UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM TO |
Commission File Number
(Exact name of Registrant as specified in its Charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
|
|
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act.
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the Registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|||
|
☒ |
|
Smaller reporting company |
|
||
|
|
|
|
|
|
|
Emerging growth company |
|
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
As of June 30, 2021, the aggregate market value of common stock held by non-affiliates of the Registrant was $
The number of shares of Registrant’s Common Stock outstanding as of March 9, 2022 was
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Company’s definitive Proxy Statement for its 2022 Annual Meeting of Shareholders are to be incorporated by reference into Part III, as specifically set forth in Part III.)
Table of Contents
|
|
Page |
PART I |
|
|
Item 1. |
3 |
|
Item 1A. |
39 |
|
Item 1B. |
84 |
|
Item 2. |
84 |
|
Item 3. |
84 |
|
Item 4. |
84 |
|
|
|
|
PART II |
|
|
Item 5. |
85 |
|
Item 6. |
85 |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
86 |
Item 7A. |
98 |
|
Item 8. |
98 |
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
99 |
Item 9A. |
99 |
|
Item 9B. |
99 |
|
|
|
|
PART III |
|
|
Item 10. |
100 |
|
Item 11. |
100 |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
100 |
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
100 |
Item 14. |
100 |
|
|
|
|
PART IV |
|
|
Item 15. |
101 |
|
Item 16 |
101 |
i
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements about us and our industry that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this Annual Report on Form 10-K, including statements regarding our future financial condition, results of operations, business strategy and plans, and objectives of management for future operations, as well as statements regarding industry trends, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potentially,” “predict,” “should,” “will” or the negative of these terms or other similar expressions.
We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. These forward-looking statements are subject to a number of risks, uncertainties, factors, and assumptions described under the section titled “Risk Factors” and elsewhere in this annual Report on Form 10-K, regarding, among other things:
1
These risks are not exhaustive. Other sections of this Annual Report on Form 10-K may include additional factors that could harm our business and financial performance. New risk factors may emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in, or implied by, any forward-looking statements.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Except as required by law, we undertake no obligation to update publicly any forward-looking statements for any reason after the date of this Annual Report on Form 10-K or to conform these statements to actual results or to changes in our expectations.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report on Form 10-K, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
You should read this Annual Report on Form 10-K and the documents that we reference and have filed as exhibits with the understanding that our actual future results, levels of activity, performance and achievements may be different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
2
PART I
Item 1. Business.
Our mission is to improve every patient’s lung care by empowering physicians with swift, comprehensive and actionable insights.
Our vision is to be a trusted business partner that the world relies on for data-driven diagnostics.
Business Overview
Biodesix, Inc. (“Biodesix”, “we,” “us,” “our” or the “Company”) is a leading data-driven diagnostic solutions company leveraging state of the art technologies with our proprietary artificial intelligence (AI) platform to discover, develop, and commercialize solutions for clinical unmet needs, with a primary focus in lung disease. By combining a multi-omic approach with a holistic view of the patient’s disease state, we believe our solutions provide physicians with greater insights to help personalize their patient’s care and meaningfully improve disease detection, evaluation, and treatment. Our unique approach to precision medicine provides timely and actionable clinical information, which we believe improves overall patient outcomes and lowers the overall healthcare cost by reducing the use of ineffective and unnecessary treatments and procedures. In addition to our diagnostic tests, we provide biopharmaceutical companies with services that include diagnostic research, clinical trial testing, and the discovery, development, and commercialization of companion diagnostics.
Our core belief is that no single technology will answer all clinical questions that we encounter. Therefore, we employ multiple technologies, including genomics, transcriptomics, proteomics, and radiomics, and leverage our proprietary AI platform, the Diagnostic Cortex®, to discover innovative diagnostic tests for clinical use. Because of this approach, we believe we are unique in the diagnostics market as this approach allows for a broader and more holistic understanding of each patient’s disease state. Our data-driven and multi-omic approach is designed to enable us to discover diagnostic tests that answer critical clinical questions faced by physicians, patients, researchers, and biopharmaceutical companies.
We operate in a single segment and derive our revenue from two sources: (i) providing diagnostic testing services associated with (a) blood-based lung tests and (b) Coronavirus Disease 2019 (COVID-19) tests (Diagnostic Tests); and (ii) providing biopharmaceutical companies with services that include diagnostic research, clinical research, development and testing services generally provided outside the clinical setting and governed by individual contracts with third parties as well as development and commercialization of companion diagnostics (Services). We derived 90% of our total revenues for fiscal 2021 and 2020 from our diagnostic testing business.
We are dedicated to continuously publishing and presenting new data on the clinical validation and utility of our diagnostic tests. Since our inception, we have performed over 500,000 tests and continue to generate a large and growing body of clinical evidence. We have participated in 27 clinical studies, five of which are ongoing, and have published over 300 peer-reviewed publications and presentations. We have over 150,000 samples and data in our biobank, including tumor profiles and immune profiles, which are used for both internal and external research and development (R&D) initiatives.
We have commercialized eight diagnostic tests which are currently on market and we perform over 30 assays for research use as part of our laboratory services that have been used by over 50 biopharmaceutical customers and academic partners.
Blood-Based Lung Tests
We have five diagnostic blood-based tests across the lung cancer continuum of care, which generated $18.7 million and $12.6 million in revenue for fiscal 2021 and 2020, respectively, an annual growth rate of 49%.
Diagnosis
Treatment & Monitoring
3
COVID-19 Tests
In response to the COVID-19 pandemic, through our partnership with Bio-Rad, we commercialized the Biodesix WorkSafe™ testing program. We generated $30.2 million and $28.3 million in revenue from COVID-19 testing during fiscal 2021 and 2020, respectively. Our scientific diagnostic expertise, technologies, and existing commercial infrastructure enabled us to rapidly commercialize two United States Food and Drug Administration (FDA) Emergency Use Authorization (EUA)-authorized tests, a part of our customizable WorkSafe™ program. Both diagnostic tests are owned and were developed by Bio-Rad and Bio-Rad has granted us permission to utilize both tests for commercial diagnostic services.
In January 2020, the U.S. Department of Health and Human Services Secretary declared a public health emergency for COVID-19 which justified the authorization of emergency use of diagnostic tests for the detection and/or diagnosis of COVID-19. The Bio-Rad SARS-CoV-2 Droplet Digital™ polymerase chain reaction (ddPCR) test and the Platelia SARS-CoV-2 Total Ab test have been granted FDA EUA pursuant to the current emergency declaration. The Bio-Rad SARS-CoV-2 ddPCR test was FDA EUA authorized on May 1, 2020, authorizing performance of the test in laboratories certified under the Clinical Laboratory Improvement Amendments (CLIA) to perform high complexity tests. The second test is the Platelia SARS-CoV-2 Total Ab test, which is an antibody test intended for detecting a B-cell immune response to SARS-CoV-2, indicating recent or prior infection. The Platelia SARS-CoV-2 Total Ab test was FDA EUA authorized on April 29, 2020. Medical products that are granted an EUA are only permitted to commercialize their products under the terms and conditions provided in the authorization. The FDA may revoke an EUA where it is determined that the underlying health emergency no longer exists or warrants such authorization, if the conditions for the issuance of the EUA are no longer met, or if other circumstances make revocation appropriate to protect the public health or safety, and we cannot predict how long the EUAs for the SARS-CoV-2 tests will remain in place. Using the Bio-Rad SARS-CoV-2 ddPCR test and the Platelia SARS-CoV-2 Total Ab tests, we operate and have commercialized the Biodesix WorkSafe testing program.
Prior to using the Bio-Rad tests as part of our WorkSafe testing program, we performed feasibility, verification, and validation studies, including developing software for process automation, sample accessioning, data management and reporting, all required to demonstrate the test operated as claimed by the manufacturer and as required by our certifying regulatory agencies for high complexity laboratory testing. We secured independent reference specimens run with EUA tests to validate these tests as fit for diagnostic use in our laboratories. Post-launch development support for these tests have included improvements in on-boarding new personnel, logistics of sample collection, sample receipt and data reporting, all required to support our testing program. Additional releases of the laboratory data management software are ongoing and planned for the foreseeable future.
In addition, during the three months ended June 30, 2021, we began partnering with GenScript Biotech Corporation to commercialize the blood-based cPass™ SARS-CoV-2 Neutralizing Antibody testing as a service. The test is the first and only surrogate neutralizing antibody test with FDA EUA and uses ELISA technology to qualitatively detect circulating neutralizing antibodies to the receptor binding domain (RBD) in the spike protein of SARS-CoV-2 that are produced in response to vaccination or previous SARS-CoV-2 infection.
These tests under the Biodesix WorkSafe testing program are utilized by healthcare providers, including hospitals and nursing homes, and are also offered to businesses and educational systems to assist in their back-to-work or back-to-school strategies, a crucial element of restarting economic activity. We have announced multiple partnerships for COVID-19 testing, and maintain an agreement with the State of Colorado to be one of the diagnostic companies to support widespread COVID-19 testing for the State. Additionally, we have overseen and managed onsite testing and validating testing for the Big Ten Conference athletic competitions through the term of our contract which expired on June 30, 2021.
COVID-19 Pandemic
The COVID-19 pandemic has disrupted, and we expect will continue to disrupt, our lung diagnostic testing operations. To protect the health and well-being of our workforce, partners, vendors and customers, we provide voluntary COVID-19 testing for employees working on-site, implemented social distance and building entry policies at work, restricted travel and facility visits, and followed the States of Colorado and Kansas’ public health orders and the guidance from the Centers for Disease Control and Prevention (CDC). Employees who can perform their duties remotely are asked to work from home and those on site are asked to follow our social distance guidelines. Our sales, marketing and business development efforts have also been constrained by our operational response to the COVID-19 pandemic due to travel restrictions. We expect to continue to adjust our operational norms to help slow the spread of COVID-19 in the coming months, including complying with government directives and guidelines as they are modified and supplemented.
The COVID-19 pandemic negatively affected, and we expect will continue to negatively affect, our lung diagnostic testing-related revenue and our clinical studies. For example, cancer patients may have more limited access to hospitals, healthcare providers and medical resources as they take steps to control the spread of COVID-19. Our biopharmaceutical customers are facing challenges in recruiting patients and in conducting clinical trials to advance their pipelines, for which our tests could be utilized. As a result of the COVID-19 pandemic, beginning in the latter half of March 2020, we saw a decline in business with existing and new biopharmaceutical customers. We began to see recovery during the fourth quarter 2020 as our delivered tests exceeded first quarter 2020 delivered tests and we continued to see the recovery extending through 2021. Further, our clinical studies, such as our ongoing INSIGHT and ALTITUDE studies, as well as our arrangements including contracted clinical trials with our biopharmaceutical customers, are expected
4
to take longer to complete than what we expected before the outbreak of the COVID-19 pandemic. Our biopharmaceutical services revenue grew by approximately 20% during 2021 as compared to 2020; however, we are continuing to experience delays in clinical trials from across the country and world due to COVID-19 restrictions. We expect further improvement in our biopharmaceutical activities during 2022 as compared to 2021.
Conversely, we experienced a significant increase in revenues related to an increase in the demand for our Biodesix WorkSafe testing program, our COVID-19 testing and services, since the onset of the pandemic. COVID-19 diagnostic testing and services contributed approximately $30.2 million and $28.3 million during fiscal years 2021 and 2020, respectively. The first quarter 2021 was our high-water mark for COVID-19 testing revenue. We experienced a steady decline in subsequent quarters as immunizations in the U.S. accelerated. We do not anticipate the need for COVID testing to be commensurate with the peak demand experienced during the first quarter 2021 and instead expect the demand to moderate as new variants and infection occur. The reduction in demand for COVID-19 diagnostic testing will be a key indicator of continued recovery and is taken as a positive sign for both our Lung Diagnostic and Biopharmaceutical Services as we head into 2022. There is no assurance that our COVID-19 testing program will continue to be accepted by the market or that other diagnostic tests will become more accepted, produce quicker results or are more accurate. Further, the longevity and extent of the COVID-19 pandemic is uncertain. If the pandemic were to dissipate, whether due to a significant decrease in new infections, due to acquiring herd immunity based on previous natural infection, and the availability and rapid distribution of vaccines, the evolution of variant strains that impact diagnostic test performance, or otherwise, the need for COVID-19 testing could decrease significantly and this could have an adverse effect on our results of operations and profitability. As a result, the increase in revenue due to any increase in demand for these diagnostic tests may not be indicative of our future revenue.
See “Risk Factors” for a description of how the COVID-19 pandemic may adversely affect our business, financial condition and results of operations.
Full Year Results for 2021
For the year ended December 31, 2021, compared to the prior year:
5
Our Market Opportunity
Diagnostic Testing Market Size and Opportunity
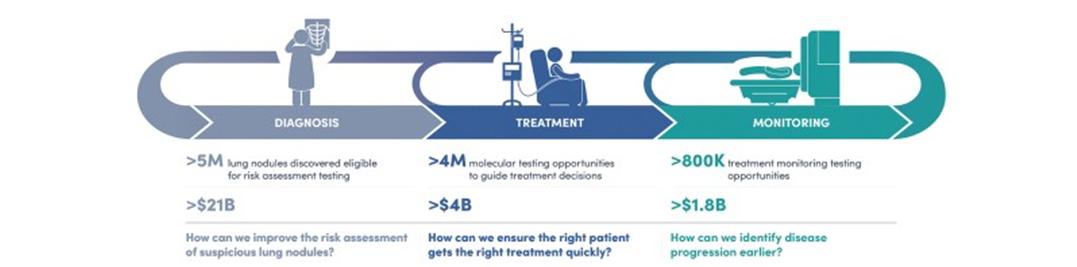
Despite significant advances over the last decade, lung cancer is still the deadliest type of cancer in both men and women in the United States. While diagnostic testing has become routinely used at certain points in the lung cancer continuum of care (diagnosis, treatment and monitoring), we believe there is substantial need for novel, advanced testing to improve on the current standard of care. We estimate that in the United States, the lung cancer continuum of care currently represents over 10 million annual testing opportunities and is over a $27 billion market annually for testing alone.
Over the last two decades, the use of biomarker testing in clinical trials has increased, with 55% of oncology trials involving the use of biomarker testing in 2018 versus 15% in 2000. We believe the field of biomarker discovery and companion diagnostic development for biopharmaceutical therapeutics is set to continue growing as biopharmaceutical companies seek to de-risk their research and development pipelines and increase chances of drug development success. We estimate that biopharmaceutical partnering and research opportunities represents over a $2 billion market annually.
Lung Cancer Continuum of Care – Clinical Unmet Needs
Standards of care in lung cancer have evolved rapidly over the past decade, along with our understanding of the disease. With the introduction of numerous treatment options, physicians need an ever-increasing amount of information in order to select the best treatment plan for each individual patient. We believe that the lung cancer continuum of care has a variety of clinical unmet needs ranging from initial diagnosis of lung cancer after discovery of a lung nodule to treatment guidance for early and advanced stage disease, and monitoring for disease progression.
6
Current Limitations in Biomarker Discovery and Companion Diagnostics
We estimate the biopharmaceutical biomarker testing and companion diagnostic market opportunity represents a $2 billion market annually. Over the last two decades, the use of biomarker testing in clinical trials has increased, with 55% of oncology trials involving the use of biomarker testing in 2018 versus 15% in 2000. From 2005 to 2015, a study identified that incorporating biomarkers into clinical development programs increased their probability of therapeutic success rate from phase 1 to FDA-approval by 570%, representing an increase from 1.6% without biomarkers to 10.7% with biomarkers. We believe the field of biomarker discovery and companion diagnostic development for biopharmaceutical therapeutics is set to continue growing as biopharmaceutical companies seek to de-risk their product development efforts and increase chances of drug development success. However, we believe as the market continues to advance, inherent limitations of both biomarker discovery and companion diagnostic development have become more apparent.
Biomarker Discovery: There are many limitations with biomarker discovery in biopharmaceutical drug development, including:
Companion Diagnostics (CDx): While developing companion diagnostics is critical to precision medicine, the promise of companion diagnostics has not been fully realized and there are multiple limitations that still need resolution. The path to co-develop a successful companion diagnostic with a corresponding drug has several challenges, including:
The Diagnostic Cortex - Our Proprietary Artificial Intelligence (AI) Platform
Our core belief is that no single technology will answer all clinical questions that we encounter. Therefore, we employ multiple technologies, including genomics, transcriptomics, proteomics, and radiomics, and leverage our proprietary AI platform, the Diagnostic
7
Cortex, to discover innovative diagnostic tests for clinical use. We focus on developing technologies that are capable of single and multi-omic research and development.
The Diagnostic Cortex is an extensively validated deep learning platform optimized for the discovery of clinical diagnostic tests, which we believe overcomes standard machine learning challenges faced in life sciences research. Researchers commonly encounter issues with machine learning-based biological discoveries that cannot be repeated or validated when assessed in additional specimen cohorts. This challenge, commonly referred to as overfitting, occurs when the machine identifies a perfect pattern in an initial training dataset but is unable to identify the same pattern in a new dataset. For over 15 years we have focused on developing our platform to overcome this challenge through proprietary computational techniques to ensure each diagnostic test that is discovered can be further developed to perform consistently in the clinical testing environment.
We are able to combine blood-based biological information related to the tumor, immune system, and host-status with clinical and radiomic data through our proprietary AI platform, which enables us to interpret the holistic disease state of each patient or clinical dataset we encounter.
We continuously evolve and improve the Diagnostic Cortex platform. These improvements range from basic code optimization to complex improvements such as the incorporation of novel computational methods for the optimization of multi-omic diagnostic tests. Any AI platform is inherently limited without the highest quality data inputs. Therefore, all of the technologies that we employ have been chosen and developed to provide high-quality data to enable our Diagnostic Cortex platform. We feel that this level of data integrity is crucial for the development of diagnostic tests that require the advanced pattern matching abilities of deep learning algorithms.
We continuously incorporate new market insights and patient data to enhance our platform through a data-driven learning loop. We regularly engage our customers, key opinion leaders, and scientific experts to stay ahead of the rapidly evolving diagnostic and therapeutic landscape and learn about biological discoveries that are clinically meaningful. Additionally, we incorporate clinical and molecular profiling data aggregated through our commercial clinical testing, research studies, clinical trials, and biopharmaceutical customers or academic partnerships, into our platform. We have over 150,000 samples and data in our biobank, including tumor profiles and immune profiles, which are used for both internal and external development initiatives. With our data-driven and multi-omic approach as data inputs into the Diagnostic Cortex, we are able to discover diagnostic tests that answer critical clinical questions faced by physicians, researchers, and biopharmaceutical companies.
The following is a diagram outlining our innovative diagnostic test discovery, development and commercialization infrastructure as outlined in the text above.
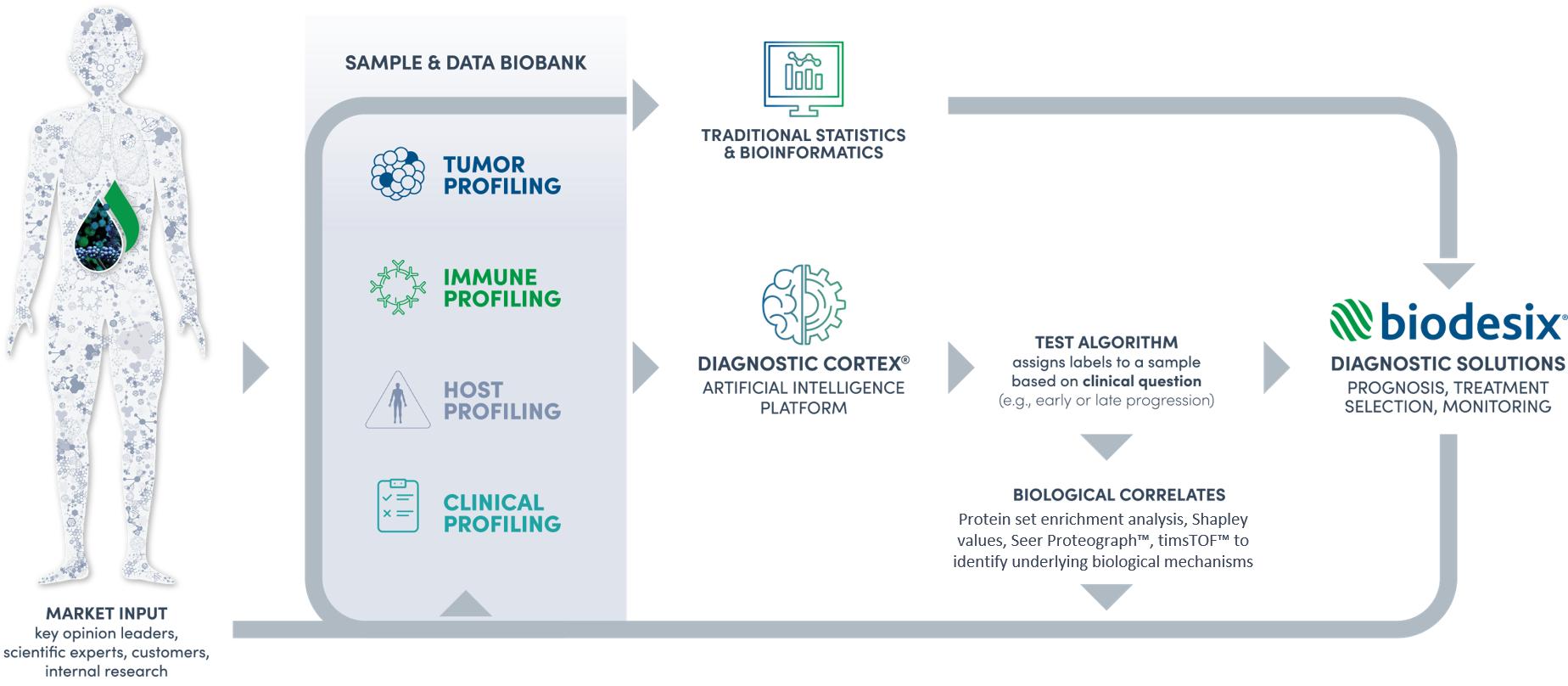
We employ multiple technologies, as illustrated below, including genomics, proteomics, transcriptomics, and radiomics, generated by different assay techniques, including ddPCR, NGS, LC-MS, ELISA, and our proprietary DeepMALDI mass spectrometry platform for the blood-based molecular analysis of the tumor, immune system, and host-status of each patient and/or clinical dataset. Through our learning loop, we continuously revisit our technology strategy and roadmap to integrate new technologies into our evolving platform, which ultimately support the addition of new service and product revenue offerings. We focus on developing technologies that are capable of single and multi-omic research and development.
8
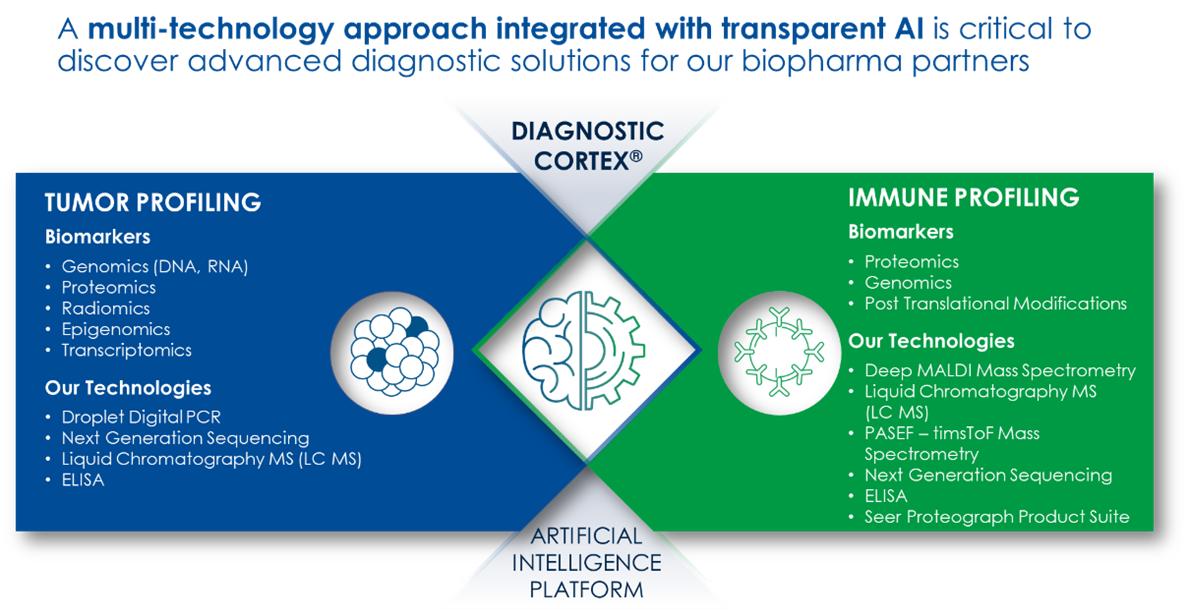
Most diagnostic companies focus their strategy on using a single technology to discover biomarkers for a broad range of clinical questions. We believe that no single technology can interrogate the complexity of the human disease state to help solve all clinical questions. For that reason, we employ a multi-omic approach to solving diagnostic challenges leveraging our proprietary transparent AI platform, the Diagnostic Cortex. Because of this approach, we believe we are unique in the diagnostics market, allowing for a broader and more holistic understanding of each patient’s disease state.
We are experts in many technologies, but we are a true market leader with over 15 years of experience in the field of clinical proteomics. For over 10 years, we have been discovering and developing proteomic-based diagnostic tests and have a deep understanding of how to incorporate technologies that can be applied to blood samples in order to extract important protein-based biological information in the form of diagnostic tests, which can aid clinicians and scientists in understanding the dynamic biology of their system of interest, such as a patient with cancer.
Our suite of technologies that assist us in discovery, development and commercialization of novel diagnostic tests includes:
DeepMALDI Mass Spectrometry
We have developed DeepMALDI, a proprietary high-density matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (MS) technology, to produce blood-based proteomic data for disease diagnosis, personalized health care, precision medicine for direct treatment options, and disease screening in lung and other disease states. DeepMALDI overcomes the limitations of conventional MALDI and other mass spectrometry methods to produce highly sensitive, stable, and reproducible data by: (1) utilizing optimized signal-to-noise reduction and signal processing algorithms; and (2) novel batch correction methods and spectral alignment methods. The combination of these improvements yields substantially higher quality data content and is thereby much better suited for the discovery of biomarkers with clinical utility.
Our current DeepMALDI methods allow us to achieve finer mass resolution, greater sensitivity, and 20-times faster imaging speeds than other instruments. Additionally, we believe recent enhancements to our DeepMALDI methods and MALDI-ToF technology evolution now allows us to measure more than 2,000 proteins, an improvement from the estimated 900 proteins we could measure a year ago. We intend to maintain our leadership role in the discovery of proteomics-based diagnostic tests. We utilize our DeepMALDI and MALDI-ToF technologies in our discovery and development efforts and as part of our collaborations with our biopharmaceutical customers and academic partners.
Liquid Chromatography Mass Spectrometry MS
We use Multiple Reaction Monitoring (MRM) MS with triple quadrupole mass spectrometers and up-front liquid chromatography (LC) sample injection in the Nodify XL2 test. This mass spectrometry method offers highly sensitive, specific, and cost-effective analysis for simultaneous quantitation of hundreds to several thousands of targeted peptides in a single experiment. We have since included the MRM technologies as part of our services for discovery and development with our biopharmaceutical customers and academic partners.
9
Enzyme-Linked Immunosorbent Assay
ELISA is the most widely used ligand binding assay platform within and outside the pharmaceutical industry. Formats include direct, indirect and sandwich assays and are typically run in manually or semi-automated modes. We use a semi-automated implementation of ELISA in clinical testing for the Nodify CDT test and the Platelia SARS-CoV-2 Total Ab test, and the cPASS neutralizing antibody test for COVID-19. The acquisition of Oncimmune USA in 2019 expanded our ability to conduct very high throughput and cost-effective ELISAs in our clinical testing laboratory. We have now included the ELISA technologies for research and development both internally and externally with our biopharmaceutical customers and academic partners.
Droplet Digital Polymerase Chain Reaction Technology (ddPCR)
We use the ddPCR technology for multiplexed, semi-automated nucleic acid detection. This allows high sensitivity, fast turn-around times, flexibility in our laboratory workflows, rapid scaling from low to moderate analyte complexity, and high-volume scalability. ddPCR is an absolute quantitation method based on the partitioning of circulating nucleic acids into up to 20,000 droplets per reaction and is used for the GeneStrat test and Bio-Rad SARS-CoV-2 ddPCR test for COVID-19. We have included the ddPCR technologies for research and development both internally and externally with our biopharmaceutical customers and academic partners.
Next Generation Sequencing Technology
We use an NGS technology for broad genomic sequencing of clinical specimens. Our strategy with NGS relies on a menu of off-the-shelf and custom research use assays, which we develop and make available as a part of our commercial pipeline and biopharmaceutical test services. The NGS technology integrates automated systems to yield high sensitivity results with a rapid turnaround time. Since adoption of this technology, we have included the NGS technologies for research and product development both internally and externally with our biopharmaceutical customers and academic partners.
Our Solutions and Products
To help address the current limitations with standard of care in lung cancer diagnosis, treatment, and monitoring, we use combinations of tumor, immune and host profiling, radiological imaging, patient clinical profiling, and our proprietary AI platform to provide a holistic view of each patient’s dynamic disease state.
We have five blood-based diagnostic tests across the lung cancer continuum of care to help address clinical unmet needs by physicians.

10
Diagnosis – Nodule Management
We believe we are the only company to offer two commercial blood-based tests to help physicians reclassify risk of malignancy in patients with suspicious lung nodules. Our blood-based nodule management offering, Nodify Lung Nodule Risk Assessment assists physicians in reclassifying a patient’s risk of lung cancer by incorporating their protein biomarker results with radiographic imaging and clinical characteristics. Nodify Lung consists of the Nodify CDT and Nodify XL2 proteomic tests, which can be ordered separately or together from a single blood draw to help reclassify risk of cancer to aid physicians in stratifying patients into distinct nodule management pathways: intervention or surveillance.
The Nodify CDT test is used to help identify lung nodules that are likely malignant and the Nodify XL2 test helps identify lung nodules that are likely benign. Nodify Lung is available for patients 40 years or older, with nodules between 8 and 30mm, and less than 65% pre-test risk of lung cancer. The testing strategy starts with the Nodify CDT test to determine if a nodule is likely malignant or at a higher risk of lung cancer. The Nodify CDT test helps physicians identify cancer more quickly by prioritizing patients with a higher risk of malignancy for a diagnostic procedure, such as biopsy or surgery. If the nodule is not identified as having a high risk of malignancy by the Nodify CDT test, then the Nodify XL2 test is performed to help determine if the patient’s nodule is likely benign or has a reduced risk of lung cancer and may be a candidate for CT imaging surveillance. The Nodify Lung testing strategy is represented graphically in the image below starting with the patient’s pre-test risk of malignancy and ending with the guideline-recommended diagnostic procedure for each risk category.
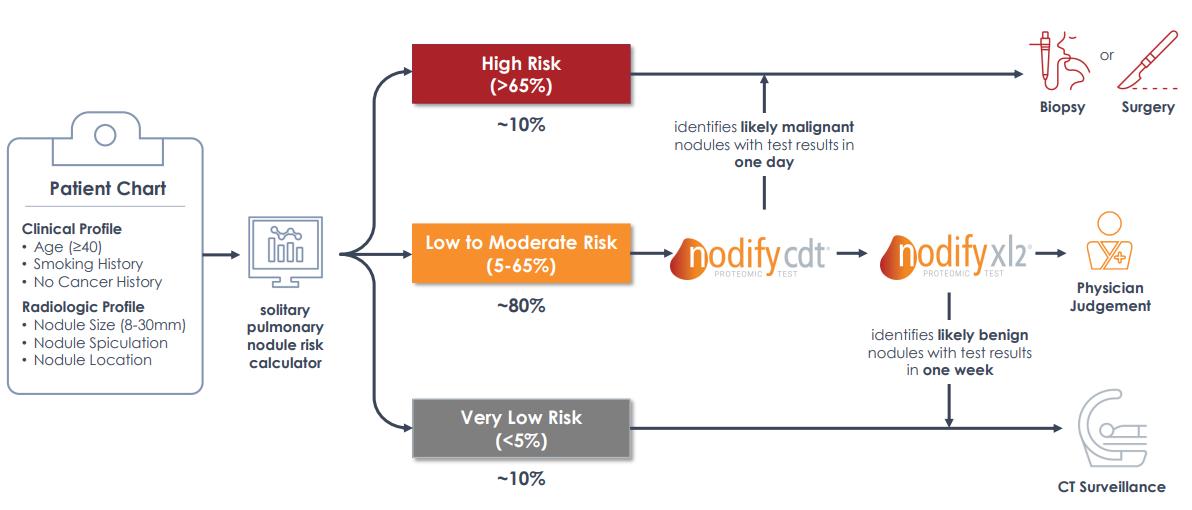
We launched the Nodify Lung combined offering of the Nodify CDT and Nodify XL2 tests in March 2020. However, the Nodify XL2 test has been available to all physicians since September 2019 and has been available to a select group of physicians since October 2018. We acquired the Nodify XL2 test from Integrated Diagnostics (Indi) in July 2018 and acquired the Nodify CDT test from Oncimmune USA in October 2019.
Nodify CDT
The Nodify CDT test is a blood-based proteomic test that helps identify patients who have a suspicious lung nodule that is likely malignant or at a higher risk of being cancerous. Results allow physicians to identify patients who may be better candidates for timely invasive diagnostic procedures such as bronchoscopy, transthoracic needle biopsy, or surgical resection, with the hope of catching cancer earlier. The Nodify CDT test enhances lung nodule risk assessment to facilitate compliance with clinical treatment guidelines such as those of the American College of Chest Physicians (ACCP). The Nodify CDT test is intended for use in patients who are 40 years or older, have nodules between 8 and 30mm, and pre-test risk of lung cancer of less than 65%.
The test measures the levels of seven circulating autoantibodies (P53, NY-ESO-1, CAGE, GBU4-5, SOX2, HuD, and MAGE A4) associated with lung cancer, combined with an algorithm to report out three potential results: High Level, Moderate Level, or No Significant Levels of Antibodies Detected (NSLAD). The seven autoantibodies have shown to be elevated for all types of lung cancer, and from the earliest stage of the disease.
Unlike the tumor antigens themselves, the autoantibody levels can be measured accurately through a blood sample, based upon the signal amplification generated by the immune response to cancer. This mechanism of action likely reflects very early events in a tumor’s evolution; as the immune system initiates a response to the cancer, it can also trigger an expansion of self-reactive antibodies that can be measured in circulation.
11
In addition to the test result of High Level, Moderate Level, or NSLAD, each test report includes the patient’s pre-test risk of malignancy as calculated by the Solitary Pulmonary Nodule (SPN) Risk Assessment calculator, and their post-test risk of cancer incorporating the result of the test. The SPN Risk Assessment calculator was developed by Stephen Swensen, M.D., of the Mayo Clinic and is designed to provide a risk of malignancy for a patient with a newly discovered incidental nodule. The model incorporates six clinical and radiologic factors into the equation: age, nodule size, smoking status, nodule location, spiculation (nodule edge characteristic), and previous history of lung cancer. Incorporating the autoantibody levels with the risk model provides physicians with a more accurate assessment of risk. The test has been studied in 14 peer reviewed published studies and presentations.
Nodify XL2
The Nodify XL2 test is a blood-based proteomic test that helps identify patients who have a suspicious lung nodule that is likely benign or at a reduced risk of being cancerous. Results allow physicians to identify patients who may be better candidates for routine CT surveillance to monitor for growth or shrinkage of the nodule over time instead of an invasive diagnostic procedure. The Nodify XL2 test is used for patients who are 40 years or older, have nodules between 8 and 30mm, and have a pre-test risk of lung cancer of less than or equal to 50%.
The Nodify XL2 test integrates peptides measured by LC-MS with clinical and radiological characteristics that are combined by an algorithm to report out three potential results: Likely Benign, Reduced Risk, or Indeterminate. Specifically, the Nodify XL2 test measures the relative abundance of two peptides (LG3BP and C163A) in circulation in the patient’s blood. The native proteins from which the peptides are derived, have been associated with an inflammatory response to cancer. The clinical factors are patient age and smoking status, and radiological factors are nodule size, location, and edge characteristics.
In addition to the test result of Likely Benign, Reduced Risk, or Indeterminate, each test report includes the patient’s pre-test risk of lung cancer as calculated by the SPN Risk Assessment calculator, and their post-Nodify XL2 risk of malignancy incorporating the result of the test. Incorporating the peptide levels with the risk model provides physicians with a revised assessment of risk incorporating the patient’s biology.
In summary, the inclusion of the Nodify Lung testing strategy into clinical practice helps physicians reclassify risk of malignancy of low to moderate risk lung nodules by incorporating the patient’s own biology into the assessment. The Nodify CDT test helps physicians identify patients with a high-risk lung nodule who may benefit from timely intervention, which can ultimately help identify lung cancer earlier. The Nodify XL2 test helps physicians identify patients with a very low risk lung nodule who may benefit from CT surveillance and could avoid unnecessary invasive procedures.
