ANNUAL INFORMATION FORM
(for the fiscal year ended November 30, 2021)
INTELLIPHARMACEUTICS INTERNATIONAL INC.
30 Worcester Road
Toronto, Ontario
M9W 5X2
CANADA
February 28, 2022
TABLE OF
CONTENTS
|
|
Page
|
|
REFERENCE
INFORMATION
|
1
|
|
FORWARD-LOOKING
INFORMATION
|
2
|
|
TRADEMARKS
|
3
|
|
CORPORATE
STRUCTURE
|
4
|
|
GENERAL
DEVELOPMENT OF THE BUSINESS
|
5
|
|
CODE
OF CONDUCT
|
31
|
|
RISK
FACTORS
|
31
|
|
DIVIDENDS
|
66
|
|
CAPITAL
STRUCTURE
|
66
|
|
MARKET
FOR SECURITIES
|
69
|
|
DIRECTORS
AND OFFICERS
|
71
|
|
AUDIT
COMMITTEE
|
74
|
|
EXTERNAL
AUDITOR SERVICE FEES
|
79
|
|
INTEREST
OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
|
80
|
|
LEGAL
PROCEEDINGS AND REGULATORY ACTIONS
|
82
|
|
TRANSFER
AGENTS AND REGISTRARS
|
85
|
|
MATERIAL
CONTRACTS
|
86
|
|
INTERESTS
OF EXPERTS
|
89
|
|
ADDITIONAL
INFORMATION
|
89
|
|
SCHEDULE
A – AUDIT COMMITTEE CHARTER
|
A-1
|
|
|
|
INTELLIPHARMACEUTICS INTERNATIONAL INC.
ANNUAL INFORMATION FORM
For the Fiscal Year Ended November 30, 2021
In this
annual information form, unless the context otherwise requires, the
terms “we”, “us”, “our”,
“Intellipharmaceutics” and the “Company”
refer to Intellipharmaceutics International Inc. and its
subsidiaries.
Unless
stated otherwise, all references to “$” are to the
lawful currency of the United States and all references to
“C$” are to the lawful currency of Canada.
Any
reference in this annual information form to our
“products” includes a reference to our product
candidates and future products we may develop. Whenever we refer to
any of our current product candidates (including additional product
strengths of products we are currently marketing and future
products we may develop), no assurances can be given that we, or
any of our strategic partners, will successfully commercialize or
complete the development of any of such product candidates or
future products under development or proposed for development, that
regulatory approvals will be granted for any such product candidate
or future product, or that any approved product will be produced in
commercial quantities or sold profitably.
In this
annual information form, we refer to information regarding
potential markets for our products, product candidates and other
industry data. We believe that all such information has been
obtained from reliable sources that are customarily relied upon by
companies in our industry. However, we have not independently
verified any such information.
We
initially named our oxycodone hydrochloride extended-release
tablets (“Oxycodone ER”) “Rexista™,”
but later changed the name of our product candidate to
“Aximris™” as the United States Food and Drug
Administration (“FDA”) did not approve the proposed
name “Rexista”. References in this annual information
form, to Oxycodone ER, Rexista™ or Aximris™ are
intended to refer to our oxycodone hydrochloride extended release
tablets product candidate.
1
Certain
statements in this annual information form constitute
“forward-looking statements” within the meaning of the
United States Private Securities Litigation Reform Act of 1995
and/or “forward-looking information” under the
Securities Act (Ontario).
These statements include, without limitation, statements expressed
or implied regarding our expectations, plans, goals and milestones,
status of developments or expenditures relating to our business,
plans to fund our current activities, and statements concerning our
partnering activities, health regulatory submissions, strategy,
future operations, future financial position, future sales,
revenues and profitability, projected costs and market penetration.
In some cases, you can identify forward-looking statements by
terminology such as “appear”, “unlikely”,
“target”, “may”, “will”,
“should”, “expects”, “plans”,
“plans to”, “anticipates”,
“believes”, “estimates”,
“predicts”, “confident”,
“prospects”, “potential”,
“continue”, “intends”, “look
forward”, “could”, “would”,
“projected”, “goals”, “set to”,
“seeking” or the negative of such terms or other
comparable terminology. We made a number of assumptions in the
preparation of our forward-looking statements. You should not place
undue reliance on our forward-looking statements, which are subject
to a multitude of known and unknown risks and uncertainties that
could cause actual results, future circumstances or events to
differ materially from those stated in or implied by the
forward-looking statements. Risks, uncertainties and other factors
that could affect our actual results include, but are not limited
to, the effects of general economic conditions, securing and
maintaining corporate alliances, our estimates regarding our
capital requirements, and the effect of capital market conditions
and other factors, including the current status of our product
development programs, capital availability, the estimated proceeds
(and the expected use of any proceeds) we may receive from any
offering of our securities, the potential dilutive effects of any
future financing, potential liability from and costs of defending
pending or future litigation, our ability to comply with the OTCQB
tier of the OTC Markets Group (“OTCQB”) and the Toronto Stock
Exchange (“TSX”)
continued listing standards, our programs regarding research,
development and commercialization of our product candidates, the
timing of such programs, the timing, costs and uncertainties
regarding obtaining regulatory approvals to market our product
candidates and the difficulty in predicting the timing and results
of any product launches, the timing and amount of profit-share
payments from our commercial partners, and the timing and amount of
any available investment tax credits, the actual or perceived
benefits to users of our drug delivery technologies, products and
product candidates as compared to others, our ability to establish
and maintain valid and enforceable intellectual property rights in
our drug delivery technologies, products and product candidates,
the scope of protection provided by intellectual property rights
for our drug delivery technologies, products and product
candidates, recent and future legal developments in the United
States and elsewhere that could make it more difficult and costly
for us to obtain regulatory approvals for our product candidates
and negatively affect the prices we may charge, increased public
awareness and government scrutiny of the problems associated with
the potential for abuse of opioid based medications, pursuing
growth through international operations could strain our resources,
our limited manufacturing, sales, marketing or distribution
capability and our reliance on third parties for such, the actual
size of the potential markets for any of our products and product
candidates compared to our market estimates, our selection and
licensing of products and product candidates, our ability to
attract distributors and/or commercial partners with the ability to
fund patent litigation and with acceptable product development,
regulatory and commercialization expertise and the benefits to be
derived from such collaborative efforts, sources of revenues and
anticipated revenues, including contributions from distributors and
commercial partners, product sales, license agreements and other
collaborative efforts for the development and commercialization of
product candidates, our ability to create an effective direct sales
and marketing infrastructure for products we elect to market and
sell directly, the rate and degree of market acceptance of our
products, delays in product approvals that may be caused by
changing regulatory requirements, the difficulty in predicting the
timing of regulatory approval and launch of competitive products,
the difficulty in predicting the impact of competitive products on
sales volume, pricing, rebates and other allowances, the number of
competitive product entries, and the nature and extent of any
aggressive pricing and rebate activities that may follow, the
inability to forecast wholesaler demand and/or wholesaler buying
patterns, seasonal fluctuations in the number of prescriptions
written for our generic Focalin XR® capsules, which may
produce substantial fluctuations in revenue, the timing and amount
of insurance reimbursement regarding our products, changes in laws
and regulations affecting the conditions required by the FDA for
approval, testing and labeling of drugs including abuse or overdose
deterrent properties, and changes affecting how opioids are
regulated and prescribed by physicians, changes in laws and
regulations, including Medicare and Medicaid, affecting among other
things, pricing and reimbursement of pharmaceutical products, the
effect of recent changes in U.S. federal income tax laws, including
but not limited to, limitations on the deductibility of business
interest, limitations on the use of net operating losses and
application of the base erosion minimum tax, on our U.S. corporate
income tax burden, the success and pricing of other competing
therapies that may become available, our ability to retain and hire
qualified employees, the availability and pricing of third-party
sourced products and materials, challenges related to the
development, commercialization, technology transfer, scale-up,
and/or process validation of manufacturing processes for our
products or product candidates, the manufacturing capacity of
third-party manufacturers that we may use for our products,
potential product liability risks, the recoverability of the cost
of any pre-launch inventory, should a planned product launch
encounter a denial or delay of approval by regulatory bodies, a
delay in commercialization, or other potential issues, the
successful compliance with FDA, Health Canada and other
governmental regulations applicable to us and our third party
manufacturers’ facilities, products and/or businesses, our
reliance on commercial partners, and any future commercial
partners, to market and commercialize our products and, if
approved, our product candidates, difficulties, delays, or changes
in the FDA approval process or test criteria for Abbreviated New
Drug Applications (“ANDAs”) and New Drug Applications
(“NDAs”),
challenges in securing final FDA approval for our product
candidates, including our Oxycodone ER product candidate, in
particular, if a patent infringement suit is filed against us with
respect to any particular product candidates (such as in the case
of Oxycodone ER), which could delay the FDA’s final approval
of such product candidates, healthcare reform measures that could
hinder or prevent the commercial success of our products and
product candidates, the risk that the FDA may not approve requested
product labeling for our product candidate(s) having
abuse-deterrent properties and targeting common forms of abuse
(oral, intra-nasal and intravenous), risks associated with
cyber-security and the potential for vulnerability of our digital
information or the digital information of a current and/or future
drug development or commercialization partner of ours, and risks
arising from the ability and willingness of our third-party
commercialization partners to provide documentation that may be
required to support information on revenues earned by us from those
commercialization partners.
2
Additional
risks and uncertainties relating to us and our business can be
found in the “Risk Factors” section of this annual
information form as well as in our reports, public disclosure
documents and other filings with the securities commissions and
other regulatory bodies in Canada and the U.S., which are available
on www.sedar.com and
www.sec.gov. The
forward-looking statements reflect our current views with respect
to future events, and are based on what we believe are reasonable
assumptions as of the date of this annual information form and we
disclaim any intention and have no obligation or responsibility,
except as required by law, to update or revise any forward-looking
statements, whether as a result of new information, future events
or otherwise.
Nothing
contained in this annual information form should be construed to
imply that the results discussed herein will necessarily continue
into the future, or that any conclusion reached herein will
necessarily be indicative of our actual operating
results.
Intellipharmaceutics™,
Hypermatrix™, Drug Delivery Engine™,
IntelliFoam™, IntelliGITransporter™,
IntelliMatrix™, IntelliOsmotics™, IntelliPaste™,
IntelliPellets™, IntelliShuttle™, nPODDDS™,
PODRAS™. Regabatin™ and Aximris XR™ are our
trademarks. These trademarks are important to our business.
Although we may have omitted the “TM” trademark
designation for such trademarks in this Annual information Form,
all rights to such trademarks are nevertheless reserved. Unless
otherwise noted, other trademarks used in this Annual Information
Form are the property of their respective holders.
We
initially named our Oxycodone ER product candidate
“Rexista™,” but later changed the name of our
product candidate to “Aximris XR™” as the FDA did
not approve the proposed name “Rexista”. References in
this Annual information Form, to Oxycodone ER, Rexista™ or
Aximris XR™ are intended to refer to our oxycodone
hydrochloride extended release tablets product
candidate.
3
CORPORATE STRUCTURE
Intellipharmaceutics
was incorporated under the Canada
Business Corporations Act by certificate and articles of
arrangement dated October 22, 2009.
The
following chart shows the corporate relationship structure of
Intellipharmaceutics and its three wholly-owned subsidiaries,
including jurisdictions of incorporation, as at November 30,
2021.
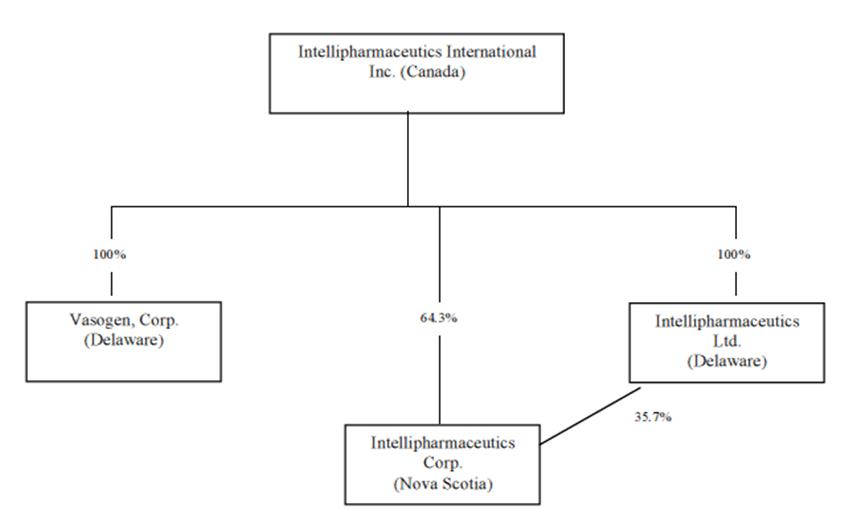
4
Our
registered and principal office is located at 30 Worcester Road,
Toronto, Ontario, Canada M9W 5X2. Our telephone number is (416)
798-3001 and our facsimile number is (416) 798-3007.
We are
currently a “reporting issuer” in all of the provinces
and territories of Canada.
Our
website is www.intellipharmaceutics.com. Any information contained
on our website is not, and will be deemed not to be, incorporated
herein by reference.
We are
a pharmaceutical company specializing in the research, development
and manufacture of novel and generic controlled-release and
targeted-release oral solid dosage drugs. Our patented
Hypermatrix™ technology is a multidimensional
controlled-release drug delivery platform that can be applied to
the efficient development of a wide range of existing and new
pharmaceuticals. Based on this technology platform, we have
developed several drug delivery systems and a pipeline of products
(some of which have received FDA or Health Canada approval) and
product candidates in various stages of development, including
ANDAs filed with the FDA . and one NDA filing, in therapeutic areas
that include neurology, cardiovascular, gastrointestinal tract
(“GIT”),
diabetes and pain.
Overview of Recent Corporate Developments
In
February 2022 the Company received marketing approval for the
Canadian market from Health Canada (notice of compliance) for
generic Pristiq (desvenlafaxine succinate extended-release tablets)
in the 50 and 100 mg strengths.
In
November 2021 the Company received final FDA approval for its
Dexmethylphenidate Extended-release Capsules in the 5 mg, 10 mg, 30
mg and 40 mg strengths. The 15 mg and 30 mg strengths were
initially approved and commercialized by Par Pharmaceutical Inc.
(“Par”) in
November 2013; the additional strengths launched at the time were
approved in a Par abbreviated new drug application.
Effective
May 5, 2021 our exclusive license agreements with Tris Pharma, Inc.
(“Tris Pharma”)
for generic Seroquel XR®, generic Pristiq® and generic
Effexor XR® for the US market were mutually terminated.
Products were never supplied nor distributed under the licenses.
Termination of the exclusive agreements may provide opportunity for
the Company to explore options of supplying the products to
multiple sources on non-exclusive bases.
5
On
April 22, 2021, the Company announced the completion of a
non-brokered private placement (the "Private Placement") of 9,414,560 common
shares of the Company (the "Common
Shares") at a price of CAD$0.41 per Common Share for total
gross proceeds of CAD$3,859,969.60, subject to the final acceptance
by the TSX. The Common Shares were subject to a four-month hold
period that expired on August 22, 2021 in accordance with
applicable securities legislation and the policies of the TSX. The
Common Shares were sold only to non-U.S. persons outside of the
United States pursuant to Regulation S under the United States
Securities Act of 1933 as amended (“U.S. Securities Act”). The Common
Shares issued in the Private Placement were not registered under
the U.S. Securities Act or the securities laws of any state in the
United States and may not be offered or sold in the United States
or to, or for the account or benefit of, U.S. persons (as defined
in Regulation S under the U.S. Securities Act) or persons in the
United States absent registration or an applicable exemption from
such registration requirements. The TSX approved the private
placement. The proceeds of the Private Placement were used to
maintain the Company’s existing operations and for general
working capital purposes and to fund research and development
activities.
On July
2, 2020 the Company had announced that the parties in the cases,
numbers 17-cv-392-RGA, 18-cv-404-RGA and 20-cv-515-RGA
(collectively, the “Litigations”) between Purdue
Pharma L.P. et al (“Purdue”) and Intellipharmaceutics
entered into a stipulated dismissal of the Litigations. On July 28,
2020 the United States District Court for the District of Delaware
signed the stipulations of dismissal into order thereby dismissing
the claims in the three cases without prejudice. The stipulated
dismissal, which was subject to approval by the bankruptcy court
presiding over Purdue’s pending chapter 11 cases, provides
for the termination of patent infringement proceedings commenced by
Purdue against the Company in the United States District Court for
the District of Delaware in respect of the Company’s NDA
filing for Aximris XR™ with the FDA. The stipulated dismissal
also provides that (i) for a thirty (30) day period following a
final approval of the Company’s Aximris XR™ NDA the
parties will attempt to resolve any potential asserted patent
infringement claims relating to the NDA and (ii) if the parties
fail to resolve all such claims during such period Purdue will have
fifteen (15) days to pursue an infringement action against the
Company. The terms of the stipulated dismissal agreement are
confidential. In consideration of the confidential stipulated
dismissal agreement and for future saved litigation expenses,
Purdue has paid an amount to the Company.
On
January 15, 2020, at a joint meeting of the Anesthetic and
Analgesic Drug Products Advisory Committee and Drug Safety and Risk
Management Advisory Committee (collectively, the
“Advisory
Committees”) of the FDA to discuss our NDA for Aximris
XR™, abuse-deterrent oxycodone hydrochloride extended-release
tablets, the Advisory Committees voted 24 to 2 against the approval
of our NDA for Aximris XR™ for the management of pain severe
enough to require daily, around-the-clock, long-term opioid
treatment and for which alternative treatment options are
inadequate. We expect the FDA to take action on our application, on
completion of their review of the NDA. To date the FDA has not
communicated any action to the Company.
History
On
October 19, 2009, the shareholders of Intellipharmaceutics Ltd.
(“IPC Ltd.”) and
Vasogen Inc. (“Vasogen”) approved a
court-approved plan of arrangement and merger (the
“IPC Arrangement
Agreement”) that resulted in the October 22, 2009
combination of IPC Ltd. and Intellipharmaceutics Corp. with 7231971
Canada Inc., a new Vasogen company that acquired substantially all
of the assets and certain liabilities of Vasogen, including the
proceeds from its non-dilutive financing transaction with Cervus LP
(the “IPC Arrangement
Transaction”). The completion of the IPC Arrangement
Transaction on October 22, 2009 resulted in the formation of the
Company, which is incorporated under the laws of Canada. The Common
Shares began trading on the TSX and Nasdaq up to March 21, 2019
when the Company was delisted from the Nasdaq. The Common Shares
are currently trading on the TSX and OTCQB.
6
Nasdaq Delisting and OTCQB Quotation
In
March 2019, we received formal notice that the Nasdaq Panel had
determined to delist our Common Shares from Nasdaq based upon our
non-compliance with the $1.00 bid price requirement, as set forth
in Nasdaq Listing Rule 5550(a)(2). The suspension of trading on
Nasdaq took effect at the open of business on March 21, 2019. Our
Common Shares began trading on the OTCQB under the symbol
“IPCIF”, commencing on March 21, 2019. Our Common
Shares are also listed on the TSX under the symbol
“IPCI” and our non-compliance with Nasdaq's
requirements did not impact our listing or trading status on the
TSX.
Our Strategy
Our
Hypermatrix™ technologies are central to the development and
manufacture of novel and generic controlled-release and
targeted-release oral solid dosage drugs. The Hypermatrix™
technologies are a multidimensional controlled-release drug
delivery platform that we believe can be applied to the efficient
development of a wide range of existing and new pharmaceuticals. We
believe that the flexibility of these technologies allows us to
develop complex drug delivery solutions within an
industry-competitive timeframe. Based on this technology platform,
we have developed several drug delivery systems and a pipeline of
products (some of which have received FDA approval) and product
candidates in various stages of development, including ANDAs filed
with the FDA (and one ANDS filed with Health Canada) and one NDA
filing, in therapeutic areas that include neurology,
cardiovascular, GIT, diabetes and pain. We expect that certain, but
not all, of the products in our pipeline may be developed from time
to time for third parties pursuant to drug development agreements
with those third parties, under which our commercialization partner
may pay certain of the expenses of development, make certain
milestone payments to us and receive a share of revenues or profits
if the drug is developed successfully to completion, the control of
which would generally be in the discretion of our drug development
partner.
The principal focus of our development activities
previously targeted difficult-to-develop controlled-release generic
drugs which follow an ANDA regulatory pathway. Later, our
development program became increasingly directed towards improved
difficult-to-develop controlled-release drugs which follow an NDA
505(b)(2) regulatory pathway. We increased emphasis towards
specialty new product development, facilitated by the 505(b)(2)
regulatory pathway, by advancing the product development program
for Oxycodone ER and RegabatinTM XR, and commencing
other projects in our 505(b)(2) pipeline. We work on these and
other product candidates as resources permit. In January 2019, we
announced that we had commenced a research and development
(“R&D”)
program of pharmaceutical cannabidiol (“CBD”)-based products. As part of
the CBD-based R&D program, we filed provisional patent
applications with the United States Patent and Trademark Office
pertaining to the delivery and application of cannabinoid-based
therapeutics. There can be no assurance that any of our provisional
patent applications will successfully mature into patents. The
Company holds a Health Canada Cannabis Drug License
(“CDL”). Under
the CDL, we are authorized to possess, produce, sell and deliver
drug products containing CBD in Canada.We had also previously
identified several additional 505(b)(2) product candidates for
development in various areas including cardiovascular, dermatology,
pulmonary disease and oncology. We are still exploring the
potential development of such product candidates when resources are
available. The technology
that is central to our abuse deterrent formulation of our Oxycodone
ER is the nPODDDS™, or novel Point of Divergence Drug
Delivery System. nPODDDS™ is designed to provide for certain
unique drug delivery features in a product. These include the
release of the active substance to show a divergence in a
dissolution and/or bioavailability profile. The divergence
represents a point or a segment in a release timeline where the
release rate, represented by the slope of the curve, changes from
an initial rate or set of rates to another rate or set of rates,
the former representing the usually higher rate of release shortly
after ingesting a dose of the drug, and the latter representing the
rate of release over a later and longer period of time, being more
in the nature of a controlled-release or sustained action. It is
applicable for the delivery of opioid analgesics in which it is
desired to discourage common methods of tampering associated with
misuse and abuse of a drug, and also dose dumping in the presence
of alcohol. It can potentially retard tampering without interfering
with the bioavailability of the product.
7
In
addition, our PODRAS™, or Paradoxical OverDose Resistance
Activating System, delivery technology was initially introduced to
enhance our Oxycodone ER (abuse deterrent oxycodone hydrochloride
extended release tablets) product candidate. The PODRAS™
delivery technology platform was designed to prevent an overdose
when more pills than prescribed are swallowed intact. Preclinical
studies of prototypes of oxycodone with PODRAS™ technology
suggest that, unlike other third-party abuse-deterrent oxycodone
products in the marketplace, if more tablets than prescribed are
deliberately or inadvertently swallowed, the amount of drug active
ingredient (“drug
active”) released over 24 hours may be substantially
less than expected. However, if the prescribed number of pills is
swallowed, the drug release should be as expected. Certain aspects
of our PODRAS™ technology are covered by U.S. Patent Nos.
9,522,119, 9,700,515, 9,700,516 and 9,801,939 and Canadian Patent
No. 2,910,865 issued by the U.S. Patent and Trademark Office and
the Canadian Intellectual Property Office in respect of
“Compositions and Methods for Reducing Overdose” in
December 2016, July 2017 and October 2017, respectively. The
issuance of these patents provides us with the opportunity to
accelerate our PODRAS™ development plan by pursuing proof of
concept studies in humans. We intend to incorporate this technology
in future product candidates, including Oxycodone ER and other
similar pain products, as well as pursuing out-licensing
opportunities. The development of an Oxycodone immediate-release
(IR) product incorporating this technology as resources permit is
in the Company’s development pipeline.
The NDA
505(b)(2) pathway (which relies in part upon the FDA’s
findings for a previously approved drug) both accelerates
development timelines and reduces costs in comparison to NDAs for
new chemical entities. An advantage of our strategy for development
of NDA 505(b)(2) drugs is that our product candidates can, if
approved for sale by the FDA, potentially enjoy an exclusivity
period which may provide for greater commercial opportunity
relative to the generic ANDA route.
The
market we operate in is created by the expiration of drug product
patents, challengeable patents and drug product exclusivity
periods. There are three ways that we employ our controlled-release
technologies, which we believe represent substantial opportunities
for us to commercialize on our own or develop products or
out-license our technologies and products:
●
For branded
immediate-release (multiple-times-per-day) drugs, we can formulate
improved replacement products, typically by developing new,
potentially patentable, controlled-release once-a-day drugs. Among
other out-licensing opportunities, these drugs can be licensed to
and sold by the pharmaceutical company that made the original
immediate-release product. These can potentially protect against
revenue erosion in the brand by providing a clinically attractive
patented product that competes favorably with the generic
immediate-release competition that arises on expiry of the original
patent(s). The regulatory pathway for this approach requires NDAs
via a 505(b)(2) application for the U.S. or corresponding pathways
for other jurisdictions where applicable.
●
Some of our
technologies are also focused on the development of abuse-deterrent
and overdose preventive pain medications. The growing abuse and
diversion of prescription “painkillers”, specifically
opioid analgesics, is well documented and is a major health and
social concern. We believe that our technologies and know-how are
aptly suited to developing abuse-deterrent pain medications. The
regulatory pathway for this approach requires NDAs via a 505(b)(2)
application for the U.S. or corresponding pathways for other
jurisdictions where applicable.
●
For existing
controlled-release (once-a-day) products whose active
pharmaceutical ingredients (“APIs”) are covered by drug
molecule patents about to expire or already expired, or whose
formulations are covered by patents about to expire, already
expired or which we believe we do not infringe, we can seek to
formulate generic products which are bioequivalent to the branded
products. Our scientists have demonstrated a successful track
record with such products, having previously developed several drug
products which have been commercialized in the U.S. by their former
employer/clients. The regulatory pathway for this approach requires
ANDAs for the U.S. and ANDSs for Canada.
8
We intend to collaborate in the development and/or marketing of one
or more products with partners, when we believe that such
collaboration may enhance the outcome of the project. We also plan
to seek additional collaborations as a means of developing
additional products. We believe that our business strategy enables
us to reduce our risk by (a) having a diverse product portfolio
that includes both branded and generic products in various
therapeutic categories, and (b) building collaborations and
establishing licensing agreements with companies with greater
resources thereby allowing us to share costs of development and to
improve cash-flow. There can be no assurance that we will be able
to enter into additional collaborations or, if we do, that such
arrangements will be commercially viable or
beneficial.
Our Drug Delivery Technologies
HypermatrixTM
Our
scientists have developed drug delivery technology systems, based
on the Hypermatrix™ platform, that facilitate
controlled-release delivery of a wide range of pharmaceuticals.
These systems include several core technologies, which enable us to
flexibly respond to a wide range of drug attributes and patient
requirements, producing a desired controlled-release effect. Our
technologies have been incorporated in drugs manufactured and sold
by major pharmaceutical companies.
This
group of drug delivery technology systems is based upon the drug
active being imbedded in, and an integral part of, a homogeneous
(uniform), core and/or coatings consisting of one or more polymers
which affect the release rates of drugs, other excipients
(compounds other than the drug active), such as for instance
lubricants which control handling properties of the matrix during
fabrication, and the drug active itself. The Hypermatrix™
technologies are the core of our current marketing efforts and the
technologies underlying our existing development
agreements.
nPODDDSTM
In
addition to continuing efforts with Hypermatrix™ as a core
technology, our scientists continue to pursue novel research
activities that address unmet needs. Oxycodone ER (abuse deterrent
oxycodone hydrochloride extended release tablets) is an NDA
candidate, with a unique long acting oral formulation of oxycodone
intended to treat moderate-to-severe pain. The formulation is
intended to present a significant barrier to tampering when
subjected to various forms of physical and chemical manipulation
commonly used by abusers. It is also designed to prevent dose
dumping when inadvertently co-administered with alcohol. The
technology that supports our abuse deterrent formulation of
oxycodone is the nPODDDS™ Point of Divergence Drug Delivery
System. The use of nPODDDS™ does not interfere with the
bioavailability of oxycodone. We intend to apply the nPODDDS™
technology platforms to other extended release opioid drug
candidates (e.g., oxymorphone, hydrocodone, hydromorphone and
morphine) utilizing the 505(b)(2) regulatory pathway.
9
PODRASTM
Our
Paradoxical OverDose Resistance Activating System (PODRAS™)
delivery technology is designed to prevent overdose when more pills
than prescribed are swallowed intact. Preclinical studies of
prototypes of oxycodone with PODRAS™ technology suggest that,
unlike other third-party abuse-deterrent oxycodone products in the
marketplace, if more tablets than prescribed are deliberately or
inadvertently swallowed, the amount of drug active released over 24
hours may be substantially less than expected. However, if the
prescribed number of pills is swallowed, the drug release should be
as expected. We started, and intend to continue, working on an
alternate Oxycodone ER product candidate incorporating our
PODRAS™ delivery technology. In April 2015, the FDA published
Guidance for Industry:
Abuse-Deterrent Opioids — Evaluation and Labeling,
which cited the need for more efficacious abuse-deterrence
technology. In this Guidance, the FDA stated, “opioid
products are often manipulated for purposes of abuse by different
routes of administration or to defeat extended-release properties,
most abuse-deterrent technologies developed to date are intended to
make manipulation more difficult or to make abuse of the
manipulated product less attractive or less rewarding. It should be
noted that these technologies have not yet proven successful at
deterring the most common form of abuse—swallowing a number
of intact capsules or tablets to achieve a feeling of
euphoria.” The FDA reviewed our request for Fast Track
designation for our abuse deterrent Oxycodone ER development
program incorporating PODRAS™, and in May 2015 notified us
that the FDA had concluded that it had met the criteria for Fast
Track designation. Fast Track is a designation assigned by the FDA
in response to an applicant’s request which meets FDA
criteria. The designation mandates the FDA to facilitate the
development and expedite the review of drugs intended to treat
serious or life threatening conditions and that demonstrate the
potential to address unmet medical needs.
In
December 2016, July 2017 and October 2017, U.S. Patent Nos.
9,522,119, 9,700,515, 9,700,516 and 9,801,939 and Canadian Patent
No. 2,910,865 were issued by the U.S. Patent and Trademark Office
and the Canadian Intellectual Property Office in respect of
“Compositions and Methods for Reducing Overdose”. The
issued patents cover aspects of the PODRAS™ delivery
technology. The issuance of these patents represents a significant
advance in our abuse deterrence technology platform. The
PODRAS™ platform has the potential to positively
differentiate our technology from others of which we are aware, and
may represent an important step toward addressing the FDA’s
concern over the ingestion of a number of intact pills or tablets.
In addition to its use with opioids, the PODRASTM platform is
potentially applicable to a wide range of drug products, inclusive
of over-the-counter drugs, that are intentionally or inadvertently
abused and cause harm by overdose to those who ingest them. We
intend to apply the PODRAS™ technology platforms to other
extended release opioid drug candidates (e.g., oxymorphone,
hydrocodone, hydromorphone and morphine) utilizing the 505(b)(2)
regulatory pathway.
The HypermatrixTM Family of
Technologies
Our
platform of Hypermatrix™ drug delivery technologies include,
but are not limited to, IntelliFoam™,
IntelliGITransporter™, IntelliMatrix™,
IntelliOsmotics™, IntelliPaste™, IntelliPellets™,
IntelliShuttle™, nPODDDS™ and PODRAS™. Some of
their key attributes are described below.
These
technologies provide a broad range of release profiles, taking into
account the physical and chemical characteristics of a drug
product, the therapeutic use of the particular drug, and the
optimal site for release of the API in the GIT. At present those
technologies have been applied in the laboratory and/or in
bioavailability/bioequivalence studies in man to such orally
administered small molecule drugs as are used in the treatment of
neurological, cardiovascular, GIT, diabetes, pain and other
significant indications.
10
IntelliFoamTM
The
IntelliFoam™ technology is based on the drug active being
embedded in, but separate from a syntactic foam substrate, the
properties of which are used to modulate the release of the drug
active. The drug actives are embedded in a resin polymer
matrix.
IntelliGITransporterTM
The
IntelliGITransporter™ technology consists of an active drug
immobilized in a homogeneous (uniform) matrix structure. A precise
choice of mix ratios, polymers, and other ingredients imparts
characteristics which protect the drug composition from mechanical
degradation due to digestion, and/or from chemical degradation in
the acidic stomach environment, and ensures that this technology
allows control of release as well as releasing the medication at
certain parts of the stomach or intestines without significant food
effects or unintentional premature release of the entire drug dose.
