As filed with the Securities and Exchange Commission on April 21, 2015
Registration No. 333-203357
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1/A
(Amendment No. 1)
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
DELMAR PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Nevada | 2834 | 99-0360497 | ||
(State
or other jurisdiction of |
(Primary Standard Industrial Classification Code Number) |
(I.R.S.
Employer |
Suite 720-999 West Broadway
Vancouver, British Columbia
Canada V5Z 1K5
(604) 629-5989
(Address, including zip code, and telephone number, including area code, of registrant's principal executive offices)
Jeffrey Bacha
Suite 720-999 West Broadway
Vancouver, British Columbia
Canada V5Z 1K5
(604) 629-5989
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Gregory Sichenzia, Esq. | Mitchell S. Nussbaum, Esq. |
| David B. Manno, Esq. Jeff Cahlon, Esq. Sichenzia Ross Friedman Ference LLP 61 Broadway, 32nd Floor New York, NY 10006 Telephone: (212) 930-9700 Facsimile: (212) 930-9725 |
Norwood P. Beveridge, Esq. Loeb & Loeb LLP 345 Park Avenue New York, NY 10154 Telephone: (212) 407-4000 Facsimile: (212) 407-4990 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. þ
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer £ | Accelerated filer £ | |
| Non-accelerated filer £ (Do not check if a smaller reporting company) | Smaller reporting company þ |
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to be Registered | Proposed Maximum Aggregate Offering Price (1) | Amount of Registration Fee | ||||||
| Common Stock, $0.001 par value per share (2)(3) | ||||||||
| Common Stock Purchase Warrants(3) | -- | -- | ||||||
| Shares of Common Stock underlying Common Stock Purchase Warrants (2) (3) | -- | -- | ||||||
| Underwriters’ Common Stock Purchase Warrants(4) | -- | -- | ||||||
| Shares of Common Stock underlying Underwriters’ Common Stock Purchase Warrants (2) (5) | ||||||||
| Total | $ | 12,650,000 | $ | 1,469.93 | (6) | |||
| (1) | Estimated solely for the purpose of calculating the amount of registration fee pursuant to Rule 457(o) under the Securities Act. |
| (2) | Pursuant to Rule 416 under the Securities Act, the securities being registered hereunder include such indeterminate number of additional shares of common stock as may be issued after the date hereof as a result of stock splits, stock dividends or similar transactions. |
| (3) | Includes securities the underwriters have the option to purchase to cover over-allotments, if any. |
| (4) | We have agreed to issue warrants exercisable within five years after the effective date of this registration statement representing an aggregate of 8% of the shares issued in the offering (the “Underwriters’ Warrants”) to certain of the Underwriters. Resales of the Underwriters’ Warrants on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended, are registered hereby. Resales of shares issuable upon exercise of the Underwriters Warrants are also being similarly registered on a delayed or continuous basis hereby. See “Underwriting.” No fee required pursuant to Rule 457(g) under the Securities Act of 1933, as amended. |
| (5) | Represents 8% of the securities to be sold in this offering including securities that may be sold upon exercise of the underwriters’ over-allotment option. The Underwriters’ Warrants are exercisable at a per share price equal to 125% of the common stock public offering price. |
| (6) | Previously paid. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to completion, dated April 21, 2015

$10,000,000 of Shares of Common Stock and
Warrants to Purchase Shares of Common Stock
DelMar Pharmaceuticals, Inc. is offering $10,000,000 of shares of our common stock and warrants to purchase shares of our common stock. The warrants will have an exercise price of $__ per share, subject to adjustment as set forth herein. The warrants are exercisable immediately and will expire __ years from the date of issuance.
Our common stock is quoted on the OTCQX under the symbol “DMPI”. There is no established trading market for the warrants. Subsequent to the closing of this offering, we intend to apply to list our common stock on the NYSE MKT or the NASDAQ Capital Market. No assurance can be given that our application will be approved. On April 20, 2015, the last reported sale price for our common stock on the OTCQX was $0.89 per share.
The warrants will be issued in book-entry form pursuant to a warrant agency agreement between us and Island Stock Transfer, as warrant agent.
Our business and an investment in our securities involves a high degree of risk. See “Risk Factors” beginning on page 6 of this prospectus for a discussion of information that you should consider before investing in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Per Share | Per Warrant | Total | ||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Underwriting discount (1) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
(1) The underwriters will receive compensation in addition to the underwriting discount. See “Underwriting” beginning on page 76 of this prospectus for a description of the compensation payable to the underwriters.
The underwriters may also purchase up to an additional shares of common stock and/or warrants to purchase shares of our common stock from us at the public offering price, less in each case the underwriting discount, within 45 days from the date of this prospectus solely to cover over-allotments, if any.
The underwriters expect to deliver the shares and warrants against payment therefor on or about , 2015.
Joint Book-Running Managers
| Maxim Group LLC | Roth Capital Partners |
Co-Manager
National Securities Corporation
The date of this prospectus is , 2015
You should rely only on the information contained in this prospectus or in any free writing prospectus that we may specifically authorize to be delivered or made available to you. We have not, and the underwriters have not, authorized anyone to provide you with any information other than that contained in this prospectus or in any free writing prospectus we may authorize to be delivered or made available to you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus may only be used where it is legal to offer and sell our securities. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of securities. Our business, financial condition, results of operations and prospects may have changed since that date. We are not, and the underwriters are not, making an offer of these securities in any jurisdiction where the offer is not permitted.
For investors outside the United States: We have not and the underwriters have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of securities and the distribution of this prospectus outside the United States.
This summary highlights information contained elsewhere in this prospectus and does not contain all of the information that you should consider in making your investment decision. Before deciding to invest in our securities, you should read this entire prospectus carefully, including the sections of this prospectus entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes contained elsewhere in this prospectus. References in this prospectus to “we,” “us,” “our” and similar words refer to the Company and its wholly-owned subsidiaries, DelMar (BC), Callco (defined below) and Exchangeco (defined below), unless the context indicates otherwise, and, prior to the effectiveness of the Reverse Acquisition, these terms refer to DelMar (BC). References to “Berry” relate to the Company prior to the Reverse Acquisition (see “Business”).
Overview
We are a clinical and commercial stage drug development company with a focus on the treatment of cancer. We are conducting clinical trials in the United States with our product candidate, VAL-083, as a potential new treatment for glioblastoma multiforme (“GBM”), the most common and aggressive form of brain cancer. We have also acquired certain exclusive commercial rights to VAL-083 in China where it is approved as a chemotherapy for the treatment of chronic myelogenous leukemia (“CML”) and lung cancer. We plan to seek marketing partnerships in China in order to generate royalty revenue.
We are developing a new drug candidate targeting orphan cancer indications. We aim to develop products that will have a high impact on patient care. In order to accelerate our development timelines and reduce technical risk, we leverage existing clinical and commercial data from a wide range of sources.
VAL-083
Our product candidate, VAL-083, represents a “first–in-class” small molecule chemotherapeutic, which means that the molecular structure of VAL-083 is not an analogue or derivative of other small molecule chemotherapeutics approved for the treatment of cancer. VAL-083 has been assessed in 42 Phase 1 and Phase 2 clinical trials sponsored by the National Cancer Institutes (“NCI”) in the United States as a treatment for various cancers including lung, brain, cervical, ovarian tumors and leukemia. Published pre-clinical and clinical data suggest that VAL-083 may be active against a range of tumor types. VAL-083 is approved as a cancer chemotherapeutic in China for the treatment of CML and lung cancer. VAL-083 has not been approved for any indications outside of China.
Upon obtaining regulatory approval, we intend to commercialize VAL-083 for the treatment of orphan cancer indications where patients have failed other therapies or have limited medical options. An orphan disease is defined in the United States under the Rare Disease Act of 2002 as “any disease or condition that affects less than 200,000 persons in the United States.” The Orphan Drug Act of 1983 is a federal law that provides financial and other incentives including a period of market exclusivity to encourage the development of new treatments for orphan diseases.
Recent Highlights
Recently, we announced important milestones demonstrating progress on our drug development programs:
| ● | In April 2015, we presented new clinical and non-clinical data at the American Association for Cancer Research (“AACR”) annual meeting related to the development of VAL-083 as a potential new therapy for the treatment of glioblastoma multiforme (“GBM”) and non-small cell lung cancer (“NSCLC”). Specifically, we reported the completion of the dose-escalation portion of our ongoing Phase I/II clinical trial with VAL-083 as a potential new therapy for the treatment of refractory GBM and we reported new non-clinical data supporting the opportunity for VAL-083 to address significant unmet medical needs in the treatment of GBM and NSCLC. | |
| ● | In April 2015, we announced that the Mayo Clinic Cancer Center in Rochester, Minnesota had been added as a clinical trial site for our ongoing, multicenter Phase I/II clinical trial study of VAL-083 in patients with refractory GBM. |
| ● | In March 2015, we announced that we received a notice of allowance
for a United States patent covering methods of use and compositions for VAL-083. As of December 31, 2014, we have filed a total of ten patent applications which are being prosecuted in the United States and in international jurisdictions; four U.S. patents and one international patent have been allowed to date. | |
| ● | In January 2015, we announced the achievement of an important milestone in our ongoing
Phase I/II clinical trial with VAL-083 as a potential new therapy for the treatment of refractory GBM. Specifically,
we reported the first observation of a dose limiting toxicity (“DLT”), which signaled the near-term completion
of dose-escalation and identification of a maximum tolerated dose (“MTD”) for advancement to a Phase II/III registration-directed
clinical trial in refractory GBM.
|
| 1 |
| In January 2015, we also announced that we had received a notice of allowance for a United States patent covering analytical methods related to the manufacturing and quality control of VAL-083 drug product. | ||
| ● | In November 2014, we presented an update on our ongoing Phase I/II clinical trial with VAL-083 as a potential new therapy for refractory glioblastoma at the Society for NeuroOncology (“SNO”) annual meeting. At SNO, we also presented new non-clinical data supporting the favorable differentiation of VAL-083 versus the standard-of-care in the treatment of GBM. | |
| ● | In October 2014, we presented new non-clinical research supporting the potential utility of VAL-083 in the treatment of NSCLC at the American Association for Cancer Research’s (“AACR”) New Horizons in Cancer Research. | |
| ● | In October 2014, we also participated in the second Brain Tumor Clinical Trial Endpoints Workshop held in Bethesda, MD. The workshops, which are sponsored by the National Brain Tumor Society, bring together private industry, leading clinicians and key members of the US Food and Drug Administration (“FDA”) staff and leaders of the NCI to discuss clinical trial design and strategies for accelerating approval of promising brain tumor therapies. | |
| ● | As part of our strategy to list our common stock on a national securities exchange in the timeliest manner possible, we also: |
| ● | Appointed Erich Mohr and Lynda Cranston to our Board of Directors and established an independent Corporate Governance and Compensation Committee. | |
| ● | Received net proceeds of $1,266,177 from the exercise of certain investor warrants at $0.65 per warrant during the six months ended December 31, 2014. The exercise of these warrants, including through a tender offer, has provided us with additional non-dilutive capital that we believe is sufficient to fund our current operations through at least the end of March 2016. | |
| ● | Issued an aggregate 945,514 shares of common stock in exchange for the surrender of certain investor warrants to purchase an aggregate of 2,836,541 shares of common stock, resulting in a reclassification of the derivative liability to equity of $706,069. | |
| ● | Entered into amendments to warrants issued as a dividend to stockholders on January 24, 2013 (the “Dividend Warrants”) such that all of the Dividend Warrants were reclassified to equity on October 31, 2014. | |
| ● | Changed our fiscal year end to June 30 from December 31. |
Our executive offices are located at Suite 720-999 West Broadway, Vancouver, British Columbia, Canada V5Z 1K5. Our clinical operations are managed at Suite R, 3475 Edison Way, Menlo Park, California, 94025. Our website is located at www.delmarpharma.com, and our telephone number is 604-629-5989. The information contained on our website is not incorporated by reference into this prospectus, and you should not consider any information contained on, or that can be accessed through, our website as part of this prospectus, or in deciding whether to purchase our securities
Risks Associated With Our Business
Our business is subject to numerous risks described in the section entitled “Risk Factors” and elsewhere in this prospectus. You should carefully consider these risks before making an investment. Some of these risks include:
| ● | We have a limited operating history and a history of operating losses and expect to incur significant additional operating losses. | |
| ● | We will need to secure additional financing. | |
| ● | We are an early-stage company with an unproven business strategy and may never achieve commercialization of our candidate product or profitability. | |
| ● | We are currently focused on the development of a single product candidate. | |
| ● | Clinical trials for our product candidate are expensive and time consuming, and their outcome is uncertain. | |
| ● | We may not receive regulatory approvals for our product candidate or there may be a delay in obtaining such approvals. | |
| ● | You will experience immediate and substantial dilution as a result of this offering and may experience additional dilution in the future. |
| 2 |
Summary of the Offering
| Securities offered by us | $10,000,000 of shares of common stock and warrants to purchase shares of common stock | |
| Common Stock to be outstanding after this offering | shares ( if the warrants are exercised in full). If the underwriters’ over-allotment option is exercised in full, the total number of shares of common stock outstanding immediately after this offering would be ( if the warrants are exercised in full). | |
| Description of Warrants | The warrants will have an exercise price equal to $ per share. The warrants are exercisable immediately and expire _____ years from the date of issuance. | |
| Over-allotment option | We have granted a 45-day option to the underwriters to purchase up to additional shares of common stock and/or warrants to purchase up to an aggregate of shares of our common stock solely to cover over-allotments, if any. | |
| Use of proceeds | We expect to use the net proceeds received from this offering to fund our research and drug development activities, including a Phase II/III registration-directed clinical trial with VAL-083 as a potential new treatment for refractory GBM and for general working capital purposes. See “Use of Proceeds”. | |
| Underwriters’ Warrants | We have agreed to issue to the underwriters, warrants to purchase up to the number of shares of our common stock equal to 8% of the aggregate number of shares sold in this offering (excluding shares or warrants, if any, sold in connection with the exercise of the over-allotment option). The warrants are exercisable at a per share price equal to $______ , at any time, and from time to time, in whole or in part, during the five -year period commencing six months from the effective date of the offering. | |
| Risk factors | See “Risk Factors” beginning on page 6 and the other information included in this prospectus for a discussion of factors you should carefully consider before investing in our securities. | |
| OTCQX trading symbol of common stock | DMPI | |
| Listing | Subsequent to the closing of this offering, we intend to apply to list our common stock on the NYSE MKT or NASDAQ Capital Market. |
Unless we indicate otherwise, all information in this prospectus:
| ● | is based on 35,199,889 shares of common stock issued and outstanding as of April 1, 2015; | |
| ● | assumes no exercise by the underwriters of their option to purchase up to an additional shares of common stock and/or warrants to cover over-allotments, if any; | |
| ● | excludes 3,595,000 shares of our common stock issuable upon exercise of outstanding stock options at a weighted average exercise price of $0.94 per share as of April 1, 2015; | |
| ● | excludes 13,472,870 shares of our common stock issuable upon exercise of outstanding warrants at a weighted average exercise price of $0.92 per share as of April 1, 2015; | |
| ● | excludes 4,256,042 shares of common stock issuable upon exchange of the Exchangeable Shares as of April 1, 2015; and | |
| ● | excludes shares of common stock underlying the warrants to be issued to the underwriters in connection with this offering. |
| 3 |
Summary Consolidated Financial Data
On July 21, 2014, the Board of Directors of the Company approved a change in the Company’s fiscal year end from December 31 to June 30. As a result of this change, the Company has prepared consolidated financial statements for the six month transition period ended June 30, 2014. References to any of the Company’s 2013 or earlier fiscal years mean the fiscal year ending December 31 of that calendar year.
The following tables set forth our (i) summary statement of operations data for the six months ended December 31, 2014 and 2013 (unaudited) and (ii) summary balance sheet data as of December 31, 2014 (unaudited and pro forma), derived from our audited and unaudited consolidated financial statements and accompanying notes appearing elsewhere in this prospectus. The unaudited financial data include all adjustments, consisting of normal recurring adjustments, necessary for a fair presentation of our financial position and results of operations for these periods.
You should read this information together with the section entitled “Capitalization,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and accompanying notes appearing elsewhere in this prospectus.
| Selected Statement of Operations Data |
| Six Months Ended | ||||||||
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Research and development | $ | 1,283,796 | $ | 1,126,295 | ||||
| General and administrative | 1,101,229 | 1,377,550 | ||||||
| Change in fair value of derivative liability | (66,606 | ) | (8,466,826 | ) | ||||
| Change in fair value of derivative liability due to change in warrant terms | (23,658 | ) | - | |||||
| Loss on exchange of warrants | 92,843 | - | ||||||
| Foreign exchange loss | 9,686 | 31,963 | ||||||
| Interest expense | 2,091 | 4,073 | ||||||
| Interest income | (261 | ) | (1,311 | ) | ||||
| Net and comprehensive loss (income) | $ | 2,399,120 | $ | (5,928,256 | ) | |||
| Basic weighted average number of shares outstanding | 37,125,074 | 31,477,137 | ||||||
| Basic loss (income) per share | $ | 0.06 | $ | (0.19 | ) | |||
| Diluted weighted average number of shares outstanding | 37,125,074 | 41,742,401 | ||||||
| Diluted loss (income) per share | $ | 0.06 | $ | 0.00 | ||||
| 4 |
Selected Balance Sheet Data
| December 31, 2014 | ||||||||
(actual) | (Pro forma) | |||||||
| Cash and cash equivalents | $ | 3,958,439 | $ | |||||
| Working capital | 3,834,192 | |||||||
| Total assets | 4,128,378 | |||||||
| Derivative liability | 1,567,291 | |||||||
| Total stockholders’ equity | 2,086,551 | |||||||
| (1) | Pro forma amounts give effect to the sale of the common stock and warrants in this offering at the assumed public offering price of $ per share and $ per warrant, and after deducting underwriting discounts and commissions and other estimated offering expenses payable by us. |
| 5 |
Any investment in our common stock involves a high degree of risk. Investors should carefully consider the risks described below and all of the information contained in this prospectus before deciding whether to purchase our common stock. Our business, financial condition or results of operations could be materially adversely affected by these risks if any of them actually occur. This prospectus also contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including the risks we face as described below and elsewhere in this prospectus.
Risks Related to Our Business
We have a limited operating history and a history of operating losses, and expect to incur significant additional operating losses.
We are an early stage company and there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We have generated net losses since we began operations, including $2,399,120, $2,798,908, $8,290,689 and $2,400,363 for the six months ended December 31, 2014 and June 30, 2014 and the years ended December 31, 2013 and 2012, respectively. We expect to incur substantial additional net expenses over the next several years as our research, development and commercial activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the preclinical and clinical development of our product candidate; obtaining necessary regulatory approvals from the Food and Drug Administration (“FDA”) and international regulatory agencies; successful manufacturing, sales and marketing arrangements; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, prospects and results of operations may be materially adversely affected.
Raising additional capital may cause dilution to our stockholders, including purchasers of our securities in this offering, restrict our operations or require us to relinquish rights to technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of public or private equity offerings, debt financings and/or license and development agreements with collaboration partners. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures or declaring dividends. In addition, debt financing would result in fixed payment obligations.
If we raise funds through collaborations, strategic partnerships or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
We are an early-stage company with an unproven business strategy and may never achieve commercialization of our candidate products or profitability.
We are at an early stage of development and commercialization of our technologies and product candidate. We have not yet begun to market any products and, accordingly, have not begun or generate revenues from the commercialization of our product. Our product will require significant additional clinical testing and investment prior to commercialization. A commitment of substantial resources by ourselves and, potentially, our partners to conduct time-consuming research and clinical trials will be required if we are to complete the development of our product candidate. There can be no assurance that our product candidate will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs or be successfully marketed. Our product candidate is not expected to be commercially available for several years, if at all.
We are currently focused on the development of a single product candidate.
Our product development efforts are currently focused on a single product, VAL-083, for which we are researching multiple indications. If VAL-083 fails to achieve clinical endpoints or exhibits unanticipated toxicity or if a superior product is developed by a competitor, our prospects for obtaining regulatory approval and commercialization may be negatively impacted. In the long term, we hope to establish a pipeline of product candidates, and we have identified additional product candidates that we may be able to acquire or license in the future. However, at this time we do not have any formal agreements granting us any rights to such additional product candidates.
| 6 |
Even if we are able to commercialize any product candidate that we develop, the product may become subject to unfavorable pricing regulations, third party payor reimbursement practices or healthcare reform initiatives that could harm our business.
The commercial success of our product candidate will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidate will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities (such as Medicare and Medicaid), private health coverage insurers and other third party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidate. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish and maintain pricing sufficient to realize a meaningful return on our investment.
There is significant uncertainty related to third party payor coverage and reimbursement of newly approved drugs. Marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some non-U.S. markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay commercial launch of the product, possibly for lengthy time periods, which may negatively impact the revenues we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
Our ability to commercialize VAL-083 or any other product candidate will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third party payors, such as private health insurers and health maintenance organizations, decide which medications they will cover and establish reimbursement levels. The healthcare industry is acutely focused on cost containment, both in the United States and elsewhere. Government authorities and third party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications, which could affect our ability to sell our product candidate profitably. These payors may not view our products, if any, as cost-effective, and coverage and reimbursement may not be available to our customers, or may not be sufficient to allow our products, if any, to be marketed on a competitive basis. Cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower than anticipated product revenues. If the prices for our products, if any, decrease or if governmental and other third party payors do not provide adequate coverage or reimbursement, our prospects for revenue and profitability will suffer.
There may also be delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the indications for which the drug is approved by the FDA or comparable non-U.S. regulatory authorities. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Reimbursement rates may vary, by way of example, according to the use of the drug and the clinical setting in which it is used. Reimbursement rates may also be based on reimbursement levels already set for lower cost drugs or may be incorporated into existing payments for other services.
In addition, increasingly, third party payors are requiring higher levels of evidence of the benefits and clinical outcomes of new technologies and are challenging the prices charged. We cannot be sure that coverage will be available for any product candidate that we, or they, commercialize and, if available, that the reimbursement rates will be adequate. Further, the net reimbursement for drug products may be subject to additional reductions if there are changes to laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. An inability to promptly obtain coverage and adequate payment rates from both government-funded and private payors for any our product candidates for which we obtain marketing approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
We are dependent on obtaining certain patents and protecting our proprietary rights.
Our success will depend, in part, on our ability to obtain patents, maintain trade secret protection and operate without infringing on the proprietary rights of third parties or having third parties circumvent our rights. We have filed and are actively pursuing patent applications for our products. The patent positions of biotechnology, biopharmaceutical and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. Thus, there can be no assurance that any of our patent applications will result in the issuance of patents, that we will develop additional proprietary products that are patentable, that any patents issued to us or those that already have been issued will provide us with any competitive advantages or will not be challenged by any third parties, that the patents of others will not impede our ability to do business or that third parties will not be able to circumvent our patents. Furthermore, there can be no assurance that others will not independently develop similar products, duplicate any of our products not under patent protection, or, if patents are issued to us, design around the patented products we developed or will develop.
| 7 |
We may be required to obtain licenses from third parties to avoid infringing patents or other proprietary rights. No assurance can be given that any licenses required under any such patents or proprietary rights would be made available, if at all, on terms we find acceptable. If we do not obtain such licenses, we could encounter delays in the introduction of products or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
A number of pharmaceutical, biopharmaceutical and biotechnology companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to or affect our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. Such conflict could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of our patent applications. In addition, if patents that cover our activities are issued to other companies, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited. In addition, we could incur substantial costs in defending ourselves in suits brought against us on patents it might infringe or in filing suits against others to have such patents declared invalid.
Patent applications in the U.S. are maintained in secrecy and not published if either: i) the application is a provisional application or, ii) the application is filed and we request no publication, and certify that the invention disclosed “has not and will not” be the subject of a published foreign application. Otherwise, U.S. applications or foreign counterparts, if any, publish 18 months after the priority application has been filed. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we or any licensor were the first creator of inventions covered by pending patent applications or that we or such licensor was the first to file patent applications for such inventions. Moreover, we might have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost to us, even if the eventual outcome were favorable to us. There can be no assurance that our patents, if issued, would be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents.
Moreover, we may be subject to third party preissuance submissions of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third party patent rights.
Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us, or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate, or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, the protection of intellectual property rights in China (where our clinical product candidate, VAL-083, is manufactured pursuant to a collaboration agreement with the only manufacturer presently licensed by the China Food and Drug Administration to produce the product for the China market, and where VAL-083 is approved for the treatment of CML and lung cancer) is relatively weak compared to the United States, which may negatively affect our ability to generate royalty revenue from sales of VAL-083 in China.
Much of our know-how and technology may not be patentable. To protect our rights, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance, however, that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, our business may be adversely affected by competitors who independently develop competing technologies, especially if we obtain no, or only narrow, patent protection.
| 8 |
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to United States patent law. The Leahy-Smith Act includes provisions that affect the way patent applications are prosecuted and affect patent litigation. The United States Patent and Trademark Office, or PTO, recently developed new regulations and procedures to govern administration of the Leahy-Smith Act. However, many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
We may be unable to protect our patents and proprietary rights.
Our future success will depend to a significant extent on our ability to:
| ● | obtain and keep patent protection for our products and technologies on an international basis; | |
| ● | enforce our patents to prevent others from using our inventions; | |
| ● | maintain and prevent others from using our trade secrets; and | |
| ● | operate and commercialize products without infringing on the patents or proprietary rights of others. |
We cannot assure you that our patent rights will afford any competitive advantages and these rights may be challenged or circumvented by third parties. Further, patents may not be issued on any of our pending patent applications in the U.S. or abroad. Because of the extensive time required for development, testing and regulatory review of a product candidate, it is possible that before a product candidate can be commercialized, any related patent may expire, or remain in existence for only a short period following commercialization, reducing or eliminating any advantage of the patent.
If we sue others for infringing our patents, a court may determine that such patents are invalid or unenforceable. Even if the validity of our patent rights is upheld by a court, a court may not prevent the alleged infringement of our patent rights on the grounds that such activity is not covered by our patent claims.
In addition, third parties may sue us for infringing their patents. In the event of a successful claim of infringement against us, we may be required to:
| ● | defend litigation or administrative proceedings; | |
| ● | pay substantial damages; | |
| ● | stop using our technologies and methods; | |
| ● | stop certain research and development efforts; | |
| ● | develop non-infringing products or methods; and | |
| ● | obtain one or more licenses from third parties. |
If required, we cannot assure you that we will be able to obtain such licenses on acceptable terms, or at all. If we are sued for infringement, we could encounter substantial delays in development, manufacture and commercialization of our product candidates. Any litigation, whether to enforce our patent rights or to defend against allegations that we infringe third party rights, will be costly, time consuming, and may distract management from other important tasks.
As is commonplace in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. To the extent our employees are involved in research areas which are similar to those areas in which they were involved at their former employers, we may be subject to claims that such employees and/or we have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of the former employers. Litigation may be necessary to defend against such claims, which could result in substantial costs and be a distraction to management and which may have a material adverse effect on us, even if we are successful in defending such claims.
We are subject to various government regulations.
The manufacture and sale of human therapeutic and diagnostic products in the U.S., Canada and foreign jurisdictions are governed by a variety of statutes and regulations. These laws require approval of manufacturing facilities, controlled research and testing of products and government review and approval of a submission containing manufacturing, preclinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current cGMP during production and storage, and control of marketing activities, including advertising and labeling.
| 9 |
The product we are currently developing will require significant development, preclinical and clinical testing and investment of substantial funds prior to their commercialization. The process of obtaining required approvals can be costly and time-consuming, and there can be no assurance that future products will be successfully developed and will prove to be safe and effective in clinical trials or receive applicable regulatory approvals. Markets other than the U.S. and Canada have similar restrictions. Potential investors and shareholders should be aware of the risks, problems, delays, expenses and difficulties which we may encounter in view of the extensive regulatory environment which controls our business.
If we are unable to keep up with rapid technological changes in our field or compete effectively, we will be unable to operate profitably.
We are engaged in a rapidly changing field. Other products and therapies that will compete directly with the products that we are seeking to develop and market currently exist or are being developed. Competition from fully integrated pharmaceutical companies and more established biotechnology companies is intense and is expected to increase. Most of these companies have significantly greater financial resources and expertise in discovery and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and marketing than us. Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and established biopharmaceutical or biotechnology companies. Many of these competitors have significant products that have been approved or are in development and operate large, well-funded discovery and development programs. Academic institutions, governmental agencies and other public and private research organizations also conduct research, seek patent protection and establish collaborative arrangements for therapeutic products and clinical development and marketing. These companies and institutions compete with us in recruiting and retaining highly qualified scientific and management personnel. In addition to the above factors, we will face competition based on product efficacy and safety, the timing and scope of regulatory approvals, availability of supply, marketing and sales capability, reimbursement coverage, price and patent position. There is no assurance that our competitors will not develop more effective or more affordable products, or achieve earlier patent protection or product commercialization, than our own.
Other companies may succeed in developing products earlier than ourselves, obtaining Health Canada, European Medicines Agency (“EMA”) and FDA approvals for such products more rapidly than we will, or in developing products that are more effective than products we propose to develop. While we will seek to expand our technological capabilities in order to remain competitive, there can be no assurance that research and development by others will not render our technology or products obsolete or non-competitive or result in treatments or cures superior to any therapy we develop, or that any therapy we develop will be preferred to any existing or newly developed technologies.
We may request priority review for our product candidate in the future. The FDA may not grant priority review for our product candidate. Moreover, even if the FDA designated such product for priority review, that designation may not lead to a faster regulatory review or approval process and, in any event, would not assure FDA approval.
We may be eligible for priority review designation for our product candidate if the FDA determines such product candidate offers major advances in treatment or provides a treatment where no adequate therapy exists. A priority review designation means that the goal for the FDA to review an application is six months, rather than the standard review period of ten months. The FDA has broad discretion with respect to whether or not to grant priority review status to a product candidate, so even if we believe a particular product candidate is eligible for such designation or status, the FDA may decide not to grant it. Thus, while the FDA has granted priority review to other oncology disease products, our product candidate, should we determine to seek priority review, may not receive similar designation. Moreover, even if our product candidate is designated for priority review, such a designation does not necessarily mean a faster regulatory review process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority review from the FDA does not guarantee approval within an accelerated timeline or thereafter.
We believe we may in some instances be able to secure approval from the FDA or comparable non-U.S. regulatory authorities to use accelerated development pathways. If we are unable to obtain such approval, we may be required to conduct additional preclinical studies or clinical trials beyond those that we contemplate, which could increase the expense of obtaining, and delay the receipt of, necessary marketing approvals.
We anticipate that we may seek an accelerated approval pathway for our product candidate. Under the accelerated approval provisions in the Federal Food, Drug, and Cosmetic Act, or FDCA, and the FDA’s implementing regulations, the FDA may grant accelerated approval to a product designed to treat a serious or life-threatening condition that provides meaningful therapeutic benefit over available therapies upon a determination that the product has an effect on a surrogate endpoint or intermediate clinical endpoint that is reasonably likely to predict clinical benefit. The FDA considers a clinical benefit to be a positive therapeutic effect that is clinically meaningful in the context of a given disease, such as irreversible morbidity or mortality. For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. An intermediate clinical endpoint is a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. The accelerated approval pathway may be used in cases in which the advantage of a new drug over available therapy may not be a direct therapeutic advantage, but is a clinically important improvement from a patient and public health perspective. If granted, accelerated approval is usually contingent on the sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’s clinical benefit. If such post-approval studies fail to confirm the drug’s clinical benefit, the FDA may withdraw its approval of the drug.
| 10 |
Prior to seeking such accelerated approval, we will seek feedback from the FDA and will otherwise evaluate our ability to seek and receive such accelerated approval. There can also be no assurance that after our evaluation of the feedback and other factors we will decide to pursue or submit a Biologics License Application, or BLA, or a New Drug Application, or NDA, for accelerated approval or any other form of expedited development, review or approval. Similarly, there can be no assurance that after subsequent FDA feedback that we will continue to pursue or apply for accelerated approval or any other form of expedited development, review or approval, even if we initially decide to do so. Furthermore, if we decide to submit an application for accelerated approval or under another expedited regulatory designation (e.g., breakthrough therapy designation), there can be no assurance that such submission or application will be accepted or that any expedited development, review or approval will be granted on a timely basis, or at all. The FDA or other non-U.S. authorities could also require us to conduct further studies prior to considering our application or granting approval of any type. A failure to obtain accelerated approval or any other form of expedited development, review or approval for any of our product candidates that we determine to seek accelerated approval for would result in a longer time period to commercialization of such product candidate, could increase the cost of development of such product candidate and could harm our competitive position in the marketplace.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome. We may incur additional costs or experience delays in completing, or ultimately be unable to complete the development and commercialization of our product candidate.
Our only current product candidate is in clinical development and the risk of failure of our product candidate is high. It is impossible to predict when or if our product candidate will prove effective or safe in humans or will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete preclinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidate in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The clinical development of our product candidate is susceptible to the risk of failure inherent at any stage of drug development, including failure to demonstrate efficacy in a clinical trial or across a broad population of patients, the occurrence of severe or medically or commercially unacceptable adverse events, failure to comply with protocols or applicable regulatory requirements and determination by the FDA or any comparable non-U.S. regulatory authority that a drug product is not safe or effective for its intended uses. It is possible that even if our product candidates has a beneficial effect, that effect will not be detected during clinical evaluation as a result of one or more of a variety of factors, including the size, duration, design, measurements, conduct or analysis of our clinical trials. Conversely, as a result of the same factors, our clinical trials may indicate an apparent positive effect of a product candidate that is greater than the actual positive effect, if any. Similarly, in our clinical trials we may fail to detect toxicity of or intolerability caused by our product candidate, or mistakenly believe that our product candidate is toxic or not well tolerated when that is not in fact the case.
The outcome of preclinical studies and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in earlier development, and we cannot be certain that we will not face additional setbacks.
The design of a clinical trial can determine whether its results will support approval of a product; however, flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced or completed. We have limited experience in designing clinical trials and may be unable to design and execute a clinical trial to support marketing approval. In addition, preclinical and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for the product candidates. Even if we believe that the results of clinical trials for our product candidate warrant marketing approval, the FDA or comparable non-U.S. regulatory authorities may disagree and may not grant marketing approval of our product candidate.
In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the clinical trial protocols and the rate of dropout among clinical trial participants. Any Phase 2, Phase 3 or other clinical trials that we may conduct may not demonstrate the efficacy and safety necessary to obtain regulatory approval to market our product candidate.
| 11 |
We have conducted, and may in the future conduct, clinical trials for certain of our product candidate at sites outside the United States, and the FDA may not accept data from trials conducted in such locations.
We have conducted and may in the future choose to conduct one or more of our clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical trials conducted outside of the United States must be representative of the population for whom we intend to seek approval in the United States. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the trials also complied with all applicable U.S. laws and regulations. There can be no assurance that the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept the data from any of our clinical trials that we determine to conduct outside the United States, it would likely result in the need for additional trials, which would be costly and time-consuming and delay or permanently halt our development of the product candidate.
In addition, the conduct of clinical trials outside the United States could have a significant impact on us. Risks inherent in conducting international clinical trials include:
| · | foreign regulatory requirements that could restrict or limit our ability to conduct our clinical trials; |
| · | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; |
| · | foreign exchange fluctuations; and |
| · | diminished protection of intellectual property in some countries. |
If clinical trials of our product candidate fail to demonstrate safety and efficacy to the satisfaction of the FDA and comparable non-U.S. regulators, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidate.
We are not permitted to commercialize, market, promote or sell any product candidate in the United States without obtaining marketing approval from the FDA. Comparable non-U.S. regulatory authorities, such as the EMA, impose similar restrictions. We may never receive such approvals. We must complete extensive preclinical development and clinical trials to demonstrate the safety and efficacy of our product candidate in humans before we will be able to obtain these approvals.
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. We have not previously submitted a BLA or an NDA to the FDA or similar drug approval filings to comparable non-U.S. regulatory authorities for any product candidate.
Any inability to successfully complete preclinical and clinical development could result in additional costs to us and impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, if (1) we are required to conduct additional clinical trials or other testing of our product candidate beyond the trials and testing that we contemplate, (2) we are unable to successfully complete clinical trials of our product candidate or other testing, (3) the results of these trials or tests are unfavorable, uncertain or are only modestly favorable, or (4) there are unacceptable safety concerns associated with our product candidate, we, in addition to incurring additional costs, may:
| · | be delayed in obtaining marketing approval for our product candidates; |
| · | not obtain marketing approval at all; |
| · | obtain approval for indications or patient populations that are not as broad as we intended or desired; |
| · | obtain approval with labeling that includes significant use or distribution restrictions or significant safety warnings, including boxed warnings; |
| · | be subject to additional post-marketing testing or other requirements; or |
| · | be required to remove the product from the market after obtaining marketing approval. |
If we experience any of a number of possible unforeseen events in connection with clinical trials of our product candidate, potential marketing approval or commercialization of our product candidates could be delayed or prevented.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent marketing approval of our product candidate, including:
| · | clinical trials of our product candidate may produce unfavorable or inconclusive results; |
| · | we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; |
| 12 |
| · | the number of patients required for clinical trials of our product candidate may be larger than we anticipate, patient enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; |
| · | data safety monitoring committees may recommend suspension, termination or a clinical hold for various reasons, including concerns about patient safety; |
| · | regulators or IRBs may suspend or terminate the trial or impose a clinical hold for various reasons, including noncompliance with regulatory requirements or concerns about patient safety; |
| · | patients with serious, life-threatening diseases included in our clinical trials may die or suffer other adverse medical events for reasons that may not be related to our product candidate; |
| · | participating patients may be subject to unacceptable health risks; |
| · | patients may not complete clinical trials due to safety issues, side effects, or other reasons; |
| · | changes in regulatory requirements and guidance may occur, which require us to amend clinical trial protocols to reflect these changes |
| · | our third party contractors, including those manufacturing our product candidate or components or ingredients thereof or conducting clinical trials on our behalf, may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner or at all; |
| · | regulators or institutional review boards, or IRBs may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| · | we may experience delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; |
| · | patients who enroll in a clinical trial may misrepresent their eligibility to do so or may otherwise not comply with the clinical trial protocol, resulting in the need to drop the patients from the clinical trial, increase the needed enrollment size for the clinical trial or extend the clinical trial’s duration; |
| · | we may have to suspend or terminate clinical trials of our product candidate for various reasons, including a finding that the participants are being exposed to unacceptable health risks, undesirable side effects or other unexpected characteristics of a product candidate; |
| · | regulators or IRBs may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or their respective standards of conduct, a finding that the participants are being exposed to unacceptable health risks, undesirable side effects or other unexpected characteristics of the product candidate or findings of undesirable effects caused by a chemically or mechanistically similar drug or drug candidate; |
| · | the FDA or comparable non-U.S. regulatory authorities may disagree with our clinical trial design or our interpretation of data from preclinical studies and clinical trials; |
| · | the FDA or comparable non-U.S. regulatory authorities may fail to approve or subsequently find fault with the manufacturing processes or facilities of third party manufacturers with which we enter into agreements for clinical and commercial supplies; |
| · | the supply or quality of raw materials or manufactured product candidate or other materials necessary to conduct clinical trials of our product candidate may be insufficient, inadequate, delayed, or not available at an acceptable cost, or we may experience interruptions in supply; and |
| · | the approval policies or regulations of the FDA or comparable non-U.S. regulatory authorities may significantly change in a manner rendering our clinical data insufficient to obtain marketing approval. |
| 13 |
Product development costs for us will increase if we experience delays in testing or pursuing marketing approvals and we may be required to obtain additional funds to complete clinical trials and prepare for possible commercialization of our product candidate. We do not know whether any preclinical tests or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant preclinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidate or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidate and may harm our business and results of operations. In addition, many of the factors that cause, or lead to, clinical trial delays may ultimately lead to the denial of marketing approval of our product candidate.
If we experience delays or difficulties in the enrollment of patients in clinical trials, we may not achieve our clinical development on our anticipated timeline, or at all, and our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for VAL-083 or any other product candidate if we are unable to locate and enroll a sufficient number of eligible patients to participate in clinical trials. Patient enrollment is a significant factor in the timing of clinical trials, and is affected by many factors, including:
| · | the size and nature of the patient population; |
| · | the severity of the disease under investigation; |
| · | the proximity of patients to clinical sites; |
| · | the eligibility criteria for the trial; |
| · | the design of the clinical trial; |
| · | efforts to facilitate timely enrollment; |
| · | competing clinical trials; and |
| · | clinicians’ and patients’ perceptions as to the potential advantages and risks of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. |
Our inability to enroll a sufficient number of patients for our clinical trials could result in significant delays or may require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our product candidate, delay or halt the development of and approval processes for our product candidate and jeopardize our ability to achieve our clinical development timeline and goals, including the dates by which we will commence, complete and receive results from clinical trials. Enrollment delays may also delay or jeopardize our ability to commence sales and generate revenues from our product candidate. Any of the foregoing could cause the value of our company to decline and limit our ability to obtain additional financing, if needed.
Positive results in previous clinical trials of VAL-083 may not be replicated in future clinical trials, which could result in development delays or a failure to obtain marketing approval.
Positive results in previous clinical studies of VAL-083 may not be predictive of similar results in future clinical trials. Also, interim results during a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early-stage development. Accordingly, the results from the completed preclinical studies and clinical trials for VAL-083 may not be predictive of the results we may obtain in later stage trials. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain FDA or EMA, or other regulatory agency, approval for their products.
FDA approval of VAL-083 may be denied.
There can be no assurance that the FDA will ultimately approve our NDA. The FDA may deny approval of VAL-083 for many reasons, including:
| · | we may be unable to demonstrate to the satisfaction of the FDA that VAL-083 is safe and effective for its intended uses; |
| · | the FDA may disagree with our interpretation of data from the clinical trials; |
| · | we may be unable to demonstrate that any clinical or other benefits of VAL-083 outweigh any safety or other perceived risks; or |
| · | we may not be able to successfully address any other issues raised by the FDA. |
| 14 |
If VAL-083 fails to receive FDA approval, our business and prospects will be materially adversely impacted.
We expect to rely on orphan drug status to develop and commercialize our product candidate, but our orphan drug designations may not confer marketing exclusivity or other expected commercial benefits as anticipated.
Market exclusivity afforded by orphan drug designation is generally offered as an incentive to drug developers to invest in developing and commercializing products for unique diseases that impact a limited number of patients. The FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. Qualification to maintain orphan drug status is generally monitored by the regulatory authorities during the orphan drug exclusivity period, currently seven years from the date of approval in the United States.
We have been granted orphan drug designation in the United States for GBM and we expect to rely on orphan drug exclusivity for our product candidate. It is possible that with the approval of VAL-083 in the United States, that the incidence and prevalence numbers for GBM could change. Should the incidence and prevalence of GBM patients materially increase, it is possible that the orphan drug designation, and related market exclusivity, in the United States could be lost. Further, while we have been granted this orphan designation, the FDA can still approve different drugs for use in treating the same indication or disease, which would create a more competitive market for us and our revenues will be diminished.
Further, for our product candidate, it is possible that another company also holding orphan drug designation for the same product candidate will receive marketing approval for the same indication before we do. If that were to happen, our applications for that indication may not be approved until the competing company’s period of exclusivity expires. Even if we are the first to obtain marketing authorization for an orphan drug indication, there are circumstances under which a competing product may be approved for the same indication during the seven-year period of marketing exclusivity, such as if the later product is shown to be clinically superior to the orphan product, or if the later product is deemed a different product than ours. Further, the seven-year marketing exclusivity would not prevent competitors from obtaining approval of the same product candidate as ours for indications other than those in which we have been granted orphan drug designation, or for the use of other types of products in the same indications as our orphan product.