Blood samples for the Nodify XL2 and Nodify CDT tests can be collected in the physician’s office, laboratory, or at home through use of mobile phlebotomy. Mobile phlebotomy options facilitate testing for patients even if they are not seen in person by the physician and instead are seen through telehealth visits. This benefits the patient as scheduling can be conveniently fit to their needs and can keep them away from a physician’s office or hospital for safety concerns, especially with the evolving coronavirus pandemic. Additionally, mobile phlebotomy benefits the physician as the logistics around a blood draw or tissue sampling are out of their hands. We have a national network of contracted Nurses and Phlebotomists to support at-home or mobile blood collection.
Both tests require a single blood sample shipped at ambient temperature to our certified, high-complexity clinical laboratory in De Soto, Kansas. Nodify CDT testing requires whole blood and Nodify XL2 testing requires either a whole blood tube, or whole blood spotted onto our proprietary Blood Collection Device (BCD). The introduction of the BCD and now the whole blood tube as qualified specimen collection methods for use with the Nodify XL2 test alleviated the need for serum separation processing steps by phlebotomists at blood draw sites such as centrifugation, and cold chain (dry ice) shipments, which has increased the market access to our proteomic-based tests. Results for the Nodify CDT test alone are typically available within one day. If both tests are ordered for the patient and Nodify CDT returns a result of NSLAD, then both test results are typically available within 4 to 5 days. All results are available through a portal, fax, hard copy, or mobile device.
12
Treatment Guidance and Monitoring
Profiling the tumor through blood-based testing can help identify mutations in genes that may be driving growth of the tumor and may be targets for therapeutics. However, tumors also suppress intrinsic mechanisms that prevent the patient’s immune system from identifying and eliminating the cancer cells. Profiling the immune system can show if the patient’s immune system may have been subverted and therefore, is less likely to be responsive to immunotherapies. Our blood-based IQLung testing strategy consists of the GeneStrat ddPCR and GeneStrat NGS tumor profiling tests and the VeriStrat immune profiling test, which can be ordered together or separately for patients with NSCLC. Together, the tests typically have a 72-hour turnaround time, providing physicians with timely results to facilitate treatment decisions.
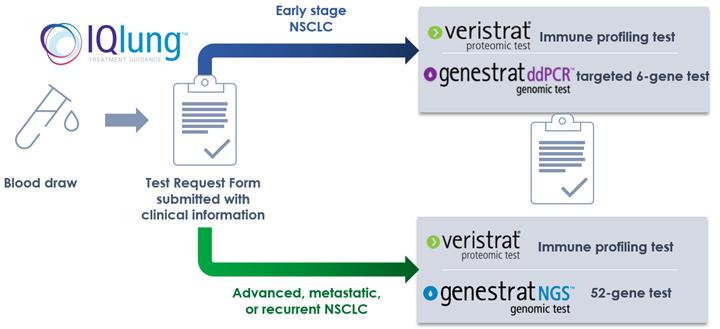
GeneStrat ddPCR
The GeneStrat test is a blood-based tumor profiling test that detects the guideline recommended, actionable mutations in lung cancer: EGFR, KRAS, BRAF, EML4-ALK, ROS-1, and RET. Physicians can order one or any combination of the gene tests, whichever they deem medically necessary for the individual patient. The presence of a mutation in one of the genes could indicate the patient is a candidate for the associated guideline-recommended targeted therapy. The GeneStrat test performance and potential clinical utility have been published in 3 peer reviewed studies.
The GeneStrat test results are typically available within 36 hours from our receipt of the sample in our Boulder, Colorado clinical laboratory. In a study at Eastern Carolina University, it was observed that blood-based testing was up to three weeks faster than tissue-based testing, with tissue-based testing taking a median of 26 days from sample collection. With GeneStrat testing, results are typically available in time for the patients first oncology visit, allowing the patient to start front-line treatment as quickly as possible. In the same study, it was observed that only 4% of patients had tissue-based molecular test results prior to start of front-line treatment. Meanwhile, after integrating Biodesix testing at the institution, 72% of patients had molecular test results available. Testing with the GeneStrat test can help physicians identify driver mutations quickly to help speed up time to treatment.
GeneStrat NGS Test
The GeneStrat NGS test is a blood-based 52-gene tumor profiling test panel that detects the guideline recommended, actionable mutations in lung cancer including five gene classes (SNV, INDELS, CNA, fusions, and exon-skipping). Specific variants of relevance to NSCLC include EGFR, KRAS, BRAF, EML4-ALK, ROS-1, RET, MET, NTRK. The GeneStrat NGS test is used for late-stage, metastatic NSCLC and Physicians can order one or any combination of the IQLung tests, whichever they deem medically necessary for the individual patient. The presence of a mutation in one of the genes could indicate the patient is a candidate for the associated guideline-recommended targeted therapy. The GeneStrat NGS test performance and potential clinical utility have been published in two published studies.
GeneStrat NGS test results are typically available within 72 hours from our receipt of the sample in our Boulder, Colorado clinical laboratory.
13
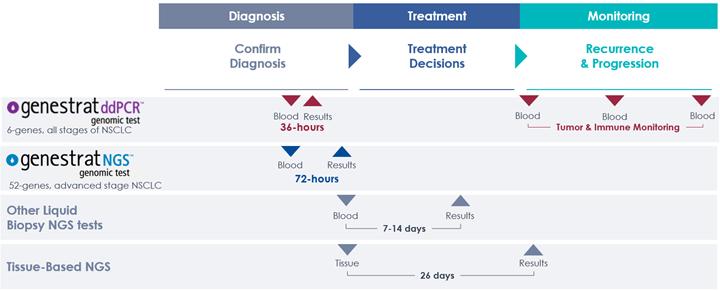
We believe that rapid, blood-based tumor profiling with the GeneStrat ddPCR and GeneStrat NGS tests can be complementary to both targeted tissue-based testing (including PD-L1) and tissue-based broad genomic sequencing. Testing with GeneStrat ddPCR and GeneStrat NGS tests at diagnosis can help quickly identify patients who are eligible for targeted therapies. Additionally, blood-based testing upfront can help save valuable tissue for diagnostic evaluation, PD-L1 testing and broad genomic profiling for rare mutations to enroll in clinical trials.
VeriStrat
The VeriStrat test is a blood-based proteomic test that provides a personalized view of each patient’s immune response to their lung cancer. Results help inform physicians whether their patient has a more aggressive cancer and can help with treatment planning. The VeriStrat test profiles the patient’s immune system by measuring eight protein features measured by mass spectrometry and interpreted by a proprietary machine learning-based algorithm to produce either a VeriStrat Good or VeriStrat Poor test result.
The presence of a VeriStrat Poor result indicates the presence of chronic inflammation and a chronic acute phase immune response. A chronic acute phase immune response can trigger the immune system to provide growth factors to the tumor to increase blood flow and tumor growth. The test has been studied in over 85 peer-reviewed and published clinical studies across many different types of therapies such as chemotherapy, targeted therapies, immune therapies, and combinations. The results consistently show the test to be predictive of outcomes, independent of other prognostic factors including PD-L1 expression and performance status. Patients who test as VeriStrat Poor, on average, have an overall survival that is less than half of those who test as VeriStrat Good, independent of treatment type, demonstrating that the test is strongly prognostic. Conversely, patients with a VeriStrat Good test result typically respond better to standard of care treatments than those patients that test as VeriStrat Poor. By using the VeriStrat test for immune profiling, physicians can help identify the patients with an immune status associated with generally poor prognosis who should be treated with alternate therapies or in clinical trials.
GeneStrat ddPCR testing is performed using PCR technology, GeneStrat NGS testing is performed using Next Generation Sequencing technology, and the protein features in VeriStrat are measured using MALDI-ToF mass spectrometry. Results are typically available within 72 hours through a portal, fax, hard copy, or mobile device.
The GeneStrat ddPCR and GeneStrat NGS tests require whole blood specimen collection tubes, and VeriStrat requires a whole blood sample spotted onto our proprietary BCD. Both sample types are shipped at ambient temperature and testing is performed in our certified, high-complexity clinical laboratory in Boulder, Colorado.
COVID-19
Biodesix WorkSafe COVID-19 Testing Program
In response to the COVID-19 pandemic, through our partnership with Bio-Rad, we commercialized the Biodesix WorkSafe testing program. Our scientific diagnostic expertise, technologies, and existing commercial infrastructure enabled us to rapidly commercialize two diagnostic tests for the SARS-CoV-2 virus that causes COVID-19. The first test is the Bio-Rad SARS-CoV-2 ddPCR test, which is a molecular test intended for detecting active SARS-CoV-2 infection. The test was FDA EUA authorized on May 1, 2020, authorizing performance of the test in laboratories certified under CLIA to perform high complexity tests. The second test is the Platelia
14
SARS-CoV-2 Total Ab test, which is an antibody test intended for detecting a B-cell immune response to SARS-CoV-2, indicating recent or prior infection. The test was FDA EUA authorized on April 29, 2020.
Within a month of initiating our development collaboration with Bio-Rad, we were able to launch two tests for commercial use. We were able to bring these tests to market as fast as possible due to our scientific diagnostic expertise, technologies and existing commercial infrastructure. In addition to our launch agility, we have been able to rapidly scale our laboratory operations for high-volume testing. We remain committed to delivering rapid test results in 24 to 48 hours on average.
Using the Bio-Rad SARS-CoV-2 ddPCR test and the Platelia SARS-CoV-2 Total Ab tests, we operate and have commercialized the Biodesix WorkSafe testing program.
Prior to using the Bio-Rad tests as part of our testing program, we performed feasibility, verification, and validation studies, including developing software for process automation, sample accessioning, data management and reporting, all required to demonstrate the test operated as claimed by the manufacturer and as required by our certifying regulatory agencies for high complexity laboratory testing. We secured independent reference specimens run with EUA tests to validate these tests as fit for diagnostic use in our laboratories. Post-launch development support for these tests have included improvements in on-boarding new personnel, logistics of sample collection, sample receipt and data reporting, all required to support our testing program. Additional releases of the laboratory data management software are ongoing and planned for the foreseeable future.
In addition, during the three months ended June 30, 2021, we began partnering with GenScript Biotech Corporation to commercialize the blood-based cPass™ SARS-CoV-2 Neutralizing Antibody testing as a service. The test is the first and only surrogate neutralizing antibody test with FDA EUA and uses ELISA technology to qualitatively detect circulating neutralizing antibodies to the RBD in the spike protein of SARS-CoV-2 that are produced in response to vaccination or previous SARS-CoV-2 infection.
These tests under the Biodesix WorkSafe testing program are utilized by healthcare providers, including hospitals and nursing homes, and are also offered to businesses and educational systems to assist in their back-to-work or back-to-school strategies, a crucial element of restarting economic activity. We have announced multiple partnerships for COVID-19 testing, and maintain an agreement with the State of Colorado to be one of the diagnostic companies to support widespread COVID-19 testing for the State. Additionally, we have overseen and managed onsite testing and validating testing for the Big Ten Conference athletic competitions through the term of our contract which expired on June 30, 2021.
Bio-Rad SARS-CoV-2 ddPCR test
The Bio-Rad SARS-CoV-2 ddPCR test, also known as a molecular or viral test, is intended for the qualitative detection of nucleic acid from SARS-CoV-2, the virus that causes COVID-19. The test targets detection of the nucleic acid from SARS-CoV-2 (not from any other viruses or pathogens) in respiratory specimens to identify and isolate infected individuals. Recent studies have shown that ddPCR-based testing is more sensitive than qPCR for detecting SARS-CoV-2, specifically in the reduction of false negative results. In one study, the ddPCR test demonstrated 95% accuracy vs. 47% for other “bulk” RT-PCR technologies used in other molecular tests Specimens are shipped at ambient temperature or dry ice depending on the viral transport media (VTM) used, and testing is performed using ddPCR in our certified, high-complexity clinical laboratory in Boulder, Colorado. Results are typically available through fax, hard copy, or encrypted email within 24 to 48 hours on average from receipt of the sample.
Platelia SARS-CoV-2 Total Ab Test
The Platelia SARS-CoV-2 Total Ab test (also known as a serology or antibody test) is intended for use as an aid in identifying individuals who have developed an adaptive immune response to the SARS-CoV-2 virus, indicating recent or prior infection. The test uses whole blood to detect circulating antibodies against the virus. The sensitivity is 98% and specificity is 99% eight days after the onset of symptoms. At this time, it is not known how long antibodies persist following infection and if the presence of antibodies confers protective immunity. The test is intended for the qualitative detection of total anti-SARS-CoV-2 nucleocapsid antibodies (IgG, IgM and IgA) in human serum or plasma specimens. The test requires a 3 mL blood draw, and samples are shipped at ambient temperature. Testing is conducted using semi-automated ELISA technology in our certified, high-complexity clinical laboratory in De Soto, Kansas. Results are typically available within 24 to 48 hours from receipt of the sample through fax, hard copy, or encrypted email.
Biopharmaceutical Diagnostic Discovery, Development and Testing Services Business
We believe our leadership in clinical proteomics and our multi-omic approach to probe the cancer disease state provides our customers with a clear and distinct advantage over other diagnostic service providers who solely focus on either genomics or proteomics. Similar to our commercial clinical testing business, our biopharmaceutical diagnostic discovery, development and testing services business leverages the Diagnostic Cortex to provide an extensively validated and deep learning approach to discovering new biomarkers, which in turn helps drive the clinical development of therapeutics. We recognize each clinical development program is complex, which is why we offer end-to-end diagnostic solutions, ranging from initial biomarker discovery and feasibility projects to commercialization of companion diagnostics.
15
To address the increasing complexity of disease biology and new drug mechanisms of action, we employ a range of genomic and proteomic technologies to uncover insights about the tumor biology and patient’s immune response to cancer for therapeutics in clinical development. With our broad technology and service offering, including the performance of over 30 assays for research use as part of our laboratory services (see diagram below), we are able to provide the depth and breadth of biomarker tools for our partners for multi-omic analyses across their product development efforts. Although we recognize the importance of a multi-omic, approach in translational research, we are experts in discovering and developing proteomic-based diagnostic tests to help interrogate the immune profile and host disease state of patients on particular therapeutics. Traditionally, oncology biomarkers have been discovered from tumor tissue, but with the increased trend in the number of programs featuring immuno-oncology agents in therapeutic development, we believe there is a clinical unmet need for blood-based tumor markers and host/immune biomarkers to complement information obtained from tissue.
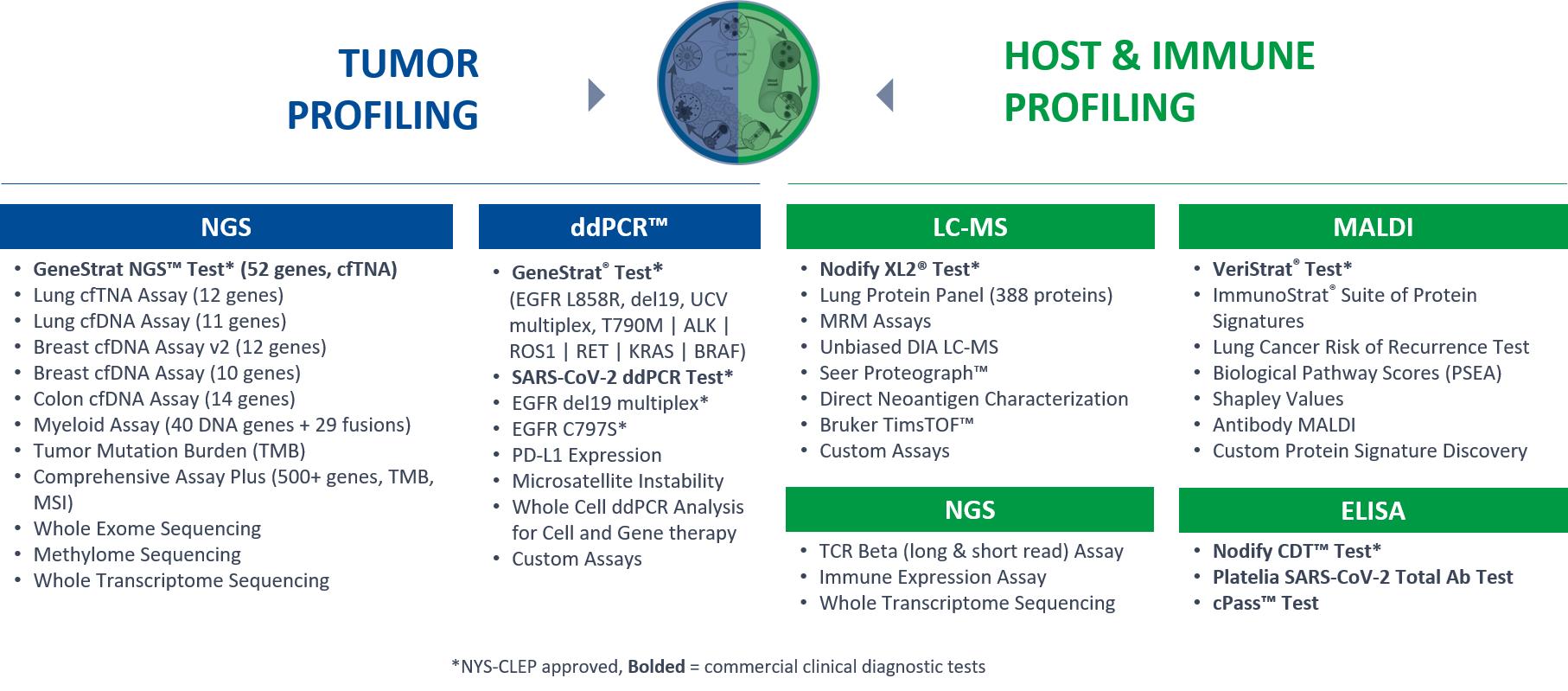
We believe we provide benefit to our biopharmaceutical customers as they integrate strategies for increasing the probability of success for pivotal clinical trials. Specifically, our diagnostic testing services may help enable quicker enrollment rates for patients in prospective clinical trials, ranging from phase 1 to phase 3, and could help identify patient populations who may experience the greatest benefit from new therapeutics. Ultimately, our goal is to help biopharmaceutical customers realize greater efficiency in their clinical development programs. Additionally, we have the ability to access and leverage our large sample and data biobank for our partners’ data mining needs, including new test discovery.
While our biopharmaceutical discovery, diagnostic development and testing revenue continues to grow, it is important to note that we benefit greatly from these partnerships in many ways that expand beyond revenue. We are continuously expanding our knowledge and biological understanding of multiple diseases and the rapidly evolving treatment landscape, while our Diagnostic Cortex continues to be powered through these biomarker analyses. Additionally, our sample and data biobank continue to grow and can be further leveraged for internal test development and external partnering. Importantly, we look to supplement our product development efforts with companion diagnostics as they are developed.
To date, we have over 50 biopharmaceutical customers and academic partners who have utilized our diagnostic tests and services. The following are a few case studies of early-stage biomarker discovery and development with our biopharmaceutical customers.
16
Our Competitive Advantages
We believe the following are our key competitive advantages:
17
Our Strategy
We strive to provide swift, comprehensive and actionable insights to improve patient outcomes across lung disease and to help answer critical clinical questions faced by physicians, researchers, and biopharmaceutical companies. To achieve this, we intend to:
18
Our Diagnostic Tests in Development
With the goal of finding solutions for clinical unmet needs related to diagnosis, treatment and monitoring in lung disease, our diagnostic tests in development include the following:
Early-Stage NSCLC—Risk of Recurrence (ROR)
Currently, surgical resection of the tumor without systemic or radiation therapy is standard of care for stage I NSCLC patients. However, 20 to 40% of surgically treated patients will suffer a recurrence within 5 years after surgery. From market research with pulmonologists, thoracic surgeons, and medical oncologists, we identified a significant clinical unmet need for a blood-based test to help identify stage I NSCLC patients who are at a higher risk of recurrence and may benefit from a more aggressive surgical procedure, or from neoadjuvant or adjuvant systemic treatment. Based on this unmet diagnostic need, we discovered the Risk of Recurrence (ROR) test, which is a pre-surgery blood-based proteomic test, designed with the Diagnostic Cortex to predict whether a stage I NSCLC patient has a higher risk of recurrence post-surgical resection. Knowing this information early and before surgery may change the surgical plan and/or support treatment decisions such as neoadjuvant or adjuvant therapy, which have the potential to reduce tumor volume and address micro-metastatic disease as early as possible. Our ROR test validated in an independent sample set, and we are currently working with major academic institutions across the United States to further validate the test.
Late-Stage NSCLC—Immunotherapy Treatment Guidance (PIR)
In 2015, the first immunotherapy-based treatment regimen was approved by the FDA for use in lung cancer. Currently, there are 9 immune checkpoint inhibitor (ICI) regimens (single agent or combinations) recommended by the NCCN guidelines for treatment of advanced NSCLC patients. For a portion of patients treated, these drugs can result in significant improvement in overall survival compared with platinum-based chemotherapy options.
The combination ICI regimens see some improvement in performance over single agent ICI, but side effect profiles are worse, and costs are higher than for single agent ICI. In addition, recent data have shown that a subset of patients experience more rapid disease progression on ICI compared with chemotherapy. We utilized the Diagnostic Cortex platform to discover our Primary Immune Response (PIR) test. PIR is a blood-based proteomic test designed to profile a potential to mount an immune response to their cancer and predict those patients likely to respond to ICI monotherapy treatment, ICI + chemotherapy combination treatment, or who would be highly resistant to ICI therapy. Our PIR test has been validated in multiple independent sample sets for advanced stage NSCLC patients treated with single agent ICI, and we are currently working with major academic institutions across the United States to further validate the test in a prospective study called BEACON-Lung.
Monitoring – Progression & Resistance
Blood-based monitoring with our ddPCR technology may offer a feasible method to non-invasively evaluate therapeutic mechanism of action, disease progression, and the emergence of resistance mutations in patients treated with targeted therapies. Our internal validation studies have shown the utility of the GeneStrat EGFR ddPCR test in all three of these indications. The test can identify disease progression up to 3 months (median) in advance of standard imaging. Using ddPCR for longitudinal blood-based monitoring of EGFR cell-free DNA mutations is a cost-effective testing method while patients are being treated with targeted therapies.
Clinical Trials
We are dedicated to continuously publishing and presenting new data on the clinical validation and utility of our diagnostic tests. We have participated in 27 clinical studies, 5 of which are ongoing, and have published over 300 peer-reviewed publications and presentations. The following are our ongoing clinical studies for our diagnostic testing solutions.
ORACLE Registry Study (NCT03766958)
The ORACLE registry study was designed to develop real-world clinical utility data for Nodify XL2 and is titled “An Observational Registry Study to Evaluate the Performance of the Nodify XL2 Test”. The study objectives are to show a reduction in invasive procedures on patients with benign nodules compared to a historical control obtained from chart review. The first patient enrolled on October 16, 2018. As of May 1, 2020, 423 patients have been enrolled and are undergoing primary endpoint analysis, with 2-year follow-up estimated to be completed by the first half of 2022.
19
ALTITUDE Clinical Utility Study (NCT04171492)
The ALTITUDE clinical utility study is designed to evaluate the performance of Nodify Lung (Nodify XL2 and Nodify CDT) in a randomized controlled study (RCT). The study is titled “A Multicenter, Randomized Controlled Trial, Prospectively Evaluating the Clinical Utility of the Nodify XL2 Proteomic Test in Incidentally Discovered Low to Moderate Risk Lung Nodules”. We received central investigational review board (IRB) approval in December 2019 and have an enrollment goal of 2,000 patients. The study objectives are to evaluate how the addition of the Nodify Lung test result impacts the clinical decision making for patients with new, incidentally identified solid lung nodules assessed as low to moderate risk of lung cancer. The trial has an adaptive study design with a blinded standard of care arm and 2:1 randomization for open-label results for Nodify XL2. The study launched in December of 2020. Phase 1 of the study with only Nodify XL2 is expected to enroll 500 patients. Phase 2 of the adaptive study design will include an open-label arm for Nodify CDT, which is aligned with our commercial testing algorithm.
INSIGHT Observational Study (NCT03289780)
The INSIGHT observational study is designed to evaluate the real-world clinical utility and performance of the IQLung (GeneStrat ddPCR, GeneStrat NGS and VeriStrat) testing strategy. The title is “Observational Study Assessing the Clinical Effectiveness of VeriStrat and Validating Immunotherapy Tests in Subjects with Non-Small Cell Lung Cancer (INSIGHT)” and the first patient enrolled was on May 11, 2016. To date, we have over 3,500 patients enrolled with a target 5,000 enrollment goal. Final analysis with 3-year follow-up is estimated to be completed by 2024. Results of an interim analysis were presented at ASCO 2020. The study rationale is to guide the adoption of VeriStrat and inform medical decision making, including treatment choice, and enable the validation of additional mass spectrometry-based proteomic tests. The primary study objective is to describe the impact of the VeriStrat test results on treatment decisions, including but not limited to the percentage change in treatment decision, differences in chosen treatments between patients classified as VeriStrat Good and those classified as VeriStrat Poor, and the percentage of patients receiving systemic therapy or supportive therapies only.
BEACON-Lung Clinical Study (Trial in Progress)
In partnership with ALCMI (Addario Lung Cancer Medical Institute), the BEACON-Lung clinical study is intended to evaluate the performance and utility of our proteomic product currently in development, PIR, in advanced stage NSCLC patients who express high PD-L1. The study title is “A Biomarker Analysis in High PD-L1 Expressing NSCLC Patients Treated with An Immune Checkpoint Inhibitor (ICI) With or Without Platinum-Based Chemotherapy.” The study design is an observational, multicenter, open-label study to assess biomarkers (serum, microbiome, radiomics and tissue) as predictive of early progression in 390 treatment-naive patients with advanced stage NSCLC and PD-L1 greater than or equal to 50% treated with two standard of care regimens, triplet therapy (platinum-based chemotherapy plus ICI regimen) and ICI monotherapy (single agent ICI). The objectives are to collect biospecimens and evaluate candidate biomarkers, with a focus on PIR, to detect early progression on ICI monotherapy versus triplet therapy.
DROPLET
Droplet is an exploratory observational study to assess the utility a novel gene linkage phenomenon as measured by the Bio-Rad SARS-CoV-2 ddPCR test, on the correlative immune response in individuals previously infected with the COVID-19 virus. The primary objective of this study (titled “ddPCR Linkage, Viral Load and Immune Response in Individuals Infected with COVID-19”) is to evaluate the correlation between linkage, viral load and the immune responses in individuals who have had a natural infection with SARS-CoV-2. Fifty patients will be enrolled and serially tested for viral load and immune responses for up to 12 months. More recently, the focus of the study has shifted to monitoring vaccine antibody durability and T cell responses.
Commercialization
For our lung cancer and nodule management tests, commercial efforts are focused on the promotion of our testing strategies to healthcare professionals actively involved in the diagnosis and treatment of lung cancer. Primarily focusing on pulmonology and oncology, the commercial team, consisting of specialty sales representatives, medical affairs, marketing and customer care representatives, works to educate and inform the entire patient care group consisting of physicians, nurses, office staff, laboratory personnel, and administration as to the appropriate use and value provided by our testing. The team’s goal is to drive test adoption through articulating the scientific and clinical evidence behind our tests, how they impact the clinical care of a patient, and how the tests can ultimately help to improve patient outcomes.
Patients with pulmonary nodules are concentrated in the pulmonology sub-specialty, where additional resources such as lung cancer screening and nodule management clinics may exist to provide an increased level of care. We are also engaging large hospital systems in a “top-down” approach, with a goal of incorporating our tests into system wide pathways and protocols.
After a physician orders our tests, blood is collected either in the physician office or laboratory, third-party “store front” patient service centers, or it can be collected in the patient’s home or workplace. We have contracted with a network of patient service centers and mobile phlebotomy services to be enable collection of blood samples outside of the physician office, at home or work for patients across the United States.
20
For our Biodesix WorkSafe testing program, we have a dedicated outreach team that works with the State of Colorado healthcare providers and hospitals, and employers looking to safely return to work across many industries, including food services, oil and gas, biotechnology and pharmaceuticals, sports teams, universities, and many small businesses. We recognize everyone’s COVID-19 situation is unique, which is why we provide end-to-end customized solutions to support testing for our different customers, such as risk assessment tools, physician ordering services, on-site testing, phlebotomy services, shipping logistics, and ongoing client support.
Our business development team is focused on selling our complete offering of tests and services to biopharmaceutical companies in the United States and internationally. We currently hold a medical device establishment license in accordance with the Health Canada Medical Devices Regulations of the Food and Drugs Act and successfully passed an international Health Canada inspection in October 2021. Our team consists of customer facing business development associates that work with our biopharmaceutical customers to identify projects, draw up statements of work and negotiate service agreements. Alliance managers help to manage the contractual obligations and scope of the project, whereas our operations team assures the project is managed with adequate resources and delivers on time. We take a two-pronged approach generating business in this segment. Primarily, we leverage existing projects and relationships to expand sales in current accounts. We also actively map ongoing drug development projects in biopharmaceutical companies and target programs best suited to our platform for new test development.
Coverage and Reimbursement
The primary source of reimbursement for our tests in the United States is from third-party payers including government payers, such as Medicare, and commercial payers, such as insurance companies. Reimbursement for laboratory tests in the United States is determined by various payers, including private third-party payers, managed care organizations, and state and federal health care programs, such as Medicare and Medicaid. In Medicare, coverage of an item or service depends on whether it is “reasonable and necessary” under the section 1862(a)(1)(A) of the Social Security Act (SSA). For single-source laboratory tests, this determination is typically made by the Medicare Administrative Contractor (MAC) with jurisdiction over the laboratory where the test is performed. Our Boulder, Colorado laboratory is currently under the jurisdiction of Novitas Solutions, Inc. Our De Soto, Kansas laboratory is under the jurisdiction of Wisconsin Physicians Service Insurance Corporation, which participates in the MolDX program (administered by another MAC, Palmetto GBA) to set coverage policy for molecular diagnostic tests.
Medicare pays for clinical diagnostic laboratory tests (CDLTs), on the Clinical Laboratory Fee Schedule (CLFS). Section 216(a) of the Protecting Access to Medicare Act of 2014 (PAMA), added section 1834A to the SSA, which established the current CLFS rate setting processes and coding provisions for CDLTs, and created a new subcategory of CDLTs called Advanced Diagnostic Laboratory Tests (ADLTs), with separate reporting and payment requirements.
Under section 1834A and its implementing regulations, clinical laboratories that receive the majority of their Medicare revenues from payments made under the CLFS and the Physician Fee Schedule report on a triennial basis (or annually for ADLTs), private payer rates and volumes for their tests with specific billing codes based on final payments made during a set data collection period. The payment rate for a test for the ensuing three-year period (or one year for ADLTs) is set at the weighted median of the rates reported under the specific billing code for that test. Newly established codes for CDLTs are priced until the next private payer rate reporting cycle either based on the payment rate of a comparable code on the CLFS, as determined by CMS ("crosswalking") or at the median of rates submitted by the individual MACs based on statutory and regulatory factors ("gapfilling"). New ADLTs are initially priced at “actual list charge” for a nine-month period, after which they are priced based on private payer rates, with a recoupment provision if actual list charge is more than 130% of the weighted median of private payer rates reported.
The various payers in the United States also determine their own billing rules. In December 2020, Medicare revised its billing rules for clinical laboratory tests to require cancer-related protein-based Multianalyte Assays with Algorithmic Analyses to be billed directly to Medicare by the performing laboratory in most cases when performed on a specimen collected from a hospital outpatient. Molecular pathology tests and most ADLTs are also generally required to be billed directly to Medicare by the laboratory under these circumstances.
For our COVID-19 testing program, the primary source of reimbursement is through contracts with hospitals, companies providing wellness testing for their employees, or direct pay from patients. We believe that our lung cancer tests can both improve patient outcomes and help guide cost-effective treatment choices for patients with and at-risk of lung cancer. Achieving broad coverage and adequate reimbursement for each of our tests is a key component of our financial success and will continue to be important over time.
Under the Families First Coronavirus Response Act (FFCRA) and Coronavirus Aid, Relief, and Economic Security Act (CARES), Congress has imposed a broad requirement that Medicare fee-for-service, Medicare Advantage, Medicaid, and commercial payers generally cover clinical diagnostic laboratory tests administered during the Public Health Emergency for the detection of SARS-CoV-2 or the diagnosis of the virus that causes COVID-19. Where applicable, the FFCRA and CARES require payers to cover COVID-19 tests without cost sharing or medical management techniques like prior authorization. Medicare fee-for-service does not require cost sharing or prior authorization for any clinical diagnostic laboratory tests.
The Department of Health & Human Services (HHS) has indicated in guidance that the coverage requirements of the FFCRA and CARES can apply both to infection and serological testing, but that they are only applicable to testing that is “diagnostic,” which “may
21
include testing of individuals with signs or symptoms compatible with COVID-19, as well as asymptomatic individuals with known or suspected recent exposure to SARS-CoV-2.” Testing that is “not primarily intended for individualized diagnosis or treatment of COVID-19 or another health condition,” including “testing conducted to screen for general workplace health and safety,” is beyond the scope of the statutory coverage requirements. We believe these coverage requirements apply to the Bio-Rad SARS-CoV-2 ddPCR test and the Platelia SARS-CoV-2 Total Ab test when performed for diagnostic purposes. In some cases, we submit claims to insurance payers for the Bio-Rad SARS-CoV-2 ddPCR test and/or the Platelia SARS-CoV-2 Total Ab test. In these cases, insurance payers may have a different position on the scope of mandatory coverage of our tests under the FFCRA and CARES. More commonly, we enter into arrangements with clients under which the client pays us directly for testing.
Compliance with applicable laws and regulations, as well as internal compliance policies and procedures adds complexity to the billing process. The Centers for Medicare & Medicaid Services (CMS) is responsible for overseeing the establishment of new Healthcare Common Procedure Coding System (HCPCS) codes for billing the Medicare program and other payers. CMS continuously evaluates and implements changes to the Medicare billing, coding, and reimbursement processes. To receive reimbursement from third-party payers, we bill our tests using a variety of HCPCS codes or Current Procedural Terminology (CPT) codes, as defined by the American Medical Association. For some of the tests we conduct, there may not be a specific CPT or HCPCS code, in which case the test may be billed under a miscellaneous code for an unlisted molecular pathology procedure or unlisted multiple analyte test with algorithmic analysis (MAAA) procedure. Because these miscellaneous codes do not describe a specific service, the third-party payer claim may be examined to determine the service provided, whether the service was appropriate and medically necessary and whether payment should be rendered. This process can result in a delay in processing the claim, a lower reimbursement amount, and/or denial of the claim.
Competitors
We primarily face competition from lung cancer diagnostic solutions companies in the United States, Europe and Asia seeking to answer clinical questions in the space, all of whom provide cancer-focused diagnostic tests to hospitals, researchers, clinicians, laboratories and other medical facilities.
Diagnosis—Nodule Management
We are not aware of any other company that offers two commercial blood-based tests to help physicians reclassify risk of malignancy in patients with suspicious lung nodules. We are aware of efforts by Veracyte, Inc. to develop and validate a test that may be competitive to the Nodify XL2 and/or Nodify CDT tests in the future. Additionally, Veracyte currently markets a test that is used post-bronchoscopy that is not competitive with our pre-bronchoscopy nodule risk assessment tests.
Prognosis, Treatment Guidance and Monitoring—NSCLC
We are unaware of any other diagnostic test available, commercially or in development, that will compete with our VeriStrat immune profiling test. There is substantial interest and activity in tumor profiling through liquid biopsy. Our genomic test offerings, the GeneStrat ddPCR and GeneStrat NGS tests, face competition from academic hospital laboratories, and companies such as Guardant Health and Foundation Medicine. We believe that there are several companies and academic research institutions in the process of developing tests for monitoring patients on or following treatment for recurrence or progression of lung cancer.
COVID-19 Testing
We believe that our competitors include university and state laboratories, national reference laboratories such as Lab Corporation and Quest Diagnostics, in-hospital laboratories, and a number of other diagnostic providers. We are aware that a number of other companies have announced development efforts to develop COVID-19 tests.
Biopharmaceutical Diagnostic Discovery, Development & Testing Services
We are aware of a number of companies who compete with our diagnostic tests and services, including diagnostic research, clinical trial testing, and the discovery, development, and commercialization of companion diagnostics. From the perspective of tumor profiling, we believe Guardant Health and Foundation Medicine are our most significant competitors. Conversely, in the immune profiling market, we believe Adaptive Biotechnologies and Personalis are our most significant competitors.
Clinical Laboratory Operations
We perform the VeriStrat, GeneStrat ddPCR, GeneStrat NGS, and COVID-19 ddPCR tests in our Boulder, Colorado high-complexity CLIA certified clinical laboratory. The laboratory is College of American Pathology (CAP) accredited, New York State Department of Health (NYSDOH)—permitted and licensed, ISO 13485:2016 Quality Management Systems—Requirements for Regulatory Purposes for Medical Devices certified, along with all other states that require licensing: California, Maryland, Pennsylvania, and Rhode Island. All aspects of the testing process from receipt of the test requisition form through to delivery of test results are performed in the Boulder, Colorado facility. The proprietary testing methods use semi-automated workflows that facilitate the successful delivery of greater than 90% of our tests within 3 days, and we believe our existing workflows will continue to successfully deliver our tests within this timeframe.