We believe that this technology is most useful for drug molecules
with characteristics such as very low or very high potency, opiate
analgesics (pain medications derived from the chemical compounds
found in opium), or susceptibility to acid degradation. It is also
useful for products where a zero-order (constant rate over time,
independent of the amount of drug available for dissolution)
release profile is desirable.
IntelliMatrixTM
The
IntelliMatrix™ technology is a proprietary blend of several
polymers. Depending on the constituents of the blend and the manner
in which these interact, the use of the blend with a drug allows
the drug to be released at predetermined rates, while imparting
protective characteristics to both the drug and the GIT. This is
most useful for drugs which require precisely controlled
first-order release profiles, where the amount released with time
is dependent on one component like the amount of drug available for
dissolution.
IntelliOsmoticsTM
The
IntelliOsmotics™ technology is based upon the inclusion of
multiple populations of polymers with distinct chemical bonding
characteristics. These set up a complex matrix of hydrophilic
(water attracting) and hydrophobic (water repelling) domains. When
the tablet or bead is in an aqueous environment, like gastric
contents, a “mixture” of water-soluble polymer and drug
core is surrounded by gel layer(s) of water-insoluble polymer.
Osmotic pressure drives the drug out when solvent passes through
the gel layer while the polymer molecules remain. This permits
control of the rate of release of the drug active by the variation
of polymer ratios. This technology is most useful for drug
molecules which require precisely controlled pseudo-first-order
release profiles, where the rate of release is proportional to the
amount available for dissolution as well as being proportional to
one other component; however the effect of the amount of drug is
overriding, so that the rate appears first-order. This type of
release control can be useful when attempting to match difficult
profiles for generic formulation.
11
IntelliPasteTM
The
IntelliPaste™ technology is comprised of blends of multiple
polymers, oils, excipients and drug active(s) which result in a
paste-in-a-capsule dosage form. The physical attributes of the
paste include that it is thixotropic, pseudoplastic and
non-Newtonian or, in layman’s terms, like toothpaste.
Typically, it is formulated as having very low solubility in water
or oil, and low solubility in alcohol. These characteristics enable
the resulting drug product to have tamper-deterrent properties, and
to resist dissolution in even high concentrations of alcohol. As a
result, IntelliPaste™ is our preferred delivery technology
for the controlled delivery of opiates, narcotics and other central
nervous system drug products which are susceptible to unlawful
diversion or abuse.
IntelliPelletsTM
The
IntelliPellets™ technology consists of one or more type
(population) of granule, bead, pellet, or tablet in a holding
chamber or reservoir, such as a hard gelatin capsule. Each type
(population) may be uniquely different from the other in the manner
or rate it releases the drug. Our IntelliPellets™ technology
is designed to control, prolong, delay or modify the release of
drugs. It is particularly useful for the delivery of multiple
drugs, for delayed, timed, pulsed or for chronotherapeutic drug
delivery, designed to mimic our internal clocks for therapeutic
optimization (the drug is delivered in the right amount for the
patient at the right time). This technology is most useful for the
delivery of multiple-drug cocktails, or in situations where the
timing of a single dose or the sequencing of multiple doses of the
same drug is important.
IntelliShuttleTM
The
IntelliShuttle™ technology provides for drug release past the
stomach, such as for drugs required for action beyond the stomach,
for drugs which could be destroyed by the stomach environment, or
for drugs which could harm the stomach itself. This technology
“shuttles” the drug past the stomach to be released at
predetermined times or sites where appropriate for optimum
therapeutic effect. This technology is most useful for acid labile
drug molecules (drugs that are destroyed in acid environment), such
as the proton pump inhibitors, of which well-known omeprazole
(Prilosec) and lansoprazole (Prevacid) are examples, or for drug
molecules which may harm the stomach, of which the well-known
aspirin is an example.
Each of
the above-noted proprietary technologies was fully developed and
ready for application to client drug delivery requirements from the
date of our inception. Each of them has been utilized and applied
to client drug delivery requirements under our existing and
previous development contracts; in several instances more than one
technology has been applied to a single drug development. We
continue to develop all of our existing technologies and to conduct
the necessary research to develop new products and
technologies.
12
Our Products and Product Candidates
The
table below shows the present status of our ANDA, ANDS and NDA
products and product candidates that have been disclosed to the
public.
|
Generic name
|
Brand
|
Indication
|
Stage of
Development(1)
|
Regulatory Pathway
|
Rights(2)
|
|
Dexmethylphenidate hydrochloride extended-release
capsules
|
Focalin XR®
|
Attention deficit hyperactivity disorder
|
Received final approval for 5, 10, 15, 20, 25, 30, 35 and 40 mg
strengths from FDA(3)
Received final approval for 5 mg, 10 mg, 30 mg and 40 mg in
Intellipharmaceutics’ ANDA
|
ANDA
|
Intellipharmaceutics and Par (US)
Philippines rights subject to licensing and distribution
agreement
|
|
Levetiracetam extended-release tablets
|
Keppra XR®
|
Partial onset seizures for epilepsy
|
Received final approval for the 500 and 750 mg strengths from
FDA
|
ANDA
|
ANDA Repository(4)
|
|
Venlafaxine hydrochloride extended-release capsules
|
Effexor XR®
|
Depression
|
Received final approval for 37.5, 75 and 150 mg strengths from
FDA
|
ANDA
|
Intellipharmaceutics.
|
|
Pantoprazole sodium delayed- release tablets
|
Protonix®
|
Conditions associated with gastroesophageal reflux
disease
|
ANDA Application for commercialization approval for 2 strengths
under review by FDA
|
ANDA
|
Intellipharmaceutics
|
|
Metformin hydrochloride extended-release tablets
|
Glucophage® XR
|
Management of type 2 diabetes
|
Received final approval for 500 and 750 mg strengths from
FDA
|
ANDA
|
Intellipharmaceutics
Philippines and Vietnamese rights subject to licensing and
distribution agreements
|
|
Quetiapine fumarate extended-release tablets
|
Seroquel XR®
|
Schizophrenia, bipolar disorder & major depressive
disorder
|
Received final FDA approval for all 5 strengths. .
|
ANDA
ANDS
|
Intellipharmaceutics.
Philippines, Malaysian and Vietnamese rights subject to licensing
and distribution agreements
..
|
|
Lamotrigine extended-release tablets
|
Lamictal® XR™
|
Anti-convulsant for epilepsy
|
ANDA application for commercialization approval for 6 strengths
under review by FDA
|
ANDA
|
Intellipharmaceutics
|
|
Desvenlafaxine extended-release tablets
|
Pristiq®
|
Depression
|
Received approval for the 50 and 100 mg strengths from
FDA.
received approval (notice of compliance) from Health Canada for the
50 mg and 100 mg strengths
|
ANDA
ANDS
|
Intellipharmaceutics.
|
|
Carvedilol phosphate extended-release capsules
|
Coreg CR®
|
Heartfailure, hypertension
|
Late-stage development
|
ANDA
|
Intellipharmaceutics
|
|
Oxycodone hydrochloride controlled-release capsules
|
|
Pain
|
NDA application accepted February 2017 and under review by FDA.
Second FDA Advisory Committees meeting held January 2020. Awaiting
action from FDA
|
NDA 505(b)(2)
|
Intellipharmaceutics
|
|
Pregabalin extended-release capsules
|
|
Neuropathic pain
|
IND application submitted in August 2015
|
NDA 505(b)(2)
|
Intellipharmaceutics
|
|
Ranolazine extended-release tablets
|
Ranexa®
|
Chronic angina
|
ANDA application for commercialization approval for 2 strengths
under review by FDA
|
ANDA
|
Intellipharmaceutics
|
|
Oxycodone hydrochloride immediate release tablets
(IPCI006)
|
|
Pain
|
IND application submitted in November 2018
|
NDA 505(b)(2)
|
Intellipharmaceutics
|
13
Notes:
(1)
There
can be no assurance as to when, or if at all, the FDA or Health
Canada will approve any product candidate for sale in the U.S. or
Canadian markets.
(2)
For
information regarding the Par agreement, the Tris Pharma agreement,
and the licensing and distribution agreements with pharmaceutical
distributors in Malaysia, Vietnam and the Philippines, see
“Business Overview” and “Other Potential Products
and Markets” sections. There can be no assurance as to when,
or if at all, any of our products or product candidates, as the
case may be, will receive regulatory approval for sale in the
Philippines, Malaysia or Vietnam. For unpartnered products, we are
seeking licensing agreement opportunities or other opportunities.
While we believe that licensing agreements are possible, there can
be no assurance that any can be secured.
(3)
Includes
a Company ANDA final approval for our 15 and 30 mg strengths, and a
Par ANDA final approval for their 5, 10, 15, 20, 25, 30, 35 and 40
mg strengths. Profit sharing payments to us under the Par agreement
are the same irrespective of the ANDA owner.
(4)
As at September 30, 2019, pursuant to an ANDA sale
agreement (the “Levetiracetam ANDA
Agreement”), we sold all
of the assets relating to our ANDA for Levetiracetam
extended-release 500mg and 750 mg tablets (collectively, the
“Transferred Levetiracetam
ANDA”) to ANDA
Repository, LLC (the “Levetiracetam ANDA
Purchaser”) in exchange
for a purchase price of $1. Additionally, pursuant to the
Levetiracetam ANDA Agreement, we agreed to pay the Levetiracetam
ANDA Purchaser an annual fee for each fiscal year equal to 50% of
the difference between the FDA Program Fee for 6 to 19 approved
ANDAs and the FDA Program Fee for 1 to 5 approved ANDAs. Under the
Levetiracetam ANDA Agreement, we have the option to repurchase the
Transferred Levetiracetam ANDA for a purchase price of $1 at any
time, provided that any outstanding fees are paid to ANDA
Repository.
Dexmethylphenidate Hydrochloride
– Generic Focalin XR® (a registered trademark of
the brand manufacturer)
Dexmethylphenidate hydrochloride, a Schedule II restricted product
(drugs with a high potential for abuse) in the U.S., is indicated
for the treatment of attention deficit hyperactivity disorder. In
November 2005, we entered into the Par agreement pursuant to which
we granted Par an exclusive, royalty-free license to make and
distribute in the U.S. all of our FDA approved strengths of our
generic Focalin XR® (dexmethylphenidate hydrochloride
extended-release) capsules for a period of 10 years from the date
of commercial launch (which was November 19, 2013). We retain the
right to make and distribute all strengths of the generic product
outside of the U.S. Calendar quarterly profit-sharing payments for
its U.S. sales of all strengths of generic Focalin XR® are
payable by Par to us as calculated pursuant to the Par
agreement.
14
We received final approval from the FDA in November 2013 under the
Company ANDA to launch the 15 and 30 mg strengths of our generic
Focalin XR® capsules. Commercial sales of these strengths were
launched immediately by our commercialization partner in the U.S.,
Par. Our 5, 10, 20 and 40 mg strengths were also then tentatively
FDA approved, subject to the right of Teva Pharmaceuticals USA,
Inc. (“Teva”) to 180 days of generic exclusivity from
the date of first launch of such products. In January 2017, Par
launched the 25 and 35 mg strengths of its generic Focalin XR®
capsules in the U.S., and in May 2017, Par launched the 10 and 20
mg strengths, complementing the 15 and 30 mg strengths of our
generic Focalin XR® marketed by Par. In November 2017, Par
launched the remaining 5 and 40 mg strengths providing us with the
full line of generic Focalin XR® strengths available in the
U.S. market. The Company has now received final FDA approval for
the 5 mg, 10 mg, 30 mg and 40 mg strengths in its
ANDA.
In November 2018, we announced that we entered into an exclusive
licensing and distribution agreement with a pharmaceutical
distributor in the Philippines pursuant to which the distributor
was granted the exclusive right, subject to regulatory approval, to
import and market our generic Focalin XR® in the Philippines.
Under the terms of the agreement, the distributor will be required
to purchase a minimum yearly quantity of our generic Focalin
XR® and we will be the exclusive supplier of such product.
This multi-year agreement is subject to early termination. There
can be no assurance as to when and if such product will receive
regulatory approval for the sale in the Philippines or that, if so
approved, the product will be successfully commercialized there and
produce significant revenues for us.
Levetiracetam – Generic
Keppra XR® (a registered trademark of the brand
manufacturer)
We received final approval from the FDA in February 2016 for the
500 and 750 mg strengths of our generic Keppra XR®
(levetiracetam extended-release) tablets. Keppra XR®, and the
drug active levetiracetam, are indicated for use in the treatment
of partial onset seizures associated with epilepsy. We are aware
that several other generic versions of this product are currently
available and serve to limit the overall market opportunity. We
have been exploring licensing and other options for this
product.
In November 2018, we announced that we entered into two exclusive
licensing and distribution agreements with pharmaceutical
distributors in Vietnam and the Philippines pursuant to which the
distributors were granted the exclusive right, subject to
regulatory approval, to import and market our generic Keppra
XR® in Vietnam and the Philippines, respectively. Under the
terms of the agreements, the distributors will be required to
purchase a minimum yearly quantity of our generic Keppra XR®.
These multi-year agreements are each subject to early termination.
There can be no assurance that the Company’s generic Keppra
XR® for the 500 and 750 mg strengths will be successfully
commercialized in the US market. Further, there can be no assurance
as to when and if such product will receive regulatory approval for
the sale in Vietnam or the Philippines or that, if so approved, the
product will ever be successfully commercialized there and produce
significant revenues for us.
On September 30, 2019, pursuant to an ANDA sale agreement (the
“Levetiracetam ANDA Agreement”), we sold all of the
assets relating to our ANDA for Levetiracetam extended-release 500
mg and 750 mg tablets (collectively, the “Transferred
Levetiracetam ANDA”) to ANDA Repository LLC (the
“Levetiracetam ANDA Purchaser”) in exchange for a
purchase price of $1. Additionally, pursuant to the Levetiracetam
ANDA sale agreement, we agreed to pay the Levetiracetam ANDA
Purchaser an annual fee for each fiscal year equal to 50% of the
difference between the FDA Program Fee for 6 to 19 approved ANDAs
and the FDA Program Fee for 1 to 5 approved ANDAs. Under the
Levetiracetam ANDA Agreement, we have the option to repurchase the
Transferred Levetiracetam ANDA for a purchase price of $1 at any
time, provided that any outstanding fees are paid to ANDA
Repository.
15
Metformin hydrochloride –
Generic Glucophage® XR (a registered trademark of the
brand manufacturer)
We received final approval from the FDA in February 2017 for the
500 and 750 mg strengths of our generic Glucophage® XR
(metformin hydrochloride extended release) tablets.
Glucophage® XR, and the drug active metformin, are indicated
for use in the management of type 2 diabetes treatment. The Company
is aware that several other generic versions of this product are
currently available and serve to limit the overall market
opportunity, however, we are continuing to evaluate options to
realize commercial returns on this product, particularly in
international markets.
In November 2018, we announced that we entered into two exclusive
licensing and distribution agreements with pharmaceutical
distributors in Vietnam and the Philippines pursuant to which the
distributors were granted the exclusive right, subject to
regulatory approval, to import and market our generic
Glucophage® XR in Vietnam and the Philippines, respectively.
Under the terms of the agreements, the distributors will be
required to purchase a minimum yearly quantity of our generic
Glucophage® XR. These multi-year agreements are each subject
to early termination.
There can be no assurance that our generic Glucophage® XR for
the 500 and 750 mg strengths will be successfully commercialized in
the US market. Further, there can be no assurance as to when and if
such product will receive regulatory approval for the sale in
Vietnam or the Philippines or that, if so approved, the product
will be successfully commercialized there and produce significant
revenues for us.
Venlafaxine hydrochloride – Generic Effexor
XR® (a registered trademark of the
brand manufacturer)
We received final approval from the FDA in November 2018 for our
ANDA for venlafaxine hydrochloride extended-release capsules in the
37.5, 75 and 150 mg strengths. The approved product is a generic
equivalent of the branded product Effexor® XR sold in the U.S.
by Wyeth Pharmaceuticals, LLC. Effexor® XR, and the drug
active venlafaxine hydrochloride, are indicated for the treatment
of major depressive disorder or MDD. We are actively exploring the
best approach to maximize our commercial returns from this
approval. On November 25, 2019, we announced that we had
entered into a license and commercial supply agreement with Tris
Pharma, by which we granted Tris Pharma an exclusive license to
market, sell and distribute in the United States, Venlafaxine
extended-release capsules in the 37.5, 75, and 150 mg strengths.
Several other generic versions of the licensed products are
currently available in the market and that this limits the overall
market opportunity. Product was never supplied nor distributed
under this license. Effective May 5, 2021 the Company and Tris
Pharma mutually terminated the license agreement. There can be no assurance that the Company’s
venlafaxine hydrochloride extended-release capsules for the 37.5
mg, 75 mg, and 150 mg strengths will be successfully commercialized
and produce significant revenue for us.
Quetiapine fumarate extended-release tablets - Generic Seroquel
XR® (a registered
trademark of the brand manufacturer)
In May
2017, we received final approval from the FDA for our ANDA for
quetiapine fumarate extended-release tablets in the 50, 150, 200,
300 and 400 mg strengths. Our approved product is a generic
equivalent for the corresponding strengths of the branded product
Seroquel XR® sold in the U.S. by AstraZeneca. Seroquel
XR®, and the drug active quetiapine fumarate, are indicated
for use in the management of schizophrenia, bipolar disorder and
major depressive disorder. The Company manufactured and shipped
commercial quantities of all strengths of generic Seroquel XR®
to our then marketing and distribution partner Mallinckrodt, and
Mallinckrodt launched all strengths in June 2017. On April 12,
2019, we and Mallinckrodt mutually agreed to terminate the
Mallinckrodt agreement, and effective August 12, 2019 the
Mallinckrodt agreement was terminated.
16
In November 2018, we announced that we entered into three exclusive
licensing and distribution agreements with pharmaceutical
distributors in Malaysia, Vietnam and the Philippines pursuant to
which the distributors were granted the exclusive right, subject to
regulatory approval, to import and market our generic Seroquel
XR® in Malaysia, Vietnam and the Philippines, respectively.
Under the terms of the agreements, the distributors will be
required to purchase a minimum yearly quantity of our generic
Seroquel XR®. The multi-year agreements are each subject to
early termination. There can be no assurance as to when and if such
product will receive regulatory approval for the sale in Malaysia,
Vietnam or the Philippines or that, if so approved, the product
will be successfully commercialized there and produce significant
revenues for us.
On August 15, 2019 we announced a license and commercial supply
agreement with Tris Pharma, granting Tris Pharma an exclusive
license to market, sell and distribute all strengths of the product
in the United States. The agreement provides for the Company to
have a profit-sharing arrangement with respect to the licensed
product. Product was never supplied nor distributed under this
license. Effective May 5, 2021 the Company and Tris Pharma mutually
terminated the license agreement. There can be no assurance that
the product will be successfully commercialized and produce
significant revenue for us.
Desvenlafaxine
succinate extended-release tablets – Generic
Pristiq® (a registered
trademark of the brand manufacturer)
In May
2019, we received approval from the FDA for our ANDA for
desvenlafaxine extended-release tablets in the 50 and 100 mg
strengths. This product is a generic equivalent of the branded
product Pristiq® sold in
the U.S. by Wyeth Pharmaceuticals, LLC. Pristiq®, and the drug active desvenlafaxine
succinate, are indicated for use in the management of depression.
We previously announced that we had entered into the Mallinckrodt
agreement, which granted Mallinckrodt, subject to its terms, an
exclusive license to market, sell and distribute in the U.S. the
Company's desvenlafaxine extended-release tablets (generic
Pristiq®). On April 12, 2019, we and Mallinckrodt mutually
agreed to terminate the Mallinckrodt agreement and effective August
12, 2019, the Mallinckrodt agreement was terminated. On September
5, 2019, we announced a license and commercial supply agreement
with Tris Pharma, granting Tris Pharma an exclusive license to
market, sell and distribute the two strengths of the product in the
United States. The agreement provides for the Company to have a
profit-sharing arrangement with respect to the licensed product.
The product was never supplied nor distributed under this license.
Effective May 5, 2021 the Company and Tris Pharma mutually
terminated the license agreement. There can be no assurance that
our desvenlafaxine extended-release tablets in the 50 and 100 mg
strengths will be successfully commercialized and produce
significant revenue for us.
In
February 2022 the Company received Health Canada marketing approval
(notice of compliance) for generic Pristiq (desvenlafaxine
extended-release tablets) in the 50 and 100 mg strengths. The
Company is discussing a licensing agreement for the
commercialization of the product.
Oxycodone ER (Abuse Deterrent Oxycodone Hydrochloride Extended
Release Tablets)
One of our non-generic products under development is our Oxycodone
ER (abuse deterrent oxycodone hydrochloride extended release
tablets) product candidate, intended as an abuse and
alcohol-deterrent controlled-release oral formulation of oxycodone
hydrochloride for the relief of pain. Our Oxycodone ER is a new
drug candidate, with a unique long acting oral formulation of
oxycodone intended to treat moderate-to-severe pain when a
continuous, around the clock opioid analgesic is needed for an
extended period of time. The formulation is intended to present a
significant barrier to tampering when subjected to various forms of
physical and chemical manipulation commonly used by abusers. It is
also designed to prevent dose dumping when inadvertently
co-administered with alcohol. Dose dumping is the rapid release of
an active ingredient from a controlled-release drug into the blood
stream that can result in increased toxicity, side effects, and a
loss of efficacy. Dose dumping can result by consuming the drug
through crushing, taking with alcohol, extracting with other
beverages, vaporizing or injecting. In addition, when crushed or
pulverized and hydrated, the proposed extended release formulation
is designed to coagulate instantaneously and entrap the drug in a
viscous hydrogel, which is intended to prevent syringing, injecting
and snorting. Our Oxycodone ER formulation is difficult to abuse
through the application of heat or an open flame, making it
difficult to inhale the active ingredient from
burning.
17
In November 2016, we filed an NDA seeking authorization to market
our Oxycodone ER in the 10, 15, 20, 30, 40, 60 and 80 mg strengths,
relying on the 505(b)(2) regulatory pathway which allowed us to
reference data from Purdue’s file for its OxyContin®. In
February 2017, the FDA accepted for filing our NDA, and set a
Prescription Drug User Fee Act (“PDUFA”), goal date of September 25, 2017. Our
submission is supported by pivotal pharmacokinetic studies that
demonstrated that Oxycodone ER is bioequivalent to OxyContin®.
The submission also includes abuse-deterrent studies conducted to
support abuse-deterrent label claims related to abuse of the drug
by various pathways, including oral, intra-nasal and intravenous,
having reference to the FDA’s “Abuse-Deterrent Opioids
- Evaluation and Labeling” guidance published in April 2015.
FDA had agreed that we would not be required to conduct Phase III
studies if bioequivalence to OxyContin® was demonstrated based
on pivotal bioequivalence studies.
Our NDA was filed under Paragraph IV of the Hatch-Waxman Act, as
amended. We certified to the FDA that we believed that our
Oxycodone ER product candidate would not infringe any of the
OxyContin® patents listed in the FDA’s Approved Drug
Products with Therapeutic Equivalence Evaluations, commonly known
as the Orange Book (the “Orange Book”), or that such patents are invalid, and so
notified all holders of the subject patents of such certification.
On April 7, 2017, we received notice that Purdue, Purdue
Pharmaceuticals L.P., The P.F. Laboratories, Inc., or collectively
the Purdue parties, Rhodes Technologies, and Grünenthal GmbH,
or collectively the Purdue litigation plaintiffs, had commenced
patent infringement proceedings, or the Purdue litigation, against
us in the U.S. District Court for the District of Delaware (docket
number 17-392) in respect of our NDA filing for Oxycodone ER,
alleging that our proposed Oxycodone ER infringes 6 out of the 16
patents associated with the branded product OxyContin®, or the
OxyContin® patents, listed in the Orange
Book.
Subsequent to the above-noted filing of lawsuit, 4 further such
patents were listed and published in the Orange Book. On March 16,
2018, we received notice that the Purdue litigation plaintiffs had
commenced further such patent infringement proceedings adding the 4
further patents. On April 15, 2020, Purdue filed a new patent
infringement suit against the Company relating to additional
Paragraph IV certifications lodged against two more listed Purdue
patents.
As a result of the commencement of the first of these legal
proceedings, the FDA is stayed for 30 months from granting final
approval to our Oxycodone ER product candidate. That time period
commenced on February 24, 2017, when the Purdue litigation
plaintiffs received notice of our certification concerning the
patents, and would expire on August 24, 2019, unless the stay is
earlier terminated by a final declaration of the courts that the
patents are invalid, or are not infringed, or the matter is
otherwise settled among the parties.
On or about June 26, 2018, the court issued an order to sever 6
“overlapping” patents from the second Purdue case, but
ordered litigation to proceed on the 4 new (2017-issued) patents.
An answer and counterclaim was filed on July 9, 2018.
On July 6, 2018, the court issued a so-called “Markman”
claim construction ruling on the first case and the October 22,
2018 trial date remained unchanged.
On July 24, 2018, the parties to the case mutually agreed to and
did have dismissed without prejudice the infringement claims
related to the Grünenthal ‘060 patent, which is one of
the six patents included in the original litigation
case.
18
On April 4, 2019, the U.S. Federal Circuit Court of Appeals
affirmed the invalidity of one Purdue OxyContin® formulation
patent, subject to further appeal to the U.S. Supreme
Court.
On October 4, 2019, we announced that following the filing of a
bankruptcy stay by Purdue Pharma L.P., the Company’s ongoing
litigation case numbers 1:17-cv-00392-RGA and 1:18-cv-00404-RGA-SRF
between Purdue Pharma L.P. et al and Intellipharmaceutics, were
stayed and the existing trial dates in both cases vacated by orders
issued in each case by the judge in the District of Delaware on
October 3, 2019. On April 24, 2019, an order had been issued,
setting a trial date of November 12, 2019 for case number 17-392 in
the District of Delaware, and also extending the 30-month stay date
for regulatory approval to March 2, 2020. With the litigation stay
order, the previous 30-month stay date of March 2, 2020 was
unchanged.
On or about July 2, 2020 the parties in the Litigations between
Purdue and Intellipharmaceutics entered into a stipulated dismissal
of the Litigations. The stipulated dismissal, provides for the
termination of patent infringement proceedings commenced by Purdue
against the Company in the United States District Court for the
District of Delaware in respect of the Company’s NDA filing
for Aximris XR™ with the FDA. The stipulated dismissal also
provides that (i) for a thirty (30) day period following a final
approval of the Company’s Aximris XR™ NDA the parties
will attempt to resolve any potential asserted patent infringement
claims relating to the NDA and (ii) if the parties fail to resolve
all such claims during such period Purdue Pharma will have fifteen
(15) days to pursue an infringement action against the Company. The
terms of the stipulated dismissal agreement are
confidential.
On July 28, 2020 the United States District Court for the District
of Delaware signed the stipulations of dismissal into order thereby
dismissing the claims in the three cases without prejudice. In
consideration of the confidential stipulated dismissal agreement
and for future saved litigation expenses, Purdue has paid an amount
of money to the Company.
In June 2017, we announced that a joint meeting of the Advisory
Committees meeting was scheduled for July 26, 2017 to review our
NDA for Oxycodone ER. The submission requested that our Oxycodone
ER product candidate include product label claims to support the
inclusion of language regarding abuse-deterrent properties for the
intravenous route of administration.
In July 2017, the Company announced that the FDA Advisory
Committees voted 22 to 1 in finding that the Company’s NDA
for Oxycodone ER should not be approved at this time. The Advisory
Committees also voted 19 to 4 that the Company had not demonstrated
that Oxycodone ER has properties that can be expected to deter
abuse by the intravenous route of administration, and 23 to 0 that
there was not sufficient data for Oxycodone ER to support inclusion
of language regarding abuse-deterrent properties in the product
label for the intravenous route of administration. The Advisory
Committees expressed a desire to review the additional safety and
efficacy data for Oxycodone ER that may be obtained from human
abuse potential studies for the oral and intranasal routes of
administration.
In September 2017, the Company received a Complete Response Letter
(“CRL”) from the FDA for the Oxycodone ER NDA,
stating that it could not approve the application at that time. In
its CRL, the FDA provided certain recommendations and requests for
information, including that Intellipharmaceutics complete studies
to assess the abuse-deterrent properties of Oxycodone ER by the
oral and nasal routes of administration, provide additional
information related to the inclusion of the blue dye in the
formulation, of the product, and submit an alternate proposed
proprietary name for Oxycodone ER. The FDA required a response
within a year of issuing the CRL but granted our request for an
extension to resubmit by February 28, 2019.
In February 2018, the Company met with the FDA to discuss the
above-referenced CRL for Oxycodone ER, including issues related to
the blue dye in the product candidate. Based on those discussions,
the product candidate will no longer include the blue dye. The blue
dye was intended to act as an additional deterrent if Oxycodone ER
is abused and serve as an early warning mechanism to flag potential
misuse or abuse. The FDA confirmed that the removal of the blue dye
is unlikely to have any impact on formulation quality and
performance. As a result, the Company will not be required to
repeat in vivo bioequivalence studies and pharmacokinetic studies
submitted in the Oxycodone ER NDA. The FDA also indicated that,
from an abuse liability perspective, Category 1 studies will not
have to be repeated on Oxycodone ER with the blue dye
removed.
The NDA resubmission in response to CRL was filed February 28,
2019. In March 2019, the FDA acknowledged receipt of our
resubmission of the Oxycodone ER NDA. The FDA had informed the
Company that it considered the resubmission a complete response to
the September 22, 2017 action letter it issued in respect of the
NDA.
19
On July
24, 2019, we announced that the Company had been advised by the FDA
that the FDA “is postponing product-specific advisory
committee meetings for opioid analgesics,” including the one
previously scheduled to discuss our NDA, “while it continues
to consider a number of scientific and policy issues relating to
this class of drugs.” According to the FDA, the reason for
the postponement was not unique to our product and the Anesthetic
and Analgesic Drug Products Advisory Committee (“AADPAC”) meeting earlier planned
by the FDA, to discuss our NDA was going to be rescheduled at a
future date. The FDA informed the Company that it would continue to
review the Company’s NDA according to the existing PDUFA
timeline, but noted that, due to the postponement of the AADPAC
meeting, it was possible that the FDA may be unable to meet the
PDUFA goal date of August 28, 2019 that it set when the
resubmission was filed. The FDA did not meet the PDUFA goal date of
August 28, 2019.
In
December 2019, we announced that a joint meeting of the Anesthetic
and Analgesic Drug Products Advisory Committee and Drug Safety and
Risk Management Advisory Committee of the FDA has been scheduled
for January 15, 2020 to review the NDA for Aximris XRTM abuse-deterrent
oxycodone hydrochloride extended-release tablets.
On
January 15, 2020, at a joint meeting of the Advisory Committees of
the FDA to review our NDA for Aximris XR™, abuse-deterrent
oxycodone hydrochloride extended-release tablets, the Advisory Committees voted 24 to 2
against the approval of our NDA for Aximris XRTM for the management of pain severe
enough to require daily, around-the-clock, long-term opioid
treatment and for which alternative treatment options are
inadequate. The
30-month stay date of March 2, 2020 has since expired. The Company
has been following up with FDA, however no action has been taken on
the NDA yet.
There can be no assurance that the studies submitted to the FDA
will be adequate, that we will not be required to conduct further
studies for Oxycodone ER, that the FDA will approve any of the
Company’s requested abuse-deterrent label claims, that the
FDA will ultimately approve our NDA for the sale of Aximris
XRTM
in the U.S. market, or that it will
ever be successfully commercialized and produce significant revenue
for us. If the Aximris XRTM
NDA is approved, there can be no
assurance that the Company and Purdue will resolve any potential
asserted patent infringement claims relating to the NDA within a
thirty (30) day period following the final approval as provided in
the stipulated settlement agreement of the Purdue litigations.
There can be no assurance that the Purdue parties will not pursue
an infringement claim against the Company
again.
In November 2018, we announced that we entered into an exclusive
licensing and distribution agreement with a pharmaceutical
distributor in the Philippines pursuant to which the distributor
was granted the exclusive right, subject to regulatory approval, to
import and market Oxycodone ER in the Philippines. Under the terms
of the agreement, the distributor will be required to purchase a
minimum yearly quantity of our Oxycodone ER and we will be the
exclusive supplier of our Oxycodone ER. This multi-year agreement
is subject to early termination. There can be no assurance as to
when and if such product candidate will receive regulatory approval
for the sale in the Philippines or that, if so approved, the
product will be successfully commercialized there and produce
significant revenues for us.
