnished with the Securities and Exchange Commission pursuant to Section 13(a) of the Exchange Act are available for review on our website and the Securities and Exchange Commission’s (“SEC”) website at www.sec.gov. Any such materials that we file with, or furnish to, the SEC in the future will be available on our website as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. The information on our website is not incorporated by reference into this Annual Report on Form 10-K.
| 17 |
| Item 1A. | Risk Factors. |
The following factors should be reviewed carefully, in conjunction with the other information contained in this Annual Report on Form 10-K. As previously discussed, our actual results could differ materially from our forward-looking statements. Our business faces a variety of risks. We describe below what we believe are currently the material risks and uncertainties we face, but they are not the only risks and uncertainties we face. Additional risks and uncertainties of which we are unaware, or that we currently believe are not material, may also become important factors that adversely affect our business. In addition, past financial performance may not be a reliable indicator of future performance and historical trends should not be used to anticipate results or trends in future periods. If any of the following risks and uncertainties develops into actual events, these events could have a material adverse effect on our business, financial condition or results of operations. In such case, the trading price of our common stock could decline, and you may lose all or part of your investment in our securities. The risk factors generally have been separated into three groups: (i) risks relating to our business, (ii) risks relating to the Separation and (iii) risks relating to our common stock. These risk factors should be read in conjunction with the other information in this Annual Report on Form 10-K.
Risks Relating To Our Business
Risks Associated with Clinical Trials and Pre-Clinical Development
The results of our clinical trials or pre-clinical development efforts may not support our product claims or may result in the discovery of adverse side effects.
Even if our pre-clinical development efforts or clinical trials are completed as planned, we cannot be certain that their results will support our product claims or that the FDA, foreign regulatory authorities or notified bodies will agree with our conclusions regarding them. Although we have obtained some positive results from the use of our scaffolds and bioreactors for trachea transplants performed to date, we also discovered that our first-generation trachea product design encountered certain body response issues that we have sought to resolve with our ongoing development of our Cellframe implant design. We cannot be certain that our Cellframe implant design or any future modifications or improvements with respect thereto will support our claims, and any such developments may result in the discovery of further adverse side effects. We also may not see positive results when our products undergo clinical testing in humans in the future. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. Our pre-clinical development efforts and any clinical trial process may fail to demonstrate that our products are safe and effective for the proposed indicated uses, which could cause us to abandon a product and may delay development of others. Also, patients receiving surgeries using our products under compassionate use or in clinical trials may experience significant adverse events following the surgeries, including serious health complications or death, which may or may not be related to materials provided by us. Our current Cellframe technology has never been used in humans. We provided a previous generation trachea implant that was used in human patients under compassionate use. To date, we believe that at least four of the six patients who received that first-generation implant have died. While we believe that none of them have died because of a failure of the first-generation implant, these and any other such adverse events have and may cause or contribute to the delay or termination of our clinical trials or pre-clinical development efforts. Any delay or termination of our pre-clinical development efforts or clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our products and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product’s profile.
Clinical trials necessary to support a biological product license or other marketing authorization for our products will be expensive and will require the enrollment of sufficient patients to adequately demonstrate safety and efficacy for the product’s target populations. Suitable patients may be difficult to identify and recruit. Delays or failures in our clinical trials will prevent us from commercializing any products and will adversely affect our business, operating results and prospects.
In the U.S., initiating and completing clinical trials necessary to support Biological License Applications (BLAs), will be time consuming, expensive and the outcome uncertain. Moreover, the FDA may not agree that clinical trial results support an application for the indications sought in the application for the product. In other jurisdictions such as the EU, the conduct of extensive and expensive clinical trials may also be required in order to demonstrate the quality, safety and efficacy of our products, depending on each specific product, the claims being studied, and the target condition or disease. The outcome of these clinical trials, which can be expensive and are heavily regulated, will also be uncertain. Moreover, the results of early clinical trials are not necessarily predictive of future results, and any product we advance into clinical trials following initial positive results in early clinical trials may not have favorable results in later clinical trials.
| 18 |
Conducting successful clinical trials will require the enrollment of a sufficient number of patients to support each trial’s claims, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population, the nature of the trial protocol, the attractiveness of, or the discomfort and risks associated with, the treatments received by enrolled subjects, the availability of appropriate clinical trial investigators, support staff, and proximity of patients to clinical sites and ability to comply with the eligibility and exclusion criteria for participation in the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and effectiveness of our products, or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomfort. Also, patients may not participate in our clinical trials if they choose to participate in contemporaneous clinical trials of competitive products. In addition, patients participating in clinical trials may die before completion of the trial or suffer adverse medical events unrelated to investigational products.
Development of sufficient and appropriate clinical protocols to demonstrate safety and efficacy are required and we may not adequately develop such protocols to support clearance and approval. Further, the FDA and foreign regulatory authorities may require us to submit data on a greater number of patients than we originally anticipated and/or for a longer follow-up period or change the data collection requirements or data analysis applicable to our clinical trials. Delays in patient enrollment or failure of patients to continue to participate in a clinical trial may cause an increase in costs and delays in the approval and attempted commercialization of our products or result in the failure of the clinical trial. In addition, despite considerable time and expense invested in our clinical trials, the FDA and foreign regulatory authorities may not consider our data adequate to demonstrate safety and efficacy. Although FDA regulations allow submission of data from clinical trials outside the U.S., there can be no assurance that such data will be accepted or that the FDA will not apply closer scrutiny to such data. Increased costs and delays necessary to generate appropriate data, or failures in clinical trials could adversely affect our business, operating results and prospects. In the U.S., clinical studies for our products will be reviewed through the Investigational New Drug, or IND, pathway for biologics or combination products.
If the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical development do not perform as contractually-required or expected, we may not be able to obtain regulatory approval for or commercialize our products.
We do not have the ability to independently conduct our pre-clinical and clinical trials for our products and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct, or assist us in conducting, such trials, including data collection and analysis. We do not have direct control over such third parties’ personnel or operations. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or any regulatory requirements, or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to seek or obtain regulatory approval for, or successfully commercialize, our products on a timely basis, if at all. Our business, operating results and prospects may also be adversely affected. Furthermore, any third-party clinical trial investigators pertaining to our products may be delayed in conducting our clinical trials for reasons outside of their control.
| 19 |
Risks Associated with Regulatory Approvals
If we fail to obtain, or experience significant delays in obtaining, regulatory approvals in the U.S. and the EU for our products, including those for the esophagus and airways, or are unable to maintain such clearances or approvals for our products, our ability to commercially distribute and market these products would be adversely impacted.
We currently do not have regulatory approval to market any of our implant products, including those for the esophagus and airways (trachea and bronchus). Our products are subject to rigorous regulation by the FDA, and numerous other federal and state governmental authorities in the U.S., as well as foreign governmental authorities. In the U.S., the FDA permits commercial distribution of new medical products only after approval of a Premarket Approval (PMA), NDA or BLA, unless the product is specifically exempt from those requirements. A PMA, NDA or BLA must be supported by extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the product for its intended use. There are similar approval processes in the EU and other foreign jurisdictions. Our failure to receive or obtain such clearances or approvals on a timely basis or at all would have an adverse effect on our results of operations.
The first bioengineered trachea implant approved in the U.S. using our first-generation trachea implant was approved under the IND pathway through CBER for a compassionate use. Such initial U.S. surgery was led by Professor Paolo Macchiarini, M.D., a surgeon pioneering tracheal replacement techniques. Dr. Macchiarini was not employed or affiliated with our company, and we did no pay him any compensation or consulting fees. In June 2014, shortly after our Chief Medical Officer joined our company, we ceased support of any human surgeries with Dr. Macchiarini. Since the time we withdrew from involvement with Dr. Macchiarini, allegations that Dr. Macchiarini had failed to obtain informed consent and accurately report patient conditions, among other things, for surgeries performed at the Karolinska Institutet in Stockholm, Sweden, were made public.
The Karolinska Institutet investigated the allegations and concluded that while in some instances Dr. Macchiarini did act without due care, his actions did not qualify as scientific misconduct. Subsequent to this investigation, further negative publicity and claims continued to be released questioning the conduct of Dr. Macchiarini, the Karolinska Institutet, the Krasnodar Regional Hospital in Krasnodar, Russia as well as our company relating to surgeries performed by Dr. Macchiarini and other surgeons at such facilities. In February 2015, the Karolinska Institutet announced that it would conduct an additional investigation into the allegations made about Dr. Macchiarini and the Karolinska Institutet’s response and actions in the earlier investigation. In March 2015, the Karolinska Institutet announced that it was terminating Dr. Macchiarini’s employment, and in December 2016 the Karolinska Institutet found Dr. Macchiarini, along with three co-authors, guilty of scientific misconduct. These allegations, the results of the investigation and any further actions that may be taken in connection with these matters, have and may continue to harm the perception of our products or company and make it difficult to recruit patients for any clinical trials.
The FDA has informed us that our first-generation trachea product and our Cellspan esophageal implant would be viewed by the FDA as a combination product comprised of a biologic (cells) and a medical device component. Nevertheless, we cannot be certain how the FDA will regulate our products. The FDA may require us to obtain marketing clearance and approval from multiple FDA centers. The review of combination products is often more complex and more time consuming than the review of products under the jurisdiction of only one center within the FDA.
While the FDA has informed us that our first-generation trachea product and our Cellspan esophageal implant would be regulated by the FDA as a combination product, we cannot be certain that any of our other products would also be regulated by the FDA as a combination product. For a combination product, the Office of Combination Products, or OCP, within FDA can determine which center or centers within the FDA will review the product and under what legal authority the product will be reviewed. Generally, the center within the FDA that has the primary role in regulating a combination product is determined based on the primary mode of action of the product. Generally, if the primary mode of action is as a device CDRH takes the lead. Alternatively, if the primary mode of action is cellular, then the Center for Biologics Evaluation and Research takes the lead. On August 29, 2013, we received written confirmation from FDA’s Office of Combination Products that FDA intends to regulate our first-generation trachea product as a combination product under the primary jurisdiction of CBER. On October 18, 2016, we also received written confirmation from FDA’s Center for Biologics Evaluation and Research, or CBER, that FDA intends to regulate our Cellspan esophageal implant as a combination product under the primary jurisdiction of CBER. We further understand that CBER may choose to consult or collaborate with CDRH with respect to the characteristics of the synthetic scaffold component of our product based on CBER’s determination of need for such assistance.
The process of obtaining FDA marketing approval is lengthy, expensive, and uncertain, and we cannot be certain that our products, including products pertaining to the esophagus, airways, or otherwise, will be cleared or approved in a timely fashion, or at all. In addition, the review of combination products is often more complex and can be more time consuming than the review of a product under the jurisdiction of only one center within the FDA.
We cannot be certain that the FDA will not elect to have our combination products reviewed and regulated by only one FDA center and/or different legal authority, in which case the path to regulatory approval would be different and could be more lengthy and costly.
| 20 |
If the FDA does not approve or clear our products in a timely fashion, or at all, our business and financial condition will be adversely affected.
In the EU, our esophagus product will likely be regulated as a combined advanced therapy medicinal product and our other products, including for the trachea or bronchus, may also be viewed as advanced therapy medicinal products, which could delay approvals and clearances and increase costs of obtaining such approvals and clearances.
On May 28, 2014, we received notice from the European Medicines Agency (EMA) that our first-generation trachea product would be regulated as a combined advanced therapy medicinal product. While we have not had any formal interaction with the EMA with respect to our Cellframe implant technology, including pertaining to the esophagus, we believe that such implant technology would likely be regulated as a combined advanced therapy medicinal product. In the event of such classification, it would be necessary to seek a marketing authorization for these products granted by the European Commission before being marketed in the EU.
Other products we may develop, including any products pertaining to the airways or otherwise, may similarly be regulated as advanced therapy medicinal products or combined advanced therapy medicinal products. The regulatory procedures leading to marketing approval of our products vary among jurisdictions and can involve substantial additional testing. Compliance with the FDA requirements does not ensure clearance or approval in other jurisdictions, and the ability to legally market our products in any one foreign country does not ensure clearance, or approval by regulatory authorities in other foreign jurisdictions. The foreign regulatory process leading to the marketing of the products may include all of the risks associated with obtaining FDA approval in addition to other risks. In addition, the time required to comply with foreign regulations and market products may differ from that required to obtain FDA approval, and we may not obtain foreign approval or clearance on a timely basis, if at all.
The United Kingdom’s vote to leave the European Union will have uncertain effects and could adversely affect us.
On June 23, 2016, eligible members of the electorate in the United Kingdom (the U.K.) decided by referendum to leave the European Union, commonly referred to as "Brexit". On March 29, 2017, the U.K. formally notified the E.U. of its intention to withdraw pursuant to the Treaty on European Union. The withdrawal of the U.K. from the E.U. will take effect either when agreed upon or, in the absence of such an agreement, two years after the U.K. provided its notice of withdrawal. It appears likely that this withdrawal will involve a process of lengthy negotiations between the U.K. and the E.U. member states to determine the terms of the withdrawal as well as the U.K.’s relationship with the E.U. going forward. The effects of Brexit will depend on any agreements the U.K. makes to retain access to E.U. markets either during a transitional period or more permanently. Since a significant proportion of the regulatory framework in the U.K. is derived from European Union directives and regulations, the referendum could materially change the regulatory regime applicable to the approval of any product candidates in the U.K. In addition, since the EMA is located in the U.K., the implications for the regulatory review process in the European Union has not been clarified and could result in relocation of the EMA or a disruption in the EMA review process.
Further, Brexit could adversely affect European and worldwide economic or market conditions and could contribute to instability in global financial markets. Brexit is likely to lead to legal uncertainty and potentially divergent national laws and regulations as the U.K. determines which E.U. laws to replace or replicate. Any of these effects of Brexit, and others we cannot anticipate, could adversely affect our business and financial condition.
Risk Associated with Product Marketing
Even if our products are cleared or approved by regulatory authorities, if we or our suppliers fail to comply with ongoing FDA or other foreign regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain clearance or approval in the U.S. or the EU, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA and other domestic and foreign regulatory authorities or notified bodies. In particular, we and our suppliers are required to comply with the FDA’s Quality System Regulations, or QSR, and Good Manufacturing Practices, or GMPs, for our medical products, and International Standards Organization, or ISO, regulations for the manufacture of our products and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain clearance or approval. Manufacturing may also be subject to controls by the FDA for parts of the system or combination products that the FDA may find are controlled by the biologics regulations. Equivalent regulatory obligations apply in foreign jurisdictions. Regulatory authorities, such as the FDA, the competent authorities of the EU Member States, the European Medicines Agency and notified bodies, enforce the QSR, GMP and other applicable regulations in the U.S. and in foreign jurisdictions through periodic inspections. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA and other regulatory authorities or notified bodies in the U.S. or in foreign jurisdictions, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in, among other things, any of the following enforcement actions:
| 21 |
| • | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | unanticipated expenditures to address or defend such actions; |
| • | customer notifications for repair, replacement, refunds; |
| • | recall, detention or seizure of our products; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | withdrawing BLA or NDA approvals that have already been granted; |
| • | withdrawal of the marketing authorization granted by the European Commission or delay in obtaining such marketing authorization; |
| • | withdrawal of the CE Certificates of Conformity granted by the notified body or delay in obtaining these certificates; |
| • | refusal to grant export approval for our products; and |
| • | criminal prosecution. |
Post-market enforcement actions can generate adverse commercial consequences.
Even if regulatory approval of a product is granted, such clearance or approval may be subject to limitations on the intended uses for which the product may be marketed and reduce our potential to successfully commercialize the product and generate revenue from the product. If the FDA or a foreign regulatory authority determines that our promotional materials, labeling, training or other marketing or educational activities constitute promotion of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with medical products reporting requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
| 22 |
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in March 2010, the Affordable Care Act, or the ACA, was passed, which substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA, among other things, subjects biological products to potential competition by lower-cost biosimilars, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations, establishes annual fees and taxes on manufacturers of certain branded prescription drugs, and creates a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (70% commencing January 1, 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Some of the provisions of the ACA have yet to be fully implemented, while certain provisions have been subject to judicial and Congressional challenges, as well as efforts by the Trump administration to repeal or replace certain aspects of the ACA. Since January 2017, President Trump has signed two Executive Orders designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA.
Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017, or TCJA, includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, or the BBA, among other things, amends the ACA, effective January 1, 2019, to reduce the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole.” The effect that the ACA and its possible repeal and replacement may have on our business remains unclear.
Other legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2027 unless additional congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers.
Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, the Middle Class Tax Relief and Job Creation Act of 2012 required that the Centers for Medicare & Medicaid Services, or CMS, the agency responsible for administering the Medicare program, reduce the Medicare clinical laboratory fee schedule by 2% in 2013, which served as a base for 2014 and subsequent years. In addition, effective January 1, 2014, CMS also began bundling the Medicare payments for certain laboratory tests ordered while a patient received services in a hospital outpatient setting. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for any product candidate we develop or complementary diagnostics or companion diagnostics or additional pricing pressures.
Additionally, there has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
Any of these regulatory changes and events could limit our ability to form collaborations and our ability to commercialize our products, and if we fail to comply with any such new or modified regulations and requirements it could adversely affect our business, operating results and prospects.
| 23 |
If we fail to complete the required IRS forms for exemptions, make timely semi-monthly payments of collected excise taxes, or submit quarterly reports as required by the Medical Device Excise Tax, we may be subject to penalties, such as Section 6656 penalties for any failure to make timely deposits.
Section 4191 of the Internal Revenue Code, enacted by Section 1405 of the Health Care and Education Reconciliation Act of 2010, Public Law 111-152 (124 Stat. 1029 (2010)), in conjunction with the Patient Protection and Affordable Care Act, Public Law 111-148 (124 Stat. 119 (2010)), imposed as of January 1, 2013, an excise tax on the sale of certain medical devices. The excise tax imposed by Section 4191 is 2.3% of the price for which a taxable medical device is sold within the U.S.
While the provision for a medical device excise tax has been suspended for 2017 and 2016, there is no guarantee that the moratorium will be approved for subsequent years. The excise tax will apply to future sales of any company medical device listed with the FDA under Section 510(j) of the Federal Food, Drug, and Cosmetic Act and 21 C.F.R. Part 807, unless the device falls within an exemption from the tax, such as the exemption governing direct retail sale of devices to consumers or for foreign sales of these devices. We will need to assess to what extent this excise tax may impact the sales price and distribution agreements under which any of our products are sold in the U.S. We also expect general and administrative expense to increase due to the medical device excise tax. We will need to submit IRS forms applicable to relevant exemptions, make semi-monthly payments of any collected excise taxes, and make timely (quarterly) reports to the IRS regarding the excise tax. To the extent we do not comply with the requirements of the Medical Device Excise Tax we may be subject to penalties.
Financial and Operating Risks
Our audited financial statements for the year ended December 31, 2017 contain a going concern qualification. Our financial status creates doubt whether we will continue as a going concern. We will need additional funds in the near future and our operations will be adversely affected if we are unable to obtain needed funding.