22
The Nodify XL2, Nodify CDT, COVID cPASS and COVID-19 total antibody tests are performed in our De Soto, Kansas high-complexity CLIA certified clinical laboratory. This clinical laboratory is also CAP-accredited, NYSDOH—permitted and licensed, ISO 13485:2016 Quality Management Systems—Requirements for Regulatory Purposes for Medical Devices certified and licensed by California, Maryland, Pennsylvania, and Rhode Island. Receipt of requisitions and testing is performed in our De Soto, Kansas clinical laboratory. Delivery of the test results is performed by personnel from our Boulder, Colorado headquarters. The proprietary testing methods use semi-automated workflows that facilitate the successful delivery of greater than 90% of our tests within 5 days, and we believe our existing workflows will continue to successfully deliver our tests within this timeframe.
Personnel in both facilities are responsible for quality assurance oversight, licensing, and regulation compliance and maintenance to ensure data integrity and consistent, validated processes.
Supply Chain
We rely on third-party suppliers, including in some instances single source suppliers, to provide us with certain components of our diagnostic tests. The number of suppliers feeding into the production of our diagnostic tests is in excess of 65 worldwide. We consider a select few of these suppliers, located in the United States, Europe and China, as critical single source providers of components. Bio-Rad, as described below, is the sole source supplier for our GeneStrat test and COVID-19 and antibody testing program. Oncimmune is also the sole source supplier for our Nodify CDT tests but there are known secondary suppliers for these materials. We have initiated the second source qualification process for the majority of these critical components.
In addition, we purchase supplies through purchase orders without long-term supply agreements with, or guaranteed commitments from, many of our suppliers, including single source suppliers. Additionally, at present, we rely on contract manufacturers for the production of our diagnostic tests. We depend on our suppliers and contract manufacturers to provide us and our customers with materials in a timely manner that meet our and their quality, quantity and cost requirements.
We entered into a nonexclusive license and supply agreement with Bio-Rad in August 2019. We rely on Bio-Rad to supply equipment and reagents used to perform ddPCR testing, a service offered by us under a variety of fee for service agreements and the core technology powering the GeneStrat test, but these supplies are able to be supplied by known suppliers. A disruption to this supply would negatively impact our ability to perform the GeneStrat and SARS-CoV-2 tests until alternatives could be validated.
While we have initiated the second source qualification process for the majority of these critical components, we may not be successful in securing second sourcing for all of them at all or on a timely basis. A disruption to this supply would negatively impact our ability to perform these tests until an alternative supplier could be validated.
All materials for our VeriStrat test and Nodify XL2 tests have alternative suppliers readily available, and a disruption in any single supplier would not materially impact our ability to deliver the test.
The COVID-19 pandemic has allowed us to pressure-test our supply chain and logistics processes as we purchased additional manufacturing capacity above our normal run rates to ensure that supply to execute on tests for the foreseeable future was available in-stock and in-house. Our suppliers have been able to allocate sufficient capacity to meet this increased demand with reasonable lead times and therefore we believe sufficient capacity exists for the expected volumes of all tests for the next 12 months.
Intellectual Property
Our success depends, in part, on our ability to obtain and maintain intellectual property and proprietary protection for our products and other know-how, to operate our business without infringing, misappropriating or otherwise violating the intellectual property and proprietary rights of others, and to defend and enforce our intellectual property and proprietary rights. We take efforts to protect our proprietary position using a variety of methods, which include pursuit of United States and foreign patent applications related to our proprietary technology, inventions and improvements that we determine are important to our business. We also may rely on trade secrets, trademarks, know-how, continuing technological innovation and potential in-licensing and acquisition opportunities to develop and maintain our proprietary position. For more information regarding risks relating to intellectual property, please see “Risk Factors—Risks Related to Our Intellectual Property.”
We have invested heavily in the protection of our key assets, namely the VeriStrat® and GeneStrat® tests, and we acquired a patent portfolio relating to the Nodify XL2® and Nodify CDT® tests in our acquisitions of Indi in June 2018, and of Oncimmune USA in October 2019 from Oncimmune Limited (Oncimmune). We own patents and patent applications as well as trade secrets relating to our products currently in development, a collection device for whole blood, our business strategy, client lists and business methods. Further, we have expanded our access to key intellectual property through license and co-development agreements, including our Non-Exclusive License Agreement with Bio-Rad (the Bio-Rad License), which allows us to use the Droplet Digital PCR™ technology developed by Bio-Rad and which we employ in our GeneStrat test.
23
Our patent strategy has focused on creating and acquiring protection for our VeriStrat and Nodify XL2 proteomic tests, while utilizing trade secret and some methods patent protection for our genomic test (the GeneStrat ddPCR and GeneStrat NGS™ tests) and ELISA test (the Nodify CDT test). We have entered into a non-exclusive license agreement with Bio-Rad, without the right to grant sublicenses, to utilize certain of Bio-Rad’s intellectual property, machinery, materials, reagents, supplies and know-how necessary for the performance of ddPCR in cancer detection testing for third parties in the United States. For more information regarding this license and supply agreement, please see “—Material Agreements—Agreements with Bio-Rad.” Bio-Rad owns patents relating to ddPCR and to which we have a non-exclusive license to utilize for the performance of ddPCR in cancer detection testing for third parties as set forth in the Bio- Rad License Agreement. In addition, we have separately been granted permission by Bio-Rad to use the Bio-Rad SARS-CoV-2 ddPCR test and Platelia SARS-CoV-2 Total Ab test for commercial diagnostic services. For more information regarding the permission granted to us by Bio-Rad with respect to such tests, please see “Business—Material Agreements—Agreements with Bio-Rad.” We are unaware of patents owned by Bio-Rad relating to its COVID- 19 antibody test but acknowledge that Bio-Rad has know-how and trade secrets relating to this test. We have patent protection in the United States and other countries around the world for the primary use of the VeriStrat test for profiling of patients with NSCLC, and various other uses of the VeriStrat test, such as breast cancer, prostate cancer, head and neck cancer have received patent protection. We have also received patent protection relating to our core classifier development program, our Diagnostic Cortex® technologies and our approaches to using MALDI-ToF technology (DeepMALDI® techniques). Additionally, our first device patent was issued in 2019 for our internally designed blood collection device.
As of December 31, 2021, our patent portfolio includes approximately 50 issued United States patents, 59 issued foreign patents, which includes 5 European patents that were nationalized in multiple European countries, and 41 pending applications (including 21 foreign patent applications). With regard to our product development efforts, new applications have been filed around developments relating to ARDS and COVID diagnostics, new analytic methodologies using Shapley values and semi-quantitative spectra analysis in MALDI, and national stage applications are now in active prosecution to protect our pipeline ROR and PIR tests.
The patent portfolio can be broken down into 5 major categories:
The patents relating to the VeriStrat test are scheduled to expire between 2026 and 2032. The patents relating to the Nodify XL2 test are scheduled to expire beginning in 2031 (excluding any patent term extension granted by the United States Patent and Trademark Office (USPTO)), and the patents relating to the Nodify CDT test are scheduled to expire in 2027. The patent related to the blood collection device is scheduled to expire in 2039. Should our current patent applications in prosecution in the United States issue, the resulting patents would be scheduled to have expiration dates between 2036 and 2040 (excluding any patent term extension(s) granted by the USPTO).
We currently have one pending Patent Cooperation Treaty (PCT) application. PCT patent applications are not eligible to become issued patents until, among other things, we file such PCT applications as national stage patent application(s) within 30 months in the countries in which we seek patent protection. If we do not timely file any national stage patent applications, we may lose our priority date with respect to any such PCT patent applications and any patent protection on the inventions disclosed in such PCT patent applications. Provisional patent applications are not eligible to become issued patents but can become the basis of PCT foreign stage and United States non-provisional patent applications, if such applications are filed within 12 months of filing the related provisional patent application. If we do not timely file any non-provisional patent applications, we will lose our priority date and any patent protection on the inventions disclosed in any such provisional patent application.
In addition, the term of individual issued patents depends upon the legal term for patents in the countries in which they are obtained. In most countries in which we have filed, including the United States, the patent term is generally 20 years from the earliest filing date of a non-provisional patent application, assuming the patent has not been terminally disclaimed over a commonly-owned patent or a patent naming a common inventor, or over a patent not commonly owned but that was disqualified as prior art as the result of activities undertaken within the scope of a joint research agreement. The life of a patent, and the protection it affords, is therefore limited and once the patent lives of our issued patents have expired, we may face competition, including from other competing technologies. In the United States, the term of a patent may also be eligible for patent term adjustment for delays within the USPTO. The term of a patent that covers a biological product may also be eligible for patent term extension when FDA approval is granted for a portion of the term effectively lost as a result of the FDA regulatory review period, subject to certain limitations and provided statutory and regulatory requirements are met. Any such patent term extension can be for no more than five years, only one patent per approved product can be extended, the extension cannot extend the total patent term beyond 14 years from approval, and only those claims covering the approved biological product, a method for using it or a method for manufacturing it may be extended. We may not receive an extension if we fail
24
to exercise due diligence during the testing phase or regulatory review process, fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. There can be no assurance that we will benefit from any patent term extension or favorable adjustment to the term of any of our patents. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Our ability to maintain and solidify our proprietary and intellectual property position will depend on our success in obtaining effective patent claims and maintaining and enforcing claims that are granted. However, our owned and licensed patents could be invalidated or narrowed or otherwise fail to adequately protect our proprietary and intellectual property position and our pending owned and licensed patent applications, and any patent applications that we may in the future file or license from third parties, may not result in the issuance of patents.
Because branding is as much a part of any intellectual property strategy as patent or trade secret protection we have a number of registered and pending trademarks relating to our company and products. We have received or filed for trademark protection in the United States for our tradename (Biodesix), the names of five of our commercial tests (namely the VeriStrat, GeneStrat ddPCR, GeneStrat NGS, Nodify XL2 and Nodify CDT tests), and a suite of research tests (ImmunoStrat), as well as having trademark protection for our core development and methodological platforms, such as our Diagnostic Cortex and DeepMALDI technologies. In all, as of December 31, 2021, we have 22 uniquely registered United States trademarks, 11 of which (including Biodesix, VeriStrat, and GeneStrat) have received foreign issuances as well, with 3 additional trademarks pending approving from the USPTO. We will continue to pursue protection in the United States and abroad for our branded assets and will continue to use branding to protect products currently in development, key Biodesix developments and non-trade secret methodologies.
We also rely on trade secrets, including know how, confidential information, unpatented technologies and other proprietary information, to strengthen or enhance our competitive position, protect and maintain aspects of our business that are not amenable to, or that we do not presently consider appropriate for, patent protection, and prevent competitors from reverse engineering or copying our technologies. We have decided that some technologies, such as our laboratory methodologies (including sample preparation and assay development), and some information (such as client and billing information) are best kept as trade secrets. However, trade secrets and confidential know-how are difficult to protect. To avoid inadvertent and improper disclosure of trade secrets, and to avoid the risks of former employees using these trade secrets to future employment, it is our policy to require employees, consultants and independent contractors to assign all rights to intellectual property they develop in connection with their employment with or services for the Company to the Company. We also protect our existing and developing intellectual property expressly through confidentiality provisions in agreements with third parties. There can be no assurance, however, that these agreements will be self-executing or otherwise provide meaningful protection for our trade secrets or other intellectual property or proprietary information, or adequate remedies in the event of unauthorized use or disclosure of such trade secrets or other intellectual property or proprietary information.
We also seek to preserve the integrity and confidentiality of our trade secrets and other confidential information by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in the measures we take to protect and preserve our trade secrets, such measures can be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
We intend to pursue additional intellectual property protection to the extent we believe it would advance our business objectives, which may include objectives within and outside the United States. Despite our efforts to protect our intellectual property rights, and despite the breadth of protection that has issued around our key assets, these rights may not be respected in the future or may be circumvented or challenged (and potentially invalidated) in a legal proceeding in any jurisdiction where we have intellectual property rights. In addition, the laws of various foreign countries where we have received intellectual property protection and where we may eventually distribute our products may not afford the same protections or assurances to the same extent as the laws in the United States. See “Risk Factors—Risks Related to Our Intellectual Property” for additional information regarding these and other risks related to our intellectual property portfolio and their potential effect on us.
Government Regulations
Clinical laboratory tests like our diagnostic tests are regulated under CLIA and State law. The FDA regulates medical devices pursuant to the FDCA, including many diagnostic test kits, such as in vitro diagnostic tests (IVDs). However, most Laboratory Developed Tests (LDTs) are not currently subject to the FDA’s regulation (although reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to such regulation) because the FDA has historically exercised enforcement discretion over LDTs. LDTs are a subset of IVDs that are intended for clinical use and developed, validated, and offered within a single laboratory for use only in that laboratory. FDA’s authority to regulate LDTs has been contested for many years, and there have been several legislative and administrative proposals regarding LDT regulation seeking to end or limit enforcement discretion and to bring LDTs under new or existing FDA regulatory frameworks:
25
26
We currently market our GeneStrat, VeriStrat, Nodify XL2 and Nodify CDT tests as LDTs in the United States. As a result, we believe our diagnostic services are not currently subject to the FDA’s enforcement of its medical device regulations and the applicable FDCA provisions. If the FDA disagrees with the LDT status of any of our tests, the FDA may consider the test to be an unapproved medical device and may subject us to FDA enforcement action, including, without limitation, requiring us to seek clearance, authorization or approval for the laboratory test. If the FDA were to begin enforcement with respect to our LDTs, we could incur substantial costs and delays associated with trying to obtain pre-market clearance or approval and costs associated with complying with post-market requirements.
To date, the FDA has not engaged in notice-and-comment rulemaking or released broad-sweeping guidance over LDTs, but it could choose to do so in the future and if pre-market review is required, our business could be negatively impacted as a result of commercial delay that may be caused by the new requirements. The cost of conducting clinical trials and otherwise developing data and information to support pre-market applications may be significant. If we are required to submit applications for our currently marketed tests, we may be required to conduct additional studies, which may be time-consuming, costly and could result in our currently-marketed tests being withdrawn from the market. Continued compliance with the FDA’s regulations would increase the cost of conducting our business, and subject us to heightened regulation by the FDA including penalties for failure to comply with these requirements. Failure to comply with applicable regulatory requirements could result in an enforcement action by the FDA, such as fines, product suspensions, warning letters, recalls, injunctions and other civil and criminal sanctions. There are other regulatory and legislative proposals that would increase general FDA oversight of clinical laboratories and LDTs. Until the FDA finalizes its regulatory position regarding LDTs, or the VALID Act or other legislation is passed reforming the federal government’s regulation of LDTs, it is unknown how the FDA may regulate our tests in the future and what testing and data may be required to support any required clearance or approval. The outcome and ultimate impact of such proposals on the business is difficult to predict at this time and we are monitoring developments and anticipate that our products will be able to comply with requirements that may be imposed by the FDA. In the meantime, we maintain our CLIA accreditation, which permits the use of LDTs for diagnostics purposes.
FDA Emergency Use Authorization
Section 564 of the FDCA allows the FDA to authorize the shipment of drugs, biological products, or medical devices that either lack required approval, licensure, or clearance (unapproved products), or are approved but are to be used for unapproved ways to diagnose, treat, or prevent serious diseases or conditions in the event of an emergency declaration by the HHS Secretary. 21 U.S.C. § 360bbb-3(a)(1)-(2).
On January 31, 2020, HHS Secretary Alex M. Azar II declared a public health emergency for COVID-19, under 21 U.S.C. § 360bbb-3(b)(1), justifying the authorization of emergency use of IVDs for detection and/or diagnosis of COVID-19. This determination was published in the Federal Register on February 7, 2020. 85 Fed. Reg. 7316 (Feb. 7, 2020). The public health emergency has been extended several times and remains in effect.
While this emergency declaration is effective, the FDA may authorize the use of an unapproved product or an unapproved use of an approved product if it concludes that:
Medical products that are granted an EUA are only permitted to commercialize their products under the terms and conditions provided in the authorization. The FDA may revoke an EUA where it is determined that the underlying health emergency no longer exists or warrants such authorization, if the conditions for the issuance of the EUA are no longer met, or if other circumstances make revocation appropriate to protect the public health or safety, and we cannot predict how long the EUAs for the SARS-CoV-2 tests will remain in place.
The Bio-Rad SARS-CoV-2 ddPCR test, the cPASS neutralization antibody test kit, and the Platelia SARS-CoV-2 Total Ab test have been granted FDA EUA pursuant to the current emergency declaration. We have completed all required performance verification studies to validate the use of the tests in our laboratories in accordance with the FDA Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency, CAP and New York State Clinical Laboratory Standards of Practice (NYS CLEP) requirements. The FDA Policy for COVID-19 tests is a guidance document that explains the FDA’s current thinking on the topic, is subject to change, and does
27
not establish any legally enforceable responsibilities. The FDA does not expect a separate notification or EUA request from high complexity CLIA laboratories that are performing testing for SARS-CoV-2 using only EUA-authorized test kits purchased from commercial manufacturers or their distributors. According to previous versions of the FDA's Policy for COVID-19 tests, a laboratory may make certain modifications to an EUA-authorized test if the modified test is validated using a bridging study without submitting an EUA amendment or formal notification. According to the current FDA Policy for COVID-19 tests (Nov. 15, 2021), a laboratory that modified an EUA authorized test pursuant to the previous policy for use of a new specimen through a bridging study when the new specimen type had been previously authorized for another test of the same technology may continue to offer the modified test without submitting an EUA amendment or formal notification.
Federal and State Laboratory Licensing Requirements
The Biodesix Boulder, Colorado clinical laboratory is a CAP-accredited clinical laboratory regulated by CMS pursuant to CLIA. CMS has granted CAP deeming authority under CLIA, which allows CAP to inspect laboratories in lieu of CMS. In addition to holding a CLIA Certificate and CAP laboratory accreditation, Biodesix’s Quality Management System (QMS) holds an ISO 13485:2016 certificate (recently passed recertification audit in December 2021). The Biodesix Boulder, Colorado clinical laboratory has received approval from the NYSDOH, NYS CLEP in Soluble Tumor Markers, and Molecular and Cellular Tumor Markers and Virology as well as holding state permits and licenses in California, Maryland, New York, Pennsylvania, and Rhode Island.
CLIA regulations establish standards for proficiency testing; facility administration; general laboratory systems; pre-analytic, analytic systems, post- analytic systems; personnel qualifications and responsibilities; quality control, quality assessment; and specific provisions for laboratories performing moderate to high complexity tests. Our Boulder, Colorado clinical laboratory is inspected biennially as part of its ongoing certification under CLIA certificate of accreditation by CAP. The Boulder, Colorado clinical laboratory most recently passed its CAP inspection in April 2021.
Under CLIA, a laboratory is any facility that performs laboratory testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease, or the impairment of or assessment of health. CLIA requires that a laboratory hold a certificate applicable to the type of laboratory examinations it performs and that it complies with, among other things, standards covering operations, personnel, facilities administration, quality systems and proficiency testing, which are intended to ensure, among other things, that clinical laboratory testing services are accurate, reliable and timely.
The Biodesix De Soto, Kansas clinical laboratory is a CAP-accredited clinical laboratory regulated by CMS pursuant to CLIA. CMS has granted CAP deeming authority under CLIA, which allows CAP to inspect laboratories in lieu of CMS. In addition to holding a CLIA Certificate and CAP laboratory accreditation, the De Soto, Kansas clinical laboratory most recently passed its first CAP inspection in June 2021. Biodesix’s QMS holds an ISO 13485:2016 certificate (recently passed surveillance audit in December 2021). The De Soto, Kansas clinical laboratory has received approval from the NYSDOH, NYS CLEP in Soluble Tumor Markers and Diagnostic Immunology as well as holding state permits and licenses in California, Maryland, New York, Pennsylvania, and Rhode Island.
The International Organization for Standardization (ISO) is an independent, non-governmental international organization that defines world-class specifications for products, services and systems, to ensure quality, safety and efficiency. ISO 13485:2016 is a harmonized, international regulatory benchmark for quality management systems that addresses most or all of the QMS requirements in markets including the United States, European Union, Australia, Japan and Canada. The ISO 13485:2016 certificate confirms that an organization operates a QMS that conforms to the standards established by ISO. The FDA has proposed a rule to harmonize and modernize its QSR, which would supplant the existing requirements with ISO 13485:2016. Although this rule has been delayed several times since spring 2018, the Office of Management and Budget (OMB) updated its website (January 2022) to indicate that the FDA’s proposed rule to harmonize the agency’s medical device Quality System Regulation with the ISO 13485 standard has been finished and is now under review by OMB.
To renew our CLIA certificate, we are subject to survey and inspection every two years to assess compliance with program standards. Laboratories such as ours, which are performing high complexity testing, are required to meet more stringent CLIA requirements than laboratories performing less complex tests, and therefore our laboratories are also subject to random, unannounced survey and inspection at any time. In addition, a laboratory that is certified as “high complexity” under CLIA may develop, manufacture, validate and use proprietary LDTs. CLIA requires analytical validation including accuracy, precision, specificity, sensitivity and establishment of a reference range for any LDT used in clinical testing. The regulatory and compliance standards applicable to the testing we perform may change over time and any such changes could have a material effect on our business.
CLIA provides that a state may adopt laboratory regulations that are more stringent than those under federal law, and a number of states have implemented their own more stringent laboratory regulatory requirements. State laws may require that out-of-state laboratories maintain an in-state laboratory license to perform tests on samples from patients who reside in that state. As a condition of licensure, certain states may require that laboratory personnel meet qualifications, quality control procedures, facility requirements, record maintenance requirements or other state-specific requirements.
Because our Boulder, Colorado clinical laboratory is located in the State of Colorado, we do not need a specific State of Colorado laboratory license, however, we maintain licenses to conduct testing in other states where nonresident laboratories are required to obtain
28
state laboratory licenses. We maintain licenses for our Boulder, Colorado and De Soto, Kansas laboratories with the NYSDOH. We also hold licenses in other states in which we operate, including California, Maryland, Pennsylvania and Rhode Island, that require licensing of out-of-state laboratories under certain circumstances. Other states may currently have or adopt similar licensure requirements in the future, which may require us to modify, delay or stop its operations in those states until such requirements are met.
Failure to comply with CLIA certification and state clinical laboratory licensure requirements may result in a range of enforcement actions, including certificate or license suspension, limitation, or revocation, directed plan of action, onsite monitoring, civil monetary penalties, criminal sanctions, and revocation of the laboratory’s approval to receive Medicare and Medicaid payment for its services, as well as significant adverse publicity.
CLIA and state laws and regulations, operating together, sometimes limit the ability of laboratories to offer consumer-initiated testing, also known as direct access testing. We do not offer direct access testing and instead require that our tests be ordered by licensed healthcare providers.
Our Boulder, Colorado and De Soto, Kansas laboratories are certified and adhere to the NYS CLEP, based on New York State Public Health Law, Article 5 Title 5. NYS CLEP is exempt from CLIA and establishes their own method of laboratory certification and test validation approval. To process New York State patient specimens a laboratory must submit a robust analytical and clinical validation package to demonstrate clinical utility of the test and receive approval prior to offering the test in the state of New York. All of our tests have obtained NYS CLEP approval including GeneStrat ddPCR, GeneStrat NGS, VeriStrat, Nodify XL2 and Nodify CDT tests. NYS CLEP requires semi-annual inspections to ensure the laboratory meets all general and specialty standards. Both Biodesix laboratories passed NYS CLEP inspections in May 2019. Due to the pandemic, routine re-inspections have been delayed by NYS CLEP.
Regulatory framework for medical devices in the United States
Pursuant to its authority under the FDCA, the FDA has jurisdiction over medical devices, which are defined to include, among other things, IVDs. The FDA regulates the research, design, development, pre-clinical and clinical testing, manufacturing, safety, effectiveness, packaging, labeling, storage, recordkeeping, pre-market clearance or approval, adverse event reporting, marketing, promotion, sales, distribution and import and export of medical devices. It is possible that one of our current, or future, tests will be subject to FDA authority and oversight as either an IVD or a CDx pursuant to the FDA’s authority to regulate medical devices under the FDCA.
Medical devices are subject to extensive regulation in the United States and elsewhere, including by the FDA and its foreign counterparts. Government regulations specific to medical devices are wide ranging and govern, among other things:
Market access, sales and marketing of medical devices in non-U.S. countries are subject to foreign regulatory requirements that vary widely from country to country. For example, in the European Economic Area (EEA), a medical device must meet the Medical Devices Directive’s (MDD)/In Vitro Medical Devices Directive’s (IVDD) Essential Requirements or, applicable on May 26, 2021, the Medical Devices Regulation’s (MDR) / applicable on May 26, 2022, In Vitro Medical Devices Regulation’s (IVDR) General Safety and Performance Requirements which apply to it, taking into account its intended purpose as defined by the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation. Before placing a medical device on the EEA market, the manufacturer must draw up a declaration of conformity, certifying that the device complies with the MDD/IVDD/MDR/IVDR, and must then affix the CE mark. For medium and high-risk devices as well as low risk devices that are placed on the market in sterile condition, have a measuring function, or are reusable surgical instruments, the manufacturer must obtain a CE Certificate from a notified body. The notified body typically audits and examines the device’s technical documentation, including the clinical evaluation, and the quality system for the manufacture, design
29
and final inspection of the relevant device before issuing a CE Certificate. Following the issuance of this CE Certificate, manufacturers may draw up the declaration of conformity and affix the CE mark to the devices covered by this CE Certificate.
Manufacturers of medical devices must document in a clinical evaluation report (CER) the evaluation of the clinical data related to the device. The CER is part of the device’s technical file. The evaluation shall document that the applicable Essential Requirements/General Safety and Performance Requirements are met and document the evaluation of the undesirable side-effects and the acceptability of the benefit-risk- ratio. The CER must be updated based on information from the post-market surveillance and vigilance activities related to the device. The CER shall consist, inter alia, of analyzed clinical data collected from a clinical investigation of the device, or the results of other studies on substantially equivalent devices. Reliance on “substantially equivalent” devices is very restrictive and requires, inter alia, that the manufacturer has full access to the technical documentation of the equivalent device on an ongoing basis and, if the “equivalent device” is not its own, that the manufacture has in place a contract with the manufacturer of the “equivalent device.”
Similar requirements apply in the UK. For access to the UK market, manufacturers must obtain a UKCA Certificate and affix a UKCA mark to their medical devices. However, the CE mark will be accepted in the UK until July 1, 2023.
Device classification
Under the FDCA, medical devices are classified into one of three classes: Class I, Class II or Class III, depending on the degree of risk to patients that is associated with each medical device and the amount of oversight needed to provide reasonable assurances with respect to safety and effectiveness of the medical device.
Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be reasonably assured by adherence to a set of FDA regulations, referred to as the General Controls for Medical Devices, which require compliance with the applicable portions of the FDA’s QSR, facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. Some Class I devices also require premarket clearance by the FDA through the 510(k) premarket notification process described below. Most Class I products are exempt from the premarket notification requirements.
Class II devices are subject to the General Controls as well as any special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, patient registries, FDA guidance documents and post-market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process, although some Class II devices are exempt from the 510(k) requirements.
Class III devices include devices deemed by the FDA to pose the greatest risk: such as life-supporting or life-sustaining devices, implantable devices, or those deemed novel and not substantially equivalent to a predicate device following the 510(k) process. CDx tests are regularly considered Class III devices.
Premarket submission process
Unless a statutory or regulatory exemption or enforcement discretion policy applies, before a new medical device, or a new intended use of, claim for, or significant modification to an existing device, can be marketed in the United States, the manufacturer must obtain the FDA’s: (1) permission for commercial distribution under section 510(k) of the FDCA (510(k) clearance); or (2) approval of a PMA; or (3) de novo classification and authorization. These processes can be resource intensive, expensive, and lengthy, and require payment of significant user fees.
Under the 510(k)-clearance process, the manufacturer must submit to the FDA a premarket notification, demonstrating that the device is “substantially equivalent” to a legally marketed predicate device. A predicate device is a legally marketed device that is not subject to a PMA, i.e., a device that was legally marketed prior to May 28, 1976 (pre-amendments device) and therefore a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was previously found substantially equivalent through the 510(k) process. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Premarket notifications typically include bench, analytical, and preclinical data. Clinical data is sometimes required to support substantial equivalence. If a manufacturer obtains a 510(k) clearance for its device and then makes a modification that could significantly affect the device’s safety or effectiveness or constitutes a major change or modification in the intended use of the device, a new clearance, authorization or approval may be required.
By statute, the FDA is required to complete its review of a 510(k) notification within 90 days of receiving the 510(k) notification. As a practical matter, clearance often takes longer, and clearance is never assured. Although many 510(k) premarket notifications are cleared without clinical data, the FDA may require further information, including clinical data, to make a determination regarding substantial equivalence, which may significantly prolong the review process. If the FDA agrees that the device is substantially equivalent, it will grant clearance to commercially market the device. If the FDA determines that the device is not “substantially equivalent” to a predicate
30
device, or if the device is automatically classified into Class III, the device sponsor must then fulfill the much more rigorous, costly, and time-consuming PMA approval process or seek reclassification of the device through the de novo process.
To obtain a PMA, the applicant must submit data and information demonstrating reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA typically includes, but is not limited to, extensive technical information regarding device design and development, pre-clinical and clinical trial data, manufacturing information, labeling, and financial disclosure information for the clinical investigators in device studies. The PMA application must provide valid scientific evidence that demonstrates to the FDA’s satisfaction a reasonable assurance of the safety and effectiveness of the device for its intended use.
Once filed as a PMA, the FDA has 180 days to review the filed PMA application, although the review of an application more often occurs over a significantly longer period of time. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA.
Prior to approval of a PMA, the FDA may conduct inspections of any clinical trial data and clinical trial sites, as well as inspections of any manufacturing facility and processes. The FDA can delay, limit or deny approval of a PMA application for many reasons, including (1) the device may not be shown safe or effective to the FDA’s satisfaction; (2) the data from pre-clinical studies and/or clinical trials may be found unreliable or insufficient to support approval; (3) the manufacturing process or facilities may not meet applicable requirements; and (4) changes in FDA approval policies or adoption of new regulations may require additional data.
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when data is available. The PMA process can be expensive, uncertain and lengthy. A number of devices for which the FDA approval has been sought by other companies have never been approved by the FDA for marketing. New PMA applications or PMA supplements are required for any modifications to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design of a device that has been approved through the PMA process.
As a condition of PMA application approval, the FDA may also require some form of post-approval study or post-market surveillance, whereby the applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer-term safety and effectiveness data for the device. The FDA may also approve a PMA application with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use.
Alternatively, the FDA also allows the submission of a direct de novo petition. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of FDASIA, a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent. FDASIA streamlined the de novo classification pathway by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination.
The 510(k), de novo or PMA processes can be expensive, lengthy and unpredictable. The FDA’s 510(k) clearance process usually takes from three to 12 months, but can last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k)-clearance process and generally takes from one to three years, or even longer, from the time the application is filed with the FDA. In addition, a PMA generally requires the performance of one or more clinical trials. Despite the time, effort and cost, a device may not be approved or cleared by the FDA. Any delay or failure to obtain necessary regulatory clearances or approvals could harm our business. Furthermore, even if we are granted regulatory clearances or approvals, they may include significant limitations on the indicated uses for the device, which may limit the market for the device.
Companion Diagnostics and the Premarket Process
We believe that one of our future product candidates may include a companion diagnostic or complementary diagnostic (collectively CDx). CDx’s can identify patients who are most likely to benefit from a particular therapeutic product; identify patients likely to be at increased risk for serious side effects as a result of treatment with a particular therapeutic product; or monitor response to treatment with a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness. The use of the CDx will be stipulated in the labeling of both the CDx and the therapeutic product. The FDA may require an application for the CDx to be separate from the drug approval process, and this could potentially delay the approval of any new drug application or the CDx, or
31
complicate the review process. CDx’s are generally regulated as Class III medical devices by the FDA and are therefore most often subject to the PMA approval process.
The FDA issued guidance in July 2016 for the co-development of CDx tests with a therapeutic product and issued another draft guidance in December 2018 specific to oncology CDx tests. The FDA finalized this draft guidance in April 2020 in “Developing and Labeling In vitro Companion Diagnostic Devices for a Specific Group of Oncology Therapeutic Products.” The guidance is meant to guide the development of CDx products, which are defined as IVDs that provide information that is essential for the safe and effective use of the therapeutic product. A CDx is often developed and approved or cleared contemporaneously with the therapeutic, and the use of the CDx is stipulated in the labeling of both the CDx and the corresponding therapeutic product. While it supports contemporaneous marketing authorizations, if there are any deficiencies in the submissions, the FDA may place a PMA review of a CDx on hold or request additional testing, which could potentially delay the approval of the corresponding new drug application or the marketing authorization of the CDx, or otherwise complicate the review process. Some oncology CDx tests can be developed in a way that results in labeling for a specific group of oncology therapeutic products, rather than a single therapeutic product.
Post-Market FDA Regulation
Even if regulatory clearance, authorization or approval of a device is granted, the FDA may impose limitations on the uses and indications for which the device may be labeled and promoted, and the device remains subject to significant regulatory requirements. Medical devices may be marketed only for the uses and indications for which they are cleared, authorized or approved. After a device, including a device exempt from FDA premarket review, is placed on the market, numerous post-market regulatory requirements apply, and the FDA has broad authority to enforce these requirements. Medical device manufacturers are subject to unannounced inspections by the FDA and other state, local and foreign regulatory authorities to assess compliance with the QSR and other applicable regulations, and these inspections may include the manufacturing facilities of any suppliers. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include sanctions such as: warning letters, fines, injunctions, consent decrees and civil penalties; unanticipated expenditures, including requirements to repair, replace, and/or refund the cost of the devices, recall or seizure of our products; operating restrictions, partial suspension or total shutdown of production; the FDA’s refusal of our requests for 510(k) clearance, de novo classification, or PMA of new products, new intended uses or modifications to existing products; the FDA’s refusal to issue certificates to foreign governments needed to export products for sale in other countries; and withdrawing 510(k) clearance or PMAs that have already been granted and criminal prosecution. In the event that a supplier fails to maintain compliance with the FDA’s or our quality requirements, we may have to qualify a new supplier and could experience manufacturing delays as a result.
Federal and State Fraud and Abuse Laws
We are subject to federal fraud and abuse laws such as the federal Anti-Kickback Statute (AKS), the federal prohibition against physician self- referral (Stark Law), the Eliminating Kickbacks in Recovery Act (EKRA), and the federal False Claims Act (FCA). We are also subject to similar state and foreign fraud and abuse laws.
The AKS (Social Security Act § 1128B(b)) prohibits knowingly and willfully offering, paying, soliciting, or receiving remuneration, directly or indirectly, overtly or covertly, in cash or in kind, in return for or to induce such person to refer an individual, or to purchase, lease, order, arrange for, or recommend purchasing, leasing or ordering, any item or service that may be reimbursable, in whole or in part, under a federal healthcare program, such as Medicare or Medicaid. There are a number of statutory exceptions and regulatory safe harbors to the AKS that provide protection from AKS liability to arrangements that fully satisfy the applicable requirements.
EKRA (18 USC § 220) prohibits knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in return for the referral of a patient to, or in exchange for an individual using the services of certain entities, including laboratories, if the services are covered by a health care benefit program. The term “health care benefit program” is broadly defined such that EKRA extends to referrals reimbursed by both governmental and commercial third-party payers. EKRA includes a number of statutory exceptions that provide protection from EKRA liability if the applicable requirements are met.
The Stark Law (Social Security Act § 1877) generally prohibits, among other things, clinical laboratories and other so-called “designated health services” entities from billing Medicare for any designated health services when the physician ordering the service, or any member of such physician’s immediate family, has a financial relationship, such as a direct or indirect investment interest in or compensation arrangement with the billing entity, unless the arrangement meets an exception to the prohibition. The Stark Law also prohibits physicians from making such referrals to a designated health services entity. There are also similar state laws that apply where Medicaid and/or commercial payers are billed.
The FCA (31 USC § 3729) imposes civil penalties against individuals or entities for, among other things, knowingly presenting, or causing to be presented, claims for payment to the government that are false or fraudulent, or knowingly making, using or causing to be made or used a false record or statement material to such a false or fraudulent claim, or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. This statute also permits a private individual acting as a “qui tam” whistleblower to bring actions on behalf of the federal government alleging violations of the FCA and
32
to share in any monetary recovery. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and mandatory penalties of $11,803 to $23,607 per false claim or statement for penalties assessed after December 13, 2021, with respect to violations occurring after November 2, 2015.
Other federal statutes pertaining to healthcare fraud and abuse include the civil monetary penalties statute, which prohibits, among other things, the offer or payment of remuneration to a Medicaid or Medicare beneficiary that the offeror or payer knows or should know is likely to influence the beneficiary to order or receive a reimbursable item or service from a particular provider, practitioner, or supplier, and contracting with an individual or entity that the person knows or should know is excluded from participation in a federal health care program. In addition, federal criminal statutes created by HIPAA prohibit, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare benefit program or obtain by means of false or fraudulent pretenses, representations or promises any money or property owned by or under the control of any healthcare benefit program in connection with the delivery of or payment for healthcare benefits, items or services.
In addition to these federal laws, there are often similar state anti-kickback and false claims laws that typically apply to arrangements involving reimbursement by a state-funded Medicaid or other health care program. Often, these laws closely follow the language of their federal law counterparts, although they do not always have the same exceptions or safe harbors. In some states, these anti-kickback laws apply with respect to all payers, including commercial payers.