Regabatin™ XR
(Pregabalin Extended-Release)
Another Intellipharmaceutics non-generic controlled-release product
under development is Regabatin™ XR, pregabalin
extended-release capsules. Pregabalin is indicated for the
management of neuropathic pain associated with diabetic peripheral
neuropathy, postherpetic neuralgia, spinal cord injury and
fibromyalgia. A controlled-release version of pregabalin should
reduce the number of doses patients take, which could improve
patient compliance, and therefore possibly enhance clinical
outcomes. Lyrica® pregabalin, twice-a-day
(“BID”) dosage and three-times-a-day
(“TID”) dosage, are drug products marketed in the
U.S. by Pfizer Inc. (“Pfizer”). In October 2017, Pfizer also received
approval for a Lyrica® CR, a controlled-release version of
pregabalin. In 2014, we conducted and analyzed the results of six
Phase I clinical trials involving a twice-a-day formulation and a
once-a-day formulation. For formulations directed to certain
indications which include fibromyalgia, the results suggested that
Regabatin™ XR 82.5 mg BID dosage was comparable in
bioavailability to Lyrica® 50 mg (immediate-release
pregabalin) TID dosage. For formulations directed to certain other
indications which include neuropathic pain associated with diabetic
peripheral neuropathy, the results suggested that Regabatin™
XR 165 mg once-a-day dosage was comparable in bioavailability to
Lyrica® 75 mg BID dosage.
20
In March 2015, the FDA accepted a Pre-Investigational New Drug or
Pre-IND meeting request for our once-a-day Regabatin™ XR
non-generic controlled release version of pregabalin under the NDA
505(b)(2) regulatory pathway, with a view to possible
commercialization in the U.S. at some time following the December
30, 2018 expiry of the patent covering the pregabalin molecule.
Regabatin™ XR is based on our controlled release drug
delivery technology platform which utilizes the symptomatology and
chronobiology of fibromyalgia in a formulation intended to provide
a higher exposure of pregabalin during the first 12 hours of
dosing. Based on positive feedback and guidance from the FDA, we
submitted an IND application for RegabatinTM XR in August 2015. The
FDA completed its review of the IND application and provided
constructive input that we will use towards further development of
the program. We believe our product candidate has significant
additional benefits to existing treatments and have been evaluating
strategic options to advance this opportunity.
There can be no assurance that any additional Phase I or other
clinical trials we conduct will meet our expectations, that we will
have sufficient capital to conduct such trials, that we will be
successful in submitting an NDA 505(b)(2) filing with the FDA, that
the FDA will approve this product candidate for sale in the U.S.
market, or that it will ever be successfully
commercialized.
Oxycodone Hydrochloride IR Tablets (“IPCI006”) (Abuse
Deterrent and Overdose Resistant Oxycodone Hydrochloride Immediate
Release Tablets) – ROXICODONE®
In November 2018, we announced that we had submitted an IND
application to the FDA for our IPCI006 oxycodone hydrochloride
immediate release tablets in the 5, 10, 15, 20 and 30 mg strengths.
This novel drug formulation incorporates the Company’s
PODRAS™, or Paradoxical OverDose Resistance Activating
System, delivery technology and its nPODDDS™, or novel Point
Of Divergence Drug Delivery System, technology. IPCI006 is designed
to prevent, delay or limit the release of oxycodone hydrochloride
when more intact tablets than prescribed are ingested, thus
delaying or preventing overdose and allowing for sufficient time
for a rescue or medical intervention to take place. It is also
intended to present a significant barrier to abuse by snorting,
“parachuting,” injecting or smoking finely crushed
oxycodone hydrochloride immediate release tablets. The data
generated from the studies conducted under this IND is expected to
form part of an NDA seeking FDA approval for IPCI006
tablets.
If approved, IPCI006 may be the first immediate release formulation
of oxycodone hydrochloride intended to simultaneously prevent or
delay overdose and prevent abuse by intranasal or intravenous
routes.
There can be no assurance that we will be successful in submitting
any NDA with the FDA, that the FDA will approve the Company’s
IPCI006 product candidate for sale in the U.S. market or any
related abuse-deterrent label claims, or that it will ever be
successfully commercialized and produce significant revenue for
us.
Other Potential Products and Markets
We are
continuing our efforts to identify opportunities internationally,
particularly in China, that could if effectuated provide product
distribution alternatives through partnerships and therefore would
not likely require an investment or asset acquisition by us.
Discussions toward establishing a partnership to facilitate future
development activities in China are ongoing. We have not at this
time entered into and may not ever enter into any such
arrangements.
In
addition, we are seeking to develop key relationships in several
other international jurisdictions where we believe there may be
substantial demand for our generic products. These opportunities
could potentially involve out-licensing of our products,
third-party manufacturing supply and more efficient access to
pharmaceutical ingredients and therefore assist with the
development of our product pipeline.
21
In
November 2018, we announced that we had entered into an exclusive
licensing and distribution agreement for our abuse resistant
Oxycodone ER product candidate and four generic drug products with
a pharmaceutical distributor in the Philippines. Under the terms of
the agreement the distributor was granted the exclusive right,
subject to regulatory approval, to import and market our first
novel drug formulation, abuse-deterrent Oxycodone ER, in the
Philippines. Additionally, this distributor was granted, subject to
regulatory approval, the exclusive right to import and market our
generics of Seroquel XR®, Focalin XR®, Glucophage®
XR, and Keppra XR® in the Philippines. Under the terms of the
agreement, the distributor will be required to purchase a minimum
yearly quantity of all products included in the agreement and we
will be the exclusive supplier of said products. The multi-year
agreement with the Philippines distributor is subject to early
termination. Financial terms of the agreement have not been
disclosed. There can be no assurance as to when or if any of our
products or product candidates will receive regulatory approval for
sale in the Philippines or that, if so approved, any such products
will be successfully commercialized there and produce significant
revenues for us. Moreover, there can be no assurance that we will
not be required to conduct further studies for Oxycodone ER, that
the FDA will approve any of our requested abuse-deterrent label
claims, that the FDA will meet its deadline for review, that the
FDA will ultimately approve the NDA for the sale of Oxycodone ER in
the U.S. market, or that it will ever be successfully
commercialized and produce significant revenue for us.
In
November 2018, we announced that we had entered into two exclusive
licensing and distribution agreements with pharmaceutical
distributors in Malaysia and Vietnam.
A
Malaysian pharmaceutical distribution company was granted the
exclusive right, subject to regulatory approval, to import and
market our generic Seroquel XR® (quetiapine fumarate
extended-release) in Malaysia. Under the terms of the agreement,
four strengths (50, 200, 300 and 400 mg) of generic Seroquel
XR® will be manufactured and supplied by us for distribution
in Malaysia. We are also in discussions to include other products
in the agreement with said distributor, who will be required to
purchase a minimum yearly quantity of all products included in the
agreement.
A
Vietnamese pharmaceutical distributor was granted the exclusive
right, subject to regulatory approval, to import and market our
generic Seroquel XR®, Glucophage® XR, and Keppra XR®
in Vietnam. Under the terms of the agreement, two strengths (500
and 750 mg) of generic Glucophage® XR, three strengths (50,
150 and 200 mg) of generic Seroquel XR® and one strength (500
mg) of generic Keppra XR® will be manufactured and supplied by
us for distribution in Vietnam. The Vietnamese distributor will be
required to purchase a minimum yearly quantity of all products
included in the agreement.
The
multi-year agreements with the Malaysian and Vietnamese
distributors are each subject to early termination. Financial terms
of the agreements have not been disclosed. There can be no
assurance as to when or if any of our products will receive
regulatory approval for sale in Malaysia or Vietnam or that, if so
approved, the products will ever be successfully commercialized
there and produce significant revenues for the
Company.
22
Additionally,
in January 2018, we announced we had commenced a research and
development program of pharmaceutical cannabidiol, or CBD, based
products. As part of this research and development program, we
filed multiple provisional patent applications with the United
States Patent and Trademark Office pertaining to the delivery and
application of cannabinoid-based therapeutics, began talks with
potential commercialization partners in the cannabidiol industry,
and identified a potential supplier of CBD. The patent filings,
together with certain of our already issued drug delivery patents,
are intended to form the basis of the development of a pipeline of
novel controlled-release product candidates with CBD as the main
active ingredient. The Company holds a Health Canada Cannabis Drug
License (CDL). Under the CDL, we are authorized to possess,
produce, sell and deliver drug products containing CBD in
Canada.
There
can be no assurance that we will be able to develop cannabis-based
products or that any cannabis-based product candidates we develop
will ever be successfully commercialized or produce significant
revenue for us. There can be no assurance that any patents
pertaining to the delivery and application of cannabinoid-based
therapeutics will be issued to the Company
Intellectual Property
Proprietary
rights are an important aspect of our business. These include
know-how, trade secrets and patents. Know-how and trade secrets are
protected by internal company policies and operating procedures,
and where necessary, by contractual provisions with development
partners and suppliers. We also seek patent protection for
inventive advances which form the basis of our drug delivery
technologies. With respect to particular products, we may seek
patent protection on the commercial composition, our methods of
production and our uses, to prevent the unauthorized marketing and
sale of competitive products.
Patents
which relate to and protect various aspects of our drug delivery
technologies include the following United States, Canadian ,
Japanese, European and Chinese patents which have been issued to
us:
|
|
|
|
|
|
|
|
|
Country
|
Issue Date
|
Issue No.
|
Title
|
|
|
|
U.S. A
|
18-Feb-20
|
10,561,602
|
Controlled Extended Release Pregabalin
|
|
|
|
U.S.A
|
21-Apr-20
|
10,624,858
|
Controlled Release Composition Using Transition Coating And Method
of Preparing Same
|
|
|
|
U.S.A.
|
28-Apr-20
|
10,632,205
|
Pharmaceutical Compositions Having Reduced Abuse
Potential
|
|
|
|
U.S.A.
|
19-May-20
|
10,653,776
|
Compositions and Methods For Reducing Overdose
|
|
|
|
U.S.A.
|
31-Oct-17
|
9,801,939
|
Compositions and Methods For Reducing Overdose
|
|
|
|
U.S.A.
|
11-Jul-17
|
9,700,516
|
Compositions and Methods For Reducing Overdose
|
|
|
|
U.S.A.
|
11-Jul-17
|
9,700,515
|
Compositions and Methods For Reducing Overdose
|
|
|
|
U.S.A.
|
14-Jul-15
|
9,078,827
|
Pharmaceutical Composition Having Reduced Abuse
Potential
|
|
|
|
U.S.A.
|
12-Aug-14
|
8,802,139
|
Proton Pump-Inhibitor-Containing Capsules Which Comprise Subunits
Differently Structured For A Delayed Release Of The Active
Ingredient
|
|
|
|
U.S.A.
|
10-Dec-13
|
8,603,520
|
Oral Multi-functional Pharmaceutical Capsule Preparations of Proton
Pump Inhibitors
|
|
|
|
U.S.A.
|
12-Mar-13
|
8,394,409
|
Controlled Extended Drug Release Technology
|
|
|
|
U.S.A.
|
28-Dec-10
|
7,858,119
|
Extended Release Pharmaceuticals
|
|
23
|
|
U.S.A.
|
5-May-17
|
9,636,306
|
Proton Pimp-Inhibitor Containing Capsules which Comprise Subunits
Differently Structured for a Delayed Release of the Active
Ingredient
|
|
|
|
U.S.A.
|
6-Nov-19
|
10,314,787
|
Controlled Release Delivery Device Comprising an Organosol
Coat
|
|
|
|
U.S.A.
|
9-Apr-18
|
10,064,828
|
Pulsed Extended-Pulsed and Extended-Pulsed Drug Delivery
Systems
|
|
|
|
U.S.A.
|
25-Dec-18
|
10,159,649
|
Controlled Release Delivery Device Comprising an Organosol
Coat
|
|
|
|
U.S.A.
|
2-Jul-17
|
9,561,188
|
Controlled Release Delivery Device Comprising an Organosol
Coat
|
|
|
|
U.S.A.
|
21-May-19
|
10,293,046
|
Compositions and Methods for Reducing Overdose
|
|
|
|
Japan
|
28-Aug-15
|
5,798,293
|
Pharmaceutical Composition Having Reduced Abuse
Potential
|
|
|
|
Japan
|
28-Jun-19
|
6,544,749
|
Compositions and Methods for Reducing Overdose
|
|
|
|
Europe
|
26-Nov-14
|
2,007,360
|
Controlled Release Delivery Device Comprising an Organosol
Coat
|
|
|
|
Canada
|
29-Nov-16
|
2,910,865
|
Compositions and Methods for Reducing Overdose
|
|
|
|
Canada
|
28-May-19
|
2,648,278
|
Drug Delivery Composition
|
|
|
|
Canada
|
26-May-15
|
2,579,382
|
Controlled Release Composition Using Transition Coating, And Method
Of Preparing Same
|
|
|
|
Canada
|
28-Jan-14
|
2,571,897
|
Controlled Extended Drug Release Technology
|
|
|
|
Canada
|
8-Apr-14
|
2,576,556
|
Drug Delivery Device
|
|
|
|
Canada
|
11-Mar-14
|
2,648,280
|
Controlled Release Delivery Device Comprising an Organosol
Coat
|
|
|
|
Canada
|
19-Jun-12
|
2,626,558
|
Pharmaceutical Composition having Reduced Abuse
Potential
|
|
|
|
Canada
|
25-Sep-12
|
2,529,984
|
Oral Multi-Functional Pharmaceutical Capsule Preparations of Proton
Pump Inhibitors
|
|
|
|
Canada
|
22-Feb-11
|
2,459,857
|
Combinatorial Type Controlled Release Drug Delivery
Device
|
|
|
|
Canada
|
15-Mar-05
|
2,435,276
|
Syntactic Deformable Foam Compositions and Methods for
Making
|
|
|
|
China
|
5-Nov-16
|
ZL 200780019665.5
|
Drug Delivery Composition
|
|
|
|
|
|
|
|
|
In
addition to these issued patents, we have several U.S. patent
applications, and corresponding foreign applications pending,
including Patent Cooperation Treaty - national stage processing and
entry applications, relating to various aspects of our
HyperMatrixTM drug delivery
technologies, including methods and compositions for coating of
tablets and beads, compositions incorporating disintegrants to
assist in controlled release, compositions incorporating multiple
drug actives, and compositions directed to classes of drug actives
designed as therapies for specific indications and compositions
intended to enhance deterrence of willful abuse of narcotic
compositions.
Government Regulation
We
focus on the development of both branded drug products (which
require NDAs) and generic drug products (which require ANDAs). The
research and development, manufacture and marketing of
controlled-release pharmaceuticals are subject to regulation by
U.S., Canadian and other governmental authorities and agencies.
Such national agencies and other federal, state, provincial and
local entities regulate the testing, manufacturing, safety and
promotion of our products. The regulations applicable to our
products may change as the currently limited number of approved
controlled-release products increases and regulators acquire
additional experience in this area.
24
United States Regulation
New Drug Application
We will
be required by the FDA to comply with NDA procedures for our
branded products prior to commencement of marketing by us or our
licensees. New drug compounds and new formulations for existing
drug compounds which cannot be filed as ANDAs, but follow a
505(b)(2) regulatory pathway, are subject to NDA
procedures.
These
procedures for a new drug compound include (a) preclinical
laboratory and animal toxicology tests; (b) scaling and testing of
production batches; (c) submission of an IND, and subsequent
approval is required before any human clinical trials can commence;
(d) adequate and well controlled replicate human clinical trials to
establish the safety and efficacy of the drug for its intended
indication; (e) the submission of an NDA to the FDA; and (f) FDA
approval of an NDA prior to any commercial sale or shipment of the
product, including pre-approval and post-approval inspections of
our manufacturing and testing facilities. If all of this data in
the product application is owned by the applicant, the FDA will
issue its approval without regard to patent rights that might be
infringed or exclusivity periods that would affect the FDA’s
ability to grant an approval if the application relied upon data
which the applicant did not own.
Preclinical
laboratory and animal toxicology tests may have to be performed to
assess the safety and potential efficacy of the product. The
results of these preclinical tests, together with information
regarding the methods of manufacture of the products and quality
control testing, are then submitted to the FDA as part of an IND
requesting authorization to initiate human clinical trials. Once
the IND notice period has expired, clinical trials may be
initiated, unless an FDA hold on clinical trials has been
issued.
A new
formulation for an existing drug compound requires a 505(b)(2)
application. This application contains full reports of
investigations of safety and effectiveness but at least some
information required for approval comes from studies not conducted
by or for the applicant and for which the applicant has not
obtained a right of reference. A 505(b)(2) application is submitted
when some specific information necessary for approval is obtained
from: (1) published literature and/or (2) the FDA findings of
safety and effectiveness for an approved drug. The FDA has
implemented this approach to encourage innovation in drug
development without requiring duplicative studies while protecting
the patent and exclusivity rights for the approved drug. A
505(b)(2) application can be submitted for a new chemical entity, a
new molecular entity or any changes to previously approved drugs
such as dosage form, strength, route of administration,
formulation, indication, or bioinequivalence where the application
may rely on the FDA’s finding on safety and effectiveness of
the previously approved drug. In addition, the applicant may also
submit a 505(b)(2) application for a change in drug product that is
eligible for consideration pursuant to a suitability petition. For
example, a 505(b)(2) application would be appropriate for a
controlled-release product that is bioinequivalent to a reference
listed drug where the proposed product is at least as bioavailable
and the pattern of release is at least as favorable as the approved
pharmaceutically equivalent product. A 505(b)(2) application may be
granted three years of exclusivity if one or more clinical
investigations, other than bioavailability/bioequivalence studies,
was essential to the approval and conducted or sponsored by the
applicant; five years of exclusivity is granted if it is for a new
chemical entity. A 505(b)(2) application may also be eligible for
orphan drug and pediatric exclusivity.
25
A
505(b)(2) application must contain the following: (1)
identification of those portions of the application that rely on
the information the applicant does not have a right of reference,
(2) identification of any or all listed drugs by established name,
proprietary name, dosage form, strength, route of administration,
name of the listed drug’s sponsor, and the application number
if application relies on the FDA’s previous findings of
safety and effectiveness for a listed drug, (3) information with
respect to any patents that claim the drug or the use of the drug
for which approval is sought, (4) patent certifications or
statement with respect to any relevant patents that claim the
listed drug, (5) if approval for a new indication, and not for the
indications approved for the listed drug, a certification so
stating, (6) a statement as to whether the listed drug has received
a period of marketing exclusivity, (7)
bioavailability/bioequivalence studies comparing the proposed
product to the listed drug (if any) and (8) studies necessary to
support the change or modification from the listed drugs or drugs
(if any). Before submitting the application, the applicant should
submit a plan to identify the types of bridging studies that should
be conducted and also the components of application that rely on
the FDA’s findings of safety and effectiveness of a
previously approved drug product. We intend to generate all data
necessary to support FDA approval of the applications we file. A
505(b)(2) application must provide notice of certain patent
certifications to the NDA holder and patent owner, and approval may
be delayed due to patent or exclusivity protections covering an
approved product.
Clinical
trials involve the administration of a pharmaceutical product to
individuals under the supervision of qualified medical
investigators who are experienced in conducting studies under
“Good Clinical Practice” guidelines. Clinical studies
are conducted in accordance with protocols that detail the
objectives of a study, the parameters to be used to monitor safety
and the efficacy criteria to be evaluated. Each protocol is
submitted to the FDA and to an institutional review board prior to
the commencement of each clinical trial. Clinical studies are
typically conducted in three sequential phases, which may overlap.
In Phase I, the initial introduction of the product into human
subjects, the compound is tested for absorption, safety, dosage,
tolerance, metabolic interaction, distribution, and excretion.
Phase II involves studies in a limited patient population with the
disease to be treated to (1) determine the efficacy of the product
for specific targeted indications, (2) determine optimal dosage and
(3) identify possible adverse effects and safety risks. In the
event Phase II evaluations demonstrate that a pharmaceutical
product is effective and has an acceptable safety profile, Phase
III clinical trials are undertaken to further evaluate clinical
efficacy of the product and to further test its safety within an
expanded patient population at geographically dispersed clinical
study sites. Periodic reports on the clinical investigations are
required.
We, or
the FDA, may suspend clinical trials at any time if either party
believes the clinical subjects are being exposed to unacceptable
health risks. The results of the product development, analytical
laboratory studies and clinical studies are submitted to the FDA as
part of an NDA for approval of the marketing and commercialization
of a pharmaceutical product.
Abbreviated New Drug Application
In
certain cases, where the objective is to develop a generic version
of an approved product already on the market in controlled-release
dosages, an ANDA may be filed in lieu of filing an NDA. Under the
ANDA procedure, the FDA waives the requirement to submit complete
reports of preclinical and clinical studies of safety and efficacy
and instead requires the submission of bioequivalency data, that
is, demonstration that the generic drug produces the same effect in
the body as its brand-name counterpart and has the same
pharmacokinetic profile, or change in blood concentration over
time. The ANDA procedure is available to us for a generic version
of a drug product approved by the FDA. In certain cases, an ANDA
applicant may submit a suitability petition to the FDA requesting
permission to submit an ANDA for a drug product that differs from a
previously approved reference drug product (the “Listed Drug”) when the change is
one authorized by statute. Permitted variations from the Listed
Drug include changes in: (1) route of administration, (2) dosage
form, (3) strength and (4) one of the active ingredients of the
Listed Drug when the Listed Drug is a combination product. The FDA
must approve the petition before the ANDA may be submitted. An
applicant is not permitted to petition for any other kinds of
changes from Listed Drugs. The information in a suitability
petition must demonstrate that the change from the Listed Drug
requested for the proposed drug product may be adequately evaluated
for approval without data from investigations to show the proposed
drug product’s safety or effectiveness. The advantages of an
ANDA over an NDA include reduced research and development costs
associated with bringing a product to market, and generally a
shorter review and approval time at the FDA.
26
The
Generic Drug User Fee Amendments of 2012 (“GDUFA”) implemented substantial
fees for new ANDAs, Drug Master Files, product and establishment
fees. In return, the program is intended to provide faster and more
predictable ANDA reviews by the FDA and more timely inspections of
drug facilities. For the FDA’s
fiscal year 2022, the annual
facility fee is $210,012 and the GUDFA fee is $153,686.
Under GDUFA, generic product companies face significant penalties
for failure to pay the new user fees, including rendering an ANDA
not “substantially complete” until the fee is paid. It
is currently uncertain the effect the new fees will have on our
ANDA process and business. However, any failure by us or our
suppliers to pay the fees or to comply with the other provisions of
GDUFA may adversely impact or delay our ability to file ANDAs,
obtain approvals for new generic products, generate revenues and
thus may have a material adverse effect on our business, results of
operations and financial condition.
Patent Certification and Exclusivity Issues
ANDAs
and/or NDAs, filed under Paragraph IV of the Hatch Waxman Act,
which seek approval by a non-brand owner to market a generic
version of a branded drug product prior to the expiry of patents
owned or listed in the Orange Book (the “Listed Patents”) as applicable to
the brand owner’s product, are required to include
certifications pursuant to Paragraph IV that either the Listed
Patents are invalid or that the applicant’s drug product does
not infringe the Listed Patents. In such circumstances, the owner
of the branded drug and/or the holder of the patents may commence
patent infringement litigation against the applicant. In such a
case, the FDA is not empowered to approve such pending ANDA or NDA
until the expiry of 30 months from the commencement of such
litigation, unless within such 30 month period the said patents are
found to be invalid, or the drug product covered by the ANDA or NDA
is finally found by a court not to infringe such
patents.
Under
the U.S. Food, Drug and Cosmetic Act (“FDC Act”), the first filer of an
ANDA (but not an NDA) with a “non-infringement”
certification is entitled, if its drug product is approved, to
receive 180 days of market exclusivity. Subsequent filers of
generic products, if non-infringing and approved by the FDA, are
entitled to market their products six months after the first
commercial marketing of the first filer’s generic product. A
company having FDA approval and permission from the original brand
owner is able to market an authorized generic at any time. The
180-day exclusivity period can be forfeited if the first applicant
withdraws its application or the FDA considers the application to
have been withdrawn, the first applicant amends or withdraws
Paragraph IV Certification for all patents qualifying for 180 day
exclusivity, or the first applicant fails to obtain tentative
approval within 30 months after the date filed, unless failure is
due to a change in review requirements. The preservation of the 180
day exclusivity period related to the first-to-file status of a
drug not approved within 30 months after the date filed, generally
requires that an application be made to the FDA for extension of
the time period where the delay has been due to a change in the
review requirements for the drug. The approval of the continued
first-to-file status in such circumstances is subject to the
discretion of the FDA. There can be no assurance that the FDA would
accede to such a request if made.
Patent
expiration refers to expiry of U.S. patents (inclusive of any
extensions) on drug compounds, formulations and uses. Patents
outside the United States may differ from those in the United
States. Under U.S. law, the expiration of a patent on a drug
compound does not create a right to make, use or sell that
compound. There may be additional patents relating to a
person’s proposed manufacture, use or sale of a product that
could potentially prohibit such person’s proposed
commercialization of a drug compound.
The FDC
Act contains other market exclusivity provisions that offer
additional protection to pioneer drug products which are
independent of any patent coverage that might also apply.
Exclusivity refers to the fact that the effective date of approval
of a potential competitor’s ANDA for a generic of the pioneer
drug may be delayed or, in certain cases, an ANDA may not be
submitted until the exclusivity period expires. Five years of
exclusivity are granted to the first approval of a “new
chemical entity”. Three years of exclusivity may apply to
products which are not new chemical entities, but for which new
clinical investigations are essential to the approval. For example,
a new indication for use, or a new dosage strength of a previously
approved product, may be entitled to exclusivity, but only with
respect to that indication or dosage strength. Exclusivity only
offers protection against a competitor entering the market via the
ANDA route, and does not operate against a competitor that
generates all of its own data and submits a full NDA.
27
If
applicable regulatory criteria are not satisfied, the FDA may deny
approval of an NDA or an ANDA or may require additional testing.
Product approvals may be withdrawn if compliance with current or
future regulatory standards is not maintained or if problems occur
after the product reaches the market. The FDA may require further
testing and surveillance programs to monitor the pharmaceutical
product that has been commercialized. Non-compliance with
applicable requirements can result in additional penalties,
including product seizures, injunction actions and criminal
prosecutions.
Canadian Regulation
The
requirements for selling pharmaceutical drugs in Canada are
substantially similar to those of the United States described
above.
Investigational New Drug Application
Before
conducting clinical trials of a new drug in Canada, we must submit
a Clinical Trial Application to the Therapeutic Products
Directorate (“TPD”). This application includes
information about the proposed trial, the methods of manufacture of
the drug and controls, preclinical laboratory and animal toxicology
tests on the safety and potential efficacy of the drug, and
information on any previously executed clinical trials with the new
drug. If, within 30 days of receiving the application, the TPD does
not notify us that our application is unsatisfactory, we may
proceed with clinical trials of the drug. The phases of clinical
trials are the same as those described above under
“United States Regulation
– New Drug Application”.
New Drug Submission
Before
selling a new drug in Canada, we must submit a New Drug Submission
(“NDS”) or
Supplemental New Drug Submission (“sNDS”) to the TPD and receive a
Notice of Compliance (“NOC”) from the TPD to sell the
drug. The submission includes information describing the new drug,
including its proper name, the proposed name under which the new
drug will be sold, a quantitative list of ingredients in the new
drug, the methods of manufacturing, processing, and packaging the
new drug, the controls applicable to these operations, the tests
conducted to establish the safety of the new drug, the tests to be
applied to control the potency, purity, stability and safety of the
new drug, the results of bio-pharmaceutics and clinical trials as
appropriate, the intended indications for which the new drug may be
prescribed and the effectiveness of the new drug when used as
intended. The TPD reviews the NDS or sNDS. If the submission meets
the requirements of Canada’s Food and Drugs Act and
Regulations, the TPD will issue an NOC for the new
drug.
Where
the TPD has already approved a drug for sale in controlled-release
dosages, we may seek approval from the TPD to sell an equivalent
generic drug through an ANDS. In certain cases, the TPD does not
require the manufacturer of a proposed drug that is claimed to be
equivalent to a drug that has already been approved for sale and
marketed, to conduct clinical trials; instead, the manufacturer
must satisfy the TPD that the drug is bioequivalent to the drug
that has already been approved and marketed.
The TPD
may deny approval or may require additional testing of a proposed
new drug if applicable regulatory criteria are not met. Product
approvals may be withdrawn if compliance with regulatory standards
is not maintained or if problems occur after the product reaches
the market. Contravention of Canada’s Food and Drugs Act and
Regulations can result in fines and other sanctions, including
product seizures and criminal prosecutions.
28
Proposals
have recently been made that, if implemented, would significantly
change Canada’s drug approval system. In general, the
recommendations emphasize the need for efficiency in Canadian drug
review. Proposals include establishment of a separate agency for
drug regulation and modeling the approval system on those found in
European Union countries. There is no assurance, however, that such
changes will be implemented or, if implemented, will expedite the
approval of new drugs.
The
Canadian government has regulations which can prohibit the issuance
of an NOC for a patented medicine to a generic competitor, provided
that the patentee or an exclusive licensee has filed a list of its
Canadian patents covering that medicine with the Minister of Health
and Welfare. After submitting the list, the patentee or an
exclusive licensee can commence a proceeding to obtain an order of
prohibition directed to the Minister prohibiting him or her from
issuing an NOC. The minister may be prohibited from issuing an NOC
permitting the importation or sale of a patented medicine to a
generic competitor until patents on the medicine expire or the
waiver of infringement and/or validity of the patent(s) in question
is resolved by litigation in the manner set out in such
regulations. There may be additional patents relating to a
company’s proposed manufacture, use or sale of a product that
could potentially prohibit such company’s proposed
commercialization of a drug compound.
Certain
provincial regulatory authorities in Canada have the ability to
determine whether the consumers of a drug sold within such province
will be reimbursed by a provincial government health plan for that
drug by listing drugs on formularies. The listing or non-listing of
a drug on provincial formularies may affect the prices of drugs
sold within provinces and the volume of drugs sold within
provinces.
Additional Regulatory Considerations
Sales
of our products by our licensees outside the United States and
Canada will be subject to regulatory requirements governing the
testing, registration and marketing of pharmaceuticals, which vary
widely from country to country.
Under
the U.S. Generic Drug Enforcement Act, ANDA applicants (including
officers, directors and employees) who are convicted of a crime
involving dishonest or fraudulent activity (even outside the FDA
regulatory context) are subject to debarment. Debarment is
disqualification from submitting or participating in the submission
of future ANDAs for a period of years or permanently. The Generic
Drug Enforcement Act also authorizes the FDA to refuse to accept
ANDAs from any company which employs or uses the services of a
debarred individual. We do not believe that we receive any services
from any debarred person.
In
addition to the regulatory approval process, pharmaceutical
companies are subject to regulations under provincial, state and
federal law, including requirements regarding occupational safety,
laboratory practices, environmental protection and hazardous
substance control, and may be subject to other present and future
local, provincial, state, federal and foreign regulations,
including possible future regulations of the pharmaceutical
industry. We believe that we are in compliance in all material
respects with such regulations as are currently in
effect.
Before
medicinal products can be distributed commercially, a submission
providing detailed information must be reviewed and approved by the
applicable government or agency in the jurisdiction in which the
product is to be marketed. The regulatory review and approval
process varies from country to country.
29
Competitive Environment
We are
engaged in a business characterized by extensive research efforts,
rapid technological developments and intense competition. Our
competitors include medical technology, pharmaceutical,
biotechnology and other companies, universities and research
institutions. All of these competitors currently engage in, have
engaged in or may engage in the future, in development,
manufacturing, marketing and commercialization of new
pharmaceuticals and existing pharmaceuticals, some of which may
compete with our present or future products and product
candidates.
Our
drug delivery technologies may compete with existing drug delivery
technologies, as well as new drug delivery technologies that may be
developed or commercialized in the future. Any of these drugs and
drug delivery technologies may receive government approval or gain
market acceptance more rapidly than our products and product
candidates. As a result, our products and product candidates may
become non-competitive or obsolete.
We
believe that our ability to successfully compete will depend on,
among other things, the efficacy, safety and reliability of our
products and product candidates, the timing and scope of regulatory
approval, the speed at which we develop product candidates, our, or
our commercialization partners’, ability to manufacture and
sell commercial quantities of a product to the market, product
acceptance by physicians and other professional healthcare
providers, the quality and breadth of our technologies, the skills
of our employees and our ability to recruit and retain skilled
employees, the protection of our intellectual property, and the
availability of substantial capital resources to fund development
and commercialization activities.
Employees
As of
November 30, 2021, we had 11 full-time employees, which is a
decrease from the 13 employees we had on November 30,
2020. Our employees are
not governed by a collective agreement. We have not experienced a
work stoppage and believe our employee relations are
satisfactory.
