
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For
the fiscal year ended
or
For the transition period from __________________________ to __________________________
Commission
file number
(Exact name of registrant as specified in its charter)
| State or other jurisdiction | (I.R.S. Employer | |
| of incorporation or organization | Identification No.) |
(Address of Principal Executive Offices) (Zip Code)
Registrant’s
telephone number, including area code:
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| The
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Smaller
reporting company | |
| Emerging
growth company |
If an emerging growth company, indicate by checkmark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No
The
aggregate market value of the common stock held by non-affiliates of the registrant as of the last business day of the registrant’s
most recently completed second fiscal quarter (June 30, 2023) was $
The registrant had shares of common stock outstanding as of April 15, 2024.
DOCUMENTS INCORPORATED BY REFERENCE
ORGENESIS INC.
2023 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
| 2 |
SPECIAL CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
The following discussion should be read in conjunction with the financial statements and related notes contained elsewhere in this Annual Report on Form 10-K. Certain statements made in this discussion are “forward-looking statements” within the meaning of 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended. These statements are based upon beliefs of, and information currently available to, the Company’s management as well as estimates and assumptions made by the Company’s management. Readers are cautioned not to place undue reliance on these forward-looking statements, which are only predictions and speak only as of the date hereof. When used herein, the words “anticipate,” “believe,” “estimate,” “expect,” “forecast,” “future,” “intend,” “plan,” “predict,” “project,” “target,” “potential,” “will,” “would,” “could,” “should,” “continue” or the negative of these terms and similar expressions as they relate to the Company or the Company’s management identify forward-looking statements. Such statements reflect the current view of the Company with respect to future events and are subject to risks, uncertainties, assumptions, and other factors, including the risks relating to the Company’s business, industry, and the Company’s operations and results of operations. Should one or more of these risks or uncertainties materialize, or should the underlying assumptions prove incorrect, actual results may differ significantly from those anticipated, believed, estimated, expected, intended, or planned.
Although the Company believes that the expectations reflected in the forward-looking statements are reasonable, the Company cannot guarantee future results, levels of activity, performance, or achievements. Except as required by applicable law, including the securities laws of the United States, the Company does not intend to update any of the forward-looking statements to conform these statements to actual results.
Our financial statements are prepared in accordance with accounting principles generally accepted in the United States (“GAAP”). These accounting principles require us to make certain estimates, judgments and assumptions. We believe that the estimates, judgments and assumptions upon which we rely are reasonable based upon information available to us at the time that these estimates, judgments and assumptions are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities as of the date of the financial statements as well as the reported amounts of revenues and expenses during the periods presented. Our financial statements would be affected to the extent there are material differences between these estimates and actual results. The following discussion should be read in conjunction with our financial statements and notes thereto appearing elsewhere in this report.
Unless otherwise indicated or the context requires otherwise, the words “we,” “us,” “our,” the “Company,” “our Company” or “Orgenesis” refer to Orgenesis Inc., a Nevada corporation, and our majority or wholly-owned subsidiaries: Orgenesis Belgium SRL, a Belgian-based entity (the “Belgian Subsidiary”); Orgenesis Ltd., an Israeli corporation (the “Israeli Subsidiary”); Orgenesis Switzerland Sarl, (the “Swiss Subsidiary”); Koligo Therapeutics Inc., a Kentucky corporation (“Koligo”); Orgenesis CA, Inc. (the “California Subsidiary”); Mida Biotech BV (“Mida”); Orgenesis Italy SRL (the “Italian Subsidiary”), Orgenesis Austria GmbH, an Austrian corporation (“Orgenesis Austria”), Octomera LLC (formerly Morgenesis LLC, a Delaware entity which was renamed to Octomera LLC during 2023) (“Octomera”) and its wholly or majority owned subsidiaries, Orgenesis Korea Co. Ltd., a Korean based entity; Orgenesis Services SRL, a Belgian-based entity; Orgenesis Maryland LLC a Maryland entity; Orgenesis Biotech Israel Ltd. (“OBI”), an Israeli entity; Tissue Genesis International LLC (“Tissue Genesis”) a Texas limited liability company; Orgenesis Germany GmbH, a German entity; Orgs POC CA Inc, a Californian entity; Orgenesis Australia PTY LTD an Australian entity, Theracell Laboratories IKE (“Theracell Laboratories”), a Greek company, and OCTO Services LLC, a Delaware limited liability company.
Forward-looking statements made in this Annual Report on Form 10-K include statements about:
Corporate and Financial
| ● | our ability to generate revenue from the commercialization of our point-of-care cell therapy (“POCare”) to reach patients and to increase such revenues; |
| ● | our ability to achieve profitability; |
| ● | our ability to manage our research and development programs that are based on novel technologies; |
| ● | our ability to grow the size and capabilities of our organization through further collaboration and strategic alliances to expand our point-of-care cell therapy business; |
| 3 |
| ● | our ability to control key elements relating to the development and commercialization of therapeutic product candidates with third parties; |
| ● | our ability to manage potential disruptions as a result of the continued impact of the coronavirus outbreak; |
| ● | our ability to manage the growth of our company; |
| ● | our ability to attract and retain key scientific or management personnel and to expand our management team; |
| ● | the accuracy of estimates regarding expenses, future revenue, capital requirements, profitability, and needs for additional financing; and |
| ● | our belief that our therapeutic related developments have competitive advantages and can compete favorably and profitably in the cell and gene therapy industry. |
Cell & Gene Therapy Business (“CGT”)
| ● | our ability to adequately fund and scale our various collaboration, license, partnership and joint venture agreements for the development of therapeutic products and technologies; |
| ● | our ability to advance our therapeutic collaborations in terms of industrial development, clinical development, regulatory challenges, commercial partners and manufacturing availability; |
| ● | our ability to implement our POCare strategy in order to further develop and advance autologous therapies to reach patients; |
| ● | expectations regarding our ability to obtain and maintain existing intellectual property protection for our technologies and therapies; |
| ● | our ability to commercialize products in light of the intellectual property rights of others; |
| ● | our ability to obtain funding necessary to start and complete such clinical trials; |
| ● | our ability to further our CGT development projects, either directly or through our JV partner agreements, and to fulfill our obligations under such agreements; |
| ● | our belief that our systems and therapies are as at least as safe and as effective as other options; |
| ● | our relationship with Tel Hashomer Medical Research Infrastructure and Services Ltd. (“THM”) and the growing risk that THM may cancel or, at the very least continue to challenge, the License Agreement with the Israeli Subsidiary; |
| ● | the outcome of certain legal proceedings that we are or may become involved in; |
| ● | our license agreements with other institutions; |
| ● | expenditures not resulting in commercially successful products; |
| ● | our dependence on the financial results of our POCare business; |
| ● | our ability to complete development, processing and then roll out Orgenesis Mobile Processing Units and Labs (“OMPULs”) generate sufficient revenue from our POCare Services; and |
| ● | our ability to grow our POCare business and to develop additional joint venture relationships in order to produce demonstrable revenues. |
These statements are only predictions and involve known and unknown risks, uncertainties and other factors, including the risks in the section entitled “Risk Factors” set forth in this Annual Report on Form 10-K for the year ended December 31, 2023, any of which may cause our Company’s or our industry’s actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. These risks may cause the Company’s or its industry’s actual results, levels of activity or performance to be materially different from any future results, levels of activity or performance expressed or implied by these forward-looking statements.
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity or performance. Moreover, neither we nor any other person assumes responsibility for the accuracy and completeness of these forward-looking statements. The Company is under no duty to update any forward-looking statements after the date of this report to conform these statements to actual results.
| 4 |
PART I
ITEM 1. BUSINESS
(All monetary amounts are expressed in thousands of US dollars, unless stated otherwise)
Business Overview
We are a global biotech company working to unlock the potential of cell and gene therapies (“CGTs”) in an affordable and accessible format. CGTs can be centered on autologous (using the patient’s own cells) or allogenic (using master banked donor cells) and are part of a class of medicines referred to as advanced therapy medicinal products (“ATMPs”). We are mostly focused on autologous therapies that can be manufactured under processes and systems that are developed for each therapy using a closed and automated approach that is validated for compliant production near the patient for treatment of the patient at the point of care (“POCare”). This approach has the potential to overcome the limitations of traditional commercial manufacturing methods that do not translate well to commercial production of advanced therapies due to their cost prohibitive nature and complex logistics to deliver such treatments to patients (ultimately limiting the number of patients that can have access to, or can afford, these therapies).
Advanced Therapy Medicinal Products and POCare Overview
ATMP means one of any of the following medicinal products that are developed and commercialized for human use:
| ● | A somatic cell therapy medicinal product (“STMP”) that contains cells or tissues that have been manipulated to change their biological characteristics or cells or tissues not intended to be used for the same essential functions in the body; |
| ● | A tissue engineered product (“TEP”) that contains cells or tissues that have been modified so that they can be used to repair, regenerate, or replace human tissue; or |
| ● | A gene therapy medicinal product (“GTMP”) that engineers genes that lead to a therapeutic, prophylactic, or diagnostic effect and, in many cases, work by inserting “recombinant” genes into the body, usually to treat a variety of diseases, including genetic disorders, cancer, or long-term diseases. In this case, a recombinant gene is a stretch of DNA that is created in the laboratory, bringing together DNA from different sources. |
It is important to note that, although STMPs and GTMPs currently dominate the market, in order to access the market potential and trends in the future, other cell products are likely to be essential in all of these categories. We believe that autologous therapies represent a substantial segment of the ATMP market. Autologous therapies are produced from a patient’s own cells versus allogeneic therapies that are mass-cultivated from donor cells via the construction of master and working cell banks and are then produced on a large scale. Developers and manufacturers of ATMPs (both autologous and allogeneic) currently rely heavily on production using traditional centralized supply chains and manufacturing sites.
CGTs are costly and complex to produce. We also refer to CGTs as “living drugs” since they are based on maintaining the cell’s vitality. Therefore, there is no possibility to sterilize the products, since such a process involves killing any living organism. Many of these therapies require sourcing of the patient’s cells, engineering them in a sterile environment and then transplanting them back to the patient (so-called “autologous” CGT). This presents multiple logistic challenges as each patient requires their own production batch, and the current processes involve complex laboratory-based types of manipulations requiring highly trained lab technicians. We are leveraging a unique approach to therapy production using our POCare Platform to potentially overcome some of the development and supply chain challenges of affordably bringing CGT to patients.
To achieve these goals, we have developed a collaborative worldwide network of research institutes and hospitals who are engaged in the POCare model (“POCare Network”), and a pipeline of licensed POCare advanced therapies that can be processed and produced under such closed and automated processes and systems (“POCare Therapies”). We are developing our pipeline of advanced therapies and with the goal of entering into out-licensing agreements for these therapies.
We believe that, for this industry to prosper, it must be based on utilizing a standardized platform. Cellular therapies, though defined as drug products, conceptually differ from other drug modalities. The way these drug products are produced is inherently different from producing existing drugs. They are based on reprogramming of cells sourced from the patient or from a donor. They are not composed of purchased chemical components such as typical pharmaceuticals, nor are they harvested in large quantities from genetically engineered cell lines and then sterilized such as typical biotech products. These “living drug” products are, in most cases, produced per patient individually in a highly sterile and controlled environment, and their efficacy is optimized when administered a short time following production as fresh product.
| 5 |
To advance the execution of our goal of bringing such therapies to market, we have designed and built our POCare Platform - a scalable infrastructure of technology and services that ensures a central quality system, replicability and standardization of infrastructure and equipment, and centralized monitoring and data management. The platform is constructed on POCare Centers that serve as hubs that implement locally our POCare quality system, Good Manufacturing Practices (“GMP”), training procedures, quality control testing and incoming supply of materials and oversee the actual production in the Orgenesis Mobile Processing Units & Labs (“OMPULs”). The POCare Platform is operated by Octomera (see below). This platform is utilized by other parties, such as biotech companies and hospitals for the supply of their products. Octomera services include adapting the process to the platform and supplying the products (“POCare Services”). These are services for third party companies and for CGTs that are not necessarily based on our POCare Therapies.
We believe that decentralized cell processing offered through our POCare Platform could potentially democratize supply, increase production capacity, simplify logistics and shorten turnaround time. These benefits may significantly lower production costs and potentially allow us to make progress toward its vision of improved access and outcomes in healthcare.
POCare Therapies
The global CGT market is growing at a rapid pace, now with over 2,000 active clinical trials (Alliance for Regenerative Medicine (ARM) H1 2022 Report), including 200+ in Phase III and 254 new clinical trials in 2022 (ARM State of the Industry Briefing). Several biotech companies developing CGTs have been acquired by large pharma (Gilead Sciences acquired Kite Pharma, Roche acquired Spark Therapeutics, Bayer acquired AskBio) for several billion dollars before generating their first revenues. According to an article by McKinsey & Company from April 2020, CGT products account for 12 percent of the industry’s clinical and 16 percent of the preclinical pipeline.
This is a relatively new field, developing quickly in the last decade. The initial development of these therapies began at clinical research centers, based on attempts of researchers and clinicians to incorporate the scientific knowledge that accumulated from the biotechnology industry, including advancements in genetic engineering of cells, cell sourcing, tissue engineering and the medical advancements of immunology. In the early years of development, it was not even clear if such therapies would be considered a clinical treatment (such as a bone marrow transplant) or drug product such as a recombinant protean. In the last decade there has been much development in the regulatory framework required to bring such products to market, but still there is vagueness in some markets and unique regulatory pathways (such as the legal framework in the EU for hospital exemption allowing hospitals who wish to provide such therapies to their patients to take responsibility for treating patients). Though the biotech industry has embraced this new modality of drug development, they face many challenges. The pharma and biotech companies are used to centralized production and providing shelf products that can be stored and made available on demand. Their development and production teams are eager to fit these therapies into the existing well-known paradigms. This has proven to be extremely challenging, and the result has been approvals of products such as CAR-Ts for blood cancers and products for treatment of genetic diseases costing hundreds of thousands of dollars, or even over a million dollars per patient. The capacity to produce such products is limited and though they are considered a breakthrough in terms of clinical results, the high cost has been prohibitive of market acceptance.
While the biotech industry struggles to determine the best way to lower cost of goods and enable CGTs to scale, the scientific community continues to advance and push the development of such therapies to new heights. Clinicians and researchers are excited by all the new tools (new generations of industrial viruses, big data analysis for genetic and molecular data) and technologies (CRISPR, mRNA, etc.) available (often at a low cost) to perform advanced research in small labs. Most new therapies arise from academic institutes or small spinouts from such institutes. Though such research efforts may manage to progress into a clinical stage, utilizing lab based or hospital-based production solutions they lack the resources to continue the development of such drugs to market approval.
| 6 |
Historically, drug/therapeutic development has required investments of hundreds of millions of dollars to be successful. One significant cause for the high cost is that each therapy often requires unique production facilities and technologies that must be subcontracted or built. Further the cost of production during the clinical stage is extremely expensive, and the cost of the clinical trial itself is very high. Given these financial restraints, researchers and institutes hope to out- license their therapeutic products to large biotech companies or spin-out new companies and raise large fundraising rounds. However, in many cases they lack the resources and the capability to de-risk their therapeutic candidates enough to be attractive for such fundings or partnership.
Our POCare Network is an alternative to the traditional pathway of drug development. Orgenesis works closely with many such institutes and is in close contact with researchers in the field. The partnerships with leading hospitals and research institutes gives us a deep insight as to the developments in the field, as well as the market potential, the regulatory landscape and optimal clinical pathway to potentially bring these products to market.
The ability to produce these products at low cost, allows for an expedited development process and the partnership with hospitals around the globe enables joint grants and lower cost of clinical development. The POCare Therapies division reviews many therapies available for out licensing and select the ones which they believe have the highest market potential, can benefit the most from a point of care approach and have the highest chance of clinical success. It assesses such issues by utilizing its global POCare Network and its internal knowhow accumulated over a decade of involvement in the field.
The goal of this in-licensing is to quickly adapt such therapies to a point-of- care approach through regional partnerships, and to out-license the products for market approval in preferred geographical regions. This approach lowers overall development cost, through minimizing pre-clinical development costs incurred by us, and through receiving of the additional funding from grants and/or payments by regional partners.
Our Therapies development subsidiaries are:
| ● | Koligo Therapeutics, Inc., a Kentucky corporation, which is a regenerative medicine company, specializing in developing personalized cell therapies. It is currently focused on commercializing its metabolic pipeline via the POCare Network throughout the United States and in international markets. |
| ● | Orgenesis CA, Inc. a Delaware corporation, which is currently focused on development of our technologies and therapies in California. |
| ● | Orgenesis Belgium SRL which is currently focused on product development. Since its incorporation the subsidiary has received grant awards of over Euro 19 million from the Walloon region for several projects (DGO6 grants). We intend to continue applying for the Walloon Region support of our future pre-clinical and clinical development plans. |
| ● | Orgenesis Switzerland Sarl, which is currently focused on providing group management services. |
| ● | MIDA Biotech BV, which is currently focused on research and development activities, was granted a 4 million Euro grant under the European Innovation Council Pathfinder Challenge Program which supports cutting-edge science and technology. The grant is for technologies enabling the production of autologous induced pluripotent stem cells (iPSCs) using microfluidic technologies and artificial intelligence (AI). |
| ● | Orgenesis Italy SRL which is currently focused on R&D activities. Orgenesis has joined an Italian consortium dedicated to the implementation of a research program in the field of gene therapy and drug development with RNA technology. The program is sponsored by the Italian national recovery and resilience plan “strengthening of research structures and creation of national R&D champions on key enabling technologies. |
| ● | Orgenesis Ltd., an Israeli subsidiary which is focused on R&D and a provider of R&D management services for out licenced products. Israel as a hub for biotech research and pioneers in this field |
| ● | Orgenesis Austria GmbH, which is currently focused on the development of the Company’s technologies and therapies. |
| 7 |
Therapies in Development
Our cell and gene therapies pipeline includes investigational therapies and next-generation technologies that have the power to transform the way cancer and other unmet clinical needs are treated. Our pipeline is predominantly comprised of personalised autologous cell therapies, meaning that patients receive cells that originate from their own body, virtually eliminating the risk of an immune response and rejection.
Our promising pipeline focuses on Advanced Therapy Medicinal Products originating from proprietary internal, joint ventures and in-licensing agreements with both biotech companies and leading research institutions. Our main therapeutic fields encompass cell-based immuno-oncology, cell-based drug delivery platforms, regenerative medicine, anti-viral and autoimmune disease.
The following table summarizes our therapies in development, which are discussed in detail below:
| Therapy | Development Stage | Indication | ||
| Immuno-Oncology | ||||
| HiCAR-T | Hospital exemption/ IND enabling studies |
B-ALL, B-cell Lymphoma | ||
| T-LOOP | IND enabling studies | Solid Tumors | ||
| MDVAC | IND enabling studies | Solid Tumors | ||
| CeCART | Pre-clinical | Solid Tumors | ||
| Intra Nasal Delivery of Cell based Immunotherapy | Pre-clinical | Drug delivery technology, Glioblastoma | ||
| Intra Nasal Delivery of Cell based Immunotherapy | Pre-clinical | Drug delivery technology, Glioblastoma | ||
| Metabolic Diseases | ||||
| KYSLECEL | Market approval in the US | TP-IAT | ||
| CellFix | Clinical use | Cartilage Defects | ||
AutoSVF |
Clinical development | Systemic ARDS, vascular disorders | ||
| MSCP | Pre-clinical | Wound healing | ||
| EVRD | Pre-clinical | CKD | ||
KT-DM-103 and KT-CP-203 (3D-Printed Pancreatic Islets) |
Pre-clinical |
Type 1 diabetes and chronic pancreatitis | ||
Bioxomes |
Pre-clinical | Drug Delivery Technology | ||
| MSPP | Pre-clinical | Urinary Incontinence | ||
| Anti-Viral | ||||
RanTop, Ranpirnase Topical Formulation |
Clinical development | Anti-viral/ Immune oncology | ||
Autovac |
Pre-clinical | Autologous viral vaccine | ||
Immuno-Oncology
HiCAR-T (CD 19)
Chimeric antigen receptor T cells (also known as CAR-T cells) are T cells that have been genetically engineered to produce an artificial T-cell receptor for use in immunotherapy. CAR-T cell therapy uses T cells engineered with CARs for cancer therapy. The premise of CAR-T immunotherapy is to modify T cells to recognize cancer cells in order to more effectively target and destroy them. Physicians harvest T cells from patients, genetically alter them, then infuse the resulting CAR-T cells into patients to attack their tumors. CAR-T cells can be either derived from T cells in a patient’s own blood (autologous) or derived from the T cells of another healthy donor (allogeneic). Once isolated from a person, these T cells are genetically engineered to express a specific CAR, which programs them to target an antigen that is present on the surface of tumors. After CAR-T cells are infused into a patient, they act as a “living drug” against cancer cells. When they come in contact with their targeted antigen on a cell, CAR-T cells bind to it and become activated, then proceed to proliferate and become cytotoxic.
We are developing a new and advanced anti-CD19 CAR-T therapy for treating B-cell Acute lymphoblastic leukemia (ALL) and other B-cell lymphoma patients. This platform is utilizing a first-in-class processing technology that enables fast delivery of this product at low cost. This CAR-T platform technology can potentially be utilized for multi-indications beyond blood cancer including for autoimmune indications. Based on what management believes to be encouraging real world clinical data generated in an investigator initiated trial, we are prioritizing cGMP production of our proprietary viral vector in order to generate clinical data to support regulatory filings in Europe and the US.
During 2023, the OMPUL production site in Israel was qualified to produce clinical batches for the CAR-T (CD 19). Agreement on conditions for initiation of clinical study, which would be under a US IND, was reached with the Israeli ministry of health. In addition, Orgenesis engaged the Paul Ehrlich Institute (PEI), which has provided scientific advice needed for initiation of trials in Belgium and Greece for potential EU approval.
CeCART
Following the success CAR-T therapy demonstrated in hematological malignancies, the therapeutic potential of CAR-T is employed for solid tumors as well.
We are developing a CAR-T therapy for the treatment of solid tumors including pancreatic and colorectal cancers. The CAR is directed against two members of the carcinoembryonic antigen-related cell adhesion molecule (CEACAM) family. These adhesion proteins are involved in tumor growth, invasion, angiogenesis and immune evasion and their expression is correlated with poor prognosis. In pancreatic cancer, these adhesion molecules are overexpressed on tumor cells while expression on healthy tissues is limited making them a promising therapeutic target.
| 8 |
The CAR binding domain is based on a humanized monoclonal antibody, that specifically binds specific CEACAM molecules. We have an exclusive license to use this proprietary antibody in CAR-T therapy. Using the humanized antibody binding domain, we have successfully completed the CAR construct optimization, engineered CAR-T cells using our platform process and demonstrated in vitro efficacy and specificity.
T-LOOP (Tumor Infiltrating Lymphocytes (TIL))
TIL therapy is a clinically validated personalized cancer treatment based on infusion of autologous TILs expanded ex vivo from tumors. Once expanded, the TILs are infused back into the patient where they attack the cancer cells with a high degree of specificity. We have developed a GMP-compliant, reproducible and efficient production approach that is performed in a fully closed system enabling the generation of functional TILs from various solid tumor biopsies. The expanded TILs lead to a more robust therapeutic response especially for solid tumors such as lung cancer.
During 2023, we have completed methods validation and qualification required for clinical batch production. Moreover, the OMPUL production site in Israel was qualified to produce clinical batches. Agreement on conditions for initiation of clinical study was reached with the Israeli Ministry of Health.
MDVAC
Dual vaccine cell-based cancer immunotherapy (MDVAC) is composed of two pre-activated APCs (DCs and Macrophages) loaded with allogenic whole cancer cell lines, which maximize repertoire of cancer antigen presentation. MDVAC harnesses the immune system’s natural ability to recognize and react to cancer neo-antigens to boost cancer immunotherapy. Parallel cancer antigen presentation promotes improved immune education and tumor recognition in the patient, leading to tumor growth arrest and metastasis decrease. This cell-based immunotherapy, licensed from Columbia University, can be a developed for a wide range of solid tumors. The GMP production process was optimized, specificity and activity tests were successfully developed. We plan to initiate interaction with regulatory authorities towards finalization of our clinical strategy.
Metabolic Diseases
KYSLECEL (Autologous Pancreatic Islets)
The
patient’s own pancreatic islets, comprised of the cells that secrete insulin to regulate blood sugar, form KYSLECEL, a minimally
manipulated autologous cell-based product produced according to current good tissue practices (cGTP). The therapy has been allowed by
the U.S. Food and Drug Administration (“FDA”) and is available in the US. The target population of KYSLECEL, as an islet
autologous transplant after total pancreatectomy (TP-IAT), is chronic or acute recurrent pancreatitis patients who are in need of insulin
secretory capacity preservation.
KT-DM-103 and KT-CP-203 (3D-Printed Pancreatic Islets)
Through the acquisition of Koligo, we have exclusively licensed patents and technology from the University of Louisville Research Foundation, related to the revascularization and 3D printing of cells and tissues intended for transplantation (“3D-V” technology platform). Utilizing this technology, potential autologous and allogeneic pancreatic islet transplants may be implemented to treat type 1 diabetes (KT-DM-103), and chronic pancreatitis (KT-CP-203). In addition to pancreatic islet transplantation, the 3D-V technology platform may also support improved transplantation of other cell and tissue types.
MSCP
We are developing a personalized cell-based therapy product for wound healing. The product is based on allogeneic Adipose-Derived Stem Cells (ADSCs). Following expansion, the ADSCs are used for the extraction of BioxomeTM. We have established a process for encapsulation of Topiramate, a well-known substrate used in other indications, during the Bioxome manufacture. The Bioxome-encapsulated Topiramate (Biox-Top) will be further formulated in commercially available hyaluronic acid (HA), a well-known dermal filler, for topical application. Pre-clinical development is ongoing following demonstration of anti-inflammatory efficacy in human skins explants.
Bioxomes as a cell-based delivery product
Exosomes are small, membrane-enclosed extracellular vesicles involved in cell-to-cell interactions. They may serve as a valuable therapeutic modality given their ability to transfer a wide variety of therapeutic payloads to cells affecting the cells in multiple ways. The exosomes may be designed to reach specific cell types.
Bioxomes are liposomes that are biocompatible and serve as cGMP/GLP-compliant exosome-like membrane nanostructures that can be produced from various cell types. To this end, we have developed a proprietary large-scale cGMP-compatible manufacturing process for preparation of Bioxomes from the following: human adipose cells, fibroblasts, blood cells, and plant cells.
| 9 |
Additionally, preliminary biodistribution studies demonstrated specific organ tropism, as well as enhanced skin penetration, when applied topically. Further biodistribution and bioavailability studies with Bioxomes, encapsulated with selected therapeutic cargos are on-going to confirm efficacy and safety. Bioxomes may be utilized as the next generation biological delivery platform for Immuno-Oncology indications. Currently, the regulatory strategy is being finalized according to US FDA requirements.
Anti viral
RanTop, Ranpirnase Topical Formulation
We are developing a novel topical gel formulation of an active RNA-degrading enzyme, called ranpirnase. Ranpirnase combats viral infections by targeting double-stranded RNA including miRNA precursors, via RNA degradation catalysis. Topical ranpirnase demonstrated good tolerability and preliminary clinical efficacy in the treatment of HPV-associated external anogenital warts (EGW) in a Phase 2a clinical study conducted in Bolivia.
Following FDA positive pre-IND feedback, preclinical development program was initiated to support human clinical studies in the US. A dermal toxicology feasibility study was conducted, showing that RanTop was well-tolerated in repeated daily topical administration. Systemic exposure following topical administration need to be assessed during preclinical and clinical studies. For this purpose, a sensitive ranpirnase blood concentration bioanalytical method was established.
In laboratory experiments, we have demonstrated the feasibility of ranpirnase encapsulation in Orgenesis Bioxome delivery platform. Bioxome encapsulation, enhanced ranpirnase anti-viral activity in an in vitro test.
Ranpirnase was originally isolated from frog oocytes. We have focused on developing of a recombinant renpirnase, aiming at avoiding use of animal and enabling a scalable cost-effective industrial process that meets regulatory requirements for biological drugs. We have successfully demonstrated feasibility of producing active recombinant ranpirnase using genetically engineered bacterial fermentation. We plan to use the recombinant ranpirnase in future development.
Orgenesis licensing partner, Okogen, Inc., has announced in October 2023 the initiation of a Phase IIb clinical trial in India evaluating OKG-0303 for acute infectious conjunctivitis (“Pink Eye”). OKG-0303 is a combination product containing ranpirnase (OKG-301) as an antiviral active component.
Autovac
AutoVac is an autologous, pan-antigenic vaccine platform for viral infections. The vaccine is based on the use of a specific target for ex vivo induction of autologous cell-based vaccine that enables rapid response in times of a viral outbreak. As initial proof of concept, we are validating this novel cell-based vaccine platform against Coronavirus disease 2019 (COVID-19). Preliminary in vitro results demonstrated successful immune cell activation, correlated with antigen expression. We have confirmed vaccine platform specificity and robustness by testing additional viral pathogens.
We are planning to complete pre-clinical immunogenicity studies and finalize product development toward clinical submissions.
Strategic CGT Therapeutics Collaborations
Collaborations, partnerships, joint ventures and license agreements are key components of our POCare strategy.
Our POCare technology collaborators and partners include Ori Biotech, Accellix, Columbia University in the City of New York, Caerus Therapeutics Corporation, UC Davis, The Johns Hopkins University, The Weizman Institute of Science and others.
| 10 |
In addition, we have collaborations and joint ventures for developing POCare Therapies in jurisdictions throughout the world, including various countries in North America, Europe, Latin America, Asia, and Australia. Such partnerships include in-licensing and out-licensing of therapies, service contracts from the partners under co-development agreements, and development and manufacturing agreements for POCare products supplied regionally. For more information, see note 12, “Collaboration and Licensing Agreements” of the “Notes to the Financial Statements” included in Item 8 of this Annual Report on Form 10-K.
Current POCare Therapies Development Facilities
Koligo
Koligo maintains commercial production facilities for KYSLECEL at an FDA-registered establishment in Indiana. Koligo is also developing new technologies such as bio-degradable 3D structure to deliver islets and other cell/tissues. Koligo also maintains development labs at its Indiana location to support continued development.
The Belgian Subsidiary
The Belgian Subsidiary specializes in developing and validating proprietary and advanced cell and gene therapies. The Belgian Subsidiary benefits both from its central position in Europe and its being in the leading Walloon biotech cluster. Located near Namur, at Novalis Science Park, the Belgian Subsidiary collaborates with leading medical and academic facilities which enables it to cover the drug product life cycle from research to clinical stage through pre-clinical and quality control.
Mida
Mida specializes in developing and validating proprietary and licensed advanced cell and gene therapies such IPS based therapies and AI in its development labs in the Netherlands.
The Israel Subsidiary
The Israel Subsidiary occupies 400 square meters of labs and offices in Nes Ziona, Israel.
POCare Services
The POCare Services that we and our affiliated entities perform include:
| ● | Process development of therapies, process adaptation, and optimization inside the OMPULs, or “OMPULization”; |
| ● | Adaptation of automation and closed systems to serviced therapies; |
| ● | Incorporation of the serviced therapies compliant with GMP in the OMPULs that we designed and built; |
| ● | Tech transfers and training of local teams for the serviced therapies at the POCare Centers; |
| ● | Processing and supply of the therapies and required supplies under GMP conditions within our POCare Network, including required quality control testing; and |
| ● | Contract Research Organization (“CRO”) services for clinical trials. |
The POCare Services are performed in decentralized hubs that provide harmonized and standardized services to customers (“POCare Centers”). We are working to expand the number and scope of our POCare Centers. We believe that this provides an efficient and scalable pathway for CGT therapies to reach patients rapidly at lowered costs. Our POCare Services are designed to allow rapid capacity expansion while integrating new technologies to bring together patients, doctors and industry partners with a goal of achieving standardized, regulated clinical development and production of therapies.
| 11 |
POCare Services Operations via Octomera
We currently conduct our core business operations ourselves and through Octomera and its subsidiaries which are all wholly owned except as otherwise stated below (collectively, the “Subsidiaries”). The following is a description of Octomera and its subsidiaries:
Octomera LLC
In connection with the investment by an affiliate of Metalmark Capital Partners (“Metalmark” or “MM”) in the Company’s subsidiary Octomera LLC (formerly Morgenesis LLC) (“Octomera” or “Morgenesis”) in November 2022 (“the Metalmark Investment”), the Company streamlined its Services related business into Octomera.
On June 30, 2023, in connection with an additional $1,000 investment in Octomera, the Company and MM entered into Amendment No. 1 to the Second Amended and Restated Limited Liability Company Agreement (the “LLC Agreement Amendment”) to change the name of Morgenesis to “Octomera LLC” and to amend Morgenesis’ board composition. Pursuant to the LLC Agreement Amendment, the board of managers of Octomera (the “Octomera Board”) became comprised of five managers, two of which were appointed by the Company, one of which was an industry expert appointed by MM, and two of which were appointed by MM. The change was effective immediately. As a result of the amendment to the composition of the Octomera Board pursuant to the LLC Agreement Amendment described above, the Company deconsolidated Octomera from its consolidated financial statements as of June 30, 2023 (“date of deconsolidation”) and recorded its equity interest in Octomera as an equity method investment.
On January 29, 2024, the Company and MM entered into a Unit Purchase Agreement (the “UPA”), pursuant to which the Company acquired all of the interests of Octomera that were owned by MM (the “Acquisition”). In consideration for such Acquisition, the Company and MM agreed to the following consideration:
Royalty Payments: If Octomera and its subsidiaries generate Net Revenue during the calendar years of 2025, 2026 and 2027, then the Company will pay 5% of Net Revenues to Seller pursuant to the UPA up to $40 million.
Milestone Payments: If the Company sells Octomera within ten years from the date of the Closing at a price that is more than $40 million excluding consideration for certain Excluded Assets as per the UPA, the Company shall pay Seller 5% of the net proceeds.
Pursuant to the acquisition, MM’s designated members of the Board of Managers of Octomera resigned and the Company amended the Second Amended and Restated Limited Liability Company Agreement of Octomera to be a single member agreement to reflect the transactions contemplated by the UPA so that MM shall no longer (i) be a party to such agreement, (ii) have a right to appoint members of the board of managers of Octomera or (iii) be a member of Octomera.
The Company currently owns 100% of Octomera.
The Octomera subsidiaries which are all wholly owned except as otherwise stated below (collectively, the “Subsidiaries”) include:
| ● | Orgenesis Maryland LLC, which is the center of POCare Services activity in North America and is currently focused on setting up and providing POCare Services and cell-processing services to the POCare Network. |
| ● | Tissue Genesis International LLC, a Texas limited liability company currently focused on development of our technologies and therapies. |
| ● | Orgenesis Services SRL, which is currently focused on expanding our POCare Network in Belgium. |
| ● | Orgenesis Germany GmbH, which is currently focused on providing CRO services to the POCare Network. |
| ● | Orgenesis Korea Co. Ltd., which is a provider of cell-processing and pre-clinical services in Korea. Octomera owns 94.12% of the Korean Subsidiary. |
| ● | Orgenesis Biotech Israel Ltd., which is a provider of process development and cell-processing services in Israel. |
| ● | Orgenesis Australia PTY LTD, which was transferred to Octomera in January 2023 and is currently focused on the development of our POC Network in Australia. |
| ● | Theracell Laboratories IKE (“Theracell Labs”), a Greek company currently focused on expanding our POCare Network. |
| ● | ORGS POC CA Inc, incorporated in 2023, which is currently focussed on expanding our POCare Network in California. |
| ● | Octo Services LLC, a Delaware entity incorporated in 2023. |
| 12 |
Integration of Custom Fit Solutions within the POCare Center
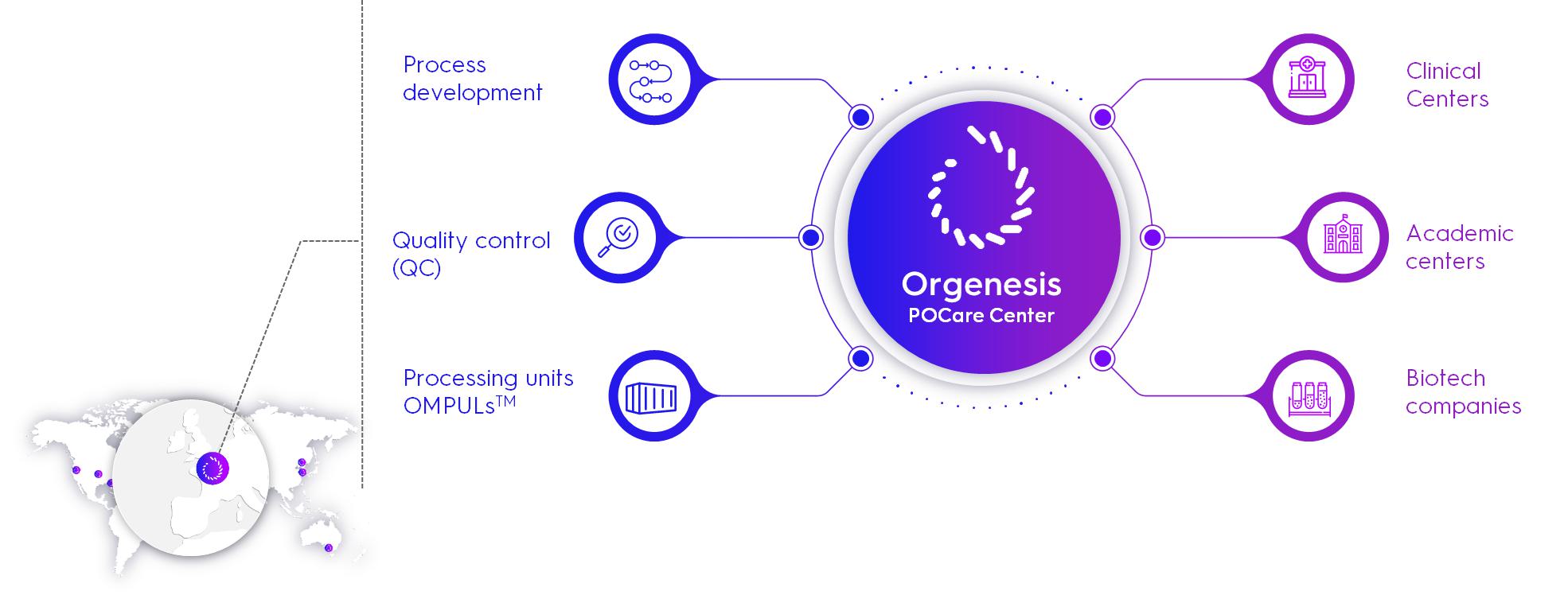
Our aim is to provide a pathway to bring ATMPs in the cell and gene therapy industry from research to patients worldwide through our POCare Platform. We define point of care as a process of collecting, processing, and administering cells as close as possible to the clinical setting. We believe that this approach is an attractive proposition for CGT during the clinical development stage and even more so upon market approval therapies. This will potentially help to minimize or eliminate the need for cell transportation, which is a high-risk and costly aspect of the supply chain, further allowing flexible production and patient treatment and reduce the cost and lengthy timelines associated with building additional clean rooms and complex tech transfers between production sites.
We believe that the existing industry paradigm in which each therapy developer invests in setting up unique infrastructure such as specialized clean rooms and production facilities is inefficient. The cost of construction, regulatory authorization and maintenance of these facilities is not only prohibitive but extremely difficult and lengthy to replicate, allowing no economies of scale. We have based the design of our POCare Platform on the concept of standardizing infrastructure by providing flexible building blocks through the POCare Centers and OMPULs, which allows for quick expansion at multiple locations.
| ● | Local Decentralization: POCare Centers are set up in preferred regions, based on nearby hospitals’ capacity needs, and support the POCare Services model by providing POCare Services. |
| ● | Global Harmonization: The POCare Platform overcomes conventional processing challenges by enabling high quality standards and sterile, scalable onsite processing of CGTs orchestrated by the POCare Centers to service local hospitals. Processing infrastructure is harmonized and reproducible using the OMPUL. The use of an OMPUL can shorten implementation time from approximately 18-24 months to approximately 3-9 months, offers a more cost-effective environment and enables local scalability by connecting additional OMPULs. The network structure is supported and connected by the centralization of the harmonized best industry practices and standards to meet the highest quality standards (“QMS”, Quality Management System). Further global harmonization is implemented through standardization of the training programs, centralized data management and a unified supply chain. |
| ● | OMPULization of Therapies: Strong process development capabilities are critical for any CGT to scale. All therapeutic candidates must undergo some level of process development to move from the discovery phase to the clinical phase, if only to establish the same protocols under GMP. The POCare Platform takes process development to the next level, implementing a process we call OMPULization. OMPULization includes unitizing the process to the exact specifications of the OMPUL so it can be rapidly implemented in OMPULs around the world. In addition, OMPULization incorporates the latest technology solutions to close and automate the process whenever possible. |
Integrated closed and automated processing systems require fewer full-time employees (“FTEs”) to produce GMP batches, resulting in lower cost of goods and a process that has the ability to scale in sync with market demand. Full automation may not be necessary for all clinical phases, but it is important to plan for future incorporation. To this end, we have invested time and capital into evaluating relevant technology for CGT processing and have developed proprietary equipment that did not exist in the marketplace.