In their audit report dated April 2, 2018, included in this Form 10-K, our independent registered public accounting firm included a “going concern” qualification as to our ability to continue as a going concern. We believe that if we do not raise additional capital from outside sources in the very near future, we may be forced to curtail or cease our operations. We believe that our existing cash resources will be sufficient to fund our planned operations into the third quarter of 2018. Our cash requirements and cash resources will vary significantly depending upon the timing, financial and other resources that will be required to complete ongoing development and pre-clinical and clinical testing of our products as well as regulatory efforts and collaborative arrangements necessary for our products that are currently under development. In addition to development and other costs, we expect to incur capital expenditures from time to time. These capital expenditures will be influenced by our regulatory compliance efforts, our success, if any, at developing collaborative arrangements with strategic partners, our needs for additional facilities and capital equipment and the growth, if any, of our business in general. We will require additional funding by the end of the third quarter of 2018 to continue our anticipated operations and support our capital needs. We may seek to raise necessary funds through a combination of public or private equity offerings, debt financings, other financing mechanisms, strategic collaborations and licensing arrangements. We may not be able to obtain additional financing on terms favorable to us, if at all. In addition, general market conditions may make it difficult for us to seek financing from the capital markets.
| 24 |
Any additional equity financings could result in significant dilution to our stockholders and possible restrictions on subsequent financings. Debt financing, if available, could result in agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, making capital expenditures or paying dividends. Other financing mechanisms may involve selling intellectual property rights, payment of royalties or participation in our revenue or cash flow. In addition, in order to raise additional funds through strategic collaborations or licensing arrangements, we may be required to relinquish certain rights to some or all of our technologies or products. If we cannot raise funds or engage strategic partners on acceptable terms when needed, we may not be able to continue our research and development activities, develop or enhance our products, take advantage of future opportunities, grow our business or respond to competitive pressures or unanticipated requirements.
We have generated insignificant revenue to date and have an accumulated deficit. We anticipate that we will incur losses for the foreseeable future. We may never achieve or sustain profitability.
We have generated insignificant revenues to date and we have generated no revenues from sales of any clinical products, and as of December 31, 2017, we had an accumulated deficit of approximately $48.2 million. We expect to continue to experience losses in the foreseeable future due to our limited anticipated revenues and significant anticipated expenses. We do not anticipate that we will achieve meaningful revenues for the foreseeable future. In addition, we expect that we will continue to incur significant operating expenses as we continue to focus on additional research and development, preclinical testing, clinical testing and regulatory review and/or approvals of our products and technologies. As a result, we cannot predict when, if ever, we might achieve profitability and cannot be certain that we will be able to sustain profitability, if achieved.
Our products are in an early stage of development. If we are unable to develop or market any of our products, our financial condition will be negatively affected, and we may have to curtail or cease our operations.
We are in the early stage of product development. One must evaluate us in light of the uncertainties and complexities affecting an early stage biotechnology company. Our products require additional research and development, preclinical testing, clinical testing and regulatory review and/or approvals or clearances before marketing. In addition, we may not succeed in developing new products as an alternative to our existing portfolio of products. If we fail to successfully develop and commercialize our products, including our esophageal or airway products, our financial condition may be negatively affected, and we may have to curtail or cease our operations.
We have a limited operating history and it is difficult to predict our future growth and operating results.
We have a limited operating history and limited operations and assets. Accordingly, one should consider our prospects in light of the costs, uncertainties, delays and difficulties encountered by companies in the early stage of development. As such, our development timelines have been and may continue to be subject to delay that could negatively affect our cash flow and our ability to develop or bring products to market, if at all. Our estimates of patient population are based on published data and analysis of external databases by third parties and are subject to uncertainty and possible future revision as they often require inference or extrapolations from one country to another or one patient condition to another.
Our prospects must be considered in light of inherent risks, expenses and difficulties encountered by all early stage companies, particularly companies in new and evolving markets, such as bioengineered organ implants, and regenerative medicine. These risks include, but are not limited to, unforeseen capital requirements, delays in obtaining regulatory approvals, failure to gain market acceptance and competition from foreseen and unforeseen sources.
If we fail to retain key personnel, we may not be able to compete effectively, which would have an adverse effect on our operations.
Our success is highly dependent on the continued services of key management, technical and scientific personnel and collaborators. Our management and other employees may voluntarily terminate their employment at any time upon short notice. The loss of the services of any member of our senior management team, including our Chief Executive Officer, James McGorry, our Chief Financial Officer, Thomas McNaughton, and our other key scientific, technical and management personnel, may significantly delay or prevent the achievement of product development and other business objectives.
If our collaborators do not devote sufficient time and resources to successfully carry out their duties or meet expected deadlines, we may not be able to advance our products in a timely manner or at all.
We are currently collaborating with multiple academic researchers and clinicians at a variety of research and clinical institutions. Our success depends in part on the performance of our collaborators. Some collaborators may not be successful in their research and clinical trials or may not perform their obligations in a timely fashion or in a manner satisfactory to us. Typically, we have limited ability to control the amount of resources or time our collaborators may devote to our programs or potential products that may be developed in collaboration with us. Our collaborators frequently depend on outside sources of funding to conduct or complete research and development, such as grants or other awards. In addition, our academic collaborators may depend on graduate students, medical students, or research assistants to conduct certain work, and such individuals may not be fully trained or experienced in certain areas, or they may elect to discontinue their participation in a particular research program, creating an inability to complete ongoing research in a timely and efficient manner. As a result of these uncertainties, we are unable to control the precise timing and execution of any experiments that may be conducted.
| 25 |
Although we have formal co-development collaboration agreements with Mayo Clinic and Connecticut Children’s Medical Center, we do not have formal agreements in place with other collaborators, and most of our collaborators retain the ability to pursue other research, product development or commercial opportunities that may be directly competitive with our programs. If any of our collaborators elect to prioritize or pursue other programs in lieu of ours, we may not be able to advance product development programs in an efficient or effective manner, if at all. If a collaborator is pursuing a competitive program and encounters unexpected financial or capability limitations, they may be motivated to reduce the priority placed on our programs or delay certain activities related to our programs. Any of these developments could harm or slow our product and technology development efforts.
Public perception of ethical and social issues surrounding the use of cell technology may limit or discourage the use of our technologies, which may reduce the demand for our products and technologies and reduce our revenues.
Our success will depend in part upon our collaborators’ ability to develop therapeutic approaches incorporating, or discovered through, the use of cells. If either bioengineered organ implant technology is perceived negatively by the public for social, ethical, medical or other reasons, governmental authorities in the U.S. and other countries may call for prohibition of, or limits on, cell-based technologies and other approaches to bioengineering and tissue engineering. Although the surgeons using our products have not, to date, used the more controversial stem cells derived from human embryos or fetuses in the human transplant surgeries using our products, claims that human-derived stem cell technologies are ineffective or unethical may influence public attitudes. The subject of cell and stem cell technologies in general has at times received negative publicity and aroused public debate in the U.S. and some other countries. Ethical and other concerns about such cells could materially harm the market acceptance of our products.
Our products will subject us to liability exposure.
We face an inherent risk of product liability claims, especially with respect to our products that will be used within the human body, including the scaffolds we manufacture. Product liability coverage is expensive and sometimes difficult to obtain. We may not be able to obtain or maintain insurance at a reasonable cost. We may be subject to claims for liabilities for unsuccessful outcomes of surgeries involving our products, which may include claims relating to patient death. We may also be subject to claims for liabilities relating to patients that suffer serious complications or death during or following transplants involving our products, including the patients who had surgeries utilizing our first-generation scaffold product or our bioreactor technology, or patients that may have surgeries utilizing any of our products in the future. Our current product liability coverage is $15 million per occurrence and in the aggregate. We will need to increase our insurance coverage if and when we begin commercializing any of our products. There can be no assurance that existing insurance coverage will extend to other products in the future. Any product liability insurance coverage may not be sufficient to satisfy all liabilities resulting from product liability claims. A successful claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable items, if at all. If claims against us substantially exceed our coverage, then our business could be adversely impacted. Regardless of whether we are ultimately successful in any product liability litigation, such litigation could consume substantial amounts of our financial and managerial resources and could result in, among others:
| • | significant awards against us; |
| • | substantial litigation costs; |
| • | injury to our reputation and the reputation of our products; |
| • | withdrawal of clinical trial participants; and |
| • | adverse regulatory action. |
Any of these results would substantially harm our business.
If restrictions on reimbursements or other conditions imposed by payers limit our customers’ actual or potential financial returns on our products, our customers may not purchase our products or may reduce their purchases.
Our customers’ willingness to use our products will depend in part on the extent to which coverage for these products is available from government payers, private health insurers and other third-party payers. These payers are increasingly challenging the price of medical products and services. Significant uncertainty exists as to the reimbursement status of newly approved treatments and products in the fields of biotechnology and regenerative medicine, and coverage and adequate payments may not be available for these treatments and products. In addition, third-party payers may require additional clinical trial data to establish or continue reimbursement coverage. These clinical trials, if required, could take years to complete and could be expensive. There can be no assurance that the payers will agree to continue reimbursement or provide additional coverage based upon these clinical trials. Failure to obtain adequate reimbursement would result in reduced sales of our products.
| 26 |
We depend upon a single-source supplier for the hardware used for our organ bioreactor control and acquisition system. The loss of this supplier, or future single-source suppliers we may rely on, or their failure to provide us with an adequate supply of their products or services on a timely basis, could adversely affect our business.
We currently have a single supplier for certain components that we use for our organ bioreactor control and acquisition systems as well as materials used in scaffolds. We may also rely on other single-source suppliers for critical components of our products in the future. If we were unable to acquire hardware or other products or services from applicable single-source suppliers, we could experience a delay in developing and manufacturing our products.
We use and generate hazardous materials in our business and must comply with environmental laws and regulations, which can be expensive.
Our research, development and manufacturing involve the controlled use of hazardous chemicals, and we may incur significant costs as a result of the need to comply with numerous laws and regulations. For example, certain volatile organic laboratory chemicals we use, such as fluorinated hydrocarbons, must be disposed of as hazardous waste. We are subject to laws and regulations enforced by the FDA, foreign health authorities and other regulatory requirements, including the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other current and potential federal, state, local and foreign laws and regulations governing the use, manufacture, storage, handling and disposal of our products, materials used to develop and manufacture our products, and resulting waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, our operations could be interrupted. Further, we could be held liable for any damages that result and any such liability could exceed our resources.
Our products are novel and will require market acceptance.
Even if we receive regulatory approvals for the commercial use of our products, their commercial success will depend upon acceptance by physicians, patients, third party payers such as health insurance companies and other members of the medical community. Market acceptance of our products is also dependent upon our ability to provide acceptable evidence and the perception of the positive characteristics of our products relative to existing or future treatment methods, including their safety, efficacy and/or other positive advantages. If our products fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business. Market acceptance of, and demand for, any product that we may develop and commercialize will depend on many factors, both within and outside of our control. If our products receive only limited market acceptance, our business, financial condition and results of operations would be materially and adversely affected.
Our long-term growth depends on our ability to develop products for other organs.
Our growth strategy includes expanding the use of our products in treatments pertaining to organs other than the esophagus and airways, such as the lungs, GI tract, among others. These other organs are more complex than the esophagus and airways. There is no assurance that we will be able to successfully apply our technologies to these other more complex organs, which might limit our expected growth.
Our success will depend partly on our ability to operate without infringing on, or misappropriating, the intellectual property or confidentiality rights of others.
We may be sued for infringing on the intellectual property or confidentiality rights of others, including the patent rights, trademarks and trade names and confidential information of third parties. We have received correspondence from legal counsel to Nanofiber Solutions, Inc., or NFS, claiming that in developing our scaffold product and related intellectual property, we may have committed misappropriation, unauthorized use and disclosure of confidential information, and possible infringement of intellectual property rights of NFS. We have received correspondence from legal counsel to UCL Business PLC, or UCLB, challenging the validity of the assignment of certain patent applications that have been assigned to us by Dr. Macchiarini. We have also received correspondence from an academic researcher implying that one of our research bioreactor products may violate an issued patent. We do not believe that our current products violate this patent. To the extent that any of such claims are valid, if we had utilized, or were to utilize, such patent applications or patents without an agreement from the owner thereof, it could result in infringement of the intellectual property rights of the respective owner. Intellectual property and related litigation is costly and the outcome is uncertain. If we do not prevail in any such intellectual property or related litigation, in addition to any damages we might have to pay, we could be required to stop the infringing activity, or obtain a license to or design around the intellectual property or confidential information in question. If we are unable to obtain a required license on acceptable terms or are unable to design around any third-party patent, we may be unable to sell some of our products and services, which could result in reduced revenue.
We may be involved in lawsuits to protect or enforce our patents that would be expensive and time consuming.
In order to protect or enforce our patent rights, we may initiate patent litigation against third parties. We may also become subject to interference proceedings conducted in the patent and trademark offices of various countries to determine the priority of inventions. The defense and prosecution, if necessary, of intellectual property suits, interference proceedings and related legal and administrative proceedings would be costly, and may divert our technical and management personnel from their normal responsibilities. We may not prevail in any of these suits should they occur. An adverse determination of any litigation or defense proceedings could put our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of being rejected and patents not being issued.
| 27 |
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. For example, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments in the litigation. Securities analysts or investors may perceive these announcements to be negative, which could cause the market price of our stock to decline.
If we are unable to effectively protect our intellectual property, third parties may use our technology, which would impair our ability to compete in our markets.
Our continued success will depend significantly on our ability to obtain and maintain meaningful patent protection for certain of our products throughout the world. Patent law relating to the scope of claims in the biotechnology, regenerative medicine, and medical device fields in which we operate is still evolving. The degree of future protection for our proprietary rights is uncertain. We may rely on patents to protect a significant part of our intellectual property and to enhance our competitive position. However, our presently pending or future patent applications may not be accepted and patents might not be issued, and any patent previously issued to us may be challenged, invalidated, held unenforceable or circumvented. Furthermore, the claims in patents which have been issued or which may be issued to us in the future may not be sufficiently broad to prevent third parties from producing competing products similar to our products. We may also operate in countries where we do not have patent rights and in those countries we would not have patent protection. We also rely on trademarks and trade names in our business. The laws of various foreign countries in which we compete may not protect our intellectual property to the same extent as do the laws of the U.S. If we fail to obtain adequate patent protection for our proprietary technology, our ability to be commercially competitive could be materially impaired. It is also possible that our intellectual property may be stolen via cyber-attacks or similar methods.
In addition to patent protection, we also rely on protection of trade secrets, know-how and confidential and proprietary information. To maintain the confidentiality of trade-secrets and proprietary information, we generally seek to enter into confidentiality agreements with our employees, consultants and strategic partners upon the commencement of a relationship. However, we may not be able to obtain these agreements in all circumstances in part due to local regulations. In the event of unauthorized use or disclosure of this information, these agreements, even if obtained, may not provide meaningful protection for our trade-secrets or other confidential information. In addition, adequate remedies may not exist in the event of unauthorized use or disclosure of this information. The loss or exposure of our trade secrets and other proprietary information would impair our competitive advantages and could have a material adverse effect on our operating results, financial condition and future growth prospects.
Our competitors and potential competitors may have greater resources than we have and may develop products and technologies that are more effective or commercially attractive than our products and technologies or may develop competing relationships with our key collaborators.
We expect to compete with multiple pharmaceutical, biotechnology, medical device and scientific research product companies. Companies working in competing areas include, among others, Aldagen, Asterias Biotherapeutics, Athersys, BioTime, Caladrius Biosciences, Celgene, Cytori Therapeutics, E. I. du Pont de Nemours and Company, InVivo Therapeutics, Mesoblast, Miramatrix Medical, Nanofiber Solutions, Neuralstem, Organovo, Osiris Therapeutics, Pluristem Therapeutics, Smiths Medical, Tissue Genesis, Inc., Tissue Growth Technologies, United Therapeutics, Vericel Corporation, and W.L. Gore and Associates. In addition, there are many academic and clinical centers that are developing bioengineered or regenerative organ technologies that may one day become competitors for us. Many of our competitors and potential competitors have substantially greater financial, technological, research and development, marketing, and personnel resources than we do. We cannot, with any accuracy, forecast when or if these companies are likely to bring bioengineered organ or regenerative medicine products to market for indications that we are also pursuing. Many of these potential competitors may be further along in the process of product development and also operate large, company-funded research and development programs.
We expect that other products will compete with our current and future products based on efficacy, safety, cost, and intellectual property positions. While we believe that these will be the primary competitive factors, other factors include obtaining marketing exclusivity under certain regulations, availability of supply, manufacturing, marketing and sales expertise and capability, and reimbursement coverage. Our competitors may develop or market products that are more effective or commercially attractive than our current or future products and may also develop competing relationships with our key collaborators. In addition, we may face competition from new entrants into the field. We may not have the financial resources, technical expertise or marketing, distribution or support capabilities to compete successfully in the future. The effects of any such actions of our competitors may have a material adverse effect on our business, operating results and financial condition.
| 28 |
If we do not successfully manage our growth, our business goals may not be achieved.
To manage growth, we will be required to continue to improve existing, and implement additional, operational and financial systems, procedures and controls, and hire, train and manage additional employees. Our current and planned personnel, systems, procedures and controls may not be adequate to support our anticipated growth and we may not be able to hire, train, retain, motivate and manage required personnel. Competition for qualified personnel in the biotechnology and regenerative medicine area is intense, and we operate in several geographic locations where labor markets are particularly competitive, including Boston, Massachusetts, where demand for personnel with these skills is extremely high and is likely to remain high. As a result, competition for qualified personnel is intense and the process of hiring suitably qualified personnel is often lengthy and expensive, and may become more expensive in the future. If we are unable to hire and retain a sufficient number of qualified employees or otherwise manage our growth effectively, our ability to conduct and expand our business could be seriously reduced.
We are exposed to a variety of risks relating to our international sales and operations, including fluctuations in exchange rates, local economic conditions and delays in collection of accounts receivable.
We intend to generate significant revenues outside the U.S. in multiple foreign currencies including Euros, British pounds, and in U.S. dollar-denominated transactions conducted with customers who generate revenue in currencies other than the U.S. dollar. For those foreign customers who purchase our products in U.S. dollars, currency fluctuations between the U.S. dollar and the currencies in which those customers do business may have a negative impact on the demand for our products in foreign countries where the U.S. dollar has increased in value compared to the local currency.
Since we have vendors and customers outside the U.S. and we may generate revenues and incur operating expenses in multiple foreign currencies, we will experience currency exchange risk with respect to any foreign currency-denominated revenues and expenses. We cannot predict the consolidated effects of exchange rate fluctuations upon our future operating results because of the number of currencies involved, the variability of currency exposure and the potential volatility of currency exchange rates. Our international activities subject us to laws regarding sanctioned countries, entities and persons, customs, import-export, laws regarding transactions in foreign countries, the U.S. Foreign Corrupt Practices Act and local anti-bribery and other laws regarding interactions with healthcare professionals. Among other things, these laws restrict, and in some cases prohibit, U.S. companies from directly or indirectly selling goods, technology or services to people or entities in certain countries. In addition, these laws require that we exercise care in structuring our sales and marketing practices in foreign countries.