The
nature of our business requires the recruitment and retention of a
highly educated and skilled workforce, including highly qualified
management, scientific and manufacturing personnel for innovation,
research and development. Typically, a high proportion of our
employees have a Bachelor’s degree or higher. For each of the
last three fiscal years, all employees of the Company were employed
at the Company’s offices in Toronto.
Facilities
On
December 1, 2015, we had entered into a lease agreement for a
25,000 square foot facility located at 30 Worcester Road Toronto,
Ontario, Canada M9W 5X2 (“30
Worcester Road Facility”), as well as a 40,000 square
foot facility on the adjoining property located at 22 Worcester
Road, Toronto, Ontario, Canada M9W 5X2, both of which are owned
indirectly by the same landlord (“22 Worcester Road Facility”, and
together with 30 Worcester Road Facility, the “Combined Properties”) for a
five-year term with a five-year renewal option. We also had an
option to purchase the Combined Properties after March 1, 2017
until November 30, 2020 based on a fair value purchase formula. On
June 21, 2020, the Company entered into a lease surrender agreement
and vacated the property at 22 Worcester Road on June 30, 2020. On
August 20, 2020, The Company extended its lease for the 30
Worcester Road premises for one year, commencing December 1, 2020,
with an option to continue on a month-to-month basis after November
30, 2021. Effective December 1, 2021, the Company extended its
lease for the premises (30 Worcester Road) for one year with an
option to renew for another year. The basic rent for the current
property is $218,750 per year.
30
We use
our 30 Worcester Road Facility as a current Good Laboratory
Practices (“cGLP”) research laboratory, office
space, and current Good Manufacturing Process (“cGMP”) scale-up and small to
medium-scale manufacturing plant for solid oral dosage forms. The
30 Worcester Road Facility consists of approximately 4,900 square
feet for administrative space, 4,300 square feet for R&D, 9,200
square feet for manufacturing, and 3,000 square feet for
warehousing.
We
continually monitor our facility requirements in the context of our
needs and we expect these requirements to change commensurately
with our activities.
Manufacturing
We have
internal manufacturing capabilities consisting of cGLP research
laboratories and a cGMP manufacturing plant for solid oral dosage
forms at our facility located at 30 Worcester Road Facility. Raw
materials used in manufacturing our products are available from a
number of commercial sources and the prices for such raw materials
are generally not particularly volatile. In October 2014, the FDA
provided us with written notification that 30 Worcester Road
Facility had received an “acceptable” classification.
Such inspections are carried out on a regular basis by the FDA and
an “acceptable” classification is necessary to permit
us to be in a position to receive final approvals for ANDAs and
NDAs and to permit manufacturing of drug products intended for
commercial sales in the United States after any such approvals. The
most recent inspections by FDA were conducted July 2017 and June
2019; both closed satisfactorily. Similarly, Health Canada
completed an inspection of 30 Worcester Road Facility in September
2015 which resulted in a “compliant” rating. The most
recent Health Canada inspection was conducted in June 2019 and a
compliance rating was issued August 15, 2019.
Our
Code of Business Conduct and Ethics (“Code of Conduct”) has been
implemented and it applies to all directors, officers and employees
of the Company and its subsidiaries. It may be viewed on our
website at www.intellipharmaceutics.com or under our company
profile at www.sedar.com. During
the year ended November 30, 2021, no waivers or requests for
exemptions from the Code of Conduct were either requested or
granted.
Prospects for companies in the pharmaceutical industry generally
may be regarded as uncertain given the research and development
nature of the industry and uncertainty regarding the prospects of
successfully commercializing product candidates and, accordingly,
investments in companies such as ours should be regarded as very
speculative. An investor should carefully consider the risks and
uncertainties described below, as well as other information
contained in this annual information form. The list of risks and
uncertainties described below is not an exhaustive list. Additional
risks and uncertainties not presently known to us or that we
believe to be immaterial may also adversely affect our business. If
any one or more of the following risks occur, our business,
financial condition and results of operations could be seriously
harmed. Further, if we fail to meet the expectations of the public
market in any given period, the market price of our common shares
could decline. If any of the following risks actually occurs, our
business, operating results, or financial condition could be
materially adversely affected.
31
Our
activities entail significant risks. In addition to the usual risks
associated with a business, the following is a general description
of certain significant risk factors which may be applicable to
us.
Risks related to our Company
Our business is capital intensive and requires significant
investment to conduct the research and development, clinical and
regulatory activities necessary to bring our products to market,
which capital may not be available in amounts or on terms
acceptable to us, if at all.
Our
business requires substantial capital investment in order to
conduct the R&D, clinical and regulatory activities and to
defend against patent litigation claims in order to bring our
products to market and to establish commercial manufacturing,
marketing and sales capabilities. As of November 30, 2021, our cash
balance was $771,945. We currently expect to meet our short-term
cash requirements from the proceeds of the
private placement financing and quarterly profit share
payments from Par and by cost savings resulting from reduced
R&D activities and staffing levels, as well as from potential
revenues for approved generic products or other collaborations.
Effective May 5, 2021 our exclusive license agreements with Tris
Pharma for generic Seroquel XR®, generic Pristiq® and
generic Effexor XR® were mutually terminated. Products were
never supplied nor distributed under the licenses. Termination of
the exclusive agreements may provide opportunity for the Company to
explore options of supplying the products to multiple sources on
non-exclusive bases. However, there can be no assurance that the
products previously licensed to Tris Pharma will be successfully
commercialized and produce significant revenues for us. We will
still need to obtain additional funding to, among other things,
further product commercialization activities and development of our
product candidates. Potential sources of capital may include, if
conditions permit, equity and/or debt financing, payments from
licensing and/or development agreements and/or new strategic
partnership agreements. The Company has funded its business
activities principally through the issuance of securities, loans
from related parties (see “Related Party Transactions”
for more information related to the terms of such loans and
applicable maturities) and funds from development agreements. There
is no certainty that such funding will be available going forward
or, if it is, whether it will be sufficient to meet our needs. Our
future operations are highly dependent upon our ability to source
additional funding to support advancing our product candidate
pipeline through continued R&D activities and to expand our
operations. Our ultimate success will depend on whether our product
candidates are approved by the FDA, Health Canada, or the
regulatory authorities of other countries in which our products are
proposed to be sold and whether we are able to successfully market
our approved products. We cannot be certain that we will receive
such regulatory approval for any of our current or future product
candidates, that we will reach the level of revenues necessary to
achieve and sustain profitability, or that we will secure other
capital sources on terms or in amounts sufficient to meet our
needs, or at all. Our cash requirements for R&D during any
period depend on the number and extent of the R&D activities we
focus on. At present, we are focused principally on the development
of 505(b)(2) product candidates, such as our Regabatin™ XR
and Oxycodone ER 505(b)(2) product candidates, and selected generic
product candidates as resources permit. Our development of
Oxycodone ER required significant expenditures, including costs to
defend against the Purdue (as defined below) litigation. Some of
these costs remain to be paid by the Company. For our
Regabatin™ XR product candidate, Phase III clinical trials
can be capital intensive, and will only be undertaken consistent
with the availability of funds and a prudent cash management
strategy.
32
The
availability of equity or debt financing will be affected by, among
other things, the results of our R&D, our ability to obtain
regulatory approvals, our success in commercializing approved
products with our commercial partners and the market acceptance of
our products, the state of the capital markets generally, our
delisting from Nasdaq, strategic alliance agreements, and other
relevant commercial considerations. In addition, if we raise
additional funds by issuing equity securities, our then existing
security holders will likely experience dilution, and the incurring
of indebtedness would result in increased debt service obligations
and could require us to agree to operating and financial covenants
that would restrict our operations. In the event that we do not
obtain sufficient additional capital, it will raise substantial
doubt about our ability to continue as a going concern, realize our
assets and pay our liabilities as they become due. Our cash
outflows are expected to consist primarily of internal and external
R&D, legal and consulting expenditures to advance our product
pipeline and selling, general and administrative expenses to
support our commercialization efforts. Depending upon the results
of our R&D programs, the impact of the litigation against us
and the availability of financial resources, we could decide to
accelerate, terminate, or reduce certain projects, or commence new
ones. Any failure on our part to successfully commercialize
approved products or raise additional funds on terms favorable to
us or at all, may require us to significantly change or curtail our
current or planned operations in order to conserve cash until such
time, if ever, that sufficient proceeds from operations are
generated, and could result in us not taking advantage of business
opportunities, in the termination or delay of clinical trials or us
not taking any necessary actions required by the FDA or Health
Canada for one or more of our product candidates, in curtailment of
our product development programs designed to identify new product
candidates, in the sale or assignment of rights to our
technologies, products or product candidates, and/or our inability
to file ANDAs, ANDSs or NDAs at all or in time to competitively
market our products or product candidates.
Delays, suspensions and terminations in our preclinical studies and
clinical trials could result in increased costs to us and delay our
ability to generate product revenues.
The
commencement of clinical trials can be delayed for a variety of
reasons, including delays in:
●
demonstrating
sufficient safety and efficacy to obtain regulatory approval to
commence a clinical trial;
●
reaching agreement
on acceptable terms with prospective contract research
organizations and clinical trial sites;
●
manufacturing
sufficient quantities of a drug candidate;
●
obtaining
institutional review board approval to conduct a clinical trial at
a prospective clinical trial site;
●
patient enrollment;
and
●
for controlled
substances, obtaining specific permission to conduct a study, and
obtaining import and export permits to ship study
samples.
33
Once a
clinical trial has begun, it may be delayed, suspended or
terminated due to a number of factors, including:
●
the number of
patients that participate in the trial;
●
the length of time
required to enroll suitable subjects;
●
the duration of
patient follow-up;
●
the number of
clinical sites included in the trial;
●
changes in
regulatory requirements or regulatory delays or clinical holds
requiring suspension or termination of the trials;
●
delays, suspensions
or termination of clinical trials due to the institutional review
board overseeing the study at a particular site;
●
failure to conduct
clinical trials in accordance with regulatory
requirements;
●
unforeseen safety
issues, including serious adverse events or side effects
experienced by participants; and
●
inability to
manufacture, through third party manufacturers, adequate supplies
of the product candidate being tested.
Based
on results at any stage of product development, we may decide to
repeat or redesign preclinical studies or clinical trials, conduct
entirely new studies or discontinue development of products for one
or all indications. In addition, our product candidates may not
demonstrate sufficient safety and efficacy in pending or any future
preclinical testing or clinical trials to obtain the requisite
regulatory approvals. Even if such approvals are obtained for our
products, they may not be accepted in the market as a viable
alternative to other products already approved or pending
approvals.
If we
experience delays, suspensions or terminations in a preclinical
study or clinical trial, the commercial prospects for our products
will be harmed, and our ability to generate product revenues will
be delayed or we may never be able to generate such
revenues.
34
We have a history of operating losses, which may continue for the
foreseeable future and there is a substantial doubt about our
ability to continue as a going concern.
To
date, we have not been profitable and have incurred significant
losses and cash flow deficits. For fiscal year ended November 30,
2021, we reported net losses of $5,145,155. As of November 30,
2021, we had an aggregate accumulated deficit of $102,241,705. We
anticipate that we will continue to report losses and negative
operating cash flow. As a result of these net losses and other
factors our independent auditors issued an audit opinion with
respect to our financial statements for the three years ended
November 30, 2021 that indicated that there is a substantial doubt
about our ability to continue as a going concern.
There
can be no assurance that we will ever be able to achieve or sustain
profitability or positive cash flows. In addition to the other
factors described in this Annual Information Form, our ultimate
success will depend on how many of our product candidates receive
approval by the FDA or Health Canada, or other jurisdictions and
whether we are able to successfully market approved products. We
cannot be certain that we will be able to receive FDA or Health
Canada approval for any of our current or future product
candidates, or that we will reach the level of sales and revenues
necessary to achieve and sustain profitability. If we are
unsuccessful in commercializing our products and/or securing
sufficient financing, we may need to cease or curtail our
operations.
Our
financial statements do not include any adjustments that might
result from the outcome of this uncertainty. These adjustments
would likely include substantial impairment of the carrying amount
of our assets and potential contingent liabilities that may arise
if we are unable to fulfill various operational commitments. In
addition, the value of our securities would be greatly impaired.
Our ability to continue as a going concern is dependent upon
generating sufficient cash flows from operations and obtaining
additional capital and financing. If our ability to generate cash
flows from operations is delayed or reduced and we are unable to
raise additional funding from other sources, we may be unable to
continue in business. For further discussion about our ability to
continue as a going concern and our plan for future liquidity, see
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations—Ability to Continue as a
Going Concern” incorporated herein by reference.
Loss of key scientists and/or failure to attract qualified
personnel could limit our growth and negatively impact our
operations.
We are
dependent upon the scientific expertise of Dr. Isa Odidi, our
Chairman, Chief Executive Officer and Co-Chief Scientific Officer,
and Dr. Amina Odidi, our President, Chief Operating Officer and
Co-Chief Scientific Officer. Although we employ other qualified
scientists, Drs. Isa and Amina Odidi are our only employees with
the knowledge and experience necessary for us to continue the
development of controlled-release products. We do not maintain
key-person life insurance on any of our officers or employees.
Although we have employment agreements with key members of our
management team, each of our employees may terminate his or her
employment at any time. The success of our business depends, in
large part, on our continued ability to attract and retain highly
qualified management, scientific, manufacturing and sales and
marketing personnel, on our ability to successfully integrate new
employees, and on our ability to develop and maintain important
relationships with leading research and medical institutions and
key distributors. If we lose the services of our executive officers
or other qualified personnel or are unable to attract and retain
qualified individuals to fill these roles or develop key
relationships, our business, financial condition and results of
operations could be materially adversely affected.
35
Our intellectual property may not provide meaningful protection for
our products and product candidates.
We hold
certain U.S., Canadian and foreign patents and have pending
applications for additional patents outstanding. We intend to
continue to seek patent protection for, or maintain as trade
secrets, all of our commercially promising drug delivery platforms
and technologies. Our success depends, in part, on our and our
collaborative partners’ ability to obtain and maintain patent
protection for products and product candidates, maintain trade
secret protection and operate without infringing the proprietary
rights of third parties. Without patent and other similar
protection, other companies could offer substantially identical
products without incurring sizeable development costs which could
diminish our ability to recover expenses of and realize profits on
our developed products. If our pending patent applications are not
approved, or if we are unable to obtain patents for additional
developed technologies, the future protection for our technologies
will remain uncertain. Furthermore, third parties may independently
develop similar or alternative technologies, duplicate some or all
of our technologies, design around our patented technologies or
challenge our issued patents. Such third parties may have filed
patent applications, or hold issued patents, relating to products
or processes competitive with those we are developing or otherwise
restricting our ability to do business in a particular area. If we
are unable to obtain patents or otherwise protect our trade secrets
or other intellectual property and operate without infringing on
the proprietary rights of others, our business, financial condition
and results of operations could be materially adversely
affected.
We may be subject to intellectual property claims that could be
costly and could disrupt our business.
Third
parties may claim we have infringed their patents, trademarks,
copyrights or other rights. We may be unsuccessful in defending
against such claims, which could result in the inability to protect
our intellectual property rights or liability in the form of
substantial damages, fines or other penalties such as injunctions
precluding our manufacture, importation or sales of products. The
resolution of a claim could also require us to change how we do
business or enter into burdensome royalty or license agreements;
provided, however, we may not be able to obtain the necessary
licenses on acceptable terms, or at all. Insurance coverage may be
denied or may not be adequate to cover every claim that third
parties could assert against us. Even unsuccessful claims could
result in significant legal fees and other expenses, diversion of
management’s time and disruptions in our business. Any of
these claims could also harm our reputation. Any of the foregoing
may have a material adverse effect upon our business and financial
condition.
We are a defendant in litigation and are at risk of additional
similar litigation in the future that could divert
management’s attention and adversely affect our business and
could subject us to significant liabilities.
We are
a defendant in the litigation matters described in this annual
information form. The defense of such litigation may increase our
expenses and divert our management’s attention and resources,
and any unfavorable outcome could have a material adverse effect on
our business and results of operations. Any adverse determination
in such litigation, or any settlement of such litigation matters
could require that we make significant payments. In addition, we
may be the target of other litigation in the future. Any negative
outcome in any ongoing or future litigation may have a material
adverse effect on our business and financial
condition.
36
Recent and future legal developments could make it more difficult
and costly for us to obtain regulatory approvals for our product
candidates and negatively affect the prices we may
charge.
In the
United States and elsewhere, recent and proposed legal and
regulatory changes to healthcare systems could prevent or delay our
receipt of regulatory approval for our product candidates, restrict
or regulate our post-approval marketing activities, and adversely
affect our ability to profitably sell our products. We do not know
whether additional legislative changes will be enacted, or whether
the FDA’s regulations, guidance or interpretations will be
changed, or what impact any such changes will have, if any, on our
ability to obtain regulatory approvals for our product candidates.
Further, the U.S. Centers for Medicare and Medicaid Services, or
CMS, frequently changes product descriptors, coverage policies,
product and service codes, payment methodologies and reimbursement
values. Also, increased scrutiny by the U.S. Congress of the
FDA’s approval process could significantly delay or prevent
our receipt of regulatory approval for our product candidates and
subject us to more stringent product labeling and post-marketing
testing and other requirements.
We operate in a highly litigious environment.
From
time to time, we may be exposed to claims and legal actions in the
normal course of business. There has been substantial litigation in
the pharmaceutical industry concerning the manufacture, use and
sale of new products that are the subject of conflicting patent
rights. When we file an ANDA or 505(b)(2) NDA for a bioequivalent
version of a drug, we may, in some circumstances, be required to
certify to the FDA that any patent which has been listed with the
FDA as covering the branded product has expired, the date any such
patent will expire, or that any such patent is invalid or will not
be infringed by the manufacture, sale or use of the new drug for
which the application is submitted. Approval of an ANDA is not
effective until each listed patent expires, unless the applicant
certifies that the patents at issue are not infringed or are
invalid and so notifies the patent holder and the holder of the
branded product. A patent holder may challenge a notice of
non-infringement or invalidity by suing for patent infringement
within 45 days of receiving notice. Such a challenge prevents FDA
approval for a period which ends 30 months after the receipt of
notice, or sooner if an appropriate court rules that the patent is
invalid or not infringed. From time to time, in the ordinary course
of business, we face and have faced such challenges and may
continue to do so in the future.
As of
the date of this Annual information Form, we are not aware of any
pending or threatened material litigation claims against us, other
than as described in this Annual Information Form under the caption
“Legal Proceedings and Regulatory Actions”. Litigation
to which we are, or may be, subject could relate to, among other
things, our patent and other intellectual property rights or such
rights of others, business or licensing arrangements with other
persons, product liability or financing activities. Such litigation
could include an injunction against the manufacture or sale of one
or more of our products or potential products or a significant
monetary judgment, including a possible punitive damages award, or
a judgment that certain of our patent or other intellectual
property rights are invalid or unenforceable or infringe the
intellectual property rights of others. If such litigation is
commenced, our business, results of operations, financial condition
and cash flows could be materially adversely affected.
37
We rely on maintaining as trade secrets our competitively sensitive
know-how and other information. Intentional or unintentional
disclosure of this information could impair our competitive
position.
As to
many technical aspects of our business, we have concluded that
competitively sensitive information is either not patentable or
that for competitive reasons it is not commercially advantageous to
seek patent protection. In these circumstances, we seek to protect
this know-how and other proprietary information by maintaining it
in confidence as a trade secret. To maintain the confidentiality of
our trade secrets, we generally enter into agreements that contain
confidentiality provisions with our employees, consultants,
collaborators, contract manufacturers and advisors upon
commencement of their relationships with us. These provisions
generally require that all confidential information developed by
the individual or made known to the individual by us during the
course of the individual’s relationship with us be kept
confidential and not disclosed to third parties. We may not have
these arrangements in place in all circumstances, and the
confidentiality provisions in our favour may be breached. We may
not become aware of, or have adequate remedies in the event of, any
such breach. In addition, in some situations, the confidentiality
provisions in our favour may conflict with, or be subject to, the
rights of third parties with whom our employees, consultants,
collaborators, contract manufacturers or advisors have previous
employment or consulting relationships. To the extent that our
employees, consultants, collaborators, contract manufacturers or
advisors use trade secrets or know-how owned by others in their
work for us, disputes may arise as to the ownership of relative
inventions. Also, others may independently develop substantially
equivalent trade secrets, processes and know-how, and competitors
may be able to use this information to develop products that
compete with our products, which could adversely impact our
business. The disclosure of our trade secrets could impair our
competitive position. Adequate remedies may not exist in the event
of unauthorized use or disclosure of our confidential
information.
Approvals for our product candidates may be delayed or become more
difficult to obtain if the FDA changes its approval
requirements.
The FDA
may institute changes to its ANDA approval requirements, which may
make it more difficult or expensive for us to obtain approval for
our new generic products. For instance, in July 2012, the Generic
Drug User Fee Amendments of 2012, or GDUFA, was enacted into law.
The GDUFA legislation implemented substantial fees for new ANDAs,
Drug Master Files, product and establishment fees. In return, the
program is intended to provide faster and more predictable ANDA
reviews by the FDA and more timely inspections of drug facilities.
For the FDA’s fiscal year 2022, the annual facility fee is $210,012 and
the GUDFA fee is $153,686. Under GDUFA, generic product companies
face significant penalties for failure to pay the new user fees,
including rendering an ANDA not “substantially
complete” until the fee is paid. Any failure by us or our
suppliers to pay the fees or to comply with the other provisions of
GDUFA may adversely impact or delay our ability to file ANDAs,
obtain approvals for new generic products and generate revenues and
thus may have a material adverse effect on our business, results of
operations and financial condition.
We cannot ensure the availability of raw materials.
Certain
raw materials necessary for the development and subsequent
commercial manufacture of our product candidates may be proprietary
products of other companies. While we attempt to manage the risk
associated with such proprietary raw materials through contractual
provisions in supply contracts, by management of inventory and by
continuing to search for alternative authorized suppliers of such
materials or their equivalents, if our efforts fail, or if there is
a material shortage, contamination, and/or recall of such
materials, the resulting scarcity, and scarcity as a result of any
other reason (such as the novel coronavirus (COVID-19)), could
adversely affect our ability to develop or manufacture our product
candidates. In addition, many third party suppliers are subject to
governmental regulation and, accordingly, we are dependent on the
regulatory compliance of, as well as on the strength,
enforceability and terms of our various contracts with, these third
party suppliers.
38
Further,
the FDA requires identification of raw material suppliers in
applications for approval of drug products. If raw materials are
unavailable from a specified supplier, the supplier does not give
us access to its technical information for our application or the
supplier is not in compliance with FDA or other applicable
requirements, FDA approval of the supplier could delay the
manufacture of the drug involved. Any inability to obtain raw
materials on a timely basis, or any significant price increases
which cannot be passed on to our customers, could have a material
adverse effect on our business, results of operations, financial
condition and cash flows.
Our product candidates may not be successfully developed or
commercialized.
Successful
development of our product candidates is highly uncertain and is
dependent on numerous factors, many of which are beyond our
control. Products that appear promising in research or early phases
of development may fail to reach later stages of development or the
market for several reasons including:
●
for ANDA
candidates, bioequivalence studies results may not meet regulatory
requirements or guidelines for the demonstration of
bioequivalence;
●
for NDA candidates,
a product may not demonstrate acceptable large-scale clinical trial
results, even though it demonstrated positive preclinical or
initial clinical trial results;
●
for NDA candidates,
a product may not be effective in treating a specified condition or
illness;
●
a product may have
harmful side effects on humans;
●
products may fail
to receive the necessary regulatory approvals from the FDA or other
regulatory bodies, or there may be delays in receiving such
approvals;
●
changes in the
approval process of the FDA or other regulatory bodies during the
development period or changes in regulatory review for each
submitted product application may also cause delays in the approval
or result in rejection of an application;
●
difficulties may be
encountered in formulating products, scaling up manufacturing
processes or in getting approval for manufacturing;
●
difficulties may be
encountered in the manufacture and/or packaging of our
products;
●
once manufactured,
our products may not meet prescribed quality assurance and
stability tests;
●
manufacturing
costs, pricing or reimbursement issues, other competitive
therapeutics, or other commercial factors may make the product
uneconomical; and
39
●
the proprietary
rights of others, and their competing products and technologies,
may prevent the product from being developed or
commercialized.
Further,
success in preclinical and early clinical trials does not ensure
that large-scale clinical trials will be successful, nor does
success in preliminary studies for ANDA candidates ensure that
bioequivalence studies will be successful. Results are frequently
susceptible to varying interpretations that may delay, limit or
prevent regulatory approvals. The length of time necessary to
complete bioequivalence studies or clinical trials and to submit an
application for marketing approval for a final decision by a
regulatory authority varies significantly and may be difficult to
predict.
As a
result, there can be no assurance that any of our product
candidates currently in development will ever be successfully
commercialized.
Near-term
revenue depends significantly on the success of our commercialized
products.
Our ability to generate significant near-term revenue will depend
upon successful commercialization of our ANDA
products.
Our ANDA product, a once daily generic Focalin XR® capsules,
for which we received final approval from the FDA in November 2013
under the Company ANDA (as defined below) to launch the 15 and 30
mg strengths. Commercial sales of these strengths were launched
immediately by our commercialization partner in the U.S., Par
Pharmaceutical, Inc. Our 5, 10, 20 and 40 mg strengths were also
then tentatively FDA approved, subject to the right of Teva
Pharmaceuticals USA, Inc. to 180 days of generic exclusivity from
the date of first launch of such products. Teva launched its own 5,
10, 20 and 40 mg strengths of generic Focalin XR® capsules on
November 11, 2014, February 2, 2015, June 22, 2015 and November 19,
2013, respectively. In January 2017, Par launched the 25 and 35 mg
strengths of its generic Focalin XR® capsules in the U.S., and
in May 2017, Par launched the 10 and 20 mg strengths, complementing
the 15 and 30 mg strengths of our generic Focalin XR® marketed
by Par. The FDA granted final approval under the Par ANDA (as
defined in Item 4.B. below) for its generic Focalin XR®
capsules in the 5, 10, 15, 20, 25, 30, 35 and 40 mg strengths. As
the first filer of an ANDA for generic Focalin XR® in the 25
and 35 mg strengths, Par had 180 days of U.S. generic marketing
exclusivity for those strengths. In November 2017, Par launched the
remaining 5 and 40 mg strengths of generic Focalin XR®,
complementing the 10, 15, 20, 25, 30 and 35 mg strengths previously
launched and marketed by Par and providing us with the full line of
general Focalin XR® strengths available in the U.S. market.
Under the Par agreement, we receive calendar quarterly profit-share
payments on Par’s U.S. sales of generic Focalin XR®.
There can be no assurance that commercialization of the product
will produce significant revenue for us. We depend significantly on
the actions of our marketing partner Par in the prosecution,
regulatory approval and commercialization of our generic Focalin
XR® capsules and on their timely payment to us of the
contracted calendar quarterly payments as they come due. The
Company has now received final FDA approval for the 5 mg, 10 mg, 30
mg and 40 mg strengths in its ANDA.
On
August 15, 2019, we announced a license and commercial supply
agreement with Tris Pharma, granting Tris Pharma the exclusive
license to market, sell and distribute all strengths of generic
Seroquel XR® (quetiapine fumarate extended-release
tablets) in the United States. In May 2019, we received approval
from the FDA for our ANDA for desvenlafaxine extended-release
tablets in the 50 and 100 mg strengths and on September 5, 2019, we
announced an agreement with Tris Pharma, granting Tris Pharma an
exclusive license to market, sell and distribute that product in
the United States. Our Venlafaxine hydrochloride extended-release
capsules received final approval from the FDA in the 37.5, 75 and
150 mg strengths in November 2018; and the Company announced an
exclusive licensing agreement with Tris Pharma to market, sell and
distribute that product in the United States in November 2019.
Product was never supplied nor distributed under this license.
Effective May 5, 2021 the Company and Tris Pharma mutually
terminated the license agreement.
40
There
can be no assurance that any strengths of products licensed to Tris
Pharma will be successfully commercialized. We depend significantly
on the actions of our marketing partner Tris Pharma in the
commercialization of these licensed products and on their timely
payment to us of the contracted payments as they come
due.
Our
near-term ability to generate significant revenue will depend upon
successful commercialization of our products in the U.S., where
the branded
products are in the market. Although we have some NDA
505(b)(2) product candidates in our pipeline, these are at early
stages of development. We have ANDAs still under review by the FDA
and commercial alternatives for our products that have been
approved by the FDA that are not licensed.
Our significant expenditures on R&D may not lead to successful
product introductions.
We
conduct R&D primarily to enable us to manufacture and market
pharmaceuticals in accordance with FDA regulations. Typically,
research expenses related to the development of innovative
compounds and the filing of NDAs are significantly greater than
those expenses associated with ANDAs. As we continue to develop new
products, our research expenses will likely increase. We are
required to obtain FDA approval before marketing our drug products
and the approval process is costly and time consuming. Because of
the inherent risk associated with R&D efforts in our industry,
particularly with respect to new drugs, our R&D expenditures
may not result in the successful introduction of FDA approved new
pharmaceuticals.
We may not have the ability to develop or license, or otherwise
acquire, and introduce new products on a timely basis.
Product
development is inherently risky, especially for new drugs for which
safety and efficacy have not been established and the market is not
yet proven. Likewise, product licensing involves inherent risks
including uncertainties due to matters that may affect the
achievement of milestones, as well as the possibility of
contractual disagreements with regard to terms such as license
scope or termination rights. The development and commercialization
process, particularly with regard to new drugs, also requires
substantial time, effort and financial resources. The process of
obtaining FDA or other regulatory approval to manufacture and
market new and generic pharmaceutical products is rigorous, time
consuming, costly and largely unpredictable. We, or a partner, may
not be successful in obtaining FDA or other required regulatory
approval or in commercializing any of the product candidates that
we are developing or licensing.
Our business and operations are increasingly dependent on
information technology and accordingly we would suffer in the event
of computer system failures, cyber-attacks or a deficiency in
cyber-security.
Our
internal computer systems, and those of our vendors and current
and/or future drug development or commercialization partners of
ours, may be vulnerable to damage from cyber-attacks, computer
viruses, malware, natural disasters, terrorism, war,
telecommunication and electrical failures. The risk of a security
breach or disruption, particularly through cyber-attacks, including
by computer hackers, foreign governments, and cyber terrorists, has
generally increased as the number, intensity and sophistication of
attempted attacks and intrusions have increased. If such an event
were to occur and cause interruptions in our operations or those of
a drug development or commercialization partner, it could result in
a material disruption of our product development programs. For
example, the loss of clinical trial data from completed or ongoing
or planned clinical trials could result in delays in our regulatory
approval efforts and significantly increase our costs to recover or
reproduce the data. To the extent that any disruption or security
breach results in a loss of or damage to our data or applications,
or inappropriate disclosure of confidential or proprietary
information, we could incur significant liability and damage to our
reputation. In addition, further development of our drug candidates
could be adversely affected.
41
In
addition, the unauthorized dissemination of sensitive personal
information could expose us or other third parties to regulatory
fines or penalties, litigation and potential liability, or
otherwise harm our business.
Our business can be impacted by wholesaler buying patterns,
increased generic competition and, to a lesser extent, seasonal
fluctuations, which may cause our operating results to
fluctuate.
We
believe that the revenues derived from our generic Focalin
XR®
capsules and other licensed products are subject to wholesaler
buying patterns, increased generic competition negatively impacting
price, margins and market share consistent with industry
post-exclusivity experience and, to a lesser extent, seasonal
fluctuations in relation to generic Focalin XR® capsules (as
these products are indicated for conditions including attention
deficit hyperactivity disorder which we expect may see increases in
prescription rates during the school term and declines in
prescription rates during the summer months). Accordingly, these
factors may cause our operating results to fluctuate.
We may not achieve our projected development goals in the time
frames we announce and expect.
We set
goals regarding the expected timing of meeting certain corporate
objectives, such as the commencement and completion of clinical
trials, anticipated regulatory approval and product launch dates.
From time to time, we may make certain public statements regarding
these goals. The actual timing of these events can vary
dramatically due to, among other things, insufficient funding,
delays or failures in our clinical trials or bioequivalence
studies, the uncertainties inherent in the regulatory approval
process, such as failure to secure appropriate product labeling
approvals, requests for additional information, delays in achieving
manufacturing or marketing arrangements necessary to commercialize
our product candidates and failure by our collaborators, marketing
and distribution partners, suppliers and other third parties to
fulfill contractual obligations. In addition, the possibility of a
patent infringement suit regarding one or more of our product
candidates could delay final FDA approval of such candidates. If we
fail to achieve one or more of these planned goals, the price of
our common shares could decline.
We have limited manufacturing, sales, marketing or distribution
capability and we must rely upon third parties for
such.
