United States
Securities and Exchange Commission
Washington, D.C. 20549
FORM 10-K
(Mark One)
| [X] | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2018
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM TO
Commission File Number 0-54402
BIORESTORATIVE THERAPIES, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 91-1835664 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
| 40 Marcus Drive, Melville, New York | 11747 | |
| (Address of principal executive offices) | (Zip Code) |
(631) 760-8100
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| None | Not applicable |
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, par value $0.001 per share
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes [ ] No [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer [ ] | Accelerated filer [ ] |
| Non-accelerated filer [ ] | Smaller reporting company [X] |
| Emerging growth company [X] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter.
As of June 30, 2018, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was $9,435,076 based on the closing sale price as reported on the OTCQB market. As of March 27, 2019, there were 14,676,794 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None
INDEX
This Annual Report contains forward-looking statements as that term is defined in the federal securities laws. The events described in forward-looking statements contained in this Annual Report may not occur. Generally these statements relate to business plans or strategies, projected or anticipated benefits or other consequences of our plans or strategies, projected or anticipated benefits from acquisitions to be made by us, or projections involving anticipated revenues, earnings or other aspects of our operating results. The words “may,” “will,” “expect,” “believe,” “anticipate,” “project,” “plan,” “intend,” “estimate,” and “continue,” and their opposites and similar expressions are intended to identify forward-looking statements. We caution you that these statements are not guarantees of future performance or events and are subject to a number of uncertainties, risks and other influences, many of which are beyond our control, that may influence the accuracy of the statements and the projections upon which the statements are based. Factors which may affect our results include, but are not limited to, the risks and uncertainties discussed in Item 7 of this Annual Report under “Factors That May Affect Future Results and Financial Condition”.
Any one or more of these uncertainties, risks and other influences could materially affect our results of operations and whether forward-looking statements made by us ultimately prove to be accurate. Our actual results, performance and achievements could differ materially from those expressed or implied in these forward-looking statements. We undertake no obligation to publicly update or revise any forward-looking statements, whether from new information, future events or otherwise.
Intellectual Property
This Annual Report includes references to our federally registered trademarks, BioRestorative Therapies, the Dragonfly Logo, brtxDISC, ThermoStem, Stem Pearls and Stem the Tides of Time. We also own allowed trademark applications for BRTX and BRTX-100. The Dragonfly Logo is also registered with the U.S. Copyright Office. This Annual Report also includes references to trademarks, trade names and service marks that are the property of other organizations. Solely for convenience, trademarks and trade names referred to in this Annual Report appear without the ®, SM or ™ symbols, and copyrighted content appears without the use of the symbol ©, but the absence of use of these symbols does not reflect upon the validity or enforceability of the intellectual property owned by us or third parties.
| 2 |
| ITEM 1. | BUSINESS. |
(a) Business Development
As used in this Annual Report on Form 10-K (the “Annual Report”), references to the “Company”, “we”, “us”, or “our” refer to BioRestorative Therapies, Inc. and its subsidiaries.
We were incorporated in Nevada on June 13, 1997. On August 15, 2011, we changed our name from “Stem Cell Assurance, Inc.” to “BioRestorative Therapies, Inc.” Effective January 1, 2015, we reincorporated in Delaware.
In January 2017, we submitted an Investigational New Drug application to the U.S. Food and Drug Administration, or the FDA, to obtain authorization to commence a Phase 2 clinical trial investigating the use of BRTX-100, our lead cell therapy candidate, in the treatment of chronic lower back pain arising from degenerative disc disease. In February 2017, we received such authorization from the FDA.
In January 2019, a United States patent related to the ThermoStem Program was issued to us.
In March 2018, we engaged Defined Health, a business development and strategy firm, to conduct an independent review of BRTX-100, our lead cell therapy candidate to treat chronic lumbar disc disease. As noted in the report presented by Defined Health, key opinion leaders interviewed by Defined Health reacted positively to the value proposition of BRTX-100.
In September 2018, we entered into a one year material transfer agreement with the University of Pennsylvania pursuant to which the university is provided access to our proprietary brown adipose tissue (brown fat) cells for research purposes.
In August 2018, the Journal of Translational Medicine published the results of our study evaluating the benefits of long-term hypoxic (low oxygen) culturing of human bone marrow-derived mesenchymal stems cells used in BRTX-100.
In March 2018, we appointed Dr. Wayne J. Olan, a member of our Scientific Advisory Board, as Clinical Director of our Regenerative Disc/Spine Program.
In October 2018, we created a Disc Advisory Committee of our Scientific Advisory Board and we appointed Dr. Jason Lipetz as Chairman of the Disc Advisory Committee. We have also appointed Dr. Olan to the Disc Advisory Committee as well as Drs. Harvinder Sandhu, Christopher Plasteras and Gerard Malanga to the Scientific Advisory Board and the Disc Advisory Committee.
In January 2018, we hired Adam D. Bergstein to serve as Senior Vice President, Planning and Business Development. See Item 10 (“Directors, Executive Officers and Corporate Governance”). Mr. Bergstein resigned his position in October 2018.
In October 2018, we hired Lance Alstodt to serve as Executive Vice President and Chief Strategy Officer. See Item 10 (“Directors, Executive Officers and Corporate Governance”).
| 3 |
During the year ended December 31, 2018, we raised an aggregate of $589,168 in connection with sales of common stock and warrants and from the exercise of warrants, and an aggregate of $4,149,173 in debt financing (net of repayments). As of December 31, 2018, our outstanding debt of $5,161,916, together with interest at rates ranging between 0% and 15% per annum, was due through December 2019.
Subsequent to December 31, 2018, we have received aggregate equity financing and debt financing of $600,000 and $3,073,918, respectively, debt (inclusive of accrued interest) of $643,900 has been exchanged for common stock, $1,254,805 of debt (inclusive of accrued interest and prepayment premiums) has been repaid, and the due date for the repayment of an aggregate of $155,000 of debt has been extended to December 2020. Giving effect to the above actions, we currently have notes payable with an aggregate principal balance of $107,500 which are past due.
(b) Business
General
We are a life sciences company focused on the development of regenerative medicine products and therapies using cell and tissue protocols, primarily involving adult (non-embryonic) stem cells. Our two core developmental programs, as described below, relate to the treatment of disc/spine disease and metabolic disorders:
| ● | Disc/Spine Program (brtxDisc). Our lead cell therapy candidate, BRTX-100, is a product formulated from autologous (or a person’s own) cultured mesenchymal stem cells, or MSCs, collected from the patient’s bone marrow. We intend that the product will be used for the non-surgical treatment of painful lumbosacral disc disorders. The BRTX-100 production process involves collecting bone marrow and whole blood from a patient, isolating and culturing (in a proprietary method) stem cells from the bone marrow and cryopreserving the cells in an autologous carrier. In an outpatient procedure, BRTX-100 is to be injected by a physician into the patient’s painful disc. The treatment is intended for patients whose pain has not been alleviated by non-surgical procedures or conservative therapies and who potentially face the prospect of highly invasive surgical procedures. In January 2017, we submitted an Investigational New Drug, or IND, application to the FDA to obtain authorization to commence a Phase 2 clinical trial investigating the use of BRTX-100 in the treatment of chronic lower back pain arising from degenerative disc disease. In February 2017, we received such authorization from the FDA. We intend to commence such clinical trial during the third quarter of 2019 (assuming the receipt of necessary funding). See “Disc/Spine Program” below. | |
| ● | Metabolic Program (ThermoStem). We are developing a cell-based therapy candidate to target obesity and metabolic disorders using brown adipose (fat) derived stem cells, or BADSC, to generate brown adipose tissue, or BAT. We refer to this as our ThermoStem Program. BAT is intended to mimic naturally occurring brown adipose depots that regulate metabolic homeostasis in humans. Initial preclinical research indicates that increased amounts of brown fat in animals may be responsible for additional caloric burning, as well as reduced glucose and lipid levels. Researchers have found that people with higher levels of brown fat may have a reduced risk for obesity and diabetes. See “Metabolic Brown Adipose (Fat) Program” below. |
| 4 |
We have also licensed an investigational curved needle device designed to deliver cells and/or other therapeutic products or material to the spine and discs (and other parts of the body). See “Curved Needle Device” below.
The patents and patent applications for the Disc/Spine Program, the ThermoStem Program and the curved needle device are listed below under “Technology; Research and Development”.
Overview
Every human being has stem cells in his or her body. These cells exist from the early stages of human development until the end of a person’s life. Throughout our lives, our body continues to produce stem cells that regenerate to produce differentiated cells that make up various aspects of the body such as skin, blood, muscle and nerves. These are generally referred to as adult (non-embryonic) stem cells. These cells are important for the purpose of medical therapies aiming to replace lost or damaged cells or tissues or to otherwise treat disorders.
Regenerative cell therapy relies on replacing diseased, damaged or dysfunctional cells with healthy, functioning ones or repairing damaged or diseased tissue. A great range of cells can serve in cell therapy, including cells found in peripheral and umbilical cord blood, bone marrow and adipose (fat) tissue. Physicians have been using adult stem cells from bone marrow to treat various blood cancers for 60 years (the first successful bone marrow transplant was performed in 1956). Recently, physicians have begun to use stem cells to treat various other diseases. We intend to develop cell and tissue products and regenerative therapy protocols, primarily involving adult stem cells, to allow patients to undergo cellular-based treatments.
We intend to concentrate initially on therapeutic areas in which risk to the patient is low, recovery is relatively easy, results can be demonstrated through sufficient clinical data, and patients and physicians will be comfortable with the procedure. We believe that there will be readily identifiable groups of patients who will benefit from these procedures.
Accordingly, we have focused our initial developmental efforts on cellular-based therapeutic products and clinical development programs in selective areas of medicine for which the treatment protocol is minimally invasive. Such areas include the treatment of the disc and spine and metabolic-related disorders. Upon regulatory approval, we will seek to obtain third party reimbursement for our products and procedures; however; patients may be required to pay for our products and procedures out of pocket in full and without the ability to be reimbursed by any governmental and other third party payers.
We have undertaken research and development efforts in connection with the development of investigational therapeutic products and medical therapies using cell and tissue protocols, primarily involving adult stem cells. See “Disc/Spine Program”, “Metabolic Brown Adipose (Fat) Program” and “Curved Needle Device” below. As a result of these programs, we have obtained two United States and two non-United States patents related to research regarding our ThermoStem Program, we have obtained licenses for two patent applications related to our Disc/Spine Program and we have obtained a license for one United States patent related to a curved needle device.
We have established a laboratory facility and will seek to further develop cellular-based treatments, products and protocols, stem cell-related intellectual property, or IP, and translational research applications. See “Laboratory” below.
| 5 |
We have not generated any significant revenues from our operations. The implementation of our business plan, as discussed below, will require the receipt of sufficient equity and/or debt financing to purchase necessary equipment, technology and materials, fund our research and development efforts, retire our outstanding debt (see “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Availability of Additional Funds”) and otherwise fund our operations. We intend to seek such financing from current shareholders and debtholders as well as from other accredited investors. We also intend to seek to raise capital through investment bankers and from biotech funds, strategic partners and other financial institutions. We anticipate that we will require approximately $20,000,000 in financing to commence and complete a Phase 2 clinical trial and we will require approximately $45,000,000 in further additional funding to complete our clinical trials using BRTX-100, as further described in this Item 1 (assuming the receipt of no revenues from operations), repay our outstanding debt ($5,161,916 as of December 31, 2018) (assuming that no debt is converted into equity) and fund general operations. We will also require a substantial amount of additional funding to implement our other programs described in this Item 1. No assurance can be given that the anticipated amounts of required funding are correct or that we will be able to accomplish our goals within the timeframes projected. In addition, no assurance can be given that we will be able to obtain any required financing on commercially reasonable terms or otherwise. We may also seek to have our debtholders convert all or a portion of their debt into equity. No assurance can be given that we will be able to convert such debt into equity on commercially reasonable terms or otherwise. If we are unable to obtain adequate funding, we may be required to significantly curtail or discontinue our proposed operations. See Item 7 (“Management’s Discussion and Analysis of Financial Condition and Results of Operations – Factors That May Affect Future Results and Financial Condition – We will need to obtain a significant amount of financing to initiate and complete our clinical trials and implement our business plan. – We will need to obtain additional financing to satisfy debt obligations”).
Disc/Spine Program
General
Among the initiatives that we are currently pursuing is our Disc/Spine Program, with our initial product candidate being called BRTX-100. We have obtained a license (see “License” below) that permits us to use technology for adult stem cell treatment of disc and spine conditions. The technology is an advanced stem cell culture and injection procedure into the intervertebral disc, or IVD, that may offer relief from lower back pain, buttock and leg pain, and numbness and tingling in the leg and foot.
Lower back pain is the most common, most disabling, and most costly musculoskeletal ailment faced worldwide. According to a recent market report, there are nearly 25 million people in the United States with chronic lower back pain of which approximately 5 million have pain caused by a protruding or injured disc. We believe that between 500,000 and 1 million of these back pain sufferers will have an invasive surgical procedure to try to alleviate the pain associated with these lower back conditions. Clinical studies have documented that the source of the pain is most frequently damage to the IVD. This can occur when forces, whether a single load or repetitive microtrauma, exceed the IVD’s inherent capacity to resist those loads. Aging, obesity, smoking, lifestyle, and certain genetic factors may predispose one to an IVD injury. Current surgical approaches to back pain are extremely invasive and costly (often altering the spine’s biomechanics unfavorably and predisposing it to further disc degeneration) and are associated with unacceptably low success rates.
| 6 |
While once thought to be benign, the natural history of lower back pain is often one of chronic recurrent episodes of pain leading to progressive disability. This is believed to be a direct result of the IVD’s poor healing capacity after injury. The IVD is the largest avascular (having few or no blood vessels) structure in the body and is low in cellularity. Therefore, its inherent capacity to heal after injury is poor. The clinical rationale of BRTX-100 is to deliver a high concentration of the patient’s own cultured MSCs into the site of pathology to promote healing and relieve pain.
We have developed a mesenchymal stem cell product candidate, BRTX-100, derived from autologous (or a person’s own) human bone marrow, cultured and formulated, in a proprietary method, specifically for introduction into a painful lumbar disc.
In January 2017, we submitted an IND application to the FDA to obtain authorization to commence a Phase 2 clinical trial investigating the use of BRTX-100, our lead cell therapy candidate, in the treatment of chronic lower back pain arising from degenerative disc disease. In February 2017, we received such authorization from the FDA. We intend to commence such clinical trial during the third quarter of 2019 (assuming the receipt of necessary funding).
In addition to developing BRTX-100, we may also seek to sublicense the technology to a strategic third party, who may assist in gaining FDA approval for a lumbar disc indication, or third parties for use in connection with cellular-based developmental programs with regard to disc and spine related conditions.
We have established a laboratory, which includes a clean room facility, to perform the production of cell products (possibly including BRTX-100) for use in our clinical trials, for third party cell products or for general research purposes. We may also use this laboratory to develop our pipeline of future products and expand our stem cell-related IP. See “Laboratory” and “Technology; Research and Development” below.
BRTX-100
Our lead product candidate, BRTX-100, is an autologous hypoxic (low oxygen) cultured mesenchymal stem cell product derived from a patient’s own bone marrow and formulated with a proprietary carrier. We have designed the cryopreserved sterile cellular product candidate to be provided in vials for injection into painful lumbar discs. We anticipate the product candidate will be delivered using a standard 20 gauge 3.5 inch introducer needle and a 25 gauge 6 inch needle that will extend into the disc center upon delivery. Upon regulatory approval, we plan to provide training to medical practitioners with regard to the approved injection procedure. It is anticipated that the delivery of the product candidate will be a 30 minute procedure.
Mesenchymal stem cells used in BRTX-100 are similar to other MSCs under development by others; however, in order to enhance the survivability of our bone marrow-derived MSCs in the avascular environment of the damaged disc, BRTX-100 is designed to expand under hypoxic conditions for a period of approximately three weeks. This process is intended to result in an approximate 40 million cell count population with enhanced viability and therapeutic potential following injection locally into injured spinal discs. Publications and scientific literature have indicated that MSCs preconditioned in hypoxic environment show enhanced skeletal muscle regeneration properties and improved impacts upon circulation and vascular formation compared to MSCs cultured under normoxic (normal oxygen) conditions.
| 7 |
In August 2018, the Journal of Translational Medicine published the results of our study evaluating the benefits of long-term hypoxic culturing of human bone marrow-derived MSCs.
Production and Delivery
The production of our product candidate, BRTX-100, begins with the physician collecting bone marrow from the patient under local anesthesia. Peripheral blood is also collected from the patient. The physician will then send the patient’s bone marrow and blood samples to our laboratory (or a contract laboratory) for culturing and formulation. The hypoxic culturing process applied is intended to result in the selection of a cell population that is suitable for an improved possibility of survival in the internal disc environment. We anticipate that the cell culturing process and product formulation will take approximately three weeks, with an additional two weeks required for quality control testing required to meet product release criteria. We will then send the therapeutic cryopreserved stem cells (BRTX-100) in a sterile vial back to the physician’s offices where it will undergo a controlled thaw prior to the procedure. The price structure for the procedure and our services has not been determined and no assurances can be given in this regard. The following illustrates the process:
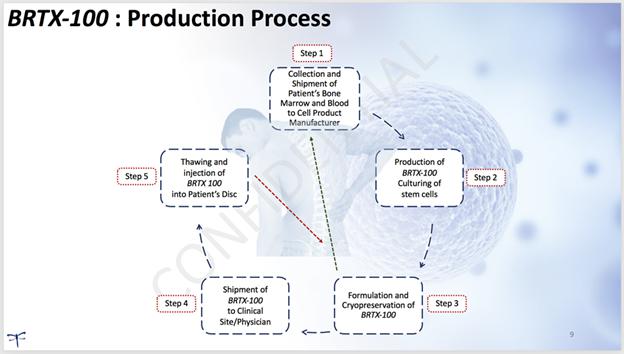
| 8 |
License
Pursuant to our license agreement with Regenerative Sciences, LLC, or Regenerative, that became effective in April 2012, we have obtained, among other things, a worldwide (excluding Asia and Argentina), exclusive, royalty-bearing license from Regenerative to utilize or sublicense a certain method for culturing cells for use in our developmental program involving disc and spine conditions, including protruding or painful discs. The investigational technology that has been licensed is an advanced stem cell culture and injection procedure that may offer relief from lower back pain, buttock and leg pain, and numbness and tingling in the leg and foot. Pursuant to the license agreement, we have also obtained a worldwide, exclusive, royalty-bearing license from Regenerative to utilize or sublicense a certain investigational curved needle device for the administration of specific cells and/or cell products to the disc and/or spine (and other parts of the body). It will be necessary to advance the design of this investigational device to facilitate the delivery of substances, including living cells, to specific locations within the body and minimize the potential for damage to nearby structures.
The license agreement provides for the requirement that we achieve certain milestones or pay certain minimum royalty amounts in order to maintain the exclusive nature of the licenses. The license agreement also provides for a royalty-bearing sublicense of certain aspects of the technology to Regenerative for use for certain purposes, including in the United States and the Cayman Islands. Further, the license agreement requires that Regenerative furnish certain training, assistance and consultation services with regard to the licensed technology.
Animal Study
The efficacy and safety of our product candidate, BRTX-100, has been tested in a degenerative intervertebral rabbit disc model. In this study, 80 rabbits underwent surgery to create a puncture in the discs. Four weeks post surgery, each rabbit had either contrast, a biomaterial carrier or BRTX-100 injected into the discs. In order to study the biodistribution and efficacy of BRTX-100, the rabbits were evaluated at day 56 and day 120.
The key safety findings of the animal study are as follows:
| ● | There was no evidence or observation of gross toxicity related to the administration of BRTX-100 at either time point. The clinical pathology across both groups and time points were within expected normal historical ranges and under the conditions of the test. No abnormalities (including fractures or overt signs of lumbar disc disease) were identified after review of the radiographic images taken at both endpoints for both groups. No toxicity or adverse finding was evident in the systemic tissues or the discs of animals receiving BRTX-100. | |
| ● | There was no detectable presence of human cells (BRTX-100) observed at the day 56 interim time point. This is consistent with the proposed mechanism of action that BRTX-100 acts through a paracrine effect of secreted growth and immunomodulation factors. |
| 9 |
The key efficacy findings of the animal study are as follows:
| ● | BRTX-100 showed a statistically significant DHI (disc height increase) over the control group at day 120. | |
| ● | BRTX-100 showed a statistically significant improvement in disc histology over the control group at day 120 as graded by a validated histology scale. BRTX-100 showed a significant improvement in the cellularity and matrix of the disc when compared to the control at day 120. |
Clinical Trial
In January 2017, we submitted an IND application to the FDA to obtain authorization to commence a Phase 2 clinical trial investigating the use of BRTX-100, our lead cell therapy candidate, in the treatment of chronic lower back pain arising from degenerative disc disease. In February 2017, we received such authorization from the FDA. We intend to commence such clinical trial during the third quarter of 2019 (assuming the receipt of necessary funding).
The following describes the Phase 2 clinical trial authorized by the FDA:
A Phase 2 Prospective, Double-Blinded, Placebo Controlled, Randomized Study
| ● | General | ||
| ● | 72 patients; randomized 2:1, BRTX-100 to control | ||
| ● | 10-20 clinical trial sites | ||
| ● | Primary efficacy endpoint at 12 months | ||
| ● | Patient safety and efficacy follow up at 24 months | ||
| ● | Primary Efficacy Endpoint | ||
| ● | Responder endpoint - % of patients that meet the improvement in function and reduction in pain threshold | ||
| ● | Improvement in function defined as at least a 30% increase in function based on the Oswestry questionnaires (ODI) | ||
| ● | Reduction of pain defined as at least a 30% decrease in pain as measured using the Visual Analogue Scale (VAS) | ||
| ● | Additional or Secondary Endpoints | ||
| ● | Quality of life assessment | ||
| ● | Evolution of affected disc(s) by magnetic resonance imaging (MRI) | ||
The FDA approval process can be lengthy, expensive and uncertain and there is no guarantee that the clinical trial(s) will be commenced or completed or that the product will ultimately receive approval or clearance. See “Government Regulation” below and Item 7 (“Management’s Discussion and Analysis of Financial Condition and Results of Operations – Factors That May Affect Future Results and Financial Condition – Risks Related to Our Cell Therapy Product Development Efforts; and – Risks Related to Government Regulation”).
As an alternative to undertaking the Phase 2 clinical trial ourselves, we are exploring the possible licensing of our rights with respect to our product candidate, BRTX-100, to a strategic partner. Such an arrangement could possibly eliminate or significantly reduce the need to raise the substantial capital needed to commence and complete the clinical trials and undertake the commercialization of BRTX-100 and would provide licensing-related revenue to us. No assurance can be given that any licensing agreement will be entered into, whether upon commercially reasonable terms or otherwise.
| 10 |
Defined Health Report
In March 2018, we engaged Defined Health, a business development and strategy consulting firm, to conduct an independent review of BRTX-100. Defined Health has worked with many of the leading companies in the pharmaceutical, biotech and healthcare industries for over 25 years.
The review was intended to collect informed, independent opinions regarding BRTX-100 among key opinion leaders, or KOLs (i.e., orthopedic surgeons specializing in back and spine surgery with experience in stem cell therapy), who, upon studying applicable clinical material, could offer opinions regarding the future therapeutic potential of BRTX-100.
As noted in the Defined Health report, the KOLs reacted positively to the value proposition of our product candidate, BRTX-100, and were optimistic that the clinical data presented to date is likely to be mirrored in future clinical investigations. Given the opportunity, the KOLs indicated that they would likely participate in a clinical trial should it be offered at their center and that they would recommend the study to appropriately eligible patients. The report indicated that, if BRTX-100 were to be granted FDA approval, the KOLs anticipate that it would be integrated into the standard of care for eligible chronic lumbar disc disease patients.
Metabolic Brown Adipose (Fat) Program
Since June 2011, we have been engaging in pre-clinical research efforts with respect to an investigational platform technology utilizing brown adipose (fat) derived stem cells, or BADSCs, for therapeutic purposes. We have labeled this initiative our ThermoStem Program.
Brown fat is a specialized adipose (fat) tissue found in the human body that plays a key role in the evolutionarily conserved mechanisms underlying thermogenesis (generation of non-shivering body heat) and energy homeostasis in mammals - long known to be present at high levels in hibernating mammals and human newborns. Recent studies have demonstrated that brown fat is present in the adult human body and may be correlated with the maintenance and regulation of healthy metabolism, thus potentially being involved in caloric regulation. The pre-clinical ThermoStem Program involves the use of a cell-based (brown adipose tissue construct) treatment for metabolic disease, such as type 2 diabetes, obesity, hypertension and other metabolic disorders, as well as cardiac deficiencies. We have had initial success in transplanting the brown adipose tissue construct in animals, and we are currently exploring ways to deliver into humans. Even though present, BAT mass is very low in healthy adults and even lower in obese populations. Therefore, it may not be sufficient to either naturally impact whole body metabolism, or to be targeted by drugs intended to increase its activity in the majority of the population. Increasing BAT mass is crucial in order to benefit from its metabolic activity and this is what our ThermoStem Program seeks to accomplish. We may also identify other naturally occurring biologics and chemically engineered molecules that may enhance brown adipose tissue performance and activity.
| 11 |
Obesity, the abnormal accumulation of white fat tissue, leads to a number of metabolic disorders and is the driving force behind the rise of type 2 diabetes and cardiovascular diseases worldwide. Pharmacological efforts to alter metabolic homeostasis through modulating central control of appetite and satiety have had limited market penetration due to significant psychological and physiological safety concerns directly attributed to modulating these brain centers. Adipose tissue is one of the largest organs in the human body and plays a key role in central energy balance and lipid homeostasis. White and brown adipose tissues are found in mammals. White adipose tissue’s function is to store energy, whereas BAT specializes in energy expenditure. Recent advancements in unraveling the mechanisms that control the induction, differentiation, proliferation, and thermogenic activity of BAT, along with the application of imaging technologies for human BAT visualization, have generated optimism that these advances may provide novel strategies for targeting BAT activation/thermogenesis, leading to efficacious and safe obesity targeted therapies.
We are developing a cell-based product candidate to target obesity and metabolic disorders using BADSCs. Our goal is to develop a bioengineered implantable brown adipose tissue construct intended to mimic ones naturally occurring in the human body. We have isolated and characterized a human multipotent stem cell population that resides within BAT depots. We have expanded these stem cells to clinically relevant numbers and successfully differentiated them into functional brown adipocytes. We intend to use adult stem cells that may be differentiated into progenitor or fully differentiated brown adipocytes, or a related cell type, which can be used therapeutically in patients. We are focusing on the development of treatment protocols that utilize allogeneic cells (i.e., stem cells from a genetically similar but not identical donor).
In order to deliver these differentiated cells into target locations in vivo, we seeded BADSCs onto 3-dimensional biological scaffolds. Pre-clinical animal models of diet-induced obesity, that were transplanted with differentiated BADSCs supported by a biological scaffold, presented significant reductions in weight and blood glucose levels compared to saline injected controls. We are identifying technology for in vivo delivery in small animal models. Having completed our proof of concept using our BAT in small animals, we are currently developing our next generation BAT. It is anticipated that this next version will contain a higher purity of BADSC and a greater percent of functional brown adipocytes, which is expected to increase the therapeutic effect compared to our first generation product. In addition, we are exploring the delivery of the therapeutic using encapsulation technology, which will only allow for reciprocal exchange of small molecules between the host circulation and the BAT implant. We expect that encapsulation may present several advantages over our current biological scaffolds, including prevention of any immune response or implant rejection that might occur in an immunocompetent host and an increase in safety by preventing the implanted cells from invading the host tissues and forming tumors. We have developed promising data on the loading of human stem cell-derived tissue engineered brown fat into an encapsulation device to be used as a cell delivery system for our metabolic platform program for the treatment of type 2 diabetes, obesity, hyperlipidemia and hypertension. This advancement may lead to successful transplantation of brown fat in humans. We are evaluating the next generation of BAT constructs that will first be tested in small animal models. No assurance can be given that this delivery system will be effective in vivo in animals or humans. Our allogeneic brown adipose derived stem cell platform potentially provides a therapeutic and commercial model for the cell-based treatment of obesity and related metabolic disorders.
| 12 |
In June 2012, we entered into an Assignment Agreement with the University of Utah Research Foundation, or the Foundation, and a Research Agreement with the University of Utah, or the Utah Research Agreement. Pursuant to the Assignment Agreement, which provides for royalty payments, we acquired the rights to two provisional patent applications that relate to human brown fat cell lines. No royalty amounts are payable to date. The applications have been converted to a utility application in the United States and several foreign jurisdictions. Pursuant to the Utah Research Agreement, the University of Utah, or the University, provided research services relating to the identification of brown fat tissue and the development and characterization of brown fat cell lines. The Utah Research Agreement provides that all inventions, discoveries, patent rights, information, data, methods and techniques, including all cell lines, cell culture media and derivatives thereof, are owned by us. In February 2019, we entered into a Services Agreement with the University of Utah pursuant to which the university has been retained to provide research services with regard to the ThermoStem Program. Pursuant to this agreement, we will initiate preclinical models to study the efficacy of our generation 2 encapsulated brown adipose tissue construct.
In February 2014, our research with regard to the identification of a population of brown adipose derived stem cells was published in Stem Cells, a respected stem cell journal.
In March 2014, we entered into a Research Agreement with Pfizer Inc., a global pharmaceutical company. Pursuant to the Pfizer Research Agreement, we were engaged to provide research and development services with regard to a joint study of the development and validation of a human brown adipose cell model. The Pfizer Research Agreement provided for an initial payment to us of $250,000 and the payment of up to an additional $525,000 during the two-year term of the Agreement, all of which has been received.
In August 2015, we entered into a one year research collaboration agreement with the University of Pennsylvania with regard to the understanding of brown adipose biology and its role in metabolic disorders. In September 2018, we entered into a one year material transfer agreement with the University of Pennsylvania pursuant to which the university is provided access to our proprietary brown adipose tissue cells for research purposes. No amounts are payable by or to us pursuant to either agreement.
In September 2015, a United States patent related to the ThermoStem Program was issued to us.
In April 2017, an Australian patent related to the ThermoStem Program was issued to us.
In December 2017, a Japanese patent related to the ThermoStem Program was issued to us.
In January 2019, a United States patent related to the ThermoStem Program was issued to us.
Following our research activities, we intend to undertake preclinical animal studies in order to determine whether our proposed treatment protocol is feasible. Such studies are planned to begin by the second quarter of 2019 (assuming the receipt of necessary financing). Following the completion of such studies, we intend to file an IND with the FDA and initiate a clinical trial. See “Government Regulation” below and Item 7 (“Management’s Discussion and Analysis of Financial Condition and Results of Operations – Factors That May Affect Future Results and Financial Condition – Risks Related to Our Cell Therapy Product Development Efforts; and – Risks Related to Government Regulation”). The FDA approval process can be lengthy, expensive and uncertain and there is no guarantee of ultimate approval or clearance.
| 13 |
We anticipate that much of our development work in this area will take place at our laboratory facility, outside core facilities at academic, research or medical institutions, or contractors. See “Laboratory” below.
Curved Needle Device
Pursuant to the Regenerative license agreement discussed under “Disc/Spine Program-License” above, we have licensed and further developed an investigational curved needle device, or CND, that is a needle system with a curved inner cannula to allow access to difficult-to-locate regions for the delivery or removal of fluids and other substances. The investigational CND is intended to deliver stem cells and/or other therapeutic products or material to the interior of a human intervertebral disc, the spine region, or potentially other areas of the body. The device is designed to rely on the use of pre-curved nested cannulae that allow the cells or material to be deposited in the posterior and lateral aspects of the disc to which direct access is not possible due to outlying structures such as vertebra, spinal cord and spinal nerves. We anticipate that the use of the investigational CND will facilitate the delivery of substances, including living cells, to specific locations within the body and minimize the potential for damage to nearby structures. The investigational device may also have more general use applications. In August 2015, a United States patent for the CND was issued to the licensor, Regenerative Sciences, LLC. We anticipate that FDA approval or clearance will be necessary for the investigational CND prior to commercialization. See “Government Regulation” below and Item 7 (“Management’s Discussion and Analysis of Financial Condition and Results of Operations – Factors That May Affect Future Results and Financial Condition – Risks Related to Our Cell Therapy Product Development Efforts; and – Risks Related to Government Regulation”). The FDA review and approval process can be lengthy, expensive and uncertain and there is no guarantee of ultimate approval or clearance.
Laboratory
We have established a laboratory in Melville, New York for research purposes and have built a cleanroom within the laboratory for the possible production of cell-based product candidates, such as BRTX-100, for use in a clinical trial, for third party cell products or general research purposes.
As operations grow, our plans include the expansion of our laboratory to perform cellular characterization and culturing, protocol and stem cell-related IP development, translational research and therapeutic outcome analysis. As we develop our business and our stem cell product candidates and obtain regulatory approval, we will seek to establish ourselves as a key provider of adult stem cells for therapies and expand to provide cells in other market areas for stem cell therapy. We may also use outside laboratories specializing in cell therapy services and manufacturing of cell products.
Technology; Research and Development
We intend to utilize our laboratory or a third party laboratory in connection with cellular research activities. We also intend to obtain cellular-based therapeutic technology licenses and increase our IP portfolio. We intend to seek to develop potential stem cell delivery systems or devices. The goal of these specialized delivery systems or devices is to deliver cells into specific areas of the body, control the rate, amount and types of cells used in a treatment, and populate these areas of the body with sufficient stem cells so that there is a successful therapeutic result.
| 14 |
We also intend to perform research to develop certain stem cell optimization compounds, media designed to enhance cellular growth and regeneration for the purpose of improving pre-treatment and post-treatment outcomes.
In our Disc/Spine Program, three patent applications have been filed with regard to technology that is the subject of the license agreement between us and Regenerative (see “Disc/Spine Program-License” above). Regenerative has been issued a patent from one of these applications with regard to its curved needle therapeutic delivery device. The other two applications remain pending.
In our ThermoStem Program, we have three pending United States patent applications within two patent families. We have been issued a United States patent in each of the two patent families. Patent applications with regard to the first patent family in the ThermoStem Program have been filed in five foreign jurisdictions (of which two applications have granted as foreign patents and one application has lapsed). Patent applications with regard to the second patent family in the ThermoStem Program have been filed in four foreign jurisdictions.
Our patent applications and those of Regenerative are currently in prosecution (i.e., we and Regenerative are seeking issued patents). A description of the patent applications and issued patents is set forth in the table below:
| Program | I.D. | Jurisdiction | Title | |||
Disc/Spine (brtxDisc) |
13/132,840* | US | Methods and compositions to facilitate repair of avascular tissue | |||
| 15/891,852 | US | Surgical methods and compositions to facilitate repair of avascular tissue | ||||
| U.S. Patent No. 9,113,950 B2** | US | Therapeutic delivery device | ||||
| Metabolic | U.S. Patent No. 9,133,438 | US | Brown fat cell compositions and methods | |||
| (ThermoStem) | 13/932,468 | US | ||||
| 15/910,625 | US | |||||
| AU Patent No. 2012275335 | Australia | |||||
| 12743811.7 | Europe | |||||
| 230237 | Israel | |||||
| JP Patent No. 6243839 | Japan | |||||
| U.S. Patent No. 10,167,449 | US | Human brown adipose derived stem cells and uses | ||||
| 16/183,370 | US | |||||
| 2014253920 | Australia | |||||
| 14729769.1 | Europe | |||||
| 242150 | Israel | |||||
| 2016-509105 | Japan |
*Patent application filed by licensor, Regenerative Sciences, LLC
**Patent issued to licensor, Regenerative Sciences, LLC
| 15 |
In March 2014, we entered into a Research and Development Agreement with Rohto Pharmaceutical Co., Ltd., a Japanese pharmaceutical company. Pursuant to the Rohto Research and Development Agreement, we were engaged to provide research and development services with regard to stem cells.
In March 2014, we entered into the Pfizer Research Agreement, as discussed above under “Metabolic Brown Adipose (Fat) Program”.
We have secured registrations in the U.S. Patent and Trademark Office for the following trademarks:
| ● |  | |
| ● |  | |
| ● |  | |
| ● | THERMOSTEM | |
| ● | STEM PEARLS, and | |
| ● | STEM THE TIDES OF TIME. |
We own a published application in the U.S. Patent and Trademark Office for the trademark BRTX and an allowed application in the U.S. Patent and Trademark Office for the trademark BRTX-100.
We also have federal common law rights in the trademark BioRestorative Therapies and other trademarks and trade names used in the conduct of our business that are not registered.
Our success will depend in large part on our ability to develop and protect our proprietary technology. We intend to rely on a combination of patent, trade secret and know-how, copyright and trademark laws, as well as confidentiality agreements, licensing agreements, non-compete agreements and other agreements, to establish and protect our proprietary rights. Our success will also depend upon our ability to avoid infringing upon the proprietary rights of others, for if we are judicially determined to have infringed such rights, we may be required to pay damages, alter our services, products or processes, obtain licenses or cease certain activities. We conduct prior rights searches before launching any new product or service to put us in the best position to avoid claims of infringement.
During the years ended December 31, 2018 and 2017, we incurred $1,513,150 and $2,152,433, respectively, in research and development expenses.
Scientific Advisors
We have established a Scientific Advisory Board whose purpose is to provide advice and guidance in connection with scientific matters relating to our business. The Scientific Advisory Board has established a Disc Advisory Committee which focuses on matters relating to our Disc/Spine Program. Our Scientific Advisory Board members are Dr. Wayne Marasco (Chairman), Dr. Naiyer Imam, Dr. Wayne Olan, Dr. Joy Cavagnaro, Dr. Jason Lipetz, Dr. Harvinder Sandhu, Dr. Christopher Plastaras and Dr. Gerard A. Malanga. The Disc Advisory Committee members are Dr. Lipetz (Chairman), Dr. Olan, Dr. Sandhu, Dr. Plastaras and Dr. Malanga. See Item 10 (“Directors, Executive Officers and Corporate Governance – Scientific Advisors”) for a listing of the principal positions for Drs. Marasco, Imam, Olan, Cavagnaro, Lipetz, Sandhu, Plastaras and Malanga.
| 16 |
Competition
We will compete with many pharmaceutical, biotechnology and medical device companies, as well as other private and public stem cell companies involved in the development and commercialization of cell-based medical technologies and therapies.
Regenerative medicine is rapidly progressing, in large part through the development of cell-based therapies or devices designed to isolate cells from human tissues. Most efforts involve cell sources, such as bone marrow, adipose tissue, embryonic and fetal tissue, umbilical cord and peripheral blood and skeletal muscle.
Many of our competitors and potential competitors have substantially greater financial, technological, research and development, marketing and personnel resources than we do. We cannot, with any accuracy, forecast when or if these companies are likely to bring their products and therapies to market in competition with those that we are pursuing.
With the enactment of the Biologics Price Competition and Innovation Act of 2009, or the BPCIA, an abbreviated pathway for the approval of biosimilar and interchangeable biological products was created. For the FDA to approve a biosimilar product, it must find that there are no clinically meaningful differences between the reference product and the proposed biosimilar product. Interchangeability requires that a product is biosimilar to the reference product, and the product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. Under the BPCIA, an application for a biosimilar product cannot be submitted to the FDA until four years following approval of the reference product, and it may not be approved by the FDA until 12 years after the original branded product is approved under a biologics license application, or BLA.
We believe that, if any of our product candidates are approved as a biological product under a BLA, it should qualify for the 12-year period of exclusivity. However, there is a risk that the FDA could permit biosimilar applicants to reference approved biologics other than our therapeutic candidates, thus circumventing our exclusivity and potentially creating the opportunity for competition sooner than anticipated. Additionally, this period of regulatory exclusivity does not apply to companies pursuing regulatory approval via their own traditional BLA, rather than via the abbreviated pathway. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
| 17 |
Customers
Upon regulatory approval, our cell product candidates are intended to be marketed to physicians, other health care professionals, hospitals, research institutions, pharmaceutical companies and the military. It is anticipated that physicians who are trained and skilled in performing spinal injections will be the physicians most likely to treat discs with injections of BRTX-100 upon regulatory approval. These physicians would include interventional physiatrists (physical medicine physicians), pain management anesthesiologists, interventional radiologists and neurosurgeons.
Governmental Regulation
U.S. Government Regulation
The health care industry is highly regulated in the United States. The federal government, through various departments and agencies, state and local governments, and private third-party accreditation organizations, regulate and monitor the health care industry, associated products, and operations. The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, approval, manufacture, distribution and marketing of medical products, including drugs, biologics, and medical devices. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, post-approval monitoring, advertising, promotion, sampling and import and export of medical products. The following is a general overview of the laws and regulations pertaining to our business.
FDA Regulation of Stem Cell Treatment and Products
The FDA regulates the manufacture of human stem cell treatments and associated products under the authority of the Public Health Service Act, or PHSA, and the Federal Food, Drug, and Cosmetic Act, or FDCA. Stem cells can be regulated under the FDA’s Human Cells, Tissues, and Cellular and Tissue-Based Products Regulations, or HCT/Ps, or may also be subject to the FDA’s drug, biologic, or medical device regulations, each as discussed below.