If the market opportunities for our product candidate are smaller than we believe they are, our revenues may be adversely affected and our business may suffer. Because the target patient populations of our product candidate are small, we must be able to successfully identify patients and capture a significant market share to achieve and maintain profitability.
We focus our research and product development on treatments for orphan cancer indications. Our projections of both the number of people who have failed other therapies or have limited medical options, are based on estimates. These estimates may prove to be incorrect and new studies may change the estimated incidence or prevalence. The number of patients in the United States, Europe and elsewhere may turn out to be lower than expected or may not be otherwise amenable to treatment with our products, or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business. Additionally, because our target patient populations are small, we will be required to capture a significant market share to achieve and maintain profitability.
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidate.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects or even death as a result of participating in our clinical trials.
We may not receive regulatory approvals for our product candidate or there may be a delay in obtaining such approvals.
Our product and our ongoing development activities are subject to regulation by regulatory authorities in the countries in which we or our collaborators and distributors wish to test, manufacture or market our products. For instance, the FDA will regulate the product in the U.S. and equivalent authorities, such as the EMA, will regulate in Europe. Regulatory approval by these authorities will be subject to the evaluation of data relating to the quality, efficacy and safety of the product for its proposed use, and there can be no assurance that the regulatory authorities will find our data sufficient to support product approval of VAL-083.
| 15 |
The time required to obtain regulatory approval varies between countries. In the U.S., for products without “Fast Track” status, it can take up to eighteen (18) months after submission of an application for product approval to receive the FDA's decision. Even with Fast Track status, FDA review and decision can take up to twelve (12) months. At present, we do not have Fast Track status for our clinical product candidate, VAL-083.
Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements as well as case load at the regulatory agency at the time.
We may fail to comply with regulatory requirements.
Our success will be dependent upon our ability, and our collaborative partners’ abilities, to maintain compliance with regulatory requirements, including cGMP, and safety reporting obligations. The failure to comply with applicable regulatory requirements can result in, among other things, fines, injunctions, civil penalties, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products, operating and production restrictions and criminal prosecutions.
Even if our product candidate receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third party payors and others in the medical community necessary for commercial success and the market opportunity for the product candidate may be smaller than we estimate.
We have never commercialized a product. Even if VAL-083 or any other product candidate is approved by the appropriate regulatory authorities for marketing and sale, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third party payors and others in the medical community. For example, physicians are often reluctant to switch their patients from existing therapies even when new and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to the therapy that they are currently taking and do not want to switch unless their physicians recommend switching products or they are required to switch therapies due to lack of reimbursement for existing therapies.
Efforts to educate the medical community and third party payors on the benefits of our product candidate may require significant resources and may not be successful. If our product candidate is approved but does not achieve an adequate level of market acceptance, we may not generate significant revenues and we may not become profitable. The degree of market acceptance of VAL-083 or any other product candidate, if approved for commercial sale, will depend on a number of factors, including:
| · | the efficacy and safety of the product; |
| · | the potential advantages of the product compared to alternative treatments; |
| · | the prevalence and severity of any side effects; |
| · | the clinical indications for which the product is approved; |
| · | whether the product is designated under physician treatment guidelines as a first-line therapy or as a second- or third-line therapy; |
| · | limitations or warnings, including distribution or use restrictions, contained in the product’s approved labeling; |
| · | our ability to offer the product for sale at competitive prices; |
| · | our ability to establish and maintain pricing sufficient to realize a meaningful return on our investment; |
| · | the product’s convenience and ease of administration compared to alternative treatments; |
| · | the willingness of the target patient population to try, and of physicians to prescribe, the product; |
| · | the strength of sales, marketing and distribution support; |
| · | the approval of other new products for the same indications; |
| · | changes in the standard of care for the targeted indications for the product; |
| · | the timing of market introduction of our approved products as well as competitive products and other therapies; |
| 16 |
| · | availability and amount of reimbursement from government payors, managed care plans and other third party payors; |
| · | adverse publicity about the product or favorable publicity about competitive products; and |
| · | potential product liability claims. |
The potential market opportunities for our product candidate are difficult to estimate precisely. Our estimates of the potential market opportunities are predicated on many assumptions, including industry knowledge and publications, third party research reports and other surveys. While we believe that our internal assumptions are reasonable, these assumptions involve the exercise of significant judgment on the part of our management, are inherently uncertain and the reasonableness of these assumptions has not been assessed by an independent source. If any of the assumptions proves to be inaccurate, the actual markets for our product candidate could be smaller than our estimates of the potential market opportunities.
If our product candidate receives marketing approval and we, or others, later discover that the drug is less effective than previously believed or causes undesirable side effects that were not previously identified, our ability to market the drug could be compromised.
Clinical trials of our product candidate are conducted in carefully defined subsets of patients who have agreed to enter into clinical trials. Consequently, it is possible that our clinical trials may indicate an apparent positive effect of a product candidate that is greater than the actual positive effect, if any, or alternatively fail to identify undesirable side effects. If, following approval of our product candidate, we, or others, discover that the drug is less effective than previously believed or causes undesirable side effects that were not previously identified, any of the following adverse events could occur:
| · | regulatory authorities may withdraw their approval of the drug or seize the drug; |
| · | we may be required to recall the drug or change the way the drug is administered; |
| · | additional restrictions may be imposed on the marketing of, or the manufacturing processes for, the particular drug; |
| · | we may be subject to fines, injunctions or the imposition of civil or criminal penalties; |
| · | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| · | we may be required to create a Medication Guide outlining the risks of the previously unidentified side effects for distribution to patients; |
| · | we could be sued and held liable for harm caused to patients; |
| · | the drug may become less competitive; and |
| · | our reputation may suffer. |
Any of these events could have a material and adverse effect on our operations and business and could adversely impact our stock price.
Any product candidate for which we obtain marketing approval, along with the manufacturing processes, qualification testing, post-approval clinical data, labeling and promotional activities for such product, will be subject to continual and additional requirements of the FDA and other regulatory authorities.
These requirements include submissions of safety and other post-marketing information, reports, registration and listing requirements, good manufacturing practices, or GMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, and recordkeeping. Even if marketing approval of our product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. The FDA closely regulates the post-approval marketing and promotion of pharmaceutical products to ensure such products are marketed only for the approved indications and in accordance with the provisions of the approved labeling.
| 17 |
In addition, later discovery of previously unknown problems with our products, manufacturing processes, or failure to comply with regulatory requirements, may lead to various adverse results, including:
| · | restrictions on such products, manufacturers or manufacturing processes; |
| · | restrictions on the labeling or marketing of a product; |
| · | restrictions on product distribution or use; |
| · | requirements to conduct post-marketing clinical trials; |
| · | requirements to institute a risk evaluation mitigation strategy, or REMS, to monitor safety of the product post-approval; |
| · | warning letters issued by the FDA or other regulatory authorities; |
| · | withdrawal of the products from the market; |
| · | refusal to approve pending applications or supplements to approved applications that we submit; |
| · | recall of products, fines, restitution or disgorgement of profits or revenue; |
| · | suspension, revocation or withdrawal of marketing approvals; |
| · | refusal to permit the import or export of our products; and |
| · | injunctions or the imposition of civil or criminal penalties. |
If we are unable to establish sales, marketing and distribution capabilities or enter into acceptable sales, marketing and distribution arrangements with third parties, we may not be successful in commercializing any product candidates that we develop, if and when those product candidates are approved.
We do not have a sales, marketing or distribution infrastructure and have limited experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product, we must either develop a sales and marketing organization, outsource these functions to third parties, or license our product candidates to others. If approved, we may seek to license VAL-083 to a large pharmaceutical company with greater resources and experience than us. We may not be able license the VAL-083 on reasonable terms, if at all. The development of sales, marketing and distribution capabilities will require substantial resources, will be time-consuming and could delay any product launch. We expect that we will commence the development of these capabilities prior to receiving approval of our product candidate. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing and distribution capabilities is delayed or does not occur for any reason, we could have prematurely or unnecessarily incurred these commercialization costs. Such a delay may be costly, and our investment could be lost if we cannot retain or reposition our sales and marketing personnel. In addition, we may not be able to hire or retain a sales force in the United States that is sufficient in size or has adequate expertise in the medical markets that we plan to target. If we are unable to establish or retain a sales force and marketing and distribution capabilities, our operating results may be adversely affected. If a potential partner has development or commercialization expertise that we believe is particularly relevant to our product candidate, then we may seek to collaborate with that potential partner even if we believe we could otherwise develop and commercialize the product independently.
We expect to seek one or more strategic partners for commercialization of our product candidate outside the United States. As a result of entering into arrangements with third parties to perform sales, marketing and distribution services, our product revenues or the profitability of these product revenues may be lower, perhaps substantially lower, than if we were to directly market and sell products in those markets. Furthermore, we may be unsuccessful in entering into the necessary arrangements with third parties or may be unable to do so on terms that are favorable to us. In addition, we may have little or no control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively.
If we do not establish sales and marketing capabilities, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidate.
We face substantial competition from other pharmaceutical and biotechnology companies and our operating results may suffer if we fail to compete effectively.
The development and commercialization of new drug products is highly competitive. We expect that we will face significant competition from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide with respect to VAL-083 and any other of our product candidates that we may seek to develop or commercialize in the future. Specifically, due to the large unmet medical need, global demographics and relatively attractive reimbursement dynamics, the oncology market is fiercely competitive and there are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of product candidates for the treatment of cancer and the immunization of infectious diseases. Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are more effective, have fewer or more tolerable side effects or are less costly than any product candidates that we are currently developing or that we may develop, which could render our product candidates obsolete and noncompetitive.
| 18 |
All of the top ten global pharmaceutical companies and most of the mid-size pharmaceutical companies have a strong research and development and commercial presence in oncology. Smaller companies also focus on oncology, including companies such as ARIAD Pharmaceuticals, Inc., Agios Pharmaceuticals, Inc., BIND Therapeutics, Inc., Clovis Oncology, Inc., Endocyte, Inc., Epizyme, Inc., ImmunoGen, Inc., Incyte Corporation, Infinity Pharmaceuticals, Inc., MacroGenics, Inc., Merrimack Pharmaceuticals, Inc., OncoMed Pharmaceuticals, Inc., Onconova Therapeutics, Inc., Pharmacyclics, Inc., Puma Biotechnology, Inc., Seattle Genetics, Inc. and TESARO, Inc.
Several companies are marketing and developing oncology immunotherapy products. Companies with approved marketed oncology products for GBM are Merck (Temodar®) and Genentech (Avastin®). Companies with oncology immunotherapy product candidates in clinical development include Northwest Biotherapeutics (DCVax-L), Celldex Therapeutics (Rindopepimut (CDX-110)) and ImmunoCellular Therapeutics (ICT-107).
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other marketing approval for their products before we are able to obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of our existing and potential future competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining marketing approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
If we are unable to or delayed in obtaining state regulatory licenses for the distribution of our product, we would not be able to sell our product candidate.
The majority of states require manufacturer and/or wholesaler licenses for the sale and distribution of drugs into that state. The application process is complicated, time consuming and requires dedicated personnel or a third party to oversee and manage. If we are delayed in obtaining these state licenses, or denied the licenses, even with FDA approval, we would not be able to sell or ship product into that state which would adversely affect our sales and revenues.
We rely on key personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to grow effectively.
We are dependent on certain members of our management, scientific and drug development staff and consultants, the loss of services of one or more of whom could materially adversely affect us.
We currently have four full-time employees, and retain the services of approximately 15 persons on an independent contractor/consultant and contract-employment basis. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel.
Our success depends in large part upon our ability to attract and retain highly qualified personnel. We compete in our hiring efforts with other pharmaceutical and biotechnology companies, as well as universities and nonprofit research organizations, and we may have to pay higher salaries to attract and retain personnel, which would be very costly.
We may be subject to foreign exchange fluctuation.
Our functional and reporting currency is the United States dollar. We maintain bank accounts in United States and Canadian dollars. A portion of our expenditures are in foreign currencies, most notably in Canadian dollars, and therefore we are subject to foreign currency fluctuations, which may, from time to time, impact our financial position and results. We may enter into hedging arrangements under specific circumstances, typically through the use of forward or futures currency contracts, to minimize the impact of increases in the value of the Canadian dollar. In order to minimize our exposure to foreign exchange fluctuations we may hold sufficient Canadian dollars to cover our expected Canadian dollar expenditures.
Product liability lawsuits against us could divert our resources, cause us to incur substantial liabilities and limit commercialization of any products that we may develop.
We face an inherent risk of product liability claims as a result of the clinical testing of our product candidates despite obtaining appropriate informed consents from our clinical trial participants. We will face an even greater risk if we commercially sell any product that we may develop. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidate. Regardless of the merits or eventual outcome, liability claims may result in:
| · | decreased demand for our product candidate or products that we may develop; |
| 19 |
| · | injury to our reputation and significant negative media attention; |
| · | withdrawal of clinical trial participants; |
| · | significant costs to defend resulting litigation; |
| · | substantial monetary awards to trial participants or patients; |
| · | loss of revenue; |
| · | reduced resources of our management to pursue our business strategy; and |
| · | the inability to commercialize any products that we may develop. |
Although we maintain general liability insurance, this insurance may not fully cover potential liabilities that we may incur. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. We will need to increase our insurance coverage if and when we begin selling any product candidate that receives marketing approval. In addition, insurance coverage is becoming increasingly expensive. If we are unable to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims, it could prevent or inhibit the development and commercial production and sale of our product candidate, which could adversely affect our business, financial condition, results of operations and prospects.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct clinical trials for our product candidates. Any failure by a third party to meet its obligations with respect to the clinical development of our product candidates may delay or impair our ability to obtain regulatory approval for our product candidate.
We rely on academic institutions and private oncology centers to conduct and sponsor clinical trials relating to VAL-083. Our reliance on third parties to conduct clinical trials could, depending on the actions of such third parties, jeopardize the validity of the clinical data generated and adversely affect our ability to obtain marketing approval from the FDA or other applicable regulatory authorities.
Such clinical trial arrangements provide us with information rights with respect to the clinical data, including access to and the ability to use and reference the data, including for our own regulatory filings, resulting from the clinical trials. If investigators or institutions breach their obligations with respect to the clinical trials of our product candidate, or if the data proves to be inadequate, then our ability to design and conduct any future clinical trials may be adversely affected.
We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
We currently rely on third party clinical research organizations, or CROs, to conduct our clinical trials. We expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to conduct our clinical trials. Our agreements with these third parties generally allow the third party to terminate the agreement at any time. If we are required to enter into alternative arrangements because of any such termination the introduction of our product candidates to market could be delayed.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we design our clinical trials and will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as good clinical practices, or GCPs, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
| 20 |
We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We may seek to enter into collaborations with third parties for the development and commercialization of our product candidate. If we fail to enter into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of our product candidate.
We may seek third-party collaborators for development and commercialization of our product candidate. Our likely collaborators for any marketing, distribution, development, licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, non-profit organizations, government agencies, and biotechnology companies. We are currently party to a limited number of such arrangements and have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidate. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidate currently pose, and will continue to pose, the following risks to us:
| · | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| · | collaborators may not pursue development and commercialization of our product candidate or may elect not to continue or renew development or commercialization programs based on preclinical or clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities; |
| · | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| · | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidate if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| · | collaborators with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; |
| · | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
| · | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
| · | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our product candidate or that result in costly litigation or arbitration that diverts management attention and resources; and |
| · | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaboration agreements may not lead to development or commercialization of our product candidate in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
If we are not able to establish collaborations, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidate will require substantial additional cash to fund expenses. We may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of our product candidate.
| 21 |
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of preclinical studies or clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under future license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of our product candidate, reduce or delay its development program, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidate or bring it to market and generate product revenue.
We manufacture our clinical supplies at a single location. Any disruption at this facility could adversely affect our business and results of operations.
We rely on our manufacturing partner, Guangxi Wuzhou Pharmaceuticals (Group) Co. Ltd., for the manufacture of clinical supply of VAL-083. If our partner’s facility were damaged or destroyed, or otherwise subject to disruption, it would require substantial lead-time to replace our clinical supply. In such event, we would be forced to rely entirely on other third-party contract manufacturers for an indefinite period of time. We have established a relationship with a back-up manufacturer, which has produced quantities of the active pharmaceutical ingredient contained in VAL-083. However, at this time no drug product has been manufactured by a third-party back-up manufacturer. Any disruptions or delays by Guangxi Wuzhou Pharmaceuticals or their failure to meet regulatory compliance could impair our ability to develop VAL-083, which would adversely affect our business and results of operations.
We may become subject to liabilities related to risks inherent in working with hazardous materials.
Our discovery and development processes involve the controlled use of hazardous and radioactive materials. We are subject to federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by such laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our resources. We are not specifically insured with respect to this liability. Although we believe that we are in compliance in all material respects with applicable environmental laws and regulations and currently do not expect to make material capital expenditures for environmental control facilities in the near-term, there can be no assurance that we will not be required to incur significant costs to comply with environmental laws and regulations in the future, or that our operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
Risks Related to Our Common Stock and This Offering
There is a limited trading market for our common stock, and you may have difficulty trading and obtaining quotations for our common stock.
Our common stock is registered under the Exchange Act and is quoted on the OTCQX. Prior to January 25, 2013, there was no reported trading in our common stock. Since January 25, 2013, there has been limited trading in our common stock. As a result, investors may find it difficult to dispose of, or to obtain accurate quotations of the price of, our securities. This severely limits the liquidity of the common stock, and may adversely affect the market price of our common stock. A limited market may also impair our ability to raise capital by selling shares of capital stock and may impair our ability to acquire other companies or assets by using common stock as consideration.
Subsequent to the closing of this offering, we intend to apply to our common stock on the NYSE MKT or NASDAQ Capital Market. There is no assurance our application will be approved. We expect that we may be required to effect a reverse split of our common stock to become eligible to be quoted on the NYSE MKT or NASDAQ Capital Market. For our common stock to remain listed on the NYSE MKT or NASDAQ Capital Market, we must meet the ongoing NYSE MKT or NASDAQ Capital Market listing requirements. If we were unable to meet these requirements, our common stock could be delisted from the NYSE MKT or NASDAQ Capital Market. If our common stock were to be delisted from the NYSE MKT or NASDAQ Capital Market, our common stock could continue to trade on the OTCQX following any delisting from the NYSE MKT or NASDAQ Capital Market. Any such delisting of our common stock could have an adverse effect on the market price of, and the efficiency of the trading market for, our common stock, not only in terms of the number of shares that can be bought and sold at a given price, but also through delays in the timing of transactions and less coverage of us by securities analysts, if any. Also, if in the future we were to determine that we need to seek additional equity capital, it could have an adverse effect on our ability to raise capital in the public or private equity markets.
| 22 |
The market price of our common stock is, and is likely to continue to be, highly volatile and subject to wide fluctuations.
The market price of our common stock is highly volatile and could be subject to wide fluctuations in response to a number of factors that are beyond our control, including:
| · | variations in our quarterly operating results; | |
| · | announcements that our revenue or income are below analysts’ expectations; | |
| · | general economic slowdowns; | |
| · | sales of large blocks of our common stock; and | |
| · | announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments. |
Because we became public by means of a reverse acquisition, we may not be able to attract, or maintain, the attention of brokerage firms.
Because we became public through a “reverse acquisition”, securities analysts of brokerage firms may not provide or continue to provide coverage of us since there is little incentive to brokerage firms to recommend the purchase of our common stock. No assurance can be given that brokerage firms will want to conduct any secondary offerings on behalf of the Company in the future.
Applicable regulatory requirements, including those contained in and issued under the Sarbanes-Oxley Act of 2002, may make it difficult for the Company to retain or attract qualified officers and directors, which could adversely affect the management of its business and its ability to obtain or retain listing of its common stock.
The Company may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by the stock exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers.
Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. The Company may have difficulty attracting and retaining directors with the requisite qualifications. If the Company is unable to attract and retain qualified officers and directors, the management of its business and its ability to obtain or retain listing of our shares of common stock on any stock exchange (assuming the Company elects to seek and are successful in obtaining such listing) could be adversely affected.
Voting power of our shareholders is highly concentrated by insiders.
Our officers and directors control, either directly or indirectly, a substantial portion of our voting securities. Therefore, our management may significantly affect the outcome of all corporate actions and decisions for an indefinite period of time including election of directors, amendment of charter documents and approval of mergers and other significant corporate transactions.
We do not intend to pay dividends for the foreseeable future.
We have paid no dividends on our common stock to date and it is not anticipated that any dividends will be paid to holders of our common stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, it is currently anticipated that any earnings will be retained to finance our future expansion and for the implementation of our business plan. As an investor, you should take note of the fact that a lack of a dividend can further affect the market value of our common stock, and could significantly affect the value of any investment in our Company.
| 23 |
Our articles of incorporation allow for our board to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. Our Board of Directors has the authority to issue up to 5,000,000 shares of our preferred stock (of which 1 share has been designated Special Voting Preferred Stock and is issued and outstanding, and 278,530 shares have been designated Series A Preferred Stock and are issued and outstanding; see “Description of Securities”) without further stockholder approval. As a result, our board of directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our common stock. In addition, our board of directors could authorize the issuance of a series of preferred stock that has greater voting power than our common stock or that is convertible into our common stock, which could decrease the relative voting power of our common stock or result in dilution to our existing stockholders. Although we have no present intention to issue any additional shares of preferred stock or to create any additional series of preferred stock, we may issue such shares in the future.
Our issuance of common stock upon exercise of warrants or options or exchange of Exchangeable Shares may depress the price of our common stock.
As of April 1, 2015, we have 35,199,889 shares of common stock issued and outstanding, 4,256,042 shares of common stock issuable upon exchange of the Exchangeable Shares, outstanding warrants to purchase 13,472,870 shares of common stock, and outstanding options to purchase 3,595,000 shares of common stock. The issuance of shares of common stock upon exercise of outstanding warrants or options or exchange of Exchangeable Shares could result in substantial dilution to our stockholders, which may have a negative effect on the price of our common stock.
The warrants are speculative in nature.
The warrants to be issued to investors in this offering do not confer any rights of common stock ownership on their holders, such as voting rights or the right to receive dividends, but rather merely represent the right to acquire shares of common stock at a fixed price for a limited period of time. Specifically, commencing on the date of issuance, holders of the warrants may exercise their right to acquire the common stock and pay an exercise price of $___ per share, prior to _____ years from the date of issuance, after which date any unexercised warrants will expire and have no further value. Moreover, following this offering, the market value of the warrants is uncertain and there can be no assurance that the market value of the warrants will equal or exceed their public offering price. The warrants will not be listed or quoted for trading on any market or exchange. There can be no assurance that the market price of the common stock will ever equal or exceed the exercise price of the warrants, and consequently, whether it will ever be profitable for holders of the warrants to exercise the warrants.
Our management will have broad discretion over the use of the net proceeds from this offering and we may use the net proceeds in ways with which you disagree.
We currently intend to use the net proceeds from this offering to fund our research and drug development activities, including a Phase II/III registration-directed clinical trial with VAL-083 as a potential new treatment for refractory GBM and for working capital and general corporate purposes. We have not allocated specific amounts of the net proceeds from this offering for any of the foregoing purposes. Accordingly, our management will have significant discretion and flexibility in applying the net proceeds of this offering. You will be relying on the judgment of our management with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that the net proceeds will be invested in a way that does not yield a favorable, or any, return for us or our stockholders. The failure of our management to use such funds effectively could have a material adverse effect on our business, prospects, financial condition, and results of operation.
Additional stock offerings in the future may dilute your percentage ownership of our company.
Given our plans and expectations that we may need additional capital and personnel, we may need to issue additional shares of common stock or securities convertible or exercisable for shares of common stock, including convertible preferred stock, convertible notes, stock options or warrants. The issuance of additional securities in the future will dilute the percentage ownership of then current stockholders.
You will experience immediate and substantial dilution as a result of this offering and may experience additional dilution in the future.
You will incur immediate and substantial dilution as a result of this offering. After giving effect to the sale by us of shares offered in this offering at an assumed public offering price of $ per share (the closing price of a share of our common stock on , 2015) and warrants at an assumed public offering price of $ per warrant, and after deducting the underwriter’s discount and estimated offering expenses payable by us, investors in this offering can expect an immediate dilution of $ per share. In addition, in the past, we issued options and warrants to acquire shares of common stock. To the extent these options or warrants are ultimately exercised, you will sustain future dilution.
| 24 |
Risks Related To Our Proposed Reverse Stock Split
We may be required to complete a reverse stock split of our outstanding common stock in order to meet the initial listing requirements of the NYSE MKT or NASDAQ Capital Market. However, we cannot assure you that we will be able to continue to comply with the minimum price requirements of the NYSE MKT or NASDAQ Capital Market.
We may be required to complete a reverse stock split in order to achieve the requisite increase in the market price of our common stock to be in compliance with the minimum price requirements of the NYSE MKT or NASDAQ Capital Market. We cannot assure you that the market price of our common stock following the reverse stock split will remain at the level required for the period of time required for listing or for continuing compliance with that requirement. It is not uncommon for the market price of a company’s common stock to decline in the period following a reverse stock split. If the market price of our common stock declines following the effectuation of a reverse stock split, the percentage decline may be greater than would occur in the absence of a reverse stock split. In any event, other factors unrelated to the number of shares of our common stock outstanding, such as negative financial or operational results, could adversely affect the market price of our common stock and jeopardize our ability to obtain or maintain the NYSE MKT or NASDAQ Capital Market’s minimum price requirements. In addition to specific listing and maintenance standards, the NYSE MKT and NASDAQ Capital Market have broad discretionary authority over the initial and continued listing of securities, which it could exercise with respect to the listing of our common stock.
Even if a reverse stock split increases the market price of our common stock, there can be no assurance that we will be able to comply with other initial or continued listing standards of the NYSE MKT or NASDAQ Capital Market.
Even if the market price of our common stock increases sufficiently so that we comply with the minimum bid price requirement, we cannot assure you that we will be able to comply with the other standards that we are required to meet in order to achieve or maintain a listing of our common stock sold in this offering on the NYSE MKT or NASDAQ Capital Market. Our failure to meet these requirements may result in our common stock sold in this offering being delisted from the NYSE MKT or NASDAQ Capital Market, irrespective of our compliance with the minimum bid price requirement.
The proposed reverse stock split may decrease the liquidity of the shares of our common stock.
The liquidity of the shares of our common stock may be affected adversely by a reverse stock split given the reduced number of shares that will be outstanding following a reverse stock split, especially if the market price of our common stock does not increase as a result of the reverse stock split.
Following a reverse stock split, the resulting market price of our common stock may not attract new investors, including institutional investors, and may not satisfy the investing requirements of those investors. Consequently, the trading liquidity of our common stock may not improve.
Although we believe that a higher market price of our common stock may help generate greater or broader investor interest, we cannot assure you that the reverse stock split will result in a share price that will attract new investors.
| 25 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
Statements in this prospectus may be “forward-looking statements.” Forward-looking statements include, but are not limited to, statements that express our intentions, beliefs, expectations, strategies, predictions or any other statements relating to our future activities or other future events or conditions. These statements are based on current expectations, estimates and projections about our business based, in part, on assumptions made by management. These statements are not guarantees of future performance and involve risks, uncertainties and assumptions that are difficult to predict. Therefore, actual outcomes and results may, and are likely to, differ materially from what is expressed or forecasted in the forward-looking statements due to numerous factors, including those described above and those risks discussed from time to time in this prospectus, including the risks described under “Risk Factors,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this prospectus and in other documents which we file with the Securities and Exchange Commission. In addition, such statements could be affected by risks and uncertainties related to:
| · | our ability to raise funds for general corporate purposes and operations, including our clinical trials; | |
| · | our ability to recruit qualified management and technical personnel; | |
| · | the success of our clinical trials; | |
| · | our ability to obtain and maintain required regulatory approvals for our products; and | |
| · | the other factors discussed in the “Risk Factors” section and elsewhere in this prospectus. |
Any forward-looking statements speak only as of the date on which they are made, and except as may be required under applicable securities laws, we do not undertake any obligation to update any forward-looking statement to reflect events or circumstances after the date of this prospectus.
| 26 |
We estimate that the net proceeds from the sale of the common stock and warrants offered pursuant to this prospectus will be approximately $ million, or approximately $ million if the underwriters exercise in full their option to purchase additional shares and/or warrants, after deducting the underwriting discount and the estimated offering expenses that are payable by us.
We currently intend to use the net proceeds from this offering to fund our research and drug development activities, including a Phase II/III registration-directed clinical trial with VAL-083 as a potential new treatment for refractory GBM and for working capital and general corporate purposes.
We have not yet determined the amount of net proceeds to be used specifically for any of the foregoing purposes. Accordingly, our management will have significant discretion and flexibility in applying the net proceeds from this offering. Pending any use as described above, we intend to invest the net proceeds in high-quality, short-term, interest-bearing securities.
Our common stock is quoted on the OTCQX, under the symbol “DMPI”. Previously, the Company’s common stock was quoted on the OTCQB. There was no reported trading in our common stock prior to January 25, 2013. Subsequent to the closing of this offering, we intend to apply for listing of our common stock on the NYSE MKT or NASDAQ Capital Market. No assurance can be given that our application will be approved.
The following table sets forth the range of high and low bid prices of our common stock as reported and summarized on the OTCQB or OTCQB, as applicable, for the periods indicated. These prices are based on inter-dealer bid and asked prices, without markup, markdown, commissions, or adjustments and may not represent actual transactions.
| Calendar Quarter | High Bid | Low Bid | ||||||
| 2013 First Quarter | $ | 2.50 | $ | 1.30 | ||||
| 2013 Second Quarter | $ | 2.48 | $ | 1.55 | ||||
| 2013 Third Quarter | $ | 2.04 | $ | 0.90 | ||||
| 2013 Fourth Quarter | $ | 1.48 | $ | 0.75 | ||||
| 2014 First Quarter | $ | 1.60 | $ | 0.79 | ||||
| 2014 Second Quarter | $ | 1.41 | $ | 0.75 | ||||
| 2014 Third Quarter | $ | 1.03 | $ | 0.62 | ||||
| 2014 Fourth Quarter | $ | 1.02 | $ | 0.73 | ||||
| 2015 First Quarter | $ | 0.99 | $ | 0.64 | ||||
| 2015 Second Quarter (through April 20, 2015) | $ | 0.89 | $ | 0.69 | ||||
As of March 31, 2015, there were approximately 194 holders of record of the Company’s common stock.
The Company has never declared or paid any cash dividends on its common stock. The Company currently intends to retain future earnings, if any, to finance the expansion of its business. As a result, the Company does not anticipate paying any cash dividends in the foreseeable future.
| 27 |
CAPITALIZATION
The following table sets forth our capitalization, as of December 31, 2014:
| ● | on an actual basis; and | |
| ● | on a pro forma to give effect to the sale of the shares and warrants in this offering at the assumed public offering price of $ per share and $ per warrant, after deducting underwriting discounts and commissions and other estimated offering expenses payable by us. |
You should consider this table in conjunction with our financial statements and the notes to those financial statements included elsewhere in this prospectus.
| December 31, 2014 | ||||||||
| (Actual) | (Pro forma) | |||||||
| Preferred Stock, $.001 par value, 5,000,000 shares authorized; 278,530 Series A shares issued and outstanding | $ | 278,530 | $ | |||||
| 1 special voting share issued and outstanding | - | |||||||
| Common stock; $0.001 par value; 200,000,000 shares authorized, 38,580,306 shares issued and outstanding actual (1), _______ shares issued and outstanding pro forma | 38,580 | |||||||
| Additional paid-in capital | 16,625,081 | |||||||
| Warrants | 6,187,805 | |||||||
| Accumulated deficit | (21,064,623 | ) | ||||||
| Accumulated other comprehensive income | 21,178 | |||||||
| Total stockholder's equity | $ | 2,086,551 | $ | |||||
| (1) | On an as-exchanged basis with respect to Exchangeable Shares. |
| 28 |
If you invest in our securities, your interest will be immediately and substantially diluted to the extent of the difference between the public offering price per share of our common stock and the pro forma net tangible book value per share of our common stock after giving effect to this offering.
Our net tangible book value as of December 31, 2014 was $2,086,551 or approximately $0.05 per share of common stock, based upon 38,580,306 shares outstanding as of December 31, 2014 (and on an-exchanged basis with respect to Exchangeable Shares). After giving effect to the sale of the shares and warrants in this offering at the assumed public offering price of $ per share and $ per warrant and after deducting underwriting discounts and commissions and other estimated offering expenses payable by us, our pro forma net tangible book value at December 31, 2014 would have been approximately $ million or $ per share. This represents an immediate increase in pro forma net tangible book value of approximately $ per share to our existing stockholders, and an immediate dilution of $ per share to investors purchasing
Dilution in pro forma net tangible book value per share represents the difference between the amount per share paid by purchasers of our common stock in this offering and the pro forma net tangible book value per share of our common stock immediately after this offering.
The following table illustrates the per share dilution to investors purchasing shares in the offering:
| Assumed public offering price per share | $ | ||||||
| Net tangible book value per share as of December 31, 2014 | $ | 0.05 | |||||
| Increase in net tangible book value per share attributable to this offering | $ | ||||||
| Pro forma net tangible book value per share after this offering | $ | ||||||
| Amount of dilution in net tangible book value per share to new investors in this offering | $ |
The information above assumes that the underwriters do not exercise their over-allotment option. If the underwriters exercise their over-allotment option in full, the pro forma net tangible book value will increase to $ per share, representing an immediate increase to existing stockholders of $ per share and an immediate dilution of $ per share to new investors. If any shares are issued upon exercise of outstanding options or warrants, new investors will experience further dilution.
| 29 |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read together with our financial statements and the related notes appearing elsewhere in this prospectus. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. See “Cautionary Note Regarding Forward-Looking Statements and Industry Data” for a discussion of the uncertainties, risks and assumptions associated with these statements. Actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under “Risk Factors” and elsewhere in this prospectus.
Overview
DelMar Pharmaceuticals, Inc. (the “Company”) is developing a new drug candidate targeting orphan cancer indications. We aim to develop products that will have a high impact in patient care and a high return for our investors. In order to accelerate our development timelines and reduce technical risk, we leverage existing clinical and commercial data from a wide range of sources.
Recent Developments
Change in Fiscal Year End
On July 21, 2014, the Board of Directors of the Company approved a change in the Company’s fiscal year end from December 31 to June 30. As a result of this change, the Company has prepared consolidated financial statements for the six month transition period ended June 30, 2014. References to any of the Company’s 2013 or earlier fiscal years mean the fiscal year ending December 31 of that calendar year.
Reverse Acquisition
On January 25, 2013 (the “Closing Date”), the Company entered into and closed an exchange agreement (the “Exchange Agreement”), with DelMar (BC), Callco, Exchangeco, and the securityholders of DelMar (BC). Pursuant to the Exchange Agreement, (i) the Company issued 4,340,417 shares of common stock (the “Parent Shares”) to the shareholders of DelMar (BC) who are United States residents (the “U.S. Holders”) in exchange for the transfer to Exchangeco of all 4,340,417 outstanding common shares of DelMar (BC) held by the U.S. Holders, (ii) the shareholders of DelMar (BC) who are Canadian residents (the “Canadian Holders”) received, in exchange for the transfer to Exchangeco of all 8,729,583 outstanding common shares of DelMar (BC) held by the Canadian Holders, 8,729,583 exchangeable shares (the “Exchangeable Shares”) of Exchangeco, and (iii) outstanding warrants to purchase 3,360,000 common shares of DelMar (BC) and outstanding options to purchase 1,020,000 common shares of DelMar (BC) were deemed to be amended such that, rather than entitling the holder to acquire common shares of DelMar (BC), such options and warrants will entitle the holders to acquire shares of common stock of the Company. The Canadian Holders will be entitled to require Exchangeco to redeem (or, at the option of the Company or Callco, to have the Company or Callco purchase) the Exchangeable Shares, and upon such redemption or purchase to receive an equal number of shares of common stock of the Company. The aggregate of 13,070,000 shares of common stock of the Company issued to the former shareholders of DelMar (BC) (on an as-exchanged basis with respect to the Exchangeable Shares) represents 80.1% of the outstanding shares of common stock of the Company following the closing of the Exchange Agreement (the “Reverse Acquisition”).
Upon completion of the Reverse Acquisition DelMar (BC) became a wholly-owned subsidiary of the Company. As a result of the shareholders of DelMar (BC) owning a controlling interest in the Company subsequent to the Reverse Acquisition, for accounting purposes the transaction is a capital transaction with DelMar (BC) being the accounting acquirer even though the legal acquirer is Berry. No goodwill is recorded with respect to the transaction as it does not constitute a business combination. For accounting purposes, the transaction is reflected as a recapitalization of DelMar (BC) and consideration for the Reverse Acquisition was deemed to be the fair value of the shares that were issued by DelMar (BC) to acquire the net liabilities of Berry on January 25, 2013. The net identifiable liabilities of Berry on the Closing Date of the Reverse Acquisition were as follows:
| Net liabilities (derivative liability) | $ | 2,041,680 | ||
The Company determined the fair value of the shares issued on the Reverse Acquisition to be $1,690,004. As a result of the Reverse Acquisition being treated as a recapitalization of DelMar (BC) the Company recognized the loss of $3,731,684 incurred upon the closing of the Reverse Acquisition as an adjustment to opening deficit in the consolidated statement of stockholder’s deficiency at December 31, 2013.
| 30 |
Unit Offering
In connection with the Reverse Acquisition, on January 25, 2013, January 31, 2013, February 8, 2013, February 21, 2013, February 28, 2013, March 1, 2013, and March 6, 2013, the Company entered into and closed a series of subscription agreements with accredited investors (the “Investors”), pursuant to which the Company issued an aggregate of 13,125,002 Units at a purchase price of $0.80 per Unit, for aggregate gross proceeds of $10,500,000 (the “Private Offering”). Each Unit consists of one share of common stock and one five-year warrant (the “Investor Warrants”) to purchase one share of common stock at an exercise price of $0.80. The exercise price of the Investor Warrants is subject to adjustment in the event that the Company sells common stock at a price lower than the exercise price, subject to certain exceptions. The Investor Warrants are redeemable by the Company at a price of $0.001 per Investor Warrant at any time subject to the conditions that (i) the Company’s common stock has traded for twenty consecutive trading days with a closing price of at least $1.60 per share with an average trading volume of 50,000 shares per day and (ii) the underlying shares of common stock are registered.
The Company retained Charles Vista, LLC (the “Placement Agent”) as the Placement Agent for the Private Offering. The Company paid the Placement Agent a cash fee of $1,050,000 (equal to 10% of the gross proceeds), a non-accountable expense allowance of $315,000 (equal to 3% of the gross proceeds), and a one-year consulting fee of $60,000. In addition, the Company incurred other closing costs of approximately $500,000 resulting in net proceeds to the Company of $8,575,000. Certain of the additional closing costs were not eligible to be treated as share issue costs and as a result they have been expensed. Net unit proceeds per the consolidated statements of cash flows include gross unit proceeds less cash issue costs attributable to the common stock only. The portion of the unit issue costs attributable to the derivative liability has been expensed.
In addition, the Company issued to the Placement Agent five-year warrants (the “Placement Agent Warrants”) to purchase 5,250,000 shares of common stock (equal to 20% of the shares of common stock (i) included as part of the Units sold in the Private Offering and (ii) issuable upon exercise of the Investor Warrants) at an exercise price of $0.80, exercisable on a cash or cashless basis.
The Company agreed to pay a warrant commission of 5% of the amount of funds raised by an agent upon the exercise of the Investor Warrants following such redemption.
In connection with the Private Offering, the Company entered into a registration rights agreement with the Investors, pursuant to which the Company agreed to file a registration statement (the “Registration Statement”) registering for resale all shares of common stock (a) included in the Units; and (b) issuable upon exercise of the Investor Warrants, no later than 90 days after the completion of the Private Offering (the “Filing Deadline”) and to use commercially reasonable efforts to cause the Registration Statement to become effective within 180 days of the Filing Deadline. The Company agreed to use commercially reasonably efforts to keep the Registration Statement effective while the Investor Warrants are outstanding.
Certain of the Private Offering costs were incurred by the Company prior to December 31, 2012. These costs of $90,771 were treated as issue costs during the year ended December 31, 2013.
Derivative Liability
The Company has issued common stock purchase warrants. Based on the terms of certain of these warrants the Company determined that the warrants were a derivative liability which is recognized at fair value at the date of the transaction and re-measured at fair value each reporting period with the changes in fair value recorded in the consolidated condensed statement of loss and comprehensive loss.
CDN $0.50 Unit Warrants
The Company issued 4,150,000 units on January 23, 2012, 560,000 on February 27, 2012 and 50,000 on May 10, 2012. In addition, during the year ended December 31, 2011 the Company issued 500,000 units on October 3, 2011, 100,000 on October 7, 2011, and 50,000 on November 11, 2011. In total, the Company issued 5,410,000 units for services in settlement of accounts payable and cash proceeds for an aggregate of $2,671,923 (CDN $2,705,000).
The proceeds from the issuance of 3,000,000 of these units were held in escrow pursuant to an exclusive option investment agreement with a strategic investor. Subsequently, the Company elected to allow the option to expire and the related units were cancelled and the funds returned from escrow to the subscriber in order for the Company to retain control over certain intellectual property and commercial rights.
During the year ended December 31, 2013, 221,000 of these warrants were exercised for no additional consideration for 221,000 shares of common stock. As a result, $241,715 of the derivative liability has been reclassified to equity. During the six months ended June 30, 2014, 20,000 of these warrants were exercised for no additional consideration for 20,000 shares of common stock with $17,600 of the derivative liability being reclassified to equity. The warrants that have been exercised were revalued at their exercise date and then the reclassification to equity was recorded.
| 31 |
On January 25, 2014 the remaining 2,169,000 of these warrants expired. All of the CDN $0.50 warrants outstanding at December 31, 2013 have either been exercised or have expired as at June 30, 2014.
The warrants that were outstanding at December 31, 2013 were re-valued at December 31, 2013 using a simulated probability valuation model using the following assumptions: dividend rate - 0%, volatility – 72.8%, risk free rate - 0.09% and a term of one month.
Investor Warrants
In connection with the Reverse Acquisition, on January 25, 2013, January 31, 2013, February 8, 2013, February 21, 2013, February 28, 2013, March 1, 2013, and March 6, 2013, the Company entered into and closed a series of subscription agreements with accredited investors (the “Investors”), pursuant to which the Company issued an aggregate of 13,125,002 Units at a purchase price of $0.80 per Unit, for aggregate gross proceeds of $10,500,000 (the “Private Offering”). Each Unit consists of one share of common stock and one five-year warrant (the “Investor Warrants”) to purchase one share of common stock at an exercise price of $0.80. The exercise price of the Investor Warrants is subject to adjustment in the event that the Company sells common stock at a price lower than the exercise price, subject to certain exceptions. The Investor Warrants are redeemable by the Company at a price of $0.001 per Investor Warrant at any time subject to the conditions that (i) the Company’s common stock has traded for twenty (20) consecutive trading days with a closing price of at least $1.60 per share with an average trading volume of 50,000 shares per day and (ii) the underlying shares of common stock are registered.
During the six months ended June 30, 2014, 277,313 warrants were exercised at $0.80 per warrant for 277,313 shares of common stock. The Company received proceeds of $221,850 from the exercise. As a result, $126,064 of the derivative liability has been reclassified to equity.
On June 6, 2014, pursuant to election to exercise warrants agreements the Company reduced the Investor Warrant exercise price from $0.80 to $0.65 per share for warrants to purchase 3,652,211 shares of the Company’s common stock and in accordance with the agreements, the holders of the Investor Warrants exercised the Investor Warrants for cash at the foregoing reduced exercise price. The Company received net proceeds of $2,255,240 after paying a 5% warrant agent fee of $118,697. As a result, $984,484 of the derivative liability has been reclassified to equity.