We aim to build value in various aspects of our company ranging from supply related processes including development and distribution systems, clinical and regulatory services, engineering and devices such as OMPULs discussed below and delivery systems. Therapies serviced include immuno-oncology, anti-aging, metabolic, dermatology, orthopedic, as well as regenerative technologies.
The POCare Platform is a unique globally harmonized and decentralized CGT-processing infrastructure that offers cost-effective processing capacities with ease for scalability and reproducibility. By producing personalized cell and gene therapies (CGTs) utilizing the POCare Platform, we are able to add new capacity within months instead of years. Over time, we have worked to develop and validate POCare Technologies that can be combined within mobile production units for advanced therapies.
We have made significant investments in the implementation of several therapy types in OMPULs and have made significant progress in the validation, risk analysis, regulatory and other related tasks relating to the OMPULs. We are setting up the OMPULs through our POCare Centers. OMPULs are designed for the purpose of validation, development, performance of clinical trials, manufacturing and/or processing of potential or approved cell and gene therapy products in a safe, reliable, and cost-effective manner at the point of care, as well as the manufacturing of such CGTs in a consistent and standardized manner in all locations. The design delivers a potential industrial solution for us to deliver CGTs to most clinical institutions at the point of care.

| 13 |


Above are diagrams of an OMPUL and partial interior for illustrative purposes only.
We have finalized or are in the process of finalizing the development of several POCare Centers and adapting to the local requirements of each POCare Center with the target of achieving a capacity to process and supply CGTs per production contracts. As we expand operations, we expect that the OMPUL setup costs will decline over time. Most of our POCare revenue to date is in support of the implementation of technologies and therapies in the OMPULs and production at the POCare Sites.
We have established POCare Centers in several locations globally, in which we perform process development and manufacturing activities for several types of CGT products. For example, in Israel, our POCare Center includes process development and QC labs, as well as OMPULs located at a hospital site in the center of Israel and an additional OMPUL in preparation for an additional hospital. In these OMPULs, we currently manufacture TILs and CAR-T therapies. In Greece, our POCare Center includes three OMPULs installed in place and a process development lab, currently servicing two customers. Our POCare Center in Maryland, USA, includes an operating process development lab. We are also establishing cleanroom-based facility funded by a government grant. In Spain we have an OMPUL producing a clinical grade product.
POCare Services Development Facilities
OBI
OBI is our specialized process and technology development wholly-owned subsidiary focused on custom-made process development, upscaling design from lab to industry innovation and automation procedures, which are extremely essential in the cell therapy industry. OBI is located in Bar-Lev Industrial Park utilizing the exclusive Israeli innovative ecosystem and highly experienced and talented associates including Ph.D. holders and biotechnology engineers. The center provides end to end solutions to cell therapy industrialization, process development capabilities and proficiency, custom-made engineering and a unique platform for creative design and process optimization. OBI occupies 1,300 square meters of labs and offices resulting in an efficient and unique environment for cell therapy development. In connection with the sales of our Masthercell Global subsidiary (“Masthercell Sale”) completed in 2020, for a period of three years in the European Union and five years in the United States and the rest of the world from the closing date of the Masthercell Sale, we agreed that OBI will not manufacture products on a contract basis for third-party customers in any jurisdiction other than the State of Israel, but it may conduct such CDMO business in the State of Israel, solely for customers located within the State of Israel or with respect to therapies intended for distribution solely within the State of Israel. The Masthercell sale agreement stipulated that OBI may also conduct, worldwide, (i) point-of-care system, point-of-care products, point-of-care systems, point-of-care processing, and point-of-care development services for the development, manufacturing or processing of therapeutics, processes, systems and technologies to treat patients in a point-of-care clinical, hospital or institutional setting, any future point-of-care services substantially related to the foregoing, and advanced therapy medicinal products either proprietary to us or our affiliates or proprietary to a third-party partner (including a joint venture partner) or collaborator, which includes research, development, systems, manufacturing and processing of therapeutic technology products, systems, and processes, methods or services and (ii) research, manufacturing, development and other activities related to the research, development, manufacturing, discovery and commercialization of therapeutic products or technologies, and processes, systems, methods or services thereof for its own account or in order to make such products or services available for the account of their third-party partners (including joint venture partners) or collaborators (including such therapeutic products, processes or technologies in which we or one of our affiliates has an economic interest or any relationship with any third-party or that are created, developed, manufactured or sold by a joint venture, partnership or collaboration between us or any of our affiliates and a third-party (individually and collectively, “Permitted Business”).
| 14 |
On February 14, 2024, following a claim for payment of past salaries due, by employees of Orgenesis Biotech Israel Limited (“OBI”), the district court in Haifa appointed a trustee to run the affairs of OBI with the intention of rehabilitating OBI to be able to operate and pay OBI’s creditors under an arrangement with them.
The Korean Subsidiary
The Korean Subsidiary has a particular focus on developing innovative cell therapies for our customers. In connection with the Masthercell Sale completed in 2020, for a period of three years in the European Union and five years in the United States and the rest of the world from the closing date of the Masthercell Sale, we agreed that the Korean Subsidiary will not manufacture cell and gene products on a contract basis for third-party customers in any jurisdiction other than South Korea, but it may conduct CDMO business in South Korea, solely for customers located within South Korea and with respect to therapies intended for distribution solely within South Korea, provided that the Korean Subsidiary may conduct Permitted Business.
Tissue Genesis International
The Tissue Genesis Icellator™ is used to isolate stromal and vascular fraction cells (“SVF”) from a patient’s own (autologous) adipose tissue (fat). The Tissue Genesis Icellators, associated disposable kits, and our proprietary enzyme Adipase™, are made by contract manufacturers and warehoused at our ISO 13485-certified and FDA-registered facility in Texas. From this facility we fill orders for our customers all around the world and maintain research and development labs to support continued product development.
Tissue Genesis International (“TGI”) has expanded its development pipeline from the Icellator to additional systems for automation of Cell and Gene Therapy and incorporation of these various platforms into the OMPULs.
On the Icellator front, in 2022 TGI continued to service our existing customers both domestically and abroad, added new customers, increased revenue from sales, extended shelf-life of existing Icellator inventory, continued Adipase development, and engaged in production of a new lot of disposables.
TGI includes the integration of our development projects, foremost among them the Control Tower for automation of cGMP cell and gene therapy inside the OMPULs. In 2022 TGI brought this project into the ISO quality system and engaged with contract engineering firms with the requisite experience and that meet our stringent quality assurance standards.
Orgenesis Services SRL
Orgenesis Services SRL specializes on developing innovative cell therapies for our customers. The subsidiary benefits both from its central position in Europe and its being in the leading Walloon biotech cluster. It occupies innovative facilities for the development and quality control of therapies in R&D and GMP grades.
| 15 |
Theracell Laboratories
Theracell Laboratories, located in Greece, specializes on developing and processing innovative cell therapies for our customers. It was designated as a “Priority Investment of Strategic National Importance” by Enterprise Greece, the official Greek national investment and trade promotion agency, which is responsible for the allocation of Greek government funding. As a result of this designation, Theracell will be inducted into Greece’s fast-track licensing and approval process. This is expected to help advance development and clinical use of our CGT at POCare, subject to regulatory requirements.
Notable 2023 POCare Services Activities
In 2023, we continued to focus on setting up our regional POCare activities. This included the setup of POCare Centers that oversee regional development and GMP services, local OMPUL deployment and supply of products to the local clinical centers. We are in the process of expanding the capacity of our POCare Centers in Maryland, Boston, California, Belgium, Greece, Slovenia, Israel, Italy, Spain and Korea. Future set-up plans include potential sites in the U.S. and EU where we already have initial activity such as in Germany and Texas, as well as in Australia and China.
As part of our POCare Services, we have developed the relevant GMP processes for a variety of therapies such as CAR-T, TILs and MSC based therapies. We have developed OMPULs with the required systems for production of CAR-T, TILs and MSC products, and are working on several other therapies intended for clinical testing. TIL, CAR-Ts and MSCs were already produced in the OMPULs for our customers. We have worked closely with technology partners to adapt various systems for closed system production of the above products and continue our collaboration efforts to develop fully automated systems for integration in the OMPULs.
We have expanded our collaboration with UC Davis having completed the first production batch of GMP grade lentivirus to be utilized for clinical-grade production of CAR-Ts and the initial engineering batch of a CAR-T based on the Lenti Virus. We intend to establish and validate the decentralized model of OMPUL placement in compliance with regulatory requirements. UC Davis has received a grant from the California Institute for Regenerative Medicine (CIRM) to validate the decentralized approach based on our platform. In addition, the parties aim to commercialize and install OMPULs at other sites within the State of California.
We have a partnership with Johns Hopkins University that already includes establishment of an analytical lab at FastForward, Johns Hopkins Technology Ventures’ (JHTV) innovation hub, and an agreed upon placement of an OMPUL.. Other activities include the provision of Kyslecel to eight hospitals in the U.S. Finally, we have deployed OMPULs at leading hospitals in Israel, Italy and Spain.
We have set up a partnerships in Greece focusing on delivering advanced therapies to Greek hospitals.
| ● | ICT-University of Patras |
Collaboration with the Institute of Cell Therapies (“ICT”), which was established as a part of the University Centre for Research and Innovation of the University of Patras. Theracell Laboratories will be responsible for the accreditation and operation of the Institute under GMP.
| ● | Manufacturing of Cell and Gene Therapies at Athens Point of Care |
A biomanufacturing unit has been set up in Athens (municipality of Koropi, Attika) The unit is staffed by experts in ATMP development, production quality control and release of medicinal products from fully operational OMPULs under GMP principles.
Pursuant to the Priority Investment of Strategic National Importance designation by Enterprise Greece, Theracell Laboratories received an investment grant covering industrial research activities associated with the development and production of Cell and Gene therapies in a decentralized manner in Greece. As of the date of this report, no funds have been received. However, once received, the operational costs of the activities described above will be covered by the grant.
| 16 |
Our POCare Services are expanding to additional geographies, and we are providing services to the U.S., EU, and Asia.
Revenue Model, Business Development and Licenses
Our POCare Platform is comprised of three enabling components: a multitude of licensed cell based POCare Therapies to be produced in closed, automated POCare Technology systems across a collaborative POCare Network. Our therapies include, but are not limited to, autologous, cell-based immunotherapies, therapeutics for metabolic diseases, anti-viral diseases, and tissue regeneration. We are establishing and positioning the business to bring point-of-care therapies to patients in a scalable way working directly with hospitals and through regional partners and organizations active in autologous cell therapy product development, including facilities in various countries in North America, Europe, Asia, the Middle East, and Australia. Our goal through the POCare Platform is to enable a rapid, globally harmonized pathway for these therapies to reach large numbers of patients at lowered costs through efficient, and decentralized production. Our POCare Network brings together industry partners, research institutes and hospitals worldwide to achieve harmonized, regulated clinical development and production of the therapies.
We are focused on technology in licensing and therapeutic collaborations, and we out-license therapies marketing rights and manufacturing rights to partners. In many cases, the partners are responsible for the preparation of clinical trials, local regulatory approvals and regional marketing activities. Such licensing includes exclusive or nonexclusive, sublicensable, royalty bearing rights and license to the Orgenesis Background IP as required to manufacture, distribute and market and sell Orgenesis products within the relevant territories. In consideration of the rights and the licenses so granted, we receive a royalty in the range of ten percent of the net sales generated by the partners and/or licensees or sublicensees (as applicable) with respect to the Orgenesis products.
Our business model of partnering with regional partners for initial clinical development of licensed POCare Therapies allows us to de-risk our clinical development plans. We have access to the development and clinical data generated by our partners based on which we can make informed decisions as to which of our assets have the most promising value for development in major markets such as the US and EU. Our goal is once we have proof of concept and clinical data from our regional partners, we can focus on developing such therapeutic products.
Further to revenues generated from out-licensing, we generate revenues from POCare Services and sales which is comprised of:
| ● | R&D development services provided to out-licensing partners |
We have signed POCare development services Master Services Agreements (“MSAs”) with our partners. In terms of the MSAs, we provide certain broadly defined development services that relate to our licensed therapies designed to develop or enhance the therapy with the objective of preparing it for clinical use. Such services, per therapy, include regulatory services, pre-clinical studies, intellectual property services, development services, and GMP process translation. We also provide support services to our customers.
| ● | Hospital supply |
Hospital services includes the sale or lease of products and the performance of processing services to our POCare hospitals or other medical providers. We either work directly with hospitals or receive payments through our regional partnerships.
| ● | Cell process development revenue |
We provide cell process development services in some regions to third party customers. Those services are unique to the customers who retain the ownership of the intellectual property created through the process.
| ● | POCare cell processing |
We provide distributed cell processing services for third party customers at POCare Centers in close proximity to patients.
| 17 |
Our POCare revenue is as follows:
| Years Ended December 31, | ||||||||
| Revenue stream: | 2023 | 2022 | ||||||
| (in thousands) | ||||||||
| POCare development services | $ | - | $ | 14,894 | ||||
| Cell process development services and hospital services | 515 | 11,212 | ||||||
| POCare cell processing | - | 9,919 | ||||||
| License fees | 15 | - | ||||||
| Total | $ | 530 | $ | 36,025 | ||||
Competition in the Cell Therapy Field
The biopharmaceutical industry is intensely competitive. There is continuous demand for innovation and speed, and as the cell-based therapies market evolves, there is always the risk that a competitor may be able to develop other compounds or drugs that are able to achieve similar or better results for indications. Potential competition includes major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, universities, and other research institutions. Many of these competitors have substantially greater financial, technical, and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations with established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
Currently, we are not aware of any other companies pursuing a business model similar to what we are developing under our POCare Platform. However, our competitors in the CGT field who are significantly larger and better capitalized than us could undertake strategies similar to what we are pursuing and even develop them at a much more rapid rate. These potential competitors include the same multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, universities, and other research institutions that are operating in the CGT field. In that respect, smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
Intellectual Property
We will be able to protect our technology and products from unauthorized use by third parties only to the extent it is covered by valid and enforceable claims of our patents or is effectively maintained as trade secrets. Patents and other proprietary rights are thus an essential element of our business.
Our success will depend in part on our ability to obtain and maintain proprietary protection for our product candidates, technology, and know-how, to operate without infringing on the proprietary rights of others, and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions, and improvements that are important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation, and in-licensing opportunities to develop and maintain our proprietary position.
In addition, we own or have exclusive rights to thirty-two (32) United States patents, eighty-seven (87) foreign-issued patents, twelve (12) pending patent applications in the United States, fifty three (53) pending patent applications in foreign jurisdictions, including Australia, Brazil, Canada, China, Europe, Hong Kong, India, Israel, Japan, Mexico, New Zealand, North Korea, Panama, Russia, Singapore, South Africa, and South Korea, and fifteen (15) international Patent Cooperation Treaty (“PCT”) patent applications. These patents and patent applications relate, among others, to (1) dendritic cell based (whole cell) vaccines, and their use for treating cancer and viral diseases; (2) compositions comprising Ranpirnase and other ribonucleases and their use for treating viral diseases; (3) tumor infiltrating lymphocytes (TILs) and their use for treating cancer; (4) compositions comprising immune cells, ribonucleases, or antibodies for treating COVID-19; (5) therapeutic compositions comprising exosomes, bioxomes, and redoxomes; (6) bioreactors for cell culture and automated devices for supporting cell therapies; (7) chimeric antigen receptors (CARs); (8) Mobile Processing Units; (9) Cell-delivery devices; and (10) skin diseases treatment and anti-aging compositions.
| 18 |
We have a granted U.S. patent and a pending U.S. patent application directed, among others, to dendritic cell-based (whole cell) vaccines, and their use for treating cancer and viral diseases. If issued, any patents based on these applications will expire in 2037. The granted U.S. patent will expire in 2037.
We have granted and pending U.S. patent applications directed, among others, to compositions comprising Ranpirnase and other ribonucleases for the treatment of viral diseases. Granted U.S. patents and if issued, any patents based on these applications will expire between 2024 and 2042. Counterpart granted patents and patents applications were filed in Australia, Canada, China, Europe, Hong Kong, Japan, Israel, Mexico, New Zealand, South Korea, Russian Federation, Singapore, and South Africa. If issued, any patents based on these applications will expire between 2035 and 2042. These expiration dates do not include any patent term extensions that might be available following the grant of marketing authorizations.
We have pending U.S. patent applications directed, among others, to therapeutic compositions comprising exosomes, bioxomes, and redoxomes. If issued, any patents based on these applications will expire between 2029 and 2041. Counterpart patents applications were filed in Australia, Brazil, Canada, China, Europe, India, Israel, Japan, Singapore and South Korea. If issued, any patents based on these applications will expire in 2039 and 2041. These expiration dates do not include any patent term extensions that might be available following the grant of marketing authorizations.
We have pending U.S. patent applications directed, among others, to compositions comprising ribonucleases and antibodies or bioxomes, and their use for treating viral diseases, including COVID-19. Counterpart patent application was also filed in Israel. If issued, any patents based on these applications will expire in 2042, without including any patent term extensions that might be available following the grant of marketing authorizations. A counterpart patent application was filed in Israel.
We have a pending International PCT application directed, among others, to compositions comprising immune cells for treating COVID-19. If converted into national phase applications and issued, any patents based on these applications will expire in 2042, without including any patent term extensions that might be available following the grant of marketing authorizations.
We have granted U.S. patents and a granted AU patent, pending U.S. patent applications, directed, among others, to bioreactors for cell culture and automated devices for supporting cell therapies. The granted U.S. patents will expire in 2027, and the granted AU patent will expire in 2026. If issued, any patents based on these applications will expire in 2042. Counterpart patent applications were filed in Australia, Europe, Israel, and Korea.
We have a pending US patent application directed, among others, to tumor infiltrating lymphocytes (TILs) and their use for treating cancer. If issued, patents will expire in 2042, without including any patent term extensions that might be available following the grant of marketing authorizations.
We have a pending U.S. patent application directed, among others, to compositions comprising mesenchymal stem cells, and their use for treating solid tumors. If issued, any patent based on this application would expire in 2040. Counterpart patent applications were filed in China, Europe, and Israel. If issued, any patents based on these applications would expire in 2040. These expiration dates do not include any patent term extensions that might be available following the grant of marketing authorizations.
We have a pending International PCT application directed, among others, to methods of treating cancer or CNS-related diseases by intranasal administration of an oncolytic virus. If converted into national phase applications and issued, any patents based on these applications will expire in 2043, without including any patent term extensions that might be available following the grant of marketing authorizations.
We have two pending U.S. patent application and a pending international patent application, directed, among others, to chimeric antigen receptors (CARs), and their use for treating malignancies. If issued, any patents based on the U.S. applications would expire in 2040 or 2042, without including any patent term extensions that might be available following the grant of marketing authorizations.
| 19 |
We have a pending International PCT application and a pending U.S. patent application directed, among others, to mobile processing laboratories configured for performing there within a cell therapy process. A counterpart patent application was filed in Europe. If issued, any patents based on these applications would expire in 2042, without including any patent term extensions that might be available following the grant of marketing authorizations.
We have a pending U.S. patent application and a pending PCT application, directed, among others, to a composition comprising topiramate and bioxome, redoxome, HA, extracellular vesicles (EV), or PRP extracellular vesicles and its use for the treatment of a dermatological condition. If converted into national phase applications and issued, any patents based on these applications would expire in 2042 and 2043, without including any patent term extensions that might be available following the grant of marketing authorizations.
The Israeli Subsidiary has exclusive rights to seven (7) United States patents, thirty (30) foreign-issued patents, and three (3) pending patent applications in foreign jurisdictions, including Brazil, Canada, and Europe. These patents and patent applications relate, among others, to the trans-differentiation of cells (including hepatic cells) to cells having pancreatic β-cell-like phenotype and function and to their use in the treatment of degenerative pancreatic disorders, including diabetes, pancreatic cancer and pancreatitis. Granted U.S. patents, which are directed to trans-differentiation to pancreatic β-cell-like phenotype and function cells and to their use in the treatment of degenerative pancreatic disorders, including diabetes, pancreatic cancer and pancreatitis, will expire between 2024 and 2040. Counterpart patents granted in Austria, Australia, Belgium, China, Eurasia, France, Germany, Greece, Israel, Switzerland, Japan, Mexico, Panama, Singapore, South Korea, and the United Kingdom, will expire between 2024 and 2035.
We also own IP and related Extracellular Vesicle (“EV”) Technology pursuant to an EV purchase agreement (the “EV Agreement”). Pursuant to the EV Agreement, we received all of the rights in EV technology purchased. In addition, we received an exclusive worldwide license to use the EV IP technology for any purpose.
Government Regulation
Development Business
We are required to comply with the regulatory requirements of various local, state, national and international regulatory bodies having jurisdiction in the countries or localities where we manufacture products, where our OMPULs are established or where we plan to supply products. In particular, we are subject to laws and regulations concerning research and development, testing, manufacturing processes, equipment and facilities, including compliance with GMPs, labeling and distribution, import and export, facility registration or licensing, and product registration and listing. As a result, our facilities are subject to regulation in Israel and South Korea. We are also required to comply with environmental, health and safety laws and regulations, as discussed below. These regulatory requirements impact many aspects of our operations, including manufacturing, developing, labeling, packaging, storage, distribution, import and export and record keeping related to customers’ products. Noncompliance with any applicable regulatory requirements can result in government refusal to approve facilities for manufacturing products or products for commercialization.
Both of our products and our customers’ products must undergo pre-clinical and clinical evaluations relating to product safety and efficacy before they are approved as commercial therapeutic products. The regulatory authorities that have jurisdiction in the countries in which our and our customers’ products are intended to be marketed may delay or put on hold clinical trials, delay approval of a product or determine that the product is not approvable. The regulatory agencies can delay approval of a drug if our manufacturing facilities or OMPULs are not able to demonstrate compliance with cGTPs, pass other aspects of pre-approval inspections (i.e., compliance with filed submissions) or properly scale up to produce commercial supplies. The government authorities having jurisdiction in the countries in which our customers intend to market their products have the authority to withdraw product approval or suspend manufacture if there are significant problems with raw materials or supplies, quality control and assurance or the product is deemed adulterated or misbranded. In addition, if new legislation or regulations are enacted or existing legislation or regulations are amended or are interpreted or enforced differently, we may be required to obtain additional approvals or operate according to different manufacturing or operating standards or pay additional fees. This may require a change in our manufacturing techniques or additional capital investments in our facilities.
| 20 |
Certain products manufactured by us involve the use, storage and transportation of toxic and hazardous materials. Our operations are subject to extensive laws and regulations relating to the storage, handling, emission, transportation and discharge of materials into the environment and the maintenance of safe working conditions. We maintain environmental and industrial safety and health compliance programs and training at our facilities.
Prevailing legislation tends to hold companies primarily responsible for the proper disposal of their waste even after transfer to third party waste disposal facilities. Other future developments, such as increasingly strict environmental, health and safety laws and regulations, and enforcement policies, could result in substantial costs and liabilities to us and could subject the handling, manufacture, use, reuse or disposal of substances or pollutants at our facilities to more rigorous scrutiny than at present.
Our development operations involve the controlled use of hazardous materials and chemicals. Although we believe that our procedures for using, handling, storing and disposing of these materials comply with legally prescribed standards, we may incur significant additional costs to comply with applicable laws in the future. Also, even if we are in compliance with applicable laws, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials or chemicals. As a result of any such contamination or injury, we may incur liability or local, city, state or federal authorities may curtail the use of these materials and interrupt our business operations. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our contract manufacturing operations, which could materially harm our business, financial condition and results of operations.
The costs associated with complying with the various applicable local, state, national and international regulations could be significant and the failure to comply with such legal requirements could have an adverse effect on our results of operations and financial condition. See “Risk Factors — Risks Related to Development and Regulatory Approval of Our Therapies and Product Candidates — Extensive industry regulation has had, and will continue to have, a significant impact on our business, especially our product development, manufacturing and distribution capabilities.” for additional discussion of the costs associated with complying with the various regulations.
POCare Therapies Portfolio
Our therapeutic product portfolio pipeline is diverse and addresses various unmet clinical needs. It is predominantly comprised of personalized autologous cell therapies, implying that patients receive cells that originate from their own body, virtually eliminating the risk of an immune response and rejection and thus easing various regulatory hurdles. In addition, by leveraging our vast experience and proven track record in developing and optimizing cell processing, these selective therapies are adapted to be produced in closed, automated systems, reducing the need for health care provider in-house, high-grade and expensive cleanroom environments. The systems enable each stage of the manufacturing process (cell sorting, expansion, genetic modifications, quality control) to be optimized in order to substantially reduce the cost burden for patients and making the therapies widely accessible. Notably, some of our therapeutic pipeline is developed by researchers from our network and is subsequently out-licensed to the researcher for its territory and validated in multi-center clinical trials conducted across point of care partner sites leveraging the robustness of our POCare Network. Having access to a portfolio of therapeutics, for the most attractive products, the Company intends to than seek additional regulatory approvals and offer the products for sale to medical institutions globally within our network In exchange, the inventors will receive a royalty.
| 21 |
Regulatory Process in the United States
Our potential product candidates are subject to regulation as a biological product under the Public Health Service Act and the Food, Drug and Cosmetic Act. The FDA generally requires the following steps for pre-market approval or licensure of a new biological product:
| ● | Pre-clinical laboratory and animal tests conducted in compliance with Good Laboratory Practice, or GLP, requirements to assess a drug’s biological activity and to identify potential safety problems, and to characterize and document the product’s chemistry, manufacturing controls, formulation, and stability; |
| ● | Submission to the FDA of an Investigational New Drug, or IND, application, which must become effective before clinical testing in humans can start; |
| ● | Obtaining approval of Institutional Review Boards, or IRBs, of research institutions or other clinical sites to introduce biologic drug candidates into humans in clinical trials; |
| ● | Conducting adequate and well-controlled clinical trials to establish the safety and efficacy of the product for its intended indication conducted in compliance with Good Clinical Practice, or GCP, requirements; |
| ● | Compliance with current GMP regulations and standards; |
| ● | Submission to the FDA of a Biologics License Application (“BLA”) for marketing that includes adequate results of pre-clinical testing and clinical trials; |
| ● | The FDA reviews the marketing application in order to determine, among other things, whether the product is safe, effective and potent for its intended uses; and |
| ● | Obtaining FDA approval of the BLA, including inspection and approval of the product manufacturing facility as compliant with GMP requirements, prior to any commercial sale or shipment of the pharmaceutical agent. The FDA may also require post marketing testing and surveillance of approved products or place other conditions on the approvals. |
Regulatory Process in Europe
In the European Union (“EU”) somatic cell and gene therapy products are called Advanced Therapy Medicinal Product (ATMPs). Since January 2022 the Clinical Trial Regulation (EU) 536/2014 regulates the application of medicinal products including ATMPs to humans immediately effective in all member states. In conjunction with Regulation 536/2014 the EU commission has released two delegated acts regulating manufacturing of investigational as well as marketed AMPs. For products that are regulated as an ATMP,
Regulation requires:
| ● | Compliance with current GMP regulations and standards, as described in the delegated acts; |
| ● | Filing a Clinical Trial Application (“CTA”); |
| ● | in EU member states and EEA countries according to regulation 536/2014 via CTIS (Clinical Trial Information System) allowing a harmonized approval process among all member states (including multinational clinical trials); |
| ● | Obtaining approval by ethic committees responsible for medical institutions; |
| ● | Adequate and well-controlled clinical trials according to GCP standards protecting the well-being of a study participant and establishing the safety and efficacy of the product for its intended use; |
| ● | Centralized submission procedure for ATMPs via EMA for Marketing Authorization; and |
| ● | Review and approval of the Marketing Authorization Application. |
Exemption from the centralized procedure was introduced into the ATMP Regulation to allow marketing of certain ATMPs in individual EU member states. The so-called “hospital exemption” can only be applied for custom-made ATMPs used in a hospital setting for a specific patient by a treating physician. In addition, a competent authority must authorize hospital exemption for ATMPs. Hospital exemption products must comply with the same national requirements concerning quality, traceability and pharmacovigilance that apply to authorized medicinal products. The “hospital exemption” has to be applied for individually in each EU member state according to national procedures and control measures.
Clinical Trials
Typically, both in the U.S. and the EU, clinical testing involves a three-phase process, although the phases may overlap. In Phase I, clinical trials are conducted with a small number of healthy volunteers or patients and are designed to provide information about product safety and to evaluate the pattern of drug distribution and metabolism within the body. In Phase II, clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. In some cases, an initial trial is conducted in diseased patients to assess both preliminary efficacy and preliminary safety and patterns of drug metabolism and distribution, in which case it is referred to as a Phase I/II trial. Phase III clinical trials are generally large-scale, multi-center, comparative trials conducted with patients afflicted with a target disease in order to provide statistically valid proof of efficacy, as well as safety and potency. In some circumstances, the FDA or EMA may require Phase IV or post-marketing trials if it feels that additional information needs to be collected about the drug after it is on the market. During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, as well as clinical trial investigators. An agency may, at its discretion, re-evaluate, alter, suspend, or terminate the testing based upon the data that have been accumulated to that point and its assessment of the risk/benefit ratio to the patient. Monitoring all aspects of the study to minimize risks is a continuing process. All adverse events must be reported to the FDA or EMA.
| 22 |
Human Capital Resources
As of December 31, 2023, we, including Octomera, had an aggregate of 146 employees working at our company and Subsidiaries. In addition, we retain the services of outside consultants for various functions including clinical work, finance, accounting and business development services. Most of our senior management and professional employees have had prior experience in pharmaceutical or biotechnology companies. None of our employees are covered by collective bargaining agreements. We believe that we have good relations with our employees.
Compensation and Benefits
We believe that our future success largely depends upon our continued ability to attract and retain highly skilled employees. Biotechnology companies both large and small compete for a limited number of qualified applicants to fill specialized positions. To attract qualified applicants, we offer a total rewards package consisting of base salary and cash target bonus, a comprehensive benefit package and equity compensation to select employees. Bonus opportunity and equity compensation increase as a percentage of total compensation based on level of responsibility. Actual bonus payout is based on performance.
Diversity, Equity and Inclusion
Much of our success is rooted in the diversity of our teams and our commitment to inclusion. We value diversity at all levels. We believe that our business benefits from the different perspectives a diverse workforce brings, and we pride ourselves on having a strong, inclusive and positive culture based on our shared mission and values. This is reflected in our numbers with our total workforce being approximately 55% women, 12% ethnically diverse and 51% over the age of 40.
Environmental, Social and Governance
Our commitment to integrating sustainability across our organization begins with our Board of Directors, or the Board. The Nominating and Governance Committee of the Board has oversight of strategy and risk management related to Environmental, Social and Governance, or ESG. All employees are responsible for upholding our core values, including to communicate, collaborate, innovate and be respectful, as well as for adhering to our Code of Ethics and Business Conduct, including our policies on bribery, corruption, conflicts of interest and our whistleblower program. We encourage employees to come to us with observations and complaints, ensuring we understand the severity and frequency of an event in order to escalate and assess accordingly. Our Chief Compliance Officer strives to ensure accountability, objectivity, and compliance with our Code of Conduct. If a complaint is financial in nature, the Audit Committee Chair is notified concurrently, which triggers an investigation, action, and report.
We are committed to protecting the environment and attempt to mitigate any negative impact of our operations. We monitor resource use, improve efficiency, and at the same time, reduce our emissions and waste. We are systematically addressing the environmental impacts of the buildings we rent as we make improvements, including adding energy control systems and other energy efficiency measures. Waste in our own operation is minimized by our commitment to reduce both single-use plastics and operating paper-free, primarily in a digital environment. We have safety protocols in place for handling biohazardous waste in our labs, and we use third-party vendors for biohazardous waste and chemical disposal.
Corporate and Available Information
Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and all amendments to those reports are available free of charge though our website (http://www.orgenesis.com) as soon as practicable after such material is electronically filed with, or furnished to, the Securities and Exchange Commission (the “SEC”). Except as otherwise stated in these documents, the information contained on our website or available by hyperlink from our website is not incorporated by reference into this report or any other documents we file, with or furnish to, the SEC.
Our common stock is listed and traded on the Nasdaq Capital Market under the symbol “ORGS.”
As used in this Annual Report on Form 10-K and unless otherwise indicated, the term “Company” refers to Orgenesis Inc. and its Subsidiaries. Unless otherwise specified, all amounts are expressed in United States Dollars.
| 23 |
ITEM 1A. RISK FACTORS
Summary of Risk Factors
Below is a summary of the principal factors that make an investment in our common stock speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in this Annual Report on Form 10-K and our other filings with the SEC, before making an investment decision regarding our common stock.
| ● | Our POCare business has a limited operating history and an unproven business model and faces significant challenges as the cell therapy industry is rapidly evolving. Our prospects may be considered speculative and any failure to execute our business strategy could adversely impact our business. |
| ● | Our management, as of December 31, 2023, and our independent registered public accounting firm, in its report on our financial statements as of and for the fiscal year ended December 31, 2023, have concluded that there is substantial doubt as to our ability to continue as a going concern. |
| ● | We are not profitable as of December 31, 2023, have limited cash flow and, unless we increase revenues and take advantage of any commercial opportunities that arise to expand our POCare business, the perceived value of our company may decrease and our stock price could be affected accordingly. |
| ● | Our research and development efforts on novel technology using cell-based therapy and our future success is highly dependent on the successful development of that technology. |
| ● | We require additional capital to support our business, and this capital may not be available on acceptable terms or at all. |
| ● | We have entered into collaborations and may form or seek collaborations or strategic alliances or enter into additional licensing arrangements in the future, and we may not realize the benefits of such alliances or licensing arrangements. |
| ● | Our success will depend on strategic collaborations with third parties to develop and commercialize therapeutic product candidates, and we may not have control over a number of key elements relating to the development and commercialization of any such product candidate. |
| ● | Our success depends on our ability to protect our intellectual property and our proprietary technologies. |
| ● | Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business. |
| ● | Our success depends on our ability to develop and grow the Octomera business. |
| ● | Our success depends on our ability to develop and roll out our OMPULs. |
| ● | If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates. |
| ● | We are increasingly dependent on information technology and our systems and infrastructure face certain risks, including cybersecurity and data storage risks. |
| 24 |
| ● | There can be no assurance that we will be able to develop in-house sales and commercial distribution capabilities or establish or maintain relationships with third-party collaborators to successfully commercialize any product in the United States or overseas, and as a result, we may not be able to generate product revenue. |
| ● | Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences. |
| ● | Our product candidates are biologics, and the manufacture of our product candidates is complex, and we may encounter difficulties in production, particularly with respect to process development or scaling-out of our manufacturing capabilities. |
| ● | Cell-based therapies rely on the availability of reagents, specialized equipment, and other specialty materials, which may not be available to us on acceptable terms or at all. For some of these reagents, equipment, and materials, we rely or may rely on sole source vendors or a limited number of vendors, which could impair our ability to manufacture and supply our products. |
| ● | We currently have no marketing and sales organization and have no experience in marketing therapeutic products. If we are unable to establish marketing and sales capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to generate product revenue. |
| ● | There can be no assurance that we will be able to develop in-house sales and commercial distribution capabilities or establish or maintain relationships with third-party collaborators to successfully commercialize any product in the United States or overseas, and as a result, we may not be able to generate product revenue. |
| ● | We face significant competition from other biotechnology and pharmaceutical companies, many of which have substantially greater financial, technical and other resources, and our operating results will suffer if we fail to compete effectively. |
| ● | We are highly dependent on key personnel who would be difficult to replace, and our business plans will likely be harmed if we lose their services or cannot hire additional qualified personnel. |
| ● | Extensive industry regulation has had, and will continue to have, a significant impact on our business, especially our product development, manufacturing and distribution capabilities. |
| ● | Third parties to whom we may license or transfer development and commercialization rights for products covered by intellectual property rights may not be successful in their efforts and, as a result, we may not receive future royalty or other milestone payments relating to those products or rights. |
| ● | Conditions in Israel, including the recent attack by Hamas and other terrorist organizations from the Gaza Strip and Israel’s war against them, may affect certain of our operations. |
| ● | We have identified a material weakness in our internal control over financial reporting. Failure to achieve and maintain effective internal controls over financial reporting could adversely affect our ability to report our results of operations and financial condition accurately and in a timely manner, which could have an adverse impact on our business. |
Risk Factors
An investment in our common stock involves a number of very significant risks. You should carefully consider the following risks and uncertainties in addition to other information in this report in evaluating our company and its business before purchasing shares of our company’s common stock. Our business, operating results and financial condition could be seriously harmed due to any of the following risks. You could lose all or part of your investment due to any of these risks.
Risks Related to Our Company and POCare Business
Our POCare business has a limited operating history and an unproven business model and faces significant challenges as the cell therapy industry is rapidly evolving. Our prospects may be considered speculative and any failure to execute our business strategy could adversely impact our operations and the price of our common stock.
| 25 |
Our POCare business has a limited operating history and an unproven business model. Our plans to continue to grow our POCare cell therapy business and to further the development of ATMPs are subject to significant challenges. Although we have sufficient capital resources for the next 12 months and the foreseeable future, we may not be able to implement our POCare business or commence clinical trials or respond to competitive pressures due to other non-financial factors beyond our control. Our failure to effectively execute our business strategy could adversely affect our ability to successfully grow our POCare business and develop cell therapy product candidates, which could cause the value of your investment in our common stock to decline.
Our management, as of December 31, 2023, and our independent registered public accounting firm, in its report on our financial statements as of and for the fiscal year ended December 31, 2023, have concluded that there is substantial doubt as to our ability to continue as a going concern.
Our audited financial statements for the fiscal year ended December 31, 2023 were prepared assuming that we will continue as a going concern. The going concern basis of the presentation assumes that we will continue in operation for the foreseeable future and will be able to realize our assets and satisfy our liabilities in the normal course of business and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or amounts and classification of liabilities that may result from our inability to continue as a going concern. As of December 31, 2023, our management concluded that, based on expected operating losses and negative cash flows, there is substantial doubt about our ability to continue as a going concern for the twelve months after the date the financial statements were issued. Our ability to continue as a going concern is subject to our ability to raise additional capital through equity offerings or debt financings. However, we may not be able to secure additional financing in a timely manner or on favorable terms, if at all. If we cannot continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our financial statements, and it is likely that our stockholders may lose some or all of their investment in us. If we seek additional financing to fund our business activities in the future and there remains substantial doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding on commercially reasonable terms or at all.
We are not profitable as of December 31, 2023, have limited cash flow and, unless we increase revenues and take advantage of any commercial opportunities that arise to expand our POCare business, the perceived value of our company may decrease and our stock price could be affected accordingly.
For the year ended December 31, 2023 and as of the date of this report, we assessed our financial condition and concluded that based on current and projected cash resources and commitments, there is a substantial doubt about the Company’s ability to continue as a going concern to meet the Company’s current operations for the next 12 months from the date of this report. Our auditor’s report for the year ended December 31, 2023 includes a going concern opinion on the matter. Management is unable to predict if and when we will be able to generate significant revenues or achieve profitability. Our plan regarding these matters is to continue improving the net results in our POCare business into fiscal year 2024. There can be no assurance that we will be successful in increasing revenues, improving our POCare results or that the perceived value of our Company will increase. In the event that we are unable to generate significant revenues in our POCare business, our stock price could be adversely affected.
Our research and development programs are based on novel technologies and are inherently risky.