Local economic conditions, legal, regulatory or political considerations, disruptions from strikes, the effectiveness of our sales representatives and distributors, local competition and changes in local medical practice could also affect our sales to foreign markets. Relationships with customers and effective terms of sale frequently vary by country, often with longer-term receivables than are typical in the U.S.
Comprehensive tax reform legislation could adversely affect our business and financial condition.
On December 22, 2017, the U.S. government enacted the TCJA, which significantly reforms the Internal Revenue Code of 1986, as amended. The TCJA, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, effective January 1, 2018; limitation of the tax deduction for interest expense; limitation of the deduction for net operating losses and elimination of net operating loss carrybacks, in each case, for losses arising in taxable years beginning after December 31, 2017 (though any such tax losses may be carried forward indefinitely); and modifying or repealing many business deductions and credits, including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions generally referred to as “orphan drugs”. The tax rate change resulted in (i) a reduction in the gross amount of our deferred tax assets recorded as of December 31, 2017, without an impact on the net amount of our deferred tax assets, which are recorded with a full valuation allowance. We continue to examine the impact this tax reform legislation may have on our business. However, the effect of the TCJA on us and our affiliates, whether adverse or favorable, is uncertain and may not become evident for some period of time. We urge investors to consult with their legal and tax advisers regarding the implications of the TCJA on an investment in our common stock.
Changes in the European regulatory environment regarding privacy and data protection regulations could have a material adverse impact on our results of operations.
The E.U. has recently adopted a comprehensive overhaul of its data protection regime in the form of the General Data Protection Regulation (GDPR), which comes into effect in May 2018. GDPR extends the scope of the existing E.U. data protection law to foreign companies processing personal data of E.U. residents. The regulation imposes a strict data protection compliance regime with severe penalties of 4% of worldwide turnover or €20 million, whichever is greater, and includes new rights such as the right of erasure of personal data. Although the GDPR will apply across the E.U., as has been the case under the current data protection regime, E.U. Member States have some national derogations and local data protection authorities (DPAs) will still have the ability to interpret the GDPR, which has the potential to create inconsistencies on a country-by-country basis. Implementation of, and compliance with the GDPR could increase our cost of doing business and/or force us to change our business practices in a manner adverse to our business. In addition, violations of the GDPR may result in significant fines, penalties and damage to our brand and business which could, individually or in the aggregate, materially harm our business and reputation.
| 29 |
Risks Related To Our Separation From Harvard Bioscience
If the Separation and related distribution of all of the shares of our common stock by Harvard Bioscience, together with certain related transactions, does not qualify as a transaction that is generally tax-free for U.S. federal income tax purposes, Harvard Bioscience could be subject to significant tax liability and, in certain circumstances, we could be required to indemnify Harvard Bioscience for material taxes pursuant to indemnification obligations under the tax sharing agreement.
Harvard Bioscience has informed us that on June 28, 2013 it received a Supplemental Ruling to the Private Letter Ruling dated March 22, 2013 from the IRS to the effect that, among other things, the Separation and related distribution of all of the shares of our common stock by Harvard Bioscience, or the Distribution, will qualify as a transaction that is tax-free for U.S. federal income tax purposes under Section 355 and 368(a)(1)(D) of the Internal Revenue Code continuing in effect. The private letter and supplemental rulings and the tax opinion that Harvard Bioscience received from Burns & Levinson LLP, special counsel to Harvard Bioscience, rely on certain representations, assumptions and undertakings, including those relating to the past and future conduct of our business, and neither the private letter and supplemental rulings nor the opinion would be valid if such representations, assumptions and undertakings were incorrect. Moreover, the private letter and supplemental rulings do not address all the issues that are relevant to determining whether the Distribution will qualify for tax-free treatment. Notwithstanding the private letter and supplemental rulings and opinion, the IRS could determine the Distribution should be treated as a taxable transaction for U.S. federal income tax purposes if, among other reasons, it determines any of the representations, assumptions or undertakings that were included in the request for the private letter and supplemental rulings are false or have been violated or if it disagrees with the conclusions in the opinion that are not covered by the IRS ruling.
If the Distribution fails to qualify for tax-free treatment, in general, Harvard Bioscience would be subject to tax as if it had sold our common stock in a taxable sale for its fair market value, and Harvard Bioscience stockholders who receive shares of our common stock in the Distribution would be subject to tax as if they had received a taxable Distribution equal to the fair market value of such shares.
Under the tax sharing agreement between Harvard Bioscience and us, we would generally be required to indemnify Harvard Bioscience against any tax resulting from the Distribution to the extent that such tax resulted from (i) an acquisition of all or a portion of our stock or assets, whether by merger or otherwise, (ii) other actions or failures to act by us, or (iii) any of our representations or undertakings being incorrect or violated. Our indemnification obligations to Harvard Bioscience and its subsidiaries, officers and directors are not limited by any maximum amount. If we are required to indemnify Harvard Bioscience or such other persons under the circumstances set forth in the tax sharing agreement, we may be subject to substantial liabilities.
| 30 |
We may have received better terms from unaffiliated third parties than the terms we received in our agreements with Harvard Bioscience.
The agreements related to the Separation, including the separation and distribution agreement, tax sharing agreement, transition services agreement and the other agreements, were negotiated in the context of the Separation while we were still part of Harvard Bioscience and, accordingly, may not reflect terms that would have resulted from arm’s-length negotiations among unaffiliated third parties. The terms of the agreements we negotiated in the context of the Separation related to, among other things, allocation of assets, liabilities, rights, indemnifications and other obligations among Harvard Bioscience and us. We may have received better terms from third parties because third parties may have competed with each other to win our business. One of the members of our Board of Directors is also a member of the Harvard Bioscience Board of Directors.
The ownership by one of our executive officers and one of our directors of shares of common stock, options, or other equity awards of Harvard Bioscience, as well as the continued role of our director with Harvard Bioscience may create, or may create the appearance of, conflicts of interest.
The ownership by one of our executive officers and one of our directors of shares of common stock, options, or other equity awards of Harvard Bioscience may create, or may create the appearance of, conflicts of interest. Because of their current or former positions with Harvard Bioscience, one of our executive officers, and one of our directors, own shares of Harvard Bioscience common stock, options to purchase shares of Harvard Bioscience common stock or other equity awards. The individual holdings of common stock, options to purchase common stock of Harvard Bioscience or our company or other equity awards, may be significant for some of these persons compared to such persons’ total assets. Ownership by our directors and officers of common stock or options to purchase common stock of Harvard Bioscience, or any other equity awards, creates, or, may create the appearance of, conflicts of interest when these directors and officers are faced with decisions that could have different implications for Harvard Bioscience than the decisions have for us.
Third parties may seek to hold us responsible for liabilities of Harvard Bioscience that we did not assume in our agreements.
In connection with the Separation, Harvard Bioscience has generally agreed to retain all liabilities that did not historically arise from our business. Third parties may seek to hold us responsible for Harvard Bioscience’s retained liabilities. Under our agreements with Harvard Bioscience, Harvard Bioscience has agreed to indemnify us for claims and losses relating to these retained liabilities. However, if those liabilities are significant and we are ultimately liable for them, we cannot assure you that we will be able to recover the full amount of our losses from Harvard Bioscience.
Any disputes that arise between us and Harvard Bioscience with respect to our past and ongoing relationships could harm our business operations.
Disputes may arise between Harvard Bioscience and us in a number of areas relating to our past and ongoing relationships, including:
| • | intellectual property, technology and business matters, including failure to make required technology transfers and failure to comply with non-compete provisions applicable to Harvard Bioscience and us; |
| • | labor, tax, employee benefit, indemnification and other matters arising from the Separation; |
| • | distribution and supply obligations; |
| • | employee retention and recruiting; |
| • | business combinations involving us; |
| • | sales or distributions by Harvard Bioscience of all or any portion of its ownership interest in us; and |
| • | business opportunities that may be attractive to both Harvard Bioscience and us. |
We may not be able to resolve any potential conflicts, and even if we do, the resolution may be less favorable than if we were dealing with a different party.
| 31 |
Risks Relating To Our Common Stock
Our principal stockholders hold a majority of voting power and will be able to exert significant control over us.
The stockholders who purchased shares of our common stock pursuant to a Securities Purchase Agreement dated December 27, 2017 collectively hold shares of common stock and Series D Convertible Preferred Stock that represent approximately 47% of all outstanding voting power, and as such may significantly influence the results of matters voted on by the Company’s shareholders. The interests of these stockholders may conflict with your interests. These stockholders have the right to nominate a majority of our Board of Directors and, therefore, effectively could control many other major decisions regarding our operations. This significant concentration of share ownership may adversely affect the trading price for our common stock because investors may perceive disadvantages in owning stock in companies with controlling stockholders.
Substantial sales of common stock have and may continue to occur, or may be anticipated, which have and could continue to cause our stock price to decline.
We expect that we will seek to raise additional capital from time to time in the future, which may involve the issuance of additional shares of common stock, or securities convertible into common stock. Since our February 2015 public offering, the holders of the shares of Series B Convertible Preferred Stock issued in that offering have converted all such shares and have sold substantially all of the common stock they received upon such conversion. We believe that the effect of these conversions and sales contributed, at that time, to a decline in the price of our common stock. On February 10, 2017, we completed a public offering of 1,000,000 shares of common stock and the issuance of warrants to purchase 1,000,000 shares of common stock. Additionally, we issued to the placement agent warrants to purchase 50,000 shares of common stock to the placement agent for the offering. The purchasers of the shares of common stock and warrants to purchase shares of common stock from that offering may sell significant quantities of our common stock in the market, which may cause a decline in the price of our common stock. Further, we cannot predict the effect, if any, that any additional market sales of common stock, or anticipation of such sales, or the availability of those shares of common stock for sale will have on the market price of our common stock. Any future sales of significant amounts of our common stock, or the perception in the market that this will occur, may result in a decline in the price of our common stock.
A trading market that will provide you with adequate liquidity may not develop for our common stock.
The current public market for our common stock has limited trading volume and liquidity. We cannot predict the extent to which investor interest in our company will lead to the development of a more active trading market in our common stock, or how liquid that market might be.
Our revenues, operating results and cash flows may fluctuate in future periods and we may fail to meet investor expectations, which may cause the price of our common stock to decline.
Variations in our quarterly and year-end operating results are difficult to predict and may fluctuate significantly from period to period. If our revenues or operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. In addition to the other factors discussed under these “Risk Factors,” specific factors that may cause fluctuations in our operating results include:
| • | demand and pricing for our products; |
| • | government or private healthcare reimbursement policies; |
| • | adverse events or publicity related to our products, our research or investigations, or our collaborators or other partners; |
| • | physician and patient acceptance of any of our current or future products; |
| • | manufacturing stoppages or delays; |
| • | introduction of competing products or technologies; |
| • | our operating expenses which fluctuate due to growth of our business; and |
| • | timing and size of any new product or technology acquisitions we may complete. |
The market price of our shares may fluctuate widely.
The market price of our common stock may fluctuate widely, depending upon many factors, some of which may be beyond our control, including:
| • | the success and costs of preclinical and clinical testing and obtaining regulatory approvals or clearances for our products; |
| • | the success or failure of surgeries and procedures involving the use our products; |
| • | a shift in our investor base; |
| • | our quarterly or annual results of operations, or those of other companies in our industry; |
| • | actual or anticipated fluctuations in our operating results due to factors related to our business; |
| • | changes in accounting standards, policies, guidance, interpretations or principles; |
| 32 |
| • | announcements by us or our competitors of significant acquisitions, dispositions or intellectual property developments or issuances; |
| • | the failure of securities analysts to cover our common stock; |
| • | changes in earnings estimates by securities analysts or our ability to meet those estimates; |
| • | the operating and stock price performance of other comparable companies; our issuance of equity, debt or other financing instruments; |
| • | overall market fluctuations; and |
| • | general macroeconomic conditions. |
Stock markets in general have experienced volatility that has often been unrelated to the operating performance of a particular company. These broad market fluctuations may adversely affect the trading price of our common stock.
Your percentage ownership will be diluted in the future.
Your percentage ownership will be diluted in the future because of equity awards that we expect will be granted to our directors, officers and employees, as well as shares of common stock, or securities convertible into common stock, we issue in connection with future capital raising or strategic transactions. Our 2013 Equity Incentive Plan provides for the grant of equity-based awards, including restricted stock, restricted stock units, stock options, stock appreciation rights and other equity-based awards to our directors, officers and other employees, advisors and consultants. In addition, your percentage ownership will be diluted by our issuance of common stock following the exercise of options, or vesting of restricted stock units, we issued pertaining to the adjustment and conversion of outstanding Harvard Bioscience equity awards as a result of the Separation. The issuance of any shares of our stock would dilute the proportionate ownership and voting power of existing security holders.
Provisions of Delaware law, of our amended and restated charter and amended and restated bylaws may make a takeover more difficult, which could cause our stock price to decline.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws and in the Delaware corporate law may make it difficult and expensive for a third party to pursue a tender offer, change in control or takeover attempt, which is opposed by management and the Board of Directors. Public stockholders who might desire to participate in such a transaction may not have an opportunity to do so. We have a staggered Board of Directors that makes it difficult for stockholders to change the composition of the Board of Directors in any one year. Any removal of directors will require a super-majority vote of the holders of at least 75% of the outstanding shares entitled to be cast on the election of directors which may discourage a third party from making a tender offer or otherwise attempting to obtain control of us. These anti-takeover provisions could substantially impede the ability of public stockholders to change our management and Board of Directors. Such provisions may also limit the price that investors might be willing to pay for shares of our common stock in the future.
Any issuance of preferred stock in the future may dilute the rights of our common stockholders.
Our Board of Directors has the authority to issue up to 2,000,000 shares of preferred stock and to determine the price, privileges and other terms of these shares. Our Board of Directors is empowered to exercise this authority without any further approval of stockholders. The rights of the holders of common stock may be adversely affected by the rights of future holders of preferred stock.
We have in the past issued, and we may at any time in the future issue, additional shares of authorized preferred stock. For example, in our December 2017 private placement transaction, we authorized 12,000 shares of Series D Convertible Preferred Stock, of which we issued 3,108 shares.
We do not intend to pay cash dividends on our common stock.
Currently, we do not anticipate paying any cash dividends to holders of our common stock. As a result, capital appreciation, if any, of our common stock will be a stockholder’s sole source of gain.
| 33 |
The JOBS Act allows us to postpone the date by which we must comply with certain laws and regulations and to reduce the amount of information provided in reports filed with the SEC. We cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are and we will remain an “emerging growth company” until the earliest to occur of (i) the last day of the fiscal year during which our total annual revenues equal or exceed $1 billion (subject to adjustment for inflation), (ii) the last day of the fiscal year following the fifth anniversary of the date of our first sale of common equity securities pursuant to an effective registration statement, (iii) the date on which we have, during the previous three-year period, issued more than $1 billion in non-convertible debt, or (iv) the date on which we are deemed a “large accelerated filer” under the Securities and Exchange Act of 1934, as amended, or the Exchange Act. For so long as we remain an “emerging growth company” as defined in the JOBS Act, we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we will rely on some or all of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. If we avail ourselves of certain exemptions from various reporting requirements, our reduced disclosure may make it more difficult for investors and securities analysts to evaluate us to a level acceptable by them and may result in less investor confidence.
Our common stock has been delisted on The NASDAQ Capital Market, which may negatively impact the trading price of our common stock and the levels of liquidity available to our stockholders.
Our common stock was suspended from trading on The NASDAQ Capital Market, prior to the opening of the market on October 6, 2017 and began quotation on the OTCQB Venture Market on that date, retaining the symbol “BSTG”. On December 7, 2017, NASDAQ filed a Form 25-NSE with the SEC to complete the delisting process. The trading of our common stock on the OTCQB Venture Market rather than The NASDAQ Capital Market may negatively impact the trading price of our common stock and the levels of liquidity available to our stockholders.
Upon such delisting, our common stock became subject to the regulations of the SEC relating to the market for penny stocks. A penny stock is any equity security not traded on a national securities exchange that has a market price of less than $5.00 per share. The regulations applicable to penny stocks may severely affect the market liquidity for our common stock and could limit the ability of shareholders to sell securities in the secondary market. Accordingly, investors in our common stock may find it more difficult to dispose of or obtain accurate quotations as to the market value of our common stock, and there can be no assurance that our common stock will continue to be eligible for trading or quotation on the OTCQB Venture Market or any other alternative exchanges or markets.
The delisting of our common stock from The NASDAQ Capital Market may adversely affect our ability to raise additional financing through public or private sales of equity securities, may significantly affect the ability of investors to trade our securities, and may negatively affect the value and liquidity of our common stock. Such delisting may also have other negative results, including the potential loss of confidence by employees, the loss of institutional investor interest and fewer business development opportunities. Furthermore, because of the limited market and low volume of trading in our common stock that could occur, the share price of our common stock could more likely be affected by broad market fluctuations, general market conditions, fluctuations in our operating results, changes in the market’s perception of our business, and announcements made by us, our competitors, parties with whom we have business relationships or third parties.
| Item 1B. | Unresolved Staff Comments. |
None.
| Item 2. | Properties. |
On November 1, 2013 we entered into a sublease of approximately 17,000 square feet of mixed use space of the facility located at 84 October Hill Road, Suite 11, Holliston, Massachusetts from Harvard Bioscience, which is our corporate headquarters. Our principal facilities incorporate manufacturing, laboratory, development, sales and marketing, and administration functions. We believe our current facilities are adequate for our needs for the foreseeable future.
| 34 |
| Item 3. | Legal Proceedings. |
On April 14, 2017, representatives for the estate of a deceased individual filed a civil lawsuit in the Suffolk Superior Court, in Boston, Massachusetts, against the Company, Harvard Bioscience and other defendants. The complaint alleges that the decedent was harmed by two tracheal implants that incorporated synthetic trachea scaffolds and a biologic component combined by the implanting surgeon with a bioreactor, and surgically implanted in the decedent in two surgeries performed in 2012 and 2013, which harm caused her injury and death. The civil complaint seeks a non-specific sum of money to compensate the plaintiffs. This civil lawsuit relates to the Company’s first-generation trachea scaffold technology for which the Company discontinued development in 2014, and not to the Company’s current Cellframe technology nor to its lead development product candidate, the Cellspan esophageal implant. The litigation is at an early stage and the Company intends to vigorously defend this case. While the Company believes that such claim lacks merit and has filed a motion seeking dismissal of the lawsuit, the Company is unable to predict the ultimate outcome of such litigation. In accordance with a separation and distribution agreement between Harvard Bioscience and the Company relating to the Separation, the Company would be required to indemnify Harvard Bioscience against losses that Harvard Bioscience may suffer as a result of this litigation. The Company has been informed by its insurance provider that the case has been accepted as an insurable claim under the Company’s product liability insurance policy.