Human Cells, Tissues, and Cellular and Tissue-Based Products Regulation
Under Section 361 of the PHSA, the FDA issued specific regulations governing the use of HCT/Ps in humans. Pursuant to Part 1271 of Title 21 of the Code of Federal Regulations, or CFR, the FDA established a unified registration and listing system for establishments that manufacture and process HCT/Ps. The regulations also include provisions pertaining to donor eligibility determinations; current good tissue practices covering all stages of production, including harvesting, processing, manufacture, storage, labeling, packaging, and distribution; and other procedures to prevent the introduction, transmission, and spread of communicable diseases.
| 18 |
The HCT/P regulations strictly constrain the types of products that may be regulated solely under these regulations. Factors considered include the degree of manipulation, whether the product is intended for a homologous function, whether the product has been combined with noncellular or non-tissue components, and the product’s effect or dependence on the body’s metabolic function. In those instances where cells, tissues, and cellular and tissue-based products have been only minimally manipulated, are intended strictly for homologous use, have not been combined with noncellular or nontissue substances, and do not depend on or have any effect on the body’s metabolism, the manufacturer is only required to register with the FDA, submit a list of manufactured products, and adopt and implement procedures for the control of communicable diseases. If one or more of the above factors has been exceeded, the product would be regulated as a drug, biological product, or medical device rather than an HCT/P.
Because we are an enterprise in the early stages of operations and have not generated significant revenues from operations, it is difficult to anticipate the likely regulatory status of the array of products and services that we may offer. We believe that some of the adult autologous (self derived) stem cells that will be used in our cellular therapy products and services, including the brown adipose (fat) tissue that we intend to use in our ThermoStem Program, may be regulated by the FDA as HCT/Ps under 21 C.F.R. Part 1271. This regulation defines HCT/Ps as articles “containing or consisting of human cells or tissues that are intended for implantation, transplantation, infusion or transfer into a human recipient.” However, the FDA may disagree with this position or conclude that some or all of our stem cell therapy products or services do not meet the applicable definitions and exemptions to the regulation. If we are not regulated solely under the HCT/P provisions, we would need to expend significant resources to comply with the FDA’s broad regulatory authority under the FDCA. Third party litigation concerning the autologous use of a stem cell mixture to treat musculoskeletal and spinal injuries has increased the likelihood that some of our products and services are likely to be regulated as a drug or biological product and require FDA approval. In past litigation, the FDA asserted that the defendants’ use of cultured stem cells without FDA approval is in violation of the FDCA, claiming that the defendants’ product is a drug. The defendants asserted that their procedure is part of the practice of medicine and therefore beyond the FDA’s regulatory authority. The District Court ruled in favor of the FDA, and in February 2014 the Circuit Court affirmed the District Court’s holding.
If regulated solely under the FDA’s HCT/P statutory and regulatory provisions, once our laboratory in the United States becomes operational, it will need to satisfy the following requirements, among others, to process and store stem cells:
| ● | registration and listing of HCT/Ps with the FDA; | |
| ● | donor eligibility determinations, including donor screening and donor testing requirements; | |
| ● | current good tissue practices, specifically including requirements for the facilities, environmental controls, equipment, supplies and reagents, recovery of HCT/Ps from the patient, processing, storage, labeling and document controls, and distribution and shipment of the HCT/Ps to the laboratory, storage, or other facility; | |
| ● | tracking and traceability of HCT/Ps and equipment, supplies, and reagents used in the manufacture of HCT/Ps; | |
| ● | adverse event reporting; | |
| ● | FDA inspection; and | |
| ● | abiding by any FDA order of retention, recall, destruction, and cessation of manufacturing of HCT/Ps. |
| 19 |
Non-reproductive HCT/Ps and non-peripheral blood stem/progenitor cells that are offered for import into the United States and regulated solely under Section 361 of the PHSA must also satisfy the requirements under 21 C.F.R. § 1271.420. Section 1271.420 requires that the importer of record of HCT/Ps notify the FDA prior to, or at the time of, importation and provide sufficient information for the FDA to make an admissibility decision. In addition, the importer must hold the HCT/P intact and under conditions necessary to prevent transmission of communicable disease until an admissibility decision is made by the FDA.
If the FDA determines that we have failed to comply with applicable regulatory requirements, it can impose a variety of enforcement actions including public warning letters, fines, consent decrees, orders of retention, recall or destruction of product, orders to cease manufacturing, and criminal prosecution. If any of these events were to occur, it could materially adversely affect us.
To the extent that our cellular therapy activities are limited to developing products and services outside the United States, as described in detail below, the products and services would not be subject to FDA regulation, but will be subject to the applicable requirements of the foreign jurisdiction. We intend to comply with all applicable foreign governmental requirements.
Drug and Biological Product Regulation
An HCT/P product that does not meet the criteria for being solely regulated under Section 361 of the PHSA will be regulated as a drug, device or biological product under the FDCA and/or Section 351 of the PHSA, and applicable FDA regulations. The FDA has broad regulatory authority over drugs and biologics marketed for sale in the United States. The FDA regulates the research, clinical testing, manufacturing, safety, effectiveness, labeling, storage, recordkeeping, promotion, distribution, and production of drugs and biological products. The FDA also regulates the export of drugs and biological products manufactured in the United States to international markets in certain situations.
The process required by the FDA before a drug or biologic may be marketed in the United States generally involves the following:
● completion of non-clinical laboratory tests, animal studies and formulation studies conducted according to Good Laboratory Practice, or GLP, or other applicable regulations;
● submission of an IND, which allows clinical trials to begin unless the FDA objects within 30 days;
● performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug or biologic for its intended use or uses conducted in accordance with FDA regulations and Good Clinical Practices, or GCP, which are international ethical and scientific quality standards meant to ensure that the rights, safety and well-being of trial participants are protected and that the integrity of the data is maintained;
| 20 |
● registration of clinical trials of FDA-regulated products and certain clinical trial information;
● preparation and submission to the FDA of a new drug application, or NDA, in the case of a drug or BLA in the case of a biologic;
● review of the product by an FDA advisory committee, where appropriate or if applicable;
● satisfactory completion of pre-approval inspection of manufacturing facilities and clinical trial sites at which the product, or components thereof, are produced to assess compliance with Good Manufacturing Practice, or cGMP, requirements and of selected clinical trial sites to assess compliance with GCP requirements; and
● FDA approval of an NDA or BLA which must occur before a drug or biologic can be marketed or sold.
Approval of an NDA requires a showing that the drug is safe and effective for its intended use and that the methods, facilities, and controls used for the manufacturing, processing, and packaging of the drug are adequate to preserve its identity, strength, quality, and purity. To obtain a BLA, a manufacturer must show that the proposed product is safe, pure, and potent and that the facility in which the product is manufactured, processed, packed, or held meets established quality control standards.
For purposes of an NDA or BLA approval by the FDA, human clinical trials are typically conducted in the following phases (which may overlap):
● Phase 1: The investigational product is initially given to healthy human subjects or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. These trials may also provide early evidence on effectiveness. During Phase 1 clinical trials, sufficient information about the investigational product’s pharmacokinetics and pharmacologic effects may be obtained to permit the design of well-controlled and scientifically valid Phase 2 clinical trials.
● Phase 2: These clinical trials are conducted in a limited number of human subjects in the target population to identify possible adverse effects and safety risks, to determine the efficacy of the investigational product for specific targeted diseases and to determine dosage tolerance and dosage levels. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more costly Phase 3 clinical trials.
● Phase 3: Phase 3 clinical trials are undertaken after Phase 2 clinical trials demonstrate that a dosage range of the investigational product appears effective and has a tolerable safety profile. The Phase 2 clinical trials must also provide sufficient information for the design of Phase 3 clinical trials. Phase 3 clinical trials are conducted to provide statistically significant evidence of clinical efficacy and to further test for safety risks in an expanded human subject population at multiple clinical trial sites. These clinical trials are intended to further evaluate dosage, effectiveness and safety, to establish the overall benefit-risk profile of the investigational product and to provide an adequate basis for product labeling and approval by the FDA. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of an investigational drug or biologic.
| 21 |
All clinical trials must be conducted in accordance with FDA regulations, GCP requirements and their protocols in order for the data to be considered reliable for regulatory purposes. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. These government regulations may delay or prevent approval of product candidates for a considerable period of time and impose costly procedures upon our business operations.
The FDA may require, or companies may pursue, additional clinical trials, referred to as Phase 4 clinical trials, after a product is approved. Such trials may be made a condition to be satisfied for continuing drug approval. The results of Phase 4 clinical trials can confirm the effectiveness of a product candidate and can provide important safety information. In addition, the FDA has authority to require sponsors to conduct post-marketing trials to specifically address safety issues identified by the agency.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA, or an NDA or BLA supplement, before the change can be implemented. An NDA or BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA and BLA supplements as it does in reviewing NDAs and BLAs.
Drug and biological products must also comply with applicable requirements, including monitoring and recordkeeping activities, manufacturing requirements, reporting to the applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling, or off-label use, limitations on industry-sponsored scientific and educational activities and requirements for promotional activities involving the internet. Although physicians may, in their independent professional medical judgment, prescribe legally available drugs for off-label uses, manufacturers typically may not market or promote such off-label uses. Modifications or enhancements to the product or its labeling, or changes of the site of manufacture, are often subject to the approval of the FDA and other regulators, who may or may not grant approval or may include a lengthy review process.
In the event that the FDA does not regulate our product candidates in the United States solely under the HCT/P regulation, our products and activities could be regulated as drug or biological products under the FDCA. If regulated as drug or biological products, we will need to expend significant resources to ensure regulatory compliance. If an IND and NDA or BLA are required for any of our product candidates, there is no assurance as to whether or when we will receive FDA approval of the product candidate. The process of designing, conducting, compiling and submitting the non-clinical and clinical studies required for NDA or BLA approval is time-consuming, expensive and unpredictable. The process can take many years, depending on the product and the FDA’s requirements.
| 22 |
In addition, even if a product candidate receives regulatory approval, the approval may be limited to specific disease states, patient populations and dosages, or might contain significant limitations on use in the form of warnings, precautions or contraindications, or in the form of onerous risk management plans, restrictions on distribution or use, or post-marketing trial requirements. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product, including safety labeling or imposition of a Risk Evaluation and Mitigation Strategy, or REMS, the requirement to conduct post-market studies or clinical trials or even complete withdrawal of the product from the market. Delay in obtaining, or failure to obtain, regulatory approval for our products, or obtaining approval but for significantly limited use, would harm our business. Further, we cannot predict what adverse governmental regulations may arise from future United States or foreign governmental action.
If the FDA determines that we have failed to comply with applicable regulatory requirements, it can impose a variety of enforcement actions from public warning letters, fines, injunctions, consent decrees and civil penalties to suspension or delayed issuance of approvals, seizure of our products, total or partial shutdown of our production, withdrawal of approvals, and criminal prosecutions. If any of these events were to occur, it could materially adversely affect us.
FDA Expedited Review Programs
The FDA is authorized to expedite the review of NDAs and BLAs in several ways. Under the Fast Track program, the sponsor of a drug or biologic product candidate may request the FDA to designate the product for a specific indication as a Fast Track product concurrent with or after the filing of the IND. Drug and biologic products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product candidate and the specific indication for which it is being studied.
In addition to other benefits, such as the ability to have greater interactions with the FDA, the FDA may initiate review of sections of a Fast Track NDA or BLA before the application is complete, a process known as rolling review.
Any product submitted to the FDA for marketing, including under a Fast Track program, may also be eligible for the following other types of FDA programs intended to expedite development and review:
● Breakthrough therapy designation. To qualify for the breakthrough therapy program, product candidates must be intended to treat a serious or life-threatening disease or condition, and preliminary clinical evidence must indicate that such product candidates may demonstrate substantial improvement on one or more clinically significant endpoints over existing therapies. The FDA will seek to ensure the sponsor of a breakthrough therapy product candidate receives intensive guidance on an efficient drug development program, intensive involvement of senior managers and experienced staff on a proactive, collaborative and cross-disciplinary review, and rolling review.
| 23 |
● Priority review. A product candidate is eligible for priority review if it treats a serious condition and, if approved, it would be a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious condition compared to marketed products. The FDA aims to complete its review of priority review applications within six months as opposed to ten months for standard review.
● Accelerated approval. Drug or biologic products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval. Accelerated approval means that a product candidate may be approved on the basis of adequate and well-controlled clinical trials establishing that the product candidate has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity and prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug or biologic product candidate receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials.
Fast Track designation, breakthrough therapy designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Further, with the passage of the 21st Century Cures Act, or the Cures Act, in December 2016, Congress authorized the FDA to accelerate review and approval of products designated as regenerative advanced therapies. A product is eligible for this designation if it is a regenerative medicine advanced therapy, or RMAT, (which may include a cell therapy) that is intended to treat, modify, reverse or cure a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition. The benefits of a RMAT designation include early interactions with the FDA to expedite development and review, benefits available to breakthrough therapies, potential eligibility for priority review and accelerated approval based on surrogate or intermediate endpoints.
Medical Device Regulation
The FDA also has broad authority over the regulation of medical devices marketed for sale in the United States. The FDA regulates the research, clinical testing, manufacturing, safety, labeling, storage, recordkeeping, premarket clearance or approval, promotion, distribution, and production of medical devices. The FDA also regulates the export of medical devices manufactured in the United States to international markets.
| 24 |
Under the FDCA, medical devices are classified into one of three classes, Class I, Class II, or Class III, depending upon the degree of risk associated with the medical device and the extent of control needed to ensure safety and effectiveness. Class I devices are subject to the lowest degree of regulatory scrutiny because they are considered low risk devices and need only comply with the FDA’s General Controls. The General Controls include compliance with the registration, listing, adverse event reporting requirements, and applicable portions of the Quality System Regulation as well as the general misbranding and adulteration prohibitions.
Class II devices are subject to the General Controls as well as certain Special Controls such as 510(k) premarket notification. Class III devices are subject to the highest degree of regulatory scrutiny and typically include life supporting and life sustaining devices and implants. They are subject to the General Controls and Special Controls that include a premarket approval application, or PMA. “New” devices are automatically regulated as Class III devices unless they are shown to be low risk, in which case they may be subject to de novo review to be moved to Class I or Class II. Clinical research of an investigational device is subject to the FDA’s Investigational Device Exemption, or IDE, regulations. Nonsignificant risk devices are subject to abbreviated requirements that do not require a submission to the FDA but must have Institutional Review Board (IRB) approval and comply with other requirements pertaining to informed consent, labeling, recordkeeping, reporting, and monitoring. Significant risk devices require the submission of an IDE application to the FDA and the FDA’s approval of the IDE application.
The FDA premarket clearance and approval process can be lengthy, expensive and uncertain. It generally takes three to twelve months from submission to obtain 510(k) premarket clearance, although it may take longer. Approval of a PMA could take one to four years, or more, from the time the application is submitted and there is no guarantee of ultimate clearance or approval. Securing FDA clearances and approvals may require the submission of extensive clinical data and supporting information to the FDA. Additionally, the FDA actively enforces regulations prohibiting marketing and promotion of devices for indications or uses that have not been cleared or approved by the FDA. In addition, modifications or enhancements of products that could affect the safety or effectiveness or effect a major change in the intended use of a device that was either cleared through the 510(k) process or approved through the PMA process may require further FDA review through new 510(k) or PMA submissions.
In the event we develop processes, products or services which qualify as medical devices subject to FDA regulation, we intend to comply with such regulations. If the FDA determines that our products are regulated as medical devices and we have failed to comply with applicable regulatory requirements, it can impose a variety of enforcement actions from public warning letters, application integrity proceedings, fines, injunctions, consent decrees and civil penalties to suspension or delayed issuance of approvals, seizure of our products, total or partial shutdown of our production, withdrawal of approvals, and criminal prosecutions. If any of these events were to occur, it could materially adversely affect us.
Current Good Manufacturing Practices and other FDA Regulations of Cellular Therapy Products
Products that fall outside of the HCT/P regulations and are regulated as drugs, biological products, or devices must comply with applicable cGMP regulations. These cGMPs and related quality standards are designed to ensure the products that are processed at a facility meet the FDA’s applicable requirements for identity, strength, quality, sterility, purity, and safety. In the event that our domestic United States operations are subject to the FDA’s drug, biological product, or device regulations, we intend to comply with the applicable cGMPs and quality regulations.
| 25 |
If the FDA determines that we have failed to comply with applicable regulatory requirements, it can impose a variety of enforcement actions from public warning letters, fines, injunctions, consent decrees and civil penalties to suspension or delayed issuance of approvals, seizure of our products, total or partial shutdown of our production, withdrawal of approvals, and criminal prosecutions. If any of these events were to occur, it could materially adversely affect us.
Promotion of Foreign-Based Cellular Therapy Treatment— “Medical Tourism”
We may establish, or license technology to third parties in connection with their establishment of, adult stem cell therapy facilities outside the United States. We also intend to work with hospitals and physicians to make the stem cell-based therapies available for patients who travel outside the United States for treatment. “Medical tourism” is defined as the practice of traveling across international borders to obtain health care.
The Federal Trade Commission, or the FTC, has the authority to regulate and police advertising of medical treatments, procedures, and regimens in the United States under the Federal Trade Commission Act, or the FTCA. The FTC has regulatory authority to prevent unfair and deceptive practices and false advertising. Specifically, the FTC requires advertisers and promoters to have a reasonable basis to substantiate and support claims. The FTC has many enforcement powers, one of which is the power to order disgorgement by promoters deemed in violation of the FTCA of any profits made from the promoted business and can order injunctions from further violative promotion. Advertising that we may utilize in connection with our medical tourism operations will be subject to FTC regulatory authority, and we intend to comply with such regulatory régime. Similar laws and requirements are likely to exist in other countries and we intend to comply with such requirements.
Federal Regulation of Clinical Laboratories
Congress passed the Clinical Laboratory Improvement Amendments, or CLIA, in 1988, which provided the Centers for Medicare and Medicaid Services, or CMS, authority over all laboratory testing, except research, that is performed on humans in the United States. The Division of Laboratory Services, within the Survey and Certification Group, under the Center for Medicaid and State Operations, or CMSO, has the responsibility for implementing the CLIA program.
The CLIA program is designed to establish quality laboratory testing by ensuring the accuracy, reliability, and timeliness of patient test results. Under CLIA, a laboratory is a facility that does laboratory testing on specimens derived from humans and used to provide information for the diagnosis, prevention, treatment of disease, or impairment of, or assessment of health. Laboratories that handle stem cells and other biologic matter are, therefore, included under the CLIA program. Under the CLIA program, laboratories must be certified by the government, satisfy governmental quality and personnel standards, undergo proficiency testing, be subject to inspections, and pay fees. To the extent that our business activities require CLIA certification, we intend to obtain and maintain such certification. If we are subject to CLIA, the failure to comply with CLIA standards could result in suspension, revocation, or limitation of a laboratory’s CLIA certificate. In addition, fines or criminal penalties could also be levied. If any of these events were to occur, it could impact our business operations.
| 26 |
Health Insurance Portability and Accountability Act—Protection of Patient Health Information
We may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. The Health Insurance Portability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, including the Final Omnibus Rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information on certain types of individuals and organizations. In addition, certain state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other and from HIPAA in significant ways and may not have the same effect, thus complicating compliance efforts. Further, we may need to also comply with additional federal or state privacy laws and regulations that may apply to certain diagnoses, such as HIV/AIDS, to the extent that they apply to us.
The Department of Health and Human Services, or HHS, through its Office for Civil Rights, investigates breach reports and determines whether administrative or technical modifications are required and whether civil or criminal sanctions should be imposed. Companies failing to comply with HIPAA and the implementing regulations may also be subject to civil money penalties or in the case of knowing violations, potential criminal penalties, including monetary fines, imprisonment, or both. In some cases, the State Attorneys General may seek enforcement and appropriate sanctions in federal court.
Other Applicable U.S. Laws
In addition to the above-described regulation by United States federal and state government, the following are other federal and state laws and regulations that could directly or indirectly affect our ability to operate the business:
| ● | state and local licensure, registration, and regulation of the development of pharmaceuticals and biologics; | |
| ● | state and local licensure of medical professionals; | |
| ● | state statutes and regulations related to the corporate practice of medicine; | |
| ● | laws and regulations administered by U.S. Customs and Border Protection related to the importation of biological material into the United States; | |
| ● | other laws and regulations administered by the FDA; |
| ● | other laws and regulations administered by HHS; | |
| ● | state and local laws and regulations governing human subject research and clinical trials; | |
| ● | the federal physician self-referral prohibition, also known as Stark Law, and any state equivalents to Stark Law; | |
| ● | the federal False Claims Act, or FCA; |
| 27 |
| ● | the federal Anti-Kickback Statute, or AKS, and any state equivalent statutes and regulations; | |
| ● | federal and state coverage and reimbursement laws and regulations; | |
| ● | state and local laws and regulations for the disposal and handling of medical waste and biohazardous material; | |
| ● | Occupational Safety and Health Administration, or OSHA, regulations and requirements; | |
| ● | the Intermediate Sanctions rules of the IRS providing for potential financial sanctions with respect to “excess benefit transactions” with tax-exempt organizations; | |
| ● | the Physician Payments Sunshine Act (in the event that our products are classified as drugs, biologics, devices or medical supplies and are reimbursed by Medicare, Medicaid or the Children’s Health Insurance Program); | |
| ● | state and other federal laws addressing the privacy of health information; and | |
| ● | state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare professionals and other potential referral sources, state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare professionals or marketing expenditures, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
Violation of any of the laws described above or any other governmental laws and regulations may result in penalties, including civil and criminal penalties, damages, fines, the curtailment or restructuring of operations, the exclusion from participation in federal and state healthcare programs and imprisonment. Furthermore, efforts to ensure that business activities and business arrangements comply with applicable healthcare laws and regulations can be costly for manufacturers of branded prescription products.
Foreign Government Regulation
In general, we will need to comply with the government regulations of each individual country in which our therapy centers are located and products are to be distributed and sold. These regulations vary in complexity and can be as stringent, and on occasion even more stringent, than FDA regulations in the United States. Due to the fact that there are new and emerging cell therapy regulations that have recently been drafted and/or implemented in various countries around the world, the application and subsequent implementation of these new and emerging regulations have little to no precedence. Therefore, the level of complexity and stringency is not always precisely understood for each country, creating greater uncertainty for the international regulatory process. Furthermore, government regulations can change with little to no notice and may result in up-regulation of our product(s), thereby creating a greater regulatory burden for our cell processing technology products. We have not yet thoroughly explored the applicable laws and regulations that we will need to comply with in foreign jurisdictions. It is possible that we may not be permitted to expand our business into one or more foreign jurisdictions.
| 28 |
We do not have any definitive plans or arrangements with respect to the establishment by us of stem cell therapy clinics in any country. We intend to explore any such opportunities as they arise.
Offices
Our principal executive offices are located at 40 Marcus Drive, Melville, New York, and our telephone number is (631) 760-8100. Our website is www.biorestorative.com. Our internet website and the information contained therein or connected thereto are not intended to be incorporated by reference into this Annual Report.
Employees
We currently have nine employees, eight of whom are full-time employees. We believe that our employee relations are good.
| ITEM 1A. | RISK FACTORS. |
Not applicable. See, however, Item 7 (“Management’s Discussion and Analysis of Financial Condition and Results of Operations - Factors That May Affect Future Results and Financial Condition”).
| ITEM 1B. | UNRESOLVED STAFF COMMENTS. |
Not applicable.
| ITEM 2. | PROPERTIES. |
Our principal executive offices and laboratory are located at 40 Marcus Drive, Melville, New York. We occupy 6,800 square feet of space at the premises pursuant to a lease that was entered into in August 2014 and expires in March 2020; we have an option to extend the term of the lease for five years. The lease provides for an annual base rental during the initial term ranging between $132,600 and $149,260. Our premises are suitable and adequate for our current operations.
| ITEM 3. | LEGAL PROCEEDINGS. |
Not applicable.
| ITEM 4. | MINE SAFETY DISCLOSURES. |
Not applicable.
| 29 |
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Market Information
Transactions in our common stock are currently reported under the symbol “BRTX” on the OTCQB market. Any over-the-counter market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not necessarily represent actual transactions.
Holders
As of March 19, 2019, there were 303 record holders of our shares of common stock.
Dividends
Not applicable.
Recent Sales of Unregistered Securities
During the three months ended December 31, 2018, we issued the following securities in transactions not involving any public offering. For each of the following transactions, we relied upon Section 4(a)(2) of the Securities Act of 1933, as amended, or the Securities Act, as transactions by an issuer not involving any public offering or Section 3(a)(9) of the Securities Act as a security exchanged by an issuer with its existing security holders exclusively where no commission or other remuneration is paid or given directly or indirectly for soliciting such exchange. For each such transaction, we did not use general solicitation or advertising to market the securities, the securities were offered to a limited number of persons, the investors had access to information regarding us (including information contained in our Annual Report on Form 10-K for the year ended December 31, 2017, Quarterly Reports on Form 10-Q for the periods ended March 31, 2018, June 30, 2018 and September 30, 2018 and Current Reports on Form 8-K filed with the Securities and Exchange Commission, and press releases made by us), and we were available to answer questions by prospective investors. We reasonably believe that each of the investors is an accredited investor. The proceeds were used to reduce our working capital deficiency and for other corporate purposes.
| 30 |
| Warrants | ||||||||||||||||||||||||
| Date Issued | Common Stock | Shares | Exercise Price | Term (Years) | Purchaser(s) | Consideration (1) | ||||||||||||||||||
| 10/4/2018 | 92,802 | - | - | - | (2 | ) | 93,800 | (3) | ||||||||||||||||
| 10/5/2018 | 100,000 | - | - | - | (2 | ) | 97,750 | (3) | ||||||||||||||||
| 10/9/2018 | 1,250 | - | - | - | (2 | ) | 922 | (4) | ||||||||||||||||
| 10/12/2018 | 113,972 | - | - | - | (2 | ) | 88,583 | (3) | ||||||||||||||||
| 10/12/2018 | 16,000 | - | - | - | (2 | ) | 12,342 | (3) | ||||||||||||||||
| 10/17/2018 | 7,000 | - | - | - | (2 | ) | 4,988 | (4) | ||||||||||||||||
| 10/18/2018 | 55,664 | - | - | - | (2 | ) | 49,750 | (3) | ||||||||||||||||
| 10/18/2018 | 111,888 | - | - | - | (2 | ) | 100,000 | (3) | ||||||||||||||||
| 10/19/2018 | 16,666 | - | - | - | (2 | ) | 12,183 | (4) | ||||||||||||||||
| 10/22/2018 | 45,000 | - | - | - | (2 | ) | 34,713 | (3) | ||||||||||||||||
| 10/22/2018 | 20,000 | - | - | - | (2 | ) | 15,428 | (3) | ||||||||||||||||
| 10/26/2018 | 50,000 | - | - | - | (2 | ) | 35,670 | (3) | ||||||||||||||||
| 10/29/2018 | 119,457 | - | - | - | (2 | ) | 85,221 | (3) | ||||||||||||||||
| 10/29/2018 | 11,000 | - | - | - | (2 | ) | 7,847 | (3) | ||||||||||||||||
| 10/29/2018 | - | 75,000 | $ | 2.00 | 5 | (5 | ) | 46,658 | (6) | |||||||||||||||
| 11/2/2018 | 50,000 | - | - | - | (2 | ) | 32,698 | (3) | ||||||||||||||||
| 11/4/2018 | 22,288 | - | - | - | (2 | ) | 14,575 | (3) | ||||||||||||||||
| 11/4/2018 | 61,538 | - | - | - | (2 | ) | 40,000 | (3) | ||||||||||||||||
| 11/8/2018 | 13,000 | - | - | - | (2 | ) | 7,992 | (3) | ||||||||||||||||
| 11/12/2018 | 769,757 | - | - | - | (2 | ) | 554,225 | (3) | ||||||||||||||||
| 11/12/2018 | 54,815 | - | - | - | (2 | ) | 32,429 | (3) | ||||||||||||||||
| 11/13/2018 | 26,984 | - | - | - | (2 | ) | 15,648 | (3) | ||||||||||||||||
| 11/19/2018 | 13,800 | - | - | - | (2 | ) | 8,003 | (3) | ||||||||||||||||
| 11/19/2018 | 72,398 | - | - | - | (2 | ) | 40,000 | (3) | ||||||||||||||||
| 11/29/2018 | 16,683 | - | - | - | (2 | ) | 7,044 | (3) | ||||||||||||||||
| 12/5/2018 | 20,500 | - | - | - | (2 | ) | 8,026 | (3) | ||||||||||||||||
| 12/6/2018 | 61,887 | - | - | - | (2 | ) | 25,142 | (3) | ||||||||||||||||
| 12/12/2018 | 56,250 | - | - | - | (5 | ) | 45,000 | (7) | ||||||||||||||||
| 12/13/2018 | 59,351 | - | - | - | (2 | ) | 20,000 | (3) | ||||||||||||||||
| 12/14/2018 | 108,590 | - | - | - | (2 | ) | 35,292 | (3) | ||||||||||||||||
| 12/14/2018 | 54,317 | - | - | - | (2 | ) | 15,752 | (3) | ||||||||||||||||
| 12/18/2018 | 30,000 | - | - | - | (2 | ) | 10,109 | (3) | ||||||||||||||||
| 12/19/2018 | 238,310 | - | - | - | (2 | ) | 93,921 | (3) | ||||||||||||||||
| 12/19/2018 | 211,164 | - | - | - | (2 | ) | 83,222 | (3) | ||||||||||||||||
| 12/20/2018 | 108,805 | - | - | - | (2 | ) | 35,362 | (3) | ||||||||||||||||
| 12/21/2018 | 54,417 | - | - | - | (2 | ) | 15,781 | (3) | ||||||||||||||||
| 12/24/2018 | 49,238 | - | - | - | (2 | ) | 16,592 | (3) | ||||||||||||||||
| 12/26/2018 | 17,593 | - | - | - | (2 | ) | 5,000 | (3) | ||||||||||||||||
| 12/28/2018 | 74,171 | - | - | - | (2 | ) | 21,079 | (3) | ||||||||||||||||
(1) The value of the non-cash consideration was estimated to be the fair value of our restricted common stock. Since our shares are thinly traded in the open market, the fair value of our equity instruments was estimated by management based on observations of the cash sales prices of both restricted shares and freely tradable shares.
(2) Accredited investor.
(3) Issued in connection with the exchange of convertible notes payable.
(4) Issued in connection with issuance of debt.
(5) Consultant.
(6) Issued in consideration of consulting services.
(7) Issued in exchange of accrued compensation.
| 31 |
Between February 28, 2019 and March 27, 2019, we issued convertible promissory notes in the aggregate principal amount of $701,250 for aggregate cash proceeds of $671,280. We used $255,218 of the net proceeds to prepay certain convertible promissory notes.
On March 13, 2019, a certain lender acquired another promissory note previously issued by us in the outstanding amount of $148,014 (inclusive of accreted interest of $23,014) from a different lender. We exchanged the acquired note for a new convertible note in the principal amount of $148,014.
The newly-issued convertible notes bear interest at the rate of 12% per annum payable at maturity with original maturity dates ranging between February 2020 and March 2020. The notes are convertible as follows: (i) $581,250 of aggregate principal is convertible into shares of our common stock at the election of the respective holder at a fixed price ranging between $1.00 to $2.00 per share for the first six months following the issue date; thereafter, principal and accrued interest are convertible at the election of the holder at a conversion price generally equal to 58% of the fair value of our common stock, (ii) $85,000 of principal and accrued interest is convertible into shares of our common stock at the election of the holder until the 180th day following the issue date at a conversion price equal to $0.25 per share; thereafter, at a conversion price equal to 58% of fair market value of our common stock, (iii) $35,000 of principal and accrued interest is convertible into shares of our common stock at the election of the holder after the 180th day following the issue date at a conversion price equal to 58% of fair market value of our common stock and (iv) $148,014 of principal and accrued interest is convertible into shares of our common stock at the election of the holder immediately following the issue date at a conversion price equal to 65% of fair market value of our common stock.
Between March 13, 2019 and March 20, 2019, we issued 157,733 shares of our common stock in exchange for outstanding indebtedness in the amount of $65,012, inclusive of accrued and unpaid interest and fees.
For each of the securities issuances, we relied upon Section 4(a)(2) of the Securities Act as transactions by an issuer not involving any public offering or Section 3(a)(9) of the Securities Act as a security exchanged by an issuer with its existing security holders exclusively where no commission or other remuneration is paid or given directly or indirectly for soliciting such exchange. For each such transaction, we did not use general solicitation or advertising to market the securities, the securities were offered to a limited number of persons, the investors had access to information regarding us (including information contained in our Annual Report on Form 10-K for the year ended December 31, 2017, Quarterly Reports on Form 10-Q for the periods ended March 31, 2018, June 30, 2018 and September 30, 2018 and Current Reports on Form 8-K filed with the Securities and Exchange Commission, and press releases made by us), and we were available to answer questions by prospective investors. We reasonably believe that each of the investors is an accredited investor.
| 32 |
Issuer Purchases of Equity Securities
During the quarter ended December 31, 2018, there were no purchases of common stock made by us or any “affiliated purchaser”.
| ITEM 6. | SELECTED FINANCIAL DATA. |
Not applicable.
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS. |
The following discussion and analysis of the results of operations and financial condition of BioRestorative Therapies, Inc. (and including its subsidiaries, “BRT” or the “Company”) as of December 31, 2018 and 2017 and for the years ended December 31, 2018 and 2017 should be read in conjunction with our financial statements and the notes to those financial statements that are included elsewhere in this Annual Report on Form 10-K following Item 16 (“Form 10-K Summary”). References in this Management’s Discussion and Analysis of Financial Condition and Results of Operations to “us,” “we,” “our,” and similar terms refer to BRT. This Annual Report contains forward-looking statements as that term is defined in the federal securities laws. The events described in forward-looking statements contained in this Annual Report may not occur. Generally these statements relate to business plans or strategies, projected or anticipated benefits or other consequences of our plans or strategies, projected or anticipated benefits from acquisitions to be made by us, or projections involving anticipated revenues, earnings or other aspects of our operating results. The words “may,” “will,” “expect,” “believe,” “anticipate,” “project,” “plan,” “intend,” “estimate,” and “continue,” and their opposites and similar expressions, are intended to identify forward-looking statements. We caution you that these statements are not guarantees of future performance or events and are subject to a number of uncertainties, risks and other influences, many of which are beyond our control, which may influence the accuracy of the statements and the projections upon which the statements are based. Reference is made to “Factors That May Affect Future Results and Financial Condition” in this Item 7 for a discussion of some of the uncertainties, risks and assumptions associated with these statements.
Overview
We develop therapeutic products and medical therapies using cell and tissue protocols, primarily involving adult (non-embryonic) stem cells. We are currently pursuing our Disc/Spine Program with our initial investigational therapeutic product being called BRTX-100. In January 2017 we submitted an IND application to the FDA to obtain authorization to commence a Phase 2 clinical trial investigating the use of BRTX-100, our lead cell therapy candidate, in the treatment of chronic lower back pain arising from degenerative disc disease. In February 2017, we received such authorization from the FDA. We intend to commence such clinical trial during the third quarter of 2019 (assuming the receipt of necessary funding). We have obtained a license to use technology for investigational adult stem cell treatment of disc and spine conditions, including protruding and bulging lumbar discs. The technology is an advanced stem cell injection procedure that may offer relief from lower back pain, buttock and leg pain, and numbness and tingling in the leg and foot. We are also developing our ThermoStem Program. This pre-clinical program involves the use of brown adipose (fat) in connection with the cell-based treatment of type 2 diabetes and obesity as well as hypertension, other metabolic disorders and cardiac deficiencies. United States patents related to the ThermoStem Program were issued in September 2015 and January 2019, an Australian patent related to the ThermoStem Program was issued in April 2017, and a Japanese patent related to the ThermoStem Program was issued in December 2017.
| 33 |
We have licensed a patented investigational curved needle device that is a needle system designed to deliver cells and/or other therapeutic products or materials to the spine and discs.
Our offices are located in Melville, New York where we have established a laboratory facility in order to increase our capabilities for the further development of possible cellular-based treatments, products and protocols, stem cell-related intellectual property and translational research applications.
As of December 31, 2018, our accumulated deficit was $63,922,256, our stockholders’ deficiency was $8,641,038 and our working capital deficiency was $9,073,901. We have historically only generated a modest amount of revenue, and our losses have principally been operating expenses incurred in research and development, marketing and promotional activities in order to commercialize our products and services, plus costs associated with meeting the requirements of being a public company. We expect to continue to incur substantial costs for these activities over at least the next year. These conditions indicate that there is substantial doubt about our ability to continue as a going concern within one year after the financial statement issuance date.
Based upon our working capital deficiency as of December 31, 2018, and our forecast for continued operating losses, we require equity and/or debt financing to continue our operations. As of December 31, 2018, our outstanding debt of $5,161,916 together with interest at rates ranging between 0% and 15% per annum, was due on various dates through December 2019. Subsequent to December 31, 2018, we have received aggregate equity financing and debt financing of $600,000 and $3,073,918, respectively, debt (inclusive of accrued interest) of $643,900 has been exchanged for common stock, $1,254,805 of debt (inclusive of accrued interest and prepayment premiums) has been repaid, and the due date for the repayment of an aggregate of $155,000 of debt has been extended to December 2020. Giving effect to the above actions, we currently have notes payable in the aggregate principal amount of $107,500 which are past due. Based upon our working capital deficiency and outstanding debt, we expect to be able to fund our operations through April 2019 while we continue to apply efforts to raise additional capital. We anticipate that we will require approximately $20,000,000 in financing to commence and complete a Phase 2 clinical trial with regard to our Disc/Spine Program. We anticipate that we will require approximately $45,000,000 in further additional funding to complete our clinical trials using BRTX-100 (assuming the receipt of no revenues). We will also require a substantial amount of additional funding if we determine to establish a manufacturing operation with regard to our Disc/Spine Program (as opposed to utilizing a third party manufacturer) and to implement our other programs, described in Item 1 (“Business”), including our metabolic ThermoStem Program. No assurance can be given that the anticipated amounts of required funding are correct or that we will be able to accomplish our goals within the timeframes projected. In addition, no assurance can be given that we will be able to obtain any required financing on commercially reasonable terms or otherwise.
| 34 |
We are currently seeking several different financing alternatives to support our future operations and are currently in the process of negotiating extensions or discussing conversions to equity with respect to our outstanding indebtedness. If we are unable to obtain such additional financing on a timely basis or, notwithstanding any request we may make, our debt holders do not agree to convert their notes into equity or extend the maturity dates of their notes, we may have to curtail our development, marketing and promotional activities, which would have a material adverse effect on our business, financial condition and results of operations, and ultimately we could be forced to discontinue our operations and liquidate. See “Liquidity and Capital Resources” below.
Consolidated Results of Operations
Year Ended December 31, 2018 Compared with Year Ended December 31, 2017
The following table presents selected items in our consolidated statements of operations for the years ended December 31, 2018 and 2017, respectively:
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Revenues | $ | 111,000 | $ | 81,000 | ||||
| Operating Expenses | ||||||||
| Marketing and promotion | 352,204 | 65,455 | ||||||
| Consulting | 1,870,829 | 2,334,212 | ||||||
| Research and development | 1,513,150 | 2,152,433 | ||||||
| General and administrative | 4,022,469 | 3,903,184 | ||||||
| Total Operating Expenses | 7,758,652 | 8,455,284 | ||||||
| Loss From Operations | (7,647,652 | ) | (8,374,284 | ) | ||||
| Other Expense | ||||||||
| Interest expense | (932,187 | ) | (468,107 | ) | ||||
| Amortization of debt discount | (2,289,591 | ) | (619,266 | ) | ||||
| Loss on extinguishment of notes payable, net | (1,415,950 | ) | (59,938 | ) | ||||
| Change in fair value of derivative liabilities | (229,323 | ) | 107,039 | |||||
| Warrant modification expense | (3,100 | ) | (30,099 | ) | ||||
| Total Other Expense | (4,870,151 | ) | (1,070,371 | ) | ||||
| Net Loss | $ | (12,517,803 | ) | $ | (9,444,655 | ) | ||
Revenues
For the year ended December 31, 2018, we generated $111,000 from royalty revenue in connection with our sublicense agreement. For the year ended December 31, 2017, we generated $81,000 from royalty revenue in connection with our sublicense agreement. The increase in our revenues for the year ended December 31, 2018 versus 2017 was due to an increase in royalty revenue in connection with our sublicense agreement.
| 35 |
Marketing and promotion
Marketing and promotion expenses include advertising and promotion, marketing and seminars, meals, entertainment and travel expenses. For the year ended December 31, 2018, marketing and promotion expenses increased by $286,749, or 438 %, to $352,204 from $65,455 for the year ended December 31, 2017. The increase is primarily due to the hiring of an advertising and promotion firm in 2018.
We expect that marketing and promotion expenses will increase in the future as we increase our marketing activities following full commercialization of our products and services.
Consulting
Consulting expenses consist of consulting fees and stock-based compensation to consultants. For the year ended December 31, 2018, consulting expenses decreased $463,383, or 20 %, to $1,870,829 from $2,334,212 for the year ended December 31, 2017. The decrease is primarily due to a decrease of approximately $642,000 in stock-based compensation expense related to options and warrants issued to directors and consultants, partially offset by an increase of approximately $168,000 in cash consulting fees.