We are subject to the risks of failure inherent in the development of products based on new technologies. The novel nature of our cell therapy technology creates significant challenges with respect to product development and optimization, manufacturing, government regulation and approval, third-party reimbursement and market acceptance. For example, the FDA and EMA have relatively limited experience with the development and regulation of cell therapy products and, therefore, the pathway to marketing approval for our cell therapy product candidates may accordingly be more complex, lengthy and uncertain than for a more conventional product candidate. The indications of use for which we choose to pursue development may have clinical effectiveness endpoints that have not previously been reviewed or validated by the FDA or EMA, which may complicate or delay our effort to ultimately obtain FDA or EMA approval. Because this is a new approach to treating diseases, developing and commercializing our product candidates subjects us to a number of challenges, including:
| ● | obtaining regulatory approval from the FDA, EMA and other regulatory authorities that have very limited experience with the commercial development of our technology for treating different diseases; |
| 26 |
| ● | developing and deploying consistent and reliable processes for removing the cells from the patient engineering cells ex vivo and infusing the engineered cells back into the patient; | |
| ● | developing processes for the safe administration of these products, including long-term follow-up for all patients who receive our products; | |
| ● | sourcing clinical and, if approved, commercial supplies for the materials used to manufacture and process our products; | |
| ● | developing a manufacturing process and distribution network with a cost of goods that allows for an attractive return on investment; | |
| ● | establishing sales and marketing capabilities after obtaining any regulatory approval to gain market acceptance; and | |
| ● | maintaining a system of post marketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug approval process. |
Our efforts to overcome these challenges may not prove successful, and any product candidate we seek to develop may not be successfully developed or commercialized.
Kyslecel may not achieve patient or market acceptance, which could have a material adverse effect on our business.
Our commercialization strategy for Kyslecel relies on medical specialists, medical facilities and patients adopting TP-IAT with Kyslecel as an accepted treatment for chronic pancreatitis. However, medical specialists are historically slow to adopt new treatments, regardless of perceived merits, when older treatments continue to be supported by established providers. Overcoming such resistance often requires significant marketing expenditure or definitive product performance and/or pricing superiority. The cost of allocating resources for such requirements might severely impact the potential for profitability of Kyslecel.
There is no guarantee that physician or patient acceptance of TP-IAT with Kyslecel will be substantial. Further, there is no guarantee that Koligo will be able to achieve patient acceptance or obtain enough customers (clinical providers) to meet its sales objectives. If we do not meet our sales objectives, our business prospects and financial performance will be materially and adversely affected.
Further, we are partially reliant on published clinical trials and scientific research conducted by third parties to justify the patient benefit and safety of TP-IAT with Kyslecel and, as such, we rely, in part, on the accuracy and integrity of those third-parties to have reported the results and correctly collected and interpreted the data from all clinical trials conducted to date. If published data turn out to later be incorrect or incomplete, our business prospects and financial performance may be materially and adversely affected.
The therapeutic efficacy of Ranpirnase and our other product candidates is unproven in humans, and we may not be able to successfully develop and commercialize Ranpirnase or any of our other product candidates.
Ranpirnase and our other product candidates are novel compounds and their potential benefit as antiviral drugs or immunotherapies is unproven. Ranpirnase and our other product candidates may not prove to be effective against the indications for which they are being designed to act and may not demonstrate in clinical trials any or all of the pharmacological effects that have been observed in preclinical studies. As a result, our clinical trial results may not be indicative of the results of future clinical trials.
Ranpirnase and our other product candidates may interact with human biological systems in unforeseen, ineffective or harmful ways. If Ranpirnase or any of our other product candidates is associated with undesirable side effects or have characteristics that are unexpected, we may need to abandon the development of such product candidate or limit development to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Because of these and other risks described herein that are inherent in the development of novel therapeutic agents, we may never successfully develop or commercialize Ranpirnase or any of our other product candidates, in which case our business will be harmed.
| 27 |
We will need to grow the size and capabilities of our organization, and we may experience difficulties in managing this growth.
As of December 31, 2023, we, including Octomera, employed 146 employees. As our development and commercialization plans and strategies develop, we must add a significant number of additional managerial, operational, sales, marketing, financial, and other personnel. Future growth will impose significant added responsibilities on members of management, including:
| ● | identifying, recruiting, integrating, maintaining, and motivating additional employees; | |
| ● | managing our internal development efforts effectively, including the clinical and FDA review process for our product candidates, while complying with our contractual obligations to contractors and other third parties; and | |
| ● | improving our operational, financial and management controls, reporting systems, and procedures. |
Our future financial performance and our ability to commercialize our product candidates will depend, in part, on our ability to effectively manage any future growth, and our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities. This lack of long-term experience working together may adversely impact our senior management team’s ability to effectively manage our business and growth.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors and consultants to provide certain services. There can be no assurance that the services of these independent organizations, advisors and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed, or terminated, and we may not be able to obtain regulatory approval of our product candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, if at all. If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop and commercialize our product candidates and, accordingly, may not achieve our research, development, and commercialization goals.
We require additional capital to support our business, and this capital may not be available on acceptable terms or at all.
We intend to continue to make investments to support our business growth and require additional funds to respond to business challenges and to grow our POCare cell therapy business and to further the development of ATMPs. Accordingly, we will need to engage in equity or debt financings to secure additional funds.
Capital and credit market conditions, adverse events affecting our business or industry, the tightening of lending standards, rising interest rates, negative actions by regulatory authorities or rating agencies, or other factors also could negatively impact our ability to obtain future financing on terms acceptable to us or at all. If we are unable to obtain adequate financing or financing on terms satisfactory to us when we require it, our ability to support our business growth and respond to business challenges could be significantly limited. In addition, the terms of any additional equity or debt issuances may adversely affect the value and price of our common stock, our results of operations, financial condition and cash flows.
If we raise additional funds through further issuances of equity or convertible debt securities, our existing stockholders could suffer significant dilution, and any new securities we issue could have rights, preferences and privileges superior to those of holders of our common stock. Any financing secured by us in the future could include restrictive covenants relating to our capital raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities, including potential acquisitions.
| 28 |
We conduct certain of our operations in Israel. Conditions in Israel, including the recent attack by Hamas and other terrorist organizations from the Gaza Strip and Israel’s war against them, may affect certain of our operations.
Because we conduct certain operations in the State of Israel, some of our business and operations may be affected by economic, political, geopolitical and military conditions in Israel. In October 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel. Following the attack, Israel’s security cabinet declared war against Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. Moreover, the clash between Israel and Hezbollah in Lebanon, may escalate in the future into a greater regional conflict.
Any hostilities involving Israel, or the interruption or curtailment of trade within Israel or between Israel and its trading partners could adversely affect certain of our operations and results of operations and could make it more difficult for us to raise capital. The conflict in Israel could also result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements. There have been travel advisories imposed relating to travel to Israel, and restriction on travel, or delays and disruptions as related to imports and exports may be imposed in the future. Additionally, certain members of our management and employees are located and reside in Israel. Shelter-in-place and work-from-home measures, government-imposed restrictions on movement and travel and other precautions taken to address the ongoing conflict may temporarily disrupt our management and employees’ ability to effectively perform their daily tasks.
The Israel Defense Force (the “IDF”), the national military of Israel, is a conscripted military service, subject to certain exceptions. Several of our employees are subject to military service in the IDF and have been, or may be, called to serve. It is possible that there will be further military reserve duty call-ups in the future, which may affect our business due to a shortage of skilled labor and loss of institutional knowledge, and necessary mitigation measures we may take to respond to a decrease in labor availability, such as overtime and third-party outsourcing, for example, may have unintended negative effects and adversely impact our results of operations, liquidity or cash flows.
It is currently not possible to predict the duration or severity of the ongoing conflict or its effects on our business, operations and financial conditions. The ongoing conflict is rapidly evolving and developing, and could disrupt certain of our business and operations, among others.
Currency exchange fluctuations may impact the results of our operations.
The results of our operations are affected by fluctuations in currency exchange rates in both sourcing and selling locations. Our results of operations may still be impacted by foreign currency exchange rates, primarily, the euro-to-U.S. dollar exchange rate. In recent years, the euro-to-U.S. dollar exchange rate has been subject to substantial volatility which may continue, particularly in light of recent political events regarding the European Union, or EU. Because we do not hedge against all of our foreign currency exposure, our business will continue to be susceptible to foreign currency fluctuations.
We have entered into collaborations and joint ventures and may form or seek collaborations or strategic alliances or enter into additional licensing arrangements in the future, and we may not realize the benefits of such alliances or licensing arrangements.
We have entered into collaborations and joint ventures and may form or seek strategic alliances, create joint ventures or collaborations, or enter into additional licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future product candidates that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing stockholders, or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners for which the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. Further, collaborations involving our product candidates, such as our collaborations with third-party research institutions, are subject to numerous risks, which may include the following:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to a collaboration; |
| 29 |
| ● | collaborators may not perform their obligations as expected; | |
| ● | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in their strategic focus due to the acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; | |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial, stop a clinical trial, abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product candidate for clinical testing; | |
| ● | collaborators could fail to make timely regulatory submissions for a product candidate; | |
| ● | collaborators may not comply with all applicable regulatory requirements or may fail to report safety data in accordance with all applicable regulatory requirements; | |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates; | |
| ● | product candidates developed in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the commercialization of our product candidates; | |
| ● | a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to their marketing and distribution; | |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; | |
| ● | disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our product candidates, or that result in costly litigation or arbitration that diverts management attention and resources; | |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; and | |
| ● | collaborators may own or co-own intellectual property covering our products that results from our collaborating with them and, in such cases, we would not have the exclusive right to commercialize such intellectual property. |
As a result, if we enter into collaboration agreements and strategic partnerships or license our products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture, which could delay our timelines or otherwise adversely affect our business. The success of our existing and future collaboration arrangements and strategic partnerships, which include research and development services by our collaborators to improve our intellectual property, will depend heavily on the efforts and activities of our collaborators and may not be successful. We also cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new collaborations or strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition, and results of operations.
Our success will depend on strategic collaborations with third parties to develop and commercialize therapeutic product candidates, and we may not have control over a number of key elements relating to the development and commercialization of any such product candidate.
A key aspect of our strategy is to seek collaborations with partners, such as a large pharmaceutical organization, that are willing to further develop and commercialize a selected product candidate. To date, we have entered into a number of collaborative arrangements with cell therapy organizations. By entering into any such strategic collaborations, we may rely on our partner for financial resources and for development, regulatory and commercialization expertise. Our partner may fail to develop or effectively commercialize our product candidate because they:
| ● | do not have sufficient resources or decide not to devote the necessary resources due to internal constraints such as limited cash or human resources; |
| 30 |
| ● | decide to pursue a competitive potential product developed outside of the collaboration; |
| ● | cannot obtain the necessary regulatory approvals; |
| ● | determine that the market opportunity is not attractive; or |
| ● | cannot manufacture or obtain the necessary materials in sufficient quantities from multiple sources or at a reasonable cost. |
We may not be able to enter into additional collaborations on acceptable terms, if at all. We face competition in our search for partners from other organizations worldwide, many of whom are larger and are able to offer more attractive deals in terms of financial commitments, contribution of human resources, or development, manufacturing, regulatory or commercial expertise and support. If we are not successful in attracting a partner and entering into a collaboration on acceptable terms, we may not be able to complete development of or commercialize any product candidate. In such event, our ability to generate revenues and achieve or sustain profitability would be significantly hindered and we may not be able to continue operations as proposed, requiring us to modify our business plan, curtail various aspects of our operations or cease operations.
Our business has been affected by the COVID-19 pandemic and may be significantly adversely affected by a resurgence of the COVID-19 pandemic or if other events out of our control disrupt our business or that of our third-party partners.
A continued and prolonged public health crisis such as the COVID-19 pandemic could have a material negative impact on our business, financial condition and operating results. We have experienced and may in the future experience disruptions from a resurgence of COVID-19 to our business in a number of ways, including:
| ● | Delays in supply chain and manufacturing, including the suspension of cell transport, limitations on transfer of technology, shutdown of manufacturing facilities and delays in delivery of supplies and reagents; |
| ● | Delays in discovery and preclinical efforts; |
| ● | Changes to procedures or shut down, or reduction in capacity, of clinical trial sites due to limited availability of clinical trial staff, reduced number of inpatient intensive care unit beds for patients receiving cell therapies, diversion of healthcare resources away from clinical trials and other business considerations; |
| ● | Limited patient access, enrollment and participation due to travel restrictions and safety concerns, as well as housing and travel difficulties for out-of-town patients and relatives; and |
| ● | Changes in regulatory and other requirements for conducting preclinical studies and clinical trials during the pandemic. |
In addition, we currently rely on third parties to, among other things, manufacture raw materials, manufacture our product candidates for our clinical trials, ship investigation drugs and clinical trial samples, perform quality testing and supply other goods and services to run our business. If any such third party in our supply chain for materials is adversely impacted by effects from a resurgence of the COVID-19 pandemic, including staffing shortages, production slowdowns and disruptions in delivery systems, our supply chain may be disrupted and our costs could be increased, limiting our ability to manufacture our product candidates for our clinical trials and planned future clinical trials and conduct our research and development operations as planned.
| 31 |
In addition, our business could be significantly adversely affected by other business disruptions to us or our third-party partners or collaborators that could seriously harm our potential future revenue and financial condition and increase our costs and expenses. Our operations, and those of our partners and collaborators, contract manufacturing organizations (CMOs) and other contractors, consultants, and third parties could be subject to other global pandemics, earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to produce and process our product candidates. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption.
Our success depends on our ability to protect our intellectual property and our proprietary technologies.
Our commercial success depends in part on our ability to obtain and maintain patent protection and trade secret protection for our product candidates, proprietary technologies, and their uses as well as our ability to operate without infringing upon the proprietary rights of others. We can provide no assurance that our patent applications or those of our licensors will result in additional patents being issued or that issued patents will afford sufficient protection against competitors with similar technologies, nor can there be any assurance that the patents issued will not be infringed, designed around or invalidated by third parties. Even issued patents may later be found unenforceable or may be modified or revoked in proceedings instituted by third parties before various patent offices or in courts. The degree of future protection for our proprietary rights is uncertain. Only limited protection may be available and may not adequately protect our rights or permit us to gain or keep any competitive advantage. Composition-of-matter patents on the biological or chemical active pharmaceutical ingredients are generally considered to offer the strongest protection of intellectual property and provide the broadest scope of patent protection for pharmaceutical products, as such patents provide protection without regard to any method of use or any method of manufacturing. While we have issued patents in the United States, we cannot be certain that the claims in our issued patent will not be found invalid or unenforceable if challenged.
We cannot be certain that the claims in our issued United States methods of use patents will not be found invalid or unenforceable if challenged.
We cannot be certain that the pending applications covering among others the bioconjugates comprising sulfated polysaccharides; Ranpirnase and other ribonucleases for treating viral diseases; therapeutic compositions comprising exosomes, bioxomes, and redoxomes; bioreactors for cell culture, automated devices for supporting cell therapies, and point-of-care systems; immune cells, ribonucleases, or antibodies for treating COVID-19; or chimeric antigen receptors (CARs); will be considered patentable by the United States Patent and Trademark Office (USPTO), and courts in the United States or by the patent offices and courts in foreign countries, nor can we be certain that the claims in our issued patents will not be found invalid or unenforceable if challenged. Even if our patent applications covering these inventions issue as patents, the patents protect specific products and may not be enforced against competitors making and marketing a product that has the same activity. Method-of-use patents protect the use of a product for the specified method or for treatment of a particular indication. These types of patents may not be enforced against competitors making and marketing a product that provides the same activity but is used for a method not included in the patent. Moreover, even if competitors do not actively promote their product for our targeted indications, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we or any of our future development partners will be successful in protecting our product candidates by obtaining and defending patents. These risks and uncertainties include the following:
| ● | the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case; |
| 32 |
| ● | patent applications may not result in any patents being issued; | |
| ● | patents that may be issued or in-licensed may be challenged, invalidated, modified, revoked, circumvented, found to be unenforceable or otherwise may not provide any competitive advantage; | |
| ● | our competitors, many of whom have substantially greater resources and many of whom have made significant investments in competing technologies, may seek or may have already obtained patents that will limit, interfere with or eliminate our ability to make, use, and sell our potential product candidates; | |
| ● | there may be significant pressure on the U.S. government and international governmental bodies to limit the scope of patent protection both inside and outside the United States for disease treatments that prove successful, as a matter of public policy regarding worldwide health concerns; and | |
| ● | countries other than the United States may have patent laws less favorable to patentees than those upheld by U.S. courts, allowing foreign competitors a better opportunity to create, develop and market competing product candidates. |
In addition, we rely on the protection of our trade secrets and proprietary know-how. Although we have taken steps to protect our trade secrets and unpatented know-how, including entering into confidentiality agreements with third parties, and confidential information and inventions agreements with employees, consultants and advisors, we cannot provide any assurances that all such agreements have been duly executed, and third parties may still obtain this information or may come upon this or similar information independently. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating its trade secrets. If any of these events occurs or if we otherwise lose protection for our trade secrets or proprietary know-how, our business may be harmed.
Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing, misappropriating or otherwise violating the intellectual property and proprietary rights of third parties. There is considerable patent and other intellectual property litigation in the pharmaceutical and biotechnology industries. We may become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our technology and product candidates, including interference proceedings, post grant review, inter partes review, and derivation proceedings before the USPTO and similar proceedings in foreign jurisdictions such as oppositions before the European Patent Office.
The legal threshold for initiating litigation or contested proceedings is low, so that even lawsuits or proceedings with a low probability of success might be initiated and require significant resources to defend. Litigation and contested proceedings can also be expensive and time-consuming, and our adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we can. The risks of being involved in such litigation and proceedings may increase if and as our product candidates near commercialization. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future, regardless of merit. We may not be aware of all such intellectual property rights potentially relating to our technology and product candidates and their uses, or we may incorrectly conclude that third party intellectual property is invalid or that our activities and product candidates do not infringe such intellectual property. Thus, we do not know with certainty that our technology and product candidates, or our development and commercialization thereof, do not and will not infringe, misappropriate or otherwise violate any third party’s intellectual property.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations or methods, such as methods of manufacture or methods for treatment, related to the discovery, use or manufacture of the product candidates that we may identify or related to our technologies. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that the product candidates that we may identify may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Moreover, as noted above, there may be existing patents that we are not aware of or that we have incorrectly concluded are invalid or not infringed by our activities. If any third-party patents were held by a court of competent jurisdiction to cover, for example, the manufacturing process of the product candidates that we may identify, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire.
| 33 |
Generally, conducting clinical trials and other development activities in the United States is not considered an act of infringement. If and when products are approved by the FDA, that certain third party may then seek to enforce its patents by filing a patent infringement lawsuit against us or our licensee(s). In such lawsuit, we or our licensees may incur substantial expenses defending our rights or our licensees’ rights to commercialize such product candidates, and in connection with such lawsuit and under certain circumstances, it is possible that we or our licensees could be required to cease or delay the commercialization of a product candidate and/or be required to pay monetary damages or other amounts, including royalties on the sales of such products. Moreover, any such lawsuit may also consume substantial time and resources of our management team and board of directors. The threat or consequences of such a lawsuit may also result in royalty and other monetary obligations being imposed on us, which may adversely affect our results of operations and financial condition.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize the product candidates that we may identify. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
We may choose to take a license or, if we are found to infringe, misappropriate or otherwise violate a third party’s intellectual property rights, we could also be required to obtain a license from such third party to continue developing, manufacturing and marketing our technology and product candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors and other third parties access to the same technologies licensed to us and could require us to make substantial licensing and royalty payments. We could be forced, including by court order, to cease developing, manufacturing and commercializing the infringing technology or product. In addition, we could be found liable for significant monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent or other intellectual property right and could be forced to indemnify our customers or collaborators. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. In addition, we may be forced to redesign our product candidates, seek new regulatory approvals and indemnify third parties pursuant to contractual agreements. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar material adverse effect on our business, financial condition, results of operations and prospects.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if our product candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our products; |
| ● | injury to our reputation; |
| ● | withdrawal of clinical trial participants and inability to continue clinical trials; |
| ● | initiation of investigations by regulators; |
| ● | costs to defend the related litigation; |
| 34 |
| ● | a diversion of management’s time and our resources; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | loss of revenue; |
| ● | exhaustion of any available insurance and our capital resources; |
| ● | the inability to commercialize any product candidate; and |
| ● | a decline in our share price. |
Because most of our products have not reached commercial stage, we do not currently need to carry clinical trial or extensive product liability insurance. In the future, our inability to obtain additional sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop, alone or with collaborators. Such insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage.
It may be difficult to enforce a U.S. judgment against us, our officers and directors and the foreign persons named in this Annual Report on Form 10-K in the United States or in foreign countries, or to assert U.S. securities laws claims in foreign countries or serve process on our officers and directors and these experts.
While we are incorporated in the State of Nevada, currently a majority of our directors and executive officers are not residents of the United States, and the foreign persons named in this Annual Report on Form 10-K are located outside of the United States. The majority of our assets are located outside the United States. Therefore, it may be difficult for an investor, or any other person or entity, to enforce a U.S. court judgment based upon the civil liability provisions of the U.S. federal securities laws against us or any of these persons in a U.S. or foreign court, or to effect service of process upon these persons in the United States. Additionally, it may be difficult for an investor, or any other person or entity, to assert U.S. securities law claims in original actions instituted in foreign countries in which we operate. Foreign courts may refuse to hear a claim based on a violation of U.S. securities laws on the grounds that foreign countries are not necessary the most appropriate forum in which to bring such a claim. Even if a foreign court agrees to hear a claim, it may determine that foreign law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by foreign countries law. There is little binding case law in foreign countries addressing the matters described above.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information, including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. In the U.S., numerous federal and state laws and regulations, including state security breach notification laws, state health information privacy laws, and federal and state consumer protection laws, govern the collection, use, disclosure, and protection of personal information. Each of these laws is subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations, we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain or disclose individually identifiable health information from a covered entity in a manner that is not authorized or permitted by the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, or HIPAA.
Numerous other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. The EU and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. In the EU, for example, effective May 25, 2018, the GDPR replaced the prior EU Data Protection Directive (95/46) that governed the processing of personal data in the European Union. The GDPR imposes significant obligations on controllers and processors of personal data, including, as compared to the prior directive, higher standards for obtaining consent from individuals to process their personal data, more robust notification requirements to individuals about the processing of their personal data, a strengthened individual data rights regime, mandatory data breach notifications, limitations on the retention of personal data and increased requirements pertaining to health data, and strict rules and restrictions on the transfer of personal data outside of the EU, including to the U.S. The GDPR also imposes additional obligations on, and required contractual provisions to be included in, contracts between companies subject to the GDPR and their third-party processors that relate to the processing of personal data. The GDPR allows EU member states to make additional laws and regulations further limiting the processing of genetic, biometric or health data.
| 35 |
Adoption of the GDPR increased our responsibility and liability in relation to personal data that we process and may require us to put in place additional mechanisms to ensure compliance. Any failure to comply with the requirements of GDPR and applicable national data protection laws of EU member states, could lead to regulatory enforcement actions and significant administrative and/or financial penalties against us (fines of up to Euro 20,000,000 or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher), and could adversely affect our business, financial condition, cash flows and results of operations.
We are increasingly dependent on information technology and our systems and infrastructure face certain risks, including cybersecurity and data storage risks.
Significant disruptions to our information technology systems or breaches of information security could adversely affect our business. In the ordinary course of business, we collect, store and transmit confidential information, and it is critical that we do so in a secure manner in order to maintain the confidentiality and integrity of such confidential information. Our information technology systems are potentially vulnerable to service interruptions and security breaches from inadvertent or intentional actions by our employees, partners, vendors, or from attacks by malicious third parties. Maintaining the secrecy of this confidential, proprietary, and/or trade secret information is important to our competitive business position. While we have taken steps to protect such information and invested in information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches in our systems or the unauthorized or inadvertent wrongful access or disclosure of confidential information that could adversely affect our business operations or result in the loss, dissemination, or misuse of critical or sensitive information. A breach of our security measures or the accidental loss, inadvertent disclosure, unapproved dissemination or misappropriation or misuse of trade secrets, proprietary information, or other confidential information, whether as a result of theft, hacking, or other forms of deception, or for any other cause, could enable others to produce competing products, use our proprietary technology and/or adversely affect our business position. Further, any such interruption, security breach, loss or disclosure of confidential information could result in financial, legal, business, and reputational harm to us and could have a material effect on our business, financial position, results of operations and/or cash flow.
There can be no assurance that we will be able to develop in-house sales and commercial distribution capabilities or establish or maintain relationships with third-party collaborators to successfully commercialize any product in the United States or overseas, and as a result, we may not be able to generate product revenue.
A variety of risks associated with operating our business internationally could materially adversely affect our business. We plan to seek regulatory approval of our product candidates outside of the United States and, accordingly, we expect that we, and any potential collaborators in those jurisdictions, will be subject to additional risks related to operating in foreign countries, including:
| ● | differing regulatory requirements in foreign countries, unexpected changes in tariffs, trade barriers, price and exchange controls, and other regulatory requirements; |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| ● | compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
| ● | foreign taxes, including withholding of payroll taxes; |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| ● | difficulties staffing and managing foreign operations; |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| ● | potential liability under the Foreign Corrupt Practices Act of 1977 or comparable foreign laws; |
| ● | challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| ● | business interruptions resulting from geo-political actions, including war, and terrorism or disease outbreaks (such as the recent outbreak of COVID-19, or the novel coronavirus). |
| 36 |
These and other risks associated with our planned international operations may materially adversely affect our ability to attain or maintain profitable operations.
If we are unable to integrate acquired businesses effectively, our operating results may be adversely affected.
From time to time, we seek to expand our business through acquisitions. We may not be able to successfully integrate acquired businesses and, where desired, their product portfolios into ours, and therefore we may not be able to realize the intended benefits. If we fail to successfully integrate acquisitions or product portfolios, or if they fail to perform as we anticipate, our existing businesses and our revenue and operating results could be adversely affected. If the due diligence of the operations of acquired businesses performed by us and by third parties on our behalf is inadequate or flawed, or if we later discover unforeseen financial or business liabilities, acquired businesses and their assets may not perform as expected. Additionally, acquisitions could result in difficulties assimilating acquired operations and, where deemed desirable, transitioning overlapping products into a single product line and the diversion of capital and management’s attention away from other business issues and opportunities. The failure to integrate acquired businesses effectively may adversely impact our business, results of operations or financial condition.
Risks Related to Our OMPULs
We may not be able to operate our OMPULs in all cities or desired locations and the sizes and use of our laboratories in such OMPULs may be restricted due to zoning, environmental, medical waste, or other licensing regulations.
We may be subject to local zoning ordinances or other similar restrictions that may limit where the OMPULs can be located and the extent of their size and use. In addition, international, federal, state and local environmental and other administrative and licensing regulations could restrict the ability of the OMPULs to connect with local power, water, sewer, and other infrastructure. Our success depends on our ability to develop and roll out our OMPULs which may become more difficult or more expensive by such applicable regulations. Changes in any of these regulations could require us to close or move our OMPULs which would affect our ability to conduct and grow our business.
If our existing OMPULs facilities become damaged or inoperable or if we are required to vacate our existing facilities, our ability to perform our tests and pursue our research and development efforts may be jeopardized.
We currently perform a majority of tests relating to our POCare Services out of our OMPULs. Our facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including war, fire, earthquake, power loss, communications failure or terrorism, which may render it difficult or impossible for us to operate for some period of time. In addition, since there is no lengthy history of use of OMPULs and the OMPULs are still in the development stage, we are unable to predict the normal wear and tear on such OMPULs or how many years each OMPUL will remain operational.
The inability to perform our tests or to reduce the backlog that could develop if our facilities are inoperable, for even a short period of time, may result in the loss of customers or harm to our reputation, and we may be unable to regain those customers or repair our reputation. Furthermore, our OMPUL facilities and the equipment we use to perform our research and development work could be unavailable or costly and time-consuming to repair or replace. It would be difficult, time-consuming and expensive to rebuild our facilities, or to locate and qualify new facilities.
We carry insurance for damage to our property and disruption of our business, but this insurance may not cover all of the risks associated with damage or disruption to our facility and business, may not provide coverage in amounts sufficient to cover our potential losses and may not continue to be available to us on acceptable terms, if at all.
| 37 |
Changes in the price and availability of our raw materials could be detrimental to our OMPUL operations.
Supply chain issues, including limited supply of certain raw material or supply interruptions, delays or shortages of material may disrupt our daily operations as the OMPULs may be unable to retain an inventory of materials required to maintain operations or to build or repair OMPULs.
We are dependent on skilled human capital for our OMPULs.
Our ability to innovate and execute is dependent on the ability to hire, replace, and train skilled personnel. The employment market suffers from shortage of candidates that may continue in future years and cause delays and inabilities to execute our plans. Additionally, based on current trends in the US labor market, there could be a shortage of available trained staff for the OMPULs in the United States. Staff retention could also be a significant operational issue.
If we are unable to successfully secure our locations and premises, we may be unable to operate out of our OMPULs or keep our employees and laboratory equipment safe.
In certain cities and urban markets, homelessness, rising crime rates and decreased police funding, could impact the security of the OMPULs and the safety of employees and patients. If we are unable to successfully secure our OMPULs, our research and development could be negatively impacted.
Our OMPULs are operated in a heavily regulated industry, and changes in regulations or violations of regulations may, directly or indirectly, reduce our revenue, adversely affect our results of operations and financial condition, and harm our business.
The clinical laboratory testing industry is highly regulated, and there can be no assurance that the regulatory environment in which we operate will not change significantly and adversely to us in the future. Areas of the regulatory environment that may affect our ability to conduct our OMPUL business include, without limitation:
| ● | federal and state laws governing laboratory testing, including CLIA, and state licensing laws; |
| ● | federal and state laws and enforcement policies governing the development, use and distribution of diagnostic medical devices, including laboratory developed tests, or LDTs; |
| ● | federal, state and local laws governing the handling and disposal of medical and hazardous waste; |
| ● | federal and state Occupational Safety and Health Administration rules and regulations; and |
| ● | European Union GMP approvals, which may be delayed because of the use OMPULs which could then delay manufacturing for clinical trials. |
Risks Related to Our Trans-Differentiation Technologies for Diabetes and the THM License Agreement
THM is entitled to cancel the THM License Agreement.
Pursuant to the terms of the THM License Agreement with THM, Orgenesis Ltd, the Israeli Subsidiary, must develop, manufacture, sell and market the products pursuant to the milestones and time schedule specified in the development plan. In the event the Israeli Subsidiary fails to fulfill the terms of the development plan under the THM License Agreement, THM shall be entitled to terminate the THM License Agreement by providing the Israeli Subsidiary with written notice of such a breach and if the Israeli Subsidiary does not cure such breach within one year of receiving the notice. THM may also terminate the THM License Agreement if the Israeli Subsidiary breaches an obligation contained in the THM License Agreement and does not cure it within 180 days of receiving notice of the breach. We also run the risk that THM may attempt cancel or, at the very least challenge, the License Agreement with the Israeli Subsidiary as we continue to expand our focus to other therapies and business activities. While we have not received any notice of cancellation of the THM License Agreement, we have received an allegation regarding the scope of the rights by THM that may present future challenges for our Israeli Subsidiary to continue to develop, manufacture, sell and market the products pursuant to the milestones and time schedule specified in the development plan of the THM License Agreement. In addition, THM has filed a complaint against us in the Tel Aviv District Court relating to the scope of such THM license and the royalties and other payments that THM is entitled to thereunder. See “Legal Proceedings” in this Annual Report on Form 10-K. Such complaint may lead to further risk of cancellation of the THM License Agreement.
| 38 |
The Israeli Subsidiary is a licensed technology that demonstrates the capacity to induce a shift in the developmental fate of cells from the liver and differentiating (converting) them into “pancreatic beta cell-like” insulin-producing cells for patients with diabetes. Our intention is to develop our technology to the clinical stage for regeneration of functional insulin-producing cells, thus enabling normal glucose regulated insulin secretion, via cell therapy. By using therapeutic agents that efficiently convert a sub-population of liver cells into pancreatic islets phenotype and function, this approach allows the diabetic patient to be the donor of his/her own therapeutic tissue and to start producing his/her own insulin in a glucose-responsive manner, thereby eliminating the need for insulin injections. Because this is a new approach to treating diabetes, developing and commercializing our product candidates subjects us to a number of challenges, including:
| ● | obtaining regulatory approval regulatory authorities that have very limited experience with the commercial development of the trans-differentiating technology for diabetes; |
| ● | developing and deploying consistent and reliable processes for engineering a patient’s liver cells ex vivo and infusing the engineered cells back into the patient; |
| ● | developing processes for the safe administration of these products, including long-term follow-up for all patients who receive our products; |
| ● | sourcing clinical and, if approved, commercial supplies for the materials used to manufacture and process our products; |
| ● | developing a manufacturing process and distribution network with a cost of goods that allows for an attractive return on investment; |
| ● | establishing sales and marketing capabilities after obtaining any regulatory approval to gain market acceptance; and |
| ● | maintaining a system of post marketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug approval process. |
Risks Related to Development and Regulatory Approval of Our Therapies and Product Candidates
Research and development of biopharmaceutical products is inherently risky.
We may not be successful in our efforts to use and enhance our technology platform to create a pipeline of product candidates and develop commercially successful products. Furthermore, we may expend our limited resources on programs that do not yield a successful product candidate and fail to capitalize on product candidates or diseases that may be more profitable or for which there is a greater likelihood of success. If we fail to develop additional product candidates, our commercial opportunity will be limited. Even if we are successful in continuing to build our pipeline, obtaining regulatory approvals and commercializing additional product candidates will require substantial additional funding and are prone to the risks of failure inherent in medical product development. Investment in biopharmaceutical product development involves significant risk that any potential product candidate will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval, and become commercially viable. We cannot provide you any assurance that we will be able to successfully advance any of these additional product candidates through the development process. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development or commercialization for many reasons, including the following:
| ● | our platform may not be successful in identifying additional product candidates; |
| ● | we may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; |
| ● | our product candidates may not succeed in preclinical or clinical testing; |
| ● | a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
| ● | competitors may develop alternatives that render our product candidates obsolete or less attractive; |
| ● | product candidates we develop may nevertheless be covered by third parties’ patents or other exclusive rights; |
| ● | the market for a product candidate may change during our program so that the continued development of that product candidate is no longer reasonable; |
| ● | a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
| ● | a product candidate may not be accepted as safe and effective by patients, the medical community or third- party payers, if applicable. |
| 39 |
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, or we may not be able to identify, discover, develop, or commercialize additional product candidates, which would have a material adverse effect on our business and could potentially cause us to cease operations.
Extensive industry regulation has had, and will continue to have, a significant impact on our business, especially our product development, manufacturing and distribution capabilities.
All pharmaceutical companies are subject to extensive, complex, costly and evolving government regulation. For the U.S., this is principally administered by the FDA and to a lesser extent by the Drug Enforcement Administration (“DEA”) and state government agencies, as well as by varying regulatory agencies in foreign countries where products or product candidates are being manufactured and/or marketed. The Federal Food, Drug and Cosmetic Act, the Controlled Substances Act and other federal statutes and regulations, and similar foreign statutes and regulations, govern or influence the testing, manufacturing, packing, labeling, storing, record keeping, safety, approval, advertising, promotion, sale and distribution of our future products. Under these regulations, we may become subject to periodic inspection of our facilities, procedures and operations and/or the testing of our future products by the FDA, the DEA and other authorities, which conduct periodic inspections to confirm that we are in compliance with all applicable regulations. In addition, the FDA and foreign regulatory agencies conduct pre-approval and post-approval reviews and plant inspections to determine whether our systems and processes are in compliance with current GMP and other regulations. Following such inspections, the FDA or other agency may issue observations, notices, citations and/or warning letters that could cause us to modify certain activities identified during the inspection. FDA guidelines specify that a warning letter is issued only for violations of “regulatory significance” for which the failure to adequately and promptly achieve correction may be expected to result in an enforcement action. We may also be required to report adverse events associated with our future products to FDA and other regulatory authorities. Unexpected or serious health or safety concerns would result in labeling changes, recalls, market withdrawals or other regulatory actions.
The range of possible sanctions includes, among others, FDA issuance of adverse publicity, product recalls or seizures, fines, total or partial suspension of production and/or distribution, suspension of the FDA’s review of product applications, enforcement actions, injunctions, and civil or criminal prosecution. Any such sanctions, if imposed, could have a material adverse effect on our business, operating results, financial condition and cash flows. Under certain circumstances, the FDA also has the authority to revoke previously granted drug approvals. Similar sanctions as detailed above may be available to the FDA under a consent decree, depending upon the actual terms of such decree. If internal compliance programs do not meet regulatory agency standards or if compliance is deemed deficient in any significant way, it could materially harm our business.
The European Medicines Agency (“EMA”) will regulate our future products in Europe. Regulatory approval by the EMA will be subject to the evaluation of data relating to the quality, efficacy and safety of our future products for its proposed use. The time taken to obtain regulatory approval varies between countries. Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators.
Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements.
Further trials and other costly and time-consuming assessments of the product may be required to obtain or maintain regulatory approval. Medicinal products are generally subject to lengthy and rigorous pre-clinical and clinical trials and other extensive, costly and time-consuming procedures mandated by regulatory authorities. We may be required to conduct additional trials beyond those currently planned, which could require significant time and expense. In addition, even after the technology approval, both in the U.S. and Europe, we will be required to maintain post marketing surveillance of potential adverse and risk assessment programs to identify adverse events that did not appear during the clinical studies and drug approval process. All of the foregoing could require an investment of significant time and expense.
| 40 |
We have generated limited revenue from therapeutic product sales, and our ability to generate any significant revenue from product sales and become profitable depends significantly on our success in a number of factors.
We have a limited number of therapeutic products approved for commercial sale, and we have generated only limited revenue from product sales. Our ability to generate revenue of more significant scale and achieve profitability depends significantly on our success in many factors, including:
| ● | completing research regarding, and nonclinical and clinical development of, our product candidates; |
| ● | obtaining regulatory approvals and marketing authorizations for product candidates for which we complete clinical studies; |
| ● | developing a sustainable and scalable manufacturing process for our product candidates, including establishing and maintaining commercially viable supply relationships with third parties and establishing our own manufacturing capabilities and infrastructure; |
| ● | launching and commercializing product candidates for which we obtain regulatory approvals and marketing authorizations, either directly or with a collaborator or distributor; |
| ● | obtaining market acceptance of our product candidates as viable treatment options; |
| ● | addressing any competing technological and market developments; |
| ● | identifying, assessing, acquiring and/or developing new product candidates; |
| ● | negotiating favorable terms in any collaboration, licensing, or other arrangements into which we may enter; |
| ● | maintaining, protecting, and expanding our portfolio of intellectual property rights, including patents, trade secrets, and know-how; and |
| ● | attracting, hiring, and retaining qualified personnel. |
Even if more of the product candidates that we develop are approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate. Our expenses could increase beyond expectations if we are required by the U.S. Food and Drug Administration, or the FDA, or other regulatory agencies, domestic or foreign, to change our manufacturing processes or assays, or to perform clinical, nonclinical, or other types of studies in addition to those that we currently anticipate. If we are successful in obtaining regulatory approvals to market more of our product candidates, our revenue will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of our addressable disease patients is not as significant as we estimate, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales of such products, even if approved. If we are not able to generate revenue from the sale of any approved products, we may never become profitable.
When we commence any clinical trials, we may not be able to conduct our trials on the timelines we expect.
Clinical testing is expensive, time consuming, and subject to uncertainty. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. We cannot be sure that we will be able to submit an IND, and we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin. Moreover, even if these trials begin, issues may arise that could suspend or terminate such clinical trials. A failure of one or more clinical studies can occur at any stage of testing, and our future clinical studies may not be successful. Events that may prevent successful or timely completion of clinical development include:
| ● | the inability to generate sufficient preclinical or other in vivo or in vitro data to support the initiation of clinical studies; |
| ● | delays in reaching a consensus with regulatory agencies on study design; |
| ● | delays in establishing CMC (Chemistry, Manufacturing, and Controls) which is a cornerstone in clinical study submission and later on, the regulatory approval; |
| ● | the FDA not allowing us to use the clinical trial data from a research institution to support an IND if we cannot demonstrate the comparability of our product candidates with the product candidate used by the relevant research institution in its clinical studies; |
| 41 |
| ● | delays in obtaining required Institutional Review Board, or IRB, approval at each clinical study site; |
| ● | imposition of a temporary or permanent clinical hold by regulatory agencies for a number of reasons, including after review of an IND application or amendment, or equivalent application or amendment; |
| ● | a result of a new safety finding that presents unreasonable risk to clinical trial participants; |
| ● | a negative finding from an inspection of our clinical study operations or study sites; |
| ● | developments on trials conducted by competitors for related technology that raises FDA concerns about risk to patients of the technology broadly; |
| ● | if the FDA finds that the investigational protocol or plan is clearly deficient to meet its stated objectives; |
| ● | delays in recruiting suitable patients to participate in our clinical studies; |
| ● | difficulty collaborating with patient groups and investigators; |
| ● | failure to perform in accordance with the FDA’s current good clinical practices, or cGCPs, requirements, or applicable regulatory guidelines in other countries; |
| ● | delays in having patients complete participation in a study or return for post-treatment follow-up; |
| ● | patients dropping out of a study; |
| ● | occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential benefits; |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; |
| ● | changes in the standard of care on which a clinical development plan was based, which may require new or additional trials; |
| ● | the cost of clinical studies of our product candidates being greater than we anticipate; |
| ● | clinical studies of our product candidates producing negative or inconclusive results, which may result in our deciding, or regulators requiring us, to conduct additional clinical studies or abandon product development programs; and |
| ● | delays in manufacturing, testing, releasing, validating, or importing/exporting sufficient stable quantities of our product candidates for use in clinical studies or the inability to do any of the foregoing. |
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required to, or we may elect to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical study delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
Our clinical trial results may also not support approval, whether accelerated approval, conditional marketing authorizations, or regular approval. The results of preclinical and clinical studies may not be predictive of the results of later-stage clinical trials, and product candidates in later stages of clinical trials may fail to show the desired safety and efficacy despite having progressed through preclinical studies and initial clinical trials. In addition, our product candidates could fail to receive regulatory approval for many reasons, including the following:
| ● | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| ● | the population studied in the clinical program may not be sufficiently broad or representative to assure safety in the full population for which we seek approval; |
| ● | we may be unable to demonstrate that our product candidates’ risk-benefit ratios for their proposed indications are acceptable; |
| ● | the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| ● | we may be unable to demonstrate that the clinical and other benefits of our product candidates outweigh their safety risks; |
| ● | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected from clinical trials of our product candidates may not be sufficient to the satisfaction of the FDA or comparable foreign regulatory authorities to obtain regulatory approval in the United States or elsewhere; |
| ● | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes, our own manufacturing facilities, or our third-party manufacturers’ facilities with which we contract for clinical and commercial supplies; and |
| ● | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
| 42 |
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences.