A number of states have enacted laws that require pharmaceutical and medical device companies to monitor and report payments, gifts and other remuneration made to physicians and other healthcare providers, and, in some states, marketing expenditures. In addition, some state statutes impose outright bans on certain manufacturer gifts to physicians or other health care professionals. Some of these laws, referred to as “aggregate spend” or “gift” laws, carry substantial fines if they are violated.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs and extensive annual trainings for all of our employees and contractors. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from participation in government-funded healthcare programs, such as Medicare and Medicaid, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, additional reporting or oversight obligations if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with the law, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. If any of the physicians or other healthcare providers or entities with whom we do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government-funded healthcare programs.
Anti-Corruption
The Foreign Corrupt Practices Act of 1977 (FCPA) and similar international bribery laws make it unlawful for persons or entities to make payments to foreign government officials to assist in obtaining and maintaining business. Specifically, the anti-bribery provisions of the FCPA prohibit any offer, payment, promise to pay, or authorizing the payment of money or anything of value to any person, while knowing that all or a portion of such money or thing of value will be offered, given or promised, directly or indirectly, to a foreign official to do or omit to do an act in violation of his or her duty, or to secure any improper advantage in order to assist in obtaining or retaining business for or with, or directing business, to any person. In addition to the anti-bribery provisions of the FCPA, the statute also contains accounting requirements designed to operate in tandem with the anti-bribery provisions. Covered companies are required to make and keep books and records that accurately and fairly reflect the transactions of the company and devise and maintain an adequate system of internal accounting controls. With our international operations through our third-party partnerships, we could incur significant fines and penalties, as well as criminal liability, if we fail to comply with either the anti-bribery or accounting requirements of the FCPA, or similar international bribery laws. Even an unsuccessful challenge of our compliance with these laws could cause us to incur adverse publicity and significant legal and related costs. We successfully passed our first FCPA compliance review in 2021 with no findings.
Privacy and Data Protection Laws
Numerous federal and state laws and regulations, including HIPAA, as amended by the HITECH Act, govern the collection, dissemination, security, use and confidentiality of protected health information (PHI) and personal information. In the course of performing our business we obtain personal information, including PHI. Laws and regulations relating to privacy, data protection, and consumer protection are evolving and, in some cases, particularly with regard to newer laws, may be subject to potentially differing interpretations. Under HIPAA and HITECH, the HHS issues regulations that establish uniform standards governing the conduct of certain electronic healthcare transactions and requirements for protecting the privacy and security of PHI, used or disclosed by covered entities (CEs) and their authorized business associates (BAs). Because we are a health care provider that electronically transmits health care information, and we also provide certain services to CEs and receive PHI from them, we are at times either a CE or a BA, as defined
33
by HIPAA. Our subcontractors that create, receive, maintain, transmit or otherwise process PHI on our behalf are HIPAA BAs and must also comply with HIPAA, as applicable.
HIPAA and HITECH include the privacy and security rules, breach notification requirements and electronic transaction standards. The privacy rule governs the use and disclosure of PHI, generally prohibits the use or disclosure of PHI except as permitted under the rule, and mandates certain safeguards to protect the privacy of PHI. The privacy rule also sets forth individual rights, such as the right to access or amend certain records containing such individual’s PHI, or to request restrictions on the use or disclosure of such individual’s PHI. The security rule requires CEs and BAs to safeguard the confidentiality, integrity, and availability of electronically transmitted or stored PHI (also referred to as ePHI) by implementing administrative, physical and technical safeguards. Under HIPPAA’s breach notification rule, a CE must notify individuals, the Secretary of HHS, and in some circumstances, the media of certain breaches of unsecured PHI or ePHI, and similar breach notification provisions apply to certain BAs under the HITECH Act.
Penalties for failure to comply with a requirement of HIPAA and HITECH vary depending on the number and nature of the violations and any history of prior violations, but can be significant and include civil monetary or criminal penalties. HIPAA is enforced by the Department of Health and Human Services, Office for Civil Rights, and HIPAA also authorizes state attorneys general to file suit on behalf of their residents for violations. Courts are able to award damages, costs and attorneys’ fees related to violations of HIPAA in such cases. While HIPAA does not create a private right of action allowing individuals to file suit in civil court for violations of HIPAA, its standards have been used as the basis for duty of care cases in state civil suits such as those for negligence or recklessness in improper use, access to or disclosure of PHI. In addition, HIPAA mandates that the Secretary of HHS conduct periodic compliance audits of HIPAA CEs, such as us, and their BAs for compliance with the HIPAA privacy and security standards and breach notification rules. It also tasks HHS with establishing a methodology whereby harmed individuals who were the victims of breaches of unsecured PHI may receive a percentage of the civil monetary penalty paid by the violator.
In addition, we may be subject to state privacy, cybersecurity, and data breach notification laws, which may govern the collection, use, disclosure and protection of health-related and other personal information. California, for example, has enacted the Confidentiality of Medical Information Act, which, in addition to HIPAA and HITECH, sets forth standards with which all California health care providers must abide. Colorado has enacted the Colorado Privacy Act, and Virginia has enacted the Consumer Data Protection Act, both of which also have standards that must be complied with that supplement Federal data protection requirements, State laws may be more stringent, broader in scope or offer greater individual rights with respect to PHI than HIPAA, and state laws may differ from each other in regards to personal information treatment, which may complicate compliance efforts. For instance, the California Consumer Privacy Act (CCPA) became effective on January 1, 2020 and was amended by the passage of the California Privacy Rights Act (CPRA) in November of 2020. The CCPA, among other things, gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used by requiring covered companies to provide new disclosures to California consumers (as that term is broadly defined) and provide such consumers new ways to opt-out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA has been amended from time to time, and it remains unclear what, if any, further modifications will be made to this legislation or how it will be interpreted. Although there are certain exemptions for PHI and clinical trial data, the CCPA’s implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future and the CCPA may increase our compliance costs and potential liability. Additionally, the CPRA will impose additional data protection obligations on companies doing business in California, including additional consumer rights processes and opt outs for certain uses of sensitive data. It would also create a new California data protection agency specifically tasked to enforce the law, which would likely result in increased regulatory scrutiny of California businesses in the areas of data protection and security. Similar laws have been proposed in other states and at the federal level, and if passed, such laws may have potentially conflicting requirements that could continue to make compliance challenging and costly.
Additionally, the Federal Trade Commission (FTC) and state attorneys general enforce consumer protection laws that prohibit unfair and deceptive acts and practices, including Section 5 of the FTC Act, which creates standards for the collection, use, dissemination and security of health-related and other personal information. Claims of unfair or deceptive trade practices regarding privacy and security can lead to significant liabilities and consequences, including regulatory investigations, penalties, fines and orders as well as civil claims, which could impact our data practices and operations or cause reputational damage.
We may also be subject to laws and regulations in foreign countries covering data privacy and other protection of health and employee information that may add additional compliance burden and complexity. For example, in the EEA, the collection and use of personal data is governed by the European Union's General Data Protection Regulation (GDPR). In the United Kingdom, the GDPR has been adopted in substantially the same form, however the UK may potentially make revisions in the coming years. The GDPR, together with national legislation, regulations and guidelines of the EU member states and the United Kingdom governing the processing of personal data, impose strict obligations and restrictions on the ability to collect, analyze, store, transfer and otherwise process personal data. European and United Kingdom data protection authorities may interpret the GDPR and national laws differently and impose additional requirements, which adds to the complexity of processing personal data in or from the EEA or United Kingdom. Guidance on implementation and compliance practices are often updated or otherwise revised. GDPR applies extra-territorially under certain circumstances and imposes stringent requirements on controllers and processors of personal data, including, for example, requirements to ensure a legal bases to process personal information, provide robust disclosures to individuals, facilitate data subject rights, provide
34
data security breach notifications within 72 hours after discovering a breach in certain circumstances, limit retention of personal information and apply enhanced protections to health data and other categories of sensitive personal information. The GDPR also has requirements around international transfers of personal data. Requirements around transfers to the United States and other jurisdictions have increased since a July 2020 decision by the Court of Justice of the European Union invalidated the Privacy Shield as a basis to transfer personal data from Europe to the United States, and added requirements for reliance on Standard Contractual Clauses. Regulatory guidance on requirements for international transfers, and other GDPR compliance matters, continues to evolve. Failure to comply with the requirements of the GDPR may result in fines of up to €20 million or up to 4% of the total worldwide annual turnover of our preceding fiscal year, whichever is higher, and other administrative penalties. To comply with the GDPR and other applicable international data protection laws and regulations, we may be required to put in place additional mechanisms ensuring compliance, which may result in other substantial expenditures.
Cybersecurity
Our business relies on secure and continuous processing of information and the availability of our IT networks and IT resources, as well as critical IT vendors that support our technology, research and other data processing operations. While we take steps to protect our systems and data, security incidents, data breaches, computer malware and computer hacking attacks have become more prevalent across industries, including the life sciences sector, and may occur on our systems or those of our third-party service providers. Unauthorized persons may in the future be able to exploit weaknesses in the security systems of our (or our third-party service providers) IT networks and gain access to PHI and other personal information, or sensitive trade secrets or other proprietary information. Any wrongful use or disclosure of PHI, other personal information, trade secrets or other proprietary information by us or our third-party service providers could subject us to regulatory fines or penalties, third-party claims or otherwise could adversely affect our business and results of operations. Although HIPAA and the regulations promulgated thereunder do not provide for a private right of action, failures to adequately protect PHI or our IT systems could be viewed as violations of the HIPAA security rule or violations of other applicable information security laws, regulations, contractual obligations or industry standards, and could further result in costly data breach notification obligations that negatively impact our reputation.
Moreover, data security incidents or data breaches, as well as attacks on our IT systems, could result in operational disruptions or data loss or corruption that could adversely impact our business and operations, result in substantial investment of resources to investigate, recover and remediate and subject us to heightened regulatory scrutiny.
International Regulations
Many countries in which we may offer any of our diagnostic tests in the future have anti-kickback regulations prohibiting providers from offering, paying, soliciting or receiving remuneration, directly or indirectly, in order to induce business that is reimbursable under any national health care program. In situations involving physicians employed by state-funded institutions or national health care agencies, violation of the local anti-kickback law may also constitute a violation of the FCPA.
The FCPA prohibits any United States individual, business entity or employee of a United States business entity to offer or provide, directly or through a third party, including any potential distributors we may rely on in certain markets, anything of value to a foreign government official with corrupt intent to influence an award or continuation of business or to gain an unfair advantage, whether or not such conduct violates local laws. In addition, it is illegal for a company that reports to the SEC to have false or inaccurate books or records or to fail to maintain a system of internal accounting controls. We will also be required to maintain accurate information and control over sales and distributors’ activities that may fall within the purview of the FCPA, its books and records provisions and its anti-bribery provisions.
The standard of intent and knowledge in anti-bribery cases is minimal. Intent and knowledge are usually inferred from that fact that bribery took place. The accounting provisions do not require intent. Violations of the FCPA’s anti-bribery provisions for corporations and other business entities are subject to a fine of up to $2 million and officers, directors, stockholders, employees, and agents are subject to a fine of up to $100,000 and imprisonment for up to five years. Other countries, including the United Kingdom and other OECD Anti-Bribery Convention members, have similar anti-corruption regulations, such as the United Kingdom Anti-Bribery Act.
When marketing our diagnostic tests outside of the United States, we may be subject to foreign regulatory requirements governing human clinical testing, prohibitions on the import of tissue necessary for us to perform our diagnostic tests or restrictions on the export of tissue imposed by countries outside of the United States or the import of tissue into the United States, and marketing approval. These requirements vary by jurisdiction, differ from those in the United States and may in some cases require us to perform additional pre-clinical or clinical testing. In many countries outside of the United States, coverage, pricing and reimbursement approvals are also required.
Market access, sales and marketing of medical devices in non-U.S. countries are subject to foreign regulatory requirements that vary widely from country to country. For example, in the EEA, a medical device must meet MDDs/IVDD Essential Requirements or, applicable on May 26, 2021, the MDR applicable on May 26, 2022, IVDR General Safety and Performance Requirements which apply to it, taking into account its intended purpose as defined by the data supplied by the manufacturer on the label, in the instructions for use
35
or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation. Before placing a medical device on the EEA market, the manufacturer must draw up a declaration of conformity, certifying that the device complies with the MDD/IVDD/MDR/IVDR, and must then affix the CE mark. For medium and high-risk devices as well as low risk devices that are placed on the market in sterile condition, have a measuring function, or are reusable surgical instruments, the manufacturer must obtain a CE Certificate from a notified body. The notified body typically audits and examines the device’s technical documentation, including the clinical evaluation, and the quality system for the manufacture, design and final inspection of the relevant device before issuing a CE Certificate. Following the issuance of this CE Certificate, manufacturers may draw up the declaration of conformity and affix the CE mark to the devices covered by this CE Certificate.
Manufacturers of medical devices must document in a CER the evaluation of the clinical data related to the device. The CER is part of the device’s technical file. The evaluation shall document that the applicable Essential Requirements/General Safety and Performance Requirements are met and document the evaluation of the undesirable side-effects and the acceptability of the benefit-risk- ratio. The CER must be updated based on information from the post-market surveillance and vigilance activities related to the device. The CER shall consist, inter alia, of analyzed clinical data collected from a clinical investigation of the device, or the results of other studies on substantially equivalent devices. Reliance on “substantially equivalent” devices is very restrictive and requires, inter alia, that the manufacturer has full access to the technical documentation of the equivalent device on an ongoing basis and, if the “equivalent device” is not its own, that the manufacture has in place a contract with the manufacturer of the “equivalent device.”
Similar requirements apply in the UK. For access to the UK market, manufacturers must obtain a UKCA Certificate and affix a UKCA mark to their medical devices. However, the CE mark will be accepted in the UK until July 1, 2023.
Healthcare Reform
In March 2010, the Patient Protection and Affordable Care Act (ACA) was enacted in the United States. The ACA made a number of substantial changes to the way healthcare is financed both by governmental and private insurers. For example, the ACA requires each medical device manufacturer to pay a sales tax equal to 2.3% of the price for which such manufacturer sells its medical devices. The medical device tax was permanently repealed at the end of 2019. The ACA also contains a number of other provisions, including provisions governing enrollment in federal and state healthcare programs, reimbursement matters, and fraud and abuse, which we expect will impact our industry and our operations in ways that we cannot currently predict.
Beginning in 2017, the Trump administration sought to modify, repeal, or otherwise invalidate all, or certain provisions of, the ACA. The Trump administration issued three executive orders and other directives designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. For example, on December 20, 2019, President Trump signed appropriations legislation for fiscal year 2020 that repealed certain ACA-mandated fees, including the so-called “Cadillac” tax on certain high-cost employer-sponsored insurance plans, for tax years beginning after December 31, 2019; the annual fee imposed on certain health insurance providers based on market share, for calendar years beginning December 31, 2020; and the medical device excise tax on non-exempt medical devices, for sales after December 31, 2019. While Congress did not pass comprehensive legislation that would repeal all or part of the ACA, two bills affecting the implementation of certain taxes under the ACA have been signed into law. Specifically, the Tax Cuts and Jobs Act of 2017 (TCJA), among other things, included a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment, or penalty, imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Beginning in 2021, the Biden administration has signaled its intent to pursue policies strengthening the ACA.
Other legislative changes have been proposed and adopted in the United States since the ACA was enacted to reduce healthcare expenditures. These changes include the Budget Control Act of 2011, which, among other things, led to aggregate reductions of Medicare payments to providers of up to 2% per fiscal year that started in 2013 and, due to subsequent statutory amendments, will remain in effect through 2030 unless additional Congressional action is taken. In 2020, the CARES Act temporarily suspended the 2% cut in Medicare payments from May 1, 2020 through December 31, 2020, and it extended the sequestration reductions through FY 2030 to offset the cost of such temporary suspension. The Consolidated Appropriations Act, 2021 further extended the temporary suspension through March 31, 2021. On April 14, 2021, Congress enacted legislation that further extended the suspension through December 31, 2021. On December 10, 2021, further legislation was enacted to extend the suspension through March 31, 2022.
The American Taxpayer Relief Act of 2012 made other changes, including reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. If federal spending is reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or the National Institutes of Health to continue to function at current levels. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve R&D, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop. Additionally, the Biden administration has already indicated a shift in direction from Trump administration policies by issuing orders and other documents rolling back regulations and Executive Orders from the Trump administration, and as noted, indicating that it will pursue policies strengthening the ACA. The recent change in presidential administration could materially impact our business in ways that are difficult to predict.
36
In December 2020, in its enactment of the Consolidated Appropriations Act, Congress enacted the No Surprises Act. This law, which took effect January 1, 2022, bars an out-of-network providers from billing patients in excess of the in-network cost sharing for services furnished with respect to a visit at certain in-network health care facilities. The law establishes an independent dispute resolution process between the provider and the payer to determine the appropriate payment rate to the provider. As written, the No Surprises Act may apply to laboratory tests furnished by an independent laboratory with respect to a hospital visit. The law establishes a notice and consent exception that generally does not apply to laboratory tests, although it allows HHS to apply this exception to certain advanced tests. Regulations and subregulatory guidance were issued by HHS, the Department of Labor, and the Department of the Treasury in 2021, with the first set of regulations was issued as an Interim Final Rule on July 1, 2021, and the second set issued as an Interim Final Rule on September 30, 2021. These regulations and subregulatory guidance have provided additional information on the applicability of the No Surprises Act, the rules governing the independent dispute resolution process, and specific provider requirements (including the obligation to furnish a “good faith estimates” of “expected charges” to uninsured or self-pay patients), as well as areas of temporary enforcement discretion.
Environmental, Health and Safety Regulations
We are subject to various federal, state, local, and foreign environmental, health and safety laws and regulations and permitting and licensing requirements. Such laws include those governing laboratory practices, the generation, storage, use, manufacture, handling, transportation, treatment, remediation, release and disposal of, and exposure to, hazardous materials and wastes and worker health and safety. Our operations involve the generation, use, storage and disposal of hazardous materials, and the risk of injury, contamination or non-compliance with environmental, health and safety laws and regulations or permitting or licensing requirements cannot be eliminated. In particular, the introduction of our Bio-Rad SARS-CoV-2 ddPCR and Platelia SARS-CoV-2 Total Ab tests requires that we maintain compliance with applicable and evolving federal and state laws and regulations relating to COVID-19, including the generation, use, storage, and disposal of testing materials and agents. Compliance with environmental laws and regulations has not had a material effect on our capital expenditures, earning or competitive position. Most recently, the Boulder, Colorado clinical laboratory passed an on-site inspection from the CDC in June 2021.
Corporate Information
We were incorporated in Delaware in 2005 as Elston Technologies, Inc. Our principal executive offices are located at 2970 Wilderness Place, Suite 100, Boulder, Colorado 80301, and our telephone number is (303) 417-0500. On June 20, 2006, we changed our name to Biodesix, Inc.
Implications of Being an Emerging Growth Company and Smaller Reporting Company
We are an “emerging growth company” within the meaning of the Jumpstart Our Business Startups Act (JOBS Act). As an emerging growth company, we may take advantage of certain exemptions from various public company reporting requirements, including the requirement that our internal control over financial reporting be audited by our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, certain requirements related to the disclosure of executive compensation in our periodic reports and proxy statements, the requirement that we hold a nonbinding advisory vote on executive compensation and any golden parachute payments. We may take advantage of these exemptions until we are no longer an emerging growth company. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended (the Securities Act), for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
We have elected to take advantage of the extended transition period to comply with new or revised accounting standards and to adopt certain of the reduced disclosure requirements available to emerging growth companies. As a result of the accounting standards election, we will not be subject to the same implementation timing for new or revised accounting standards as other public companies that are not emerging growth companies, which may make comparison of our financials to those of other public companies more difficult. Additionally, because we have taken advantage of certain reduced reporting requirements, the information contained herein may be different from the information you receive from other public companies in which you hold stock.
We will remain an emerging growth company until the earliest to occur of (i) the last day of the fiscal year in which we have more than $1.07 billion in annual revenue; (ii) the date we qualify as a “large accelerated filer,” with at least $700 million of equity securities held by non-affiliates; (iii) the date on which we have issued, in any three-year period, more than $1.0 billion in non-convertible debt securities; and (iv) the last day of the fiscal year ending after the fifth anniversary of the completion of our IPO.
Additionally, we are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of the fiscal year in which (i) the market value of our common stock held by non-affiliates exceeds $250 million as of the end of that year’s second fiscal quarter, or (ii) our annual revenues exceeded $100 million during such completed fiscal year and the market value of our common stock held by non-affiliates exceeds $700 million
37
as of the end of that year’s second fiscal quarter. To the extent we take advantage of such reduced disclosure obligations, it may also make comparison of our financial statements with other public companies difficult or impossible.
For certain risks related to our status as an emerging growth company, see “Risk Factors—Risks Related to our Common Stock—We are an “emerging growth company” and a “smaller reporting company,” and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our common stock less attractive to investors.”
Human Capital Resources
Our culture is underpinned by our cultural beliefs including an unwavering commitment to inclusion and diversity. We are committed to fostering a diverse and inclusive workplace that attracts and retains exceptional talent offering opportunities for our team members to grow and develop in their careers, supported by strong compensation, benefits and health and wellness programs. We regularly engage our team members in monthly all-hands meetings to align and focus on the current state of affairs of our business, our partnerships, new products, clinical trials, and other pertinent information about our business. We engage our team members in programs such as peer recognition, recruitment and our newly introduced Diversity, Equity and Inclusion Council that focus on recognizing outstanding individual contributions to company performance, cultural fit of new team members and acknowledging the diversity of our team to ensure our team members feel valued and can do their best work. We have a national annual community service initiative, “Biodesix Gives Back” that allows each team member to invest ten hours of paid community service to organizations of their choosing.
As of December 31, 2021, we had approximately 218 full-time and part-time employees, all of which are located in the United States. Of our 218 full-time and part-time team members, the majority of our team member base is located in proximity to our corporate office and testing facilities located in Boulder, Colorado and our laboratory in De Soto, Kansas.
Diversity, Equity and Inclusion
We believe that a diverse employee population, including cultural background, gender, ethnicity, sexual orientation and lived experiences, is critical to our success. Our employees are encouraged to leverage their personal strengths and experiences to continually innovate and contribute to the development of new ideas and process improvements that drive better experiences for our partners.
Employee Engagement
Our company culture emphasizes the satisfaction and well-being of our team members and a diverse, engaged workforce. We solicit the opinion and views of our team members through surveys and peer focus groups. We have an established and valued Peer Recognition Culture. Teammates recognize other teammates publicly for their support and contributions fostering collaboration, engagement and retention. We regularly review feedback we receive on our current cultural beliefs to determine if any modifications are needed. During the year ended December 31, 2021, we updated our cultural beliefs to align with our core values that reflect our current focus on Teamwork, Innovation, Making an Impact and Excel. Additionally, the Company annually celebrates our top sales performers through our President’s Club and other top company performers as nominated by fellow team members through our four (4) Performance Excellence Awards that recognize creativity and innovation, an entrepreneurial spirit, a strategic impact on the success of the Company and lastly, embodying the Biodesix cultural beliefs and goes “above and beyond” daily.
Training and Development
We invest in our team members career growth and provide team members with a wide range of development opportunities, including face-to-face, virtual, and self-directed learning, mentoring, coaching, and external development.
Health, Safety and Wellness
The physical health, financial stability, life balance and mental health of each of our team members is vital to our success. We sponsor several cancer awareness activities in our local communities to bring engagement and awareness of health, safety and wellness to positively impact lives. We provide an Employee Assistance Program through our professional employer organization (PEO) to enhance physical, financial, and mental well-being for all our team members. Since the onset of the COVID-19 pandemic, we have successfully transitioned a significant portion of our workforce to a remote working environment and implemented strict safety and social distancing measures in our sites to protect the health and safety of our team members. We also have provided extraordinary safety protocols and standards for all of “essential” team members who work in our Laboratories and our field sales organization including regularly scheduled serial testing for COVID-19. We also have an active “Friends and Family” COVID-19 testing program for our team members, their families and close friends.
Pay Equity
The main objective of our compensation program is to provide a compensation package that will attract, retain, motivate, and reward superior team members who operate in a highly competitive and technologically challenging environment. We emphasize overall Company performance and provide equity incentives for all team members to align their financial interests with the interests of shareholders. In addition, we offer an employee stock purchase plan to all employees in which participants are eligible to purchase shares at a discount to market. We think like customers and act like owners.
38
Biodesix seeks fairness in total compensation. We benchmark with external comparisons, internal comparisons and look at the relationship between team member roles within the organization. We also review our compensation practices, both in terms of our overall workforce and individual team members, to ensure our pay is fair and equitable. We currently have no pay disparities based on gender, race, or ethnicity.
Available Information
We file with, or furnish to, the SEC reports including our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to those reports pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended. These reports are available free of charge on our corporate website (www.biodesix.com) as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. Copies of any materials we file with the SEC can be obtained at www.sec.gov. The foregoing website addresses are provided as inactive textual references only. The information provided on our website (or any other website referred to in this report) is not part of this report and is not incorporated by reference as part of this Annual Report on Form 10-K.
Item 1A. Risk Factors.
Risk Factors Summary
The following is a summary of the principal risks that could adversely affect our business, operations and financial results. This summary does not address all of the risk that we face and should be read in conjunction with the entire Risk Factors section below beginning at “Risks Related to Our Business and Industry” within this Item 1A. Risk Factors.
39
Risk Factors
Our operations and financial results are subject to various risks and uncertainties that could adversely affect our business, financial condition, and results of operations and cash flows. All of the risks described below should be carefully considered together with the other information contained and incorporated by reference in this report.
Risks Related to our Business and Industry
We have a history of net losses, and we expect to continue to incur losses for the foreseeable future. If we achieve profitability, we may not be able to sustain it.
We have incurred losses since our inception and expect to continue to incur losses for the foreseeable future. We reported net losses of $43.2 million and $31.4 million the years ended December 31, 2021 and 2020, respectively. As a result of these losses, as of December 31, 2021, we had an accumulated deficit of approximately $302.0 million.
We expect that our sales and marketing, research and development, regulatory and other expenses will continue to increase as we expand our marketing efforts for our diagnostic tests and services, expand existing relationships with our customers, obtain regulatory clearances or approvals or certifications for future enhancements to our existing diagnostic tests and services and conduct further clinical trials. In addition, we expect our general and administrative expenses to increase due to the additional costs associated with scaling our business operations and testing capacity, particularly with respect to our COVID-19 testing capacity, as well as being a public company, including due to legal, accounting, insurance, exchange listing and compliance, investor relations and other expenses. As a result, we expect to continue to incur operating losses and may never achieve profitability. We will need to generate significant additional revenue in order to achieve and sustain profitability. Even if we achieve profitability, we cannot be sure that we will remain profitable for any substantial period of time. If we do not achieve or sustain profitability, it will be more difficult for us to finance our business and accomplish our strategic objectives, either of which would have a material adverse effect on our business, financial condition and results of operations.
The continuing spread of COVID-19 and the impact to our business may continue to have a material and adverse effect on our ability to generate revenues, maintain compliance with our financial covenants and result in our inability to continue as a going concern. Any such impacts could have a material and adverse effect on the price of our common stock.
Our financial statements as of and for the period ended December 31, 2021 were prepared on the assumption that we would continue as a going concern. These financial statements did not include any adjustments that might result from the outcome of this uncertainty. As of December 31, 2021, we maintained cash and cash equivalents of $32.7 million and we have $10 million in principal balance remaining on our 2021 Term Loan. We have incurred significant losses since inception and, as a result, we have funded our operations to date primarily through the sale of common stock in our IPO in October 2020, the issuance of notes payable, and from our two primary revenue sources: (i) diagnostic testing, which include lung diagnostic testing and COVID-19 testing, and (ii) providing biopharmaceutical companies with development and testing services. Our ability to meet our obligations as they come due may be impacted by our ability to remain compliant with financial covenants in our 2021 Term Loan or to obtain waivers or amendments that impact the related covenants. Due to the continued uncertainty caused by the COVID-19 pandemic, significant risks remain with respect to our ability to meet these thresholds and any material adverse effect on our revenues, income and expenses could impact our ability to maintain compliance with these covenants. As a result of the items mentioned above, our management has determined that there is a substantial doubt about our ability to continue as a going concern over the next twelve months from the date these financial statements were issued. Although we have taken steps to improve our liquidity through the recent amendments to our 2021 Term Loan to modify certain financial covenants and have taken measures to reduce planned capital expenditures and operating expenses, these actions alone will not be sufficient to mitigate our liquidity concerns in the wake of the material impact to our revenues, operating results and cash flows from the continued spread of COVID-19. In addition, if we are not able to improve our operating results, we may need to limit our operations substantially. We will need to raise additional capital in the form of equity or debt to increase our liquidity but there is no assurance that we will be able to secure any such funding in a sufficient amount or on terms that are acceptable to us. If we do raise additional capital through public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our existing stockholders’ rights. Furthermore, the reaction of investors to the inclusion of a going concern statement in this report, and our potential inability to continue as a going concern, could materially adversely affect the price of our common stock.
The commercial success of our current and future diagnostic tests and services and our revenue growth depends upon attaining significant market acceptance among payers, providers, clinics, patients, and biopharmaceutical companies.
Our commercial success depends, in part, on the acceptance of our diagnostic tests and services as being safe and relatively simple for medical personnel to learn and use, clinically flexible, operationally versatile and, with respect to providers and payers, cost effective. We cannot predict how quickly, if at all, payers, providers, clinics and patients will accept future diagnostic tests and services or, if accepted, how frequently they will be used. These constituents must believe that our diagnostic tests offer benefits over other available alternatives.
40
The degree of market acceptance of our current and future diagnostic tests and services depends on a number of factors, including:
Additionally, even if our diagnostic tests achieve widespread market acceptance, they may not maintain that market acceptance over time if competing diagnostic tests or technologies, which are more cost effective or are received more favorably, are introduced. Failure to achieve or maintain market acceptance and/or market share would limit our ability to generate revenue and would have a material adverse effect on our business, financial condition and results of operations.
There is no assurance that our COVID-19 and antibody testing program will continue to be accepted by the market or that other diagnostic tests will become more accepted, produce quicker results or are more accurate. Further, the longevity and extent of the COVID-19 pandemic is uncertain. If the pandemic were to dissipate, whether due to a significant decrease in new infections, due to the availability of vaccines, or otherwise, the need for a COVID-19 test could decrease significantly and this could have an adverse effect on our results of operation and profitability.
We may encounter difficulties in managing our growth, which could disrupt our operations.
As of December 31, 2021, we had approximately 218 full and part-time employees. Over the next several years, we expect to increase significantly the number of our employees and the scope of our operations, particularly in the areas of sales, marketing and reimbursement, product development, regulatory affairs and other functional areas, including finance, accounting, quality and legal. Additionally, we expect to expand our testing capacity as we commercialize additional diagnostic tests. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational quality and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources, we may not be able to manage the expansion of our operations or recruit and train additional qualified personnel in an effective manner. Any inability to manage growth could delay the execution of our business plans or disrupt our operations and have a material and adverse effect on our prospects.
Since our inception, we have experienced multiple cycles of growth and anticipate further growth in our business operations. This future growth could create strain on our organizational, administrative and operational infrastructure, including laboratory operations, quality control, customer service and sales organization management. We expect to continue to increase headcount and to hire more specialized personnel in the future as we grow our business. We will need to continue to hire, train and manage additional qualified scientists, laboratory personnel, client and account services personnel, and sales and marketing staff and improve and maintain our technology to properly manage our growth. If our new hires perform poorly, if we are unsuccessful in hiring, training, managing and integrating these new employees or if we are not successful in retaining our existing employees, our business may be harmed.
We may not be able to maintain the quality or expected turnaround times of our diagnostic tests and services, or satisfy customer demand as it grows. We may not be able to expand our COVID-19 testing capacity rapidly enough to meet the current and anticipated demand. Our ability to manage our growth properly will require us to continue to improve our operational, financial and management controls, as well as our reporting systems and procedures. The time and resources required to implement these new systems and procedures is uncertain, and failure to complete this in a timely and efficient manner could materially adversely affect our operations. Additionally, if we are required to reduce expenses substantially to sustain our operations, we may not have the human resources to maintain growth in our business operations.
41
If we fail to retain sales and marketing personnel and, as we grow, fail to increase our sales and marketing capabilities or develop broad awareness of our diagnostic tests in a cost-effective manner, we may not be able to generate revenue growth.
We have limited experience marketing and selling our diagnostic tests. We currently rely on our direct sales force to sell our diagnostic tests in the United States, and any failure to maintain and grow our direct sales force will negatively affect our business, financial condition and results of operations. The members of our direct sales force are highly trained and possess substantial technical expertise, which we believe is critical in increasing adoption of our diagnostic tests. The members of our United States sales force are at-will employees. The loss of these personnel to competitors, or otherwise, will negatively affect our business, financial condition and results of operations. If we are unable to retain our direct sales force personnel or replace them with individuals of equivalent technical expertise and qualifications, or if we are unable to successfully instill such technical expertise in replacement personnel, it may negatively affect our business, financial condition and results of operations.
In order to generate future growth, we plan to continue to expand and leverage our sales and marketing infrastructure. Identifying and recruiting qualified sales and marketing personnel and training them on how to promote our diagnostic tests, on applicable federal and state laws and regulations and on our internal policies and procedures requires significant time, expense and attention. It often takes several months or more before a sales representative is fully trained and productive. Our sales force may subject us to higher fixed costs than those of companies with competing techniques or diagnostic tests that utilize independent third parties, which could place us at a competitive disadvantage. It will negatively affect our business, financial condition and results of operations if our efforts to expand and train our sales force do not generate a corresponding increase in revenue, and our higher fixed costs may slow our ability to reduce costs in the face of a sudden decline in demand for our diagnostic tests. Any failure to hire, develop and retain talented sales personnel, to achieve desired productivity levels in a reasonable period of time, or timely reduce fixed costs, could negatively affect our business, financial condition and results of operations. Our ability to increase our customer base and achieve broader market acceptance of our diagnostic tests will depend to a significant extent on our ability to expand our marketing efforts. We plan to dedicate significant resources to our marketing programs. It will negatively affect our business, financial condition and results of operations if our marketing efforts and expenditures do not generate a corresponding increase in revenue. In addition, we believe that developing and maintaining broad awareness of our diagnostic tests in a cost-effective manner is critical to achieving broad acceptance of our diagnostic tests. Promotion activities may not generate patient or physician awareness or increase revenue, and even if they do, any increase in revenue may not offset the costs and expenses we incur in building our brand. If we fail to successfully promote, maintain and protect our brand, we may fail to attract or retain the physician acceptance necessary to realize a sufficient return on our brand building efforts, or to achieve the level of brand awareness that is critical for broad use of our diagnostic tests, which in turn could have a material adverse effect on our business, financial condition and results of operations.
If we cannot maintain our current relationships, or enter into new relationships, with biopharmaceutical companies, our revenue prospects could be reduced.
We collaborate with biopharmaceutical companies to analyze patient samples for multiple applications primarily to support clinical trials, including patient identification, companion or complementary diagnostics and retrospective testing. For fiscal years 2021 and 2020, revenue from our top biopharmaceutical customer accounted for 2% and 3% of our total revenue, respectively. The revenue attributable to our biopharmaceutical customers may also fluctuate in the future, which could have a material adverse effect on our financial condition and results of operations. In addition, the termination of these relationships could result in a temporary or permanent loss of revenue.
Our future success depends in part on our ability to maintain these relationships and to establish new relationships. Many factors have the potential to impact such collaborations, including the type of biomarker support required and our ability to deliver it and our biopharmaceutical customers’ satisfaction with our tests or services and other factors that may be beyond our control. Furthermore, our biopharmaceutical customers may decide to decrease or discontinue their use of our tests due to changes in research and product development plans, failures in their clinical trials, financial constraints, or utilization of internal testing resources or tests performed by other parties, or other circumstances outside of our control. In addition to reducing our revenue, the loss of one or more of these relationships may reduce our exposure to research and clinical trials that facilitate the collection and incorporation of new information into our biobank and proprietary AI platform.
We engage in conversations with biopharmaceutical companies regarding potential commercial opportunities on an ongoing basis. There is no assurance that any of these conversations will result in a commercial agreement, or if an agreement is reached, that the resulting relationship will be successful or that clinical or research studies conducted as part of the engagement will produce successful outcomes. Speculation in the industry about our existing or potential relationships with biopharmaceutical companies can also be a catalyst for adverse speculation about us, our tests and our technology, which can adversely affect our reputation and our business.
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or any guidance we may provide.
Our quarterly and annual revenue and operating results may fluctuate significantly, which makes it difficult for us to predict our future operating results. Our quarterly and annual operating results may fluctuate as a result of a variety of factors, many of which are outside
42
our control and, as a result, may not fully reflect the underlying performance of our business. These fluctuations may occur due to a variety of factors, including, but not limited to:
In addition, we can provide no assurances that the demand for our COVID-19 and antibody testing program will be sustained, and even if it is, the period of time for which it would be sustained. As vaccines for COVID-19 become more available and widespread in the future, we expect the demand for our COVID-19 diagnostic and antibody tests will decrease. As a result, the increase in revenue due to any increase in demand for our COVID-19 and antibody testing program is not indicative of results expected for any future period.