All Investor Warrants that have been exercised were revalued at their exercise date and then the reclassification to equity was recorded.
Tender offer - Investor Warrant exercise price reduction
On June 9, 2014, as amended on June 26, 2014, July 10, 2014, and July 29, 2014, the Company filed a tender offer statement with the Securities and Exchange Commission with respect to certain warrants to purchase common stock of the Company issued to investors (the “Investor Warrants”) to provide the holders thereof with the opportunity to amend and exercise their warrants, upon the terms and subject to the conditions set forth in the Company’s tender offer statement. Pursuant to the tender offer, the Company offered to amend Investor Warrants to purchase an aggregate of 9,195,478 shares of common stock (the “Offer to Amend and Exercise”). There was no minimum participation requirement with respect to the Offer to Amend and Exercise.
Pursuant to the Offer to Amend and Exercise, the Investor Warrants subject to the tender offer were amended (the “Amended Warrants”) to: (i) reduce the exercise price of the Investor Warrants from $0.80 per share to $0.65 per share of common stock in cash, (ii) shorten the exercise period of the Investor Warrants so that they expire concurrently with the expiration of the Offer to Amend and Exercise at 5:00 p.m. (Pacific Time) on August 8, 2014, as may be extended by the Company in its sole discretion (“Expiration Date”), (iii) delete the price-based anti-dilution provisions contained in the Investor Warrants, (iv) restrict the ability of the holder of shares issuable upon exercise of the Amended Warrants to sell, make any short sale of, loan, grant any option for the purchase of, or otherwise dispose of any of such shares without the prior written consent of the Company for a period of time twenty (20) days after the Expiration Date (the “ Lock-Up Period ”); and (v) provide that a holder, acting alone or with others, will agree not to effect any purchases or sales of any securities of the Company in any “short sales” as defined in Rule 200 promulgated under Regulation SHO under the Exchange Act, or any type of direct and indirect stock pledges, forward sale contracts, options, puts, calls, short sales, swaps, “put equivalent positions” (as defined in Rule 16a-1(h) under the Exchange Act) or similar arrangements, or sales or other transactions through non-U.S. broker dealers or foreign regulated brokers through the expiration of the Lock-Up Period.
Upon the expiration of the Offer to Amend and Exercise on August 8, 2014, 762,227 Amended Warrants were exercised for net proceeds of $470,676 after payment by the Company of a 5% warrant agent fee of $24,772.
Investor Warrant exercises
In addition, during the six months ended December 31, 2014, 1,223,847 Investor Warrants were exercised at $0.65 per warrant for 1,223,847 shares of common stock. The Company received proceeds of $795,501 from these exercises.
| 32 |
All Investor Warrants that have been exercised during the period, including those exercised under the tender offer, were revalued at their respective exercise dates and then a reclassification to equity was recorded. As a result of all of the Investor Warrant exercises for the six months ended December 31, 2014 an aggregate $391,422 of the derivative liability has been reclassified to equity.
To date, including Investor Warrants exercised prior to June 30, 2014, a total of 5,195,598 Investor Warrants have been exercised for cash for total gross proceeds of $3,886,736.
Investor Warrant exchange
On December 31, 2014, the Company issued 414,889 shares of common stock in exchange for 1,244,666 Investor Warrants. The Investor Warrants that have been exchanged were revalued at their exchange date and then a reclassification to equity was recorded. The reclassification to equity upon the exchange was $305,112. The Company recognized a loss of $92,843 at the time of the exchange.
On January 8, 2015, the Company filed a tender offer statement with the Securities and Exchange Commission, and on January 23, 2015, the Company filed an amendment thereto, with respect to certain Investor Warrants to purchase common stock of the Company. The tender offer provided the holders of the Investor Warrants with the opportunity to receive one share of common stock for every three Investor Warrants that are tendered. The tender offer was available to all 5,964,738 Investor Warrants outstanding at December 31, 2014. If all outstanding Investor Warrants were tendered, the Company would have issued 1,988,246 shares of common stock. To participate in the tender offer the Investor Warrant holders were required to deliver completed exchange documents to the Company, prior to the expiration of the tender offer, which was 5:00 p.m. (Pacific Time) on February 9, 2015.
The tender offer expired on February 9, 2015. A total of 1,591,875 Investor Warrants were exchanged for 530,625 shares of common stock resulting in a net increase in stockholder’s equity of approximately $400,957.
The remaining 5,964,738 Investor Warrants outstanding at December 31 , 2014 have been re-valued at December 31, 2014 using a simulated probability valuation model using the following assumptions: dividend rate - 0%, volatility - 73%, risk free rate - 1.34% and a term of approximately 3.0 years.
All 5,964,738 Investor warrants outstanding at December 31, 2014 have an exercise price of $0.80.
Dividend Warrants
In connection with the reverse acquisition, effective January 24, 2013, the Company effected a warrant dividend (the “Warrant Dividend”) pursuant to which the Company issued one five-year warrant to purchase one share of common stock at an exercise price of $1.25 for each outstanding share of common stock (the “Dividend Warrants”). Pursuant to the Warrant Dividend, the Company issued an aggregate of 3,250,007 Dividend Warrants.
On October 31, 2014, the Company and all of its Dividend Warrant holders entered into amendments to the Dividend Warrants such that the Company’s redemption rights and certain provisions of the Dividend Warrant agreements relating to potential cash settlement of the Dividend Warrants were removed. The Dividend Warrants were revalued to the date of the amendment on October 31, 2013 which resulted in a reduction in the derivative liability and a corresponding reclassification to equity of $975,278.
Warrants issued for services
The Company has issued 300,000 warrants for services. The warrants were issued on September 12, 2013 and are exercisable on a cashless basis at an exercise price of $1.76 for five years. The warrants have been measured at December 31, 2014 using a simulated probability valuation model using the following assumptions: dividend rate - 0%, volatility - 74%, risk free rate - 1.49% and a term of approximately 3.5 years.
| 33 |
The Company’s derivative liability is summarized as follows:
| December 31, 2014 $ | June 30, 2014 $ | |||||||
| Opening balance | 3,329,367 | 4,402,306 | ||||||
| Change in fair value of warrants | (66,606 | ) | 166,388 | |||||
| Change in fair value due to change in warrant terms | (23,658 | ) | (111,179 | ) | ||||
| Reclassification to equity upon amendment of warrants | (975,278 | ) | - | |||||
| Reclassification to equity upon exchange of warrants | (305,112 | ) | - | |||||
| Reclassification to equity upon exercise of warrants | (391,422 | ) | (1,128,148 | ) | ||||
| Closing balance | 1,567,291 | 3,329,367 | ||||||
| June 30, 2014 $ | December 31, 2013 $ | December 31, 2012 $ | ||||||||||
| Opening balance | 4,402,306 | 121,000 | 106,146 | |||||||||
| Issuance of units | - | 3,681,372 | 333,356 | |||||||||
| Dividend warrant liability acquired on reverse acquisition | - | 2,041,680 | - | |||||||||
| Warrants issued for services | - | 124,020 | - | |||||||||
| Change in fair value of unexercised warrants | 166,388 | (1,324,051 | ) | (318,502 | ) | |||||||
| Change in fair value due to tender offer | (111,179 | ) | - | - | ||||||||
| Reclassification to equity upon exercise of warrants | (1,128,148 | ) | (241,715 | ) | - | |||||||
| Closing balance | 3,329,367 | 4,402,306 | 121,000 | |||||||||
Selected Quarterly Information
The financial information reported here in has been prepared in accordance with accounting principles generally accepted in the United States. The Company’s functional currency at December 31, 2014 is the USD. The following table represents selected financial information for the Company as of December 31, 2014 and June 30, 2014.
Selected Balance Sheet Data
| December 31, 2014 $ | June 30, 2014 $ | |||||||
| Cash and cash equivalents | 3,958,439 | 4,759,711 | ||||||
| Working capital | 3,834,192 | 4,704,044 | ||||||
| Total Assets | 4,128,378 | 5,003,910 | ||||||
| Derivative liability | 1,567,291 | 3,329,367 | ||||||
| Total stockholders’ equity | 2,086,551 | 880,479 | ||||||
Selected Statement of Operations Data
For the three months ended:
| December 31, | December 31, | |||||||
| 2014 | 2013 | |||||||
| $ | $ | |||||||
| Research and development | 612,169 | 566,060 | ||||||
| General and administrative | 656,229 | 636,182 | ||||||
| Change in fair value of derivative liability | (435,200 | ) | (372,487 | ) | ||||
| Change in fair value of derivative liability due to change in warrant terms | 143,532 | - | ||||||
| Loss on exchange of warrants | 92,843 | - | ||||||
| Foreign exchange loss | 7,295 | 34,797 | ||||||
| Interest expense | - | 2,044 | ||||||
| Interest income | (109 | ) | (620 | ) | ||||
| Net and comprehensive loss | 1,076,759 | 865,976 | ||||||
| Weighted average number of shares outstanding | 37,798,183 | 31,523,732 | ||||||
| Loss per share | 0.03 | 0.03 | ||||||
| 34 |
| For the six months ended: | ||||||||
| December 31, | December 31, | |||||||
| 2014 | 2013 | |||||||
| $ | $ | |||||||
| Research and development | 1,283,796 | 1,126,295 | ||||||
| General and administrative | 1,101,229 | 1,377,550 | ||||||
| Change in fair value of derivative liability | (66,606 | ) | (8,466,826 | ) | ||||
| Change in fair value of derivative liability due to change in warrant terms | (23,658 | ) | - | |||||
| Loss on exchange of warrants | 92,843 | - | ||||||
| Foreign exchange loss | 9,686 | 31,963 | ||||||
| Interest expense | 2,091 | 4,073 | ||||||
| Interest income | (261 | ) | (1,311 | ) | ||||
| Net and comprehensive loss (income) | 2,399,120 | (5,928,256 | ) | |||||
| Basic weighted average number of shares outstanding | 37,125,074 | 31,477,137 | ||||||
| Basic loss (income) per share | 0.06 | (0.19 | ) | |||||
| Diluted weighted average number of shares outstanding | 37,125,074 | 41,742,401 | ||||||
| Diluted loss (income) per share | 0.06 | 0.00 | ||||||
Expenses net of share-based payments
The following table discloses research and development, and general and administrative expenses net of share-based payment expenses.
For the three months ended:
| December 31, 2014 $ | December 31, 2013 $ | |||||||
| Research and development | 612,169 | 566,060 | ||||||
| Share-based payments included in research and development | 8,077 | (163,514 | ) | |||||
| Research and development net of share-based compensation | 620,246 | 402,546 | ||||||
| General and administrative | 656,229 | 636,182 | ||||||
| Share-based payments included in general and administrative | (219,647 | ) | (192,391 | ) | ||||
| General and administrative net of share-based compensation | 436,582 | 443,791 | ||||||
For the six months ended:
| December 31, 2014 $ | December 31, 2013 $ | |||||||
| Research and development | 1,283,796 | 1,126,295 | ||||||
| Share-based payments included in research and development | (13,056 | ) | (259,589 | ) | ||||
| Research and development net of share-based compensation | 1,270,740 | 866,706 | ||||||
| General and administrative | 1,101,229 | 1,377,550 | ||||||
| Share-based payments included in general and administrative | (247,454 | ) | (490,710 | ) | ||||
| General and administrative net of share-based compensation | 853,775 | 886,840 | ||||||
| 35 |
Comparison of the three months ended December 31, 2014 and December 31, 2013
| Three Months Ended | ||||||||||||||||
| December 31, 2014 $ | December 31, 2013 $ | Change $ | Change % | |||||||||||||
| Research and development | 612,169 | 566,060 | 46,109 | 8 | ||||||||||||
| General and administrative | 656,229 | 636,182 | 20,047 | 3 | ||||||||||||
| Change in fair value of derivative liability | (435,200 | ) | (372,487 | ) | (62,713 | ) | 17 | |||||||||
| Change in fair value of derivative liability due to change in warrant terms | 143,532 | - | 143,532 | -- | ||||||||||||
| Loss on exchange of warrants | 92,843 | - | 92,843 | -- | ||||||||||||
| Foreign exchange loss | 7,295 | 34,797 | (27,502 | ) | (79 | ) | ||||||||||
| Interest expense | - | 2,044 | (2,044 | ) | (100 | ) | ||||||||||
| Interest income | (109 | ) | (620 | ) | 511 | (82 | ) | |||||||||
| Net and comprehensive loss | 1,076,759 | 865,976 | 210,783 | |||||||||||||
Research and Development
Research and development expenses increased to $612,169 for the three months ended December 31, 2014 from $566,060 for the three months ended December 31, 2013. The increase was largely attributable to an increase in clinical development, pre-clinical research, and intellectual property costs partially offset by a decrease in share-based compensation expenses. Clinical development costs have increased due to higher support costs related to regulatory activities and submissions to the FDA, drug manufacturing as the Company prepares for its registration trial, and activities relating to the preparation of protocols for the lung cancer and GBM studies in China. Pre-clinical research has increased due to the Company undertaking mechanism of action and lung cancer studies that had not yet been initiated at December 31, 2013. Intellectual property costs have increased in the three months ended December 31, 2014 compared to the three months ended December 31, 2013 as the Company has now been granted three patents and has recently submitted additional patent applications. Partially offsetting the impact of higher costs in these areas are lower share-based payments of a reversal of expense of ($8,077) in the current quarter compared to an expense of $163,514 for the corresponding 2013 period. In relation to research and development expenses during the three months ended December 31, 2014 and 2013 the Company incurred share-based payments relating to stock option expense only. The decrease in stock option expense in the current quarter was due to a decrease in the Company’s share price in the current quarter compared to the prior quarter.
General and Administrative
General and administrative expenses were $656,229 for the three months ended December 31, 2014 compared to $636,182 for the three months ended December 31, 2013. Overall, an increase in share-based payments and facilities costs was largely offset by decreases in professional fees in the current period compared to the corresponding period in 2013. Share-based payments increased to $219,646 in the three months ended December 31, 2014 from $192,391 for the three months ended December 31, 2013. In relation to general and administrative expenses during the three months ended December 31, 2014, the Company incurred share-based payments related to stock option expense and shares issued for services while during the three months ended December 31, 2013 the Company incurred share-based payments relating to stock options and for warrants issued for services. The decrease in stock option expense in the current quarter was due to a decrease in the Company’s share price in the current quarter compared to the corresponding quarter in the prior year.
Excluding the impact of share-based payments, general and administrative expenses remained relatively consistent decreasing slightly to $436,582 during the three months ended December 31, 2014 from $443,791 for the three months ended December 31, 2013. The principal reasons for the decrease were lower professional fees partially offset by higher facilities costs. Professional fees decreased during the three months ended December 31, 2014 compared the three months ended December 31, 2013 due to lower business development and investor relations costs. Facilities costs increased for the three months ended December 31, 2014 compared to the three months ended December 31, 2013 largely due an increase in promotion and press releases, and filing and related fees. The filings fees related to the Company listing its common stock on the OTCQX.
Change in fair value of derivative liability
Based on the terms of certain warrants issued by the Company, the Company determined that the warrants were a derivative liability which is recognized at fair value at the date of the transaction and re-measured at fair value every reporting period with gains or losses on the changes in fair value recorded in the consolidated condensed interim statement of loss and comprehensive loss. The balances recognized during the three months ended December 31, 2014 and 2013 were largely due to a decrease in the Company’s common stock price between the date the warrants were last valued and December 31, 2014 and 2013, respectively, which were the current revaluation dates presented for the three months ended December 31, 2014 and 2013.
| 36 |
The Company recognized gains of $435,200 and $372,487 from the change in fair value of the derivative liability for the three months ended December 31, 2014 and 2013, respectively. In addition, as a result of amending the Investor Warrants and Dividend Warrants during the quarter ended December 31, 2014, the Company also recognized a loss of $143,532. All warrants that have been exercised or amended were revalued at their respective exercise or amendment dates and then the reclassification to equity was recorded. Also, during the quarter ended December 31, 2014, the Company exchanged certain Investor Warrants for shares of common stock resulting in the recognition of a loss of $92,843 on the exchange. The Company consummated a tender offer in relation to the 5,964,738 Investor Warrants outstanding at December 31, 2014. The Investor Warrant holders could elect to exchange three Investor Warrants for one share of common stock of the Company until the expiration of the tender offer on February 9, 2015.
Changes in the Company’s common stock price can result in significant volatility in the Company’s reported net loss due to its impact on the fair value of the derivative liability. As a result of revaluation gains and losses, the Company expects that its reported net income or loss will continue to fluctuate significantly.
Foreign Exchange Gain
The Company’s functional currency at December 31, 2014 is the USD but the Company incurs a portion of its expenses in CDN. The foreign exchange gains and losses are reported in other (income) loss in the Consolidated Condensed Interim Statement of Loss and Comprehensive Loss.
The Company recognized a foreign exchange loss of $7,295 for the quarter ended December 31, 2014 compared to a loss of $34,797 for the quarter ended December 31, 2013. The change was due to changes in the exchange rate between the CDN and the USD and to varying levels of CDN accounts payable.
Interest Expense
Pursuant to a loan agreement dated February 3, 2011, the Company received a loan from Valent in the amount of $250,000 for the purchase of the prototype drug product. The loan was payable on demand, unsecured and bore interest at 3% per year. Effective September 30, 2014 the loan balance, including accumulated interest to September 30, 2014, was exchanged for 278,530 shares of the Company’s Series A Preferred Stock. The Series A Preferred Stock pays an annual dividend of 3%.
For the three-months ended December 31, 2014, the Company accrued $2,089 related to the dividend payable to Valent. The dividend has been recorded as a direct increase in accumulated deficit and was paid subsequent to December 31, 2014. For the three-months ended December 31, 2013 the Company accrued $2,044 in interest on its loan payable with Valent.
Comparison of the six months ended December 31, 2014 and December 31, 2013
| Six Months Ended | ||||||||||||||||
| December 31, 2014 $ | December 31, 2013 $ | Change $ | Change % | |||||||||||||
| Research and development | 1,283,796 | 1,126,295 | 157,501 | 14 | ||||||||||||
| General and administrative | 1,101,229 | 1,377,550 | (276,321 | ) | (20 | ) | ||||||||||
| Change in fair value of derivative liability | (66,606 | ) | (8,466,826 | ) | 8,400,220 | (99 | ) | |||||||||
| Change in fair value of derivative liability due to change in warrant terms | (23,658 | ) | - | (23,658 | ) | -- | ||||||||||
| Loss on exchange of warrants | 92,843 | - | 92,843 | -- | ||||||||||||
| Foreign exchange loss | 9,686 | 31,963 | (22,277 | ) | (70 | ) | ||||||||||
| Interest expense | 2,091 | 4,073 | (1,982 | ) | (49 | ) | ||||||||||
| Interest income | (261 | ) | (1,311 | ) | 1,050 | (80 | ) | |||||||||
| Net and comprehensive loss (income) | 2,399,120 | (5,928,256 | ) | 8,327,376 | ||||||||||||
| 37 |
Research and Development
Research and development expenses increased to $1,283,796 for the six months ended December 31, 2014 from $1,126,295 for the six months ended December 31, 2013. The increase was largely attributable to an increase in clinical development, pre-clinical research, and intellectual property costs partially offset by a decrease in share-based compensation expenses. Clinical development costs have increased due to higher support costs related to regulatory activities and submissions to the FDA, drug manufacturing as the Company prepares for its registration trial, and activities relating to the preparation of protocols for the lung cancer and GBM studies in China. Pre-clinical research has increased due to the Company undertaking mechanism of action and lung cancer studies that had not yet been initiated at December 31, 2013. Intellectual property costs increased in the six months ended December 31, 2014 compared to the six months ended December 31, 2013 as the Company has now been granted three patents and is has recently submitted additional patent applications. Partially offsetting the impact of higher costs in these areas are lower share-based payments of $13,056 in the current six month period compared to $259,589 for the corresponding 2013 period. In relation to research and development expenses during the six months ended December 31, 2014 the Company incurred share-based payments relating to stock option expense only. During the six months ended December 31, 2013 the Company incurred expenses for stock options and the issuance of shares for services. The decrease in stock option expense in the current period was due to a decrease in the Company’s share price in the six month period in 2014 compared to the corresponding period in 2013.
General and Administrative
General and administrative expenses were $1,101,229 for the six months ended December 31, 2014 compared to $1,377,550 for the six months ended December 31, 2013. The decrease was partially attributable to a decrease in share-based payments to $247,454 in the six months ended December 31, 2014 from $490,710 for the six months ended December 31, 2013. In relation to general and administrative expenses during the six months ended December 31, 2014, the Company incurred share-based payments related to stock options and shares issued for services while during the six months ended December 31, 2013 the Company incurred share-based payments relating to stock options and for warrants issued for services. The decrease in stock option expense in the current period was due to a decrease in the Company’s share price in the current period compared to the corresponding period in 2013.
Excluding the impact of share-based payments, general and administrative expenses remained relatively consistent decreasing slightly to $853,775 during the six months ended December 31, 2014 from $886,840 for the six months ended December 31, 2013. The principal reasons for the decrease were lower professional fees partially offset by higher personnel and facilities costs. Professional fees were lower during the six months ended December 31, 2014 compared to the six months ended December 31, 2013 due to lower business development and investor relations costs. Personnel costs increased due to higher management fees and benefits in the current six months compared to the corresponding period in 2013. Facilities costs increased for the six months ended December 31, 2014 compared to the six months ended December 31, 2013 largely due an increase in promotion and press releases, and filing and related fees. The filings fees related to the Company listing its common stock on the OTCQX.
Change in fair value of derivative liability
Based on the terms of certain warrants issued by the Company, the Company determined that the warrants were a derivative liability which is recognized at fair value at the date of the transaction and re-measured at fair value every reporting period with gains or losses on the changes in fair value recorded in the consolidated condensed interim statement of loss and comprehensive loss. The balances recognized during the six months ended December 31, 2014 and 2013 were primarily due to a changes in the Company’s common stock price between the date the warrants were last valued and December 31, 2014 and 2013, respectively, which were the current revaluation dates presented for the six months ended December 31, 2014 and 2013.
The Company recognized gains of $66,606 and $8,466,826 from the change in fair value of the derivative liability for the six months ended December 31, 2014 and 2013, respectively. In addition, as a result of amending the Investor Warrants and Dividend Warrants during the period ended December 31, 2014, the Company also recognized a gain of $23,658. All warrants that have been exercised or amended were revalued at their respective exercise or amendment dates and then the reclassification to equity was recorded. Also, during the six months ended December 31, 2014, the Company exchanged certain Investor Warrants for shares of common stock resulting in the recognition of a loss of $92,843 on the exchange. The Company consummated a tender offer in relation to the 5,964,738 Investor Warrants outstanding at December 31, 2014. The Investor Warrant holders could elect to exchange three Investor Warrants for one share of common stock of the Company until the expiration of the tender offer on February 9, 2015.
Changes in the Company’s common stock price can result in significant volatility in the Company’s reported net loss due to its impact on the fair value of the derivative liability. As a result of revaluation gains and losses, the Company expects that its reported net income or loss will continue to fluctuate significantly.
Foreign Exchange Gain
The Company’s functional currency at December 31, 2014 is the USD but the Company incurs a portion of its expenses in CDN. The foreign exchange gains and losses are reported in other (income) loss in the Consolidated Condensed Interim Statement of Loss and Comprehensive Loss.
The Company recognized a foreign exchange loss of $9,686 for the six months ended December 31, 2014 compared to a loss of $31,963 for the six months ended December 31, 2013. The change was due to changes in the exchange rate between the CDN and the USD and to varying levels of CDN accounts payable.
| 38 |
Interest Expense
Pursuant to a loan agreement dated February 3, 2011, the Company received a loan from Valent in the amount of $250,000 for the purchase of the prototype drug product. The loan was payable on demand, unsecured and bore interest at 3% per year. Effective September 30, 2014 the loan balance, including accumulated interest to September 30, 2014, was exchanged for 278,530 shares of the Company’s Series A Preferred Stock. The Series A Preferred Stock pays an annual dividend of 3%.
For the six-months ended December 31, 2014, the Company accrued $2,089 related to the dividend payable to Valent and $2,091 related to interest from June 30, 2014 to September 30, 2014 when the loan was converted to preferred shares. The dividend has been recorded as a direct increase in accumulated deficit and the $2,091 has been recognized as interest expense. For the six-months ended December 31, 2013 the Company accrued $4,073 in interest on its loan payable with Valent.
Selected Annual Information
The financial information reported here in has been prepared in accordance with US GAAP. The Company’s functional currency at June 30, 2014 is the USD. The following table represents selected financial information for the Company as of June 30, 2014, December 31, 2013 and December 31, 2012.
Selected Balance Sheet Data
| June 30, 2014 $ | December 31, 2013 $ | December 31, 2012 $ | ||||||||||
| Cash and cash equivalents | 4,759,711 | 4,136,803 | 17,782 | |||||||||
| Working capital (deficiency) | 4,704,044 | 4,069,261 | (942,562 | ) | ||||||||
| Total Assets | 5,003,910 | 4,318,748 | 182,830 | |||||||||
| Derivative liability | 3,329,367 | 4,402,306 | 121,000 | |||||||||
| Total shareholders’ equity (deficiency) | 880,479 | (817,978 | ) | (1,327,914 | ) | |||||||
Selected Statement of Operations Data
For the six months ended:
| June 30, | June 30, | |||||||
| 2014 | 2013 | |||||||
| $ | $ | |||||||
| Research and development | 992,922 | 1,216,359 | ||||||
| General and administrative | 1,756,859 | 2,574,757 | ||||||
| Change in fair value of derivative liability | 166,388 | 7,142,775 | ||||||
| Change in fair value of derivative liability due to tender offer | (111,179 | ) | - | |||||
| Derivative issuance costs | - | 2,713,220 | ||||||
| Foreign exchange (gain) loss | (9,382 | ) | (28,933 | ) | ||||
| Shares issued for Valent royalty reduction | - | 598,000 | ||||||
| Interest expense | 4,067 | 3,947 | ||||||
| Interest income | (767 | ) | (1,180 | ) | ||||
| Net and comprehensive loss | 2,798,908 | 14,218,945 | ||||||
| Weighted average number of shares outstanding | 32,468,861 | 27,727,845 | ||||||
| Loss per share | (0.09 | ) | (0.51 | ) | ||||
For the years ended:
| December 31, 2013 $ | December 31, 2012 $ | |||||||
| Research and development | 2,342,654 | 1,550,490 | ||||||
| General and administrative | 3,952,307 | 1,154,604 | ||||||
| Change in fair value of derivative liability | (1,324,051 | ) | (318,502 | ) | ||||
| Derivative issuance costs | 2,713,220 | 24,742 | ||||||
| Foreign exchange (gain) loss | 3,030 | (18,492 | ) | |||||
| Shares issued for Valent royalty reduction | 598,000 | - | ||||||
| Interest expense | 8,020 | 7,521 | ||||||
| Interest income | (2,491 | ) | - | |||||
| Loss from operations | 8,280,689 | 2,400,363 | ||||||
| Weighted average number of shares outstanding | 29,667,324 | 13,232,349 | ||||||
| Loss per share | (0.28 | ) | (0.18 | ) | ||||
| 39 |
Expenses net of share-based payments
The following table discloses research and development, and general and administrative expenses net of share-based payment expenses.
| June 30, 2014 $ | June 30, 2013 $ | December 31, 2013 $ | December 31, 2012 $ | |||||||||||||
| Research and development | 992,922 | 1,216,359 | 2,342,654 | 1,550,490 | ||||||||||||
| Share-based payments included in research and development | (144,587 | ) | (309,136 | ) | (568,725 | ) | (866,111 | ) | ||||||||
| Research and development net of share-based compensation | 848,335 | 907,223 | 1,773,929 | 684,379 | ||||||||||||
| General and administrative | 1,756,859 | 2,574,757 | 3,952,307 | 1,154,604 | ||||||||||||
| Share-based payments included in general and administrative | (749,078 | ) | (1,211,351 | ) | (1,702,061 | ) | (493,652 | ) | ||||||||
| General and administrative net of share-based compensation | 1,007,781 | 1,363,406 | 2,250,246 | 660,952 | ||||||||||||
Comparison of the six months ended June 30, 2014 and June 30, 2013
| Six Months Ended | ||||||||||||||||
| June 30, 2014 $ | June 30 2013 $ | Change $ | Change % | |||||||||||||
| Research and development | 992,922 | 1,216,359 | (223,437 | ) | (18 | ) | ||||||||||
| General and administrative | 1,756,859 | 2,574,757 | (817,898 | ) | (32 | ) | ||||||||||
| Change in fair value of derivative liability | 166,388 | 7,142,775 | (6,976,387 | ) | (98 | ) | ||||||||||
| Change in fair value of derivative liability due to tender offer | (111,179 | ) | - | (111,179 | ) | (100 | ) | |||||||||
| Shares issued to Valent for future royalty reduction | - | 598,000 | (598,000 | ) | (100 | ) | ||||||||||
| Derivative issue costs | - | 2,713,220 | (2,713,220 | ) | (100 | ) | ||||||||||
| Foreign exchange (gain) loss | (9,382 | ) | (28,933 | ) | 19,551 | (68 | ) | |||||||||
| Interest expense | 4,067 | 3,947 | 120 | 3 | ||||||||||||
| Interest income | (767 | ) | (1,180 | ) | 413 | (35 | ) | |||||||||
| Net and comprehensive loss | 2,798,908 | 14,218,945 | (11,420,037 | ) | ||||||||||||
Research and Development
Research and development expenses decreased to $992,922 for the six months ended June 30, 2014 from $1,216,359 for the six months ended June 30, 2013. Share-based payments attributable to research and development were $144,587 in the six months ended June 30, 2014 compared to $309,136 for the six months ended June 30, 2013. In relation to research and development expenses during the six months ended June 30, 2014 and 2013 the Company incurred share-based payments relating to stock options expense only. The decrease in share-based payments in relation to research and development was due to a reduction in the Company’s share price between periods.
After considering the impact of share-based payments, research and development expenses decreased in the six months ended June 30, 2014 to $848,335 from $907,223 for the six months ended June 30, 2013.
The largest component of research and development for both the six months ended June 30, 2014 and 2013 was clinical development costs as the Company continued with its Phase I/II clinical trial with VAL-083 in GBM. The clinical development costs were slightly lower in the current period compared to the prior period due to several factors. Due to the timing of patient enrollment and due to the fact cohorts 2 and 3 completed in 2013 were expanded beyond the planned three patients, direct clinical costs were lower in the current period than the prior period. In addition, the Company began a back-up manufacturing campaign during the six months ended June 30, 2013 which resulted in higher costs in the prior period compared to the current period. Partially offsetting these items were higher costs in the current period for protocol development as we plan for our registration trial. Intellectual property expenses were lower in the six months ended June 30, 2104 compared to the six months ended June 30, 2013 due to timing as intellectual property costs vary considerably depending on where we are in the patent process. During the six months ended June 30, 2014 the Company commenced several nonclinical studies resulting in higher expenses compared to the prior period.
| 40 |
General and Administrative
General and administrative expenses were $1,756,859 for the six months ended June 30, 2014 compared to $2,574,757 for the six months ended June 30, 2013. The decrease was partially attributable to a decrease in share-based payments to $749,078 in the six months ended June 30, 2014 compared to $1,211,351 for the six months ended June 30, 2013. In relation to general and administrative expenses during the six months ended June 30, 2014 and 2013 the Company incurred share-based payments relating to stock options and to shares issued for services. The decrease in share-based payments in relation to general and administrative was due to a reduction in the Company’s share price between periods.
After considering the impact of share-based payments, general and administrative expenses decreased in the six months ended June 30, 2014 to $1,007,781 from $1,363,406 for the six months ended June 30, 2013. The principal reason for the decrease was due to professional fees. During the six months ended June 30, 2013 the Company incurred professional fees related to the Company’s Reverse Acquisition and the preparation and filing of the Company’s Registration Statement on Form S-1. A significant portion of the accounting and legal fees related to the Reverse Acquisition were expensed as they did not quality as direct share issue costs. The fees and expenses for professional fees for the Reverse Acquisition were one-time fees that have not been incurred in the current period. Personnel costs have also decreased in the six months ended June 30, 2014 compared to the six months ended June 30, 2013 due to lower amounts paid to Company management. Partially offsetting the impact of lower professional fees and personnel costs are higher costs related to insurance and investor relations activities related to press releases, attendance at conferenced and other public awareness activities.
Change in fair value of derivative liability
Based on the terms of certain warrants issued by the Company, the Company determined that the warrants were a derivative liability which is recognized at fair value at the date of the transaction and re-measured at fair value every reporting period with gains or losses on the changes in fair value recorded in the consolidated condensed interim statement of loss and comprehensive loss. The balance recognized during the six months ended June 30, 2014 was due to a decrease in the Company’s share price between the date the warrants were issued and June 30, 2014 which was the revaluation date. In addition, 3,929,524 Investor Warrants were exercised during the period resulting in fewer Investor Warrants being subjected to revaluation.
The Company recognized a loss of $166,388 for the six months ended June 30, 2014 compared to a loss of $7,142,775 from the change in fair value of the derivative liability at June 30, 2013. In addition, the Company recognized a gain of $111,179 in relation to the revaluation due to the terms of the tender offer.
Issuance of Shares to Valent for future royalty reduction
On January 25, 2013, in connection with the Reverse Acquisition, the Company issued to Valent 1,150,000 shares of common stock in exchange for Valent reducing certain future royalties under the Assignment Agreement. As a result of the share issuance the Company has recognized an expense of $598,000 for the six months ended June 30, 2013.
Derivative issue costs
The proceeds from the $0.80 unit offering have been allocated between common stock and derivative liability based on the respective fair values of the shares of common stock and the warrants on the issuance date. Additionally, the unit issue costs have also been allocated between common stock and derivative liability on the same pro rata basis as the proceeds. The portion of the issue costs allocated to the derivative liability has been expensed in the consolidated statement of operations and comprehensive loss. The Company recognized $2,713,220 in derivative issue costs for the six months ended June 30, 2013. There was no derivative issue costs recognized for the six months ended June 30, 2014.
Foreign Exchange Gain
The Company’s functional currency at June 30, 2013 is the USD but the Company incurs a portion of its expenses in CDN. The translation gains and losses are reported in other comprehensive loss/income.
The Company recognized a foreign exchange gain of $9,382 for the six months ended June 30, 2014 compared to a gain of $28,933 for the six months ended June 30, 2013. The change was due to changes in the exchange rates between the CDN and the USD and to varying levels of CDN accounts payable.
| 41 |
Interest Expense
Pursuant to a loan agreement dated February 3, 2011, the Company has received a loan from Valent in the amount of $250,000 for the purchase of the prototype drug product. The loan was previously payable on demand but is now a five year term loan due, along with all accrued interest, on June 30, 2019. As a result of the loan payable the Company recognized $4,067 and $3,947 respectively in accrued interest for the six months ended June 30, 2014 and 2013.
Comparison of the year ended December 31, 2013 and 2012
| Years Ended | ||||||||||||||||
| December 31, 2013 $ | December 31, 2012 $ | Change $ | Change % | |||||||||||||
| Research and development | 2,342,654 | 1,550,490 | 792,164 | 51 | ||||||||||||
| General and administrative | 3,952,307 | 1,154,604 | 2,797,703 | 242 | ||||||||||||
| Change in fair value of derivative liability | (1,324,051 | ) | (318,502 | ) | (1,005,549 | ) | 316 | |||||||||
| Shares issued to Valent for future royalty reduction | 598,000 | - | 598,000 | 100 | ||||||||||||
| Derivative issue costs | 2,713,220 | 24,742 | 2,688,478 | 10,866 | ||||||||||||
| Foreign exchange (gain) loss | 3,030 | (18,492 | ) | 21,522 | (116 | ) | ||||||||||
| Interest expense | 8,020 | 7,521 | 499 | 7 | ||||||||||||
| Interest income | (2,491 | ) | - | (2,491 | ) | 100 | ||||||||||
| Net loss | 8,290,689 | 2,400,363 | 5,890,326 | |||||||||||||
Research and Development
Research and development expenses increased to $2,342,654 for the year ended December 31, 2013 from $1,550,490 for the year ended December 31, 2012. Share-based payments attributable to research and development were $568,725 in the year ended December 31, 2013 compared to $866,111 for the year ended December 31, 2012. In regards to research and development expenses during the year ended December 31, 2013 the Company incurred share-based payments relating to stock options and the issuance of shares for services. For the year ended December 31, 2012 the Company recognized the fair value of shares issued from the DelMar Employee Share Purchase Trust (the “Trust”) to employees and consultants for services rendered to the Company, stock option expense as the Company’s first grant of stock options occurred in February 2012, and the fair value amount recognized for units issued for services. All of the shares had been issued from the Trust at December 31, 2012 and as a result no additional expense was recognized during the year ended December 31, 2013.
After considering the impact of share-based payments research and development expenses increased in the year ended December 31, 2013 to $1,773,929 from $684,379 for the year ended December 31, 2012. The largest component of research and development for the year ended December 31, 2013 was clinical development costs as the Company continued with its Phase I/II clinical trial with VAL-083. The clinical development costs were higher in 2013 compared to the prior period largely due to the timing of patient enrollment. Additionally, personnel, intellectual property, and travel costs were all higher during the year ended December 31, 2013 compared to the year ended December 31, 2012.
Personnel costs have increased due to the officers and directors of the Company being compensated with cash during the year ended December 31, 2013 while during the year ended December 31, 2012 a portion of management compensation was in the form of units. Intellectual property costs increased in 2013 as a result of the Company becoming more active in filing and advancing its patents compared to the prior year. Travel increased in 2013 compared to 2012 as a result of increased travel to scientific and medical conferences to present data and meet with potential collaborators.
General and Administrative
General and administrative expenses were $3,952,307 for the year ended December 31, 2013 compared to $1,154,604 for the year ended December 31, 2012. The increase was partially attributable to an increase in share-based payments to $1,702,061 for the year ended December 31, 2013 compared to $493,652 for the year ended December 31, 2012. In relation to general and administrative expenses during the year ended December 31, 2013 the Company incurred share-based payments relating to stock options, shares issued for services, and warrants issued for services. For the year ended December 31, 2012 the Company recognized the fair value of shares issued from the Trust to employees and consultants for services rendered to the Company, stock option expense as the Company’s first grant of stock options occurred in February 2012, and the fair value amount recognized for warrants and units issued for services. All of the shares had been issued from the Trust at December 31, 2012 and as a result no additional expense was recognized during the year ended December 31, 2013.
| 42 |
After considering the impact of share-based payments, general and administrative expenses increased in the year ended December 31, 2013 to $2,250,246 from $660,952 for the year ended December 31, 2012.
The principal reason for the increase was due to professional fees related to the Company’s Reverse Acquisition and the preparation and filing of the Company’s Registration Statement on Form S-1. A significant portion of the accounting and legal fees related to the Reverse Acquisition were expensed as they did not quality to be recognized as direct share issue costs. The fees and expenses for professional fees for the Reverse Acquisition and the S-1 were one-time fess that will not be incurred in subsequent periods. Additionally, as a result of the Company becoming public due to its Reverse Acquisition, the Company has incurred investor relations fees which it did not incur during the year ended December 31, 2012. The Company becoming a public reporting entity has also led to higher travel costs due to the need to attend more investor and business development conferences.
Personnel, and office and sundry increased in 2013 compared to the prior year. Personnel costs increased due to the officers and directors being compensated with cash in the year ended December 31, 2013 while in the year ended December 31, 2012 a portion of management compensation was in the form of units. In addition, as a result of the Company becoming a public entity, additional officers joined the Company during 2013. Office and sundry increased for the year ended December 31, 2013 compared to the year ended December 31, 2012 largely due an increase in filing and related fees. As a result of the Reverse Acquisition the Company become a public company and began filing obligations with various regulatory authorities.
Change in fair value of derivative liability
Based on the terms of certain warrants issued by the Company, the Company determined that the warrants were a derivative liability which is recognized at fair value at the date of the transaction and re-measured at fair value every reporting period with gains or losses on the changes in fair value recorded in the consolidated statement of loss and comprehensive loss. The balance recognized during the year ended December 31, 2013 was due to a reduction in the Company’s share price between the date the warrants were issued and December 31, 2013 which was the revaluation date.
The Company recognized a gain of $1,324,051 from the change in fair value of the derivative liability for the year ended December 31, 2013 compared to a gain of $318,502 for the year ended December 31, 2012. Changes in the Company’s common stock price can result in significant volatility in the Company’s reported net loss due to its impact on the fair value of the derivative liability. As a result of revaluation gains and losses, the Company expects that its reported net income or loss will continue to experience large fluctuations.
Issuance of Shares to Valent for future royalty reduction
On January 25, 2013, in connection with the Reverse Acquisition, the Company issued to Valent 1,150,000 shares of common stock in exchange for Valent reducing certain future royalties under the Assignment Agreement. As a result of the share issuance the Company has recognized an expense of $598,000 for the year ended December 31, 2013.
Derivative issue costs
The proceeds from the $0.80 unit offering have been allocated between common stock and derivative liability based on the respective fair values of the shares of common stock and the warrants on the issuance date. Additionally, the unit issue costs have also been allocated between common stock and derivative liability on the same pro rata basis as the proceeds. The portion of the issue costs allocated to the derivative liability has been expensed in the consolidated statement of loss and comprehensive loss. The Company recognized $2,713,220 in derivative issue costs for the year ended December 31, 2013. Derivative issue costs of $24,742 related to the issuance of the CDN $0.50 units were recognized for the year ended December 31, 2012.
Foreign Exchange Gain
The Company’s functional currency at December 31, 2013 is the USD but the Company incurs a portion of its expenses in CDN. The foreign exchange gains and losses are reported in other (income) loss in the consolidated statement of loss and comprehensive loss.
The Company recognized a foreign exchange loss of $3,030 for the year ended December 31, 2013 compared to a gain of $18,492 for the year ended December 31, 2012. The change was due to changes in the exchange rate between the CDN and the USD and to varying levels of CDN cash and accounts payable.
Interest Expense
Pursuant to a loan agreement dated February 3, 2011, the Company has received a loan from Valent in the amount of $250,000 for the purchase of the prototype drug product. The loan is payable on demand, unsecured and bears interest at 3.00% per year. As a result of the loan payable the Company recognized $8,020 and $7,521 respectively in accrued interest for the years ended December 31, 2013 and 2012.
| 43 |
Related Party Transactions
The Company acquired its VAL-083 prototype drug, patents and technology rights from Valent. In addition, Valent incurred a significant portion of the Company’s clinical expenses during the periods ended December 31, 2011 and 2012 and in turn invoiced the Company for those expenses. One of the Company’s officers and directors is a principal of Valent and as result Valent is a related party to the Company.
The following related party transactions and balances have been recorded by the Company.
During the six months ended December 31, 2014
Effective September 30, 2014, the Company entered into and closed an agreement with Valent to exchange its loan with Valent for 278,530 shares of preferred stock of the Company.
Included in accounts payable at December 31, 2014 is an aggregate amount of $37,659 (June 30, 2014 - $54,960) owed to the Company’s officers and directors for fees and expenses. The Company pays related party payables incurred for fees and expenses under normal commercial terms.
During the six months ended June 30, 2014
Included in accounts payable at June 30, 2014 is an aggregate amount owing of $54,960 to the Company’s officers and directors for fees and expenses. The Company pays related party payables incurred for fees and expenses under normal commercial terms.
The Company also has a loan payable of $276,439, including aggregate accrued interest of $26,439, due to Valent. The Company accrued $4,067 of interest on this loan during the six months ended June 30, 2014. The loan was previously payable on demand but is now a five year term loan due, along with all accrued interest, on June 30, 2019. One of the directors and officers of the Company is also a Principal of Valent. As a result of the Company not expecting to repay Valent within the next twelve months, the balance of the loan and accrued interest has been disclosed as a long-term liability.