From time to time, we may be involved in various claims and legal proceedings arising in the ordinary course of business. Other than the above matter, there are no such matters pending that we expect to be material in relation to our business, financial condition, and results of operations or cash flows.
| Item 4. | Mine Safety Disclosures. |
Not Applicable.
| 35 |
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Reverse Stock Split
On December 22, 2017, we effected a reverse stock split of its shares of common stock at a ratio of 1-for-20. Our common stock began trading on the OTCQB marketplace on a reverse stock split-adjusted basis at the open of the market on December 22, 2017. The Reverse Split was previously authorized at the annual meeting of our stockholders on April 26, 2017, and our Board of Directors approved the ratio and timing of the Reverse Split on December 11, 2017. All references to numbers of common shares and per-share information in this Annual Report have been adjusted retroactively to reflect the 1-for-20 reverse stock split.
Unregistered Sales of Equity Securities
Aspire Capital, LLC Transaction
On December 15, 2015, we entered into a Common Stock Purchase Agreement (the “Aspire Purchase Agreement”) with Aspire Capital Fund, LLC, an Illinois limited liability company (“Aspire Capital”), which provided that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital was committed to purchase up to an aggregate of $15.0 million of shares of our common stock over the 30-month term set forth in the Aspire Purchase Agreement.
On December 15, 2015, we issued 7,500 shares of our common stock to Aspire Capital in consideration for entering into the Aspire Purchase Agreement and sold 25,000 shares to Aspire Capital for an aggregate purchase price of $1,000,000.
On May 12, 2016, we issued 7,500 shares of common stock under this arrangement in exchange for gross proceeds of approximately $371,000. We terminated the Aspire Purchase Agreement effective as of May 17, 2016. The shares of common stock were issued to Aspire Capital in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act as transactions not involving a public offering and in reliance on similar exemptions under applicable state laws.
May 2016 Offering
On May 15, 2016, we entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain investors (the “Investors”) for the sale by us of 141,844 registered shares of our common stock at a purchase price of $35.25 per share. Concurrently with the sale of the shares of our common stock, pursuant to the Purchase Agreement we also sold unregistered warrants to purchase 70,922 shares of our common stock. The aggregate gross proceeds for the sale of the shares of common stock and the warrants was approximately $5.0 million. Additionally, the Company issued to the placement agent warrants to purchase 7,092 shares of unregistered common stock for the offering at an exercise price of $35.25 per warrant. Subject to certain ownership limitations, the warrants are initially exercisable commencing six months from the issuance date at an exercise price equal to $35.25 per share of common stock, subject to adjustments as provided under the terms of the warrants. The warrants are exercisable for five years from the initial exercise date. The closing of the sales of these securities under the Purchase Agreement occurred on May 19, 2016. The warrants and placement agent warrants were issued in reliance on the exemptions provided by Section 4(a)(2) of the Securities Act as transactions not involving a public offering and Rule 506 promulgated under the Securities Act as sales to accredited investors, and in reliance on similar exemptions under applicable state laws.
December 2017 Private Placement
On December 27, 2017 we entered into a Securities Purchase Agreement and we issued to investors (i) 518,000 shares of our common stock, (ii) 3,108 shares of Series D Convertible Preferred Stock (the “Series D Preferred Stock”), and (ii) warrants to purchase 3,108,000 shares of common stock (the “Warrants”). The Warrants have an exercise price of $2.00 per share, subject to adjustments as provided under the terms of the Warrants, and are immediately exercisable. The Warrants are exercisable for five years from the issuance date.
The Series D Preferred Stock ranks on parity to the common stock, and is entitled to vote on any matters to which shares of the common stock are entitled to vote, on an as-if-converted basis. The Series D Preferred Stock includes an ownership limitation that limits the Investors and their affiliates to owning no more than 49.99% of the common stock. The shares of common stock, the warrants (including the shares of common stock issuable upon exercise of the warrants) and the shares of Series D Preferred Stock (including the shares of common stock issuable upon conversion of the Series D Preferred Stock) were issued in reliance on the exemptions provided by Section 4(a)(2) of the Securities Act as transactions not involving a public offering and Rule 506 promulgated under the Securities Act as sales to accredited investors, and in reliance on similar exemptions under applicable state laws.
| 36 |
Connecticut Children’s Medical Center Private Placement
On December 29, 2017 we entered into a Securities Purchase Agreement with Connecticut Children’s Medical Center (“Connecticut Children’s”) pursuant to which Connecticut Children’s agreed to purchase in a private placement (the “Connecticut Children’s Private Placement”), and we agreed to issue, 50,000 shares of common stock at a purchase price of $2.00 per share and warrants to purchase 75,000 shares of common stock, with an exercise price of $2.00 per warrant in exchange for an aggregate of $0.1 million. The Connecticut Children’s Private Placement closed on January 3, 2018. The shares of common stock and the warrants (including the shares of common stock issuable upon exercise of the warrants) were issued in reliance on the exemptions provided by Section 4(a)(2) of the Securities Act as transactions not involving a public offering and Rule 506 promulgated under the Securities Act as sales to accredited investors, and in reliance on similar exemptions under applicable state laws.
February 2018 Private Placement
On February 20, 2018, we entered into a Securities Purchase Agreement with an investor pursuant to which the investor agreed in a private placement to purchase 302,115 shares of common stock at a purchase price of $3.31 per share for net proceeds of $1.0 million.
Price Range of Common Stock
Our common stock started trading on the OTCQB Venture Market at the opening of business on October 6, 2017 under the symbol “BSTG.” Prior to October 6, 2017, our common stock traded on The NASDAQ Capital Market also under the symbol “BSTG.” Prior to April 1, 2016, in connection with our name change, our common stock traded on The NASDAQ Capital Market under the symbol “HART” since October 21, 2013. The following table sets forth the range of the high and low sales prices per share of our common stock as reported on the OTCQB Venture Market or The NASDAQ Capital Market for the quarterly periods indicated.
| Fiscal Year Ended December 31, 2017 | High | Low | ||||||
| First Quarter | $ | 19.00 | $ | 5.86 | ||||
| Second Quarter | 9.78 | 4.42 | ||||||
| Third Quarter | 13.00 | 5.02 | ||||||
| Fourth Quarter | 6.66 | 0.60 | ||||||
| Fiscal Year Ended December 31, 2016 | High | Low | ||||||
| First Quarter | $ | 46.80 | $ | 24.00 | ||||
| Second Quarter | 50.00 | 20.60 | ||||||
| Third Quarter | 23.00 | 18.60 | ||||||
| Fourth Quarter | 22.20 | 14.96 | ||||||
On March 26, 2018, the closing sale price of our common stock on the OTCQB Venture Market was $3.20 per share. There were 128 holders of record of our common stock as of March 26, 2018. We believe that the number of beneficial owners of our common stock at that date was substantially greater.
Dividend Policy
We have never declared or paid cash dividends on our common stock in the past and do not intend to pay cash dividends on our common stock in the foreseeable future. Any future determination to pay cash dividends will be at the discretion of our Board of Directors and will depend on our financial condition, results of operations, capital requirements and other factors our Board of Directors deems relevant.
| Item 6. | Selected Financial Data |
Not Applicable.
| 37 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
NOTE REGARDING FORWARD-LOOKING STATEMENTS
Forward-Looking Statements
The following section of this Annual Report on Form 10-K entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contains statements that are not statements of historical fact and are forward-looking statements within the meaning of federal securities laws. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties.
In some cases, you can identify forward-looking statements by terms such as “believe,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “could,” “would,” “target,” “seek,” “aim,” “believe,” “predicts,” “think,” “objectives,” “optimistic,” “new,” “goal,” “strategy,” “potential,” “is likely,” “will,” “expect,” “plan” “project,” “permit” and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events, are based on assumptions and are subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We discuss many of these risks in greater detail in Item 1A.“Risk Factors” of this Annual Report on Form 10-K. You should carefully review all of these factors, as well as the comprehensive discussion of forward-looking statements on page 1 of this Annual Report on Form 10-K.
Overview
We are a biotechnology company developing bioengineered organ implants based on our novel Cellframe technology. Our Cellframe technology is comprised of a biocompatible scaffold that is seeded with the recipient’s own stem cells. This technology is being developed to treat life-threatening conditions of the esophagus, trachea or bronchus with the objective of dramatically improving the treatment paradigm for those patients.
We believe that our Cellframe technology will provide surgeons with new ways to address damage to the esophagus, bronchus, and trachea due to congenital abnormalities, cancer, infection or trauma. Products being developed based on our Cellframe technology for those indications are called Cellspan products.
We announced favorable preliminary pre-clinical results of large-animal studies for the esophagus, trachea and bronchus in November 2015. Since then, the Cellspan esophageal implant product candidates have been our lead development product candidates. We are pursuing two development programs that address conditions of the esophagus: esophageal atresia in pediatric patients and esophageal cancer in adult patients. Our Cellspan esophageal product candidates are each intended to provide a surgical solution to stimulate regeneration of a segment of the esophagus missing due to a congenital abnormality or following surgical removal, to establish or reestablish the organ’s continuity and integrity.
| 38 |
Approximately one in 2,500 babies in the U.S. is born with esophageal atresia, a congenital condition where the child’s esophagus is underdeveloped and does not extend completely from the mouth to the stomach. When a long segment of the esophagus is lacking, the current standard of care is a series of surgical procedures where surgical sutures are applied to both ends of the esophagus in an attempt to stretch them together so they can be connected at a later date. This process can take weeks and the procedure can result in serious complications and may carry high rates of failure. Such approach also requires, in time, at least two separate surgical interventions. Other options include the use of the child’s stomach that would be pulled up, or a piece of the patient’s intestine that would be moved to the gap, to allow a connection to the mouth. We are working to develop a Cellspan esophageal implant product candidate to address newborns’ esophageal atresia, to provide a simpler, more effective and potentially organ-sparing solution.
A portion of all patients diagnosed with esophageal cancer are treated via a surgical procedure known as an esophagectomy. The current standard of care for an esophagectomy requires a complex surgical procedure that involves moving the patient’s stomach or a portion of their colon into the chest to replace the portion of esophagus resected by the removal of the tumor. These current procedures have high rates of complications and can lead to a severely diminished quality of life and require costly ongoing care. Our Cellspan esophageal implants aim to simplify the procedure, reduce complications, result in a better quality of life and reduce the overall cost of these patients to the healthcare system.
In May 2016, we reported an update of results from pre-clinical large-animal studies. We disclosed that the study had demonstrated in a predictive large-animal model the ability of Biostage Cellspan organ implants to successfully stimulate the regeneration of sections of esophagus that had been surgically removed for the study. This study and its results were published in an article in Nature Scientific Reports in March 2018. Cellspan esophageal implants, consisting of a proprietary biocompatible synthetic scaffold seeded with the recipient animal’s own stem cells, were surgically implanted in place of the esophagus section that had been removed.
Study animals were returned to a solid diet two weeks after implantation surgery. The scaffolds, which are intended to be in place only temporarily, were later retrieved via the animal’s mouth in a non-surgical endoscopic procedure. After two and a half months post-surgery, a complete epithelium and other specialized esophagus tissue layers were regenerated. Animals in the study demonstrated weight gain and appeared healthy and free of any significant side effects, including two that were studied for almost two years and received no specialized care.
In November 2016, we were granted Orphan Drug Designation for our Cellspan esophageal implant by the U.S. Food and Drug Administration, or the FDA to restore the structure and function of the esophagus subsequent to esophageal damage due to cancer, injury or congenital abnormalities. Orphan drug status provides market exclusivity in the U.S. for seven years from the date of the product’s approval for marketing. This exclusivity is in addition to any exclusivity we may obtain due to our patents. Additionally, orphan designation provides certain incentives, including tax credits and a waiver of the Biologics License Application or BLA fee. We also intend to apply for orphan drug designation for our Cellspan esophageal implant in Europe in the future. Orphan drug status in Europe provides market exclusivity there for ten years from the date of the product’s approval for marketing.
We are conducting Good Laboratory Practice or GLP studies to demonstrate that our technology, personnel, systems and practices are sufficient for advancing into clinical trials. GLP safety studies are required to advance to an Investigational New Drug or IND application with the FDA, which would seek approval to initiate clinical trials for Biostage Cellspan esophageal implants in humans.
In October 2016, we announced a regulatory update following our planned pre-Investigational New Drug, or pre-IND, meeting with the FDA, for the advancement of our lead product candidate, a Cellspan Esophageal Implant to be used to stimulate esophageal regeneration following surgery to address esophageal cancer in adults, into human clinical studies. We subsequently announced our expectation to file an IND application with the FDA in the third quarter of 2017 based on our election to extend the duration of our ongoing GLP animal studies following the feedback provided by the FDA.
On August 7, 2017, we announced the use of our Cellspan Esophageal Implant product candidate in a patient at a major U.S. hospital via an FDA-approved single-use expanded access application. The patient was a 75-year old male with a life-threatening cancerous mass in his chest that spanned his heart, a lung and his esophagus. The surgery was performed in May 2017 to remove the tumor, repair the heart, part of one lung, and a section of the esophagus. The Cellspan Esophageal Implant was inserted into the gap in the esophagus created by the removal of the tumor. In February 2018 the patient’s surgeon informed us that the patient had died after living approximately eight months after surgery. The surgeon stated that the cause of death was stroke, and that the stroke was unrelated to the esophageal implant. The surgeon also informed us that a preliminary autopsy had shown that the esophageal implant resulted in a regenerated esophageal tube in the patient, except for a very small (approximately 5mm) hole on the lateral wall that was right up against a synthetic graft inserted as part of the patient’s heart repair on the pericardium in that same surgery. The synthetic graft on the pericardium was not related to our esophageal implant product and may have acted as an irritant to esophageal regeneration where it contacted the esophageal implant. The surgeon also informed us that the esophageal regeneration in this patient was consistent with the regeneration previously observed in our large-animal studies.
| 39 |
In August 2017, we announced that we were reprioritizing our product development program based on greatest unmet medical need, analysis of existing surgical options, and physician validation. We believe that, of our two current programs, the Cellspan Esophageal Implant program to treat pediatric esophageal atresia provides a shorter time to a commercial product and the greater overall potential value. We also believe that the pediatric esophageal atresia program needs to advance in the first position with the FDA to ensure eligibility for the pediatric rare disease accelerated review voucher program. Additionally, receipt of such a voucher, if achieved, could potentially provide significant value to the company in the future. As a result, we elevated the pediatric program to our lead program. We plan to continue to advance the Cellspan Esophageal Implant adult program, but have not filed an IND for that product candidate at this time. Our current plan for that product candidate is to update the FDA on the progress and status of our preclinical testing, including our GLP studies, for the adult esophagus program in the future. Based on the FDA’s feedback, we may amend our preclinical testing plan and continue toward the filing of an IND.
Following the failure to receive the funding with respect to a securities purchase agreement in August 2017, and in an effort to conserve cash, we completed a reduction in headcount of 20 persons during October and November 2017. In addition, our officers agreed to a temporary reduction in their cash salaries by 50% effective November 2017. Following the capital raises in December 2017 and January 2018 described below we re-hired five of our former employees into key positions in January 2018. We believe that our new staffing level after those hires is sufficient to pursue both of our esophageal programs and we anticipate our 2018 cash burn needs to be approximately 60% of our 2017 burn.
Our products are currently in development and have not yet received regulatory approval for sale anywhere in the world.
We were incorporated and commenced operations on November 1, 2013 as a result of a spin-off from Harvard Bioscience. On that date, the Company became an independent company that operates the regenerative medicine business previously owned by Harvard Bioscience. The spin-off was completed through the distribution of all the shares of common stock of Biostage to Harvard Bioscience stockholders.
Since our incorporation, we have devoted substantially all of our resources to developing our programs, building our intellectual property portfolio, business planning, raising capital and providing general and administrative support for these operations. To date, we have financed our operations with proceeds from the sales of common stock and preferred stock. We generated limited revenues from the sale of our research bioreactor equipment through December 31, 2016 to Harvard Bioscience. We did not recognize any revenues from research bioreactors during the year ended December 31, 2017. In December 2017, we sold the inventory and rights to manufacture and sell research-only versions of our bioreactors to Harvard Bioscience. We expect to continue to incur operating losses and negative cash flows from operations for the remainder of 2018 and in future years.
Between December 27 and December 29, 2017, we entered into Securities Purchase Agreements with new investors for the sale of our capital stock. This agreement resulted in the following transactions:
| · | On December 22, 2017, we effected a reverse stock split of our shares of common stock at a ratio of 1-for-20. All share and per share amounts of common stock in this Annual Report have been retroactively adjusted to reflect the reverse stock split. |
| · | Our Common stock commenced trading on the OTCQB Venture Market on a reverse stock split basis on December 22, 2017. The Company had delisted from The NASDAQ Capital Market in October 2017 and commenced trading on the OTCQB Venture Market at that time. |
| · | On December 27, 2017, we issued 518,000 shares of our Common stock at $2.00 per share, 3,108 shares of our Series D Convertible Preferred Stock at $1000 per share, and warrants to purchase 3,108,000 shares of common stock at an exercise price of $2.00 per share, in exchange for aggregate gross proceeds of approximately $4.1 million in a private placement transaction of unregistered shares with a new investor. The warrants were immediately exercisable and expire in December 2022. |
| · | On January 3, 2018, we issued 50,000 shares of our Common stock at $2.00 per share and warrants to purchase 75,000 shares of common stock at an exercise price of $2.00 per share, in exchange for aggregate gross proceeds of $100,000 in a private placement transaction of unregistered shares with Connecticut Children’s Medical Center. The warrants were immediately exercisable and expire in January 2023. |
Additionally, on February 20, 2018, we completed a private placement of 302,115 shares of common stock at a purchase price of $3.31 per share for net proceeds of $1.0 million.
On March 28, 2018, we were awarded a Fast-Track Small Business Innovation Research (SBIR) grant by the Eunice Kennedy National Institute of Child Health and Human Development. The award for Phase I was $225,000 and the SBIR grant has the potential to provide a total award of $1.7 million. If Phase I is successful, and funding is available, a Phase II award of up to approximately $1.5 million would support pre-clinical testing of pediatric Cellspan™ Esophageal Implants planned to begin later in 2017. The Phase II funds, if awarded, would be spent over an estimated two years.
We have incurred substantial operating losses since our inception, and as of December 31, 2017 have an accumulated deficit of approximately $48.2 million and will require additional financing to fund future operations. We expect that our cash on hand at December 31, 2017 of $4.0 million, along with net proceeds of approximately $1.1 million from the issuance of common stock in private placement transactions in January and February 2018, will enable us to fund our operating expenses and capital expenditure requirements into the third quarter of 2018.
We are currently investing significant resources in development of products for use by clinicians in the field of regenerative medicine. We will need to raise additional funds in future periods to fund our operations. In the event that we do not raise additional capital from outside sources in the near future, we may be forced to further curtail or cease our operations. Cash requirements and cash resource needs will vary significantly depending upon the timing of clinical and animal studies and other resource needs that will be required to complete ongoing development and pre-clinical and clinical testing of products as well as regulatory efforts and collaborative arrangements necessary for our products that are currently under development. We will seek to raise necessary funds through a combination of public or private equity offerings, debt financings, other financing mechanisms, or strategic collaborations and licensing arrangements. We may not be able to obtain additional financing on terms favorable to us, if at all.
| 40 |
Results of Operations
Components of Operating Loss
Research and development expense. Research and development expense consists of salaries and related expenses, including stock-based compensation, for personnel and contracted consultants and various materials and other costs to develop our new products, primarily: synthetic organ scaffolds, including investigation and development of materials and investigation and optimization of cellularization, and 3D organ bioreactors, as well as studies of cells and cell behavior. Other research and development expenses include the costs of outside service providers and material costs for prototype and test units and outside laboratories and testing facilities performing cell growth and materials experiments, as well as the costs of all other preclinical research and testing including animal studies and expenses related to potential patents. We expense research and development costs as incurred.