The cumulative effects of these factors could result in large fluctuations and unpredictability in our quarterly and annual financial results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. Further, our historical results are not necessarily indicative of results expected for any future period, and quarterly results are not necessarily indicative of the results to be expected for the full year or any other period, and accordingly should not be relied upon as indicative of future performance.
This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any guidance we may provide, or if the guidance we provide is below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any publicly stated guidance we may provide, and could in turn negatively impact our business, financial condition and results of operations.
We need to ensure strong product performance and reliability to maintain and grow our business.
We need to maintain and continuously improve the performance and reliability of our diagnostic tests, including our Biodesix WorkSafe testing program, the Nodify XL2 and Nodify CDT tests, and the GeneStrat and VeriStrat tests, to achieve our profitability objectives. Poor product performance and reliability could lead to customer dissatisfaction, adversely affect our reputation and revenues, and increase our service and distribution costs and working capital requirements. Our diagnostic tests may contain errors or defects, and while we have made efforts to test them extensively, we cannot assure that our current diagnostic tests, or those developed in the future,
43
will not have performance problems. Performance issues with our diagnostic tests will increase our costs in the near-term and accordingly adversely affect our business, financial condition and results of operations.
We depend upon third-party suppliers, including contract manufacturers and single source suppliers, making us vulnerable to supply problems and price fluctuations.
We rely on third-party suppliers, including in some instances single source suppliers, to provide us with certain components of our diagnostic tests. The number of suppliers feeding into the production of our diagnostic tests is in excess of 65 worldwide. We consider a select few of these suppliers, located in the United States, Europe and China, as critical single source providers of components. Bio-Rad Laboratories, as described below, is the sole source supplier for our GeneStrat tests and COVID-19 diagnostic and antibody testing program. Oncimmune is also the sole source supplier for our Nodify CDT tests. While we have initiated the second source qualification process for the majority of these critical components, we may not be successful in securing second sourcing for all of them at all or on a timely basis.
In addition, we may purchase supplies through purchase orders and may not have long-term supply agreements with, or guaranteed commitments from, many of our suppliers, including single source suppliers. Additionally, at present, we rely on contract manufacturers for the production of our supplies for our diagnostic test. Many of our suppliers and contract manufacturers are not obligated to perform services or supply diagnostic testing materials for any specific period, in any specific quantity or at any specific price, except as may be provided in a particular purchase order. We depend on our suppliers and contract manufacturers to provide us and our customers with materials in a timely manner that meet our and their quality, quantity and cost requirements. These suppliers and contract manufacturers may encounter problems during manufacturing for a variety of reasons, any of which could delay or impede their ability to meet our demand. These suppliers and contract manufacturers may cease producing the components we purchase from them or otherwise decide to cease doing business with us. Further, we maintain limited volumes of inventory from most of our suppliers and contract manufacturers. If we inaccurately forecast demand for finished goods, we may be unable to meet customer demand which could harm our competitive position and reputation. In addition, if we fail to effectively manage our relationships with our suppliers and contract manufacturers, we may be required to change suppliers or contract manufacturers. While we believe replacement suppliers exist for all materials, components and services necessary to manufacture our diagnostic tests, establishing additional or replacement suppliers for any of these materials, components or services, if required, could be time-consuming and expensive, may result in interruptions in our operations and product delivery, may affect the performance of our diagnostic tests or could require that we modify their processes. Even if we are able to find replacement suppliers, we will be required to verify that the new supplier maintains facilities, procedures and operations that comply with our quality expectations and applicable regulatory requirements. Any of these events could require that we obtain a new regulatory authority approval before we implement the change, which we may not obtain on a timely basis or at all.
If our third-party suppliers fail to deliver the required commercial quantities of materials on a timely basis and at commercially reasonable prices, and we are unable to find one or more replacement suppliers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality on a timely basis, the continued commercialization of our diagnostic tests, the supply of our diagnostic tests to customers and the development of any future diagnostic tests will be delayed, limited or prevented, which could have material adverse effect on our business, financial condition and results of operations.
We entered into a nonexclusive license and supply agreement with Bio-Rad in August 2019. We rely on Bio-Rad to supply equipment and reagents used to perform ddPCR testing, a service offered by us under a variety of fee for service agreements and the core technology powering the GeneStrat test. Under the terms of this arrangement, we were granted non-exclusive rights to utilize the intellectual property, machinery, materials, reagents, supplies and know-how necessary for the performance of ddPCR in cancer detection testing for third parties in the United States. We agreed to purchase all of the necessary supplies and reagents for such testing exclusively from Bio-Rad. As further consideration for the non-exclusive license, we agreed to pay a royalty of two and one half percent (2.5%) on net service fees (such fees are defined in the Non-Exclusive License Agreement with Bio-Rad) collected from contracted third parties who receive ddPCR services from us. In addition, we have separately been granted permission by Bio-Rad to use the Bio-Rad SARS-CoV-2 ddPCR test and Platelia SARS-CoV-2 Total Ab test for commercial diagnostic services. On May 24, 2021, we entered into the First Amendment to the Non-Exclusive License Agreement between Bio-Rad Laboratories, Inc., and Biodesix, Inc. which amended the original agreement such that, effective May 1, 2021, we are not required to pay a royalty of two and one half percent (2.5%) on net service fees. For more information regarding this license and supply agreement and the permission granted to us by Bio-Rad with respect to such tests, please see “Business—Material Agreements—Agreements with Bio-Rad” previously filed in our Form S-1 on October 23, 2020 and “First Amendment to the Non-Exclusive License Agreement between Bio-Rad Laboratories, Inc., and Biodesix, Inc., dated May 24, 2021” previously filed in our Form 10-Q on August 10, 2021.
This relationship may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing stockholders or disrupt our management and business. We cannot be certain that, following the realization of this relationship, we will achieve the revenue or specific net income that justifies our entry into it. Any termination of this relationship, or delays in entering into new strategic partnership agreements with Bio-Rad, could delay our sales and marketing efforts, which would harm our business prospects, financial condition and results of operations.
44
We may not be able to sufficiently reduce costs in the performance, manufacturing and production of our diagnostic tests to achieve sustainable gross margins.
We partner with contract manufacturers in the development and production of supplies for our diagnostic tests. While we are undertaking a number of initiatives designed to reduce the cost of performing our diagnostic tests, including reducing the costs of supplies, there is no guarantee that we will be able to achieve planned cost reductions from our various cost savings initiatives. There may also be unforeseen occurrences that increase our costs, such as increased prices of the components of our diagnostic tests, changes to labor costs or less favorable terms with third-party suppliers or contract manufacturing partners. If we are unable to reduce our costs, or if cost reductions are less significant or less timely than projected, we will not be able to achieve sustainable gross margins, which would adversely affect our ability to invest in and grow our business and adversely impact our business, financial condition and results of operations.
A pandemic, epidemic or outbreak of an infectious disease in the United States or worldwide, including the outbreak of the novel strain of coronavirus disease, COVID-19, and its variants could adversely affect our business.
If a pandemic, epidemic or outbreak of an infectious disease occurs in the United States or worldwide, our business may be adversely affected. COVID-19 has spread throughout the United States and to most countries globally. Numerous U.S. state and local jurisdictions have imposed, and others in the future may impose, “shelter-in-place” orders, quarantines, executive orders and similar government orders and restrictions for their residents to control the spread of COVID-19. In March 2020, the Governor of Colorado, where our headquarters are located, issued “stay at home” orders limiting non-essential activities, travel and business operations. Such orders or restrictions have resulted in reduced operations at our headquarters, work stoppages, slowdowns and delays, travel restrictions and cancellation of events. Other disruptions or potential disruptions include the inability of our suppliers to manufacture components and parts and to deliver these to us on a timely basis, or at all; disruptions in our production schedule and ability to assemble diagnostic tests; inventory shortages or obsolescence; delays in actions of regulatory bodies; diversion of or limitations on employee resources that would otherwise be focused on the operations of our business; delays in growing or reductions in our sales organization, including through delays in hiring, lay-offs, furloughs or other losses of sales representatives; business adjustments or disruptions of certain third parties, including suppliers, medical institutions and clinical investigators with whom we conduct business; and additional government requirements or other incremental mitigation efforts that may further impact our or our suppliers’ capacity to manufacture our diagnostic tests.
The COVID-19 pandemic also has negatively affected our non-COVID-19 testing-related revenue and our clinical studies. For example, cancer patients may have more limited access to hospitals, healthcare providers and medical resources as they take steps to control the spread of COVID-19. Our biopharmaceutical customers are facing challenges in recruiting patients and in conducting clinical trials to advance their pipelines, for which our tests could be utilized. As a result of the COVID-19 pandemic, beginning in the latter half of March 2020, we have been receiving fewer samples for non-COVID-19 testing on a daily average basis from our clinical and biopharmaceutical customers than before the outbreak of the COVID-19 pandemic. Further, our clinical studies, such as our ongoing INSIGHT study and our recently launched ALTITUDE study, as well as our arrangements with our biopharmaceutical customers, are expected to take longer to complete than what we expected before the outbreak of the COVID-19 pandemic.
The COVID-19 pandemic has also created an opportunity for our diagnostic tests and we have commercialized two diagnostic tests to test for the presence of COVID-19 and antibodies. We are expecting to increase our testing capacity for our COVID-19 diagnostic and antibody testing program in the near term to meet the rising demand for rapid and accurate testing. We expect that the revenue we generate from this expansion will comprise a significant portion of our revenue for the first quarter of 2021. However, there is no assurance that our COVID-19 diagnostic and antibody testing program will continue to be accepted by the market or that other diagnostic tests will become more accepted, produce quicker results or be accurate. Further, the longevity and extent of the COVID-19 pandemic is uncertain. If the pandemic were to dissipate, whether due to a significant decrease in new infections, due to the availability of vaccines, or otherwise, the need for a COVID-19 test could decrease significantly and this could have an adverse effect on our results of operations and profitability. As a result, the increase in revenue due to any increase in demand for these diagnostic tests may not be indicative of our future revenue.
The extent to which the COVID-19 pandemic impacts our business will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity and spread of COVID-19 and the actions to contain COVID-19 or treat its impact, among others. Furthermore, there is no assurance that our diagnostic tests will continue to be effective against the virus in the future.
While the potential economic impact brought by, and the duration of, any pandemic, epidemic or outbreak of an infectious disease, including COVID-19, may be difficult to assess or predict, the widespread COVID-19 pandemic has resulted in, and may continue to result in, significant disruption of global financial markets and a reduction in our ability to access capital, which could adversely affect our liquidity. In addition, a recession or market correction resulting from the spread of an infectious disease, including COVID-19, could materially affect our business. Such economic recession could have a material adverse effect on our long-term business. To the extent the COVID-19 pandemic adversely affects our business and financial results, it may also have the effect of heightening many of the other risks described in this “Risk Factors” section.
45
Natural or man-made disasters and other similar events, including the COVID-19 pandemic, may significantly disrupt our business, and negatively impact our business, financial condition and results of operations.
A significant portion of our employee base, operating facilities and infrastructure are centralized in Boulder, Colorado and we operate a laboratory facility in De Soto, Kansas. Any of our facilities may be harmed or rendered inoperable by natural or man-made disasters, including earthquakes, wildfires, floods, nuclear disasters, riots, acts of terrorism or other criminal activities, infectious disease outbreaks or pandemic events, including the COVID-19 pandemic, power outages and other infrastructure failures, which may render it difficult or impossible for us to operate our business for some period of time. Our facilities would likely be costly to repair or replace, and any such efforts would likely require substantial time. Any disruptions in our operations could adversely affect our business, financial condition and results of operations and harm our reputation. Moreover, although we have disaster recovery plans, they may prove inadequate. We may not carry sufficient business insurance to compensate for losses that may occur. Any such losses or damages could have a material adverse effect on our business, financial condition and results of operations. In addition, the facilities of our suppliers and manufacturers may be harmed or rendered inoperable by such natural or man-made disasters, which may cause disruptions, difficulties or otherwise materially and adversely affect our business.
Any failure to offer high-quality support for our diagnostic tests and services may adversely affect our relationships with providers and negatively impact our reputation among patients and providers, which may adversely affect our business, financial condition and results of operations.
In implementing and using our diagnostic tests and services, providers depend on our support to resolve issues in a timely manner. We may be unable to respond quickly enough to accommodate short-term increases in demand for customer support. Increased customer demand for support could increase costs and adversely affect our business, financial condition and results of operations. Our sales are highly dependent on our reputation and on positive recommendations from our existing patients, care partners, providers and clinics. Any failure to maintain high-quality customer support, or a market perception that we do not maintain high-quality customer support, could adversely affect our reputation, our ability to sell our diagnostic tests and services, and in turn our business, financial condition and results of operations.
The sizes of the markets for our diagnostic tests and services and any future diagnostic tests and services may be smaller than we estimate and may decline.
Our estimates of the annual total addressable market for our diagnostic tests and services are based on a number of internal and third-party estimates and assumptions, including, without limitation, the assumed prices at which we can sell our diagnostic tests and services in the market. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and estimates may not be correct and the conditions supporting our assumptions or estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors.
As a result, our estimates of the annual total addressable market for our diagnostic tests and services in different market segments may prove to be incorrect. If the actual number of patients who would benefit from our diagnostic tests, the price at which we can sell them or the annual total addressable market for them is smaller than we have estimated, it may impair our sales growth and negatively affect our business, financial condition and results of operations.
Our industry is subject to rapid change, which could make our solutions and the diagnostic tests we develop and services we offer, obsolete. If we are unable to continue to innovate and improve our diagnostic tests and services we offer, we could lose customers or market share.
Our industry is characterized by rapid changes, including technological and scientific breakthroughs, frequent new product introductions and enhancements and evolving industry standards, all of which could make our current diagnostic tests and others we are developing obsolete. Our future success will depend on our ability to keep pace with the evolving needs of our customers on a timely and cost-effective basis and to pursue new market opportunities that develop as a result of scientific and technological advances. In recent years, there have been numerous advances in technologies relating to the diagnosis and treatment of cancer. There have also been advances in methods used to analyze very large amounts of molecular information. We must continuously enhance our offerings and develop new and improved diagnostic tests to keep pace with evolving standards of care. If we do not leverage or scale our sample and data biobank to discover new diagnostic tests or applications or update our diagnostic tests to reflect new scientific knowledge, including about lung cancer biology, information about new cancer therapies or relevant clinical trials, our diagnostic tests could become obsolete and sales of our current diagnostic tests and any new tests we develop could decline or fail to grow as expected. This failure to make continuous improvements to our diagnostic tests to keep ahead of those of our competitors could result in the loss of customers or market share that would adversely affect our business, financial condition and results of operations.
We may face additional costs, loss of revenue, significant liabilities, harm to our brand, decreased use of our products or services and business disruption if there are any security or data privacy breaches or other unauthorized or improper access.
In connection with various facets of our business, we collect and use a variety of personal data, such as names, mailing addresses, email addresses, mobile phone numbers, location information, prescription information and other medical information. Any failure to prevent or mitigate security breaches or improper access to, use, disclosure or other misappropriation of our data or consumers’ personal data
46
could result in significant liability under state, (e.g., state breach notification and privacy laws such as the CCPA) federal (e.g., HIPAA), and the HITECH Act and laws in other jurisdictions (e.g., the GDPR). Such an incident may also cause a material loss of revenue from the potential adverse impact to our reputation and brand, affect our ability to retain or attract new users of our diagnostic tests and services and potentially disrupt our business.
Unauthorized disclosure of sensitive or confidential patient or employee data, including personally identifiable information, whether through a breach of computer systems, systems failure, employee negligence, fraud or misappropriation, or otherwise, or unauthorized access to or through our information systems and networks, whether by our employees or third parties, could result in negative publicity, legal liability and damage to our reputation. Unauthorized disclosure of personally identifiable information could also expose us to sanctions for violations of data privacy laws and regulations around the world. To the extent that any disruption or security breach resulted in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed. For example, the loss of or damage to clinical trial data, such as from completed or ongoing clinical trials, for any of our product candidates would likely result in delays in our marketing approval efforts and significantly increased costs in an effort to recover or reproduce the data.
As we become more dependent on information technologies to conduct our operations, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, may increase in frequency and sophistication. These threats pose a risk to the security of our systems and networks, the confidentiality and the availability and integrity of our data and these risks apply both to us, and to third parties on whose systems we rely for the conduct of our business. Because the techniques used to obtain unauthorized access, disable or degrade service or sabotage systems change frequently and often are not recognized until launched against a target, we and our partners may be unable to anticipate these techniques or to implement adequate preventative measures. We have in the past experienced, and may in the future, experience security incidents. While no security incidents in the past have had a material adverse effect on our business, financial condition and results of operations, we cannot predict the impact of any such future events. Further, we do not have any control over the operations of the facilities or technology of our cloud and service providers, including any third-party vendors that collect, process and store personal data on our behalf. Our systems, servers and platforms and those of our service providers may be vulnerable to computer viruses or physical or electronic break-ins that our or their security measures may not detect. Individuals able to circumvent such security measures may misappropriate our confidential or proprietary information, disrupt our operations, damage our computers or otherwise impair our reputation and business. We may need to expend significant resources and make significant capital investments to protect against security breaches or to mitigate the impact of any such breaches. In addition, to the extent that our cloud and other service providers, experience security breaches that result in the unauthorized or improper use of confidential data, employee data or personal data, we may not be indemnified for any losses resulting from such breaches. There can be no assurance that we or our third-party providers will be successful in preventing cyber-attacks or successfully mitigating their effects. Recent cyber-attacks purportedly originated by Russian controlled entities have exacerbated in the wake of Russia’s invasion of Ukraine and our systems may be infiltrated by foreign actors. If we are unable to prevent or mitigate the impact of such security breaches, our ability to attract and retain new customers, patients and other partners could be harmed as they may be reluctant to entrust their data to us, and we could be exposed to litigation and governmental investigations, which could lead to a potential disruption to our business or other adverse consequences.
We have significant payer concentration, with a limited number of customers accounting for a substantial portion of our revenues.
For year ended December 31, 2021, Medicare reimbursed to us 18% of our diagnostic test revenue and two customers accounted for 45% of our total revenue. For the year ended December 31, 2020, Medicare reimbursed to us 17% of our diagnostic test revenue and two customers accounted for 47% of our total revenue. There are risks whenever a large percentage of total revenues are concentrated with a limited number of payers and customers. It is not possible for us to predict the level of demand for our diagnostic tests and services that will be generated by any of these customers in the future. In addition, revenues from these larger customers may fluctuate from time to time based on these customers’ business needs, the timing of which may be affected by market conditions or other factors outside of our control. These payers and customers could also potentially pressure us to reduce the prices we charge for our diagnostic tests and services, which could have an adverse effect on our margins and financial position and could negatively affect our revenues and results of operations. If any of our largest payers terminates its relationship with us or our tests are no longer reimbursable by such payer, such termination could negatively affect our revenues and results of operations.
Our results of operations will be materially harmed if we are unable to accurately forecast customer demand for, and utilization of, our diagnostic tests and manage our inventory.
To ensure adequate inventory supply, we must forecast inventory needs and manufacture our diagnostic tests based on our estimates of future demand for our diagnostic tests. Our ability to accurately forecast demand for them could be negatively affected by many factors, including our failure to accurately manage our expansion strategy, product introductions by competitors, an increase or decrease in customer demand for our diagnostic tests or for those of our competitors, our failure to accurately forecast customer acceptance of new diagnostic tests, unanticipated changes in general market conditions or regulatory matters and weakening of economic conditions or consumer confidence in future economic conditions. Inventory levels in excess of customer demand may result in inventory write-downs or write-offs, which would cause our gross margin to be adversely affected and could impair the strength of our brand. Conversely, if we underestimate customer demand for our diagnostic tests, our supply chain, manufacturing partners and/or internal manufacturing
47
team may not be able to deliver components and diagnostic tests to meet our requirements, and this could result in damage to our reputation, sales growth and customer relationships. In addition, if we experience a significant increase in demand, such as we are currently experiencing with respect to our COVID-19 and antibody testing program, additional supplies of raw materials or additional manufacturing capacity may not be available when required on terms that are acceptable to us, or at all, or suppliers may not be able to allocate sufficient capacity in order to meet our increased requirements, which will adversely affect our business, financial condition and results of operations.
If we experience significant disruptions in our information technology systems, our business may be adversely affected.
We depend on our information technology systems for the efficient functioning of our business, including the performance, distribution and maintenance of our diagnostic tests and services, as well as for accounting, data storage, compliance, purchasing and inventory management. We do not have redundant information technology in all aspects of our systems at this time. Our information technology systems may be subject to computer viruses, ransomware or other malware, attacks by computer hackers, failures during the process of upgrading or replacing software, databases or components thereof, power outages, damage or interruption from fires or other natural disasters, hardware failures, telecommunication failures and user errors, among other malfunctions. We could be subject to an unintentional event that involves a third party gaining unauthorized access to our systems, which could disrupt our operations, corrupt our data or result in release of our confidential information. Technological interruptions would disrupt our operations, including our ability to timely ship and track diagnostic test orders and results, project inventory requirements, manage our supply chain and otherwise adequately service our customers or disrupt our customers’ ability to use our diagnostic tests. In the event we experience significant disruptions, we may be unable to repair our systems in an efficient and timely manner. Accordingly, such events may disrupt or reduce the efficiency of our entire operation and have a material adverse effect on our business, financial condition and results of operations.
Currently, we carry business interruption coverage to mitigate certain potential losses but this insurance is limited in amount, and we cannot be certain that such potential losses will not exceed our policy limits. The successful assertion of one or more large claims against us that exceed or are not covered by our insurance coverage or changes in our insurance policies, including premium increases or the imposition of large deductible or co-insurance requirements, could have a material adverse effect on our business, financial condition and results of operations. We are increasingly dependent on complex information technology to manage our infrastructure. Our information systems require an ongoing commitment of significant resources to maintain, protect and enhance our existing systems. Failure to maintain or protect our information systems and data integrity effectively could have a material adverse effect on our business, financial condition and results of operations.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit or halt the marketing and sale of our diagnostic tests and services. The expense and potential unavailability of insurance coverage for liabilities resulting from issues with our diagnostic tests and services could harm us and negatively impact sales.
We face an inherent risk of product liability as a result of the marketing and sale of our diagnostic tests and services. For example, we may be sued if our diagnostic tests or services cause or are perceived to cause injury or are found to be otherwise unsuitable during manufacturing, marketing or sale. Any such product liability claim may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. In addition, we may be subject to claims against us even if the apparent injury is due to the actions of others or the pre-existing health of the patient. For example, medical personnel, care partners and patients collect samples for our diagnostic tests. If these medical personnel, care partners or patients are not properly trained, are negligent or use our diagnostic tests incorrectly, the capabilities of such tests may be diminished or the patient may suffer critical injury. We may also be subject to claims that are caused by the activities of our suppliers, such as those who provide us with components and sub-assemblies for our diagnostic tests.
If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit or halt the marketing and sale of our diagnostic tests and services. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
48
We believe we have adequate product liability insurance, but it may not prove to be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain or obtain insurance at a reasonable cost or in an amount adequate to satisfy any liability that may arise. Our insurance policy contains various exclusions, and we may be subject to a product liability claim for which we have no coverage. The potential inability to obtain sufficient product liability insurance at an acceptable cost to protect against product liability claims could prevent or inhibit the marketing and sale of our diagnostic tests and services. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts, which would have a material adverse effect on our business, financial condition and results of operations. In addition, any product liability claims brought against us, with or without merit, could increase our product liability insurance rates or prevent us from securing continuing coverage, harm our reputation in the industry, significantly increase our expenses and reduce product sales.
We face competition from many sources, including larger companies, and we may be unable to compete successfully.
There are a number of lung cancer diagnostic solutions companies in the United States, Europe and Asia. Notable competitors in the United States include Veracyte, Inc., Guardant Health, Inc., and Foundation Medicine, Inc. These competitors all provide cancer-focused diagnostic tests to hospitals, researchers, clinicians, laboratories and other medical facilities. Many of these organizations are significantly larger with greater financial and personnel resources than us, and enjoy significantly greater market share and have greater resources than we do. As a consequence, they may be able to spend more on product development, marketing, sales and other product initiatives than we can. Some of our competitors have:
Our continued success depends on our ability to:
In addition, competitors with greater financial resources than ours could acquire other companies to gain enhanced name recognition and market share, as well as new technologies or diagnostic tests that could effectively compete with our existing diagnostic tests, which may cause our revenue to decline and would harm our business.
Our competitors also compete with us in recruiting and retaining qualified scientific, management and commercial personnel, as well as in acquiring technologies complementary to, or necessary for, development of our diagnostic tests. Because of the complex and technical nature of diagnostic testing and the dynamic market in which we compete, any failure to attract and retain a sufficient number of qualified employees could materially harm our ability to develop and commercialize our diagnostic tests, which would have a material adverse effect on our business, financial condition and results of operations.
As we attain greater commercial success, our competitors are likely to develop diagnostic tests that offer features and functionality similar to our diagnostic tests that are currently on the market. Improvements in existing competitive diagnostic tests or the introduction of new competitive diagnostic tests may make it more difficult for us to compete for sales, particularly if those competitive diagnostic tests demonstrate better reliability, convenience or effectiveness or are offered at lower prices.
Performance issues, service interruptions or price increases by our shipping carriers and warehousing providers could adversely affect our business and harm our reputation and ability to provide our services on a timely basis.
Expedited, reliable shipping and delivery services and secure warehousing are essential to our operations. We rely heavily on providers of transport services for reliable and secure point-to-point transport of our diagnostic tests to our customers and for tracking of these shipments, and from time to time require warehousing for our diagnostic tests, sample collection kits and supplies. Should a carrier encounter delivery performance issues such as loss, damage or destruction of any systems, it would be costly to replace such systems in
49
a timely manner and such occurrences may damage our reputation and lead to decreased demand for our diagnostic tests and increased cost and expense to our business. In addition, any significant increase in shipping or warehousing rates could adversely affect our operating margins and results of operations. Similarly, strikes, severe weather, natural disasters, civil unrest and disturbances or other service interruptions affecting delivery or warehousing services we use would adversely affect our ability to process orders for our diagnostic tests on a timely basis.
We rely on commercial courier delivery services to transport samples to our laboratory facility in a timely and cost-efficient manner and if these delivery services are disrupted, our business will be harmed. Our business depends on our ability to quickly and reliably deliver test results to our customers. Blood samples are typically received within days from the United States and outside the United States for analysis at our Boulder, Colorado and De Soto, Kansas facilities. Disruptions in delivery service, whether due to labor disruptions, bad weather, natural disaster, civil unrest or disturbances, terrorist acts or threats or for other reasons could adversely affect specimen integrity and our ability to process samples in a timely manner and to service our customers, and ultimately our reputation and our business. In addition, if we are unable to continue to obtain expedited delivery services on commercially reasonable terms, our operating results may be adversely affected.
Cost-containment efforts of our customers, purchasing groups and governmental purchasing organizations could have a material adverse effect on our sales and profitability.
In an effort to reduce costs, many hospitals in the United States have become members of GPOs and Integrated Delivery Networks (IDNs). GPOs and IDNs negotiate pricing arrangements with medical device companies and distributors on behalf of their members, which may include hospitals and other providers. GPOs and IDNs typically award contracts on a category-by-category basis through a competitive bidding process. Bids are generally solicited from multiple providers with the intention of driving down pricing or reducing the number of vendors. Due to the highly competitive nature of the GPO and IDN contracting processes, we may not be able to obtain new, or maintain existing, contract positions with major GPOs and IDNs. Furthermore, the increasing leverage of organized buying groups may reduce market prices for our diagnostic tests, thereby reducing our revenue and margins.
While having a contract with a GPO or IDN for a given product category can facilitate sales to members of that GPO or IDN, such contract positions can offer no assurance that any level of sales will be achieved, as sales are typically made pursuant to individual purchase orders. Even when a provider is the sole contracted supplier of a GPO or IDN for a certain product category, members of the GPO or IDN are generally free to purchase from other suppliers. Furthermore, GPO and IDN contracts typically are terminable without cause by the GPO or IDN upon 60 to 90 days’ notice. Accordingly, the members of such groups may choose to purchase alternative diagnostic tests due to the price or quality offered by other companies, which could result in a decline in our revenue.
Pricing and reimbursement of medical devices is not harmonized at European level, but is the exclusive competence of the EU Member States. In Europe, pricing and reimbursement decisions are generally made by regional or centralized bodies based on an assessment of the efficacy and clinical effectiveness of the devices or broad device types or procedures. There is a general trend for EU Member States to adopt cost containment measures to control public spend on medical devices. Due to the competitive nature of product offers and prices, we may not be able to obtain new, or maintain existing, contract positions with the EU Member States.
Litigation and other legal proceedings may adversely affect our business.
From time to time, we may become involved in legal proceedings relating to patent and other intellectual property matters, product liability claims, employee claims, tort or contract claims, federal regulatory investigations, securities class action and other legal proceedings or investigations, which could have an adverse impact on our reputation, business and financial condition and divert the attention of our management from the operation of our business. Litigation is inherently unpredictable and can result in excessive or unanticipated verdicts and/or injunctive relief that affect how we operate our business. We could incur judgments or enter into settlements of claims for monetary damages or for agreements to change the way we operate our business, or both. There may be an increase in the scope of these matters or there may be additional lawsuits, claims, proceedings or investigations in the future, which could have a material adverse effect on our business, financial condition and results of operations. Adverse publicity about regulatory or legal action against us could damage our reputation and brand image, undermine our customers’ confidence and reduce long-term demand for our diagnostic tests and services, even if the regulatory or legal action is unfounded or not material to our operations.
We maintain product and professional liability insurance, but this insurance may not fully protect us from the financial impact of defending against product liability or professional liability claims. Any product liability or professional liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future.
General economic and financial market conditions may exacerbate our business risks.
Global macroeconomic conditions and the world’s financial markets remain susceptible to significant stresses, resulting in reductions in available credit and government spending, economic downturn or stagnation, foreign currency fluctuations and volatility in the valuations of securities generally. As a result of uncertainties with respect to financial institutions and the global credit markets and other macroeconomic challenges such as inflationary pressures currently or potentially affecting the economy of the United States and other parts of the world, customers and distributors may experience serious cash flow problems and other financial difficulties,
50
decreasing demand for our products. Our customers and distributors may respond to such economic pressures by reducing or deferring their capital spending or reducing staff.
In addition, events in the United States or foreign markets, such as the United Kingdom’s exit from the European Union, the worldwide effects from the spread of COVID-19 and political and social unrest in various countries around the world, can impact the global economy and capital markets. Additionally, if our customers and distributors are not successful in generating sufficient revenue or are precluded from securing financing, their businesses will suffer, which may materially and adversely affect our business, financial condition and results of operations.
We may not realize the benefits or costs of our Co-Development and Collaboration Agreement with AVEO Oncology.
In 2014, we entered into a Co-Development and Collaboration Agreement with AVEO Oncology (formerly known as AVEO Pharmaceuticals, Inc.) (AVEO) whereby the two parties agreed to various terms and conditions necessary for the co-development of AVEO’s compound ficlatuzumab (the Collaboration Agreement).
We were granted a limited legal interest in ficlatuzumab and may not have the right to control the development and exploitation of ficlatuzumab. As consideration for the grant, we agreed to cover the first $15.0 million of ficlatuzumab’s clinical development costs, with both parties then sharing all costs equally after the cap was reached.
In October of 2016, the Collaboration Agreement was amended to eliminate the requirement that we cover all of the initial costs. Under the amended terms, we agreed to allow AVEO to recapture its cost that it otherwise would not have been responsible for said recapture to occur out of any royalties or revenues eventually derived from the Collaboration Agreement. As part of the Collaboration Agreement, unless we or AVEO exercise our right to opt-out of co-development, we equally share in any income received from licensing rights to ficlatuzumab to any third parties. In September 2020, we exercised our opt-out right for the payment of half of the development and regulatory costs for ficlatuzumab. This opt-out was effective as of December 2, 2020 with remaining obligations estimated to be $0.1 million. Following the effective date, we will be entitled to a 10% royalty of net sales of ficlatuzumab and 25% of license income generated from the licensing of ficlatuzumab. Ficlatuzumab is currently being evaluated in squamous cell carcinoma of the head and neck (SCCHN), metastatic pancreatic ductal cancer (PDAC), and acute myeloid leukemia (AML). For more information regarding this Collaboration Agreement, please see “Business —Drug Co-Development.”
Our relationship with AVEO may require us to incur non-recurring and other charges, increase our near and long-term expenditures, or disrupt our management and business. We cannot be certain that, following the realization of this relationship, we will achieve the revenue or specific net income that justifies our entry into it. Any termination of this relationship, or delays in entering into new strategic partnership agreements with AVEO, could delay our sales and marketing efforts, which would harm our business prospects, financial condition and results of operations.
We are exposed to significant future payments and other obligations associated with our acquisitions of Integrated Diagnostics and Oncimmune, U.S.A., and may not realize the advantages we expect from these acquisitions.
In 2018, we purchased select assets and liabilities from Integrated Diagnostics, Inc. and IND Funding, LLC (collectively, the Seller or Indi) which included the CLIA lab in Seattle, Washington, and all rights to the Nodify XL2 test and intellectual property rights related to that test. The purchase was made for total consideration of $27.6 million, consisting of $8.0 million (10,649,604 shares) of our Series G Preferred Stock and contingent consideration with an initial fair market value of $19.6 million.
The acquisition of Indi included a contingent consideration arrangement that requires additional consideration to be paid by us to the Seller based on the milestone of the attainment of a three consecutive month gross margin target of $2 million within a seven-year period. Under the terms of the original agreement, when the gross margin target was met, the Company was required to issue 2,520,108 shares of common stock. For the six months following the achievement of the milestone, the Indi had the option to require the Company to redeem the common shares for $37.0 million in cash over eight equal installments. If Indi elected not to exercise this option, we had 12 months to repurchase the common stock in two equal quarterly cash installments totaling $37 million.
The Company met the gross margin target of $2.0 million for three consecutive months during the three months ended June 30, 2021. The Company entered into an amendment to the original agreement in August 2021 in which all parties agreed to forgo the issuance of common stock and agreed that the Company will in lieu thereof make six quarterly installments of approximately $4.6 million each beginning in January 2022 and a final payment of approximately $9.3 million in July 2023 for a total of $37.0 million. The aggregate amount of payments owed by the Company under this amendment is the same as if Indi had exercised the put right or the Company had exercised the call right provided for in the original agreement. The amendment to the original agreement is subject to consent from our lender under the 2021 Term Loan and related amendments. As of December 31, 2021, we have not made any payments in connection with the contingent consideration. We obtained consent and subsequently made the first milestone payment of $4.6 million in January 2022 and we are in discussions with our lender to obtain consents for future payments.
In addition, on October 31, 2019 we completed an acquisition of United Kingdom-based Oncimmune, Ltd.’s (Oncimmune) United States operations including its CLIA lab in De Soto, Kansas and its incidental pulmonary nodule (IPN) malignancy test, then marketed
51
in the United States as the EarlyCDT®-Lung. We renamed the test and relaunched the test on February 28, 2020 as the Nodify CDT test and the De Soto, Kansas lab will be the sole United States provider of the Nodify CDT test.
As part of the acquisition, we and Oncimmune entered into several agreements to govern the relationship between the parties and to allow us to provide the Nodify CDT test. The overarching umbrella Purchase and Commercialization Agreement (PCA) defines the general relationship between the parties. Included under the PCA was (a) an APA whereby we acquired all of the United States assets associated with the De Soto, Kansas clinical laboratory, as well as the trademarks and patent application associated with the test; (b) an intellectual property license granting us the rights necessary under Oncimmune’s background intellectual property rights to perform the Nodify CDT test; (c) a supply agreement for supplying us with the necessary materials and reagents needed to run the Nodify CDT test; and (d) a development agreement where Oncimmune agrees to assist us in further developing the Nodify CDT test. We were also granted an option through December 31, 2020 to acquire the rights to expand the field of use of the Nodify CDT test to include lung cancer screening, which we elected to not exercise.
We agreed to a revenue share payment of 8% of recognized revenue for non-screening tests up to an annual minimum volume and 5% thereafter, with an escalating minimum through the first four years of sales. An insignificant amount of royalty payments was paid for the year ended December 31, 2020. In September 2020, we notified Oncimmune that we would not exercise this option for expansion of the field of use.
Our acquisitions may require us to incur non-recurring and other charges, increase our near and long-term expenditures, or disrupt our management and business. We cannot be certain that, following the realization of these acquisitions, we will achieve the revenue or specific net income that justifies our entry into them. This could delay our sales and marketing efforts, which would harm our business prospects, financial condition and results of operations.
We are highly dependent on our senior management team and key personnel, and our business could be harmed if we are unable to attract and retain personnel necessary for our success.
We are highly dependent on our senior management and other key personnel. Our success will depend on our ability to retain senior management and to attract and retain qualified personnel in the future, including sales and marketing professionals, scientists, clinical specialists, and other highly skilled personnel and to integrate current and additional personnel in all departments. The loss of members of our senior management, sales and marketing professionals, scientists, clinical and regulatory specialists could result in delays in product development and harm our business. If we are not successful in attracting and retaining highly qualified personnel, it would have a material adverse effect on our business, financial condition and results of operations.
Our research and development programs and laboratory operations depend on our ability to attract and retain highly skilled scientists and technicians.