As with most biological drug products, use of our product candidates could be associated with side effects or adverse events which can vary in severity from minor reactions to death and in frequency from infrequent to prevalent. Any of these occurrences may materially and adversely harm our business, financial condition and prospects.
Our product candidates are biologics, and the manufacture of our product candidates is complex, and we may encounter difficulties in production, particularly with respect to process development or scaling-out of our manufacturing capabilities.
If we encounter such difficulties, our ability to provide supply of our product candidates for clinical trials or our products for patients, if approved, could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure. Our product candidates are biologics and the process of manufacturing our products is complex, highly regulated and subject to multiple risks. As a result of the complexities, the cost to manufacture biologics is generally higher than traditional small molecule chemical compounds, and the manufacturing process is less reliable and is more difficult to reproduce.
Our manufacturing process will be susceptible to product loss or failure due to logistical issues associated with the collection of liver cells, or starting material, from the patient, shipping such material to the manufacturing site, shipping the final product back to the patient, and infusing the patient with the product, manufacturing issues associated with the differences in patient starting materials, interruptions in the manufacturing process, contamination, equipment or reagent failure, improper installation or operation of equipment, vendor or operator error, inconsistency in cell growth, failures in process testing and variability in product characteristics. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects, and other supply disruptions. If for any reason we lose a patient’s starting material or later-developed product at any point in the process, the manufacturing process for that patient will need to be restarted and the resulting delay may adversely affect that patient’s outcome. If microbial, viral, or other contaminations are discovered in our product candidates or in the manufacturing facilities in which our product candidates are made, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. Because our product candidates are manufactured for each particular patient, we will be required to maintain a chain of identity and tractability of all reagents and viruses involved in the process with respect to materials as they move from the patient to the manufacturing facility, through the manufacturing process, and back to the patient. Maintaining such a chain of identity is difficult and complex, and failure to do so could result in adverse patient outcomes, loss of product, or regulatory action including withdrawal of our products from the market. Further, as product candidates are developed through preclinical to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials.
Although we are working to develop commercially viable processes, doing so is a difficult and uncertain task, and there are risks associated with scaling to the level required for advanced clinical trials or commercialization, including, among others, cost overruns, potential problems with process scale-out, process reproducibility, stability issues, lot consistency, and timely availability of reagents or raw materials. We may ultimately be unable to reduce the cost of goods for our product candidates to levels that will allow for an attractive return on investment if and when those product candidates are commercialized.
| 43 |
In addition, the manufacturing process for any products that we may develop is subject to FDA and foreign regulatory authority approval process, and we will need to contract with manufacturers who can meet all applicable FDA and foreign regulatory authority requirements on an ongoing basis. If we are unable to reliably produce products to specifications acceptable to the FDA or other regulatory authorities, we may not obtain or maintain the approvals we need to commercialize such products. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our subsidiaries and joint ventures will be able to manufacture the approved product to specifications acceptable to the FDA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidate, impair commercialization efforts, increase our cost of goods, and have an adverse effect on our business, financial condition, results of operations and growth prospects.
The manufacture of biological drug products is complex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of biologic products often encounter difficulties in production, particularly in scaling up or out, validating the production process, and assuring high reliability of the manufacturing process (including the absence of contamination). These problems include logistics and shipping, difficulties with production costs and yields, quality control, including stability of the product, product testing, operator error, availability of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Furthermore, if contaminants are discovered in our supply of our product candidates or in the manufacturing facilities, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. We cannot assure you that any stability failures or other issues relating to the manufacture of our product candidates will not occur in the future. Additionally, our manufacturers may experience manufacturing difficulties due to resource constraints or as a result of labor disputes or unstable political environments. If our manufacturers were to encounter any of these difficulties, or otherwise fail to comply with their contractual obligations, our ability to provide our product candidate to patients in clinical trials would be jeopardized. Any delay or interruption in the supply of clinical trial supplies could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to begin new clinical trials at additional expense or terminate clinical trials completely.
Cell-based therapies rely on the availability of reagents, specialized equipment, and other specialty materials, which may not be available to us on acceptable terms or at all. For some of these reagents, equipment, and materials, we rely or may rely on sole source vendors or a limited number of vendors, which could impair our ability to manufacture and supply our products.
Manufacturing our product candidates will require many reagents and viruses, which are substances used in our manufacturing processes to bring about chemical or biological reactions, and other specialty materials and equipment, some of which are manufactured or supplied by small companies with limited resources and experience to support commercial biologics production. We currently depend on a limited number of vendors for certain materials and equipment used in the manufacture of our product candidates. Some of these suppliers may not have the capacity to support commercial products manufactured under GMP by biopharmaceutical firms or may otherwise be ill-equipped to support our needs. We also do not have supply contracts with many of these suppliers and may not be able to obtain supply contracts with them on acceptable terms or at all. Accordingly, we may experience delays in receiving key materials and equipment to support clinical or commercial manufacturing.
For some of these reagents, viruses, equipment, and materials, we rely and may in the future rely on sole source vendors or a limited number of vendors. An inability to continue to source product from any of these suppliers, which could be due to regulatory actions or requirements affecting the supplier, adverse financial or other strategic developments experienced by a supplier, labor disputes or shortages, unexpected demands, or quality issues, could adversely affect our ability to satisfy demand for our product candidates, which could adversely and materially affect our product sales and operating results or our ability to conduct clinical trials, either of which could significantly harm our business.
As we continue to develop and scale our manufacturing process, we expect that we will need to obtain rights to and supplies of certain materials and equipment to be used as part of that process. We may not be able to obtain rights to such materials on commercially reasonable terms, or at all, and if we are unable to alter our process in a commercially viable manner to avoid the use of such materials or find a suitable substitute, it would have a material adverse effect on our business.
| 44 |
There can be no assurance that we will be able to further develop in-house sales and commercial distribution capabilities or establish or maintain relationships with third-party collaborators to successfully commercialize any product in the United States or overseas, and as a result, we may not be able to generate product revenue.
A variety of risks associated with operating our business internationally could materially adversely affect our business. We plan to seek regulatory approval of our product candidates outside of the United States and, accordingly, we expect that we, and any potential collaborators in those jurisdictions, will be subject to additional risks related to operating in foreign countries, including:
| ● | differing regulatory requirements in foreign countries, unexpected changes in tariffs, trade barriers, price and exchange controls, and other regulatory requirements; |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| ● | compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
| ● | foreign taxes, including withholding of payroll taxes; |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| ● | difficulties staffing and managing foreign operations; |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| ● | potential liability under the Foreign Corrupt Practices Act of 1977 or comparable foreign laws; |
| ● | challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| ● | business interruptions resulting from geo-political actions, including war and terrorism. |
These and other risks associated with our planned international operations may materially adversely affect our ability to attain or maintain profitable operations.
We face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively.
The biopharmaceutical industry, and the rapidly evolving market for developing cell-based therapies is characterized by intense competition and rapid innovation. Our competitors may be able to develop other compounds or drugs that are able to achieve similar or better results. Our potential competitors include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, universities, and other research institutions. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations as well as established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors, either alone or with collaborative partners, may succeed in developing, acquiring or licensing on an exclusive basis drug or biologic products that are more effective, safer, more easily commercialized, or less costly than our product candidates or may develop proprietary technologies or secure patent protection that we may need for the development of our technologies and products.
We are highly dependent on our key personnel, and if we are not successful in attracting, motivating and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract, motivate and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our senior management, particularly our Chief Executive Officer, Vered Caplan. The loss of the services of any of our executive officers, other key employees, and other scientific and medical advisors, and our inability to find suitable replacements, could result in delays in product development and harm our business. Competition for skilled personnel is intense and the turnover rate can be high, which may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all.
| 45 |
To induce valuable employees to remain at our company, in addition to salary and cash incentives, we have provided stock option grants that vest over time. The value to employees of these equity grants that vest over time may be significantly affected by movements in our stock price that are beyond our control and may at any time be insufficient to counteract more lucrative offers from other companies. Although we have employment agreements with our key employees, most these employment agreements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. We do not maintain “key man” insurance policies on the lives of all of these individuals or the lives of any of our other employees.
Risks Related to our Common Stock
We may fail to comply with the continued listing requirements of the Nasdaq Capital Market, such that our common stock may be delisted and the price of our common stock and our ability to access the capital markets could be negatively impacted.
Our common stock is listed for trading on the Nasdaq Capital Market. We must satisfy Nasdaq’s continued listing requirements, including, among other things, a minimum closing bid price requirement of $1.00 per share for 30 consecutive business days (the “Minimum Bid Price Requirement”). If a company trades for 30 consecutive business days below the $1.00 minimum closing bid price requirement, Nasdaq will send a deficiency notice to the company advising that it has been afforded a “compliance period” of 180 calendar days to regain compliance with the applicable requirements. We received such a notice on September 27, 2023 and thus risk delisting unless we are able to regain compliance in a timely fashion.
In accordance with Nasdaq Listing Rules, we were provided an initial period of 180 calendar days to regain compliance with the Minimum Bid Price Requirement. The initial compliance period ended on March 25, 2024 and we did not evidence compliance with the Minimum Bid Price Requirement during the initial compliance period. On March 26, 2024, we received a new letter from the Staff stating that it had determined to grant the Company an extension through September 23, 2024 to evidence compliance with the Minimum Bid Price Requirement. If at any time before September 23, 2024, the closing bid price of our common stock is at least $1.00 per share for a minimum of 10 consecutive business days, the Staff will provide written notification that we have achieved compliance with the Minimum Bid Price Requirement and the common stock will continue to be eligible for listing on the Nasdaq Capital Market. If, however, compliance with the Minimum Bid Price Requirement cannot be demonstrated by September 23, 2024, the Staff will provide written notification that our common stock will be subject to delisting. At that time, we may appeal the Staff’s delisting determination to a Panel. There can be no assurance that, if we do appeal the Staff’s delisting determination to the Panel, such appeal would be successful.
There can be no assurance that we will regain compliance with the Minimum Bid Price Requirement, that we will maintain compliant with other Nasdaq listing requirements or that we will be granted a second compliance period. A delisting of our common stock from Nasdaq could materially reduce the liquidity of our common stock and result in a corresponding material reduction in the price of our common stock. In addition, delisting could harm our ability to raise capital through alternative financing sources on terms acceptable to us, or at all, and may result in the potential loss of confidence by investors, employees and fewer business development opportunities.
If we issue additional shares in the future, it will result in the dilution of our existing stockholders.
Our articles of incorporation authorizes the issuance of up to 145,833,334 shares of our common stock with a par value of $0.0001 per share. Our Board of Directors may choose to issue some or all of such shares to acquire one or more companies or products and to fund our overhead and general operating requirements. The issuance of any such shares will reduce the book value per share and may contribute to a reduction in the market price of the outstanding shares of our common stock. If we issue any such additional shares, such issuance will reduce the proportionate ownership and voting power of all current stockholders. Further, such issuance may result in a change of control of our company.
| 46 |
Our stock price and trading volume may be volatile, which could result in losses for our stockholders.
The equity trading markets have recently experienced high volatility resulting in highly variable and unpredictable pricing of equity securities. If the turmoil in the equity trading markets continues, the market for our common stock could change in ways that may not be related to our business, our industry or our operating performance and financial condition. In addition, the trading volume in our common stock may fluctuate and cause significant price variations to occur. Some of the factors that could negatively affect our share price or result in fluctuations in the price or trading volume of our common stock include:
| ● | actual or anticipated quarterly variations in our operating results; |
| ● | changes in expectations as to our future financial performance or changes in financial estimates, if any; |
| ● | announcements relating to our business; |
| ● | conditions generally affecting the biotechnology industry; |
| ● | the success of our operating strategy; and |
| ● | the operating and stock performance of other comparable companies. |
Many of these factors are beyond our control, and we cannot predict their potential effects on the price of our common stock. In addition, the stock market is subject to extreme price and volume fluctuations. During the 52 weeks ended December 31, 2023, our stock price has fluctuated from a low of $1.23 to a high of $3.74. This volatility has had a significant effect on the market price of securities issued by many companies for reasons unrelated to their operating performance and could have the same effect on our common stock.
No assurance can be provided that a purchaser of our common stock will be able to resell their shares of common stock at or above the price that they acquired those shares. We can provide no assurances that the market price of common stock will increase or that the market price of common stock will not fluctuate or decline significantly.
We do not intend to pay dividends on any investment in the shares of stock of our company.
We have never paid any cash dividends, and currently do not intend to pay any dividends for the foreseeable future. The Board of Directors has not directed the payment of any dividends and does not anticipate paying dividends on the shares for the foreseeable future and intends to retain any future earnings to the extent necessary to develop and expand our business. Payment of cash dividends, if any, will depend, among other factors, on our earnings, capital requirements, and the general operating and financial condition, and will be subject to legal limitations on the payment of dividends out of paid-in capital. Because we do not intend to declare dividends, any gain on an investment in our company will need to come through an increase in the stock’s price. This may never happen, and investors may lose all of their investment in our company.
We have identified a material weakness in our internal control over financial reporting. Failure to achieve and maintain effective internal controls over financial reporting could adversely affect our ability to report our results of operations and financial condition accurately and in a timely manner, which could have an adverse impact on our business.
Ensuring that we have adequate internal financial and accounting controls and procedures in place to produce accurate financial statements on a timely basis has been, and will continue to be, costly and a time-consuming effort. In addition, the rapid changes in our operations and corporate structure have created a need for additional resources within the accounting and finance functions in order to produce timely financial information and to ensure the level of segregation of duties customary for a U.S. public company.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States (“GAAP”). Our management is also required, on a quarterly basis, to evaluate the effectiveness of our internal controls and to disclose any changes and material weakness identified. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement in our annual or interim consolidated financial statements might not be prevented or detected on a timely basis, as occurred with certain of our interim consolidated financial statements in 2023, which were then restated and corrected in amended Quarterly Reports on Form 10-Q prior to the filing of this Annual Report on Form 10-K. As described in Item 9A of this Annual Report on Form 10-K, there was a material weakness identified in our internal control over financial reporting.
We are working to remediate our material weakness as soon as practicable. Our remediation plan, which is continuing to be developed, can only be accomplished over time, and these initiatives may not accomplish their intended effects. Failure to maintain our internal control over financial reporting could adversely impact our ability to report our financial position and results from operations on a timely and accurate basis or result in misstatements. Likewise, if our financial statements are not filed on a timely basis, we could be subject to regulatory actions, legal proceedings or investigations by Nasdaq, the SEC or other regulatory authorities, which could result in a material adverse effect on our business and/or we may not be able to maintain compliance with certain of our agreements. Ineffective internal controls could also cause investors to lose confidence in our financial reporting, which could have a negative effect on our stock price, business strategies and ability to raise capital.
Even after the remediation of our material weakness, our management does not expect that our internal controls will ever prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. No evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, within the business will have been detected.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
Cybersecurity
We recognize the critical importance of maintaining the trust and confidence of customers, clients, patients, business partners and employees toward our business and are committed to protecting the confidentiality, integrity and availability of our business operations and systems. Our board of directors is actively involved in oversight of our risk management activities, and cybersecurity represents an important element of our overall approach to risk management. Our cybersecurity policies, standards, processes and practices are based on recognized frameworks established by our cybersecurity consultants and other applicable industry standards. In general, we seek to address cybersecurity risks through a comprehensive, cross-functional approach that is focused on preserving the confidentiality, security and availability of the information that we collect and store by identifying, preventing and mitigating cybersecurity threats and effectively responding to cybersecurity incidents when they occur.
| 47 |
Cybersecurity Risk Management and Strategy; Effect of Risk
We face risks related to cybersecurity such as unauthorized access, cybersecurity attacks and other security incidents, including as perpetrated by hackers and unintentional damage or disruption to hardware and software systems, loss of data, and misappropriation of confidential information. To identify and assess material risks from cybersecurity threats, we maintain a comprehensive cybersecurity program to ensure our systems are effective and prepared for information security risks, including regular oversight of our programs for security monitoring for internal and external threats to ensure the confidentiality and integrity of our information assets. We consider risks from cybersecurity threats alongside other company risks as part of our overall risk assessment process. We employ a range of tools and services, including regular network and endpoint monitoring, audits, vulnerability assessments, penetration testing, threat modeling and tabletop exercises to inform our risk identification and assessment. As discussed in more detail under “Cybersecurity Governance” below, our board of directors provides oversight of our cybersecurity risk management and strategy processes, which are led by our Chief Executive Officer.
We also identify our cybersecurity threat risks by comparing our processes to standards set by the Center for Internet Security (CIS) as well as by engaging experts to attempt to infiltrate our information systems. To provide for the availability of critical data and systems, maintain regulatory compliance, manage our material risks from cybersecurity threats, and protect against and respond to cybersecurity incidents, we undertake the following activities:
● monitor emerging data protection laws and implement changes to our processes that are designed to comply with such laws and implement latest Center for Internet Security benchmarks to comply with up-to-date requirements;
● through our policies, practices and contracts (as applicable), require employees, as well as third parties that provide services on our behalf, to treat confidential information and data with care, including using policies “right to know” and “right to access” with granular access to confidential information;
● employ technical safeguards that are designed to protect our information systems from cybersecurity threats, including firewalls, intrusion prevention and detection systems, anti-malware functionality and access controls, which are evaluated and improved through vulnerability assessments and cybersecurity threat intelligence, including active threat hunting and alerts monitoring by cyber security operators, threat analytics, endpoint management and application evaluations;
● provide regular, mandatory training for our employees and contractors regarding cybersecurity threats as a means to equip them with effective tools to address cybersecurity threats, and to communicate our evolving information security policies, standards, processes and practices;
● conduct regular phishing email simulations for all employees and contractors with access to our email systems to enhance awareness and responsiveness to possible threats, including built-in tools for phishing campaigns and attack simulators and usage of sandbox environment to evaluate threats;
● conduct annual cybersecurity management and incident training for employees involved in our systems and processes that handle sensitive data;
● run tabletop exercises to simulate a response to a cybersecurity incident and use the findings to improve our processes and technologies;
● leverage the NIST incident handling framework to help us identify, protect, detect, respond and recover when there is an actual or potential cybersecurity incident; and
● carry information security risk insurance that provides protection against the potential losses arising from a cybersecurity incident.
Our processes also address cybersecurity threat risks associated with our use of third-party service providers, including our suppliers and manufacturers or who have access to patient and employee data or our systems. In addition, cybersecurity considerations affect the selection and oversight of our third-party service providers. We perform diligence on third parties that have access to our systems, data or facilities that house such systems or data, and continually monitor cybersecurity threat risks identified through such diligence. Additionally, we generally require those third parties that could introduce significant cybersecurity risk to us to agree by contract to manage their cybersecurity risks in specified ways, and to agree to be subject to cybersecurity audits, which we conduct as appropriate.
We describe whether and how risks from identified cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations, or financial condition, under the heading “We are increasingly dependent on information technology and our systems and infrastructure face certain risks, including cybersecurity and data storage risks.” which disclosures are incorporated by reference herein.
| 48 |
In the last three fiscal years, we have not experienced any material cybersecurity incidents and the expenses we have incurred from cybersecurity incidents were immaterial. This includes penalties and settlements, of which there were none.
Cybersecurity Governance; Management
Cybersecurity is an important part of our risk management processes and an area of focus for our board of directors and management. In general, our board of directors oversees risk management activities designed and implemented by our management, and considers specific risks, including, for example, risks associated with our strategic plan, business operations, and capital structure. Our board of directors executes its oversight responsibility for risk management both directly and through delegating oversight of certain of these risks to its committees, and our board of directors has authorized our audit committee to oversee risks from cybersecurity threats.
Our board of directors receives an annual update, and more often if required, from management of our cybersecurity threat risk management and strategy processes covering topics such as data security posture, results from third-party assessments, progress towards pre-determined risk-mitigation-related goals, our incident response plan, and material cybersecurity threat risks or incidents and developments, as well as the steps management has taken to respond to such risks. In such sessions, our board of directors generally receives a report that details includes cybersecurity details and other materials discussing current and emerging material cybersecurity threat risks, and describing our ability to mitigate those risks, as well as recent developments, evolving standards, technological developments and information security considerations arising with respect to our peers and third parties, and discusses such matters with our Chief Executive Officer and also receive prompt and timely information regarding any cybersecurity incident that meets establishing reporting thresholds, as well as ongoing updates regarding any such incident until it has been addressed.
Members of board of directors are also encouraged to regularly engage in conversations with management on cybersecurity-related news events and discuss any updates to our cybersecurity risk management and strategy programs. Material cybersecurity threat risks are also considered during separate board meeting discussions of important matters like enterprise risk management, operational budgeting, business continuity planning, mergers and acquisitions, brand management, and other relevant matters.
Our cybersecurity risk management and strategy processes, which are discussed in greater detail above, are led by our Chief Executive Officer and our external cybersecurity consultants. These are informed about and monitor the prevention, mitigation, detection, and remediation of cybersecurity incidents through their management of, and participation in, the cybersecurity risk management and strategy processes described above, including the operation of our incident response plan. As discussed above, these consultants report to management about cybersecurity threat risks, among other cybersecurity related matters, at least annually.
| 49 |
ITEM 2. PROPERTIES
We do not own any real property. A description of the leased premises we utilize in several of our facilities is as follows:
Entity |
Property Description | ||
|
Orgenesis Inc. |
● |
Our principal office is located at 20271 Goldenrod Lane, Germantown, MD 20876. | |
| Orgenesis Maryland LLC | ● | FastForward laboratory and office located at 1812 Ashland Ave, Baltimore, Maryland 21205. | |
Orgenesis Korea Co. Ltd |
● | Operational production laboratory and office area located at Gwanggyo business centre 156, Gwanggyo-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do, Republic of Korea. | |
| Orgenesis Ltd. | ● | Laboratory and office located in Nes Ziona, Israel | |
| Koligo Therapeutics Inc. | ● | Production facility and development labs in New Albany, Indiana. | |
| Tissue Genesis International LLC | ● | Production facility and development labs in Leander, Texas | |
| Orgenesis Biotech Israel Ltd. | ● | Laboratories and offices located in the Bar Lev Industrial Park M.P. MISGAV, Israel. | |
| Mida Biotech BV | ● | Laboratories and offices located in Leiden, The Netherlands | |
Orgenesis Belgium and Orgenesis Services SRL
|
● | Laboratories and offices located near Namur, at Novalis Science Park, Belgium | |
| Theracell Laboratories | ● | Laboratory and offices located Koropi, Greece |
We believe that our facilities are generally in good condition and suitable to carry on our business. We also believe that, if required, suitable alternative or additional space will be available to us on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS
See note 22 of Item 8 of this Annual Report on Form 10-K for details of pending legal proceedings.
Except as described therein, we are not involved in any pending material legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Since March 13, 2018, our common stock has been listed for trading on the Nasdaq Capital Market (“Nasdaq CM”) under the symbol “ORGS.”
As of April 12, 2024, there were 346 holders of record of our common stock, and the last reported sale price of our common stock on the NasdaqCM on April 12, 2024 was $0.49. A significant number of shares of our common stock are held in either nominee name or street name brokerage accounts, and consequently, we are unable to determine the total number of beneficial owners of our common stock.
| 50 |
Dividend Policy
To date, we have paid no dividends on our common stock and do not expect to pay cash dividends in the foreseeable future. We plan to retain all earnings to provide funds for the operations of our company. In the future, our Board of Directors will decide whether to declare and pay dividends based upon our earnings, financial condition, capital requirements, and other factors that our Board of Directors may consider relevant. We are not under any contractual restriction as to present or future ability to pay dividends.
Unregistered Sales of Equity Securities
None.
Issuer Purchases of Equity Securities
None.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to provide information necessary to understand our audited consolidated financial statements for the years ended December 31, 2023 and December 31, 2022 and highlight certain other information which, in the opinion of management, will enhance a reader’s understanding of our financial condition, changes in financial condition and results of operations. In particular, the discussion is intended to provide an analysis of significant trends and material changes in our financial position and the operating results of our business during the year ended December 31, 2023, as compared to the year ended December 31, 2022.
This discussion should be read in conjunction with our consolidated financial statements for the years ended December 31, 2023 and December 31, 2022 and related notes included elsewhere in this Annual Report on Form 10-K. These historical financial statements may not be indicative of our future performance. This Management’s Discussion and Analysis of Financial Condition and Results of Operations contains numerous forward-looking statements, all of which are based on our current expectations and could be affected by the uncertainties and risks described throughout this filing, particularly in “Item 1A. Risk Factors.” (All monetary amounts are expressed in thousands of US dollars, unless stated otherwise)
Corporate Overview
We are a global biotech company working to unlock the potential of CGTs in an affordable and accessible format. CGTs can be centered on autologous (using the patient’s own cells) or allogenic (using master banked donor cells) and are part of a class of medicines referred to as advanced therapy medicinal products, or ATMPs. We are mostly focused on autologous therapies that can be manufactured under processes and systems that are developed for each therapy using a closed and automated approach that is validated for compliant production near the patient for treatment of the patient at the point of care, or POCare. This approach has the potential to overcome the limitations of traditional commercial manufacturing methods that do not translate well to commercial production of advanced therapies due to their cost prohibitive nature and complex logistics to deliver such treatments to patients (ultimately limiting the number of patients that can have access to, or can afford, these therapies).
| 51 |
To achieve these goals, we have developed a collaborative worldwide network of research institutes and hospitals who are engaged in the POCare model, or our POCare Network, and a pipeline of licensed POCare advanced therapies that can be processed and produced under such closed and automated processes and systems, or POCare Therapies. We are developing our pipeline of advanced therapies and with the goal of entering into out-licensing agreements for these therapies.
Following the Metalmark Investment in November 2022, we separated our operations into two operating segments namely 1) Octomera and 2) Therapies. Prior to that, we conducted all of our operations as one single segment. The Octomera operations includes mainly POCare Services, and include the results of the subsidiaries transferred to Octomera. The Therapies segment includes our therapeutic development operations. The segment information presented in note 5 of Item 8 of this Annual Report on Form 10-K reflects the results of the subsidiaries that were transferred to Octomera.
Therapies segment (POCare Therapies)
While the biotech industry struggles to determine the best way to lower cost of goods and enable CGTs to scale, the scientific community continues to advance and push the development of such therapies to new heights. Clinicians and researchers are excited by all the new tools (new generations of industrial viruses, big data analysis for genetic and molecular data) and technologies (CRISPR, mRNA, etc.) available, often at a low cost, to perform advanced research in small labs. Most new therapies arise from academic institutes or small spinouts from such institutes. Though such research efforts may manage to progress into a clinical stage, utilizing lab based or hospital-based production solutions they lack the resources to continue the development of such drugs to market approval.
Historically, drug/therapeutic development has required investments of hundreds of millions of dollars to be successful. One significant cause for the high cost is that each therapy often requires unique production facilities and technologies that must be subcontracted or built. Further the cost of production during the clinical stage is extremely expensive, and the cost of the clinical trial itself is very high. Given these financial restraints, researchers and institutes hope to out-license their therapeutic products to large biotech companies or spin-out new companies and raise large fundraising rounds. However, in many cases they lack the resources and the capability to de-risk their therapeutic candidates enough to be attractive for such fundings or partnership.
Our POCare Network is an alternative to the traditional pathway of drug development. We collaborate with academic institutions and entities that have been spun out from such institutions. We are in close contact with researchers who are experts in the field of the drug and also partners with leading hospitals and research institutes. Based on such collaborations, we enter into in-licensing agreements with relevant institutions for promising therapies with the aim of adapting them to a point-of-care setting through regional or strategic biological partnerships. Based on the results of the collaboration, we are then able to out-license our own therapeutic developments, as well as those therapies developed from in-licensing agreements to out-licensing partners at preferred geographical regions.
The ability to produce these products at low cost allows for an expedited development process, and the partnership with hospitals around the globe enables joint grants and lower cost of clinical development. The POCare Therapies division reviews many therapies available for out licensing and select the ones which they believe have the highest market potential, can benefit the most from a point of care approach and have the highest chance of clinical success. It assesses such issues by utilizing its global POCare Network and its internal knowhow accumulated over a decade of involvement in the field.
The goal of this in-licensing is to quickly adapt such therapies to a point-of-care approach through regional partnerships, and to out-license the products for market approval in preferred geographical regions. This approach lowers overall development cost, through minimizing pre-clinical development costs incurred by us, and through receiving of the additional funding from grants and/or payments by regional partners.
Our therapies development subsidiaries are:
| ● | Koligo Therapeutics, Inc., a Kentucky corporation, which is a regenerative medicine company, specializing in developing personalized cell therapies. It is currently focused on commercializing its metabolic pipeline via the POCare Network throughout the United States and in international markets. |
| 52 |
| ● | Orgenesis CA, Inc. a Delaware corporation, which is currently focused on development of technologies and therapies in California. | |
| ● | Orgenesis Belgium SRL which is currently focused on product development. Since its incorporation, the subsidiary has been awarded grants in excess of 18,000 Euro from the Walloon region for several projects (DGO6 grants). | |
| ● | Orgenesis Switzerland Sarl, which is currently focused on providing group management services. | |
| ● | MIDA Biotech BV, which is currently focused on research and development activities, was granted a 4,000 Euro grant under the European Innovation Council Pathfinder Challenge Program which supports cutting-edge science and technology. The grant is for technologies enabling the production of autologous induced pluripotent stem cells (iPSCs) using microfluidic technologies and artificial intelligence (AI). | |
| ● | Orgenesis Italy SRL which is currently focused on R&D activities. | |
| ● | Orgenesis Ltd., an Israeli subsidiary which is focused on R&D and a provider of R&D management services for out licenced products. Israel is a hub for biotech research and pioneers in this field. | |
| ● | Orgenesis Austria GmbH, an Austrian subsidiary, which is focused on R&D activities. |
Octomera segment (mainly POCare Services)
Octomera LLC (“Octomera” or “Morgenesis”) is responsible for most of our POCare services platform. The POCare Services platform is utilized by parties such as biotech companies and hospitals for the supply of their products. Octomera’s services include adapting the process to the platform and supplying the products, or POCare Services. These are services for third party companies and for CGTs that are not necessarily based on our POCare Therapies. POCare services that we and our affiliated entities perform include:
| ● | Process development of therapies, process adaptation, and optimization inside the OMPULs, or “OMPULization”; |
| ● | Adaptation of automation and closed systems to serviced therapies; |
| ● | Incorporation of the serviced therapies compliant with GMP in the OMPULs that we design and built; |
| ● | Tech transfers and training of local teams for the serviced therapies at the POCare Centers; |
| ● | Processing and supply of the therapies and required supplies under GMP conditions within our POCare Network, including required quality control testing; and |
| ● | Contract Research Organization services for clinical trials. |
The POCare Services are performed in decentralized hubs that provide harmonized and standardized services to customers, or POCare Centers. We are working to expand the number and scope of our POCare Centers with the intention of providing an efficient and scalable pathway for CGT therapies to reach patients rapidly at lowered costs. Our POCare Services are designed to allow rapid capacity expansion while integrating new technologies to bring together patients, doctors and industry partners with a goal of achieving standardized, regulated clinical development and production of therapies.
POCare Services Operations Subsidiaries
We conduct our core POCare operations through our wholly-owned subsidiary Octomera which was a consolidated subsidiary of the Company until June 30, 2023 and which became a consolidated subsidiary again effective January 29, 2024. Octomera’s subsidiaries which are all wholly owned except as otherwise stated below (collectively, the “Subsidiaries”) include:
| ● | Orgenesis Maryland LLC, which is the center of POCare Services activity in North America and is currently focused on setting up and providing POCare Services and cell-processing services to the POCare Network. |
| ● | Tissue Genesis International LLC, a Texas limited liability company currently focused on development of our technologies and therapies. |
| ● | Orgenesis Services SRL, which is currently focused on expanding our POCare Network in Belgium. |
| ● | Orgenesis Germany GmbH, which is currently focused on providing CRO services to the POCare Network. |
| ● | Orgenesis Korea Co. Ltd., which is a provider of cell-processing and pre-clinical services in Korea. Octomera owns 94.12% of the Korean Subsidiary. |
| 53 |
| ● | Orgenesis Biotech Israel Ltd., which is a provider of process development and cell-processing services in Israel. | |
| ● | Orgenesis Australia PTY LTD, which was transferred to Octomera in January 2023 and is currently focused on the development of our POC Network in Australia. | |
| ● | Theracell Laboratories IKE (“Theracell Labs”), a Greek company currently focused on expanding our POCare Network. | |
| ● | ORGS POC CA Inc, a Californian entity, is currently focussed on expanding our POCare Network in California. | |
| ● | Octo Services LLC, a Delaware entity focussed on expanding our POCare network. |
During 2023, we and MM invested $660 and $6,500 respectively into Octomera in exchange for Octomera preferred shares.
During 2023, we and MM loaned $276 and $2,475 respectively to Octomera’s subsidiary, Orgenesis Maryland LLC. The loans bear 10% annual interest and were originally scheduled to be repaid during 2024. Pursuant to an extension agreement signed between us and MM on January 28, 2024, the maturity date of the MM loans was extended to January 28, 2034.
.
Significant Developments During Fiscal 2023
Financing Activities
Equity
On February 23, 2023, we entered into a securities purchase agreement with certain institutional and accredited investors (the “Purchaser”) relating to the issuance and sale of 1,947,368 shares of our common stock, and warrants to purchase up to 973,684 shares of common stock (the “Warrants”) at a purchase price of $1.90 per share of common stock and accompanying Warrants in a registered direct offering (the “February 2023 Offering”). The February 2023 Offering closed on February 27, 2023.
The Warrants have an exercise price of $1.90 per share, were exercisable immediately and will expire five years following the date of issuance. The Warrants had an alternate cashless exercise option (beginning on or after the earlier of (a) the thirty-day anniversary of the date of the Purchase Agreement and (b) the date on which the aggregate composite trading volume of Common Stock following the public announcement of the pricing terms exceeds 13,600,000 shares), to receive an aggregate number of shares equal to the product of (x) the aggregate number of shares of Common Stock that would be issuable upon a cash exercise and (y) 1.0. The aggregate gross proceeds to us from the February 2023 Offering were $3,700, before deducting placement agent cash fees equal to 7.0% of the gross proceeds received and other expenses from the Offering payable by us.
As of September 30, 2023, all of the Warrants were exercised using the alternate cashless exercise option described above.
On August 31, 2023, we entered into a Securities Purchase Agreement with a certain accredited investor, pursuant to which we agreed to issue and sell, in a private placement (the “August 2023 Offering”), 2,000,000 shares of our common stock at a purchase price of $0.50 per share. We received proceeds of $1,000. The August 2023 Offering closed on August 31, 2023.
On November 8, 2023, we entered into a Securities Purchase Agreement with an institutional investor named therein, pursuant to which we agreed to issue and sell, in a registered direct offering directly to the investor (the “November 2023 Offering”), (i) 1,410,256 shares of our common stock and (ii) warrants exercisable for 1,410,256 shares of common stock. The combined offering price for each share and accompanying warrant was $0.78. The warrants were exercisable immediately following the date of issuance and may be exercised for a period of five years from the initial exercisability date at an exercise price of $0.78 per share. We received proceeds of $1,100. The November 2023 Offering closed on November 9, 2023.
| 54 |
Loans
On July 25, 2023, the Israeli subsidiary received a loan from an offshore investor in the amount of $175. The loan bears 8% annual interest and is repayable on January 1, 2024. During 2024, the maturity date of the loan was extended by a year.
On August 15, 2023, the Company received a loan from an investor in the amount of $250. The loan bears 8% annual interest and is repayable on January 1, 2024.
During October and November 2023, the Israeli subsidiary received loans in the amount of $150. The loans are interest free and repayable between November 30, 2023 and January 1, 2024. During 2024, the maturity date of the loans was extended by a year.
During October through December 2023, Orgenesis Maryland, LLC received $2,726 of loans which bear 10% annual interest and were originally scheduled to be repaid during 2024. Pursuant to an extension agreement signed between us and MM on January 28, 2024, the maturity dates of the MM loans were extended to January 28, 2034
License Agreements
In addition, during 2023, we continued the development of license agreements previously entered into, as described more fully in notes 12 and 13 to our consolidated financial statements included in Item 8 of this Annual Report on Form 10-K.
Results of Operations
Comparison of the Year Ended December 31, 2023 to the Year Ended December 31, 2022.
Our financial results for the year ended December 31, 2023 are summarized as follows in comparison to the year ended December 31, 2022:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Revenues | $ | 530 | $ | 34,741 | ||||
| Revenues from related party | - | 1,284 | ||||||
| Total revenues | $ | 530 | $ | 36,025 | ||||
| Cost of sales | 6,255 | 5,133 | ||||||
| Gross profit | $ | (5,725 | ) | $ | 30,892 | |||
| Cost of development services and research and development expenses | 10,623 | 21,933 | ||||||
| Amortization of intangible assets | 721 | 911 | ||||||
| Selling, general and administrative expenses included credit losses of $24,367 for the year ended December 31, 2023 | 35,134 | 15,589 | ||||||
| Share in loss of associated company | 734 | 1,508 | ||||||
| Impairment of investment | 699 | - | ||||||
| Impairment expenses | - | 1,061 | ||||||
| Operating loss | $ | 53,636 | $ | 10,110 | ||||
Loss from deconsolidation of Octomera (see Note 3) | 5,343 | - | ||||||
| Other income | (4 | ) | (173 | ) | ||||
| Credit loss on convertible loan receivable | 2,688 | - | ||||||
| Loss from extinguishment in connection with convertible loan (see note 7 a of Item 8) | 283 | 52 | ||||||
| Financial expense, net | 2,499 | 1,971 | ||||||
| Loss before income taxes | $ | 64,445 | $ | 11,960 | ||||
| Tax expense | 473 | 209 | ||||||
| Net loss | $ | 64,918 | $ | 12,169 | ||||
| 55 |
Revenues
The following table shows our revenues by major revenue streams:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Revenue stream: | ||||||||
| POCare development services | $ | - | $ | 14,894 | ||||
| Cell process development services and hospital services | 515 | 11,212 | ||||||
| POCare cell processing | - | 9,919 | ||||||
| License fees | 15 | - | ||||||
| Total | $ | 530 | $ | 36,025 | ||||
Our revenues for the year ended December 31, 2023 were $530, as compared to $36,025 for the year ended December 31, 2022, representing a decrease of 99%. This was attributable failure of customers to timely pay for services received and to the deconsolidation of Octomera at June 30, 2023. Almost all of our potential revenues was from Octomera. During the year ended December 31, 2023, the Octomera segment completed revenue performance obligations but did not recognize revenue for such completed performance obligations because certain revenue recognition conditions under ASC 606 were not satisfied.
