Decellularization of Porcine Liver

Figure: These images show: (i) a porcine liver prior to being decellularized, (ii) a porcine liver eight hours into the decellularization process and (iii) a porcine liver twenty-four hours into the decellularization process. These images have been provided to demonstrate the decellularization process, and are not protypes for our BEL product candidate, and do not represent results from a clinical trial.
Recellularization
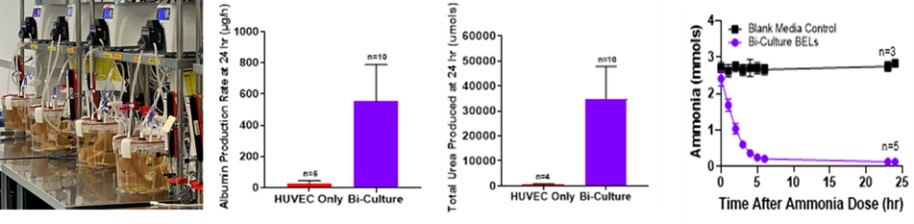
Recellularization is a process that incorporates perfusion to re-seed the ECM with unmodified human cells inside individual bioreactors. We source the human cells from human kidneys and livers not placed for transplant. We have agreements with several Organ Procurement Organizations (“OPOs”) to secure our supplies of human kidneys and livers. We have successfully demonstrated the ability to isolate the required cells from even highly compromised organs and these isolated cells are either directly seeded, or cultured and expanded in proprietary media, which we believe will enable us to recellularize multiple ECMs from one human organ. The recellularized organ matures in a bioreactor where it is continuously perfused with nutrient-rich media and oxygenated under normal physiological conditions. Our entire recellularization process can be completed in two to four weeks depending on the organ type. Our finished bioengineered organs will be able to be transplanted utilizing the same techniques and equipment as current organ transplantation procedures, which we believe could prove favorable in the adoption of our products.
Our Bioengineered Organ Pipeline
All of our bioengineered organ transplant programs utilize our proprietary technology platform. Our product pipeline was developed by leveraging data and learnings from prior programs to inform our development decisions for future programs. The first programs we developed are Miromesh and Miroderm, which are commercialized for hernia repair and wound care applications, respectively. Both products received 510(k) clearances from the FDA and are not classified or regulated as xenotransplants. Both products are derived from porcine livers and utilize our proprietary perfusion decellularization process to create an ECM implant. These products have been on the market for over seven years and have been implanted in thousands of patients since then. Over that time, we have completed three post-market clinical studies and we believe this provides valuable safety data to leverage in the development of our current pipeline. In June 2019, we decided to focus all of our resources on bioengineering human organ transplants, so we licensed Miromesh and Miroderm to a business named Reprise Biomedical, Inc (“Reprise”). In March 2021, we divested the entirety of our equity ownership in Reprise and continue to collect royalties relating to the licensing of these products.
As Miromesh and Miroderm utilize our decellularization process and are not regulated as xenotransplants, and our recellularization process incorporates unmodified human cells, we believe our bioengineered organs should not be regulated as xenotransplants. Our current expectation for our bioengineered organs is that they will be regulated as biologics or potentially combination products. We believe that either classification will lead to a more efficient regulatory pathway than if they were to be classified as xenotransplants.
The FDA defines xenotransplantation as any procedure that involves the transplantation, implantation or infusion into a human recipient of either (a) live cells, tissues, or organs from a nonhuman animal source, or (b) human body fluids, cells, tissues or organs that have had ex vivo contact with live nonhuman animal cells, tissues or organs. The guidance also specifically states that xenotransplantation does not include transplantation, implantation or other use of acellular animal tissues. During our decellularization process, we remove all animal cells and the acellular scaffold is recellularized with unmodified human cells, which we believe will lead regulators to determine that our bioengineered organs are not
3