During the six months ended June 30, 2013
On January 25, 2013, in connection with the Reverse Acquisition, the Company issued to Valent 1,150,000 shares of common stock in exchange for Valent reducing certain future royalties under the Assignment Agreement. As a result of the share issuance the Company has recognized an expense of $598,000 for the six months ended June 30, 2013.
During the year ended December 31, 2013
Included in accounts payable at December 31, 2013 is an aggregate amount owing of $65,023 to the Company’s officers and directors for fees and expenses. The Company pays related party payables incurred for fees and expenses under normal commercial terms.
Included in related party payables at December 31, 2013 is an amount of $44,007 relating to clinical development costs incurred by Valent on behalf of the Company. Additionally, the Company also has a loan payable of $272,372, including accrued interest of $22,372, due to Valent. One of the directors and officers of the Company is also a Principal of Valent. As a result of the Company not expecting to repay Valent within the next twelve months, the balance of the loan and accrued interest has been disclosed as a long-term liability.
On January 25, 2013, in connection with the Reverse Acquisition, the Company issued to Valent 1,150,000 shares of common stock of the Company in exchange for Valent reducing certain future royalties under the Assignment Agreement. As a result of the share issuance the Company has recognized an expense of $598,000 for the year ended December 31, 2013.
During the year ended December 31, 2012
Included in related party payables December 31, 2012 is an amount of $314,119 relating to clinical development costs incurred by Valent on behalf of the Company. On April 30, 2012, Valent was issued 500,000 common shares for partial settlement of the Company’s accounts payable balance with Valent. The total settlement amount was $253,050. Additionally, the Company has a loan payable, including accrued interest, of $264,352 due to Valent.
| 44 |
Through a Company owned by one of the Company’s directors, a $25,000 retainer was paid pursuant to the unit financing completed by the Company. The $25,000 is included in accounts payable at December 31, 2012.
The Company transferred a total of 1,390,625 shares from the DelMar Employee Share Purchase Trust to the Company’s directors.
Liquidity and Capital Resources
Six months ended December 31, 2014 compared to the six months ended December 31, 2013
| December 31, 2014 $ | December 31, 2013 $ | Change $ | Change % | |||||||||||||
| Cash used in operating activities | (2,065,360 | ) | (2,146,189 | ) | 80,829 | (4 | ) | |||||||||
| Cash flows from financing activities | 1,264,088 | - | 1,264,088 | -- | ||||||||||||
Operating Activities
Net cash used in operating activities decreased to $2,065,360 for the six months ended December 31, 2014 from $2,146,189 for the six months ended December 31, 2013. During the six months ended December 31, 2014 the Company reported a loss of $2,399,120 compared to income of $5,928,256 for the six months ended December 31, 2013. However, included in the net income in 2013 was a gain of $8,466,826 attributable to changes in the fair value of the derivative liability. During the six months ended December 31, 2014, the Company recognized a gain of $66,606 from changes in the fair value of the derivative liability. Excluding the impact of changes in the fair value of the derivative liability, non-cash items relating to accrued interest, gains from amending the terms of certain warrants, losses from the exchange of warrants, and stock-based compensation totaled $331,786 for the six months ended December 31, 2014. Non-cash items relating to accrued interest, warrants issued for services, and share-based compensation totaled $754,372 for the six months ended December 31, 2013. The most significant changes in non-cash working capital for the six months ended December 31, 2014 were from a decrease in prepaid expenses of $78,770. In the six months ended December 31, 2013 the most significant items were due to reductions in accounts payable and accrued liabilities and related party payables of $290,770 and $127,432 respectively.
Financing Activities
The Company received net proceeds of $1,266,177 from the exercise of warrants during the six months ended December 31, 2014. In addition, the Company recognized $2,089 in dividends on the Series A preferred stock issued to Valent. There were no financing activities in the prior period.
Operating Capital and Capital Expenditure Requirements
For the six month period ended December 31, 2014, the Company reported a loss of $2,399,120 and an accumulated deficit of $21,064,623 at that date. As at December 31, 2014, the Company has cash and cash equivalents on hand of $3,958,439. The Company does not have the prospect of achieving revenues in the near future and the Company will require additional funding to maintain its research and development projects and for general operations. There is a great degree of uncertainty with respect to the expenses the Company will incur in executing its business plan. In addition, the Company has not begun to commercialize or generate revenues from any product candidate.
For the six-month period ended June 30, 2014, the Company reported a loss of $2,798,908 and an accumulated deficit of $18,663,414 at that date. As at June 30, 2014, the Company had cash and cash equivalents on hand of $4,759,711.
Consequently, management is pursuing various financing alternatives to fund the Company’s operations so it can continue as a going concern in the medium to longer term. During the six months ended December 31, 2014 the Company received an aggregate $1,266,177 in net proceeds from the exercise of 1,986,074 warrants. In addition, management is pursuing an underwritten common stock offering for up to $12,000,000 in gross proceeds assuming no overallotment proceeds. If management is successful in completing the proposed financing, we believe, based on our current estimates, that we would be able to fund our operations until at least the end of June 2017, including costs associated with a planned Phase II/III registration-directed clinical trial in the United States with VAL-083 as a potential new treatment for refractory GBM.
| 45 |
There is no assurance that our cost estimates will prove to be accurate or that unforeseen events, problems or delays will not occur that would require us to seek additional debt and/or equity funding. The ability of the Company to meet its obligations and continue the research and development of its product candidate is dependent on its ability to continue to raise adequate financing. There can be no assurance that such financing will be available to the Company in the amount required at any time or for any period or, if available, that it can be obtained on terms satisfactory to the Company. The Company may tailor its drug candidate program based on the amount of funding the Company raises.
Our future funding requirements will depend on many factors, including but not limited to:
| ● | the rate of progress and cost of our clinical trials, preclinical studies and other discovery and research and development activities; | |
| ● | the costs associated with establishing manufacturing and commercialization capabilities; | |
| ● | the costs of acquiring or investing in businesses, product candidates and technologies; | |
| ● | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; | |
| ● | the costs and timing of seeking and obtaining FDA and other regulatory approvals; | |
| ● | the effect of competing technological and market developments; and | |
| ● | the economic and other terms and timing of any collaboration, licensing or other arrangements into which we may enter. |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never do, we expect to finance future cash needs primarily through public or private equity offerings, debt financings or strategic collaborations. Although we are not reliant on institutional credit finance and therefore not subject to debt covenant compliance requirements or potential withdrawal of credit by banks, the current economic climate has also impacted the availability of funds and activity in equity markets. We do not know whether additional funding will be available on acceptable terms, or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs or make changes to our operating plan. In addition, we may have to seek a partner for one or more of our product candidate programs at an earlier stage of development, which would lower the economic value of those programs to us.
Six months ended June 30, 2014 compared to the six months ended June 30, 2013
| June 30, 2014 $ | June 30, 2013 $ | Change $ | Change % | |||||||||||||
| Cash used in operating activities | (1,857,842 | ) | (3,374,310 | ) | 1,516,468 | (45 | ) | |||||||||
| Cash flows from financing activities | 2,480,750 | 9,639,520 | (7,158,770 | ) | (74 | ) | ||||||||||
Operating Activities
Net cash used in operating activities decreased to $1,857,842 for the six months ended June 30, 2014 from $3,374,310 for the six months ended June 30, 2013. The decrease was largely the result of a decrease in the net loss to $2,798,908 for the six months ended June 30, 2014 compared to $14,218,945 for the six months ended June 30, 2013. The lower net loss in the current period was partially offset by several non-cash items incurred during the six months ended June 30, 2013 totaling $11,466,217 relating to accrued loan interest, change in fair value of the derivative liability, shares issued to Valent for a future royalty reduction, non-cash derivative issue costs and share-based payments. The non-cash items for the six months ended June 30, 2014 totaled $952,941 and consisted of accrued loan interest, change in fair value of the derivative liability, and share-based payments. The most significant changes in non-cash working capital for the six months ended June 30, 2014 were an outflow of $63,744 related to an increase in prepaid expenses, an outflow of $54,070 for a reduction in related party payables, and an inflow of $104,449 related to an increase in accounts payable and accrued liabilities. For the six months ended June 30, 2013 there were outflows of $246,388 and $211,315 from the payment of accounts payable and accrued liabilities, and related party payables respectively. In addition, in the prior period there was an outflow due to an increase in prepaid expenses of $192,484.
As a result of the Company’s expectations as to the timing of the repayment of the Valent loan, the Company has presented the full loan and accrued interest balance as a long-term liability at June 30, 2014, and December 31, 2013 and 2012.
| 46 |
Financing Activities
The Company received gross proceeds of $2,599,447 from the exercise of warrants during the six months ended June 30, 2014. The Company paid a 5% warrant agent commission of $118,697 on the exercise of certain Investor Warrants resulting in net proceeds of $2,480,750. During the six months ended June 30, 2013 the Company received $9,639,520 in net proceeds from the issuance of units in relation to the Reverse Acquisition. The net cash proceeds from the issuance of units were $8,575,000. However, as a result of a portion of the unit proceeds and issue costs being accounted for as a derivative liability the net proceeds on the consolidated statement of cash flows is $9,639,520. During the six months ended June 30, 2013 certain of the additional closing costs related to the issuance of units were not eligible to be treated as share issue costs and as a result they have been expensed. Net unit proceeds per the consolidated statement of cash flows for the six months ended June 30, 2013 include gross unit proceeds less cash issue costs attributable to the shares only. The portion of the unit issue costs attributable to the derivative liability has been expensed.
Year ended December 31, 2013 compared to the year ended December 31, 2012
| December 31, 2013 $ |
December 31, 2012 $ |
Change $ |
Change % |
|||||||||||||
| Cash used in operating activities | (5,520,499 | ) | (578,035 | ) | (4,942,464 | ) | 855 | |||||||||
| Cash flows from financing activities | 9,639,520 | 580,799 | 9,058,721 | 1,560 | ||||||||||||
Operating Activities
Net cash used in operating activities increased to $5,520,499 for the year ended December 31, 2013 from $578,035 for the year ended December 31, 2012. The increase was largely the result of an increase in the net loss to $8,290,689 for the year ended December 31, 2013 compared to $2,400,363 for the year ended December 31, 2012. Partially offsetting the impact on cash of the higher net loss were non-cash items totaling $3,753,763 incurred in the current year consisting of accrued loan interest, change in fair value of the derivative liability, warrants issued for services, shares issued to Valent for a future royalty reduction, non-cash derivative issue costs and share-based payments. The non-cash items for the year ended December 31, 2012 totaled $1,048,782 and consisted of accrued loan interest, change in fair value of the derivative liability, units issued for services, warrants issued for services, and share-based payments. The largest changes in non-cash working capital for the year ended December 31, 2013 were outflows of $546,889 and $329,016 from the payment of accounts payable and accrued liabilities, and related party payables respectively. In the year ended December 31, 2012 there was an inflow of $865,007 from an increase accounts payable and accrued liabilities and an outflow of $70,183 from a reduction in related party payables. Additionally, during the year ended December 31, 2013 and 2012 there were respective outflows of $142,105 and $14,581 from increases in prepaid expenses.
As a result of the Company’s expectations as to the timing of the repayment of the Valent loan, the Company has presented the full loan and accrued interest balance as a long-term liability at December 31, 2013 and December 31, 2012.
Financing Activities
The Company received $9,639,520 in net proceeds from the issuance of units during the year ended December 31, 2013 compared to $671,570 in net proceeds from the issuance of units during the year ended December 31, 2012. Also in 2012, the Company incurred $90,771 in deferred financing costs that were treated as share issue costs in 2013. The net proceeds from units issued in 2013 were $8,575,000. However, as a result of a portion of the unit proceeds and issue costs being accounted for as a derivative liability the net proceeds on the consolidated statement of cash flows is $9,639,520. During the year ended December 31, 2013 certain of the additional closing costs were not eligible to be treated as share issue costs and as a result they have been expensed. Net unit proceeds per the consolidated statements of cash flows include gross unit proceeds less cash issue costs attributable to the shares only. The portion of the unit issue costs attributable to the derivative liability has been expensed.
The units issued in the year ended December 31, 2013 were the $0.80 units issued in conjunction with the Reverse Acquisition while in the prior period the units issued were the CDN $0.50 units.
Critical Accounting Policies
The preparation of financial statements, in conformity with generally accepted accounting principles in the United States, requires companies to establish accounting policies and to make estimates that affect both the amount and timing of the recording of assets, liabilities, revenues and expenses. Some of these estimates require judgments about matters that are inherently uncertain and therefore actual results may differ from those estimates.
A detailed summary of all of the Company’s significant accountings policies and the estimates derived therefrom is included in Note 3 to the Company’s consolidated financial statements for the period ended June 30, 2014. While all of the significant accounting policies are important to the Company’s consolidated financial statements, the following accounting policies and the estimates derived therefrom have been identified as being critical:
| ● | Shares for services | |
| ● | Stock options | |
| ● | Derivative liability |
| 47 |
Shares for services
The Company has issued equity instruments for services provided by employees and nonemployees. The equity instruments are valued at the fair value of the instrument granted (see notes 8 and 9 of the consolidated financial statements for assumptions).
In prior periods the Company transferred shares from the DelMar Employee Share Purchase Trust (the “Trust”) to consultants and management in exchange for services rendered to the Company. The Company recognizes the fair value of the shares transferred as an expense with a corresponding increase in common stock. The shares reserved for issuance to consultants and management that are held by the Trust are included in the financial statements at year end. There are no other assets in the Trust. The number of shares outstanding for issue from the Trust at December 31, 2014, June 30, 2014, December 31, 2013 and 2012 is nil.
The shares transferred from the Trust in prior periods have been valued using the fair value of the shares transferred. The Company has used recent share transactions in order to determine the fair value of the shares transferred from the Trust.
Stock options
The Company accounts for these awards under ASC 718, “Compensation - Stock Compensation” (“ASC 718”). ASC 718 requires measurement of compensation cost for all stock-based awards at fair value on the date of grant and recognition of compensation over the requisite service period for awards expected to vest. Compensation expense for unvested options to non-employees is revalued at each period end and is being amortized over the vesting period of the options. The determination of grant-date fair value for stock option awards is estimated using the Black-Scholes model, which includes variables such as the expected volatility of the Company’s share price, the anticipated exercise behavior of its grantee, interest rates, and dividend yields. These variables are projected based on the Company’s historical data, experience, and other factors. Changes in any of these variables could result in material adjustments to the expense recognized for share-based payments. Such value is recognized as expense over the requisite service period, net of estimated forfeitures, using the straight-line attribution method. The estimation of stock awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from current estimates, such amounts are recorded as a cumulative adjustment in the period estimates are revised. The Company considers many factors when estimating expected forfeitures, including type of awards granted, employee class, and historical experience. Actual results and future estimates may differ substantially from current estimates.
Derivative liability
The Company accounts for certain warrants under the authoritative guidance on accounting for derivative financial instruments indexed to, and potentially settled in, a company’s own stock, on the understanding that in compliance with applicable securities laws, the warrants require the issuance of securities upon exercise and do not sufficiently preclude an implied right to net cash settlement. The Company classifies warrants in its balance sheet as a derivative liability which is fair valued at each reporting period subsequent to the initial issuance. As quoted prices for the derivative liability are not available, the Company uses a simulated probability valuation model to value the warrants. Determining the appropriate fair-value model and calculating the fair value of warrants requires considerable judgment. Any change in the estimates used may cause the value to be higher or lower than that reported. The estimated volatility of the Company’s common stock at the date of issuance, and at each subsequent reporting period, is based on the historical volatility of similar life sciences companies. The risk-free interest rate is based on rates published by the government for bonds with a maturity similar to the expected remaining life of the warrants at the valuation date. The expected life of the warrants is assumed to be equivalent to their remaining contractual term.
Off-Balance Sheet Arrangements
We do not have any off balance sheet arrangements.
| 48 |
Background
We are a clinical and commercial stage drug development company with a focus on the treatment of cancer. We are conducting clinical trials in the United States with VAL-083 as a potential new treatment for glioblastoma multiforme (“GBM”), the most common and aggressive form of brain cancer. We have also acquired certain exclusive commercial rights to VAL-083 in China where it is approved as a chemotherapy for the treatment of chronic myelogenous leukemia (“CML”) and lung cancer. We plan to seek marketing partnerships in China in order to generate royalty revenue.
Our mission is to benefit patients and create shareholder value by rapidly developing and commercializing anti-cancer therapies in orphan cancer indications where patients have failed or are unlikely to respond to modern therapy. VAL-083 represents a “first-in-class” small molecule chemotherapeutic, which means that the molecular structure of VAL-083 is not an analogue or derivative of other small molecule chemotherapeutics approved for the treatment of cancer. VAL-083 has been assessed in 42 Phase 1 and Phase 2 clinical trials sponsored by the National Cancer Institute (“NCI”) in the United States as a treatment against various cancers including lung, brain, cervical, ovarian tumors and leukemia. Published pre-clinical and clinical data suggest that VAL-083 may be active against a range of tumor types. VAL-083 is approved as a cancer chemotherapeutic in China for the treatment of CML and lung cancer. VAL-083 has not been approved for any indication outside of China.
Upon obtaining regulatory approval, we intend to commercialize VAL-083 for the treatment of orphan and other cancer indications where patients have failed other therapies or have limited medical options. Orphan diseases are defined in the United States under the Rare Disease Act of 2002 as “any disease or condition that affects fewer than 200,000 persons in the United States”. The Orphan Drug Act of 1983 is a federal law that provides financial and other incentives including a period of market exclusivity to encourage the development of new treatments for orphan diseases. In February 2012, we announced that VAL-083 has been granted protection under the Orphan Drug Act by the U.S. Food and Drug Administration (“FDA”) for the treatment of glioma, including GBM. In January 2013, the European Medicines Agency (“EMA”) also granted orphan drug protection to VAL-083 for the treatment of glioma.
In October 2011 we initiated clinical trials with VAL-083 as a potential new treatment for GBM, the most common and aggressive form of brain cancer. We have presented interim data from our clinical trial at peer reviewed scientific meetings demonstrating that VAL-083 can shrink or halt the growth of tumors in some brain cancer patients who have failed other approved treatments. Currently, there is no approved therapy for these patients.
In addition to our clinical development activities in the United States, we have obtained certain exclusive commercial rights to VAL-083 in China. In October 2012, we announced that we had entered into a collaboration agreement with the only manufacturer presently licensed by the China Food and Drug Administration (“CFDA”) to produce the product for the China market. This agreement potentially positions us to generate revenue through product sales or royalties for its approved indications in China while we seek global approval in new indications.
VAL-083 was originally discovered in the 1960’s. We have filed a broad portfolio of patent applications to protect our intellectual property. Our patent applications claim compositions and methods related to the use of VAL-083 and related compounds as well as methods of synthesis and quality controls for the manufacturing process of VAL-083. In October 2013, our first patent was granted by the United States Patent and Trademark Office, U.S. Patent 8,563,758. The patent expiration date is August 17, 2031. In addition, VAL-083 has been granted orphan drug status by the FDA and the EMA. We believe that our portfolio of intellectual property rights provides a defensible market position for the commercialization of VAL-083 and other anti-cancer products.
Our drug discovery research focuses on identifying well-validated clinical and commercial-stage compounds and establishing a scientific rationale for development in modern orphan drug indications. Through our relationship with Valent Technologies, LLC (“Valent”), a company owned by Dr. Dennis Brown, our Chief Scientific Officer, we are able to utilize Valent’s proprietary ChemEstate™ bioinformatics tools which are used to screen and identify potential candidates. Promising candidates are further researched through our network of consultants and contract research organizations. This approach allows us to rapidly identify and advance potential drug candidates without significant investment in “wet lab” infrastructure. Based on this strategy, we acquired the initial VAL-083 intellectual property and prototype drug product from Valent and have identified additional drug candidates that we may have the opportunity to license or acquire in the future.
We also believe the experience of our clinical development team will position us to acquire or license additional product candidates to establish a pipeline of product opportunities. We have secured four non-refundable financial contributions from the National Research Council of Canada for total financial contributions of approximately CDN $327,000 to date.
VAL-083
Our product candidate, VAL-083, represents a “first-in-class” small molecule chemotherapeutic, which means that the molecular structure of VAL-083 is not an analogue or derivative of other small molecule chemotherapeutics approved for the treatment of cancer.
VAL-083 has been assessed in multiple clinical studies sponsored by the NCI in the United States as a treatment for various cancers including lung, brain, cervical, ovarian tumors and leukemia. Published pre-clinical and clinical data suggest that VAL-083 may be active against a range of tumor types. VAL-083 is approved as a cancer chemotherapeutic in China for the treatment of chronic CML and lung cancer. VAL-083 has not been approved for any indications outside of China.
Upon obtaining regulatory approval, we intend to commercialize VAL-083 for the treatment of orphan cancer indications where patients have failed other therapies or have limited medical options. An orphan disease is defined in the United States under the Rare Disease Act of 2002 as “any disease or condition that affects less than 200,000 persons in the United States”. The Orphan Drug Act of 1983 is a federal law that provides financial and other incentives including a period of market exclusivity to encourage the development of new treatments for orphan diseases.
| 49 |
We research the mechanism of action of our product candidate to determine the clinical indications best suited for therapy and attempt to rapidly advance our product candidate into human clinical trials and toward commercialization. The mechanism of action of VAL-083 is understood to be a bi-functional alkylating agent. Alkylating agents are a commonly used class of chemotherapy drugs. They work by binding to DNA and interfering with normal processes within the cancer cell, which prevents the cell from making the proteins needed to grow and survive. After exposure to alkylating agents, the cancer cell becomes dysfunctional and dies. There are a number of alkylating agents on the market that are used by physicians to treat different types of cancer.
Based on published research and our own data, the cytotoxic functional groups and the mechanism of action of VAL-083 are understood to be functionally different from alkylating agents commonly used in the treatment of cancer. VAL-083 has previously demonstrated activity in cell-lines that are resistant to other types of chemotherapy. No evidence of cross-resistance has been reported in published clinical studies. Therefore, we believe that VAL-083 may be effective in treating tumors that have failed or become resistant to other chemotherapies.
We have presented new research at peer-reviewed scientific meetings demonstrating that VAL-083 is active in patient-derived tumor cell lines and cancer stem cells that are resistant to other chemotherapies.
VAL-083 readily crosses the blood brain barrier where it maintains a long half-life in comparison to the plasma. Published pre-clinical and clinical research demonstrates that VAL-083 is selective for brain tumor tissue.
The main dose-limiting toxicity (“DLT”) related to the administration of VAL-083 in previous NCI-sponsored clinical studies was myelosuppression. Myelosuppression is the decrease in cells responsible for providing immunity, carrying oxygen, and those responsible for normal blood clotting. Myelosuppression is a common side effect of chemotherapy. There is no evidence of lung, liver or kidney toxicity even with prolonged treatment by VAL-083. Commercial data from the Chinese market where the drug has been approved for more than 15 years supports the safety findings of the NCI studies.
We note that the DLT of VAL-083 was established prior to the development of medicines now available to manage myelosuppression. Various types of medications and other forms of therapy are now available for management of myelosuppressive side effects. We believe this offers the potential of increasing the dose of VAL-083 in the modern patient population thereby providing a potential opportunity to improve the drugs already established efficacy profile.
VAL-083 in GBM
Worldwide, there are an estimated 240,000 new cases of brain and central nervous system (“CNS”) tumors each year.1 Gliomas are a type of CNS tumor that arises from glial cells in the brain or spine. Glial cells are the cells surrounding nerves. Their primary function is to provide support and protection for neurons in the CNS.
Glioblastoma multiforme (“GBM”), also known as Grade IV astrocytoma, is the most common and the most lethal form of glioma. According to the World Health Organization, GBM occurs with an incidence of 3.17 per 100,000 person-years.2 Approximately 15,000 new cases of GBM are expected to be diagnosed in the United States during 2015.3
GBM progresses quickly and patients deteriorate rapidly. Common symptoms include headaches, seizures, nausea, weakness, paralysis and personality or cognitive changes such as loss of speech or difficulty in thinking clearly.
The majority of GBM patients do not survive for more than two years following diagnosis, and the median survival in newly diagnosed patients with best available treatments is 14.6 months.
Standard treatment following diagnosis includes surgical resection to remove as much of the tumor as possible (debulking) followed by radiotherapy with concomitant and adjuvant chemotherapy with Temodar® (temozolomide, “TMZ”). Nearly all patients diagnosed with GBM will relapse following first-line treatment, with a 1-year survival rate of approximately 25% following failure of front-line therapy, with average 5-year survival rate less than 3%.4
| 1 | WHO, IARC, Globocan Cancer Incidence and Mortality Worldwide in 2008. Last updated June 2012. globocan.iarc.fr/ |
| 2 | CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2006. Hinsdale, IL: Central Brain Tumor Registry of the United States; 2010. |
| 3 | Ostrom QT, Gittleman H, L iao P, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007-2011. Neuro Oncol. 2014;16(s5):iv1-iv63 |
| 4 | Johnson, Derek R.; O'Neill, Brian Patrick (2011). "Glioblastoma survival in the United States before and during the temozolomide era". Journal of Neuro-Oncology 107 (2): 359–64 |
| 50 |
Avastin® (bevacizumab – an anti-VEGF antibody) is approved as a single agent for patients with recurrent GBM following prior therapy as an alternative to corticosteroids to relieve disease symptoms in the US, Canada, Australia and Japan. Avastin carries a “black-box warning” related to severe, sometimes fatal, side effects related to gastrointestinal perforations, wound healing complications and hemorrhage. There are no data demonstrating an improvement in disease-related symptoms or increased survival in refractory GBM with Avastin. 5
TMZ and the nitrosoureas, including– carmustine, lomustine, and– nimustine, are alkylating agents that readily cross the blood-brain-barrier (“BBB”) and are used in the treatment of central nervous system (“CNS”) cancers, including GBM. Alkylating agents are among the oldest type of cancer chemotherapies in use today. Alkylating agents bind to DNA to cause damage to cancer cells. Their anti-tumor mechanism is via alkylation of DNA resulting in base-pair mismatch or strand-mediated cross links between base pairs. The DNA damage caused by alkylating agents mimics naturally occurring errors, resulting in apoptosis and tumor cell death.
The primary anti-cancer mechanism of TMZ and the nitrosoureas is to attack the tumor’s DNA via alkylation of the O6 position of the DNA base residue, guanine. TMZ treatment causes DNA damage mainly by methylation at the O6 position of guanine resulting in guanine-thymine base pair mismatches during replication. Nitrosoureas mediate their cytotoxic effect by ethylation at the O6 position of guanine which produces a cross-link to cytosine residues resulting in double-strand DNA breaks during mitosis.
Most GBM patients tumors are resistant to TMZ or nitrosourea therapy due to high expression of a naturally occurring enzyme called O6 -DNA methylguanine methyl-transferase (“MGMT”) enzyme which repairs O6 -guanine lesions. MGMT repair mechanism in turn prevents TMZ and introsoureas and allows a patients’ GBM tumor to survive and grow in spite of treatment.
Consistent with the importance of its repair activity, high expression of MGMT is strongly correlated with poor patient outcomes. Several clinical studies have established that MGMT is an important prognostic indicator of response to TMZ and patient survival. 6
Probability of GBM Patient Survival Correlated to Expression of MGMT Enzyme
(Unmethylated promoter = High MGMT Expression and Significantly Shorter Survival)
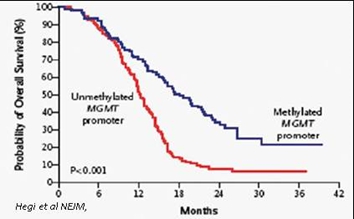
VAL-083 is an alkylating agent which readily crosses the BBB. Its primary cytotoxic mechanism, epoxide derived DNA cross-links at the N7 position of guanine, is distinct from TMZ or the nitrosoureas.
Our research demonstrates that VAL-083’s N7 targeting mechanism retains cytotoxic activity independent of MGMT expression in vitro . We have presented new research at peer-reviewed scientific meetings demonstrating that VAL-083 is active in patient-derived tumor cell lines and cancer stem cells that are resistant to other chemotherapies. Of particular importance is resistance to Temodar® due to activity of the repair enzyme known as MGMT, which results in chemoresistance in many GBM patients. At AACR in 2012, we presented data demonstrating that VAL-083 is active independent of MGMT resistance in laboratory studies. VAL-083 has more potent activity against brain tumor cells in comparison to TMZ and overcome resistance associated with MGMT suggesting the potential to surpass the current standard-of-care in the treatment of GBM. 7
A Summary of Our Data Demonstrating that VAL-083’s Anti-Tumor Mechanism is Distinct from and can Overcome MGMT-Related Chemoresistance in the Treatment of GBM:
| 5 | As stated in Avastin Prescribing Information: http://www.gene.com/download/pdf/avastin_prescribing.pdf |
| 6 | Hegi et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N Engl J Med 2005;352:997-1003 |
| 7 | Hu et al. VAL083, a novel N7 alkylating agent, surpasses temozolomide activity and inhibits cancer stem cells providing a new potential treatment option for glioblastoma multiforme. AACR Annual Meeting (2012) Experimental & Molecular Therapeutics; AACR Permanent Abstract No. 811 |
| 51 |
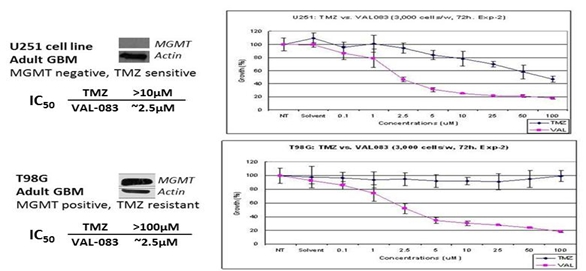
VAL-083 has been assessed in multiple historical NCI-sponsored clinical studies as chemotherapy in the treatment of newly diagnosed and recurrent brain tumors and other cancers. In general, tumor regression in brain cancer was achieved following therapy in greater than 40% of patients treated and stabilization was achieved in an additional 20% to 30%. In published clinical studies VAL-083 has previously been shown to have a statistically significant impact on median survival in high grade glioma brain tumors when combined with radiation versus radiation alone with results similar or superior to other chemotherapies approved for use in GBM.
A Summary of Published Data adapted from Separate Sources Comparing the Efficacy of VAL-083 and Other Therapies in the Treatment of GBM:
| Comparative Therapy | Median Survival Benefit | |||||
| Chemotherapy | Radiation (XRT) | Radiation + Chemotherapy | vs. XRT alone | |||
| Temodara | 12.1 months |
58 weeks (14.6 months) |
2.5 months | |||
| VAL-083b | 8.8 months |
67 weeks (16.8 months) |
8.0 months | |||
| Lomustinec | 52 weeks | |||||
| Carmustinec | 40-50 weeks | |||||
| Semustinec | 35 weeks | |||||
| Avastind | n.a. | |||||
aStupp (2005); bEagan (1979); cHauch(2005); dAvastin Label
Additional support for the differentiated profile of VAL-083 and TMZ comes from the results of studies with GBM cancer stem cells (“CSCs”). GBM CSCs display strong resistance to TMZ, even where MGMT expression is low; however, our data demonstrates that GBM CSCs are susceptible to VAL-083 independent of MGMT expression. 8
Based on historical data and our own research, we believe that VAL-083 has the potential to offer a physicians and patients a new paradigm in the treatment of GBM that will address significant to address unmet medical needs. In addition, the profile of VAL-083 offers the potential of additive or synergistic benefit as a future combination therapy with existing chemotherapeutic agents or novel vaccine or immunotherapy approaches currently under investigation.
We filed an investigational new drug (“IND”) application with the FDA and initiated human clinical trials with VAL-083 as a potential treatment for GBM in 2011. Details of the study, including enrollment estimates, are available at http://www.clinicaltrials.gov/ct2/show/NCT01478178?term=VAL-083&rank=1 )
| 8 | Fause et al. The novel N7 alkylating agent, VAL-083, inhibits the growth of primary glioma stem and non-stem cultures, including those that are temozolomide-resistant. Society for NeuroOncology Annual Meeting (2014) . Abstract No. ET-18 |
| 52 |
Our clinical trial is a Phase I/II an open-label, single arm dose-escalation study designed to evaluate the safety, tolerability, pharmacokinetics and anti-cancer activity of VAL-083 in patients with GBM. To be eligible for our clinical trial, patients must have been previously treated for GBM with surgery and/or radiation, if appropriate, and must have failed both bevacizumab (Avastin®) and temozolomide (Temodar®), unless either or both are contra-indicated.
Response to treatment with VAL-083 is measured prior to each treatment cycle. An initial phase of the study involves dose escalation cohorts until a maximum tolerated dose (“MTD”) is established in the context of modern care. The goal of our Phase I/II clinical trial is to determine a modernized dosing regimen for advancement into a registration directed clinical trial.
In August 2013, we received a notice of allowance from the FDA enabling the Company to implement a more rapid dose-escalation scheme in our Phase I/II clinical trial. The revised dosing regimen was allowed by the FDA following an extensive safety review of patients treated prior to that date. In comparison to the original dose-escalation scheme, the revised plan enabled us to skip two interim doses, which allowed the trial to reach higher doses than originally contemplated.
We have presented interim data from our Phase I/II clinical trial at peer-reviewed scientific meetings including most recently at the annual meetings of the American Association of Cancer Research (“AACR”) in April 2015 and SNO in November 2014 and previously during the year at the annual meetings of AACR and the American Society of Clinical Oncology (“ASCO”). We anticipate presenting additional data at upcoming scientific meetings during 2015.
In summary, at doses tested to date, our interim clinical data is as follows:
| · | We completed dose escalation cohorts up to 40mg/m2 without observation of dose limiting toxicity and subsequently filed a protocol amendment to allow for exploration of VAL-083 at doses up to 60mg/m2; |
| · | In cohort 8 (50mg/m2), we observed evidence of DLT, as defined by Grade 4 thrombocytopenia (low platelets), in one patient and trends toward DLT, as defined by Grade 3 thrombocytopenia in three additional patients, which suggests that we have reached the MTD. Based on a review of the totality of data from this cohort, we intend to confirm the dose for expansion of the trial and prepare for advancement into registration-directed Phase II/III clinical trials. One of three GBM patients in cohort 7 (40mg/m 2) and one of three GBM patients in cohort 6 (30 mg/m 2) exhibited stable disease after one or two cycles of treatment. In earlier cohorts, we reported that two patients exhibited a response (stable disease or partial response) with a maximum response of 84 weeks and improved clinical signs prior to discontinuing due to adverse events unrelated to the study; |
| · | Pharmacokinetics are linear and consistent with previous published data suggesting that concentrations of VAL-083 are achieving tissue levels in the central nervous system that are effective against glioma cell lines in vitro; |
| · | We presented additional data demonstrating that the cytotoxic activity of VAL-083 is distinct from standard-of-care in GBM. Specifically, the tumor-killing activity of VAL-083 has been demonstrated to be independent of MGMT, the enzyme believed to cause resistance to the current front-line therapy in the treatment of GBM. |
While these data are interim in nature, we believe they support the further development of VAL-083. We are currently conducting our clinical trial at four centers: the Mayo Clinic in Rochester, Minnesota (“Mayo”), the Brain Tumor Center at University of California, San Francisco (“UCSF”), the Sarah Cannon Cancer Research Center (“SCRI”) in Nashville, Tennessee and the SCRI affiliate site at the Florida Cancer Specialist Research Institute in Sarasota, Florida. We plan to add additional clinical sites in order to accelerate enrollment as the trial progresses.
We are now delivering doses of VAL-083 that are substantially higher than were achieved in the original NCI-sponsored clinical trials. Our modernized dosing regimen takes advantage of improved side-effect management and new knowledge of the pharmacokinetic and toxicity profile of VAL-083. Our strategy to “hit the tumor harder more often” allows us to achieve higher levels of drug at the tumor-site, which we believe will result in significant clinical benefit for GBM patients who currently have no viable treatment options.
A summary of our current dose escalation scheme including doses completed to date is as follows:
| Dosing
Regimen & Study |
Single Dose | Acute
Regimen (single cycle) |
Comparative
Cumulative Dose (@ 35 days) |
Dose
Intensity (dose per week) |
Status | |||||||
NCI GBM historical regimen (Eagan etal) daily x 5 q 5wks (cycle = 35 days) |
25 mg/m2 | x5 days = | 125 mg/m2 | 125 mg/m2 | 25mg/m2/wk | Historical Studies: Myelosuppression observed |
||||||
DelMar VAL-083 regimen daily x 3 q 3wks (cycle = 21 days) |
30 mg/m2 40 mg/m2 50 mg/m2 |
x3 days = | 90 mg/m2 120 mg/m2 150 mg/m2 |
180 mg/m2 |
30mg/m2/wk 40mg/m2/wk 50mg/m2/wk |
No DLT No DLT DLT observed |
||||||
Daily x 5 q 5wks refers to a dosing regimen of once per day for five consecutive days every five weeks (35 day cycle); while daily x 3 q 3wks refers to a dosing regimen of once per day for three consecutive days every three weeks (21 day cycle)
| 53 |
Patients being enrolled in our current Phase I/II clinical trial have a growing brain tumor that has failed to respond to any other approved treatment. The correlation between tumor progression and impending death in this patient population is well-documented. Therefore, our interim results demonstrating that VAL-083 can either stabilize disease progression by halting tumor growth or shrinking the tumor is expected to result in longer patient survival and improved quality of life.
We plan to continue our clinical trials with VAL-083 as a potential treatment for GBM patients who have failed other therapies. Currently, there is no approved therapy for these patients. The goal of our current Phase I/II clinical trial is to establish a modernized dosing regimen for advancement into registration directed trials in the United States as a potential new therapy for the treatment of refractory GBM.
We have had our first observation of a DLT which signals VAL-083's potential advancement toward registration-directed clinical trials in refractory GBM. We are currently studying a dose of 50 mg/m 2 and six patients have been enrolled at this dose in accordance with the protocol. The Company's clinical protocol requires acquisition of safety data for 35 days following initial treatment with VAL-083. At this dose, one patient completed the required 35 day follow-up period without observation of a DLT. The second patient in the 50 mg/m 2 cohort experienced myelosuppressive DLT as defined by grade four thrombocytopenia (low platelet counts). Three additional patients undergoing treatment have not experienced a DLT to-date, but did show a strong trend toward DLT as defined by grade three thrombocytopenia. The patients’ symptoms resolved rapidly and spontaneously returned to normal without concomitant medication or transfusion.
Our goal is to maximize the amount of VAL-083 that can safely reach the tumor. Our protocol currently allows dosing up to 60 mg/m 2. However, based on the observation of, and strong trends toward DLT, we have determined that we will not continue dose-escalation beyond 50 mg/m 2.
In accordance with the protocol that has been filed with the FDA, we plan to expand enrollment by up to an additional 14 GBM patients at a dose determined to be at or below the MTD, to obtain additional safety and preliminary activity data. During this period, we plan to request a guidance meeting with the FDA to discuss our proposed Phase II/III registration trial design.
The final decision on the dose chosen for trial expansion and advancement to Phase II/III registration directed studies will be determined by the analysis of safety and tolerability of our modernized dosing regimen when all six patients enrolled in the 50 mg/m2 cohort have completed the required follow-up period. Based on our current enrollment and timelines, we believe it is likely that we will initiate Phase II/III registration directed studies during the second half of calendar 2015.
We anticipate that the Phase II/III registration trial will be an open-label trial with radiographic response and overall survival as the primary endpoints. The dose chosen, size, design and timing of initiation of the registration-directed clinical trial will depend on review of the data from the current Phase I/II dose-escalation study and discussions with the FDA and our clinical advisors. We will provide a formal update, including any adjustment to our projected timelines based on our discussions with the FDA and our clinical advisors.
Based on historical development of other products in GBM, we believe that we may be able to obtain FDA approval to commercialize VAL-083 to treat patients who have failed other therapies from an open-label Phase II/III registration-directed clinical trial, which will save significant costs of a large randomized Phase III clinical trial. We also believe that the FDA may grant Breakthrough Therapy, Fast Track, Accelerated Approval and/or Priority Review status to VAL-083, which will enable us to begin filing for commercial approval during the clinical trial process. Breakthrough Therapy, Fast Track, Accelerated Approval and Priority Review are approaches established by the FDA that are intended to make therapeutically important drugs available at an earlier time. (See “Government Regulation and Product Approval”)
Data from the Phase II/III registration-directed trial will form the basis of our application for FDA approval. Our overall goal remains to complete registration-directed clinical trial with VAL-083 and to seek FDA approval as a new therapy for refractory glioblastoma in the timeliest manner possible. Based on our current financial resources, initiation of the registration trial will require additional funding to support the expanded clinical operations necessary to conduct and manage the study.
We also believe that VAL-083 may be a potentially superior alternative to currently approved chemotherapies used in the treatment of newly diagnosed GBM. Subject to the availability of financial resources, we plan to investigate VAL-083 in clinical trials for newly diagnosed GBM patients whose tumors exhibit molecular features suggesting that they are unlikely to respond to currently available chemotherapies.
| 54 |
In February 2012, VAL-083 was granted protection under the Orphan Drug Act by the FDA for the treatment of glioma. In January 2013, the European Union also granted orphan drug protection to VAL-083. Orphan drugs generally follow the same regulatory development path as any other pharmaceutical product. However, incentives such as scientific advice and reduction or waiver of registration fees and access to specialized grant funding may be available to support and accelerate development of orphan drug candidates. In addition, we may sell VAL-083 as a treatment for glioma without competition for seven years in the U.S. and for ten years in the EU following market approval, due to the orphan drug protection afforded – meaning that the neither the FDA nor the EU regulatory authority will approve a medicinal product containing a similar active substance for the same indication during that time.
As part of our ASCO presentation on June 1, 2013, we also announced that we plan to split our current Phase I/II clinical trial protocol into two separate studies: one focusing solely on refractory GBM and the other focusing on secondary brain cancers caused by other tumors that have spread to the brain. Due to prior chemotherapy and radiation therapy, patients with secondary brain tumors are likely more prone to myelosuppression and may have a different toxicity and MTD than patients with GBM. We believe the strategy of splitting the trial into two separate studies will enable us to focus on accelerating the development of VAL-083 as a potential new treatment for GBM while appropriately exploring the potential of the drug to treat patients with solid tumors that have spread to the brain. In the future, we may develop a separate protocol for the continued exploration of VAL-083 in patients with secondary brain cancer caused by a solid tumor spreading to the brain.
VAL-083 in Lung Cancer
Lung cancer is a leading cause of cancer-related mortality around the world and effective treatment for lung cancer remains a significant global unmet need despite advances in therapy. In general, prognosis for lung cancer patients remains poor, with 5-year relative survival less than 14% among males and less than 18% among females in most countries.[9] Globally, the market for lung cancer treatment may exceed $7 billion by 2019 according to report published by Transparency Market research.
Non-small cell lung cancer (“NSCLC”) is the most common type of lung cancer. There are three common forms of NSCLC: adenocarcinomas are often found in an outer area of the lung; squamous cell carcinomas are usually found in the center of the lung next to an air tube (bronchus); and large cell carcinomas, which can occur in any part of the lung and tend to grow and spread faster than adenocarcinoma. NSCLC accounts for 85% of all lung cancer cases in the United States and approximately 90% of lung cancer cases diagnosed in China.[10]
Smoking is the most important risk factor in the development of lung cancer. According to the World Cancer Report (2008), 21% of cancer deaths are related to smoking, especially lung cancer. Additionally, high levels of air pollution have been implicated as significant causes of lung cancer. Incidence of lung cancer in the United States is approximately 59 per 100,000 with the majority (52:100,000) being NSCLC.
According to The Nationwide Nutrition and Health Survey (2002), China has the world’s largest smoking population, with a smoking rate of 24.0% on average (50.2% for men and 2.8% for women), and a total number of 350 million smokers. The World Health Organization reports that the incidence of lung cancer in China is 34 per 100,000 population. However, some estimates are much higher exceeding 120 per 100,000 population for males aged 55-60 in urban areas.