Research and development
Research and development expenses include cash and non-cash compensation of (a) our Vice President of Research and Development; (b) our Scientific Advisory Board members; (c) our President, Disc/Spine Division (who resigned in July 2017); and (d) laboratory staff and costs related to our brown fat and disc/spine initiatives. Research and development expenses are expensed as they are incurred. For the year ended December 31, 2018, research and development expenses decreased by $639,283, or 30%, to $1,513,150 from $2,152,433 for the year ended December 31, 2017. The decrease was primarily the result of a decrease of approximately $295,000 in payroll and payroll-related costs due to the resignation of the former President of our Disc/Spine Division and a lab employee in 2017, a decrease of approximately $150,000 in cash compensation related to the termination of our Chief Medical Advisor for Spine Medicine in February 2018, and a decrease of approximately $141,000 in stock-based compensation expense related to options issued to our Scientific Advisory Board members.
We expect that our research and development expenses will increase with the continuation of the aforementioned initiatives.
General and administrative
General and administrative expenses consist primarily of salaries, bonuses, payroll taxes, severance costs and stock-based compensation to employees (excluding any cash or non-cash compensation of (a) our Vice President of Research and Development; (b) our President, Disc/Spine Division; and (c) our laboratory staff) as well as corporate support expenses such as legal and professional fees, investor relations and occupancy related expenses. For the year ended December 31, 2018, general and administrative expenses increased by $119,285, or 3%, to $4,022,469 from $3,903,184 for the year ended December 31, 2017.
| 36 |
We expect that our general and administrative expenses will increase as we expand our staff, develop our infrastructure and incur additional costs to support the growth of our business.
Interest expense
For the year ended December 31, 2018, interest expense increased by $464,080, or 99%, to $932,187, as compared to $468,107 during the year ended December 31, 2017. The increase was due to an increase in interest-bearing short-term borrowings as compared to the year ended December 31, 2017.
Amortization of debt discount
For the year ended December 31, 2018, amortization of debt discount increased by $1,670,325, or 270%, to $2,289,591, as compared to $619,266 during the year ended December 31, 2017. The increase was primarily due to the timing of the recognition of expense related to the bifurcated embedded conversion options of convertible notes and the recognition of the debt discount expense.
Loss on extinguishment of notes payable, net
For the year ended December 31, 2018, we recorded a loss on extinguishment of notes payable, net, of $1,415,950, which is associated with debtholders’ exchange of debt into equity securities, as compared to a loss on extinguishment of notes payable of $59,938 for the year ended December 31, 2017.
Change in fair value of derivative liabilities
For the year ended December 31, 2018, we recorded a loss related to the change in fair value of derivative liabilities of $229,323 due to the increase in time value of embedded conversion options within certain convertible notes payable, as compared to a gain related to the change in fair value of derivative liabilities of $107,039 for the year ended December 31, 2017.
Warrant modification expense
During the year ended December 31, 2018, we recorded expense related to the modification of an exercise price of an outstanding warrant of $3,100, as compared to expense related to the modification of the expiration dates and exercise prices of outstanding warrants of $30,099 for the year ended December 31, 2017.
| 37 |
Liquidity and Capital Resources
Liquidity
We measure our liquidity in a number of ways, including the following:
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Cash | $ | 117,523 | $ | 451,680 | ||||
| Working Capital Deficiency | $ | 9,073,901 | $ | (7,833,592 | ) | |||
| Notes Payable (Gross) | $ | 5,161,916 | $ | 3,999,335 | ||||
Availability of Additional Funds
Based upon our working capital and stockholders’ deficiencies of $9,073,901 and $8,641,038, respectively, as of December 31, 2018, we require additional equity and/or debt financing to continue our operations. These conditions raise substantial doubt about our ability to continue as a going concern within one year after the issuance date of our audited financial statements as of December 31, 2018 and 2017 and for the years then ended, which are included in this Annual Report following Item 16 (“Form 10-K Summary”).
As of December 31, 2018, our outstanding debt of $5,161,916, together with interest at rates ranging between 0% and 15% per annum, was due on various dates through December 2019. Subsequent to December 31, 2018, we have received aggregate equity financing and debt financing of $600,000 and $3,073,918 respectively, debt (inclusive of accrued interest) of $643,900 has been exchanged for common stock, $1,254,805 of debt (inclusive of accrued interest and prepayment premiums) has been repaid, and the due date for the repayment of an aggregate of $155,000 of debt has been extended to December 2020. Giving effect to the above actions, we currently have notes payable in the aggregate principal amount of $107,500 which are past due. As of the date of filing, our outstanding debt was as follows:
| Maturity Date | Principal Amount | |||
| Past Due | $ | 107,500 | ||
| QE 3/31/19 | 350,000 | |||
| QE 6/30/19 | 1,070,000 | |||
| QE 9/30/19 | 1,950,023 | |||
| QE 12/31/19 | 1,725,000 | |||
| QE 3/31/20 | 1,528,014 | |||
| $ | 6,730,537 | |||
| 38 |
Based upon our working capital deficiency, outstanding debt and forecast for continued operating losses we expect that the cash we currently have available will fund our operations through April 2019 while we continue to apply efforts to raise additional capital. Thereafter, we will need to raise further capital, through the sale of additional equity or debt securities, to support our future operations and to repay our debt (unless, if requested, the debt holders agree to convert their notes into equity or extend the maturity dates of their notes). Our operating needs include the planned costs to operate our business, including amounts required to fund working capital and capital expenditures. Our future capital requirements and the adequacy of our available funds will depend on many factors, including our ability to successfully commercialize our products and services, competing technological and market developments, and the need to enter into collaborations with other companies or acquire other companies or technologies to enhance or complement our product and service offerings.
We may be unable to raise sufficient additional capital when we need it or raise capital on favorable terms. Debt financing may require us to pledge certain assets and enter into covenants that could restrict certain business activities or our ability to incur further indebtedness, and may contain other terms that are not favorable to our stockholders or us. If we are unable to obtain adequate funds on reasonable terms, we may be required to significantly curtail or discontinue operations or obtain funds by entering into financing agreements on unattractive terms.
Our consolidated financial statements included elsewhere in this Annual Report on Form 10-K have been prepared in conformity with accounting principles generally accepted in the United States of America, or U.S. GAAP, which contemplate our continuation as a going concern and the realization of assets and satisfaction of liabilities in the normal course of business. The carrying amounts of assets and liabilities presented in the financial statements do not necessarily purport to represent realizable or settlement values. The financial statements do not include any adjustment that might result from the outcome of this uncertainty.
During the year ended December 31, 2018, our sources and uses of cash were as follows:
Net Cash Used in Operating Activities
We experienced negative cash flow from operating activities for the years ended December 31, 2018 and 2017 in the amounts of $5,104,629 and $3,853,821, respectively. The net cash used in operating activities for the year ended December 31, 2018 was primarily due to cash used to fund a net loss of $12,517,803, adjusted for non-cash expenses in the aggregate amount of $7,458,950 and $45,776 of cash used in changes in the levels of operating assets and liabilities, primarily as a result of a decrease in accounts payable, partially offset by increases in accrued interest, expenses and other current liabilities. The net cash used in operating activities for the year ended December 31, 2017 was primarily due to cash used to fund a net loss of $9,444,655, adjusted for non-cash expenses in the aggregate amount of $4,769,506 partially offset by $821,328 of cash provided by changes in the levels of operating assets and liabilities, primarily as a result of increases in accrued interest, expenses and other current liabilities, increases in accounts payable, a decrease in security deposit and decreases in prepaid expenses and other current assets, partially offset by increases in accounts receivable, primarily due to cash constraints during the period.
Net Cash Used in Investing Activities
During the year ended December 31, 2018, net cash used in investing activities was $12,869 used for the purchase of office and computer equipment. During the year ended December 31, 2017, net cash used in investing activities was $3,617 used for the purchase of computer equipment.
| 39 |
Net Cash Provided by Financing Activities
Net cash provided by financing activities during the years ended December 31, 2018 and 2017 was $4,783,341 and $4,277,296, respectively. During the year ended December 31, 2018, $4,194,173 of net proceeds were from debt financings and other borrowings and $589,168 of proceeds were from equity financings (including proceeds received in connection with the exercise of common stock purchase warrants). During the year ended December 31, 2017, $2,197,046 of net proceeds were from debt financings and other borrowings and $2,080,250 of proceeds were from equity financings (including proceeds received in connection with the exercise of common stock purchase warrants).
Critical Accounting Policies and Estimates
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at dates of the financial statements and the reported amounts of revenue and expenses during the periods. Our significant estimates and assumptions include the recoverability and useful lives of long-lived assets, the fair value of our common stock, stock-based compensation, warrants issued in connection with notes payable, derivative liabilities and the valuation allowance related to our deferred tax assets. Certain of our estimates, including the carrying amount of the intangible assets, could be affected by external conditions, including those unique to us and general economic conditions. It is reasonably possible that these external factors could have an effect on our estimates and could cause actual results to differ from those estimates.
Intangible Assets
Intangible assets are comprised of trademarks and licenses with original estimated useful lives of 10 and 17.7 years, respectively. Once placed into service, we amortize the cost of the intangible assets over their estimated useful lives on a straight-line basis.
Impairment of Long-lived Assets
We review for the impairment of long-lived assets whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment loss would be recognized when estimated future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. While our near term liquidity is tight, historically we have been successful in raising capital as needed (although there can be no assurance that we will continue to be successful in raising capital as needed). We continue to progress our scientific agenda and meet related milestones. We have not identified any impairment losses.
Income Taxes
We recognize deferred tax assets and liabilities for the expected future tax consequences of items that have been included or excluded in our financial statements or tax returns. Deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts, or temporary differences, at enacted tax rates in effect for the years in which the temporary differences are expected to reverse.
| 40 |
We adopted the provisions of Accounting Standards Codification, or ASC, Topic 740-10, which prescribes a recognition threshold and measurement process for financial statements recognition and measurement of a tax position taken or expected to be taken in a tax return.
Stock-Based Compensation
We measure the cost of services received in exchange for an award of equity instruments based on the fair value of the award. For employees and directors, the fair value of the award is measured on the grant date and for non-employees, the fair value of the award is generally re-measured on vesting dates and interim financial reporting dates until the service period is complete. The fair value amount is then recognized over the period during which services are required to be provided in exchange for the award, usually the vesting period. Since the shares underlying our 2010 Equity Participation Plan, or the Plan, were registered on May 27, 2014, we estimate the fair value of the awards granted under the Plan based on the market value of our freely tradable common stock as reported on the OTCQB market. The fair value of our restricted equity instruments was estimated by management based on observations of the cash sales prices of both restricted shares and freely tradable shares. Awards granted to directors are treated on the same basis as awards granted to employees.
Recently Issued Accounting Pronouncements
See Note 3 to our consolidated financial statements for the year ended December 31, 2018.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Factors That May Affect Future Results and Financial Condition
The risk factors listed in this section provide examples of risks, uncertainties and events that may cause our actual results to differ materially from the expectations we describe in our forward-looking statements. Readers should be aware that the occurrence of any of the events described in these risk factors could have a material adverse effect on our business, results of operations and financial condition. We undertake no obligation to update or revise publicly any forward-looking statements, whether as a result of new information, future events, or otherwise.
| 41 |
Risks Related to Our Business Generally
We have a limited operating history; we have incurred substantial losses since inception; we expect to continue to incur losses for the near term; we have a substantial working capital deficiency and a stockholders’ deficiency; we believe these conditions indicate that there is substantial doubt about our ability to continue as a going concern within the next twelve months from the date of this filing; the report of our independent registered public accounting firm contains an explanatory paragraph that expresses substantial doubt about our ability to continue as a going concern.
We have a limited operating history. Since our inception, we have incurred net losses. As of December 31, 2018, we had a working capital deficiency of $9,073,901 and stockholders’ deficiency of $8,641,038. The report of our independent registered public accounting firm with respect to our financial statements as of December 31, 2018 and 2017 and for the years then ended indicates that our financial statements have been prepared assuming that we will continue as a going concern. The report states that, since we have incurred net losses since inception and we need to raise additional funds to meet our obligations and sustain our operations, there is substantial doubt about our ability to continue as a going concern within one year after the issuance date of our audited financial statements as of December 31, 2018 and 2017 and for the years then ended, which are included in this Annual Report following Item 16 (“Form 10-K Summary”). Our plans in regard to these matters are described in footnote 2 to such financial statements. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We will need to obtain a significant amount of financing to initiate and complete our clinical trials and implement our business plan.
Since our inception, we have not generated significant revenues from our operations and have funded our operations through the sale of our equity securities (approximately $16,000,000) and debt securities (approximately $23,000,000). The implementation of our business plan, as discussed in Item 1 (“Business”), will require the receipt of sufficient equity and/or debt financing to purchase necessary equipment, technology and materials, fund our research and development efforts, retire our outstanding debt and otherwise fund our operations. We anticipate that we will require approximately $20,000,000 in financing to commence and complete a Phase 2 clinical trial using BRTX-100. We anticipate that we will require approximately $45,000,000 in further additional funding to complete our clinical trials using BRTX-100 (assuming the receipt of no revenues). We will also require a substantial amount of additional funding if we determine to establish a manufacturing operation with regard to our Disc/Spine Program (as opposed to utilizing a third party manufacturer) and to implement our other programs described in Item 1 (“Business”), including our metabolic ThermoStem Program. No assurance can be given that the anticipated amounts of required funding are correct or that we will be able to accomplish our goals within the timeframes projected. In addition, no assurance can be given that we will be able to obtain any required financing on commercially reasonable terms or otherwise. In the event we do not obtain the financing required for the above purposes, we may have to curtail our development, marketing and promotional activities, which would have a material adverse effect on our business, financial condition and results of operations, and ultimately we could be forced to discontinue our operations and liquidate.
We will need to obtain additional financing to satisfy debt obligations.
As described in Item 7 (“Management’s Discussion and Analysis of Financial Condition and Results of Operations – Liquidity and Capital Resources – Availability of Additional Funds”), as of December 31, 2018, our outstanding debt of $5,161,916, together with interest at rates ranging between 0% and 15% per annum, are due on various dates through December 2019. Subsequent to December 31, 2018, we have received aggregate equity financing and debt financing of $600,000 and $3,073,918, respectively, debt (inclusive of accrued interest) of $643,900 has been exchanged for common stock, $1,254,805 of debt (inclusive of accrued interest and prepayment premiums) has been repaid, and the due date for the repayment of $155,000 of debt has been extended to December 2020. Giving effect to the above actions, we currently have notes payable in the aggregate principal amount of $107,500 which are past due. As of March 19, 2019, the outstanding balance of our debt of $6,375,537, together with accrued interest, was due and payable either on demand or on various dates through March 2020. Unless we obtain additional financing or, upon our request, the debt holders agree to convert their debt into equity or extend the maturity dates of the debt, we will not be able to repay such debt. Based upon our working capital deficiency and outstanding debt, we expect to be able to fund our operations through April 2019, while we continue to apply efforts to raise additional capital. Even if we are able to satisfy our debt obligations, our cash balance and the revenues for the foreseeable future from our anticipated operations will not be sufficient to fund the development of our business plan.
| 42 |
Our business strategy is high risk.
We are focusing our resources and efforts primarily on the development of cellular-based products and services which will require extensive cash for research, development and commercialization activities. This is a high-risk strategy because there is no assurance that our products and services, including our Disc/Spine Program and our ThermoStem metabolic brown fat research initiative, will ever become commercially viable (commercial risk), that we will prevent other companies from depriving us of market share and profit margins by offering services and products based on our inventions and developments (legal risk), that we will successfully manage a company in a new area of business, regenerative medicine, and on a different scale than we have operated in the past (operational risk), that we will be able to achieve the desired therapeutic results using stem and regenerative cells (scientific risk), or that our cash resources will be adequate to develop our products and services until we become profitable, if ever (financial risk). We are using our cash in one of the riskiest industries in the economy (strategic risk). This may make our stock an unsuitable investment for many investors.
We will need to enter into agreements in order to implement our business strategy.
Except for a certain license agreement described in Item 1 (“Business”), we do not have any material agreements or understandings in place with respect to the implementation of our business strategy. No assurances can be given that we will be able to enter into any necessary agreements with respect to the development of our business. Our inability to enter into any such agreements would have a material adverse effect on our results of operations and financial condition.
We depend on our executive officers and on our ability to attract and retain additional qualified personnel; we do not currently have a Chief Financial Officer.
Our performance is substantially dependent on the performance of Mark Weinreb, our Chief Executive Officer. We rely upon him for strategic business decisions and guidance. Mr. Weinreb is subject to an employment agreement with us that is scheduled to expire on December 31, 2019. We are also dependent on the performance of Lance Alstodt, our Executive Vice President and Chief Strategy Officer, and Francisco Silva, our Vice President of Research and Development. Messrs. Alstodt and Silva are also subject to employment agreements with us. We do not have any key-man insurance policies on the lives of any of our executive officers. We do not currently have a Chief Financial Officer. Pending the hiring of a Chief Financial Officer, we are utilizing financial consultants with regard to the preparation of our financial statements. We believe that our future success in developing marketable products and services and achieving a competitive position will depend in large part upon whether we can attract and retain additional qualified management and scientific personnel, including a Chief Financial Officer. Competition for such personnel is intense, and there can be no assurance that we will be able to attract and retain such personnel. The loss of the services of Mr. Weinreb, Mr. Alstodt and/or Mr. Silva or the inability to attract and retain additional personnel, including a Chief Financial Officer, and develop expertise as needed would have a substantial negative effect on our results of operations and financial condition.
| 43 |
Continued turmoil in the economy could harm our business.
Negative trends in the general economy, including, but not limited to, trends resulting from an actual or perceived recession, tightening credit markets, increased cost of commodities, actual or threatened military action by the United States and threats of terrorist attacks in the United States and abroad, could cause a reduction of investment in and available funding for companies in certain industries, including ours. Our ability to raise capital has been and may in the future be adversely affected by downturns in current credit conditions, financial markets and the global economy.
Risks Related to Our Cell Therapy Product Development Efforts
Our future success is significantly dependent on the timely and successful development and commercialization of BRTX-100, our lead product candidate for the treatment of chronic lumbar disc disease; if we encounter delays or difficulties in the development of this product candidate, as well as any other product candidates, our business prospects would be significantly harmed.
We are dependent upon the successful development, approval and commercialization of our product candidates. Before we are able to seek regulatory approval of our product candidates, we must conduct and complete extensive clinical trials to demonstrate their safety and efficacy in humans. Our lead product candidate, BRTX-100, is in early stages of development and we have not commenced a Phase 2 clinical trial using BRTX-100 to investigate the use of the candidate in treating chronic lower back pain due to degenerative disc disease related to protruding/bulging discs.
Clinical testing is expensive, difficult to design and implement, and can take many years to complete. Importantly, a failure of one or more of these or any other clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to complete our clinical studies, receive regulatory approval or commercialize our cell therapy product candidates, including the following:
| ● | suspensions, delays or changes in the design, initiation, enrollment, implementation or completion of required clinical trials; adverse changes in our financial position or significant and unexpected increases in the cost of our clinical development program; changes or uncertainties in, or additions to, the regulatory approval process that require us to alter our current development strategy; clinical trial results that are negative, inconclusive or less than desired as to safety and/or efficacy, which could result in the need for additional clinical studies or the termination of the product’s development; delays in our ability to manufacture the product in quantities or in a form that is suitable for any required clinical trials; |
| 44 |
| ● | intellectual property constraints that prevent us from making, using, or commercializing any of our cell therapy product candidates; | |
| ● | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of these product candidates may be insufficient or inadequate; inability to generate sufficient pre-clinical, toxicology, or other in vivo or in vitro data to support the initiation of clinical studies; | |
| ● | delays in reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical study sites; | |
| ● | delays in obtaining required Institutional Review Board, or IRB, approval at each clinical study site; | |
| ● | imposition of a temporary or permanent clinical hold by regulatory agencies for a number of reasons, including after review of an IND application or amendment, or equivalent application or amendment; as a result of a new safety finding that presents unreasonable risk to clinical trial participants; a negative finding from an inspection of our clinical study operations or study sites; developments on trials conducted by competitors or approved products post-market for related technology that raises FDA concerns about risk to patients of the technology broadly; or if the FDA finds that the investigational protocol or plan is clearly deficient to meet its stated objectives; | |
| ● | difficulty collaborating with patient groups and investigators; | |
| ● | failure by our CROs, other third parties, or us to adhere to clinical study requirements; | |
| ● | failure to perform in accordance with the FDA’s current Good Clinical Practices, or GCP, requirements, or applicable regulatory guidelines in other countries; | |
| ● | delays in having patients qualify for or complete participation in a study or return for post-treatment follow-up; | |
| ● | patients dropping out of a study; | |
| ● | occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential benefits; | |
| ● | changes in the standard of care on which a clinical development plan was based, which may require new or additional trials; |
| 45 |
| ● | transfer of manufacturing processes from any academic collaborators to larger-scale facilities operated by either a contract manufacturing organization, or CMO, or by us, and delays or failure by our CMOs or us to make any necessary changes to such manufacturing process; | |
| ● | delays in manufacturing, testing, releasing, validating, or importing/exporting sufficient stable quantities of our product candidates for use in clinical studies or the inability to do any of the foregoing; and | |
| ● | the FDA may not accept clinical data from trials that are conducted at clinical sites in countries where the standard of care is potentially different from the United States. |
Any inability to successfully complete pre-clinical and clinical development could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required, or we may elect, to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical study delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
Even if we are able to successfully complete our clinical development program for our product candidates, and ultimately receive regulatory approval to market one or more of the products, we may, among other things:
| ● | obtain approval for indications that are not as broad as the indications we sought; | |
| ● | have the product removed from the market after obtaining marketing approval; | |
| ● | encounter issues with respect to the manufacturing of commercial supplies; | |
| ● | be subject to additional post-marketing testing requirements; and/or | |
| ● | be subject to restrictions on how the product is distributed or used. |
We anticipate that we will not be able to commercialize our BRTX-100 product candidate for at least five years.
We may experience delays and other difficulties in enrolling a sufficient number of patients in our clinical trials which could delay or prevent the receipt of necessary regulatory approvals.
We may not be able to initiate or complete as planned any clinical trials if we are unable to identify and enroll a sufficient number of eligible patients to participate in the clinical trials required by the FDA or other regulatory authorities. We also may be unable to engage a sufficient number of clinical trial sites to conduct our trials.
| 46 |
We may face challenges in enrolling patients to participate in our clinical trials due to the novelty of our cell-based therapies, the size of the patient populations and the eligibility criteria for enrollment in the trial. In addition, some patients may have concerns regarding cell therapy that may negatively affect their perception of therapies under development and their decision to enroll in the trials. Furthermore, patients suffering from diseases within target indications may enroll in competing clinical trials, which could negatively affect our ability to complete enrollment of our trials. Enrollment challenges in clinical trials often result in increased development costs for a product candidate, significant delays and potentially the abandonment of the clinical trial.
We may have other delays in completing our clinical trials and we may not complete them at all.
We have not commenced the clinical trials necessary to obtain FDA approval to market our product candidate, BRTX-100, or any of our other product candidates in development. While our management lacks significant experience in completing clinical trials and bringing a drug through commercialization, we have hired outside consultants with such experience. Clinical trials for BRTX-100 and other product candidates in development may be delayed or terminated as a result of many factors, including the following:
| ● | patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons; | |
| ● | failure by regulators to authorize us to commence a clinical trial; | |
| ● | suspension or termination by regulators of clinical research for many reasons, including concerns about patient safety, the failure of study sites and/or investigators in our clinical research program to comply with Good Clinical Practices, or GCP, requirements, or our failure, or the failure of our contract manufacturers, to comply with current cGMP requirements; | |
| ● | delays or failure to obtain clinical supply for our products necessary to conduct clinical trials from contract manufacturers; | |
| ● | treatment candidates demonstrating a lack of efficacy during clinical trials; | |
| ● | treatment candidates demonstrating significant safety signals | |
| ● | inability to continue to fund clinical trials or to find a partner to fund the clinical trials; | |
| ● | competition with ongoing clinical trials and scheduling conflicts with participating clinicians; and | |
| ● | delays in completing data collection and analysis for clinical trials. |
Any delay or failure to complete clinical trials and obtain FDA approval for our product candidates could have a material adverse effect on our cost to develop and commercialize, and our ability to generate revenue from, a particular product candidate.
| 47 |
The development of our cell therapy product candidates is subject to uncertainty because autologous cell therapy is inherently variable.
When manufacturing an autologous cell therapy, the number and the composition of the cell population varies from patient to patient. Such variability in the number and composition of these cells could adversely affect our ability to manufacture autologous cell therapies in a cost-effective or profitable manner and meet acceptable product release specifications for use in a clinical trial or, if approved, for commercial sale. As a consequence, the development and regulatory approval process for autologous cell therapy products could be delayed or may never be completed.
Any disruption to our access to the media (including cell culture media) and reagents we are using in the clinical development of our cell therapy product candidates could adversely affect our ability to perform clinical trials and seek future regulatory submissions.
Certain media (including cell culture media) and reagents, as well as devices, materials and systems, that we intend to use in our planned clinical trials, and that we may need or use in commercial production, are provided by unaffiliated third parties. Any lack of continued availability of these media, reagents, devices, materials and systems for any reason would have a material adverse effect on our ability to complete these studies and could adversely impact our ability to achieve commercial manufacture of our planned therapeutic products. Although other available sources for these media, reagents, devices, materials and systems may exist in the marketplace, we have not evaluated their cost, effectiveness, or intellectual property foundation and therefore cannot guarantee the suitability or availability of such other potential sources.
Products that appear promising in research and development may be delayed or may fail to reach later stages of clinical development.
The successful development of cellular based products is highly uncertain. Product candidates that appear promising in preclinical and early research and development may be delayed or fail to reach later stages of development. Decisions regarding the further development of product candidates must be made with limited and incomplete data, which makes it difficult to ensure or even accurately predict whether the allocation of limited resources and the expenditure of additional capital on specific product candidates will result in desired outcomes. Pre-clinical and clinical data can be interpreted in different ways, and negative or inconclusive results or adverse events during a clinical trial could delay, limit or prevent the development of a product candidate. Positive preclinical data may not continue or occur for future subjects in our clinical studies and may not be repeated or observed in ongoing or future studies involving our product candidates. Furthermore, our product candidates may also fail to show the desired safety and efficacy in later stages of clinical development despite having successfully advanced through initial clinical studies. In addition, regulatory delays or rejections may be encountered as a result of many factors, including changes in regulatory policy during the period of product development.
| 48 |
Our clinical trials may fail to demonstrate adequately the safety and efficacy of our product candidates, which would prevent or delay regulatory approval and commercialization.
The clinical trials of our product candidates are, and the manufacturing and marketing of our products will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and market our product candidates. Before obtaining regulatory approvals for the commercial sale of any of our product candidates, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that our product candidates are both safe and effective for use in each target indication. In particular, because our product candidates are subject to regulation as biological drug products, we will need to demonstrate that they are safe, pure, and potent for use in their target indications. Each product candidate must demonstrate an adequate risk versus benefit profile in its intended patient population and for its intended use. The risk/benefit profile required for product licensure will vary depending on these factors and may include decrease or elimination of pain, adequate duration of response, a delay in the progression of the disease, an improvement in function and/or decrease in disability.
In addition, even if such trials are successfully completed, we cannot guarantee that the FDA will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. To the extent that the results of the trials are not satisfactory to the FDA for support of a marketing application, we may be required to expend significant resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates.
Even if we complete the necessary clinical trials, we cannot predict when, or if, we will obtain regulatory approval to commercialize a product candidate, and the approval may be for a narrower indication than we seek.
We cannot commercialize a product candidate until the appropriate regulatory authorities have reviewed and approved the product candidate. Even if our product candidates meet their safety and efficacy endpoints in clinical trials, the regulatory authorities may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions or conditions on approval. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory authority policy during the period of product development, clinical trials and the review process. Regulatory authorities also may approve a product candidate for more limited indications than requested or they may impose significant limitations in the form of narrow indications, contraindications or a REMS. These regulatory authorities may require warnings or precautions with respect to conditions of use or they may grant approval subject to the performance of costly post-marketing clinical trials. In addition, regulatory authorities may not approve the labeling claims or allow the promotional claims that are necessary or desirable for the successful commercialization of our product candidates. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates and materially and adversely affect our business, financial condition, results of operations and prospects.
| 49 |
We may never obtain FDA approval for any of our product candidates in the United States, and even if we do, we may never obtain approval for or commercialize any of our product candidates in any foreign jurisdiction, which would limit our ability to realize our full market potential.
In order to eventually market any of our product candidates in any particular foreign jurisdiction, we must establish and comply with numerous and varying regulatory requirements regarding safety and efficacy on a jurisdiction-by-jurisdiction basis. Approval by the FDA in the United States, if obtained, does not ensure approval by regulatory authorities in other countries or jurisdictions. In addition, preclinical studies and clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not guarantee regulatory approval in any other country.
Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approval could result in difficulties and costs for us and require additional preclinical studies or clinical trials which could be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our product candidates in those countries. The foreign regulatory approval process involves similar risks to those associated with FDA approval. We do not have any product candidates approved for sale in any jurisdiction, including international markets, nor have we attempted to obtain such approval. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approvals in international markets are delayed, our target market will be reduced and our ability to realize the full market potential of our products may be unrealized.
We presently lack manufacturing capabilities to produce our product candidates at commercial scale quantities and do not have an alternate manufacturing supply, which could negatively impact our ability to meet any future demand for the products.
Currently, we expect our laboratory (or a contract laboratory) to provide the cell processing services necessary for clinical production of BRTX-100 for our disc clinical trial. To date, we have not produced any products at our laboratory. We expect that we would need to significantly expand our manufacturing capabilities to meet potential commercial demand for BRTX-100 and any other of our product candidates, if approved, as well as any of our other product candidates that might attain regulatory approval. Such expansion would require additional regulatory approvals. Even if we increase our manufacturing capabilities, it is possible that we may still lack sufficient capacity to meet demand. Ultimately, if we are unable to supply our products to meet commercial demand, whether because of processing constraints or other disruptions, delays or difficulties that we experience, sales of the products and their long-term commercial prospects could be significantly damaged.
We do not presently have a third-party manufacturer for BRTX-100 or any of our other product candidates. If our facilities at which these product candidates would be manufactured or our equipment were significantly damaged or destroyed, or if there were other disruptions, delays or difficulties affecting manufacturing capacity, our planned and future clinical studies and commercial production for these product candidates would likely be significantly disrupted and delayed. It would be both time consuming and expensive to replace this capacity with third parties, particularly since any new facility would need to comply with the regulatory requirements.
Ultimately, if we are unable to supply our cell therapy product candidates to meet commercial demand (assuming commercial approval is obtained), whether because of processing constraints or other disruptions, delays or difficulties that we experience, our production costs could dramatically increase and sales of the product and its long-term commercial prospects could be significantly damaged.
| 50 |
The commercial potential and profitability of our products are unknown and subject to significant risk and uncertainty.
Even if we successfully develop and obtain regulatory approval for our cell therapy product candidates, the market may not understand or accept the products, which could adversely affect both the timing and level of future sales. Ultimately, the degree of market acceptance of our product candidates (or any of our future product candidates) will depend on a number of factors, including:
| ● | the clinical effectiveness, safety and convenience of the product particularly in relation to alternative treatments; | |
| ● | our ability to distinguish our products (which involve adult cells) from any ethical and political controversies associated with stem cell products derived from human embryonic or fetal tissue; and | |
| ● | the cost of the product, the reimbursement policies of government and third-party payors and our ability to obtain sufficient third-party coverage or reimbursement. |
Even if we are successful in achieving sales of our product candidates, it is not clear to what extent, if any, the products will be profitable. The costs of goods associated with production of cell therapy products are significant. In addition, some changes in manufacturing processes or procedures generally require FDA or foreign regulatory authority review and approval prior to implementation. We may need to conduct additional pre-clinical studies and clinical trials to support approval of any such changes. Furthermore, this review process could be costly and time-consuming and could delay or prevent the commercialization of product candidates.
We may have difficulties in sourcing brown adipose (fat) tissue.
We use brown adipose (fat) tissue to identify and characterize brown adipose derived stem cells for use in our pre-clinical ThermoStem Program. There is no certainty that we will be able to continue to collect brown adipose samples through any relationships that we have, have had or may establish with potential sources of brown adipose tissue. The inability to procure brown fat tissue would have a material adverse effect upon our ability to advance our ThermoStem Program.
We are required to complete a certain milestone or pay a certain royalty amount to maintain our exclusive license rights with regard to the disc/spine technology. The loss of such exclusive rights would have a material adverse effect upon us.
Pursuant to our license agreement with Regenerative Sciences, LLC, we must complete a certain milestone or pay a certain royalty amount in order to maintain our exclusive rights with regard to the disc/spine technology. No assurances can be given that we will achieve such milestone or have the funds, if necessary, to pay such royalty amount. Any loss of such exclusive rights would have a material adverse effect upon our business, results of operations and financial condition.
| 51 |
If safety problems are encountered by us or others developing new stem cell-based therapies, our stem cell initiatives could be materially and adversely affected.
The use of stem cells for therapeutic indications is still in the very early stages of development. If an adverse event occurs during clinical trials related to one of our proposed products and/or services or those of others, the FDA and other regulatory authorities may halt clinical trials or require additional studies. The occurrence of any of these events would delay, and increase the cost of, our development efforts and may render the commercialization of our proposed products and/or services impractical or impossible.
Ethical and other concerns surrounding the use of stem cell therapy may negatively impact the public perception of our stem cell products and/or services, thereby suppressing demand for our products and/or services.
Although our contemplated stem cell business pertains to adult stem cells only, and does not involve the more controversial use of embryonic stem cells, the use of adult human stem cells for therapy could give rise to similar ethical, legal and social issues as those associated with embryonic stem cells, which could adversely affect its acceptance by consumers and medical practitioners. Additionally, it is possible that our business could be negatively impacted by any stigma associated with the use of embryonic stem cells if the public fails to appreciate the distinction between adult and embryonic stem cells. Delays in achieving public acceptance may materially and adversely affect the results of our operations and profitability.
We are vulnerable to competition and technological change, and also to physicians’ inertia.
We will compete with many domestic and foreign companies in developing our technology and products, including biotechnology, medical device and pharmaceutical companies. Many current and potential competitors have substantially greater financial, technological, research and development, marketing, and personnel resources. There is no assurance that our competitors will not succeed in developing alternative products and/or services that are more effective, easier to use, or more economical than those which we may develop, or that would render our products and/or services obsolete and non-competitive. In general, we may not be able to prevent others from developing and marketing competitive products and/or services similar to ours or which perform similar functions or which are marketed before ours.
Competitors may have greater experience in developing products, therapies or devices, conducting clinical trials, obtaining regulatory clearances or approvals, manufacturing and commercialization. It is possible that competitors may obtain patent protection, approval or clearance from the FDA or achieve commercialization earlier than we can, any of which could have a substantial negative effect on our business.
We will compete against cell-based therapies derived from alternate sources, such as bone marrow, adipose tissue, umbilical cord blood and potentially embryos. Doctors historically are slow to adopt new technologies like ours, whatever the merits, when older technologies continue to be supported by established providers. Overcoming such inertia often requires very significant marketing expenditures or definitive product performance and/or pricing superiority.
We expect that physicians’ inertia and skepticism will also be a significant barrier as we attempt to gain market penetration with our future products and services. We may need to finance lengthy time-consuming clinical studies (so as to provide convincing evidence of the medical benefit) in order to overcome this inertia and skepticism.
| 52 |
We may form or seek collaborations or strategic alliances or enter into additional licensing arrangements in the future, and we may not realize the benefits of such alliances or licensing arrangements.
We may form or seek strategic alliances, create joint ventures or collaborations, or enter into additional licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future product candidates that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute the shares of our existing stockholders, or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. To date, such efforts have not been successful.
Further, collaborations involving our product candidates, such as our collaborations with third-party research institutions, are subject to numerous risks, which may include the following:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to a collaboration; | |
| ● | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in their strategic focus due to the acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; | |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial, stop a clinical trial, abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product candidate for clinical testing; | |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates; | |
| ● | a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to their marketing and distribution; | |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; | |
| ● | disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our product candidates, or that result in costly litigation or arbitration that diverts management attention and resources; | |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; and | |
| ● | collaborators may own or co-own intellectual property covering our products that results from our collaborating with them, and in such cases, we would not have the exclusive right to commercialize such intellectual property. |
| 53 |
As a result, if we enter into collaboration agreements and strategic partnerships or license our products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture, which could delay our timelines or otherwise adversely affect our business. We also cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new collaborations or strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition, and results of operations.
We have limited experience in the development and marketing of cell therapies and may be unsuccessful in our efforts to establish a profitable business.
Over the past eight years, our business plan has been focused on capturing a piece of the burgeoning field of cell therapy. We have limited experience in the areas of cell therapy product development and marketing, and in the related regulatory issues and processes. Although we have recruited a team that has experience with designing and conducting clinical trials and hired contract research organizations, contract manufacturing organizations and FDA consultants, as a company, we have limited experience in conducting clinical trials and no experience in conducting clinical trials through to regulatory approval of any product candidate. In part because of this lack of experience, we cannot be certain that planned clinical trials will begin or be completed on time, if at all. We cannot assure that we will successfully achieve our clinical development goals or fulfill our plans to capture a piece of the cell therapy market.
Our cell therapy business is based on novel technologies that are inherently expensive, risky and may not be understood by or accepted in the marketplace, which could adversely affect our future value.
The clinical development, commercialization and marketing of cell and tissue-based therapies are at an early-stage, substantially research-oriented, and financially speculative. To date, very few companies have been successful in their efforts to develop and commercialize a cell therapy product. In general, cell-based or tissue-based products may be susceptible to various risks, including undesirable and unintended side effects, unintended immune system responses, inadequate therapeutic efficacy, or other characteristics that may prevent or limit their approval or commercial use. In addition, BRTX-100 is a cell-based candidate that is produced by using a patient’s own stem cells derived from bone marrow. Regulatory approval of novel product candidates such as BRTX-100, which is manufactured using novel manufacturing processes, can be more complex and expensive and take longer than other, more well-known or extensively studied pharmaceutical or biopharmaceutical products, due to the FDA’s lack of experience with them. To our knowledge, the FDA has not yet approved a disc related stem cell therapy product. This lack of experience may lengthen the regulatory review process, require us to conduct additional studies or clinical trials, which would increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these product candidates or lead to significant post-approval limitations or restrictions. Furthermore, the number of people who may use cell or tissue-based therapies is difficult to forecast with accuracy. Our future success is dependent on the establishment of a large global market for cell- and tissue-based therapies and our ability to capture a share of this market with our product candidates.
| 54 |
Our cell therapy product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
The enactment of the Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated regulatory pathway for the approval of products demonstrated to be biosimilar, or “highly similar,” to or “interchangeable” with an FDA-approved innovator (original) biologic product. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing reference product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product is approved under a biologics license application, or BLA. Although the FDA has approved several biosimilar products, complex provisions of the law are still being implemented by the FDA and interpreted by the federal courts. As a result, the ultimate impact, implementation, and meaning of the BPCIA are still subject to some uncertainty and FDA actions and court decisions concerning the law could have a material adverse effect on the future commercial prospects for our biological products.
We believe that, if any of our product candidates are approved as a biological product under a BLA, it should qualify for the 12-year period of exclusivity. However, there is a risk that the FDA could approve biosimilar applicants for other reference products that no longer have such exclusivity, thus potentially creating the opportunity for greater competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
The FDA’s regulation of regenerative medicine products remains unpredictable and we are not certain what impact this will have on the potential approval of our products.
The FDA’s regulation of therapies derived from stem cell products and technologies is evolving and may continue to evolve. In December 2016, the 21st Century Cures Act, or the Cures Act, was signed into law in the United States to advance access to medical innovations. Among other things, the Cures Act established a new FDA RMAT designation. This designation offers a variety of benefits to product candidates, including enhanced FDA support during clinical development, priority review on application filing, accelerated approval based on potential surrogate endpoints, and the potential use of patient registry data and other forms of real world evidence for post-approval confirmatory studies. There is no certainty that any of our product candidates will receive RMAT designation or any other type of expedited review program designation from the FDA. In any event, the receipt of an FDA RMAT designation or other expedited review program designation may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA.
| 55 |
We may be subject to significant product liability claims and litigation, including potential exposure from the use of our product candidates in human subjects, and our insurance may be inadequate to cover claims that may arise.
Our business exposes us to potential product liability risks inherent in the testing, processing and marketing of cell therapy products. Such liability claims may be expensive to defend and result in large judgments against us. We face an inherent risk of product liability exposure related to the testing of our current and any future product candidates in human clinical trials and will face an even greater risk with respect to any commercial sales of our products should they be approved. No product candidate has been widely used over an extended period of time, and therefore safety data is limited. Cell therapy companies derive the raw materials for manufacturing of product candidates from human cell sources, and therefore the manufacturing process and handling requirements are extensive, which increases the risk of quality failures and subsequent product liability claims.
We will need to maintain insurance coverage adequate to cover our clinical trials and increase that coverage before commercializing product candidates, if ever. At any time during our clinical trials or after commercialization, if that occurs, we may not be able to obtain or maintain product liability insurance on acceptable terms with adequate coverage or at all, or if claims against us substantially exceed our coverage, then our financial position could be significantly impaired.
Whether or not we are ultimately successful in any product liability litigation that may arise, such litigation could consume substantial amounts of our financial and managerial resources, result in decreased demand for our products and injure our reputation.