We may not be able to attract or retain qualified scientists and technicians in the future due to the competition for qualified personnel among life science businesses, particularly near our headquarters in Boulder, Colorado and our laboratory facility in De Soto, Kansas. We also face competition from universities and public and private research institutions in recruiting and retaining highly qualified scientific personnel. We may have difficulties locating, recruiting, or retaining qualified salespeople. Recruiting and retention difficulties can limit our ability to support our research and development and sales programs. To induce valuable employees to remain at our company, in addition to salary and cash incentives, we have issued and may continue to issue equity awards that vest over time. The value to employees of equity awards that vest over time may be significantly affected by movements in our stock price that are beyond our control and may at any time be insufficient to counteract more lucrative offers from other companies. Despite our efforts to retain valuable employees, members of our management, scientific and development teams may terminate their employment with us on short notice. Our employment arrangements with our employees provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. We also do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees.
Our corporate culture has contributed to our success, and if we cannot maintain this culture as we grow, we could lose the innovation, creativity and teamwork fostered by our culture and our business may be harmed.
We believe that our culture has been and will continue to be a critical contributor to our success. We expect to continue to hire aggressively as we expand, and we believe our corporate culture has been crucial in our success and our ability to attract highly skilled personnel. If we do not continue to develop our corporate culture or maintain and preserve our core values as we grow and evolve, we may be unable to foster the innovation, curiosity, creativity, focus on execution, teamwork and the facilitation of critical knowledge transfer and knowledge sharing we believe we need to support our growth. Moreover, liquidity available to our employee securityholders could lead to disparities of wealth among our employees, which could adversely impact relations among employees and our culture in general. Our anticipated headcount growth and our transition from a private company to a public company may result in a change to our corporate culture, which could harm our business.
52
Our ability to utilize our net operating loss carryforwards and research and development credit may be limited.
In general, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (the Code) a corporation that undergoes an ownership change, generally defined as a greater than 50% change by value in its equity ownership by certain shareholders over a three-year period, is subject to limitations on its ability to utilize its pre-change net operating losses (NOLs) and its research and development credit carryforwards to offset future taxable income. The applicable rules generally operate by focusing on changes in ownership among stockholders considered by the rules as owning, directly or indirectly, 5% or more of the stock of a company, as well as changes in ownership arising from new issuances of stock by the company. We believe that our NOLs are currently not subject to limitation under these rules. However, if we undergo an ownership change now or in the future, our ability to utilize NOLs and research and development credit carryforwards could be limited by Sections 382 and 383 of the Code. Future changes in stock ownership may be beyond our control. In addition, our ability to deduct net interest expense may be limited if we have insufficient taxable income for the year during which the interest is incurred, and any carryovers of such disallowed interest would be subject to the limitation rules similar to those applicable to NOLs and other attributes. For these reasons, in the event we experience a change of control, we may not be able to utilize a material portion of the NOLs, research and development credit carryforwards or disallowed interest expense carryovers, even if we attain profitability.
The terms of our secured credit agreement require us to meet certain operating and financial covenants and place restrictions on our operating and financial flexibility. If we raise additional capital through debt financing, the terms of any new debt could further restrict our ability to operate our business.
On March 19, 2021 (Effective Date), we entered into an agreement with Silicon Valley Bank, a California corporation (SVB or Lender) to refinance long-term debt (2021 Term Loan). The initial amount borrowed under the 2021 Term Loan was $30 million and provides for an “interest-only” period from the Effective Date through February 28, 2023, with interest due and payable monthly on the first calendar day of each month. However, the Company achieved a revenue milestone of at least $65 million on a trailing twelve-month basis during the three months ended March 31, 2021 which automatically extended the interest-only period through February 28, 2024. Further, we granted the Lender a security interest in substantially all of our assets.
The 2021 Term Loan may be prepaid by us at any time, subject to a prepayment penalty of up to 3% of the outstanding principal amount, depending on the date of prepayment. The 2021 Term Loan contains customary affirmative and negative covenants for a loan, requires us to comply with a minimum liquidity ratio covenant, and has a trailing six month rolling minimum revenue requirement. Failure to comply with the covenants and loan requirements may result in an event of default.
On December 31, 2021, we entered into the Consent and Second Amendment to Loan and Security Agreement (Second Amendment) to, among other things, amend our 2021 Term Loan and First Amendment to obtain consent for the $4.6 million January 2022 milestone payment under the Indi APA, repay $20 million in outstanding principal on December 31, 2021, waive the $600,000 prepayment fee on the $20 million Term Loan repayment, and waive the minimum revenue covenant as of December 31, 2021 and modify the minimum revenue requirement to not less 75% for the three months ended March 31, 2022 and not less than 75% on a trailing six month rolling basis for each quarter thereafter of the Company’s projected revenue performed at the end of each reporting period.
The 2021 Term Loan also contains certain covenants limiting the ability of the Company to, among other things, incur future debt, transfer assets except for the ordinary course of business, make acquisitions, pay dividends or make other certain restricted payments, or sell assets, subject to certain exceptions, without the prior written consent of the Lender. These covenants may restrict our ability to pursue new business opportunities and access additional capital.
In the event of a default, including, among other things, our failure to make any payment when due or our failure to comply with any covenant under the 2021 Term Loan, the Lender may elect to declare all amounts outstanding to be immediately due and payable, and may proceed against the collateral granted to them to secure such indebtedness, including a royalty-free license or other right to use all of our intellectual property, which could have a material adverse effect on our business, financial condition, and results of operations.
We will need to raise additional capital to fund our existing operations, develop our platform, commercialize new diagnostic tests or expand our operations.
We will need to raise additional capital in the future to expand our business, to pursue strategic investments, to take advantage of financing opportunities or for other reasons, including to:
53
Our present and future funding requirements will depend on many factors, including:
The various ways we could raise additional capital carry potential risks. If we raise funds by issuing equity securities, our stockholders could experience dilution. Any preferred equity securities issued also could provide for rights, preferences or privileges senior to those of holders of our common stock. If we raise funds by issuing debt securities, those debt securities would have rights, preferences and privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings pursuant to a credit agreement could impose significant restrictions on our operations. If we raise funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our platform technologies or diagnostic tests, pay a portion of our royalties, or grant licenses on terms that are not favorable to us.
We may acquire other businesses, which could require significant management attention, disrupt our business, dilute stockholder value and adversely affect our results of operations.
As part of our business strategy, we may in the future make additional acquisitions or investments in complementary companies, diagnostic tests or technologies that we believe fit within our business model and can address the needs of our customers and potential customers. In the future, we may not be able to acquire and integrate other companies, diagnostic tests or technologies in a successful manner. We may not be able to find suitable acquisition candidates, and we may not be able to complete such acquisitions on favorable terms, if at all. In addition, the pursuit of potential acquisitions may divert the attention of management and cause us to incur additional expenses in identifying, investigating, and pursuing suitable acquisitions, whether or not they are consummated. If we do complete acquisitions, we may not ultimately strengthen our competitive position or achieve our goals, including increases in revenue, and any acquisitions we complete could be viewed negatively by our customers, investors and industry analysts.
Future acquisitions may reduce our cash available for operations and other uses and could result in amortization expense related to identifiable assets acquired. We may have to pay cash, incur debt or issue equity securities to pay for any such acquisition, each of which could adversely affect our financial condition or the value of our common stock. The sale or issuance of equity to finance any such acquisitions would result in dilution to our stockholders. The incurrence of indebtedness to finance any such acquisition would result in fixed obligations and could also include covenants or other restrictions that could impede our ability to manage our operations. In addition, our future results of operations may be adversely affected by the dilutive effect of an acquisition, performance earn-outs or contingent bonuses associated with an acquisition. Furthermore, acquisitions may require large, onetime charges and can result in increased debt or contingent liabilities, adverse tax consequences, additional stock-based compensation expenses and the recording and subsequent amortization of amounts related to certain purchased intangible assets, any of which items could negatively affect our future results of operations. We may also incur goodwill impairment charges in the future if we do not realize the expected value of any such acquisitions.
Also, the anticipated benefit of any strategic alliance, joint venture or acquisition may not materialize, or such strategic alliance, joint venture or acquisition may be prohibited. For example, our 2021 Term Loan restricts our ability to pursue certain mergers, acquisitions, amalgamations or consolidations that we may believe to be in our best interest. Additionally, future acquisitions or dispositions could result in potentially dilutive issuances of our equity securities, the incurrence of debt, contingent liabilities or amortization expenses or write-offs of goodwill, any of which could harm our financial condition. We cannot predict the number, timing or size of future joint ventures or acquisitions, or the effect that any such transactions might have on our operating results.
54
Risks Related to our Governmental Regulation
The insurance coverage and reimbursement status of newly approved diagnostic tests, particularly in a new category of diagnostics and therapeutics, is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for current or future diagnostic tests could limit our ability, and that of our collaborators, to fully commercialize our diagnostic tests and decrease our ability to generate revenue.
The availability and extent of reimbursement by governmental and private payers is essential for most patients to be able to afford the clinical diagnostic tests and cellular therapeutics that we and our collaborators currently or in the future plan to develop and sell. In addition, because our clinical diagnostics and diagnostic tests represent new approaches to the research, diagnosis, detection and treatment of diseases, we cannot accurately estimate how our diagnostic tests, and those jointly created with our collaborators, would be priced, whether reimbursement could be obtained or any potential revenue generated. Sales of our diagnostic tests will depend substantially, both domestically and internationally, on the extent to which the costs of our diagnostic tests are paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payers. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize some of our diagnostic tests or services. Even if coverage is provided, the available reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize an adequate return on our investment in any of our diagnostic tests or services. Changes in the reimbursement landscape may occur, which are outside of our control, and may impact the commercial viability of our diagnostic tests.
There is significant uncertainty related to the insurance coverage and reimbursement of newly launched, cleared, authorized or approved diagnostic tests. In the United States, many significant decisions about reimbursement for new diagnostics and medicines are typically made by the CMS, an agency within the HHS. CMS decides whether and to what extent a new diagnostic or medicine will be covered and reimbursed under Medicare, although it frequently delegates this authority to local Medicare Administrative Contractors (MACs). Private payers tend to follow Medicare to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for novel diagnostic tests such as ours. Additionally, reimbursement authorities or bodies in Europe may be more conservative than CMS. For example, a number of cancer drugs have been approved for reimbursement in the United States and have not been approved for reimbursement, or have been approved under restricted conditions, in certain European countries.
Outside the United States, the reimbursement process and timelines vary significantly. In Europe, pricing and reimbursement of medical devices is the exclusive competence of the EU Member States. However, the European Commission is facilitating a voluntary corporation between the EU Member States on health technology assessments (HTA) which consists of a network of the EU Member States’ national authorities and bodies responsible for HTA and a joint action to support cooperation at scientific and technical level between the HTA bodies. We cannot be sure that such prices and reimbursement decisions will be acceptable to us or our collaborators. If the regulatory authorities in these foreign jurisdictions set prices or make reimbursement criteria that are not commercially attractive for us or our collaborators, our revenues and the potential profitability of our products in those countries would be negatively affected. An increasing number of countries are taking initiatives to control the healthcare budget by focusing cost-cutting efforts on medicinal products, and to a lesser extent, medical devices, provided under their state-run healthcare systems. These price control efforts have impacted all regions of the world, but have been most prominent in the EU. Additionally, some countries require approval of the sale price of a product before it can be marketed or mandatory discounts or profit caps may be applied. Further, after the sale price is approved, it remains subject to review during the product lifecycle. In many countries, the pricing review period begins after marketing or product licensing approval is granted or the CE mark is obtained. As a result, we or our collaborators might obtain marketing approval for a product or service in a particular country, but then may experience delays in the reimbursement approval or be subject to price regulations that would delay the commercial launch of our product or service, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of that product or service in that particular country.
Moreover, increasing efforts by governmental and third-party payers, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for newly cleared, authorized, certified or approved devices and medicines and, as a result, they may not cover or provide adequate payment for our clinical diagnostics to be sold by us or our collaborators. For example, in May 2018 the United States government released a “blueprint,” or plan, to reduce the cost of drugs. This blueprint contains certain measures that HHS has been working to implement, although it is possible that HHS’s regulatory priorities may change under the new Biden administration. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, which are, in some cases, designed to encourage importation from other countries and bulk purchasing.
We expect to experience pricing pressures on our clinical diagnostics sold by us and our collaborators due to the trend toward value-based pricing and coverage, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs, surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new diagnostic tests.
55
Measures to reduce healthcare costs may hurt our business.
The majority of our customers are healthcare providers who depend upon reimbursement by government and commercial insurance payers for lung cancer diagnostic solutions services. With a majority of United States patients with lung cancer covered by Medicare, the Medicare reimbursement rate is an important factor in a customer’s decision to use our diagnostic tests and limits the prices we may charge for them. Commercial insurance payers may also exert downward pressure on payment rates for lung cancer treatment services. A reduction in reimbursement rates for lung cancer treatments may adversely affect our customers’ businesses and cause them to enact cost reduction measures that may include reducing the scope of their programs, thereby potentially reducing demand for our diagnostic tests.
Healthcare reform measures could hinder or prevent the commercial success of our diagnostic tests.
In the United States, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system in ways that may harm our future revenues and profitability and the demand for our diagnostic tests. Federal and state lawmakers regularly propose and, at times, enact legislation that would result in significant changes to the healthcare system, some of which are intended to contain or reduce the costs of medical products and services. Current and future legislative proposals to further reform healthcare or reduce healthcare costs may limit coverage of or lower reimbursement for the procedures associated with the use of our diagnostic tests. The effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our diagnostic tests. For example, the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the ACA), contains a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse measures, all of which will impact existing government healthcare programs and will result in the development of new programs.
There have been judicial challenges to certain aspects of the ACA, as well as efforts by the Trump administration and Congress to repeal, replace or alter the implementation of certain aspects of the ACA. For example, as part of the Tax Cuts and Jobs Act of 2017 (TCJA), Congress eliminated the tax penalty, starting January 1, 2019, for not complying with the ACA’s individual mandate to carry health insurance. The Further Consolidated Appropriations Act of 2020, Pub. L. No. 116-94, signed into law December 20, 2019, fully repealed the ACA’s “Cadillac Tax” on certain high-cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share (repeal effective in 2021), and the medical device excise tax on non-exempt medical devices. On December 14, 2018, a Texas District Court Judge invalidated the ACA in its entirety because he concluded that the individual mandate, which was repealed by Congress as noted above, is unconstitutional and cannot be severed from the remainder of the ACA. The Fifth Circuit Court of Appeals affirmed the district court’s ruling that the individual mandate was unconstitutional, but it remanded the case back to the district court for further analysis of whether the mandate could be severed from the ACA; that is, whether the entire ACA was therefore also invalid. The Supreme Court of the United States granted certiorari on March 2, 2020 and heard oral argument on November 10, 2020; the case is expected to be decided by mid-2021. It is unclear how this decision, subsequent appeals, and other efforts to challenge, repeal, or replace, or alter the implementation of the ACA will affect our business, financial condition and results of operations.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011, among other things, included reductions to CMS payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030 unless additional Congressional action is taken, with the exception of a temporary suspension of the 2% cut in Medicare payments from May 1, 2020 through March 31, 2021. Additionally, the American Taxpayer Relief Act of 2012, among other things, reduced CMS payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover Medicare overpayments to providers from three to five years.
The Biden administration and Congress may continue to pursue significant changes to the current healthcare laws, and the Biden administration has indicated its intent to strengthen the ACA and focus on reducing the cost of healthcare. We face uncertainties that might result from modifications or repeal of any of the provisions of the ACA, including as a result of current and future executive orders and legislative actions. The impact of those changes on us and potential effect on the medical device industry as a whole is currently unknown. Any changes to the ACA are likely to have an impact on our results of operations, and may negatively affect our business, financial condition and results of operations. We cannot predict what other healthcare programs and regulations will ultimately be implemented at the federal or state level or the effect of any future legislation or regulation in the United States on our business, financial condition and results of operations.
The continuing efforts of the government, insurance companies, managed care organizations and other payers of healthcare services to contain or reduce costs of healthcare may harm:
56
The ACA substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacts our industry. Future changes in healthcare policy could increase our costs and subject us to additional regulatory requirements that may interrupt commercialization of our current and future solutions. Future changes in healthcare policy could also decrease our revenue and impact sales of and reimbursement for our current and future diagnostic tests.
We must comply with anti-corruption, anti-bribery, anti-money laundering and similar laws.
We are subject to the FCPA, which generally prohibits companies in the United States from engaging in bribery or other prohibited payments to foreign officials for the purpose of obtaining or retaining business and requires companies to maintain accurate books and records and internal controls. We are also subject to requirements under the United States Treasury Department’s Office of Foreign Assets Control, United States domestic bribery laws and other anti-corruption, anti-bribery and anti-money laundering laws. While we have policies and procedures in place designed to promote compliance with such laws, our employees or other agents may nonetheless engage in prohibited conduct under these laws for which we or our executives might be held responsible. If our employees or other agents are found to have engaged in such practices, we could suffer severe penalties and other consequences that may have an adverse effect on our business, financial condition and results of operations.
Furthermore, international customers may currently order our diagnostic tests, either directly from us or through a potential joint venture, and we are subject to the FCPA, which prohibits companies and their intermediaries from making payments in violation of law to non-United States government officials for the purpose of obtaining or retaining business or securing any other improper advantage. Our reliance on independent distributors to sell our diagnostic tests internationally demands a high degree of vigilance in maintaining our policy against participation in corrupt activity, because these distributors could be deemed to be our agents and we could be held responsible for their actions. Other United States companies in the medical device and biopharmaceutical field have faced criminal penalties under the FCPA for allowing their agents to deviate from appropriate practices in doing business with these individuals. We are also subject to similar anti-bribery laws in the jurisdictions in which we operate, including laws promulgated by OECD countries in which we operate, such as Israel. These laws are complex and far-reaching in nature, and, as a result, we cannot assure you that we would not be required in the future to alter one or more of our practices to be in compliance with these laws or any changes in these laws or the interpretation thereof. Any violations of these laws, or allegations of such violations, could disrupt our operations, involve significant management distraction, involve significant costs and expenses, including legal fees and could result in a material adverse effect on our business, prospects, financial condition and results of operations. We could also suffer severe penalties, including criminal and civil penalties, disgorgement and other remedial measures.
We must comply with healthcare fraud and abuse laws.
Various federal and state laws, as well as the laws of foreign countries, prohibit payments to induce the referral, purchase, order or use of healthcare products or services and require medical device companies to limit prevent, and/or monitor, and report certain payments to third-party payers, health care professionals, and other individuals. These healthcare fraud and abuse, anti-kickback, public reporting and aggregate spend laws affect our sales, marketing and other promotional activities by limiting the kinds of financial arrangements, including sales programs, we may have with lung cancer treatment providers, hospitals, physicians or other potential purchasers or users, including patients, of medical devices and services. They also impose additional administrative and compliance burdens on us. In particular, these laws influence, among other things, how we structure our sales offerings, including discount practices, customer support, education and training programs and physician consulting and other service arrangements. These laws prohibit certain marketing initiatives that are commonplace in other industries. If we were to offer or pay inappropriate inducements for the purchase, order or use of our diagnostic tests or our services, or our arrangements are perceived as inappropriate inducements, we could be subject to claims under various healthcare fraud and abuse laws.
Restrictions under applicable United States federal and state healthcare laws and regulations include the following:
57
Other federal and state laws, as well as the laws of foreign countries, generally prohibit individuals or entities from knowingly presenting, or causing to be presented, claims for payments to government or commercial payers that are false or fraudulent, or for items or services that were not provided as claimed. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of product candidates and medical devices from government-funded healthcare programs, such as Medicare and Medicaid, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations. Moreover, any investigation into our practices could cause adverse publicity and require a costly and time-consuming response. If any physicians or other healthcare providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government-funded healthcare programs.
Manufacturers can also be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by providing inaccurate billing or coding information to customers, by providing improper financial inducements, or through certain other activities. We attempt to ensure that any billing and coding information we provide for our diagnostic tests emphasizes the need for physicians and other providers to make independent judgments, use accurate and appropriate billing and coding that complies with all applicable payer policies, and document the medical need for their patients as appropriate. Nevertheless, the government may not regard any billing errors that may be made by our customers as inadvertent and may examine our role in providing information to our customers, physicians and patients concerning the benefits and potential coverage of more frequent therapy.
FDA regulation of our industry generally or our tests specifically could be disruptive to our business.
Our operations are subject to extensive federal, state, local and foreign laws and regulations, including FDA laws and regulations, all of which are subject to change. These laws and regulations are complex and are subject to interpretation by the courts and by government agencies. We believe that we are in material compliance with all statutory and regulatory requirements applicable to us, but there is a risk that one or more government agencies could take a contrary position, or that a private party could file suit under the qui tam provisions of the federal False Claims Act or a similar state law. Such occurrences, regardless of their outcome, could damage our reputation and adversely affect important business relationships with third parties, including managed care organizations, and other private third-party payers.
The FDA has recently increased its attention to marketing of pharmacogenetic tests. For example, in late 2018, the FDA issued a safety communication regarding genetic laboratory tests with claims to predict a patient’s response to specific medications that have not been reviewed by the FDA and may not be supported by clinical evidence. Among other tests, the FDA notice cited genetic tests that claim results can be used to help physicians identify which antidepressant medication would have increased effectiveness or side effects compared to other antidepressant medications. As explained by the FDA in its update to this safety communication, the FDA sent notices to several firms marketing such pharmacogenetic tests where the FDA believes the relationship between genetic variations and the medication’s effects has not been established, including a warning letter sent to a laboratory, in part, for failing to obtain premarket review of its test. HHS recently issued an announcement stating that the FDA cannot require premarket review of any LDT without engaging in formal notice-and-comment rulemaking, as opposed to through guidance documents, compliance manuals, website statements, or other informal issuances.
The HHS announcement was made on the Department’s website and we can provide no assurances that the HHS statement will not be rescinded or revised or that it would preclude the FDA from renewing its attention on diagnostic tests, including those that we provide. If this were to happen, it may impact our marketing practices relating to the relevant tests, which in turn may have an adverse impact on our business, financial condition and results of operations.
The SARS-CoV-2 tests we perform are currently the subject of EUAs, which permit the use of unapproved medical products or unapproved uses of medical products to be used in an emergency to diagnose, treat, or prevent serious or life-threatening diseases or
58
conditions when there are no adequate, approved, and available alternatives, as provided under section 564 for the Federal Food, Drug, and Cosmetic Act (FDCA). EUAs are temporary authorizations that are revoked at the end of the public health emergency, when there is an adequate, approved, or available alternative, or when there are performance or safety concerns. These EUAs also set out conditions for laboratories who are authorized to perform the particular test. The HHS statement mentioned above did not affect EUAs for COVID-19 laboratory-developed tests that were already in effect at the time the statement was released. The HHS statement did not affect EUAs issued for commercially developed COVID-19 IVD tests, which apply to test developers and authorized laboratories.
The EUA for Bio-Rad’s SARS-CoV-2 Droplet Digital PCR™ test provides several conditions for authorized laboratories, including that the test result reports will include Fact Sheets that are authorized as part of the EUA, deviations from the authorized procedures, including specimen types, are not permitted, notification of public health authorities of intent to run the test prior to initiating testing, collection and reporting of performance data to the FDA, including false positives, false negatives, and significant deviations from the established performance characteristics, and appropriate training and protective equipment for laboratory staff. This EUA also states that authorized laboratories must maintain records associated with the EUA and be made available to the FDA for inspection upon request. Printed materials, advertising, and promotion related to use of the test must be consistent with the authorized labeling and Fact Sheets, as well as other terms set forth in the EUA and any applicable requirements under the FDCA and its implementing regulations, and conspicuously bear the following statements:
Other statements that appear in advertising and promotional materials must not represent or suggest that this test is safe or effective for the detection of SARS-CoV-2.
The EUA for Bio-Rad’s serological test for the antibodies associated with SARS-CoV-2, also sets out several conditions for authorized laboratories that mirror the conditions for the PCR test described above, except that the printed materials, advertising, and promotion of the test must conspicuously bear the following statements:
Failure to comply with federal, state and foreign laboratory licensing requirements and the applicable requirements of the FDA or any other regulatory authority, could cause us to lose the ability to perform our tests, experience disruptions to our business, or become subject to administrative or judicial sanctions.
The diagnostic testing industry is subject to extensive laws and regulations, many of which have not been interpreted by the courts, including the application of the FDA’s EUA authority. As noted above, the EUAs for the COVID-19 tests that are part of our Biodesix WorkSafe testing program set out certain conditions for authorized laboratories using the tests, which have not received premarket clearance, approval, or a de novo from the FDA. If we fail to meet these conditions, the FDA may take enforcement action, such as issuing a warning letter, seeking an injunction, seizure, fines, or criminal penalties. Pursuant to the August 19, 2020 statement by HHS, the FDA cannot require any LDT to undergo premarket approval until it has engaged in notice-and-comment rulemaking. Laboratory tests that have already received an EUA to detect the COVID-19 virus were “unaffected” by the announcement. Tests without FDA clearance, approval, or authorization would not be considered covered countermeasures under the Public Readiness and Emergency Preparedness Act (PREP Act). The HHS statement also did not affect EUAs issued for commercially developed COVID-19 IVD tests, which apply to test developers and authorized laboratories.
We are also subject to CLIA, a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA requires virtually all laboratories to be certified by the federal government and mandates compliance with various operational, personnel, facilities administration, quality and proficiency testing requirements intended to ensure that testing services are accurate, reliable and timely. CLIA certification is also
59
a prerequisite to be eligible to bill state and federal health care programs, as well as many private third-party payers, for laboratory testing services. As a condition of CLIA certification, each of our laboratories is subject to survey and inspection every other year, in addition to being subject to additional random inspections. The biennial survey is conducted by CMS, a CMS agent (typically a state agency), or, if the laboratory holds a CLIA certificate of accreditation, a CMS-approved accreditation organization.
Sanctions for failure to comply with CLIA requirements, including proficiency testing violations, may include suspension, revocation, or limitation of a laboratory’s CLIA certificate, which is necessary to conduct our business, as well as the imposition of significant fines or criminal penalties.
Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing licensure, or our failure to renew a CLIA certificate, a state or foreign license, or accreditation, could have a material adverse effect on our business. If the CLIA certificate of any one of our laboratories is revoked, CMS could seek revocation of the CLIA certificates of our other laboratories based on their common ownership or operation, even though they are separately certified.
In addition, we are subject to state laws and regulations governing laboratory licensure. Some states have enacted state licensure laws that are more stringent than CLIA. Although we have obtained licenses from states where we believe we are required to be licensed, we may become aware of other states that require out-of-state laboratories to obtain licensure in order to accept specimens from the state, and it is possible that other states currently have such requirements or will have such requirements in the future.
We may also be subject to regulation in foreign jurisdictions as we seek to expand international utilization of our tests or such jurisdictions adopt new licensure requirements, which may require review of our tests in order to offer them or may have other limitations that may limit our ability to make our tests available outside of the United States. Complying with licensure requirements in new jurisdictions may be expensive, time-consuming and subject us to significant and unanticipated delays. Changes in state or foreign licensure laws that affect our ability to offer and provide diagnostic services across state or foreign country lines could materially and adversely affect our business. In addition, state and foreign requirements for laboratory certification may be costly or difficult to meet and could affect our ability to receive specimens from certain states or foreign countries.
Failure to comply with applicable clinical laboratory licensure requirements may result in a range of enforcement actions, including suspension, limitation or revocation of our CLIA certificate and/or state licenses, imposition of a directed plan of action, onsite monitoring, civil monetary penalties, criminal sanctions and revocation of the laboratory’s approval to receive Medicare and Medicaid payment for its services, as well as significant adverse publicity. Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing clinical laboratory licensure or our failure to renew our CLIA certificate, a state or foreign license or accreditation, could have a material adverse effect on our business, financial condition and results of operations. Even if we were able to bring our laboratory back into compliance, we could incur significant expenses and potentially lose revenue in doing so.
Our Boulder, Colorado and De Soto, Kansas laboratories are both CAP-accredited clinical laboratories regulated by CMS pursuant to CLIA. We also have a current CLIA certificate for each facility. To maintain these certificates, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make random inspections of our laboratory from time to time. Furthermore, our diagnostic tests are categorized as LDTs and are not currently subject to FDA regulation, although certain components provided by third parties and used to create and/or administer the test may be. LDTs are a subset of IVDs that are intended for clinical use and developed, validated, and offered within a single laboratory for use only in that laboratory. The FDA’s authority to regulate LDTs has been frequently contested, and HHS recently issued a public statement purporting to rescind the FDA’s policies regarding the premarket review of LDTs. According to the HHS statement, the FDA will not require premarket review of LDTs unless it engages in notice-and-comment rulemaking. There is no guarantee, however, that the HHS statement will not be revised or rescinded, that legislation reforming the federal government’s regulation of LDTs will not be passed, or that LDTs will otherwise continue to be able to operate without first receiving FDA premarket review. Failure to adhere to any new FDA regulation would result in fines, product suspensions, warning letters, recalls, injunctions and other civil and criminal penalties.
Changes in the way that the FDA regulates tests performed by laboratories like ours could result in delay or additional expense in offering our tests and tests that we may develop in the future.
Historically, the FDA has exercised enforcement discretion with respect to most LDTs and has not required laboratories that furnish LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, premarket clearance or premarket approval, and post-market controls). In recent years, however, the FDA publicly announced its intention to regulate certain LDTs and issued two draft guidance documents that set forth a proposed phased-in risk-based regulatory framework that would apply varying levels of FDA oversight to LDTs. However, these guidance documents were withdrawn at the end of the Obama administration and replaced by an informal discussion paper reflecting some of the feedback that the FDA had received on LDT regulation. The FDA acknowledged that the discussion paper in January 2017 does not represent the formal position of the FDA and is not enforceable. Nevertheless, the FDA wanted to share its synthesis of the feedback that it had received in the hope that it might advance public discussion on future LDT oversight. HHS has since issued a statement purporting to rescind FDA’s policies regarding the premarket review of LDTs, stating that FDA must engage in notice and comment rulemaking (as opposed to through guidance documents, compliance manuals, website statements, or other informal issuances) prior to requiring premarket review of LDTs. HHS issued this statement on its website and it may be subject to change. Also, it is possible that Congress will pass legislation to reform
60
the federal government’s regulation of LDTs or that FDA will engage in notice-and-comment rulemaking to require premarket review of LDTs, which could result in delay or additional expense in offering our tests and tests that we may develop in the future.
Our current line of diagnostic tests is covered under CLIA and CMS, although our COVID testing program and select partnerships we may enter may cause us to be subject to additional FDA requirements.
The laws and regulations governing the marketing of diagnostic products are evolving, extremely complex and in many instances, there are no significant regulatory or judicial interpretations of these laws and regulations. Pursuant to its authority under the FDCA, the FDA has jurisdiction over medical devices, including in vitro diagnostics and, therefore, potentially our clinical laboratory tests.
Pursuant to the FDCA and its implementing regulations, the FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, premarket clearance or approval, marketing and promotion, and sales and distribution of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. Although the FDA has asserted that it has authority to regulate the development and use of LDTs, such as our and many other laboratories’ tests, as medical devices, it has generally exercised enforcement discretion and is not otherwise regulating most tests developed and performed within a single high complexity CLIA-certified laboratory. On August 19, 2020, HHS issued a statement purporting to rescind the FDA’s policies regarding the premarket review of LDTs, absent notice-and-comment rulemaking. the FDA’s policies to date have been articulated through guidance documents, compliance manuals, website statements, and other informal issuances. The FDA could, at any time, engage in notice-and-comment rulemaking, or Congress could take action to amend the law to change the current regulatory framework for in vitro diagnostics and LDTs. Further, the HHS statement was issued on its website and may be subject to change, particularly given the reasoning that additional flexibility is needed due to the COVID-19 pandemic.
We believe that our tests, as utilized in our clinical laboratory, are and would be considered LDTs and that as a result, the FDA does not require that we obtain regulatory clearances or approvals for our LDTs or their components pursuant to the FDA’s current policies and guidance. Although we believe that our tests and test components are either exempt from FDA medical device regulations or are subject to an enforcement discretion policy, it is possible that the FDA would not agree with our determinations or that the FDA will change its regulations and policies such that our products become regulated as medical devices.
In contrast with our LDTs, the FDA has regulatory jurisdiction over the two FDA EUA-authorized COVID-19 tests that were developed by Bio-Rad, which we offer as part of our Biodesix WorkSafe testing programs.
Our operations, therefore, are or may become subject to extensive regulation by the FDA in the United States. Government regulations specific to medical devices are wide ranging and govern, among other things:
The FDA classifies medical devices into one of three classes on the basis of the intended use of the device, the risk associated with the use of the device for that indication, as determined by the FDA, and on the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices, which have the lowest level of risk associated with them, are subject to general controls. Class II devices are subject to general controls and special controls, including performance standards. Class III devices, which have the highest level of risk associated with them, are subject to general controls and premarket approval.
Before a new medical device or service, or a new intended use for an existing product or service, can be marketed in the United States, a company must first submit and receive either 510(k) clearance, de-novo authorization, or premarket approval (PMA) from the FDA, unless an exemption applies. Most Class I devices and some Class II devices are exempt from these requirements. In the 510(k) clearance process, before a device may be marketed, the FDA must determine that a proposed device is substantially equivalent to a legally-marketed predicate device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (preamendments device), a device that was originally on the United States market pursuant to an approved PMA and later down-classified, or a 510(k)-exempt device. To be substantially equivalent, the proposed device must have the
61
same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence.
In the process of obtaining PMA approval, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices.
The FDA also allows the submission of a direct de-novo petition. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA), a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent. FDASIA streamlined the de novo classification pathway by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination.
The 510(k), de-novo or PMA process can be expensive, lengthy and unpredictable. The FDA can delay, limit, or deny clearance or approval of a device for many reasons, including:
The FDA and state authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by any such agency, which may include any of the following sanctions:
As discussed above, we believe that our current line of diagnostic tests and their components are LDTs, subject to state licensing requirements and federal regulation by CMS under CLIA, although our COVID-19 testing program and select partnerships we may enter may cause us to be subject to additional FDA regulations discussed above.
While we believe that we are currently in material compliance with applicable laws and regulations, it is possible that the FDA, or other regulatory agencies, would not agree with our determinations. If our products became become subject to 510(k) or other similar FDA regulations, we would need to comply with the applicable regulations or face significant civil and criminal penalties. In addition, IVDs and CDx tests are widely considered to be Class III devices, and it is possible that in the future, we may develop tests that fall into this category. CDx tests in particular may require further administrative procedures in the PMA process. Exposure to these additional regulatory requirements would also affect our business, financial condition and results of operations.
62
Our future success depends on our ability to develop, receive regulatory clearance or approval or certification for, and introduce new diagnostic tests or enhancements to existing diagnostic tests that will be accepted by the market in a timely manner. There is no guarantee that the FDA will grant 510(k) clearance or PMA approval of our future diagnostic tests and failure to obtain necessary clearances or approvals for our future diagnostic tests would adversely affect our ability to grow our business.
It is important to our business that we build a pipeline of diagnostic test offerings that address limitations of current lung disease diagnostic tests. As such, our success will depend in part on our ability to develop and introduce new diagnostic tests. However, we may not be able to successfully develop and obtain regulatory clearance or approval or certification for enhancements to our existing diagnostic tests, or new diagnostic tests for any number of reasons, including due to the cost associated with certain regulatory approval requirements, or these diagnostic tests may not be accepted by physicians or users.
The success of any new diagnostic test or enhancement to an existing diagnostic test will depend on a number of factors, including our ability to, among others:
If we do not develop new diagnostic tests or enhancements to our existing diagnostic tests in time to meet market demand or if there is insufficient demand for these diagnostic tests or enhancements, or if our competitors introduce new diagnostic tests with functionalities that are superior to ours, our results of operations will suffer.
Some of our future diagnostic tests may require FDA clearance of a 510(k) submission. Other diagnostic tests may require the approval of a PMA. In addition, some of our future diagnostic tests may require clinical trials to support regulatory approval and we may not successfully complete these clinical trials. The FDA may not approve or clear these diagnostic tests for the indications that are necessary or desirable for successful commercialization. Indeed, the FDA may refuse our requests for 510(k) clearance or premarket approval of new diagnostic tests. Failure to receive clearance or approval for our new diagnostic tests would have an adverse effect on our ability to expand our business.
Modifications to our marketed tests may require new 510(k) clearances or PMA approvals, or may require us to cease marketing or recall the modified tests until clearances or approvals are obtained.
Modifications to our diagnostic tests may require new regulatory approvals or clearances, including 510(k) clearances or premarket approvals, or require us to recall or cease marketing the modified systems until these clearances or approvals are obtained. The FDA requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance. A manufacturer may determine that a modification could not significantly affect safety or efficacy and does not represent a major change in its intended use, so that no new 510(k) clearance is necessary. However, the FDA can review a manufacturer’s decision and may disagree. The FDA may also on its own initiative determine that a new clearance or approval is required. We have made modifications to our diagnostic tests in the past and may make additional modifications in the future that we believe do not or will not require additional clearances or approvals. If the FDA disagrees and requires new clearances or approvals for the modifications, we may be required to recall and to stop marketing our diagnostic tests as modified, which could require us to redesign our diagnostic tests and harm our operating results. In these circumstances, we may be subject to significant enforcement actions.