While
we have our own manufacturing facility in Toronto, we rely on
third-party manufacturers to supply pharmaceutical ingredients, and
we will be reliant upon a third-party manufacturer to produce
certain of our products and product candidates. Third-party
manufacturers may not be able to meet our deadlines or adhere to
quality standards and specifications. Our reliance on third parties
for the manufacture of pharmaceutical ingredients and finished
products creates a dependency that could severely disrupt our
research and development, our clinical testing, and ultimately our
sales and marketing efforts if such third party manufacturers fail
to perform satisfactorily, or do not adequately fulfill their
obligations. If our manufacturing operation or any contracted
manufacturing operation is unreliable or unavailable, we may not be
able to move forward with our intended business operations and our
entire business plan could fail. There is no assurance that our
manufacturing operation or any third-party manufacturers will be
able to meet commercialized scale production requirements in a
timely manner or in accordance with applicable standards or current
Good Manufacturing Practices or cGMP.
42
If our manufacturing facility is unable to manufacture our
product(s) or the manufacturing process is interrupted due to
failure to comply with regulations or for other reasons, it could
have a material adverse impact on our business.
If our
manufacturing facility fails to comply with regulatory requirements
or encounter other manufacturing difficulties, it could adversely
affect our ability to supply products. All facilities and
manufacturing processes used for the manufacture of pharmaceutical
products are subject to inspection by regulatory agencies at any
time and must be operated in conformity with the current cGMP
regulations. Compliance with FDA and Health Canada cGMP
requirements applies to both drug products seeking regulatory
approval and to approved drug products. In complying with cGMP
requirements, pharmaceutical manufacturing facilities must
continually expend significant time, money and effort in
production, record-keeping and quality assurance and control so
that their products meet applicable specifications and other
requirements for product safety, efficacy and quality. Failure to
comply with applicable legal requirements subjects our
manufacturing facility to possible legal or regulatory action,
including shutdown, which may adversely affect our ability to
manufacture product. Were we not able to manufacture products at
our manufacturing facility because of regulatory, business or any
other reasons, the manufacture and marketing of these products
would be interrupted. This could have a material adverse impact on
our business, results of operations, financial condition, cash
flows and competitive position.
The use of legal and regulatory strategies by competitors with
innovator products, including the filing of citizen petitions, may
delay or prevent the introduction or approval of our product
candidates, increase our costs associated with the introduction or
marketing of our products, or significantly reduce the profit
potential of our product candidates.
Companies
with innovator drugs often pursue strategies that may serve to
prevent or delay competition from alternatives to their innovator
products. These strategies include, but are not limited
to:
●
filing
“citizen petitions” with the FDA that may delay
competition by causing delays of our product
approvals;
●
seeking to
establish regulatory and legal obstacles that would make it more
difficult to demonstrate a product’s bioequivalence or
“sameness” to the related innovator
product;
●
filing suits for
patent infringement that automatically delay FDA approval of
products seeking approval based on the Section 505(b)(2)
pathway;
●
obtaining
extensions of market exclusivity by conducting clinical trials of
innovator drugs in pediatric populations or by other
methods;
●
persuading the FDA
to withdraw the approval of innovator drugs for which the patents
are about to expire, thus allowing the innovator company to develop
and launch new patented products serving as substitutes for the
withdrawn products;
●
seeking to obtain
new patents on drugs for which patent protection is about to
expire; and
●
initiating
legislative and administrative efforts in various states to limit
the substitution of innovator products by pharmacies.
43
These
strategies could delay, reduce or eliminate our entry into the
market and our ability to generate revenues from our products and
product candidates.
Our products and product candidates, if approved for sale, may not
gain acceptance among physicians, patients and the medical
community, thereby limiting our potential to generate
revenue.
Even if
we are able to obtain regulatory approvals for our product
candidates, the success of any of our products will be dependent
upon market acceptance by physicians, healthcare professionals and
third-party payers and our profitability and growth will depend on
a number of factors, including:
●
demonstration of
safety and efficacy;
●
changes in the
practice guidelines and the standard of care for the targeted
indication;
●
relative
convenience and ease of administration;
●
the prevalence and
severity of any adverse side effects;
●
the availability of
alternative products from competitors;
●
the prices of our
products relative to those of our competitors;
●
pricing,
reimbursement and cost effectiveness, which may be subject to
regulatory control;
●
the number of
competitive product entries, and the nature and extent of any
aggressive pricing and rebate activities that may
follow;
●
the timing of our
market entry;
●
the ability to
market our products effectively at the retail level;
●
the acceptance of
our products by government and private formularies;
and
●
the availability of
adequate third-party insurance coverage or
reimbursement.
44
If any
product candidate that we develop does not provide a treatment
regimen that is as beneficial as, or is perceived as being as
beneficial as, the current standard of care or otherwise does not
provide patient benefit, that product candidate, if approved for
commercial sale by the FDA or other regulatory authorities, likely
will not achieve market acceptance. Our ability to effectively
promote and sell any approved products will also depend on pricing
and cost-effectiveness, including our ability to produce a product
at a competitive price and our ability to obtain sufficient
third-party coverage or reimbursement. If any product candidate is
approved but does not achieve an adequate level of acceptance by
physicians, patients and third-party payers, our ability to
generate revenues from that product would be substantially reduced.
In addition, our efforts to educate the medical community and
third-party payers on the benefits of our product candidates may
require significant resources, may be constrained by FDA rules and
policies on product promotion, and may never be
successful.
The risks and uncertainties inherent in conducting clinical trials
could delay or prevent the development and commercialization of our
own branded products, which could have a material adverse effect on
our results of operations, liquidity, financial condition, and
growth prospects.
There
are a number of risks and uncertainties associated with clinical
trials, which may be exacerbated by our relatively limited
experience in conducting and supervising clinical trials and
preparing NDAs. The results of initial clinical trials may not be
indicative of results that would be obtained from large scale
testing. Clinical trials are often conducted with patients having
advanced stages of disease and, as a result, during the course of
treatment these patients can die or suffer adverse medical effects
for reasons that may not be related to the pharmaceutical agents
being tested, but which nevertheless affect the clinical trial
results. In addition, side effects experienced by the patients may
cause delay of approval of our product candidates or a limited
application of an approved product. Moreover, our clinical trials
may not demonstrate sufficient safety and efficacy to obtain FDA
approval.
Failure
can occur at any time during the clinical trial process and, in
addition, the results from early clinical trials may not be
predictive of results obtained in later and larger clinical trials,
and product candidates in later clinical trials may fail to show
the desired safety or efficacy despite having progressed
successfully through earlier clinical testing. A number of
companies in the pharmaceutical industry have suffered significant
setbacks in clinical trials, even in advanced clinical trials after
showing positive results in earlier clinical trials. In the future,
the completion of clinical trials for our product candidates may be
delayed or halted for many reasons, including those relating to the
following:
●
delays in patient
enrollment, and variability in the number and types of patients
available for clinical trials;
●
regulators or
institutional review boards may not allow us to commence or
continue a clinical trial;
●
our inability, or
the inability of our partners, to manufacture or obtain from third
parties materials sufficient to complete our clinical
trials;
●
delays or failures
in reaching agreement on acceptable clinical trial contracts or
clinical trial protocols with prospective clinical trial
sites;
●
risks associated
with trial design, which may result in a failure of the trial to
show statistically significant results even if the product
candidate is effective;
●
difficulty in
maintaining contact with patients after treatment commences,
resulting in incomplete data;
●
poor effectiveness
of product candidates during clinical trials;
●
safety issues,
including adverse events associated with product
candidates;
●
the failure of
patients to complete clinical trials due to adverse side effects,
dissatisfaction with the product candidate, or other
reasons;
45
●
governmental or
regulatory delays or changes in regulatory requirements, policy and
guidelines; and
●
varying
interpretation of data by the FDA or other applicable foreign
regulatory agencies.
In
addition, our product candidates could be subject to competition
for clinical study sites and patients from other therapies under
development by other companies which may delay the enrollment in or
initiation of our clinical trials. Many of these companies have
significantly more resources than we do.
The FDA
or other foreign regulatory authorities may require us to conduct
unanticipated additional clinical trials, which could result in
additional expense and delays in bringing our product candidates to
market. Any failure or delay in completing clinical trials for our
product candidates would prevent or delay the commercialization of
our product candidates. There can be no assurance our expenses
related to clinical trials will lead to the development of
brand-name drugs which will generate revenues in the near future.
Delays or failure in the development and commercialization of our
own branded products could have a material adverse effect on our
results of operations, liquidity, financial condition, and our
growth prospects.
We rely on third parties to conduct clinical trials for our product
candidates, and if they do not properly and successfully perform
their legal and regulatory obligations, as well as their
contractual obligations to us, we may not be able to obtain
regulatory approvals for our product candidates.
We
design the clinical trials for our product candidates, but rely on
contract research organizations and other third parties to assist
us in managing, monitoring and otherwise carrying out these trials,
including with respect to site selection, contract negotiation and
data management. We do not control these third parties and, as a
result, they may not treat our clinical studies as their highest
priority, or in the manner in which we would prefer, which could
result in delays. Although we rely on third parties to conduct our
clinical trials, we are responsible for confirming that each of our
clinical trials is conducted in accordance with our general
investigational plan and protocol. Moreover, the FDA and foreign
regulatory agencies require us to comply with regulations and
standards, commonly referred to as good clinical practices,
(“good clinical practices”) for conducting, recording
and reporting the results of clinical trials to ensure that the
data and results are credible and accurate and that the trial
participants are adequately protected. Our reliance on third
parties does not relieve us of these responsibilities and
requirements. The FDA enforces good clinical practices through
periodic inspections of trial sponsors, principal investigators and
trial sites. If we, our contract research organizations or our
study sites fail to comply with applicable good clinical practices,
the clinical data generated in our clinical trials may be deemed
unreliable and the FDA may require us to perform additional
clinical trials before approving our marketing applications. There
can be no assurance that, upon inspection, the FDA will determine
that any of our clinical trials comply with good clinical
practices. In addition, our clinical trials must be conducted with
product manufactured under the FDA’s cGMP regulations. Our
failure, or the failure of our contract manufacturers, if any are
involved in the process, to comply with these regulations may
require us to repeat clinical trials, which would delay the
regulatory approval process.
If
third parties do not successfully carry out their duties under
their agreements with us; if the quality or accuracy of the data
they obtain is compromised due to failure to adhere to our clinical
protocols or regulatory requirements; or if they otherwise fail to
comply with clinical trial protocols or meet expected deadlines,
our clinical trials may not meet regulatory requirements. If our
clinical trials do not meet regulatory requirements or if these
third parties need to be replaced, such clinical trials may be
extended, delayed, suspended or terminated. If any of these events
occur, we may not be able to obtain regulatory approval of our
product candidates, which could have a material adverse effect on
our results of operations, financial condition and growth
prospects.
46
Competition in our industry is intense, and developments by other
companies could render our products and product candidates
obsolete.
Many of
our competitors, including medical technology, pharmaceutical or
biotechnology and other companies, universities, government
agencies, or research organizations, have substantially greater
financial and technical resources and production and marketing
capabilities than we have. They also may have greater experience in
conducting bioequivalence studies, preclinical testing and clinical
trials of pharmaceutical products, obtaining FDA and other
regulatory approvals, and ultimately commercializing any approved
products. Therefore, our competitors may succeed in developing and
commercializing technologies and products that are more effective
than the drug delivery technologies we have developed or we are
developing or that will cause our technologies or products to
become obsolete or non-competitive. In addition, such competitors
may obtain FDA approval for products faster than us. Any of the
foregoing could render our products obsolete and uncompetitive,
which would have a material adverse effect on our business,
financial condition and results of operations. Even if we commence
further commercial sales of our products, we will be competing
against the greater manufacturing efficiency and marketing
capabilities of our competitors, areas in which we have limited or
no experience.
We rely
on collaborative arrangements with third parties that provide
manufacturing and/or marketing support for some or all of our
products and product candidates. Even if we find a potential
partner, we may not be able to negotiate an arrangement on
favourable terms or achieve results that we consider satisfactory.
In addition, such arrangements can be terminated under certain
conditions and do not assure a product’s success. We also
face intense competition for collaboration arrangements with other
pharmaceutical and biotechnology companies.
Although
we believe that our ownership of patents for some of our drug
delivery products will limit direct competition for such products,
we must also compete with established existing products and other
technologies, products and delivery alternatives that may be more
effective than our products and proposed products. In addition, we
may not be able to compete effectively with other commercially
available products or drug delivery technologies.
We require regulatory approvals for any products that use our drug
delivery technologies.
Our
drug delivery technologies can be quite complex, with many
different components. The development required to take a technology
from its earliest stages to its incorporation in a product that is
sold commercially can take many years and cost a substantial amount
of money. Significant technical challenges are common as additional
products incorporating our technologies progress through
development.
Any
particular technology such as our abuse-deterrent technology may
not perform in the same manner when used with different therapeutic
agents, and therefore this technology may not prove to be as useful
or valuable as originally thought, resulting in additional
development work.
47
If our
efforts do not repeatedly lead to successful development of product
candidates, we may not be able to grow our pipeline or to enter
into agreements with marketing and distribution partners or
collaborators that are willing to distribute or develop our product
candidates. Delays or unanticipated increases in costs of
development at any stage, or failure to solve a technical
challenge, could adversely affect our operating
results.
If
contract manufacturers fail to devote sufficient time and resources
to our concerns, or if their performance is substandard, the
commercialization of our products could be delayed or prevented,
and this may result in higher costs or deprive us of potential
product revenues.
We rely
on contract manufacturers for certain components and ingredients of
our clinical trial materials, such as APIs, and we may rely on such
manufacturers for commercial sales purposes as well. Our reliance
on contract manufacturers in these respects will expose us to
several risks which could delay or prevent the commercialization of
our products, result in higher costs, or deprive us of potential
product revenues, including:
●
Difficulties in
achieving volume production, quality control and quality assurance,
or technology transfer, as well as with shortages of qualified
personnel;
●
The failure to
establish and follow cGMP and to document adherence to such
practices;
●
The need to
revalidate manufacturing processes and procedures in accordance
with FDA and other nationally mandated cGMPs and potential prior
regulatory approval upon a change in contract
manufacturers;
●
Failure to perform
as agreed or to remain in the contract manufacturing business for
the time required to produce, store and distribute our products
successfully;
●
The potential for
an untimely termination or non-renewal of contracts;
and
●
The potential for
us to be in breach of our collaboration and marketing and
distribution arrangements with third parties for the failure of our
contract manufacturers to perform their obligations to
us.
In
addition, drug manufacturers are subject to ongoing periodic
unannounced inspection by the FDA and corresponding state and
foreign agencies to ensure strict compliance with cGMP and other
government regulations. While we may audit the performance of
third-party contractors, we will not have complete control over
their compliance with these regulations and standards. Failure by
either our third-party manufacturers or by us to comply with
applicable regulations could result in sanctions being imposed on
us, including fines, injunctions, civil penalties, failure of
applicable regulatory authorities to grant review of submissions or
market approval of drugs, delays, suspension or withdrawal of
approvals, product seizures or recalls, operating restrictions,
facility closures and criminal prosecutions, any of which could
harm our business.
48
We are subject to currency rate fluctuations that may impact our
financial results.
Although
our financial results are reported in U.S. dollars and our revenues
are payable in U.S. dollars, a majority of our expenses are payable
in Canadian dollars. Our financial condition may be affected by
movements of the U.S. dollar against the Canadian dollar. There may
be instances where we have net foreign currency exposure. Any
fluctuations in exchange rates may have an adverse effect on our
financial results.
We are exposed to risks arising from the ability and willingness of
our third-party commercialization partners to provide documentation
that may be required to support information on revenues earned by
us from those commercialization partners.
If our
third-party commercialization partners, from whom we receive
revenues, are unable or unwilling to supply necessary or sufficient
documentation to support the revenue numbers in our financial
statements in a timely manner to the satisfaction of our auditors,
this may lead to delays in the timely publication of our financial
results, our ability to obtain an auditor’s report on our
financial statements and our possible inability to access the
financial markets during the time our results remain
unpublished.
We rely on commercial partners, and may rely on future commercial
partners, to market and commercialize our products and, if
approved, our product candidates, and one or more of those
commercial partners may fail to develop and effectively
commercialize our current, and any future, products.
Our
core competency and strategic focus is on drug development and we
now, and may in the future, utilize strategic commercial partners
to assist in the commercialization of our products and our product
candidates, if approved by the FDA. If we enter into strategic
partnerships or similar arrangements, we will rely on third parties
for financial resources and for commercialization, sales and
marketing. Our commercial partners may fail to develop or
effectively commercialize our current, and any future products, for
a variety of reasons, including, among others, intense competition,
lack of adequate financial or other resources or focus on other
initiatives or priorities. Any failure of our third-party
commercial partners to successfully market and commercialize our
products and product candidates would diminish our
revenues.
We have limited sales, marketing and distribution
experience.
We have
limited experience in the sales, marketing, and distribution of
pharmaceutical products. There can be no assurance that, if
required, we would be able to establish sales, marketing, and
distribution capabilities or make arrangements with our
collaborators, licensees, or others to perform such activities or
that such efforts would be successful. If we fail to establish
successful marketing and sales capabilities or to make arrangements
with third parties, our business, financial condition and results
of operations will be materially adversely affected.
49
Our effective tax rate may vary.
Various
internal and external factors may have favorable or unfavorable
effects on our future effective tax rate. These factors include,
but are not limited to, changes in tax laws, regulations and/or
rates, changing interpretations of existing tax laws or
regulations, future levels of R&D spending, the availability of
tax credit programs for the reimbursement of all or a significant
proportion of R&D spending, and changes in overall levels of
pre-tax earnings. At present, we qualify in Canada for certain
research tax credits for qualified scientific research and
experimental development pertaining to our drug delivery
technologies and drug products in research stages. If Canadian tax
laws relating to research tax credits were substantially negatively
altered or eliminated, or if a substantial portion of our claims
for tax credits were denied by the relevant taxing authorities,
pursuant to an audit or otherwise, it would have a material adverse
effect upon our financial results.
The
effect of U.S. federal income tax law changes enacted in 2017 on
the U.S. corporate income tax burden on our future U.S. operations
cannot be predicted. Although such legislation reduced the maximum
corporate income tax rate from 35% to 21%, it also introduced
several changes that could increase our effective rate of tax to a
rate in excess of 21% on any net operating income we earn in the
future. For example, if our operations are highly leveraged, the
new limitations on business interest deductions may prevent us from
being able to reduce our corporate income tax base by a significant
amount of interest incurred on debt necessary to fund operations.
In addition, newly enacted limitations on a corporation’s
ability to reduce its taxable income by net operating loss
carryovers may prevent us from using prior year accumulated losses
fully to offset taxable income earned in profitable years. Finally,
if we make significant payments for interest, royalties, services
and otherwise deductible items to our foreign affiliates, the base
erosion minimum tax enacted in 2017 may apply to increase our
effective rate of U.S. corporate income tax.
Our significant shareholders have the ability to exercise
significant influence over certain corporate actions.
Drs.
Amina and Isa Odidi, our President, Chief Operating Officer and
Co-Chief Scientific Officer and our Chairman, Chief Executive
Officer and Co-Chief Scientific Officer, respectively, and
shareholders of our Company, and Odidi Holdings Inc., a
privately-held company controlled by Drs. Amina and Isa Odidi, own
in the aggregate approximately 1.75% of our issued and outstanding
Common Shares as of February 28, 2022 (and collectively
beneficially owned in the aggregate approximately 15.51% of our
Common Shares, including Common Shares issuable upon the exercise
of outstanding options and the conversion of the 2018 Debenture (as
herein defined), May 2019 Debenture (as herein defined) and the
November 2019 Debenture (as herein defined) (collectively, the
“Debentures”).
As a result, these shareholders have the ability to exercise
significant influence over matters submitted to our shareholders
for approval.
Stockholder ownership interest in the Company may be diluted as a
result of future financings and acquisitions.
The
Company may seek to raise funds from time to time in public or
private issuances of equity in the near future or over the longer
term. Sales of the Company’s securities offered through
future equity offerings may result in substantial dilution to the
interests of the Company’s current shareholders. The sale of
a substantial number of securities to investors, or anticipation of
such sales, could make it more difficult for the Company to sell
equity or equity-related securities in the future at a time and at
a price that the Company might otherwise wish to effect sales. In
addition, the Company may issue its Common Shares for various
acquisitions in the future, which may also result in substantial
dilution to the interests of the Company’s current
shareholders.
Authorized capital consists of an unlimited number of shares of one
class designated as Common Shares.
The
Company’s authorized capital consists of an unlimited number
of shares of one class designated as Common Shares. The directors
may create any class or series of shares by resolution but may not
make any modification to the provisions attaching to our Common
Shares without the affirmative vote of two-thirds of the votes cast
by the holders of the Common Shares. The Company’s Common
Shares do not have pre-emptive rights to purchase additional
shares.
50
We may lose our foreign private issuer status in the future, which
could result in significant additional costs and
expenses.
We are
a “foreign private issuer,” as such term is defined
under the U.S. Securities Act, and, therefore, we are not required
to comply with all the periodic disclosure and current reporting
requirements of the U.S. Securities Exchange Act of 1934, as
amended (the “U.S. Exchange
Act”) and related rules and regulations. Under the
U.S. Securities Act, the determination of foreign private issuer
status is made annually on the last business day of an
issuer’s most recently completed second fiscal quarter and,
accordingly, the next determination will be made with respect to us
on May 30, 2021.
In the
future, we would lose our foreign private issuer status if a
majority of our shares are owned by U.S residents and a majority of
our directors or executive officers are U.S. citizens or residents
or we fail to meet additional requirements necessary to avoid loss
of foreign private issuer status. Although we have elected to
comply with certain U.S. regulatory provisions, our loss of foreign
private issuer status would make such provisions mandatory. If we
are not a foreign private issuer, we will be required to file
periodic reports and registration statements on U.S. domestic
issuer forms with the Securities and Exchange Commission
(“SEC”), which
are more detailed and extensive than the forms available to a
foreign private issuer. For example, the annual report on Form 10-K
requires domestic issuers to disclose executive compensation
information on an individual basis with specific disclosure
regarding the domestic compensation philosophy, objectives, annual
total compensation (base salary, bonus and equity compensation) and
potential payments in connection with change in control,
retirement, death or disability, while the annual report on Form
20-F permits foreign private issuers to disclose compensation
information on an aggregate basis. We would also have to
mandatorily comply with U.S. federal proxy requirements, and our
executive officers, directors and principal shareholders would
become subject to the short-swing profit disclosure and recovery
provisions of Section 16 of the U.S. Exchange Act. We may also
be required to modify certain of our policies to comply with good
governance practices associated with U.S. domestic issuers. In
addition, we may lose our ability to rely upon exemptions from
certain corporate governance requirements on U.S. stock exchanges
that are available to foreign private issuers. Such transition and
modifications would involve additional costs and may divert our
management’s attention from other business concerns, which
could have a material adverse effect on our business, financial
condition and results of operations.
Future issuances of our shares could adversely affect the trading
price of our Common Shares and could result in substantial dilution
to shareholders.
We may
need to issue substantial amounts of Common Shares in the future.
There can be no assurance that we will be able to sell any
additional shares. To the extent that the market price of our
Common Shares declines, we will need to issue an increasing number
of Common Shares per dollar of equity investment. In addition to
our Common Shares issuable in connection with the exercise of our
outstanding warrants, our employees, and directors will hold rights
to acquire substantial amounts of our Common Shares. In order to
obtain future financing if required, it is likely that we will
issue additional Common Shares or financial instruments that are
exchangeable for or convertible into Common Shares. Also, in order
to provide incentives to employees and induce prospective employees
and consultants to work for us, we may offer and issue options to
purchase Common Shares and/or rights exchangeable for or
convertible into Common Shares. Future issuances of shares could
result in substantial dilution to shareholders. Capital raising
activities, if available, and dilution associated with such
activities could cause our share price to decline. In addition, the
existence of Common Share purchase warrants may encourage short
selling by market participants. Also, in order to provide
incentives to current employees and directors and induce
prospective employees and consultants to work for us, we have
historically granted options and DSUs, and intend to continue to do
so or offer and issue other rights exchangeable for or convertible
into Common Shares. Future issuances of shares could result in
substantial dilution to all our shareholders. In addition, future
public sales by holders of our Common Shares could impair our
ability to raise capital through any future equity
offerings.
51
Risks related to our Industry
Generic drug manufacturers will increase competition for certain
products and may reduce our expected royalties.
Part of
our product development strategy includes making NDA filings
relating to product candidates involving the novel reformulation of
existing drugs with active ingredients that are off-patent. Such
NDA product candidates, if approved, are likely to face competition
from generic versions of such drugs in the future. Regulatory
approval for generic drugs may be obtained without investing in
costly and time consuming clinical trials. Because of substantially
reduced development costs, manufacturers of generic drugs are often
able to charge much lower prices for their products than the
original developer of a new product. If we face competition from
manufacturers of generic drugs on products we may commercialize,
such as our once-daily Oxycodone ER product candidate, the prices
at which such of our products are sold and the revenues we may
receive could be reduced.
Revenues from generic pharmaceutical products typically decline as
a result of competition, both from other pharmaceutical companies
and as a result of increased governmental pricing
pressure.
Our
generic drugs face intense competition. Prices of generic drugs
typically decline, often dramatically, especially as additional
generic pharmaceutical companies (including low-cost generic
producers based in China and India) receive approvals and enter the
market for a given product and competition intensifies.
Consequently, our ability to sustain our sales and profitability on
any given product over time is affected by the number of new
companies selling such product and the timing of their
approvals.
In
addition, intense pressure from government healthcare authorities
to reduce their expenditures on prescription drugs could result in
lower pharmaceutical pricing, causing decreases in our
revenues.
Furthermore,
brand pharmaceutical companies continue to defend their products
vigorously. For example, brand companies often sell or license
their own generic versions of their products, either directly or
through other generic pharmaceutical companies (so-called
“authorized
generics”). No significant regulatory approvals are
required for authorized generics, and brand companies do not face
any other significant barriers to entry into such market. Brand
companies may seek to delay introductions of generic equivalents
through a variety of commercial and regulatory tactics. These
actions may increase the costs and risks of our efforts to
introduce generic products and may delay or prevent such
introduction altogether.
Market acceptance of our products will be limited if users of our
products are unable to obtain adequate reimbursement from
third-party payers.
Government
health administration authorities, private health insurers and
other organizations generally provide reimbursement for products
like ours, and our commercial success will depend in part on
whether appropriate reimbursement levels for the cost of our
products and related treatments are obtained from government
authorities, private health insurers and other organizations, such
as health maintenance organizations and managed care organizations.
Even if we succeed in bringing any of our products to market,
third-party payers may not provide reimbursement in whole or in
part for the use of such products.
52
Significant
uncertainty exists as to the reimbursement status of newly approved
health care products. Some of our product candidates, such as our
once-daily Oxycodone ER, are intended to replace or alter existing
therapies or procedures. These third-party payers may conclude that
our products are less safe, less effective or less economical than
those existing therapies or procedures. Therefore, third-party
payers may not approve our products for reimbursement. We may be
required to make substantial pricing concessions in order to gain
access to the formularies of large managed-care organizations. If
third party payers do not approve our products for reimbursement or
fail to reimburse them adequately, sales will suffer as some
physicians or their patients may opt for a competing product that
is approved for reimbursement or is adequately reimbursed. Even if
third-party payers make reimbursement available, these
payers’ reimbursement policies may adversely affect our
ability and our potential marketing and distribution
partners’ ability to sell our products on a profitable
basis.
We are subject to significant costs and uncertainties related to
compliance with the extensive regulations that govern the
manufacturing, labeling, distribution, cross-border imports and
promotion of pharmaceutical products as well as environmental,
safety and health regulations.
Governmental
authorities in the United States and Canada regulate the research
and development, testing and safety of pharmaceutical products. The
regulations applicable to our existing and future products may
change. Regulations require extensive clinical trials and other
testing and government review and final approval before we can
market our products. The cost of complying with government
regulation can be substantial and may exceed our available
resources, causing delay or cancellation of our product
introductions.
Some
abbreviated application procedures for controlled-release drugs and
other products, including those related to our ANDA filings, or to
the ANDA filings of unrelated third parties in respect of drugs
similar to or chemically related to those of our ANDA filings, are
or may become the subject of petitions filed by brand-name drug
manufacturers or other ANDA filers seeking changes from the FDA in
the interpretation of the statutory approval requirements for
particular drugs as part of their strategy to thwart or advance
generic competition. We cannot predict whether the FDA will make
any changes to its interpretation of the requirements applicable to
our ANDA applications as a result of these petitions, or whether
unforeseen delays will occur in our ANDA filings while the FDA
considers such petitions or changes or otherwise, or the effect
that any changes may have on us. Any such changes in FDA
interpretation of the statutes or regulations, or any legislated
changes in the statutes or regulations, may make it more difficult
for us to file ANDAs or obtain further approval of our ANDAs and
generate revenues and thus may materially harm our business and
financial results.
Any
failure or delay in obtaining regulatory approvals could make it so
that we are unable to market any products we develop and therefore
adversely affect our business, results of operations, financial
condition and cash flows. Even if product candidates are approved
in the United States or Canada, regulatory authorities in other
countries must approve a product prior to the commencement of
marketing the product in those countries. The time required to
obtain any such approval may be longer than in the United States or
Canada, which could cause the introduction of our products in other
countries to be cancelled or materially delayed.
The
manufacturing, distribution, processing, formulation, packaging,
labeling, cross-border importation and advertising of our products
are subject to extensive regulation by federal agencies, including
the FDA, Drug Enforcement Administration, Federal Trade Commission,
Consumer Product Safety Commission and Environmental Protection
Agency in the United States, and Health Canada and Canada Border
Services Agency in Canada, among others. We are also subject to
state and local laws, regulations and agencies. Compliance with
these regulations requires substantial expenditures of time, money
and effort in such areas as production and quality control to
ensure full technical compliance. Failure to comply with FDA and
Health Canada and other governmental regulations can result in
fines, disgorgement, unanticipated compliance expenditures, recall
or seizure of products, total or partial suspension of production
or distribution, suspension of the FDA’s or Health
Canada’s review of NDAs, ANDAs or ANDSs, as the case may be,
enforcement actions, injunctions and civil or criminal
prosecution.
Environmental
laws have changed in recent years and we may become subject to
stricter environmental standards in the future and face larger
capital expenditures in order to comply with environmental laws. We
are subject to extensive federal, state, provincial and local
environmental laws and regulations which govern the discharge,
emission, storage, handling and disposal of a variety of substances
that may be used in, or result from, our operations. We are also
subject periodically to environmental compliance reviews by
environmental, safety, and health regulatory agencies and to
potential liability for the remediation of contamination associated
with both present and past hazardous waste generation, handling,
and disposal activities. We cannot accurately predict the outcome
or timing of future expenditures that we may be required to make in
order to comply with the federal, state, local and provincial
environmental, safety, and health laws and regulations that are
applicable to our operations and facilities.
53
There has been an increased public awareness of the problems
associated with the potential for abuse of opioid-based
medications.
There
has been increasing legislative attention to opioid abuse in the
U.S., including passage of the 2016 Comprehensive Addiction and
Recovery Act and the 21st Century Cures Act, which, among other
things, strengthens state prescription drug monitoring programs and
expands educational efforts for certain populations. These laws
could result in fewer prescriptions being written for opioid drugs,
which could impact future sales of our Oxycodone ER and related
opioid product candidates.
Federal,
state and local governmental agencies have increased their level of
scrutiny of commercial practices of companies marketing and
distributing opioid products, resulting in investigations,
litigation and regulatory intervention affecting other companies. A
number of counties and municipalities have filed lawsuits against
pharmaceutical wholesale distributors, pharmaceutical manufacturers
and retail chains related to the distribution of prescription
opioid pain medications. Policy makers and regulators are seeking
to reduce the impact of opioid abuse on families and communities
and are focusing on policies aimed at reversing the potential for
abuse. In furtherance of those efforts, the FDA has developed an
action plan and has committed to enhance safety labeling, require
new data, strengthen post-market requirements, update the Risk
Evaluation and Mitigation Strategy program, expand access to and
encourage the development of abuse-deterrent formulations and
alternative treatments, and re-examine the risk-benefit profile of
opioids to consider the wider public health effects of opioids,
including the risk of misuse. Several states also have passed laws
and have employed other clinical and public health strategies to
curb prescription drug abuse, including prescription limitations,
increased physician education requirements, enhanced monitoring
programs, tighter restrictions on access, and greater oversight of
pain clinics. This increasing scrutiny and related governmental and
private actions, even if not related to a product that we intend to
manufacture and commercialize, could have an unfavorable impact on
the overall market for opioid-based products such as our Oxycodone
ER product candidate, or otherwise negatively affect our
business.