We seek to maintain errors and omissions, directors and officers, workers’ compensation and other insurance at levels we believe to be appropriate to our business activities. If, however, we were subject to a claim in excess of this coverage or to a claim not covered by our insurance and the claim succeeded, we would be required to pay the claim from our own limited resources, which could have a material adverse effect on our financial condition, results of operations and business. Additionally, liability or alleged liability could harm our business by diverting the attention and resources of our management and damaging our reputation.
| 56 |
Our internal computer systems, or those that are expected to be used by our clinical investigators, clinical research organizations or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of development programs for our product candidates.
We rely on information technology systems to keep financial records, maintain laboratory and corporate records, communicate with staff and external parties and operate other critical functions. Any significant degradation or failure of these computer systems could cause us to inaccurately calculate or lose data. Despite the implementation of security measures, these internal computer systems and those used by our clinical investigators, clinical research organizations, and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. The techniques that could be used by criminal elements or foreign governments to attack these computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. While we have not experienced any such system failure, theft of information, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our clinical development activities. For example, the loss of clinical trial data from historical or future clinical trials could result in delays in regulatory approval efforts and significantly increase costs to recover or reproduce the data. To the extent that any disruption, theft of information, or security breach were to result in a loss of or damage to data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the clinical development and the future development of our product candidates could be delayed.
To operate and sell in international markets carries great risk.
We intend to market our products and services both domestically and in foreign markets. A number of risks are inherent in international transactions. In order for us to market our products and services in non-U.S. jurisdictions, we need to obtain and maintain required regulatory approvals or clearances in these countries and must comply with the country specific regulations regarding safety, manufacturing processes and quality. These regulations, including the requirements for approvals or clearances to market, may differ from the FDA regulatory scheme. International operations and sales also may be limited or disrupted by political instability, price controls, trade restrictions and changes in tariffs. Additionally, fluctuations in currency exchange rates may adversely affect demand for our services and products by increasing the price of our products and services in the currency of the countries in which the products and services are offered.
There can be no assurance that we will obtain regulatory approvals or clearances in all of the countries where we intend to market our products and services, or that we will not incur significant costs in obtaining or maintaining foreign regulatory approvals or clearances, or that we will be able to successfully commercialize our products and services in various foreign markets. Delays in receipt of approvals or clearances to market our products and services in foreign countries, failure to receive such approvals or clearances or the future loss of previously received approvals or clearances could have a substantial negative effect on our results of operations and financial condition.
Our inability to obtain reimbursement for our products and services from private and governmental insurers could negatively impact demand for our products and services.
Market acceptance and sales of our product candidates may depend on coverage and reimbursement policies and health care reform measures. Decisions about formulary coverage as well as levels at which government authorities and third-party payors, such as private health insurers and health maintenance organizations, reimburse patients for the price they pay for our product candidates, as well as levels at which these payors pay directly for our product candidates, where applicable, could affect whether we are able to successfully commercialize these products. We cannot guarantee that reimbursement will be available for any of our product candidates. We also cannot guarantee that coverage or reimbursement amounts will not reduce the demand for, or the price of, our product candidates.
| 57 |
If coverage and reimbursement are not available or are available only at limited levels, we may not be able to successfully commercialize our products. The Patient Protection and Affordable Care Act, or PPACA, and other health reform proposals include measures that would limit or prohibit payments for certain medical treatments or subject the pricing of drugs to government control. In addition, in many foreign countries, particularly the countries of the European Union, or the EU, the pricing of drugs and biologics is subject to government control. If our products are or become subject to government regulation that limits or prohibits payment for our products, or that subjects the price of our products to government control, we may not be able to generate revenue, attain profitability or commercialize our products.
In addition, third-party payors are increasingly limiting both coverage and the level of reimbursement of new drugs and biologics. They may also impose strict prior authorization requirements and/or refuse to provide any coverage of uses of approved products for medical indications other than those for which the FDA has granted market approvals. As a result, significant uncertainty exists as to whether and how much third-party payors will reimburse patients for their use of newly-approved drugs and biologics. If we are unable to obtain adequate levels of reimbursement for our product candidates, our ability to successfully market and sell our product candidates will be harmed.
Risks Related to Our Intellectual Property
We may not be able to protect our proprietary rights.
Our commercial success will depend in large part upon our ability to protect our proprietary rights. There is no assurance, for example, that any additional patents will be issued based on our or our licensor’s pending applications or, if issued, that such patents will not become the subject of a re-examination, will provide us with competitive advantages, will not be challenged by any third parties, or that the patents of others will not prevent the commercialization of products and services incorporating our technology. Furthermore, there can be no guarantee that others will not independently develop similar products and services, duplicate any of our products and services, or design around any patents we obtain.
Our commercial success will also depend upon our ability to avoid infringing patents issued to others. If we were judicially determined to be infringing on any third-party patent, we could be required to pay damages, alter our products, services or processes, obtain licenses, or cease certain activities. If we are required in the future to obtain any licenses from third parties for some of our products and/or services, there can be no guarantee that we would be able to do so on commercially favorable terms, if at all. United States and foreign patent applications are not immediately made public, so we might be surprised by the grant to someone else of a patent on a technology we are actively using. Although we conducted a freedom to operate, or FTO, search on the licensed technology associated with our Disc/Spine Program, modifications made, and/or further developments that may be made, to that technology may not be covered by the initial FTO. No FTO has been undertaken with respect to our ThermoStem brown fat initiative.
| 58 |
Litigation, which would result in substantial costs to us and the diversion of effort on our part, may be necessary to enforce or confirm the ownership of any patents issued or licensed to us, or to determine the scope and validity of third-party proprietary rights. If our competitors claim technology also claimed by us and prepare and file patent applications in the United States, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office, or the Patent Office, or a foreign patent office to determine priority of invention, which could result in substantial costs and diversion of effort, even if the eventual outcome is favorable to us. Any such litigation or interference proceeding, regardless of outcome, could be expensive and time-consuming.
Successful challenges to our patents through oppositions, re-examination proceedings or interference proceedings could result in a loss of patent rights in the relevant jurisdiction. If we are unsuccessful in actions we bring against the patents of other parties, and it is determined that we infringe upon the patents of third parties, we may be subject to litigation, or otherwise prevented from commercializing potential products and/or services in the relevant jurisdiction, or may be required to obtain licenses to those patents or develop or obtain alternative technologies, any of which could harm our business. Furthermore, if such challenges to our patent rights are not resolved in our favor, we could be delayed or prevented from entering into new collaborations or from commercializing certain products and/or services, which could adversely affect our business and results of operations.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential or sensitive information could be compromised by disclosure in the event of litigation. In addition, during the course of litigation there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
In addition to patents, we intend to also rely on unpatented trade secrets and proprietary technological expertise. Some of our intended future cell-related therapeutic products and/or services may fit into this category. We intend to rely, in part, on confidentiality agreements with our partners, employees, advisors, vendors, and consultants to protect our trade secrets and proprietary technological expertise. There can be no guarantee that these agreements will not be breached, or that we will have adequate remedies for any breach, or that our unpatented trade secrets and proprietary technological expertise will not otherwise become known or be independently discovered by competitors.
Failure to obtain or maintain patent protection, failure to protect trade secrets, third-party claims against our patents, trade secrets, or proprietary rights or our involvement in disputes over our patents, trade secrets, or proprietary rights, including involvement in litigation, could divert our efforts and attention from other aspects of our business and have a substantial negative effect on our results of operations and financial condition.
We may not be able to protect our intellectual property in countries outside of the United States.
Intellectual property law outside the United States is uncertain and, in many countries, is currently undergoing review and revisions. The laws of some countries do not protect our patent and other intellectual property rights to the same extent as United States laws. Third parties may attempt to oppose the issuance of patents to us in foreign countries by initiating opposition proceedings. Opposition proceedings against any of our patent filings in a foreign country could have an adverse effect on our corresponding patents that are issued or pending in the United States. It may be necessary or useful for us to participate in proceedings to determine the validity of our patents or our competitors’ patents that have been issued in countries other than the United States. This could result in substantial costs, divert our efforts and attention from other aspects of our business, and could have a material adverse effect on our results of operations and financial condition.
| 59 |
Changes to United States patent law may have a material adverse effect on our intellectual property rights.
The Leahy-Smith America Invents Act, or AIA, which was signed into law in 2011, significantly changes United States patent law. It may take some time to establish what the law means, since it is just being interpreted by the lower courts, Federal Circuit Courts of Appeal, and the Supreme Court. The effects of these decisions are still not known. The first major change is that AIA switches the United States patent system from a “first to invent” system to a “first to file” system. Now that the first to file system is in effect, there is a risk that another company may independently develop identical or similar patents at approximately the same time, and be awarded the patents instead of us. Further, for the second major change, AIA abolished interference proceedings, and establishes derivation proceedings to replace interference proceedings in all cases in which the time period for instituting an interference proceeding has not lapsed where an inventor named in an earlier application derived the claimed invention from a named inventor. Now that the derivation proceedings are in effect, there is a risk that the inventorship of any pending patent application can be challenged for reasons of derivation. The third major change is that AIA established post-grant opposition proceedings that will apply only to patent applications filed after “first to file” became effective. Post-grant opposition will enable a person who is not the patent owner to initiate proceedings in the Patent Office within nine months after the grant of a patent that can result in cancellation of a patent as invalid. In addition to AIA, recent court decisions have created uncertainty with regard to our ability to obtain and maintain patents. Therefore there is a risk that any of our patents once granted may be subject to post-grant opposition, which will increase uncertainty on the validity of any newly granted patent or may ultimately result in cancellation of the patent.
In addition, the Supreme Court has recently taken more limiting positions as to what constitutes patentable subject matter. As a result, many patents covering what were previously patentable inventions are now determined to cover inventions which are deemed non-statutory subject matter and are now invalid. As a result of this and subsequent opinions by the Court of Appeals for the Federal Circuit, the Patent Office is now applying more stringent limitations to claims in patent applications and is refusing to grant patents in areas of technology where patents were previously deemed available. Therefore there is a risk that we will be unable to acquire patents to cover our products and if such patents are granted they may subsequently be found to be invalid.
In certain countries, patent holders may be required to grant compulsory licenses, which would likely have a significant and detrimental effect on any future revenues in such country.
Many countries, including some countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, most countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may be limited to monetary relief and may be unable to enjoin infringement, which could materially diminish the value of the patent. Compulsory licensing of life-saving products is also becoming increasingly common in developing countries, either through direct legislation or international initiatives. Such compulsory licenses could be extended to our product candidates, which may limit our potential revenue opportunities, including with respect to any future revenues that may result from our product candidates.
| 60 |
Risks Related to Government Regulation
Even if we obtain regulatory approval for a product candidate, our products will remain subject to regulatory oversight.
Our product candidates for which we obtain regulatory approval will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information. Any regulatory approvals that we receive for our product candidates also may be subject to a REMS or the specific obligations imposed as a condition for marketing authorization by equivalent authorities in a foreign jurisdiction, limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the quality, safety and efficacy of the product. For example, in the United States, the holder of an approved NDA or BLA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the NDA or BLA. The holder of an approved NDA or BLA also must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Advertising and promotional materials must comply with the FDCA and implementing regulations and are subject to FDA oversight and post-marketing reporting obligations, in addition to other potentially applicable federal and state laws.
In addition, product manufacturers and their facilities may be subject to payment of application and program fees and are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP requirements and adherence to commitments made in the NDA, BLA or foreign marketing application. If we or a regulatory authority discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or if a regulatory authority disagrees with the promotion, marketing or labeling of our product, a regulatory authority may impose restrictions relative to that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements for any product candidate following approval, a regulatory authority may:
| ● | issue a warning or untitled letter asserting that we are in violation of the law; | |
| ● | seek an injunction or impose administrative, civil or criminal penalties or monetary fines; | |
| ● | suspend or withdraw regulatory approval; | |
| ● | suspend any ongoing clinical trials; | |
| ● | refuse to approve a pending BLA or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners; | |
| ● | restrict the marketing or manufacturing of the product; |
| 61 |
| ● | seize or detain the product or otherwise demand or require the withdrawal or recall of the product from the market; | |
| ● | refuse to permit the import or export of products; | |
| ● | request and publicize a voluntary recall of the product; or | |
| ● | refuse to allow us to enter into supply contracts, including government contracts. |
Any government enforcement action or investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and adversely affect our business, financial condition, results of operations and prospects.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
In the United States, the research, manufacturing, distribution, sale, and promotion of drugs and biologic products are subject to regulation by various federal, state, and local authorities, including the FDA, CMS, other divisions HHS (e.g., the Office of Inspector General), the United States Department of Justice offices of the United States Attorney, the Federal Trade Commission and state and local governments. Our operations are directly, or indirectly through our prescribers, customers and purchasers, subject to various federal and state fraud and abuse laws and regulations, including the federal AKS, the federal civil and criminal FCA, the Physician Payments Sunshine Act and regulations and equivalent provisions in other countries. In addition, we may be subject to patient privacy laws by both the federal government and the states in which we conduct our business.
State and federal regulatory and enforcement agencies continue actively to investigate violations of health care laws and regulations, and the United States Congress continues to strengthen the arsenal of enforcement tools. Most recently, the Bipartisan Budget Act of 2018 increased the criminal and civil penalties that can be imposed for violating certain federal health care laws, including the AKS. Enforcement agencies also continue to pursue novel theories of liability under these laws. Government agencies have recently increased regulatory scrutiny and enforcement activity with respect to programs supported or sponsored by pharmaceutical companies, including reimbursement and co-pay support, funding of independent charitable foundations and other programs that offer benefits for patients. Several investigations into these programs have resulted in significant civil and criminal settlements.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the laws described above or any other government regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which could harm our financial condition and divert the attention of our management from operating our business.
| 62 |
Further, in the event we determine to operate in foreign jurisdictions, including conducting clinical trials, we will need to comply with the United States Foreign Corrupt Practices Act of 1977, or FCPA. The FCPA prohibits a corporation, including its subsidiaries, third-party contractors, distributors, consultants and employees, from corruptly making or offering to make payments to foreign officials for the purpose of obtaining or enhancing business. Under the law, “foreign officials” include employees of health systems operated by government entities. The FCPA also establishes specific record-keeping and internal accounting controls. Violations of the FCPA can result in the imposition of civil penalties or criminal prosecution. Failure to comply with the FCPA will adversely affect our business.
In addition to the FCPA, we will also need to comply with the foreign government laws and regulations of each individual country in which any therapy centers that we may establish are located and products are to be distributed and sold. These regulations vary in complexity and can be as stringent, and on occasion even more stringent, than FDA regulations in the United States. Due to the fact that there are new and emerging stem cell and cell therapy regulations that have recently been drafted and/or implemented in various countries around the world, the application and subsequent implementation of these new and emerging regulations have little to no precedence. Therefore, the level of complexity and stringency is not always precisely understood today for each country, creating greater uncertainty for the international regulatory process. Furthermore, there can be no guarantee that laws and regulations will not be implemented, amended and/or reinterpreted in a way that will negatively affect our business. Likewise, there can be no assurance that we will be able, or will have the resources, to maintain compliance with all such healthcare laws and regulations. Failure to comply with such healthcare laws and regulations, as well as the costs associated with such compliance or with enforcement of such healthcare laws and regulations, may have a material adverse effect on our operations or may require restructuring of our operations or impair our ability to operate profitably.
Our employees, principal investigators, medical institutions, consultants, advisors and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, medical institutions, consultants, advisors and commercial partners, including contract laboratories, and CROs. Misconduct by these parties could include intentional failures to comply with FDA regulations or the regulations applicable in other jurisdictions, provide accurate information to the FDA and other regulatory authorities, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us.
We may not independently conduct all aspects of our product candidate research and preclinical and clinical testing and product candidate manufacturing. If we rely on third parties, including CROs, medical institutions, and contract laboratories to monitor and manage data for our ongoing preclinical and clinical programs, we will still maintain responsibility for ensuring their activities are conducted in accordance with the applicable study protocol, legal, regulatory and scientific standards. We and our third-party vendors will be required to comply with current cGMP, GCP, and GLP requirements, which are a collection of laws and regulations enforced by the FDA, the EU and comparable foreign authorities for all of our product candidates in clinical development.
| 63 |
In addition, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct also could involve the improper use of information obtained in the course of clinical trials or interactions with the FDA or other regulatory authorities, which could result in regulatory sanctions and cause serious harm to our reputation.
The precautions we take to detect and prevent employee and third-party misconduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from government investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, financial condition, results of operations and prospects, including the imposition of significant fines or other sanctions.
The failure to receive regulatory approvals for our cell therapy product candidates would likely have a material and adverse effect on our business and prospects.
To date, we have not received regulatory approval to market any of our product candidates in any jurisdiction. If we seek approval of any of our cell therapy product candidates, we will be required to submit to the FDA and potentially other regulatory authorities extensive pre-clinical and clinical data supporting its safety and efficacy, as well as information about the manufacturing process and to undergo inspection of our manufacturing facility or other contract manufacturing facilities, among other things. The process of obtaining FDA and other regulatory approvals is expensive, generally takes many years and is subject to numerous risks and uncertainties, particularly with complex and/or novel product candidates such as our cell-based product candidates. Changes in regulatory approval requirements or policies may cause delays in the approval or rejection of an application or may make it easier for our competitors to gain regulatory approval to enter the marketplace. Ultimately, the FDA and other regulatory agencies have substantial discretion in the approval process and may refuse to accept any application or may decide that our product candidate data are insufficient for approval without the submission of additional preclinical, clinical or other studies. In addition, varying agency interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate. Any difficulties or failures that we encounter in securing regulatory approval for our product candidates would likely have a substantial adverse impact on our ability to generate product sales, and could make any search for a collaborative partner more difficult. Similarly, any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
| 64 |
If we are unable to conduct clinical studies in accordance with regulations and accepted standards, we may be delayed in receiving, or may never receive, regulatory approvals of our product candidates from the FDA and other regulatory authorities.
To obtain marketing approvals for our product candidates in the United States and abroad, we must, among other requirements, complete adequate and well-controlled clinical trials sufficient to demonstrate to the FDA and other regulatory bodies that the product candidate is safe and effective for each indication for which approval is sought. If the FDA finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury, due to, among other things, occurrence of a serious adverse event in an ongoing clinical trial, the FDA can place one or more of our clinical trials on hold. If safety concerns develop, we may, or the FDA or an institutional review board may require us to, stop the affected trials before completion.
The completion of our clinical trials also may be delayed or terminated for a number of other reasons, including if:
| ● | third-party clinical investigators do not perform the clinical trials on the anticipated schedule or consistent with the clinical trial protocol, good clinical practices required by the FDA and other regulatory requirements, or other third parties do not perform data collection and analysis in a timely or accurate manner; | |
| ● | inspections of clinical trial sites by the FDA or other regulatory authorities reveal violations that require us to undertake corrective action, suspend or terminate one or more sites, or prohibit use of some or all of the data in support of marketing applications; or | |
| ● | the FDA or one or more institutional review boards suspends or terminates the trial at an investigational site, or precludes enrollment of additional subjects. |
Our development costs will increase if there are material delays in our clinical trials, or if we are required to modify, suspend, terminate or repeat a clinical trial. If we are unable to conduct our clinical trials properly, we may never receive regulatory approval to market our product candidates.
Health care companies have been the subjects of federal and state investigations, and we could become subject to investigations in the future.
Both federal and state government agencies have heightened civil and criminal enforcement efforts. There are numerous ongoing investigations of health care companies, as well as their executives and managers. In addition, amendments to the federal FCA, including under healthcare reform legislation, have made it easier for private parties to bring “qui tam” (or whistleblower) lawsuits against companies under which the whistleblower may be entitled to receive a percentage of any money paid to the government. The FCA provides, in part, that an action can be brought against any person or entity that has knowingly presented, or caused to be presented, a false or fraudulent request for payment from the federal government, or who has made a false statement or used a false record to get a claim approved. The government has taken the position that claims presented in violation of the federal AKS, Stark Law or other healthcare-related laws, including laws enforced by the FDA, may be considered a violation of the FCA. Penalties include substantial fines for each false claim, plus three times the amount of damages that the federal government sustained because of the act of that person or entity and/or exclusion from the Medicare program. In addition, a majority of states have adopted similar state whistleblower and false claims provisions.
| 65 |
We are not aware of any government investigations involving any of our facilities or management. While we believe that we are in compliance with applicable governmental healthcare laws and regulations, any future investigations of our business or executives could cause us to incur substantial costs, and result in significant liabilities or penalties, as well as damage to our reputation.
It is uncertain to what extent the government, private health insurers and third-party payors will approve coverage or provide reimbursement for the therapies and products to which our services relate. Availability for such reimbursement may be further limited by reductions in Medicare, Medicaid and other federal healthcare program funding in the United States.
To the extent that health care providers cannot obtain coverage or reimbursement for our products and therapies, they may elect not to provide such products and therapies to their patients and, thus, may not need our services. Further, as cost containment pressures are increasing in the health care industry, government and private payors may adopt strategies designed to limit the amount of reimbursement paid to health care providers.
Similarly, the trend toward managed health care and bundled pricing for health care services in the United States, could significantly influence the purchase of healthcare products and services, resulting in lower prices and reduced demand for our therapeutic products under development.
We may directly or indirectly receive revenues from federal health care programs, such as Medicare. Federal health care programs are subject to changes in coverage and reimbursement rules and procedures, including retroactive rate adjustments. These contingencies could materially decrease the range of services covered by such programs or the reimbursement rates paid directly or indirectly for our products and services. To the extent that any health care reform favors the reimbursement of other therapies over our therapeutic products under development, such reform could affect our ability to sell our services, which may have a material adverse effect on our revenues.
The limitation on reimbursement available from private and government payors may reduce the demand for, or the price of, our products and services, which could have a material adverse effect on our revenues. Additional legislation or regulation relating to the health care industry or third-party coverage and reimbursement may be enacted in the future which could adversely affect the revenues generated from the sale of our products and services.
Furthermore, there has been a trend in recent years towards reductions in overall funding for Medicare, Medicaid and other federal health care programs. There has also been an increase in the number of people who are not eligible for or enrolled in Medicare, Medicaid or other governmental programs. The reduced funding of governmental programs could have a negative impact on the demand for our services to the extent it relates to products and services which are reimbursed by government and private payors.
| 66 |
Unintended consequences of healthcare reform in the United States may adversely affect our business.
The healthcare industry is undergoing fundamental changes resulting from political, economic and regulatory influences. In the United States, the PPACA was signed into law in 2010 under the Obama administration. By implementing comprehensive reforms, the law seeks to, among other things, increase access to healthcare for the uninsured and control the escalation of healthcare expenditures within the economy. While we do not believe this law will have a direct impact on our business, the law requires the adoption of various implementing regulations, which may have unintended consequences or indirectly impact our business.
In addition, other legislative changes have been adopted since the PPACA was enacted. These changes include aggregate reductions in Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, following passage of the Bipartisan Budget Act of 2018, will remain in effect through 2027 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These laws may result in additional reductions in Medicare and other health care funding, which could impact our business.
Healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and decreased reimbursement. Under the Trump administration, Congress has passed certain legislation to alter the PPACA. In addition, Congress and select states have proposed legislation to alter and/or repeal the PPACA and/or transform certain aspects of existing federal and state health programs. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates. It is difficult to predict how enforcement initiatives under the PPACA and/or additional legislation or regulation enacted in the future may impact our business. If the PPACA and/or additional legislation or regulation enacted in the future cause such unintended consequences or indirect impact, they could have a material adverse effect on our business, financial condition and results of operations.
Competitor companies or hospitals in the European Union, or EU, may be able to take advantage of EU rules permitting sales of unlicensed medicines for individual patients to sell competing products without a marketing authorization.
The EU medicines rules allow individual member states to permit the supply of a medicinal product without a marketing authorization to fulfill special needs, where the product is supplied in response to a bona fide unsolicited order, formulated in accordance with the specifications of a healthcare professional and for use by an individual patient under his direct personal responsibility. This may, in certain countries, also apply to products manufactured in a country outside the EU and imported to treat specific patients or small groups of patients. In addition, advanced therapy medicinal products do not need a marketing authorization if they are prepared on a non-routine basis and are used within the same EU member state in a hospital in accordance with a medical prescription for an individual patient.
These exemptions could allow our competitors to make sales in the EU without having obtained a marketing authorization and without undergoing the expense of clinical trials, especially if those competitors have cell processing facilities in the relevant EU member state. Similarly, certain hospitals may be able to compete with us on the basis of these rules.
| 67 |
Risks Related to Our Common Stock
We pay no dividends.
We have never paid cash dividends in the past, and currently do not intend to pay any cash dividends in the foreseeable future.
There is at present only a limited market for our common stock and there is no assurance that an active trading market for our common stock will develop.
Although our common stock is quoted on the OTCQB market from time to time, the market for our common stock is extremely limited. Trading prices and volumes on the OTCQB market are thin and erratic. We cannot predict at what price our shares will trade and there can be no assurance that an active market for our shares will develop or, if developed, will be sustained. The volume traded at any one time can be limited, and as a result, there may not be a liquid trading market for our shares. In addition, although there have been market makers in our shares, we cannot assure that these market makers will continue to make a market in our shares or that other factors outside of our control will not cause them to stop market making in our shares. Making a market in shares involves maintaining bid and ask quotations and being able to effect transactions in reasonable quantities at those quoted prices, subject to various securities laws and other regulatory requirements. Furthermore, the development and maintenance of a public trading market depends upon the existence of willing buyers and sellers, the presence of which is not within our control or that of any market maker. Market makers are not required to maintain a continuous two-sided market, are required to honor firm quotations for only a limited number of shares, and are free to withdraw firm quotations at any time. Even with a market maker, factors such as our past losses from operations and the small size of our company mean that there can be no assurance of an active and liquid market for our shares developing in the foreseeable future. Even if a market develops, we cannot assure that a market will continue, or that stockholders will be able to resell their shares at any price.
Our common stock is classified as a “penny stock”; the restrictions of the penny stock regulations of the Securities and Exchange Commission, or SEC, may result in less liquidity for our common stock.
The SEC has adopted regulations which define a “penny stock” to be any equity security that has a market price (as therein defined) of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exceptions. Based upon the last reported sale price of our common stock on the OTCQB market on March 27, 2019, as of such date, our common stock was a “penny stock”. For any transactions involving a penny stock, unless exempt, the rules require the delivery, prior to any transaction involving a penny stock by a retail customer, of a disclosure schedule prepared by the SEC relating to the penny stock market. Disclosure is also required to be made about commissions payable to both the broker/dealer and the registered representative and current quotations for the securities. Finally, monthly statements are required to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks. If the market price for shares of our common stock remains below $5.00, and we do not satisfy any of the exceptions to the SEC’s definition of penny stock, our common stock will continue to be classified as a penny stock. If such classification should remain in place, as a result of the penny stock restrictions, brokers or potential investors may be reluctant to trade in our securities, which may result in less liquidity for our common stock.
| 68 |
Because state securities laws may limit secondary trading, stockholders may be restricted as to the states in which they can sell their shares.
Because state securities laws may limit secondary trading, stockholders may be restricted as to the states in which they can sell their shares. Stockholders may not be able to resell them in any state unless and until the shares are qualified for secondary trading under the applicable securities laws of such state or there is confirmation that an exemption, such as listing in certain recognized securities manuals, is available for secondary trading in such state. There can be no assurance that we will be successful in registering or qualifying our shares for secondary trading, or identifying an available exemption for secondary trading in such shares in every state. If we fail to register or qualify, or to obtain or verify an exemption for the secondary trading of, our shares in any particular state, the shares could not be offered or sold to, or purchased by, a resident of that state. In the event that a significant number of states refuse to permit secondary trading in our shares, the market for the shares will be limited, which could drive down the market price of the shares and reduce the liquidity of the shares and a stockholder’s ability to resell the shares at all or at current market prices.
Stockholders who hold unregistered shares of our common stock are subject to resale restrictions pursuant to Rule 144 due to our former status as a “shell company”.
We previously were a “shell company” pursuant to Rule 144, promulgated under the Securities Act of 1933, as amended, or Rule 144, and, as such, sales of our securities pursuant to Rule 144 cannot be made unless, among other things, we continue to remain subject to Section 13 or 15(d) of the Securities Exchange Act of 1934, or the Exchange Act, and we file all of our required periodic reports with the SEC under the Exchange Act. Because our unregistered securities cannot be sold pursuant to Rule 144 unless we continue to meet such requirements, any unregistered securities we sell in the future or issue to consultants or employees, in consideration for services rendered or for any other purpose, will have no liquidity unless we continue to comply with such requirements. As a result, it may be more difficult for us to obtain financing to fund our operations and pay our consultants and employees with our securities instead of cash.
We have incurred, and will continue to incur, increased costs as a result of being an SEC reporting company.
The Sarbanes-Oxley Act of 2002, as well as a variety of related rules implemented by the SEC, have required changes in corporate governance practices and generally increased the disclosure requirements of public companies. As a reporting company, we incur significant legal, accounting and other expenses in connection with our public disclosure and other obligations. Based upon SEC regulations currently in effect, we are required to establish, evaluate and report on our internal control over financial reporting. We believe that compliance with the myriad of rules and regulations applicable to reporting companies and related compliance issues will require a significant amount of time and attention from our management.
| 69 |
Our stock price may fluctuate significantly and be highly volatile and this may make it difficult for a stockholder to resell shares of our common stock at the volume, prices and times the stockholder finds attractive.
The market price of our common stock may be subject to significant fluctuations and be highly volatile, which may make it difficult for a stockholder to resell shares of our common stock at the volume, prices and times the stockholder finds attractive. There are many factors that will impact our stock price and trading volume, including, but not limited to, the factors listed above under “Risks Related to Our Business Generally”, “Risks Related to Our Cell Therapy Product Development Efforts”, “Risks Related to Our Intellectual Property”, “Risks Related to Government Regulation”, and “Risks Related to Our Common Stock.”
Stock markets, in general, experience significant price and volume volatility, and the market price of our common stock may continue to be subject to such market fluctuations that may be unrelated to our operating performance and prospects. Increased market volatility and fluctuations could result in a substantial decline in the market price of our common stock.
There may be future issuances or resales of our common stock which may materially and adversely dilute stockholders’ ownership interest and affect the market price of our common stock.
We are not restricted from issuing additional shares of our common stock in the future, including securities convertible into, or exchangeable or exercisable for, shares of our common stock. Our issuance of additional shares of common stock in the future will dilute the ownership interests of our then existing stockholders.
We have effective registration statements on Form S-8 under the Securities Act registering an aggregate of 10,000,000 shares of our common stock issuable under our 2010 Equity Participation Plan, or the Plan. As of March 20, 2019, options to purchase 4,750,868 shares of our common stock were outstanding under the Plan. In addition, as of such date, 45,000 shares of common stock were issued as restricted stock pursuant to the Plan and 5,204,132 shares were reserved for future grants under the Plan. The shares issuable pursuant to the registration statements on Form S-8 will be freely tradable in the public market, except for shares held by affiliates.
The sale of a substantial number of shares of our common stock or securities convertible into, or exchangeable or exercisable for, shares of our common stock, whether directly by us in future offerings or by our existing stockholders in the secondary market, the perception that such issuances or resales could occur or the availability for future issuances or resale of shares of our common stock or securities convertible into, or exchangeable or exercisable for, shares of our common stock could materially and adversely affect the market price of our common stock and our ability to raise capital through future offerings of equity or equity-related securities on attractive terms or at all.
In addition, our Board of Directors is authorized to designate and issue preferred stock without further stockholder approval, and we may issue other equity and equity-related securities that are senior to our common stock in the future for a number of reasons, including, without limitation, to support operations and growth, and to comply with any future changes in regulatory standards.
| 70 |
Our principal stockholders currently own a substantial number of shares of our common stock and have the power to significantly influence the vote on all matters submitted to a vote of our stockholders.
As of March 19, 2019, Dale Broadrick beneficially owned 3,161,452 shares of our common stock (including 1,000,000 shares of our common stock issuable pursuant to a currently exercisable warrant), representing 20.2% of the outstanding shares of our common stock. In addition, as of March 19, 2019, John M. Desmarais, one of our directors, beneficially owned 2,029,574 shares of our common stock (including 1,536,176 shares of our common stock issuable pursuant to currently exercisable options and warrants), representing 12.6% of the outstanding shares of our common stock. Further, as of March 19, 2019, SCG Capital, LLC, or SCG, and Steven Geduld beneficially owned 1,537,817 shares of common stock (including 768,060 shares of our common stock issuable pursuant to currently exercisable warrants and a currently convertible note), representing 9.99% of the outstanding shares of our common stock. Moreover, as of March 19, 2019, Westbury (Bermuda), Ltd., or Westbury, beneficially owned 1,151,661 shares of our common stock (including 199,182 shares of our common stock issuable pursuant to currently exercisable warrants), representing 7.8% of the outstanding shares of our common stock.
Mr. Broadrick, Mr. Desmarais, SCG/Mr. Geduld and Westbury, through their beneficial ownership of our common stock, have the power to significantly influence the vote on all matters submitted to a vote of our stockholders, including the election of directors, amendments to our certificate of incorporation or bylaws, mergers or other business combination transactions and certain sales of assets outside the usual and regular course of business. The interests of Mr. Broadrick, Mr. Desmarais, SCG/Steven Geduld and Westbury may not coincide with the interests of our other stockholders, and they could take actions that advance their own interests to the detriment of our other stockholders.
Anti-takeover provisions and the regulations to which we may be subject may make it more difficult for a third party to acquire control of us, even if the change in control would be beneficial to our stockholders.
We are incorporated in Delaware. Anti-takeover provisions in Delaware law and our certificate of incorporation and bylaws could make it more difficult for a third party to acquire control of us and may prevent stockholders from receiving a premium for their shares of common stock. Our certificate of incorporation provides that our Board of Directors may issue up to 20,000,000 shares of preferred stock, in one or more series, without stockholder approval and with such terms, preferences, rights and privileges as the Board of Directors may deem appropriate. These provisions and other factors may hinder or prevent a change in control, even if the change in control would be beneficial to, or sought by, our stockholders.
In the event that a significant amount of our outstanding debt is converted into equity, the percentage ownership of existing stockholders will be substantially diluted.
As of March 19, 2019, we had outstanding indebtedness in the amount of $6,375,537. All or a significant amount of such debt may be converted into equity. In the event of any such conversion, the percentage ownership of existing stockholders will be substantially diluted.
| 71 |
We are an emerging growth company, and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an emerging growth company, as defined in The Jumpstart Our Business Startups Act, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in this 10-K and our periodic reports and proxy statements and exemptions from the requirements of holding nonbinding advisory votes on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could remain as an emerging growth company until December 31, 2020, although circumstances could cause us to lose that status earlier.
Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company” which would allow us to take advantage of many of the same exemptions from disclosure requirements including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation. We cannot predict whether investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and warrants and the prices for our securities may be more volatile.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
Not applicable.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
The financial statements required by this Item 8 are included in this Annual Report following Item 16 (“Form 10-K Summary”). As a smaller reporting company, we are not required to provide supplementary financial information.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES. |
Disclosure Controls and Procedures
Disclosure controls are procedures that are designed with the objective of ensuring that information required to be disclosed in our reports filed under the Exchange Act, such as this Annual Report, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls are also designed with the objective of ensuring that such information is accumulated and communicated to our management, including the Principal Executive and Financial Officer, as appropriate to allow timely decisions regarding required disclosure. Internal controls are procedures which are designed with the objective of providing reasonable assurance that (1) our transactions are properly authorized, recorded and reported; and (2) our assets are safeguarded against unauthorized or improper use, to permit the preparation of our consolidated financial statements in conformity with United States generally accepted accounting principles.
| 72 |
In connection with the preparation of this Annual Report, management, with the participation of our Principal Executive and Financial Officer, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e) and 15d-15(e)). Based upon that evaluation, our Principal Executive and Financial Officer concluded that, as of December 31, 2018, our disclosure controls and procedures were effective.
Internal Control over Financial Reporting
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Exchange Act. Internal control over financial reporting is a process designed by, or under the supervision of, our Principal Executive and Financial Officer, and effected by the Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP including those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and the disposition of our assets, (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP and that receipts and expenditures are being made only in accordance with authorizations of our management and Board of Directors, and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies and procedures may deteriorate.
Management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the 2013 framework in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2018.
Changes in Internal Controls
There were no changes in our internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act) during the quarter ended December 31, 2018 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 73 |
Limitations of the Effectiveness of Control
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations of any control system, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected.
No Attestation Report of Registered Public Accounting Firm
This Annual Report does not contain an attestation report of our independent registered public accounting firm regarding internal control over financial reporting since the rules for smaller reporting companies provide for this exemption.
| ITEM 9B. | OTHER INFORMATION. |
None.
| 74 |
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
Directors and Executive Officers
Information regarding our directors and executive officers is set forth below. Each of our officers devotes his or her full business time in providing services on our behalf.
| Name | Age | Positions Held | ||
| Mark Weinreb | 66 | Chief Executive Officer, President and Chairman of the Board | ||
| Lance Alstodt | 47 | Executive Vice President and Chief Strategy Officer | ||
| Francisco Silva | 44 | Vice President of Research and Development | ||
| Robert Paccasassi | 50 | Vice President of Quality and Compliance | ||
| Mandy D. Clyde | 37 | Vice President of Operations and Secretary | ||
| Robert B. Catell | 82 | Director | ||
| John M. Desmarais | 55 | Director | ||
| A. Jeffrey Radov | 67 | Director | ||
| Charles S. Ryan | 54 | Director | ||
| Paul Jude Tonna | 60 | Director |
Mark Weinreb
Mark Weinreb has served as our Chief Executive Officer since October 2010, as our President since February 2012 and as our Chairman of the Board since April 2011. From February 2003 to January 2006, he served as the Chief Executive Officer and Chairman of the Board of Directors of Phase III Medical, Inc. where he orchestrated the acquisition of an adult stem cell collection company that became NeoStem, Inc. (now Caladrius Biosciences, Inc. - NASDAQ: CLBS), a public international biopharmaceutical company engaged in, among other things, adult stem cell-related science. From June 2006 through October 2009, Mr. Weinreb served as President and a director of NeoStem. In 1976, Mr. Weinreb joined Bio Health Laboratories, Inc., a state-of-the-art medical diagnostic laboratory providing clinical testing services for physicians, hospitals, and other medical laboratories. He became the laboratory administrator in 1978 and then an owner and the laboratory’s Chief Operating Officer in 1982. In such capacity, he oversaw all technical and business facets, including finance and laboratory science technology. In 1989, Bio Health Laboratories was acquired by Enzo Biochem (NYSE: ENZ). From 1992 to 2002, Mr. Weinreb pursued entrepreneurial and consulting interests. Mr. Weinreb received a Bachelor of Arts degree from Northwestern University and a Master of Science degree in Medical Biology from C.W. Post, Long Island University. We believe that Mr. Weinreb’s executive-level management experience, his extensive experience in the adult stem cell sector and his service on our Board since October 2010 give him the qualifications and skills to serve as one of our directors.
| 75 |
Lance Alstodt
Lance Alstodt has served as our Executive Vice President and Chief Strategy Officer since October 2018. From 2013 until 2018, Mr. Alstodt served as Chief Executive Officer of MedVest Consulting Corporation, an advisory and capital firm that focused exclusively on the healthcare industry. Prior to MedVest, he was an investment banker with over 20 years of experience with respect to healthcare investment banking, including mergers and acquisitions. From 2011 to 2013, Mr. Alstodt was a Managing Director at Lerrink Partners where he helped lead its medical technology sector. From 2009 to 2011, he was a Managing Director and Head of Medical Technology at Oppenheimer & Co. From 2000 to 2009, Mr. Alstodt was a Managing Director in the Healthcare Group and Global Mergers and Acquisitions Group at Bank of America Merrill Lynch. He previously spent seven years as a Vice President in the Global Mergers and Acquisitions Group at J.P. Morgan Chase, where he worked extensively on acquisitions, leveraged buyouts, private and public financings, exclusive sales and general advisory assignments. Mr. Alstodt received a degree in Economics from the State University of New York at Albany, with a secondary concentration in Finance and Marketing.
Francisco Silva
Francisco Silva has served as our Vice President of Research and Development since March 2013, having also previously served in such position from April 2011 until March 2012. He served as our Research Scientist from March 2012 to June 2012 and as our Chief Scientist from June 2012 to March 2013. From 2007 to 2011, Mr. Silva served as Chief Executive Officer of DV Biologics LLC, and as President of DaVinci Biosciences, LLC, companies engaged in the commercialization of human based biologics for both research and therapeutic applications. From 2003 to 2007, Mr. Silva served as Vice President of Research and Development for PrimeGen Biotech LLC, a company engaged in the development of cell based platforms. From 2002 to 2003, he was a Research Scientist with PrimeGen Biotech and was responsible for the development of experimental designs that focused on germ line reprogramming stem cell platforms. Mr. Silva has taught courses in biology, anatomy and advanced tissue culture at California State Polytechnic University. He has obtained a number of patents relating to stem cells and has had numerous articles published with regard to stem cell research. Mr. Silva graduated from California State Polytechnic University with a degree in Biology. He also obtained a Graduate Presidential Fellowship and MBRS Fellowship from California State Polytechnic University.