If a manufacturer determines that a modification to an FDA-cleared device could significantly affect its safety or efficacy, or would constitute a major change in its intended use, then the manufacturer must file for a new 510(k) clearance or possibly a premarket approval application. Where we determine that modifications to our diagnostic tests require a new 510(k) clearance or premarket approval application, we may not be able to obtain those additional clearances or approvals for the modifications or additional indications in a timely manner, or at all. Obtaining clearances and approvals can be a time-consuming process, and delays in obtaining required future clearances or approvals would adversely affect our ability to introduce new or enhanced diagnostic tests in a timely manner, which in turn would harm our future growth.
63
If we or our suppliers fail to comply with ongoing FDA or other domestic and foreign regulatory authority or conformity assessment body requirements, or if we experience unanticipated problems with our diagnostic tests, they could be subject to restrictions or withdrawal from the market.
Any medical device that we manufacture, including those for which we obtain regulatory clearance or approval or certification, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such diagnostic test, will be subject to continued regulatory review, oversight, and periodic inspections by the FDA and other domestic and foreign regulatory bodies or conformity assessment bodies. In particular, we and our suppliers may be required to comply with FDA’s Quality System Regulations (QSR codified at 21 C.F.R. § 820) for medical devices and ISO regulations for the manufacture of our diagnostic tests and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any diagnostic test for which we obtain clearance or approval. Regulatory bodies, such as the FDA, and conformity assessment bodies enforce the QSR and other regulations through periodic inspections and audits. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA and other regulatory bodies or conformity assessment bodies, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in, among other things, one or more of the following enforcement actions:
If any of these actions were to occur it would harm our reputation and cause our diagnostic test sales and profitability to suffer and may prevent us from generating revenue. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements which could result in our failure to produce our diagnostic tests on a timely basis and in the required quantities, if at all.
In addition, we are required to conduct surveillance to monitor the safety or effectiveness of our diagnostic tests, and we must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to our diagnostic tests. Later discovery of previously unknown problems with our diagnostic tests, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such diagnostic tests or manufacturing processes, withdrawal of the diagnostic tests from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
Our diagnostic tests and services may in the future be subject to product recalls that could harm our reputation, business and financial results.
Medical devices can experience performance problems in the field that require review and possible corrective action. The occurrence of component failures, manufacturing errors, software errors, design defects or labeling inadequacies affecting a medical device could lead to a government-mandated or voluntary recall by the device manufacturer, in particular when such deficiencies may endanger health. The FDA requires that certain classifications of recalls be reported to the FDA within 10 working days after the recall is initiated. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls involving our diagnostic tests and services in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. Product recalls may divert management attention and financial resources, expose us to product liability or other claims, harm our reputation with customers and adversely impact our business, financial condition and results of operations. Other jurisdictions have similar recall requirements.
64
Legislative or regulatory reforms may make it more difficult and costly for us to obtain regulatory clearance or approval or certification of any future diagnostic tests and to manufacture, market and distribute our diagnostic tests after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. For example, the Verifying Accurate, Leading-edge IVCT Development (VALID) Act recently introduced in Congress would codify into law the term “in vitro clinical test” in order to create a new medical product category separate from medical devices that would include products currently regulated as in vitro diagnostics as well as LDTs.
In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our diagnostic tests. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of planned or future diagnostic tests. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be.
Any change in the laws or regulations that govern the clearance and approval processes relating to our current, planned and future diagnostic tests could make it more difficult and costly to obtain clearance or approval for new diagnostic tests or to produce, market and distribute existing diagnostic tests. Significant delays in receiving clearance or approval or the failure to receive clearance or approval for any new diagnostic tests would have an adverse effect on our ability to expand our business.
Clinical trials may be necessary to support future product submissions to FDA. These clinical trials are expensive and will require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Delays or failures in our clinical trials will prevent us from commercializing any modified or new diagnostic tests and will adversely affect our business, operating results and prospects.
Initiating and completing clinical trials necessary to support any future PMA applications, and additional safety and efficacy data beyond that typically required for a 510(k) clearance, for our possible future product candidates, will be time consuming and expensive and the outcome uncertain. Moreover, the results of early clinical trials are not necessarily predictive of future results, and any product we advance into clinical trials may not have favorable results in later clinical trials.
Conducting successful clinical studies will require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population, the nature of the trial protocol, the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects, the availability of appropriate clinical trial investigators, support staff, and proximity of patients to clinical sites and ability to comply with the eligibility and exclusion criteria for participation in the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and effectiveness of our diagnostic tests or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts.
Development of sufficient and appropriate clinical protocols to demonstrate safety and efficacy are required and we may not adequately develop such protocols to support clearance and approval. Further, the FDA may require us to submit data on a greater number of patients than we originally anticipated and/or for a longer follow-up period or change the data collection requirements or data analysis applicable to our clinical trials. Delays in patient enrollment or failure of patients to continue to participate in a clinical trial may cause an increase in costs and delays in the approval and attempted commercialization of our diagnostic tests or result in the failure of the clinical trial. In addition, despite considerable time and expense invested in our clinical trials, the FDA may not consider our data adequate to demonstrate safety and efficacy. Such increased costs and delays or failures could adversely affect our business, operating results and prospects.
If the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical development do not perform as contractually required or expected, we may not be able to obtain regulatory clearance or approval or certification for or commercialize our diagnostic tests and services.
We may not have the ability to independently conduct our pre-clinical and clinical trials for our future diagnostic tests and services and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct such trials. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, our diagnostic tests and services on a timely basis, if at all, and our business, operating results and prospects may be adversely affected. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control.
65
Our use, disclosure, and other processing of personally identifiable information, including health information, is subject to HIPAA and other federal, state, and foreign privacy and security regulations, and our failure to comply with those regulations or to adequately secure the information we hold could result in significant liability or reputational harm and, in turn, a material adverse effect on our business, operating results and prospects.
We maintain and process, and our third-party vendors, collaborators, contractors and consultants maintain and process on our behalf, a large quantity of sensitive information, including confidential business, personal and patient health information in connection with our clinical studies and our employees, and are subject to data privacy and protection laws and regulations that apply to the collection, transmission, storage and use of personally identifying information, which among other things, impose certain requirements relating to the privacy, security and transmission of personal information. Failure by us or our third-party vendors, collaborators, contractors and consultants to comply with any of these laws and regulations could result in notification obligations or enforcement actions against us, which could result in fines, imprisonment of company officials and public censure, claims for damages by affected individuals, damage to our reputation and loss of goodwill, any of which could have a material adverse effect on our business, financial condition, results of operations or prospects. These laws, rules and regulations evolve frequently and their scope may continually change, through new legislation, amendments to existing legislation and changes in enforcement, and may be inconsistent from one jurisdiction to another. The interpretation and application of consumer, health-related and data protection laws, especially with respect to genetic samples and data, in the United States, the European Union (EU) and elsewhere, are often uncertain, contradictory and in flux. As a result, implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future.
In the United States, numerous federal and state laws and regulations, including federal health information privacy laws, state data breach notification laws, state health information privacy laws and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our collaborators.
Domestic laws in this area are complex and developing rapidly. Many state legislatures have adopted legislation relating to privacy, data security and data breaches. Laws in all 50 states require businesses to provide notice to customers whose personally identifiable information has been disclosed as a result of a data breach. The laws are not consistent, and compliance in the event of a widespread data breach is costly. States are also frequently amending existing laws, requiring attention to frequently changing regulatory requirements. For example, California recently enacted the CCPA, which became effective on January 1, 2020. The CCPA, among other things, requires new disclosures to California consumers and affords such consumers new abilities to access and delete their personal information, opt-out of certain sales of personal information and receive detailed information about how their personal information is used. The CCPA provides for fines of up to $7,500 per violation, as well as a private right of action for data breaches that is expected to increase the frequency of data breach litigation. While the CCPA has already been amended multiple times, it is unclear how this legislation will be further modified or how it will be interpreted. Interpretations of the CCPA may continue to evolve with regulatory guidance and the CCPA continue to be amended, including through a ballot initiative, the CPRA. That passed in November 2020. The CPRA will impose additional data protection obligations on companies doing business in California, including additional consumer rights, including regarding certain uses of sensitive data. It also creates a new California data protection agency specifically tasked to enforce the law, which may likely result in increased regulatory scrutiny of California businesses in the areas of data protection and security. The effects of this legislation potentially are far-reaching, however, and may require us to modify our data processing practices and policies and incur substantial compliance-related costs and expenses. The CCPA and other changes in state and federal laws or regulations relating to privacy, data protection and information security, particularly any new or modified laws or regulations that require enhanced protection of certain types of data or new obligations with regard to data retention, transfer or disclosure, could increase the cost of providing our offerings, require significant changes to our operations or even prevent us from providing certain offerings in jurisdictions in which we currently operate and in which we may operate in the future.
Because of the breadth of these data protection laws and the narrowness of their exceptions and safe harbors, it is possible that our business or data protection policies could be subject to challenge under one or more of such laws. The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of heightened regulatory focus on data privacy and security issues. Although we endeavor to comply with our published policies and documentation and ensure their compliance with current laws, rules and regulations, we may at times fail to do so or be alleged to have failed to do so. The publication of our privacy policy and other documentation that provide promises and assurances about privacy and security can subject us to potential state and federal action in the United States if they are found to be deceptive, unfair, or misrepresentative of our actual practices. Any failure by us or other parties with whom we do business to comply with this documentation or with federal, state, local or international regulations could result in proceedings against us by governmental entities, private parties or others. In many jurisdictions, enforcement actions and consequences for noncompliance are rising.
If our operations are found to be in violation of any of the data protection laws described above or any other laws that apply to us, we may be subject to penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, individual imprisonment, possible exclusion from participation in government healthcare programs, injunctions, private qui tam actions brought by individual whistleblowers in the name of the government, class action litigation and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corrective action plan or other agreement
66
to resolve allegations of non-compliance with these laws, any of which could adversely affect our ability to operate our business and our results of operations.
In addition, numerous state and federal laws and regulations govern the collection, dissemination, use, privacy, confidentiality, security, availability, integrity, and other processing of PHI and PII. These laws and regulations include HIPAA. HIPAA establishes a set of national privacy and security standards for the protection of protected health information (as defined in HIPAA, PHI) by health plans, healthcare clearinghouses and certain healthcare providers, referred to as covered entities, and the business associates with whom such covered entities contract for services. We are a covered entity under HIPAA when we are conducting our clinical trials. We are a covered entity with regard to our observational studies and clinical trials, and also a business associate under HIPAA for certain other business activities, and we execute business associate agreements with our clients.
HIPAA requires covered entities and business associates, such as us, to develop and maintain policies with respect to the protection of, use and disclosure of electronic PHI, including the adoption of administrative, physical and technical safeguards to protect such information, and certain notification requirements in the event of a data breach.
HIPAA imposes mandatory penalties for certain violations. Penalties for violations of HIPAA and its implementing regulations start at $119 per violation and are subject to a cap of $1,785,651 for violations of the same standard in a single calendar year. However, a single breach incident can result in violations of multiple standards. HIPAA also authorizes state attorneys general to file suit on behalf of their residents. Courts may award damages, costs and attorneys’ fees related to violations of HIPAA in such cases. While HIPAA does not create a private right of action allowing individuals to sue us in civil court for violations of HIPAA, its standards have been used as the basis for duty of care in state civil suits such as those for negligence or recklessness in the misuse or breach of PHI.
In addition, HIPAA mandates that the Secretary of HHS conduct periodic compliance audits of HIPAA covered entities and business associates. With regard to business associates, those audits assess the business associate’s compliance with the HIPAA Privacy and Security Standards. Such audits are conducted randomly and after an entity experiences a breach affecting more than 500 individuals’ data. Undergoing an audit can be costly, can result in fines or onerous obligations, and can damage a business associate’s reputation.
In addition to HIPAA, numerous other federal, state, and foreign laws and regulations protect the confidentiality, privacy, availability, integrity and security of PHI and other types of PII. Some of these laws and regulations may be preempted by HIPAA with respect to PHI, or may exclude PHI from their scope but impose obligations with regard to PII that is not PHI, and in some cases, can impose additional obligations with regard to PHI. These laws and regulations are often uncertain, contradictory, and subject to changing or differing interpretations, and we expect new laws, rules and regulations regarding privacy, data protection, and information security to be proposed and enacted in the future. HHS is also proposing amendments to the HIPPA Privacy Rule to modernize certain data sharing provisions and enhance patient access to their information. This complex, dynamic legal landscape regarding privacy, data protection, and information security creates significant compliance issues for us and our clients and potentially exposes us to additional expense, adverse publicity and liability. While we have implemented data privacy and security measures in an effort to comply with applicable laws and regulations relating to privacy and data protection, some PHI and other PII or confidential information is transmitted to us by third parties, who may not implement adequate security and privacy measures, but it is possible that laws, rules and regulations relating to privacy, data protection, or information security may be interpreted and applied in a manner that is inconsistent with our practices or those of third parties who transmit PHI and other PII or confidential information to us. If we or these third parties are found to have violated such laws, rules or regulations, it could result in government-imposed fines, orders requiring that we or these third parties change our or their practices, or criminal charges, which could adversely affect our business.
Complying with these various laws and regulations could cause us to incur substantial costs or require us to change our business practices, systems and compliance procedures in a manner adverse to our business.
We may eventually operate in a number of countries outside of the United States whose laws, including data privacy laws, may in some cases be more stringent than the requirements in the United States. For example, EU and UK data privacy laws have specific requirements relating to cross-border transfers of personal data to certain jurisdictions, including to the United States, have strict requirements relating to personal data collection, use or sharing, and have more stringent requirements relating to organizations’ privacy programs and provide stronger individual rights. Moreover, we may also be subject to evolving international privacy and data security regulations which could result in greater compliance costs and in turn lead to penalties, where such compliance programs are not implemented correctly.
Certain of our processing activities are subject to the EU General Data Protection Regulation and the UK General Data Protection Regulation (collectively, the “GDPR”) – including, those involving pseudonymised / key-coded data - as the GDPR applies extra-territorially. The GDPR imposes strict requirements on controllers and processors processing personal data, including, for example, requirements to: (i) identify a legal basis for the processing of personal data, (ii) provide robust disclosures to individuals, (iii) respond to requests from individuals to exercise their data subject rights, (iv) provide personal data breach notifications within 72 hours after discovering the breach, (v) limit the collection and retention of personal data, (vi) impose specific contractual obligations on processors engaged to process personal data on the instructions of the controller, and (vii) apply enhanced protections to health data and other special categories of personal data.
67
The EU GDPR also provides that EU Member States may make their own further laws and regulations limiting the processing of personal data, including genetic, biometric or health data, which could limit our ability to use and share such personal data and cause our costs to increase and harm our financial condition.
Failure to comply with the requirements of the GDPR may result in fines of up to €20 million (£17.5 million in the case of the UK GDPR) or up to 4% of the total worldwide annual turnover of our preceding fiscal year, whichever is higher, and other administrative penalties. GDPR compliance may require us to put in place additional mechanisms, which may result in compliance costs and other substantial expenditures. This may be onerous and adversely affect our business, financial condition, results of operations and the profitability of our platform of diagnostic tests. Failure to comply with the GDPR and other countries’ privacy or data security-related laws, rules or regulations could result in material penalties imposed by regulators, affect our compliance with contracts entered into with our collaborators and other third-party payers, and have an adverse effect on our business and financial condition. Currently, the GDPR is only applicable to us as a processor, but as we continue to expand into the European market, the GDPR will have direct applicability to us as a controller.
The GDPR also prohibits the transfer of personal data from the EEA/UK to a country outside of the EEA/UK (e.g., the United States) unless made to a country deemed to have adequate data privacy laws by the European Commission (or UK Government in case of the UK GDPR) or a data transfer mechanism has been put in place. Until recently, one such data transfer mechanism was the EU-US Privacy Shield. However, in July 2020 the Court of Justice of the European Union (CJEU) declared the Privacy Shield to be invalid. The CJEU upheld the validity of standard contractual clauses (SCCs) as a legal mechanism to transfer personal data but companies relying on SCCs will need to carry out a transfer privacy impact assessment, which among other things, assesses laws governing access to personal data in the recipient country and considers whether supplementary measures that provide privacy protections additional to those provided under SCCs will need to be implemented to ensure an essentially equivalent level of data protection to that afforded in the EEA. In turn, the findings of the CJEU will have significant implications for cross-border data flows and may lead to increased transaction, compliance, and technological costs to support international data transfers.
Organizations operating in Canada and covered by the Personal Information Protection and Electronic Documents Act (PIPEDA), or equivalent Canadian provincial laws, must obtain an individual’s consent when they collect, use or disclose that individual’s personal information. Individuals have the right to access and challenge the accuracy of their personal information held by an organization, and personal information may only be used for the purposes for which it was collected. If an organization intends to use personal information for another purpose, it must again obtain that individual’s consent.
We regularly monitor, defend against and respond to attacks to our networks and other information security incidents. Despite our information security efforts, our facilities, systems, and data, as well as those of our third-party service providers, may be vulnerable to privacy and information security incidents such as data breaches, viruses or other malicious code, coordinated attacks, data loss, phishing attacks, ransomware, denial of service attacks, or other security or IT incidents caused by threat actors, technological vulnerabilities or human error. If we, or any of our vendors that support our IT or have access to our data, including any third-party vendors that collect, process and store personal data on our behalf, fail to comply with laws requiring the protection of personal information, or fail to safeguard and defend personal information or other critical data assets or IT systems, we may be subject to regulatory enforcement and fines as well as private civil actions. We may be required to expend significant resources in the response, containment, mitigation of cybersecurity incidents as well as in defense against claims that our information security was unreasonable or otherwise violated applicable laws or contractual obligations.
Our employees, collaborators, independent contractors and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, collaborators, independent contractors and consultants may engage in fraudulent or other illegal activity with respect to our business. Misconduct by these employees could include intentional, reckless and/or negligent conduct or unauthorized activity that violates:
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by these parties could also involve individually identifiable information, including, without limitation, the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. Any incidents or any other conduct that leads to an employee, contractor, or other agent, or our company, receiving an FDA debarment or exclusion by the HHS Office of Inspector General (OIG) could result in penalties, a loss of business from third parties, and severe reputational harm.
68
We have adopted a Code of Business Conduct and Ethics and compliance policies to govern and deter such behaviors, but it is not always possible to identify and deter misconduct by our employees and other agents, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, treble damages, monetary fines, disgorgement, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and curtailment of our operations.
Clinical development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Our ongoing research and development and clinical trial activities are subject to extensive regulation and review by numerous governmental authorities both in the United States and abroad. We are currently conducting pre-and post-market clinical studies of some of our tests. In the future we may conduct clinical trials to support approval of new diagnostic tests and services, or new indications. Clinical studies may need to be conducted in compliance with FDA regulations or the FDA may take enforcement action. The data collected from these clinical studies may ultimately be used to support marketing authorization for these diagnostic tests and services. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product candidate claims or that the FDA or foreign authorities and conformity assessment bodies will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our tests are safe and effective for the proposed indicated uses, which could cause us to abandon development of our tests and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, may impact our ability to commercialize our tests and generate revenues.
Many of the factors that may cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to delay or denial of regulatory clearance or approval or certification. The commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial.
We may find it necessary to engage contract research organizations to perform data collection and analysis and other aspects of our clinical trials, which might increase the cost and complexity of our trials. We may also depend on clinical investigators, medical institutions, and contract research organizations to perform the trials, and would control only certain aspects of their activities. Nevertheless, we would be responsible for ensuring that each of our trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on these third parties would not relieve us of our regulatory responsibilities. We and our third-party contractors are required to comply with good clinical practices (GCPs) which are regulations and guidelines enforced by the FDA, and comparable regulations enforced by foreign regulatory authorities for products in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any third-party contractor fails to comply with applicable GCPs, the clinical data generated in clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities or conformity assessment bodies may require us to perform additional clinical trials before clearing or approving our marketing applications. A failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory clearance or approval or certification process. In addition, if these parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality, completeness or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our clinical trials may have to be extended, delayed or terminated.
Many of these factors could be beyond our control. We may not be able to undertake additional trials, repeat trials or enter into new arrangements with third parties without undue delays or considerable expenditures. If there are delays in testing or clearances or approvals as a result of the failure to perform by third parties, our research and development costs would increase and we may not be able to obtain regulatory clearance or approval or certification for our tests. In addition, we may not be able to establish or maintain relationships with these parties on favorable terms, if at all. Each of these outcomes would harm our ability to market our tests, or to achieve sustained profitability.
The results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
We cannot be certain that the results of our future clinical trials will support our future product claims or that the FDA or comparable foreign regulatory authorities or conformity assessment bodies will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to
69
commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the future product’s profile.
Our billing, collections and claims processing activities are complex and time-consuming, and any delay in transmitting and collecting claims or failure to comply with applicable billing requirements, could have an adverse effect on our future revenue.
Billing for our tests is complex, time-consuming and expensive. Depending on the billing arrangement and applicable law, we bill various payers, such as government payers, insurance companies and patients, all of which may have different billing requirements. We may face increased risk in our collection efforts, including long collection cycles and the risk that we may never collect at all, either of which could adversely affect our business, financial condition and results of operations. Several factors make the billing process complex, including:
These billing complexities and the related uncertainty in obtaining payment for our tests could negatively affect our revenue and cash flow, our ability to achieve profitability and the consistency and comparability of our results of operations. In addition, if claims for our tests are not submitted to payers on a timely basis, or if we fail to comply with applicable billing requirements, it could have an adverse effect on our revenue and our business.
Third-party payers require us to identify the test for which we are seeking reimbursement using a Current Procedural Terminology (CPT) code. The CPT code set is maintained by the American Medical Association (AMA). In cases where there is not a specific CPT code to describe a test, such as with Nodify CDT and GeneStrat, the test may be billed under an unlisted molecular pathology procedure code or through the use of a combination of single gene CPT codes, depending on the payer. The Protecting Access to Medicare Act of 2014 (PAMA) authorized the adoption of new, temporary billing codes and unique test identifiers for FDA-cleared or approved tests as well as advanced diagnostic laboratory tests. The AMA has created a new section of CPT codes, Proprietary Laboratory Analyses codes to facilitate implementation of this section of PAMA. In addition, CMS may assign unique level II Healthcare Common Procedure Coding System codes to tests that are not already described by a unique CPT code. VeriStrat and Nodify XL2 both have test specific CPT codes, but the GeneStrat and Nodify CDT tests do not at this time.
In the instance where a code used does not describe a specific test, the insurance claim must be examined to determine what test was provided, whether the test was appropriate and medically necessary, and whether payment should be rendered, which may require a letter of medical necessity from the ordering physician. This process can result in a delay in processing the claim, a lower reimbursement amount or denial of the claim. As a result, obtaining approvals from third-party payers to cover our tests and establishing adequate reimbursement levels is an unpredictable, challenging, time-consuming and costly process and we may never be successful.
We and our third-party manufacturers and suppliers must comply with environmental, health and safety laws and regulations, which can be expensive and restrict how we do, or interrupt our, business.
Our research and development activities and our third-party manufacturers’ and suppliers’ activities involve the generation, use, storage and disposal of hazardous materials. We work with materials, including chemicals, biological agents and compounds and samples that could be hazardous to human health and safety or the environment. Our operations also produce hazardous and biological waste products. Accordingly, we and our third-party manufacturers and suppliers are subject to federal, state, local and foreign environmental, health and safety laws and regulations, and permitting and licensing requirements, including those governing the generation, use, manufacture, storage, handling, transportation, release and disposal of, and exposure to, these materials, and worker health and safety.
We cannot eliminate the risk of contamination or injury resulting from such hazardous materials. We also cannot guarantee that the procedures utilized by our third-party manufacturers for handling and disposing of hazardous materials and wastes comply with all applicable environmental, health and safety laws and regulations. As a result, we may be held liable for any resulting damages, costs or liabilities, including cleanup costs and liabilities, which could be significant, or our commercialization, research and development efforts and business operations may be restricted or interrupted.
Environmental, health and safety laws and regulations are complex, change frequently and have tended to become more stringent. Compliance with such laws and regulations is expensive, and current or future environmental, health and safety laws and regulations
70
may restrict our operations. If we do not comply with applicable environmental health and safety laws and regulations, and permitting and licensing requirements, we may be subject to fines, penalties, a suspension of our business or other sanctions.
Risks Related to our Intellectual Property
Our success may be impaired if we are unable to obtain, maintain and protect our intellectual property rights.
Our commercial success will depend in part on our ability to obtain and maintain patent and other intellectual property protection in the United States and other countries with respect to our diagnostic tests, products and services and technology. We rely on patent protection, as well as a combination of copyright, trade secret and trademark laws, to protect our proprietary technology and prevent others from duplicating our suite of diagnostic tests and products. However, these means may afford only limited protection and may not:
Any of our patents, including those we may license, may be challenged, invalidated, rendered unenforceable or circumvented. Consequently, we do not know whether any of our diagnostic tests, products and services will be protectable or remain protected by valid and enforceable patents. We may not prevail if our patents are challenged by competitors or other third parties. The United States federal courts or equivalent national courts or patent offices elsewhere may invalidate our patents, find them unenforceable, or narrow their scope. Furthermore, competitors may be able to design around our patents by developing similar or alternative technologies or products in a non-infringing manner, or obtain patent protection for more effective technologies, designs or methods, including for treating lung cancer. If these developments were to occur, our diagnostic tests and products may become less competitive and sales may decline.
We have filed numerous patent applications seeking protection of diagnostic tests and other inventions originating from our research and development. Our patent applications may not result in issued patents, and any patents that are issued may not provide meaningful protection against competitors or competitive technologies. Further, the examination process may require us to narrow the claims for our pending patent applications, which may limit the scope of patent protection that may be obtained if these applications issue. The scope of a patent may also be reinterpreted and significantly reduced after issuance. Even if patent applications we license or own currently or in the future issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with the protection or competitive advantages we are seeking.
Moreover, some of our owned and in-licensed patents and patent applications are, and may in the future be, co-owned with third parties. If we are unable to obtain or maintain an exclusive license to any such third-party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing products and technology. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us. Any of the foregoing could have a material adverse effect on our competitive position, business, financial condition, results of operations, and prospects.
The patent position of biotechnology companies generally is highly uncertain, involves complex legal and factual questions, and has been the subject of much litigation in recent years. No consistent policy regarding the breadth of claims allowed in biotechnology and pharmaceutical patents has emerged to date in the United States or in many foreign jurisdictions. In addition, the determination of patent rights with respect to pharmaceutical compounds and technologies commonly involves complex legal and factual questions, which has in recent years been the subject of much litigation. Various courts, including the United States Supreme Court have rendered decisions that affect the scope of patentability of certain inventions or discoveries relating to biotechnology. These decisions state, among other things, that a patent claim that recites an abstract idea, natural phenomenon, or law of nature (for example, the relationship between particular genetic variants and cancer) are not themselves patentable. Precisely what constitutes a law of nature or abstract idea is uncertain, and it is possible that certain aspects of our technology could be considered unpatentable under applicable law. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. Depending on decisions by the United States Congress, the federal courts and the United States Patent and Trademark Office (USPTO), the laws and regulations governing patents could change in unpredictable ways that would weaken our and our licensors’ ability to obtain new patents or to enforce our existing owned or in-licensed patents and patents that we might obtain or in-license in the future. Additionally, our pending and future patent applications may not result in patents being issued which protect our technology or products or which effectively prevent others from commercializing competitive technologies and products. The scope of patent protection outside of the United States is also uncertain. Changes in either the patent laws or their interpretation in the United States and other countries may diminish our
71
ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property rights or narrow the scope of our owned and licensed patents.
If we are unable to obtain and maintain patent protection for our technology, or if the scope of the patent protection obtained is not sufficient, our competitors could develop and commercialize diagnostic tests, products and services similar or superior to ours, and our competitive position may be adversely affected. It is also possible that we will fail to identify patentable aspects of inventions made in the course of our development and commercialization activities before it is too late to obtain patent protection on them. Therefore, we may miss potential opportunities to strengthen our patent position. In addition, the patent prosecution process is expensive, time-consuming and complex, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patent applications at a reasonable cost or in a timely manner. Although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, consultants, advisors and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
Additionally, while software and other of our proprietary works may be protected under copyright law, we have chosen not to register any copyrights in these works, and instead, primarily rely on protecting our software as a trade secret. In order to bring a copyright infringement lawsuit in the United States, the copyright must be registered. Accordingly, the remedies and damages available to us for unauthorized use of our copyrights may be limited.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position may be harmed.
In addition to seeking patent protection for the patents underlying our diagnostic tests, products and services, we also rely upon unpatented trade secrets, know-how and continuing technological innovation to develop and maintain a competitive position. Trade secrets and know-how can be difficult to protect. We seek to protect such proprietary information, in part, through confidentiality agreements with our employees, collaborators, contractors, advisors, consultants and other third parties and invention assignment agreements with our employees. We also have agreements with some of our consultants that require them to assign to us any inventions created as a result of their working with us. The confidentiality agreements are designed to protect our proprietary information and, in the case of agreements or clauses containing invention assignment, to grant us ownership of technologies that are developed through a relationship with employees or third parties.
We cannot guarantee that we have entered into such agreements with each party that has or may have had access to our trade secrets or proprietary information. Additionally, despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to, or independently developed by, a competitor or other third party, our competitive position would be materially and adversely harmed. Furthermore, we expect these trade secrets, know-how and proprietary information to over time be disseminated within the industry through independent development, the publication of journal articles describing the methodology and the movement of personnel from academic to industry scientific positions. Consequently, we may be unable to prevent our proprietary technology from being exploited in the United States and abroad, which could affect our ability to expand in domestic and international markets or require costly efforts to protect our technology.
We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known, or be independently discovered by, competitors. To the extent that our employees, consultants, contractors or collaborators use intellectual property rights owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions, which could have a material adverse effect on our business, financial condition and results of operations.
We may be subject to claims that we or our employees have misappropriated the intellectual property rights of a third party, including trade secrets or know-how, or are in breach of non-competition or non-solicitation agreements with our competitors, and third parties may claim an ownership interest in intellectual property we regard as our own.
Many of our employees and consultants were previously employed at or engaged by universities or other medical device, diagnostic, biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these employees, consultants and contractors, may have executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees, consultants and independent contractors do not use the intellectual property rights, proprietary information, know-how or trade secrets of others in their work for us, we may be subject to claims that we or these individuals have, inadvertently or otherwise, used, infringed, misappropriated or otherwise violated the intellectual property rights or disclosed the alleged trade secrets or other proprietary information, of these former employers, competitors or other third parties, or to
72
claims that we have improperly used or obtained such trade secrets. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Any litigation or the threat of litigation may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize potential diagnostic tests, products and services, which could harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management and other employees.
Additionally, we may be subject to claims from third parties challenging our ownership interest in intellectual property rights we regard as our own, based on claims that our employees or consultants have breached an obligation to assign inventions to another employer, to a former employer, or to another person or entity. Litigation may be necessary to defend against any other claims, and it may be necessary or we may desire to enter into a license to settle any such claim; however, there can be no assurance that we would be able to obtain a license on commercially reasonable terms, if at all. If our defense to those claims fails, in addition to paying monetary damages, a court could prohibit us from using technologies or features that are essential to our diagnostic tests or products, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers.
An inability to incorporate technologies or features that are important or essential to our diagnostic tests or products could have a material adverse effect on our business, financial condition and results of operations, and may prevent us from selling our rights to our COVID-19 testing program, either of the Nodify XL2 and Nodify CDT tests, or the VeriStrat and GeneStrat tests.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property rights to execute agreements assigning such intellectual property rights to us, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property rights that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property rights. Such claims could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our existing and future diagnostic tests, products and services.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. In 2011, the Leahy-Smith America Invents Act (Leahy-Smith Act) was signed into law. The Leahy-Smith Act includes a number of significant changes to United States patent law. These include provisions that affect the way patent applications are prosecuted and also may affect patent litigation. These also include provisions that switched the United States from a first-to-invent system to a first-inventor-to-file system, allow third-party submission of prior art to the USPTO during patent prosecution and set forth additional procedures to attack the validity of a patent by the USPTO administered post grant proceedings, including post-grant review, inter partes review and derivation proceedings. Under a first-inventor-to-file system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor was the first to invent the claimed invention. The USPTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective in 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition and results of operations.
In addition, patent reform legislation may pass in the future that could lead to additional uncertainties and increased costs surrounding the prosecution, enforcement and defense of our patents and applications. Furthermore, the United States Supreme Court and the United States Court of Appeals for the Federal Circuit have made, and will likely continue to make, changes in how the patent laws of the United States are interpreted. Recent United States Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. This combination of events has created uncertainty with respect to the validity and enforceability of patents, once obtained. Similarly, foreign courts have made, and will likely continue to make, changes in how the patent laws in their respective jurisdictions are interpreted. We cannot predict future changes in the interpretation of patent laws or changes to patent laws that might be enacted into law by United States and foreign legislative bodies. Those changes may materially affect our patents or patent applications and our ability to obtain additional patent protection in the future.
If our trademarks and tradenames are not adequately protected, then we may not be able to build name recognition in our markets and our business may be adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented, declared generic or determined to be violating or infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these trademarks or trade names, which we need to build name recognition among potential partners and customers in our markets of interest. At times, competitors or other third parties may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement or
73
dilution claims brought by owners of other trademarks. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected.
We have not yet registered certain of our trademarks in all of our potential markets, although we have registered several connected to our diagnostic tests, products and services in the United States. If we apply to register these and trademarks in the United States and other countries, our applications may not be allowed for registration in a timely fashion or at all, and our registered trademarks may not be maintained or enforced. During trademark registration proceedings, we may receive rejections. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademark applications and registrations, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
Our efforts to enforce or protect our rights related to trademarks, trade secrets, domain names or other intellectual property rights may be ineffective, could result in substantial costs and diversion of resources and could adversely affect our business, financial condition and results of operations.
We may become involved in lawsuits to protect or enforce our patents, the patents of our licensors or other intellectual property rights, which could be expensive, time consuming and unsuccessful.
Competitors or other third parties may infringe, misappropriate or otherwise violate our patents, the patents of our licensors or other intellectual property rights, or we may be required to defend against claims of infringement, misappropriation or other violations. In addition, our patents also may become involved in inventorship, priority or validity disputes. To counter or defend against such claims can be expensive and time-consuming. Any claims we assert against perceived infringers could provoke those parties to assert counterclaims against us alleging that we infringe their patents or other intellectual property. In any such proceeding, a court or other administrative body may decide that a patent or other intellectual property right owned by us is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover such technology. Grounds for a validity challenge could include an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, written description, non-enablement or failure to claim patent-eligible subject matter. Grounds for an unenforceability assertion could include an allegation that someone connected with prosecution of the patent withheld information material to patentability from the USPTO, or made a misleading statement, during prosecution. Third parties also may raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include reexamination, post-grant review, inter partes review, interference proceedings, derivation proceedings and equivalent proceedings in foreign jurisdictions, including opposition proceedings. Such proceedings could result in the revocation or cancellation of or amendment to our patents in such a way that they no longer cover our diagnostic tests, products and services or prevent third parties from competing with our diagnostic tests, products and services. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which the patent examiner and we or our licensing partners were unaware during prosecution. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we could lose at least part, and perhaps all, of the patent protection on our diagnostic tests, products and services. An adverse result in any litigation or other proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during litigation.
Moreover, some of our owned and in-licensed patents and patent applications are, and may in the future be, co-owned with third parties. If we are unable to obtain an exclusive license to any such third-party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing diagnostic tests, products, services or technology. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us. Any of the foregoing could have a material adverse effect on our business, financial condition or results of operations.
Even if resolved in our favor, litigation or other proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our management and other personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on our common stock price. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from
74
the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Any of the foregoing could have a material adverse effect on our business, financial condition or results of operations.
Third parties may initiate legal proceedings alleging that we are infringing, misappropriating, or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
The intellectual property landscape in the field of precision oncology is in flux, and it may remain uncertain for the coming years. There may be significant intellectual property related litigation and proceedings relating to our owned and in-licensed, and other third party, intellectual property and proprietary rights in the future. As we move into new markets and applications for our diagnostic tests, products or services, incumbent participants in such markets may assert their patents and other intellectual property rights against us as a means of slowing our entry into such markets or as a means to extract substantial license and royalty payments from us. Our competitors and others may now and, in the future, have significantly larger and more mature patent portfolios than we currently have. In addition, future litigation may involve patent holding companies or other adverse patent owners who have no relevant product or service revenue and against whom our own patents may provide little or no deterrence or protection. Therefore, our commercial success depends in part on our non-infringement of the patents or other intellectual property rights of third parties.
However, we may in the future be subject to claims that we, or other parties we have agreed to indemnify, infringe, misappropriate or otherwise violate patents or other intellectual property rights owned or controlled by third parties. Because patent applications are published sometime after filing, and because applications can take several years to issue, there may be additional currently pending third-party patent applications that are unknown to us, which may later result in issued patents. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. We may not have sufficient resources to bring these actions to a successful conclusion.