Healthcare reform measures could hinder or prevent the commercial
success of our products and product candidates.
In the
United States, there have been, and we expect there will continue
to be, a number of legislative and regulatory changes to the
healthcare system that could affect our future revenues and
potential profitability. Federal and state lawmakers regularly
propose and, at times, enact legislation that results in
significant changes to the healthcare system, some of which are
intended to contain or reduce the costs of medical products and
services. An example of this is the Patient Protection and
Affordable Care Act, as amended by the Health Care and Education
Reconciliation Act, or, collectively, the Affordable Care Act. In
addition, other legislative changes have been proposed and adopted
in the U.S. since the Affordable Care Act was enacted.
The
cost of prescription pharmaceuticals has also been the subject of
considerable discussion in the U.S. Members of Congress have
indicated that they will address such costs through new legislative
and administrative measures. To date, there have been several U.S.
Congressional inquiries and proposed and enacted federal and state
legislation designed to, among other things, bring more
transparency to drug pricing, review the relationship between
pricing and manufacturer patient programs, reduce the costs of
drugs under Medicare and reform government program reimbursement
methodologies for drug products.
54
Individual
state legislatures in the U.S. have become increasingly aggressive
in passing legislation and implementing regulations designed to
control pharmaceutical and biological product pricing. Some of
these measures include price or patient reimbursement constraints,
discounts, restrictions on certain product access, marketing cost
disclosure and transparency measures, and, in some cases, measures
designed to encourage importation from other countries and bulk
purchasing. In addition, regional health care authorities and
individual hospitals are increasingly using bidding procedures to
determine what pharmaceutical products and which suppliers will be
included in their prescription drug and other health care programs.
These measures could reduce the ultimate demand for our products,
once approved, or put pressure on our product pricing.
We
expect that additional state and federal healthcare reform measures
will be adopted in the future, any of which could limit the amounts
that federal and state governments will pay for healthcare products
and services, and which could result in reduced demand for our
products once approved or additional pricing pressures, and may
adversely affect our operating results.
Our ability to market and promote our Oxycodone ER product
candidate and its abuse-deterrent features will be determined by
FDA-approved labeling requirements.
The
commercial success of our Oxycodone ER product candidate will
depend upon our ability to obtain requested FDA-approved labeling
describing its abuse-deterrent features. Our failure to achieve FDA
approval of requested product labeling containing such information
will prevent us from advertising and promoting the abuse-deterrent
features of our product candidate in a way to differentiate it from
competitive products. This would make our product candidate less
competitive in the market. Moreover, FDA approval is required in
order to make claims that a product has an abuse-deterrent
effect.
In
April 2015, the FDA published final guidance with respect to the
evaluation and labeling of abuse-deterrent opioids. The guidance
provides direction as to the studies and data required for
obtaining abuse-deterrent claims in a product label. If a product
is approved by the FDA to include such claims in its label, the
applicant may use the approved labeling information about the
abuse-deterrent features of the product in its marketing efforts to
physicians.
Although
we intend to provide data to the FDA to support approval of
abuse-deterrence label claims for Oxycodone ER, there can be no
assurance that Oxycodone ER or any of our other product candidates
will receive FDA-approved labeling that describes the
abuse-deterrent features of such products. The FDA may find that
our studies and data do not support our requested abuse-deterrent
labeling or that our product candidate does not provide substantial
abuse-deterrence benefits because, for example, its deterrence
mechanisms do not address the way it is most likely to be abused.
Furthermore, the FDA could change its guidance, which could require
us to conduct additional studies or generate additional data. If
the FDA does not approve our requested abuse-deterrent labeling, we
will be limited in our ability to promote Oxycodone ER based on its
abuse-deterrent features and, as a result, our business may
suffer.
We may be subject to product liability claims for which we may not
have or be able to obtain adequate insurance coverage.
The
testing and marketing of pharmaceutical products entails an
inherent risk of product liability. Liability exposures for
pharmaceutical products can be extremely large and pose a material
risk. In some instances, we may be or may become contractually
obligated to indemnify third parties for such liability. Our
business may be materially and adversely affected by a successful
product liability claim or claims in excess of any insurance
coverage that we may have. Further, even if claims are not
successful, the costs of defending such claims and potential
adverse publicity could be harmful to our business.
55
While
we currently have, and in some cases are contractually obligated to
maintain, insurance for our business, property and our products as
they are administered in bioavailability/bioequivalence studies,
first and third party insurance is increasingly costly and narrow
in scope. Therefore, we may be unable to meet such contractual
obligations or we may be required to assume more risk in the
future. If we are subject to third party claims or suffer a loss or
damage in excess of our insurance coverage, we may be required to
bear that risk in excess of our insurance limits. Furthermore, any
first or third party claims made on our insurance policy may impact
our ability to obtain or maintain insurance coverage at reasonable
costs or at all in the future. Any of the foregoing may have a
material adverse effect on our business and financial
condition.
Our products involve the use of hazardous materials and waste, and
as a result we are exposed to potential liability claims and to
costs associated with complying with laws regulating hazardous
waste.
Our
R&D activities involve the use of hazardous materials,
including chemicals, and are subject to Canadian federal,
provincial and local laws and regulations governing the use,
manufacture, storage, handling and disposal of hazardous materials
and waste products. It is possible that accidental injury or
contamination from these materials may occur. In the event of an
accident, we could be held liable for any damages, which could
exceed our available financial resources. Further, we may not be
able to maintain insurance to cover these costs on acceptable
terms, or at all. In addition, we may be required to incur
significant costs to comply with environmental laws and regulations
in the future.
Our operations may be adversely affected by risks associated with
international business.
We may
be subject to certain risks that are inherent in an international
business, including:
●
varying regulatory
restrictions on sales of our products to certain markets and
unexpected changes in regulatory requirements;
●
tariffs, customs,
duties, and other trade barriers;
●
difficulties in
managing foreign operations and foreign distribution
partners;
●
longer payment
cycles and problems in collecting accounts receivable;
●
political
risks;
●
foreign exchange
controls that may restrict or prohibit repatriation of
funds;
●
export and import
restrictions or prohibitions, and delays from customs brokers or
government agencies;
56
●
seasonal reductions
in business activity in certain parts of the world;
and
●
potentially adverse
tax consequences.
Depending
on the countries involved, any or all of the foregoing factors
could materially harm our business, financial condition and results
of operations.
In the event we pursue growth through international operations,
such growth could strain our resources, and if we are unable to
manage any growth we may experience, we may not be able to
successfully implement our business plan.
In
connection with any geographic expansion we may pursue,
international operations would involve substantial additional
risks, including, among others: difficulties complying with the
U.S. Foreign Corrupt Practices Act and other applicable
anti-bribery laws. difficulties maintaining compliance with the
various laws and regulations of multiple jurisdictions that may be
applicable to our business, many of which may be unfamiliar to us.
more complexity in our regulatory and accounting compliance.
differing or changing obligations regarding taxes, duties or other
fees. limited intellectual property protection in some
jurisdictions. risks associated with currency exchange and
convertibility, including vulnerability to appreciation and
depreciation of foreign currencies. uncertainty related to
developing legal and regulatory systems and standards for economic
and business activities in some jurisdictions. trade restrictions
or barriers, including tariffs or other charges and import-export
regulations, changes in applicable laws or policies. the impact of
and response to natural disasters. and the potential for war, civil
or political unrest and economic and financial instability. The
occurrence of any of these risks could limit our ability to pursue
international expansion, increase our costs or expose us to fines
or other legal sanctions, any of which could negatively impact our
business, reputation and financial condition.
Our business could be adversely affected by the current novel
coronavirus (COVID-19) outbreak.
In
December 2019, COVID-19, a novel strain of the coronavirus was
first identified in Wuhan, Hubei Province, China. COVID-19 spread
to other parts of the world, including Canada as well as the United
States, India and Europe from where we obtain services and
supplies. As COVID-19 has spread, we and third parties with which
we contract are having to ask employees to temporarily work from
home, which could adversely impact the productivity of our
workforce or the workforce of third parties on which we rely for
supplies and services required for our operations. The result is
generally, potential interruptions or delays in our business
operations. The limitations on travel and interruption in global
shipping could affect the transport of supplies and raw materials.
Any disruption of our suppliers would likely impact our ability to
conduct R&D and commercial operations, and ultimately
materially adversely affect our operating results.
The
extent to which COVID-19 impacts our results will depend on future
developments, which are still uncertain and cannot be predicted,
including the duration of the outbreak, new information which may
emerge concerning the severity of COVID-19 and the actions to
contain COVID-19 or treat its impact, among others.
57
Risks related to our Common Shares
Trading on the OTC Markets is volatile and sporadic, which could
depress the market price of the Company’s Common Shares and
make it difficult for the Company’s stockholders to resell
their shares.
The
Company’s Common Shares are quoted on the OTCQB tier of the
OTC Markets. Trading in stock quoted on the OTC Markets is often
thin and characterized by wide fluctuations in trading prices, due
to many factors, some of which may have little to do with the
Company’s operations or business prospects. This volatility
could depress the market price of the Company’s Common Shares
for reasons unrelated to operating performance. Moreover, the OTC
Markets is not a stock exchange, and trading of securities on the
OTC Markets is often more sporadic than the trading of securities
listed on a quotation system like Nasdaq or a stock exchange like
the New York Stock Exchange. These factors may result in investors
having difficulty reselling any shares of the Company’s
Common Shares.
We may on occasion be unable to timely
file certain periodic reports and other documents with
the regulatory bodies
in Canada and the United States.
We may
not be able to timely file with the regulatory bodies in Canada and
the United States our year-end financial statements, management
discussion and analysis, Annual Information Form and 20F within the
required 120 days of our fiscal year end. If we are not able to
file our current and periodic reports and other documents in the
future in the times specified by the Securities Exchange Act,
we will continue to lose our eligibility to use Form F-1 for future
capital raises, and that could impair our ability to conduct more
efficient and expeditious public offerings of our stock. Our
inability to timely file current and periodic reports in the future
could materially and adversely affect our financial condition and
results of operations.
Our share price has been highly volatile and our shares could
suffer a further decline in value.
The
trading price of our Common Shares has been highly volatile and
could continue to be subject to wide fluctuations in price in
response to various factors, many of which are beyond our control,
including:
●
sales of our Common
Shares, including any sales made in connection with future
financings;
●
announcements
regarding new or existing corporate relationships or
arrangements;
●
announcements by us
of significant acquisitions, joint ventures, or capital
commitments;
●
actual or
anticipated period-to-period fluctuations in financial
results;
58
●
clinical and
regulatory development regarding our product
candidates;
●
litigation or
threat of litigation;
●
failure to achieve,
or changes in, financial estimates by securities
analysts;
●
comments or
opinions by securities analysts or members of the medical
community;
●
announcements
regarding new or existing products or services or technological
innovations by us or our competitors;
●
conditions or
trends in the pharmaceutical and biotechnology
industries;
●
additions or
departures of key personnel or directors;
●
economic and other
external factors or disasters or crises;
●
limited daily
trading volume; and
●
developments
regarding our patents or other intellectual property or that of our
competitors.
In
addition, the stock market in general and the market for drug
development companies in particular have experienced significant
price and volume fluctuations that have often been unrelated or
disproportionate to the operating performance of those companies.
Further, there has been significant volatility in the market prices
of securities of life science companies. In the past, following
periods of volatility in the market price of a company’s
securities, securities class action litigation has often been
instituted. Litigation of this type has been instituted against us
could result in substantial costs, potential liabilities, and the
diversion of management’s attention and
resources.
A large number of our Common Shares could be sold in the market in
the near future, which could depress our stock price.
As of
February 28, 2022, we had approximately 33,092,665 Common Shares
outstanding. In order to raise additional capital, we intend to
offer additional Common Shares or other securities convertible into
or exchangeable for our Common Shares. In addition, a substantial
portion of our shares are currently freely trading without
restriction under the U.S. Securities Act, having been registered
for resale or held by their holders for over six months and are
eligible for sale under Rule 144.
59
On July
17, 2017, the Company’s most recent registration statement on
Form F-3 (the “Shelf
Registration Statement”) was declared effective by the
SEC. The Shelf Registration Statement allows for, subject to
securities regulatory requirements and limitations, the potential
offering of up to an aggregate of US$100 million of the
Company’s Common Shares, preference shares, warrants,
subscription receipts, subscription rights and units, or any
combination thereof, from time to time in one or more offerings,
and are intended to give the Company the flexibility to take
advantage of financing opportunities when, and if, market
conditions are favorable to the Company. The specific terms of such
future offerings, if any, would be established, subject to the
approval of the Company’s board of directors (the
“Board”), at the
time of such offering and will be described in detail in a
prospectus supplement filed at the time of any such offering. To
the extent any securities of the Company are issued by the Company
under the Shelf Registration Statement or the shelf prospectus, a
shareholder’s percentage ownership will be diluted and our
stock price could be further adversely affected. As of February 28,
2022, the Company has issued 1,246,969 Common Shares using the
Shelf Registration Statement, and there can be no assurance that
any additional securities will be sold under the Shelf Registration
Statement or the shelf prospectus. Notwithstanding the foregoing,
currently the Company does not meet the requirements to utilize its
Form F-3 to issue any further securities under the Form
F-3.
As of
February 28, 2022, there are currently Common Shares issuable upon
the exercise of outstanding options and warrants and the conversion
of the outstanding Debentures for an aggregate of approximately
26,679,474 Common Shares. To the extent any of our options and
warrants are exercised and the Debenture are converted, a
shareholder’s percentage ownership will be diluted and our
stock price could be further adversely affected. Moreover, as the
underlying shares are sold, the market price could drop
significantly if the holders of these restricted shares sell them
or if the market perceives that the holders intend to sell these
shares.
We have no history or foreseeable prospect of paying cash
dividends.
We have
not paid any cash dividends on our Common Shares and do not intend
to pay cash dividends in the foreseeable future. We intend to
retain future earnings, if any, for reinvestment in the development
and expansion of our business. Dividend payments in the future may
also be limited by loan agreements or covenants contained in other
securities we may issue. Any future determination to pay cash
dividends will be at the discretion of our Board and depend on our
financial condition, results of operations, capital and legal
requirements and such other factors as our Board deems
relevant.
There may not be an active, liquid market for our Common
Shares.
There
is no guarantee that an active trading market for our Common Shares
will be maintained on OTCQB or TSX. Investors may not be able to
sell their shares quickly or at the latest market price if trading
in our Common Shares is not active.
There
may be future sales or other dilution of our equity, which may
adversely affect the market price of our Common
Shares.
60
The
Company may, from time to time, issue additional Common Shares,
including any securities that are convertible into or exchangeable
for, or that represent the right to receive, Common Shares. The
market price of our Common Shares could decline as a result of
sales of Common Shares or securities that are convertible into or
exchangeable for, or that represent the right to receive, Common
Shares after this offering or the perception that such sales could
occur.
Future sales of our Common Shares may cause the prevailing market
price of our Common Shares to decrease.
We have
registered a substantial number of outstanding Common Shares and
Common Shares that are issuable upon the exercise of outstanding
warrants. If the holders of our registered Common Shares choose to
sell such shares in the public market or if holders of our warrants
exercise their purchase rights and sell the underlying Common
Shares in the public market, or if holders of currently restricted
Common Shares choose to sell such shares in the public market, the
prevailing market price for our Common Shares may decline. The sale
of shares issued upon the exercise of our warrants (and options)
could also further dilute the holdings of our then existing
shareholders. In addition, future public sales by holders of our
Common Shares could impair our ability to raise capital through
equity offerings.
Future issuances of our shares could adversely affect the trading
price of our Common Shares and could result in substantial dilution
to shareholders.
We may
need to issue substantial amounts of Common Shares in the future.
There can be no assurance that we will be able to sell any
additional shares. To the extent that the market price of our
Common Shares declines, we will need to issue an increasing number
of Common Shares per dollar of equity investment. In addition to
our Common Shares issuable in connection with the exercise of our
outstanding warrants, our employees, and directors will hold rights
to acquire substantial amounts of our Common Shares. In order to
obtain future financing if required, it is likely that we will
issue additional Common Shares or financial instruments that are
exchangeable for or convertible into Common Shares. Also, in order
to provide incentives to employees and induce prospective employees
and consultants to work for us, we may offer and issue options to
purchase Common Shares and/or rights exchangeable for or
convertible into Common Shares. Future issuances of shares could
result in substantial dilution to shareholders. Capital raising
activities, if available, and dilution associated with such
activities could cause our share price to decline. In addition, the
existence of common share purchase warrants may encourage short
selling by market participants. Also, in order to provide
incentives to current employees and directors and induce
prospective employees and consultants to work for us, we have
historically granted options and DSUs, and intend to continue to do
so or offer and issue other rights exchangeable for or convertible
into common shares. Future issuances of shares could result in
substantial dilution to all our shareholders. In addition, future
public sales by holders of our common shares could impair our
ability to raise capital through any future equity
offerings.
We may in the future issue preference shares which could adversely
affect the rights of holders of our Common Shares and the value of
such shares.
Our
Board has the ability to authorize the issue of an unlimited number
of preference shares in series, and to determine the price, rights,
preferences and privileges of those shares without any further vote
or action by the holders of our common shares. Although we have no
preference shares issued and outstanding, preference shares issued
in the future could adversely affect the rights and interests of
holders of our Common Shares.
61
Our Common Shares may not continue to be listed on the
TSX.
Failure
to maintain the applicable continued listing requirements of the
TSX could result in our common shares being delisted from the TSX.
The TSX will normally consider the delisting of securities if, in
the opinion of the exchange, it appears that the public
distribution, price, or trading activity of the securities has been
so reduced as to make further dealings in the securities on TSX
unwarranted. For example, participating securities may be delisted
from the TSX if, among other things, the market value of an
issuer’s securities that are listed on the TSX is less than
C$3,000,000 over any period of 30 consecutive trading days. In such
circumstances, the TSX may notify an issuer that it is under
delisting review and the issuer will normally be given up to 120
days from the date of such notification to correct the fall in
market value and such other deficiencies noted by the TSX. At any
time prior to the end of the delisting review period, the TSX will
provide the issuer with an opportunity to be heard where the issuer
may present submissions to satisfy the TSX that all deficiencies
identified in the TSX’s notice have been rectified. If at the
conclusion of the hearing the issuer cannot satisfy the TSX that
the deficiencies identified have been rectified and that no other
delisting criteria are then applicable to the issuer, the TSX will
determine whether to delist the issuer’s
securities.
If the
market price of our Common Shares declines further or we are unable
to maintain other listing requirements, the TSX may determine to
delist our Common Shares. If our Common Shares are no longer listed
on the TSX, they may be eligible for listing on the TSX Venture
Exchange. In the event that we are not able to maintain a listing
for our Common Shares on the TSX or the TSX Venture Exchange, it
may be extremely difficult or impossible for shareholders to sell
their common shares in Canada. Moreover, if we are delisted from
the TSX, but obtain a substitute listing for our Common Shares on
the TSX Venture Exchange, our Common Shares will likely have less
liquidity and more price volatility than experienced on the
TSX.
Shareholders
may not be able to sell their Common Shares on any such substitute
exchange in the quantities, at the times, or at the prices that
could potentially be available on a more liquid trading market. As
a result of these factors, if our Common Shares are delisted from
the TSX, the price of our common shares is likely to
decline.
Our
Common Shares are currently a “penny stock” under SEC
rules. It may be more difficult to resell shares of Common Shares
classified as “penny stock.” Our Common Shares are
a “penny stock” under applicable SEC rules.
Transactions in securities that are traded in the United States by
companies with net tangible assets of $5,000,000 or less and a
market price per share of less than $5.00 that are not traded on
Nasdaq or on other securities exchanges may be subject to the
“penny stock” rules promulgated under the U.S. Exchange
Act. Under these rules, broker-dealers who recommend such
securities to persons other than institutional investors
must:
●
make a
special written suitability determination for the
purchaser;
●
receive the
purchaser’s written agreement to a transaction prior to
sale;
●
provide the
purchaser with risk disclosure documents which identify risks
associated with investing in “penny stocks” and which
describe the market for these “penny stocks” as well as
a purchaser’s legal remedies; and
●
obtain a signed and
dated acknowledgment from the purchaser demonstrating that the
purchaser has actually received the required risk disclosure
document before a transaction in a “penny stock” can be
completed.
As a
result of these requirements, since our Common Shares are subject
to the “penny stock” rules, broker-dealers may find it
difficult to effectuate customer transactions and trading activity
in these shares in the United States may be significantly limited.
Accordingly, the market price of the shares may be depressed, and
investors may find it more difficult to sell the
shares.
62
As long as our stock price remains below $5.00 per share, our
shareholders will face restrictions in using our shares as
collateral for margin accounts.
The
closing price of our Common Shares on the OTCQB on February 25,
2022 was $0.12 per share. If the market price of our Common Shares
remains below $5.00 per share, under Federal Reserve regulations
and account maintenance rules of many brokerages, our shareholders
will face restrictions in using such shares as collateral for
borrowing in margin accounts. These restrictions on the use of our
Common Shares as collateral may lead to sales of such shares
creating downward pressure on and increased volatility in, the
market price of our Common Shares. In addition, many institutional
investors will not invest in stocks whose prices are below $5.00
per share.
Our shareholders may face significant restrictions on the resale of
our Common Shares due to state “Blue Sky”
laws.
Each
state has its own securities laws, often called “blue
sky” laws, which (i) limit sales of securities to a
state’s residents unless the securities are registered in
that state or qualify for an exemption from registration, and (ii)
govern the reporting requirements for broker-dealers doing business
directly or indirectly in the state. Before a security is sold in a
state, there must be a registration in place to cover the
transaction, or the transaction must be exempt from registration.
The applicable broker must be registered in that
state.
Absent
compliance with such individual state laws, our Common Shares may
not be traded in such jurisdictions. Because the securities have
not been registered for resale under the Blue Sky laws of any
state, the holders of such shares and persons who desire to
purchase them in any trading market that might develop in the
future, should be aware that there may be significant state Blue
Sky law restrictions upon the ability of investors to sell the
securities and of purchasers to purchase the securities.
Accordingly, investors may not be able to liquidate their
investments and should be prepared to hold Common Shares for an
indefinite period of time. You should therefore consider the resale
market for our Common Shares to be limited, as you may be unable to
resell your shares without the significant expense of state
registration or qualification.
As a foreign private issuer in the United States, we are subject to
different U.S. securities laws and rules than a domestic U.S.
issuer.
As a
foreign private issuer under U.S. securities laws we are not
required to comply with all the periodic disclosure requirements of
the U.S. Exchange Act applicable to domestic United States
companies and therefore the publicly available information about us
may be different or more limited than if we were a United States
domestic issuer. In addition, our officers, directors, and
principal shareholders are exempt from the “real time”
reporting and “short swing” profit recovery provisions
of Section 16 of the U.S. Exchange Act and the rules thereunder.
Although under Canadian rules, our officers, directors and
principal shareholders are generally required to file on SEDI
(www.sedi.ca) reports of transactions involving our common shares
within five calendar days of such transaction, our principal
shareholders may not know when our officers, directors and
principal shareholders purchase or sell our common shares as timely
as they would if we were a United States domestic
issuer.
63
We are exposed to risks if we are unable to comply with laws and
future changes to laws affecting public companies, including the
Sarbanes-Oxley Act of 2002 (“SOX”), and also to
increased costs associated with complying with such
laws.
Any
future changes to the laws and regulations affecting public
companies, as well as compliance with existing provisions of SOX in
the United States and applicable Canadian securities laws,
regulations, rules and policies, may cause us to incur increased
costs to comply with such laws and requirements, including, among
others, hiring additional personnel and increased legal, accounting
and advisory fees. Delays, or a failure to comply with applicable
laws, rules and regulations could result in enforcement actions,
the assessment of other penalties and civil suits. The new laws and
regulations may increase potential costs to be borne under
indemnities provided by us to our officers and directors and may
make it more difficult to obtain certain types of insurance,
including liability insurance for directors and officers; as such,
we may be forced to accept reduced policy limits and coverage or
incur substantially higher costs to obtain the same or similar
coverage. The impact of these events could also make it more
difficult to attract and retain qualified persons to serve on our
Board, or as executive officers.
We are
required annually to review and report on the effectiveness of our
internal control over financial reporting in accordance with SOX
Section 404 and Multilateral Instrument 52-109 –
Certification of Disclosure in Issuer’s Annual and Interim
Filings of the Canadian Securities Administrators. The results of
this review are reported in our Annual Report on Form 20-F and in
our Management Discussion and Analysis.
Management’s
review is designed to provide reasonable, not absolute, assurance
that all material weaknesses in our internal controls are
identified. Material weaknesses represent deficiencies in our
internal controls that may not prevent or detect a misstatement
occurring which could have a material adverse effect on our
quarterly or annual financial statements. In addition, there can be
no assurance that any remedial actions we take to address any
material weaknesses identified will be successful, nor can there be
any assurance that further material weaknesses will not be
identified in future years. Material errors, omissions or
misrepresentations in our disclosures that occur as a result of our
failure to maintain effective internal control over financial
reporting could have a material adverse effect on our business,
financial condition, results of operations, and the value of our
common shares.
We may be classified as a “passive foreign investment
company” or PFIC for U.S. income tax purposes, which could
have significant and adverse tax consequences to U.S.
investors.
The
possible classification of our Company as a passive foreign
investment company, or PFIC, for U.S. federal income tax purposes
could have significant and adverse tax consequences for U.S.
Holders (as defined below) with respect to the sale or other
disposition of our Common Shares acquired through the exercise of
certain warrants. It may be possible for U.S. Holders of Common
Shares to mitigate certain of these consequences by making an
election (a so-called “QEF Election”) to treat us as a
“qualified electing fund” or “QEF” under
Section 1295 of the Internal Revenue Code (the “Code”); or a mark-to-market
election under Section 1296 of the Code. A non-U.S. corporation
generally will be a PFIC if, for a taxable year (a) 75% or more of
the gross income of such corporation for such taxable year consists
of specified types of passive income or (b) on average, 50% or more
of the assets held by such corporation either produce passive
income or are held for the production of passive income, based on
the fair market value of such assets (or on the adjusted tax basis
of such assets, if such non-U.S. corporation is not publicly traded
and either is a “controlled foreign corporation” under
Section 957(a) of the Code, or makes an election to determine
whether it is a PFIC based on the adjusted basis of the
assets).
64
The
determination of whether we are, or will be, a PFIC for a taxable
year depends, in part, on the application of complex U.S. federal
income tax rules, which are subject to various interpretations. We
believe that there is a substantial basis for concluding that we
were not a PFIC during our 2019 taxable year and will not likely be
a PFIC during our 2020 taxable year, although that conclusion is
not free from doubt. Because PFIC status is based on our income,
assets and activities for the entire taxable year, and our market
capitalization, it is not possible to determine whether we will be
characterized as a PFIC for the 2020 taxable year until after the
close of the taxable year. The tests for determining PFIC status
are subject to a number of uncertainties. These tests are applied
annually, and it is difficult to accurately predict future income,
assets and activities relevant to this determination. In addition,
because the market price of our Common Shares is likely to
fluctuate, the market price may affect the determination of whether
we will be considered a PFIC. There can be no assurance that we
will not be considered a PFIC for any taxable year (including our
2020 taxable year). Absent one of the elections described above, if
we are a PFIC for any taxable year during which a U.S. Holder holds
our Common Shares, we generally will continue to be treated as a
PFIC regardless of whether we cease to meet the PFIC tests in one
or more subsequent years. Accordingly, no assurance can be given
that we will not constitute a PFIC in the current (or any future)
tax year or that the Internal Revenue Service (the
“IRS”) will not
challenge any determination made by us concerning our PFIC
status.
If we
are a PFIC, the U.S. federal income tax consequences to a U.S.
Holder of the ownership and disposition of our Common Shares will
depend to some extent on whether such U.S. Holder makes a QEF or
mark-to-market election after acquisition of such shares through
the exercise of warrants. Unless otherwise provided by the IRS, a
U.S. holder of our Common Shares is generally required to file an
informational return annually to report its ownership interest in
the Company during any year in which we are a PFIC.
The
foregoing only speaks to the United States federal income tax
considerations as to the Code in effect on the date of this Annual
information Form.
The foregoing does not purport to be a complete enumeration or
explanation of the tax risks involved in an investment in our
Company. Prospective investors should read this entire annual
report and consult with their own legal, tax and financial advisors
before deciding to invest in our company.
It may be difficult to obtain and enforce judgments against us
because of our Canadian residency.
We are
governed by the laws of Canada. All of our directors and officers
are residents of Canada and all or a substantial portion of our
assets and the assets of such persons may be located outside of the
United States. As a result, it may be difficult for shareholders to
effect service of process upon us or such persons within the United
States or to realize in the United States on judgments of courts of
the United States predicated upon the civil liability provisions of
the U.S. federal securities laws or other laws of the United
States. In addition, there is doubt as to the enforceability in
Canada of liabilities predicated solely upon U.S. federal
securities law against us, our directors, controlling persons and
officers who are not residents of the United States, in original
actions or in actions for enforcements of judgments of U.S.
courts.
65
Other Risks
There are other unidentified risks.
The
risks set forth above are not a complete list of the risks facing
purchasers of our securities. We acknowledge that there
may exist significant risks yet to be recognized or encountered to
which we may not be able to effectively respond. There
can be no assurance that we will succeed in addressing these risks
or future potential risks, and any failure to do so could have a
material adverse effect on our business, financial condition and
results of operations.
We have
not paid any cash dividends on our common shares and do not intend
to pay cash dividends in the foreseeable future. We intend to
retain future earnings, if any, for reinvestment in the development
and expansion of our business. Dividend payments in the future may
also be limited by loan agreements or covenants contained in other
securities we may issue. Any future determination to pay cash
dividends will be at the discretion of our board of directors and
will depend on our financial condition, results of operations,
capital and legal requirements and such other factors as our board
of directors deems relevant.
Our
authorized share capital consists of an unlimited number of Common
Shares, all without nominal or par value and an unlimited number of
preference shares issuable in series. At November 30, 2021, there
were 33,092,665 Common Shares and no preference shares issued and
outstanding. As of the date of this annual information form there
were 33,092,665 Common Shares and no preference shares issued and
outstanding.
Common Shares
Each of
our Common Shares entitles the holder thereof to one vote at any
meeting of shareholders of the Company, except meetings at which
only holders of a specified class of shares are entitled to vote.
Subject to the prior rights of the holders of any preference
shares, the holders of Common Shares are entitled to receive, as
and when declared by the board of directors, dividends in such
amounts as shall be determined by the board of directors of the
Company. The holders of common shares of the Company have the right
to receive the remaining property of the Company in the event of
liquidation, dissolution, or winding-up of the Company, whether
voluntary or involuntary.
66
Preference Shares
The
preference shares may at any time and from time to time be issued
in one or more series. The board of directors will, by resolution,
from time to time, before the issue thereof, fix the rights,
privileges, restrictions and conditions attaching to the preference
shares of each series. Except as required by law, the holders of
any series of preference shares will not as such be entitled to
receive notice of, attend or vote at any meeting of the
shareholders of the Company. Holders of preference shares will be
entitled to preference with respect to payment of dividends and the
distribution of assets in the event of liquidation, dissolution or
winding-up of the Company, whether voluntary or involuntary, or any
other distribution of the assets of the Company among its
shareholders for the purpose of winding up its affairs, on such
shares over the common shares and over any other shares ranking
junior to the preference shares.
Warrants
At November 30, 2021, an aggregate of 21,160,314 Common Shares were
issuable upon the exercise of outstanding common share purchase
warrants, with a weighted average exercise price of $0.76 per
common share. As of the date of this annual information form, an
aggregate of 21,160,314 Common Shares were issuable upon the
exercise of outstanding common share purchase warrants, with a
weighted average exercise price of $0.76 per common
share.
Options
At November 30, 2021, an aggregate of 1,489,500 Common
Shares were issuable upon the exercise of outstanding options, with
a weighted average exercise price of $2.40 per common share and up
to 1,819,767 additional Common Shares were reserved for issuance
under our stock option plan.
From
November 30, 2021 to the date of this annual information form, no
options to purchase our Common Shares were granted, no options to
purchase our Common Shares were exercised. At the date of this
annual information form, up to 1,819,767 additional Common Shares
were reserved for issuance under our stock option
plan.