Robert Paccasassi
Robert Paccasassi has served as our Vice President of Quality and Compliance since August 2016, having previously served as our Director of Quality and Compliance from September 2015. Mr. Paccasassi has over 20 years of experience in highly regulated product operations, with specific expertise in GMP (large and small molecule) clinical and commercial quality assurance and regulatory compliance leadership roles. He was the Director of Quality Systems (GMP) at Merck KGaA (Dermstadt, Germany) from 2011 to 2014. In this role, Mr. Paccasassi was responsible for leading the ongoing development and implementation of the Corporate Quality Unit’s global GMP policies, processes and directives. He held key quality and compliance management roles at EMD Serono, Biogen Idec, Millennium Pharmaceuticals and Regeneron Pharmaceuticals. Mr. Paccasassi was a Chief Technologist/Site Head overseeing all day to day technical and quality operations of two cGMP biologic production laboratories for Curative Health Services. He was also a Medical Technologist working in the field of immunohematology at Brigham & Women’s Hospital, Boston, Massachusetts. Mr. Paccasassi received a Masters in Business Administration (MBA) degree from Johnson & Wales University and a Bachelor of Science degree in Medical Technology/Biology from the University of Rhode Island.
| 76 |
Mandy D. Clyde
Mandy D. Clyde has been our Vice President of Operations since August 2009. She has served as our Secretary since December 2010 and served on our Board from September 2010 to April 2011. From 2006 to 2009, Ms. Clyde served as Educational Envoy and then CME/CE Coordinator for Professional Resources in Management Education, an accredited provider of continuing medical education. She conducted needs assessments nationally to determine in which areas clinicians most needed current education. She also oversaw onsite educational meetings and analyzed data for outcomes reporting. From 2005 to 2006, Ms. Clyde served as surgical coordinator for Eye Surgery Associates and the Rand Eye Institute, two prominent physician practices in Florida. Ms. Clyde has experience in medical editing for educational programs and is a published author of advanced scientific and clinical content on topics including Alzheimer’s disease, breast cancer, sleep apnea and adult learning. She received a degree in Biology from Mercyhurst College.
Robert B. Catell
Robert B. Catell became a member of our Board of Directors in February 2016. Mr. Catell served as Chairman and Chief Executive Officer of KeySpan Corporation and KeySpan Energy Delivery, the former Brooklyn Union Gas, from 1998 to 2007. His career with Brooklyn Union Gas started in 1958. Following National Grid’s acquisition of KeySpan Corporation in 2007, Mr. Catell became Chairman of National Grid, U.S. and Deputy Chairman of National Grid plc. Mr. Catell currently serves as Chairman of the Board of the Advanced Energy Research and Technology Center (AERTC) at Stony Brook University, New York State Smart Grid Consortium, Cristo Rey Brooklyn High School, Futures in Education Endowment Fund, and the National Offshore Wind Research and Development Consortium. He also serves on the Board of Directors of Applied DNA Sciences, Inc., a company that uses biotechnology as a forensic foundation in creating unique security solutions addressing the challenges of modern commerce. In addition, Mr. Catell serves as a board member of a number of other business, governmental and not-for-profit organizations. Mr. Catell holds both a Master’s and Bachelor’s degree in Mechanical Engineering from City College of New York. We believe that Mr. Catell’s executive-level management experience and his extensive experience in the Long Island community give him the qualifications and skills to serve as one of our directors.
John M. Desmarais
John M. Desmarais became a member of our Board of Directors in December 2015. Mr. Desmarais is the founding partner of Desmarais LLP, an intellectual property trial boutique established in 2010, and the founder and owner of Round Rock Research LLC and Sound View Innovations LLC, patent licensing companies. From 1997 to 2009, he was a partner at the international law firm of Kirkland & Ellis LLP and served as a member of the firm’s Management Committee from 2004 to 2009. Prior to joining Kirkland, and after practicing in the area of intellectual property litigation and counseling for several years, he left private practice to serve as an Assistant United States Attorney in the Southern District of New York, where for three years he represented the federal government in criminal jury trials. Mr. Desmarais is a member of the bars of California, New York and Washington, D.C., the United States Supreme Court, the Federal Circuit Court of Appeals, and various other federal district courts and courts of appeal. He is also registered to practice before the United States Patent and Trademark Office. Mr. Desmarais has been recognized by numerous publications as one of the nation’s leading intellectual property litigators. Mr. Desmarais obtained a degree in Chemical Engineering from Manhattan College and a law degree from New York University. We believe that Mr. Desmarais’ business and legal experience, including his extensive experience in the area of intellectual property, give him the qualifications and skills to serve as one of our directors.
| 77 |
A. Jeffrey Radov
A. Jeffrey Radov became a member of our Board and Chair of our Audit Committee in April 2011. Mr. Radov is an entrepreneur and businessman with more than 35 years of experience in media, communications and financial endeavors. Since 2002, he has served as the Managing Partner of Walworth Group, which provides consulting and advisory services to a variety of businesses, including hedge funds, media, entertainment and Internet companies, financial services firms and early stage ventures. Mr. Radov is also a registered representative of Young America Capital, LLC, a broker-dealer. From 2008 to 2010, Mr. Radov was a Principal and Chief Operating Officer at Aldebaran Investments, LLC, a registered investment advisor. From 2005 to 2008, Mr. Radov was Chief Operating Officer at EagleRock Capital Management, a group of hedge funds. Prior to joining EagleRock, Mr. Radov was a founding investor in and Board member of Edusoft, Inc., an educational software company. From 2001 to 2002, Mr. Radov was a Founder-in-Residence at SAS Investors, an early-stage venture fund. From 1999 to 2001, Mr. Radov was CEO and co-founder of VocaLoca, Inc., an innovator in consumer-generated audio content on the Internet. Mr. Radov was a founding executive of About.Com, Inc., an online information source, and was its EVP of Business Development and Chief Financial Officer from its inception. In 1996, prior to founding About.Com, Mr. Radov was a Director at Prodigy Systems Company, a joint venture of IBM and Sears. Mr. Radov was also a principal in the management of a series of public limited partnerships that invested in the production and distribution of more than 130 major motion pictures. From 1982 to 1984, Mr. Radov was the Director of Finance at Rainbow Programming Enterprises, a joint venture among Cablevision Systems Corporation, Cox Broadcasting and Daniels & Associates. From 1977 to 1981, Mr. Radov was Director of Marketing at Winklevoss & Associates. Mr. Radov earned a Masters of Business Administration from The Wharton School of the University of Pennsylvania and holds a Bachelor of Arts degree from Cornell University. We believe that Mr. Radov’s executive-level management experience and his extensive experience in the finance industry give him the qualifications and skills to serve as one of our directors.
Charles S. Ryan
Dr. Charles S. Ryan became a member of our Board in April 2015 and has served as Chair of our Nominating Committee since August 2018. Since February 2018, Dr. Ryan has served as Chief Executive Officer of Neurotrope Bioscience, Inc., a company that develops novel therapies for the treatment of neurodegenerative diseases and developmental disorders. From October 2016 to February 2018, Dr. Ryan served as Chief Executive Officer of Orthobond, Inc., a company that seeks to improve the performance and safety of medical devices through the use of proprietary non-polymer technology. From March 2015 to May 2016, Dr. Ryan served as Vice President, General Counsel of Cold Spring Harbor Laboratory, a not-for-profit research and education institution at the forefront of molecular biology and genetics, with research programs focusing on cancer, neuroscience, plant biology, genomics and quantitative biology. From 2003 to 2014, he served as Senior Vice President and Chief Intellectual Property Counsel at Forest Laboratories, Inc., a New York Stock Exchange company that developed and marketed pharmaceutical products in a variety of therapeutic categories including central nervous system, cardiovascular, anti-infective, respiratory, gastrointestinal, and pain management medicine. Dr. Ryan has over 20 years experience in managing all aspects of intellectual property litigation, conducting due diligence investigations and prosecuting patent and trademark applications in the pharmaceutical and biotechnology industries. He also serves as a director of Applied DNA Sciences, Inc., a company that uses biotechnology as a forensic foundation in creating unique security solutions addressing the challenges of modern commerce. Dr. Ryan earned a doctorate in Oral Biology and Pathology from Stony Brook University and a law degree from Western New England University. We believe that Dr. Ryan’s executive-level management and legal experience give him the qualifications and skills to serve as one of our directors.
| 78 |
Paul Jude Tonna
Paul Jude Tonna became a member of our Board and Chair of our Compensation Committee in June 2014. Mr. Tonna is a highly regarded community leader and an accomplished businessman with an extensive history of public service. From 1994 to 2005 he served as a Suffolk County, New York Legislator, and from 2000 through 2002 was its Presiding Officer. He currently serves as Executive Director and a member of the Board of Advisors for The Energeia Partnership at Molloy College, a leadership academy based in Rockville Centre, New York, dedicated to identifying and addressing the serious, complex and multi-dimensional issues challenging the Long Island region. Mr. Tonna is a former Adjunct Professor in Theology & Religious Studies at St. John’s University. He served as Chairman of the Suffolk County Industrial Development Agency, and currently serves as Trustee of the Long Island State Parks & Recreation Commission and as Public Trustee of the Stationary Engineers Industry Stabilization Fund. Mr. Tonna is a board member of The Advanced Energy Research & Technology Center at Stony Brook University, The Long Island Index Advisory Board and Erase Racism’s College of Advisors. He also serves as the Executive Director of the Suffolk County Village Officials Association and the United States Green Building Council-Long Island Chapter. Mr. Tonna is a founding director of Empire National Bank and Chairman and Commissioner of the South Huntington Water District. Mr. Tonna holds an undergraduate degree in Philosophy from New York University and a Master’s degree in Theology from Immaculate Conception Seminary, and he conducted doctoral studies in Systemic Theology at Fordham University. We believe that Mr. Tonna’s executive-level management experience and his extensive experience in the Long Island community give him the qualifications and skills to serve as one of our directors.
Scientific Advisors
Scientific Advisory Board
The following persons are the members of our Scientific Advisory Board:
| Name | Principal Positions | |
Wayne Marasco, M.D., Ph.D. Chairman |
Professor, Department of Cancer Immunology & AIDS, Dana-Farber Cancer Institute; Professor of Medicine, Harvard Medical School; Principal Faculty Member, Harvard Stem Cell Institute |
| 79 |
| Naiyer Imam, M.D. | Chairman and President, First Medicine, Inc., an international telemedicine corporation dedicated to virtual physician services and chronic disease management | |
| Wayne J. Olan, M.D. | Director, Interventional and Endovascular Neurosurgery; Associate Professor, Neurosurgery and Radiology, George Washington University Medical Center; Consulting Physician, Department of Radiology, National Institutes of Health | |
| Joy Cavagnaro, Ph.D., DABT, RAC | President and Founder, Access BIO, L.C.; Fellow, Academy of Toxicological Sciences and the Regulatory Professional Society; Formerly Senior Pharmacologist and Director of Quality Assurance, Food and Drug Administration’s Center for Biologics Evaluation and Research | |
Jason Lipetz, M.D. Chairman, Disc Advisory Committee |
Founder, Long Island Spine Rehabilitation Medicine; Chief of Spine Medicine, Northwell Health Spine Center; Assistant Professor of Rehabilitation Medicine, Hofstra University School of Medicine | |
| Harvinder Sandhu, M.D. | Orthopedic Spine Surgeon, Hospital for Special Surgery; Formerly Chief of Spinal Surgery Service, UCLA Medical Center | |
| Christopher Plastaras, M.D. | Clinical Director of Musculoskeletal Spine and Sports Rehabilitation Medicine and Physiatrist, MossRehab; Formerly Director of The Penn Spine and Rehabilitation Center; Formerly Director of Spine, Sports and Musculoskeletal Medicine Fellowship, University of Pennsylvania | |
| Gerard A. Malanga, M.D. | Founder, Partner and Physiatrist, New Jersey Sports Medicine, LLC and New Jersey Regenerative Institute; Chair, American Academy of Physical Medicine and Rehabilitation Task Force on Regenerative Medicine; President Elect, Interventional Orthopedic Foundation |
| 80 |
Clinical Director, Regenerative Disc/Spine Program
Dr. Olan also serves as Clinical Director of our Regenerative Disc/Spine Program.
Family Relationships
There are no family relationships among any of our executive officers and directors.
Term of Office
We have a classified Board of Directors. The directors will hold office until the respective annual meetings of stockholders indicated below and until their respective successors are elected and qualified or until their earlier resignation or removal.
| Name | Class | Term Expires | ||
| Mark Weinreb | III | 2020 | ||
| Robert B. Catell | I | 2021 | ||
| John M. Desmarais | II | 2019 | ||
| A. Jeffrey Radov | III | 2020 | ||
| Charles S. Ryan | I | 2021 | ||
| Paul Jude Tonna | II | 2019 |
Each executive officer will hold office until the initial meeting of the Board of Directors following the next annual meeting of stockholders and until his or her successor is elected and qualified or until his or her earlier resignation or removal.
Audit Committee
The Audit Committee of the Board of Directors is responsible for overseeing our accounting and financial reporting processes and the audits of our financial statements. The members of the Audit Committee are Messrs. Radov (Chair), Ryan and Tonna.
Audit Committee Financial Expert
Our Board of Directors has determined that Mr. Radov is an “audit committee financial expert,” as that is defined in Item 407(d)(5) of Regulation S-K. Mr. Radov is an “independent director” based on the definition of independence in Listing Rule 5605(a)(2) of The Nasdaq Stock Market.
| 81 |
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16 of the Exchange Act requires that reports of beneficial ownership of common stock and changes in such ownership be filed with the Securities and Exchange Commission by Section 16 “reporting persons,” including directors, certain officers, holders of more than 10% of the outstanding common stock and certain trusts of which reporting persons are trustees. We are required to disclose in this Annual Report each reporting person whom we know to have failed to file any required reports under Section 16 on a timely basis during the fiscal year ended December 31, 2018. To our knowledge, based solely on a review of copies of Forms 3, 4 and 5 filed with the Securities and Exchange Commission and written representations that no other reports were required, during the fiscal year ended December 31, 2018, our officers, directors and 10% stockholders complied with all Section 16(a) filing requirements applicable to them.
Code of Ethics for Senior Financial Officers
Our Board of Directors has adopted a Code of Ethics for our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A copy of the Code of Ethics is posted on our website, www.biorestorative.com. We intend to satisfy the disclosure requirement under Item 5.05(c) of Form 8-K regarding an amendment to, or a waiver from, our Code of Ethics by posting such information on our website, www.biorestorative.com.
| ITEM 11. | EXECUTIVE COMPENSATION. |
Summary Compensation Table
The following Summary Compensation Table sets forth all compensation earned in all capacities during the fiscal years ended December 31, 2018 and 2017 by (i) our principal executive officer, (ii) our two most highly compensated executive officers, other than our principal executive officer, who were serving as executive officers as of December 31, 2018 and whose total compensation for the 2018 fiscal year, as determined by Regulation S-K, Item 402, exceeded $100,000, and (iii) a person who would have been included as one of our two most highly compensated executive officers, other than our principal executive officer, but for the fact that he was not serving as one of our executive officers as of December 31, 2018 (the individuals falling within categories (i), (ii) and (iii) are collectively referred to as the “Named Executive Officers”):
| Name and Principal Position | Year | Salary | Bonus (1) | Option Awards Earned (2) | All Other Compensation | Total | |||||||||||||||||
| Mark Weinreb, | 2018 | $ | 400,000 | $ | - | $ | 291,800 | (3) | $ | 7,200 | (4) | $ | 699,000 | (4) | |||||||||
| Chief Executive Officer | 2017 | $ | 400,000 | $ | 32,000 | $ | 784,700 | (5) | $ | 7,200 | (6) | $ | 1,223,900 | (6) | |||||||||
| Adam Bergstein | 2018 | $ | 264,538 | $ | - | $ | 1,491,300 | (7) | $ | - | $ | 1,755,838 | |||||||||||
| Senior VP, Planning and Business Development(8) | 2017 | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
| Francisco Silva, | 2018 | $ | 287,500 | $ | 23,000 | $ | 107,800 | (9) | $ | - | $ | 418,300 | |||||||||||
| VP, Research and Development | 2017 | $ | 250,000 | $ | 6,000 | $ | 200,400 | (10) | $ | - | $ | 456,400 | |||||||||||
| Robert Paccasassi, | 2018 | $ | 201,250 | $ | 32,703 | $ | 53,900 | (11) | $ | - | $ | 287,853 | |||||||||||
| VP, Quality and Compliance(12) | 2017 | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
| (1) | Represents bonus amounts earned pursuant to the achievement of certain performance goals. |
| 82 |
| (2) | The amounts reported in this column represent the grant date fair value of the option awards granted during the years ended December 31, 2018 and 2017, calculated in accordance with FASB ASC Topic 718. For a detailed discussion of the assumptions used in estimating fair values, see Note 10 – Stockholders’ Deficiency in the notes that accompany our consolidated financial statements included in this Annual Report following Item 16 (“Form 10-K Summary”). | |
| (3) | During 2018, Mr. Weinreb was granted a ten-year option under our 2010 Equity Participation Plan, or the Plan, for the purchase of 275,000 shares of common stock at an exercise price of $1.23 per share. Such option is exercisable to the extent of 91,667 shares as of each of the date of grant and the first anniversary of the date of grant and 91,666 shares as of the second anniversary of the date of grant. See “Employment Agreements” below for a discussion of certain provisions relating to the options granted to Mr. Weinreb. | |
| (4) | Of the aggregate $699,000 earned during 2018, $291,800 represents the grant date value of non-cash stock-based compensation awards, irrespective of the vesting period of those awards. Of the $407,200 earned cash compensation, $406,761and $439 were paid in cash during 2018 and 2019 (prior to the date of the filing of this Annual Report), respectively, and none remains unpaid for 2018. All Other Compensation represents an automobile allowance paid to Mr. Weinreb in 2018. | |
| (5) | During 2017, Mr. Weinreb was granted a ten-year option under the Plan for the purchase of 275,000 shares of common stock at an exercise price of $3.35 per share. Such option is exercisable to the extent of 91,667 shares as of each of the date of grant and the first anniversary of the date of grant and 91,666 shares as of the second anniversary of the date of grant. See “Employment Agreements” below for a discussion of certain provisions relating to the options granted to Mr. Weinreb. | |
| (6) | Of the aggregate $1,223,900 earned during 2017, $784,700 represents the grant date value of non-cash stock-based compensation awards, irrespective of the vesting period of those awards. Of the $439,200 earned cash compensation, $178,113 and $261,087 were paid in cash during 2017 and 2018, respectively, and none remains unpaid for 2017. All Other Compensation represents an automobile allowance paid to Mr. Weinreb in 2017. | |
| (7) | During 2018, Mr. Bergstein was granted a ten-year option under the Plan for the purchase of 500,000 shares of common stock at an exercise price of $3.40 per share. Such option was exercisable upon the satisfaction of a certain condition. As a result of Mr. Bergstein’s resignation on October 28, 2018, such option has terminated. | |
| (8) | Mr. Bergstein resigned as an officer on October 28, 2018. | |
| (9) | During 2018, Mr. Silva was granted a ten-year option under the Plan for the purchase of 100,000 shares of common stock at an exercise price of $1.23 per share. Such option is exercisable to the extent of 33,334 shares as of the first anniversary of the date of the grant and 33,333 shares as of each of the second and third anniversaries of the date of grant. |
| 83 |
| (10) | During 2017, Mr. Silva was granted a ten-year option under the Plan for the purchase of 80,000 shares of common stock at an exercise price of $2.80 per share. Such option is exercisable to the extent of 26,667 shares as of each of the first and second anniversaries of the date of grant and 26,666 shares as of the third anniversary of the date of grant. | |
| (11) | During 2018, Mr. Paccasassi was granted a ten-year option under the Plan for the purchase of 50,000 shares of common stock at an exercise price of $1.23 per share. Such option is exercisable to the extent of 16,667 shares as of each of the first and second anniversaries of the date of grant and 16,666 shares as of the third anniversary of the date of grant. | |
| (12) | Mr. Paccasassi was not a Named Executive Officer during 2017. |
Outstanding Equity Awards at Fiscal Year-End
The following table provides information on outstanding equity awards as of December 31, 2018 to the Named Executive Officers:
| Option Awards | Stock Awards | |||||||||||||||||||||||||||||||||
| Name | Number of securities underlying unexercised options exercisable | Number of securities underlying unexercised options unexercisable | Equity incentive plan awards: Number of securities underlying unexercised unearned options | Option exercise price | Option expiration date | Number of shares or units of stock that have not vested | Market value of shares of units that have not vested | Equity incentive plan awards: Number of unearned shares, units or other rights that have not vested | Equity incentive plan awards: Market or payout value of unearned shares, units or other rights that have not vested | |||||||||||||||||||||||||
| Mark Weinreb | 4,000 | - | - | $ | 4.70 | 12/14/2020 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 50,000 | - | - | $ | 4.70 | 2/10/2022 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 20,000 | - | - | $ | 4.70 | 12/7/2022 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 12,500 | - | - | $ | 4.70 | 10/4/2023 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 50,000 | - | - | $ | 4.70 | 2/18/2024 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 150,000 | - | - | $ | 4.70 | 10/23/2024 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 208,000 | - | - | $ | 4.70 | 9/4/2025 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 275,000 | - | - | $ | 3.73 | 6/10/2026 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Mark Weinreb | 183,334 | 91,666 | (1) | - | $ | 3.35 | 6/23/2027 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Mark Weinreb | 91,667 | 183,333 | (2) | - | $ | 1.23 | 10/29/2028 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Adam D. Bergstein | - | 500,000 | (3) | - | $ | 3.40 | 1/19/2028 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Francisco Silva | 4,000 | - | - | $ | 4.70 | 4/4/2021 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 150 | - | - | $ | 4.70 | 6/23/2021 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 1,000 | - | - | $ | 4.70 | 11/16/2021 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 2,000 | - | - | $ | 4.70 | 2/10/2022 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 4,500 | - | 3,000 | (4) | $ | 4.70 | 5/2/2022 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Francisco Silva | 4,000 | - | - | $ | 4.70 | 12/7/2022 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 5,000 | - | - | $ | 4.70 | 10/4/2023 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 12,500 | - | - | $ | 4.70 | 2/18/2024 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 2,000 | - | - | $ | 4.70 | 3/12/2024 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 37,500 | - | - | $ | 4.70 | 10/23/2024 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 25,000 | - | - | $ | 4.70 | 9/4/2025 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Francisco Silva | 40,000 | 20,000 | (5) | - | $ | 3.73 | 6/10/2026 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Francisco Silva | 26,667 | 53,333 | (6) | - | $ | 2.80 | 7/12/2027 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Francisco Silva | - | 100,00 | (7) | - | $ | 1.23 | 10/29/2028 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Robert Paccasassi | 5,000 | - | - | $ | 4.70 | 8/13/2025 | - | $ | - | - | $ | - | ||||||||||||||||||||||
| Robert Paccasassi | 10,000 | 5,000 | (8) | - | $ | 3.73 | 6/10/2026 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Robert Paccasassi | 13,334 | 26,666 | (9) | - | $ | 2.80 | 7/12/2027 | - | $ | - | - | $ | - | |||||||||||||||||||||
| Robert Paccasassi | - | 50,000 | (10) | - | $ | 1.23 | 10/29/2028 | - | $ | - | - | $ | - | |||||||||||||||||||||
| 84 |
| (1) | Option is exercisable on June 23, 2019. |
| (2) | Option is exercisable to the extent of 91,667 shares on October 29, 2019 and 91,666 shares on October 29, 2020. |
| (3) | Option was exercisable subject to the satisfaction of a certain condition. As a result of Mr. Bergstein’s resignation on October 28, 2018, such option has terminated. |
| (4) | Options are exercisable commencing on the date (provided that such date is during Mr. Silva’s employment with us), if any, on which either (i) the FDA approves a biologics license application made by us with respect to any biologic product or (ii) a 510(k) Premarket Notification submission is made by us to the FDA with respect to a certain device. |
| (5) | Option is exercisable on June 10, 2019. |
| (6) | Option is exercisable to the extent of 26,667 shares on July 12, 2019 and 26,666 shares on July 12, 2020. |
| (7) | Option is exercisable to the extent of 33,334 shares on October 29, 2019 and 33,333 shares on each of October 29, 2020 and October 29, 2021. |
| (8) | Option is exercisable on June 10, 2019. |
| (9) | Option is exercisable to the extent of 13,333 shares on each of July 12, 2019 and July 12, 2020. |
| (10) | Option is exercisable to the extent of 16,667 shares on each of October 29, 2019 and October 29, 2020 and 16,666 shares on October 29, 2021. |
| 85 |
Employment Agreements
In March 2015, we entered into an employment agreement with Mark Weinreb, our Chief Executive Officer. Pursuant to the employment agreement, which expires on December 31, 2019, Mr. Weinreb is entitled to receive a salary of $400,000 per annum. Mr. Weinreb was entitled to receive an annual bonus for 2015 equal to 50% of his annual base salary and was entitled to receive an annual bonus for 2016, 2017 and 2018 of up to 40%, 50% and 50%, respectively, of his annual base salary, in the event certain performance goals, as determined by our Compensation Committee, were satisfied. Mr. Weinreb is entitled to receive an annual bonus for 2019 of up to 50% of his annual base salary in the event certain performance goals, as determined by our Compensation Committee, are satisfied. Pursuant to the employment agreement, in the event that Mr. Weinreb’s employment is terminated by us without “cause”, or Mr. Weinreb terminates his employment for “good reason” (each as defined in the employment agreement), Mr. Weinreb would be entitled to receive severance in an amount equal to one time his then annual base salary and certain benefits, plus $100,000 (in lieu of bonus). In addition, pursuant to the employment agreement, Mr. Weinreb would be entitled to receive such severance in the event that the term of his employment agreement is not extended beyond December 31, 2019 and, within three months of such expiration date, his employment is terminated by us without “cause” or if Mr. Weinreb terminates his employment for any reason. Further, in the event that Mr. Weinreb’s employment is terminated by us without “cause”, or Mr. Weinreb terminates his employment for “good reason”, following a “change in control” (as defined in the employment agreement), Mr. Weinreb would be entitled to receive severance in an amount equal to one and one-half times his then annual base salary and certain benefits, plus $300,000 (in lieu of bonus). Pursuant to the employment agreement, with respect to options granted to Mr. Weinreb during the term of his employment with us, such options shall vest and become exercisable if Mr. Weinreb is entitled to receive severance based upon a termination of his employment as set forth above. In addition, pursuant to the employment agreement, to the extent that an option granted to Mr. Weinreb during his term of his employment with us becomes exercisable (whether due to the passage of time or otherwise), such option shall remain exercisable until its expiration date notwithstanding any termination of employment with us.
Effective January 16, 2018, we entered into an at will employment agreement with Adam D. Bergstein, Senior Vice President, Planning and Business Development. Pursuant to the employment agreement, Mr. Bergstein was entitled to receive a base annual salary of $250,000 during the initial three months of employment. Thereafter, his base salary increased to $350,000. In addition, pursuant to the employment agreement, Mr. Bergstein was entitled to receive an annual bonus of up to 30% of his annual salary based on the satisfaction of certain performance goals. The employment agreement also provided for the payment of three months severance under certain circumstances. Mr. Bergstein resigned his employment with us effective October 28, 2018.
Effective April 5, 2011, we entered into an at will employment agreement with Francisco Silva, our Vice President of Research and Development. Pursuant to the employment agreement, as amended, Mr. Silva is currently entitled to receive a salary of $287,500 per annum. In addition, pursuant to the employment agreement, as amended, Mr. Silva is entitled to receive an annual bonus of up to 20% of his annual salary (up to 16% of his annual salary for 2016) based on the satisfaction of certain performance goals. Further, pursuant to the employment agreement, as amended, in the event that Mr. Silva’s employment with us is terminated without cause, Mr. Silva would be entitled to receive severance in an amount equal to 50% of his then annual base salary.
| 86 |
Effective September 2, 2015, we entered into an at will employment agreement with Robert Paccasassi, our Vice President of Quality and Compliance. Pursuant to the employment agreement, as amended, Mr. Paccasassi is currently entitled to receive a salary of $201,250 per annum. In addition, pursuant to the employment agreement, Mr. Paccasassi is entitled to receive an annual bonus of up to 25% of his annual salary based upon the satisfaction of certain performance goals.
Director Compensation
The following table sets forth certain information concerning the compensation of our non-employee directors for the fiscal year ended December 31, 2018:
| Name | Fees Earned or Paid in Cash | Stock Awards | Option Awards (1) | Non-Equity Incentive Plan Compensation | Nonqualified Deferred Compensation Earnings | All Other Compensation | Total | |||||||||||||||||||||
| Robert B. Catell | $ | 40,000 | $ | - | $ | 79,600 | (2) | $ | - | $ | - | $ | - | $ | 119,600 | |||||||||||||
| John M. Desmarais | $ | 40,000 | $ | - | $ | 79,600 | (3) | $ | - | $ | - | $ | - | $ | 119,600 | |||||||||||||
| A. Jeffrey Radov | $ | 40,000 | $ | - | $ | 79,600 | (4) | $ | - | $ | - | $ | - | $ | 119,600 | |||||||||||||
| Charles S. Ryan | $ | 40,000 | $ | - | $ | 79,600 | (5) | $ | - | $ | - | $ | - | $ | 119,600 | |||||||||||||
| Paul Jude Tonna | $ | 40,000 | $ | - | $ | 79,600 | (6) | $ | - | $ | - | $ | - | $ | 119,600 | |||||||||||||
| (1) | The amounts reported in this column represent the grant date fair value of the option awards granted during the year ended December 31, 2018, calculated in accordance with FASB ASC Topic 718. For a detailed discussion of the assumptions used in estimating fair values, see Note 10 – Stockholders’ Deficiency in the notes that accompany our consolidated financial statements in this Annual Report following Item 16 (“Form 10-K Summary”). | |
| (2) | As of December 31, 2018. Mr. Catell held options for the purchase of 219,000 shares of common stock. | |
| (3) | As of December 31, 2018, Mr. Desmarais held options for the purchase of 250,000 shares of common stock. | |
| (4) | As of December 31, 2018, Mr. Radov held options for the purchase of 566,000 shares of common stock. | |
| (5) | As of December 31, 2018, Mr. Ryan held options for the purchase of 256,000 shares of common stock. | |
| (6) | As of December 31, 2018, Mr. Tonna held options for the purchase of 364,000 shares of common stock. |
Each of Messrs. Catell, Desmarais, Radov, Ryan and Tonna, our non-employee directors, is entitled to receive, as compensation for his services as a director, $30,000 per annum plus $10,000 per annum for all committee service, in each case payable quarterly (subject to our cash needs). Our non-employee directors also receive stock options, from time to time, in consideration of their services.
| 87 |
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
Principal Stockholders
The following table sets forth certain information regarding the beneficial ownership of our common stock, as of March 19, 2019, known by us, through transfer agent records, to be held by: (i) each person who beneficially owns 5% or more of the shares of common stock then outstanding; (ii) each of our directors; (iii) each of our Named Executive Officers (as defined above); and (iv) all of our directors and executive officers as a group.
The information in this table reflects “beneficial ownership” as defined in Rule 13d-3 of the Exchange Act. To our knowledge, and unless otherwise indicated, each shareholder has sole voting power and investment power over the shares listed as beneficially owned by such shareholder, subject to community property laws where applicable. Percentage ownership is based on 14,625,503 shares of common stock outstanding as of March 19, 2019.
| Beneficial Owner | Number of Shares Beneficially Owned | Approximate Percent of Class | ||||||
| Dale Broadrick 3003 Brick Church Pike Nashville, Tennessee | 3,161,452 | (1) | 20.2 | % | ||||
| John M. Desmarais 230 Park Avenue New York, New York | 2,029,574 | (2) | 12.6 | % | ||||
SCG Capital, LLC | 1,537,817 | (3) | 9.99 | %(3) | ||||
| Westbury (Bermuda) Ltd. Westbury Trust Victoria Hall 11 Victoria Street Hamilton, HMEX Bermuda | 1,151,661 | (4) | 7.8 | % | ||||
| Mark Weinreb 40 Marcus Drive, Suite One Melville, New York | 1,124,501 | (5) | 7.2 | % | ||||
| A. Jeffrey Radov | 486,834 | (6) | 3.2 | % | ||||
| Paul Jude Tonna | 339,834 | (7) | 2.3 | % | ||||
| Robert B. Catell | 305,399 | (8) | 2.1 | % | ||||
| Charles S. Ryan | 251,001 | (9) | 1.7 | % | ||||
| Francisco Silva | 168,562 | (10) | 1.1 | % | ||||
| Robert Paccasassi | 28,334 | (11) | * | |||||
| Adam Bergstein | 2 | * | ||||||
| All directors and executive officers as a group (10 persons) | 4,931,423 | (12) | 26.2 | % | ||||
| * | Less than 1% |
| 88 |
| (1) | Based upon Schedule 13D filed with the Securities and Exchange Commission (the “SEC”). Includes 1,000,000 shares of common stock issuable upon the exercise of currently exercisable warrants. |
| (2) | Based upon Schedule 13D filed with the SEC and other information known to us. Includes 1,536,176 shares of common stock issuable upon the exercise of currently exercisable options and warrants (including warrants for the purchase of 40,000 shares of common stock held by a trust for which Mr. Desmarais and his wife serve as the trustees and which was established for the benefit of his immediate family). |
| (3) | Based upon Schedule 13G filed with the SEC and other information known to us. Includes 150,000 shares of common stock issuable upon the exercise of currently exercisable warrants. The shares and warrants are owned directly by SCG Capital, LLC, or SCG. Steven Geduld as President of SCG has an indirect beneficial ownership in the securities held by SCG. SCG has rights, under a convertible promissory note, to own an aggregate number of shares of our common stock which, except for a contractual cap on the amount of outstanding shares of the common stock that SCG may own, could exceed such a cap. SCG’s ownership cap under the convertible promissory note is 9.99% of the outstanding shares of our common stock. Therefore, based on 14,625,503 shares of common stock outstanding as of March 19, 2019 (15,393,563 shares of common stock outstanding giving effect to the shares issuable pursuant to the warrants and the convertible note, subject to the cap), the number of shares of our common stock beneficially owned by SCG as of March 19, 2019 was 1,537,817 shares of common stock. |
| (4) | Based upon Schedule 13D filed with the SEC and other information known to us. Includes 199,182 shares of common stock issuable upon the exercise of currently exercisable warrants. The shares and warrants are owned directly by Westbury (Bermuda) Ltd. which is 100% owned by Westbury Trust. |
| (5) | Includes 1,044,501 shares of common stock issuable upon the exercise of currently exercisable options. |
| (6) | Includes 474,334 shares of common stock issuable upon the exercise of currently exercisable options. |
| (7) | Represents (i) 1,500 shares of common stock held jointly with Mr. Tonna’s wife and (ii) 303,834 shares of common stock issuable upon the exercise of currently exercisable options and warrants. |
| (8) | Includes 224,533 shares of common stock issuable upon the exercise of currently exercisable options and warrants. |
| (9) | Includes 208,084 shares of common stock issuable upon the exercise of currently exercisable options and warrants. |
| (10) | Includes (i) 170 shares of common stock held in an individual retirement account for the benefit of Mr. Silva and (ii) 164,317 shares of common stock issuable upon the exercise of currently exercisable options. |
| (11) | Represents shares of common stock issuable upon the exercise of currently exercisable options. |
| (12) | Includes 4,181,497 shares of common stock issuable upon the exercise of currently exercisable options and warrants. |
Securities Authorized for Issuance Under Equity Compensation Plans
The following table sets forth information as of December 31, 2018 with respect to compensation plans (including individual compensation arrangements) under which our common stock are authorized for issuance, aggregated as follows:
| ● | All compensation plans previously approved by security holders; and | |
| ● | All compensation plans not previously approved by security holders. |
| 89 |
EQUITY COMPENSATION PLAN INFORMATION
| Number of securities to be issued upon exercise of outstanding options (a) | Weighted-average exercise price of outstanding options (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | ||||||||||
| Equity compensation plans approved by security holders | 4,703,785 | $ | 3.21 | 5,251,215 | ||||||||
| Total | 4,703,785 | $ | 3.21 | 5,251,215 | ||||||||
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
John M. Desmarais
In June 2016, we borrowed $500,000 from Tuxis Trust, a trust for which John M. Desmarais, one of our non-employee directors and principal stockholders, and his wife serve as the trustees and which was established for the benefit of Mr. Desmarais’s immediate family. The promissory note evidencing the loan, or the Tuxis Trust Note, provided for the payment of the principal amount, together with interest at the rate of 10% per annum, in July 2017. In July 2017, we and Tuxis Trust agreed to amend the Tuxis Trust Note to provide for a maturity date of December 1, 2017 and an increase in the interest rate to 15% per annum effective July 1, 2017. In the event that, prior to the maturity date of the Tuxis Trust Note, we receive net proceeds of $10,000,000 from a single equity or debt financing (as opposed to a series of related or unrelated financings), Tuxis Trust has the right to require that we prepay the amount due under the Tuxis Trust Note (subject to the consent of the party that provided the particular financing). In consideration of the loan, we issued to Tuxis Trust a five-year warrant for the purchase of 40,000 shares of our common stock at an exercise price of $4.00 per share.
In December 2016, we borrowed $60,000 from Mr. Desmarais. The promissory note evidencing the loan provided for the payment of $65,000 on January 31, 2017. In consideration of the loan, we further extended the expiration dates of the warrants held by Mr. Desmarais for the purchase of 444,444 and 400,000 shares of our common stock to December 31, 2018. In February 2017, we entered into an exchange agreement with Mr. Desmarais pursuant to which Mr. Desmarais exchanged the principal amount of the promissory note, together with accrued interest, for 21,731 shares of our common stock at an exchange price of $3.00 per share and, in consideration thereof, received a five year warrant for the purchase of 21,731 shares of our common stock at an exercise price of $4.00 per share.
In July 2017, we borrowed $175,000 from Mr. Desmarais. The promissory note evidencing the loan, or the Desmarais Note, provides for the payment of the principal amount, together with interest at the rate of 15% per annum, on December 1, 2017. In the event that, prior to the maturity date of the Desmarais Note, we receive net proceeds of $10,000,000 from a single equity or debt financing (as opposed to a series of related or unrelated financings), Mr. Desmarais has the right to require that we prepay the amount due under the Desmarais Note (subject to the consent of the party that provided the particular financing). The payment of the Desmarais Note is secured by the grant of a security interest in our equipment and intellectual property. Concurrently, we agreed that the payment of the Desmarais Trust Note is also secured by the grant of such security interest.
| 90 |
In November 2017, we and Mr. Desmarais agreed that the due date for the payment of the Desmarais Note was extended to December 1, 2018. Concurrently, we agreed with Tuxis Trust that the due date for the payment of the Tuxis Trust Note was also extended to December 1, 2018. In consideration of the note extensions by Mr. Desmarais and Tuxis Trust, we agreed to reduce the exercise prices of certain warrants held by Mr. Desmarais for the purchase of an aggregate of 775,000 shares of our common stock from $5.00 per share to $4.00 per share and the exercise price of a certain warrant held by Mr. Desmarais for the purchase of 444,444 shares of our common stock from $4.50 per share to $4.00 per share.
In November 2018, we and Mr. Desmarais agreed that the due date for the payment of the Desmarais Note was extended to December 31, 2019. Concurrently, we agreed with Tuxis Trust that the due date for the payment of the Tuxis Trust Note was also extended to December 31, 2019. In consideration of the note extensions by Mr. Desmarais and Tuxis Trust, we agreed to reduce the exercise prices of certain warrants held by Mr. Desmarais for the purchase of an aggregate of 844,444 shares of our common stock from $4.00 per share to $1.50 per share and to extend the expiration date of such warrants from December 31, 2018 to December 31, 2019.
Others
In August 2016, we borrowed $100,000 from Robert B. Catell, one of our non-employee directors. The promissory note evidencing the loan, or the Catell Note, provided for the payment of the principal amount, together with interest at the rate of 10% per annum, on February 5, 2017. In consideration of the loan, we issued to Mr. Catell a five-year warrant for the purchase of 8,000 shares of our common stock at an exercise price of $4.00 per share. In August 2017, we and Mr. Catell agreed that the outstanding principal amount of the Catell Note of $50,000 will be payable on February 5, 2018. In consideration of such extension of the maturity date, we issued to Mr. Catell a five-year warrant for the purchase of 5,000 shares of our common stock at an exercise price of $4.00 per share. In April 2018, we and Mr. Catell agreed that the outstanding principal amount of the Catell Note of $45,000 will be payable on December 31, 2018. On February 8, 2019, we and Mr. Catell agreed that the outstanding principal amount of the Catell Note of $30,000 will be payable on December 31, 2019. In the event that, prior to the maturity date of the Catell Note, we receive net proceeds of $10,000,000 from a single equity or debt financing (as opposed to a series of related or unrelated financings), Mr. Catell has the right to require that we prepay the amount due under the Catell Note (subject to the consent of the party that provided the particular financing).