A breakdown of the revenues per customer that constituted at least 10% of revenues is as follows:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Revenue earned: | ||||||||
| Customer A (United States) | $ | 280 | $ | - | ||||
| Customer B (United States) | 90 | - | ||||||
| Customer C (United States) | 130 | - | ||||||
| Customer D (Greece) | - | 8,936 | ||||||
| Customer E (United States) | - | 8,316 | ||||||
| Customer F (United Arab Emirates) | - | 5,271 | ||||||
| Customer G (Korea) | - | 3,873 | ||||||
Expenses
Cost of Revenues
| Year Ended | ||||||||
| December 31, 2023 | December 31, 2022 | |||||||
| Salaries and related expenses | $ | 2,387 | $ | 1,689 | ||||
| Stock-based compensation | 4 | 36 | ||||||
| Professional fees and consulting services | 1,917 | 968 | ||||||
| Raw materials | 731 | 1,173 | ||||||
| Depreciation expenses, net | 481 | 354 | ||||||
| Other expenses | 735 | 913 | ||||||
| Total | $ | 6,255 | $ | 5,133 | ||||
| 56 |
Cost of revenues for the year ended December 31, 2023 were $6,255, as compared to $5,133 for the year ended December 31, 2022, representing an increase of 22%. This was due to increased costs including additional salaries, professional fees, and depreciation expenses incurred as a result of increased process development and cell processing revenues mainly in the Octomera segment, which were incurred until the date of deconsolidation.
Cost of development services and research and development expenses
| Year Ended | ||||||||
| December 31, 2023 | December 31, 2022 | |||||||
| Salaries and related expenses | $ | 4,800 | $ | 9,517 | ||||
| Stock-based compensation | 210 | 580 | ||||||
| Subcontracting, professional and consulting services | 3,662 | 4,687 | ||||||
| Lab expenses | 377 | 1,512 | ||||||
| Depreciation expenses, net | 312 | 663 | ||||||
| Other research and development expenses | 1,542 | 5,097 | ||||||
| Less – grant | (280 | ) | (123 | ) | ||||
| Total | $ | 10,623 | $ | 21,933 | ||||
Cost of development services and research and development for the year ended December 31, 2023 were $10,623, as compared to $21,933 for the year ended December 31, 2022, representing a decrease of 52%. The decrease was mainly attributable to the deconsolidation of the Octomera segment at June 30, 2023, and our decision to reduce investing in subcontracting, professional and consulting service fees and other research and development expenses this year.
Selling, General and Administrative Expenses
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Salaries and related expenses | $ | 2,825 | $ | 4,008 | ||||
| Stock-based compensation | 249 | 362 | ||||||
| Accounting and legal fees | 3,355 | 5,527 | ||||||
| Professional fees | 1,891 | 3,080 | ||||||
| Rent and related expenses | 161 | 199 | ||||||
| Business development | 464 | 474 | ||||||
| Depreciation expenses, net | 46 | 50 | ||||||
| Other general and administrative expenses | 26,143 | 1,889 | ||||||
| Total | $ | 35,134 | $ | 15,589 | ||||
Selling, general and administrative expenses for the year ended December 31, 2023 were $35,134, as compared to $15,589 for the year ended December 31, 2022, representing an increase of 125%.
The increase was mainly due to increased expenses in the Octomera segment, where selling, general and administrative expense (excluding depreciation) for the year ended December 31, 2023 were $37,878 as compared to $7,762 for the year ended December 31, 2022, representing an increase of 388%. The increase was mainly as a result of an increase of credit losses in the amount of $29,774 included in other general and administrative expenses.
| 57 |
Share in Net Loss of Associated Company
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Share of Net Loss of Associated Company | $ | 734 | $ | 1,508 | ||||
| Total | $ | 734 | $ | 1,508 | ||||
Share in net loss of associated company for the year ended December 31, 2023 was $ 734, as compared to $ 1,508 for the year ended December 31, 2022, representing an decrease of 51%. The decrease in Share in net loss of associated company in the year ended December 31, 2023 compared to the year ended December 31, 2022 is primarily attributable to a decline in Octomera revenues, and credit losses in the year ended December 31, 2023 resulting from higher than previously expected credit losses related to a group of customers that are significantly overdue in Octomera.
Impairment Expenses
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Impairment expenses | $ | 699 | $ | 1,061 | ||||
Impairment expenses for the year ended December 31, 2023 were $699, as compared to $1,061 for the year ended December 31, 2022. These were attributable to the write-off of assets purchased in previous years.
Credit Loss on Convertible loan receivable
| Years Ended December 31 | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Credit loss on convertible loan receivable | $ | 2,688 | $ | - | ||||
The credit loss for the year ended December 31, 2023 was $2,688 compared to $0 for the year ended December 31, 2022. This was attributable to a provision created for a credit loss on a loan.
Financial Expenses, net
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Interest expense on convertible loans and loans | 2,167 | 1,824 | ||||||
| Foreign exchange loss, net | 325 | 145 | ||||||
| Other income | 7 | 2 | ||||||
| Total | $ | 2,499 | $ | 1,971 | ||||
Financial expenses, net for the year ended December 31, 2023 were $2,499, as compared to $1,971 for the year ended December 31, 2022, representing an increase of 27%. The increase was mainly attributable to increased interest and related expenses on new and existing convertible loans.
Tax expense
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Tax expense | $ | 473 | $ | 209 | ||||
| Total | $ | 473 | $ | 209 | ||||
Tax income, net for the year ended December 31, 2023 were $473, as compared to $209 for the year ended December 31, 2022, representing an increase of 126%. The increase is mainly attributable to increased tax liabilities in the U.S. Effective for years beginning after December 31, 2021, Internal Revenue Code Section 174 changed the tax treatment of research and experimentation (R&E) expenditures. While companies have historically deducted such costs for federal income tax purposes, these new rules require capitalization and prescribe cost recovery over a period of five years for research and development paid or incurred in the United States and 15 years for R&E paid or incurred outside of the United States.
| 58 |
Working Capital
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Current assets | $ | 4,076 | $ | 46,318 | ||||
| Current liabilities | $ | 16,407 | $ | 15,910 | ||||
| Working capital | $ | (12,331 | ) | $ | 30,408 | |||
Current assets decreased by $42,242 between December 31, 2022 and December 31, 2023, due mainly to the deconsolidation of Octomera. The majority of cash and cash equivalents, restricted cash, and accounts receivable at December 31, 2022 were part of Octomera. Receivables from related parties in the amount of $458 are receivables from Octomera subsidiaries, that were consolidated at December 31, 2022 but not at December 31, 2023. Octomera became a consolidated subsidiary again effective January 29, 2024. In addition we provided a credit loss for a convertible loan in the amount of $2,688 that was not yet repaid to us.
Current liabilities increased by $497 between December 31, 2022 and December 31, 2023, primarily due to an increase in accounts payable in the amount of $2,022 as a result of a shortage of funds; an increase in tax payable in the amount of $451 as a result of increased tax in the US; and grants payable in the amount of $602 as a result of a grant received. These increases were offset by a decline in short-term and current maturities of convertible loans in the amount of $1,834 due to the extension of the maturity date of convertible loans to 2026 (see Note 10)
Liquidity and Capital Resources
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Net loss | $ | (64,918 | ) | $ | (12,169 | ) | ||
| Net cash used in operating activities | (14,837 | ) | (24,924 | ) | ||||
| Net cash used in investing activities | (3,707 | ) | (14,133 | ) | ||||
| Net cash provided by financing activities | 13,618 | 39,578 | ||||||
| Net change in cash and cash equivalents and restricted cash | $ | (4,926 | ) | $ | 521 | |||
During year ended December 31, 2023, we funded our operations from operations as well as from proceeds raised from equity and debt offerings.
Net cash used in operating activities for the year ended December 31, 2023 was approximately $14,837, as compared to net cash used in operating activities of approximately $24,924 for the year ended December 31, 2022. The decline was mainly as a result of
a loss of $64,918 for the year ended December 31, 2023 compared to a loss of $12,169 for the year ended December 31, 2022, which is mainly related a decline in activity in Octomera.
Net cash used in investing activities for the year ended December 31, 2023 was approximately $3,707, as compared to net cash used in investing activities of approximately $14,133 for the year ended December 31, 2022. The decrease was mainly due to loans granted to associated entities last year not granted this year, reduced investments in OMPULs, and the deconsolidation of Octomera.
| 59 |
Net cash provided by financing activities for the year ended December 31, 2023 was approximately $13,618, as compared to net cash provided by financing activities of approximately $39,578 for the year ended December 31, 2022. During the year ended December 31, 2023 we raised equity investments in the net amount of 5,732, raised proceeds from loans in the amount of 635, raised proceeds from MM in the amount of $5,000 and repaid convertible loans in the amount of $3,000.
Liquidity and Capital Resources Outlook
As of December 31, 2023, we had an accumulated deficit of $176,622 and for the year ended December 31, 2023 incurred negative operating cashflows of $14,837. Our activities have been funded by generating revenue, through offerings of our securities, and through proceeds from loans. There is no assurance that our business will generate sustainable positive cash flows to fund our business.
We will need to use mitigating actions such as to seek additional financing, refinance or amend the terms of existing loans or postpone expenses that are not based on firm commitments. In order to fund our operations until such time that we can generate sustainable positive cash flows, we will need to raise additional funds. For the year ended December 31, 2023 and as of the date of this report, we assessed our financial condition and concluded that based on our current and projected cash resources and commitments, as well as other factors mentioned above, there is a substantial doubt about our ability to continue as a going concern. We are planning to raise additional capital to continue our operations and to repay our outstanding loans when they become due, as well as to explore additional avenues to increase revenues and reduce expenditures. There can be no assurance that we will be able to raise additional capital on acceptable terms, or at all.
Our common stock is listed for trading on the Nasdaq Capital Market. We must satisfy Nasdaq’s continued listing requirements, including, among other things, a minimum closing bid price requirement of $1.00 per share for 30 consecutive business days (the “Minimum Bid Price Requirement”). If a company trades for 30 consecutive business days below the $1.00 minimum closing bid price requirement, Nasdaq will send a deficiency notice to the company advising that it has been afforded a “compliance period” of 180 calendar days to regain compliance with the applicable requirements. We received such a notice on September 27, 2023 and thus risk delisting unless we are able to regain compliance in a timely fashion.
During January 2024, we received extensions on our loan payments as follows:
| ● | Israeli subsidiary loan from an offshore investor in the amount of $175 originally repayable on January 1, 2024: The maturity date of the loan was extended by a year. | |
| ● | Israeli subsidiary loans in the amount of $150 repayable between November 30, 2023 and January 1, 2024. During 2024, the maturity date of the loans was extended by a year. | |
| ● | During October through December 2023, Orgenesis Maryland, LLC received $2,726 of loans which were originally scheduled to be repaid during 2024. Pursuant to an extension agreement signed between us and MM on January 28, 2024, the maturity dates of the MM loans were extended to January 28, 2034. |
On March 3, 2024, we entered into a Securities Purchase Agreement with certain accredited investors, pursuant to which we agreed to issue and sell, in a private placement, 2,272,719 shares of our common stock, par value $0.0001 per share, at a purchase price of $1.03 per share and warrants to purchase up to 2,272,719 shares of Common Stock at an exercise price of $1.50 per share and warrants to purchase up to 2,272,719 shares of Common Stock at an exercise price of $2.00 per share (collectively, the “Warrants”). We received gross proceeds of approximately $2.3 million before deducting related offering expenses.
On April 5, 2024, we entered into an Asset Purchase and Strategic Collaboration Agreement (the “Purchase Agreement”) with Griffin Fund 3 BIDCO, Inc., (“Germfree”), for the sale by us of five OMPULs to Germfree, which will be incorporated into Germfree’s lease fleet and leased back to us or third-party lessees designated by Orgenesis. Pursuant to the Purchase Agreement, and upon the terms and subject to the conditions set forth therein, in consideration for the purchase of the OMPULs, the Orgenesis Quality Management Systems Framework (“OQMSF”) and related intellectual property rights, Germfree will pay us an aggregate purchase price of $8,340 subject to any final adjustment through the verification mechanism as set forth in the Purchase Agreement.
Pursuant to the Agreement, Germfree paid us $750 on February 27, 2024 and $5,538 during April 2024.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to stockholders.
Critical Accounting Policies and Estimates
Our significant accounting policies are more fully described in the notes to our financial statements included in this Annual Report on Form 10-K for the year ended December 31, 2023. We believe that the accounting policies below are critical for one to fully understand and evaluate our financial condition and results of operations.
Income Taxes
Deferred income tax assets and liabilities are computed for differences between the financial statement and tax basis of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable or refundable for the period plus or minus the change during the period in deferred tax assets and liabilities.
In addition, our management performs an evaluation of all uncertain income tax positions taken or expected to be taken in the course of preparing our income tax returns to determine whether the income tax positions meet a “more likely than not” standard of being sustained under examination by the applicable taxing authorities. This evaluation is required to be performed for all open tax years, as defined by the various statutes of limitations, for federal and state purposes.
| 60 |
Revenue from Contracts with Customers
Our agreements are primarily service contracts that range in duration. We recognize revenue when control of these services is transferred to the customer for an amount, referred to as the transaction price, which reflects the consideration to which we are expected to be entitled in exchange for those goods or services.
A contract with a customer exists only when:
| ● | the parties to the contract have approved it and are committed to perform their respective obligations; |
| ● | we can identify each party’s rights regarding the distinct goods or services to be transferred (“performance obligations”); |
| ● | we can determine the transaction price for the goods or services to be transferred; and |
| ● | the contract has commercial substance, and it is probable that we will collect the consideration to which it will be entitled in exchange for the goods or services that will be transferred to the customer. |
Nature of Revenue Streams
We have three main revenue streams, which are POCare development services, cell process development services, including hospital supplies, and POCare cell processing.
POCare Development Services
Revenue recognized under contracts for POCare development services may, in some contracts, represent multiple performance obligations (where promises to the customers are distinct) in circumstances in which the work packages are not interrelated or the customer is able to complete the services performed.
For arrangements that include multiple performance obligations, the transaction price is allocated to the identified performance obligations based on their relative standalone selling prices.
We recognize revenue when, or as, it satisfies a performance obligation. At contract inception, we determine whether the services are transferred over time or at a point in time. Performance obligations that have no alternative use and that we have the right to payment for performance completed to date, at all times during the contract term, are recognized over time. All other Performance obligations are recognized as revenues by us at point of time (upon completion).
Significant Judgement and Estimates
Significant judgment is required to identifying the distinct performance obligations and estimating the standalone selling price of each distinct performance obligation and identifying which performance obligations create assets with alternative use to us, which results in revenue recognized upon completion, and which performance obligations are transferred to the customer over time.
Cell Process Development Services
Revenue recognized under contracts for cell process development services may, in some contracts, represent multiple performance obligations (where promises to the customers are distinct) in circumstances in which the work packages and milestones are not interrelated or the customer is able to complete the services performed independently or by using our competitors. In other contracts when the above circumstances are not met, the promises are not considered distinct, and the contract represents one performance obligation. All performance obligations are satisfied over time, as there is no alternative use to the services it performs, since, in nature, those services are unique to the customer, which retain the ownership of the intellectual property created through the process.
For arrangements that include multiple performance obligations, the transaction price is allocated to the identified performance obligations based on their relative standalone selling prices. For these contracts, the standalone selling prices are based on our normal pricing practices when sold separately with consideration of market conditions and other factors, including customer demographics and geographic location.
| 61 |
We measure the revenue to be recognized over time on a contract-by-contract basis, determining the use of either a cost-based input method or output method, depending on whichever best depicts the transfer of control over the life of the performance obligation.
Included in Cell Process Development Services is hospital supplies revenue which is derived principally from the sale or lease of products and the performance of services to hospitals or other medical providers. Revenue is earned and recognized when product and services are received by the customer.
Revenue from POCare Cell processing
Revenues from POCare Cell processing represent performance obligations which are recognized either over, or at a point of time. The progress towards completion will continue to be measured on an output measure based on direct measurement of the value transferred to the customer (units produced).
Concentration of Credit Risk
Financial instruments that potentially subject us to concentration of credit risk consist of principally cash and cash equivalents, bank deposits and certain receivables. We held these instruments with highly rated financial institutions, and we have not experienced any significant credit losses in these accounts and does not believe the we are exposed to any significant credit risk on these instruments, except for accounts receivable. We perform ongoing credit evaluations of its customers for the purpose of determining the appropriate allowance for doubtful accounts.
Our accounts receivable accounting policy until December 31, 2022, prior to the adoption of the new Current Expected Credit Losses (“CECL”) standard, created bad debts when objective evidence existed of inability to collect all sums owed it under the original terms of the debit balances. Material customer difficulties, the probability of their going bankrupt or undergoing economic reorganization and insolvency, material delays in payments and other objective considerations by management that indicate expected risk of payment were all considered indicative of reduced debtor balance value. Effective January 1, 2023, we adopted the new CECL standard.
We maintain the allowance for estimated losses resulting from the inability of our customers to make required payments. We consider historical collection experience for each of its customers and when revenue and accounts receivable are recorded. We also recognize estimated expected credit losses over the life of the accounts receivables. The estimate of expected credit losses considers not only historical information, but also current and future economic conditions and events.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information called for by Item 8 is included following the “Index to Financial Statements” on page F-1 contained in this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Securities Exchange Act of 1934, as amended (the “Exchange Act”)) that are designed to ensure that information required to be disclosed in our reports under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosures. In designing disclosure controls and procedures, our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible disclosure controls and procedures. The design of any disclosure controls and procedures also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Any controls and procedures, no matter how well designed and operated, can provide only reasonable, not absolute, assurance of achieving the desired control objectives.
Our management, with the participation of our principal executive officer and principal financial officer, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this Annual Report. Based upon that evaluation and subject to the foregoing, our principal executive officer and principal financial officer concluded that, as of the end of the period covered by this Annual Report, the design and operation of our disclosure controls and procedures were not effective due to the material weakness in our internal control over financial reporting described below.
Management’s Report on Internal Control over Financial Reporting
Our management, under the supervision of the Chief Executive Officer and Chief Financial Officer, is responsible for establishing and maintaining adequate internal control over financial reporting for our company. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP and includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of our company are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our company’s assets that could have a material effect on the financial statements.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this evaluation, our management used the criteria set forth in the Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
| 62 |
Management has determined that we had the following material weakness in our internal control over financial reporting as of December 31, 2023:
We did not perform appropriate analyses related to our internal control over financial reporting in the accounting for whether it is probable we will collect substantially all the consideration to which we are entitled for revenue services provided, as well as our estimated credit losses during 2023. As a result, we identified a deficiency in the operating effectiveness of our internal control over financial reporting related to our accounting for revenues, credit losses and the related impacts related to that, which resulted in the restatement of our unaudited condensed consolidated financial statements for the three months ended March 31, 2023, the three and six months ended June 30, 2023 and the three and nine months ended September 30, 2023.
As of December 31, 2023, such weakness has not been remediated. Management’s plans for remediation, which will occur during 2024, include a thorough credit assessment of all new customers, analysis of payment history for existing customers as well as an analysis on expected credit losses by customer.
This Annual Report on Form 10-K does not include an attestation report of our registered public accounting firm on internal control over financial reporting because we are a smaller reporting company and non-accelerated filer.
Changes in Internal Control Over Financial Reporting
Except as described above, there were no changes in our internal control over financial reporting that occurred during the fourth quarter of the year ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
None.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The following table sets forth certain information regarding our each of our current Directors and Executive Officers as of April 15, 2024.
| Name | Age | Position | ||
| Vered Caplan | 55 | Chief Executive Officer and Chairperson of the Board of Directors | ||
| Victor Miller | 54 | Chief Financial Officer, Secretary and Treasurer | ||
| David Sidransky (1) (2) (4) | 63 | Director | ||
| Guy Yachin (1) (2) (3) (4) | 56 | Director | ||
| Yaron Adler (2) (3) | 53 | Director | ||
| Ashish Nanda (3) | 58 | Director | ||
Mario Philips (1) |
54 |
Director |
| (1) | A member on the audit committee. |
| (2) | A member on the compensation committee. |
| (3) | A member on the nominating and corporate governance committee. |
| (4) | A member of the research and development committee. |
| 63 |
Our Executive Officers
Vered Caplan – Chief Executive Officer and Chairperson of the Board of Directors
Ms. Caplan has served as our CEO and Chairperson of the Board of Directors since August 14, 2014, prior to which she served as Interim President and CEO commencing on December 23, 2013. She joined our Board of Directors in February 2012. She has 26 years of industry experience, previously holding positions as CEO of Kamedis Ltd. from 2009 to 2014, CEO of GammaCan International Inc. from 2004 to 2007. She also served as a director of the following companies: Opticul Ltd., Inmotion Ltd., Nehora Photonics Ltd., Ocure Ltd., Eve Medical Ltd., and Biotech Investment Corp. Ms. Caplan holds a M.Sc. in biomedical engineering from Tel Aviv University specializing in signal processing; management for engineers from Tel Aviv University specializing in business development; and a B.Sc. in mechanical engineering from the Technion– Israel Institute of Technology specialized in software and cad systems.
Victor Miller – Chief Financial Officer, Secretary and Treasurer
On December 28, 2023, we appointed Victor Miller as our Chief Financial Officer, Secretary and Treasurer effective January 2, 2024. Mr. Miller previously served as Chief Financial Officer and Secretary at Hycor Biomedical LLC. (“HYCOR”), an in vitro allergy diagnostic company, from 2014 to May 2023. Mr. Miller has over 30 years of healthcare and finance industry experience, including 14 years leading finance functions at early-stage life science companies. From 2009 to 2014, prior to joining HYCOR, Mr. Miller led the Finance function at Neos Therapeutics, an early-stage specialty pharmaceutical company. From 2000 to 2009, Mr. Miller developed broad healthcare functional experience with roles in Corporate Development, Business Development, Marketing and Strategy while working for Baxter Healthcare and Giles & Associates. From 1996 to 2000, Mr. Miller gained significant transaction experience as an investment banker in London for Bankers Trust and Merrill Lynch. Mr. Miller holds a Bachelor of Science in Economics from The Wharton School, University of Pennsylvania and is a Chartered Financial Analyst.
Our Directors
Dr. David Sidransky – Director
Dr. Sidransky has served as a director since his appointment on July 18, 2013. Dr. Sidransky is a renowned oncologist and research scientist named and profiled by TIME magazine in 2001 as one of the top physicians and scientists in America, recognized for his work with early detection of cancer. Since 1994, Dr. Sidransky has been the Director of the Head and Neck Cancer Research Division at Johns Hopkins University School of Medicine’s Department of Otolaryngology and Professor of Oncology, Cellular & Molecular Medicine, Urology, Genetics, and Pathology at the John Hopkins University School of Medicine. Dr. Sidransky is one of the most highly cited researchers in clinical and medical journals in the world in the field of oncology during the past decade, with over 600 peer reviewed publications. Dr. Sidransky is a founder of a number of biotechnology companies and holds numerous biotechnology patents. Dr. Sidransky has served as Vice Chairman of the board of directors, and was, until the merger with Eli Lilly, a director of ImClone Systems, Inc., a global biopharmaceutical company committed to advancing oncology care. He is currently on the board of Directors of Ascentage Pharma, Galmed and Champions Oncology. and chairs the board of directors of Advaxis and Ayala. Dr. Sidransky served as Director from 2005 until 2008 of the American Association for Cancer Research (AACR). He was the chairperson of AACR International Conferences during the years 2006 and 2007 on Molecular Diagnostics in Cancer Therapeutic Development: Maximizing Opportunities for Personalized Treatment. Dr. Sidransky is the recipient of a number of awards and honors, including the 1997 Sarstedt International Prize from the German Society of Clinical Chemistry, the 1998 Alton Ochsner Award Relating Smoking and Health by the American College of Chest Physicians, and the 2004 Richard and Hinda Rosenthal Award from the American Association of Cancer Research. Dr. Sidransky received his BS in Chemistry from Brandies University and his medical degree from Baylor College of medicine where he also completed his residency in internal medicine. His specialty in Medical Oncology was completed at Johns Hopkins University and Hospital.
| 64 |
We believe Dr. Sidransky is qualified to serve on our Board of Directors because of his education, medical background, experience within the life science industry and his business acumen in the public markets.
Guy Yachin – Director
Mr. Yachin has served as a director since his appointment on April 2, 2012. Mr. Yachin serves, since November 2020, as the executive chairman of Xerient Pharma which develops a drug for the treatment of abdominal cancers. He served as the President and CEO of Serpin Pharma, a clinical stage Virginia-based company focused on the development of anti-inflammatory drugs, from April 2013 until October 2020. Prior to that, Mr. Yachin was the CEO of NasVax Ltd., a company focused on the development of improved immunotherapeutics and vaccines. Prior to joining NasVax, Mr. Yachin served as CEO of MultiGene Vascular Systems Ltd (a.k.a. Vessl), a cell therapy company focused on blood vessels disorders, leading the company through clinical studies in the U.S. and Israel, financial rounds, and a keystone strategic agreement with Teva Pharmaceuticals Industries Ltd. He was CEO and founder of Chiasma Inc., a biotechnology company focused on the oral delivery of macromolecule drugs, where he built the company’s presence in Israel and the U.S., concluded numerous financial rounds, and guided the company’s strategy and operation for over six years. Earlier, he was CEO of Naiot Technological Center Ltd., and provided seed funding and guidance to more than a dozen biomedical startups such as Remon Medical Technologies Ltd., Enzymotec Ltd. and NanoPass Technologies Ltd. He holds a BSc. in Industrial Engineering and Management and an MBA from the Technion – Israel Institute of Technology.
We believe Mr. Yachin is qualified to serve on our Board of Directors because of his education, experience within the life science industry and his business acumen in the public markets.
Yaron Adler – Director
Mr. Adler has served as a director since his appointment on April 17, 2012. Mr. Adler is the co-founder of a startup incubator, We Group Ltd. In 1999, Mr. Adler co-founded IncrediMail Ltd. and served as its CEO until 2008 and President until 2009. After IncrediMail, Mr. Adler consulted Israeli startup companies regarding Internet products, services and technologies. Mr. Adler served as a product manager from 1997 to 1999, and as a software engineer from 1994 to 1997, at Tecnomatix Technologies Ltd., a software company that develops and markets production engineering solutions to complex automated manufacturing lines that fill the gap between product design and production, and which was acquired by UGS Corp. in April 2005. In 1993, Mr. Adler held a software engineer position at Intel Israel Ltd. He has a B.A. in computer sciences and economics from Tel Aviv University.
We believe Mr. Adler is qualified to serve on our Board of Directors because of his education, success with early-stage enterprises and his business acumen in the public markets.
Ashish Nanda – Director
Mr. Nanda has served as a director since his appointment on February 22, 2017. Since 1998, Mr. Nanda has been the Managing Director of Innovations Group, one of the largest outsourcing companies in the financial sector that employs close to 14,000 people working across various financial sectors. Since 1992, Mr. Nanda has served as the Managing Partner of Capstone Insurance Brokers LLC and, since 2009, has served as Managing Partner of Dive Tech Marine Engineering Services L.L.C. From 1991 to 1994, Mr. Nanda held the position of Asst. Manager Corporate Banking at Emirates Banking Group where he was involved in establishing relationships with business houses owned by UAE nationals and expatriates in order to set up banking limits and also where he managed portfolios of USD $26 billion. Mr. Nanda holds a Chartered Accountancy from the Institute of Chartered Accountants from India.
We believe that Mr. Nanda is qualified to serve on our Board of Directors because of his business experience and strategic understanding of advancing the valuation of companies in emerging industries.
| 65 |
Pursuant to an agreement entered into between us and Image Securities fzc. (“Image”), for so long as Image’s ownership of our company is 10% or greater, it was granted the right to nominate a director to our Board of Directors. Mr. Nanda was nominated for a directorship at the 2017 annual meeting in compliance with our contractual undertakings. Although Image is no longer a beneficial owner of 10% or greater of our common stock, Mr. Nanda remains as a member of our Board of Directors.
Mario Philips – Director
Mr. Philips has served as a director since his appointment on January 9, 2020. Since November 2020, Mr. Philips has been Chief Executive Officer of Polyplus, a leading Biotech supplier of transfection reagents for cell & gene therapy as well as the research life sciences market. He is also chairmen of the Board of PLL Therapeutics, a drug company based in France that has developed a diagnostic platform technology for neurodegenerative diseases in combination with a therapy to cure neurodegenerative diseases such as ALS and Parkinson’s.
Prior to that, Mr. Philips acted as VP/GM for Danaher Pall Biotech business with full P&L responsibility for a $1.3 billion business unit. Mr. Philips joined Pall in February 2014, as part of the Pall acquisition of ATMI Life Sciences, and was appointed to Vice President and General Manager to lead the Single-Use Technologies BU. In this role he was responsible for leading and executing an aggressive investment and growth strategy.
Mr. Philips joined ATMI in 1999 with ATMI’s acquisition of MST Analytics, Inc., serving as European Sales Manager for ATMI Analytical Systems. In 2004, he was appointed to General Manager of ATMI Packaging, a role he held through 2010 when he was promoted to the position of Senior Vice President and General Manager, ATMI Life Sciences. In that role, he was responsible for developing and executing all business strategies, including the introduction of new products and service solutions for the Life Sciences industry. Mr. Philips also held in the past several board member positions in the life sciences industry with Clean Biologics, Austar Life Sciences (China), Disposable Lab (France) and Artelis (Belgium).
We believe that Mr. Philips is qualified to serve on our Board of Directors because of his business experience and strategic understanding of advancing the valuation of companies in emerging industries.
There are no family relationships between any of the above executive officers or directors or any other person nominated or chosen to become an executive officer or a director.
Board of Directors
Our Board of Directors currently consists of six (6) members. All directors hold office until the next annual meeting of stockholders. At each annual meeting of stockholders, the successors to directors whose terms then expire are elected to serve from the time of election and qualification until the next annual meeting following election.
Management has been delegated the responsibility for meeting defined corporate objectives, implementing approved strategic and operating plans, carrying on our business in the ordinary course, managing cash flow, evaluating new business opportunities, recruiting staff and complying with applicable regulatory requirements. The Board of Directors exercises its supervision over management by reviewing and approving long-term strategic, business and capital plans, material contracts and business transactions, and all debt and equity financing transactions and stock issuances.
Director Independence
Our Board of Directors is comprised of a majority of independent directors. In determining director independence, we use the definition of independence in Rule 5605(a)(2) of the listing standards of The Nasdaq Stock Market.
The Board has concluded that each of Dr. Sidransky, and Messrs. Yachin, Adler, Philips and Nanda is “independent” based on the listing standards of the Nasdaq Stock Market, having concluded that any relationship between such director and our company, in its opinion, does not interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
| 66 |
Board Committees
Our Board of Directors has established an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee, with each comprised of independent directors in accordance with the rules of The Nasdaq Stock Market and applicable federal securities laws and regulations. The members of the Audit Committee are Dr. Sidransky and Messrs. Yachin and Philips. The members of the Compensation Committee are Dr. Sidransky and Messrs. Adler and Yachin. The members of the Nominating and Corporate Governance Committee are Messrs. Nanda, Adler and Yachin. We have also established a Research and Development Committee. The members of the Research and Development Committee are Mr. Yachin and Dr. Sidransky.
Each committee operates under a written charter that has been approved by our Board of Directors. Copies of our committee charters are available on the investor relations section of our website, which is located at http://www.orgenesis.com.
Audit Committee
The Audit Committee (a) assists the Board of Directors in fulfilling its oversight of: (i) the quality and integrity of our financial statements; (ii) our compliance with legal and regulatory requirements relating to our financial statements and related disclosures; (iii) the qualifications and independence of our independent auditors; and (iv) the performance of our independent auditors; and (b) prepares any reports that the rules of the SEC require be included in our proxy statement for our annual meeting.
The Audit Committee held 4 meetings in 2023. In addition, the Audit Committee reviewed and approved various corporate items by way of written consent during the year 2023. The Board has determined that each member of the Audit Committee is an independent director in accordance with the rules of The Nasdaq Stock Market and applicable federal securities laws and regulations. In addition, the Board has determined that Dr. Sidransky is an “audit committee financial expert” within the meaning of Item 407(d)(5) of Regulation S-K and has designated him to fill that role. See “Directors, Executive Officers and Corporate Governance – Directors” above for descriptions of the relevant education and experience of each member of the Audit Committee.
At no time since the commencement of our most recently completed fiscal year was a recommendation of the Audit Committee to nominate or compensate an external auditor not adopted by the Board of Directors.
The Audit Committee is responsible for the oversight of our financial reporting process on behalf of the Board of Directors and such other matters as specified in the Audit Committee’s charter or as directed by the Board of Directors. Our Audit Committee is directly responsible for the appointment, compensation, retention and oversight of the work of any registered public accounting firm engaged by us for the purpose of preparing or issuing an audit report or performing other audit, review or attest services for us (or to nominate the independent registered public accounting firm for stockholder approval), and each such registered public accounting firm must report directly to the Audit Committee. Our Audit Committee must approve in advance all audit, review and attest services and all non-audit services (including, in each case, the engagement and terms thereof) to be performed by our independent auditors, in accordance with applicable laws, rules and regulations.
Compensation Committee
The Compensation Committee (i) assists the Board of Directors in discharging its responsibilities with respect to compensation of our executive officers and directors, (ii) evaluates the performance of our executive officers, and (iii) administers our stock and incentive compensation plans and recommends changes in such plans to the Board as needed.
The Compensation Committee held 4 meetings in 2023. In addition, the Compensation Committee reviewed and approved various corporate items by way of written consent during the year ended December 31, 2023. The Board of Directors has determined that each member of the Compensation Committee is an independent director in accordance with the rules of The Nasdaq Stock Market and applicable federal securities laws and regulations.
| 67 |
Nominating and Corporate Governance Committee
The Nominating and Corporate Governance Committee assists the Board in (i) identifying qualified individuals to become directors, (ii) determining the composition of the Board and its committees, (iii) developing succession plans for executive officers, (iv) monitoring a process to assess Board effectiveness, and (v) developing and implementing our corporate governance procedures and policies.
The Nominating and Corporate Governance Committee held 2 meetings in 2023. In addition, the Nominating and Corporate Governance Committee reviewed and approved various corporate items by way of written consent during the year ended December 31, 2023. The Board has determined that each member of the Nominating and Corporate Governance Committee is an independent director in accordance with the rules of The Nasdaq Stock Market and applicable federal securities laws and regulations.
Research and Development Committee
The Research and Development Committee assists the Board in fulfilling the Board’s responsibilities to oversee our research and development programs, and strategies.
The Research and Development Committee was established in January 2021. The Research and Development approved various corporate items by way of written consent during the year ended December 31, 2023.
DELINQUENT SECTION 16(a) REPORTS
Section 16(a) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), requires our officers and directors and persons who beneficially own more than ten percent (10%) of the Common Stock outstanding to file initial statements of beneficial ownership of Common Stock (Form 3) and statements of changes in beneficial ownership of Common Stock (Forms 4 or 5) with the SEC. Officers, directors and greater than 10% stockholders are required by SEC regulation to furnish us with copies of all such forms they file.
Our records reflect that all reports which were required to be filed pursuant to Section 16(a) of the Securities Exchange Act of 1934, as amended, were filed on a timely basis.
CORPORATE CODE OF CONDUCT AND ETHICS
Our Board of Directors has adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Copies of our corporate code of conduct and ethics are available, without charge, upon request in writing to Orgenesis Inc., 20271 Goldenrod Lane, Germantown, MD, 20876, Attn: Secretary and are posted on the investor relations section of our website, which is located at www.orgenesis.com. The inclusion of our website address in this Annual Report on Form 10-K does not include or incorporate by reference the information on our website into this Annual Report on Form 10-K. We also intend to disclose any amendments to the Corporate Code of Conduct and Ethics, or any waivers of its requirements, on our website.
ITEM 11. EXECUTIVE COMPENSATION
The following table shows the total compensation paid or accrued during the years ended December 31, 2023 and 2022. Our named executive officers consist of (1) our Chief Executive Officer, (2) our former Chief Financial Officer and (3) our former Chief Development Officer. As of December 31, 2023, there were no other executive officers who earned more than $100,000 during the year ended December 31, 2023 and were serving as executive officers as of such date. The table includes two additional executive officers who would have been among the three most highly compensated executive officers except for the fact that they were not serving as executive officers of the Company as of the end of 2023.
| 68 |
Summary Compensation Table
Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards ($) (1) | Non-Equity Incentive Plan Compensa- tion ($) | Non-qualified Deferred Compensation Earnings ($) | All Other Compensa- tion ($) (2) | Total ($) | |||||||||
| Vered Caplan | 2023 | 259,029 | - | - | - | - | - | 82,355 | 341,384 | |||||||||
| CEO | 2022 | 243,868 | - | - | 107,941 | - | - | 92,100 | 443,909 | |||||||||
Elliot Maltz Former CFO, Treasurer & Secretary(3) | 2023 | 111,667 | - | - | 81,883 | - | - | - | 193,550 | |||||||||
| Efrat Assa-Kunik, | 2023 | 129,633 | - | - | - | - | - | 18,690 | 148,323 | |||||||||
| Former Chief Development Officer(4) | 2022 | 162,316 | - | - | 19,048 | - | - | 44,467 | 225,831 |
| (1) | In accordance with SEC rules, the amounts in this column reflect the fair value on the grant date of the option awards granted to the named executive, calculated in accordance with ASC Topic 718. Stock options were valued using the Black-Scholes model. The grant-date fair value does not necessarily reflect the value of shares which may be received in the future with respect to these awards. The grant-date fair value of the stock options in this column is a non-cash expense for us that reflects the fair value of the stock options on the grant date and therefore does not affect our cash balance. The fair value of the stock options will likely vary from the actual value the holder receives because the actual value depends on the number of options exercised and the market price of our Common Stock on the date of exercise. For a discussion of the assumptions made in the valuation of the stock options, see Note 15 to this Annual Report on Form 10-K for the year ended December 31, 2023. No executive officers received options awards in the year ended December 31, 2023. See below for a summary of options awarded in previous years. |
| (2) | For 2023 and 2022, represents the compensation as described under the caption “All Other Compensation” below. |
| (3) | Mr. Maltz resigned from his position at the Company effective December 31, 2023.
|
| (4) | Ms. Assa Kunik resigned from her position at the Company effective August 8, 2023. |
All Other Compensation
The following table provides information regarding each component of compensation for the years ended December 31, 2023 and 2022 included in the All Other Compensation column in the Summary Compensation Table above. Represents amounts paid in New Israeli Shekels (NIS) or Swiss Franks and converted at average exchange rates for the year.
| Name | Year | Automobile and Communication Related Expenses $ | Social Benefits $ (1) | Total $ | ||||||||||||
| Vered Caplan | 2023 | 2,627 | 79,728 | 82,355 | ||||||||||||
| 2022 | 2,536 | 89,564 | 92,100 | |||||||||||||
| Efrat Assa Kunik | 2023 | 377 | 18,313 | 18,690 | ||||||||||||
| 2022 | 436 | 44,031 | 44,467 | |||||||||||||
| (1) | These are comprised of contributions by us to savings, health, severance, pension, disability and insurance plans generally provided in Israel and Switzerland, including health, education, managerial insurance funds, and redeemed vacation pay. This amount represents Israeli and Swiss severance fund payments, managerial insurance funds, disability insurance, supplemental education fund contribution and social securities. See discussion below under “Narrative Disclosure to Summary Compensation Table – Vered Caplan.” |
| 69 |
Outstanding Equity Awards at December 31, 2023
The following table summarizes the outstanding equity awards held by each named executive officer of our company as of December 31, 2023.
| Name | Grant Date | Number of Shares Underlying Unexercised Options (#) Exercisable | Number of Shares Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||||||||
| Vered Caplan | 22-Aug-14(1) | 230,189 | - | 0.0012 | 22-Aug-24 | |||||||||||
| 09-Dec-16(1) | 166,667 | - | 4.80 | 09-Dec-26 | ||||||||||||
| 06-Jun-17(1) | 83,334 | - | 7.20 | 06-Jun-27 | ||||||||||||
| 28-Jun-18(1) | 250,001 | - | 8.36 | 28-Jun-28 | ||||||||||||
| 22-Oct-18(1) | 85,000 | - | 5.99 | 22-Oct-28 | ||||||||||||
| 19-Mar-20(1) | 85,000 | - | 2.99 | 18-Mar-30 | ||||||||||||
| 14-Jun-22(2) | 63,750 | 21,250 | 2.00 | 13-Jun-32 | ||||||||||||
| Elliot Maltz | 04-Sep-23 | 25,000 | - | 2.00 | 13-Jun-32 | |||||||||||
| Efrat Assa Kunik | 09-Dec-16(1) | 16,667 | - | 4.8 | 09-Dec-26 | |||||||||||
| 22-Oct-18(1) | 15,000 | - | 5.99 | 22-Oct-28 | ||||||||||||
| 19-Mar-20(1) | 15,000 | - | 2.99 | 18-Mar-30 | ||||||||||||
| 14-Jun-22(2) | 7,500 | - | 2.00 | 13-Jun-32 | ||||||||||||
| (1) | The options were fully vested as of December 31, 2023. |
| (2) | The options vest on a quarterly basis over a period of two years from the date of grant. |
Option Exercises and Stock Vested in 2023
The following table shows information regarding exercises of options to purchase our common stock and vesting of stock awards held by each executive officer named in the Summary Compensation Table during the year ended December 31, 2023.
| Option Awards | Stock Awards | |||||||||||||||
| Name | Number of Shares Acquired on Exercise (#) | Value Realized on Exercise ($) (1) | Number of Shares Acquired on Vesting (#) | Value Realized on Vesting ($) | ||||||||||||
| (a) | (b) | (c) | (d) | (e) | ||||||||||||
| Vered Caplan | - | - | - | - | ||||||||||||
(1) Amounts shown in this column do not necessarily represent actual value realized from the sale of the shares acquired upon exercise of options because in many cases the shares are not sold on exercise but continue to be held by the executive officer exercising the option. The amounts shown represent the difference between the option exercise price and the market price on the date of exercise, which is the amount that would have been realized if the shares had been sold immediately upon exercise.
| 70 |
Narrative Disclosure to Summary Compensation Table and Employment Agreements
Vered Caplan
On August 14, 2014, our Board of Directors confirmed that Ms. Vered Caplan, who had served as our President and Chief Executive Officer on an interim basis since December 23, 2013, was appointed as our President and Chief Executive Officer.