Convertible Debenture
In
January 2013, the Company completed the private placement financing
of an unsecured debenture in the original principal amount of $1.5
million (the “2013
Debenture”). The 2013 Debenture bore interest at a
rate of 12% per annum, payable monthly, was pre-payable at any time
at the option of the Company and was convertible at any time into
Common Shares at a conversion price of $30.00 per Common Share at
the option of the holder. Drs. Isa and Amina Odidi, who are
directors, executive officers and shareholders of our Company,
provided us with the original $1.5 million of the proceeds for the
2013 Debenture. In December 2016, a principal repayment of $150,000
was made on the 2013 Debenture and the maturity date was extended
until April 1, 2017. Effective March 28, 2017, the maturity date of
the 2013 Debenture was extended to October 1, 2017. Effective
September 28, 2017, the maturity date of the 2013 Debenture was
further extended to October 1, 2018. Effective October 1, 2018, the
maturity date for the 2013 Debenture was further extended to April
1, 2019. Effective April 1, 2019, the maturity date for the 2013
Debenture was further extended to May 1, 2019. In December 2018, a
principal repayment of $300,000 was made on the 2013 Debenture. On
April 4, 2019, a tentative approval from TSX was received for a
proposed refinancing of the 2013 Debenture, subject to certain
conditions being met. As a result of the proposed refinancing, the
principal amount owing under the 2013 Debenture was refinanced by
the 2019 Debenture. On May 1, 2019, the 2019 Debenture was issued
with a principal amount of $1,050,000, was originally scheduled to
mature on November 1, 2019, bear interest at a rate of 12% per
annum and be convertible into 1,779,661 common shares of the
Company at a conversion price of $0.59 per common share. Dr. Isa
Odidi and Dr. Amina Odidi, who are shareholders, directors, and
executive officers of the Company, are the holders of the 2019
Debenture. The maturity date for the May 2019 Debenture has been
extended from time to time and the maturity date for the May 2019
Debenture is now February 28, 2022. No interest was paid on the May
2019 Debenture for the ended November 30, 2021.
67
In
September 2018, the Company completed a private placement financing
of an unsecured convertible debenture in the principal amount of
$500,000 (the “2018
Debenture”). The 2018 Debenture bears interest at a
rate of 10% per annum, is payable monthly, may be prepaid at any
time at our option, and is convertible into Common Shares at any
time prior to the maturity date at a conversion price of $3.00 per
Common Share at the option of the holder. Drs. Isa and Amina Odidi,
who are directors, executive officers and shareholders of our
Company, provided us with the original $500,000 of proceeds for the
2018 Debenture. The net proceeds of the 2018 Debenture were used
for working capital and general corporate purposes. The maturity
date for the 2018 Debenture was September 1, 2020. Effective November 30,
2020, the maturity date for the 2018 Debenture was extended to
February 28, 2022. No interest was paid on the 2018 Debenture for
the year ended November 30, 2021.
On
August 26, 2019, the Company completed a private placement
financing of the unsecured August 2019 Debenture in the principal
amount of $140,800. The August 2019 Debenture was originally
scheduled to mature on August 26, 2020, bore interest at a rate of
8% per annum, was pre-payable at any time at the option of the
Company up to 180 days from date of issuance with pre-payment
penalties ranging from 5% - 30% and was convertible at the option
of the holder into common shares after 180 days at a conversion
price which was equal to 75% of the market price (defined as the
average of the lowest three (3) trading prices for the common
shares during the twenty (20) trading day period prior to the
conversion date). The Company incurred $15,800 in debt issuance
costs. In November 2019, the August 2019 Debenture was fully
paid.
On
November 15, 2019, we issued to Drs. Isa and Amina Odidi, by way of
a private placement, an unsecured convertible debenture of the
Company in consideration for, and in the aggregate principal amount
of, USD$250,000 (the "November 2019
Debenture"). The principal amount owing under the November
2019 Debenture is convertible at any time and from time to time
into Common Shares at a conversion price equal to U.S. $0.12 per
Common Share. Up to an aggregate of 2,083,333 Common Shares may be
issued upon conversion of the principal amount owing under the
November 2019 Debenture. The November 2019 Debenture bears interest
at a rate of 12% per annum (calculated monthly) and, subject to our
right to prepay the November 2019 Debenture in whole or in part at
any time without penalty, and was originally scheduled to mature on
December 31, 2019. The maturity date for the November 2019
Debenture has been extended from time to time and the maturity date
for the November 2019 Debenture is now February 28, 2022. No
interest was paid on the November 2019 Debenture for the year ended
November 30, 2021. Dr. Isa Odidi is our Chairman, Chief Executive
Officer and Co-Chief Scientific Officer, and Dr. Amina Odidi is our
President, Chief Operating Officer and Co-Chief Scientific
Officer.
Deferred Share Units
At November 30, 2021, there were no DSUs issued and
outstanding. At February 28, 2022, 11,000 additional DSUs are
reserved for issuance under our DSU plan. A description of the principal aspects of our DSU
plan is contained in our May 2019 management information circular
available under our company profile on SEDAR at www.sedar.com.
Restricted Share Units
At November 30, 2021, there were no restricted share units
(“RSUs”) issued and outstanding. From November 30,
2021 to the date of this annual information form, no RSUs have been
issued. At the date of this annual information form, 33,000 RSUs
are reserved for issuance under our RSU Plan. A description
of the principal aspects of our RSU plan is contained in our latest
management information circular available under our company profile
on SEDAR at www.sedar.com.
68
Trading Price and Volume
Our
common shares are currently listed on the TSX and quoted for
trading on OTCQB, under the symbols “IPCI” and
“IPCIF”, respectively. Our shares began trading on
October 22, 2009, when the transaction with Vasogen was
completed.
In
March 2019, we received formal notice that the Nasdaq Panel had
determined to delist our shares from Nasdaq based upon our
non-compliance with the $1.00 bid price requirement, as set forth
in Nasdaq Listing Rule 5550(a)(2). The suspension of trading on
Nasdaq took effect at the open of business on March 21, 2019. Our
shares began trading on the OTCQB under the symbol
“IPCIF”, commencing on March 21, 2019. Our shares also
are listed on the TSX under the symbol “IPCI” and our
non-compliance with Nasdaq's requirements did not impact our
listing or trading status on that exchange.
The
following table sets forth the monthly trading history for the
fiscal year ended November 30, 2021, the reported high, low and
closing prices (in Canadian dollars) and total volume traded of our
common shares on the TSX and reported high, low and closing prices
(in United States dollars) and total volume of our common shares
traded on OTCQB.
|
|
TSX
|
OTCQB
|
||||||
|
|
(C$ per
share)
|
($ per
share)
|
||||||
|
Date
|
High
|
Low
|
Close
|
Volume
|
High
|
Low
|
Close
|
Volume
|
|
Dec-20
|
0.22
|
0.17
|
0.19
|
406,200
|
0.17
|
0.11
|
0.14
|
851,700
|
|
Jan-21
|
0.23
|
0.18
|
0.22
|
602,400
|
0.18
|
0.14
|
0.16
|
1,172,300
|
|
Feb-21
|
0.60
|
0.20
|
0.36
|
2,118,500
|
0.48
|
0.15
|
0.27
|
2,892,400
|
|
Mar-21
|
0.44
|
0.21
|
0.24
|
1,138,000
|
0.30
|
0.17
|
0.19
|
1,414,900
|
|
Apr-21
|
0.35
|
0.23
|
0.27
|
372,300
|
0.24
|
0.16
|
0.23
|
345,200
|
|
May-21
|
0.27
|
0.22
|
0.22
|
169,600
|
0.22
|
0.17
|
0.18
|
178,100
|
|
Jun-21
|
0.28
|
0.20
|
0.22
|
263,400
|
0.23
|
0.16
|
0.18
|
320,000
|
|
Jul-21
|
0.22
|
0.19
|
0.22
|
108,500
|
0.18
|
0.14
|
0.18
|
157,900
|
|
Aug-21
|
0.22
|
0.19
|
0.20
|
86,900
|
0.18
|
0.14
|
0.15
|
144,200
|
|
Sep-21
|
0.20
|
0.16
|
0.16
|
86,900
|
0.16
|
0.12
|
0.14
|
54,000
|
|
Oct-21
|
0.20
|
0.16
|
0.19
|
47,700
|
0.16
|
0.12
|
0.15
|
59,800
|
|
Nov-21
|
0.19
|
0.14
|
0.14
|
237,600
|
0.15
|
0.11
|
0.12
|
345,400
|
69
Prior Sales
During
the 12-month period prior to the date of this Annual Information
Form, we have issued Common Shares, or securities convertible into
Common Shares, as follows:
In
January 2013, the Company completed the private placement financing
of the unsecured convertible 2013 Debenture in the original
principal amount of $1.5 million. The 2013 Debenture bears interest
at a rate of 12% per annum, payable monthly, is pre-payable at any
time at the option of the Company and is convertible at any time
into Common Shares at a conversion price of $30.00 per Common Share
at the option of the holder. Drs. Isa and Amina Odidi, who are
directors, executive officers and shareholders of our Company,
provided us with the original $1.5 million of the proceeds for the
2013 Debenture. In December 2016, a principal repayment of $150,000
was made on the 2013 Debenture and the maturity date was extended
until April 1, 2017. Effective March 28, 2017, the maturity date of
the 2013 Debenture was extended to October 1, 2017. Effective
September 28, 2017, the maturity date of the 2013 Debenture was
further extended to October 1, 2018. Effective October 1, 2018, the
maturity date for the 2013 Debenture was further extended to April
1, 2019. Effective April 1, 2019, the maturity date for the 2013
Debenture was further extended to May 1, 2019. In December 2018, a
principal repayment of $300,000 was made on the 2013
Debenture.
On
April 4, 2019, a tentative approval from TSX was received for a
proposed refinancing of the 2013 Debenture, subject to certain
conditions being met. As a result of the proposed refinancing, the
principal amount owing under the 2013 Debenture was refinanced by
the May 2019 Debenture. On May 1, 2019, the May 2019 Debenture was
issued with a principal amount of $1,050,000, that will mature on
November 1, 2019, bear interest at a rate of 12% per annum and be
convertible into 1,779,661 common shares of the Company at a
conversion price of $0.59 per common share. Dr. Isa Odidi and Dr.
Amina Odidi, who are shareholders, directors, and executive
officers of the Company, are the holders of the May 2019 Debenture.
The maturity date for the May 2019 Debenture has been extended from
time to time and the maturity date for the May 2019 Debenture is
now February 28, 2022.
On
September 10, 2018, the Company issued the 2018 Debenture. The 2018
Debenture bears interest at a rate of 10% per annum, payable
monthly, may be prepaid at any time at our option, and is
convertible into Common Shares at any time prior to the maturity
date at a conversion price of $3.00 per Common Share at the option
of the holder. Drs. Isa and Amina Odidi, who are directors,
executive officers and shareholders of our Company, provided us
with the original $500,000 of proceeds for the 2018 Debenture. The
maturity date for the 2018 Debenture was originally September 1,
2020. Effective September 1, 2020, the maturity date for the 2018
Debenture was extended to November 30, 2020. Effective November 30,
2020, the maturity date for the 2018 Debenture was further extended
to February 28, 2022.
On
November 15, 2019, we issued to Drs. Isa and Amina Odidi, by way of
a private placement, an unsecured convertible debenture of the
Company in consideration for, and in the aggregate principal amount
of, USD$250,000. The principal amount owing under the November 2019
Debenture is convertible at any time and from time to time into
Common Shares at a conversion price equal to U.S. $0.12 per Common
Share. Up to an aggregate of 2,083,333 Common Shares may be issued
upon conversion of the principal amount owing under the November
2019 Debenture. The November 2019 Debenture bears interest at a
rate of 12% per annum (calculated monthly) and, subject to our
right to prepay the November 2019 Debenture in whole or in part at
any time without penalty, and matures on December 31, 2019.
Effective January 31, 2020, the December 31, 2019 maturity date was
extended to March 31, 2020. The maturity date for the November 2019
Debenture has been extended from time to time and the maturity date
for the November 2019 Debenture is now February 28,
2022.
70
In
April 2021, the Company completed a private placement offering of
an aggregate of 9,414,560 Common Shares at a price of CAD$0.41 per
Common Share.
Dr. Isa
Odidi is our Chairman, Chief Executive Officer and Co-Chief
Scientific Officer, and Dr. Amina Odidi is our President, Chief
Operating Officer and Co-Chief Scientific Officer.
During
the 12-month period ended November 30, 2021, no warrants (including Pre-Funded
Warrants) were exercised.
During
the 12-month period ended November 30, 2021, no options were
granted, and no options were exercised.
During
the 12-month period ended November 30, 2021, no DSUs were granted,
no DSUs were exercised.
The
name and province of residence of each of our directors and
officers as at the date hereof, the office presently held,
principal occupation, and the year each director first became a
director of the Company or its predecessor, IPC Ltd., are set out
below. Each director is elected to serve until the next annual
meeting of our shareholders or until his or her successor is
elected or appointed. Officers are appointed annually and serve at
the discretion of the Board.
|
Name and Province of Residence
|
Position held with the Company
|
Principal Occupations During the Last 5 Years
|
Other Public Company Boards
|
Director Since
|
|
Dr. Isa Odidi Ontario, Canada
|
Chairman
of the Board and Chief Executive Officer
|
Officer
of the Company
|
None
|
September
2004
|
|
Dr. Amina Odidi Ontario, Canada
|
President,
Chief Operating Officer and Director
|
Officer
of the Company
|
None
|
September
2004
|
|
Norman Betts(1)(2)(3)New
Brunswick, Canada
|
Director(4)
|
Associate
Professor and Professor, University of New Brunswick, Faculty of
Business Administration
|
Tanzanian
Royalty Exploration Inc.;
Adex
Mining Inc.;
49
North Resources Inc.;
Biotricity
Inc.;
Canada
House Wellness Group Inc.
|
January
2019
|
|
Shawn Graham(1)(2)(3)New
Brunswick, Canada
|
Director
|
Associate
Professor and Professor, University of New Brunswick, Faculty of
Business Administration
|
None
|
May
2018
|
|
Kenneth Keirstead(4) New Brunswick,
Canada
|
Director
|
Executive
Manager of Lyceum Group, a consulting business
|
None
|
January
2006
|
|
Bahadur Madhani(1)(2)(3)Ontario,
Canada
|
Director
|
Chief
Executive Officer of Equiprop Management Limited, a consulting
business
|
None
|
March
2006
|
|
Dr. Patrick Yat Ontario, Canada
|
Vice-President,
Chemistry and Analytical Services
|
Officer
of the Company
|
None
|
N/A
|
71
Notes:
(1)
Member of the Audit
Committee.
(2)
Member of the
Compensation Committee.
(3)
Member of the
Corporate Governance Committee.
(4)
Kenneth Keirstead
is deceased January 6, 2022
Drs.
Isa Odidi and Amina Odidi are spouses to each other.
Cease Trade Order
Except
as noted below, no director or executive officer of the Company is,
as at the date of this annual information form, or has been within
10 years before the date of this annual information form, a
director, chief executive officer or chief financial officer of any
company that was the subject of a cease trade or similar order, or
an order that denied the other issuer access to any statutory
exemptions, for a period of more than thirty consecutive days (i)
while that person was acting as a director, chief executive officer
or chief financial officer; or (ii) after that person ceased acting
as a director, chief executive officer or chief financial officer
which resulted from an event that occurred while that person was
acting in that capacity.
72
From
March 2006 until June 2013, Dr. Norman Betts served as a director
of Starfield Resources Inc. (TSX: SRU) (“Starfield”). On August 22, 2013,
Starfield was the subject of a cease trade order issued by the
Ontario Securities Commission as a result of Starfield’s
failure to file, inter
alia, its audited annual financial statements, related
management’s discussion and analysis and officer
certifications for the year ended February 28, 2013. The order is
still in effect. On April 18, 2013, Starfield’s shares were
delisted from the TSX.
Bankruptcies
Except
as noted below, no director or executive officer or a shareholder
holding a sufficient number of securities of the Company to affect
materially the control of the Company (i) is, as at the date of
this annual information form, or has been, within 10 years before
the date of this annual information form, a director or executive
officer of any company that, while that person was acting in that
capacity, or within a year of that person ceasing to act in that
capacity, became bankrupt, made a proposal under any legislation
relating to bankruptcy or insolvency, or was subject to or
instituted any proceedings, arrangement or compromise with
creditors or had a receiver, receiver manager or trustee appointed
to hold its assets, or (ii) has, within 10 years before the date of
this annual information form, become bankrupt, made a proposal
under any legislation relating to bankruptcy or insolvency or (iii)
was subject to or instituted any proceedings, arrangement or
compromise with creditors or had a receiver, receiver manager or
trustee appointed to hold its assets.
From
March 2006 until June 2013, Dr. Norman Betts served as a director
of Starfield. On July 2, 2013, Starfield announced that it was
deemed to have made an assignment in bankruptcy, effective at the
close of business on June 28, 2013 for failure to file a proposal
before the time for doing so had past pursuant to the provisions of
the Bankruptcy and Insolvency
Act (Canada) (the “BIA”). Starfield had previously
filed a Notice of Intention to Make a Proposal (“Notice of Intention”) pursuant to
the provisions of Part III of the BIA. Pursuant to the Notice of
Intention, PriceWaterhouseCoopers Inc. (“PwC”) was appointed as the trustee
(“Proposal
Trustee”) in Starfield’s proposal proceedings.
Pursuant to a Order of the Ontario Superior Court of Justice
(Commercial List), the time for Starfield to file a proposal
expired at the end of the day on June 28, 2013. Starfield completed
a sale of substantially all of its assets related to its Ferguson
Lake Project in early June 2013. However, in consultation with the
Proposal Trustee, Starfield determined that it would not be able to
put forward a viable proposal and would not be filing a proposal by
the deadline. As a result, Starfield was deemed to have made an
assignment in bankruptcy at the end of the day on June 28, 2013.
PwC acted as the trustee in bankruptcy for Starfield.
Conflicts of Interest
Certain
of the directors and officers of the Company and its subsidiaries
are also directors, officers and shareholders of other companies
and conflicts may arise between their duties as directors or
officers of the Company and its subsidiaries and as directors,
officers or shareholders of other companies. All such possible
conflicts are required to be disclosed in accordance with the
requirements of the Canada
Business Corporations Act and the Company’s Code of
Business Conduct and Ethics and those concerned are required to
govern themselves in accordance with the obligations imposed upon
them by law and such code.
73
The
Audit Committee of the Board monitors our financial activities,
policies, and internal control procedures. The Audit Committee
assists the Board in fulfilling its oversight responsibility to
shareholders, potential shareholders, the investment community, and
others with respect to the Company’s financial statements,
financial reporting process, systems of internal accounting and
disclosure controls, performance of the external auditors, and risk
assessment and management. The Audit Committee has the power to
conduct or authorize investigations into any matters within its
scope of responsibilities, with full access to all books, records,
facilities and personnel of the Company, its auditors and its legal
advisors. In connection with such investigations or otherwise in
the course of fulfilling its responsibilities under the Audit
Committee Charter, the Audit Committee has the authority to
independently retain special legal, accounting, or other
consultants to advise it.
Audit Committee Charter
The
text of the Audit Committee Charter is set out in Schedule A
hereto.
Composition of the Audit Committee
Our
Audit Committee is comprised of Norman Betts, Shawn Graham and
Bahadur Madhani (Chair), each of whom is considered independent and
financially literate (as such terms are defined under National
Instrument 52-110 – Audit
Committee). The members of the Audit Committee have selected
a Chair from amongst themselves, being
Mr. Madhani.
Under
the SEC rules implementing the Sarbanes-Oxley Act of 2002, Canadian
issuers filing reports in the United States must disclose whether
their audit committees have at least one “audit committee
financial expert”. Additionally, under Nasdaq Listing Rule
5605(c)(2)(A), Nasdaq requires that one member of the audit
committee be financially sophisticated, meaning that they must have
“past employment experience in finance or accounting,
requisite professional certification in accounting, or any other
comparable experience or background which results in the
individual’s financial sophistication, including being or
having been a chief executive officer, chief financial officer, or
other senior officer with financial oversight
responsibilities.” The Board has determined that Mr. Madhani
qualifies as an Audit Committee financial expert under the SEC
rules and as financially sophisticated under the applicable Nasdaq
rules.
Relevant Education and Experience
Norman
Betts is a Professor, Faculty of Business Administration,
University of New Brunswick, a Chartered Professional Accountant
Fellow (FCPA) and a member of the Institute of Corporate Directors
(ICD). Dr. Betts currently serves as a director and member of the
audit committees of Tanzanian Royalty Exploration Corporation, 49
North Resources, Biotricity Inc and Adex Mining Inc. He has
extensive public company and Crown Corporation experience including
having served on boards including Tembec Inc, New Brunswick Power
Corporation, and the Bank of Canada. He is also co-chair of the
board of trustees of the University of New Brunswick Pension Plan
for Academic Employees. Dr. Betts is a former Finance Minister and
Minister of Business New Brunswick with the Province of New
Brunswick. He was awarded a Ph.D. in Management from the School of
Business at Queens University in 1992.
74
Shawn
Graham is a member of the advisory board of the Faculty of
Business, University of New Brunswick, volunteers as a national
board member to Ducks Unlimited Canada and has been a director of
the Company since May 2018. Mr. Graham has been awarded an Honorary
Doctor of Laws degree from the University of New Brunswick. He
served as the 31st Premier of New
Brunswick when he effectively steered the province through the
worst recession in generations. He was Co-Chair of the Council of
the Federation, Co-Chair if Northeastern Governors and Eastern
Canadian Premiers, and Co-Chair of a Pan-Canadian Trade Mission to
China. He is currently President and CEO of G&R Holdings Inc.,
a company involved in the development and implementation of global
projects and business alliance strategies with a special focus on
globalising with China.
Bahadur
Madhani is a chartered accountant who has been a director of the
Company since March 31, 2006. He was a member of the advisory board
of Quebecor Ontario and former Chairman of United Way of Toronto,
former Chair of YMCA of Greater Toronto, former Chair of Nelson
Mandela Children’s Fund Canada, former Chair of YMCA Canada
and former Chair, Toronto Grants Review Team of the Ontario
Trillium Foundation. He was awarded membership in the Order of
Canada in 2001. Since 1983, Mr. Madhani’s principal
occupation has been as President and CEO of Equiprop Management
Limited, a Canadian property management company of which Mr.
Madhani is the principal shareholder.
Pre-Approval Policies and Procedures
The
Audit Committee reviewed with the independent auditor (who is
responsible for expressing an opinion on the conformity of the
Company’s audited financial statements with accounting
principles generally accepted in the United States of America)
their judgments as to the quality, not just the acceptability, of
the Company’s accounting principles and such other matters as
are required to be discussed with the Audit Committee under
Canadian and United States generally accepted auditing standards.
In addition, the Audit Committee has discussed with the independent
auditor the auditor’s independence from management and the
Company including the matters in the written disclosures provided
to the Audit Committee by the independent auditor, and considered
the compatibility of non-audit services with the auditor’s
independence.
The
Company’s independent auditor is accountable to the Board and
to the Audit Committee. The Board, through the Audit Committee, has
the ultimate responsibility to evaluate the performance of the
independent auditor, and through the shareholders, to appoint,
replace and compensate the independent auditor. Under the
Sarbanes-Oxley Act of 2002, the independent auditor of a public
company is prohibited from performing certain non-audit services.
The Audit Committee has adopted procedures and policies for the
pre-approval of non-audit services, as described in the Audit
Committee Charter. Under the terms of such policies and procedures,
the Audit Committee has adopted a list of pre-approved services,
including audit and audit-related services and tax services, and a
list of prohibited non-audit services deemed inconsistent with an
auditor’s independence.
75
The
list of pre-approved services includes:
1.
Audit
Services
●
Audits of the
Company’s consolidated financial statements; and
●
Statutory audits of
the financial statements of the Company’s
subsidiaries.
2.
Audit-Related
Services
●
Reviews of the
quarterly consolidated financial statements of the
Company;
●
Services associated
with registration statements, prospectuses, periodic reports and
other documents filed with securities regulatory bodies (such as
the SEC and the Ontario Securities Commission) or other documents
issued in connection with securities offerings (e.g., comfort
letters and consent letters) and assistance in responding to
comment letters from securities regulatory bodies;
●
Special attest
services as required by regulatory and statutory
requirements;
●
Regulatory
attestation of management reports on internal controls as required
by the regulators;
●
Consultations with
the Company’s management as to the accounting or disclosure
treatment of transactions or events and/or the actual or potential
impact of final or proposed rules, standards or interpretations by
the securities regulatory authorities, accounting standard setting
bodies (such as the Financial Accounting Standards Board or
Chartered Professional Accountants of Canada), or other regulatory
or standard setting bodies;
●
Presentations or
training on accounting or regulatory pronouncements;
and
●
Due diligence
services related to accounting and tax matters in connection with
potential acquisitions / dispositions.
76
3.
Tax
Services
a.
Compliance Services
●
Assistance with the
preparation of corporate income tax returns and related schedules
for the Company and its subsidiaries;
●
Assistance with the
preparation of Scientific Research & Experimental Development
investment tax credit claims and amended tax returns of the
Company; and
●
Assistance in
responding to Canada Revenue Agency or Internal Revenue Service on
proposed reassessments and other matters.
b.
Canadian & International Planning Services
●
Advice with respect
to cross-border/transfer pricing tax issues;
●
Advice related to
the ownership of corporate intellectual property in jurisdictions
outside of Canada;
●
Assistance in
interpreting and understanding existing and proposed domestic and
international legislation, and the administrative policies followed
by various jurisdictions in administering the law, including
assisting in applying for and requesting advance tax rulings or
technical interpretations;
●
Assistance in
interpreting and understanding the potential impact of domestic and
foreign judicial tax decisions;
●
Assistance and
advising on routine planning matters; and
●
Assistance in
advising on the implications of the routine financing of domestic
and foreign operations, including the tax implications of using
debt or equity in structuring such financing, the potential impact
of non-resident withholding tax and the taxation of the
repatriation of funds as a return of capital, a payment of a
dividend, or a payment of interest.
77
c.
Commodity Tax Services
●
Assistance
regarding Harmonized Sales Tax/Goods and Services Sales
Tax/Provincial Sales Tax/Customs/Property Tax filings and
assessments;
●
Commodity tax
advice and compliance assistance with business
reorganizations;
●
Advice and
assistance with respect to government
audits/assessments;
●
Advice with respect
to other provincial tax filings and assessments; and
●
Assistance with
interpretations or rulings.
4.
All Other
Services
●
Advice and
documentation assistance with respect to internal controls over
financial reporting and disclosure controls and procedures of the
Company.
The
list of prohibited services includes:
●
Bookkeeping or
other services related to the preparation of accounting records or
financial statements;
●
Financial
information systems design and implementation;
●
Appraisal or
valuation services for financial reporting purposes;
●
Actuarial services
for items recorded in the financial statements;
●
Internal audit
outsourcing services;
78
●
Management
functions;
●
Human
resources;
●
Certain corporate
finance and other services;
●
Legal services;
and
●
Certain expert
services unrelated to the audit.
The
Audit Committee also discusses with the Company’s independent
auditor the overall scope and plans for their audit. The Audit
Committee meets with the independent auditor, with and without
management present, to discuss the results of their examination,
their evaluations of the Company’s internal controls, and the
overall quality of the Company’s financial reporting. The
Audit Committee held 4 meetings during the period from December 1,
2020 to November 30, 2021.
In
reliance on the reviews and discussions referred to above, the
Audit Committee recommended to the Board (and the Board approved)
that the audited consolidated financial statements be included in
the Annual Report for the year ended November 30, 2021 for filing
with the Canadian provincial securities commissions and the
SEC.
The
following table summarizes the total fees paid or accrued by the
Company for audit and other services provided by MNP LLP, the
Company’s external auditor since July 27, 2016, in relation
to the fiscal year ended November 30, 2021 and 2020 in Canadian
dollars:
|
|
2021
MNP
LLP
|
2020
MNP
LLP
|
|
Audit
Fees(1)
|
$C128,400
|
$C144,450
|
|
Audit-Related
Fees(2)
|
$C101,650
|
$C108,070
|
|
Tax
Fees(3)
|
2,782
|
$C78,431
|
|
All Other
Fees(4)
|
-
|
-
|
|
Total
Fees
|
$C232,832
|
$C330,951
|
79
Notes:
(1)
Audit fees consist
of fees related to the audit of the Company’s consolidated
financial statements.
(2)
Audit-related fees
consist of consultation on accounting and disclosure matters and
reviews of quarterly interim financial statements, prospectus and
base shelf activities and Form 20-F reviews.
(3)
Tax fees consist of
fees for tax consultation, tax advice and tax compliance services
for the Company and its subsidiaries.
(4)
All other fees
related to internal control reviews.
To our
knowledge, except as set forth below, no director or officer of the
Company or any other insider of the Company, or any associate or
affiliate thereof, has or had any material interest, direct or
indirect, in any transaction within the three most recently
completed fiscal years or during the current fiscal year that has
materially affected or is reasonably expected to materially affect
the Company.
In
January 2013, the Company completed the private placement financing
of an unsecured debenture in the original principal amount of $1.5
million. The 2013 Debenture bore interest at a rate of 12% per
annum, payable monthly, was pre-payable at any time at the option
of the Company and was convertible at any time into Common Shares
at a conversion price of $30.00 per Common Share at the option of
the holder. Drs. Isa and Amina Odidi, who are directors, executive
officers and shareholders of our Company, provided us with the
original $1.5 million of the proceeds for the 2013 Debenture. In
December 2016, a principal repayment of $150,000 was made on the
2013 Debenture and the maturity date was extended until April 1,
2017. Effective March 28, 2017, the maturity date of the 2013
Debenture was extended to October 1, 2017. Effective September 28,
2017, the maturity date of the 2013 Debenture was further extended
to October 1, 2018. Effective October 1, 2018, the maturity date
for the 2013 Debenture was further extended to April 1, 2019.
Effective April 1, 2019, the maturity date for the 2013 Debenture
was further extended to May 1, 2019. In December 2018, a principal
repayment of $300,000 was made on the 2013 Debenture. On April 4,
2019, a tentative approval from TSX was received for a proposed
refinancing of the 2013 Debenture, subject to certain conditions
being met. As a result of the proposed refinancing, the principal
amount owing under the 2013 Debenture was refinanced by the 2019
Debenture. On May 1, 2019, the May 2019 Debenture was issued with a
principal amount of $1,050,000, that was originally scheduled to
mature on November 1, 2019, bear interest at a rate of 12% per
annum and be convertible into 1,779,661 common shares of the
Company at a conversion price of $0.59 per common share. Dr. Isa
Odidi and Dr. Amina Odidi, who are shareholders, directors, and
executive officers of the Company, are the holders of the 2019
Debenture. The maturity date for the May 2019 Debenture has been
extended from time to time and the maturity date for the May 2019
Debenture is now February 28, 2022. No interest was paid on the May
2019 Debenture for the ended November 30, 2021.
80
In
September 2018, the Company completed a private placement financing
of an unsecured convertible debenture in the principal amount of
$500,000. The 2018 Debenture bears interest at a rate of 10% per
annum, is payable monthly, may be prepaid at any time at our
option, and is convertible into Common Shares at any time prior to
the maturity date at a conversion price of $3.00 per Common Share
at the option of the holder. Drs. Isa and Amina Odidi, who are
directors, executive officers and shareholders of our Company,
provided us with the original $500,000 of proceeds for the 2018
Debenture. The original maturity date for the 2018 Debenture was
September 1, 2020. Effective November 30,
2020, the maturity date for the 2018 Debenture was extended to
February 28, 2022. No interest was paid on the 2018 Debenture for
the year ended November 30, 2021. The net proceeds of the 2018
Debenture were used for working capital and general corporate
purposes.
In
September 2019, the Company issued two unsecured, non-interest
bearing promissory notes, with no fixed repayment terms, in the
amounts of US$6,500 and CDN$203,886, to Dr. Isa Odidi and Dr. Amina
Odidi, our principal stockholders, directors and executive officers
of the Company. The proceeds from such notes were used for working
capital and general corporate purposes. The amounts are still
outstanding.
On
November 15, 2019, we issued to Drs. Isa and Amina Odidi, by way of
a private placement, an unsecured convertible debenture of the
Company in consideration for, and in the aggregate principal amount
of, USD$250,000. The principal amount owing under the November 2019
Debenture is convertible at any time and from time to time into
Common Shares at a conversion price equal to U.S. $0.12 per Common
Share. Up to an aggregate of 2,083,333 Common Shares may be issued
upon conversion of the principal amount owing under the November
2019 Debenture. The November 2019 Debenture bears interest at a
rate of 12% per annum (calculated monthly) and, subject to our
right to prepay the November 2019 Debenture in whole or in part at
any time without penalty, and was scheduled to mature on December
31, 2019. The maturity date for the November 2019 Debenture has
been extended from time to time and the maturity date for the
November 2019 Debenture is now February 28, 2022. No interest was
paid on the November 2019 Debenture for the year ended November 30,
2021. Dr. Isa Odidi is our Chairman, Chief Executive Officer and
Co-Chief Scientific Officer, and Dr. Amina Odidi is our President,
Chief Operating Officer and Co-Chief Scientific
Officer.