In December 2016, we borrowed $30,000 from Mr. Catell. The promissory note evidencing the loan provided for the payment of $32,500 on January 31, 2017. In February 2017, we entered into an exchange agreement with Mr. Catell pursuant to which Mr. Catell exchanged the principal amount of the promissory note, together with accrued interest, for 10,866 shares of our common stock at an exchange price of $3.00 per share and, in consideration thereof, received a five year warrant for the purchase of 10,866 shares of our common stock at an exercise price of $4.00 per share.
| 91 |
In February 2017, the Compensation Committee of our Board of Directors reduced the exercise price of outstanding options for the purchase of an aggregate of 1,219,450 shares of our common stock (with exercise prices ranging between $5.70 and $30.00 per share) to $4.70 per share, which was the closing price for our common stock on the day prior to the determination, as reported by the OTCQB. The exercise price reduction related to options held by, among others, our Named Executive Officers and directors with respect to the following number of shares: (i) Mark Weinreb, our President, Chief Executive Officer and Chairman of the Board: 494,500 shares, (ii) A. Jeffrey Radov, one of our directors: 238,000 shares, (iii) Paul Jude Tonna, one of our directors: 100,000 shares, (iv) Dr. Charles S. Ryan, one of our directors: 35,000 shares, (v) Francisco Silva, our Vice President of Research and Development: 100,650 shares, and (vi) Edward L. Field, then President of our Disc/Spine Division: 50,000 shares.
In March 2017, we entered into exchange agreements with Messrs. Catell, Desmarais, Ryan and Tonna, each a non-employee director, pursuant to which accrued director fees were exchanged for our common stock at an exchange price of $3.00 per share and, in consideration thereof, we issued to them five year warrants for the purchase of our common stock at an exercise price of $4.00 per share as follows: (i) Mr. Catell: $45,000 for 15,000 shares and 15,000 warrants; (ii) Mr. Desmarais: $50,000 for 16,667 shares and 16,667 warrants; (iii) Mr. Ryan: $80,000 for 26,667 shares and 26,667 warrants; and (iv) Mr. Tonna: $90,000 for 30,000 shares and 30,000 warrants.
In August 2017, we borrowed $125,000 from Robert Austin Sperling, Jr., one of our then principal stockholders. The promissory note evidencing the loan provided for the payment of the principal amount, together with interest at the rate of 12% per annum, on May 23, 2018. In the event that, prior to the maturity date of the note, we receive net proceeds of $10,000,000 from a single equity or debt financing (as opposed to a series of related or unrelated financings), Mr. Sperling has the right to require that we prepay the amount due under the note (subject to the consent of the party that provided the particular financing). In consideration of the loan, we issued to Mr. Sperling a five-year warrant for the purchase of 15,000 shares of our common stock at an exercise price of $4.00 per share.
In February 2019, we borrowed $450,000 from Harvey P. Alstodt and Melody Alstodt. The convertible promissory note issued to the lenders provides for the payment of the principal amount, together with interest at the rate of 15% per annum, six months from the date of issuance. The note is convertible, at the option of the lenders, into shares of our common stock at a conversion price of $0.60 per share, subject to adjustment, and a five year warrant for the purchase of a number of shares equal to the number of shares issued upon the conversion of the principal amount of the note. The warrant provides for an exercise price of $0.80 per share, subject to adjustment. The lenders are the parents of Lance Alstodt, our Executive Vice President and Chief Strategy Officer.
In March 2019, our Board of Directors reduced the exercise price of outstanding options for the purchase of an aggregate of 4,631,700 shares of our common stock (with exercise prices ranging between $1.00 and $4.70 per share) to $0.75 per share, which was the closing price for our common stock on the day prior to the determination, as reported by the OTCQB market. The exercise price reduction related to options held by, among others, our Named Executive Officers and directors with respect to the following number of shares: (i) Mark Weinreb, our President, Chief Executive Officer and Chairman of the Board: 1,319,500 shares, (ii) A. Jeffrey Radov, one of our directors: 566,000 shares, (iii) Paul Jude Tonna, one of our directors: 364,000 shares, (iv) Dr. Charles S. Ryan, one of our directors: 256,000 shares, (v) Mr. Desmarais: 250,000 shares, (vi) Mr. Catell: 219,000 shares, (vii) Francisco Silva, our Vice President of Research and Development: 340,650 shares, and (viii) Robert Paccasassi, our Vice President of Quality and Compliance: 110,000 shares.
| 92 |
Director Independence
Board of Directors
Our Board of Directors is currently comprised of Mark Weinreb (Chair), Robert B. Catell, John M. Desmarais, A. Jeffrey Radov, Charles S. Ryan and Paul Jude Tonna. Each of Messrs. Catell, Desmarais, Radov, Ryan and Tonna is currently an “independent director” based on the definition of independence in Listing Rule 5605(a)(2) of The Nasdaq Stock Market.
Audit Committee
The members of our Board’s Audit Committee currently are Messrs. Radov (Chair), Ryan and Tonna, each of whom is an “independent director” based on the definition of independence in Listing Rule 5605(a)(2) The Nasdaq Stock Market and Rule 10A-3(b)(1) under the Exchange Act.
Nominating Committee
The members of our Board’s Nominating Committee currently are Messrs. Ryan (Chair), Radov and Tonna, each of whom is an “independent director” based on the definition of independence in Listing Rule 5605(a)(2) of The Nasdaq Stock Market.
Compensation Committee
The members of our Board’s Compensation Committee currently are Messrs. Tonna (Chair), Catell and Radov, each of whom is an “independent director” based on the definition of independence in Listing Rule 5605(a)(2) of The Nasdaq Stock Market.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES. |
Marcum LLP has served as our independent registered public accountants for the years ended December 31, 2018 and 2017.
The following is a summary of the fees billed or expected to be billed to us by Marcum LLP, our independent registered public accountants, for professional services rendered with respect to the fiscal years ended December 31, 2018 and 2017:
| 2018 | 2017 | |||||||
| Audit fees (1) | $ | 110,846 | $ | 144,551 | ||||
| Audit-related fees (2) | - | - | ||||||
| Tax fees (3) | 10,000 | 9,000 | ||||||
| All other fees (4) | - | - | ||||||
| $ | 120,846 | $ | 153,551 | |||||
| 93 |
| (1) | Audit Fees consist of fees billed and expected to be billed for services rendered for the audit of our consolidated financial statements for the fiscal years ended December 31, 2018 and 2017 and in connection with the filing of Forms S-1 and S-8 registration statements. |
| (2) | Audit-Related Fees consist of fees billed for assurance and related services that are reasonably related to the performance of the audit of our financial statements and are not reported under “Audit Fees.” |
| (3) | Tax Fees consist of fees billed for professional services related to preparation of our U.S. federal and state income tax returns and tax advice. |
| (4) | All Other Fees consist of fees billed for products and services provided by our independent registered public accountants, other than those disclosed above. |
The Audit Committee is responsible for the appointment, compensation and oversight of the work of the independent registered public accountants, and approves in advance any services to be performed by the independent registered public accountants, whether audit-related or not. The Audit Committee reviews each proposed engagement to determine whether the provision of services is compatible with maintaining the independence of the independent registered public accountants. The fees shown above were pre-approved either by our Board or our Audit Committee.
| 94 |
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES. |
| 95 |
| 96 |
| 97 |
| 98 |
| 99 |
| 100 |
| 101 |
| 102 |
* Filed herewith
** Furnished herewith
| ITEM 16. | FORM 10-K SUMMARY. |
Not applicable.
| 103 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| BIORESTORATIVE THERAPIES, INC. | ||
| Dated: March 29, 2019 | By: | /s/ Mark Weinreb |
|
Mark Weinreb Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Capacity | Date | ||
| /s/ Mark Weinreb | Chief Executive Officer, President, Chairman of the Board and Director | March 29, 2019 | ||
| Mark Weinreb | (Principal Executive Officer, Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Robert B. Catell | Director | March 29, 2019 | ||
| Robert B. Catell | ||||
| /s/ John M. Desmarais | Director | March 29, 2019 | ||
| John M. Desmarais | ||||
| /s/ A. Jeffrey Radov | Director | March 29, 2019 | ||
| A. Jeffrey Radov | ||||
| /s/ Charles S. Ryan | Director | March 29, 2019 | ||
| Charles S. Ryan | ||||
| /s/ Paul Jude Tonna | Director | March 29, 2019 | ||
| Paul Jude Tonna |
| 104 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
| 105 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
BioRestorative Therapies, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of BioRestorative Therapies, Inc. (the “Company”) as of December 31, 2018 and 2017, the related consolidated statements of operations, changes in stockholders’ deficiency and cash flows for each of the two years in the period ended December 31, 2018, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2018, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 2, the Company has a significant working capital deficiency, has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Marcum LLP
Marcum llp
We have served as the Company’s auditor since 2011.
New York, NY
March 29, 2019
| F-1 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Consolidated Balance Sheets
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash | $ | 117,523 | $ | 451,680 | ||||
| Accounts receivable | 29,000 | 38,000 | ||||||
| Prepaid expenses and other current assets | 34,464 | 30,030 | ||||||
| Total Current Assets | 180,987 | 519,710 | ||||||
| Property and equipment, net | 175,235 | 327,847 | ||||||
| Intangible assets, net | 814,059 | 888,950 | ||||||
| Security deposit | 22,100 | 22,100 | ||||||
| Total Assets | $ | 1,192,381 | $ | 1,758,607 | ||||
| Liabilities and Stockholders’ Deficiency | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 1,893,827 | $ | 2,454,944 | ||||
| Accrued expenses and other current liabilities | 2,302,176 | 1,885,551 | ||||||
| Accrued interest | 338,619 | 329,166 | ||||||
| Current portion of notes payable, net of debt discount of $936,866 and $336,229 at December 31, 2018 and 2017, respectively | 3,625,659 | 3,467,568 | ||||||
| Derivative liabilities | 1,094,607 | 216,073 | ||||||
| Total Current Liabilities | 9,254,888 | 8,353,302 | ||||||
| Accrued expenses, non-current portion | 36,500 | 38,000 | ||||||
| Accrued interest, non-current portion | 18,137 | 9,591 | ||||||
| Notes payable, non-current portion, net of debt discount of $75,497 and $1,256 at December 31, 2018 and 2017, respectively | 523,894 | 194,282 | ||||||
| Total Liabilities | 9,833,419 | 8,595,175 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ Deficiency: | ||||||||
| Preferred stock, $0.01 par value; Authorized, 20,000,000 shares; None issued and outstanding at December 31, 2018 and 2017 | - | - | ||||||
| Common stock, $0.001 par value; Authorized, 75,000,000 shares; Issued and outstanding 11,728,394 and 6,112,473 shares at December 31, 2018 and 2017, respectively | 11,728 | 6,112 | ||||||
| Additional paid-in capital | 55,269,490 | 44,561,773 | ||||||
| Accumulated deficit | (63,922,256 | ) | (51,404,453 | ) | ||||
| Total Stockholders’ Deficiency | (8,641,038 | ) | (6,836,568 | ) | ||||
| Total Liabilities and Stockholders’ Deficiency | $ | 1,192,381 | $ | 1,758,607 | ||||
See Notes to these Consolidated Financial Statements
| F-2 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Consolidated Statements of Operations
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Revenues | $ | 111,000 | $ | 81,000 | ||||
| Operating Expenses | ||||||||
| Marketing and promotion | 352,204 | 65,455 | ||||||
| Consulting | 1,870,829 | 2,334,212 | ||||||
| Research and development | 1,513,150 | 2,152,433 | ||||||
| General and administrative | 4,022,469 | 3,903,184 | ||||||
| Total Operating Expenses | 7,758,652 | 8,455,284 | ||||||
| Loss From Operations | (7,647,652 | ) | (8,374,284 | ) | ||||
| Other Expense | ||||||||
| Interest expense | (932,187 | ) | (468,107 | ) | ||||
| Amortization of debt discount | (2,289,591 | ) | (619,266 | ) | ||||
| Loss on extinguishment of notes payable, net | (1,415,950 | ) | (59,938 | ) | ||||
| Change in fair value of derivative liabilities | (229,323 | ) | 107,039 | |||||
| Warrant modification expense | (3,100 | ) | (30,099 | ) | ||||
| Total Other Expense | (4,870,151 | ) | (1,070,371 | ) | ||||
| Net Loss | $ | (12,517,803 | ) | $ | (9,444,655 | ) | ||
| Net Loss Per Share | ||||||||
| - Basic and Diluted | $ | (1.64 | ) | $ | (1.74 | ) | ||
| Weighted Average Number of Common Shares Outstanding | ||||||||
| - Basic and Diluted | 7,630,112 | 5,422,389 | ||||||
See Notes to these Consolidated Financial Statements
| F-3 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Consolidated Statements of Changes in Stockholders’ Deficiency
For the Years Ended December 31, 2018 and 2017
| Additional | ||||||||||||||||||||
| Common Stock | Paid-In | Accumulated | ||||||||||||||||||
| Shares | Amount | Capital | Deficit | Total | ||||||||||||||||
| Balance - December 31, 2016 | 4,699,035 | $ | 4,699 | $ | 36,954,817 | $ | (41,959,798 | ) | $ | (5,000,282 | ) | |||||||||
| Shares and warrants issued for cash | 361,335 | 361 | 1,083,639 | - | 1,084,000 | |||||||||||||||
| Exercise of warrants for purchase of common stock | 460,625 | 461 | 995,789 | - | 996,250 | |||||||||||||||
| Conversion of notes payable and accrued interest into common stock | 243,441 | 243 | 524,291 | - | 524,534 | |||||||||||||||
| Shares and warrants issued in exchange for notes payable and accrued interest | 132,082 | 132 | 421,170 | - | 421,302 | |||||||||||||||
| Shares and warrants issued in satisfaction of accrued services | 165,002 | 165 | 588,427 | - | 588,592 | |||||||||||||||
| Shares and warrants issued or modified and recorded as debt discount in connection with notes payable | 40,953 | 41 | 257,015 | - | 257,056 | |||||||||||||||
| Reclassification of derivative liabilities to equity | - | - | 9,019 | - | 9,019 | |||||||||||||||
| Beneficial conversion features related to convertible notes payable | - | - | 11,991 | - | 11,991 | |||||||||||||||
| Warrant modifications | - | - | 114,821 | - | 114,821 | |||||||||||||||
| Stock-based compensation: | ||||||||||||||||||||
| - common stock | 10,000 | 10 | 19,990 | - | 20,000 | |||||||||||||||
| - options and warrants | - | - | 3,580,804 | - | 3,580,804 | |||||||||||||||
| Net loss | - | - | - | (9,444,655 | ) | (9,444,655 | ) | |||||||||||||
| Balance - December 31, 2017 | 6,112,473 | $ | 6,112 | $ | 44,561,773 | $ | (51,404,453 | ) | $ | (6,836,568 | ) | |||||||||
| Shares and warrants issued for cash | 70,000 | 70 | 99,930 | - | 100,000 | |||||||||||||||
| Exercise of warrants for purchase of common stock | 207,084 | 207 | 413,961 | - | 414,168 | |||||||||||||||
| Shares and warrants issued in satisfaction of accrued services | 75,250 | 75 | 80,113 | - | 80,188 | |||||||||||||||
| Conversion of notes payable and accrued interest into common stock | 97,424 | 97 | 110,539 | - | 110,636 | |||||||||||||||
| Shares issued in exchange of notes payable and accrued interest | 5,031,914 | 5,033 | 7,210,333 | - | 7,215,366 | |||||||||||||||
| Shares and warrants issued or modified and recorded as debt discount in connection with notes payable issuances or extensions | 99,249 | 99 | 384,336 | - | 384,435 | |||||||||||||||
| Beneficial conversion features related to convertible notes payable | - | - | 69,394 | - | 69,394 | |||||||||||||||
| Warrant modifications | - | - | 3,100 | - | 3,100 | |||||||||||||||
| Reclassification of derivative liabilities to equity | - | - | 105,187 | - | 105,187 | |||||||||||||||
| Stock-based compensation: | ||||||||||||||||||||
| - common stock | 35,000 | 35 | 52,465 | - | 52,500 | |||||||||||||||
| - options and warrants | - | - | 2,178,359 | - | 2,178,359 | |||||||||||||||
| Net loss | - | - | - | (12,517,803 | ) | (12,517,803 | ) | |||||||||||||
| Balance - December 31, 2018 | 11,728,394 | $ | 11,728 | $ | 55,269,490 | $ | (63,922,256 | ) | $ | (8,641,038 | ) | |||||||||
See Notes to these Consolidated Financial Statements
| F-4 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Consolidated Statements of Cash Flows
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Cash Flows From Operating Activities | ||||||||
| Net loss | $ | (12,517,803 | ) | $ | (9,444,655 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization of debt discount | 2,289,591 | 619,266 | ||||||
| Accretion of interest expense | 624,041 | 206,284 | ||||||
| Depreciation and amortization | 240,372 | 259,259 | ||||||
| Stock-based compensation | 2,399,385 | 3,600,804 | ||||||
| Loss on extinguishment of note payables, net | 1,415,950 | 59,938 | ||||||
| Gain (loss) on settlement of payables | (2,812 | ) | 100,895 | |||||
| Change in fair value of derivative liabilities | 229,323 | (107,039 | ) | |||||
| Consulting services provided in exchange for notes payable | 260,000 | - | ||||||
| Warrant modification expense | 3,100 | 30,099 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | 9,000 | (32,000 | ) | |||||
| Prepaid expenses and other current assets | (4,434 | ) | 5,548 | |||||
| Security deposit | - | 12,076 | ||||||
| Accounts payable | (516,117 | ) | 185,963 | |||||
| Accrued interest, expenses and other current liabilities | 465,775 | 649,741 | ||||||
| Total Adjustments | 7,413,174 | 5,590,834 | ||||||
| Net Cash Used In Operating Activities | (5,104,629 | ) | (3,853,821 | ) | ||||
| Cash Flows From Investing Activities | ||||||||
| Purchases of property and equipment | (12,869 | ) | (3,617 | ) | ||||
| Net Cash Used In Investing Activities | (12,869 | ) | (3,617 | ) | ||||
| Cash Flows From Financing Activities | ||||||||
| Proceeds from notes payable | 5,057,475 | 2,542,222 | ||||||
| Repayments of notes payable | (863,302 | ) | (330,176 | ) | ||||
| Advances from officers and a family member of an officer | 38,500 | 43,515 | ||||||
| Repayments of advances from officers, a director and a family member of an officer | (38,500 | ) | (58,515 | ) | ||||
| Proceeds from exercise of warrants | 414,168 | 996,250 | ||||||
| Sales of common stock and warrants for cash | 175,000 | 1,084,000 | ||||||
| Net Cash Provided By Financing Activities | 4,783,341 | 4,277,296 | ||||||
| Net (Decrease) Increase In Cash | (334,157 | ) | 419,858 | |||||
| Cash - Beginning | 451,680 | 31,822 | ||||||
| Cash - Ending | $ | 117,523 | $ | 451,680 | ||||
See Notes to these Consolidated Financial Statements
| F-5 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Consolidated Statements of Cash Flows - Continued
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Supplemental Disclosures of Cash Flow Information: | ||||||||
| Cash paid during the period for: | ||||||||
| Interest | $ | 44,787 | $ | 17,538 | ||||
| Non-cash investing and financing activities: | ||||||||
| Warrant modifications | $ | 3,100 | $ | 114,821 | ||||
| Shares and warrants issued or modified and recorded as debt discount in connection with notes payable issuances or extensions | $ | 384,435 | $ | 257,056 | ||||
| Shares issued in exchange for notes payable and accrued interest | $ | 7,215,366 | $ | 421,302 | ||||
| Conversion of notes payable and accrued interest into common stock | $ | 110,636 | $ | 524,534 | ||||
| Shares and warrants issued in satisfaction of accrued consulting and director services | $ | 80,188 | $ | 588,592 | ||||
| Reclassification of derivative liabilities to equity | $ | 105,187 | $ | 9,019 | ||||
| Bifurcated embedded conversion options recorded as debt discount | $ | 3,181,376 | $ | 332,131 | ||||
| Beneficial conversion features recorded as debt discount | $ | 69,394 | $ | 11,991 | ||||
| Consulting services provided in exchange for notes payable | $ | 260,000 | $ | - | ||||
| Sale of warrants recorded as derivative liabilities | $ | 75,000 | $ | - | ||||
| Warrants and options issued for consulting services recorded as derivative liabilities | $ | 168,526 | $ | - | ||||
| Write-offs of fully depreciated property recorded as derivative liabilities and equipment | $ | 101,423 | $ | - | ||||
See Notes to these Consolidated Financial Statements
| F-6 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 1 – Business Organization and Nature of Operations
BioRestorative Therapies, Inc. has one wholly-owned subsidiary, Stem Pearls, LLC (“Stem Pearls”). Stem Cell Cayman Ltd. (“Cayman”), which was formed in the Cayman Islands as a wholly-owned subsidiary of the Company, was dissolved in March 2017. BioRestorative Therapies, Inc. and its subsidiaries are referred to collectively as “BRT” or the “Company” (See Note 3 – Summary of Significant Accounting Policies – Principles of Consolidation). BRT develops therapeutic products and medical therapies using cell and tissue protocols, primarily involving adult stem cells. BRT’s website is at www.biorestorative.com. BRT is currently developing a Disc/Spine Program referred to as “brtxDISC”. Its lead cell therapy candidate, BRTX-100, is a product formulated from autologous (or a person’s own) cultured mesenchymal stem cells collected from the patient’s bone marrow. The product is intended to be used for the non-surgical treatment of painful lumbosacral disc disorders. BRT is also engaging in research efforts with respect to a platform technology utilizing brown adipose (fat) for therapeutic purposes to treat type 2 diabetes, obesity and other metabolic disorders and has labeled this initiative its ThermoStem Program. Further, BRT has licensed a patented curved needle device that is a needle system designed to deliver cells and/or other therapeutic products or material to the spine and discs or other potential sites.
Note 2 – Going Concern and Management’s Plans
As of December 31, 2018, the Company had a working capital deficiency and a stockholders’ deficiency of $9,073,901 and $8,641,038, respectively. During the years ended December 31, 2018 and 2017, the Company incurred net losses of $12,517,803 and $9,444,655, respectively. These conditions indicate that there is substantial doubt about the Company’s ability to continue as a going concern within one year after the financial statement issuance date.
The Company’s primary source of operating funds since inception has been equity and debt financings. The Company intends to continue to raise additional capital through debt and equity financings. There is no assurance that these funds will be sufficient to enable the Company to fully complete its development activities or attain profitable operations. If the Company is unable to obtain such additional financing on a timely basis or, notwithstanding any request the Company may make, the Company’s debt holders do not agree to convert their notes into equity or extend the maturity dates of their notes, the Company may have to curtail its development, marketing and promotional activities, which would have a material adverse effect on the Company’s business, financial condition and results of operations, and ultimately the Company could be forced to discontinue its operations and liquidate.
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”), which contemplate continuation of the Company as a going concern and the realization of assets and satisfaction of liabilities in the normal course of business. The carrying amounts of assets and liabilities presented in the financial statements do not necessarily purport to represent realizable or settlement values. The consolidated financial statements do not include any adjustment that might result from the outcome of this uncertainty.
Subsequent to December 31, 2018, the Company has received aggregate equity financings and debt financings of $600,000 and $3,073,918, respectively, debt (inclusive of accrued interest) of $643,900 has been exchanged for common stock, $1,254,805 of debt (inclusive of accrued interest and prepayment premiums) has been repaid, and the due date for the repayment of an aggregate $155,000 of debt has been extended to December 2020. As a result, the Company expects to have the cash required to fund its operations through April 2019 while it continues to apply efforts to raise additional capital. While there can be no assurance that it will be successful, the Company is in negotiations to raise additional capital. As of the filing date of this report, the Company has notes payable with an aggregate principal balance of $107,500 which are past due. The Company is currently in the process of negotiating repayments or discussing conversions to equity with respect to one of these notes. However, there can be no assurance that the Company will be successful in repaying or converting the note. See Note 12 – Subsequent Events for additional details.
| F-7 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements of the Company include the accounts of Cayman and Stem Pearls. All significant intercompany transactions have been eliminated in the consolidation. As discussed above, Cayman, which had no material assets, liabilities or operations (other than intercompany balances) and is no longer needed to facilitate certain financings, was dissolved in March 2017.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at the dates of the financial statements and the reported amounts of revenue and expenses during the periods. The Company’s significant estimates and assumptions include the recoverability and useful lives of long-lived assets, the fair value of the Company’s stock, stock-based compensation, warrants issued in connection with notes payable, derivative liabilities and the valuation allowance related to the Company’s deferred tax assets. Certain of the Company’s estimates, including the carrying amount of the intangible assets, could be affected by external conditions, including those unique to the Company and general economic conditions. It is reasonably possible that these external factors could have an effect on the Company’s estimates and could cause actual results to differ from those estimates.
Concentrations
One license and the related royalties comprised all of the Company’s revenue during the years ended December 31, 2018 and 2017. See “Revenue Recognition” below.
During the year ended December 31, 2018, 23.1% of the Company’s debt financings were from one lender. During the year ended December 31, 2017, 30.9% and 13.7% respectively, of the Company’s debt financings were from two lenders.
Cash
The Company maintains cash in bank accounts, which, at times, may exceed Federal Deposit Insurance Corporation (“FDIC”) insured limits. The Company has not experienced any losses in such accounts, periodically evaluates the creditworthiness of the financial institutions and has determined the credit exposure to be negligible. As of December 31, 2018, the Company did not have cash balances in excess of FDIC insured limits. As of December 31, 2017, the Company had cash balances in excess of FDIC insured limits of $205,302. The Company considers all highly liquid investments with an original maturity of three months or less when purchased to be cash equivalents. As of December 31, 2018, and 2017 the Company did not have any cash equivalents.
Property and Equipment, net
Property and equipment are stated at cost, net of accumulated depreciation which is recorded commencing at the in-service date using the straight-line method at rates sufficient to charge the cost of depreciable assets to operations over their estimated useful lives, which range from 3 to 5 years. Leasehold improvements are amortized over the lesser of (a) the useful life of the asset; or (b) the remaining lease term. Maintenance and repairs are charged to operations as incurred. The Company capitalizes cost attributable to the betterment of property and equipment when such betterment extends the useful life of the assets.
Intangible Assets
Intangible assets are comprised of patents and trademarks and licenses with original estimated useful lives of 10 and 17.7 years, respectively. Once placed into service, the Company amortizes the cost of the intangible assets over their estimated useful lives on a straight-line basis.
| F-8 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies – Continued
Impairment of Long-lived Assets
The Company reviews for the impairment of long-lived assets whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company measures the carrying amount of the asset against the estimated undiscounted future cash flows associated with it. Should the sum of the expected future net cash flows be less than the carrying value of the asset being evaluated, an impairment loss would be recognized for the amount by which the carrying value of the asset exceeds its fair value. The evaluation of asset impairment requires the Company to make assumptions about future cash flows over the life of the asset being evaluated. These assumptions require significant judgment and actual results may differ from assumed and estimated amounts. While the Company’s near-term liquidity is tight, historically the Company has been successful in raising capital as needed (although there can be no assurance that the Company will continue to be successful in raising capital as needed). The Company continues to progress its scientific agenda and meet related milestones. The Company has not identified any impairment losses at December 31, 2018 and 2017.
Revenue Recognition
The Company recognizes sublicensing and royalty revenue when all of the following have occurred: (i) persuasive evidence of an arrangement exists, (ii) the service is completed without further obligation, (iii) the sales price to the customer is fixed or determinable, and (iv) collectability is reasonably assured. In November 2015, the Company and a stem cell treatment company (“SCTC”) entered into an amendment to a January 27, 2012 license agreement between them. Pursuant to the amendment, effective November 30, 2015, the Company granted to the SCTC (i) a non-exclusive sublicense to use certain of the licensed intellectual property in one location outside the United States and (ii) a non-exclusive sublicense to use, and the right to sublicense to third parties the right to use, in certain locations in the United States, certain of the licensed intellectual property. In consideration of the sublicenses, the SCTC has agreed to pay the Company royalties on a per disc procedure basis. During the years ended December 31, 2018 and 2017, the Company recognized $111,000 and $81,000, respectively, of revenue related to the Company’s sublicense agreement.
Income Taxes
The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of items that have been included or excluded in the financial statements or tax returns. Deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (“temporary differences”) at enacted tax rates in effect for the years in which the temporary differences are expected to reverse.
The Company utilizes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return.
Management has evaluated and concluded that there were no material uncertain tax positions requiring recognition in the Company’s consolidated financial statements as of December 31, 2018 and 2017. The Company does not expect any significant changes in its unrecognized tax benefits within twelve months of the reporting date.
The Company’s policy is to classify assessments, if any, for tax related interest as interest expense and penalties as general and administrative expenses in the consolidated statements of operations.
| F-9 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies – Continued
Net Loss Per Common Share
Basic loss per common share is computed by dividing net loss by the weighted average number of vested common shares outstanding during the period. Diluted earnings per share reflects the potential dilution that could occur if securities or other instruments to issue common stock were exercised or converted into common stock.
The following securities are excluded from the calculation of weighted average dilutive common shares because their inclusion would have been anti-dilutive:
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Options | 4,703,785 | 3,122,202 | ||||||
| Warrants | 3,483,403 | 3,435,134 | ||||||
| Convertible notes | 9,200,062 | [1] | 1,411,762 | |||||
| Total potentially dilutive shares | 17,387,250 | 7,969,098 | ||||||
| [1] | As of December 31, 2018, many of the convertible notes had variable conversion prices and the shares issuable were estimated based on market conditions. Pursuant to the note agreements, there were 57,019,880 shares of common stock reserved for future note conversions. |
Stock-Based Compensation
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award. For employees, the fair value of the award is measured on the grant date and for non-employees, the fair value of the award is generally re-measured on vesting dates and interim financial reporting dates until the service period is complete. The fair value amount is then recognized over the period during which services are required to be provided in exchange for the award, usually the vesting period. Since the shares underlying the Company’s 2010 Equity Participation Plan (the “Plan”) are registered, the Company estimates the fair value of the awards granted under the Plan based on the market value of its freely tradable common stock as reported on the OTCQB market. The fair value of the Company’s restricted equity instruments was estimated by management based on observations of the cash sales prices of both restricted shares and freely tradable shares. Awards granted to directors are treated on the same basis as awards granted to employees. Upon the exercise of an option or warrant, the Company issues new shares of common stock out of its authorized shares.
Advertising
Advertising costs are charged to operations as incurred. For the years ended December 31, 2018 and 2017, the Company incurred advertising costs of $288,986 and $26,840, respectively. Advertising expense is reflected in marketing and promotion expenses in the consolidated statements of operations.
Research and Development
Research and development expenses are charged to operations as incurred. For the years ended December 31, 2018 and 2017, the Company incurred research and development expenses of $1,513,150 and $2,152,433, respectively.
| F-10 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies – Continued
Fair Value of Financial Instruments
The Company measures the fair value of financial assets and liabilities based on the guidance of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 820 “Fair Value Measurements and Disclosures” (“ASC 820”).
ASC 820 defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. ASC 820 also establishes a fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. ASC 820 describes three levels of inputs that may be used to measure fair value:
Level 1 — quoted prices in active markets for identical assets or liabilities
Level 2 — quoted prices for similar assets and liabilities in active markets or inputs that are observable
Level 3 — inputs that are unobservable (for example, cash flow modeling inputs based on assumptions)
The carrying amounts of accrued liabilities approximate fair value due to the short-term nature of these instruments. The carrying amounts of our short–term credit obligations approximate fair value because the effective yields on these obligations, which include contractual interest rates, taken together with other features such as concurrent issuance of warrants, are comparable to rates of returns for instruments of similar credit risk.
See Note 11 – Derivative Liabilities for additional details regarding the valuation technique and assumptions used in valuing Level 3 inputs.
Derivative Financial Instruments
The Company evaluates its convertible instruments to determine if those contracts or embedded components of those contracts qualify as derivative financial instruments to be separately accounted for in accordance with FASB ASC 815 “Derivatives and Hedging” (“ASC 815”). The accounting treatment of derivative financial instruments requires that the Company record embedded conversion options (“ECOs”) and any related freestanding instruments at their fair values as of the inception date of the agreement and at fair value as of each subsequent balance sheet date. Any change in fair value is recorded as non-operating, non-cash income or expense for each reporting period at each balance sheet date. Conversion options are recorded as a discount to the host instrument and are amortized as amortization of debt discount on the consolidated statements of operations over the life of the underlying instrument. The Company reassesses the classification of its derivative instruments at each balance sheet date. If the classification changes as a result of events during the period, the contract is reclassified as of the date of the event that caused the reclassification.
The Multinomial Lattice Model and Black-Scholes Model were used to estimate the fair value of the ECOs of convertible notes payable, warrants and stock options that are classified as derivative liabilities on the consolidated balance sheets. The models include subjective input assumptions that can materially affect the fair value estimates. The expected volatility is estimated based on the actual volatility during the most recent historical period of time equal to the weighted average life of the instruments.
Sequencing Policy
Under ASC 815-40-35, the Company follows a sequencing policy whereby, in the event that reclassification of contracts from equity to assets or liabilities is necessary pursuant to ASC 815 due to the Company’s inability to demonstrate it has sufficient authorized shares as a result of certain securities with a potentially indeterminable number of shares, shares will be allocated on the basis of the earliest issuance date of potentially dilutive instruments, with the earliest grants receiving the first allocation of shares. Pursuant to ASC 815, issuance of securities to the Company’s employees or directors are not subject to the sequencing policy.
| F-11 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies – Continued
Convertible Instruments
The Company bifurcates conversion options from their host instruments and accounts for them as free standing derivative financial instruments according to certain criteria. The criteria include circumstances in which (a) the economic characteristics and risks of the embedded derivative instrument are not clearly and closely related to the economic characteristics and risks of the host contract, (b) the hybrid instrument that embodies both the embedded derivative instrument and the host contract is not re-measured at fair value under otherwise applicable generally accepted accounting principles with changes in fair value reported in earnings as they occur and (c) a separate instrument with the same terms as the embedded derivative instrument would be considered a derivative instrument. An exception to this rule is when the host instrument is deemed to be conventional.
When the Company has determined that the embedded conversion options should not be bifurcated from their host instruments, the Company records, when necessary, discounts to convertible notes for the intrinsic value of conversion options embedded in debt instruments (the beneficial conversion feature) based upon the differences between the fair value of the underlying common stock at the commitment date of the note transaction and the effective conversion price embedded in the note. Debt discounts under these arrangements are amortized over the term of the related debt to their stated date of redemption.
Reclassification
Certain amounts in prior periods have been reclassified to conform to the current period presentation. These reclassifications had no effect on previously reported net loss.
Subsequent Events
The Company evaluates events that have occurred after the balance sheet date but before the financial statements are issued. Based upon the evaluation, the Company did not identify any recognized or non-recognized subsequent events that would have required adjustment or disclosure in the consolidated financial statements, except as disclosed.
Recently Issued Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update (“ASU”) No. 2014-09, “Revenue from Contracts with Customers (Topic 606)” (“ASU 2014-09”). ASU 2014-09 supersedes the revenue recognition requirements in ASC 605 - Revenue Recognition (“ASC 605”) and most industry-specific guidance throughout ASC 605. The FASB has issued numerous updates that provide clarification on a number of specific issues as well as requiring additional disclosures. The core principle of ASU 2014-09 requires that an entity recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. ASU 2014-09 defines a five-step process to achieve this core principle and, in doing so, it is possible more judgment and estimates may be required within the revenue recognition process than required under existing U.S. GAAP including identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. The guidance also requires enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue and cash flows arising from an entity’s contracts with customers. The guidance may be adopted through either retrospective application to all periods presented in the financial statements (full retrospective approach) or through a cumulative effect adjustment to retained earnings at the effective date (modified retrospective approach). The Company expects to adopt ASU 2014-09 using a modified retrospective approach effective as of January 1, 2019. The Company has completed an analysis and concluded that the adoption of ASU 2014-09 will not have an impact on the Company’s financial statements.
| F-12 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies – Continued
Recently Issued Accounting Pronouncements - Continued
In February 2016, the FASB issued ASU 2016-02, “Leases (Topic 842)” (“ASU 2016-02”). ASU 2016-02 requires that a lessee recognize the assets and liabilities that arise from operating leases. A lessee should recognize in the statement of financial position a liability to make lease payments (the lease liability) and a right-of-use asset representing its right to use the underlying asset for the lease term. For leases with a term of 12 months or less, a lessee is permitted to make an accounting policy election by class of underlying asset not to recognize lease assets and lease liabilities. In transition, lessees and lessors are required to recognize and measure leases at the beginning of the earliest period presented using a modified retrospective approach. This amendment will be effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. The FASB issued ASU No. 2018-10 “Codification Improvements to Topic 842, Leases” (“ASU 2018-10”), ASU No. 2018-11 “Leases (Topic 842) Targeted Improvements” (“ASU 2018-11”) in July 2018, and ASU No. 2018-20 “Leases (Topic 842) - Narrow Scope Improvements for Lessors” (“ASU 2018-20”) in December 2018. ASU 2018-10 and ASU 2018-20 provide certain amendments that affect narrow aspects of the guidance issued in ASU 2016-02. ASU 2018-11 allows all entities adopting ASU 2016-02 to choose an additional (and optional) transition method of adoption, under which an entity initially applies the new leases standard at the adoption date and recognizes a cumulative-effect adjustment to the opening balance of retained earnings in the period of adoption. The Company is currently evaluating these ASUs and their impact on its consolidated financial statements.
In March 2016, the FASB issued ASU No. 2016-09, “Compensation – Stock Compensation (Topic 718)” (“ASU 2016-09”). ASU 2016-09 requires an entity to simplify several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. ASU 2016-09 is effective for fiscal years beginning after December 15, 2017, with early adoption permitted. The Company adopted this guidance on January 1, 2017. The adoption of this standard did not have a material impact on the Company’s financial statement disclosures.
In August 2016, the FASB issued ASU 2016-15, “Statement of Cash Flows (Topic 230) Classification of Certain Cash Receipts and Cash Payments” (“ASU 2016-15”). The new standard will make eight targeted changes to how cash receipts and cash payments are presented and classified in the statement of cash flows. The new standard is effective for fiscal years beginning after December 15, 2018. The Company will require adoption on a retrospective basis unless it is impracticable to apply, in which case the Company would be required to apply the amendments prospectively as of the earliest date practicable. The Company does not believe the adoption of ASU 2016-15 will have a material impact on its consolidated financial statements or disclosures.
In May 2017, the FASB issued ASU No. 2017-09, “Compensation—Stock Compensation (Topic 718)” (“ASU 2017-09”). ASU 2017-09 provides clarity on the accounting for modifications of stock-based awards. ASU 2017-09 requires adoption on a prospective basis in the annual and interim periods for the Company’s fiscal year ending December 31, 2017 for share-based payment awards modified on or after the adoption date. The adoption of this standard did not have a material impact on the Company’s financial statement disclosures.
In July 2017, the FASB issued ASU No. 2017-11, “Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815)”: (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception (“ASU 2017-11”). ASU 2017-11 allows companies to exclude a down round feature when determining whether a financial instrument (or embedded conversion feature) is considered indexed to the entity’s own stock. As a result, financial instruments (or embedded conversion features) with down round features may no longer be required to be accounted for as derivative liabilities. A company will recognize the value of a down round feature only when it is triggered and the strike price has been adjusted downward. For equity-classified freestanding financial instruments, an entity will treat the value of the effect of the down round as a dividend and a reduction of income available to common shareholders in computing basic earnings per share. For convertible instruments with embedded conversion features containing down round provisions, entities will recognize the value of the down round as a beneficial conversion discount to be amortized to earnings. ASU 2017-11 is effective for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years. Early adoption is permitted. The guidance in ASU 2017-11 can be applied using a full or modified retrospective approach. The Company does not believe the adoption of ASU 2017-11 will have a material impact on its consolidated financial statements or disclosures.
| F-13 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 3 – Summary of Significant Accounting Policies – Continued
Recently Issued Accounting Pronouncements - Continued
In June 2018, the FASB issued ASU No. 2018-07, “Compensation — Stock Compensation (Topic 718)” (“ASU 2018-07”). ASU 2018-07 is intended to reduce cost and complexity and to improve financial reporting for nonemployee share-based payments. Currently, the accounting requirements for nonemployee and employee share-based payment transactions are significantly different. ASU 2018-07 expands the scope of Topic 718, Compensation — Stock Compensation (which currently only includes share-based payments to employees) to include share-based payments issued to nonemployees for goods or services. Consequently, the accounting for share-based payments to nonemployees and employees will be substantially aligned. This ASU supersedes Subtopic 505-50, Equity — Equity-Based Payments to Nonemployees. The amendments in this ASU are effective for fiscal years beginning after December 15, 2019, and including interim periods within that fiscal year. Early adoption is permitted, but no earlier than a company’s adoption date of Topic 606, Revenue from Contracts with Customers. The Company is currently evaluating ASU 2018-07 and its impact on its consolidated financial statements.
In July 2018, the FASB issued ASU No. 2018-09, “Codification Improvements” (“ASU 2018-09”). These amendments provide clarifications and corrections to certain ASC subtopics including the following: Income Statement - Reporting Comprehensive Income – Overall (Topic 220-10), Debt - Modifications and Extinguishments (Topic 470-50), Distinguishing Liabilities from Equity – Overall (Topic 480-10), Compensation - Stock Compensation - Income Taxes (Topic 718-740), Business Combinations - Income Taxes (Topic 805-740), Derivatives and Hedging – Overall (Topic 815-10), and Fair Value Measurement – Overall (Topic 820-10). The majority of the amendments in ASU 2018-09 will be effective in annual periods beginning after December 15, 2019. The Company is currently evaluating and assessing the impact this guidance will have on its consolidated financial statements.