On November 19, 2020, we and Ms. Caplan entered into an executive directorship agreement, effective as of October 1, 2020 (the “Executive Directorship Agreement”), that superseded and replaced a previous employment agreement (the “Prior Agreement”). Pursuant to the Executive Directorship Agreement, Ms. Caplan will continue to serve the Company as its Chairperson of the Board of Directors (the “Board”) and shall receive in consideration for her serving as Chairperson of the Board an annual regular Board fee in the amount of $75,000 payable by the Company in equal quarterly installments in advance. In addition, Ms. Caplan may be eligible for non-recurring special Board fees as reviewed and approved by the Compensation Committee of the Board (the “Compensation Committee”) and then reviewed and ratified by the Board. In addition, Ms. Caplan may be granted option awards from time to time at the discretion of the Compensation Committee.
Ms. Caplan’s position as Chairperson of the Board under the Executive Directorship Agreement may be terminated for any reason by either Ms. Caplan or the Company upon 90 days prior written notice (the “Notice Period”), provided that the Company may terminate such appointment as Chairperson at any time during the Notice Period subject to certain conditions. Such termination as Chairperson of the Board will be deemed a termination even if Ms. Caplan remains as a regular director of the Board. Upon termination by the Company of Ms. Caplan’s employment other than for cause or by Ms. Caplan for any reason whatsoever, in addition to any Accrued Obligations (as defined therein) she shall be entitled to receive a lump sum payment equal to the sum of (i) the annual regular Board fee (the “Board Fee”) and (ii) the greater of actual or target annual performance bonus to which she may have been entitled to as of the termination date (in each case, less all customary and required taxes and related deductions).
Ms. Caplan’s position under the Executive Directorship Agreement may be terminated in the event of a Change of Control (as defined therein) by the Company other than for cause or by Ms. Caplan for any reason whatsoever. In the event of a Change of Control and if, within one year following such Change of Control, employment under the Executive Directorship Agreement is terminated by the Company other than for cause or by Ms. Caplan for any reason whatsoever, in addition to any Accrued Obligations, she shall be entitled to receive a lump sum payment equal to one and a half times the sum of (i) the Board Fee and (ii) the target annual performance remuneration to which she may have been entitled as of the termination date (in each case, less all customary and required taxes and related deductions).
In addition, on November 19, 2020, Orgenesis Services Sàrl, a Swiss corporation and wholly-owned, direct subsidiary of the Company (“Orgenesis Services”), and Ms. Caplan entered into a personal employment agreement (the “Swiss Employment Agreement” and together with the Executive Directorship Agreement, the “Agreements”), pursuant to which Ms. Caplan will serve as Chief Executive Officer, President and Chairperson of the Board of Directors of Orgenesis Services and will be a material provider of services to the Company pursuant to a services agreement between the Company and Orgenesis Services. The Swiss Employment Agreement provides that Ms. Caplan is entitled to a monthly base salary of CHF 13,345.05 (equivalent to $14,583 based on the current exchange rate at signing), and an annual representation fee of CHF 24,000 (equivalent to $26,226 based on the current exchange rate at signing), payable in monthly installments of CHF 2,000. Ms. Caplan is eligible to receive a bonus at the absolute discretion of Orgenesis Services and its compensation committee. Ms. Caplan may also be granted option awards from time to time, as per the recommendation of the compensation committee of Orgenesis Services as reviewed and approved by the Compensation Committee. Under the Swiss Employment Agreement, Ms. Caplan is entitled to be paid annual vacation days, monthly travel allowance, sick leave, expenses reimbursement and a mobile phone. The Swiss Employment Agreement had an effective date as of October 1, 2020.
| 71 |
Employment under the Swiss Employment Agreement may be terminated for any reason by Ms. Caplan or by Orgenesis Services other than for just cause (as defined therein) upon six months prior written notice or by Orgenesis Services other than for just cause in the event of a Change of Control (as defined therein) of the Company upon at least 12 months prior written notice. Upon termination by Orgenesis Services of Ms. Caplan’s employment without just cause or by Ms. Caplan for any reason whatsoever, in addition to any Accrued Obligations (as defined therein), she shall be entitled to receive a lump sum payment equal to the sum of (i) her Base Salary (as defined therein) at the rate in effect as of the termination date and (ii) the greater of actual or target annual performance bonus to which she may have been entitled to for the year in which employment terminates (in each case, less all customary and required taxes and employment-related deductions). In the event of a Change of Control and if, within one year following such Change of Control, employment is terminated by Orgenesis Services other than for cause or by Ms. Caplan for any reason whatsoever, in addition to any Accrued Obligations she shall be entitled to receive a lump sum payment equal to one and a half times the sum of (i) her Base Salary and (ii) the target annual performance bonus to which she may have been entitled to for the year in which employment terminates (in each case, less all customary and required taxes and employment-related deductions).
The Swiss Employment Agreement provides for customary protections of Orgenesis’ confidential information and intellectual property.
Ms. Caplan received an aggregate salary and board fee of $259,029 during 2023. As of December 31, 2023, the $150,000 chairperson fee for 2022 and 2023 was unpaid, but accrued, per agreement by Ms. Caplan. In addition, in 2022 Ms. Caplan was awarded options to purchase 85,000 shares of common stock.
Ms. Caplan received reimbursement for automobile and communication related expenses in the amount of $2,627 in 2023 and $2,536 in 2022. In addition, the Company contributed to savings, health, severance, pension, disability and insurance plans generally provided in Switzerland, including health, education, managerial insurance funds, and redeemed vacation pay in an amount equivalent to $79,728 in 2023 and $89,564 in 2022. These amounts represent Swiss severance fund payments, managerial insurance funds, disability insurance, supplemental education fund contribution and social securities.
Elliot Maltz, former CFO, Secretary and Treasurer
Mr. Maltz was appointed Chief Financial Officer, Treasurer and Secretary on September 1, 2023. Pursuant to Mr. Maltz’s personal employment agreement (the “Employment Agreement”) with the Company he is entitled to receive an annual base salary of $335,000 and an annual cash bonus of up to 40% of his then-current base salary (the “Annual Performance Bonus”). The Annual Performance Bonus, if any, will be based upon the achievement of certain corporate and individual performance objectives. Additionally, pursuant to the Employment Agreement Mr. Maltz was granted 200,000 stock options (the “Stock Award”). The Stock Award will vest quarterly from the grant date over four years subject to Mr. Maltz’s continued employment through each such vesting date. Mr. Maltz resigned his position at the Company effective December 31, 2023. Mr. Maltz base salary of $111,667 earned during 2023 was paid to him as per his employment and we have no further obligations due to him.
Efrat Assa-Kunik
Ms. Assa-Kunik was appointed Chief Development Officer in December 2021. According to the terms of Ms. Assa-Kunik’s Employment Agreement, Ms. Assa Kunik is entitled to a monthly salary of 45 thousand New Israeli Shekels, customary contributions to a pension and training fund, participation in cellphone expenses, and annual leave of 24 days. In 2022, Ms. Assa-Kunik was awarded options to purchase 15,000 shares of common stock. Ms. Assa Kunik resigned her position at the Company effective August 2023.
| 72 |
Ms. Assa-Kunik received an aggregate salary of $126,933 during 2023 and $162,316 in 2022. In addition, in 2022 Ms. Assa-Kunik was awarded options to purchase 15,000 shares of common stock.
Ms. Assa-Kunik received reimbursement for automobile and communication related expenses in the amount of $377 in 2023 and $436 in 2022. In addition, the Company contributed to savings, health, severance, pension, disability and insurance plans generally provided in Israel, including health, education, managerial insurance funds, and redeemed vacation pay in an amount equivalent to $18,313 in 2023 and $44,031 in 2022. These amounts represent Israeli severance fund payments, managerial insurance funds, disability insurance, supplemental education fund contribution and social securities.
Potential Payments upon Change of Control or Termination following a Change of Control
Our employment agreements with our named executive officers provide incremental compensation in the event of termination, as described above.
Due to the factors that may affect the amount of any benefits provided upon the events described below, any actual amounts paid or payable may be different than those shown in this table. Factors that could affect these amounts include the basis for the termination, the date the termination event occurs, the base salary of an executive on the date of termination of employment and the price of our common stock when the termination event occurs.
The following table sets forth the compensation that would have been received by each of our executive officers had they been terminated as of December 31, 2023.
Name |
Salary Continuation |
|||
| Vered Caplan | $ | * | ||
(*) Termination by Company without cause: $250,000
Termination without cause following a change in control: $375,000
Director Compensation
The following table sets forth for each non-employee director that served as a director during the year ended December 31, 2023:
Year Ended December 31, 2023
| Name | Fees Earned or Paid in Cash ($) | Stock Awards ($) | Option Awards ($) (1) | Non-equity Incentive Plan Compensation ($) | Nonqualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||
| Guy Yachin | 100,000 | - | 6,067 | (2) | - | - | - | 106,067 | ||||||||||||||||||||
| Yaron Adler | 60,000 | - | 4,643 | (3) | - | - | - | 64,643 | ||||||||||||||||||||
| Dr. David Sidransky | 105,000 | - | 6,330 | (4) | - | - | - | 111,330 | ||||||||||||||||||||
| Ashish Nanda | 65,000 | - | 4,907 | (5) | - | - | - | 69,907 | ||||||||||||||||||||
| Mario Philips | 50,000 | - | 4,256 | (6) | - | - | - | 54,256 | ||||||||||||||||||||
| (1) | In accordance with SEC rules, the amounts in this column reflect the fair value on the grant date of the option awards granted to the named executive, calculated in accordance with ASC Topic 718. Stock options were valued using the Black-Scholes model. The grant-date fair value does not necessarily reflect the value of shares which may be received in the future with respect to these awards. The grant-date fair value of the stock options in this column is a non-cash expense for us that reflects the fair value of the stock options on the grant date and therefore does not affect our cash balance. The fair value of the stock options will likely vary from the actual value the holder receives because the actual value depends on the number of options exercised and the market price of our common stock on the date of exercise. For a discussion of the assumptions made in the valuation of the stock options, see Note 15 (Stock Based Compensation) to our financial statements, which are included in this Annual Report on Form 10-K. |
| 73 |
| (2) | In respect of 19,600 options which will vest on December 12, 2024. |
| (3) | In respect of 15,000 options which will vest on December 12, 2024. |
| (4) | In respect of 20,450 options which will vest on December 12, 2024. |
| (5) | In respect of 15,850 options which will vest on December 12, 2024. |
| (6) | In respect of 13,750 options which will vest on December 12, 2024. |
All directors receive reimbursement for reasonable out of pocket expenses in attending Board of Directors meetings and for participating in our business.
Compensation Policy for Non-Employee Directors.
In January 2021, the Board of Directors adopted an updated compensation policy for non-employee directors which replaced the previous non-employee director compensation terms, and which became effective January 2021. Under the policy, each director is to receive an annual cash compensation of $40,000 and the Chairman or lead director is paid an additional $20,000 per annum. Each committee member will be paid an additional $10,000 per annum and the committee chairman of the Audit and Research and Development committees is to receive $20,000 per annum while the chairman of the other committees is to receive $15,000 per annum. Cash compensation will be made on a quarterly basis.
All newly appointed directors also receive options to purchase up to 6,250 shares of our common stock. All directors are entitled to an annual bonus of options for 12,500 shares and each committee member is entitled to a further option to purchase up to 1,250 shares of common stock and each committee chairperson to options for an additional 2,100 shares of common stock. In addition, the Chairman and Vice Chairman shall be granted an option to purchase 4,200 shares of our common stock. In all cases, the options are granted at a per share exercise price equal to the closing price of our publicly traded stock on the date of grant and the vesting schedule is determined by the compensation committee at the time of grant.
Compensation Committee Interlocks and Insider Participation
None of our executive officers has served as a member of the Board of Directors, or as a member of the compensation or similar committee, of any entity that has one or more executive officers who served on our Board of Directors or Compensation Committee during the year ended December 31, 2023.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of April 15, 2024 for (a) the named executive officers, (b) each of our directors, (c) all of our current directors and executive officers as a group and (d) each stockholder known by us to own beneficially more than 5% of our common stock. Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. We deem shares of common stock that may be acquired by an individual or group within 60 days of April 15, 2024 pursuant to the exercise of options or warrants to be outstanding for the purpose of computing the percentage ownership of such individual or group but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table. Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them based on information provided to us by these stockholders. Percentage of ownership is based on 34,338,782 shares of common stock outstanding on April 15, 2024.
| 74 |
Security Ownership of Greater than 5% Beneficial Owners
Name and Address of Beneficial Owner | Amount and Nature of Beneficial Ownership (1) | Percent(1) | ||||||
| Jacob Safier c/o The Wolfson Group, One State Street Plaza, 29th Floor New York, NY 10004 | 4,988,000 | (2) | 14.53 | % | ||||
| Yehuda Nir c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 11,297,179 | (3) | 24.75 | % | ||||
Security Ownership of Directors and Executive Officers
Name and Address of Beneficial Owner | Amount and Nature of Beneficial Ownership (1) | Percent(1) | ||||||
| Vered Caplan c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 1,252,757 | (4) | 3.55 | % | ||||
| Elliot Maltz c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 25,000 | (5) | <1% | |||||
| Efrat Assa Kunik c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 54,167 | (7) | <1% | |||||
| Guy Yachin c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 150,867 | (8) | <1% | |||||
| Dr. David Sidransky c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 153,467 | (9) | <1% | |||||
| Yaron Adler c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 203,721 | (10) | <1% | |||||
| Ashish Nanda c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 98,400 | (11) | <1% | |||||
| Mario Philips c/o Orgenesis Inc. 20271 Goldenrod Lane Germantown, MD 20876 | 60,000 | (12) | <1% | |||||
| Directors & Executive Officers as a Group (8 persons) | 1,998,379 | 5.82 | % | |||||
| 75 |
Notes:
| (1) | Percentage of ownership is based on 34,338,782 shares of our common stock outstanding as of April 15, 2024. Except as otherwise indicated, we believe that the beneficial owners of the common stock listed above, based on information furnished by such owners, have sole investment and voting power with respect to such shares, subject to community property laws where applicable. Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock subject to options, warrants or convertible debt currently exercisable, or convertible or exercisable or convertible within 60 days, are deemed outstanding for purposes of computing the percentage ownership of the person holding such options, warrants or convertible debt but are not deemed outstanding for purposes of computing the percentage ownership of any other person. |
| (2) | Consists of 4,988,000 shares of common stock. |
| (3) | Consists of (i) 10,016 shares of common stock, (ii) 453,294 shares of common stock issuable upon exercise of outstanding warrants at a price of $6.24 per share, exercisable until, January 31, 2026, (iii) 277,778 shares of common stock issuable upon exercise of outstanding warrants at a price of $4.50 per share, exercisable until, January 31, 2026, (iv) 1,111,111 shares of common stock issuable upon exercise of outstanding warrants at a price of $2.50 per share, exercisable until, January 31, 2026, (v) 840,000 shares of common stock issuable upon exercise of outstanding warrants at a price of $0.85 per share, exercisable until, December 31, 2026, (vi) 218,750 shares of common stock issuable upon exercise of outstanding warrants at a price of $0.80 per share, exercisable until, October 4, 2024, (vii) 7,375,100 shares of common stock issuable upon conversion of convertible debt at a conversion price of $2.50 per share, and (viii) 936,477 shares of common stock issuable upon conversion of convertible debt at a conversion price of $0.85 per share. |
| (4) | Consists of (i) 278,191 shares of common stock, (ii) 230,189 shares of common stock issuable upon exercise of outstanding options at a price of $0.0012 per share, (iii) 166,667 shares of common stock issuable upon exercise of outstanding options at a price of $4.80 per share, (iv) 83,334 shares of common stock issuable upon exercise of outstanding options at a price of $7.20 per share, (v) 250,001 shares of common stock issuable upon exercise of outstanding options at a price of $8.36 per share, (vi) 85,000 shares of common stock issuable upon exercise of outstanding options at a price of $5.99 per share, (vii) 85,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, and (viii) 74,375 shares of common stock issuable upon exercise of outstanding options at a price of $2.00 per share. Does not include option for 10,625 shares of common stock with an exercise price of $2.00 per share that are exercisable quarterly after June 24, 2024. |
| (5) | Consists of 25,000 shares of common stock issuable upon exercise of outstanding options at a price of $0.58 per share. |
| (6) | |
| (6) | Consists of (i) 16,667 shares of common stock issuable upon exercise of outstanding options at a price of $4.80 per share, (ii) 15,000 shares of common stock issuable upon exercise of outstanding options at a price of $5.99 per share, (iii) 15,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, and (iv) 7,500 shares of common stock issuable upon exercise of outstanding options at a price of $2.00 per share. |
| 76 |
| (7) | Consists of (i) 41,667 shares of common stock issuable upon exercise of outstanding options at a price of $4.80 per share, (ii) 28,750 shares of common stock issuable upon exercise of outstanding options at a price of $5.99 per share, (iii) 25,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, (iv) 16,250 shares of common stock issuable upon exercise of outstanding options at a price of $4.60 per share, (v) 19,600 shares of common stock issuable upon exercise of outstanding options at a price of $2.89 per share. and(v) 19,600 shares of common stock issuable upon exercise of outstanding options at a price of $1.86 per share. Does not include option for 19,600 shares of common stock with an exercise price of $0.45 per share that are exercisable on December 13, 2024. |
| (8) | Consists of (i) 41,667 shares of common stock issuable upon exercise of outstanding options at a price of $4.80 per share, (ii)29,200 shares of common stock issuable upon exercise of outstanding options at a price of $5.99 per share, (iii) 25,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, (iv) 16,700 shares of common stock issuable upon exercise of outstanding options at a price of $4.60 per share, (v) 20,450 shares of common stock issuable upon exercise of outstanding options at a price of $2.89 per share, and (vi) 20,450 shares of common stock issuable upon exercise of outstanding options at a price of $1.86 per share. Does not include option for 20,450 shares of common stock with an exercise price of $0.45 per share that are exercisable on December 13, 2024. |
| (9) | Consists of (i) 63,304 shares of common stock, (ii) 41,667 shares of common stock issuable upon exercise of outstanding options at a price of $4.80 per share, (iii) 28,750 shares of common stock issuable upon exercise of outstanding options at a price of $5.99 per share, (iv) 25,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, (v) 15,000 shares of common stock issuable upon exercise of outstanding options at a price of $4.60 per share,(vi) 15,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.89 per share, and (vi) 15,000 shares of common stock issuable upon exercise of outstanding options at a price of $1.86 per share. Does not include option for 15,000 shares of common stock with an exercise price of $0.45 per share that are exercisable on December 13, 2024. |
| (10) | Consists of (i) 27,100 shares of common stock issuable upon exercise of outstanding options at a price of $5.99 per share, (ii) 25,000 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, (iii) 14,600 shares of common stock issuable upon exercise of outstanding options at a price of $4.60 per share, (iv) 15,850 shares of common stock issuable upon exercise of outstanding options at a price of $2.89 per share, and (iv) 15,850 shares of common stock issuable upon exercise of outstanding options at a price of $1.86 per share. Does not include option for 15,850 shares of common stock with an exercise price of $0.45 per share that are exercisable on December 13, 2024. |
| (11) | Consists of (i) 6,250 shares of common stock issuable upon exercise of outstanding options at a price of $4.70 per share, (ii) 12,500 shares of common stock issuable upon exercise of outstanding options at a price of $2.99 per share, (iii) 13,750 shares of common stock issuable upon exercise of outstanding options at a price of $4.60 per share, (iv) 13,750 shares of common stock issuable upon exercise of outstanding options at a price of $2.89 per share, and (iv) 13,750 shares of common stock issuable upon exercise of outstanding options at a price of $1.86 per share. Does not include option for 13,750 shares of common stock with an exercise price of $0.45 per share that are exercisable on December 13, 2024. |
| 77 |
Securities Authorized for Issuance Under Existing Equity Compensation Plans
The following table summarizes certain information regarding our equity compensation plans as of December 31, 2023:
Plan Category | Number of Securities to be Issued Upon Exercise of Outstanding Options | Weighted-Average Exercise Price of Outstanding Options and RSUs | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) | |||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders (1) | 2,944,865 | 3.66 | 2,046,646 | |||||||||
| Equity compensation plans not approved by security holders | 491,671 | 4.80 | - | |||||||||
| Total | 3,436,536 | 3.82 | 2,046,646 | |||||||||
| (1) | Consists of the 2017 Equity Incentive Plan and the Global Share Incentive Plan (2012). For a short description of those plans, see Note 15 to our 2022 Consolidated Financial Statements included in this Annual Report on Form 10-K for the year ended December 31, 2023. |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with Related Persons
Except as set out below, as of December 31, 2023, there have been no transactions, or currently proposed transactions, in which we were or are to be a participant and the amount involved exceeds the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years, and in which any of the following persons had or will have a direct or indirect material interest:
| ● | any director or executive officer of our company; |
| ● | any person who beneficially owns, directly or indirectly, shares carrying more than 5% of the voting rights attached to our outstanding shares of common stock; |
| ● | any promoters and control persons; and |
| ● | any member of the immediate family (including spouse, parents, children, siblings and in laws) of any of the foregoing persons. |
Pursuant to our Audit Committee charter adopted in March 2017, the Audit Committee is responsible for reviewing and approving, prior to our entry into any such transaction, all transactions in which we are a participant and in which any parties related to us have or will have a direct or indirect material interest.
Named Executive Officers and Current Directors
For information regarding compensation for our named executive officers and current directors, see “Executive Compensation.”
Director Independence
See “Directors, Executive Officers and Corporate Governance – Director Independence” and “Directors, Executive Officers and Corporate Governance – Board Committees” in Item 10 above.
| 78 |
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Our Board of Directors has appointed Kesselman & Kesselman, a member firm of PricewaterhouseCoopers International Limited (“PwC”) as our independent registered public accounting firm for the years ended December 31, 2023 and 2022. The following table sets forth the fees billed to us for professional services rendered by PwC for the years ended December 31, 2023 and December 31, 2022:
| Years Ended December 31, | ||||||||
| Services: | 2023 | 2022 | ||||||
| Audit Fees (1) | $ | 225,000 | $ | 288,705 | ||||
| Audit-Related Fees (2) | 42,000 | 6,405 | ||||||
| Total fees | $ | 267,000 | $ | 295,110 | ||||
| (1) | Audit fees consisted of audit work performed in the preparation of financial statements, as well as work generally only the independent registered public accounting firm can reasonably be expected to provide, such as statutory audits. |
| (2) | Audit related fees consisted principally of audits of employee benefit plans and special procedures related to regulatory filings in 2023. |
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-audit Services of Independent Public Accountant
Consistent with SEC policies regarding auditor independence, the Audit Committee has responsibility for appointing, setting compensation and overseeing the work of our independent registered public accounting firm. In recognition of this responsibility, the Audit Committee has established a policy to pre-approve all audit and permissible non-audit services provided by our independent registered public accounting firm.
Prior to engagement of an independent registered public accounting firm for the next year’s audit, management will submit an aggregate of services expected to be rendered during that year for each of four categories of services to the Audit Committee for approval.
1. Audit services include audit work performed in the preparation of financial statements, as well as work that generally only an independent registered public accounting firm can reasonably be expected to provide, including comfort letters, statutory audits, and attest services and consultation regarding financial accounting and/or reporting standards.
2. Audit-Related services are for assurance and related services that are traditionally performed by an independent registered public accounting firm, including due diligence related to mergers and acquisitions, employee benefit plan audits, and special procedures required to meet certain regulatory requirements.
3. Tax services include all services performed by an independent registered public accounting firm’s tax personnel except those services specifically related to the audit of the financial statements, and includes fees in the areas of tax compliance, tax planning, and tax advice.
4. Other Fees are those associated with services not captured in the other categories. We generally do not request such services from our independent registered public accounting firm.
Prior to engagement, the Audit Committee pre-approves these services by category of service. The fees are budgeted, and the Audit Committee requires our independent registered public accounting firm and management to report actual fees versus the budget periodically throughout the year by category of service. During the year, circumstances may arise when it may become necessary to engage our independent registered public accounting firm for additional services not contemplated in the original pre-approval. In those instances, the Audit Committee requires specific pre-approval before engaging our independent registered public accounting firm.
The Audit Committee may delegate pre-approval authority to one or more of its members. The member to whom such authority is delegated must report, for informational purposes only, any pre-approval decisions to the Audit Committee at its next scheduled meeting.
| 79 |
PART IV
ITEM 15. EXHIBIT AND FINANCIAL STATEMENT SCHEDULES
| (a) |
| c. | Financial Statements |
Our consolidated financial statements are set forth in Part II, Item 8 of this Annual Report on Form 10-K and are incorporated herein by reference.
| d. | Financial Statement Schedules |
No financial statement schedules have been filed as part of this Annual Report on Form 10-K because they are not applicable or are not required or because the information is otherwise included herein.
| e. | Exhibits required by Regulation S-K |
| 80 |
| 81 |
*Filed herewith
**Furnished herewith
ITEM 16. FORM 10-K SUMMARY
Not applicable.
| 82 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ORGENESIS INC.
| By: | /s/ Vered Caplan | |
| Vered Caplan | ||
Chief Executive Officer and Chairperson of the Board of Directors (Principal Executive Officer) |
||
| Date: | April 15, 2024 | |
| By: | /s/ Victor Miller | |
| Victor Miller | ||
Chief Financial Officer, Treasurer and Secretary (Principal Financial and Accounting Officer) |
||
| Date: | April 15, 2024 |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| By: | /s/ Vered Caplan | |
| Vered Caplan | ||
| Chief Executive Officer and Chairperson of the Board of Directors (Principal Executive Officer) | ||
| Date: | April 15, 2024 | |
| By: | /s/ Victor Miller | |
| Victor Miller | ||
| Chief Financial Officer, Treasurer and Secretary (Principal Financial and Accounting Officer) | ||
| Date: | April 15, 2024 | |
| By: | /s/ Guy Yachin | |
| Guy Yachin | ||
| Director | ||
| Date: | April 15, 2024 | |
| By: | /s/ David Sidransky | |
| David Sidransky | ||
| Director | ||
| Date: | April 15, 2024 | |
| By: | /s/ Yaron Adler | |
| Yaron Adler | ||
| Director | ||
| Date: | April 15, 2024 | |
| By: | /s/ Ashish Nanda | |
| Ashish Nanda | ||
| Director | ||
| Date: | April 15, 2024 | |
| By: | /s/ Mario Philips | |
| Mario Philips | ||
| Director | ||
| Date: | April 15, 2024 |
| 83 |
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
ORGENESIS INC.
CONSOLIDATED FINANCIAL STATEMENTS AS OF DECEMBER 31, 2023
TABLE OF CONTENTS
| Page | |
| REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM (PCAOB name: Kesselman & Kesselman C.P.A.s; PCAOB ID: |
F-2 |
| CONSOLIDATED FINANCIAL STATEMENTS: | |
| Consolidated Balance Sheets | F-4 |
| Consolidated Statements of Comprehensive Loss (Income) | F-6 |
| Consolidated Statements of Changes in Equity | F-7 |
| Consolidated Statements of Cash Flows | F-9 |
| Notes to Consolidated Financial Statements | F-10 |
| F-1 |

Report of Independent Registered Public Accounting Firm
To the Board of Directors and shareholders of Orgenesis Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Orgenesis Inc and its subsidiaries (the “Company”) as of December 31, 2023 and 2022, and the related consolidated statements of comprehensive loss (income), changes in equity and cash flows for the years then ended, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years then ended in conformity with accounting principles generally accepted in the United States of America.
Changes in Accounting Principle
As discussed in note 2(x) to the consolidated financial statements, the Company changed the manner in which it accounts for credit losses.
Substantial Doubt about the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1b to the consolidated financial statements, the Company has suffered recurring losses from operations and has incurred cash outflows from operating activities that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1b. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| F-2 |
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that (i) relates to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Revenue recognition and accounts receivables – collectability criteria
As described in note 2 and 17 of the consolidated financial statements, total revenue recognized for the year ended December 31, 2023 was $530 thousand. The Company’s account receivable balance as of December 31, 2023 was $88 thousand and the related credit losses for the year then ended was $24,388 thousand. The Company recognizes revenue from services to its customers when control of the services is transferred to the customer for an amount, referred to as the transaction price, which reflects the consideration to which the Company is expected to be entitled in exchange for those goods or services. The Company applies the revenue guidance to contracts when it is probable that the Company will collect substantially all of the consideration to which it is entitled to in exchange for the goods and services it transfers to the customer. The Company considers historical collection experience for each of its customers and when revenue and accounts receivable are recorded. The Company also recognizes estimated expected credit losses over the life of the accounts receivables. The estimate of expected credit losses considers not only historical information, but also current and future economic conditions and events.
The principal considerations for our determination that performing procedures relating to revenue recognition and accounts receivables – collectability criteria are a critical audit matter are the high degree of auditor judgement and effort in performing procedures to evaluate management’s assumptions of the collectability criteria.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included, among others, testing management’s process for evaluating the collectability criteria, and the relevance of historical billing and collection data as an input to the analysis as well as current and future economic conditions and events; testing the accuracy of a sample of revenue transactions and a sample of cash collections from the historical billing data and the historical collection which is used in management’s analysis; and performing a retrospective comparison of actual cash collected to the prior year estimate of net accounts receivable.
| Certified Public Accountants (Isr.) | |
| A member of PricewaterhouseCoopers International Limited |
April 15, 2024
We have served as the Company’s auditor since 2012.
Kesselman
& Kesselman, Building 25, MATAM, P.O BOX 15084 Haifa, 3190500, Israel,
Telephone: +972 -4- 8605000, Fax: +972 -4- 8605001, www.pwc.com/il
| F-3 |
ORGENESIS INC.
CONSOLIDATED BALANCE SHEETS
(U.S. Dollars, in thousands, except share and per share amounts)
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Assets | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash | ||||||||
| Accounts receivable, net of credit losses of $ | ||||||||
| Prepaid expenses and other receivables | ||||||||
| Receivables from related parties | ||||||||
| Convertible loan | ||||||||
| Inventory | ||||||||
| Total current assets | ||||||||
| NON CURRENT ASSETS: | ||||||||
| Deposits | $ | $ | ||||||
| Equity investees | ||||||||
| Loans to associates | ||||||||
| Property, plants and equipment, net | ||||||||
| Intangible assets, net | ||||||||
| Operating lease right-of-use assets | ||||||||
| Goodwill | ||||||||
| Deferred tax | ||||||||
| Other assets | ||||||||
| Total non-current assets | ||||||||
| TOTAL ASSETS | $ | $ | ||||||
| F-4 |
ORGENESIS INC.
CONSOLIDATED BALANCE SHEETS
(U.S. Dollars, in thousands, except share and per share amounts)
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Liabilities and equity | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accounts payable related Parties | ||||||||
| Accrued expenses and other payables | ||||||||
| Income tax payable | ||||||||
| Employees and related payables | ||||||||
| Other payable related parties | ||||||||
| Advance payments on account of grant | ||||||||
| Short-term loans | ||||||||
| Current maturities of finance leases | ||||||||
| Current maturities of operating leases | ||||||||
| Short-term and current maturities of convertible loans | ||||||||
| TOTAL CURRENT LIABILITIES | ||||||||
| LONG-TERM LIABILITIES: | ||||||||
| Non-current operating leases | $ | $ | ||||||
| Convertible loans | ||||||||
| Retirement benefits obligation | ||||||||
| Finance leases | ||||||||
| Other long-term liabilities | ||||||||
| TOTAL LONG-TERM LIABILITIES | ||||||||
| TOTAL LIABILITIES | ||||||||
| REDEEMABLE NON-CONTROLLING INTEREST | ||||||||
| EQUITY (CAPITAL DEFICIENCY): | ||||||||
| Common stock of $ par value: Authorized at December 31, 2023 and December 31, 2022: shares; Issued at December 31, 2023 and December 31, 2022: and shares, respectively; Outstanding at December 31, 2023 and December 31, 2022: and shares, respectively. | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated other comprehensive income (loss) | ( | ) | ||||||
| Treasury stock shares as of December 31, 2023 and December 31, 2022 | ( | ) | ( | ) | ||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Equity attributable to Orgenesis Inc. | ( | ) | ||||||
| Non-controlling interests | ||||||||
| TOTAL EQUITY (CAPITAL DEFICIENCY) | ( | ) | ||||||
| TOTAL LIABILITIES, REDEEMABLE NON-CONTROLLING INTEREST AND EQUITY (CAPITAL DEFICIENCY) | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
ORGENESIS INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS (INCOME)
(U.S. Dollars, in thousands, except share and per share amounts)
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Revenues | $ | $ | ||||||
| Revenues from related party | ||||||||
| Total revenues | $ | $ | ||||||
| Cost of revenues | ||||||||
| Gross (loss) profit | $ | ( | ) | $ | ||||
| Cost of development services and research and development expenses | ||||||||
| Amortization of intangible assets | ||||||||
| Selling, general and administrative expenses included credit losses of $ | ||||||||
| Share in net loss of associated companies | ||||||||
| Impairment of investment | ||||||||
| Impairment of intangible assets | ||||||||
| Operating loss | $ | $ | ||||||
| Loss from deconsolidation of Octomera (see Note 3) | ||||||||
| Other income, net | ( | ) | ( | ) | ||||
| Credit loss on convertible loan receivable | ||||||||
| Loss from extinguishment in connection with convertible loan | ||||||||
| Financial expenses, net | ||||||||
| Loss before income taxes | $ | $ | ||||||
| Tax expense | ||||||||
| Net loss | $ | $ | ||||||
| Net (loss) income attributable to non-controlling interests | ( | ) | ||||||
| Net loss attributable to Orgenesis Inc. | $ | $ | ||||||
| Loss per share: | ||||||||
| Basic and diluted | $ | $ | ||||||
| Weighted average number of shares used in computation of Basic and Diluted loss per share: | ||||||||
| Basic and diluted | ||||||||
| Comprehensive loss: | ||||||||
| Net loss | $ | $ | ||||||
| Other Comprehensive loss – Translation adjustment | ||||||||
| Release of translation adjustment due to deconsolidation of Octomera | ( | ) | ||||||
| Comprehensive loss | $ | $ | ||||||
| Comprehensive (loss) income attributed to non-controlling interests | ( | ) | ||||||
| Comprehensive loss attributed to Orgenesis Inc. | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
ORGENESIS INC.
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY (CAPITAL DEFICIENCY)
(U.S. Dollars, in thousands, except share amounts)
| Common Stock | Accumulated | Equity Attributable | ||||||||||||||||||||||||||||||||||
| Number | Par Value | Additional Paid-in Capital | Other Comprehensive Income (loss) | Treasury Shares | Accumulated Deficit | to | Non- Controlling Interest | Total | ||||||||||||||||||||||||||||
Balance at January 1, 2023 | $ | $ | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | $ | $ | ||||||||||||||||||||||
| Changes during the Year ended December 31, 2023: | ||||||||||||||||||||||||||||||||||||
| Stock-based compensation to employees and directors | - | |||||||||||||||||||||||||||||||||||
| Stock-based compensation to service providers | - | |||||||||||||||||||||||||||||||||||
| Issuance of shares and warrants net of issuance costs | * | |||||||||||||||||||||||||||||||||||
| Issuance of Shares due to exercise of warrants | * | |||||||||||||||||||||||||||||||||||
| Issuance of warrants with respect to convertible loans | - | |||||||||||||||||||||||||||||||||||
| Extinguishment in connection with convertible loan restructuring | - | |||||||||||||||||||||||||||||||||||
| Deconsolidation of Octomera | - | ( | ) | |||||||||||||||||||||||||||||||||
| Adjustment to redemption value of redeemable non-controlling interest | - | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||||||||
| Comprehensive income (loss) for the period | - | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||||
| Balance at December 31, 2023 | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||||
| * |
The accompanying notes are an integral part of these consolidated financial statement
| F-7 |
ORGENESIS INC.
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
(U.S. Dollars, in thousands, except share amounts)
| Common Stock | Accumulated Other | Equity Attributable | ||||||||||||||||||||||||||||||||||
| Number | Par Value | Additional Paid-in Capital | Comprehensive Income (loss) | Treasury Shares | Accumulated Deficit | to | Non- Controlling Interest | Total | ||||||||||||||||||||||||||||
Balance at January 1, 2022 | $ | $ | $ | $ | ( | ) | $ | ( | ) | $ | $ | $ | ||||||||||||||||||||||||
| Changes during the Year ended December 31, 2022: | ||||||||||||||||||||||||||||||||||||
| Stock-based compensation to employees and directors | - | |||||||||||||||||||||||||||||||||||
| Stock-based compensation to service providers | - | |||||||||||||||||||||||||||||||||||
| Exercise of options | * | |||||||||||||||||||||||||||||||||||
| Issuance and modification of warrants with respect to convertible loans | ||||||||||||||||||||||||||||||||||||
| Extinguishment in connection with convertible loan restructuring | - | |||||||||||||||||||||||||||||||||||
| Issuance of Shares | * | |||||||||||||||||||||||||||||||||||
| Issuance of shares related to acquisition of Mida | * | |||||||||||||||||||||||||||||||||||
| Non- Controlling Interest arising from a business combination | - | ( | ) | ( | ) | |||||||||||||||||||||||||||||||
| Comprehensive income (loss) for the period | - | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||||||
| Balance at December 31, 2022 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||||||
| * |
The accompanying notes are an integral part of these consolidated financial statements.
| F-8 |
ORGENESIS INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS(*)
(U.S. Dollars, in thousands)
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments required to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation | ||||||||
| Capital gain, net | ( | ) | ||||||
| Loss from deconsolidation of Octomera | ||||||||
| Share in loss of associated companies, net | ||||||||
| Depreciation and amortization expenses | ||||||||
| Credit loss on convertible loan receivable | ||||||||
| Impairment of investment | ||||||||
| Impairment expenses of intangible assets | ||||||||
| Effect of exchange differences on inter-company balances | ||||||||
| Net changes in operating leases | ( | ) | ( | ) | ||||
| Interest expense accrued on loans and convertible loans | ||||||||
| Loss from extinguishment in connection with convertible loan restructuring | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | ( | ) | ||||||
| Prepaid expenses, other accounts receivable | ||||||||
| Inventory | ( | ) | ( | ) | ||||
| Other assets | ||||||||
| Related parties, net | ( | ) | ||||||
| Accounts payable | ( | ) | ||||||
| Accrued expenses and other payable | ||||||||
| Employee and related payables | ( | ) | ||||||
| Deferred taxes, net | ( | ) | ||||||
| Net cash used in operating activities | $ | ( | ) | $ | ( | ) | ||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Repayment of convertible loan to related party partners | ||||||||
| Decrease in loan to associate entities | ||||||||
| Increase in loan to associate entities | ( | ) | ||||||
| Repayment of loan granted | ||||||||
| Sale of property, plants and equipment | ||||||||
| Purchase of property, plants and equipment | ( | ) | ( | ) | ||||
| Investment in associated company | ( | ) | ||||||
| Cash acquired from acquisition of Mida | ||||||||
| Impact to cash resulting from deconsolidation (see Note 3) | ( | ) | ||||||
| Increase in cash from business combinations of TLABS and Orgenesis Austria | ||||||||
| Investment in long-term deposits | ( | ) | ( | ) | ||||
| Net cash used in investing activities | $ | ( | ) | $ | ( | ) | ||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from issuance of shares due to exercise of options and warrants (net of transaction costs) | ||||||||
| Proceeds from issuance of convertible loans | ||||||||
| Proceeds from transaction with redeemable non-controlling interest that do not result in a loss of control, see note 3 | ||||||||
| Repayment of convertible loans and convertible bonds | ( | ) | ( | ) | ||||
| Repayment of short and long-term debt | ( | ) | ( | ) | ||||
| Proceeds from issuance of loans payable | ||||||||
| Grant received in respect of third party | ||||||||
| Transfer of the grant received to third party | ( | ) | ||||||
| Net cash provided by financing activities | $ | $ | ||||||
| NET CHANGE IN CASH AND CASH EQUIVALENTS AND RESTRICTED CASH | ( | ) | ||||||
| EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS | $ | $ | ( | ) | ||||
| CASH AND CASH EQUIVALENTS AND RESTRICTED CASH AT BEGINNING OF YEAR | $ | $ | ||||||
| CASH, CASH EQUIVALENTS AND RESTRICTED CASH AT END OF YEAR | $ | $ | ||||||
| SUPPLEMENTAL NON-CASH FINANCING AND INVESTING ACTIVITIES | ||||||||
| Right-of-use assets obtained in exchange for new finance lease liabilities | $ | $ | ||||||
| Right-of-use assets obtained in exchange for new operation lease liabilities | $ | $ | ||||||
| Increase (decrease) in accounts payable related to purchase of property, plant and equipment | $ | $ | ( | ) | ||||
| Loan conversion for Redeemable non-controlling interest (See note 3) | $ | $ | ||||||
| Issuance of common stocks in connection with the acquisition of Mida | $ | $ | ||||||
| Extinguishment in connection with convertible loan restructuring | $ | |||||||
| CASH PAID DURING THE YEAR FOR: | ||||||||
| Interest | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-9 |
ORGENESIS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(US Dollars in Thousands)
NOTE 1 – DESCRIPTION OF BUSINESS
| a. | General |
Orgenesis Inc. (the “Company”) is a global biotech company working to unlock the potential of Cell and Gene Therapies (“CGTs”) in an affordable and accessible format. CGTs can be centered on autologous (using the patient’s own cells) or allogenic (using master banked donor cells) and are part of a class of medicines referred to as advanced therapy medicinal products (“ATMPs”). The Company is mostly focused on the development of autologous therapies that can be manufactured under processes and systems that are developed for each therapy using a closed and automated approach that is validated for compliant production near the patient for treatment of the patient at the point of care (“POCare”).