Selling, general and administrative expense. Selling, general and administrative expense consists primarily of salaries and other related expenses, including stock-based compensation, for personnel in executive, accounting, information technology and human resources roles. Other costs include professional fees for legal and accounting services, insurance, investor relations and facility costs. In 2017 and 2016, our sales and marketing expenses were immaterial given our focus on research and development.
Changes in fair value of warrant liability, net of issuance costs. Changes in fair value of warrant liability, net of issuance costs, represent the change in the fair value of common stock warrants from the date of issuance to the end of the reporting period during the years ended December 31, 2017 and 2016, and in subsequent quarterly periods, the change in the fair value of common stock warrants from the date between each reporting period until the liability is settled. We use the Black-Scholes pricing model to value the related warrant liability. The costs associated with the issuance of the warrants have been recorded as an expense upon issuance.
Other expense
Other expense represented a loss on disposal of equipment.
Critical Accounting Estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with Generally Accepted Accounting Principles in the United States (“U.S. GAAP”). The preparation of these consolidated financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenues, expenses and related disclosures. We believe the following policies to be critical to the judgments and estimates used in the preparation of our financial statements.
Share-based Compensation
We account for our share-based compensation in accordance with the fair value recognition provisions of current authoritative guidance. Share-based awards, including stock options, are measured at fair value as of the grant date and recognized as expense over the requisite service period (generally the vesting period), which we have elected to amortize on a straight-line basis. Expense on share-based awards for which vesting is performance or milestone based is recognized on a straight-line basis from the date when we determine the achievement of the milestone is probable to the vesting/milestone achievement date. Since share-based compensation expense is based on awards ultimately expected to vest, it has been reduced by an estimate for future forfeitures. We estimate forfeitures at the time of grant and revise our estimate, if necessary, in subsequent periods. We estimate the fair value of options granted using the Black-Scholes option valuation model. Significant judgment is required in determining the proper assumptions used in these models. The assumptions used include the risk-free interest rate, expected term, expected volatility and expected dividend yield. We base our assumptions on historical data when available or when not available, on a peer group of companies. However, these assumptions consist of estimates of future market conditions, which are inherently uncertain and subject to our judgment, and therefore any changes in assumptions could significantly impact the future grant date fair value of share-based awards
Total share-based compensation expense for the years ended December 31, 2017 and 2016 was $0.7 million and $1.3 million, respectively. Share based compensation is further described in Note 11 to the Consolidated Financial Statements.
Warrant Liability
We classify a warrant to purchase shares of our common stock as a liability in our consolidated balance sheets when the warrant is a free-standing financial instrument that may require us to transfer cash consideration upon exercise and that cash transfer event would be out of our control. Such a “liability warrant” is initially recorded at fair value on the date of grant using the Black-Scholes model and net of issuance costs, and it is subsequently re-measured to fair value at each subsequent balance sheet date. Changes in fair value of the warrant are recognized as a component of other income (expense) in the consolidated statement of operations and comprehensive loss. We will continue to adjust the liability for changes in fair value until the earlier of the exercise or expiration of the warrant.
| 41 |
Results of Operations
Year Ended December 31, 2017 Compared to Year Ended December 31, 2016
Revenues
We did not recognize any revenues during the year ended December 31, 2017. Revenues amounted to $82 thousand for the year ended December 31, 2016 and represented the sale of research bioreactor equipment through Harvard Bioscience, to end users working on organ regeneration research. We do not expect to have any additional sales of research bioreactor equipment in the future.
Cost of revenues
There were no cost of revenues during the year ended December 31, 2017. Cost of revenues amounted to $116 thousand during the year ended December 31, 2016 and represented labor, materials and allocated overhead costs for our research bioreactor equipment sales.
Research and Development Expense
Research and development expense was virtually unchanged at $7.6 million for the year ended December 31, 2017 as compared to the year ended December 31, 2016. This similar level of expense was primarily due to an increase in research and development associated with our programs offset by a decrease in share-based compensation in the amount of $0.4 million. The Company’s research and development costs decreased during the fourth quarter of 2017 compared to the same period in 2016 due to headcount reductions.
Selling, General and Administrative Expense
Selling, general and administrative expense decreased $0.6 million, or 13.3% to $3.9 million for the year ended December 31, 2017 compared from $4.5 million for the year ended December 31, 2016. This decrease was due to primarily a decrease in share-based compensation costs in the amount of $0.2 million related to general and administrative personnel and a decrease in investor relations costs of $0.2 million, partially offset by an increase in professional fees primarily related to legal fees for potential financings during the year, as well as the Company’s private placement in the fourth quarter of 2017.
Change in fair value of warrant liability, net of issuance costs
During the year ended December 31, 2017, the change in fair value of our warrant liability, net of $0.4 million of issuance costs resulted in $0.3 million of expense compared to $0.5 million of income for the year ended December 31, 2016. The $0.3 million of expense represented a $0.1 million decrease in the fair value of the warrants, which was more than offset by $0.4 million of issuance costs associated with the warrants issued in 2017. Holders agreed to the modification of 952,184 warrants in the second and third quarters of 2017 such that the modified warrants meet the definition of an equity instrument. The exercise of warrants to purchase 132,367 shares of common stock during the third quarter of 2017 included 83,616 warrants that had not been modified. The modification and exercise of the liability warrants resulted in a $4.3 million reclassification to additional paid in capital from warrant liability. This modification and exercise resulted in a relatively small amount of warrants subject to liability accounting, as reflected by the warrant liability balance of approximately $16,000 at December 31, 2017.
The $0.5 million of income, which resulted from the change in the fair value of our warrant liability, for the year ended December 31, 2016 was due to a decrease in the fair value of the warrants, specifically with respect to the value of the underlying common shares.
Other expense
During the year ended December 31, 2017, other expense represented a loss on the disposal of equipment of approximately $0.1 million.
There was no other expense for the year ended December 31, 2016.
| 42 |
Liquidity and Capital Resources
Sources of liquidity. We have incurred operating losses since inception, and as of December 31, 2017 we had an accumulated deficit of approximately $48.2 million. We are currently investing significant resources in the development and commercialization of our products for use by clinicians and researchers in the field of regenerative medicine. As a result, we expect to incur operating losses and negative operating cash flow for the foreseeable future.
Operating activities. Net cash used in operating activities of $11.0 million for the year ended December 31, 2017 was primarily a result of our $11.9 million net loss and $0.7 million of unfavorable changes in working capital due primarily to timing of payment of accrued and other current liabilities, which were partially offset by a $1.4 million add-back of non-cash expenses of stock-based compensation, depreciation, and change in fair value of warrant liability.
Net cash used in operating activities of $9.1 million for the year ended December 31, 2016 was primarily a result of our $11.6 million net loss, offset by a $1.3 million add-back of non-cash expenses of stock-based compensation, depreciation, change in fair value of warrant liability and favorable changes in working capital of $1.2 million.
Investing activities. Net cash used in investing activities for the years ended December 31, 2017 and 2016 totaled $0.1 million and $0.3 million, respectively, and represented additions to property, plant and equipment. The decrease of $0.2 million was due to lower levels of capital expenditures.
Financing activities. Net cash generated from financing activities during the year ended December 31, 2017 of $12.2 million consisted primarily of the net proceeds in the amount of $6.8 million from the issuance of 1,000,000 shares of common stock and warrants to purchase 1,000,000 shares of common stock in February 2017, net proceeds of $1.1 million from the exercise of warrants during 2017, and net proceeds of $4.1 million from the issuance of Series D Preferred Stock, common stock and warrants in a private placement transaction in December 2017. Net cash generated from cash flows from financing activities also included a deposit of $0.3 million from an investor related to the private placement transaction that was subsequently repaid in January 2018.
Net cash generated from financing activities during the year ended December 31, 2016 in the amount of $4.8 million consisted of net proceeds in the amount of $4.5 million from the issuance of 141,844 shares of our common stock at a purchase price of $35.25 per share and the issuance of warrants to purchase 70,992 shares of common stock at an exercise price of $35.25 per warrant, as well as net proceeds in the amount of $0.3 million from the issuance of 7,500 shares of common stock under the Aspire Purchase Agreement.
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our consolidated financial statements appearing at the end of this Annual Report.
| 43 |
Off - Balance Sheet Arrangements
We did not have, during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under applicable Securities and Exchange Commission rules.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk. |
Not Applicable.
| Item 8. | Financial Statements and Supplementary Data. |
The information required by this item is contained in the consolidated financial statements filed as part of this Annual Report on Form 10-K listed under Item 15 of Part IV below.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. |
None.
| Item 9A. | Controls and Procedures. |
This Report includes the certifications of our Chief Executive Officer and Chief Financial Officer required by Rule 13a-14 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). See Exhibits 31.1 and 31.2. This Item 9A includes information concerning the controls and control evaluations referred to in those certifications.
| (a) | Evaluation of Disclosure Controls and Procedures |
Disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) are designed to ensure that information required to be disclosed in reports filed or submitted under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms and that such information is accumulated and communicated to management, including the Chief Executive Officer and the Chief Financial Officer, to allow timely decisions regarding required disclosures.
| 44 |
In connection with the preparation of this Annual Report on the Form 10-K, our management, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures as of December 31, 2017. Our disclosure controls and procedures are designed to provide reasonable assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and our management necessarily was required to apply its judgment in evaluating and implementing our disclosure controls and procedures. Based upon the evaluation described above, our Chief Executive Officer and Chief Financial Officer have concluded that they believe that our disclosure controls and procedures were effective, as of the end of the period covered by this report, in providing reasonable assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosures, and is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms.
| (b) | Management’s Annual Report on Internal Control Over Financial Reporting |
Our management, under the supervision of the Chief Executive Officer and the Chief Financial Officer, is responsible for establishing and maintaining an adequate system of internal control over financial reporting. Internal control over financial reporting (as defined in Rules 13a-15(f) and 15d(f) under the Exchange Act) is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”).
A company’s internal control over financial reporting includes those policies and procedures that: (a) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (b) provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with U.S. GAAP; (c) provide reasonable assurance that receipts and expenditures are being made only in accordance with appropriate authorization of management and the board of directors; and (d) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In connection with the preparation of this report, our management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2017 based on the criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). As a result of that evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2017.
As an “emerging growth company” under the Jumpstart Our Business Startups Act, and as a smaller reporting company, we are exempt from the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002. As a result, KPMG LLP, our independent registered public accounting firm, has not audited or issued an attestation report with respect to the effectiveness of our internal control over financial reporting as of December 31, 2017.
| (c) | Changes in Internal Controls Over Financial Reporting |
Our management, with the participation of the Chief Executive Officer and the Chief Financial Officer, has evaluated whether any change in our internal control over financial reporting occurred during the fourth quarter ended December 31, 2017. Based on that evaluation, management concluded that there were no changes in our internal controls over financial reporting during the quarter ended December 31, 2017 that materially affected, or are reasonably likely to materially affect our internal controls over financial reporting.
| Item 9B. | Other Information. |
None.
| 45 |
| Item 10. | Directors, Executive Officers and Corporate Governance. |
Incorporated by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act, in connection with our 2018 Annual Meeting of Stockholders. Information concerning executive officers of our Company is included in Part I of this Annual Report on Form 10-K as Item 1. Business-Executive Officers of the Registrant and incorporated herein by reference.
| Item 11. | Executive Compensation. |
Incorporated by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act in connection with our 2018 Annual Meeting of Stockholders.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
Incorporated by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act in connection with our 2018 Annual Meeting of Stockholders.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
Incorporated by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act in connection with our 2018 Annual Meeting of Stockholders
| Item 14. | Principal Accounting Fees and Services. |
Incorporated by reference to our definitive Proxy Statement to be filed pursuant to Regulation 14A under the Exchange Act in connection with our 2018 Annual Meeting of Stockholders.
| 46 |
| Item 15. | Exhibits, Financial Statement Schedules. |
| (a) | Documents Filed. The following documents are filed as part of this Annual Report on Form 10-K: |
| (1) | Financial Statements. The consolidated financial statements of Biostage, Inc. and its subsidiaries filed under this Item 15: |
| Page | ||
| Index to Consolidated Financial Statements | F-1 | |
| Report of Independent Registered Public Accounting Firm | F-2 | |
| Consolidated Balance Sheets as of December 31, 2017 and 2016 | F-3 | |
| Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2017 and 2016 | F-4 | |
| Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2017 and 2016 | F-5 | |
| Consolidated Statements of Cash Flows for the years ended December 31, 2017 and 2016 | F-6 | |
| Notes to Consolidated Financial Statements | F-7 |
| (2) | Financial Statement Schedules: None. Financial statement schedules have been omitted since the required information is included in our consolidated financial statements contained elsewhere in this Annual Report on Form 10-K. |
| (3) | Exhibits. The exhibits listed in the accompanying Exhibit Index are filed as a part of this Annual Report on Form 10-K. |
| (b) | Exhibits: The exhibits listed in the accompanying Exhibit Index are filed as a part of this Annual Report on Form 10-K. |
| (c) | Separate Financial Statements and Schedules: None. Financial statement schedules have been omitted since the required information is included in our consolidated financial statements contained elsewhere in this Annual Report on Form 10-K. |
| 47 |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
BIOSTAGE, INC.
| F-1 |
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
Biostage, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Biostage, Inc. and subsidiaries (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the years then ended, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the years then ended, in conformity with U.S. generally accepted accounting principles.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has suffered recurring losses from operations and will require additional financing to fund future operations which raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ KPMG LLP | |
| We have served as the Company’s auditor since 2012. | |
| Cambridge, Massachusetts | |
| April 2, 2018 | |
| F-2 |
CONSOLIDATED BALANCE SHEETS
(in thousands, except par value and share data)
| December 31, | December 31, | |||||||
| 2017 | 2016 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 4,038 | $ | 2,941 | ||||
| Accounts receivable | - | 42 | ||||||
| Prepaid expenses | 289 | 291 | ||||||
| Other current assets | 86 | 212 | ||||||
| Total current assets | 4,413 | 3,486 | ||||||
| Property, plant and equipment, net | 632 | 1,065 | ||||||
| Total non-current assets | 632 | 1,065 | ||||||
| Total assets | $ | 5,045 | $ | 4,551 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 923 | $ | 962 | ||||
| Accrued and other current liabilities | 383 | 1,210 | ||||||
| Due to related party | 300 | - | ||||||
| Warrant liability | 16 | 605 | ||||||
| Total current liabilities | 1,622 | 2,777 | ||||||
| Total liabilities | 1,622 | 2,777 | ||||||
| Commitments and contingencies (note 6) | ||||||||
| Stockholders' equity: | ||||||||
| Undesignated preferred stock, $0.01 par value; 984,000 and 996,000 shares authorized at December 31, 2017 and 2016, respectively; none issued and outstanding | - | - | ||||||
| Series D convertible preferred stock, par value $0.01 per share, 12,000 and 0 shares authorized at December 31, 2017 and 2016, respectively; 3,108 and 0 shares issued and outstanding at December 31, 2017 and 2016, respectively (aggregate liquidation value of $3,108,000 at December 31, 2017) | 1,475 | - | ||||||
| Common stock, par value $0.01 per share, 120,000,000 and 60,000,000 shares authorized at December 31, 2017 and 2016, respectively; 2,507,304 and 855,448 issued and outstanding at December 31 2017 and 2016, respectively | 25 | 9 | ||||||
| Additional paid-in capital | 50,157 | 38,083 | ||||||
| Accumulated deficit | (48,234 | ) | (36,318 | ) | ||||
| Total stockholders' equity | 3,423 | 1,774 | ||||||
| Total liabilities and stockholders' equity | $ | 5,045 | $ | 4,551 | ||||
See accompanying notes to consolidated financial statements.
| F-3 |
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except per share data)
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Revenues | $ | - | $ | 82 | ||||
| Cost of revenues | - | 116 | ||||||
| Gross loss | - | (34 | ) | |||||
| Operating expenses: | ||||||||
| Research and development | 7,588 | 7,603 | ||||||
| Selling, general and administrative | 3,880 | 4,489 | ||||||
| Total operating expenses | 11,468 | 12,092 | ||||||
| Operating loss | (11,468 | ) | (12,126 | ) | ||||
| Other income (expense): | ||||||||
| Change in fair value of warrant liability, net of issuance costs | (337 | ) | 547 | |||||
| Other expense | (111 | ) | - | |||||
| (448 | ) | 547 | ||||||
| Loss before income taxes | (11,916 | ) | (11,579 | ) | ||||
| Income taxes | - | - | ||||||
| Net loss | $ | (11,916 | ) | $ | (11,579 | ) | ||
| Basic and diluted net loss per share | $ | (6.63 | ) | $ | (14.49 | ) | ||
| Weighted average common shares, basic and diluted | 1,797 | 799 | ||||||
| Comprehensive loss: | ||||||||
| Net loss | $ | (11,916 | ) | $ | (11,579 | ) | ||
| Foreign currency translation adjustments | - | 8 | ||||||
| Total comprehensive loss | $ | (11,916 | ) | $ | (11,571 | ) | ||
See accompanying notes to consolidated financial statements.