According to a survey conducted by the Chinese Ministry of Health and the Ministry of Science and Technology, smoking, poor diet, water pollution and environmental problems have caused the nation's cancer death rate to rise 80 percent in the past 30 years and cancer is now accountable for 25 percent of all urban deaths and 21 percent of all rural deaths. Based on these trends, the World Health Organization projects that the incidence of lung cancer in China is expected to exceed one million (1,000,000) new cases per year by 2025.
The activity of VAL-083 against solid tumors, including lung cancer, has been established in both pre-clinical and human clinical trials conducted by the NCI. VAL-083 has been approved by the CFDA for the treatment of lung cancer. However, sales of VAL-083 in China have been limited by a lack of modern data, poor distribution, and preference for targeted therapies such as tyrosine kinase inhibitors (“TKIs”) in the modern era.
Standard for newly diagnosed NSCLC is platinum-based combination therapy or TKI therapy for patients whose cancer exhibit epidermal growth factor receptor (“EGFR”) mutations. Patients exhibiting EGFR mutations have shown an initial response rate to TKIs which exceeds the response rate for conventional chemotherapy. However, TKI resistance has emerged as an important unmet medical need.
| 9 | Youlden et al. The International Epidemiology of Lung Cancer: geographical distribution and secular trends, J Thorac Oncol. 2008 Aug;3(8):819-31. |
| 10 | Molona et al. Non–Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. as: Mayo Clin Proc. 2008 May; 83(5): 584–594. |
| 55 |
We believe VAL-083’s unique bi-functional alkylating mechanism of action could make it a valuable drug of choice in NSCLC patients who are or become resistant to TKI therapy. In addition, VAL-083 readily crosses the blood brain barrier suggesting that it may be possible for VAL-083 to treat patients whose lung cancer has spread to the brain.
Based on these beliefs, we have acquired certain commercial rights to VAL-083 in China where it is approved for the treatment of lung cancer. We have begun to establish a strong scientific and clinical rationale to support the development of VAL-083 as a potential treatment for NSCLC in the modern era.
We plan to work with leading oncologists to develop new clinical and non-clinical data which will demonstrate the clinical utility of VAL-083 in NSCLC patients who are resistant to TKIs. We believe this strategy will result in sales growth for VAL-083 in China and generate near-term revenue for our company through sales and marketing partnerships as well as position VAL-083 for global development in lung cancer.
In April 2014 at AACR we announced results of a pre-clinical study designed to evaluate the activity of VAL-083 in in vivo models of drug-resistant NSCLC in comparison to cisplatin. In an established murine xenograft model of NSCLC, the activity of VAL-083 was compared to standard platinum-based therapy with cisplatin against human NSCLC cell lines A549 (TKI-sensitive) and H1975 (TKI-resistant). In the study, VAL-083 demonstrated superior efficacy and safety in the treatment of TKI-susceptible (A549) tumors and in TKI-resistant (H1975) tumors.
| ● |
Treatment of TKI-sensitive (A549) NSCLC with 3 mg/kg of VAL-083 resulted in tumor growth delay of 26 days compared to untreated controls. Cisplatin (5 mg/kg) resulted in tumor growth delay of just four days. In addition, mean tumor volume on day 68 was significantly reduced in animals treated with 3 mg/kg VAL-083 (p=0.001) compared to untreated controls.
| |
| ● | Treatment of TKI-resistant (H1975) NSCLC with 4 mg/kg of VAL-083 resulted in a statistically significant reduction in tumor volume (p=0.01) versus untreated control after 27 days. In the same model, treatment with 5 mg/kg of cisplatin failed to achieve statistically significant reduction in tumor volume (p=0.23) versus untreated control after 27 days. Longer-term safety assessments are ongoing in this model. |
In April 2015, we presented new non-clinical data at the AACR annual meeting. These data demonstrated that VAL-083’s mechanism is distinct from platinum-based chemotherapy, the current standard of care for NSCLC. VAL-083 retains its high level of anti-cancer activity in p53 mutated NSCLC cell lines compared to cisplatin or oxaliplatin.
The p53 gene plays a central role in the protection of the human body from cancer and is responsible for initiating the process of programmed cell death, or apoptosis, which directs a cell to commit suicide if it becomes damaged or cancerous. The p53 pathway is also integral to the activity of many chemotherapy drugs. p53 is frequently mutated in NSCLC and p53 mutations are highly correlated with resistance to chemotherapy and poor patient outcomes in NSCLC.1
In addition, we demonstrated that the combination of VAL-083 with either cisplatin or oxaliplatin demonstrated a superadditive (synergistic) effect against NSCLC cell lines, including those resistant to TKI therapy in vitro.
In October 2014, we presented non-clinical data at the AACR New Horizon’s in Cancer Research Meeting. These data also support superior activity of VAL-083 compared to standard platinum-based treatment in both TKI-sensitive and TKI-resistant tumor models. Further, our data demonstrate that VAL-083 may have a synergistic effect in combination with cisplatin. These data suggest the potential of VAL-083 to be used in combination with platinum-based chemotherapy and to address modern unmet medical needs in the treatment of TKI-resistant NSCLC, especially where platinum-based therapy has already failed or is predicted to give sub-optimal outcomes.
These results may have immediate implications in the treatment of NSCLC in China, where VAL-083 is approved for as a chemotherapy for the treatment of lung cancer. The data also support exploring future clinical development of VAL-083 as a lung cancer therapy in the rest of the world thereby providing DelMar with a potential opportunity to expand our clinical development focus beyond glioblastoma.
As a next step in the investigation of VAL-083 as a potential treatment for NSCLC, we have developed a protocol for a post-market clinical study to be conducted by a leading cancer clinician in the context of the current approval in China.
We plan to conduct this trial in collaboration with Guangxi Wuzhou Pharmaceutical Group Co. Ltd. (Guangxi Wuzhou Pharma). Under the terms of our collaboration agreement with Guangxi Wuzhou Pharma, we are responsible for establishing protocols for and conducting clinical trials and Guangxi Wuzhou Pharma is responsible for the costs associated with clinical trials conducted in China. Our goal is to initiate this clinical trial by mid calendar 2015, with the aim develop new data to support product growth in China and to establish clinical proof of concept to expand our drug development efforts with VAL-083.
Conducting this clinical trial in China under our collaboration agreement with Guangxi Wuzhou Pharma will allow us to enhance the potential value of VAL-083 without significantly increasing our own planned cash expenditures. We also believe that these new data will support the potential to establish global partnerships and collaborations with larger pharmaceutical companies who have the resources and commercial infrastructure to effectively develop and commercialize VAL-083 as a treatment for NSCLC on a world-wide basis.
VAL-083 in Leukemia and Hematologic Cancers
The NCI studied VAL-083 extensively in laboratory and animal models of hematological malignancies (blood cancers). VAL-083 has been approved for the treatment of chronic myeloid leukemia, or CML, in China.
| 1 | Mitsudomi et al. Prognostic Significance of p53 Alterations in Patients with Non-Small Cell Lung Cancer: A Meta-Analysis |
| 56 |
CML, also known as chronic myeloid leukemia, is a cancer of the white blood cells. The incidence of CML in the United States is approximately two per 100,000 of population. 11
CML is characterized by three progressive phases: chronic, aggressive and blast, each corresponding with poorer prognosis. Approximately 85% of patients with CML are in the chronic phase at the time of diagnosis. Chronic phase patients are usually asymptomatic or have only mild symptoms such as fatigue or no symptoms at all. The duration of chronic phase is variable and depends on how early the disease was diagnosed as well as type of treatment. Without treatment, CML progresses to an accelerated phase and eventually to blast crisis. Blast crisis is the final phase in the evolution of CML and behaves like an acute leukemia with rapid progression and short expected survival.
While VAL-083 maintains labeling for CML in China, use of the drug in the modern era has been limited by a preference for targeted therapies such as TKIs.
TKIs have become the standard of care for CML and certain types of lung cancer. TKI therapy has resulted in vastly improved outcomes. However, patients often develop resistance to TKI therapy. Recent evidence proposes unique mechanisms of resistance in patients of East Asian descent who experience significantly inferior responses to TKIs.
We believe that data from NCI-sponsored studies and commercial evidence from the Chinese market support that there exists a substantive clinical benefit of VAL-083 in CML. We also believe that the unique mechanism of action of VAL-083, in combination with newly developed data positions the drug as a valuable therapy for patients who have failed other treatments, including TKIs. This represents a significant clinical and commercial opportunity for large subsets of patient populations in the existing-approved China market as well as for global development in CML.
Based on these beliefs, we have acquired certain commercial rights to VAL-083 in China, where it is approved for the treatment of CML and lung cancer. We have also developed new non-clinical data demonstrating that VAL-083 is active against TKI-resistant CML.
We have begun to establish a network of leading oncologists to develop new clinical and non-clinical data which will demonstrate the clinical utility of VAL-083 in CML patients who are resistant to TKIs. We believe this strategy may result in sales growth for VAL-083 in China and has the potential to generate revenue for our company through sales and marketing partnerships as well as position VAL-083 for global development in CML.
In addition to CML and subject to availability of funds, we plan to investigate VAL-083 as a potential treatment for other types of blood cancer. Acute Myeloid Leukemia (“AML”) and Acute Lymphoblastic Leukemia (“ALL”) are of particular interest based on published data and lack of effective therapeutic options. We have initiated preliminary discussions with leading cancer centers regarding the development of a clinical strategy for the development of VAL-083 in other types of blood cancer.
Additional Indications
In historical studies sponsored by the NCI in the United States, VAL-083 exhibited clinical activity against a range of tumor types including central nervous system tumors, solid tumors and hematologic malignancies. We have established new non-clinical data supporting the activity of VAL-083 in different types of cancer that are resistant to modern targeted therapies and we believe that the unique cytotoxic mechanism of VAL-083 may provide benefit to patients in a range of indications. We intend to continue to research these opportunities, and if appropriate, expand our clinical development efforts to include additional indications.
VAL-083 Target Markets
We are targeting cancer indications which we believe represent market opportunities in the hundreds of millions of dollars in North America and potentially in the billions of dollars worldwide. The pharmaceutical industry, in general, is a highly profitable, highly innovative industry. According to a report published by Statistic, the global pharmaceutical industry generated nearly one trillion dollars in revenue during 2013. According to published reports, global pharmaceutical sales are highly stratified by region, with North America, the European Union and Japan accounting for 55% of global pharmaceutical sales in 2009. However, the most rapid growth in the sector is from developing countries, particularly China.
| 11 | Cortes et al. Chronic Mylogenous Leukemia. Cancer Management 2014 |
| 57 |
|
Glioblastoma Multiforme: Newly diagnosed patients suffering from GBM are initially treated through invasive brain surgery, although disease progression following surgical resection is nearly 100%. Temozolomide (Temodar®) in combination with radiation is the front-line therapy for GBM following surgery. Temodar® currently generates more than $950 million annually[12] in global revenues even though most patients fail to gain long-term therapeutic benefits. Approximately 60% of GBM patients treated with Temodar® experience tumor progression within one year.
Bevacizumab (Avastin®) has been approved for the treatment of GBM in patients failing Temodar®. In clinical studies, only about 20% of patients failing Temodar® respond to Avastin® therapy. In spite of these low efficacy results, treatment of GBM in North America alone is projected to add $200 million annually to the revenues of Avastin® with projected growth in GBM to $650 million by 2016.[13]
Approximately 48% of patients who are diagnosed with GBM will fail both front-line therapy and Avastin®. Based on disease incidence, we believe the market for treating GBM patients the post-Avastin® failure exceeds $200 million annually in North America. Subject to successfully completing clinical trials and obtaining approval by the FDA and other applicable regulatory agencies globally, we also believe that VAL-083 could potentially generate sales in excess of $1 billion worldwide as a potential front-line therapy for GBM. |
|
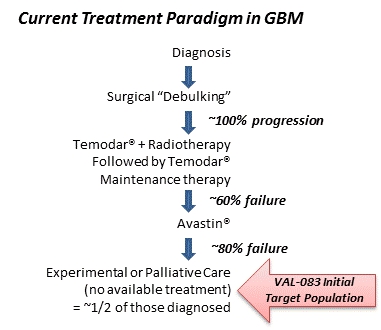
| Lung Cancer: The potential of VAL-083 in the treatment of NSLSC has been established in both human clinical trials conducted by the NCI and by the drug’s commercial approval in China. Lung cancer is the most common cancer in the world with 1.8 million cases in 2012, representing 13% of all cancers according to a report published by the World Cancer Research Fund International. Lung cancer has a higher mortality rate than next top three cancers combined and it is responsible for 1.6 million deaths annually, representing 19% of all cancer deaths. NSCLC represents approximately 90% of newly diagnosed lung cancers. Published reports indicate that the global NSCLC drug market will increase from $4.3 billion in 2009 to $6.9 billion in 2019 and the market is growing with a CAGR of 4.84% during 2009 to 2019. |
| Leukemia: The potential of VAL-083 in the treatment of CML has been established in both human clinical trials conducted by the NCI and by the drug’s commercial approval in China. The Tyrosine Kinase Inhibitor Gleevec® is currently used as front-line therapy in the treatment of CML achieved global revenue in excess of $4.7 billion annually in 2012.[14] We believe that VAL-083 has potential to capture a portion of the CML market through demonstration of activity in TKI-resistant CML patients. We also believe that VAL-083 may offer significant commercial opportunities through the treatment of other types of blood cancer such as AML or ALL. |
VAL-083 Manufacturing
VAL-083 is currently manufactured in accordance with CFDA and Chinese Pharmacopoeia guidelines to ensure drug quality control, drug use safety, and drug efficacy. Approval by the FDA will require VAL-083 and other products developed by us to be manufactured in accordance with United States Pharmacopeia (“USP”) in accordance with Good Manufacturing Practices (“cGMP”) regulations. cGMP provides for systems that assure proper design, monitoring, and control of manufacturing processes and facilities. Adherence to the cGMP regulations assures the identity, strength, quality, and purity of drug products by requiring that manufacturers of medications adequately control manufacturing operations.
We have established an exclusive purchasing relationship with a Chinese manufacturer that has enabled us to obtain drug product for human clinical trials in the United States and certain commercial rights in China. The Chinese manufacturer has established a commercial-scale manufacturing process based on the North American process originally developed for the NCI.
Ensuring a viable long-term supply of the VAL-083 drug product suitable for registration and commercialization in North America and Europe will require investment in improved manufacturing and quality controls. We will seek to build upon our expertise and our intellectual property related to the existing manufacturing processes for VAL-083 in collaboration with the current manufacturer to allow compliance with cGMP. In addition, we have identified third party contract manufacturers with the capabilities to establish the processes, procedures and quality systems necessary to meet U.S., Canadian, E.U. and other international cGMP manufacturing requirements. Such requirements include strong quality management systems, obtaining appropriate quality raw materials, establishing robust operating procedures, detecting and investigating product quality deviations, and maintaining reliable testing laboratories.
| 12 | EvaluatePharma reports |
| 13 | EvaluatePharma reports |
| 14 | Schiffer et al. "BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia". N. Engl. J. Med. 357 (3): 258–6 |
| 58 |
Patents and Proprietary Rights
Our success will depend in part on our ability to protect our existing product candidate and the products we acquire or license by obtaining and maintaining a strong proprietary position. To develop and maintain our position, we intend to continue relying upon patent protection, orphan drug status, Hatch-Waxman exclusivity, trade secrets, know-how, continuing technological innovations and licensing opportunities.
We have filed patent applications covering VAL-083 where we have claimed the use of and improvements related to VAL-083 and other novel aspects of our proposed treatment regimen, manufacturing process improvements and the formulation and composition of the active pharmaceutical ingredient and finished dosage form of VAL-083 products. We are prosecuting our patent applications in the United States and in international jurisdictions which we deem important for the potential commercial success of VAL-083.
Our patents and patent applications can be summarized in eight series as follows:
| · | Series I is generally directed to synthesis of VAL-083. |
| Patent or Patent Application No. | Title | Expiry | ||
| United States Patent No. 8,563,758 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||
| United States Patent No. 8,921,585 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||
|
United States Patent Application Serial No. 14/072,603 Notice of Allowance Received 1/22/15 |
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||
| United States Patent Application Serial No. 14/550,131 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||
| Mexican Patent No. 323310 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||
| PCT Patent Application Serial No. PCT/US2011/048032 |
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol National phase applications have published in countries including: Australia, Canada, Chile, China, European Union, Japan, Singapore and South Korea |
|||
| Additional Applications in Series I Not Yet Published |
| · | Series II is generally directed to use of VAL-083 to treat a range of diseases and conditions, including but not limited to malignancies. |
| Patent or Patent Application No. | Title | Expiry | ||
|
United States Patent Application Serial No. 13/817,096 Notice of Allowance Received 2/25/15 |
Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol | 2031 | ||
| PCT Patent Application Serial No. PCT/US2011/048031 |
Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol National phase applications have published in countries including: Australia, Canada, Chile, China, European Union, Japan, Mexico, Singapore and South Korea
Additional Applications in Series II Not Yet Published |
| · | Series III is generally directed to analytical methods for VAL-083. |
| 59 |
| Patent or Patent Application No. | Title | Expiry | ||
| United States Patent Application Serial No. 13/933,844 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol | 2032 | ||
|
United States Patent Application Serial No. 14/083,135 Notice of Allowance Received 1/7/15 |
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol | 2032 | ||
| PCT Patent Application Serial No. PCT/IB2013/000793 |
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol National phase applications have published in countries including: Australia, Canada, European Union, and South Korea |
|||
| Additional Applications in Series III Not Yet Published |
| · | Series IV is generally directed to the use of VAL-083 to treat GBM or medulloblastoma. |
| Patent or Patent Application No. | Title | Expiry | ||
| United States Patent Application Serial No. 14/373,552 | Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma | 2033 | ||
| United States Patent Application Serial No. 14/245,738 | Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma | 2033 | ||
| PCT Patent Application Serial No. PCT/US2013/022505 |
Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma National phase applications have published in countries including: Australia, Canada, European Union, and South Korea |
|||
| Additional Applications in Series IV Not Yet Published |
| · | Series V is generally directed to the veterinary use of VAL-083. |
| Patent or Patent Application No. | Title | Expiry | ||
| United States Patent Application Serial No. 14/400,271 | Veterinary Use Of Dianhydrogalactitol, Diacetyldianhydrogalactitol, And Dibromodulcitol To Treat Malignancies | 2033 | ||
| PCT Patent Application Serial No. PCT/US2013/039549 |
Veterinary Use Of Dianhydrogalactitol, Diacetyldianhydrogalactitol, And Dibromodulcitol To Treat Malignancies |
|||
| Additional Applications in Series V Not Yet Published |
| · | Series VI is generally directed to the use of VAL-083 to treat tyrosine-kinase-inhibitor-resistant malignancies. |
| Patent or Patent Application No. | Title | Expiry | ||
| United States Patent Application Serial No. 14/409,909 | Methods For Treating Tyrosine-Kinase-Inhibitor-Resistant Malignancies In Patients With Genetic Polymorphisms Or Ahi1 Dysregulations Or Mutations Employing Dianhydrogalactitol, Diacetyldianhydrogalactitol, Dibromodulcitol, Or Analogs Or Derivatives Thereof | 2033 | ||
| PCT Patent Application Serial No. PCT/US2013/047320 |
Methods For Treating Tyrosine-Kinase-Inhibitor-Resistant Malignancies In Patients With Genetic Polymorphisms Or Ahi1 Dysregulations Or Mutations Employing Dianhydrogalactitol, Diacetyldianhydrogalactitol, Dibromodulcitol, Or Analogs Or Derivatives Thereof National phase applications have published in countries including: Australia, Canada and Israel |
|||
Additional Applications in Series VI Not Yet Published |
| · | Series VII is generally directed to the use of VAL-083 to treat recurrent malignant glioma and progressive secondary brain tumor. |
| 60 |
| Patent or Patent Application No. | Title | Expiry | ||
| PCT Application Serial No. PCT/US2014/040461 | Use Of Dianhydrogalactitol And Analogs And Derivatives Thereof To Treat Recurrent Malignant Glioma Or Progressive Secondary Brain Tumor |
| · | Series VIII is generally directed to the use of VAL-083 to treat non-small-cell lung cancer. |
| Patent or Patent Application No. | Title | Expiry | ||
| Two provisional U.S. patent applications have been filed. No patent application in Series VIII has been published |
One of the inventors listed in one of our Series VIII provisional applications is an employee of the University of California, San Francisco. If a patent issues from that provisional application with a claim that the University of California employee conceived of, in whole or in part, than the Regents of the University of California will share ownership of any such patent with us. Our research agreements with the University of California address this issue by providing the Company with an exclusive option, for a limited period of time, to negotiate a royalty-bearing exclusive license for commercialization of the invention covered by that patent.
In addition to patent protection, we may also seek orphan drug status whenever it is available. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and Canada, and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for a different clinical indication.
In February 2012, we announced that the FDA has granted orphan drug status to VAL-083. In January 2013, the EMA also granted orphan drug protection to VAL-083 for the treatment of glioma.
Under the Hatch-Waxman Amendments, newly approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. These amendments provide five-year data exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active ingredient. The Hatch-Waxman Amendments prohibit the submission of an abbreviated new drug application, also known as an ANDA or generic drug application, during the five-year exclusive period if no patent is listed. If there is a patent listed and the ANDA applicant certifies that the NDA holder’s listed patent for the product is invalid or will not be infringed, the ANDA can be submitted four years after NDA approval. Protection under the Hatch-Waxman Amendments will not prevent the filing or approval of another full NDA; however, the applicant would be required to conduct its own pre-clinical studies and adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Amendments also provide three years of data exclusivity for the approval of NDAs with new clinical trials for previously approved drugs and supplemental NDAs, for example, for new indications, dosages or strengths of an existing drug, if new clinical investigations were conducted by or on behalf of the sponsor and were essential to the approval of the application. This three-year exclusivity covers only the new changes associated with the supplemental NDA and does not prohibit the FDA from approving ANDAs for drugs containing the original active ingredient. We intend to rely on the Hatch-Waxman Amendments for five years of data exclusivity for VAL-083.
We also rely on trade secret protection for our confidential and proprietary information. We believe that the substantial costs and resources required to develop technological innovations, such as the manufacturing processes associated with VAL-083, will help us to protect the competitive advantage of our product candidate.
The protection of intellectual property rights in China (where our clinical product candidate, VAL-083, is manufactured pursuant to a collaboration agreement with the only manufacturer presently licensed by the CFDA to produce the product for the China market, and where VAL-03 is approved for the treatment of CML and lung cancer) is relatively weak compared to the United States, which may negatively affect our ability to generate revenue from VAL-083 in China.
Our policy is to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual shall be our exclusive property.
Government Regulation and Product Approval
Regulation by governmental authorities in the U.S. and other countries is a significant factor, affecting the cost and time of our research and product development activities, and will be a significant factor in the manufacture and marketing of any approved products. All of our products require regulatory approval by governmental agencies prior to commercialization. In particular, our products are subject to rigorous pre-clinical and clinical testing and other approval requirements by the FDA and similar regulatory authorities in other countries. Various statutes and regulations also govern or influence the manufacturing, safety, reporting, labeling, transport and storage, record keeping and marketing of our products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, the necessary regulatory approvals could harm our business.
The regulatory requirements relating to the testing, manufacturing and marketing of our products may change from time to time and this may impact our ability to conduct clinical trials and the ability of independent investigators to conduct their own research with support from us.
| 61 |
The clinical development, manufacturing and marketing of our products are subject to regulation by various authorities in the U.S., the E.U. and other countries, including, in the U.S., the FDA, in Canada, Health Canada, and, in the E.U., the EMA. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act in the U.S. and numerous directives, regulations, local laws and guidelines in Canada and the E.U. govern the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. Product development and approval within these regulatory frameworks takes a number of years and involves the expenditure of substantial resources.
Regulatory approval will be required in all the major markets in which we seek to develop our products. At a minimum, approval requires the generation and evaluation of data relating to the quality, safety, and efficacy of an investigational product for its proposed use. The specific types of data required and the regulations relating to this data will differ depending on the territory, the drug involved, the proposed indication and the stage of development.
In general, new chemical entities are tested in animals until adequate evidence of safety is established to support the proposed clinical study protocol designs. Clinical trials for new products are typically conducted in three sequential phases that may overlap. In Phase I, the initial introduction of the pharmaceutical into either healthy human volunteers or patients with the disease (20 to 50 subjects), the emphasis is on testing for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase II involves studies in a limited patient population (50 to 200 patients) to determine the initial efficacy of the pharmaceutical for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a compound shows preliminary evidence of some effectiveness and is found to have an acceptable safety profile in Phase II evaluations, Phase III trials are undertaken to more fully evaluate clinical outcomes in a larger patient population in adequate and well-controlled studies designed to yield statistically sufficient clinical data to demonstrate efficacy and safety.
In the U.S., specific pre-clinical data, manufacturing and chemical data, as described above, need to be submitted to the FDA as part of an IND application, which, unless the FDA objects, will become effective 30 days following receipt by the FDA. Phase I studies in human volunteers may commence only after the application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the E.U. Currently, in each member state of the E.U., following successful completion of Phase I studies, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase II studies. The regulatory authorities in the E.U. typically have between one and three months in which to raise any objections to the proposed study, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase I studies, further submissions to regulatory authorities are necessary in relation to Phase II and III studies to update the existing IND. Authorities may require additional data before allowing the studies to commence and could demand that the studies be discontinued at any time if there are significant safety issues. In addition to the regulatory review, a study involving human subjects has to be approved by an independent body. The exact composition and responsibilities of this body will differ from country to country. In the U.S., for example, each study will be conducted under the auspices of an independent institutional review board at each institution at which the study is conducted. This board considers among other things, the design of the study, ethical factors, the privacy of protected health information as defined under the Health Insurance Portability and Accountability Act, the safety of the human subjects and the possible liability risk for the institution. Equivalent rules to protect subjects’ rights and welfare apply in each member state of the E.U. where one or more independent ethics committees, which typically operate similarly to an institutional review board, will review the ethics of conducting the proposed research. Other regulatory authorities around the rest of the world have slightly differing requirements involving both the execution of clinical trials and the import/export of pharmaceutical products. It is our responsibility to ensure we conduct our business in accordance with the regulations of each relevant territory.
By leveraging existing pre-clinical and clinical data, we are seeking build upon an existing pre-clinical and clinical safety and efficacy database to accelerate our research. In addition, our focus on end-stage population which has no current treatment options, commercialization may be achieved in an accelerated manner. Approval by the FDA in this category generally has been based on objective response rates and duration of responses rather than demonstration of survival benefit. As a result, trials of drugs to treat end-stage refractory cancer indications have historically involved fewer patients and generally have been faster to complete than trials of drugs for other indications. We are aware that the FDA and other similar agencies are regularly reviewing the use of objective endpoints for commercial approval and that policy changes may impact the size of trials required for approval, timelines and expenditures significantly.
In order to gain marketing approval we must submit a dossier to the relevant authority for review, which is known in the U.S. as an NDA and in the E.U. as a marketing authorization application, or MAA. The format is usually specific and laid out by each authority, although in general it will include information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product as well as the non-clinical and clinical data. Once the submitted NDA is accepted for filing by the FDA, it undertakes the review process that takes 10 months, unless an expedited priority review is granted which takes six months to complete. Approval can take several months to several years, if multiple 10-month review cycles are needed before final approval is obtained, if at all.
| 62 |
The approval process can be affected by a number of factors. The NDA may be approvable requiring additional pre-clinical, manufacturing data or clinical trials which may be requested at the end of the 10 month NDA review cycle, thereby delaying marketing approval until the additional data are submitted and may involve substantial unbudgeted costs. The regulatory authorities usually will conduct an inspection of relevant manufacturing facilities, and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. Further inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor for adverse effects or other additional studies as deemed appropriate. After approval for the initial indication, further clinical studies are usually necessary to gain approval for any additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs in the indications on which we are focusing our efforts. These include accelerated approval under Subpart H of the agency’s NDA approval regulations, fast track drug development procedures and priority review. At this time, we have not determined whether any of these approval procedures will apply to our current drug candidate.
The U.S., E.U. and other jurisdictions may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which, in the U.S., is generally a disease or condition that affects no more than 200,000 individuals. In the E.U., orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than 50 in 100,000 persons in the E.U.; without incentive it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. Orphan drug designation must be requested before submitting an NDA or MAA. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process. However, this designation provides an exemption from marketing and authorization (NDA) fees.
We are also subject to numerous environmental and safety laws and regulations, including those governing the use and disposal of hazardous materials. The cost of compliance with and any violation of these regulations could have a material adverse effect on our business and results of operations. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. Compliance with laws and regulations relating to the protection of the environment has not had a material effect on our capital expenditures or our competitive position. However, we are not able to predict the extent of government regulation, and the cost and effect thereof on our competitive position, which might result from any legislative or administrative action pertaining to environmental or safety matters.
Competition
The development and commercialization of new drugs is highly competitive and we may face competition established pharmaceutical and biotechnology companies, as well as from academic institutions, government agencies and private and public research institutions worldwide.
Various products currently are marketed for the treatment of GBM and other cancers that we may target with our product candidate and a number of companies are developing new treatments. Companies also developing products for GBM include but are not limited to Celgene Corp., Celldex Therapeutics, Northwest Biotherapeutics, Inc., Immunocellular Therapeutics Ltd., and many major pharmaceutical companies. Our success will be based in part on our ability to build and actively manage a portfolio of drugs that addresses unmet medical needs and create value in patient therapy.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunity will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than products that we may develop. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
We expect that our ability to compete effectively will depend upon our ability to:
| ● | successfully and rapidly complete adequate and well-controlled clinical trials that demonstrate statistically significant safety and efficacy and to obtain all requisite regulatory approvals in a cost-effective manner; | |
| ● | maintain a proprietary position for our manufacturing processes and other technology; |
| 63 |
| ● | produce our products in accordance with United States FDA and international regulatory guidelines; | |
| ● | attract and retain key personnel; and | |
| ● | build an adequate sales and marketing infrastructure for any approved products. |
Failure to do one or more of these activities could have an adverse effect on our business, financial condition or results of operations.
Corporate History
DelMar Pharmaceuticals, Inc. (the “Company”) is a Nevada corporation formed on June 24, 2009 under the name Berry Only Inc. Prior to the Reverse Acquisition (discussed below), Berry did not have any significant assets or operations. On January 21, 2013, the Company changed its name to DelMar Pharmaceuticals, Inc.
On January 25, 2013 (the “Closing Date”), the Company entered into and closed an exchange agreement (the “Exchange Agreement”), with DelMar (BC), 0959454 B.C. Ltd., a British Columbia corporation and a wholly-owned subsidiary of the Company (“Callco”), 0959456 B.C. Ltd., a British Columbia corporation and a wholly-owned subsidiary of the Company (“Exchangeco”), and securityholders of DelMar (BC). Pursuant to the Exchange Agreement, (i) the Company issued 4,340,417 shares of common stock (the “Parent Shares”) to the shareholders of DelMar (BC) who are United States residents (the “U.S. Holders”) in exchange for the transfer to Exchangeco of all 4,340,417 outstanding common shares of DelMar (BC) held by the U.S. Holders, (ii) the shareholders of DelMar (BC) who are Canadian residents (the “Canadian Holders”) received, in exchange for the transfer to Exchangeco of all 8,729,583 outstanding common shares of DelMar (BC) held by the Canadian Holders, 8,729,583 exchangeable shares (the “Exchangeable Shares”) of Exchangeco, and (iii) outstanding warrants to purchase 3,360,000 common shares of DelMar (BC) and outstanding options to purchase 1,020,000 common shares of DelMar (BC) were deemed to be amended such that, rather than entitling the holder to acquire common shares of DelMar (BC), such options and warrants (as amended, the “Exchange Agreement Warrants”) will entitle the holders to acquire shares of common stock of the Company. The Canadian Holders will be entitled to require Exchangeco to redeem (or, at the option of the Company or Callco, to have the Company or Callco purchase) the Exchangeable Shares, and upon such redemption or purchase to receive an equal number of shares of common stock of the Company.
Effective on the Closing Date, pursuant to the Exchange Agreement, DelMar (BC) became (indirectly through Exchangeco) a wholly-owned subsidiary of the Company. The acquisition of DelMar (BC) is treated as a reverse acquisition (the “Reverse Acquisition”), and the business of DelMar (BC) became the business of the Company. At the time of the Reverse Acquisition, Berry was not engaged in any active business.
Employees
We have four full-time employees and retain the services of approximately 15 persons on an independent contractor/consultant and contract-employment or full-time employee basis. As such, we currently operate in a “virtual” corporate structure in order to minimize fixed personnel costs. Over time, we plan to establish a base of full time employees and corporate infrastructure.
Legal Proceedings
There are no legal proceedings to which the Company or any of its property is the subject.
Properties
Our corporate headquarters are located at Suite 720-999 West Broadway, Vancouver, British Columbia, Canada. Our clinical operations are managed at 3475 Edison Way, Suite R, Menlo Park, California, 94025. Our current monthly base rent for our corporate headquarters is $2,440 (CDN $3,050) under a one-year lease which will expire in November 2015. In addition, Valent, which is owned by Dr. Dennis Brown, our Chief Scientific Officer, leases facilities in California and we have access to such facilities pursuant to an informal unwritten arrangement with Valent. Our leased premises, academic relationships, and access to the Valent facility are sufficient to meet the immediate needs of our business, research and operations.
| 64 |
Executive Officers, Directors and Key Employees
Below are the names and certain information regarding the Company’s executive officers and directors.
| Name | Age | Position | ||
| Jeffrey Bacha | 47 | President, Chief Executive Officer and Director | ||
| Dennis Brown | 65 | Chief Scientific Officer and Director | ||
| Scott Praill | 49 | Chief Financial Officer | ||
| John K. Bell | 67 | Director | ||
| Lynda Cranston | 67 | Director | ||
| William Garner | 48 | Director | ||
| Erich Mohr | 60 | Director | ||
| Robert J. Toth, Jr. | 51 | Director |
Jeffrey Bacha, BSc, MBA has been Chief Executive Officer and President of the Company since January 25, 2013, and director of the Company since February 11, 2013. Mr. Bacha is one of our founders and has been President, Chief Executive Officer and director of DelMar (BC) since inception. Mr. Bacha is a seasoned executive leader with nearly twenty years of life sciences experience in the areas of operations, strategy and finance. His background includes successful public and private company building from both a start-up and turn around perspective; establishing and leading thriving management and technical teams; and raising capital in both the public and private markets. From July 2006 to August 2009, Mr. Bacha was Executive Vice President Corporate Affairs and Chief Operating Officer at Clera, Inc. From March 2005 to July 2006 Mr. Bacha was Consultant and held various positions at Clera Inc., Urigen Holdings Inc. and XBiotech, Inc. From 1999 through 2004, Mr. Bacha served as President & CEO of Inimex Pharmaceuticals, a venture-capital funded drug discovery and development company and is a former Senior Manager and Director of KPMG Health Ventures. Mr. Bacha holds an MBA from the Goizueta Business School at Emory University and a degree in BioPhysics from the University of California, San Diego. Mr. Bacha’s experience as one of our founder and Chief Executive Officer qualifies him to serve on the Board of Directors.
Dr. Dennis M. Brown, PhD, has been Chief Scientific Officer of the Company since January 25, 2013 and director of the Company since February 11, 2013. Dr. Brown is one of our founders and has served as Chief Scientific Officer and director of DelMar (BC) since inception. Dr. Brown has more than thirty years of drug discovery and development experience. He has served as Chairman of Mountain View Pharmaceutical's Board of Directors since 2000 and is the President of Valent. In 1999 he founded ChemGenex Therapeutics, which merged with a publicly traded Australian company in 2004 to become ChemGenex Pharmaceuticals (ASX: CXS/NASDAQ: CXSP), of which he served as President and a Director until 2009. He was previously a co-founder of Matrix Pharmaceutical, Inc., where he served as Vice President (VP) of Scientific Affairs from 1985-1995 and as VP, Discovery Research, from 1995-1999. He also previously served as an Assistant Professor of Radiology at Harvard University Medical School and as a Research Associate in Radiology at Stanford University Medical School. He received his B.A. in Biology and Chemistry (1971), M.S. in Cell Biology (1975) and Ph.D. in Radiation and Cancer Biology (1979), all from New York University. Dr. Brown is an inventor of about 34 issued U.S. patents and applications, many with foreign counterparts. Dr. Brown’s scientific knowledge and experience qualifies him to serve on our Board of Directors.
Scott Praill, CPA, BSc. has been Chief Financial Officer of the Company since January 29, 2013 and previously served as a consultant to DelMar (BC). Since 2004, Mr. Praill has been an independent consultant providing accounting and administrative services to companies in the resource industry. Mr. Praill served as CFO of Strata Oil & Gas, Inc. from June 2007 to September 2008. From November 1999 to October 2003 Mr. Praill was Director of Finance at Inflazyme Pharmaceuticals Inc. Mr. Praill completed his articling at Price Waterhouse (now PricewaterhouseCoopers LLP) and obtained his Chartered Professional Accountant designation in 1996. Mr. Praill obtained his Certified Public Accountant (Illinois) designation in 2001. Mr. Praill received a Financial Management Diploma (Honors), from British Columbia Institute of Technology in 1993, and a Bachelor of Science from Simon Fraser University in 1989.
| 65 |
John K. Bell, FCPA, FCA, ICD.D has served as a director of the Company since February 11, 2013. John K. Bell is Chairman of Onbelay Capital Inc, a Canadian based private equity company with principal investments in Telematics and auto parts manufacturing (for past 5 years). Prior to that, from 1996 to 2005, Mr. Bell was CEO and owner of Polymer Technologies Inc., an automotive parts manufacturer. Prior to that, from 1977 to 1995, Mr. Bell was founder and owner of Shred-Tech Limited a global manufacturer and supplier of industrial shredders and mobile document shredders. Mr. Bell served as interim CEO and director of ATS Automation Tooling Systems (TSX-ATA) in 2007. Mr. Bell is a director of Strongco Corporation (TSX-SQP), Tweed Marijuana Inc.(TSX-V-TWD), and the Royal Canadian Mint (TSX-MNT). Mr. Bell is the past National secretary and board member of The Crohns and Colitis Foundation of Canada. Mr. Bell is also the past Chairman of Waterloo Regional Police, Cambridge Memorial Hospital, Canada’s Technology Triangle accelerator network and The Region of Waterloo prosperity counsel. Mr. Bell is a graduate of Western University Ivey School of Business, a Fellow of the institute of Chartered Accountants of Ontario, a graduate of the Institute of Directors Program of Canada and the owner’s president program at Harvard and International marketing program at Oxford. Mr. Bell’s financial and executive business experience qualifies him to serve on our board of directors.
Lynda Cranston BScN, MScN, ICD.D has served as a director of the Company since February 5, 2015 and serves as the Chair of our Governance and Compensation Committee. Mrs. Cranston recently retired from healthcare where she had been a CEO for over 20 years. Her last appointment prior to her retirement was as the first CEO of the Provincial Health Services Authority (2002 to 2013). Prior to this appointment Mrs. Cranston had been the 1st CEO of the Canadian Blood Services in Ottawa, ON (1998-2001). Before moving to Ottawa, Mrs. Cranston, as the CEO of BC Women's Hospital and Healthcare Centre had merged the organization with the BC Children's Hospital and the Sunny Hill Health Centre for Children to become the Children's and Women's Healthcare Centre of BC. Following the merger Mrs. Cranston became the first CEO. Mrs. Cranston also sits on the national board of the Gastrointestinal Society as its chair. In 2013, Mrs. Cranston was identified as a member of Diversity 50 by the Canadian Board Diversity Council as being one of Canada's most Board ready candidates. Mrs. Cranston was awarded the Board Chair Award of Excellence by the HealthCare Leaders' Association of British Columbia in 2008. In 2007, she was inducted into Canada's Most Powerful Women Top 100 Hall of Fame after having been identified in '04,'05 & '06 as one of Canada's Most Powerful Women Top 100. Mrs. Cranston is a recipient of the YWCA Women of Distinction Award, the 125th Anniversary of the Confederation of Canada Commemorative Medal for community contributions, and the Queen's Golden Jubilee Medal for contribution to Canada and community. Ms. Cranston’s healthcare industry and executive knowledge and experience qualify her to serve on the Company’s board of directors.
Dr. William Garner, MD, MPH has served as a director of the Company since February 11, 2013. Dr. Garner is one of our founders and has served as a director of DelMar (BC) since inception. Dr. Garner is an experienced entrepreneur and investor. He is founder and managing director of EGB Advisors, LLC ("EGB"), a pharmaceutical commercialization boutique. Through this entity, Dr. Garner has worked on a number of pharmaceutical business transactions and has raised financing for several drug development companies including Update Pharma, Inc. where he is currently Executive Chairman. Other EGB companies include Urigen Pharmaceuticals, Inc., and Inverseon, Inc., which is developing a novel therapy for smoking cessation, asthma and other pulmonary diseases. In 2012, he merged Inverseon with another company to form Invion Ltd. (ASX:IVX), serving as CEO until May of 2013. He also served as President and Chief Executive Officer of Urigen Pharmaceuticals, Inc. (URGP.PK) from December 2005 to December 2010 where he moved a procedure-based drug from a university license to a phase II multi-center clinical trial which achieved statistical significance on all end points in Painful Bladder Syndrome/Interstitial Cystitis. Before this, Dr. Garner worked in medical affairs at Hoffmann LaRoche in oncology. Prior to Roche, Dr. Garner was in the venture capital department at Paramount Capital Investments in New York City. He serves on the boards of ImmunoGenetix in Kansas City and the Innovation Angel Foundation in San Francisco. Dr. Garner has a Master of Public Health from Harvard and received his M.D. degree from New York Medical College. Dr. Garner did residency training in Anatomic Pathology at Columbia-Presbyterian and is currently a licensed physician in the State of New York. Dr. Garner’s medical and scientific knowledge and experience qualifies him to serve on our board of directors.
Dr. Erich Mohr, Ph.D., R. Psych., has served as a director of the Company since March 31, 2015. Dr. Mohr has nearly two decades of biotechnology experience in executive leadership roles as co-founder, chief scientific officer, chief executive officer and board member. Dr. Mohr has overseen and participated in dozens of clinical development programs and regulatory advisory panels. He is currently Chairman, Chief Executive Officer and Founder of MedGenesis Therapeutix Inc., a privately-held biopharmaceutical company committed to developing and commercializing innovative therapeutics to provide life-enhancing treatments to patients with serious neurologic diseases. Formerly, he was Chairman and Chief Executive Officer of CroMedica Global Inc., which merged with PRA International in 2002 to form one of the major contract research organizations in the world. In addition to his industry experience, Dr. Mohr has over 30 years of experience in experimental therapeutics of CNS disorders including eight years at the University of Ottawa, ultimately as a Professor of Medicine (Neurology) and Psychology. Dr. Mohr is the author of over 150 publications, books, book chapters and abstracts. Currently, he is the Chair of the Board of Governors of the University of Victoria, British Columbia, having previously served as a member and as Vice Chair. He earned his Masters of Science and Ph.D. in Neuropsychology at the University of Victoria, British Columbia, and his Bachelors of Arts in Psychology and dual Bachelors of Science in Chemistry and Biology from the University of the Pacific in Stockton, California. Dr. Mohr’s scientific and business executive knowledge and experience qualify him to serve on the Company’s board of directors.
| 66 |
Robert J. Toth, Jr., MBA has served as a director of the Company since August 20, 2013. Since 2005, Mr. Toth has primarily been managing his personal investment portfolio. From 2004-2005, Mr. Toth served as a consulting analyst to Narragansett Asset Management, a New York-based healthcare-focused hedge fund, where he advised the firm on biotechnology investments. From 2001-2003, he was the Senior Portfolio Manager for San Francisco-based EGM Capital’s Medical Technology hedge fund, where he was responsible for managing and maintaining a dedicated medical technology portfolio. Mr. Toth began his Wall Street career in 1996 as an Equity Research Associate for Vector Securities International, a healthcare-focused brokerage firm. From 1997–1999 he served as Senior Biotechnology Analyst. He joined Prudential Securities as Senior Vice President and Biotechnology Analyst where he served from 1999-2001 following Prudential’s acquisition of Vector. His responsibilities included the analysis of commercial, clinical and scientific fundamentals of oncology- and genomics-based biotechnology companies on behalf of institutional investors. Mr. Toth was named to the Wall Street Journal’s Allstar List for stock picking in 1999. Mr. Toth received an MBA from the University of Washington and Bachelor of Science degrees in Biological Sciences and Biochemistry from California Polytechnic State University, San Luis Obispo. Mr. Toth’s financial and biotechnology industry knowledge and experience quality him to serve on the Company’s board of directors.