In August 2018, the FASB issued ASU No. 2018-13, “Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement (“ASU 2018-13”). The amendments in ASU 2018-13 modify the disclosure requirements associated with fair value measurements based on the concepts in the Concepts Statement, including the consideration of costs and benefits. The amendments on changes in unrealized gains and losses, the range and weighted average of significant unobservable inputs used to develop Level 3 fair value measurements, and the narrative description of measurement uncertainty should be applied prospectively for only the most recent interim or annual period presented in the initial fiscal year of adoption. All other amendments should be applied retrospectively to all periods presented upon their effective date. The amendments are effective for all entities for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years. Early adoption is permitted, including adoption in an interim period. The Company is currently evaluating ASU 2018-13 and its impact on its consolidated financial statements.
Note 4 – Property and Equipment, net
Property and equipment include the following:
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Medical equipment | $ | 345,963 | $ | 446,506 | ||||
| Furniture and fixtures | 120,925 | 121,625 | ||||||
| Computer software and equipment | 80,748 | 78,190 | ||||||
| Office equipment | 12,979 | 2,848 | ||||||
| Leasehold improvements | 304,661 | 304,661 | ||||||
| 865,276 | 953,830 | |||||||
| Less: accumulated depreciation | (690,041 | ) | (625,983 | ) | ||||
| Property and equipment, net | $ | 175,235 | $ | 327,847 | ||||
During the years ended December 31, 2018 and 2017, depreciation expense amounted to $165,481 and $184,365, respectively. Depreciation expense is reflected in general and administrative expenses and research and development expenses in the consolidated statements of operations. During the year ended December 31, 2018, the Company disposed of an aggregate of $101,423 of fully depreciated property and equipment.
| F-14 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 5 – Intangible Assets, net
The Company is a party to a license agreement with the SCTC (as amended) (the “SCTC Agreement”). Pursuant to the SCTC Agreement, the Company obtained, among other things, a worldwide, exclusive, royalty-bearing license from the SCTC to utilize or sublicense a certain medical device patent for the administration of specific cells and/or cell products to the disc and/or spine (and other parts of the body) and a worldwide (excluding Asia and Argentina), exclusive, royalty-bearing license to utilize or sublicense a certain method for culturing cells. Pursuant to the license agreement with the SCTC, unless certain performance milestones had been or are satisfied, the Company would have been required to pay to the SCTC $150,000 by April 2017 and will be required to pay to the SCTC an additional $250,000 by April 2019 in order to maintain its exclusive rights with regard to the disc/spine technology. In February 2017, the Company received authorization from the Food and Drug Administration (the “FDA”) to proceed with a Phase 2 clinical trial. Based upon such authorization, the Company has satisfied a performance milestone such that the Company was not required to pay to the SCTC a minimum amount of $150,000 by April 2017 to retain exclusive rights with regard to the disc/spine technology. In addition, the Company believes that it has until February 2022 to complete the Phase 2 clinical trial in order to satisfy the final performance milestone such that the Company would not be required to pay the additional $250,000 by April 2019 pursuant to the SCTC Agreement to maintain its exclusive rights.
Intangible assets consist of the following:
| Patents and Trademarks | Licenses | Accumulated Amortization | Total | |||||||||||||
| Balance as of January 1, 2017 | $ | 3,676 | $ | 1,301,500 | $ | (341,331 | ) | $ | 963,845 | |||||||
| Amortization expense | - | - | (74,895 | ) | (74,895 | ) | ||||||||||
| Balance as of December 31, 2017 | 3,676 | 1,301,500 | (416,226 | ) | 888,950 | |||||||||||
| Amortization expense | - | - | (74,891 | ) | (74,891 | ) | ||||||||||
| Balance as of December 31, 2018 | $ | 3,676 | $ | 1,301,500 | $ | (491,117 | ) | $ | 814,059 | |||||||
| Weighted average remaining amortization period at December 31, 2018 (in years) | 2.0 | 10.9 | ||||||||||||||
Amortization of intangible assets consists of the following:
| Patents and Trademarks | Licenses | Accumulated Amortization | ||||||||||
| Balance as of January 1, 2017 | $ | 2,208 | $ | 339,123 | $ | 341,331 | ||||||
| Amortization expense | 368 | 74,527 | 74,895 | |||||||||
| Balance as of December 31, 2017 | 2,576 | 413,650 | 416,226 | |||||||||
| Amortization expense | 368 | 74,523 | 74,891 | |||||||||
| Balance as of December 31, 2018 | $ | 2,944 | $ | 488,173 | $ | 491,117 | ||||||
Amortization expense is reflected in general and administrative expenses in the consolidated statements of operations. Based upon the current intangible assets as of December 31, 2018, amortization expense is projected to be approximately $75,000 per annum through 2029.
| F-15 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 6 – Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities are comprised of the following:
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Accrued payroll | $ | 91,560 | $ | 349,163 | ||||
| Accrued research and development expenses | 646,175 | 636,175 | ||||||
| Accrued general and administrative expenses | 1,084,831 | 605,318 | ||||||
| Accrued director compensation | 482,500 | 282,500 | ||||||
| Deferred rent | 33,610 | 50,395 | ||||||
| Total accrued expenses | 2,338,676 | 1,923,551 | ||||||
| Less: accrued expenses, current portion | 2,302,176 | 1,885,551 | ||||||
| Accrued expenses, non-current portion | $ | 36,500 | $ | 38,000 | ||||
During the year ended December 31, 2018, the Company received non-interest bearing advances in the aggregate amount of $38,500 from officers and a family member of an officer of the Company and repaid an aggregate of $38,500 of non-interest bearing advances from officers and a family member of an officer of the Company. During the year ended December 31, 2017, the Company received non-interest bearing advances in the amount of $43,515 from an officer and a family member of an officer of the Company and repaid an aggregate of $58,515, of which $15,000 was in accounts payable at December 31, 2016, of non-interest bearing advances from a director, an officer and a family member of an officer of the Company.
Effective March 1, 2017, the Company entered into an exchange agreement with the Chairman of the Company’s Scientific Advisory Board, pursuant to which an aggregate of $175,000 of accrued consulting fees were exchanged for 58,334 shares of common stock of the Company and, in consideration thereof, the Company issued to such person an immediately vested five-year warrant for the purchase of 58,334 shares of common stock of the Company at an exercise price of $4.00 per share. The common stock and warrants had an aggregate grant date value of $211,752 and, as a result, the Company recorded a loss on settlement of payables of $36,752 which is reflected within general and administrative expenses in the consolidated statements of operations.
Effective March 1, 2017, the Company entered into exchange agreements with four non-employee directors of the Company, pursuant to which an aggregate of $265,000 of accrued director fees were exchanged for an aggregate of 88,334 shares of common stock of the Company and, in consideration thereof, the Company issued to the directors immediately vested five-year warrants for the purchase of an aggregate of 88,334 shares of common stock of the Company at an exercise price of $4.00 per share. The aggregate value of the shares and warrants was $320,652, and accordingly the Company recorded a loss on settlement of payables of $55,652 which is reflected within general and administrative expenses in the consolidated statements of operations.
Effective July 18, 2017, the Company entered into an exchange agreement with a certain vendor of the Company, pursuant to which $17,697 of accounts payable were exchanged for 8,334 shares of common stock of the Company. In consideration thereof, the Company issued to the vendor immediately vested five-year warrants for the purchase of 2,000 shares of common stock of the Company at an exercise price of $4.00 per share. The aggregate value of the shares and warrants was $19,888, and accordingly the Company recorded a loss on settlement of payables of $2,191 which is reflected within general and administrative expenses in the consolidated statements of operations.
Effective December 12, 2018, the Company entered into an exchange agreement with a certain consultant of the Company, pursuant to which $45,000 of accounts payable were exchanged for 56,250 shares of common stock of the Company. The value of the shares was $42,188, and accordingly the Company recorded a gain on settlement of payables of $2,812 which is reflected within general and administrative expenses in the consolidated statements of operations.
See Note 9 – Commitments and Contingencies – Consulting Agreements and Note 12 –
Subsequent Events for details regarding additional exchanges of accrued consulting fees for shares of common stock and warrants.
| F-16 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable
A summary of the notes payable activity during the years ended December 31, 2018 and 2017 is presented below:
| Related Party | Convertible | Other | Debt | |||||||||||||||||
| Notes | Notes | Notes | Discount | Total | ||||||||||||||||
| Outstanding, December 31, 2016 | $ | 827,500 | $ | 390,000 | $ | 1,119,065 | $ | (179,964 | ) | $ | 2,156,601 | |||||||||
| Issuances | 175,000 | 1,612,333 | 1,033,900 | - | 2,821,233 | |||||||||||||||
| Indebtedness satisfied via settlement | - | 637,250 | [1] | (637,250 | ) | - | - | |||||||||||||
| Exchanges for equity | (97,500 | ) | (50,000 | ) | (203,750 | ) | - | (351,250 | ) | |||||||||||
| Conversions to equity | - | (495,197 | ) | - | - | (495,197 | ) | |||||||||||||
| Repayments | (60,000 | ) | (69,176 | ) | (201,000 | ) | - | (330,176 | ) | |||||||||||
| Recognition of debt discount | - | - | - | (964,911 | ) | (964,911 | ) | |||||||||||||
| Accretion of interest expense | - | 4,660 | 13,500 | 188,124 | 206,284 | |||||||||||||||
| Amortization of debt discount | - | - | - | 619,266 | 619,266 | |||||||||||||||
| Outstanding, December 31, 2017 | $ | 845,000 | $ | 2,029,870 | [2] | $ | 1,124,465 | $ | (337,485 | ) | $ | 3,661,850 | ||||||||
| Issuances | - | 6,357,286 | [3] | 128,000 | - | 6,485,286 | ||||||||||||||
| Exchanges for equity | (95,000 | ) | (2,739,926 | ) | (1,047,247 | ) | 681,281 | (3,200,892 | ) | |||||||||||
| Conversions to equity | - | (105,000 | ) | - | - | (105,000 | ) | |||||||||||||
| Repayments | (30,000 | ) | (833,302 | ) | - | 61,001 | (802,301 | ) | ||||||||||||
| Extinguishment of notes payable | - | (407,295 | )[3] | (318,493 | )[3] | - | (725,788 | ) | ||||||||||||
| Recognition of debt discount | - | - | - | (4,077,234 | ) | (4,077,234 | ) | |||||||||||||
| Accretion of interest expense | - | 7,782 | 245,776 | 370,483 | 624,041 | |||||||||||||||
| Amortization of debt discount | - | - | - | 2,289,591 | 2,289,591 | |||||||||||||||
| Outstanding, December 31, 2018 | $ | 720,000 | $ | 4,309,415 | [2] | $ | 132,501 | $ | (1,012,363 | ) | $ | 4,149,553 | ||||||||
| Outstanding, December 31, 2017 | $ | 845,000 | $ | 2,029,870 | [2] | $ | 1,124,465 | $ | (337,485 | ) | $ | 3,661,850 | ||||||||
| Less: current portion, December 31, 2017 | (845,000 | ) | (1,834,332 | ) | (1,124,465 | ) | 336,229 | (3,467,568 | ) | |||||||||||
| Non-current portion, December 31, 2017 [4] | $ | - | $ | 195,538 | $ | - | $ | (1,256 | ) | $ | 194,282 | |||||||||
| Outstanding, December 31, 2018 | $ | 720,000 | $ | 4,309,415 | [2] | $ | 132,501 | $ | (1,012,363 | ) | $ | 4,149,553 | ||||||||
| Less: current portion, December 31, 2018 | (720,000 | ) | (3,710,024 | ) | (132,501 | ) | 936,866 | (3,625,659 | ) | |||||||||||
| Non-current portion, December 31, 2018 [4] | $ | - | $ | 599,391 | $ | - | $ | (75,497 | ) | $ | 523,894 | |||||||||
| F-17 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
| [1] | In connection with certain note extensions during the year ended December 31, 2017, the Company and a certain lender agreed to add embedded conversion options, permitting principal and the respective accrued interest to be convertible into shares of the Company’s common stock at the election of the lender any time until the balance has been paid in full. See Note 7 – Notes Payable – Convertible Notes and Note 11 – Derivative Liabilities for additional details regarding the embedded conversion options. | |
| [2] | As of December 31, 2018 and 2017, a portion of convertible notes with an aggregate principal balance of $2,374,415 and $1,777,788, respectively, was convertible into shares of common stock at the election of the holder any time immediately until the balance has been paid in full. As of December 31, 2018 and 2017, a portion of convertible notes with an aggregate principal balance of $0 and $252,082, respectively, was convertible into shares of common stock at the election of the Company near maturity. In the event the Company exercised that conversion right, the respective holder had the right to accelerate the conversion of up to $0 and $196,666 of principal into shares of common stock at December 31, 2018 and 2017, respectively, at the same conversion price. As of December 31, 2018, a portion of convertible notes with an aggregate principal balance of $1,935,000, which were not yet convertible, will become convertible into shares of the Company’s common stock at the election of the respective holder subsequent to December 31, 2018. | |
| [3] | During the year ended December 31, 2018, convertible notes in the aggregate principal amount of $725,788 were issued concurrently with the extinguishment of certain notes payable in the same aggregate principal amount. See below within Note 7 – Notes Payable – Conversions, Exchanges and Other for additional details. | |
| [4] | As of December 31, 2018 and 2017, the Company reclassified principal in the aggregate amount of $523,894 and $194,282, respectively (net of debt discount of $75,497 and $1,256, respectively), and accrued interest in the aggregate amount of $18,137 and $9,591, respectively, to notes payable, non-current portion, net of debt discount and accrued interest, non-current portion, respectively, on the consolidated balance sheets related to outstanding notes payable that were converted into or exchanged for shares of common stock subsequent to December 31, 2018 and 2017, respectively. See Note 12 – Subsequent Events for additional details regarding notes payable. |
Related Party Notes
As of December 31, 2018 and 2017, related party notes consisted of notes payable issued to certain directors of the Company, family members of an officer of the Company, and the Tuxis Trust (the “Trust”). A director and principal shareholder of the Company (the “Director/Principal Shareholder”) serves as a trustee of Trust, which was established for the benefit of his immediate family.
During the year ended December 31, 2017, the Company issued to the Director/Principal Shareholder a note in the principal amount of $175,000, which bears interest at a rate of 15% per annum payable and provided for a maturity date of December 1, 2017. In November 2017, the maturity date of the note was extended to December 1, 2018 as described below (subject to a Financing Acceleration, as defined below). The note is secured by the grant of a security interest in the Company’s equipment and intellectual property. In connection with the borrowing, the Company agreed that the payment of a note in the principal amount of $500,000 issued to the Trust (the “Trust Note”) is also secured by such security interest.
During the year ended December 31, 2017, the Company and the Trust agreed to extend the maturity date of the Trust Note from July 2017 to December 2017. As consideration of the extension, the Company increased the interest rate payable on the Trust Note from 10% to 15% per annum. During the year ended December 31, 2017, the maturity date of the Trust Note was further extended to December 1, 2018 as described below. In the event that, prior to maturity, the Company receives net proceeds of $10,000,000 from a single equity or debt financing (as opposed to a series of related or unrelated financings), the Trust has the right to require that the Company prepay the amount due under the Trust Note (subject to the consent of the party that provided the particular financing) (a “Financing Acceleration”).
| F-18 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
Related Party Notes - Continued
During the year ended December 31, 2017, the Company, the Trust and the Director/Principal Shareholder agreed to extend the maturity dates of the above notes payable with an aggregate principal balance of $675,000, that were near maturity, to December 1, 2018 (subject to a Financing Acceleration). In consideration of the note extensions, the Company reduced the exercise prices for an aggregate of 1,219,444 previously issued five-year warrants to purchase the Company’s common stock at prices ranging from $4.50 to $5.00 per share to a reduced exercise price of $4.00 per share. The incremental modification expense of $84,722 has been recorded as debt discount and is being amortized over the extended term of the notes. During the year ended December 31, 2018, the Company, the Trust and the Director/Principal Shareholder agreed to further extend the maturity dates of the above notes payable with an aggregate principal balance of $675,000, that were near maturity, to December 31, 2019 (subject to a Financing Acceleration). In consideration of one of the note extensions, the Company reduced the exercise prices for an aggregate of 844,444 previously issued five-year warrants to purchase the Company’s common stock from an exercise price of $4.00 per share to a reduced exercise price of $1.50 per share. The incremental modification expense of $244,889 has been recorded as debt discount and is being amortized over the extended term of the respective note. See Note 10 – Stockholders’ Deficiency for additional details regarding the warrant modification.
During the year ended December 31, 2017, the Company and a director of the Company agreed to extend the maturity date of a note payable with a principal balance of $50,000 from February 2017 to February 2018. In connection with the extension, the Company issued the director a five-year, immediately vested warrant to purchase 5,000 shares of common stock at an exercise price of $4.00 per share. The grant date fair value of the warrant of $8,050 was recorded as debt discount and is being amortized over the remaining term of the note.
During the year ended December 31, 2018, the Company and certain related parties agreed to further extend the maturity dates of notes payable with an aggregate principal balance of $140,000 from maturity dates ranging between August 2016 to February 2018 to new maturity dates ranging from July 2018 to December 2018. As of December 31, 2018, a certain related party note in the outstanding principal amount of $45,000 was past maturity.
During the year ended December 31, 2017, the Company and certain related party lenders agreed to exchange certain related party notes with an aggregate principal balance of $97,500 and aggregate accrued interest of $288 into an aggregate of 32,597 shares of common stock and immediately vested five-year warrants to purchase an aggregate of 32,597 shares of common stock at an exercise price of $4.00 per share. The common stock and warrants had an aggregate exchange date value of $118,328 and, as a result, the Company recorded a loss on extinguishment of notes payable of $20,540.
During the year ended December 31, 2018, the Company and certain related parties agreed to exchange certain notes with an aggregate principal balance of $95,000 for an aggregate of 76,000 shares of the Company’s common stock. The common stock had an aggregate exchange date value of $114,000 and, as a result, the Company recorded a loss on extinguishment of notes payable of $19,000.
During the years ended December 31, 2018 and 2017, the Company partially repaid certain related party notes in the aggregate principal amount of $30,000 and $60,000, respectively.
| F-19 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
Convertible Notes
Issuances
During the year ended December 31, 2017, the Company issued lenders convertible notes in the aggregate principal amount of $1,554,000, for aggregate gross proceeds of $1,415,970. The difference of $138,030 was recorded as an original issue discount and is being amortized over the terms of the respective notes. The convertible notes bore interest at rates ranging between 6% to 10% per annum payable at maturity with maturity dates ranging between November 2017 through July 2018. In connection with the issuance of these convertible notes, the Company issued a certain lender 8,000 shares of common stock. Additionally, in connection with the issuance of certain convertible notes, the Company issued certain lenders five-year warrants to purchase an aggregate 62,019 shares of the Company’s common stock at exercise prices ranging from $4.00 to $4.15 per share, subject to a mandatory redemption provision depending on the warrant. The aggregate relative fair value of the common stock and warrants was $104,402, which was recorded as a debt discount and is being amortized over the terms of the respective convertible notes. See Note 11 – Derivative Liabilities for details regarding the mandatory redemption provision. In connection with certain convertible notes, the Company incurred $13,750 of debt issuance costs which is being amortized over the terms of the respective notes.
During the year ended December 31, 2017, the Company issued a lender a note payable in the principal amount of $83,333 of which $25,000 of principal bore no interest and $58,333 of principal bore interest at 10% per annum and was convertible into common stock. In connection with the issuance of the note, the Company received gross proceeds of $75,000, and the difference of $8,333 has been recorded as an original issue discount and will be amortized over the term of the note. The note provided for payment as follows: (i) $25,000 of principal (classified as an other note herein), which bore no interest and was not convertible into common stock, was payable three weeks from the issuance date, (ii) $11,667 of principal and the respective interest on such principal was payable six months from the issuance date (the “First Maturity Date”), (iii) $11,667 of principal and the respective interest on such principal was payable two weeks following the First Maturity Date, (iv) $11,667 of principal and the respective accrued interest on such principal was payable four weeks following the First Maturity Date, (v) $11,667 of principal and the respective interest on such principal was payable six weeks following the First Maturity Date, and (vi) $11,667 of principal and the respective interest on such principal was payable eight weeks following the First Maturity Date. In connection with the issuance of this note, the Company issued the lender 3,500 shares of common stock with a relative fair value of $6,458 which was recorded as an original issue discount and is being amortized over the term of the note.
During the year ended December 31, 2018, the Company issued certain lenders and a consultant convertible notes payable in the aggregate principal amount of $5,631,498 for aggregate cash proceeds of $4,947,475. The difference of $684,025 was recorded as follows: (i) $424,023 was recorded as a debt discount and will be amortized over the terms of the respective notes and (ii) $260,000 was recognized as consulting expense in the consolidated financial statements for services performed during the period. See Note 9 – Commitments and Contingencies for additional details regarding convertible notes issued in connection with consulting services. The convertible notes bear interest at rates ranging between 6% and 15% per annum payable at maturity with original maturity dates ranging between June 2018 through December 2019. In connection with the issuance of certain convertible notes, the Company issued the lenders an aggregate of 53,249 shares of the Company’s common stock and the relative fair value of $60,925 was recorded as debt discount and is being amortized over the terms of the respective notes. See below within Note 7 – Notes Payable – Conversions, Exchanges and Other and Note 11 – Derivative Liabilities for additional details regarding the ECOs of the convertible notes.
During the year ended December 31, 2018, convertible notes in the aggregate principal amount of $725,788 were issued concurrently with the extinguishment of certain convertible and other notes payable in the same aggregate principal amount. See below within Note 7 – Notes Payable – Conversions, Exchanges and Other for additional details.
| F-20 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
Convertible Notes - Continued
Embedded Conversion Options and Note Provisions
As of December 31, 2018, outstanding convertible notes in the aggregate principal amount of $2,374,415 were convertible into shares of common stock of the Company as follows: (i) $920,000 of aggregate convertible notes were convertible at a fixed price ranging from $1.00 to $2.00 per share for the first six months following the respective issue date, thereafter, at a conversion price equal to 58% of the fair value of the Company’s stock, subject to adjustment, until the respective note has been paid in full, (ii) $350,000 of convertible notes were convertible at a fixed conversion price of $2.15 per share, (iii) $100,000 of convertible notes were convertible at the greater of (a) 60% of the fair value of the Company’s stock or (b) $1.00 per share, (iv) $904,415 of aggregate convertible notes were convertible at a range of 58% to 65% of the fair value of the Company’s stock (subject to adjustment), depending on the note, and (v) $100,000 of convertible notes were convertible into shares of common stock of the Company at a conversion price of $0.60 per share, subject to adjustment, and a five year warrant (the “Warrant”) for the purchase of a number of shares equal to the number of shares issued upon the conversion of the principal amount of the Note. The Warrant provides for an exercise price of $0.80 per share, subject to adjustment. The Company analyzes the ECOs of its convertible notes at issuance to determine whether the ECO should be bifurcated and accounted for as a derivative liability or if the ECO contains a beneficial conversion feature. See below within Note 7 – Notes Payable – Convertible Notes and Note 11 – Derivative Liabilities for additional details regarding the embedded conversion options of the convertible notes.
As of December 31, 2018, a portion of convertible notes with an aggregate principal balance of $1,935,000, which were not yet convertible, will become convertible into shares of the Company’s common stock subsequent to December 31, 2018, as follows: (i) $1,835,000 of aggregate convertible notes will generally become convertible at a conversion price equal to 58% of the fair value of the Company’s stock, subject to adjustment, until the respective note has been paid in full and (ii) $100,000 of convertible notes will become convertible at the greater of (a) 58% of the fair value of the Company’s stock or (b) $1.50 per share.
As of December 31, 2018, outstanding convertible notes in the aggregate principal amount of $69,978 have mandatory prepayment terms at the option of the holder (“MPOs”). Convertible notes issued with MPOs permit the respective holder to demand prepayment of the note, in cash, at a premium of 35% of the then outstanding principal balance and accrued interest during the period between 150 days to 179 days following the respective issuance date.
As of December 31, 2018, outstanding convertible notes in the aggregate principal amount of $2,798,493 have prepayment premiums, whereby, in the event that the Company elects to prepay certain notes during the first ninety-day period following the issue date, the respective holder is entitled to receive a prepayment premium of up to 35%, depending on the note, on the then outstanding principal balance including accrued interest. In the event that the Company prepays any of the notes during the second ninety-day period following the issue date, the respective holder is entitled to receive a prepayment premium of up to 40%, depending on the note, on the then outstanding principal balance including accrued interest. In the event that the Company prepays a certain note after the 180th day period following the issue date and prior to maturity, the holder is entitled to receive a prepayment premium of 50% on the then outstanding principal balance including accrued interest.
As of December 31, 2018, outstanding convertible notes in the aggregate principal amount of $1,849,978 have most favored nation (“MFN”) provisions, whereby, so long as such respective note is outstanding, upon any issuance by the Company of any security with certain identified provisions more favorable to the holder of such security, then at the respective holder’s option, those more favorable terms shall become a part of the transaction documents with the holder. As of December 31, 2018, notes with applicable MFN provisions were convertible using MFN conversion prices equal to 58% of the fair market value of the Company’s stock, as defined.
During the year ended December 31, 2018, the Company determined that certain ECOs of issued or extended convertible notes were derivative liabilities. The aggregate issuance date value of the bifurcated ECOs was $3,631,702, of which $3,181,376 was recorded as a debt discount and is being amortized over the terms of the respective convertible notes and $450,326 was recognized as part of an extinguishment loss as described below. See Note 11 – Derivative Liabilities for additional details.
| F-21 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
Convertible Notes - Continued
Embedded Conversion Options and Note Provisions - Continued
During the years ended December 31, 2018 and 2017, the contingently adjustable non-bifurcated, beneficial conversion features associated with certain convertible notes were resolved and such notes became convertible during the period. The Company estimated the intrinsic value of the beneficial conversion features based upon the difference between the fair value of the underlying common stock at the commitment date of the note transaction and the adjusted conversion price embedded in the convertible note. During the years ended December 31, 2018 and 2017, the Company recognized $69,394 and $11,991, respectively, related to the beneficial conversion feature as debt discount which was immediately amortized.
Conversions, Exchanges and Other
During the year ended December 31, 2017, the Company and a certain lender agreed to exchange a certain convertible note with a principal balance of $50,000 and accrued interest of $2,712 into 29,280 shares of common stock. The common stock had an exchange date value of $58,560 and, as a result, the Company recorded a loss on extinguishment of notes payable of $5,848.
During the year ended December 31, 2017, certain convertible notes with an aggregate principal balance of $495,197 and aggregate accrued interest of $29,338 were converted into an aggregate of 243,441 shares of common stock at conversion prices ranging from $1.75 to $2.77 per share at the election of either the Company or the respective lender.
During the year ended December 31, 2017, the Company and a lender agreed to multiple extensions of the maturity dates of notes payable with an aggregate principal balance of $637,250 with maturity dates that were near or at maturity to maturity dates ranging from December 1, 2017 through February 10, 2018. In connection with one of the note extensions, the Company issued the lender 2,500 shares of common stock. The issuance date fair value of the common stock of $5,000 has been recorded as a debt discount and is being amortized over the term of the note. Additionally, in connection with one of the extensions, the Company incurred an extension fee in the amount $8,500 which was accreted as interest expense and added to the principal balance of the note. Also, in connection with the note extensions, the Company increased the effective rate at which the notes bore interest from 0% to 8% on dates effective between August 2, 2017 and September 7, 2017. Furthermore, in connection with certain extensions, the Company and the lender agreed to add an aggregate $4,660 of incurred interest to the principal of the respective notes. Further, in connection with the note extensions, the Company added embedded conversion options, pursuant to which each payment of principal and the respective accrued interest is convertible into shares of the Company’s common stock at the election of the lender at any time until the balance has been paid in full at a conversion price equal to 80% of the fair market value of the Company’s stock (subject to reduction to 70% under certain circumstances); however, generally the conversion price could not be less than $1.00 per share. The embedded conversion options of the notes were determined to be derivative liabilities. The aggregate issuance date value of the embedded conversion options was $252,117, which was recorded as a debt discount and is being amortized over the terms of the respective convertible notes. See Note 11 – Derivative Liabilities for additional details.
During the year ended December 31, 2017, the Company repaid an aggregate principal amount of $69,176 of convertible notes.
During the year ended December 31, 2018, the Company and certain lenders exchanged certain convertible notes with bifurcated ECOs with an aggregate net carrying amount of $5,144,063 (including an aggregate of $2,058,645 of principal net of debt discount, $166,022 of accrued interest and $2,919,396 related to the separated ECOs accounted for as derivative liabilities) for an aggregate of 3,734,664 shares of the Company’s common stock at conversion prices ranging from $0.28 to $2.38 per share. The common stock had an aggregate exchange date value of $5,846,809 and, as a result, the Company recorded a loss on extinguishment of notes payable of $702,746. See Note 11 – Derivative Liabilities for additional details.
During the year ended December 31, 2018, the Company elected to convert certain convertible notes with an aggregate principal balance of $105,000 and aggregate accrued interest of $5,636 into an aggregate of 97,424 shares of the Company’s common stock at conversion prices ranging from $0.82 to $2.02 per share.
| F-22 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
Convertible Notes - Continued
Conversions, Exchanges and Other - Continued
During the year ended December 31, 2018, the Company repaid an aggregate principal amount of $833,302 of convertible notes payable, $44,787 of the respective aggregate accrued interest and an aggregate of $238,808 of prepayment premiums. As a result of the repayments, the Company recorded a loss on extinguishment of notes payable of $299,809 and an aggregate of $61,001 of the related debt discounts were extinguished.
During the year ended December 31, 2018, the Company and certain lenders agreed to multiple extensions of the maturity dates of notes payable with an aggregate principal balance of $681,445 from maturity dates ranging between December 2017 to July 2018 to new maturity dates ranging from April 2018 to September 2018. In consideration of the extensions, the Company issued a lender 4,500 shares of the Company’s common stock. The issuance date fair value of the common stock of $9,000 was recorded as debt discount and is being amortized over the remaining term of the note. See below within this Note 7 – Notes Payable – Conversions, Exchanges and Other and Note – 11 Derivative Liabilities for additional details regarding the ECOs of the convertible notes. As of December 31, 2018, there were no convertible notes payable past due.
During the year ended December 31, 2018, certain lenders to the Company acquired other promissory notes issued by the Company in the aggregate outstanding amount of $725,788 (inclusive of accreted interest of $76,272) from different lenders to the Company. The Company exchanged the acquired notes for new convertible notes in the aggregate principal amount of $725,788 which accrue interest at rates ranging between 8% to 12% per annum, payable on the respective maturity date ranging between August 2019 and November 2019. The ECOs of the notes were subject to sequencing and their issuance date fair value of $450,326 was accounted for as derivative liabilities (see Note 11 – Derivative Liabilities for additional details). Since the fair value of the new ECOs exceeded 10% of the respective principal amounts of the new notes, the note exchanges were accounted for as extinguishments, and accordingly the Company recognized a net loss on extinguishment of $248,891 in connection with the derecognition of the net carrying amount of $927,223 of the extinguished debt ($725,788 of aggregate principal and interest and the derivative liability carrying value of their ECOs of an aggregate of $201,435) and the issuance of the new convertible notes in the aggregate principal amount $725,788 plus the fair value of the new notes’ ECOs of an aggregate of $450,326.
Other Notes
Issuances
During the year ended December 31, 2017, the Company issued lenders other notes in the aggregate principal amount of $1,033,900 for aggregate gross proceeds of $915,000, and the difference of $118,900 has been recorded as an original issue discount and will be amortized over the terms of the respective notes (inclusive of $25,000 of principal of a note payable as discussed above in Note 7 – Notes Payable – Convertible Notes). The other notes bear interest at rates between 0% to 12% per annum payable at maturity. The other notes matured between dates in May 2017 to July 2018. In connection with the issuance of these other notes, the Company issued to certain lenders 22,653 shares of common stock and certain other lenders five-year warrants to purchase an aggregate of 55,000 shares of common stock at an exercise price of $4.00 per share. The aggregate relative fair value of the common stock and warrants of $116,248 was recorded as an original issue discount and is being amortized over the terms of the respective notes.
During the year ended December 31, 2018, the Company issued a lender three-month notes payable in the aggregate principal amount of $128,000, which bear no interest, for aggregate cash proceeds of $110,000. The $18,000 difference was recorded as debt discount and is being amortized over the terms of the respective notes. In connection with the issuances of the promissory notes, the Company issued the lender an aggregate of 6,500 shares of the Company’s common stock. The issuance date fair value of the common stock of $9,627 was recorded as debt discount and is being amortized over the terms of the respective notes.
| F-23 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 7 – Notes Payable – Continued
Other Notes - Continued
Exchanges and Other
During the year ended December 31, 2017, the Company and certain lenders agreed to exchange certain other notes with an aggregate principal balance of $203,750 and aggregate accrued interest of $7,114 into an aggregate of 70,205 shares of common stock and immediately vested five-year warrants to purchase an aggregate of 63,205 shares of common stock at an exercise price of $4.00 per share. In addition, in consideration of the exchange by certain lenders, the Company agreed to extend the expiration dates of certain warrants held by the lenders for the purchase of an aggregate of 18,000 shares of common stock of the Company at an exercise price of $4.00 per share, from expiration dates ranging from April 27, 2021 to January 31, 2022 to a new expiration date of February 8, 2022. The common stock, warrants, and warrant modification (which represents the incremental value of the modified warrant as compared to the original warrant value, both valued as of the modification date) had an aggregate exchange date value of $244,414 and, as a result, the Company recorded a loss on extinguishment of notes payable of $33,550.
During the year ended December 31, 2017, the Company and certain lenders agreed to extend other notes with an aggregate principal balance of $984,063, that were near or at maturity, to various dates through October 2018. In consideration of the extensions, the Company issued certain lenders an aggregate 4,300 shares of the Company’s common stock. Also, in connection with the extensions, the Company issued certain lenders five-year, immediately vested warrants to purchase an aggregate of 56,118 shares of the Company’s common stock at exercise prices ranging between $4.00 to $5.00 per share. The aggregate grant date fair value of the common stock and warrants of $96,910 has been recorded as debt discount and is being amortized over the terms of the respective notes. Additionally, in connection with one of the extensions, the Company incurred debt issuance costs in the amount $5,000 which was accreted as interest expense and added to the principal balance of the note.
During the year ended December 31, 2017, the Company and a lender agreed to extend other notes with an aggregate principal balance of $637,250 such that the notes also became convertible into shares of the Company’s common stock. See Note 7 – Notes Payable – Convertible Notes for additional details.
During the year ended December 31, 2017, the Company repaid an aggregate principal amount of $201,000 of other notes.
During the year ended December 31, 2018, the Company and certain lenders agreed to exchange certain notes with an aggregate principal balance of $1,047,247 and aggregate accrued interest of $61,802 for an aggregate of 1,221,250 shares of the Company’s common stock at exchange prices ranging from $0.72 to $1.50 per share. The common stock had an aggregate exchange date value of $1,254,557 and, as a result, the Company recorded a loss on extinguishment of notes payable of $145,508.
During the year ended December 31, 2018, the Company and certain lenders agreed to multiple extensions of the maturity dates of notes payable with an aggregate principal balance of $1,309,747 from maturity dates ranging between December 2017 to October 2018 to new maturity dates ranging from March 2018 to January 2019. In consideration of the extensions, the Company issued certain lenders an aggregate of 35,000 shares of the Company’s common stock. The aggregate issuance date fair value of the common stock of $60,000 was recorded as debt discount and is being amortized over the remaining terms of the respective notes. Additionally, in connection with a certain extension, the Company increased the stated rate at which the note bears interest, from 0% to 8% per annum, effective June 2018. Furthermore, in connection with certain of the extensions, the Company accreted an aggregate of $177,286 as interest expense to the principal balance of the respective note. As of December 31, 2018, principal of $7,500 of a certain other note payable was past due.
During the year ended December 31, 2018, a convertible promissory note in the principal amount of $318,493 was issued concurrently with the extinguishment of a certain other note payable in the same principal amount. See above within Note 7 – Notes Payable – Conversions, Exchanges and Other for additional details.
| F-24 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 8 – Income Taxes
The tax effects of temporary differences that give rise to deferred tax assets and liabilities are presented below:
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Deferred Tax Assets: | ||||||||
| Net operating loss carryforwards | $ | 4,401,000 | $ | 2,176,000 | ||||
| Stock-based compensation | 3,433,000 | 2,873,000 | ||||||
| Accruals | 6,000 | 48,000 | ||||||
| Research & development tax credits | 358,000 | 340,000 | ||||||
| Other | 1,000 | 1,000 | ||||||
| Gross deferred tax assets | 8,199,000 | 5,438,000 | ||||||
| Deferred Tax Liabilities: | ||||||||
| Fixed assets | (2,000 | ) | (34,000 | ) | ||||
| Intangible assets | (19,000 | ) | (16,000 | ) | ||||
| Gross deferred tax liabilities | (21,000 | ) | (50,000 | ) | ||||
| Net deferred tax assets | 8,178,000 | 5,388,000 | ||||||
| Valuation allowance | (8,178,000 | ) | (5,388,000 | ) | ||||
| Deferred tax asset, net of valuation allowance | $ | - | $ | - | ||||
| Changes in valuation allowance | $ | 2,790,000 | $ | (1,291,000 | ) | |||
The income tax provision (benefit) consists of the following:
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Federal: | ||||||||
| Current | $ | - | $ | - | ||||
| Deferred | (2,253,000 | ) | 1,385,000 | |||||
| State and local: | ||||||||
| Current | - | - | ||||||
| Deferred | (537,000 | ) | (94,000 | ) | ||||
| (2,790,000 | ) | 1,291,000 | ||||||
| Change in valuation allowance | 2,790,000 | (1,291,000 | ) | |||||
| Income tax provision (benefit) | $ | - | $ | - | ||||
| F-25 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 8 – Income Taxes – Continued
A reconciliation of the statutory federal income tax rate to the Company’s effective tax rate is as follows:
| For The Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Tax benefit at federal statutory rate | (21.0 | )% | (34.0 | )% | ||||
| State income taxes, net of federal benefit | (5.0 | )% | (4.0 | )% | ||||
| Permanent differences | 3.8 | % | 0.0 | % | ||||
| Change in tax rates | 0.0 | % | 24.7 | % | ||||
| Research & development tax credits | (0.1 | )% | (1.6 | )% | ||||
| Impact of Section 382 limits | 0.0 | % | 28.3 | % | ||||
| True-ups and other | 0.0 | % | 0.3 | % | ||||
| Change in valuation allowance | 22.3 | % | (13.7 | )% | ||||
| Effective income tax rate | 0.0 | % | 0.0 | % | ||||
The Company assesses the likelihood that deferred tax assets will be realized. To the extent that realization is not likely, a valuation allowance is established. Based upon the Company’s history of losses since inception, management believes that it is more likely than not that future benefits of deferred tax assets will not be realized.
At December 31, 2018 and 2017, the Company had approximately $16,900,000 and $8,400,000, respectively, of federal and state net operating losses that may be available to offset future taxable income. At December 31, 2018 approximately $8,400,000 of federal net operating losses will expire from 2029 to 2037 and approximately $8,500,000 have no expiration. In accordance with Section 382 of the Internal Revenue Code, the usage of the Company’s net operating loss carry forwards are subject to annual limitations due to several greater than 50% ownership changes. The Section 382 limitations result in approximately $28,200,000 of federal NOLs not being realizable as of December 31, 2018 and the cumulative reversal of approximately $9,600,000 of net operating loss deferred tax assets.
The Company files income tax returns in the U.S. federal jurisdiction and the state of New York (also formerly Florida where the Company filed its final return in 2015), which remain subject to examination by the various taxing authorities beginning with the tax year ended December 31, 2015 (or the tax year ended December 31, 2009 if the Company were to utilize its NOLs). No tax audits were commenced or were in process during the years ended December 31, 2018 and 2017.
The Tax Cuts and Jobs Act tax reform legislation (the “Act”) was enacted in December 2017 making significant changes to the Internal Revenue Code. Changes include but are not limited to (a) the reduction of the U.S. corporate income tax rate from 35% to 21% for tax years beginning after December 31, 2017; (b) the transition of U.S. international taxation from a worldwide tax system to a territorial system; and (c) a one-time transition tax on the mandatory deemed repatriation of foreign earnings. The latter two changes are not expected to impact the Company as its Cayman subsidiary generated cumulative losses and was dissolved in March 2017. The change in tax law required the Company to remeasure existing net deferred tax assets using the lower rate in the period of enactment resulting in an income tax expense of approximately $2.3 million which is fully offset by the corresponding tax benefit of $2.3 million from the reduction in the valuation allowance in the year ended December 31, 2017. There were no specific impacts of the Act that could not be reasonably estimated which the Company accounted for under the prior tax law.
| F-26 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 9 – Commitments and Contingencies
Operating Lease
The Company is a party to a lease for 6,800 square feet of space located in Melville, New York (the “Melville Lease”) with respect to its corporate and laboratory operations. The Melville Lease expires in March 2020 (subject to extension at the option of the Company for a period of five years) and calls for an annual base rental during the initial term ranging between $132,600 and $149,260. The aggregate base rent payable over the lease term will be recognized on a straight-line basis. In connection with the operating lease, the Company paid the landlord a security deposit of $45,900, of which $12,076 and $11,724 were applied as rent payments in 2018 and 2017, respectively.