| F-4 |
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands)
| Number of Common Shares Outstanding | Common Stock | Number of Series D Convertible Preferred Shares | Series D Convertible Preferred Stock | Additional Paid-in Capital | Accumulated Deficit | Accumulated Other Comprehensive Income (Loss) | Total Stockholders' Equity | |||||||||||||||||||||||||
| Balance at December 31, 2015 | 705 | $ | 7 | $ | - | $ | 33,042 | $ | (24,739 | ) | $ | (8 | ) | $ | 8,302 | |||||||||||||||||
| Net loss | - | - | - | - | - | (11,579 | ) | - | (11,579 | ) | ||||||||||||||||||||||
| Share based compensation | - | - | - | - | 1,327 | - | - | 1,327 | ||||||||||||||||||||||||
| Issuance of common stock under employee stock purchase plan | 1 | - | - | - | 22 | - | - | 22 | ||||||||||||||||||||||||
| Issuance of common stock, net of offering costs | 149 | 2 | - | - | 3,692 | - | - | 3,694 | ||||||||||||||||||||||||
| Other comprehensive income | - | - | - | - | - | - | 8 | 8 | ||||||||||||||||||||||||
| Balance at December 31, 2016 | 855 | 9 | - | - | 38,083 | (36,318 | ) | $ | - | 1,774 | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | (11,916 | ) | - | (11,916 | ) | ||||||||||||||||||||||
| Share based compensation | - | - | - | - | 693 | - | - | 693 | ||||||||||||||||||||||||
| Issuance of common stock under employee stock purchase plan | 1 | - | - | - | 13 | - | - | 13 | ||||||||||||||||||||||||
| Issuance of common stock, net of offering costs | 1,518 | 15 | - | - | 3,876 | - | - | 3,891 | ||||||||||||||||||||||||
| Reclassification of warrant liability upon modification | - | - | - | - | 3,746 | - | - | 3,746 | ||||||||||||||||||||||||
| Reclassification of warrant liability upon warrant exercise | - | - | - | - | 581 | - | - | 581 | ||||||||||||||||||||||||
| Issuance of common stock upon warrant exercises | 133 | 1 | - | - | 1,059 | - | - | 1,060 | ||||||||||||||||||||||||
| Issuance of series D convertible preferred, net of offering costs | - | - | 3 | 1,475 | - | - | - | 1,475 | ||||||||||||||||||||||||
| Issuance of warrants to purchase common stock in connection with issuance of Series D preferred and common stock above | - | - | - | - | 2,106 | - | - | 2,106 | ||||||||||||||||||||||||
| Balance at December 31, 2017 | 2,507 | $ | 25 | 3 | $ | 1,475 | $ | 50,157 | $ | (48,234 | ) | $ | - | $ | 3,423 | |||||||||||||||||
| F-5 |
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Cash flows used in operating activities: | ||||||||
| Net loss: | $ | (11,916 | ) | $ | (11,579 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense | 693 | 1,327 | ||||||
| Depreciation | 413 | 454 | ||||||
| Loss on sale of equipment | 160 | - | ||||||
| Change in fair value of warrant liability, net of issuance costs | 337 | (547 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | 42 | (21 | ) | |||||
| Inventories | - | 75 | ||||||
| Prepaid expenses | 2 | 39 | ||||||
| Other current assets | 126 | (212 | ) | |||||
| Accounts payable | (43 | ) | 472 | |||||
| Accrued and other current liabilities | (828 | ) | 934 | |||||
| Net cash used in operating activities | (11,014 | ) | (9,058 | ) | ||||
| Cash flows used in investing activities: | ||||||||
| Additions to property, plant and equipment | (136 | ) | (302 | ) | ||||
| Net cash used in investing activities | (136 | ) | (302 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Advance from related party | 300 | - | ||||||
| Proceeds from issuance of Series D convertible preferred stock, common stock and warrants, net | 4,086 | - | ||||||
| Proceeds from issuance of common stock and warrants, net of offering costs | 6,801 | 4,496 | ||||||
| Proceeds from issuance of common stock, net of offering costs | - | 349 | ||||||
| Proceeds from exercise of warrants | 1,060 | - | ||||||
| Net cash provided by financing activities | 12,247 | 4,845 | ||||||
| Net (decrease) increase in cash | 1,097 | (4,515 | ) | |||||
| Cash at the beginning of the year | 2,941 | 7,456 | ||||||
| Cash at the end of the year | $ | 4,038 | $ | 2,941 | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Reclassification of warrant liability upon modification | $ | 3,746 | $ | - | ||||
| Reclassification of warrant liability upon exercise of options | $ | 581 | $ | - | ||||
| Receivable for sale of equipment included in loss on sale of equipment | $ | 49 | ||||||
| Equipment purchases included in accounts payable | $ | 4 | $ | 133 | ||||
| Fair value of liability warrants issued in connection with issuance of common stock | $ | 3,787 | $ | 116 | ||||
See accompanying notes to consolidated financial statements.
| F-6 |
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years Ended December 31, 2017 and 2016
1. Organization
Overview
Biostage, Inc., formerly Harvard Apparatus Regenerative Technology, Inc. (“Biostage” or the “Company”) is a biotechnology company developing bioengineered organ implants based on the Company’s novel Cellframe TM technology. The Company’s Cellframe technology is comprised of a biocompatible scaffold that is seeded with the recipient’s own stem cells. The Company believes that this technology may prove to be effective for treating patients across a number of life-threatening medical indications who currently have unmet medical needs. The Company is currently developing its Cellframe technology to treat life-threatening conditions of the esophagus, bronchus or trachea with the objective of dramatically improving the treatment paradigm for those patients. Since inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, and acquiring operating assets.
The Company changed its name from Harvard Apparatus Regenerative Technology, Inc. to Biostage, Inc. on March 31, 2016. All references to the Company have been changed to Biostage in the accompanying consolidated financial statements and notes thereto.
On December 11, 2017, the Company entered into Securities Purchase Agreements (“SPAs”) with new investors for the sale of capital stock of the Company. This agreement resulted in the Company effecting a reverse stock split of its shares of common stock on December 22, 2017 at a ratio of 1-for-20. All share and per share amounts of common stock in the accompanying financial statements have been retroactively adjusted to reflect the reverse stock split. At that time, the Company’s common stock commenced trading on the OTCQB Venture Market on a reverse stock split basis. The Company had delisted from The NASDAQ Capital Market in October 2017 and began trading on the OTCQB Venture Market at that time. In connection with the SPAs, on December 27, 2017, the Company issued 518,000 shares of its common stock at $2.00 per share, 3,108 shares of its Series D Convertible Preferred Stock at $1,000 per share, and warrants to purchase 3,108,000 shares of common stock at an exercise price of $2.00 per share, in exchange for aggregate gross proceeds of approximately $4.1 million in a private placement transaction of unregistered shares with the new investors. The warrants were immediately exercisable and expire in December 2022.
Basis of Presentation
The financial statements reflect the Company’s financial position, results of operations and cash flows in conformity with generally accepted accounting principles in the United States (“GAAP”).
Going Concern
The Company has incurred substantial operating losses since its inception, and as of December 31, 2017 has an accumulated deficit of approximately $48.2 million and will require additional financing to fund future operations. The Company expects that its cash at December 31, 2017 of $4.0 million, along with net proceeds of approximately $1.1 million from the issuance of common stock in private placement transactions in January and February 2018, will enable it to fund its operating expenses and capital expenditure requirements into the third quarter of 2018. Therefore, these conditions raise substantial doubt about the Company’s ability to continue as a going concern.
The Company will need to raise additional funds in future periods to fund its operations. In the event the Company does not raise additional capital from outside sources in the near future, it may be forced to curtail or cease its operations. Cash requirements and cash resource needs will vary significantly depending upon the timing and the financial and other resource needs that will be required to complete ongoing development and pre-clinical and clinical testing of products as well as regulatory efforts and collaborative arrangements necessary for the Company’s products that are currently under development. The Company will seek to raise necessary funds through a combination of public or private equity offerings, debt financings, other financing mechanisms, research grants, or strategic collaborations and licensing arrangements. The Company may not be able to obtain additional financing on terms favorable to us, if at all.
The Company’s operations will be adversely affected if it is unable to raise or obtain needed funding and may materially affect the Company’s ability to continue as a going concern. The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern and therefore, the financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amount and classifications of liabilities that may result from the outcome of this uncertainty.
| F-7 |
2. Summary of Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements include the accounts of Biostage and its three wholly-owned subsidiaries, Harvard Apparatus Regenerative Technology GmbH (Germany), Harvard Apparatus Regenerative Technology AB (Sweden) and Biostage Limited (UK), which are currently dormant. All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The process of preparing financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Such estimates include, but are not limited to, stock-based compensation, valuation of warrant liability, accruals, depreciation and income taxes. Actual results could differ from those estimates and changes in estimates may occur.
Segment
The Company has one business segment and does not have significant costs or assets outside the United States.
Property, Plant and Equipment
Property, plant and equipment are carried at cost and depreciated using the straight-line method over the estimated useful lives of the assets as follows:
| Leasehold improvements | Shorter of expected useful life or lease term | |
| Furniture, machinery and equipment, computer equipment and software | 3- 7 years |
Maintenance and repairs are charged to expense as incurred, while any additions or improvements are capitalized.
Impairment of Long-Lived Assets
Long-lived assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. An asset, or group of assets, are considered to be impaired when the undiscounted estimated net cash flows expected to be generated by the asset, or group of assets, are less than its carrying amount. The impairment recognized is the amount by which the carrying amount exceeds the fair market value of the impaired asset, or group of assets.
Revenue Recognition
The Company follows the provisions of FASB ASC 605, “Revenue Recognition”. The Company recognizes product revenue when persuasive evidence of a sales arrangement exists, the price to the buyer is fixed or determinable, delivery has occurred, and collectability of the sales price is reasonably assured. To date, the Company has recognized revenues only for sales of its research bioreactor systems. Sales of some of the Company’s products include additional services such as installation and training. Revenues on these products are recognized when the additional services have been performed. Service agreements on its equipment are typically sold separately from the sale of the equipment.
The Company accounts for shipping and handling fees and costs in accordance with the provisions of FASB ASC 605-45-45, “Revenue Recognition - Principal Agent Considerations”, which requires all amounts charged to customers for shipping and handling to be classified as revenues. Costs related to shipping and handling are classified as cost of revenues. Provisions for warranties and product returns are estimated and accrued at the time sales are recorded. The Company has no obligations to customers after the date products are shipped or installed, if applicable, other than pursuant to warranty obligations. The Company provides for the estimated amount of future returns upon shipment of products or installation, if applicable, based on historical experience.
The Company did not recognize any revenue during the year ended December 31, 2017.
Research and Development
Research and development costs are expensed as incurred.
Stock-based Compensation
The Company measures all stock options and restricted stock awards granted to employees and directors based on the fair value on the date of the grant and recognizes compensation expense of those awards, net of forfeitures, over the requisite service period, which is generally the vesting period of the respective award. Generally, the Company issues stock options and restricted stock awards with only service-based vesting conditions on a straight-line basis over the requisite service period for the entire award (that is, over the requisite service period of the last separately vesting portion of the award).
| F-8 |
The Company measures share-based awards granted to consultants and non-employees based on the fair value of the award on the date at which the related service is complete. Compensation expense is recognized over the period during which services are rendered by such consultants and non-employees until completed. At the end of each financial reporting period prior to completion of the service, the fair value of these awards is re-measured using the then-current fair value of our ordinary shares and updated assumption inputs in the Black-Scholes option-pricing model
The Company elected to use the Black-Scholes option-pricing model for valuation of stock-based payment awards. The determination of fair value of stock-based payment awards on the date of grant using the Black-Scholes option-pricing model is affected by its stock price as well as assumptions regarding a number of subjective variables. These variables include, but are not limited to, its expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors. When performance-based grants are issued, the Company recognizes no expense until achievement of the performance requirement is deemed probable.
Share-based compensation expense is based on awards ultimately expected to vest and has been reduced for annualized estimated forfeiture where the minimum amount of expense recorded is at least equal to the percent of an award vested. Forfeitures were estimated based on historical experience and weighting of various employee classes under the respective Plans at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
The fair values of Restricted Stock Units (RSU) are based on the number of shares granted and market price of the stock on the date of grant and are recorded as compensation expense ratably over the applicable service period, which is generally four years. Unvested restricted stock units and vested and unvested stock options are forfeited in the event of termination of employment.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to be applied to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
A valuation allowance is recorded when it is more likely than not that some or all of the deferred tax assets will not be realized. Accordingly, the Company provides a valuation allowance, if necessary, to reduce deferred tax assets to amounts that are expected to be realizable.
Tax positions taken or expected to be taken in the course of preparing our tax returns are required to be evaluated to determine whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authority. Tax positions not deemed to meet a more-likely-than-not threshold would be recorded as a tax expense in the current year.
Net Loss per Share
Basic net loss per share is computed using the weighted average number of common shares outstanding during the period. Diluted net loss per share is computed using the sum of the weighted average number of common shares outstanding during the period and, if dilutive, the weighted average number of potential shares of common stock, including the assumed exercise of stock options, warrants, and the impact of unvested restricted stock.
The Company applies the two-class method to calculate basic and diluted net loss per share attributable to common stockholders as its warrants to purchase common stock are participating securities.
The two-class method is an earnings allocation formula that treats a participating security as having rights to earnings that otherwise would have been available to common stockholders. However, the two-class method does not impact the net loss per share of common stock as the Company has been in a net loss position and the warrant holders do not participate in losses.
Basic and diluted shares outstanding are the same for each period presented as all common stock equivalents would be antidilutive due to the net losses incurred.
Foreign Currency Translation
The functional currency of the Company’s foreign subsidiaries is their local currency. All assets and liabilities of its foreign subsidiaries are translated at exchange rates in effect at period-end. Income and expenses are translated at rates which approximate those in effect on the transaction dates. The resulting translation adjustment is recorded as a separate component of stockholders’ equity in accumulated other comprehensive loss in the consolidated balance sheets. Gains and losses resulting from foreign currency transactions are included in net loss. There was no cumulative translation adjustment at December 31, 2017 and 2016. The translation adjustment amount to approximately $8,000 for the year ended December 31, 2016. There was no translation adjustment for the year ended December 31, 2017 or foreign exchange currency gains or losses for the year ended December 31, 2017 and 2016.
| F-9 |
Comprehensive Loss
Comprehensive loss is comprised of net loss and other comprehensive loss. The Company follows the provisions of Financial Accounting Standards Board Accounting Standards Codification 220, “Comprehensive Income” (“FASB ASC 220”). FASB ASC 220 requires companies to report all changes in equity during a period, resulting from net income (loss) and transactions from non-owner sources, in a financial statement in the period in which they are recognized. The Company has chosen to disclose comprehensive loss, which encompasses net loss, foreign currency translation adjustments, net of tax, in the consolidated statements of operations and comprehensive loss.
Warrant Liability
The Company classifies a warrant to purchase shares of its common stock as a liability on its consolidated balance sheets when the warrant is a free-standing financial instrument that may require the Company to transfer cash consideration upon exercise and that cash transfer event would be out of the Company’s control. Such a “liability warrant” is initially recorded at fair value on date of grant using the Black-Scholes model and net of issuance costs, and it is subsequently re-measured to fair value at each subsequent balance sheet date. Changes in fair value of the warrant are recognized as a component of other income (expense), net in the consolidated statement of operations and comprehensive loss. The Company will continue to adjust the liability for changes in fair value until the earlier of the exercise or expiration of the warrant.
For warrants that do not meet the criteria of a liability warrant, the Company measures the value assigned to the warrant using the relative value method to allocate value to the common stock, preferred stock and warrants. The Company uses the Black-Scholes model to measure the value of the warrants at issuance and then applies the relative fair-value of the equity transaction between common stock, preferred stock and warrants. Common stock, preferred stock, and warrants each are considered permanent equity and any potential difference in fair value of each instrument would be reallocated within permanent equity with no periodic remeasurement.
Concentration of Credit Risk
Sales to Harvard Bioscience, the Company’s former distributor of research bioreactor systems, accounted for 100% of the trade receivables and revenues as of and for the year ended December 31, 2016. The Company did not record any revenues or accounts receivable during the year ended December 31, 2017.
Recently Adopted Accounting Pronouncements
In March 2016, the Financial Accounting Standards Board (“FASB”) issued ASU 2016-09, Stock Compensation - Improvements to Employee Share-Based Payment Accounting, (“ASU 2016-09”), which simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, classification on the statement of cash flows and policy elections on the impact for forfeitures. ASU 2016-09 is effective for fiscal years beginning after December 15, 2016 and interim periods within those annual periods. The Company has adopted ASU 2016-09 in 2017 and adoption did not have a significant impact on the Company’s consolidated financial statements or related disclosures.
In August 2014, the FASB issued Accounting Standards Update (“ASU”) No. 2014-15, “Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern,” (“ASU 2014-15”) to provide guidance on management’s responsibility in evaluating whether there is substantial doubt about a company’s ability to continue as a going concern and to provide related footnote disclosures. The Company adopted ASU 2014-15 during the year ended December 31, 2016 and has included the required disclosures in Note 1 to the consolidated financial statements.
| F-10 |
Recently Issued Accounting Pronouncements
In February 2016, the Financial Accounting Standards Board (“FASB”), issued ASU, 2016-02- Leases (Topic 842) (“ASU 2016-02”). The ASU requires companies to recognize on the balance sheet the assets and liabilities for the rights and obligations created by leased assets. ASU 2016-02 will be effective for the Company in the first quarter of 2019, with early adoption permitted. The Company is currently evaluating the impact that the adoption of ASU 2016-02 will have on the Company’s consolidated financial statements or related disclosures.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments (“ASU 2016-15”). This amendment addresses eight classification issues related to the statement of cash flows. For public business entities, the amendments in ASU 2016-15 are effective for public business entities for annual and interim periods in fiscal years beginning after December 15, 2017. The Company does not expect the adoption of this standard will have a material impact on its consolidated financial statements and related disclosures.
In November 2016, the FASB issued ASU 2016-18 Statement of Cash Flows (“ASU 2016-18”) which requires that amounts generally described as restricted cash and restricted cash equivalents should be included with cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. ASU 2016-18 is effective for fiscal years beginning after December 15, 2018 and should be applied using a retrospective transition method to each period presented. Early adoption is permitted. The Company does not have any restricted cash balances at December 31, 2017 and therefore, it does not expect the adoption of this standard will have a material impact on its consolidated financial statements.
Other accounting standards that have been issued or proposed by the FASB or other standards-setting bodies that do not require adoption until a future date are not expected to have a material impact on the Company’s financial statements upon adoption.
3. Fair Value Measurements
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date.
The Company utilizes a valuation hierarchy for disclosure of the inputs to the valuations used to measure fair value. This hierarchy prioritizes the inputs into three broad levels as follows. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on the Company’s own assumptions used to measure assets and liabilities at fair value. A financial asset or liability’s classification within the hierarchy is determined based on the lowest level input that is significant to the fair value measurement.
The Company had no assets or liabilities classified as Level 1 or Level 2. The Company has concluded that warrants to purchase common stock, which are accounted for as liabilities as discussed in Note 5 are classified as Level 3.
| F-11 |
The following fair value hierarchy table presents information about the Company’s financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2017 and 2016:
| Fair Value Measurement as of December 31, 2017 | ||||||||||||||||
| (In thousands) | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Warrant liability | $ | - | $ | - | $ | 16 | $ | 16 | ||||||||
| Total | $ | - | $ | - | $ | 16 | $ | 16 | ||||||||
| Fair Value Measurement as of December 31, 2016 | ||||||||||||||||
| (In thousands) | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Warrant liability | $ | - | $ | - | $ | 605 | $ | 605 | ||||||||
| Total | $ | - | $ | - | $ | 605 | $ | 605 | ||||||||
There were no transfers between Level 1 and Level 2 in either of the years ended December 31, 2017 and December 31, 2016.
4. Property, Plant and Equipment, Net
Property, plant and equipment, net consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Leasehold improvements | $ | 584 | $ | 467 | ||||
| Furniture, machinery and equipment | 1,350 | 1,563 | ||||||
| Computer equipment and software | 464 | 447 | ||||||
| Construction in progress | - | 108 | ||||||
| 2,398 | 2,585 | |||||||
| Less: accumulated depreciation | (1,766 | ) | (1,520 | ) | ||||
| Property, plant and equipment, net | $ | 632 | $ | 1,065 | ||||
Depreciation expense amounted to $413,000 and $454,000 for the years ended December 31, 2017 and 2016, respectively.