The Company’s directors are elected at the annual meeting of shareholders to hold office until the annual meeting of shareholders for the ensuing year or until their successors have been duly elected and qualified. Officers are elected annually by the Board of Directors and serve at the discretion of the Board.
The Company’s executive officers are not full-time employees, but are engaged by us on an independent contractor or contract-employment basis. Mr. Bacha and Mr. Praill each devote 100% of their business time to us, and Dr. Brown devotes approximately 80% of his business time to us. See “Executive Compensation”.
Board Leadership Structure and Role in Risk Oversight
Mr. Bacha serves as Chairman and Chief Executive Officer. Due to the small size and early stage of the Company, we believe it is currently most effective to have the Chairman and Chief Executive Officer positions combined.
Our board of directors is primarily responsible for overseeing our risk management processes. The board of directors receives and reviews periodic reports from management, auditors, legal counsel, and others, as considered appropriate regarding our company’s assessment of risks. The board of directors focuses on the most significant risks facing our company and our company’s general risk management strategy, and also ensures that risks undertaken by our Company are consistent with the board’s appetite for risk. While the board oversees our company’s risk management, management is responsible for day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks facing our company and that our board leadership structure supports this approach.
Involvement in Certain Legal Proceedings
To our knowledge, our directors and executive officers have not been involved in any of the following events during the past ten years:
| 1. | any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; | |
| 2. | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); | |
| 3. | being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities; | |
| 4. | being found by a court of competent jurisdiction in a civil action, the SEC or the Commodity Futures Trading Commission to have violated a Federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; | |
| 5. | being subject of, or a party to, any Federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of any Federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or | |
| 6. | being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
Board Committees
The Board has formed an audit committee, which currently consists of John K. Bell, Chair, and Robert Toth. The Board intends to expand the audit committee at such time as the Board has additional independent members. The Board has also recently formed a Corporate Governance and Compensation Committee which consists of Lynda Cranston, Chair, John K. Bell and Robert Toth.
| 67 |
Advisors
The Company also has the following advisors:
| Name | Current Title or Prior Experience | |
| Victor Levin, MD | Prof. Emeritus MD Anderson Cancer Center (Neuro-Oncology) | |
Susan Chang, MD |
Chair, NeuroOncology Department UCSF | |
James Perry, MD |
Chair, Canadian Brain Tumor Consortium | |
Howard Burris, MD |
Director, Sarah Cannon Cancer Research Institute | |
Bill Bodell, PhD |
Prof. Emeritus UC Berkeley (DNA Damage & Repair) | |
Dan Zhang, MD |
SFDA Oncology Advisory Panel (China FDA) | |
Christine Charette |
Former Biotech Analyst, BMO Nesbitt Burns | |
| Sol Barer, PhD | Founder, Celgene |
EXECUTIVE COMPENSATION
During its last two fiscal years, Berry did not pay any compensation to its officers or directors.
The following table sets forth all compensation paid in respect of the Company’s principal executive officers and those individuals who received compensation in excess of $100,000 per year for the six months ended June 30, 2014 and the years ended December 31, 2013 and 2012.
| Name and Principal Position | Period | Salary ($) | Option Awards ($) | Total ($) | ||||||||||
| Jeffrey Bacha, Chief Executive Officer | Six Months Ended June 30, 2014 | 72,000 | - | 72,000 | ||||||||||
| Year Ended December 31, 2013 | 139,871 | 199,850 | (2) | 339,721 | ||||||||||
| Year Ended December 31, 2012 | 144,072 | 45,832 | (1) | 189,904 | ||||||||||
| Dennis Brown, Chief Scientific Officer | Six Months Ended June 30, 2014 | 60,000 | - | 60,000 | ||||||||||
| Year Ended December 31, 2013 | 120,000 | 199,850 | (2) | 319,850 | ||||||||||
| Year Ended December 31, 2012 | 120,000 | 45,832 | (1) | 165,832 | ||||||||||
| Scott Praill, Chief Financial Officer | Six Months Ended June 30, 2014 | 60,000 | - | 60,000 | ||||||||||
| Year Ended December 31, 2013 | 136,399 | 199,850 | (3) | 336,249 | ||||||||||
| (1) | Represents the grant date fair value of 150,000 options with an exercise price of CDN $0.50 issued on February 1, 2012. The options vested over a 12 month period and expire 10 years from the date of grant. Please see Note 9 to the financial statements. |
| (2) | Represents the grant date fair value of 350,000 options with an exercise price of $1.05 issued on August 15, 2013. The options vested over a 12 month period and expire 10 years from the date of grant. Please see Note 8 to the financial statements. |
| (3) | Represents the grant date fair value of 350,000 options with an exercise price of $1.05 issued on August 15, 2013. The options vested over a 36 month period and expire 10 years from the date of grant. Please see Note 8 to the financial statements. |
| 68 |
Pursuant to consulting agreements dated August 1, 2011 with certain of DelMar (BC)’s then-officers and directors, DelMar (BC) agreed to compensate its officers and directors for services rendered to it, in the amount of an aggregate of CDN $27,000 ($12,000 for Mr. Bacha, $10,000 for Dr. Brown, and $5,000 for Dr. Garner) per month commencing August 1, 2011 and ending December 31, 2012. Under the consulting agreements, DelMar (BC) and the respective officer or director mutually agreed that a portion of the compensation payable under the respective agreement for the year ended December 31, 2011 would be deemed to have been invested in the unit offering of DelMar (BC) completed on October 3, 2011.
The consulting agreements between DelMar (BC) and each of the three executive officers and directors expired on December 31, 2012. Commencing January 2013 when we became a public company, Dr. Garner’s compensation was adjusted to be consistent with the compensation of our other independent directors. Until July 1, 2014 we continued to compensate Mr. Bacha and Dr. Brown at the rates set forth in their respective consulting agreements except that commencing January 1, 2014 we began paying compensation in USD rather than CDN. Commencing July 1, 2014, the Company began to compensate Mr. Bacha USD $15,000 on a monthly basis and Dr. Brown at USD $12,500 on a monthly basis. The Company recently completed consulting agreements with Mr. Bacha and Dr. Brown that are retroactive to January 1, 2015. The terms of the new agreements are substantially the same as the previously expired agreements except that they reflect the current compensation noted above.
Mr. Bacha and Dr. Brown have continued to provide services to us as Chief Executive Officer and Chief Scientific Officer, respectively. Mr. Bacha devotes 100% of his business time to us and Dr. Brown devotes approximately 80% of his business time to us. The consulting agreements between DelMar (BC) and Mr. Bacha and Dr. Brown, respectively, do not specify the amount of time Mr. Bacha and Dr. Brown are required to devote to us, but did require that Mr. Bacha and Dr. Brown each provide us with the full benefit of their respective knowledge, expertise and ingenuity, and prohibit Mr. Bacha and Dr. Brown from engaging in any business, enterprise or activity contrary to or that would detract from our business.
Under two of these agreements for the year ended December 31, 2012, the directors elected to receive a portion of their aggregate compensation in the form of units. During the year ended December 31, 2012 DelMar (BC) issued 360,000 units for a total amount of CDN $180,000. The units issued relate to an amount of CDN $15,000 per month from January to December 2012 inclusive.
We were party to a consulting agreement, dated February 1, 2013, with Scott Praill, our Chief Financial Officer. Pursuant to the consulting agreement, we agreed to pay Mr. Praill a fee of CDN$10,000 per month and a one-time fee of CDN $30,000 for services rendered to that date. The consulting agreement did not specify the amount of time Mr. Praill is required to devote to us, but did require that Mr. Praill provide us with the full benefit of his knowledge, expertise and ingenuity, and prohibited Mr. Praill from engaging in any business, enterprise or activity contrary to or that would detract from our business. The consulting agreement expired on December 31, 2013. Mr. Praill devotes 100% of his business time to us. Since the expiration of the consulting agreement, we have continued to compensate Mr. Praill under the terms of the original agreement except that commencing January 1, 2014 we began paying compensation in USD rather than CDN. Commencing July 1, 2014, the Company began to compensate Mr. Praill USD $12,500 on a monthly basis. Mr. Praill continues to serve as our Chief Financial Officer. The Company recently completed a consulting agreement with Mr. Praill that is retroactive to January 1, 2015. The terms of the new agreements are substantially the same as the previously expired agreements except that it reflects the current compensation noted above.
As a result of the Company establishing a Corporate Governance and Compensation Committee, the Company anticipates entering into employment agreements with Mr. Bacha, Mr. Praill, and Dr. Brown in the near future. The contemplated employment agreements will replace the existing consulting agreements.
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth outstanding equity awards to our named executive officers as of June 30, 2014.
| Option awards | ||||||||||||||||||
| Name | Number of securities underlying unexercised options (#) Exercisable | Number of securities underlying unexercised options (#) unexercisable | Equity incentive plan
awards: number of securities underlying unexercised unearned options (#) | Option exercise price (US$) | Option expiration date | |||||||||||||
| Jeffrey Bacha (1) | 150,000 | - | - | 0.47 | 2/1/2022 | |||||||||||||
| 306,250 | 43,750 | - | 1.05 | 8/15/2023 | ||||||||||||||
| Dennis Brown (1) | 150,000 | - | - | 0.47 | 2/1/2022 | |||||||||||||
| 306,250 | 43,750 | - | 1.05 | 8/15/2023 | ||||||||||||||
| Scott Praill (1) | 40,231 | 9,769 | - | 0.47 | 2/1/2022 | |||||||||||||
| 102,083 | 247,917 | - | 1.05 | 8/15/2023 | ||||||||||||||
| (1) | Actual exercise price is CDN $0.50. Price disclosed is U.S. dollar equivalent as of June 30, 2014. Options were granted on February 1, 2012 and expire on February 1, 2022. |
| 69 |
Director Compensation
The following table sets forth director compensation for the six months ended June 30, 2014 paid by the Company (excluding any compensation included in the summary compensation table above).
| Fees | ||||||||||||||||||||||||||
| Earned | Nonqualified | |||||||||||||||||||||||||
| or | Non-Equity | Deferred | ||||||||||||||||||||||||
| Paid in | Stock | Option | Incentive Plan | Compensation | All Other | |||||||||||||||||||||
| Cash | Awards | Awards | Compensation | Earnings | Compensation | Total | ||||||||||||||||||||
| Name | ($) | ($) | ($) | ($) | ($) | ($) | ($) | |||||||||||||||||||
| William Garner | 14,500 | - | - | - | - | - | 14,500 | |||||||||||||||||||
| John K. Bell | 18,000 | - | - | - | - | - | 18,000 | |||||||||||||||||||
| Robert J. Toth, Jr. | 16,000 | - | - | - | - | - | 16,000 | |||||||||||||||||||
Risk Management
The Company does not believe risks arising from its compensation policies and practices for its employees are reasonably likely to have a material adverse effect on the Company.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table sets forth the aggregate information of our equity compensation plans in effect as of June 30, 2014:
| Number of securities to be issued upon exercise of outstanding options and rights | Weighted- average exercise price of outstanding options and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in first column | ||||||||||
| Equity compensation plans approved by security holders | - | - | - | |||||||||
| Equity compensation plans not approved by security holders – Amended and Restated 2003 Employee Stock Option Plan | 3,187,214 | 0.96 | 1,149,289 | |||||||||
| Totals | 3,187,214 | 1,149,289 | ||||||||||
| 70 |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information, as of April 1, 2015, with respect to the beneficial ownership of the outstanding common stock by (i) any holder of more than five (5%) percent; (ii) each of the Company’s executive officers and directors; and (iii) the Company’s directors and executive officers as a group. Except as otherwise indicated, each of the stockholders listed below has sole voting and investment power over the shares beneficially owned.
| Name of Beneficial Owner (1) | Common Stock Beneficially Owned |
Percentage of Common Stock (2) | ||||||
| Directors and Officers: | ||||||||
| Jeffrey Bacha | 4,062,027 | (3) | 10.4 | % | ||||
| Dennis Brown | 3,952,542 | (4) | 10.9 | % | ||||
| William Garner | 2,668,541 | (5) | 7.5 | % | ||||
| John K. Bell | 307,000 | (6) | * | |||||
| Scott Praill | 510,000 | (7) | 1.4 | % | ||||
| Robert J. Toth, Jr. | 163,500 | (8) | * | |||||
| Lynda Cranston | 40,000 | (9) | * | |||||
| Erich Mohr | 20,000 | (9) | * | |||||
| All officers and directors as a group (8 persons) | 11,723,610 | 28.5 | % | |||||
| Beneficial owners of more than 5%: | ||||||||
| Valent Technologies LLC | 2,150,000 | (10) | 6.0 | % | ||||
| Howard K. Fuguet (11) | 2,500,000 | 7.1 | % | |||||
* Less than 1%
| (1) | Except as otherwise indicated, the address of each beneficial owner is c/o DelMar Pharmaceuticals, Inc., Suite 720 - 999 West Broadway, Vancouver, British Columbia, Canada V5Z 1K5. |
| (2) | Applicable percentage ownership is based on 35,199,889 shares of common stock outstanding as of April 1, 2015, together with securities exercisable or convertible into shares of common stock within 60 days of April 1, 2015 for each stockholder. Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock that are currently exercisable or exercisable within 60 days of April 1, 2015 are deemed to be beneficially owned by the person holding such securities for the purpose of computing the percentage of ownership of such person, but are not treated as outstanding for the purpose of computing the percentage ownership of any other person. |
| (3) | Includes 3,498,542 shares issuable upon exchange of Exchangeable Shares (including 660,000 shares held in trust) and 500,000 shares issuable upon exercise of options exercisable within 60 days. |
| (4) | Includes 1,650,000 shares held by Valent, 500,000 shares issuable upon exercise of warrants held by Valent, and 500,000 shares issuable upon exercise of options. |
| (5) | Consists of 2,398,541 outstanding shares and 270,000 shares issuable upon exercise of options. Does not include 50,000 shares issuable upon exchange of Exchangeable Shares, which are held in trust for Dr. Garner by Jeffrey Bacha. |
| (6) | Includes 100,000 shares issuable upon exchange of Exchangeable Shares held by Onbelay Capital, Inc., 87,000 shares owned by Onbelay Capital, Inc., and 120,000 shares issuable upon exercise of options. |
| (7) | Includes 400,000 shares issuable upon exercise of options. |
| (8) | Includes 120,000 shares issuable upon exercise of options. |
| (9) | Represents shares issuable upon exercise of vested options. |
| (10) | Includes 500,000 shares issuable upon exercise of warrants. Valent is owned by Dennis Brown, the Company’s Chief Scientific Officer. |
| (11) | The address of the shareholder is Ropes & Gray LLP, 800 Boylston Street, Boston MA, 02199-3600. |
| 71 |
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Certain Relationships and Related Transactions
On September 12, 2010, DelMar (BC) entered into a Patent Assignment Agreement (the “Assignment”) with Valent Technologies LLC pursuant to which Valent assigned to DelMar (BC) its rights to patent applications and the prototype drug product related to VAL-083. In accordance with the Assignment the consideration paid by DelMar (BC) was $250,000 to acquire the prototype drug product. In accordance with the terms of the Assignment, Valent is entitled to receive a future royalty (in the single digits) on certain revenues derived from the development and commercialization of VAL-083. In the event that DelMar (BC) terminates the agreement, DelMar (BC) may be entitled to receive royalties from Valent’s subsequent development of VAL-083 depending on the development milestones DelMar (BC) has achieved prior to the termination of the Assignment. The Assignment has a term (on a country-by-country basis), of the later of ten years or until patent rights covered by the Assignment no longer exist, subject to earlier termination in the event DelMar (BC) breaches its payment obligations and fails to remedy such breach within 60 days, or if either party materially beaches any of its obligations and does not cure such breach within 30 days after receipt of notice thereof.
On January 25, 2013, the Company issued to Valent 1,150,000 shares of common stock, in exchange for Valent agreeing to reduce certain royalties payable to it under the Assignment.
Pursuant to a loan agreement dated February 3, 2011, between DelMar (BC) and Valent, Valent loaned DelMar $250,000 for the purchase of the prototype drug product under the Assignment. The loan is unsecured, bears interest at 3% per year, and is payable on demand. Effective September 30, 2014, the Company entered into and closed an agreement with Valent to exchange its loan, including accrued interest to September 30, 2014, with Valent for 278,530 shares of preferred stock of the Company. The preferred stock has an annual 3% dividend.
In addition, under the terms of the Assignment, DelMar issued to Valent warrants to acquire 500,000 common shares at an exercise price of CDN $0.50 per upon the completion of the financing transaction that closed in February 2012.
On April 30, 2012, DelMar (BC) issued 500,000 common shares in partial settlement of accounts payable in the amount of CDN $250,000 (U.S. $253,050) owed to Valent.
Valent, which is owned by Dr. Dennis Brown, our Chief Scientific Officer, leases facilities in California and we have access to such facilities pursuant to an informal unwritten arrangement with Valent.
Included in accounts payable at December 31, 2014 is an aggregate amount of $37,659 (June 30, 2014 - $54,960) owed to the Company’s officers and directors for fees and expenses. The Company pays related party payables incurred for fees and expenses under normal commercial terms.
Included in related party payables at December 31, 2013 is an amount of $44,007 relating to clinical development costs incurred by Valent on behalf of the Company.
For additional information see note 10 to the financial statements.
Director Independence
William J. Garner, John K. Bell, Robert J. Toth, Jr., Lynda Cranston and Erich Mohr are independent as that term is defined under the Nasdaq Marketplace Rules.
Common Stock
The Company’s authorized capital stock consists of 200,000,000 shares of common stock, par value of $0.001 per share, and 5,000,000 shares of preferred stock, par value $0.001 per share, of which 1 share has been designated Special Voting Preferred Stock. As of April 1, 2015, there were 35,199,889 shares of the Company’s common stock, 1 share of Special Voting Preferred Stock, and 278,530 shares of Series A Preferred Stock issued and outstanding.
Holders of the Company’s common stock are entitled to one vote for each share on all matters submitted to a stockholder vote. Holders of common stock do not have cumulative voting rights. Therefore, holders of a majority of the shares of common stock voting for the election of directors can elect all of the directors. Holders of the Company’s common stock representing a majority of the voting power of the Company’s capital stock issued, outstanding and entitled to vote, represented in person or by proxy, are necessary to constitute a quorum at any meeting of stockholders. A vote by the holders of a majority of the Company’s outstanding shares is required to effectuate certain fundamental corporate changes such as liquidation, merger or an amendment to the Company’s certificate of incorporation.
| 72 |
Holders of the Company’s common stock are entitled to share in all dividends that the board of directors, in its discretion, declares from legally available funds. In the event of a liquidation, dissolution or winding up, each outstanding share entitles its holder to participate pro rata in all assets that remain after payment of liabilities and after providing for each class of stock, if any, having preference over the common stock. The Company’s common stock has no pre-emptive rights, no conversion rights and there are no redemption provisions applicable to the Company’s common stock.
Preferred Stock
The Company’s articles of incorporation authorize the issuance of 5,000,000 shares of “blank check” preferred stock, par value $0.001 per share, in one or more series, subject to any limitations prescribed by law, without further vote or action by the stockholders. Each such series of preferred stock shall have such number of shares, designations, preferences, voting powers, qualifications, and special or relative rights or privileges as shall be determined by our board of directors, which may include, among others, dividend rights, voting rights, liquidation preferences, conversion rights and preemptive rights.
Pursuant to the Certificate of Designation of the Company’s Special Voting Preferred Stock, one share of the Company’s blank check preferred stock has been designated as Special Voting Preferred Stock. The Special Voting Preferred Stock votes as a single class with the common stock and is entitled to a number of votes equal to the number of Exchangeable Shares of Exchangeco outstanding as of the applicable record date (i) that are not owned by the Company or any affiliated companies and (ii) as to which the holder has received voting instructions from the holders of such Exchangeable Shares in accordance with the Trust Agreement.
The Special Voting Preferred Stock is not entitled to receive any dividends or to receive any assets of the Company upon any liquidation, and is not convertible into common stock of the Company.
The voting rights of the Special Voting Preferred Stock will terminate pursuant to and in accordance with the Trust Agreement. The Special Voting Preferred Stock will be automatically cancelled at such time as the share of Special Voting Preferred Stock has no votes attached to it.
Common stock issued upon exchange of Exchangeable Shares and upon the exercise of Exchange Agreement Warrants held by Canadian residents may be subject to statutory hold periods in accordance with applicable Canadian securities laws.
Pursuant to the Company’s Certificate of Designation of Series A Preferred Stock, the Company designated 278,530 shares of preferred stock as Series A Preferred Stock. The shares of Series A Preferred Stock have a stated value of $1.00 per share and are not convertible into common stock. The holder of the Series A Preferred Stock will be entitled to dividends at the rate of 3% of the Stated Value per year, payable quarterly in arrears. Upon any liquidation of the Company, the holder of the Series A Preferred Stock will be entitled to be paid, out of any assets of the Company available for distribution to stockholders, the Stated Value of the shares of Series A Preferred Stock held by such holder, plus any accrued but unpaid dividends thereon, prior to any payments being made with respect to the common stock.
Warrants
The following summary of certain terms and provisions of the warrants offered hereby is not complete and is subject to, and qualified in its entirety by the provisions of the form of the warrant, which is filed as an exhibit to the registration statement of which this prospectus is a part of. Prospective investors should carefully review the terms and provisions set forth in the form of warrant.
The warrants will be issued in book-entry form pursuant to a warrant agency agreement between us and Island Stock Transfer, as warrant agent.
Exercisability. Holders may exercise the warrants beginning ____ after the closing of this offering and at any time up to the date that is the ____ anniversary of the closing of the offering. The Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice accompanied by payment in full for the number of shares of Common Stock purchased upon such exercise.
The Warrants provide for cashless exercise, at the holder’s option, in the event that, any time after the earlier of (i) the one year anniversary of the offering and (ii) the completion of any then-applicable holding period required by Rule 144, a registration statement covering shares of common stock underlying the Warrants is not available for the resale of such shares of common stock underlying the Warrants. In such event, the holder may, in its sole discretion, exercise the warrant in whole or in part and, in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, elect instead to receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the Warrant.
| 73 |
Exercise Price. The initial exercise price per share of common stock purchasable upon exercise of the warrants is $ per share. The exercise price and the number of shares issuable upon exercise are subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock.
Fundamental Transactions. If, at any time while the warrants are outstanding, (1) we consolidate or merge with or into another corporation and we are not the surviving corporation, (2) we sell, lease, license, assign, transfer, convey or otherwise dispose of all or substantially all of our assets, (3) any purchase offer, tender offer or exchange offer (whether by us or another individual or entity) is completed pursuant to which holders of our shares of common stock are permitted to sell, tender or exchange their shares of common stock for other securities, cash or property and has been accepted by the holders of 50% or more of our outstanding shares of common stock, (4) we effect any reclassification or recapitalization of our shares of common stock or any compulsory share exchange pursuant to which our shares of common stock are converted into or exchanged for other securities, cash or property, or (5) we consummate a stock or share purchase agreement or other business combination with another person or entity whereby such other person or entity acquires more than 50% of our outstanding shares of common stock, each referred to as a Fundamental Transaction, then upon any subsequent exercise of the warrants, the holders thereof will have the right to receive the same amount and kind of securities, cash or property as it would have been entitled to receive upon the occurrence of such Fundamental Transaction if it had been, immediately prior to such Fundamental Transaction, the holder of the number of shares of common stock then issuable upon exercise of the Warrant, and any additional consideration payable as part of the Fundamental Transaction.
Transferability. Subject to applicable laws, the warrants may be transferred at the option of the holders upon surrender of the warrants to us together with the appropriate instruments of transfer.
Rights as a Stockholder. Except as otherwise provided in the warrants or by virtue of such holder's ownership of shares of our common stock, the holder of a warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the warrant.
Stock Options
As of April 1, 2015, we had outstanding options to purchase an aggregate of 3,595,000 shares of common stock with a weighted average exercise price of approximately $0.94 per share.
Other Warrants
As of April 1, 2015 we had outstanding warrants to purchase an aggregate of 13,472,870 shares of common stock, with a weighted average exercise price of approximately $0.92 per share.
Exchangeable Shares
As of April 1, 2015, there were 4,256,042 shares of our common stock issuable upon exchange of Exchangeable Shares.
Underwriter’s Warrants
Please see “Underwriting-Underwriters’ Warrants” for a description of the warrants we have agreed to issue to the underwriters in this offering, subject to the completion of the offering.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Island Stock Transfer located at 15500 Roosevelt Boulevard, Suite 301, Clearwater, FL 33760, phone number 727-289-0010.
Listing
Our shares of common stock are quoted on the OTCQX under the symbol “DMPI”.
| 74 |
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES ACT LIABILITIES
Neither our Articles of Incorporation nor Bylaws prevent us from indemnifying our officers, directors and agents to the extent permitted under the Nevada Revised Statute ("NRS"). NRS Section 78.7502 provides that a corporation shall indemnify any director, officer, employee or agent of a corporation against expenses, including attorneys' fees, actually and reasonably incurred by him in connection with any the defense to the extent that a director, officer, employee or agent of a corporation has been successful on the merits or otherwise in defense of any action, suit or proceeding referred to Section 78.7502(1) or 78.7502(2), or in defense of any claim, issue or matter therein.
NRS 78.7502(1) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative, except an action by or in the right of the corporation, by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses, including attorneys' fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with the action, suit or proceeding if he: (a) is not liable pursuant to NRS 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful.
NRS Section 78.7502(2) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses, including amounts paid in settlement and attorneys' fees actually and reasonably incurred by him in connection with the defense or settlement of the action or suit if he: (a) is not liable pursuant to NRS 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation. Indemnification may not be made for any claim, issue or matter as to which such a person has been adjudged by a court of competent jurisdiction, after exhaustion of all appeals there from, to be liable to the corporation or for amounts paid in settlement to the corporation, unless and only to the extent that the court in which the action or suit was brought or other court of competent jurisdiction determines upon application that in view of all the circumstances of the case, the person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
NRS Section 78.747 provides that except as otherwise provided by specific statute, no director or officer of a corporation is individually liable for a debt or liability of the corporation, unless the director or officer acts as the alter ego of the corporation. The court as a matter of law must determine the question of whether a director or officer acts as the alter ego of a corporation.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed hereby in the Securities Act and we will be governed by the final adjudication of such issue.
| 75 |
Maxim Group LLC (“Maxim”), Roth Capital Partners, LLC (“Roth”), are acting as the joint book-running managers, and National Securities Corporation is acting as the co-manager of this offering. We have entered into an underwriting agreement dated , 2015 with the underwriters. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to each underwriter named below and each underwriter named below has severally agreed to purchase, at the public offering price less the underwriting discounts and commissions set forth on the cover page of this prospectus, the number of shares of common stock and warrants listed next to its name in the following table:
| Name of Underwriter | Number of Shares of Common Stock | Number of Warrants | ||||||
| Maxim Group LLC | ||||||||
| Roth Capital Partners, LLC | ||||||||
National Securities Corporation | ||||||||
| Total | ||||||||
The underwriters are committed to purchase all the shares of common stock and warrants offered by us other than those covered by the option to purchase additional shares and/or warrants described below, if they purchase any shares and warrants. The obligations of the underwriters may be terminated upon the occurrence of certain events specified in the underwriting agreement. Furthermore, pursuant to the underwriting agreement, the underwriters’ obligations are subject to customary conditions, representations and warranties contained in the underwriting agreement, such as receipt by the underwriters of officers’ certificates and legal opinions.
We have agreed to indemnify the underwriters against specified liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect thereof.
The underwriters are offering the shares and warrants, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel and other conditions specified in the underwriting agreement. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 45 days after the date of this prospectus, permits the underwriters to purchase a maximum of additional shares and/or warrants (15% of the shares and/or warrants sold in this offering) from us to cover over-allotments, if any. If the underwriters exercise all or part of this option, they will purchase shares and/or warrants covered by the option at the public offering price that appears on the cover page of this prospectus, less the underwriting discount. If this option is exercised in full, the total price to the public will be $ and the total net proceeds, before expenses, to us will be $ .
Discounts and Commissions
The following table shows the public offering price, underwriting discount and proceeds, before expenses, to us. The information assumes either no exercise or full exercise by the underwriters of their over-allotment option.
| Per Share of Common Stock | Per Warrant | Total | Total | |||||||||||||||||||||
| Without Over-allotment Option | With Over-allotment Option | Without Over-allotment Option | With Over-allotment Option | Without Over-allotment Option | With Over-allotment Option | |||||||||||||||||||
| Public offering price | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| Underwriting discounts and commissions payable by us (8%) | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| Proceeds, before expenses to us | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 76 |
The underwriters propose to offer the shares and warrants offered by us to the public at the public offering price set forth on the cover of this prospectus. In addition, the underwriters may offer some of the shares to other securities dealers at such price less a concession of $ per share. If all of the shares and warrants offered by us are not sold at the public offering price, the underwriters may change the offering price and other selling terms by means of a supplement to this prospectus.
We have paid an expense deposit of $40,000 to Maxim, which will be applied against the out-of-pocket accountable expenses that will be paid by us to Maxim in connection with this offering. The underwriting agreement, however, provides that in the event the offering is terminated, Maxim shall return any portion of the $40,000 out-of-pocket expense deposit paid to it to the extent such expenses are not actually incurred in accordance with FINRA Rule 5110(f)(2)(C).
We have also agreed to pay the underwriters’ expenses relating to the offering, including (a) all reasonable fees, expenses and disbursements relating to background checks of our officers and directors; (b) all fees incurred in clearing this offering with FINRA; (c) the costs associated with the underwriters’ use of book-building, prospectus tracking and compliance software for this offering; (d) the costs of all mailing and printing of the underwriting documents, registration statements, prospectuses and all amendments, supplements and exhibits thereto and as many preliminary and final prospectuses as the underwriters may reasonably deem necessary; (e) all fees and expenses and disbursements relating to the registration or qualification of the securities under the “blue sky” securities laws of such states and other jurisdictions as the underwriters may reasonably designate for an initial payment of $15,000 to such counsel and a payment to cover all filing fees upon the commencement of “blue sky” work by such counsel, with the balance of such counsel fees and expenses to be due on the closing of the offering; (f) the fees and expenses of the underwriters’ legal counsel and (g) the underwriters’ actual accountable “road show” expenses for the offering. The maximum amount of fees, costs and expenses incurred by the underwriters (inclusive of legal fees, disbursements and costs) that we shall be responsible for shall not exceed $110,000, without our prior written authorization.
We estimate that the total expenses of the offering payable by us, excluding the total underwriting discount and expense reimbursement, will be approximately $ .
Discretionary Accounts
The underwriters do not intend to confirm sales of the securities offered hereby to any accounts over which they have discretionary authority.
Lock-Up Agreements
Pursuant to certain “lock-up” agreements, (a) our executive officers and directors as of the pricing date of the offering, will agree, subject to certain exceptions, not to offer, issue, sell, contract to sell, encumber, grant any option for the sale of or otherwise dispose of any securities of the company without the prior written consent of the underwriters, for a period of 90 days from the date of the pricing of the offering, and (b) we, and any successor, will agree, subject to certain exceptions, not to for a period of 90 days from the date of the pricing of the offering (1) offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, directly or indirectly, any shares of our capital stock; (2) file or caused to be filed any registration statement with the SEC relating to the offering of any shares of our capital stock or any securities convertible into or exercisable or exchangeable for shares of our capital stock; or (3) enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of our capital stock, whether any such transaction described in (1), (2), or (3) above is to be settled by delivery of shares of our capital stock or such other securities, in cash or otherwise.
This lock-up provision applies to common stock and to securities convertible into or exchangeable or exercisable for common stock. It also applies to common stock owned now or acquired later by the person executing the agreement or for which the person executing the agreement later acquires the power of disposition. The exceptions permit, among other things, (1) the issuance by us of stock options pursuant to our existing stock incentive plans, or (2) the issuance of common stock upon the exercise of outstanding stock options and warrants.
Underwriters’ Warrants
We have agreed to issue to the underwriters warrants, or the underwriters’ warrants, to purchase up to a total of shares of common stock (8% of the shares of common stock sold in this offering (excluding the over-allotment and the warrants or the common stock underlying the warrants)). The warrants are exercisable at a per share price equal to $______ , at any time, and from time to time, in whole or in part, during the five -year period commencing six months from the effective date of the offering, which period shall not extend further than five years from the effective date of the offering in compliance with FINRA Rule 5110(f)(2)(H)(i). The warrants have been deemed compensation by FINRA and are therefore subject to a 180-day lock-up pursuant to Rule 5110(g)(1) of FINRA. The underwriters (or permitted assignees under Rule 5110(g)(1)) will not sell, transfer, assign, pledge, or hypothecate these warrants or the securities underlying these warrants, nor will they engage in any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the warrants or the underlying securities for a period of 180 days from the effective date of the offering. We will bear all fees and expenses attendant to registering the securities issuable on exercise of the warrants other than underwriting commissions incurred and payable by the holders. The exercise price and number of shares issuable upon exercise of the warrants may be adjusted in certain circumstances including in the event of a stock dividend, extraordinary cash dividend or our recapitalization, reorganization, merger or consolidation. However, the warrant exercise price or underlying shares will not be adjusted for issuances of shares of common stock at a price below the warrant exercise price.
| 77 |
Right of First Refusal
Until twelve (12) months from the effective date of the offering, Maxim and Roth shall have a right of first refusal to act as joint book-running underwriters with at least 80% of the economics (to be split equally between Maxim and Roth) or at least 40% of the economics if only one of the underwriters exercises its right of first refusal, for any and all future public and private equity and public debt offerings (excluding commercial bank debt) during such twelve (12) month period. Maxim and Roth will not have more than one opportunity to waive or terminate the right of first refusal in consideration of any payment or fee.
Electronic Offer, Sale and Distribution of Shares and Warrants
A prospectus in electronic format may be made available on the websites maintained by one or more of the underwriters or selling group members, if any, participating in this offering and one or more of the underwriters participating in this offering may distribute prospectuses electronically. The underwriters may agree to allocate a number of shares and warrants to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make internet distributions on the same basis as other allocations. Other than the prospectus in electronic format, the information on these websites is not part of, nor incorporated by reference into, this prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or any underwriter in its capacity as underwriter, and should not be relied upon by investors.
Stabilization
In connection with this offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate-covering transactions, penalty bids and purchases to cover positions created by short sales.
| ● | Stabilizing transactions permit bids to purchase shares so long as the stabilizing bids do not exceed a specified maximum, and are engaged in for the purpose of preventing or retarding a decline in the market price of the shares while the offering is in progress. |
| ● | Over-allotment transactions involve sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase. This creates a syndicate short position which may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any short position by exercising their over-allotment option and/or purchasing shares in the open market. |
| ● | Syndicate covering transactions involve purchases of shares in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared with the price at which they may purchase shares through exercise of the over-allotment option. If the underwriters sell more shares than could be covered by exercise of the over-allotment option and, therefore, have a naked short position, the position can be closed out only by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that after pricing there could be downward pressure on the price of the shares in the open market that could adversely affect investors who purchase in the offering. |
| ● | Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the shares originally sold by that syndicate member are purchased in stabilizing or syndicate covering transactions to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our shares or common stock or preventing or retarding a decline in the market price of our shares or common stock. As a result, the price of our common stock in the open market may be higher than it would otherwise be in the absence of these transactions. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on the over-the-counter market or otherwise and, if commenced, may be discontinued at any time.
| 78 |
Passive market making
In connection with this offering, underwriters and selling group members may engage in passive market making transactions in our common stock on the OTCQB in accordance with Rule 103 of Regulation M under the Exchange Act, during a period before the commencement of offers or sales of the shares and extending through the completion of the distribution. A passive market maker must display its bid at a price not in excess of the highest independent bid of that security. However, if all independent bids are lowered below the passive market maker’s bid, then that bid must then be lowered when specified purchase limits are exceeded.
Other Relationships
Except as disclosed in this prospectus, we have no present arrangements with any of the underwriters for any further services.
Offer restrictions outside the United States
Other than in the United States, no action has been taken by us or the underwriter that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to the offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Australia
This prospectus is not a disclosure document under Chapter 6D of the Australian Corporations Act, has not been lodged with the Australian Securities and Investments Commission and does not purport to include the information required of a disclosure document under Chapter 6D of the Australian Corporations Act. Accordingly, (i) the offer of the securities under this prospectus is only made to persons to whom it is lawful to offer the securities without disclosure under Chapter 6D of the Australian Corporations Act under one or more exemptions set out in section 708 of the Australian Corporations Act, (ii) this prospectus is made available in Australia only to those persons as set forth in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that by accepting this offer, the offeree represents that the offeree is such a person as set forth in clause (i) above, and, unless permitted under the Australian Corporations Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer to the offeree under this prospectus.
China
The information in this document does not constitute a public offer of the securities, whether by way of sale or subscription, in the People’s Republic of China (excluding, for purposes of this paragraph, Hong Kong Special Administrative Region, Macau Special Administrative Region and Taiwan). The securities may not be offered or sold directly or indirectly in the PRC to legal or natural persons other than directly to “qualified domestic institutional investors.”
European Economic Area — Belgium, Germany, Luxembourg and Netherlands
The information in this document has been prepared on the basis that all offers of securities will be made pursuant to an exemption under the Directive 2003/71/EC (“Prospectus Directive”), as implemented in Member States of the European Economic Area (each, a “Relevant Member State”), from the requirement to produce a prospectus for offers of securities.
An offer to the public of securities has not been made, and may not be made, in a Relevant Member State except pursuant to one of the following exemptions under the Prospectus Directive as implemented in that Relevant Member State:
| (a) | to legal entities that are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
| (b) | to any legal entity that has two or more of (i) an average of at least 250 employees during its last fiscal year; (ii) a total balance sheet of more than €43,000,000 (as shown on its last annual unconsolidated or consolidated financial statements) and (iii) an annual net turnover of more than €50,000,000 (as shown on its last annual unconsolidated or consolidated financial statements); |
| (c) | to fewer than 100 natural or legal persons (other than qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive) subject to obtaining our prior consent or the prior consent of any underwriter for any such offer; or |
| (d) | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall result in a requirement for the publication by us of a prospectus pursuant to Article 3 of the Prospectus Directive. |
| 79 |
France
This document is not being distributed in the context of a public offering of financial securities (offre au public de titres financiers) in France within the meaning of Article L.411-1 of the French Monetary and Financial Code (Code monétaire et financier) and Articles 211-1 et seq. of the General Regulation of the French Autorité des marchés financiers (“AMF”). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France.
This document and any other offering material relating to the securities have not been, and will not be, submitted to the AMF for approval in France and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in France.
Such offers, sales and distributions have been and shall only be made in France to (i) qualified investors (investisseurs qualifiés) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-1 to D.411-3, D. 744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation and/or (ii) a restricted number of non-qualified investors (cercle restreint d’investisseurs) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-4, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation. Pursuant to Article 211-3 of the General Regulation of the AMF, investors in France are informed that the securities cannot be distributed (directly or indirectly) to the public by the investors otherwise than in accordance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 to L.621-8-3 of the French Monetary and Financial Code.
Ireland
The information in this document does not constitute a prospectus under any Irish laws or regulations and this document has not been filed with or approved by any Irish regulatory authority as the information has not been prepared in the context of a public offering of securities in Ireland within the meaning of the Irish Prospectus (Directive 2003/71/EC) Regulations 2005 (the “Prospectus Regulations”). The securities have not been offered or sold, and will not be offered, sold or delivered directly or indirectly in Ireland by way of a public offering, except to (i) qualified investors as defined in Regulation 2(l) of the Prospectus Regulations and (ii) fewer than 100 natural or legal persons who are not qualified investors.
Israel
The securities offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority (the ISA), or ISA, nor have such securities been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with the offering or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the securities being offered. Any resale in Israel, directly or indirectly, to the public of the securities offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
Italy
The offering of the securities in the Republic of Italy has not been authorized by the Italian Securities and Exchange Commission (Commissione Nazionale per le Società e la Borsa, “CONSOB”) pursuant to the Italian securities legislation and, accordingly, no offering material relating to the securities may be distributed in Italy and such securities may not be offered or sold in Italy in a public offer within the meaning of Article 1.1(t) of Legislative Decree No. 58 of 24 February 1998 (“Decree No. 58”), other than:
| ● | to Italian qualified investors, as defined in Article 100 of Decree no. 58 by reference to Article 34-ter of CONSOB Regulation no. 11971 of 14 May 1999 (“Regulation no. 1197l”) as amended (“Qualified Investors”); and |
| ● | in other circumstances that are exempt from the rules on public offer pursuant to Article 100 of Decree No. 58 and Article 34-ter of Regulation No. 11971 as amended. |
Any offer, sale or delivery of the securities or distribution of any offer document relating to the securities in Italy (excluding placements where a Qualified Investor solicits an offer from the issuer) under the paragraphs above must be:
| ● | made by investment firms, banks or financial intermediaries permitted to conduct such activities in Italy in accordance with Legislative Decree No. 385 of 1 September 1993 (as amended), Decree No. 58, CONSOB Regulation No. 16190 of 29 October 2007 and any other applicable laws; and |
| ● | in compliance with all relevant Italian securities, tax and exchange controls and any other applicable laws. |
| 80 |
Any subsequent distribution of the securities in Italy must be made in compliance with the public offer and prospectus requirement rules provided under Decree No. 58 and the Regulation No. 11971 as amended, unless an exception from those rules applies. Failure to comply with such rules may result in the sale of such securities being declared null and void and in the liability of the entity transferring the securities for any damages suffered by the investors.
Japan
The securities have not been and will not be registered under Article 4, paragraph 1 of the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948), as amended (the “FIEL”) pursuant to an exemption from the registration requirements applicable to a private placement of securities to Qualified Institutional Investors (as defined in and in accordance with Article 2, paragraph 3 of the FIEL and the regulations promulgated thereunder). Accordingly, the securities may not be offered or sold, directly or indirectly, in Japan or to, or for the benefit of, any resident of Japan other than Qualified Institutional Investors. Any Qualified Institutional Investor who acquires securities may not resell them to any person in Japan that is not a Qualified Institutional Investor, and acquisition by any such person of securities is conditional upon the execution of an agreement to that effect.
Portugal
This document is not being distributed in the context of a public offer of financial securities (oferta pública de valores mobiliários) in Portugal, within the meaning of Article 109 of the Portuguese Securities Code (Código dos Valores Mobiliários). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in Portugal. This document and any other offering material relating to the securities have not been, and will not be, submitted to the Portuguese Securities Market Commission (Comissăo do Mercado de Valores Mobiliários) for approval in Portugal and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in Portugal, other than under circumstances that are deemed not to qualify as a public offer under the Portuguese Securities Code. Such offers, sales and distributions of securities in Portugal are limited to persons who are “qualified investors” (as defined in the Portuguese Securities Code). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Sweden
This document has not been, and will not be, registered with or approved by Finansinspektionen (the Swedish Financial Supervisory Authority). Accordingly, this document may not be made available, nor may the securities be offered for sale in Sweden, other than under circumstances that are deemed not to require a prospectus under the Swedish Financial Instruments Trading Act (1991:980) (Sw. lag (1991:980) om handel med finansiella instrument). Any offering of securities in Sweden is limited to persons who are “qualified investors” (as defined in the Financial Instruments Trading Act). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering material relating to the securities may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering material relating to the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority (FINMA). This document is personal to the recipient only and not for general circulation in Switzerland.