During the years ended December 31, 2018 and 2017, the Company received a credit of $12,991 and $21,237, respectively, towards its rent payments in connection with a tax rebate received by the landlord. The Company’s rent expense amounted to $122,739 and $115,885 for the years ended December 31, 2018 and 2017, respectively. Rent expense is reflected in general and administrative expenses and research and development expenses in the consolidated statements of operations.
Future minimum payments under this operating lease agreement is as follows:
| For the Years Ending | ||||
| December 31, | Amount | |||
| 2019 | $ | 147,257 | ||
| 2020 | 37,315 | |||
| $ | 184,572 | |||
Consulting Agreements
In March 2017, a previously expired agreement for business advisory services was further amended and the agreement was reinstated effective January 1, 2017. The agreement provided for an expiration date of December 31, 2017 (the “New Business Advisory Extended Term”). In consideration of the extension of the term of the consulting agreement, the Company issued to the consultant an immediately vested five-year warrant for the purchase of 25,000 shares of common stock of the Company. See Note 10 – Stockholders’ Deficiency – Stock Warrants for details associated with the issuance of warrants as compensation. Concurrently, the Company entered into an exchange agreement with the consultant pursuant to which $30,000 of accrued consulting fees were exchanged for 10,000 shares of common stock of the Company and, in consideration thereof, the Company issued to the consultant an immediately vested five-year warrant for the purchase of 10,000 shares of common stock of the Company at an exercise price of $4.00 per share. The aggregate value of the shares and warrant was $36,300, and accordingly the Company recorded a loss on settlement of payables of $6,300 which is reflected within general and administrative expenses in the consolidated statements of operations. During each of the years ended December 31, 2018 and 2017, the Company recorded cash consulting fee expense of $180,000 related to the business advisory agreement. In January 2018, the term of the business advisory agreement was extended to December 31, 2018. In consideration of the extension of the term of the business advisory agreement, the Company issued to the consultant an immediately vested five-year warrant for the purchase of 30,000 shares of common stock of the Company at an exercise price of $4.00 per share. The aggregate grant date value of the warrant of $48,192 was recognized immediately as stock-based compensation expense which is reflected as consulting expense in the consolidated financial statements. Concurrently, the Company and the consultant agreed to exchange $38,000 of accrued consulting fees for 19,000 shares of common stock of the Company and a two-year warrant for the purchase of 4,750 shares of common stock of the Company at an exercise price of $4.00 per share, whose combined value is consistent with the carrying value of the liabilities being satisfied.
| F-27 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 9 – Commitments and Contingencies – Continued
Consulting Agreements - Continued
On July 10, 2018, as further amended on August 22, 2018 and October 25, 2018, the Company entered into a consulting agreement with a consultant for services through March 31, 2019. In consideration of the consulting services, the Company issued the consultant convertible notes in the aggregate principal amount of $260,000 which will be earned and recognized ratably over their respective consulting agreement term. During the year ended December 31, 2018, the Company recorded an aggregate $260,000 of marketing and promotion expense for services rendered with a corresponding credit to notes payable. The notes mature at dates between January 2019 and April 2019 and bear interest at the rate of 10% per annum, payable at maturity. Pursuant to the notes, the holder has the right, from time to time following the respective issue date, at its election, to convert all or part of the outstanding and earned principal and accrued interest into shares of common stock of the Company, at a price generally equal to the lesser of (i) $1.27 or $1.75 per share, depending on the note, and (ii) 65% of the fair market value of the Company’s common stock, as defined. The Company may prepay the notes prior to the maturity date provided the principal is prepaid in full, plus interest, plus a prepayment premium of 25% on the principal.
In July 2018, the Company and a consultant agreed to further extend a previously expired consulting agreement from May 2018 to December 2018. In consideration of the extension of the term of the consulting agreement, the Company issued to the consultant an immediately vested five-year warrant for the purchase of 35,000 shares of common stock of the Company at an exercise price of $4.00 per share. The aggregate grant date value of the warrant of $43,106 was recognized immediately as stock-based compensation expense which is reflected as consulting expense in the consolidated financial statements.
See Note 10 – Stockholders’ Deficiency – Warrant and Option Valuation and Note 10 – Stockholders’ Deficiency – Stock Warrants regarding details for the valuation of warrants and the Black-Scholes valuation assumptions.
Scientific Advisory Services
In July 2018 and December 2018, the Company entered into agreements with certain consultants to serve as members of its Scientific Advisory Board and provide advice and guidance in connection with scientific matters relating to the Company’s business. The agreements will continue until terminated by either the Company or the respective party for any reason upon ten days written notice. In connection with the agreements, the Company issued the advisors five-year and ten-year options to purchase up to an aggregate 100,000 shares of the Company’s common stock at exercise prices ranging between $1.25 to $1.70 per share. The options vest as follows: (i) an aggregate 50,000 options vested immediately and (ii) an aggregate 50,000 options vest on the one-year anniversary of the grant date. The options had an aggregate grant date value of $92,100 which is being amortized over the vesting term of the respective options. The options were subject to the Company’s sequencing policy and, as a result, were recorded as derivative liabilities. In addition, on each one-year anniversary of the respective agreement date (as long as the consultant remains engaged), options to purchase an additional 5,000 shares are to be granted to the respective consultant which shall be exercisable for a period of five years from the respective dates of grant at exercise prices equal to the fair market value of the Company’s common stock.
In October 2018, the Company entered into an agreement with a consultant to serve as Chairman of the Disc Advisory Committee of its Scientific Advisory Board (the “Disc Committee Chairman”) and provide advice and guidance in connection with scientific matters relating to the Company’s business. The agreement will continue until terminated by either party for any reason upon thirty days written notice. In connection with the agreement, the Company issued the Disc Committee Chairman a ten-year option to purchase up to 75,000 shares of the Company’s common stock at an exercise price of $1.80 per share. The option vests as follows: (i) 25,000 options vested immediately and (ii) 50,000 options vest upon the achievement of certain performance conditions. The option had a grant date value of $129,800 which is being recognized over the respective expected vesting period. The option was subject to the Company’s sequencing policy and, as a result, was recorded as a derivative liability.
See Note 10 - Stockholders’ Deficiency – Options and Note 11 – Derivative Liabilities for additional details.
| F-28 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 9 – Commitments and Contingencies – Continued
Litigations, Claims and Assessments
In the normal course of business, the Company may be involved in legal proceedings, claims and assessments arising in the ordinary course of business, and as of December 31, 2018, none are expected to materially impact the Company’s financial position.
The Company records legal costs associated with loss contingencies as incurred and accrues for all probable and estimable settlements.
Employment Agreements
Chief Executive Officer
The Company and its Chief Executive Officer (“CEO”) are parties to an employment agreement that expires on December 31, 2019. Pursuant to the employment agreement, as amended, in the event that (a) the CEO’s employment is terminated by the Company without cause, or (b) the CEO terminates his employment for “good reason” (each as defined in the employment agreement), or (c) the term of the CEO’s employment agreement is not extended beyond December 31, 2019 and within three months of such expiration date, his employment is terminated by the Company without “cause” or the CEO terminates his employment for any reason, the CEO would be entitled to receive severance in an amount equal to his then annual base salary and certain benefits, plus $100,000 (in lieu of bonus). Further, in the event that the CEO’s employment is terminated by the Company without cause, or the CEO terminates his employment for “good reason”, following a “change in control” (as defined in the employment agreement), the CEO would be entitled to receive severance in an amount equal to one and one-half times his then annual base salary and certain benefits, plus $300,000 (in lieu of bonus). Additionally, as part of the amended employment agreement, the CEO is entitled to new performance-based cash bonuses payable for the years ending December 31, 2018 and 2019, such that an aggregate of up to 50% of the CEO’s then annual base salary per annum could be earned for such year pursuant to the satisfaction of such goals. See below Note 9 – Commitments and Contingencies – Employment Agreements – Other for details regarding the CEO’s bonus accruals.
Former Senior VP
In January 2018, the Company entered into an employment agreement with its then Senior Vice President of Planning and Business Development (the “Former Senior VP”). In October 2018, the Former Senior VP resigned from the Company. The Former Senior VP was entitled to any accrued unpaid salary and unused vacation days that was payable to him through his termination date pursuant to his employment agreement. As of December 31, 2018, the Company paid such liability due to the Former Senior VP. Additionally, the Former Senior VP’s unvested option to purchase 500,000 shares was forfeited as of the termination date. See Note 10 – Stockholders’ Deficiency – Stock Options for additional details.
Executive Vice President
In October 2018, the Company entered into an employment agreement with its new Executive Vice President and Chief Strategy Officer (the “Executive VP”). Pursuant to the employment agreement, in the event of the termination of the Executive VP’s employment by the Company without “cause” or the resignation by the Executive VP for “good reason” (each as defined in the employment agreement), the Executive VP would be entitled to receive severance in an amount equal to six months of his then annual base salary. Additionally, in connection with the employment agreement, the Executive VP was granted a ten-year option to purchase up to 500,000 shares of the Company’s common stock at an exercise price of $1.42 per share. The option vests as follows: (i) 100,000 options vested immediately, (ii) 150,000 options vest upon the earlier of (a) the achievement of a certain performance condition or (b) the first anniversary of the date of grant, and (iii) 250,000 options vest on the second anniversary of the date of grant. The option had a grant date value of $677,200 which is being recognized over the respective expected vesting period.
| F-29 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 9 – Commitments and Contingencies – Continued
Employment Agreements - Continued
Other
In February 2017 and March 2017, the Company’s Compensation Committee and Board of Directors, respectively, approved the following associated with performance-based cash bonuses for certain of the Company’s officers and current employees: (i) new performance-based cash bonuses payable for the year ending December 31, 2017 such that an aggregate of up to $402,500 could be earned for such year pursuant to the satisfaction of such goals; and (ii) the amendment of the performance-based cash bonuses for the year ended December 31, 2016 such that an aggregate of up to $322,000 could be earned for such year pursuant to the satisfaction of such goals. Also, pursuant to the amendment of the performance-based cash bonuses, the Company’s officers and certain employees’ achievement date of 2016 milestones was extended from January 31, 2017 to July 31, 2017. As of December 31, 2018 and 2017, the Company accrued approximately $35,000 and $87,000, respectively, for 2016 bonus milestones which were achieved and $0 for 2017 bonus milestones since such milestones were deemed not probable to be achieved.
In May 2018, the Company’s Compensation Committee and Board of Directors, respectively, approved new performance-based cash bonuses payable for the year ending December 31, 2018 for certain of the Company’s officers and employees, such that, an aggregate of up to $400,938 could be earned for 2018 pursuant to the satisfaction of such goals. As of December 31, 2018, the Company accrued approximately $56,000 for 2018 bonus milestones which were achieved but remain unpaid.
As of December 31, 2018, three employees other than the CEO have “at-will” employment agreements with the Company that provide for aggregate cash severance payments of $368,750, payable over twelve months, upon involuntary termination. As of December 31, 2017, two employees other than the CEO have “at-will” employment agreements with the Company that provide for aggregate cash severance payments of $175,000, payable over twelve months, upon involuntary termination.
Note 10 – Stockholders’ Deficiency
Authorized Capital
As of December 31, 2018, the Company was authorized to issue 75,000,000 shares of common stock, $0.001 par value, and 20,000,000 shares of preferred stock, $0.01 par value. The holders of the Company’s common stock are entitled to one vote per share. Subject to the rights of holders of preferred stock, if any, the holders of common stock are entitled to receive ratably such dividends, if any, as may be declared by the Board of Directors out of legally available funds. Subject to the rights of holders of preferred stock, if any, upon liquidation, dissolution or winding up of the Company, holders of common stock are entitled to share ratably in all assets of the Company that are legally available for distribution. No preferred stock has been issued through December 31, 2018.
2010 Equity Participation Plan
During the year ended December 31, 2018, the Compensation Committee and the Company’s stockholders, respectively, approved an increase in the number of shares authorized to be issued pursuant to the Company’s 2010 Equity Participation Plan from 4,250,000 to 10,000,000. As of December 31, 2018, 5,204,132 shares were reserved for future grant under the Company’s 2010 Equity Participation Plan.
| F-30 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency - Continued
Warrant and Option Valuation
The Company has computed the fair value of warrants and options granted using the Black-Scholes option pricing model. Option forfeitures are estimated at the time of valuation and reduce expense ratably over the vesting period. This estimate will be adjusted periodically based on the extent to which actual option forfeitures differ, or are expected to differ, from the previous estimate, when it is material. The Company estimated forfeitures related to option grants at an annual rate ranging from 0% to 5% for options granted during the years ended December 31, 2018 and 2017. The expected term used for warrants and options issued to non-employees is the contractual life and the expected term used for options issued to employees and directors is the estimated period of time that options granted are expected to be outstanding. The Company utilizes the “simplified” method to develop an estimate of the expected term of “plain vanilla” employee option grants. The Company is utilizing an expected volatility figure based on a review of the historical volatilities, over a period of time, equivalent to the expected life of the instrument being valued, of similarly positioned public companies within its industry. The risk-free interest rate was determined from the implied yields from U.S. Treasury zero-coupon bonds with a remaining term consistent with the expected term of the instrument being valued.
Common Stock and Warrant Offerings
During the year ended December 31, 2017, the Company issued an aggregate of 361,335 shares of common stock of the Company and five-year immediately vested warrants to purchase an aggregate of 371,335 shares of common stock of the Company at an exercise price of $4.00 per share to investors for aggregate gross proceeds of $1,084,000. The warrants had an aggregate grant date fair value of $601,595.
During the year ended December 31, 2018, the Company issued an aggregate of 70,000 shares of common stock of the Company and five-year immediately vested warrants to purchase an aggregate of 70,000 shares of common stock of the Company at an exercise price of $3.50 per share to investors for aggregate gross proceeds of $175,000. The warrants had an aggregate grant date fair value of $87,300.
Compensatory Common Stock Issuances
See Note 6 – Accrued Expenses and Other Current Liabilities for details regarding exchanges of accrued expenses for shares of common stock and warrants to a consultant and certain directors of the Company. See Note 9 – Commitments and Contingencies for details regarding an exchange of accrued consulting fees for shares of common stock and warrants.
During the year ended December 31, 2017, the Company issued 10,000 shares of immediately vested common stock valued at $20,000 to a consultant for services rendered during the year.
During the year ended December 31, 2018, the Company issued 35,000 shares of immediately vested common stock valued at $52,500 to a consultant for services rendered during the year.
Stock Warrants
Warrant Compensation
During the year ended December 31, 2017, the Company extended a previously expired agreement with a consultant from January 1, 2017 to December 31, 2017. In connection with this extension, the Company issued to the consultant an immediately vested five-year warrant to purchase 25,000 shares of common stock at an exercise price of $4.00 per share. The issuance date fair value of $40,763 was immediately recognized as stock-based compensation expense which is reflected in consulting expense in the consolidated statements of operations.
During the year ended December 31, 2017, the Company extended a previously expired agreement with a consultant from January 1, 2017 to June 30, 2017. In connection with this extension, the Company issued a five-year immediately vested warrant to purchase 20,000 shares of common stock at an exercise price of $4.50 per share. The warrant grant date fair value of $30,440 was recognized immediately as stock-based compensation expense which is reflected as consulting expense in the consolidated statements of operations.
| F-31 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency - Continued
Stock Warrants - Continued
Warrant Compensation - Continued
During the year ended December 31, 2017, the Company issued an immediately vested five-year warrant to purchase 25,000 shares of common stock at an exercise price of $4.00 per share to a consultant for services rendered. The warrant grant date fair value of $40,275 was recognized immediately as stock-based compensation expense and is reflected as consulting expense in the consolidated statements of operations.
During the year ended December 31, 2017, the Company extended a previously expired agreement with a consultant from October 1, 2017 to May 31, 2018. In connection with this extension, the Company issued a five-year immediately vested warrant to purchase 35,000 shares of common stock at an exercise price of $4.00 per share. The warrant grant date fair value of $56,434 was recognized immediately as stock-based compensation expense which is reflected as consulting expense in the consolidated statements of operations.
During the year ended December 31, 2018, the Company issued an immediately vested five-year warrant to purchase 75,000 shares of common stock of the Company at an exercise price of $2.00 per share to a consultant for services rendered. The warrant grant date fair value of $46,658 was recognized immediately as stock-based compensation expense and is reflected as consulting expense in the consolidated statements of operations with a corresponding credit to derivative liabilities as a result of the warrant being subject to the Company’s sequencing policy. See Note 11 – Derivative Liabilities for additional details.
See Note 9 - Commitments and Contingencies for additional details associated with the issuance of common stock and warrants in connection with consulting agreement extensions.
The Company recorded stock–based compensation expense of $137,956 and $167,912 during the years ended December 31, 2018 and 2017, respectively, related to stock warrants issued as compensation, which is reflected as consulting expense in the consolidated statements of operations. As of December 31, 2018, there was no unrecognized stock-based compensation expense related to stock warrants.
Warrant Modifications and Exercises
During the year ended December 31, 2017, the Company issued an aggregate of 410,625 shares of common stock pursuant to the exercise of warrants for aggregate gross proceeds of $821,250. The shares were issued pursuant to a warrant repricing program under which the exercise price for certain outstanding and exercisable warrants for the purchase of shares of common stock of the Company was reduced to $2.00 per share (reduced from exercises prices ranging from $4.00 to $30.00 per share). In connection with the share issuances, the Company issued to the purchasers of such shares additional two-year warrants for the purchase of an aggregate of 102,656 shares of common stock of the Company at an exercise price of $4.00 per share. The Company recognized a warrant modification charge of $6,618 during the year ended December 31, 2017, which represents the incremental value of the modified warrants and additional warrants issued as compared to the original warrants, both valued as of the respective modification dates.
During the year ended December 31, 2017, with respect to a warrant held by an investor, the Company agreed that (i) the conditions to the exercisability of the warrant for tranches to purchase an aggregate of 35,000 shares were eliminated, such that the entire warrant to purchase 50,000 shares of common stock was exercisable, and (ii) the exercise price of the warrant was reduced from an exercise price of $30.00 per share to $3.50 per share. Concurrent with the modification of the warrant, the investor exercised the warrant in full for aggregate gross proceeds to the Company of $175,000. The Company recognized a warrant modification charge of $4,500 during the year ended December 31, 2017, which represents the incremental value of the modified warrant as compared to the original warrant, both valued as of the respective modification dates which is reflected in warrant modification expense in the consolidated statement of operations.
| F-32 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency – Continued
Stock Warrants - Continued
Warrant Modifications and Exercises - Continued
During the year ended December 31, 2017, with respect to warrants held by certain lenders, the Company agreed to extend the expiration dates of certain warrants to purchase an aggregate of 53,291 shares of the Company’s common stock and reduce the exercise price of certain warrants to purchase an aggregate of 1,233,931 shares of the Company’s common stock. The expiration dates of the warrants were extended from dates ranging between December 31, 2017 through December 29, 2021 to new expiration dates ranging between December 31, 2019 and June 28, 2022. The exercise price of certain warrants was reduced from an exercise price ranging between $4.50 and $10.00 per share to $4.00 per share. The Company recognized a warrant modification charge of $18,962 during the year ended December 31, 2017, which represents the incremental value of the modified warrants as compared to the original warrants, both valued as of the respective modification dates. The charge is reflected in warrant modification expense in the consolidated statements of operations. Of the warrants with the reduced exercise prices to purchase an aggregate 1,233,931 shares of the Company’s common stock, 1,219,444 of the warrants to purchase the Company’s common stock were reduced as consideration of extending the maturity dates of certain related party notes payable and are reflected as debt discount, net of notes payable in the consolidated balance sheet. See Note 7 – Notes Payable – Related Party Notes for details.
During the year ended December 31, 2018, the Company issued an aggregate of 207,084 shares of common stock pursuant to the exercise of warrants for aggregate gross proceeds of $414,168. The shares were issued pursuant to a warrant repricing program under which the exercise price for certain outstanding and exercisable warrants for the purchase of shares of common stock of the Company was reduced to $2.00 per share (reduced from exercises prices ranging from $4.00 to $5.00 per share). In connection with the share issuances, the Company issued to the purchasers of such shares additional two-year warrants for the purchase of an aggregate of 51,771 shares of common stock of the Company at an exercise price of $4.00 per share. The Company did not recognize a warrant modification charge as there was no incremental value of the modified warrants and additional warrants issued as compared to the original warrants, both valued as of the respective modification dates.
During the year ended December 31, 2018, the Company reduced the exercise price and extended the expiration date of a certain warrant held by an investor for the purchase of 10,000 shares of common stock of the Company. The exercise price of the warrant was reduced from $5.00 per share to $4.00 per share and the expiration date of the warrant was extended from May 2021 to May 2023. The Company recognized a warrant modification charge of $3,100, which represents the incremental value of the modified warrants as compared to the original warrants, both valued as of the respective modification dates which is reflected in warrant modification expense in the consolidated statements of operations.
During the year ended December 31, 2018, with respect to warrants held by a certain related party, the Company agreed to extend the expiration dates and reduce the exercise price of certain warrants to purchase an aggregate 844,444 shares of the Company’s common stock as consideration of extending the maturity dates of certain notes payable. The expiration dates of the warrants were extended from December 2018 to December 2019. The exercise prices of the warrants were reduced from $4.00 per share to $1.50 per share. The Company recognized a warrant modification charge of $244,889 during the year ended December 31, 2018, which represents the incremental value of the modified warrants as compared to the original warrants, both valued as of the respective modification dates. The incremental modification expense has been recorded as debt discount and is being amortized over the remaining extended term of the respective note. See Note 7 – Notes Payable – Related Party Notes for details.
| F-33 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency – Continued
Stock Warrants - Continued
Warrant Activity Summary
In applying the Black-Scholes option pricing model to warrants granted, the Company used the following assumptions:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Risk free interest rate | 1.92% - 2.91 | % | 1.74% - 2.07 | % | ||||
| Contractual term (years) | 1.98 - 5.00 | 2.00 - 5.00 | ||||||
| Expected volatility | 128% - 141 | % | 120% - 132 | % | ||||
| Expected dividends | 0.00 | % | 0.00 | % | ||||
The weighted average estimated fair value of the warrants granted during the years ended December 31, 2018 and 2017 was approximately $1.06 and $1.54 per share, respectively.
See Note 6 – Accrued Expenses and Other Current Liabilities for details regarding exchanges of accrued expenses for shares of common stock and warrants to a consultant and certain directors of the Company. See Note 7 – Notes Payable for details associated with the issuance of warrants in connection with note issuances and the exchange of notes payable. See Note 9 – Commitments and Contingencies – Consulting Agreements for details associated with the issuance of warrants as compensation. See Note 10 – Stockholders’ Deficiency – Common Stock and Warrant Offerings for details associated with the issuance of warrants in connection with common stock and warrant offerings.
A summary of the warrant activity during the year ended December 31, 2018 is presented below:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | Aggregate | ||||||||||||||
| Number of | Exercise | Life | Intrinsic | |||||||||||||
| Warrants | Price | In Years | Value | |||||||||||||
| Outstanding, December 31, 2017 | 3,435,134 | $ | 4.47 | |||||||||||||
| Issued | 266,521 | 3.31 | ||||||||||||||
| Exercised | (207,084 | ) | 2.00 | |||||||||||||
| Expired | (11,168 | ) | 42.72 | |||||||||||||
| Outstanding, December 31, 2018 | 3,483,403 | $ | 3.63 | 2.1 | $ | - | ||||||||||
| Exercisable, December 31, 2018 | 3,483,403 | $ | 3.63 | 2.1 | $ | - | ||||||||||
| F-34 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency – Continued
Stock Warrants - Continued
Warrant Activity Summary - Continued
The following table presents information related to stock warrants at December 31, 2018:
| Warrants Outstanding | Warrants Exercisable | |||||||||||
| Weighted | ||||||||||||
| Outstanding | Average | Exercisable | ||||||||||
| Exercise | Number of | Remaining Life | Number of | |||||||||
| Price | Warrants | In Years | Warrants | |||||||||
| $1.50 - $1.99 | 844,444 | 1.0 | 844,444 | |||||||||
| $2.00 - $2.99 | 75,000 | 4.8 | 75,000 | |||||||||
| $3.00 - $3.99 | 70,000 | 4.5 | 70,000 | |||||||||
| $4.00 - $4.99 | 2,179,635 | 2.4 | 2,179,635 | |||||||||
| $5.00 - $5.99 | 195,989 | 2.5 | 195,989 | |||||||||
| $6.00 - $7.99 | 40,000 | 1.6 | 40,000 | |||||||||
| $8.00 - $9.99 | 2,500 | 0.9 | 2,500 | |||||||||
| $10.00 - $14.99 | 40,400 | 1.2 | 40,400 | |||||||||
| $15.00 - $19.99 | 35,435 | 0.7 | 35,435 | |||||||||
| 3,483,403 | 2.1 | 3,483,403 | ||||||||||
Stock Options
In applying the Black-Scholes option pricing model to stock options granted, the Company used the following assumptions:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Risk free interest rate | 2.44% - 3.15 | % | 1.77% - 1.88 | % | ||||
| Expected term (years) | 5.00 - 10.00 | 5.50 - 6.00 | ||||||
| Expected volatility | 129% - 141 | % | 120% - 130 | % | ||||
| Expected dividends | 0.00 | % | 0.00 | % | ||||
The weighted average estimated fair value of the stock options granted during the years ended December 31, 2018 and 2017 was approximately $1.60 and $2.75 per share, respectively.
On February 14, 2017, the Compensation Committee reduced the exercise price of outstanding options for the purchase of an aggregate of 1,219,450 shares of common stock of the Company (with exercise prices ranging between $5.70 and $30.00 per share) to $4.70 per share, which was the closing price for the Company’s common stock on February 13, 2017, as reported by the OTCQB. The exercise price reduction related to options held by, among others, the Company’s executive officers and directors. The incremental value of the modified options compared to the original options, both valued as of the respective modification date, of $430,394 is being recognized over the vesting term of the options.
During the year ended December 31, 2017, the Company issued ten-year options to employees, directors, and an advisor of the Company to purchase an aggregate of 1,117,000 shares of common stock at exercise prices ranging between $2.80 to $3.35 per share. The options vest as follows: (i) options for the purchase of 283,336 shares vested immediately, (ii) options for the purchase of 372,338 shares vested on the one-year anniversary of the issuance date, (iii) options for the purchase of 372,332 shares vest on the two-year anniversary of the issuance date and (iv) options for the purchase of 88,994 shares vest on the three-year anniversary of the issuance date. The options had an aggregate grant date value of $3,070,600 which is being amortized over the vesting term of the respective options.
| F-35 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency – Continued
Stock Options - Continued
In January 2018, the Company granted a ten-year option to a consultant of the Company to purchase 10,000 shares of the Company’s common stock at an exercise price of $3.20 per share. The option vested ratably over three years on the issuance date anniversaries. The option had an aggregate grant date value of $33,700. During the year ended December 31, 2018, the option was forfeited in connection with the consultant’s termination and accordingly, no expense related to the option was recognized.
In January 2018, the Company granted the Former Senior VP a ten-year option to purchase 500,000 shares of the Company’s common stock at an exercise price of $3.40 per share. The option grant provided for vesting based upon the achievement of a certain performance condition. The grant date value of the option was $1,491,300, which was recognizable to the extent such milestone was deemed probable to occur. See Note 9 – Commitments and Contingencies for additional details regarding the Former Senior VP’s resignation and termination of the option.
In October 2018, the Company issued ten-year options to employees and directors of the Company to purchase an aggregate of 885,000 shares of common stock at an exercise price of $1.23 per share. The options vest as follows: (i) options for the purchase of 216,667 shares vested immediately, (ii) options for the purchase of 295,002 shares vest on the one-year anniversary of the issuance date, (iii) options for the purchase of 295,000 shares vest on the two-year anniversary of the issuance date and (iv) options for the purchase of 78,331 shares vest on the three-year anniversary of the issuance date. The options had an aggregate grant date value of $943,100 which is being amortized over the vesting term of the respective options.
In October 2018 and December 2018, the Company entered into agreements with certain members of its Scientific Advisory Board to provide advice and guidance in connection with scientific matters relating to the Company’s business. In connection with the agreements, the Company issued the advisors ten-year options to purchase up to an aggregate 110,000 shares of the Company’s common stock at an exercise price of $1.23 per share. The options vest ratably over three years on the issuance date anniversaries. The options had an aggregate grant date value of $125,800. The Company recognizes the fair value of the options as consulting expenses over the respective vesting terms of the options. The options were subject to the Company’s sequencing policy and, as a result, were recorded as derivative liabilities. The Company See Note 11 – Derivative Liabilities for additional details.
See Note 9 – Commitments and Contingencies for details regarding the issuance of options to certain Scientific Advisory Board members and the Executive VP.
A summary of the option activity during the year ended December 31, 2018 is presented below:
| Weighted | ||||||||||||||||
| Weighted | Average | |||||||||||||||
| Average | Remaining | Aggregate | ||||||||||||||
| Number of | Exercise | Life | Intrinsic | |||||||||||||
| Options | Price | In Years | Value | |||||||||||||
| Outstanding, December 31, 2017 | 3,122,202 | $ | 4.25 | |||||||||||||
| Granted | 2,180,000 | 1.81 | ||||||||||||||
| Forfeited | (598,417 | ) | 3.50 | |||||||||||||
| Outstanding, December 31, 2018 | 4,703,785 | $ | 3.21 | 8.0 | $ | - | ||||||||||
| Exercisable, December 31, 2018 | 2,952,460 | $ | 4.03 | 7.1 | $ | - | ||||||||||
| F-36 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 10 – Stockholders’ Deficiency – Continued
Stock Options - Continued
The following table presents information related to stock options at December 31, 2018:
| Options Outstanding | Options Exercisable | |||||||||||
| Weighted | ||||||||||||
| Outstanding | Average | Exercisable | ||||||||||
| Exercise | Number of | Remaining Life | Number of | |||||||||
| Price | Options | In Years | Options | |||||||||
| $1.00 - $1.99 | 1,670,000 | 9.6 | 391,667 | |||||||||
| $2.00 - $2.99 | 187,834 | 8.2 | 64,503 | |||||||||
| $3.00 - $3.99 | 1,615,334 | 7.9 | 1,268,673 | |||||||||
| $4.00 - $4.99 | 1,153,117 | 5.5 | 1,150,117 | |||||||||
| $5.00 - $5.99 | 5,000 | 5.5 | 5,000 | |||||||||
| $6.00 - $19.99 | 37,500 | 5.0 | 37,500 | |||||||||
| $20.00 - $30.00 | 35,000 | 3.2 | 35,000 | |||||||||
| 4,703,785 | 7.1 | 2,952,460 | ||||||||||
The following table presents information related to stock option expense:
| Weighted | ||||||||||||||||
| Average | ||||||||||||||||
| Remaining | ||||||||||||||||
| For the Years Ended | Unrecognized at | Amortization | ||||||||||||||
| December 31, | December 31, | Period | ||||||||||||||
| 2018 | 2017 | 2018 | (Years) | |||||||||||||
| Consulting | $ | 965,916 | $ | 1,558,392 | $ | 502,144 | 0.6 | |||||||||
| Research and development | 340,471 | 481,041 | 551,073 | 1.5 | ||||||||||||
| General and administrative | 902,542 | 1,373,459 | 963,729 | 1.0 | ||||||||||||
| $ | 2,208,929 | $ | 3,412,892 | $ | 2,016,946 | 1.0 | ||||||||||
| F-37 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 11 – Derivative Liabilities
The following table sets forth a summary of the changes in the fair value of Level 3 derivative liabilities that are measured at fair value on a recurring basis:
| Beginning balance as of January 1, 2017 | $ | - | ||
| Issuance of derivative liabilities | 332,131 | |||
| Reclassification of derivative liabilities to equity | (9,019 | ) | ||
| Change in fair value of derivative liabilities | (107,039 | ) | ||
| Ending balance as of December 31, 2017 | $ | 216,073 | ||
| Issuance of derivative liabilities | 3,875,231 | |||
| Extinguishment of derivative liabilities in connection with convertible note repayments and exchanges | (3,120,833 | ) | ||
| Change in fair value of derivative liabilities | 229,323 | |||
| Reclassification of derivative liabilities to equity | (105,187 | ) | ||
| Ending balance as of December 31, 2018 | $ | 1,094,607 |
In applying the Multinomial Lattice and Black-Scholes option pricing models to derivatives issued and outstanding during the years ended December 31, 2018 and 2017, the Company used the following assumptions:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Risk free interest rate | 1.22% - 2.94 | % | 1.22% - 2.07 | % | ||||
| Expected term (years) | 0.01 - 5.00 | 0.00 - 5.00 | ||||||
| Expected volatility | 100% - 208 | % | 123% - 130 | % | ||||
| Expected dividends | 0.00 | % | 0.00 | % | ||||
During the year ended December 31, 2018, the Company recorded new derivative liabilities in the aggregate amounts of $3,631,705, $121,657 and $121,869 related to the ECOs of certain convertible notes payable, warrants and stock options subject to sequencing, respectively. During the year ended December 31, 2017, the Company recorded new derivative liabilities in the aggregate amounts of $252,117 and $80,014 related to the ECOs of certain convertible notes payable and warrants, respectively. See Note 7 – Notes Payable – Convertible Notes and Other Notes for additional details. See Note 9 – Commitments and Contingencies for a stock option issued and deemed to be a derivative liability. See Note 10 – Stockholders’ Deficiency for warrants issued and deemed to be derivative liabilities.
During the year ended December 31, 2018, the Company extinguished an aggregate of $3,120,833 of derivative liabilities in connection with repayments and exchanges of certain convertible notes payable into shares of the Company’s common stock. See Note 7 – Notes Payable – Convertible Notes and Other Notes for additional details.
During the year ended December 31, 2017, the Company reclassified $9,019 of derivative liabilities to equity in connection with the conversion of convertible notes payable into shares of common stock.
During the year ended December 31, 2018, the Company reclassified an aggregate of $105,187 of derivative liabilities to equity as a result of a change in the sequencing status.
On December 31, 2018, the Company recomputed the fair value of ECOs recorded as derivative liabilities to be $852,454. The Company recorded a loss on the change in fair value of these derivative liabilities of $310,710 for the year ended December 31, 2018.
| F-38 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 11 – Derivative Liabilities - Continued
On December 31, 2018, the Company recomputed the fair value of the derivative liabilities related to outstanding warrants to be $120,284. These warrants are either redeemable for cash equal to the Black-Scholes value, as defined, at the election of the warrant holder upon a fundamental transaction pursuant to the warrant terms or were issued subsequent to the commencement of sequencing. The Company recorded a gain on the change in fair value of these derivative liabilities of $81,387 for the year ended December 31, 2018.
On December 31, 2018, the Company recomputed the fair value of the derivative liabilities related to outstanding consultant stock options to be $121,869. The stock options were issued subsequent to the commencement of sequencing and the fair value of the options are being recorded in consulting expenses in the consolidated statements of operations over the respective expected vesting period with a corresponding credit to derivative liabilities. See Note 10 – Stockholders’ Deficiency -Stock Options for additional details.
Note 12 – Subsequent Events
Stock Options
Subsequent to December 31, 2018, the Company issued a ten-year option to a certain Scientific Advisory Board member of the Company to purchase 70,000 shares of common stock of the Company at an exercise price of $1.00 per share. The option vests as follows: (i) an option for the purchase of 23,334 shares vested immediately, (ii) an option for the purchase of 23,333 shares will vest on the one-year anniversary of the issuance date, and (iii) an option for the purchase of 23,333 shares will vest on the two-year anniversary of the issuance date. The fair value of the option will be recognized over the vesting period.
Subsequent to December 31, 2018, the Board of Directors reduced the exercise price of outstanding stock options for the purchase of an aggregate of 4,631,700 shares of common stock of the Company (with exercise prices ranging between $1.00 and $4.70 per share) to $0.75 per share, which was the closing price for the Company’s common stock on the day prior to determination, as reported by the OTCQB market. The exercise price reduction related to options held by, among others, the Company’s directors, advisors and employees. The incremental value of the modified options compared to the original options, both valued as of the respective modification date, will be recognized over the vesting term of the options.
Consulting Agreement
Subsequent to December 31, 2018, the Company and a consultant agreed to further extend a previously expired consulting agreement from January 2019 to December 2019. In connection with the extension, the Company issued to the consultant a five-year, immediately vested warrant for the purchase of 100,000 shares of the Company’s common stock at an exercise price of $1.00 per share.
Settlement Agreement
Subsequent to December 31, 2018, the Company entered into a settlement agreement with a certain consultant, pursuant to which $46,500 of previously recorded consulting fees were exchanged for 10,000 shares of the Company’s common stock and a $10,000 cash payment.
Common Stock and Warrant Offering
Subsequent to December 31, 2018, the Company issued 1,000,000 shares of common stock of the Company, a five-year immediately vested warrant to purchase 500,000 shares of common stock of the Company at an exercise price of $0.85 per share and a one-year immediately vested warrant to purchase 500,000 shares of common stock of the Company at an exercise price of $0.70 per share to an investor for gross proceeds of $600,000.
| F-39 |
BIORESTORATIVE THERAPIES, INC. & SUBSIDIARIES
Notes to Consolidated Financial Statements
Note 12 - Subsequent Events – Continued
Notes Payable
Subsequent to December 31, 2018, the Company issued a convertible promissory note in the principal amount of $450,000 to certain related parties. The convertible note bears interest at the rate of 15% per annum, payable at maturity, with an original maturity date in August 2019. The note is convertible, at the option of the lenders, into shares of common stock of the Company at a conversion price of $0.60 per share, subject to adjustment, and a five-year warrant for the purchase of a number of shares equal to the number of shares issued upon the conversion of the principal amount of the note. The warrant provides for an exercise price of $0.80 per share, subject to adjustment.
Subsequent to December 31, 2018, the Company issued convertible promissory notes in the aggregate principal amount of $575,000 to certain lenders for aggregate cash proceeds of $575,000. The convertible notes bear interest at the rate of 15% per annum, payable at maturity, with original maturity dates in July 2019. Each note is convertible, at the option of the lender, into shares of common stock of the Company at a conversion price of $0.60 per share, subject to adjustment, and a five-year warrant for the purchase of a number of shares equal to the number of shares issued upon the conversion of the principal amount of the respective note. The warrant provides for an exercise price of $0.80 per share, subject to adjustment.
Subsequent to December 31, 2018, the Company issued convertible promissory notes in the aggregate principal amount of $2,205,000 for aggregate cash proceeds of $2,048,918. The convertible notes bear interest at rates ranging from 8% to 12% per annum, payable at maturity, with original maturity dates ranging between July 2019 to March 2020. The convertible notes are convertible as follows: (i) $805,000 of aggregate principal and the respective accrued interest is convertible into shares of the Company’s common stock at the election of the holder after the 180th day following the issue date at a conversion price generally equal to 58% of the fair value of the Company’s common stock, (ii) $170,000 of aggregate principal and the respective accrued interest is convertible into shares of the Company’s common stock at the election of the holder at any time immediately on or after the issue date until the 180th day following issuance at a conversion price equal to $0.25 per share or after the 180th day following issuance at a conversion price equal to 58% of the fair value of the Company’s common stock, and (iii) $1,230,000 of aggregate principal and the respective accrued interest is convertible into shares of the Company’s common stock at the election of the holder for the first six months at a fixed conversion price ranging from $1.00 to $2.00 per share, and thereafter, at a conversion price generally equal to 58% of the fair value of the Company’s common stock. In connection with the issuance of a certain convertible promissory note, the Company issued to the lender a five-year, immediately vested warrant for the purchase of 40,000 shares of the Company’s common stock at an exercise price of $1.00 per share. The grant date fair value of the warrant will be recorded as a debt discount and will be amortized over the term of the note. In the event that the Company elects to prepay any of the respective notes during the first ninety-day period following the issue date, the holder is entitled to receive a prepayment premium of up to 30%, depending on the note, of the then outstanding principal balance plus accrued interest. In the event that the Company elects to prepay any of the notes during the second ninety-day period following the issue date, the holder is entitled to receive a prepayment premium of up to 35%, depending on the note, of the then outstanding principal balance plus accrued interest.
Subsequent to December 31, 2018, a certain lender to the Company acquired another promissory note issued by the Company in the outstanding amount of $148,014 (inclusive of accreted interest of $23,014) from a different lender to the Company. The Company exchanged the acquired note for a new convertible note in the principal amount of $148,014 which accrues interest at a rate of 12% per annum, payable on the maturity date in March 2020.
Subsequent to December 31, 2018, the Company and a certain related party agreed to extend the maturity date of a certain promissory note with a principal balance of $30,000 that was past maturity from December 2018 to December 2019.
Subsequent to December 31, 2018, the Company and a certain lender agreed to extend the maturity date of a certain promissory note with a principal balance of $125,000 that was past maturity from January 2019 to December 2019. In connection with the extension, the Company issued the lender 10,000 shares of the Company’s common stock. The issuance date fair value of the common stock will be recorded as debt discount and will be amortized over the term of the note.
Subsequent to December 31, 2018, the Company and certain lenders agreed to exchange an aggregate principal amount of $619,391 and aggregate accrued interest of $24,509 of certain convertible notes payable for an aggregate of 1,928,400 shares of the Company’s common stock at exchange prices ranging from $0.28 to $0.42 per share.
Subsequent to December 31, 2018, the Company repaid an aggregate principal amount of $1,065,000 of notes payable, $55,169 of the respective aggregate accrued interest and an aggregate of $134,636 of prepayment premiums.
| F-40 |