5. Warrant Liability
On May 19, 2016 and February 10, 2017, the Company closed on the sale of shares of the Company’s common stock, the issuance of warrants to purchase shares of common stock, and the issuance of warrants to the placement agent for each transaction. See Note 10
Due to a cash put provision within the warrant agreement, which could be enacted in certain change in control events, a liability associated with those warrants was initially recorded at fair value in the Company’s consolidated balance sheets upon issuance, and subsequently re-measured each fiscal quarter. The changes in the fair value between issuance and the end of each reporting period is recorded as a component of other income (expense), net in the consolidated statement of operations and comprehensive income.
During 2017, warrant holders of 952,184 warrants agreed to the modification of the terms of their warrants, which resulted in placing all situations that would allow the warrant holder to put the warrant for cash fully in control of the Company. As a result of the modification, the modified warrants are no longer liability classified and do not need to be re-measured. These modifications resulted in a $3.7 million value of those warrants being reclassified from Warrant Liabilities to Additional Paid in Capital. There were 132,367 warrants exercised during 2017. The remaining un-modified 92,213 warrants will continue to be re-measured at each reporting period as long as they are outstanding and un-modified.
| F-12 |
The Company has re-measured the liability to estimated fair value at inception, prior to modification and at each reporting date using the Black-Scholes option pricing model with the following weighted average assumptions:
| Assumptions for estimating fair value of warrants | Assumptions for estimating fair value | |||||||||||||||
| modified during the three months ended | on reporting dates of | |||||||||||||||
| September 30, 2017 | June
30, | December 31, 2017 | December 31, 2016 | |||||||||||||
| Risk-free interest rate | 1.89 | % | 1.77 | % | 2.09 | % | 1.93 | % | ||||||||
| Expected volatility | 82.3 | % | 82.4 | % | 85.0 | % | 72.7 | % | ||||||||
| Expected term (in years) | 4.6 | 4.6 | 4.1 | 4.9 | ||||||||||||
| Expected dividend yield | - | - | - | - | ||||||||||||
| Exercise price | $ | 8.00 | $ | 10.00 | $ | 8.00 | $ | 35.25 | ||||||||
| Market value of common stock | $ | 8.20 | $ | 6.60 | $ | 0.87 | $ | 17.80 | ||||||||
| Warrants to purchase shares of common stock | 100,101 | 824,442 | 92,212 | 78,014 | ||||||||||||
The following table presents a reconciliation of the Company’s warrant liabilities for the years ended December 31, 2017 and 2016:
| Warrant Liability | ||||
| (in thousands) | ||||
| Balance at December 31, 2015 | $ | - | ||
| Issuance of warrants | 1,281 | |||
| Change in fair value upon re-measurement (1) | (676 | ) | ||
| Balance at December 31, 2016 | 605 | |||
| Issuance of warrants | 3,787 | |||
| Reclassification of warrant liability to additional paid in capital upon modification | (3,746 | ) | ||
| Reclassification of warrant liability to additional paid in capital upon exercise | (581 | ) | ||
| Change in fair value upon re-measurement (2) | (49 | ) | ||
| Balance at December 31, 2017 | $ | 16 | ||
| (1) | Issuance costs allocated to the warranty liability issued in 2016 amounted to $129 and have been included in the change in fair value of the warranty liability in the accompanying consolidated statements of operations and comprehensive loss. |
| (2) | Issuance costs allocated to the warranty liability issued in 2017 amounted to $385 and have been included in the change in fair value of the warranty liability in the accompanying consolidated statements of operations and comprehensive loss. |
| F-13 |
6. Commitments and Contingent Liabilities
Lease Arrangement
First Pecos Breach Notice
In June, 2017, the Company entered into a binding Memorandum of Understanding with First Pecos, LLC (“First Pecos”), pursuant to which the Company agreed to issue to First Pecos in a private placement 9,700,000 shares of its common stock at a purchase price of $0.315 per share or, to the extent First Pecos, following the transaction, would own more than 19.9% of the Company’s common stock, shares of a new class of preferred stock of the Company with a per-share purchase price of $1,000.
In October 2017, as a result of First Pecos failure to deliver the Purchase Price to the Company following satisfaction of all closing conditions in the Purchase Agreement, the Company delivered a notice to First Pecos and its manager, Leon “Chip” Greenblatt III, stating that First Pecos was in breach of the Purchase Agreement. None of the shares of common stock, shares of Preferred Stock or Warrants were issued to First Pecos. Also in October 2017, First Pecos delivered a notice to the Company stating that, as a result of alleged breaches by the Company of its obligations pursuant to the Purchase Agreement, First Pecos terminated the Purchase Agreement and demanded that the Company pay a $500,000 termination fee pursuant to the terms of the Purchase Agreement.
The Company believes that it was not in breach of the Purchase Agreement at any time, and that First Pecos’ notice was unjustified and without any legal merit or factual basis. Accordingly, the Company believes that First Pecos was not entitled to terminate the Purchase Agreement, and is not entitled to any termination fee thereunder, as the failure to consummate the Pecos Placement resulted from First Pecos’ breach of the Purchase Agreement. The Company has not accrued for this liability as the Company believes the claim to be without merit.
In October 2013, the Company entered into a sublease with Harvard Bioscience effective November 1, 2013 for its headquarters, offices, manufacturing, and research and development facilities located in Holliston, Massachusetts. The operating lease was non-cancelable for an initial eighteen-month period. The sublease was automatically extended in 2017 through May 31, 2018 and will renew annually unless the Company or Harvard Bioscience provides a notice of termination within one hundred and eighty days prior to May 31 of each year. Total rent expense was $0.1 million and $0.1 million for the years ended December 31, 2017 and 2016, respectively. Future minimum lease payments for operating leases with initial or remaining terms in excess of one year at December 31, 2017 amounts to $102 thousand in 2018 and $43 thousand in 2019. See Note 15.
Other
On April 14, 2017, representatives for the estate of a deceased individual filed a civil lawsuit in the Suffolk Superior Court, in Boston, Massachusetts, against the Company, Harvard Bioscience and other defendants. The complaint alleges that the decedent was harmed by two tracheal implants that incorporated synthetic trachea scaffolds and a biologic component combined by the implanting surgeon with a bioreactor, and surgically implanted in the decedent in two surgeries performed in 2012 and 2013, which harm caused her injury and death. The civil complaint seeks a non-specific sum of money to compensate the plaintiffs. This civil lawsuit relates to the Company’s first-generation trachea scaffold technology for which the Company discontinued development in 2014, and not to the Company’s current Cellframe technology nor to its lead development product candidate, the Cellspan esophageal implant. The litigation is at an early stage and the Company intends to vigorously defend this case. While the Company believes that such claim lacks merit, the Company is unable to predict the ultimate outcome of such litigation. In accordance with a separation and distribution agreement between Harvard Bioscience and the Company relating to the Separation, the Company would be required to indemnify Harvard Bioscience against losses that Harvard Bioscience may suffer as a result of this litigation. The Company has been informed by its insurance provider that the case has been accepted as an insurable claim under the Company’s product liability insurance policy.
From time to time, the Company may be involved in various claims and legal proceedings arising in the ordinary course of business. Other than the above matter, there are no such matters pending that the Company expects to be material in relation to its business, financial condition, and results of operations or cash flows.
7. Income Taxes
Income taxes for the years ended December 31, 2017 and 2016 differed from the amount computed by applying the U.S. federal income tax rate of 34% to pre-tax loss as a result of the following:
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Computed “expected” income tax benefit | $ | (4,051 | ) | $ | (3,937 | ) | ||
| Increase (decrease) in income taxes resulting from: | ||||||||
| Foreign tax rate and regulation differential | - | 1 | ||||||
| State income tax benefit, net of federal income tax benefit | (716 | ) | (694 | ) | ||||
| Permanent items, primarily change in fair value of warrants | 143 | (185 | ) | |||||
| Tax credits | (452 | ) | (400 | ) | ||||
| Adjustment of prior year income tax | 729 | - | ||||||
| Change in deferred income tax rate | 5,652 | - | ||||||
| Change in valuation allowance allocated to income tax expense | (1,305 | ) | 5,215 | |||||
| Total income taxes | $ | - | $ | - | ||||
The Company has incurred pre-tax losses for the years ended December 31, 2017 and 2016:
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Domestic | $ | (11,916 | ) | $ | (11,569 | ) | ||
| Foreign | - | (10 | ) | |||||
| Total | $ | (11,916 | ) | $ | (11,579 | ) | ||
| F-14 |
The components of the Company’s deferred tax asset are as follows:
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Deferred tax assets: | ||||||||
| Operating loss and credit carryforwards | $ | 6,819 | $ | 7,207 | ||||
| Capitalized research and development | 4,939 | 5,282 | ||||||
| Stock-based compensation | 2,021 | 2,683 | ||||||
| Other | 56 | 63 | ||||||
| Total deferred tax assets | 13,835 | 15,235 | ||||||
| Less: valuation allowance | (13,835 | ) | (15,140 | ) | ||||
| Deferred tax assets | - | 95 | ||||||
| Deferred tax liabilities: | ||||||||
| Accrued expenses | - | 95 | ||||||
| Deferred tax assets, net | - | - | ||||||
On December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act (“TCJA”) that significantly reforms the Internal Revenue Code of 1986, as amended. The TCJA, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, effective as of January 1, 2018; limitation of the tax deduction for interest expense; limitation of the deduction for net operating losses to 80% of annual taxable income and elimination of net operating loss carrybacks, in each case, for losses arising in taxable years beginning after December 31, 2017 (though any such tax losses may be carried forward indefinitely); and modifying or repealing many business deductions and credits, including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions generally referred to as “orphan drugs”.
The staff of the Securities and Exchange Commission issued Staff Accounting Bulletin No. 118 to address the application of GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the TCJA. The Company has reevaluated its assets and liabilities associated with such future tax benefits in the current year and recognized a decrease in its deferred tax asset of $5.8 million. This reduction in the deferred tax asset from the change in the deferred tax rate has been offset by a coinciding reduction in the associated valuation allowance, creating a $0 net impact.
The Company has recorded a valuation allowance against its deferred tax assets for the years ended December 31, 2017 and 2016, because the Company’s management believes that it is more likely than not that these assets will not be realized. The valuation allowance increased by approximately $5.2 million for the year ended December 31, 2016 primarily as a result of operating losses generated with no corresponding financial statement benefit. The valuation allowance decreased by approximately $1.1 million for the year ended December 31, 2017 due to the decrease in the corporate tax rate from 34% to 21%, which was enacted on December 22, 2017, partially offset by an increase in net operating losses.
As of December 31, 2017, the Company had federal net operating loss carryforwards (“NOLs”) of approximately $19.8 million to offset future federal taxable income. As of December 31, 2017, the Company had state net operating loss carryforwards of approximately $19.4 million to offset future state taxable income. The federal and state NOLs begin to expire in 2033. As of December 31, 2017, the Company also has federal and state tax research and development credit carryforwards of approximately $0.9 million and $0.6 million, respectively, to offset future income taxes. The federal and state research and development tax credit carryforwards begin to expire in 2033 and 2029, respectively
Under the provisions of the Internal Revenue Code, the net operating loss and tax credit carryforwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. Net operating loss and tax credit carryforwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three-year period in excess of 50 percent, as defined under Sections 382 and 383 of the Internal Revenue Code, respectively, as well as similar state provisions. This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. The amount of the annual limitation is determined based on the value of the Company immediately prior to the ownership change. Subsequent ownership changes may further affect the limitation in future years. The Company has recently completed several equity financings transactions which have either individually or cumulatively resulted in a change in control as defined by Sections 382 and 383 of the Internal Revenue Code, or could result in a change in control in the future. The Company does not believe the impact of any limitation on the use of its net operating loss or credit carryforwards will have a material impact on the Company’s consolidated financial statements since the Company has a full valuation allowance against its deferred tax assets due to the uncertainty regarding future taxable income for the foreseeable future.
| F-15 |
For all years through December 31, 2017, the Company generated research credits but has not conducted a study to document the qualified activities. This study may result in an adjustment to the Company's research and development credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been provided against the Company's research and development credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the deferred tax asset established for the research and development credit carryforwards and the valuation allowance.
Harvard Bioscience received a Supplemental Ruling to the Private Letter Ruling dated March 22, 2013 from the IRS to the effect that, among other things, the Separation and related distribution of all of the shares of the Company’s common stock by Harvard Bioscience will qualify as a transaction that is tax-free for U.S. federal income tax purposes under Section 355 and 368(a)(1)(D) of the Internal Revenue Code continuing in effect. The private letter and supplemental rulings and the tax opinion that Harvard Bioscience received from legal counsel to Harvard Bioscience rely on certain representations, assumptions and undertakings, including those relating to the past and future conduct of the Biostage business, and neither the private letter and supplemental rulings nor the opinion would be valid if such representations, assumptions and undertakings were incorrect. Moreover, the private letter and supplemental rulings do not address all the issues that are relevant to determining whether the Distribution will qualify for tax-free treatment. Notwithstanding the private letter and supplemental rulings and opinion, the IRS could determine the Distribution should be treated as a taxable transaction for U.S. federal income tax purposes if, among other reasons, it determines any of the representations, assumptions or undertakings that were included in the request for the private letter and supplemental rulings are false or have been violated or if it disagrees with the conclusions in the opinion that are not covered by the IRS ruling.
To preserve the tax-free treatment to Harvard Bioscience of the Separation and Distribution, for the two-year period following the Distribution, which such period ended November 1, 2015, the Company was limited, except in specified circumstances, from entering into certain transactions pursuant to which all or a portion of the Company’s stock would be acquired, whether by merger or otherwise; issuing equity securities beyond certain thresholds; repurchasing the Company’s common stock; and ceasing to actively conduct the Company’s regenerative medicine business. In addition, at all times, including during and following such two-year period, the Company may not take or fail to take any other action that prevents the Separation and Distribution and related transactions from being tax-free.
If the Distribution fails to qualify for tax-free treatment, in general, Harvard Bioscience would be subject to tax as if it had sold the Company’s common stock in a taxable sale for its fair market value, and Harvard Bioscience stockholders who receive shares of Biostage common stock in the Distribution would be subject to tax as if they had received a taxable Distribution equal to the fair market value of such shares.
Under the tax sharing agreement between Harvard Bioscience and the Company, the Company would generally be required to indemnify Harvard Bioscience against any tax resulting from the Distribution to the extent that such tax resulted from (i) an acquisition of all or a portion of our stock or assets, whether by merger or otherwise, (ii) other actions or failures to act by the Company, or (iii) any of the Company’s representations or undertakings being incorrect or violated. The Company’s indemnification obligations to Harvard Bioscience and its subsidiaries, officers and directors are not limited by any maximum amount. If the Company is required to indemnify Harvard Bioscience or such other persons under the circumstances set forth in the tax sharing agreement, the Company may be subject to substantial liabilities.
All deferred tax assets prior to the Separation remained with Harvard Bioscience, Inc.
The Company has determined that any uncertain tax positions would have no material impact on the consolidated financial statements of the Company and there are no unrecognized tax benefits or related interest and penalties accrued for the period for the years ended December 31, 2017 and 2016. When necessary, the Company will recognize interest and penalties related to uncertain tax positions in income tax expense.
The Company is subject to U.S. federal income tax and Massachusetts state income tax. The statute of limitations for assessment by the IRS and state tax authorities is open for all periods from inception through December 31, 2017; currently, no federal or state income tax returns are under examination by the respective taxing authorities.
| F-16 |
8. Employee Benefit Plans
The Company sponsors a retirement plan for their U.S. employees, which includes employee savings plans established under Section 401(k) of the U.S. Internal Revenue Code (the “401(k) Plan”). The 401(k) Plan covers substantially all full-time employees who meet certain eligibility requirements. Contributions to the retirement plan are at the discretion of management. The Company’s matching contributions to the plan were approximately $0.1 million for each of the years ended December 31, 2017 and 2016.
9. Preferred Stock
The Company had the following categories of Preferred Stock, $0.01 par value, at December 31, 2017:
| Authorized | Issued | Outstanding | ||||||||||
| Undesignated Preferred Stock | 984,000 | - | - | |||||||||
| Series B Convertible Preferred Stock (1) | 1,000,000 | 695,857 | - | |||||||||
| Series C Convertible Preferred Stock (2) | 4,000 | - | - | |||||||||
| Series D Convertible Preferred Stock | 12,000 | 3,108 | 3,108 | |||||||||
The Company had the following categories of Preferred Stock, $0.01 par value, at December 31, 2016:
| Authorized | Issued | Outstanding | ||||||||||
| Undesignated Preferred Stock | 996,000 | - | - | |||||||||
| Series B Convertible Preferred Stock (1) | 1,000,000 | 695,857 | - | |||||||||
| Series C Convertible Preferred Stock (2) | 4,000 | - | - | |||||||||
| (1) | The Company issued 695,857 shares of its Series B Convertible Preferred Stock in February 2015, which were subsequently converted into common stock during the year ended December 31, 2015. |
| (2) | The Company designated the Series Convertible Preferred Stock in August 2017, but did not issue any Series C convertible preferred shares. |
Undesignated Preferred Stock
The Board of Directors may exercise its authority to issue undesignated preferred shares and determine the price, privileges and other terms of the shares without any further approval of stockholders.
Series D Convertible Preferred Stock
On December 27, 2017, the Company issued 518,000 shares of its common stock at $2.00 per share, 3,108 shares of its Series D Convertible Preferred Stock (the “Series D Preferred Stock”) at $1,000 per share, and warrants to purchase 3,108,000 shares of common stock at an exercise price of $2.00 per share, in exchange for aggregate gross proceeds of approximately $4.1 million in a private placement transaction of unregistered shares. The warrants were immediately exercisable and expire in December 2022. The Company has allocated $2.1 million of consideration to the warrants using the relative fair-value method and included such amount in additional paid in capital.
The relevant features of Series D Preferred Stock are as follows:
Liquidation Rights
In the event of any voluntary or involuntary liquidation, dissolution, or winding-up of the affairs of the Company (a “Deemed Liquidation Event”), each holder of a share of the Series D Preferred Stock is entitled to receive on a pari-passu basis with common stockholders any distribution of any of the assets or surplus funds of the Company as if all of the shares of Series D Preferred Stock been converted to common stock prior to the Deemed Liquidation Event.
| F-17 |
Conversion
Each share of Series D Preferred Stock is convertible at the option of the holder, at any time after the date of issuance and without the payment of any additional consideration, into that number of shares of common stock as is determined by dividing the stated value of each share of Series D Preferred Stock of $1,000 by a conversion price of $2.00 per share. The shares of Series D Preferred Stock were convertible into 1,554,000 shares of common stock at December 31, 2017.
Voting Rights
The holders of Series D Preferred Stock are entitled to vote on all matters on which shares of common stock are entitled to vote (on an as-if-converted to common stock basis).
Dividend Rights
Holders of Series D Preferred Stock are entitled to receive, and the Company will pay, dividends on shares of Series D Preferred Stock equal (on an as-if-converted-to-common stock basis) to and in the same form as dividends actually paid on shares of the common stock when, as and if such dividends are paid on shares of the common stock. No other dividends shall be paid on shares of Series D Preferred Stock.