In connection with the investment by an affiliate of Metalmark Capital Partners (“Metalmark” or “MM”) in the Company’s subsidiary Octomera LLC (formerly Morgenesis LLC) (“Octomera” or “Morgenesis”) in November 2022 (“the Metalmark Investment”), the Company separated its operations into two operating segments: the operations of Octomera (the “Morgenesis” or “Octomera” segment) and therapies related activities (the “Therapies” segment).
On
June 30, 2023, in connection with an additional $
On
January 29, 2024, the Company and MM entered into a Unit Purchase Agreement (the “UPA”), pursuant to which the Company acquired
all of the preferred units of Octomera owned by MM (the “Acquisition”). Accordingly, the Company currently owns
These consolidated financial statements include the accounts of Orgenesis Inc. and its subsidiaries.
The
Company’s common stock, par value $ per share (the “Common Stock”), is listed and traded on the Nasdaq Capital
Market under the symbol “ORGS.” The Company must satisfy Nasdaq’s continued listing requirements, including, among
other things, a minimum closing bid price requirement of $
As used in this report and unless otherwise indicated, the term “Company” refers to Orgenesis Inc. and its Subsidiaries. Unless otherwise specified, all amounts are expressed in United States Dollars.
| b. | Liquidity |
Through
December 31, 2023, the Company had an accumulated deficit of $
| F-10 |
If there are further reductions in revenues or increases in operating costs for facilities expansion, research and development, commercial and clinical activity or decreases in revenues from customers, the Company will need to use mitigating actions such as to seek additional financing or postpone expenses that are not based on firm commitments. In addition, in order to fund the Company’s operations until such time that the Company can generate sustainable positive cash flows, the Company will need to raise additional funds.
The Company expects its current and projected cash resources and commitments will not be sufficient to meet the Company’s obligations for the next 12 months, raising a substantial doubt about the Company’s ability to continue as a going concern. Management plans include raising additional capital to fund the Company’s operations and to repay the Company’s outstanding loans when they become due, as well as exploring additional avenues to increase revenue and reduce capital expenditures. The Company’s ability to fund the completion of its ongoing and planned activities may be substantially dependent upon whether the Company can obtain sufficient funding at acceptable terms. If the Company is unable to raise sufficient additional capital or meet revenue targets, it may have to reduce or eliminate certain activities and reduce its headcount.
The estimation and execution uncertainty regarding the Company’s future cash flows and management’s judgments and assumptions in estimating these cash flows is a significant estimate. Those assumptions include reasonableness of the forecasted revenue, operating expenses, and uses and sources of cash.
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES
The
consolidated financial statements are prepared in accordance with accounting principles generally
accepted in the United States (“U.S. GAAP”).
a. Use of Estimates in the Preparation of Financial Statements
The preparation of the Company’s consolidated financial statements in conformity with U.S. GAAP requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, equity, revenues and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, the Company evaluates its estimates, judgments and methodologies. The Company bases its estimates on historical experience and on various other assumptions that it believes are reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity, the amount of revenues and expenses, determination of loss on deconsolidation, valuation of investments, goodwill impairment, and assessment of credit losses. Actual results could differ from those estimates.
b. Business Combination
The Company allocates the fair value of consideration transferred in a business combination to the assets acquired, liabilities assumed, and non-controlling interests in the acquired business based on their fair values at the acquisition date. All assets and liabilities are recognized in fair value. The purchase price allocation process requires management to make significant estimates and assumptions, especially at the acquisition date with respect to intangible assets. Direct transaction costs associated with the business combination are expensed as incurred. The excess of the fair value of the consideration transferred plus the fair value of any non-controlling interest in the acquiree over the fair value of the assets acquired, liabilities assumed in the acquired business is recorded as goodwill. The allocation of the consideration transferred in certain cases may be subject to revision based on the final determination of fair values during the measurement period, which may be up to one year from the acquisition date. The cumulative impact of revisions during the measurement period is recognized in the reporting period in which the revisions are identified. The Company includes the results of operations of the business that it has acquired in its consolidated results prospectively from the date of acquisition.
If the business combination is achieved in stages, the acquisition date carrying value of the acquirer’s previously held equity interest in the acquire is re-measured to fair value at the acquisition date; any gains or losses arising from such re-measurement are recognized in profit or loss.
| F-11 |
c. Cash Equivalents
The Company considers cash equivalents to be all short-term, highly liquid investments, which include money market instruments, that are not restricted as to withdrawal or use, and short-term bank deposits with original maturities of three months or less from the date of purchase that are not restricted as to withdrawal or use and are readily convertible to known amounts of cash.
d. Cost of development services and research and development expenses
Cost of development services and research and development expenses include costs directly attributable to the conduct of research and development activities, including the cost of salaries, stock-based compensation expenses, payroll taxes and other employees’ benefits, lab expenses, consumable equipment, courier fees, travel expenses, professional fees and consulting fees. All costs associated with research and developments are expensed as incurred. Participation from government departments and from research foundations for development of approved projects is recognized as a reduction of expense as the related costs are incurred. Research and development in-process acquired as part of an asset purchase, which has not reached technological feasibility and has no alternative future use, is expensed as incurred.
e. Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its Subsidiaries. All intercompany transactions and balances have been eliminated in consolidation.
f. Non-Marketable Equity Investments
The Company’s investments in certain non-marketable equity securities in which it has the ability to exercise significant influence, but it does not control through variable interests or voting interests. These are accounted for under the equity method of accounting and presented as Investment in associates, net, in the Company’s consolidated balance sheets. Under the equity method, the Company recognizes its proportionate share of the comprehensive income or loss of the investee. The Company’s share of income and losses from equity method investments is included in share in losses of associated company.
The Company periodically reviews equity method investments for impairment in value whenever events or changes in circumstances indicate that the carrying amount of such investments may not be recoverable. The Company will record an impairment charge to the extent that the estimated fair value of an investment is less than its carrying value and the Company determines the impairment is other-than-temporary. Impairment charges, if applicable, are recorded in “Share in net (losses) profits of associated companies”.
For other investments, the Company applies the measurement alternative upon the adoption of ASU 2016-01 and elected to record equity investments without readily determinable fair values at cost, less impairment, adjusted for subsequent observable price changes. In this measurement alternative method, changes in the carrying value of the equity investments are reflected in current earnings. Changes in the carrying value of the equity investment are required to be made whenever there are observable price changes in orderly transactions for the identical or similar investment of the same issuer.
g. Fair value measurement
The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below: Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. Level 2: Observable inputs that are based on inputs not quoted on active markets, but corroborated by market data. Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value.
| F-12 |
h. Functional Currency
The currency of the primary economic environment in which the operations of the Company and part of its Subsidiaries are conducted is the U.S. dollar (“$” or “dollar”). The functional currency of the Belgian Subsidiary is the Euro (“€” or “Euro”). Most of the Company’s expenses are incurred in dollars, and the source of the Company’s financing has been provided in dollars. Thus, the functional currency of the Company and its other subsidiaries is the dollar. Transactions and balances originally denominated in dollars are presented at their original amounts. Balances in foreign currencies are translated into dollars using historical and current exchange rates for nonmonetary and monetary balances, respectively. For foreign transactions and other items reflected in the statements of operations, the following exchange rates are used: (1) for transactions – exchange rates at transaction dates or average rates and (2) for other items (derived from nonmonetary balance sheet items such as depreciation) – historical exchange rates. The resulting transaction gains or losses are recorded as financial income or expenses. The financial statements of the Belgian Subsidiary is included in the consolidated financial statements, translated into U.S. dollars. Assets and liabilities are translated at year-end exchange rates, while revenues and expenses are translated at yearly average exchange rates during the year. Differences resulting from translation of assets and liabilities are presented as other comprehensive income.
i. Inventory
The Company’s inventory consists of raw material for use for the services provided. The Company periodically evaluates the quantities on hand. Cost of the raw materials is determined using the weighted average cost method. The inventory is recorded at the lower of cost or net realizable value.
j. Property, Plants and Equipment
Property, plants and equipment are recorded at cost and depreciated by the straight-line method over the estimated useful lives of the related assets.
Annual rates of depreciation are presented in the table below:
Weighted Average Useful Life (Years) |
||||
| Production facility | ||||
| Laboratory equipment | ||||
| Office equipment and computers |
k. Intangible assets
Intangible assets and their useful lives are as follows:
| Useful Life (Years) | Amortization Recorded at Comprehensive Loss Line Item | |||
| Technology | Amortization of intangible assets |
Intangible assets are recorded at acquisition less accumulated amortization and impairment. Definite lived intangible assets are amortized over their estimated useful life using the straight-line method, which is determined by identifying the period over which the cash flows from the asset are expected to be generated. The Company capitalizes IPR&D projects acquired as part of a business combination. On successful completion of each project, IPR&D assets are reclassified to developed technology and amortized over their estimated useful lives.
| F-13 |
l. Goodwill
Goodwill represents the excess of consideration transferred over the value assigned to the net tangible and identifiable intangible assets of businesses acquired. Goodwill is allocated to reporting units expected to benefit from the business combination. Goodwill is not amortized but rather tested for impairment at least annually in the fourth quarter, or more frequently if events or changes in circumstances indicate that goodwill may be impaired. Before the Octomera deconsolidation, the Company reallocated its goodwill into two identified operating units: Octomera and Therapies. Subsequent to the Octomera deconsolidation, the goodwill allocated to Octomera was derecognized. As of December 31, 2023 - goodwill is solely allocated to Therapies operating unit. Goodwill impairment is recognized when the quantitative assessment results in the carrying value exceeding the fair value, in which case an impairment charge is recorded to the extent the carrying value exceeds the fair value.
There
were
m. Impairment of Long-lived Assets
The Company reviews its property, plants and equipment, intangible assets subject to amortization and other long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset class may not be recoverable. Indicators of potential impairment include: an adverse change in legal factors or in the business climate that could affect the value of the asset; an adverse change in the extent or manner in which the asset is used or is expected to be used, or in its physical condition; and current or forecasted operating or cash flow losses that demonstrate continuing losses associated with the use of the asset. If indicators of impairment are present, the asset is tested for recoverability by comparing the carrying value of the asset to the related estimated undiscounted future cash flows expected to be derived from the asset. If the expected cash flows are less than the carrying value of the asset, then the asset is considered to be impaired and its carrying value is written down to fair value, based on the related estimated discounted cash flows. For indefinite life intangible assets, the Company performs an impairment test annually in the fourth quarter and whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. The Company determines the fair value of the asset based on discounted cash flows and records an impairment loss if its book value exceeds fair value.
Impairment
charges of IPR&D during the year ended December 31, 2022 were $
Impairment
charges of other investment during the year ended December 31, 2023 were $
n. Income Taxes
1) With respect to deferred taxes, income taxes are computed using the asset and liability method. Under the asset and liability method, deferred income tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and are measured using the currently enacted tax rates and laws. A valuation allowance is recognized to the extent that it is more likely than not that the deferred taxes will not be realized in the foreseeable future.
2)
The Company follows a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax
position for recognition by determining if the available evidence indicates that it is more likely than not that the position will
be sustained on examination. If this threshold is met, the second step is to measure the tax position as the largest amount that is
3) Taxes that would apply in the event of disposal of investment in Subsidiaries and associated companies have not been taken into account in computing the deferred income taxes, as it is the Company’s intention to hold these investments and not realize them.
The Company recognizes stock-based compensation for the estimated fair value of share-based awards. The Company measures compensation expense for share-based awards based on estimated fair values on the date of grant using the Black-Scholes option-pricing model. This option pricing model requires estimates as to the option’s expected term and the price volatility of the underlying stock. The Company amortizes the value of share-based awards to expense over the vesting period on a straight-line basis.
| F-14 |
p. Redeemable Non-controlling Interest
Non-controlling interests with embedded redemption features, whose settlement is not at the Company’s discretion, are considered redeemable non-controlling interest. Redeemable non-controlling interests are considered to be temporary equity and are therefore presented as a mezzanine section between liabilities and equity on the Company’s consolidated balance sheets. Redeemable non-controlling interests are measured at the greater of the initial carrying amount adjusted for the non-controlling interest’s share of comprehensive income or loss or its redemption value. Subsequent adjustment of the amount presented in temporary equity is required only if the Company’s management estimates that it is probable that the instrument will become redeemable. Adjustments of redeemable non-controlling interest to its redemption value are recorded through additional paid-in capital.
Basic net loss (income) per share is computed by dividing the net loss (income) for the period by the weighted average number of shares of common stock outstanding for each period. Diluted net income per share is based upon the weighted average number of common shares and of common shares equivalents outstanding when dilutive. Common share equivalents include: (i) outstanding stock options, RSUs and warrants which are included under the treasury share method when dilutive, and (ii) common shares to be issued under the assumed conversion of the Company’s outstanding convertible loans and debt, which are included under the if-converted method when dilutive (See Note 14).
r. Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist of principally cash and cash equivalents, bank deposits and certain receivables. The Company held these instruments with highly rated financial institutions and the Company has not experienced any significant credit losses in these accounts and does not believe the Company is exposed to any significant credit risk on these instruments apart of accounts receivable. The Company performs ongoing credit evaluations of its customers for the purpose of determining the appropriate allowance for doubtful accounts.
The Company’s accounts receivable accounting policy until December 31, 2022, prior to the adoption of the new Current Expected Credit Losses (“CECL”) standard, created bad debts when objective evidence existed of inability to collect all sums owed it under the original terms of the debit balances. Material customer difficulties, the probability of their going bankrupt or undergoing economic reorganization and insolvency, material delays in payments and other objective considerations by management that indicate expected risk of payment were all considered indicative of reduced debtor balance value. Effective January 1, 2023, the Company adopted the new CECL standard.
The Company maintains the allowance for estimated losses resulting from the inability of the Company’s customers to make required payments. The Company considers historical collection experience for each of its customers and when revenue and accounts receivable are recorded. The Company also recognizes estimated expected credit losses over the life of the accounts receivables. The estimate of expected credit losses considers not only historical information, but also current and future economic conditions and events.
The Company repurchases its common stock from time to time on the open market and holds such shares as treasury stock. The Company presents the cost to repurchase treasury stock as a reduction of shareholders’ equity. The Company did not reissue nor cancel treasury shares during the year ended December 31, 2023 and December 31, 2022.
| F-15 |
t. Other Comprehensive Loss
Other comprehensive loss represents adjustments of foreign currency translation.
u. Revenue from Contracts with Customers
The Company’s agreements are primarily service and processing contracts, the performance obligations of which range in duration from a few months to one year. The Company applies the revenue guidance to contracts when control of the services is transferred to the customer for an amount, referred to as the transaction price, which reflects the consideration to which the Company is expected to be entitled in exchange for those goods or services and when it is probable that the Company will collect substantially all of the consideration to which it is entitled in exchange for the goods and services it transfers to the customer.
The Company does not adjust the promised amount of consideration for the effects of a significant financing component since the Company expects, at contract inception, that the period between the time of transfer of the promised goods or services to the customer and the time the customer pays for these goods or services to be generally one year or less. The Company’s credit terms to customers are in average between thirty and one hundred and fifty days.
Nature of Revenue Streams
The Company has four main revenue streams, which are License fees, POCare development services, cell process development services, including hospital supplies, and POCare cell processing.
License fees
Revenue recognized under license fees are recognized upon the confirmation of licensee of milestones completed and certainty of payment of the license fee.
POCare Development Services
Revenue recognized under contracts for POCare development services may, in some contracts, represent multiple performance obligations (where promises to the customers are distinct) in circumstances in which the work packages are not interrelated or the customer is able to complete the services performed.
For arrangements that include multiple performance obligations, the transaction price is allocated to the identified performance obligations based on their relative standalone selling prices.
The Company recognizes revenue when, or as, it satisfies a performance obligation. At contract inception, the Company determines whether the services are transferred over time or at a point in time. Performance obligations that have no alternative use and that the Company has the right to payment for performance completed to date, at all times during the contract term, are recognized over time. All other performance obligations are recognized as revenues by the Company at a point of time (upon completion). Revenues from support services provided to the Company’s customers are recognized as and when the services are provided, because the customer simultaneously receives and consumes the benefits provided.
Significant Judgement and Estimates
Significant judgment is required to identifying the distinct performance obligations and estimating the standalone selling price of each distinct performance obligation and identifying which performance obligations create assets with alternative use to the Company, which results in revenue recognized upon completion, and which performance obligations are transferred to the customer over time, and the estimate of credit losses.
| F-16 |
Cell Process Development Services
Revenue recognized under contracts for cell process development services may, in some contracts, represent multiple performance obligations (where promises to the customers are distinct) in circumstances in which the work packages and milestones are not interrelated or the customer is able to complete the services performed independently or by using competitors of the Company. In other contracts when the above circumstances are not met, the promises are not considered distinct, and the contract represents one performance obligation. All performance obligations are satisfied over time, as there is no alternative use to the services it performs, since, in nature, those services are unique to the customer, which retain the ownership of the intellectual property created through the process.
For arrangements that include multiple performance obligations, the transaction price is allocated to the identified performance obligations based on their relative standalone selling prices. For these contracts, the standalone selling prices are based on the Company’s normal pricing practices when sold separately with consideration of market conditions and other factors, including customer demographics and geographic location.
The Company measures the revenue to be recognized over time on a contract-by-contract basis, determining the use of either a cost-based input method or output method, depending on whichever best depicts the transfer of control over the life of the performance obligation.
Included in cell process development services is hospital supplies revenue, which is derived principally from the performance of services to hospitals or other medical providers. Revenue is earned and recognized when product and services are received by the customer.
POCare Cell Processing
Revenues from POCare Cell processing represent performance obligations which are recognized either over, or at a point of time. The progress towards completion is measured on an output measure based on direct measurement of the value transferred to the customer (units produced).
Change Orders
Changes in the scope of work are common and can result in a change in transaction price, equipment used and payment terms. Change orders are evaluated on a contract-by-contract basis to determine if they should be accounted for as a new contract or as part of the existing contract. Generally, services from change orders are not distinct from the original performance obligation. As a result, the effect that the contract modification has on the contract revenue, and measure of progress, is recognized as an adjustment to revenue when they occur.
v. Leases
The
Company determines if an arrangement is a lease at inception. Lease classification is governed by five criteria in ASC 842-10-25-2. If
any of these five criteria is met, The Company classifies the lease as a finance lease; otherwise, the Company classifies the lease as
an operating lease.
Operating leases are included in operating lease right-of-use (“ROU”) assets and operating lease liabilities in the consolidated balance sheet.
Finance leases are included in property, plants and equipment, net and finance lease liabilities in the consolidated balance sheet.
ROU assets represent Orgenesis’ right to use an underlying asset for the lease term and lease liabilities represent its obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at the commencement date based on the present value of lease payments over the lease term. The Company uses its incremental borrowing rate based on the information available at the commencement date to determine the present value of the lease payments.
| F-17 |
The standard also provides practical expedients for an entity’s ongoing accounting. The Company elected the short-term lease recognition exemption for all leases with a term shorter than 12 months. This means that for those leases, the Company does not recognize ROU assets or lease liabilities but recognizes lease expenses over the lease term on a straight-line basis.
Lease terms will include options to extend or terminate the lease when it is reasonably certain that Orgenesis will exercise or not exercise the option to renew or terminate the lease.
w. Segment reporting
Since the Metalmark Investment, the Company’s business includes two reporting segments: Octomera and Therapies. See note 5.
x. Recently adopted accounting pronouncements
In June 2016, the FASB issued ASU 2016-13 “Financial Instruments—Credit Losses—Measurement of Credit Losses on Financial Instruments.” This guidance replaces the current incurred loss impairment methodology with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. The guidance will be effective for Smaller Reporting Companies (SRCs, as defined by the SEC) for the fiscal year beginning on January 1, 2023, including interim periods within that year. The adoption of this guidance did not have a material impact on the Company’s consolidated financial statements.
In October 2021, the FASB issued ASU 2021-08 “Business Combinations (Topic 805), Accounting for Contract Assets and Contract Liabilities from Contracts with Customers”, which requires contract assets and contract liabilities acquired in a business combination to be recognized and measured by the acquirer on the acquisition date in accordance with ASC 606, Revenue from Contracts with Customers. The guidance will result in the acquirer recognizing contract assets and contract liabilities at the same amounts recorded by the acquiree. The guidance should be applied prospectively to acquisitions occurring on or after the effective date. The guidance is effective for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years. Early adoption is permitted, including in interim periods, for any financial statements that have not yet been issued. The adoption of this guidance did not have a material impact on the Company’s consolidated financial statements.
y. Recently issued accounting pronouncements, not yet adopted
On August 23, 2023, the FASB issued guidance requiring a joint venture to initially measure all contributions received upon its formation at fair value. This accounting will largely be consistent with ASC 805, Business Combinations, although there are some specific exceptions. Before the ASU, there was no authoritative guidance in US GAAP that addressed how a joint venture should recognize contributions received. As a result, there has been diversity in practice, with some joint ventures accounting for contributions received at carry over basis and others at fair value. This new guidance is intended to reduce diversity in practice and provide users of the joint venture’s financial statements with more decision-useful information. It may also reduce the amount of basis differences that an investor in a joint venture needs to track. The new guidance should be applied prospectively and is effective for all newly-formed joint venture entities with a formation date on or after January 1, 2025, with early adoption permitted. The adoption of this guidance will not have a material impact on the Company’s consolidated financial statements.
In November 2023, the FASB issued ASU 2023-07 “Segment Reporting: Improvements to Reportable Segment Disclosures”. This guidance expands public entities’ segment disclosures primarily by requiring disclosure of significant segment expenses that are regularly provided to the chief operating decision maker and included within each reported measure of segment profit or loss, an amount and description of its composition for other segment items, and interim disclosures of a reportable segment’s profit or loss and assets. The guidance is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024, with early adoption permitted. The amendments are required to be applied retrospectively to all prior periods presented in an entity’s financial statements. The Company is currently evaluating this guidance to determine the impact it may have on its consolidated financial statements related disclosures.
| F-18 |
In December 2023, the FASB issued ASU 2023-09 “Income Taxes (Topic 740): Improvements to Income Tax Disclosures”. This guidance is intended to enhance the transparency and decision-usefulness of income tax disclosures. The amendments in ASU 2023-09 address investor requests for enhanced income tax information primarily through changes to disclosure regarding rate reconciliation and income taxes paid both in the U.S. and in foreign jurisdictions. ASU 2023-09 is effective for fiscal years beginning after December 15, 2024 on a prospective basis, with the option to apply the standard retrospectively. Early adoption is permitted. The Company is currently evaluating this guidance to determine the impact it may have on its consolidated financial statements disclosures.
NOTE 3 – REDEEMABLE NON-CONTROLLING INTEREST AND DECONSOLIDATION
Metalmark Investment in Octomera LLC
On November 4, 2022, the Company and MM OS Holdings, L.P. (“MM”), an affiliate of Metalmark Capital Partners (“Metalmark”), entered into a series of definitive agreements (“MM agreement”) intended to finance, strengthen and expand the Company’s POCare Services business (the “Metalmark Investment”).
Pursuant
to the Unit Purchase Agreement (the “UPA”), MM agreed to purchase Class A Preferred Units of Octomera (the “Class
A Units”), which represented
If
(a) Octomera and its subsidiaries generate Net Revenue (as defined in the UPA) equal to or greater than $
The
Company’s stockholders approved the LLC agreement terms at its annual meeting of stockholders held in June 2023. However, Octomera
and its subsidiaries did not meet the Net Revenue milestones for either of the years ended December 31, 2022 and 2023. During 2023, the
Company and MM entered into various amendments to the Unit Purchase Agreement, dated November 4, 2022 (the “UPA”). Pursuant
to such amendments, MM or the Company as the case may be, agreed to pay certain amounts in exchange for Class A Preferred Units of Octomera
to support the continued expansion of Orgenesis’ POCare Services (the “Subsequent Investment”). In the case of MM investments,
the investment amount of the First Future Investment (as defined in the UPA) was reduced by the amount of the Subsequent Investment.
MM invested $
| F-19 |
The Preferred Units have voting rights, may be converted into ordinary shares, and are prioritized over ordinary shares in case of dividend or redemption. The Company considers the provisions of Accounting Standards Codification Distinguishing Liabilities from Equity (“ASC 480”) in order to determine whether the Preferred Units should be classified as a liability. If the instrument is not within the scope of ASC 480, the Company further analyzes the instrument’s characteristics in order to determine whether it should be classified within temporary equity (mezzanine) or within permanent equity in accordance with the provisions of ASC 480-10-S99. The preferred units are not mandatorily or currently redeemable. However, they include a liquidation or deemed liquidation event that would constitute a redemption event that is outside of the Company’s control. As such, all redeemable preferred units have been presented outside of permanent equity as a redeemable non-controlling interest.
The Company further analyzed and concluded that the future Preferred Units investments are considered embedded in the initial Preferred Units that were issued and are considered clearly and closely related to the host instrument and therefore should not be bifurcated.
As
a result of the deconsolidation (see Note 1a), the Company recorded a net loss of $
| (in thousands) | ||||
| Fair value of the retained interest in Octomera | $ | |||
Net assets deconsolidated | ||||
| Release of translation adjustment | ||||
| Net profit | $ |
The change in board composition does not constitute a strategic shift from the Company’s perspective and therefore the Company did not treat the deconsolidation as a discontinued operation.
Following
the Amendment No. 2, the Company accounted for its investment in Octomera according to the equity method in accordance with ASC Topic
323, as it has retained the ability to exercise significant influence but does not control the entity. The Company thus recognized an
equity method investment in a total amount of $
In evaluating the fair value of the Octomera Equity Investment under the income approach, the Company used a discounted cash flow model of the business, adjusted to the Company’s share in the investment. Key assumptions used to determine the estimated fair value included: (a) internal cash flows forecasts for 5 years following the assessment date, including expected revenue growth, costs to produce, operating profit margins and estimated capital needs; (b) an estimated terminal value using a terminal year long-term future growth determined based on the growth prospects of the reporting units; and (c) a discount rate which reflects the weighted average cost of capital adjusted for the relevant risk associated with the Company’s reporting unit operations and the uncertainty inherent in the Company’s internally developed forecasts. The allocation of the purchase price to the net assets acquired and liabilities assumed resulted in the recognition of other intangible assets, net, which comprised of technology. The useful life of the technology for amortization purposes was determined by considering the period of expected cash flows generated by the assets used to measure the fair value of the intangible assets, adjusted as appropriate for the entity-specific factors including legal, regulatory, contractual, competitive, economic, or other factors that may limit the useful life of intangible assets.
| F-20 |
The following table represents the deconsolidated amounts from the Company’s Balance Sheet at the date of deconsolidation:
| (in thousands) | ||||
| ASSETS: | ||||
| Cash and cash equivalents | ||||
| Other current assets | ||||
| Non-current assets | ||||
| TOTAL ASSETS | ||||
| LIABILITIES: | ||||
| Current liabilities | ||||
| Long-term liabilities | ||||
| TOTAL LIABILITIES | ||||
| REDEEMABLE NON-CONTROLLING INTEREST | ||||
NON-CONTROLLING INTEREST | ||||
| NET ASSETS DECONSOLIDATED |
On January 29, 2024, the Company and MM entered into a Unit Purchase Agreement (the “UPA”), pursuant to which the Company acquired all of the equity interests of Octomera that were owned by MM (the “Acquisition”). In consideration for such Acquisition, the Company and MM agreed to the following consideration:
Royalty
Payments: If Octomera and its subsidiaries generate Net Revenue during the three year period (2025-2027), then the Company will pay
Milestone
Payments: If the Company sells Octomera within ten years from the date of the Closing at a price that is more than $
Pursuant to the acquisition, MM’s designated members of the Board of Managers of Octomera resigned and the Company amended the Second Amended and Restated Limited Liability Company Agreement of Octomera to be a single member agreement to reflect the transactions contemplated by the UPA so that MM shall no longer (i) be a party to such agreement, (ii) have a right to appoint members of the board of managers of Octomera or (iii) be a member of Octomera.
In
addition, the outstanding indebtedness payable from Orgenesis Maryland LLC to MM pursuant to an aggregate of 10 secured promissory notes
(the “Notes”) with a collective original principal amount of $
NOTE 4 – EQUITY INVESTMENTS AND LOANS TO ASSOCIATES
As
of December 31, 2023, and December 31, 2022, the balances of our equity-method investments were $
| a. | Octomera LLC |
The
Company owned approximately
As
of December 31, 2023, the balance of our equity-method investment related to Octomera was approximately $
| F-21 |
The following table presents summarized results of operations for the six months since the date of deconsolidation:
| Six-Months Ended | ||||
| December 31, 2023 | ||||
| Total revenue | $ | |||
| Gross loss | $ | |||
| Net loss | $ | |||
| b. | Butterfly Biosciences Sarl |
During
2020, the Company and Kidney Cure (“KC”), pursuant to the Kidney Cure JVA incorporated the KC JV Entity known as Butterfly
Biosciences Sarl (“BB”) in Switzerland. BB will be involved in the (i) implementation of a point-of-care strategy; (ii) assessment
of the options for development and manufacture of various cell-based types (including kidney derived cells, MSC cells, exosomes, gene
therapies) development; and (iii) development of protocols and tests for kidney therapies. The Company holds a
| c. | RevaCel |
During
2021, the Company and Revatis S.A (“Revatis”), pursuant to the Revatis JVA (See Note 11) incorporated the Revatis JV Entity
known as RevaCel Srl (“RevaCel”) in Belgium. RevaCel will develop products in the field of muscle-derived mesenchymal stem/progenitor
cells. The Company holds a
The table below sets forth a summary of the changes in the investments and loans for the years ended December 31, 2023 and December 31, 2022
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Opening balance | $ | $ | ||||||
| Investments during the period | ||||||||
| Loan granted to associates | ||||||||
| Repayment of loan | ( | ) | ||||||
| Business Combinations | ( | ) | ||||||
| Fair value of the retained interest in Octomera (see Note 3) | ||||||||
| Interest from loans to associates | ||||||||
| Share in net loss of associated companies | ( | ) | ( | ) | ||||
| Exchange rate differences | ( | ) | ||||||
| Total | $ | $ | ||||||
| F-22 |
NOTE 5 – SEGMENT INFORMATION
The Octomera operations segment includes mainly POCare Services, while the Therapies segment includes the Company’s therapeutic development operations. The segment information includes all the results of the Octomera segment up to the effective date of deconsolidation.
Because the Company conducted all its operations as one segment prior to the Metalmark Investment, the above changes were reflected through retroactive revision of prior period segment information based on the subsidiaries that were transferred to Octomera. Certain activities of these subsidiaries have changed after they were transferred to Octomera operations segment.
The Company’s Chief Executive Officer (“CEO”), who is the chief operating decision maker (“CODM”), reviews financial information prepared on a consolidated basis, accompanied by disaggregated information about revenues and contributed profit by the two identified reportable segments, namely Octomera and Therapies, to make decisions about resources to be allocated to the segments and assess their performance.
The Company does not review assets by segment. Therefore, the measure of assets has not been disclosed for each segment.
Segment data for the year ended December 31, 2023 is as follows:
| Octomera | Therapies | Eliminations | Consolidated | |||||||||||||
| (in thousands) | ||||||||||||||||
| Revenues | $ | $ | $ | ( | ) | $ | ||||||||||
| Cost of revenues* | ( | ) | ( | ) | ( | ) | ||||||||||
| Gross profit (loss) | ( | ) | ( | ) | ( | ) | ||||||||||
| Cost of development services and research and development expenses* | ( | ) | ( | ) | ( | ) | ||||||||||
| Operating expenses* | ( | ) | ( | ) | ( | ) | ||||||||||
| Impairment of investment | ( | ) | ( | ) | ||||||||||||
| Share in net income of associated companies | ( | ) | ( | ) | ( | ) | ||||||||||
Profit from deconsolidation | ( | ) | ( | ) | ||||||||||||
| Other income, net | ||||||||||||||||
| Depreciation and amortization | ( | ) | ( | ) | ( | ) | ||||||||||
| Credit loss on convertible loan receivable | ( | ) | ( | ) | ||||||||||||
| Loss from extinguishment in connection with convertible loan | ( | ) | ( | ) | ||||||||||||
| Financial Expenses, net | ( | ) | ( | ) | ( | ) | ||||||||||
| Income (loss) before income taxes | $ | ( | ) | $ | ( | ) | $ | $ | ( | ) | ||||||
| * |
| F-23 |
Segment data for the year ended December 31, 2022 is as follows:
| Octomera | Therapies | Eliminations | Consolidated | |||||||||||||
| (in thousands) | ||||||||||||||||
| Revenues | $ | $ | $ | ( | ) | $ | ||||||||||
| Revenues from related party | ||||||||||||||||
| Total revenues | ( | ) | ||||||||||||||
| Cost of revenues* | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Gross profit (loss) | ( | ) | ||||||||||||||
| Cost of development services and research and development expenses* | ( | ) | ( | ) | ( | ) | ||||||||||
| Operating expenses* | ( | ) | ( | ) | ( | ) | ||||||||||
| Impairment expenses | ( | ) | ( | ) | ( | ) | ||||||||||
| Share in net income of associated companies | ( | ) | ( | ) | ( | ) | ||||||||||
| Other income, net | ||||||||||||||||
| Depreciation and amortization | ( | ) | ( | ) | ( | ) | ||||||||||
| Loss from extinguishment in connection with convertible loan | ( | ) | ( | ) | ||||||||||||
| Financial Expenses, net | ( | ) | ( | ) | ( | ) | ||||||||||
| Income (loss) before income taxes | $ | $ | ( | ) | $ | $ | ( | ) | ||||||||
| * |
NOTE 6 – EQUITY
| a. | Financings |
In March 2022, the Company entered into a Securities Purchase Agreement (the “Purchase Agreement) with certain
investors (collectively, the “Investors”), pursuant to which the Company agreed to issue and sell to the Investors, in a private
placement (the “Offering”), shares of the Company’s Common Stock at a purchase price of $ per share and warrants
to purchase shares of Common Stock at an exercise price of $
On
February 23, 2023, the Company entered into a securities purchase agreement with certain institutional and accredited investors relating
to the issuance and sale of shares of Common Stock and warrants to purchase up to
All of the Warrants were exercised using the alternate cashless exercise option described above.
On
August 31, 2023, the Company entered into a Securities Purchase Agreement with a certain accredited investor, pursuant to which the Company
agreed to issue and sell, in a private placement (the “August 2023 Offering”), shares of the Company’s Common
Stock at a purchase price of $ per share. The Company received proceeds of $
On
November 8, 2023, the Company entered into a Securities Purchase Agreement with an institutional investor, pursuant to which the Company
agreed to issue and sell, in a registered direct offering by the Company directly to the investor (the “November 2023 Offering”),
(i) shares of Common Stock, and (ii) warrants exercisable for
| F-24 |
On
March 8, 2024, the institutional investor exercised
| b. | Warrants |
A summary of the Company’s warrants granted to investors and as finder’s fees as of December 31, 2023, and December 31, 2022 and changes for the periods then ended is presented below:
| December 31, | ||||||||||||||||
| 2023 | 2022 | |||||||||||||||
Number of Warrants | Weighted Average Exercise Price $ | Number of Warrants | Weighted Average Exercise Price $ | |||||||||||||
Warrants outstanding at the beginning of the period | ||||||||||||||||
| Changes during the period: | ||||||||||||||||
| Issued | ||||||||||||||||
| Exercised | ( | ) | ||||||||||||||
| Expired | ( | ) | ( | ) | ||||||||||||
| Warrants outstanding and exercisable at end of the period* | ||||||||||||||||
Amendment, Consent and Waiver Agreement
In
October and November 2022, the Company and certain investors that were parties to the Securities Purchase Agreement of March 2022 (the
“SPA”) and the Registration Rights Agreement of March 2022 (the “RRA”), entered into an Amendment, Consent and
Waiver Agreement (the “RRA Amendment”). Pursuant to the RRA Amendment, the Company and the investors agreed to an extension
of the date for filing the Registration Statement to register the Registrable Securities (as defined in the RRA) to April 3, 2023 and
the effective date of such Registration Statement as provided for in the RRA Amendment; and (to) waive any potential damages or claims
under the RRA with respect to the Company’s obligations under the RRA or SPA and release the Company therefrom. In consideration
for such consent, agreement, waiver and release, the Company agreed to issue additional warrants to purchase an aggregate of
shares of Common Stock to the investors (the “Additional PIPE Warrants”) and such Additional PIPE Warrants shall have an
exercise price of $
| c. | Purchase of Mida Biotech BV |
During February 2022, pursuant to the joint venture agreement between the
Company and Mida Biotech BV (“Mida”), the Company purchased all the issued shares of Mida for a consideration of $
NOTE 7 – PROPERTY, PLANTS AND EQUIPMENT
The following table represents the components of property, plants and equipment:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Cost: | ||||||||
| Production facility | $ | $ | ||||||
| Office furniture and computers | ||||||||
| Lab equipment | ||||||||
| Advance payment | ||||||||
| Subtotal | ||||||||
| Less – accumulated depreciation | ( | ) | ( | ) | ||||
| Total | $ | $ | ||||||
| F-25 |
Depreciation
expense for the years ended December 31, 2023 and December 31, 2022 were $
Property, plants and equipment, net by geographical location were as follows:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Belgium | $ | $ | ||||||
| Greece | ||||||||
| Netherlands | ||||||||
| Korea | ||||||||
| Israel | ||||||||
| U.S. | ||||||||
| Total | $ | $ | ||||||
NOTE 8 – INTANGIBLE ASSETS AND GOODWILL
Changes in the carrying amount of the Company’s goodwill for the years ended December 31, 2023 and 2022 are as follows:
| (in thousands) | ||||
| Goodwill as of December 31, 2021 | $ | |||
| Translation differences | ( | ) | ||
| Goodwill as of December 31, 2022 | $ | |||
| Deconsolidation of Octomera | ( | ) | ||
| Translation differences | ( | ) | ||
| Goodwill as of December 31, 2023 | $ | |||
Goodwill impairment assessment for the year ended December 31, 2023
As of December 31, 2022, the Company performed an impairment analysis for its reporting units. Based on the Company’s assessment, it was concluded that the fair value of each of the Octomera and Therapies reporting units exceeded their carrying amounts and therefore no goodwill impairment was required. As of December 31, 2023 the fair value of the Therapies reporting unit exceeded its carrying amount and therefore no goodwill impairment was required.