United Arab Emirates
Neither this document nor the securities have been approved, disapproved or passed on in any way by the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates, nor have we received authorization or licensing from the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates to market or sell the securities within the United Arab Emirates. This document does not constitute and may not be used for the purpose of an offer or invitation. No services relating to the securities, including the receipt of applications and/or the allotment or redemption of such shares, may be rendered within the United Arab Emirates by us. No offer or invitation to subscribe for securities is valid or permitted in the Dubai International Financial Centre.
| 81 |
United Kingdom
Neither the information in this document nor any other document relating to the offer has been delivered for approval to the Financial Services Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as amended (“FSMA”)) has been published or is intended to be published in respect of the securities. This document is issued on a confidential basis to “qualified investors” (within the meaning of section 86(7) of FSMA) in the United Kingdom, and the securities may not be offered or sold in the United Kingdom by means of this document, any accompanying letter or any other document, except in circumstances which do not require the publication of a prospectus pursuant to section 86(1) FSMA. This document should not be distributed, published or reproduced, in whole or in part, nor may its contents be disclosed by recipients to any other person in the United Kingdom.
Any invitation or inducement to engage in investment activity (within the meaning of section 21 of FSMA) received in connection with the issue or sale of the securities has only been communicated or caused to be communicated and will only be communicated or caused to be communicated in the United Kingdom in circumstances in which section 21(1) of FSMA does not apply to us.
In the United Kingdom, this document is being distributed only to, and is directed at, persons (i) who have professional experience in matters relating to investments falling within Article 19(5) (investment professionals) of the Financial Services and Markets Act 2000 (Financial Promotions) Order 2005 (“FPO”), (ii) who fall within the categories of persons referred to in Article 49(2)(a) to (d) (high net worth companies, unincorporated associations, etc.) of the FPO or (iii) to whom it may otherwise be lawfully communicated (together “relevant persons”). The investments to which this document relates are available only to, and any invitation, offer or agreement to purchase will be engaged in only with, relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
Canada
This prospectus is not and under no circumstances is to be construed as a prospectus, advertisement or public offering of our common stock or warrants under Canadian securities laws. The securities being registered hereunder have not been and will not be qualified for offer or sale in Canada under applicable Canadian securities laws. The securities being registered hereunder are not being offered and may not be offered or sold, directly or indirectly, in Canada or to or for the account of any resident of Canada in contravention of the securities laws of any province or territory thereof.
The validity of the securities being offered by this prospectus has been passed upon for us by Sichenzia Ross Friedman Ference LLP, New York, New York. Certain legal matters in connection with this offering will be passed upon for the underwriters by Loeb & Loeb LLP, New York, New York.
The balance sheets of DelMar Pharmaceuticals, Inc. as of June 30, 2014, December 31, 2013 and December 31, 2012 and the related statements of operations and comprehensive loss, changes in stockholders’ equity (deficiency), and cash flows for the six months ended June 30, 2014 and 2013 and for the years ended December 31, 2012 and December 31, 2013 included in this registration statement on Form S-1 have been so included in reliance on the report of PricewaterhouseCoopers LLP, an independent registered public accounting firm, given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We are a reporting company and file annual, quarterly and special reports, and other information with the Securities and Exchange Commission. Copies of the reports and other information may be read and copied at the SEC's Public Reference Room at 100 F Street NE, Washington, D.C. 20549. You can request copies of such documents by writing to the SEC and paying a fee for the copying cost. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a web site at http://www.sec.gov that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC.
This prospectus is part of a registration statement on Form S-1 that we filed with the SEC. Certain information in the registration statement has been omitted from this prospectus in accordance with the rules and regulations of the SEC. We have also filed exhibits and schedules with the registration statement that are excluded from this prospectus. For further information you may:
| ● | read a copy of the registration statement, including the exhibits and schedules, without charge at the SEC's Public Reference Room; or |
| ● | obtain a copy from the SEC upon payment of the fees prescribed by the SEC. |
| 82 |
DelMar Pharmaceuticals, Inc.
Consolidated Condensed Interim Financial Statements
(Unaudited)
For the six months ended December 31, 2014
(expressed in US dollars unless otherwise noted)
| F-1 |
|
|
DelMar Pharmaceuticals, Inc.
Consolidated Condensed Interim Balance Sheets
(Unaudited)
(expressed in US dollars unless otherwise noted)
Note | December 31, 2014 $ | June 30, 2014 $ | ||||||||||
| Assets | ||||||||||||
| Current assets | ||||||||||||
| Cash and cash equivalents | 3,958,439 | 4,759,711 | ||||||||||
| Taxes and other receivables | 14,082 | 9,572 | ||||||||||
| Prepaid expenses | 155,857 | 234,627 | ||||||||||
| 4,128,378 | 5,003,910 | |||||||||||
| Liabilities | ||||||||||||
| Current liabilities | ||||||||||||
| Accounts payable and accrued liabilities | 256,527 | 244,906 | ||||||||||
| Related party payables | 4 | 37,659 | 54,960 | |||||||||
| 294,186 | 299,866 | |||||||||||
| Loan payable to Valent | 3 | - | 276,439 | |||||||||
| Stock option liability | 6 | 180,350 | 217,759 | |||||||||
| Derivative liability | 5 | 1,567,291 | 3,329,367 | |||||||||
| 2,041,827 | 4,123,431 | |||||||||||
| Stockholders’ Equity | ||||||||||||
| Preferred stock | ||||||||||||
| Authorized | ||||||||||||
| 5,000,000 shares, $0.001 par value | ||||||||||||
| Issued and outstanding | ||||||||||||
| 278,530 Series A shares at December 31, 2014 | ||||||||||||
| (June 30, 2014 - none) | 3 | 278,530 | - | |||||||||
| 1 special voting share at December 31, 2014 | ||||||||||||
| (June 30, 2014 - 1) | 6 | - | - | |||||||||
| Common stock | ||||||||||||
| Authorized | ||||||||||||
| 200,000,000 shares, $0.001 par value | ||||||||||||
| Issued and outstanding | ||||||||||||
| 38,580,306 at December 31, 2014 (June 30, 2014 – 35,992,343) | 6 | 38,580 | 35,992 | |||||||||
| Additional paid-in capital | 6 | 16,625,081 | 13,286,278 | |||||||||
| Warrants | 6 | 6,187,805 | 6,200,445 | |||||||||
| Accumulated deficit | (21,064,623 | ) | (18,663,414 | ) | ||||||||
| Accumulated other comprehensive income | 21,178 | 21,178 | ||||||||||
| 2,086,551 | 880,479 | |||||||||||
| 4,128,378 | 5,003,910 | |||||||||||
| Nature of operations and liquidity risk (note 1) | ||||||||||||
| Subsequent event (note 8) |
| F-2 |
DelMar Pharmaceuticals, Inc.
Consolidated Condensed Interim Statement of Loss and Comprehensive Loss
(Unaudited)
(expressed in US dollars unless otherwise noted)
|
|
Note
| Three months ended December 31, 2014 $ | Three months ended December 31, 2013 $ | Six months ended December 31, 2014 $ | Six months ended December 31, 2013 $ | ||||||||||||||||
| Expenses | ||||||||||||||||||||
| Research and development | 612,169 | 566,060 | 1,283,796 | 1,126,295 | ||||||||||||||||
| General and administrative | 656,229 | 636,182 | 1,101,229 | 1,377,550 | ||||||||||||||||
| 1,268,398 | 1,202,242 | 2,385,025 | 2,503,845 | |||||||||||||||||
| Other (income) loss | ||||||||||||||||||||
| Change in fair value of derivative liability | 5 | (435,200 | ) | (372,487 | ) | (66,606 | ) | (8,466,826 | ) | |||||||||||
| Change in fair value of derivative liability due to change in warrant terms | 5 | 143,532 | - | (23,658 | ) | - | ||||||||||||||
| Loss on exchange of warrants | 5 | 92,843 | - | 92,843 | - | |||||||||||||||
| Foreign exchange loss | 7,295 | 34,797 | 9,686 | 31,963 | ||||||||||||||||
| Interest expense | - | 2,044 | 2,091 | 4,073 | ||||||||||||||||
| Interest income | (109 | ) | (620 | ) | (261 | ) | (1,311 | ) | ||||||||||||
| (191,639 | ) | (336,266 | ) | 14,095 | (8,432,101 | ) | ||||||||||||||
| Net and comprehensive loss (income) for the period | 1,076,759 | 865,976 | 2,399,120 | (5,928,256 | ) | |||||||||||||||
| Basic loss (income) per share | 0.03 | 0.03 | 0.06 | (0.19 | ) | |||||||||||||||
| Diluted loss (income) per share | 0.03 | 0.03 | 0.06 | 0.00 | ||||||||||||||||
| Basic weighted average number of shares | 37,798,183 | 31,523,732 | 37,125,074 | 31,477,137 | ||||||||||||||||
| Diluted weighted average number of shares | 37,798,183 | 31,523,732 | 37,125,074 | 41,742,401 | ||||||||||||||||
| F-3 |
DelMar Pharmaceuticals, Inc.
Consolidated Condensed Interim Statement of Cash Flows
(Unaudited)
(expressed in US dollars unless otherwise noted)
Six months ended December 31, | ||||||||
2014 $ | 2013 $ | |||||||
| Cash flows from operating activities | ||||||||
| (Loss) income for the period | (2,399,120 | ) | 5,928,256 | |||||
| Items not affecting cash | ||||||||
| Accrued interest | 2,091 | 4,073 | ||||||
| Change in fair value of derivative liability | (66,606 | ) | (8,466,826 | ) | ||||
| Change in fair value of derivative liability due to change in warrant terms | (23,658 | ) | - | |||||
| Loss on exchange of warrants | 92,843 | - | ||||||
| Warrants issued for services | - | 124,020 | ||||||
| Share-based compensation | 260,510 | 626,279 | ||||||
| (2,133,940 | ) | (1,784,198 | ) | |||||
| Changes in non-cash working capital | ||||||||
| Taxes and other receivables | (4,510 | ) | 5,832 | |||||
| Prepaid expenses | 78,770 | 50,379 | ||||||
| Accounts payable and accrued liabilities | 11,621 | (290,770 | ) | |||||
| Related party payables | (17,301 | ) | (127,432 | ) | ||||
| 68,580 | (361,991 | ) | ||||||
| (2,065,360 | ) | (2,146,189 | ) | |||||
| Cash flows from financing activities | ||||||||
| Net proceeds from the exercise of warrants | 1,266,177 | - | ||||||
| Series A preferred stock dividend | (2,089 | ) | - | |||||
| 1,264,088 | - | |||||||
| Increase in cash and cash equivalents | (801,272 | ) | (2,146,189 | ) | ||||
| Cash and cash equivalents - beginning of period | 4,759,711 | 6,282,992 | ||||||
| Cash and cash equivalents - end of period | 3,958,439 | 4,136,803 | ||||||
| Supplementary information | ||||||||
| Issuance of preferred shares for the settlement of the loan payable to Valent (note 3) | 278,530 | - | ||||||
| Reclassification of derivative liability to equity upon the exercise of Investor Warrants (note 5) | 391,422 | - | ||||||
| Reclassification of derivative liability to equity upon the exchange of Investor Warrants (note 5) | 305,112 | - | ||||||
| Reclassification of derivative liability to equity upon the amendment of Dividend Warrants (note 5) | 975,278 | - | ||||||
| Reclassification of stock option liability upon the forfeiture of stock options (note 6) | 38,038 | - | ||||||
| F-4 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
| 1 | Nature of operations and liquidity risk |
Nature of operations
DelMar Pharmaceuticals, Inc. (the “Company”) is a Nevada corporation formed on June 24, 2009 under the name Berry Only, Inc. Prior to a reverse acquisition undertaken on January 25, 2013 Berry did not have any significant assets or operations. The Company is the parent company of Del Mar Pharmaceuticals (BC) Ltd. (“DelMar (BC)”), a British Columbia, Canada corporation incorporated on April 6, 2010, which is an early stage company with a focus on the development of drugs for the treatment of cancer. The Company is also the parent company of 0959454 B.C. Ltd., a British Columbia corporation (“Callco”), and 0959456 B.C. Ltd., a British Columbia corporation (“Exchangeco”). Callco and Exchangeco were formed to facilitate the reverse acquisition.
Pursuant to the reverse acquisition, the Company acquired (either directly or indirectly (through Exchangeco)) all of the issued and outstanding shares of DelMar (BC) on January 25, 2013. As a result of the shareholders of DelMar (BC) owning a controlling interest in the Company subsequent to the reverse acquisition, for accounting purposes the transaction is a capital transaction with DelMar (BC) being the accounting acquirer even though the legal acquirer is Berry. Therefore, the historic financial statements of DelMar (BC) are presented as the comparative balances for the periods prior to the reverse acquisition.
References to the Company, “we”, “us”, and “our” refer to the Company and its wholly-owned subsidiaries, DelMar (BC), Callco and Exchangeco. References to Berry relate to the Company prior to the reverse acquisition.
The Company is focused on the discovery and development of new medicines with the potential to treat cancer patients who have failed modern targeted or biologic therapy. The Company has initiated a clinical trial with its drug candidate VAL-083 for the treatment of refractory glioblastoma multiforme (“GBM”). The Phase I/II study is an open-label, single arm dose-escalation study designed to evaluate the safety, tolerability, pharmacokinetics and anti-tumor activity of VAL-083 in patients with histologically confirmed initial diagnosis of primary WHO Grade IV malignant glioma, now recurrent. Patients with prior low-grade glioma or anaplastic glioma are eligible to participate in the study, if histologic assessment of their condition demonstrates transformation to GBM.
The address of the Company’s administrative offices is Suite 720 - 999 West Broadway, Vancouver, British Columbia, V5Z 1K5 with clinical operations located at 3485 Edison Way, Suite R, Menlo Park, California, 94025.
| F-5 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
Liquidity risk
For the six-month period ended December 31, 2014, the Company reported a loss of $2,399,120 and an accumulated deficit of $21,064,623 at that date. As at December 31, 2014, the Company had cash and cash equivalents on hand of $3,958,439. The Company does not have the prospect of achieving revenues in the near future and the Company will require additional funding to maintain its research and development projects and for general operations. There is a great degree of uncertainty with respect to the expenses the Company will incur in executing its business plan. In addition, the Company has not begun to commercialize or generate revenues from any product candidate.
Consequently, management is pursuing various financing alternatives to fund the Company’s operations so it can continue as a going concern (note 5) in the medium to longer term. During the six months ended December 31, 2014 the Company received an aggregate $1,266,177 in net proceeds from the exercise of 1,986,074 warrants. We believe, based on our current estimates, that we will be able to fund our operations until at least the end of first quarter of calendar 2016.
There is no assurance that our cost estimates will prove to be accurate or that unforeseen events, problems or delays will not occur that would require us to seek additional debt and/or equity funding. The ability of the Company to meet its obligations and continue the research and development of its product candidate is dependent on its ability to continue to raise adequate financing. There can be no assurance that such financing will be available to the Company in the amount required at any time or for any period or, if available, that it can be obtained on terms satisfactory to the Company. The Company may tailor its drug candidate program based on the amount of funding the Company raises.
| 2 | Significant accounting policies |
Basis of presentation
The consolidated condensed interim financial statements of the Company have been prepared in accordance with United States Generally Accepted Accounting Principles (“U.S. GAAP”) and are presented in United States dollars. The Company’s functional currency is the United States dollar.
In the quarter ended March 31, 2013, the Company’s functional currency changed from Canadian dollars to United States dollars as a result of various objective factors. Therefore translation of goods and services in a foreign currency are re-measured to the functional currency of the Company with gains and losses on re-measurement recorded in the consolidated condensed interim statement of loss. Any gains and losses that were previously recorded in accumulated other comprehensive income are unchanged from the date of the change of functional currency which was January 1, 2013.
| F-6 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
The accompanying consolidated condensed interim financial statements include the accounts of the Company and its wholly-owned subsidiaries, DelMar BC, Callco, and Exchangeco. All intercompany balances and transactions have been eliminated.
The principal accounting policies applied in the preparation of these financial statements are set out below and have been consistently applied to all periods presented.
Unaudited interim financial data
The accompanying unaudited December 31, 2014 consolidated condensed interim balance sheet, the consolidated condensed interim statements of loss and comprehensive loss for the three and six months ended December 31, 2014 and 2013, and consolidated condensed cash flows for the six months ended December 31, 2014 and 2013, and the related interim information contained within the notes to the consolidated condensed interim financial statements have been prepared in accordance with the rules and regulations of the Securities and Exchange Commission for interim financial information. Accordingly, they do not include all of the information and the notes required by U.S. GAAP for complete financial statements. These consolidated condensed interim financial statements should read in conjunction with the audited financial statements of the Company as at June 30, 2014 and December 31, 2013 filed in our Form 10-KT filed with the Securities and Exchange Commission on August 28, 2014. In the opinion of management, the unaudited consolidated condensed interim financial statements reflect all adjustments, consisting of normal and recurring adjustments, necessary for the fair statement of the Company’s financial position at December 31, 2014 and results of its operations for the three and six months ended December 31, 2014 and 2013, and its cash flows for the six months ended December 31, 2014 and 2013. The results for three and six months ended December 31, 2014 are not necessarily indicative of the results to be expected for the fiscal year ending June 30, 2015 or for any other future annual or interim period.
Use of estimates
The preparation of consolidated condensed interim financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions about future events that affect the reported amounts of assets, liabilities, expenses, contingent assets and contingent liabilities as at the end or during the reporting period. Actual results could significantly differ from those estimates. Significant areas requiring management to make estimates include the derivative liability and the valuation of equity instruments, including stock options, issued for services. We have updated our estimates and models for the issuance of any new awards issued during the period.
Loss per share
Loss per share is calculated based on the weighted average number of common shares outstanding. For the three and six month periods ended December 31, 2014 and for the three months ended December 31, 2013 diluted loss per share does not differ from basic loss per share since the effect of the Company’s warrants and stock options are anti-dilutive. At December 31, 2014, potential common shares of 15,409,745 (December 31, 2013 – 24,864,009) relating to warrants and 3,415,000 (December 31, 2013 – 3,240,000) relating to stock options were excluded from the calculation of net loss per common share because their inclusion would be anti-dilutive.
| F-7 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
For the six months ended December 31, 2013 diluted income per share has also been presented. Diluted income per share is calculated using the treasury stock method which uses the weighted average number of common shares outstanding during the period and also includes the dilutive effect of potentially issuable common shares from outstanding stock options and warrants.
Recent accounting pronouncements
The Company reviews new accounting standards as issued. The accounting pronouncements issued subsequent to the date of these financial statements that were considered significant by management were evaluated for the potential effect on these financial statements. Management does not believe any of the subsequent pronouncements will have a material effect on these financial statements as presented and does not anticipate the need for any future restatement of these financial statements because of the retro-active application of any accounting pronouncements issued subsequent to December 31, 2014 through the date these financial statements were issued.
Accounting Standards Update (“ASU”) 2014-15 - Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern
The objective of the guidance is to require management to explicitly assess an entity's ability to continue as a going concern, and to provide related footnote disclosures in certain circumstances. In connection with each annual and interim period, management will assess if there is substantial doubt about an entity's ability to continue as a going concern within one year after the issuance date of an entity’s financial statements. The new standard defines substantial doubt and provides examples of indicators thereof. The definition of substantial doubt incorporates a likelihood threshold of "probable" similar to the current use of that term in U.S. GAAP for loss contingencies. The new standard will be effective for all entities in the first annual period ending after December 15, 2016 (December 31, 2016 for calendar year-end entities). Earlier application is permitted. The Company is currently assessing this standard for its impact on future reporting periods.
| 3 | Valent Technologies LLC agreement |
On September 30, 2014, the Company entered into an exchange agreement (the “Exchange Agreement”) with Valent Technologies, LLC (“Valent”), an entity owned by Dr. Dennis Brown, the Company’s Chief Scientific Officer and director, and DelMar (BC). Pursuant to the Exchange Agreement, Valent exchanged its loan payable in the outstanding amount of $278,530 (including aggregate accrued interest to September 30, 2014 of $28,530), issued to Valent by DelMar (BC), for 278,530 shares of the Company’s Series A Preferred Stock.
Effective September 30, 2014, the Company filed a Certificate of Designation of Series A Preferred Stock (the “Series A Certificate of Designation”) with the Secretary of State of Nevada. Pursuant to the Series A Certificate of Designation, the Company designated 278,530 shares of preferred stock as Series A Preferred Stock. The shares of Series A Preferred Stock have a stated value of $1.00 per share and are not convertible into common stock. The holder of the Series A Preferred Stock will be entitled to dividends at the rate of 3% of the Stated Value per year, payable quarterly in arrears. Upon any liquidation of the Company, the holder of the Series A Preferred Stock will be entitled to be paid, out of any assets of the Company available for distribution to stockholders, the Stated Value of the shares of Series A Preferred Stock held by such holder, plus any accrued but unpaid dividends thereon, prior to any payments being made with respect to the common stock.
| F-8 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
For the three months ended December 31, 2014, the Company accrued $2,089 related to the dividend payable to Valent. The dividend has been recorded as a direct increase in accumulated deficit and was paid subsequent to December 31, 2014. For the three months ended December 31, 2013 the Company accrued $2,044 in interest on its loan payable with Valent.
For the six months ended December 31, 2014, the Company accrued $2,089 related to the dividend payable to Valent and $2,091 related to interest from June 30, 2014 to September 30, 2014 when the loan was converted to preferred shares. The dividend of $2,089 has been recorded as a direct increase in accumulated deficit while the $2,091 has been recorded as interest expense. For the six months ended December 31, 2013 the Company accrued $4,073 in interest expense on its loan payable with Valent.
| 4 | Related party transactions |
During the six months ended December 31, 2014
Effective September 30, 2014, the Company entered into and closed an agreement with Valent to exchange its loan with Valent for 278,530 shares of preferred stock of the Company (note 3).
Pursuant to consulting agreements with the Company’s officers the Company recognized a total of $265,000 in compensation expense for the six months ended December 31, 2014.
Included in accounts payable at December 31, 2014 is an aggregate amount of $37,659 (June 30, 2014 - $54,960) owed to the Company’s officers and directors for fees and expenses. The Company pays related party payables incurred for fees and expenses under normal commercial terms.
The Company recognized $48,500 in directors’ fees during the six months ended December 31, 2014.
During the six months ended December 31, 2013
Pursuant to consulting agreements with the Company’s officers the Company recognized a total of $215,000 in compensation expense for the six months ended December 31, 2013.
The Company recognized $29,333 in directors’ fees during the six months ended December 31, 2013.
| 5 | Derivative liability |
The Company has issued common stock purchase warrants. Based on the terms of certain of these warrants the Company determined that the warrants were a derivative liability which is recognized at fair value at the date of the transaction and re-measured at fair value each reporting period with the changes in fair value recorded in the consolidated condensed statement of loss and comprehensive loss.
| F-9 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
Investor Warrants
Tender offer – Investor Warrant exercise price reduction
On June 9, 2014, as amended on June 26, 2014, July 10, 2014, and July 29, 2014, the Company filed a tender offer statement with the Securities and Exchange Commission with respect to certain warrants to purchase common stock of the Company issued to investors (the “Investor Warrants”) to provide the holders thereof with the opportunity to amend and exercise their warrants, upon the terms and subject to the conditions set forth in the Company’s tender offer statement. Pursuant to the tender offer, the Company offered to amend Investor Warrants to purchase an aggregate of 9,195,478 shares of common stock (the “Offer to Amend and Exercise”). There was no minimum participation requirement with respect to the Offer to Amend and Exercise.
Pursuant to the Offer to Amend and Exercise, the Investor Warrants subject to the tender offer were amended (the “Amended Warrants”) to: (i) reduce the exercise price of the Investor Warrants from $0.80 per share to $0.65 per share of common stock in cash, (ii) shorten the exercise period of the Investor Warrants so that they expire concurrently with the expiration of the Offer to Amend and Exercise at 5:00 p.m. (Pacific Time) on August 8, 2014, as may be extended by the Company in its sole discretion (“Expiration Date”), (iii) delete the price-based anti-dilution provisions contained in the Investor Warrants, (iv) restrict the ability of the holder of shares issuable upon exercise of the Amended Warrants to sell, make any short sale of, loan, grant any option for the purchase of, or otherwise dispose of any of such shares without the prior written consent of the Company for a period of time twenty (20) days after the Expiration Date (the “ Lock-Up Period ”); and (v) provide that a holder, acting alone or with others, will agree not to effect any purchases or sales of any securities of the Company in any “short sales” as defined in Rule 200 promulgated under Regulation SHO under the Exchange Act, or any type of direct and indirect stock pledges, forward sale contracts, options, puts, calls, short sales, swaps, “put equivalent positions” (as defined in Rule 16a-1(h) under the Exchange Act) or similar arrangements, or sales or other transactions through non-U.S. broker dealers or foreign regulated brokers through the expiration of the Lock-Up Period.
Upon the expiration of the Offer to Amend and Exercise on August 8, 2014, 762,227 Amended Warrants were exercised for net proceeds of $470,676 after payment by the Company of a 5% warrant agent fee of $24,772.
Investor Warrant exercises
In addition, during the six months ended December 31, 2014, 1,223,847 Investor Warrants were exercised at $0.65 per warrant for 1,223,847 shares of common stock. The Company received proceeds of $795,501 from these exercises.
All Investor Warrants that have been exercised during the period, including those exercised under the tender offer, were revalued at their respective exercise dates and then a reclassification to equity was recorded. As a result of all of the Investor Warrant exercises, for the six months ended December 31, 2014 an aggregate $391,422 of the derivative liability has been reclassified to equity.
To date, including Investor Warrants exercised prior to June 30, 2014, a total of 5,195,598 Investor Warrants have been exercised for cash for total gross proceeds of $3,886,736.
| F-10 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
Investor Warrant exchange
On December 31, 2014, the Company issued 414,889 shares of common stock in exchange for 1,244,666 Investor Warrants. The Investor Warrants that have been exchanged were revalued at their exchange date and then a reclassification to equity was recorded. The reclassification to equity upon the exchange was $305,112. The Company recognized a loss of $92,843 at the time of the exchange.
The remaining 5,964,738 Investor Warrants outstanding at December 31 , 2014 have been re-valued at December 31, 2014 using a simulated probability valuation model using the following assumptions: dividend rate - 0%, volatility - 73%, risk free rate – 1.34% and a term of approximately 3.0 years.
All 5,964,738 Investor Warrants outstanding at December 31, 2014 have an exercise price of $0.80.
Dividend Warrants
In connection with the reverse acquisition, effective January 24, 2013, the Company effected a warrant dividend (the “Warrant Dividend”) pursuant to which the Company issued one five-year warrant to purchase one share of common stock at an exercise price of $1.25 for each outstanding share of common stock (the “Dividend Warrants”). Pursuant to the Warrant Dividend, the Company issued an aggregate of 3,250,007 Dividend Warrants.
On October 31, 2014, the Company and all of its Dividend Warrant holders entered into amendments to the Dividend Warrants such that the Company’s redemption rights and certain provisions of the Dividend Warrant agreements relating to potential cash settlement of the Dividend Warrants were removed. The Dividend Warrants were revalued to the date of the amendment on October 31, 2014 which resulted in a reclassification to equity of $975,278.
Warrants issued for services
The Company has issued 300,000 warrants for services. The warrants were issued on September 12, 2013 and are exercisable on a cashless basis at an exercise price of $1.76 for five years. The warrants have been measured at December 31, 2014 using a simulated probability valuation model using the following assumptions: dividend rate - 0%, volatility - 74%, risk free rate – 1.49% and a term of approximately 3.5 years.
| F-11 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
The Company’s derivative liability is summarized as follows:
December 31, 2014 $ | June 30, 2014 $ | |||||||
| Opening balance | 3,329,367 | 4,402,306 | ||||||
| Change in fair value of warrants | (66,606 | ) | 166,388 | |||||
| Change in fair value due to change in warrant terms | (23,658 | ) | (111,179 | ) | ||||
| Reclassification to equity upon amendment of warrants | (975,278 | ) | - | |||||
| Reclassification to equity upon exchange of warrants | (305,112 | ) | - | |||||
| Reclassification to equity upon exercise of warrants | (391,422 | ) | (1,128,148 | ) | ||||
| Closing balance | 1,567,291 | 3,329,367 | ||||||
| 6 | Stockholders’ equity |
Preferred stock
Authorized
5,000,000 preferred shares, $0.001 par value
Issued and outstanding
Special voting shares – at December 31 and June 30, 2014 – 1
Series A shares – at December 31, 2014 – 278,530 (June 30, 2014 – none)
Effective September 30, 2014 pursuant to the Company’s Exchange Agreement with Valent (note 3), the Company filed the Series A Certificate of Designation with the Secretary of State of Nevada. Pursuant to the Series A Certificate of Designation, the Company designated 278,530 shares of preferred stock as Series A Preferred Stock. The shares of Series A Preferred Stock have a stated value of $1.00 per share and are not convertible into common stock. The holder of the Series A Preferred Stock will be entitled to dividends at the rate of 3% of the Stated Value per year, payable quarterly in arrears. Upon any liquidation of the Company, the holder of the Series A Preferred Stock will be entitled to be paid, out of any assets of the Company available for distribution to stockholders, the Stated Value of the shares of Series A Preferred Stock held by such holder, plus any accrued but unpaid dividends thereon, prior to any payments being made with respect to the common stock.
Common stock
Authorized
200,000,000 common shares, $0.001 par value
Issued and outstanding
December 31, 2014 – 38,580,306 (June 30, 2014 – 35,992,343)
| F-12 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
The issued and outstanding common shares at December 31, 2014 include 4,256,042 shares of common stock on an as-exchanged basis with respect to the shares of Exchangeco that can be exchanged for shares of common stock of the Company.
Shares of common stock outstanding | Common stock | Additional paid-in capital |
Warrants | |||||||||||||
| Balance – June 30, 2014 | 35,992,343 | 35,992 | 13,286,278 | 6,200,445 | ||||||||||||
| Exercise of Investor Warrants – net of issue costs | 1,986,074 | 1,986 | 1,264,191 | - | ||||||||||||
| Reclassification of derivative liability to equity on exercise of warrants | - | - | 391.422 | - | ||||||||||||
| Shares issued upon warrant exchange | 414,889 | 415 | 397,540 | - | ||||||||||||
| Reclassification of derivative liability to equity on amendment of warrant terms | - | - | 975,278 | - | ||||||||||||
| Shares issued for services | 187,000 | 187 | 181,000 | - | ||||||||||||
| Expiration of Broker Warrants | - | - | 12,640 | (12,640 | ) | |||||||||||
| Reclassification of stock option liability upon forfeiture of stock options | - | - | 38,038 | - | ||||||||||||
| Stock-based compensation | - | - | 78,694 | - | ||||||||||||
| Balance – December 31, 2014 | 38,580,306 | 38,580 | 16,625,081 | 6,187,805 |
| a) | Expiration of Broker Warrants |
During the six months ended December 31, 2014 92,000 warrants issued for certain broker services (“Broker Warrants”) exercisable at a price of CDN $0.50 per warrant expired.
| F-13 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
Stock Options
The following table sets forth the options outstanding:
Number of stock options outstanding | Weighted average exercise price $ | |||||||||
| Balance – June 30, 2014 | 3,187,214 | 0.96 | ||||||||
| Issued | 300,000 | 1.00 | ||||||||
| Forfeited | (72,214 | ) | 0.58 | |||||||
| Balance – December 31, 2014 | 3,415,000 | 0.97 |
The following table summarizes stock options currently outstanding and exercisable at December 31, 2014:
Exercise price $ | Number outstanding at December 31, 2014 | Weighted average remaining contractual life (years) | Weighted average exercise price $ | Number exercisable at December 31, 2014 | Exercise price $ | |||||||||||||||||
| 0.43 | 825,000 | 7.12 | 0.43 | 803,417 | 0.45 | |||||||||||||||||
| 1.00 | 300,000 | 4.75 | 1.00 | 50,000 | 1.00 | |||||||||||||||||
| 1.05 | 1,990,000 | 8.62 | 1.05 | 1,616,889 | 1.05 | |||||||||||||||||
| 1.54 | 180,000 | 8.25 | 1.54 | 180,000 | 1.54 | |||||||||||||||||
| 2.30 | 120,000 | 8.42 | 2.30 | 120,000 | 2.30 | |||||||||||||||||
| 3,415,000 | 0.97 | 2,770,306 | 0.96 | |||||||||||||||||||
Included in the number of stock options outstanding are 825,000 stock options granted at an exercise price of CDN $0.50. The exercise prices shown in the above table have been converted to $0.43 USD using the period ending closing exchange rate. Certain stock options have been granted to non-employees and will be revalued at each reporting date until they have fully vested. The stock options have been re-valued using a Black-Scholes pricing model using the following assumptions:
December 31, 2014 | ||||
| Dividend rate | 0 | % | ||
| Volatility | 73.8% to 91.9 | % | ||
| Risk-free rate | 1.25 | % | ||
| Term – years | 0.25 to 2.0 |
| F-14 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
The Company has recognized the following amounts as stock-based compensation expense for the periods noted:
| Three months ended December 31, | Six months ended December 31, | |||||||||||||||
2014 $ | 2013 $ | 2014 $ | 2013 $ | |||||||||||||
| Research and development | (8,077 | ) | 163,514 | 13,056 | 213,589 | |||||||||||
| General and administrative | 38,460 | 176,889 | 66,267 | 366,690 | ||||||||||||
| 30,383 | 340,403 | 79,323 | 580,279 | |||||||||||||
Of the total stock option expense of $79,323 (December 31, 2013 - $580,279) for the six months ended December 31, 2014, $78,694 (December 31, 2013 - $678,975) has been recognized as additional paid in capital and $629 (December 31, 2013 – a reduction of $(98,696)) has been recognized as a stock option liability. The aggregate intrinsic value of stock options outstanding at December 31, 2014 was $312,675 (December 31, 2013 - $422,910) and the aggregate intrinsic value of stock options exercisable at December 31, 2014 was $304,495 (December 31, 2013 - $341,304). As of December 31, 2014 there was $80,089 in unrecognized compensation expense that will be recognized over the next 1.50 years. No stock options granted under the Plan have been exercised to December 31, 2014. Upon the exercise of stock options new shares will be issued.
A summary of the status of the Company’s unvested stock options under all plans is presented below:
Number of Options | Weighted average exercise price $ | Weighted average grant date fair value $ | ||||||||||||
| Unvested at June 30, 2014 | 735,681 | 0.98 | 0.54 | |||||||||||
| Issued | 300,000 | 1.00 | 0.25 | |||||||||||
| Vested | (318,773 | ) | 1.03 | 0.57 | ||||||||||
| Forfeited | (72,214 | ) | 0.58 | 0.36 | ||||||||||
| Unvested at December 31, 2014 | 644,694 | 1.01 | 0.63 |
Certain of the Company’s warrants have been recognized as a derivative liability (note 5). The following table summarizes all of the Company’s outstanding warrants as of December 31, 2014:
| Description | Number | |||
| Balance – June 30, 2014 | 18,732,485 | |||
| Broker Warrants (i) | (92,000 | ) | ||
| Investor Warrants exercised (ii) | (1,986,074 | ) | ||
| Investor Warrants exchanged (iii) | (1,244,666 | ) | ||
| Balance - December 31, 2014 | 15,409,745 |
| F-15 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
| i) | During the six months ended December 31, 2014, 92,000 Broker Warrants expired. |
| ii) | During the six months ended December 31, 2014, 1,986,074 Investor Warrants were exercised for 1,986,074 shares of common stock (note 5). |
| iii) | During the six months ended December 31, 2014, 1,244,666 Investor Warrants were exchanged for 414,889 shares of common stock (notes 5 and 8). |
| 7 | Financial instruments |
The Company has financial instruments that are measured at fair value. To determine the fair value, we use the fair value hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs market participants would use to value an asset or liability and are developed based on market data obtained from independent sources. Unobservable inputs are inputs based on assumptions about the factors market participants would use to value an asset or liability. The three levels of inputs that may be used to measure fair value are as follows:
| · | Level one - inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities; |
| · | Level two - inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly such as interest rates, foreign exchange rates, and yield curves that are observable at commonly quoted intervals; and |
| · | Level three - unobservable inputs developed using estimates and assumptions, which are developed by the reporting entity and reflect those assumptions that a market participant would use. |
Assets and liabilities are classified based on the lowest level of input that is significant to the fair value measurements. Changes in the observability of valuation inputs may result in a reclassification of levels for certain securities within the fair value hierarchy.
The Company’s financial instruments consist of cash and cash equivalents, other receivables, accounts payable, related party payables and derivative liability. The carrying values of cash and cash equivalents, other receivables, accounts payable and related party payables approximate their fair values due to the immediate or short-term maturity of these financial instruments.
As quoted prices for the derivative liability are not readily available, the Company has used a simulated probability valuation model, as described in note 2 to estimate fair value. The derivative liability utilizes Level 3 inputs as defined above.
| F-16 |
DelMar Pharmaceuticals, Inc.
Notes to Consolidated Condensed Interim Financial Statements
(Unaudited)
December 31, 2014
(expressed in US dollars unless otherwise noted)
The Company has the following liabilities under the fair value hierarchy:
| December 31, 2014 | ||||||
| Liability | Level 1 | Level 2 | Level 3 | |||
| Derivative liability | - | - | 1,567,291 | |||
| June 30, 2014 | ||||||
| Liability | Level 1 | Level 2 | Level 3 | |||
| Derivative liability | - | - | 3,329,367 | |||
| 8 | Subsequent events |
Tender offer warrant exchange
On January 8, 2015 (note 5) , the Company filed a tender offer statement with the Securities and Exchange Commission, and on January 23, 2015, the Company filed an amendment thereto , with respect to certain Investor Warrants to purchase common stock of the Company. The tender offer provided the holders of the Investor Warrants with the opportunity to receive one share of common stock for every three Investor Warrants that are tendered. The tender offer was available to all 5,964,738 Investor Warrants outstanding at December 31, 2014. If all outstanding Investor Warrants were tendered, the Company would have issued 1,988,246 shares of common stock. To participate in the tender offer the Investor Warrant holders were required to deliver completed exchange documents to the Company, prior to the expiration of the tender offer, which was 5:00 p.m. (Pacific Time) on February 9, 2015.