Redemption Rights
The holders of Series D Preferred Stock are not entitled to any redemption rights, other than those under their liquidation rights.
Protective Rights
Holders of Series D Preferred Shares are entitled to certain protective rights whereby the Company cannot make certain decisions without the consent of a majority of the outstanding holders of the Series D Preferred Stock.
Classification
The Company has classified the Series D Preferred Stock as permanent equity since the ability to control redemption is not outside of the Company’s control.
10. Common Stock
During 2017, the Company increased the number of common shares authorized from 60 million to 120 million.
The following represent the Company’s common stock transactions during December 31, 2017 and 2016:
December 2017 Private Placement
On December 27, 2017, the Company issued 518,000 shares of its common stock at $2.00 per share in connection with its Series D Preferred Stock issuance in a private placement transaction. See Note 9.
February 2017 Shares Offering
On February 10, 2017, the Company completed a public offering of 1,000,000 shares of common stock at a purchase price of $8.00 per share and the issuance of warrants to purchase 1,000,000 million shares of common stock at an exercise price of $8.00 per warrant for gross proceeds of $8.0 million or approximately $6.8 million net of issuance costs. Additionally, the Company issued to the placement agent warrants to purchase 50,000 shares of common stock for the offering at an exercise price of $10.00 per warrant.
May 2016 Share Offerings
On May 19, 2016, the Company closed on a Securities Purchase Agreement (the “Purchase Agreement”) for the sale of 141,844 shares of common stock at a purchase price of $35.25 per share and the issuance of warrants to purchase 70,922 shares of common stock at an exercise price of $35.25 per warrant for gross proceeds of $5.0 million. Additionally, the Company issued to the placement agent warrants to purchase 7,092 shares of common stock to the placement agent for the offering at an exercise price of $35.25 per warrant. The warrants are exercisable and expire May 19, 2021. As of December 31, 2017, all 78,041 warrants remain outstanding.
In addition, in a separate transaction the Company issued 7,500 shares of common stock under the common stock purchase agreement with Aspire Capital Fund, LLC in exchange for gross proceeds of $371,000, or $349,000 net of issuance costs on May 12, 2016. The transaction was the final issuance under a common stock purchase agreement with Aspire Capital Fund, LLC which commenced in December 2015 and was terminated without penalty on May 17, 2016.
| F-18 |
Employee Stock Purchase Plan
In 2013, the Company approved the 2013 Employee Stock Purchase Plan (the “ESPP Plan”). Under the ESPP Plan, participating employees can authorize the Company to withhold a portion of their base pay during consecutive six-month payment periods for the purchase of shares of the Company’s common stock. At the conclusion of the period, participating employees can purchase shares of the Company’s common stock at 85% of the lower of the fair market value of the Company’s common stock at the beginning or end of the period. Shares are issued under the plan for the six-month periods ending June 30 and December 31. Under this plan, 7,500 shares of common stock are authorized for issuance of which 4,534 shares were issued as of December 31, 2017. There are 2,966 shares available for issuance as of December 31, 2017.
Shareholder Rights Plan
The Company had adopted a Shareholder Rights Plan and declared a dividend distribution of one preferred stock purchase right for each outstanding share of the Company’s common stock. Initially, these rights would not be exercisable and traded with the shares of the Company’s common stock. The Company’s Board of Directors canceled the Shareholder Right Plan in August 2017.
11. Share-Based Compensation
Biostage 2013 Equity Incentive Plan
The Company maintains the 2013 Equity Incentive Plan (the “Plan”) for the benefit of certain of its officers, employees, non-employee directors, and other key persons (including consultants and advisory board members). All options and awards granted under the Plan consist of the Company’s shares of common stock. The Company’s policy is to issue stock available from its registered but unissued stock pool through its transfer agent to satisfy stock option exercises and vesting of the restricted stock units. The vesting period for awards is generally four years and the contractual life is ten years. In March 2016, the Company’s board of directors approved an increase of 100,000 shares from 198,000 shares of its common stock authorized to be issued under the Plan to 298,000 shares.
On October 31, 2013, Harvard Bioscience, Inc. (“Harvard Bioscience”) contributed its regenerative medicine business assets, plus $15 million of cash, into Biostage (the “Separation”). On November 1, 2013, the spin-off of the Company from Harvard Bioscience was completed. On that date, the Company became an independent company that operates the regenerative medicine business previously owned by Harvard Bioscience. The spin-off was completed through the distribution to Harvard Bioscience stockholders of all the shares of common stock of Biostage (the “Distribution”).
The Company also issued equity awards under the Plan at the time of the Distribution to all holders of Harvard Bioscience equity awards as part of an adjustment (the “Adjustment”) to prevent a loss of value due to the Distribution. Compensation expense recognized under the Plan relates to service provided by employees, board members and a non-employee of the Company. There was no required compensation associated with the Adjustment awards to employees who remained at Harvard Bioscience. During 2017 and 2016, no options or restricted stock units were granted to Harvard Bioscience employees or directors, and the Company does not anticipate issuing any to Harvard Bioscience employees in the future.
Stock option activity under the Plan for the year ended December 31, 2017 was as follows:
| Amount | Weighted-average exercise price | Weighted-average contractual life | ||||||||||
| Outstanding at December 31, 2016 | 193,884 | $ | 56.10 | 8.37 | ||||||||
| Granted | 94,825 | 7.94 | ||||||||||
| Canceled | (121,235 | ) | ||||||||||
| Outstanding at December 31, 2017 | 167,474 | $ | 45.84 | 6.99 | ||||||||
| Options exercisable | 100,188 | $ | 63.28 | 5.99 | ||||||||
| Options vested and expected to vest | 152,117 | $ | 47.89 | |||||||||
| F-19 |
Aggregate intrinsic value for outstanding options and exercisable options for the year ended December 31, 2017 amounted to $0 and was based on the Company’s closing stock price of $0.87 per share as of December 31, 2017. As of December 31, 2017, unrecognized compensation cost related to unvested awards amounted to $0.7 million, which will be recognized over a weighted average period of 1.79 years.
The Company uses the Black- Scholes model to value its stock options. The weighted average assumptions for valuing those options granted were as follows:
| Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Volatility | 79.21 | % | 74.18 | % | ||||
| Risk-free interest rate | 2.27 | % | 1.53 | % | ||||
| Expected holding period | 6.31 years | 6.14 years | ||||||
| Dividend yield | - | % | - | % | ||||
The Company used the volatility of comparable companies, as management did not believe that our trading history was of a sufficient duration to provide an accurate estimate of expected volatility. The risk-free interest rate assumption is based upon observed Treasury bill interest rates (risk-free) appropriate for the expected term of the Company’s employee stock options. The simplified method of estimating expected term was used.
The Company also estimated the fair value of non-employee share options using the Black-Scholes option pricing model reflecting the same assumptions as applied to employee and director options in each of the reporting periods, other than the expected life, which is assumed to be the remaining contractual life of the options.
The weighted average estimated fair value of stock options granted using the Black-Scholes model was $5.56 per share during 2017 and $20.26 per share during 2016.
The Company also has issued restricted stock units under the Plan. Unvested shares of restricted common stock may not be sold or transferred by the holder. The following table summarizes the Company’s unvested restricted stock unit activity under the Plan for the year ended December 31, 2017:
| Amount | Grant date fair value | |||||||
| Unvested at December 31, 2016 | 13 | $ | 120.00 | |||||
| Granted | 20,237 | 7.68 | ||||||
| Canceled | (5,362 | ) | 7.68 | |||||
| Vested | (13 | ) | 120.00 | |||||
| Unvested at December 31, 2017 | 14,875 | $ | 7.68 | |||||
| F-20 |
Harvard Bioscience Stock Option and Incentive Plan
Harvard Bioscience maintains the Third Amended and Restated 2000 Stock Option and Incentive Plan (as amended, the “Harvard Bioscience Plan”) for the benefit of certain of its officers, directors and employees. All awards that were granted to the Company’s employees and directors were at exercise prices equal to or greater than fair market value of the Harvard Bioscience’s common stock on the date of grant. In connection with the Separation, those employees of Harvard Bioscience who became employees of Biostage were allowed to continue vesting in their stock-based awards of stock options and restricted stock units granted under the Harvard Bioscience Plan. Accordingly, the Company recognizes compensation expense as services are provided by those employees. The vesting period is generally four years and the contractual life is ten years.
As of December 31, 2017, there was no unrecognized compensation costs as all awards were fully vested.
Share-based compensation expense related to both the Plan and the Harvard Bioscience Plan for the years ended December 31, 2017 and 2016 was allocated as follows:
| Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Research and development | $ | 276 | $ | 668 | ||||
| Selling, general and administrative | 417 | 659 | ||||||
| Total stock-based compensation | $ | 693 | $ | 1,327 | ||||
12. Net loss per Share
Basic and diluted net loss per share was calculated as follows:
| Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands, except per share data) | ||||||||
| Net loss | $ | (11,916 | ) | $ | (11,579 | ) | ||
| Weighted average shares outstanding | 1,797 | 799 | ||||||
| Net loss per share – basic and diluted | $ | (6.63 | ) | $ | (14.49 | ) | ||
The Company’s potential dilutive securities, which include stock options, unvested restricted common stock units, Series D convertible preferred stock and warrants, have been excluded from the computation of diluted net loss per share whenever the effect of including them would be to reduce the net loss per share. In periods where there is a net loss, the weighted average number of common shares outstanding used to calculate both basic and diluted net loss per share attributable to common stockholders is the same.
| F-21 |
The following potential common shares were excluded from the calculation of diluted net loss per share attributable to common stockholders for the years ended December 31, 2017 and 2016 because including them would have had an anti-dilutive effect:
| Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Series D convertible preferred stock | 1,554,000 | - | ||||||
| Unvested restricted common stock units | 14,875 | 13 | ||||||
| Warrants to purchase common stock | 4,103,647 | 78,014 | ||||||
| Options to purchase common stock | 167,474 | 193,884 | ||||||
| Total | 5,839,996 | 271,911 | ||||||
13. Headcount Reduction
During October and November 2017 and in effort to conserve cash, the Company completed a reduction in headcount of 20 of its employees. In addition, officers of the Company agreed to a temporary reduction in their salaries by 50% effective November 2017. The Company incurred charges for termination benefits in connection with the headcount reduction of approximately $99,000 for employee severance and related costs. The Company did not make any payments during the year ended December 31, 2017. All amounts were paid in January and February 2018.
14. Other Expense
Other income (expense), net consisted of the following for the years ended December 31, 2017 and 2016:
| Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Other expense: | ||||||||
| Loss on disposal of equipment | $ | (111 | ) | $ | - | |||
| Total other expense | $ | (111 | ) | $ | - | |||
15. Related Party Transactions
Relationship with Harvard Bioscience
On October 31, 2013, Harvard Bioscience, Inc. contributed its regenerative medicine business assets, plus $15 million of cash, into Biostage pursuant to the Separation. On November 1, 2013, the spin-off of the Company from Harvard Bioscience was completed. On that date, the Company became an independent company that operates the regenerative medicine business previously owned by Harvard Bioscience. The spin-off was completed through the distribution of all the shares of common stock of Biostage to Harvard Bioscience stockholders pursuant to the Distribution.
At the time of the Separation, the Company entered into a 10-year product distribution agreement with Harvard Bioscience under which each company will become the exclusive distributor for the other party for products such other party develops for sale in the markets served by the other. In addition, Harvard Bioscience has agreed that except for certain existing activities of its German subsidiary, to the extent that any Harvard Bioscience business desires to resell or distribute any bioreactor that is then manufactured by the Company, the Company will be the exclusive manufacturer of such bioreactors and Harvard Bioscience will purchase such bioreactors from the Company. Since inception of the Company, sales to Harvard Bioscience accounted for 100% of the Company’s revenues and receivables.
| F-22 |
On November 3, 2017, in exchange for settlement of approximately $0.1 million of outstanding rent and operating expenses due to Harvard Bioscience, Biostage sold all of its current stock of research bioreactor parts, a royalty free perpetual sublicensable and transferable right and license to use the intellectual property, including but not limited to certain patents covering research bioreactors, and relinquished exclusive manufacturing or distribution rights with respect to research bioreactors to Harvard Bioscience. The Company had ceased the manufacture of research bioreactors in late 2016, to concentrate its efforts solely development of its clinical product candidates. This settlement only covers research bioreactors, not to be used for clinical purposes. The Company retains full exclusive rights to all assets and rights associated with the clinical bioreactor used in the development of the Company’s current Cellframe technology.
Due to Related Party
In connection with the Company’s private placement transaction in December 2017, an investor placed a deposit in the amount of $0.3 million with the Company, which was subsequently repaid in January 2018.
16. Quarterly Financial Information (Unaudited)
Statement of Operations Data:
| First | Second | Third | Fourth | Fiscal | ||||||||||||||||
| 2017 | Quarter | Quarter | Quarter | Quarter | Year | |||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Revenues | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||
| Gross profit | - | - | - | - | - | |||||||||||||||
| Net loss | (3,841 | ) | (3,603 | ) | (3,243 | ) | (1,229 | ) | (11,916 | ) | ||||||||||
| Basic and diluted net loss per share | $ | (2.83 | ) | $ | (1.94 | ) | $ | (1.66 | ) | $ | (0.61 | ) | $ | (6.63 | ) | |||||
| First | Second | Third | Fourth | Fiscal | ||||||||||||||||
| 2016 | Quarter | Quarter | Quarter | Quarter | Year | |||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Revenues | $ | - | $ | 28 | $ | 26 | $ | 28 | $ | 82 | ||||||||||
| Gross (loss) profit | - | (16 | ) | 13 | (31 | ) | (34 | ) | ||||||||||||
| Net loss | (2,472 | ) | (2,712 | ) | (3,053 | ) | (3,342 | ) | (11,579 | ) | ||||||||||
| Basic and diluted net loss per share | $ | (3.50 | ) | $ | (3.49 | ) | $ | (3.57 | ) | $ | (3.91 | ) | $ | (14.49 | ) | |||||
17. Subsequent events
On March 28, 2018, the Company was awarded a Fast-Track Small Business Innovation Research (SBIR) grant by the Eunice Kennedy National Institute of Child Health and Human Development. The award for Phase I was $225,000 and the SBIR grant has the potential to provide a total award of $1.7 million. If Phase I is successful, and funding is available, a Phase II award of up to approximately $1.5 million would support pre-clinical testing of pediatric Cellspan™ Esophageal Implants planned to begin later in 2017. The Phase II Funds, if awarded, would be spent over an estimated two years.
On February 20, 2018, the Company completed a private placement of 302,115 shares of common stock at a purchase price of $3.31 per share for net proceeds of $1.0 million.
On January 3, 2018, the Company issued 50,000 shares of its common stock at a price of $2.00 per share and warrants to purchase 75,000 shares of common stock at an exercise price of $2.00 per share. The Company received net proceeds from the sale of common stock of $0.1 million.
| F-23 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Biostage, Inc. | ||
| Date: April 2, 2018 | ||
| By: | /s/ James McGorry | |
| James McGorry | ||
| Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
| Signature | Title | Date | ||
| /s/ James McGorry | Chief Executive Officer and Director | April 2, 2018 | ||
| James McGorry | (Principal Executive Officer) | |||
| /s/ Thomas McNaughton | Chief Financial Officer | April 2, 2018 | ||
| Thomas McNaughton | (Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Jason Jing Chen | Chairman | April 2, 2018 | ||
| Jason Jing Chen | ||||
| /s/ John Canepa | Director | April 2, 2018 | ||
| John Canepa | ||||
| /s/ Blaine McKee | Director | April 2, 2018 | ||
| Blaine McKee | ||||
| /s/ Thomas Robinson | Director | April 2, 2018 | ||
| Thomas Robinson |
| 48 |
EXHIBIT INDEX
The following exhibits are filed as part of this Annual Report on Form 10-K. Where such filing is made by incorporation by reference to a previously filed document, such document is identified.
| 49 |
| (1) | Previously filed as an exhibit to the Company’s Registration Statement on Form 10-12B (filed July 31, 2013) and incorporated by reference thereto. |
| (2) | Previously filed as an exhibit to the Company’s Registration Statement on Form 8-A (filed October 31, 2013) and incorporated by reference thereto. |
| (3) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on November 6, 2013) and incorporated by reference thereto. |
| (4) | Previously filed as an exhibit to the Company’s Amendment No. 2 to Form S-1 Registration Statement (filed on February 15, 2013) and incorporated by reference thereto. |
| 50 |
| (5) | Previously filed as Exhibit 10.19 to the Registrant's Amendment No. 2 to Form S-1 Registration Statement (filed on February 15, 2013) and incorporated by reference thereto. |
| (6) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on February 12, 2015) and incorporated by reference thereto. |
| (7) | Previously filed as an exhibit to the Company’s Annual Report on Form 10-K (filed on March 27, 2015) and incorporated by reference thereto. |
| (8) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on July 6, 2015) and incorporated by reference thereto. |
| (9) | Previously filed as an exhibit to the Company’s Quarterly Report on Form 10-Q (filed on August 14, 2015) and incorporated by reference thereto. |
| (10) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on December 15, 2015) and incorporated by reference thereto. |
| (11) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on March 24, 2016) and incorporated by reference thereto. |
| (12) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on May 16, 2016) and incorporated by reference thereto. |
| (13) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on May 20, 2016) and incorporated by reference thereto. |
| (14) | Previously filed as an exhibit to the Company’s Current Report on Form 8-K (filed on March 31, 2016) and incorporated by reference thereto. |
| (15) | Previously filed as an exhibit to the Company's Amendment No. 2 to Form S-1 Registration Statement (filed on February 7, 2017) and incorporated by reference thereto. |
| (16) | Previously filed as an exhibit to the Company’s Annual Report on Form 10-K (filed on March 17, 2017) and incorporated by reference thereto. |
| (17) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on April 27, 2017) and incorporated by reference thereto. |
| (18) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on June 27, 2017) and incorporated by reference thereto. |
| (19) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on August 17, 2017) and incorporated by reference thereto. |
| (20) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on December 22, 2017) and incorporated by reference thereto. |
| (21) | Previously filed as an exhibit to the Company's Form S-1 Registration Statement (filed on August 15, 2017) and incorporated by reference thereto. |
| (22) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on December 14, 2017) and incorporated by reference thereto. |
| (23) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on January 3, 2018) and incorporated by reference thereto. |
| (24) | Previously filed as an exhibit to the Company's Current Report on Form 8-K (filed on February 8, 2018) and incorporated by reference thereto. |
| 51 |
| * | Filed herewith. |
| ** | This certification shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, or otherwise subject to the liability of that section, nor shall it be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934. |
| # | Management contract or compensatory plan or arrangement. |
| § | The schedules and exhibits to the Separation and Distribution Agreement have been omitted. A copy of any omitted schedule or exhibit will be furnished to the SEC supplementally upon request. The Company will furnish to stockholders a copy of any exhibit without charge upon written request. |
| † | Confidential portions of this exhibit have been redacted and filed separately with the SEC pursuant to a confidential treatment request in accordance with Rule 24b-2 of the Securities Exchange Act of 1934, as amended. |
| 52 |