Since
the beginning of the Company’s preceding three financial
years to the date hereof, other than discussed above in this
section, there have been no transactions or proposed transactions
which are material to the Company or to any associate, holder of
10% of the Company’s outstanding shares, director or officer
or any transactions that are unusual in their nature or conditions
to which the Company or any of its subsidiaries was a
party.
The
Company’s Corporate Governance Committee, made up of
independent directors, oversees any potential transaction and
negotiation that could give rise to a related party transaction or
create a conflict of interest, and conducts an appropriate
review.
81
From time to time, we may be
exposed to claims and legal actions in the normal course of
business. As at November 30, 2021, and continuing as at February
28, 2022, we are not aware of any pending or threatened material
litigation claims against us, other than the following as described
below.
In
November 2016, we filed an NDA for our Oxycodone ER product
candidate, relying on the 505(b)(2) regulatory pathway, which
allowed us to reference data from Purdue's file for its
OxyContin® extended
release oxycodone hydrochloride. Our Oxycodone ER application was
accepted by the FDA for further review in February 2017. We
certified to the FDA that we believed that our Oxycodone ER product
candidate would not infringe any of the OxyContin® patents
listed in the Orange Book, or that such patents are invalid, and so
notified Purdue and the other owners of the subject patents listed
in the Orange Book of such certification.
On
April 7, 2017, we received notice that Purdue, Purdue
Pharmaceuticals L.P., The P.F. Laboratories, Inc., or collectively
the Purdue parties, Rhodes Technologies, and Grünenthal GmbH,
or collectively the “Purdue litigation plaintiffs”, had
commenced patent infringement proceedings, or the Purdue
litigation, against us in the U.S. District Court for the District
of Delaware (docket number 17-392) in respect of our NDA filing for
Oxycodone ER, alleging that our proposed Oxycodone ER infringes 6
out of the 16 patents associated with the branded product
OxyContin®, or the OxyContin® patents, listed in the
Orange Book. Subsequent to the above-noted filing of lawsuit, 4
further such patents were listed and published in the Orange Book.
On March 16, 2018, we received notice that the Purdue litigation
plaintiffs had commenced further such patent infringement
proceedings adding the 4 further patents. On April 15, 2020, Purdue
filed a new patent infringement suit against the Company relating
to additional Paragraph IV certifications lodged against two more
listed Purdue patents.As a result of the commencement of the first
of these legal proceedings, the FDA was stayed for 30 months from
granting final approval to our Oxycodone ER product candidate. That
time period commenced on February 24, 2017, when the Purdue
litigation plaintiffs received notice of our certification
concerning the patents, and would expire on August 24, 2019, unless
the stay is earlier terminated by a final declaration of the courts
that the patents are invalid, or are not infringed, or the matter
is otherwise settled among the parties. On April 24, 2019, an order
was issued, setting a trial date of November 12, 2019 for case
number 17-392 in the District of Delaware, and also extending the
30-month stay date for regulatory approval to March 2,
2020.
On or
about June 26, 2018, the court issued an order to sever 6
“overlapping” patents from the second Purdue case, but
ordered litigation to proceed on the 4 new (2017-issued) patents.
An answer and counterclaim was filed on July 9, 2018. On July 6,
2018, the court issued a so-called “Markman” claim
construction ruling on the first case. On July 24, 2018, the
parties to the case mutually agreed to and did have dismissed
without prejudice the infringement claims related to the
Grünenthal ‘060 patent, which is one of the six patents
included in the original litigation case.
82
On
October 4, 2018, the parties mutually agreed to postpone the
scheduled court date pending a case status conference scheduled for
December 17, 2018. At that time, further trial scheduling and other
administrative matters were postponed pending the Company’s
resubmission of the Oxycodone ER NDA to the FDA, which was made on
end of February 28, 2019. The trial in the 17-392 case was
scheduled for November 12, 2019. On January 17, 2019, the court
issued a scheduling order in which the remaining major portions are
scheduled. The trial was scheduled for June 2020,.
On
April 4, 2019, the U.S. Federal Circuit Court of Appeals affirmed
the invalidity of one Purdue OxyContin® formulation patent,
subject to further appeal to the U.S. Supreme Court.
Following
the filing of a bankruptcy stay by Purdue Pharma L.P., the
Company’s ongoing litigation case numbers 1:17-cv-00392-RGA
and 1:18-cv-00404-RGA-SRF between Purdue Pharma L.P. et al and
Intellipharmaceutics were stayed and the existing trial dates in
both cases vacated by orders issued in each case by the judge in
the District of Delaware on October 4, 2019. With the litigation
stay order, the previous 30-month stay date of March 2, 2020 was
unchanged.
On or
about July 2, 2020 the parties in the cases, numbers 17-cv-392-RGA,
18-cv-404-RGA and 20-cv-515-RGA between Purdue Pharma L.P. et al
and Intellipharmaceutics entered into a stipulated dismissal of the
Litigations. The stipulated dismissal, which was subject to
approval by the bankruptcy court presiding over Purdue
Pharma’s pending chapter 11 cases, provides for the
termination of the patent infringement proceedings. The stipulated
dismissal also provides that (i) for a thirty (30) day period
following a final approval of the Company’s Aximris
XRTM NDA
the parties will attempt to resolve any potential asserted patent
infringement claims relating to the NDA and (ii) if the parties
fail to resolve all such claims during such period Purdue Pharma
will have fifteen (15) days to pursue an infringement action
against the Company. The terms of the stipulated dismissal
agreement are confidential.
On July
28, 2020 the United States District Court for the District of
Delaware signed the stipulations of dismissal into order thereby
dismissing the claims in the three cases without prejudice. In
consideration of the confidential stipulated dismissal agreement
and for future saved litigation expenses, Purdue paid an amount to
the Company.
In July
2017, three complaints were filed in the U.S. District Court for
the Southern District of New York that were later consolidated
under the caption Shanawaz v. Intellipharmaceutics Int’l
Inc., et al., No. 1:17-cv-05761 (S.D.N.Y.). The lead
plaintiffs filed a consolidated amended complaint on January 29,
2018. In the amended complaint, the lead plaintiffs assert
claims on behalf of a putative class consisting of purchasers of
our securities between May 21, 2015 and July 26, 2017. The
amended complaint alleges that the defendants violated Sections
10(b) and 20(a) of the U.S. Securities Exchange Act of 1934, as
amended,and Rule 10b-5 promulgated thereunder by making allegedly
false and misleading statements or failing to disclose certain
information regarding our NDA for Oxycodone ER abuse-deterrent
oxycodone hydrochloride extended release tablets. The
complaint seeks, among other remedies, unspecified damages,
attorneys’ fees and other costs, equitable and/or injunctive
relief, and such other relief as the court may find just and
proper.
83
In an
order entered at the parties request on May 9, 2019, the Court
stayed proceedings in the action to permit the parties time to
conduct a mediation. As a result of subsequent extensions,
the stay was extended through October 10, 2019. The parties
participated in a mediation on August 1, 2019, during which the
parties tentatively agreed to the terms of a settlement of the
action subject to the satisfaction of certain financial conditions
by the Company.
On
November 7, 2019 the Company announced that the parties reached a
settlement that is subject to the approval of the court following
notice to class members. The stipulation of settlement provides for
a settlement payment of US$1.6 million, which Intellipharmaceutics
anticipates will be funded by available insurance coverage. As part
of the settlement, the Company also agreed to contribute to the
settlement fund specific anticipated Canadian tax refunds of up to
US$400,000 to the extent received within 18 months after the entry
of final judgment. The stipulation of settlement acknowledges that
the Company and the other defendants continue to deny that they
committed any violation of the U.S. securities laws or engaged in
any other wrongdoing and that they are entering into the settlement
at this time based on the burden, expense, and inherent uncertainty
of continuing the litigation.
On
December 7, 2020 the court approved the settlement and entered an
order and final judgement to that effect, thereby concluding the
case.
On
February 21, 2019, we and our CEO, Dr. Isa Odidi, were served with
a Statement of Claim filed in the Superior Court of Justice of
Ontario for a proposed class action under the Ontario Class
Proceedings Act. The Action was brought by Victor Romita, the
proposed representative plaintiff, on behalf of a class of Canadian
persons who traded shares of the Company during the period from
February 29, 2016 to July 26, 2017. The Statement of Claim, under
the caption Victor Romita v. Intellipharmaceutics International
Inc. and Isa Odidi, asserted that the defendants knowingly or
negligently made certain public statements during the relevant
period that contained or omitted material facts concerning
Oxycodone ER abuse-deterrent oxycodone hydrochloride extended
release tablets. The plaintiff alleges that he and the class
suffered loss and damages as a result of their trading in our
shares during the relevant period. The plaintiff seeks, among other
remedies, unspecified damages, legal fees and court and other costs
as the Court may permit. On February 26, 2019, the plaintiff
delivered a Notice of Motion seeking the required approval from the
Court, in accordance with procedure under the Ontario Securities
Act, to allow the statutory claims under the Ontario Securities Act
to proceed with respect to the claims based upon the acquisition or
disposition of our shares on the TSX during the relevant period. On
June 28, 2019, the Court endorsed a timetable for the exchange of
material leading to the hearing of the Motion scheduled for January
27-28, 2020. On October 28, 2019, plaintiff’s counsel advised
the court that the Plaintiff intended to amend his claim and could
not proceed with the Leave Motion scheduled for January 27-28,
2020. As such the Court released those dates. On January 28, 2020
the plaintiff served a Notice of Motion for leave to amend the
Statement of Claim. On April 2, 2020 the plaintiff delivered an
Amended Motion Record and Amended Notice of Motion seeking an order
for leave to issue a fresh as Amended Statement of Claim including
the addition of Christopher Pearce as a Plaintiff (“Amendment
Motion”). On May 1, 2020, the court granted the
plaintiff’s Amendment Motion. An order for leave to proceed
for settlement approval has now been scheduled for purposes was
granted on 25 June 25, 2021.A tentative settlement has been reached
in this proceeding and an order for leave to proceed for settlement
purposes was granted on 25 June 2021. At a hearing on 12 October
2021, the Court approved the settlement. The stipulation of
settlement provides for a settlement payment of CAD$266,000 by the
Company, CAD$226,000 was paid from insurance coverage while the
Company paid CAD$40,000.
84
On
October 7, 2019, a complaint was filed in the U.S. District Court
for the Southern District of New York by Alpha Capital Anstalt
(“Alpha”)
against the Company, two of its existing officers and directors and
its former Chief Financial Officer. In the complaint, Alpha alleges
that the Company and the executive officers/directors named in the
complaint violated Sections 11, 12(a)(2) and 15 of the U.S.
Securities Act of 1933, as amended, by allegedly making false and
misleading statements in the Company’s Registration Statement
on Form F-1 filed with the U.S. Securities and Exchange Commission
on September 20, 2018, as amended, by failing to disclose certain
information regarding the resignation of the Company’s then
Chief Financial Officer, which was announced several weeks after
such registration statement was declared effective. In the
complaint, Alpha seeks unspecified damages, rescission of its
purchase of the Company’s securities in the relevant
offering, attorneys’ fees and other costs and further relief
as the court may find just and proper. On December 12, 2019, the
Company and the other defendants in the action filed a motion to
dismiss for failure to state a claim. The plaintiff filed an
opposition to that motion on February 4, 2020 and a reply brief in
further support of the motion to dismiss the action was filed March
6, 2020. In addition, the Court scheduled a mandatory settlement
conference with the Magistrate Judge for April 23, 2020 which the
Company and its counsel attended. On June 18, 2020, the court
largely denied the Company’s motion to dismiss the action.
Briefing on these motions was completed on February 19, 2021.In a
court order filed July 9, 2021, the District Court issued an
opinion and order granting summary judgment in the Company’s
favor and ordered the case closed. The judgment was entered on July
12, 2021. On August 10, 2021, the Plaintiff filed a notice of
appeal. On October 1, 2021, the Plaintiff filed a notice of
voluntary dismissal of the appeal with prejudice, stipulated to by
the Company. The Court of Appeals “so ordered” the
voluntary dismissal stipulation and the appeal was dismissed. As a
result, the matter has been fully resolved in favor of the Company
and the named individual Defendants.
On or
about August 5, 2020 a former employee filed a claim against the
Company for wrongful dismissal of employment plus loss of benefits,
unpaid vacation pay, interest and costs. The parties have agreed to
settlement terms in the matter. The Company has fulfilled the terms
and has received a release and consent to dismiss. A dismissal
order is pending from the court.
TRANSFER AGENTS AND REGISTRARS
Our Canadian transfer agent and registrar is TSX Trust Company, 1
Toronto Street, Suite 1200, Toronto, Ontario, M5C 2V6. Our
United States co-transfer agent and registrar is American Stock
Transfer & Trust Co., LLC, 59 Maiden Lane, Plaza Level, New
York, NY 10038.
85
Except
for contracts entered into in the ordinary course of business and
not required to be filed under Canadian securities laws, the only
contracts which are regarded as material and which were entered
into by the Company in the period subsequent to the recently
completed fiscal year, within the most recently completed fiscal
year or before the most recently completed fiscal year, but are
still in effect, are the following:
●
On November 21,
2005, the Company entered into the Par agreement (as amended on
August 12, 2011 and September 24, 2013), pursuant to which the
Company granted Par an exclusive, royalty-free license to make and
distribute in the United States all strengths of our generic
Focalin XR®
(dexmethylphenidate hydrochloride extended-release) capsules for a
period of 10 years from the date of commercial launch (which was
November 19, 2013). Under the Par agreement, we made a filing with
the FDA for approval to market generic Focalin XR® capsules in
various strengths in the U.S. (the “Company ANDA”), and are the owner
of that Company ANDA, as approved in part by the FDA. We retain the
right to make and distribute all strengths of the generic product
outside of the U.S. Calendar quarterly profit-sharing payments for
its U.S. sales under the Company ANDA are payable by Par to us as
calculated pursuant to the Par agreement. Within the purview of the
Par agreement, Par also applied for and owns an ANDA pertaining to
all marketed strengths of generic Focalin XR® (the
“Par ANDA”) and
is now approved by the FDA, to market generic Focalin
XR®
capsules in all marketed strengths in the U.S. As with the Company
ANDA, calendar quarterly profit-sharing payments are payable by Par
to us for its U.S. sales of generic Focalin XR® under the Par
ANDA as calculated pursuant to the Par agreement. The Company is
responsible under the Par agreement for the development of the
product and most related costs which, with the applications to and
recent approvals by the FDA, the Company now considers to be
completed.
●
In January 2013,
the Company completed the private placement financing of an
unsecured debenture in the original principal amount of $1.5
million. The 2013 Debenture bore interest at a rate of 12% per
annum, payable monthly, was pre-payable at any time at the option
of the Company and was convertible at any time into Common Shares
at a conversion price of $30.00 per Common Share at the option of
the holder. Drs. Isa and Amina Odidi, who are directors, executive
officers and shareholders of our Company, provided us with the
original $1.5 million of the proceeds for the 2013 Debenture. In
December 2016, a principal repayment of $150,000 was made on the
2013 Debenture and the maturity date was extended until April 1,
2017. Effective March 28, 2017, the maturity date of the 2013
Debenture was extended to October 1, 2017. Effective September 28,
2017, the maturity date of the 2013 Debenture was further extended
to October 1, 2018. Effective October 1, 2018, the maturity date
for the 2013 Debenture was further extended to April 1, 2019.
Effective April 1, 2019, the maturity date for the 2013 Debenture
was further extended to May 1, 2019. In December 2018, a principal
repayment of $300,000 was made on the 2013 Debenture.
●
On April 4, 2019, a
tentative approval from TSX was received for a proposed refinancing
of the 2013 Debenture, subject to certain conditions being met. As
a result of the proposed refinancing, the principal amount owing
under the 2013 Debenture was refinanced by the May 2019 Debenture.
On May 1, 2019, the May 2019 Debenture was issued with a principal
amount of $1,050,000, that would mature on November 1, 2019, bear
interest at a rate of 12% per annum and be convertible into
1,779,661 common shares of the Company at a conversion price of
$0.59 per common share. Dr. Isa Odidi and Dr. Amina Odidi, who are
shareholders, directors, and executive officers of the Company, are
the holders of the May 2019 Debenture. The maturity date for the
May 2019 Debenture has been extended from time to time and the
maturity date for the May 2019 Debenture is now February 28,
2022.
86
●
In September 2018,
the Company completed a private placement financing of an unsecured
convertible debenture in the principal amount of $500,000. The 2018
Debenture bears interest at a rate of 10% per annum, is payable
monthly, may be prepaid at any time at our option, and is
convertible into Common Shares at any time prior to the maturity
date at a conversion price of $3.00 per Common Share at the option
of the holder. Drs. Isa and Amina Odidi, who are directors,
executive officers and shareholders of our Company, provided us
with the original $500,000 of proceeds for the 2018 Debenture. The
net proceeds of the 2018 Debenture were used for working capital
and general corporate purposes. The 2018 Debenture was scheduled to
mature on September 1, 2020. The maturity date for the May 2019
Debenture has been extended from time to time and the maturity date
for the September 2018 Debenture is now February 28,
2022.
●
On November 15,
2019, we issued to Drs. Isa and Amina Odidi, by way of a private
placement, an unsecured convertible debenture of the Company in
consideration for, and in the aggregate principal amount of,
USD$250,000. The principal amount owing under the November 2019
Debenture is convertible at any time and from time to time into
Common Shares at a conversion price equal to U.S. $0.12 per Common
Share. Up to an aggregate of 2,083,333 Common Shares may be issued
upon conversion of the principal amount owing under the November
2019 Debenture, representing approximately 9.43% of the issued and
outstanding Common Shares. The November 2019 Debenture bears
interest at a rate of 12% per annum (calculated monthly) and,
subject to our right to prepay the November 2019 Debenture in whole
or in part at any time without penalty, and was originally
scheduled to mature on December 31, 2019. The maturity date for the
November 2019 Debenture has been extended from time to time and the
maturity date for the November 2019 Debenture is now February 28,
2022. We used the proceeds from the November 2019 Debenture for
working capital and general corporate purposes. Dr. Isa Odidi is
our Chairman, Chief Executive Officer and Co-Chief Scientific
Officer, and Dr. Amina Odidi is our President, Chief Operating
Officer and Co-Chief Scientific Officer.
●
Pursuant to the
March 2018 Wainwright Agreements, the Company completed, in March
2018, two registered direct offerings. The first offering consisted
of 583,333 common shares at a price of $6.00 per share for gross
proceeds of approximately $3.5 million. We also issued to the
investors unregistered warrants to purchase an aggregate of 291,666
common shares at an exercise price of $6.00 per share. The warrants
became exercisable six months following the closing date and will
expire 30 months after the date they became exercisable. After
commissions and offering expenses, we received net proceeds of
approximately $3.0 million. We also issued to the placement agents
warrants to purchase 29,166 common shares at an exercise price of
$7.50 per share. In the second registered direct offering, we
issued 300,000 common shares at a price of $6.00 per share for
gross proceeds of $1.8 million. We also issued to the investors
unregistered warrants to purchase an aggregate of 150,000 common
shares at an exercise price of $6.00 per share. The warrants became
exercisable six months following the closing date and will expire
30 months after the date they became exercisable. After commissions
and offering expenses, we received net proceeds of approximately
$1.6 million. We also issued to the placement agents warrants to
purchase 15,000 common shares at an exercise price of $7.50 per
share.
87
●
The Company entered
into an engagement letter (the “August 2018 Engagement Letter”)
with Wainwright on August 15, 2018, pursuant to which Wainwright
agreed to serve as (i) exclusive placement agent or underwriter for
any offering in the United States of the securities of the Company
to take place within the following 5 months, and (ii) exclusive
agent or advisor in connection with the solicitation in respect of
the Company’s outstanding warrants. The Company agreed to pay
Wainwright a cash fee, or as to an underwritten offering an
underwriter discount, equal to a maximum of 8% of the aggregate
gross proceeds raised by the Company from the sale of securities in
each offering during the term of the engagement. The Company also
agreed to grant to Wainwright, or its designees, warrants to
purchase up to a maximum of 6% of the aggregate number of shares
sold in the offering and issued on each closing. The August 2018
Engagement Letter provides that such warrants should have
substantially the same terms as the other warrants sold in the
offering, except that their exercise price should equal 125% of the
offering price per share. The August 2018 Engagement Letter has
indemnity and other customary provisions for transactions of this
nature. The Company agreed to pay Wainwright a management fee equal
to 1% of the gross proceeds raised in the offering, a reimbursement
for non-accountable expenses of $35,000 and for up to $100,000 for
fees and expenses of legal counsel and other out-of-pocket
expenses, as well as a reimbursement for up to $10,000 for the
out-of-pocket costs of clearing agent settlement and financing. In
addition, the Company granted Wainwright, for a period of 10 months
from the closing of an offering, a right of first refusal to act as
sole book-running manager or sole placement agent for every future
public or private equity or debt offering using a manager or agent
by the Company, or any of its successors or subsidiaries. The
Company also agreed to a tail fee equal to the cash and warrant
compensation provided in connection an offering if any investor to
which Wainwright introduced the Company, or that Wainwright
contacted, with respect to an offering during the term of the
engagement provides the Company with capital in a public or private
offering, or financing or capital raising transaction during the 12
month period following termination of the Company’s
engagement of Wainwright.
●
In October 2018, we
completed an underwritten public offering in the United States,
resulting in the sale to the public of 827,970 units at $0.75 per
unit, which were comprised of one common share and one 2018 Unit
Warrant exercisable at $0.75 per share. We concurrently sold an
additional 1,947,261 common shares and 2018 Option Warrants to
purchase 2,608,695 common shares exercisable at $0.75 per share
pursuant to the over-allotment option exercised in part by the
underwriter. The price for the common shares issued in connection
with exercise of the overallotment option was $0.74 per share and
the price for the warrants issued in connection with the exercise
of the overallotment option was $0.01 per warrant, less in each
case the underwriting discount. In addition, we issued 16,563,335
2018 Pre-Funded Units, each 2018 Pre-Funded Unit consisting of one
2018 Pre-Funded Warrant to purchase one common share and one 2018
Warrant to purchase one common share. The 2018 Pre-Funded Units
were offered to the public at $0.74 each, and a 2018 Pre-Funded
Warrant is exercisable at $0.01 per share. Each 2018 Firm Warrant
is exercisable immediately and has a term of five years and each
2018 Pre-Funded Warrant is exercisable immediately and until all
2018 Pre-Funded Warrants are exercised. We also issued October 2018
Placement Agent Warrants to the placement agents to purchase
1,160,314 common shares at an exercise price of $0.9375 per share,
which were exercisable immediately upon issuance. In aggregate, the
Company issued 2,775,231 common shares, 16,563,335 2018 Pre-Funded
Warrants and 20,000,000 2018 Firm Warrants in addition to 1,160,314
October 2018 Placement Agent Warrants. During the year ended
November 30, 2018, 12,153,334 2018 Pre-Funded Warrants were
exercised for proceeds of $121,553.
88
Copies
of the above agreements have been filed on SEDAR at www.sedar.com.
Our
auditor is MNP LLP (“MNP”), Chartered Professional
Accountants, 111 Richmond Street West, Suite 300, Toronto, ON M5H
2G4. MNP is independent with respect to the Company within the
meaning of the Rules of Professional Conduct of the Chartered
Professional Accountants of Ontario.
Additional
information relating to the Company may be found under the
Company’s profile on SEDAR at www.sedar.com.
Additional information relating to directors’ and
officers’ remuneration and indebtedness, principal holders of
securities, and securities authorized for issuance under equity
compensation plans, is contained in the latest management
information circular of the Company filed on SEDAR at www.sedar.com.
Additional
financial information is provided in the consolidated financial
statements and the accompanying Management Discussion and Analysis
for our fiscal year ended November 30, 2021. Copies of such
documents are filed on SEDAR at www.sedar.com and may
be obtained upon request from our Chief Financial Officer or
President at 30 Worcester Road, Toronto, Ontario, Canada M9W
5X2.
89
SCHEDULE A
INTELLIPHARMACEUTICS INTERNATIONAL INC.
AUDIT COMMITTEE CHARTER
I.
Mandate
and Purpose of the Committee
The
Audit Committee (the “Committee”) of the board of
directors (the “Board”) of Intellipharmaceutics
International Inc. (the “Company”) is a standing committee
of the Board whose primary function is to assist the Board in
fulfilling its oversight responsibilities relating to:
(a)
the integrity of
the Company’s financial statements;
(b)
the Company’s
compliance with legal and regulatory requirements, as they relate
to the Company’s financial statements;
(c)
the qualifications,
independence and performance of the Company’s
auditor;
(d)
internal controls
and disclosure controls;
(e)
the performance of
the Company’s internal audit function; and
(f)
performing the
additional duties set out in this Charter or otherwise delegated to
the Committee by the Board.
II.
Authority
The
Committee has the authority to:
(a)
engage and
compensate independent counsel and other advisors as it determines
necessary or advisable to carry out its duties; and
(b)
communicate
directly with the Company’s auditor.
A-1
The
Committee has the authority to delegate to individual members or
subcommittees of the Committee.
III.
Composition
and Expertise
The
Committee shall be composed of a minimum of three members, each
whom is a director of the Company. Each Committee member must be
“independent” and “financially literate” as
such terms are defined in applicable securities
legislation.
Committee
members shall be appointed annually by the Board at the first
meeting of the Board following each annual meeting of shareholders.
Committee members hold office until the next annual meeting of
shareholders or until they are removed by the Board or cease to be
directors of the Company.
The
Committee shall appoint one of its members to act as Chair of the
Committee. If the Chair of the Committee is absent from any
meeting, the Committee shall select one of the other members of the
Committee to preside at that meeting.
IV.
Meetings
Any
member of the Committee or the auditor may call a meeting of the
Committee. The Committee shall meet at least four times per year
and as many additional times as the Committee deems necessary to
carry out its duties. The Chair shall develop and set the
Committee’s agenda, in consultation with other members of the
Committee, the Board and senior management.
Notice
of the time and place of every meeting shall be given in writing to
each member of the Committee, at least 72 hours (excluding
holidays) prior to the time fixed for such meeting. The
Company’s auditor shall be given notice of every meeting of
the Committee and, at the expense of the Company, shall be entitled
to attend and be heard thereat. If requested by a member of the
Committee, the Company’s auditor shall attend every meeting
of the Committee held during the term of office of the
Company’s auditor.
A
majority of the Committee shall constitute a quorum. No business
may be transacted by the Committee except at a meeting of its
members at which a quorum of the Committee is present in person or
by means of such telephonic, electronic or other communications
facility that permits all persons participating in the meeting to
communicate adequately with each other during the
meeting.
The
Committee may invite such directors, officers and employees of the
Company and advisors as it sees fit from time to time to attend
meetings of the Committee.
The
Committee shall meet without management present whenever the
Committee deems it appropriate.
The
Committee shall appoint a Secretary who need not be a director or
officer of the Company. Minutes of the meetings of the Committee
shall be recorded and maintained by the Secretary and shall be
subsequently presented to the Committee for review and
approval.
V.
Committee
and Charter Review
The
Committee shall conduct an annual review and assessment of its
performance, effectiveness and contribution, including a review of
its compliance with this Charter. The Committee shall conduct such
review and assessment in such manner as it deems appropriate and
report the results thereof to the Board.
The
Committee shall also review and assess the adequacy of this Charter
on an annual basis, taking into account all legislative and
regulatory requirements applicable to the Committee, as well as any
guidelines recommended by securities regulators, the Toronto Stock
Exchange or any other stock exchange or market on which the
Company’s shares are listed or posted for trading, and shall
recommend changes to the Board thereon.
VI.
Reporting
to the Board
The
Committee shall report to the Board in a timely manner with respect
to each of its meetings held. This report may take the form of
circulating copies of the minutes of each meeting
held.
VII.
Duties
and Responsibilities
(a)
Financial
Reporting
The
Committee is responsible for reviewing and recommending approval to
the Board of the Company’s annual and interim financial
statements, MD&A and related news releases, before they are
released.
The
Committee is also responsible for:
(i)
being satisfied
that adequate procedures are in place for the review of the
Company’s public disclosure of financial information
extracted or derived from the Company’s financial statements,
other than the public disclosure referred to in the preceding
paragraph, and for periodically assessing the adequacy of those
procedures;
(ii)
engaging the
Company’s auditor to perform a review of the interim
financial statements and receiving from the Company’s auditor
a formal report on the auditor’s review of such interim
financial statements;
(iii)
discussing with
management and the Company’s auditor the quality of generally
accepted accounting principles (“GAAP”), not just acceptability of
GAAP;
(iv)
discussing with
management any significant variances between comparative reporting
periods; and
(v)
in the course of
discussion with management and the Company’s auditor,
identifying problems or areas of concern and ensuring such matters
are satisfactorily resolved.
(b)
Auditor
The
Committee is responsible for recommending to the
Board:
(i)
the auditor to be
nominated for the purpose of preparing or issuing an
auditor’s report or performing other audit, review or attest
services for the Company; and
(ii)
the compensation of
the Company’s auditor.
The
Company’s auditor reports directly to the Committee. The
Committee is directly responsible for overseeing the work of the
Company’s auditor engaged for the purpose of preparing or
issuing an auditor’s report or performing other audit, review
or attest services for the Company, including the resolution of
disagreements between management and the Company’s auditor
regarding financial reporting.
(c)
Relationship
with the Auditor
The
Committee is responsible for reviewing the proposed audit plan and
proposed audit fees. The Committee is also responsible
for:
(i)
establishing
effective communication processes with management and the
Company’s auditor so that it can objectively monitor the
quality and effectiveness of the auditor’s relationship with
management and the Committee;
(ii)
receiving and
reviewing regular feedback from the auditor on the progress against
the approved audit plan, important findings, recommendations for
improvements and the auditor’s final report;
(iii)
reviewing, at least
annually, a report from the auditor on all relationships and
engagements for non-audit services that may be reasonably thought
to bear on the independence of the auditor; and
(iv)
meeting in camera
with the auditor whenever the Committee deems it
appropriate.
(d)
Accounting
Policies
The
Committee is responsible for:
(i)
reviewing the
Company’s accounting policy note to ensure completeness and
acceptability with GAAP as part of the approval of the financial
statements;
(ii)
discussing and
reviewing the impact of proposed changes in accounting standards or
securities policies or regulations;
(iii)
reviewing with
management and the auditor any proposed changes in major accounting
policies and key estimates and judgments that may be material to
financial reporting;
(iv)
discussing with
management and the auditor the acceptability, degree of
aggressiveness/conservatism and quality of underlying accounting
policies and key estimates and judgments; and
(v)
discussing with
management and the auditor the clarity and completeness of the
Company’s financial disclosures.
(e)
Risk
and Uncertainty
The
Committee is responsible for reviewing, as part of its approval of
the financial statements:
(i)
uncertainty notes
and disclosures; and
(ii)
MD&A
disclosures.
The
Committee, in consultation with management, will identify the
principal business risks and decide on the Company’s
“appetite” for risk. The Committee is responsible for
reviewing related risk management policies and recommending such
policies for approval by the Board. The Committee is then
responsible for communicating and assigning to the applicable Board
committee such policies for implementation and ongoing
monitoring.
The
Committee is responsible for requesting the auditor’s opinion
of management’s assessment of significant risks facing the
Company and how effectively they are managed or
controlled.
(f)
Controls
and Control Deviations
The
Committee is responsible for reviewing:
(i)
the plan and scope
of the annual audit with respect to planned reliance and testing of
controls; and
(ii)
major points
contained in the auditor’s management letter resulting from
control evaluation and testing.
The
Committee is also responsible for receiving reports from management
when significant control deviations occur.
(g)
Compliance
with Laws and Regulations
The
Committee is responsible for reviewing regular reports from
management and others (e.g. auditors) concerning the
Company’s compliance with financial related laws and
regulations, such as:
(i)
tax and financial
reporting laws and regulations;
(ii)
legal withholdings
requirements;
(iii)
environmental
protection laws; and
(iv)
other matters for
which directors face liability exposure.
VIII.
Non-Audit
Services
All
non-audit services to be provided to the Company or its subsidiary
entities by the Company’s auditor must be pre-approved by the
Committee.
IX.
Submission
Systems and Treatment of Complaints
The
Committee is responsible for establishing procedures
for:
(a)
the receipt,
retention and treatment of complaints received by the Company
regarding accounting, internal accounting controls, or auditing
matters; and
(b)
the confidential,
anonymous submission by employees of the Company of concerns
regarding questionable accounting or auditing matters.
X.
Hiring
Policies
The
Committee is responsible for reviewing and approving the
Company’s hiring policies regarding partners, employees and
former partners and employees of the present and former auditor of
the Company.