The tender offer expired on February 9, 2015. A total of 1,591,875 Investor Warrants were exchanged for 530,625 shares of common stock resulting in a net increase in stockholder’s equity of approximately $400,957.
| F-17 |
| F-18 |
| F-19 |
|
Note
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
|||||||||||||
|
Assets
|
||||||||||||||||
|
Current assets
|
||||||||||||||||
|
Cash and cash equivalents
|
4,759,711 | 4,136,803 | 17,782 | |||||||||||||
|
Taxes and other receivables
|
6 | 9,572 | 11,062 | 45,499 | ||||||||||||
|
Prepaid expenses
|
234,627 | 170,883 | 28,778 | |||||||||||||
|
Deferred costs
|
9 | (f) | - | - | 90,771 | |||||||||||
| 5,003,910 | 4,318,748 | 182,830 | ||||||||||||||
|
Liabilities
|
||||||||||||||||
|
Current liabilities
|
||||||||||||||||
|
Accounts payable and accrued liabilities
|
7 | 244,906 | 140,457 | 677,615 | ||||||||||||
|
Related party payables
|
10 | 54,960 | 109,030 | 447,777 | ||||||||||||
| 299,866 | 249,487 | 1,125,392 | ||||||||||||||
|
Loan payable to Valent
|
5 | 276,439 | 272,372 | 264,352 | ||||||||||||
|
Stock option liability
|
9 | 217,759 | 212,561 | - | ||||||||||||
|
Derivative liability
|
8 | 3,329,367 | 4,402,306 | 121,000 | ||||||||||||
| 4,123,431 | 5,136,726 | 1,510,744 | ||||||||||||||
|
Stockholders’ Equity (Deficiency)
|
||||||||||||||||
|
Preferred stock
|
||||||||||||||||
|
Authorized
|
||||||||||||||||
|
5,000,000 shares, $0.001 par value
|
||||||||||||||||
|
1 share outstanding at June 30, 2014
|
||||||||||||||||
|
(December 31, 2013 - 1; December 31, 2012 - nil)
|
9 | - | - | - | ||||||||||||
|
Common stock
|
||||||||||||||||
|
Authorized
|
||||||||||||||||
|
200,000,000 shares, $0.001 par value
|
||||||||||||||||
|
35,992,343 issued at June 30, 2014 (December 31, 2013 - 31,534,819; December 31, 2012 - 13,050,000)
|
9 | 35,992 | 31,535 | 13,050 | ||||||||||||
|
Additional paid-in capital
|
9 | 13,286,278 | 8,791,715 | 2,326,885 | ||||||||||||
|
Warrants
|
9 | 6,200,445 | 6,202,100 | 153,106 | ||||||||||||
|
Accumulated deficit
|
(18,663,414 | ) | (15,864,506 | ) | (3,842,133 | ) | ||||||||||
|
Accumulated other comprehensive income
|
21,178 | 21,178 | 21,178 | |||||||||||||
| 880,479 | (817,978 | ) | (1,327,914 | ) | ||||||||||||
| 5,003,910 | 4,318,748 | 182,830 | ||||||||||||||
|
Nature of operations and liquidity risk (note 2)
|
||||||||||||||||
|
Commitments and contingencies (note 12)
|
||||||||||||||||
|
Subsequent events (note 14)
|
||||||||||||||||
|
The accompanying notes are an integral part of these consolidated financial statements.
|
| F-20 |
|
Note
|
Six months
ended
June 30,
2014
$
|
Six months
ended
June 30,
2013
$
|
Year ended
December 31,
2013
$
|
Year ended
December 31,
2012
$
|
||||||||||||||||
|
Expenses
|
||||||||||||||||||||
|
Research and development
|
992,922 | 1,216,359 | 2,342,654 | 1,550,490 | ||||||||||||||||
|
General and administrative
|
1,756,859 | 2,574,757 | 3,952,307 | 1,154,604 | ||||||||||||||||
| 2,749,781 | 3,791,116 | 6,294,961 | 2,705,094 | |||||||||||||||||
|
Other (income) loss
|
||||||||||||||||||||
|
Change in fair value of derivative liability
|
8 | 166,388 | 7,142,775 | (1,324,051 | ) | (318,502 | ) | |||||||||||||
|
Issuance of shares to Valent for future royalty reduction
|
- | 598,000 | 598,000 | - | ||||||||||||||||
|
Change in fair value of derivative liability due to tender offer
|
8 | (111,179 | ) | - | - | - | ||||||||||||||
|
Derivative issuance costs
|
- | 2,713,220 | 2,713,220 | 24,742 | ||||||||||||||||
|
Foreign exchange (gain) loss
|
(9,382 | ) | (28,933 | 3,030 | (18,492 | ) | ||||||||||||||
|
Interest expense
|
5 | 4,067 | 3,947 | 8,020 | 7,521 | |||||||||||||||
|
Interest income
|
(767 | ) | (1,180 | ) | (2,491 | ) | - | |||||||||||||
| 49,127 | 10,427,829 | 1,995,728 | (304,731 | ) | ||||||||||||||||
|
Loss for the period
|
2,798,908 | 14,218,945 | 8,290,689 | 2,400,363 | ||||||||||||||||
|
Basic and diluted loss per share
|
(0.09 | ) | (0.51 | ) | (0.28 | ) | (0.18 | ) | ||||||||||||
|
Weighted average number of shares
|
32,468,861 | 27,727,845 | 29,667,324 | 13,232,349 | ||||||||||||||||
|
Comprehensive loss
|
||||||||||||||||||||
|
Net loss
|
2,798,908 | 14,218,945 | 8,290,689 | 2,400,363 | ||||||||||||||||
|
Other comprehensive loss (income)
|
||||||||||||||||||||
|
Translation to US dollar presentation currency
|
- | - | - | 21,121 | ||||||||||||||||
|
Comprehensive loss
|
2,798,908 | 14,218,945 | 8,290,689 | 2,421,484 | ||||||||||||||||
|
The accompanying notes are an integral part of these consolidated financial statements.
|
| F-21 |
|
Number
of
shares
|
Common
stock
$
|
Additional
paid-in
capital
$
|
Accumulated
other
comprehensive
income
$
|
Warrants
$
|
Accumulated
deficit
$
|
Stockholders'
equity
(deficiency)
$
|
||||||||||||||||||||||
|
Balance - December 31, 2011
|
9,059,375 | 9,059 | 513,279 | 42,299 | - | (1,441,770 | ) | (877,133 | ) | |||||||||||||||||||
|
Issuance of units net of cash issue costs (note 8)
|
4,400,000 | 4,400 | 1,358,172 | - | - | - | 1,362,572 | |||||||||||||||||||||
|
Issuance of units for services (notes 8 and 10)
|
360,000 | 360 | 116,915 | - | - | - | 117,275 | |||||||||||||||||||||
|
Units cancelled (note 8)
|
(3,000,000 | ) | (3,000 | ) | (938,813 | ) | - | - | - | (941,813 | ) | |||||||||||||||||
|
Reclassification from additional paid-in capital to warrants upon the issuance of warrants
(note 9)
|
- | - | (103,727 | ) | - | 103,727 | - | - | ||||||||||||||||||||
|
Issuance of warrants for services (note 9)
|
- | - | - | - | 49,379 | - | 49,379 | |||||||||||||||||||||
|
Issuance of shares for settlement of accounts payable (notes 9 and 10)
|
500,000 | 500 | 252,550 | - | - | - | 253,050 | |||||||||||||||||||||
|
Shares issued from Del Mar Employee Share Purchase Trust for services - net (note 9(b))
|
1,590,625 | 1,591 | 780,255 | - | - | - | 781,846 | |||||||||||||||||||||
|
Shares issued for services (note 9(h))
|
140,000 | 140 | 75,660 | - | - | - | 75,800 | |||||||||||||||||||||
|
Stock-based compensation (note 9)
|
- | - | 272,594 | - | - | - | 272,594 | |||||||||||||||||||||
|
Comprehensive income for the year
|
- | - | - | (21,121 | ) | - | - | (21,121 | ) | |||||||||||||||||||
|
Loss for the year
|
- | - | - | - | - | (2,400,363 | ) | (2,400,363 | ) | |||||||||||||||||||
|
Balance - December 31, 2012
|
13,050,000 | 13,050 | 2,326,885 | 21,178 | 153,106 | (3,842,133 | ) | (1,327,914 | ) | |||||||||||||||||||
|
Effect of the Reverse Acquisition (note 4)
|
3,250,007 | 3,250 | 1,686,754 | - | - | (3,731,684 | ) | (2,041,680 | ) | |||||||||||||||||||
|
Issuance of units at $0.80 per unit from January 25 to March 6, 2013, net of cash issue costs (note 9(f))
|
13,125,002 | 13,125 | 5,854,252 | - | - | - | 5,867,377 | |||||||||||||||||||||
|
Issuance of placement agent warrants as issue costs for the $0.80 unit issuance
(note 9(f))
|
- | - | (4,087,586 | ) | - | 6,288,594 | - | 2,201,008 | ||||||||||||||||||||
|
Issuance of common shares to Valent for future royalty reduction (note 9 (g))
|
1,150,000 | 1,150 | 596,850 | - | - | - | 598,000 | |||||||||||||||||||||
|
Exercise of placement agent warrants (note 9)
|
123,810 | 124 | 239,476 | - | (239,600 | ) | - | - | ||||||||||||||||||||
|
Exercise of CA$0.50 unit warrants (notes 8 and 9)
|
221,000 | 221 | 241,494 | - | - | - | 241,715 | |||||||||||||||||||||
|
Shares issued for services
(note 9(h))
|
615,000 | 615 | 1,042,942 | - | - | - | 1,043,557 | |||||||||||||||||||||
|
Stock-based compensation
(note 9)
|
- | - | 890,648 | - | - | - | 890,648 | |||||||||||||||||||||
|
Loss for the year
|
- | - | - | - | - | (8,290,689 | ) | (8,290,689 | ) | |||||||||||||||||||
|
Balance - December 31, 2013
|
31,534,819 | 31,535 | 8,791,715 | 21,178 | 6,202,100 | (15,864,506 | ) | (817,978 | ) | |||||||||||||||||||
|
Exercise of Investor Warrants net of cash issue costs (note 9)
|
3,929,524 | 3,929 | 2,473,161 | - | - | - | 2,477,090 | |||||||||||||||||||||
|
Reclassification of derivative liability to equity upon exercise of Investor Warrants (note 8)
|
- | - | 1,110,548 | - | - | - | 1,110,548 | |||||||||||||||||||||
|
Exercise of CA$0,50 broker warrants
(note 8)
|
8,000 | 8 | 4,751 | - | (1,099 | ) | - | 3,660 | ||||||||||||||||||||
|
Exercise of CA$0.50 unit warrants (notes 8 and 9)
|
20,000 | 20 | 17,580 | - | - | - | 17,600 | |||||||||||||||||||||
|
Expiry of broker warrants (note 9)
|
- | - | 556 | - | (556 | ) | - | - | ||||||||||||||||||||
|
Shares issued for services
(note 9(h))
|
500,000 | 500 | 587,000 | - | - | - | 587,500 | |||||||||||||||||||||
|
Stock-based compensation
(note 9)
|
- | - | 300,967 | - | - | - | 300,967 | |||||||||||||||||||||
|
Loss for the period
|
- | - | - | - | - | (2,798,908 | ) | (2,798,908 | ) | |||||||||||||||||||
|
Balance - June 30, 2014
|
35,992,343 | 35,992 | 13,286,278 | 21,178 | 6,200,445 | (18,663,414 | ) | 880,479 | ||||||||||||||||||||
| F-22 |
|
Six months
ended
June 30,
2014
$
|
Six months
ended
June 30,
2013
$
|
Year ended
December 31,
2013
$
|
Year ended
December 31,
2012
$
|
|||||||||||||
|
Cash flows from operating activities
|
||||||||||||||||
|
Loss for the period
|
(2,798,908 | ) | (14,218,945 | ) | (8,290,689 | ) | (2,400,363 | ) | ||||||||
|
Items not affecting cash
|
||||||||||||||||
|
Accrued interest
|
4,067 | 3,947 | 8,020 | 7,521 | ||||||||||||
|
Change in fair value of derivative liability
|
166,388 | 7,142,775 | (1,324,051 | ) | (318,502 | ) | ||||||||||
|
Change in fair value of derivative liability due to tender offer
|
(111,179 | ) | - | - | - | |||||||||||
|
Shares issued to Valent for royalty reduction
|
- | 598,000 | 598,000 | - | ||||||||||||
|
Non-cash derivative issue costs
|
- | 2,201,008 | 2,201,008 | - | ||||||||||||
|
Units issued for services
|
- | - | - | 180,144 | ||||||||||||
|
Warrants issued for patents
|
- | - | - | - | ||||||||||||
|
Warrants issued for services
|
- | - | 124,020 | 49,379 | ||||||||||||
|
Stock-based compensation
|
893,665 | 1,520,487 | 2,146,766 | 1,130,240 | ||||||||||||
|
Prototype drug product
|
- | - | - | - | ||||||||||||
| (1,845,967 | ) | (2,752,728 | ) | (4,536,926 | ) | (1,351,581 | ) | |||||||||
|
Changes in non-cash working capital
|
||||||||||||||||
|
Taxes and other receivables
|
1,490 | 28,605 | 34,437 | (6,697 | ) | |||||||||||
|
Prepaid expenses
|
(63,744 | ) | (192,484 | ) | (142,105 | ) | (14,581 | ) | ||||||||
|
Accounts payable and accrued liabilities
|
104,449 | (246,388 | ) | (537,158 | ) | 865,007 | ||||||||||
|
Related party payables
|
(54,070 | ) | (211,315 | ) | (338,747 | ) | (70,183 | ) | ||||||||
| (1,857,842 | ) | (3,374,310 | ) | (5,520,499 | ) | (578,035 | ) | |||||||||
|
Cash flows from financing activities
|
||||||||||||||||
|
Net proceeds from the exercise of warrants
|
2,480,750 | - | - | - | ||||||||||||
|
Net proceeds from the issuance of units
|
- | 9,639,520 | 9,639,520 | 671,570 | ||||||||||||
|
Deferred costs
|
- | - | - | (90,771 | ) | |||||||||||
|
Net proceeds from the issuance of common shares
|
- | - | - | - | ||||||||||||
| 2,480,750 | 9,639,520 | 9,639,520 | 580,799 | |||||||||||||
|
Increase (decrease) in cash and cash equivalents
|
622,908 | 6,265,210 | 4,119,021 | 2,764 | ||||||||||||
|
Cash and cash equivalents - beginning of period
|
4,136,803 | 17,782 | 17,782 | 15,018 | ||||||||||||
|
Cash and cash equivalents - end of period
|
4,759,711 | 6,282,992 | 4,136,803 | 17,782 | ||||||||||||
|
Supplementary information
|
||||||||||||||||
|
Issuance of shares for the settlement of accounts payable (notes 7 and 10)
|
- | - | - | 253,050 | ||||||||||||
|
Issuance of units for the settlement of accounts payable (notes 8 and 10)
|
- | - | - | - | ||||||||||||
|
Non-cash share issuance costs (note 9)
|
- | 6,288,594 | 6,288,594 | - | ||||||||||||
|
Cashless exercise of Placement Agent Warrants (note 9)
|
- | - | 239,600 | - | ||||||||||||
|
Settlement of accounts payable with a loan payable (note 5)
|
- | - | - | - | ||||||||||||
|
Exercise of CA$0.50 warrants for no additional consideration (note 9)
|
17,600 | - | 241,715 | - | ||||||||||||
|
Deferred costs (note 9(f))
|
- | 90,771 | 90,771 | - | ||||||||||||
| F-23 |
|
1
|
Change in fiscal year end
|
|
2
|
Nature of operations and liquidity risk
|
| F-24 |
|
3
|
Significant accounting policies
|
| F-25 |
|
a)
|
Fair value of derivative liability
|
| F-26 |
| F-27 |
|
·
|
Level one - inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities;
|
|
·
|
Level two - inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly such as interest rates, foreign exchange rates, and yield curves that are observable at commonly quoted intervals; and
|
|
·
|
Level three - unobservable inputs developed using estimates and assumptions, which are developed by the reporting entity and reflect those assumptions that a market participant would use.
|
|
June 30, 2014
|
||||||||||||
|
Liability
|
Level 1
|
Level 2
|
Level 3
|
|||||||||
|
Derivative liability
|
- | - | 3,329,367 | |||||||||
|
December 31, 2013
|
||||||||||||
|
Liability
|
Level 1
|
Level 2
|
Level 3
|
|||||||||
|
Derivative liability
|
- | - | 4,402,306 | |||||||||
| F-28 |
|
December 31, 2012
|
||||||||||||
|
Liability
|
Level 1
|
Level 2
|
Level 3
|
|||||||||
|
Derivative liability
|
- | - | 121,000 | |||||||||
| F-29 |
| F-30 |
| F-31 |
| F-32 |
|
4
|
Reverse acquisition
|
| $ | ||||
|
Net liabilities (derivative liability)
|
2,041,680 | |||
| F-33 |
|
5
|
Valent Technologies LLC agreement
|
| F-34 |
|
6
|
Taxes and other receivables
|
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
||||||||||
|
Government grants
|
562 | - | 34,168 | |||||||||
|
Other receivables
|
9,010 | 11,062 | 11,331 | |||||||||
| 9,572 | 11,062 | 45,499 | ||||||||||
|
7
|
Accounts payable and accrued liabilities
|
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
||||||||||
|
Trade payables
|
244,906 | 140,457 | 677,615 | |||||||||
|
Payable to related parties (note 10)
|
54,960 | 109,030 | 447,777 | |||||||||
| 299,866 | 249,487 | 1,125,392 | ||||||||||
| F-35 |
|
8
|
Derivative liability
|
| F-36 |
| F-37 |
| F-38 |
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
||||||||||
|
Opening balance
|
4,402,306 | 121,000 | 106,146 | |||||||||
|
Issuance of units
|
- | 3,681,372 | 333,356 | |||||||||
|
Dividend warrant liability acquired on reverse acquisition
|
- | 2,041,680 | - | |||||||||
|
Warrants issued for services
|
- | 124,020 | - | |||||||||
|
Change in fair value of unexercised warrants
|
166,388 | (1,324,051 | ) | (318,502 | ) | |||||||
|
Change in fair value due to tender offer
|
(111,179 | ) | - | - | ||||||||
|
Reclassification to equity upon exercise of warrants
|
(1,128,148 | ) | (241,715 | ) | - | |||||||
|
Closing balance
|
3,329,367 | 4,402,306 | 121,000 | |||||||||
|
9
|
Stockholders’ equity (deficiency)
|
| F-39 |
|
a)
|
Shares issued to founders
|
| F-40 |
|
b)
|
Shares issued to the DelMar Employees Share Purchase Trust
|
|
Number of
shares held
in Trust
|
||||
|
Balance - April 6, 2010
|
- | |||
|
Shares issued to the DelMar Employee Share Purchase Trust
|
2,000,000 | |||
|
Shares transferred to employees and consultants for services
|
(325,000 | ) | ||
|
Founders shares acquired by the Trust
|
68,750 | |||
|
Balance - December 31, 2010
|
1,743,750 | |||
|
Shares transferred to employees and consultants for services
|
(200,000 | ) | ||
|
Founders shares acquired by the Trust
|
46,875 | |||
|
Balance - December 31, 2011
|
1,590,625 | |||
|
Shares transferred to employees and consultants for services
|
(1,590,625 | ) | ||
|
Balance - June 30, 2014, December 31, 2013 and 2012
|
- | |||
| F-41 |
|
c)
|
Shares issued in private placements
|
|
d)
|
Shares issued to Valent for settlement of accounts payable
|
|
e)
|
Shares issued for the Reverse Acquisition
|
| F-42 |
|
f)
|
$0.80 Unit offering
|
| F-43 |
|
g)
|
Shares issued to Valent for future royalty reduction
|
|
h)
|
Shared issued for services
|
| F-44 |
|
Number of
stock
options
outstanding
|
Weighted
average
exercise
price
|
|||||||
|
Balance - December 31, 2011
|
- | - | ||||||
|
Granted
|
1,020,000 | 0.47 | ||||||
|
Balance - December 31, 2012
|
1,020,000 | 0.47 | ||||||
|
Granted
|
2,340,000 | 1.15 | ||||||
|
Cancelled
|
(120,000 | ) | 0.47 | |||||
|
Balance - December 31, 2013
|
3,240,000 | 0.96 | ||||||
|
Forfeited
|
(52,786 | ) | 0.47 | |||||
|
Balance - June 30, 2014
|
3,187,214 | 0.96 | ||||||
| F-45 |
|
Exercise price
$
|
Number
Outstanding at
June 30,
2014
|
Weighted
average
remaining
contractual
life
(years)
|
Weighted
average
exercise
price
$
|
Number
exercisable
at
June 30,
2014
|
Exercise
price
$
|
|||||||||||||||||
| 0.47 | 883,324 | 7.73 | 0.47 | 798,893 | 0.47 | |||||||||||||||||
| 1.05 | 2,003,890 | 9.13 | 1.05 | 1,352,640 | 1.05 | |||||||||||||||||
| 1.54 | 180,000 | 8.75 | 1.54 | 180,000 | 1.54 | |||||||||||||||||
| 2.30 | 120,000 | 8.92 | 2.30 | 120,000 | 2.30 | |||||||||||||||||
| 3,187,214 | 0.96 | 2,451,533 | 0.96 | |||||||||||||||||||
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
||||||||||
|
Dividend rate
|
0 | % | 0 | % | 0 | % | ||||||
|
Volatility
|
73% to 76
|
% |
73% to 85
|
% | 74 | % | ||||||
|
Risk-free rate
|
1.25 | % | 1.00 | % | 1.25 | % | ||||||
|
Term - years
|
0.5 to 2.5
|
1 to 3
|
2.1 | |||||||||
|
Six months ended June 30,
|
Years ended December 31,
|
|||||||||||||||
| 2014 | 2013 | 2013 | 2012 | |||||||||||||
| $ | $ | $ | $ | |||||||||||||
|
Research and development
|
144,587 | 309,136 | 522,725 | 196,281 | ||||||||||||
|
General and administrative
|
161,578 | 213,794 | 580,484 | 76,313 | ||||||||||||
| 306,165 | 522,930 | 1,103,209 | 272,594 | |||||||||||||
| F-46 |
|
Number of
options
|
Weighted
average
exercise
price
$
|
Weighted
average
grant date
fair value
$
|
||||||||||
|
Unvested at December 31, 2011
|
- | - | - | |||||||||
|
Granted
|
1,020,000 | 0.47 | 0.30 | |||||||||
|
Vested
|
(575,500 | ) | 0.47 | 0.30 | ||||||||
|
Unvested at December 31, 2012
|
444,500 | 0.47 | 0.30 | |||||||||
|
Granted
|
2,340,000 | 1.15 | 0.63 | |||||||||
|
Cancelled
|
(120,000 | ) | 0.47 | 0.30 | ||||||||
|
Vested
|
(985,129 | ) | 1.05 | 0.58 | ||||||||
|
Unvested at December 31, 2013
|
1,679,371 | 1.08 | 0.59 | |||||||||
|
Forfeited
|
(52,786 | ) | 0.87 | 0.49 | ||||||||
|
Vested
|
(890,904 | ) | 1.09 | 0.60 | ||||||||
|
Unvested at June 30, 2014
|
735,681 | 0.98 | 0.54 | |||||||||
| F-47 |
|
Number of
warrants
|
Amount
$
|
|||||||
|
Balance - December 31, 2011
|
-
|
-
|
||||||
|
Warrants issued for patents (i)
|
500,000
|
89,432
|
||||||
|
Warrants issued as unit issue costs (ii)
|
105,000
|
14,295
|
||||||
|
Warrants issued for services (iii)
|
345,000
|
49,379
|
||||||
|
Balance - December 31, 2012
|
950,000
|
153,106
|
||||||
|
Warrants issued as unit issue costs (iv)
|
5,250,000
|
6,288,594
|
||||||
|
Warrants exercised on a cashless basis (v)
|
(200,000
|
)
|
(239,600
|
)
|
||||
|
Balance - December 31, 2013
|
6,000,000
|
6,202,100
|
||||||
|
Expiry of broker warrants (ii)
|
(5,000
|
)
|
(556
|
)
|
||||
|
Exercise of broker warrants (ii)
|
(8,000
|
)
|
(1,099
|
)
|
||||
|
Balance - June 30, 2014
|
5,987,000
|
6,200,445
|
||||||
|
i)
|
At
December 31, 2011, the Company recognized the fair value of the 500,000 contingent Valent warrants (note 5).
The contingent warrants were recognized in additional paid in capital at December 31, 2011 and have been reclassified to
warrants when the warrants were issued on February 1, 2012. The warrants have an exercise price of CA$0.50 per warrant and
expire February 1, 2017.
|
|
ii)
|
The Company has issued broker warrants as finder’s fees in relation to the issuance of certain Units. All of the warrants were issued on March 1, 2012 and have an exercise price of CA$0.50 per warrant. Of the total, 100,000 expire March 1, 2015 and 5,000 expired March 1, 2014. During the six months ended June 30, 2014, 8,000 broker warrants were exercised for proceeds of $3,660 (CA$4,000).
|
|
iii)
|
The Company has issued 345,000 warrants for investor relations services. The warrants were issued on February 1, 2012 and they vest in 12 equal installments over a 12-month period commencing on March 1, 2012. The warrants have an exercise price of CA$0.50 per warrant and expire February 1, 2015.
|
| F-48 |
|
iv)
|
As part of the Company’s unit offering the Company has issued 5,250,000 Placement Agent Warrants (note 9 (f)). The Placement Agent Warrants have been recognized as non-cash issue costs and the costs have been allocated to common stock and derivative liability. The portion allocated to additional paid in capital was $4,087,586 and the portion allocated to derivative liability was $2,201,008. The Placement Agent warrants have been valued using a simulated probability valuation model using the following assumptions: dividend rate - 0%, volatility - 104%, risk free rate - 1.0% and a term of five years.
|
|
v)
|
During the year ended December 31, 2013, 200,000 Placement Agent Warrants were exercised on a cashless basis for 123,810 shares of common stock.
|
|
Description
|
Number
|
|||
|
CA$0.50 warrants (i)
|
- | |||
|
Issued as broker warrants (ii)
|
92,000 | |||
|
Issued for patents (iii)
|
500,000 | |||
|
Issued for services (iv)
|
345,000 | |||
|
Investor Warrants (v)
|
9,195,478 | |||
|
Dividend warrants (vi)
|
3,250,007 | |||
|
Placement Agent (vii)
|
5,050,000 | |||
|
Issued for services (viii)
|
300,000 | |||
|
Closing balance - June 30, 2014
|
18,732,485 | |||
|
i)
|
Of the balance of 2,189,000 outstanding at December 31, 2013, 20,000 were exercised for no additional consideration and 2,169,000 expired on January 25, 2014 (note 8).
|
|
ii)
|
The Company has issued broker warrants as finder’s fees in relation to the issuance of certain of the CA$0.50 units issued during the years ended December 31, 2011 and 2012. All of the warrants were issued on March 1, 2012 and have an exercise price of CA$0.50 per warrant. Of the total, 100,000 expire March 1, 2015 and 5,000 expired March 1, 2014. During the six months ended June 30, 2014, 8,000 warrants were exercised for proceeds of $3,660 (CA$0.50).
|
|
iii)
|
The Company issued 500,000 warrants to Valent (note 5). The warrants have an exercise price of CA$0.50 per warrant and expire February 1, 2017.
|
| F-49 |
|
iv)
|
The Company has issued 345,000 warrants for investor relations services. The warrants were issued on February 1, 2012 and they vested in 12 equal installments over a 12-month period commencing on March 1, 2012. The warrants have an exercise price of CA$0.50 per warrant and expire February 1, 2015.
|
|
v)
|
The Investor Warrants were issued as part of the Company’s $0.80 unit offering. They were issued in tranches on January 25, 2013, January 31, 2013, February 8, 2013, February 21, 2013, February 28, 2013, March 1, 2013, and March 6, 2013 respectively (note 9 (f)). Of the initial number issued of 13,125,002, 277,313 have been exercised at $0.80, 3,652,211 have been exercised at $0.65 and the remaining balances of 9,195,478 were the subject of a tender offer (note 8).
|
|
vi)
|
The Dividend Warrants are exercisable at $1.25 per warrant until January 24, 2018.
|
|
vii)
|
The Placement Agent Warrants are exercisable at $0.80 per warrant until March 6, 2018 but can be exercised on a cashless basis. The Placement Agent Warrants were all issued on March 6, 2013.
|
|
viii)
|
The warrants are exercisable on a cashless basis at a price of $1.76 per warrant until September 12, 2018.
|
|
10
|
Related party transactions
|
| F-50 |
| F-51 |
|
11
|
Current and future income taxes
|
|
Expiry date
|
$ | ||||
|
2029
|
70,637 | ||||
|
2030
|
1,131,510 | ||||
|
2031
|
1,229,147 | ||||
|
2033
|
4,121,312 | ||||
|
2034
|
2,398,163 | ||||
| F-52 |
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
||||||||||
|
Non-capital losses carried forward
|
2,686,530 | 1,822,341 | 323,910 | |||||||||
|
Financing costs
|
7,737 | 4,115 | 4,302 | |||||||||
|
Scientific research and development
|
144,235 | 121,490 | 11,193 | |||||||||
| 2,838,502 | 1,947,946 | 339,405 | ||||||||||
|
Valuation allowance
|
(2,838,502 | ) | (1,947,946 | ) | (339,405 | ) | ||||||
|
Net future tax assets
|
- | - | - | |||||||||
|
June 30,
2014
$
|
December 31,
2013
$
|
December 31,
2012
$
|
||||||||||
|
Tax recovery at statutory income tax rates
|
(989,430 | ) | (2,818,834 | ) | (324,049 | ) | ||||||
|
Permanent differences
|
110,113 | 979,359 | 133,365 | |||||||||
|
Effect of rate differentials between jurisdictions
|
149,219 | 320,965 | - | |||||||||
|
Other
|
(8,713 | ) | - | 13,087 | ||||||||
|
Effect of tax rate changes on future taxes
|
- | (305,647 | ) | - | ||||||||
|
Change in valuation allowance
|
738,811 | 1,824,157 | 177,597 | |||||||||
| - | - | - | ||||||||||
|
12
|
Commitments and contingencies
|
| F-53 |
|
13
|
Financial risk management
|
|
a)
|
Foreign exchange risk
|
|
June 30,
2014
balances
CA$
|
December 31,
2013
balances
CA$
|
December 31,
2012
balances
CA$
|
||||||||||
|
Trade payables
|
136,825 | 95,835 | 359,088 | |||||||||
|
Cash
|
65,513 | 75,474 | 17,873 | |||||||||
|
b)
|
Interest rate risk
|
| F-54 |
|
Cash and
cash
equivalents
$
|
Insured
amount
$
|
Non-insured
amount
$
|
||||||||||
| 4,759,711 | 61,366 | 4,698,345 | ||||||||||
|
14
|
Subsequent events
|
| F-55 |
$10,000,000 of Shares of Common Stock and
Warrants to Purchase Shares of Common Stock
PROSPECTUS
Joint Book-Running Managers
| Maxim Group LLC | Roth Capital Partners |
Co-Manager
National Securities Corporation
, 2015
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table provides information regarding the various actual and anticipated expenses (other than underwriters' discounts) payable by us in connection with the issuance and distribution of the securities being registered hereby. All amounts shown are estimates except the Securities and Exchange Commission registration fee and the FINRA filing fee.
| Nature of Expense | Amount | |||
| SEC registration fee | $ | 1,470 | ||
| FINRA filing fee | * | |||
| Accounting fees and expenses | * | |||
| Legal fees and expenses | * | |||
| Transfer agent’s fees and expenses | * | |||
| Printing and related fees | * | |||
| Miscellaneous | * | |||
| Total | $ | |||
* To be completed by amendment.
Item 14. Indemnification of Directors and Officers.
Neither our Articles of Incorporation nor Bylaws prevent us from indemnifying our officers, directors and agents to the extent permitted under the Nevada Revised Statute ("NRS"). NRS Section 78.7502 provides that a corporation shall indemnify any director, officer, employee or agent of a corporation against expenses, including attorneys' fees, actually and reasonably incurred by him in connection with any the defense to the extent that a director, officer, employee or agent of a corporation has been successful on the merits or otherwise in defense of any action, suit or proceeding referred to Section 78.7502(1) or 78.7502(2), or in defense of any claim, issue or matter therein.
NRS 78.7502(1) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative, except an action by or in the right of the corporation, by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses, including attorneys' fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with the action, suit or proceeding if he: (a) is not liable pursuant to NRS 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful.
NRS Section 78.7502(2) provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses, including amounts paid in settlement and attorneys' fees actually and reasonably incurred by him in connection with the defense or settlement of the action or suit if he: (a) is not liable pursuant to NRS 78.138; or (b) acted in good faith and in a manner which he reasonably believed to be in or not opposed to the best interests of the corporation. Indemnification may not be made for any claim, issue or matter as to which such a person has been adjudged by a court of competent jurisdiction, after exhaustion of all appeals there from, to be liable to the corporation or for amounts paid in settlement to the corporation, unless and only to the extent that the court in which the action or suit was brought or other court of competent jurisdiction determines upon application that in view of all the circumstances of the case, the person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
NRS Section 78.747 provides that except as otherwise provided by specific statute, no director or officer of a corporation is individually liable for a debt or liability of the corporation, unless the director or officer acts as the alter ego of the corporation. The court as a matter of law must determine the question of whether a director or officer acts as the alter ego of a corporation.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by us is against public policy as expressed hereby in the Securities Act and we will be governed by the final adjudication of such issue.
| II-1 |
Item 15. Recent Sales of Unregistered Securities.
DelMar (BC) issued 4,150,000 units on January 23, 2012, 560,000 units on February 27, 2012, and 50,000 units on May 10, 2012, at a purchase price CDN $0.50 per unit. In addition, during the year ended December 31, 2011, the Company issued 500,000 units, on October 3, 2011, 100,000 units on October 7, 2011, and 50,000 units on November 11, 2011. In total, the Company issued 5,410,000 units for services in settlement of accounts payable and cash proceeds for an aggregate of $2,671,923 (CDN $2,705,000). The proceeds from the issuance of 3,000,000 of these units were held in escrow pursuant to an exclusive option investment agreement. Subsequently, DelMar (BC) elected to allow the option to expire and the related units were cancelled and the funds returned to the subscriber in order for the Company to retain control over certain intellectual property and commercial rights.
Between June 1, 2012 and January 1, 2013 inclusive DelMar (BC) issued 160,000 common shares for services.
During the six months ended June 30, 2013, DelMar (BC) issued 500,000 common shares in settlement of accounts payable in the amount of $253,050 (CDN $250,000) owed to Valent.
On January 25, 2013 the Company entered into and closed an exchange agreement, with DelMar BC, 0959454 B.C. Ltd., Callco, Exchangeco, and securityholders of DelMar (BC). Pursuant to the Exchange Agreement, (i) the Company issued 4,340,417 shares of common stock to the U.S. Holders in exchange for the transfer to Exchangeco of all 4,340,417 outstanding common shares of DelMar (BC) held by the U.S. Holders, (ii) the Canadian Holders received, in exchange for the transfer to Exchangeco of all 8,729,583 outstanding common shares of DelMar (BC) held by the Canadian Holders, 8,729,583 Exchangeable Shares of Exchangeco, and (iii) outstanding warrants to purchase 3,360,000 common shares of DelMar (BC) and outstanding options to purchase 1,020,000 common shares of DelMar (BC) were deemed to be amended such that, rather than entitling the holder to acquire common shares of DelMar (BC), such options and warrants will entitle the holders to acquire shares of common stock of the Company. The Canadian Holders will be entitled to require Exchangeco to redeem (or, at the option of the Company or Callco, to have the Company or Callco purchase) the Exchangeable Shares, and upon such redemption or purchase to receive an equal number of shares of common stock of the Company.
On January 25, 2013, January 31, 2013, February 8, 2013, February 21, 2013, February 28, 2013, March 1, 2013, and March 6, 2013, the Company entered into and closed a series of subscription agreements with accredited investors, pursuant to which the Company issued an aggregate of 13,125,002 Units at a purchase price of $0.80 per Unit, for aggregate gross proceeds of $10,500,000. Each Unit consists of one share of common stock and one five-year warrant to purchase one share of common stock at an exercise price of $0.80.
The Company retained Charles Vista, LLC as the Placement Agent for the Private Offering. The Company paid the Placement Agent a cash fee of $1,050,000 (equal to 10% of the gross proceeds), a non-accountable expense allowance of $315,000 (equal to 3% of the gross proceeds), and a one-year consulting fee of $60,000. Also, the Company issued to the Placement Agent five-year warrants to purchase 5,250,000 shares of common stock (equal to 20% of the shares of common stock (i) included as part of the Units sold in the Private Offering and (ii) issuable upon exercise of the Investor Warrants) at an exercise price of $0.80, exercisable on a cash or cashless basis.
On January 25, 2013, the Company, Callco, Exchangeco and Computershare Trust Company of Canada (the “Trustee”) entered into a voting and exchange trust agreement (the “Trust Agreement”). Pursuant to the Trust Agreement, Company issued one share of Special Voting Preferred Stock (the “Special Voting Share”) to the Trustee, and the parties created a trust for the Trustee to hold the Special Voting Share for the benefit of the holders of the Exchangeable Shares (other than the Company and any affiliated companies) (the “Beneficiaries”). Pursuant to the Trust Agreement, the Beneficiaries will have voting rights in the Company equivalent to what they would have had they received shares of common stock in the same amount as the Exchangeable Shares held by the Beneficiaries.
On January 25, 2013, the Company issued 1,150,000 shares of common stock to Valent, a company owned by Dennis Brown, the Company’s Chief Scientific Officer, in exchange for Valent agreeing to reduce certain royalties payable to it pursuant to a patent assignment agreement between Valent and DelMar (BC).
On April 17, 2013, the Company issued an aggregate of 555,000 shares of restricted common stock to consultants for services.
| II-2 |
During the three months ended September 30, 2013, the Company issued an aggregate of 1,469,810 shares of common stock, including 40,000 shares of common stock for services, 206,000 shares upon the exercise of 206,000 warrants for no additional consideration, 123,810 shares upon the cashless exercise of 200,000 warrants, and 1,100,000 shares upon the exchange of Exchangeable Shares. On September 12, 2013, the Company issued five-year warrants to purchase 300,000 shares of common stock for services. The warrants are exercisable on a cashless basis at an exercise price of $1.76.
During the three months ended December 31, 2013, the Company issued 15,000 shares of common stock upon the exercise of 15,000 warrants for no additional consideration and 255,000 shares of common stock upon the exchange of 255,000 Exchangeable Shares.
During the period from January 1, 2014 to April 14, 2014 the Company issued 305,313 shares of common stock upon the exercise of warrants, including 20,000 shares for no additional consideration, 8,000 shares for proceeds of CDN $4,000, and 277,313 shares for proceeds of $221,850. In addition, during the period from January 1, 2014 to April 14, 2014 the Company issued 250,000 shares of common stock for services, and 205,000 shares of common stock upon the exchange of 205,000 Exchangeable Shares.
During the three months ended June 30, 2014, the Company issued 250,000 shares of common stock for services, 125,000 shares of common stock upon the exchange of 125,000 Exchangeable Shares, and 8,000 shares of common stock upon exercise of warrants at an exercise price of $0.46 per share.
On August 8, 2014, the Company consummated its offer (the “Warrant Tender Offer”) to amend certain of its outstanding warrants held by investors who participated in the Company’s private placement financings that closed on January 25, 2013, January 31, 2013, February 8, 2013, February 21, 2013, February 28, 2013, March 1, 2013, and March 6, 2013 (the “Investor Warrants”). Pursuant to the Warrant Tender Offer, an aggregate of 762,227 Investor Warrants were tendered by their holders and were amended and exercised in connection therewith for an aggregate exercise price of approximately $495,448. The Company received net proceeds of approximately $470,676 after paying a 5% warrant agent fee of approximately $24,772.
Effective October 31, 2014, the Company entered into election to exercise warrants agreements (the “Investor Warrant Amendments”) with certain holders of warrants to purchase shares of common stock issued to investors (the “Investor Warrants”), pursuant to which the Company reduced the exercise price from $0.80 to $0.65 per share for 1,136,347 Investor Warrants, and in accordance with the agreements, the holders of such Investor Warrants exercised the Investor Warrants for cash at the foregoing reduced exercise price, such that the Company received aggregate gross proceeds from such exercises of $738,626. Previously, in September 2014, pursuant to election to exercise warrant agreements, the Company reduced the exercise price from $0.80 to $0.65 of 87,500 Investor Warrants, and the holders of such Investor Warrants exercised such Investor Warrants for cash in accordance with such agreements, such that the Company received aggregate gross proceeds of $56,875 from such exercises.
On December 31, 2014, the Company entered into separate warrant exchange agreements (collectively, the “Exchange Agreements”) with each of certain holders of the Company’s outstanding warrants to purchase common stock issued in connection with its private placement financings that closed on January 25, 2013, January 31, 2013, February 8, 2013, February 21, 2013, February 28, 2013, March 1, 2013, and March 6, 2013. Pursuant to the Exchange Agreements, on December 31, 2014, the Company issued an aggregate of 414,889 shares of Common Stock to these holders in exchange for the surrender of warrants to purchase an aggregate of 1,244,666 shares of common stock.
During the three months ended December 31, 2014, the Company issued 187,000 shares of common stock for services.
On February 9, 2015, the Company issued an aggregate of 530,625 shares of common stock in exchange for the cancellation of 1,591,875 warrants pursuant to an exchange offer.
The transactions described above were exempt from registration under Section 4(a)(2) of the Securities Act and/or under Regulation S promulgated by the SEC.
Item 16. Exhibits and Financial Statement Schedules.
(a) Exhibits.
A list of exhibits filed with this registration statement on Form S-1 is set forth on the Exhibit Index and is incorporated herein by reference.
(b) Financial statement schedule.
All schedules have been omitted because either they are not required, are not applicable or the information is otherwise set forth in the financial statements and related notes thereto.
| II-3 |
Item 17. Undertakings.
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by section 10(a)(3) of the Securities Act of 1933, as amended (the “Securities Act”);
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purposes of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of the securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) For the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities, other than the payment by the registrant of expenses incurred and paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding, is asserted by such director, officer or controlling person in connection with the securities being registered hereby, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
| II-4 |
(c) The undersigned Registrant hereby undertakes that it will:
(1) for determining any liability under the Securities Act, treat the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant under Rule 424(b)(1), or (4) or 497(h) under the Securities Act as part of this registration statement as of the time the Commission declared it effective.
(2) for determining any liability under the Securities Act, treat each post-effective amendment that contains a form of prospectus as a new registration statement for the securities offered in the registration statement, and that offering of the securities at that time as the initial bona fide offering of those securities.
| II-5 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of Vancouver, British Columbia, Canada, on April 21, 2015.
| DELMAR PHARMACEUTICALS, INC. | |||
| By: | /s/ Jeffrey Bacha | ||
| Jeffrey Bacha | |||
| Its: | CEO and Chairman | ||
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| /s/ Jeffrey Bacha | April 21, 2015 | |||
| Jeffrey Bacha | ||||
| Chief Executive Officer and Director (Principal Executive Officer) | ||||
| /s/ Scott Praill | April 21, 2015 | |||
Scott Praill Chief Financial Officer (Principal Financial and Accounting Officer) |
||||
| /s/ Dennis Brown | April 21, 2015 | |||
| Dennis Brown | ||||
| Director | ||||
| /s/ William Garner | April 21, 2015 | |||
| William Garner | ||||
| Director | ||||
| /s/ John K. Bell | April 21, 2015 | |||
| John K. Bell | ||||
| Director |
| /s/ Robert J. Toth, Jr. | April 21, 2015 | |||
| Robert J. Toth, Jr. | ||||
| Director |
| /s/ Lynda Cranston | April 21, 2015 | |||
| Lynda Cranston | ||||
| Director |
| /s/ Erich Mohr | April 21, 2015 | |||
| Erich Mohr | ||||
| Director |
| II-6 |
EXHIBIT INDEX
| 1.1 | Form of Underwriting Agreement+ | |
| 2.1 | Exchange Agreement, dated January 25, 2013, among the Company, Exchangeco, Callco, DelMar (BC) and securityholders of DelMar (BC) (1) | |
| 3.1 | Articles of Incorporation of the Company (2) | |
| 3.2 | Articles of Merger of the Company (3) | |
| 3.3 | Certificate of Designation of Special Voting Preferred Stock of the Company (3) | |
| 3.4 | Bylaws of the Company (2) | |
| 3.5 | Amendment to Bylaws of the Company (4) | |
| 3.6 | Certificate of Designation of Series A Preferred Stock (7) | |
| 4.1 | Form of Warrant + | |
| 5.1 | Opinion of Sichenzia Ross Friedman Ference LLP + | |
| 10.1 | Intercompany Funding Agreement, dated January 25, 2013, between the Company and Exchangeco (1) | |
| 10.2 | Support Agreement, dated January 25, 2013, among the Company, Exchangeco and Callco (1) | |
| 10.3 | Voting and Exchange Trust Agreement, dated January 25, 2013, among the Company, Callco, Exchangeco, and the Trustee (1) | |
| 10.4 | Form of Subscription Agreement (1) | |
| 10.5 | Form of Registration Rights Agreement (1) | |
| 10.6 | Form of Investor Warrant (1) | |
| 10.7 | Form of Dividend Warrant (1) | |
| 10.8 † | Memorandum of Understanding and Collaboration Agreement between Guangxi Wuzhou Pharmaceutical (Group) Co. Ltd. and DelMar (BC) (1) | |
| 10.9 † | Patent Assignment Agreement, dated September 12, 2010, between DelMar (BC) and Valent (5) | |
| 10.10 | Amendment, dated January 21, 2013, to Patent Assignment Agreement, dated September 12, 2010, between DelMar (BC) and Valent (5) | |
| 10.11 | Loan Agreement, dated February 3, 2011, between DelMar (BC) and Valent (5) | |
| 10.12 | Form of Election to Exercise Warrants (6) | |
| 10.13 | Form of Investor Warrant Amendment (8) | |
| 10.14 | Form of Dividend Warrant Amendment (8) | |
| 10.15 | Form of Exchange Agreement (9) | |
| 10.16 | Consulting Agreement, effective January 1, 2015 between DelMar (BC) and Jeffrey Bacha (previously filed) | |
| 10.17 | Consulting Agreement, effective January 1, 2015 between DelMar (BC) and Dennis Brown (previously filed) | |
| 10.18 | Consulting Agreement, effective January 1, 2015 between DelMar (BC) and Scott Praill (previously filed) | |
| 10.19 | Warrant Agency Agreement+ | |
| 16 | Letter from John Kinross-Kennedy (1) | |
| 21 | Subsidiaries (10) | |
| 23.1 | Consent of PricewaterhouseCoopers LLP* | |
| 23.2 | Consent of Sichenzia Ross Friedman Ference LLP (included in Exhibit 5.1) + | |
| 99.1 | Pro forma financial information (1) |
| EX-101.INS | XBRL Instance Document + | |
| EX-101.SCH | XBRL Taxonomy Extension Schema Document + | |
| EX-101.CAL | XBRL Taxonomy Extension Calculation Linkbase + | |
| EX-101.DEF | XBRL Taxonomy Extension Definition Linkbase + | |
| EX-101.LAB | XBRL Taxonomy Extension Labels Linkbase + | |
| EX-101.PRE | XBRL Taxonomy Extension Presentation Linkbase + |
† Confidential treatment has been granted for certain confidential portions of this exhibit pursuant to Rule 24b-2 under the Exchange Act. In accordance with Rule 24b-2, these confidential portions have been omitted from this exhibit and filed separately with the Commission
* Filed herewith.
+ To be filed by amendment.
| (1) | Filed as exhibit to 8-K filed on January 31, 2013 and incorporated herein by reference. |
| (2) | Filed as an exhibit to S-1 filed August 17, 2010 and incorporated herein by reference. |
| (3) | Filed as an exhibit to 8-K filed January 23, 2013 and incorporated herein by reference. |
| (4) | Filed as an exhibit to 8-K filed February 14, 2013 and incorporated herein by reference. |
| (5) | Filed as exhibit to 8-K/A filed on March 14, 2103 and incorporated herein by reference. |
| (6) | Filed as an exhibit to 8-K filed June 9, 2014 and incorporated herein by reference. |
| (7) | Filed as an exhibit to 8-K filed October 3, 2014 and incorporated herein by reference. |
| (8) | Filed as an exhibit to 8-K filed November 6, 2014 and incorporated herein by reference. |
| (9) | Filed as an exhibit to 8-K filed January 7, 2015 and incorporated herein. |
| (10) | Filed as an exhibit to S-1 (Registration No. 333-189337) filed June 14, 2013 and incorporated herein by reference. |
II-7