In evaluating the fair value of reporting units under the income approach, the Company used a discounted cash flow model. Key assumptions used to determine the estimated fair value included: (a) internal cash flows forecasts for 5 years following the assessment date, including expected revenue growth, costs to produce, operating profit margins and estimated capital needs; (b) an estimated terminal value using a terminal year long-term future growth determined based on the growth prospects of the reporting units; and (c) a discount rate which reflects the weighted average cost of capital adjusted for the relevant risk associated with the Company’s reporting unit operations and the uncertainty inherent in the Company’s internally developed forecasts.
Actual results may differ from those assumed in the Company’s valuation method. It is reasonably possible that the Company’s assumptions described above could change in future periods. If any of these were to vary materially from the Company’s plans, it may record impairment of goodwill allocated to any of these reporting units in the future.
| F-26 |
Other Intangible Assets
Other intangible assets consisted of the following:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Gross Carrying Amount: | ||||||||
| Know How | $ | $ | ||||||
| Customer relationships | ||||||||
| Technology | ||||||||
| Subtotal | ||||||||
| Less – Accumulated amortization | ( | ) | ( | ) | ||||
| Net carrying amount of other intangible assets | $ | $ | ||||||
Intangible
assets amortization expenses were approximately $
Estimated aggregate amortization expenses for the five succeeding years ending on December 31st are as follows:
| 2024 | 2025 to 2028 | |||||||
| (in thousands) | ||||||||
| Amortization expenses | $ | $ | ||||||
NOTE 9 –LOANS
On
July 25, 2023, the Israeli subsidiary received a loan from an offshore investor in the amount of $
On
August 15, 2023, the Company received a loan from an investor in the amount of $
During
October and November 2023, the Koligo subsidiary received loans in the amount of $
In February 2024, Koligo received a loan from a lender in the amount of the $
On
March 26, 2024, Koligo received a loan from a lender in the amount of $
During
April 2024 Koligo and the Israeli subsidiary received one month
| F-27 |
NOTE 10 – CONVERTIBLE LOANS
| a. | Long-Term Convertible Loans |
The tables below summarize the Company’s outstanding convertible loans as of December 31, 2023 and December 31, 2022 respectively:
Principal | Issuance Date | Current Interest Rate | Current Maturity | Current Conversion Price of loan into equity | ||||||||||||||
| Amount | (Year) | % | (Year) | $ | ||||||||||||||
| Convertible Loans Outstanding as of December 31, 2023 | ||||||||||||||||||
| $ | % | |||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ** | |||||||||||||||||
| % | ||||||||||||||||||
| % | * | |||||||||||||||||
| $ | ||||||||||||||||||
| * | ||
| ** |
Convertible Loans Outstanding as of December 31, 2022
| $ | % | |||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| $ |
Convertible Loans repaid during the year ended December 31, 2023
| Principal Amount | Issuance Year | Interest Rate | Maturity Period | Exercise Price | ||||||||||||||
| % | $ | |||||||||||||||||
Convertible Loans repaid during the year ended December 31, 2022
| Principal Amount | Issuance Year | Interest Rate | Maturity Period | Exercise Price | ||||||||||||||
| % | $ | |||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| % | ||||||||||||||||||
| F-28 |
Convertible Loans Entered into in 2023
On
January 10, 2023 (the “Effective Date”), the Company entered into the following agreements: (i) a convertible loan agreement
(the “NewTech Convertible Loan Agreement”) with NewTech Investment Holdings, LLC (the “NewTech Lender”), pursuant
to which the NewTech Lender loaned the Company $
The
terms of the NewTech Convertible Loan Agreement and the Malik Loan Agreement are identical. Interest is calculated at
At
any time prior to or on the Maturity Date, any Lender may provide the Company with written notice to convert all or part of the Outstanding
Amount into shares of our Common Stock equal to the quotient obtained by dividing (x) the Outstanding Amount by (y) a price equal to
$
Under the terms of the Convertible Loan Agreements, the Company used the proceeds from the Loan Amount to (i) redeem the loan amount from the previously disclosed Convertible Loan Agreement, dated as of May 19, 2022 between Orgenesis and Ricky Steven Neumann, as amended by the previously disclosed certain Convertible Loan Extension Agreement, dated as of October 23, 2022, by and between Orgenesis and Ricky Steven Neumann, and (ii) for general corporate purposes. Pursuant to the terms, the Company repaid said loan upon receipt of the Loan Amount.
In
connection with such loan, the Company agreed to issue the NewTech Lender warrants representing the right to purchase
Koligo Convertible Loan
On
March 27, 2023, the Company’s subsidiary Koligo Therapeutics Inc. (“Borrower”), entered into a convertible loan agreement
(the “Convertible Loan Agreement”) with Yehuda Nir (the “Lender,” and together with the Borrower, the “Parties”),
pursuant to which the Lender agreed to loan the Borrower up to $
| F-29 |
The
Parties agreed that the Lender shall have the option to assign $
Under
the terms of the Koligo Convertible Loan Agreement, the Borrower agreed to use the Loan Amount to fund working capital and ongoing operations
and for no other purposes unless the Lender agrees in writing. As of December 31, 2023, Koligo received $
In
January 2024, the Company and Lender agreed to extend the maturity date of the loan amount to December 31, 2026. The Company awarded
warrants to purchase
On
September 29, 2023, Borrower entered into another convertible loan agreement (the “Sai Convertible Loan Agreement”) with
Sai Traders (the “Lender,” and together with the Borrower, the “Parties”), pursuant to which the Lender agreed
to loan the Borrower up to $
Under
the terms of the Sai Convertible Loan Agreement, at the option of the Lender at the Maturity Date or any time prior, the Outstanding
Amount may be convertible, in whole or in part, into the number of shares of Common Stock of the Company equal to the quotient obtained
by dividing (x)
As of the date of this report, no part of the Loan was received, and was therefore not reflected in the Consolidated Balance sheet of December 31, 2023.
| F-30 |
Extension of Existing Loan Agreements
On
January 12, 2023, the Company entered into (i) a Convertible Credit Line and Unsecured Convertible Note Extension #2 Agreement with Yosef
Dotan (the “Dotan Extension Agreement”), (ii) a Convertible Credit Line Extension Agreement with Aharon Lukach (the “Lukach
Extension Agreement”) and (iii) a Convertible Loans and Unsecured Convertible Notes Extension #2 Agreement with Yehuda Nir (the
“Nir Extension Agreement”), each which extended the maturity date of the convertible loans under their respective loan agreements
(as described below) to January 31, 2026. The aggregate principal amount of loans extended was $
The
Dotan Extension Agreement related to a Convertible Credit Line Agreement dated as of October 3, 2019, as amended, of which $
The
Lukach Extension Agreement related to a Convertible Credit Line Agreement dated as of October 3, 2019, as amended, of which $
The
Nir Extension Agreement related to
On
March 6, 2024, the Israeli subsidiary and Koligo each received loans in the amount of $
NOTE 11 – LEASES
The Company leases research and development facilities, equipment and offices under finance and operating leases. For leases with terms greater than 12 months, the Company records the related asset and obligation at the present value of lease payments over the term. Many of the leases include rental escalation clauses, renewal options and/or termination options that are factored into the determination of lease payments when appropriate.
The Company’s leases do not provide a readily determinable implicit rate. Therefore, the Company estimated the incremental borrowing rate to discount the lease payments based on information available at lease commencement.
Manufacturing facilities
The
Company leases space for its manufacturing facilities under operating lease agreements. The leasing contracts are for a period of
| F-31 |
Research and Development facilities
The
Company leases space for its research and development facilities under operating lease agreements. The leasing contracts are for a period
of
Offices
The
Company leases space for offices under operating leases. The leasing contracts are valid for terms of
Lease Position
The table below presents the lease-related assets and liabilities recorded on the balance sheet:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Assets | ||||||||
| Operating Leases | ||||||||
| Operating lease right-of-use assets | $ | $ | ||||||
| Finance Leases | ||||||||
| Property, plants and equipment, gross | ||||||||
| Accumulated depreciation | ( | ) | ( | ) | ||||
| Property and equipment, net | $ | $ | ||||||
| Liabilities | ||||||||
| Current liabilities | ||||||||
| Current maturities of operating leases | $ | $ | ||||||
| Current maturities of long-term finance leases | $ | $ | ||||||
| Long-term liabilities | ||||||||
| Non-current operating leases | $ | $ | ||||||
| Long-term finance leases | $ | $ | ||||||
| Weighted Average Remaining Lease Term | ||||||||
| Operating leases | | | ||||||
| Finance leases | | | ||||||
Weighted Average Discount Rate | ||||||||
| Operating leases | % | % | ||||||
| Finance leases | % | % | ||||||
Lease Costs
The table below presents certain information related to lease costs and finance and operating leases:
| Years ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Operating lease cost: | $ | |||||||
| Finance lease cost: | ||||||||
| Amortization of leased assets | ||||||||
| Interest on lease liabilities | ||||||||
| Total finance lease cost | $ | |||||||
| F-32 |
The table below presents supplemental cash flow information related to lease:
| Years ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in Thousands) | ||||||||
| Cash paid for amounts included in the measurement of leases liabilities: | ||||||||
| Operating leases | $ | $ | ||||||
| Finance leases | $ | $ | ||||||
| Right-of-use assets obtained in exchange for lease obligations: | ||||||||
| Operating leases | $ | $ | ||||||
| Finance leases | ||||||||
Undiscounted Cash Flows
The table below reconciles the undiscounted cash flows for each of the first five years and total of the remaining years to the finance lease liabilities and operating lease liabilities recorded on the balance sheet.
Operating Leases | Finance Leases | |||||||
| Year ended December 31, | ||||||||
| 2024 | $ | $ | ||||||
| 2025 | ||||||||
| 2026 | ||||||||
| 2027 | ||||||||
| 2028 | ||||||||
| Thereafter | ||||||||
| Total minimum lease payments | ||||||||
| Less: amount of lease payments representing interest | ( | ) | ||||||
| Present value of future minimum lease payments | ||||||||
| Less: Current leases obligations | ( | ) | ( | ) | ||||
| Long-term leases obligations | $ | $ | ||||||
Operating lease right-of-use assets by geographical location were as follows:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Greece | $ | $ | ||||||
| Korea | ||||||||
| Israel | ||||||||
| U.S. | ||||||||
| Total | $ | $ | ||||||
| F-33 |
NOTE 12 – COMMITMENTS AND LICENSE AGREEMENTS
See Note 13 for additional commitments related to Collaborations.
| a. | Tel Hashomer Medical Research, Infrastructure and Services Ltd (“THM”) |
On February 2, 2012, the Company’s Israeli Subsidiary entered into a licensing agreement with THM. According to the agreement, the Israeli Subsidiary was granted a worldwide, royalty bearing, exclusive license to trans-differentiation of cells to insulin producing cells, including the population of insulin producing cells, methods of making this population, and methods of using this population of cells for cell therapy or diabetes treatment developed by Dr. Sarah Ferber of THM.
As consideration for the license, the Israeli Subsidiary will pay the following to THM:
| 1) | A
royalty of | |
| 2) | ||
| 3) | An
annual license fee of $ | |
| 4) | Milestone payments as follows: |
| a. | $ | |
| b. | $ | |
| c. | $ | |
| d. | $ | |
| e. | $ |
As of December 31, 2023, the Israeli Subsidiary had not reached any of these milestones.
In the event of closing of an acquisition of all of the issued and outstanding share capital of the Israeli Subsidiary and/or consolidation of the Israeli Subsidiary or the Company into or with another corporation (“Exit”), the THM shall be entitled to choose whether to receive from the Israeli Subsidiary a one-time payment based, as applicable, on the value of either shares of common stock of the Company at the time of the Exit or the value of shares of common stock of the Israeli Subsidiary at the time of the Exit.
| b. | Department De La Gestion Financiere Direction De L’analyse Financiere (“DGO6”) |
(1)
On November 17, 2014, the Belgian Subsidiary received the formal approval from the DGO6 for a Euro
(2)
In April 2016, the Belgian Subsidiary received the formal approval from DGO6 for a Euro
| F-34 |
(3) On October 8, 2016, the Belgian Subsidiary received the formal approval from the DGO6 for a Euro
(4)
In December 2020, the Belgian Subsidiary received the formal approval from DGO6 for a Euro
| c. | Israel-U.S. Binational Industrial Research and Development Foundation (“BIRD”) |
On
September 9, 2015, the Israeli Subsidiary entered into a pharma Cooperation and Project Funding Agreement (CPFA) with BIRD and Pall Corporation,
a U.S. company. BIRD awarded a conditional grant of up to $
| d. | Korea-Israel Industrial Research and Development Foundation (“KORIL”) |
On
May 26, 2016, the Israeli Subsidiary and the Orgenesis Korean (an Octomera subsidiary), entered into a pharma Cooperation and Project
Funding Agreement (CPFA) with KORIL. KORIL will make a conditional grant of up to $
| e. | BIRD Secant |
On July 30, 2018, Orgenesis Inc and OBI entered into a collaboration agreement with Secant Group LLC (“Secant”). Under the agreement, Secant will engineer and prototype 3D scaffolds based on novel biomaterials and technologies involving bioresorbable polymer microparticles, while OBI will provide expertise in cell coatings, cell production, process development and support services. Under the agreement, Orgenesis is authorized to utilize the jointly developed technology for its autologous cell therapy platform, including its Autologous Insulin Producing (“AIP”) cell technology for patients with Type 1 Diabetes, acute pancreatitis and other insulin deficient diseases. In 2018, OBI entered into a Cooperation and Project Funding Agreement (CPFA) with the BIRD fund, which provided certain grant funding, and Secant.
As
of December 31, 2023, OBI had received a total amount of $
| f. | Sponsored Research and Exclusive License Agreement with Columbia University |
Effective April 2, 2019, the Company and The Trustees of Columbia University in the City of New York, a New York corporation, (“Columbia”) entered into a Sponsored Research Agreement (the “SRA”) whereby the Company will provide financial support for studying the utility of serological tumor marker for tumor dynamics monitoring.
Effective
April 2, 2019, the Company and Columbia entered into an Exclusive License Agreement (the “Columbia License Agreement”) whereby
Columbia granted to the Company an exclusive license to discover, develop, manufacture, sell, and otherwise distribute certain product
in the field of cancer therapy. In consideration of the licenses granted under the Columbia License Agreement, the Company shall pay
to Columbia (i) a royalty of
| F-35 |
| g. | Regents of the University of California |
In
December 2019, the Company and the Regents of the University of California (“University”) entered into a joint research agreement
in the field of therapies and processing technologies according to an agreed upon work plan. According to the agreement, the Company
will pay the University royalties of up to
| h. | Caerus Therapeutics Inc |
In
October 2019, the Company and Caerus Therapeutics (“Caerus”), a Virginia company, concluded a license agreement whereby Caerus
granted the Company an exclusive license to all Caerus IP relating to Advance Chemeric Antigen Vectors for Targeting Tumors for the development
and/or commercialization of certain licensed products. In consideration for the License granted to the Company under this Agreement,
the Company shall pay Caerus annual maintenance fees and royalties of sales of up to
| i. | Tissue Genesis LLC |
Included
in the Koligo acquisition of 2020 were the assets of Tissue Genesis LLC. The Company is committed to paying the previous owners of Tissue
Genesis LLC or their assignees up to $
| j. | University of Louisville research foundation (“ULRF”) |
Koligo had exclusively licensed patents and technology from the ULRF related to the revascularization and 3D printing of cell and tissue for transplant (“ULRF licensed products”). The Company is committed to utilizing commercial reasonable efforts to achieving certain milestones regarding the ULRF licensed products.
| k. | Savicell |
During
2021, the Company and Savicell Ltd (“Savicell”) entered into a collaboration agreement (the “Savicell Agreement”)
to collaborate in the evaluation, continued development, validation, and use of Savicell’s platform designed for the early detection
and diagnosis of diseases and conditions and for quality control and monitoring purposes, in conjunction with the Company’s systems.
Pursuant to the Savicell Agreement, the Company will provide to Savicell funding for the performance of certain tasks agreed upon by
the parties in a work plan. In consideration for such funding, Savicell will supply the Company with products developed under the Savicell
Agreement at preferential rates and grant to the Company a worldwide exclusive licence to sell such products in the Company’s point-of-care
network of hospitals, clinics and institutions for quality control and monitoring of manufacturing and processing of autologous immune
cells manipulated by cell and gene therapies. The Company will be required to pay a
| l. | Stromatis Pharma |
During
2021, the Company and Stromatis Pharma Inc. (“Stromatis”) entered into a Collaboration and Sublicense Agreement (the “Stromatis
Agreement”) to collaborate in refining methods for GMP manufacturing of CAR-T/CAR-NK CT109; and the development and validation
of the Stromatis technology as it relates to the CAR-T/CAR-NK CT109 antibody up to and inclusive of filing of Investigational New Drug
Application relating to Stromatis’ CAR-T/CAR-NK CT109 antibody (“Licensed Product”), in accordance with the agreed
project plan (“Project”). The Company will fund the Project by providing Stromatis an amount of $
| F-36 |
Stromatis
has the option to convert the exclusive Manufacturing Rights to non-exclusive rights subject to repayment by Stromatis of an amount equal
to funding provided by the Company and an additional payment by Stromatis of an ongoing revenue share of five percent () of revenues
of any kind received by Stromatis or its affiliates from the sale or transfer of Licensed Products or license of rights under the licensed
technology in relation to the Licensed Products. The Company shall pay Stromatis in consideration for the Marketing Rights and royalties
equal to
| m. | Helmholtz Zentrum München Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH)) (“HMGU”)- |
During
2021, HMGU granted an exclusive licence under HGMU owned patent rights and non-exclusive license under HGMU know how and licensed materials,
to the Company in the field of certain human stem cells. In addition, payments will be due by the Company upon certain milestones. The
agreement also includes payment of royalties of between
| n. | License and research agreement with Yeda Research and Development Company Limited |
On January 25, 2022, the Company and Yeda Research and Development Company Limited (“Yeda”), an Israeli company, entered into a license and research agreement. No royalty bearing sales were made and the Company terminated this agreement during 2023.
| o. | European Innovation Council and SMEs Executive Agency (“EISMEA”) |
During
the year ended December 31, 2022, the Dutch Subsidiary, together with a consortium of other entities (“Consortium”) and EISMEA
entered into a grant funding agreement for the funding of the development of an artificial intelligence guided microfluidic device that
standardizes the GMP production of autologous induced pluripotent stem cells (iSPSCs) at greatly reduced costs (“iPSC project”).
The total grant amount is Euro
| p. | Walloon region ATMP PIT |
In
December 2023, the Belgian Subsidiary received Euro
| F-37 |
NOTE 13 – COLLABORATIONS
| a. | Adva Biotechnology Ltd. |
On January 28, 2018, the Company and Adva Biotechnology Ltd. (“Adva”), entered into a Master Services Agreement (“MSA”), pursuant to which the Company and/or its affiliates provided certain services relating to development of products for Adva.
In consideration for and subject to the fulfillment by the Company of certain funding commitments which were completed in 2019, Adva agreed that upon completion of the development of the products, the Company and/or its affiliates and Adva shall enter into a supply agreement pursuant to which for a period of eight (8) years following execution of such supply agreement, the Company and/or its affiliates (as applicable) is entitled (on a non-exclusive basis) to purchase the products from Adva at a specified discount pricing from their then standard pricing. The Company and/or its affiliates were also granted a non-exclusive worldwide right to distribute such products, directly or indirectly. The MSA shall remain in effect for 10 years unless earlier terminated in accordance with its terms.
| b. | Revised and restated joint venture agreements |
In January 2023the Company entered into updated joint venture (JV) agreements (JVAs) with Theracell Advanced Biotechnology SA, Broaden Bioscience and Technology Corp, Image Securities FZC, Cure Therapeutics, and Med Centre for Gene and Cell Therapy FZ-LLC and assigned certain rights and obligations under its JVAs to Texas Advanced Therapies LLC, a Delaware Limited Liability company (“Texas AT”) not related to the Company. Texas AT will receive the Company’s option to require the incorporation of the JV entity, Company’s share in the JV Entity, if and when the latter are incorporated, an option to invest additional funding in the JV Entity, and board and veto rights on certain critical decisions in the JV Entity. The Company has retained the call option to acquire the JV partner’s share in the JVE, to receive a royalty and a right to conclude the Manufacturing and Service Agreement with the JV entity. Pursuant to the JVAs, the Company will no longer be entitled to the additional share of fifteen percent of the JVE’s GAAP profit after tax granted as per the previous version of the JVAs. The Company also has no further obligation to provide any additional funding to the JV entities. As of December 31, 2023, no JV entities were incorporated pursuant to the JVAs.
| c. | Mircod |
On
July 25, 2023, the Company and Mircod LLC (“Mircod”) entered into a settlement and release agreement pursuant to which they
agreed to terminate the joint venture and loan agreement between themselves. Also, pursuant to the agreement, Mircod agreed to deliver
all the related deliverables to the Company, and the Company agreed to pay Mircod consideration in the amount of $
| d. | Sub-license agreement |
On July 25, 2023, the Company, a Sub-licensee, and the equity interest owner of that Sub-licensee (“Sub-licensee Owner”), entered into agreements whereby:
| 1) | the Company sub-licensed certain of its therapies to Sub-licensee in return for royalties on future sales and payments upon the successful completion of certain milestones; | |
| 2) | subject
to the fulfilment certain conditions and milestones, none of which have been fulfilled to
date, the Sub-licensee Owner granted the Company a call option to purchase his interests
in Sub-licensee at a valuation to be determined by a third-party valuation firm of not less
than $ | |
| 3) | subject
to the fulfilment of certain conditions and milestones, none of which have been fulfilled
to date, the Sub-licensee Owner was granted a put option to cause the Company to purchase
his equity interest in Sub-licensee at a valuation to be determined by a third-party valuation
firm of not less than $ |
The
Company has received $
| F-38 |
| e. | Deep Med Joint Venture agreement (JVA) |
In
November 2021, Deep Med IO Ltd (“Deep Med”) and Company entered into a JVA. The Parties agreed to collaborate in the development
and commercialization of an AI-powered system to be used in the manufacturing and/or quality control of CGTs. The Company has the right
to finance its activities under the Deep Med JVA by procuring services, advancing funds under a convertible loan agreement, or by an
equity investment. The Deep Med convertible loan bears interest at the annual rate of
| Years ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands, except per share data) | ||||||||
| Basic and diluted: | ||||||||
| Net loss attributable to Orgenesis Inc | $ | $ | ||||||
| Weighted average number of common shares outstanding | ||||||||
| Net loss per share | $ | $ | ||||||
For the year ended December 31, 2023, and December 31, 2022, all outstanding convertible notes, options, RSUs and warrants have been excluded from the calculation of the diluted net loss per share since their effect was anti-dilutive. Diluted loss per share does not include shares underlying outstanding options, RSUs and warrants and shares upon conversion of convertible loans for the year ended December 31, 2023, because the effect of their inclusion in the computation would be anti-dilutive. Diluted loss per share does not include shares underlying outstanding options and warrants and shares upon conversion of convertible loans for the year ended December 31, 2022, because the effect of their inclusion in the computation would be antidilutive.
| a. | Global Share Incentive Plan |
The Company’s stockholders have approved the 2017 Equity Incentive Plan (the “2017 Plan”) under which, the Company had reserved a pool of shares of the Company’s common stock, which may be issued at the discretion of the Company’s board of directors from time to time. Under this Plan, each option is exercisable into one share of common stock of the Company. The options may be exercised after vesting and in accordance with the vesting schedule that will be determined by the Company’s board of directors for each grant. The maximum contractual life term of the options is years. As of December 31, 2023, total options available for grants under this plan are .
On May 23, 2012, the Company’s board of directors adopted the Global Share Incentive Plan 2012 (the “2012 Plan”) under which, the Company had reserved a pool of shares of the Company’s common stock, which may be issued at the discretion of the Company’s board of directors from time to time. Under this plan, each option is exercisable into one share of common stock of the Company. The options may be exercised after vesting and in accordance with the vesting schedule that will be determined by the Company’s board of directors for each grant. The maximum contractual life term of the options is years. As of December 31, 2023, total options available for grants under this plan are .
| F-39 |
| b. | Options Granted to Employees and Directors |
Below is a table summarizing all of the options grants to employees and Directors made during the years ended December 31, 2023, and December 31, 2022:
| Year Ended | No. of options granted | Exercise price | Vesting period | Fair value at grant (in thousands) | Expiration period | |||||||||||||||||
| Employees | December 31, 2023 |
$-$ | 51% Quarterly over a period of two years and the rest quarterly over a period of | years | ||||||||||||||||||
| Directors | December 31, 2023 |
$ | years | |||||||||||||||||||
| Employees | December 31, 2022 |
$-$ | Quarterly over a period of | $ | years | |||||||||||||||||
| Directors | December 31, 2022 |
$ | years | |||||||||||||||||||
The fair value of each stock option grant is estimated at the date of grant using a Black Scholes option pricing model. The volatility is based on the historical volatility of the Company, by statistical analysis of the weekly share price for past periods based on expected term. The expected option term is calculated using the simplified method, as the Company concludes that its historical share option exercise experience does not provide a reasonable basis to estimate its expected option term. The fair value of each option grant is based on the following assumptions:
| Years Ended December 31, | ||||
| 2023 | 2022 | |||
| Value of one common share | $-$ | $-$ | ||
| Dividend yield | ||||
| Expected stock price volatility | %-% | %-% | ||
| Risk free interest rate | %-% | %-% | ||
| Expected term (years) | - | - | ||
| Years Ended December 31 | ||||||||||||||||
| 2023 | 2022 | |||||||||||||||
Number of Options | Weighted Average Exercise Price $ | Number of Options | Weighted Average Exercise Price $ | |||||||||||||
| Options outstanding at the beginning of the period | ||||||||||||||||
| Changes during the period: | ||||||||||||||||
| Granted | ||||||||||||||||
| Exercised* | ( | ) | ||||||||||||||
| Expired | ( | ) | ( | ) | ||||||||||||
| Forfeited | ( | ) | ( | )- | ||||||||||||
| Options outstanding at end of the period | ||||||||||||||||
| Options exercisable at end of the period | ||||||||||||||||
| * |
| F-40 |
Exercise Price $ | Number of Outstanding Options | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value $ |
Number of Exercisable Options | Aggregate Exercisable Options Value $ | |||||||||||||||||
| (in thousands) | (in thousands) | |||||||||||||||||||||
Costs incurred with respect to stock-based compensation for employees and directors for the years ended December 31, 2023 and December 31, 2022 were $ thousand and $ thousand, respectively. As of December 31, 2023, there was $ thousand of unrecognized compensation costs related to non-vested employees and directors stock options, to be recorded over the next years.
| c. | Options Granted to Consultants and service providers |
| Year of grant | No. of options granted | Exercise price | Vesting period | Fair value at grant (in thousands) | Expiration period | |||||||||||||
| Non-employees | 2023 | $ | Annually over a period of years | $ | years | |||||||||||||
| Non-employees | 2022 | $ | Quarterly over a period of years | $ | years | |||||||||||||
| F-41 |
The fair value of options granted during 2023 and 2022 to consultants and service providers, was computed using the Black-Scholes model. The fair value of each stock option grant is estimated at the date of grant using a Black Scholes option pricing model. The volatility is based on the historical volatility of the Company, by statistical analysis of the weekly share price for past periods based on the expected term period, the expected term is the contractual term of each grant. The underlying data used for computing the fair value of the options are as follows:
Years Ended December 31, | ||||
2023 |
2022 | |||
| Value of one common share | $ | $ | ||
| Dividend yield | ||||
| Expected stock price volatility | % | % | ||
| Risk free interest rate | % | %-% | ||
| Expected term (years) | ||||
| Years Ended December 31, | ||||||||||||||||
| 2023 | 2022 | |||||||||||||||
Number of Options | Weighted Average Exercise Price $ | Number of Options | Weighted Average Exercise Price $ | |||||||||||||
| Options outstanding at the beginning of the year | ||||||||||||||||
| Changes during the year: | ||||||||||||||||
| Granted | ||||||||||||||||
| Expired | ( | ) | ( | ) | ||||||||||||
| Forfeited | ( | ) | ||||||||||||||
| Options outstanding at end of the year | ||||||||||||||||
| Options exercisable at end of the year | ||||||||||||||||
Exercise Price $ | Number of Outstanding Options | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value $ |
Number of Exercisable Options | Aggregate Exercisable Options Value $ | |||||||||||||||||
| (in thousands) | (in thousands) | |||||||||||||||||||||
Costs incurred with respect to options granted to consultants and service providers for the years ended December 31, 2023 and December 31, 2022 were $ and $, respectively. As of December 31, 2023, there was $ of unrecognized compensation costs related to non-vested consultants and service providers, to be recorded over the next years.
| F-42 |
| d. | Restricted Stock Units (“RSUs”) Granted to Employees |
| Year Ended | No. of options granted | Vesting period | Fair value at grant (in thousands) | |||||||||
| Employees | December 31, 2023 | Quarterly over a period of years | $ | |||||||||
The fair value of each RSUs grant is estimated based on the market value of the underlying stock at the date of grant.
A summary of the Company’s RSUs granted to employees as of December 31, 2023 is presented below:
| Years Ended December 31 | ||||
| 2023 | ||||
Number of RSUs | ||||
| Options outstanding at the beginning of the period | ||||
| Changes during the period: | ||||
| Granted | ||||
| Expired | ||||
| Forfeited | ||||
| Options outstanding at end of the period | ||||
Number of Outstanding RSUs | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value $ |
Number of Exercisable RSUs | Aggregate Exercisable RSUs Value $ | ||||||||||||||
| (in thousands) | (in thousands) | |||||||||||||||||
No costs incurred with respect to RSUs compensation for employees for the years ended December 31, 2023. As of December 31, 2023, there was $ of unrecognized compensation costs related to non-vested employees RSUs, to be recorded over the next years.
| F-43 |
NOTE 16 – TAXES
| a. | Corporate taxation in the U.S. |
The
corporate U.S. Federal Income tax rate applicable to the Company and its US subsidiaries is
As
of December 31, 2023, the Company has an accumulated tax loss carryforward of approximately $
For U.S. federal income tax purposes, net operating losses (“NOLs”) arising in tax years beginning after December 31, 2017, the Internal Revenue Code of 1986, as amended (the “Code”) limits the ability to utilize NOL carryforwards to 80% of taxable income in tax years beginning after December 31, 2020. In addition, NOLs arising in tax years ending after December 31, 2017 can be carried forward indefinitely, but carryback is generally prohibited. NOLs generated in tax years beginning before January 1, 2018 will not be subject to the taxable income limitation, and NOLs generated in tax years ending before January 1, 2018 will continue to have a two-year carryback and twenty-year carryforward period. Deferred tax assets for NOLs will need to be measured at the applicable tax rate in effect when the NOL is expected to be utilized. The changes in the carryforward/carryback periods as well as the new limitation on use of NOLs may significantly impact the Company’s valuation allowance assessments for NOLs generated after December 31, 2017.
In addition, utilization of the NOLs may be subject to substantial annual limitation under Section 382 of the Code due to an “ownership change” within the meaning of Section 382(g) of the Code. An ownership change subjects pre-ownership change NOL carryforwards to an annual limitation, which significantly restricts the ability to use them to offset taxable income in periods following the ownership change. In general, the annual use limitation equals the aggregate value of the Company’s stock at the time of the ownership change multiplied by a specified tax-exempt interest rate.
| b. | Corporate taxation in Israel |
The
Israeli Subsidiaries are taxed in accordance with Israeli tax laws. The corporate tax rate applicable to 2023 and 2022 are
As
of December 31, 2023, the Israeli Subsidiary has an accumulated tax loss carryforward of approximately $
| c. | Corporate taxation in Belgium |
The
Belgian Subsidiary is taxed according to Belgian tax laws. The corporate tax rates applicable to 2023, 2022 are
As
of December 31, 2023, the Belgian Subsidiary has an accumulated tax loss carryforward of approximately $
| F-44 |
| e. | Deferred Taxes |
The following table presents summary of information concerning the Company’s deferred taxes as of the years ending December 31, 2023 and December 31, 2022:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (U.S. dollars in thousands) | ||||||||
| Deferred tax assets (liabilities), net: | ||||||||
| Net operating loss carry forwards | $ | $ | ||||||
| Research and development expenses | ||||||||
| Equity compensation | ||||||||
| Employee benefits | ||||||||
| Property, plants and equipment | ( | ) | ( | ) | ||||
| Leases asset | ||||||||
| Lease liability | ( | ) | ( | ) | ||||
| Loans | ||||||||
| Partnership Investment | ||||||||
| Intangible assets | ( | ) | ( | ) | ||||
| Bad debt allowance | ||||||||
| Other | ||||||||
| Valuation allowance | ( | ) | ( | ) | ||||
| Net deferred tax liabilities | $ | $ | ||||||
Realization of deferred tax assets is contingent upon sufficient future taxable income during the period that deductible temporary differences and carry forwards losses are expected to be available to reduce taxable income. As the achievement of required future taxable income is not considered more likely than not achievable, the Company and all its subsidiaries have recorded full valuation allowance.
The changes in valuation allowance are comprised as follows:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (U.S dollars in thousands) | ||||||||
| Balance at the beginning of year | $ | ( | ) | $ | ( | ) | ||
| Deconsolidation of Octomera | ||||||||
| Change during the year | ( | ) | ( | ) | ||||
| Balance at end of year | $ | ( | ) | $ | ( | ) | ||
| f. | Reconciliation of the Theoretical Tax Expense to Actual Tax Expense |
The main reconciling item between the statutory tax rate of the Company and the effective rate is the provision for valuation allowance with respect to tax benefits from carry forward tax losses.
| g. | Uncertain Tax Provisions |
ASC Topic 740, “Income Taxes” requires significant judgment in determining what constitutes an individual tax position as well as assessing the outcome of each tax position. Changes in judgment as to recognition or measurement of tax positions can materially affect the estimate of the effective tax rate and consequently, affect the operating results of the Company. As of December 31, 2023, the Company has not accrued a provision for uncertain tax positions.
| F-45 |
NOTE 17 – REVENUES
Disaggregation of Revenue
The following table disaggregates the Company’s revenues by major revenue streams.
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Revenue stream: | ||||||||
| POCare development services | $ | $ | ||||||
| Cell process development services and hospital services | ||||||||
| POCare cell processing | ||||||||
| License fees | ||||||||
| Total | $ | $ | ||||||
A breakdown of the revenues per customer what constituted at least 10% of revenues is as follows:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Revenue earned: | ||||||||
| Customer A (United States) | ||||||||
| Customer B (United States) | ||||||||
| Customer C (United States) | ||||||||
| Customer D (Greece) | ||||||||
| Customer E (United States) | ||||||||
| Customer F (United Arab Emirates) | ||||||||
| Customer G (Korea) | ||||||||
Contract Assets and Liabilities
Contract assets are mainly comprised of accounts receivable net of allowance for doubtful debts, which includes amounts billed and currently due from customers.
The activity for accounts receivable is comprised of:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Balance as of beginning of period | $ | $ | ||||||
| Deconsolidation of Octomera | ( | ) | ||||||
| Business combination TLABS | ( | ) | ||||||
| Additions | ||||||||
| Collections | ( | ) | ( | ) | ||||
| Allowances for credit losses | ( | ) | ||||||
| Exchange rate differences | ( | ) | ( | ) | ||||
| Balance as of end of period | $ | $ | ||||||
The activity of the related party included in the accounts receivable activity above is comprised of:
| Years Ended December 31, | ||||
| 2022 | ||||
| Balance as of beginning of period | $ | |||
| Additions | ||||
| Collections | ( | ) | ||
| Ceased to be a related party | ( | ) | ||
| Balance as of end of period | $ | |||
| F-46 |
The activity for contract liabilities is comprised of:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Balance as of beginning of period | $ | $ | ||||||
| Deconsolidation of Octomera | ( | ) | ||||||
| Additions | ||||||||
| Balance as of end of period | $ | $ | ||||||
NOTE 18 – COST OF DEVELOPMENT SERVICES AND RESEARCH AND DEVELOPMENT EXPENSES
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Salaries and related expenses | $ | $ | ||||||
| Stock-based compensation | ||||||||
| Subcontracting, professional and consulting services | ||||||||
| Lab expenses | ||||||||
| Depreciation expenses | ||||||||
| Other research and development expenses | ||||||||
| Less – grant | ( | ) | ( | ) | ||||
| Total | $ | $ | ||||||
NOTE 19 – FINANCIAL EXPENSES, NET
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Interest expense on convertible loans | $ | $ | ||||||
| Foreign exchange loss, net | ||||||||
| Other expense | ||||||||
| Total | $ | $ | ||||||
| F-47 |
NOTE 20 – RELATED PARTIES TRANSACTIONS
| a. | Related Parties presented in the consolidated statements of comprehensive loss |
| Years ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Stock-based compensation expenses to executive officers | $ | $ | ||||||
| Stock-based compensation expenses to Board Members | $ | $ | ||||||
| Compensation of executive officers | $ | $ | ||||||
| Management and consulting fees to Board Members | $ | $ | ||||||
| Revenues from customer | $ | $ | ||||||
| Financial income | $ | $ | ||||||
| b. | Related Parties presented in the consolidated balance sheets |
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| (in thousands) | ||||||||
| Executive officers’ payables | $ | $ | ||||||
| Non-executive directors’ payable | $ | $ | ||||||
NOTE 21 – LEGAL PROCEEDINGS
On
January 18, 2022, a complaint (the “Complaint”) was filed in the Tel Aviv District Court (the “Court”)
against the Company, Orgenesis LTD (“the Israeli Subsidiary”), Prof. Sarah Ferber, Vered Caplan and Dr. Efrat Asa Kunik
(collectively, the “defendants”) by plaintiffs the State of Israel, as the owner of Chaim Sheba Medical Center at Tel
Hashomer (“Sheba”), and Tel Hashomer Medical Research, Infrastructure and Services Ltd. (collectively, the
“plaintiffs”).
| F-48 |
On
September 6, 2023, a claim (the “Claim”) was filed in the Tel Aviv District Court (the “Court”) against the
Company, the Israeli Subsidiary, Octomera LLC, Orgenesis Biotech Israel LTD, Theracell Laboratories Private Company and Vered Caplan
(collectively, the “defendants”) by Ehud Almon (Plaintiff) for certain finders’ fees and / or royalties related to
sales made by an Octomera subsidiary to a Greek entity in the amount of $
On
October 26, 2023, a complaint was filed in the Supreme Court of the State of New York by plaintiffs Southern Israel Bridging Fund
Two LP and Mr. Amir Hasidim, against the Company, seeking the payment of $
On
November 1, 2023, a claim (the “Claim”) was filed in the Tel Aviv District Court (the “Court”) against the
Company, the Israeli Subsidiary, and Vered Caplan (collectively, the “defendants”) by Fidelity Venture Capital Ltd. and
Dror Atzmon (together – “the Plaintiffs”). The claim is based on two agreements the Company entered into with the
Plaintiffs on November 2, 2016: (a) an unsecured convertible note agreements for an aggregate amount of NIS
On February 14, 2024, following a claim for payment of past salaries due, by employees of Orgenesis Biotech Israel Limited (“OBI”), the district court in Haifa appointed a trustee to run the affairs of OBI with the intention of rehabilitating OBI to be able to operate and pay OBI’s creditors under an arrangement with them.
Except as described above, the Company is not involved in any pending material legal proceedings.
| F-49 |
NOTE 22 – SUBSEQUENT EVENTS
Private Placement Offering
On
March 3, 2024, the Company entered into a Securities Purchase Agreement with certain accredited investors, pursuant to which the
Company agreed to issue and sell, in a private placement,
shares of the Company’s common stock, par value $
per share, at a purchase price of $
per share and warrants to purchase up to
The
Warrants entitle the holders to purchase up to an aggregate of
Asset purchase agreement
On April 5, 2024, Orgenesis Maryland entered into an Asset Purchase
and Strategic Collaboration Agreement (the “Purchase Agreement”) with Griffin Fund 3 BIDCO, Inc., (“Germfree”),
for the sale by Orgenesis Maryland of five OMPUL to Germfree, which will be incorporated into Germfree’s lease fleet and leased
back to Orgenesis Maryland or third-party lessees designated by Orgenesis. Pursuant to the Purchase Agreement, and upon the terms and
subject to the conditions set forth therein, in consideration for the purchase of the OMPULs, the Orgenesis Quality Management Systems
Framework (“OQMSF”) and related intellectual property rights, Germfree will pay Orgenesis Maryland an aggregate purchase price
of $
Pursuant
to the Agreement, Germfree paid Orgenesis Maryland $
Strategic Advisor Agreement
On
March 7, 2024 (the “Effective Date”), the Company entered into a strategic advisor agreement with an individual relating
to the provision of strategic advice and assistance to the Company for a term of 12 months, subject to earlier termination or extension
for an additional 12 months at the request of the advisor. In consideration for such services, the Company agreed to (i) pay such individual
$
| F-50 |