There is a substantial amount of litigation and other patent challenges, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology industry, including patent infringement lawsuits, interferences, oppositions and inter partes review proceedings before the USPTO, and corresponding foreign patent offices. Numerous United States and foreign issued patents and pending patent applications, which are owned by third parties, including our competitors, exist in the fields in which we are developing diagnostic tests and in which we may develop future diagnostic tests, products and services. As the precision oncology industry expands and more patents are issued, the risk increases that our diagnostic tests may be subject to claims of infringement of the patent rights of third parties. Numerous significant intellectual property issues have been litigated, are being litigated and will likely continue to be litigated, between existing and new participants in our existing and targeted markets, and competitors have and may assert that our diagnostic tests or services infringe their intellectual property rights as part of a business strategy to impede our successful entry into or growth in those markets.
We could incur substantial costs and divert the attention of our management and technical personnel in defending against any of these claims. Parties making claims against us may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources.
Because of the inevitable uncertainty in intellectual property litigation, we could lose a patent infringement or other action asserted against us regardless of our perception of the merits of the case. There is no assurance that a court would find in our favor on questions of infringement, validity, enforceability, or priority. A court of competent jurisdiction could hold that these third-party patents are valid, enforceable, and infringed, which could materially and adversely affect our ability to commercialize any product candidates we may develop and any other product candidates or technologies covered by the asserted third-party patents. In order to successfully challenge the validity of any such United States patent in federal court, we would need to overcome a presumption of validity. As this burden is a high one requiring us to present clear and convincing evidence as to the invalidity of any such United States patent claim, there is no assurance that a court of competent jurisdiction would invalidate the claims of any such United States patent.
Parties making claims against us may be able to obtain injunctive or other relief, which could block our ability to develop, commercialize and sell diagnostic tests, products or services, and could result in the award of substantial damages against us, including treble damages, attorney’s fees, costs, and expenses if we are found to have willfully infringed. In the event of a successful claim of infringement against us, we may be required to pay damages and ongoing royalties, which could be significant, and obtain one or more licenses from third parties, or be prohibited from selling certain diagnostic tests, products or services. We may not be able to obtain these licenses on acceptable or commercially reasonable terms, if at all, or these licenses may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. In addition, we could encounter delays in diagnostic test introductions while we attempt to develop alternative diagnostic tests, products or services to avoid infringing third-party patents or intellectual property rights. Defense of any lawsuit or failure to obtain any of these licenses could prevent us from commercializing diagnostic tests, products or services, and the prohibition of sale of any of our diagnostic tests, products or services could materially affect our business and our ability to gain market acceptance for our diagnostic tests, products and services.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or
75
developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
In addition, our agreements with some of our customers, suppliers or other entities with whom we do business require us to defend or indemnify these parties to the extent they become involved in infringement claims, including the types of claims described above. We could also voluntarily agree to defend or indemnify third parties in instances where we are not obligated to do so if we determine it would be important to our business relationships. If we are required or agree to defend or indemnify third parties in connection with any infringement claims, we could incur significant costs and expenses that could adversely affect our business, operating results or financial condition.
We may be subject to claims challenging the priority or inventorship of our patents and other intellectual property rights.
We or our licensors may be subject to claims that former employees, collaborators or other third parties have an interest in our owned or in-licensed patents, trade secrets or other intellectual property rights as an inventor or co-inventor. For example, we or our licensors may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or our or our licensors’ ownership of our owned or in-licensed patents, trade secrets or other intellectual property rights. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property rights that are important to our product candidates.
If we or our licensors are unsuccessful in any interference proceeding or other priority or inventorship dispute, we may be required to obtain and maintain licenses from third parties, including parties involved in any such interference proceedings or other priority or inventorship disputes. Such licenses may not be available on commercially reasonable terms or at all, or may be non-exclusive. If we are unable to obtain and maintain such licenses, we may need to cease the development, manufacture, and commercialization of one or more of our diagnostic tests, products or services. The loss of exclusivity or the narrowing of our owned and licensed patent claims could limit our ability to stop others from using or commercializing similar or identical technology and products. Any of the foregoing could result in a material adverse effect on our business, financial condition, results of operations, or prospects. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
Obtaining and maintaining our patent protection depends on compliance with various required procedures, document submissions, fee payments and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States at several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-United States patent agencies. We are also dependent on our licensors to take the necessary action to comply with these requirements with respect to our licensed intellectual property rights. The USPTO and various non-US governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in some cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors may be able to enter the market without infringing our patents and this circumstance would have a material adverse effect on our business.
Issued patents covering our diagnostic tests and any other or future diagnostic tests, products or services could be found invalid or unenforceable if challenged.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability and some of our patents or patent applications, including licensed patents, may be challenged, in courts or patent offices in the United States and abroad, in opposition, derivation, reexamination, inter partes review, post-grant review or interference. Additionally, if we and our licensing partners initiate or become involved in legal proceedings against a third party to enforce a patent covering one of our diagnostic tests, products, services or technologies, the defendant could counterclaim that the patent covering our diagnostic tests, products or services is invalid or unenforceable. In patent litigation in the United States, counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including patent eligible subject matter, lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement during prosecution. In addition, the United States now awards patent priority to the first party to file a patent application, and others may submit patent claims covering our inventions prior to us. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art of which we and the patent examiner were unaware during prosecution. If a third party were to prevail on a legal assertion of invalidity or
76
unenforceability, we would lose at least part, and perhaps all, of the patent protection on our diagnostic tests or any diagnostic tests, products and services that we may develop.
A successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights, which could have a material adverse impact on our business. Furthermore, if the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future diagnostic tests, products or services.
We may not be aware of all third-party intellectual property rights potentially relating to our current or future diagnostic tests, products or services.
Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until approximately 18 months after filing or, in some cases, not until such patent applications issue as patents. We, or our current or future license partners or collaborators, might not have been the first to make the inventions covered by each of our pending patent applications and we might not have been the first to file patent applications for these inventions. To determine the priority of these inventions, we may have to participate in interference proceedings, derivation proceedings or other post-grant proceedings declared by the USPTO. The outcome of such proceedings is uncertain, and other patent applications may have priority over our patent applications. Such proceedings could also result in substantial costs to us and divert our management’s attention and resources.
We rely on licenses from third parties in relation to certain diagnostic tests, products and services and if we lose these licenses then we may be subjected to future litigation.
We are a party to license agreements that grant us rights to use certain intellectual property rights, including patents and patent applications, typically in certain specified fields of use, in connection with our diagnostic tests, products and services. Some of those licensed rights could provide us with freedom to operate for aspects of our diagnostic tests, products and services. We may need to obtain additional licenses from others to advance our research, development and commercialization activities.
The in-licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to in-license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
Furthermore, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. In addition, we expect that competition for the in-licensing or acquisition of third-party intellectual property rights for product candidates that are attractive to us may increase in the future, which may mean fewer suitable opportunities for us as well as higher acquisition or licensing costs. We may be unable to in-license or acquire the third-party intellectual property rights for product candidates on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain rights to suitable product candidates, our business, financial condition, results of operations and prospects for growth could suffer.
Our existing license agreements impose, and we expect that our future license agreements will impose, various diligence, royalty payment, milestone payment, insurance and other obligations on us. If we fail to comply with these obligations or other obligations in our license agreements, our licensors may have the right to terminate these agreements, in which event we may not be able to develop and market any product or use any technology that is covered by these agreements. If our license agreements terminate, or we experience a reduction or elimination of licensed rights under these agreements, we may have to negotiate new or reinstated licenses with less favorable terms or we may not have sufficient intellectual property rights to operate our business. The occurrence of such events could materially harm our business.
Our success may depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property rights. Our licensors may not successfully prosecute the patent applications we license. Even if patents issue in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents or may pursue such litigation less aggressively than we would. Without protection for the intellectual property rights we license, other companies might be able to offer substantially identical diagnostic tests for sale, which could adversely affect our competitive business position and harm our business prospects.
Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
77
Moreover, disputes may also arise between us and our current or future licensors regarding intellectual property rights subject to a license agreement, including those relating to:
If we do not prevail in such disputes, we may lose any or all of our rights under such license agreements.
In addition, the agreements under which we currently license intellectual property rights or technology from third parties are complex and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property rights or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property rights that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, or are insufficient to provide us the necessary rights to use the intellectual property rights, we may be unable to successfully develop and commercialize any affected diagnostic tests, products or services, which could have a material adverse effect on our business, financial conditions, results of operations and prospects.
Absent the license agreements, we may infringe patents subject to those agreements, and if the license agreements are terminated, we may be subject to litigation by the licensor. Litigation could result in substantial costs to us and distract our management. If we do not prevail, we may be required to pay damages, including treble damages, attorneys’ fees, costs and expenses and royalties or be enjoined from selling our diagnostic tests, products or services, which could adversely affect our ability to offer diagnostic tests, products or services, our ability to continue operations and our financial condition.
Some intellectual property that we in-license may have been developed through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for companies based in the United States. Compliance with such regulations may limit our exclusive rights, and limit our ability to contract with manufacturers that are not based in the United States.
Certain of the intellectual property that we license may have been developed through the use of United States government funding and therefore may be subject to certain federal regulations. As a result, the United States government may have certain rights to intellectual property embodied in our diagnostic tests, products and services pursuant to the Bayh-Dole Act of 1980 (Bayh-Dole Act). These United States government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the United States government has the right to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third party if it determines that: (i) adequate steps have not been taken to commercialize the invention; government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). To date, none of our commercialized products are subject to march-in rights. The United States government also has the right to take title to these inventions if we, or the applicable licensor, fail to disclose the invention to the government and fail to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us or the applicable licensor to expend substantial resources. In addition, the United States government requires that any products of the subject invention or produced through the use of the subject invention be manufactured substantially in the United States. The manufacturing preference requirement can be waived if the owner of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for United States manufacturers may limit our ability to contract
78
with product manufacturers outside of the United States for products covered by such intellectual property. To the extent any of our current or future owned or licensed intellectual property is generated through the use of United States government funding, the provisions of the Bayh-Dole Act may similarly apply. Any failure by us to comply with federal regulations regarding intellectual property rights that were developed through the use of United States government funding could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Patent terms may be inadequate to protect our competitive position on our diagnostic tests, products and services for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest United States non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited.
Even if patents covering our diagnostic tests, products and services are obtained, once the patent life has expired, we may be open to competition from competitive diagnostic tests, products and services. Given the amount of time required for the development, testing and regulatory review of potential new diagnostic tests, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing diagnostic tests, products or services similar or identical to ours.
We may not be able to protect our intellectual property rights throughout the world.
Third parties may attempt to commercialize competitive diagnostic tests, products or services in foreign countries where we do not have any patents or patent applications and/or where legal recourse may be limited. This may have a significant commercial impact on our foreign business operations.
Filing, prosecuting and defending patents on our diagnostic tests, products and services in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing diagnostic tests or products made using our inventions in and into the United States or other jurisdictions. Competitors may use our diagnostic tests, products, services and technologies in jurisdictions where we have not obtained patent protection to develop their own diagnostic tests and, further, may export otherwise infringing diagnostic tests or products to territories where we have patent protection but enforcement is not as strong as that in the United States. These diagnostic tests and products may compete with our diagnostic tests, products or services and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing diagnostic tests, products and services in violation of our intellectual property rights generally. Proceedings to enforce our intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Many countries, including India, China, and certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our current or future licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition and results of operations may be adversely affected.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
79
Any of the foregoing could have a material adverse effect on our business, financial condition and results of operations.
General Risk Factors
We expect that the price of our common stock will fluctuate substantially and you may not be able to sell your shares at or above the price you paid for them.
The market price of our common stock is likely to be highly volatile and may fluctuate substantially due to many factors, including:
In recent years, the stock markets generally have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors may significantly affect the market price of our common stock, regardless of our actual operating performance, and you may not realize any return on your investment in us and may lose some or all of your investment.
In addition, in the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Securities litigation brought against us following volatility in our stock price, regardless of the merit or ultimate results of such litigation, could result in substantial costs, which would hurt our financial condition and operating results and divert management’s attention and resources from our business.
Securities analysts may not publish favorable research or reports about our business or may publish no information at all, which could cause our stock price or trading volume to decline.
If a trading market for our common stock develops, the trading market will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We do not control these analysts. As a newly public company, we may be slow to attract research coverage and the analysts who publish information about our common stock will have had relatively little
80
experience with us, which could affect their ability to accurately forecast our results and could make it more likely that we fail to meet their estimates. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding our stock price, our stock price could decline. If one or more of these analysts cease coverage of us or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price or trading volume to decline.
Shares of our common stock may be at risk for delisting from the Nasdaq Global Select Market in the future and delisting may adversely affect the value of your investment in the Company.
Shares of our common stock may be at risk of delisting from the Nasdaq Global Select Market if the bid price per share of our common stock does not remain above the applicable listing standards in the future. Moreover, there is no assurance that we will be able to take action to meet these listing standards if such bid price falls below the minimum required price. If our common stock were to be delisted, the liquidity of our common stock would be adversely affected and the market price of our common stock could decrease further. Our failure to be listed on NASDAQ or another established securities trading market or quotation system could have a material adverse effect on the value of your investment in the Company.
We are an “emerging growth company” and a “smaller reporting company,” and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act. We may take advantage of certain exemptions and relief from various public reporting requirements, including the requirement that our internal control over financial reporting be audited by our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 (the Sarbanes-Oxley Act). We will be exempt from any rules that could be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotations or a supplement to the auditor’s report on financial statements; we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements; and we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved.
Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
We have elected to take advantage of the extended transition period to comply with new or revised accounting standards and to adopt certain of the reduced disclosure requirements available to emerging growth companies. As a result of the accounting standards election, we will not be subject to the same implementation timing for new or revised accounting standards as other public companies that are not emerging growth companies, which may make comparison of our financials to those of other public companies more difficult. Additionally, because we have taken advantage of certain reduced reporting requirements, the information contained herein may be different from the information you receive from other public companies in which you hold stock.
We will remain an “emerging growth company” until the earliest to occur of (i) the last day of the fiscal year in which we have more than $1.07 billion in annual revenue; (ii) the date we qualify as a “large accelerated filer,” with at least $700 million of equity securities held by non-affiliates; (iii) the date on which we have issued, in any three-year period, more than $1.0 billion in non-convertible debt securities; and (iv) the last day of the fiscal year ending after the fifth anniversary of the completion of our IPO.
We are also a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of the fiscal year in which (1) the market value of our common shares held by non-affiliates exceeds $250 million as of the end of that year’s second fiscal quarter, or (2) our annual revenues exceeded $100 million during such completed fiscal year and the market value of our common shares held by nonaffiliates exceeds $700 million as of the end of that year’s second fiscal quarter. To the extent we take advantage of such reduced disclosure obligations, it may also make comparison of our financial statements with other public companies difficult or impossible.
Investors may find our common stock less attractive to the extent we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline or become more volatile.
If our estimates or judgments relating to our critical accounting policies are based on assumptions that change or prove to be incorrect, our operating results could fall below the expectations of securities analysts and investors, resulting in a decline in the market price of our common stock.
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in our financial statements and accompanying notes. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets, liabilities, equity, revenue and expenses that are not readily apparent from other sources. It is possible that interpretation, industry practice and guidance may evolve over time. If our assumptions change or if actual circumstances differ
81
from our assumptions, our operating results may be adversely affected and could fall below the expectations of securities analysts and investors, resulting in a decline in the market price of our common stock.
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
Our officers, directors and principal stockholders each holding more than 5% of our common stock collectively control approximately 66.3% of our outstanding common stock as of December 31, 2021. As a result, these stockholders, if they act together, will be able to control the management and affairs of our company and most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change of control and might adversely affect the market price of our common stock. This concentration of ownership may not be in the best interests of our other stockholders.
We expect to incur significant additional costs as a result of being a public company, which may adversely affect our business, financial condition and results of operations.
As a result of our IPO completed in October 2020, we expect to incur costs associated with corporate governance requirements that are applicable to us as a public company, including rules and regulations of the SEC, under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, and the Securities Exchange Act of 1934, as amended (the Exchange Act), as well as the rules of Nasdaq. These rules and regulations are expected to significantly increase our accounting, legal and financial compliance costs and make some activities more time-consuming. We also expect these rules and regulations to make it more expensive for us to maintain directors’ and officers’ liability insurance. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our Board of Directors or as executive officers. Accordingly, increases in costs incurred as a result of becoming a publicly traded company may adversely affect our business, financial condition and results of operations.
If we experience material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately report our financial condition or results of operations, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
As a result of becoming a public company, we are required, under Section 404 of the Sarbanes-Oxley Act to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting beginning with our Annual Report on Form 10-K for the year ended December 31, 2021. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency or combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of a company’s annual and interim financial statements will not be detected or prevented on a timely basis.
During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal controls are effective. The effectiveness of our controls and procedures may be limited by a variety of factors, including:
When we cease to be an “emerging growth company” under the federal securities laws, our auditors will be required to express an opinion on the effectiveness of our internal controls. If we are unable to confirm that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, which could cause the price of our common stock to decline.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. We have designed our disclosure controls and procedures to provide reasonable assurance that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
82
Provisions in our corporate charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team.
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation specifies that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for most legal actions involving actions brought against us by stockholders. Notwithstanding the foregoing, the exclusive forum provision will not apply to any claim to enforce any liability or duty created by the Exchange Act or the Securities Act and for which the federal courts have exclusive jurisdiction. We believe this exclusive forum provision benefits us by providing increased consistency in the application of Delaware law by chancellors particularly experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums and protection against the burdens of multi-forum litigation. However, the provision may have the effect of discouraging lawsuits against our directors and officers. The enforceability of similar choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is possible that, in connection with any applicable action brought against us, a court could find the choice of forum provisions contained in our amended and restated certificate of incorporation to be inapplicable or unenforceable in such action.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful third-party claims against us and may reduce the amount of money available to us.
Our amended and restated articles of incorporation provide that we will indemnify our directors and officers to the fullest extent permitted by Section 145 of the Delaware General Corporate Law.
In addition, as permitted by the Delaware General Corporate Law, our amended and restated articles of incorporation and our indemnification agreements that we have entered into with our directors and officers provide that:
83
1B. Unresolved Staff Comments.
None.
Item 2. Properties.
Our headquarters is located in Boulder, Colorado, where we lease office and laboratory space, under an operating lease agreement that expires in January 2023, which has recently been amended to extend to January 2024. We also lease office and laboratory space located in De Soto, Kansas, under an operating lease agreement that expires in October 2023. A portion of our employees are located outside of Colorado and Kansas, and others work from home. We believe our existing facilities meet our current needs. We will need additional office space in the future as we continue to build and expand our development, commercial and support teams. We believe we can find suitable additional space in the future on commercially reasonable terms.
Location |
|
Use |
|
Square Feet |
|
|
Expiration |
|
Boulder, Colorado |
|
Office and laboratory |
|
|
29,722 |
|
|
January 14, 2024 |
De Soto, Kansas |
|
Office and laboratory |
|
|
9,066 |
|
|
October 31, 2023 |
Item 3. Legal Proceedings.
From time to time, we may become involved in legal proceedings or investigations which could have an adverse impact on our reputation, business and financial condition and divert the attention of our management from the operation of our business. In September 2021, we reached a settlement agreement with the plaintiffs, which received preliminary approval from the Circuit Court of the City of St. Louis, State of Missouri (the Court) on November 10th, regarding a dispute involving the Telephone Consumer Protection Act (TCPA). On January 31, 2022, the Court approved the final settlement payment to third parties of approximately $210,000 which was accrued as a legal contingency during the year ended December 31, 2021. We are not presently a party to any other legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition, or cash flows.
Item 4. Mine Safety Disclosures.
None.
84
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock is traded on The NASDAQ Global Market under the symbol "BDSX." The high and low closing prices for our common stock on The NASDAQ Global Market from January 1, 2021 to December 31, 2021 were $31.21 and $4.09, respectively.
Holders of our Common Stock
As of March 9, 2022, there were approximately 204 holders of our common stock.
Dividend Policy
We have never declared or paid dividends on our common stock and do not expect to pay dividends on our common stock for the foreseeable future. We anticipate that all of our liquidity in the foreseeable future will be used for the operation and growth of our business. Any future determination to declare dividends will be subject to the discretion of our Board of Directors and will depend on various factors, including applicable laws, our results of operations, financial condition, future prospects, and any other factors deemed relevant by our Board of Directors. In addition, the terms of our term loan restrict our ability to pay dividends on our common stock, and we may also enter into credit agreements or other borrowing arrangements in the future that will further restrict our ability to declare or pay dividends on our common stock.
Use of Proceeds
On December 30, 2021, the Company raised approximately $16.3 million in gross proceeds from the sale of 3,756,994 common shares in an at-the-market offering. The Company received net proceeds of $15.7 million after deducting underwriting discounts and commissions and offering expenses payable by the Company. The net proceeds received were used to, among other things, fund the partial repayment of the 2021 Term Loan (see – Part II - Item 8, Financial Statements and Supplementary Data).
We registered these shares under the Securities Act on a Registration Statement on Form S-3 (Registration No. 333-261095), which was filed on November 24, 2021 and declared effective on November 29, 2021.
Recent Sales of Unregistered Securities
There were no sales of unregistered securities for the year ended December 31, 2021. From January 1, 2020 through December 31, 2020, we granted stock options under our 2016 and 2006 Stock Plans to purchase 0.8 million shares of our common stock at exercise prices ranging from $0.77 to $6.83 per share, and issued an aggregate of 0.4 million shares of common stock pursuant to the exercise of stock options with aggregate proceeds of approximately $1.3 million. These issuances were undertaken in reliance upon an exemption from registration under Rule 701 of the Securities Act of 1933.
Item 6. [Reserved]
85
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion of our financial condition and results of operations should be read together with our audited financial statements and related notes and other financial information included elsewhere in this Annual Report on Form 10-K.
In addition to historical financial information, this discussion and other parts of this report contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, based upon current expectations that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth in the section titled “Risk Factors” under Part II, Item 1A below. Forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties that could cause actual results and events to differ from those anticipated.
These statements are based upon information available to us as of the date of this Annual Report on Form 10-K, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements, like all statements in this report, speak only as of their date, and we undertake no obligation to update or revise these statements in light of future developments. These statements are inherently uncertain, and investors are cautioned not to unduly rely upon these statements.
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A), is provided to supplement the audited financial statements and the related notes in Part II - Item 8 of this Annual Report on Form10-K. We intend for this discussion to provide you with information that will assist you in understanding our financial statements, the changes in key items in those financial statements from year to year and the primary factors that accounted for those changes. Data for the years ended December 31, 2021 and 2020 has been derived from our audited financial statements included in this Annual Report on Form 10-K.
Overview
We are a leading data-driven diagnostic solutions company leveraging state of the art technologies with our proprietary AI platform to discover, develop, and commercialize solutions for clinical unmet needs, with a primary focus in lung disease. By combining a technology multi-omic approach with a holistic view of the patient’s disease state, we believe our solutions provide physicians with greater insights to help personalize their patient’s care and meaningfully improve disease detection, evaluation, and treatment. Our unique approach to precision medicine provides timely and actionable clinical information, which we believe helps improve overall patient outcomes and lowers the overall healthcare cost by reducing the use of ineffective and unnecessary treatments and procedures. In addition to our diagnostic tests, we provide biopharmaceutical companies with services that include diagnostic research, clinical trial testing, and the discovery, development, and commercialization of companion diagnostics.
Our core belief is that no single technology will answer all clinical questions that we encounter. Therefore, we employ multiple technologies, including genomics, transcriptomics, proteomics, and radiomics, and leverage our proprietary AI-based Diagnostic Cortex® platform to discover innovative diagnostic tests for clinical use. The Diagnostic Cortex is an extensively validated deep learning platform optimized for the discovery of diagnostic tests, which we believe overcomes standard machine learning challenges faced in life sciences research. Our data-driven and multi-omic approach is designed to enable us to discover diagnostic tests that answer critical clinical questions faced by physicians, researchers, and biopharmaceutical companies.
We continuously incorporate new market insights and patient data to enhance our platform through a data-driven learning loop. We regularly engage with our customers, key opinion leaders, and scientific experts to stay ahead of the rapidly evolving diagnostic treatment landscape to identify additional clinical unmet needs where a diagnostic test could help improve patient care. Additionally, we incorporate clinical and molecular profiling data from our commercial clinical testing, research studies, clinical trials, and biopharmaceutical customers or academic partnerships, to continue to advance our platform. We have over 150,000 samples and data in our biobank, including tumor profiles and immune profiles, which are used for both internal and external research and development initiatives.
We have commercialized eight diagnostic tests which are currently available for use by physicians. Our Nodify XL2 and Nodify CDT tests, marketed as part of the Nodify Lung Nodule Risk Assessment testing strategy, assess the risk of lung cancer to help identify the most appropriate treatment pathway. We believe we are the only company to offer two commercial blood-based tests to help physicians reclassify risk of malignancy in patients with suspicious lung nodules. Our GeneStrat ddPCR, GeneStrat NGS, and VeriStrat tests, marketed as the IQLung testing strategy, are used following diagnosis of lung cancer to measure the presence of mutations in the tumor and the state of the patient’s immune system to establish the patient’s prognosis and help guide treatment decisions. The GeneStrat targeted tumor profiling test and the VeriStrat immune profiling test now have a less than 36-hour average turnaround time, down from the previous 72-hour average turnaround time, providing physicians with timely results to facilitate treatment decisions. In response to the COVID-19 pandemic, through our partnership with Bio-Rad, we commercialized the Biodesix WorkSafe™ testing program. Our scientific diagnostic expertise, technologies, and existing commercial infrastructure enabled us to rapidly commercialize two FDA EUA authorized tests, a part of our customizable program. Both diagnostic tests are owned and were developed by Bio-Rad and Bio-Rad has granted us permission to utilize both tests for commercial diagnostic services. Then U.S. Health and Human Services Secretary Azar
86
declared a public health emergency for COVID-19 in February 2020 which justified the authorization of emergency use of diagnostic tests for the detection and/or diagnosis of COVID-19. The Bio-Rad SARS-CoV-2 ddPCR test and the Platelia SARS-CoV-2 Total Ab test have been granted FDA EUA pursuant to the current emergency declaration. The Bio-Rad SARS-CoV-2 ddPCR test was FDA EUA authorized on May 1, 2020, authorizing performance of the test in laboratories certified under CLIA to perform high complexity tests. The second test is the Platelia SARS-CoV-2 Total Ab test, which is an antibody test intended for detecting a B-cell immune response to SARS-CoV-2, indicating recent or prior infection. The Platelia SARS-CoV-2 Total Ab test was FDA EUA authorized on April 29, 2020. Medical products that are granted an EUA are only permitted to commercialize their products under the terms and conditions provided in the authorization. The FDA may revoke an EUA where it is determined that the underlying health emergency no longer exists or warrants such authorization, if the conditions for the issuance of the EUA are no longer met, or if other circumstances make revocation appropriate to protect the public health or safety, and we cannot predict how long the EUAs for the SARS-CoV-2 tests will remain in place. In addition to the seven tests currently available to physicians, the Company launched the GeneStrat NGS™ (NGS) test, our 72-hour blood-based NGS test, in November 2021 to a select group of physicians, with broad US launch in the first quarter 2022.
Prior to using the Bio-Rad SARS-CoV-2 tests as part of our testing program, we performed feasibility, verification, and validation studies, including developing software for process automation, sample accessioning, data management and reporting, all required to demonstrate the test operated as claimed by the manufacturer and as required by our certifying regulatory agencies for high complexity laboratory testing. We secured independent reference specimens run with EUA tests to validate these tests as fit for diagnostic use in our laboratories. Post-launch development support for these tests have included improvements in on-boarding new personnel, logistics of sample collection, sample receipt and data reporting, all required to support our testing program. Additional releases of the laboratory data management software are ongoing and planned for the foreseeable future.
Beginning in the quarter ended June 30, 2021, we began partnering with GenScript Biotech Corporation to commercialize the blood-based cPass SARS-CoV-2 Neutralizing Antibody testing as a service. The test is the first surrogate neutralizing antibody test with FDA EUA and uses ELISA technology to qualitatively detect circulating neutralizing antibodies to the RBD in the spike protein of SARS-CoV-2 that are produced in response to vaccination or previous SARS-CoV-2 infection.
These tests under the Biodesix WorkSafe testing program are utilized by healthcare providers, including hospitals and nursing homes, and are also offered to businesses and educational systems to assist in their back-to-work or back-to-school strategies. We have announced multiple partnerships for COVID-19 testing, and maintain an agreement with the State of Colorado to be one of the diagnostic companies to support widespread COVID-19 testing for the State. Additionally, we have overseen and managed onsite testing and validating testing for the Big Ten Conference athletic competitions through the term of our contract which expired on June 30, 2021.
In addition to the eight diagnostic tests currently on the market, we perform over 30 assays for research use as part of our laboratory services that have been used by over 50 biopharmaceutical companies and academic partners. All of our diagnostic testing is performed at one of our two accredited, high-complexity clinical laboratories in Boulder, Colorado and De Soto, Kansas.
Since our inception, we have performed over 500,000 tests and continue to generate a large and growing body of clinical evidence consisting of over 300 clinical and scientific peer-reviewed publications, presentations, and abstracts. Through ongoing study of each of our tests, we continue to grow our depth of understanding of disease biology and the broad utility of each of our tests. We believe we are poised for rapid growth by leveraging our scientific development and laboratory operations expertise along with our commercial infrastructure which includes sales, marketing, reimbursement, and regulatory affairs.
In the United States, we market our tests to clinical customers through our targeted sales organization, which includes sales representatives that are engaged in sales efforts and promotional activities primarily to pulmonologists, oncologists, cancer centers and nodule clinics. We market our tests and services to biopharmaceutical companies globally through our targeted business development team, which promotes the broad utility of our tests and testing capabilities throughout drug development and commercialization which is of value to pharmaceutical companies and their drug-development process.
We have funded our operations to date principally from net proceeds from the sales of our common stock through both an IPO and issuances from our Form S-3 shelf, the sale of convertible preferred stock, revenue from diagnostic testing and services, and the incurrence of indebtedness. We had cash and cash equivalents of $32.7 million and $62.1 million as of December 31, 2021 and 2020, respectively.
Factors Affecting Our Performance
We believe there are several important factors that have impacted our operating performance and results of operations, including:
87
Historically, we have experienced situations where commercial payers proactively reduced the amounts they were willing to reimburse for our tests, and in other situations, commercial payers have determined that the amounts they previously paid were too high and have sought to recover those perceived excess payments by deducting such amounts from payments otherwise being made. When we contract to serve as a participating provider, reimbursements are made pursuant to a negotiated fee schedule and are limited to only covered indications. Becoming a participating provider generally results in higher reimbursement for covered indications and lack of reimbursement for non-covered indications. As a result, the impact of becoming a participating provider with a specific payer will vary. If we are not able to obtain or maintain coverage and adequate reimbursement from third-party payers, we may not be able to effectively increase our testing volume and revenue as expected. Additionally, retrospective reimbursement adjustments can negatively impact our revenue and cause our financial results to fluctuate.
Our clinical research has resulted in approximately 90 peer-reviewed publications for our tests. In addition to clinical studies, we are collaborating with investigators from multiple academic cancer centers. We believe these studies are critical to gaining physician adoption and driving favorable coverage decisions by payers and expect our investments in research and development to increase. Further we also expect to increase our research and development expenses to fund further innovation and develop new clinically relevant tests.
While each of these areas present significant opportunities for us, they also pose significant risks and challenges that we must address. See Part II, Item 1A. “Risk Factors” for more information.
COVID-19 Pandemic
The COVID-19 pandemic has disrupted, and we expect will continue to disrupt, our lung diagnostic testing operations. To protect the health and well-being of our workforce, partners, vendors and customers, we provide voluntary COVID-19 testing for employees
88
working on-site, implemented social distance and building entry policies at work, restricted travel and facility visits, and followed the States of Colorado and Kansas’ public health orders and the guidance from the CDC. Employees who can perform their duties remotely are asked to work from home and those on site are asked to follow our social distance guidelines. Our sales, marketing and business development efforts have also been constrained by our operational response to the COVID-19 pandemic. We expect to continue to adjust our operational norms to help slow the spread of COVID-19 in the coming months, including complying with government directives and guidelines as they are modified and supplemented.
On September 9, 2021, President Biden signed two new Executive Orders that would require vaccinations for all federal workers and contractors. In addition, it was also announced that the Department of Labor’s Occupational Safety and Health Administration (OSHA) and the CMS would each be releasing emergency rules that would respectively require (i) employers with 100 or more employees and (ii) workers in health care setting that receive Medicare and Medicaid reimbursement to implement vaccination and testing protocols. In January 2022, the United States Supreme Court blocked the Biden administration from enforcing its sweeping vaccine-or-test requirements for large private companies, but allowed a vaccine mandate to stand for medical facilities that take Medicare or Medicaid payments. We are currently a party to several federal contracts and in order to remain compliant with President Biden’s Executive Orders, all Biodesix teammates were required to have received an effective dose of the COVID-19 vaccine no later than January 4, 2022. Exemptions were requested for qualifying medical reasons or sincerely held religious belief, subject to review and approval.
The COVID-19 pandemic and the surge associated with the Delta and Omicron variants have negatively affected, and we expect will continue to negatively affect, our lung diagnostic testing-related revenue and our clinical studies. For example, cancer patients may have more limited access to hospitals, healthcare providers and medical resources as they take steps to control the spread of COVID-19. Our biopharmaceutical customers are facing challenges in recruiting patients and in conducting clinical trials to advance their pipelines, for which our tests could be utilized. As a result of the COVID-19 pandemic, beginning in the latter half of March 2020, we saw a decline in business with existing and new biopharmaceutical customers. We began to see recovery during the fourth quarter 2020 as our delivered tests exceeded first quarter 2020 delivered tests and we expect to continue to see the recovery extending into 2022; however, the rate of growth experienced since we began to see recovery is below our expectations and has been impacted by ongoing surges, such as the surge in the Delta and Omicron variants, which have negatively impacted patients willingness or ability to gain access to healthcare providers and medical resources. Further, our clinical studies, such as our ongoing INSIGHT and ALTITUDE studies, as well as our arrangements including contracted clinical studies with our biopharmaceutical customers, are expected to take longer to complete than what we expected before the outbreak of the COVID-19 pandemic. Our biopharmaceutical services revenue grew by approximately 20% during 2021 as compared to 2020; however, we are continuing to experience delays in clinical trials from across the country and world due to COVID-19 restrictions. We expect further improvement in our biopharmaceutical activities during 2022 as compared to 2021.
Conversely, we experienced a significant increase in revenues related to an increase in the demand for our Biodesix WorkSafe testing program, our COVID-19 testing program, since the onset of the pandemic. COVID-19 diagnostic services contributed approximately $30.2 million and $28.3 million during fiscal years 2021 and 2020, respectively. The first quarter 2021 was our high-water mark for COVID-19 testing revenue. We experienced a steady decline in subsequent quarters as immunizations in the U.S. accelerated. We do not anticipate the need for COVID testing to be commensurate with the peak demand experienced during the first quarter 2021 and instead expect the demand to moderate as new variants and infection occur. The reduction in demand for COVID-19 diagnostic testing will be a key indicator of continued recovery and is taken as a positive sign for both our Lung Diagnostic and Biopharmaceutical Services as we head into 2022. There is no assurance that our COVID-19 testing program will continue to be accepted by the market or that other diagnostic tests will become more accepted, produce quicker results or are more accurate. Further, the longevity and extent of the COVID-19 pandemic is uncertain. If the pandemic were to dissipate, whether due to a significant decrease in new infections, due to acquiring herd immunity based on previous natural infection, and the availability and rapid distribution of vaccines, the evolution of variant strains that impact diagnostic test performance, or otherwise, the need for COVID-19 testing could decrease significantly and this could have an adverse effect on our results of operations and profitability. As a result, the increase in revenue due to any increase in demand for these diagnostic tests may not be indicative of our future revenue. See Part II., Item 1A. “Risk Factors” for a description of how the COVID-19 pandemic may adversely affect our business, financial condition and results of operations.
Fourth Quarter and Full Year 2021 Financial and Operational Highlights
The Company generated record revenue in fiscal year 2021 of $54.5 million, representing an increase of 20% as compared to fiscal year 2020. The following were significant developments affecting our business, capital structure and liquidity during the year ended December 31, 2021 as compared to the same period in 2020 unless otherwise noted:
89
Components of Operating Results
Revenues
We derive our revenue from two primary sources: (i) providing diagnostic testing in the clinical setting (Diagnostic Tests); and (ii) providing biopharmaceutical companies with services that include diagnostic research, clinical research, clinical trial testing, development and testing services generally provided outside the clinical setting and governed by individual contracts with third parties as well as development and commercialization of companion diagnostics (Services).
Diagnostic Tests
Diagnostic test revenue is generated from delivery of results from our diagnostic tests. In the United States, we performed tests as both an in-network and out-of-network service provider depending on the test performed and the contracted status of the insurer. We provide diagnostic tests in two primary categories: (i) core lung diagnostics testing and (ii) COVID-19 testing.
We consider diagnostic testing to be completed upon the delivery of test results to our customer, either the prescribing physician or third-party to which we contracted for services to be performed, which is considered the performance obligation. The fees for such services are billed either to a third party such as Medicare, medical facilities, commercial insurance payers, or to the patient. We determine the transaction price related to our contracts by considering the nature of the payer, the historical amount of time until payment by a payer and historical price concessions granted to groups of customers.
Services
Services revenue is generated from the delivery of our on-market tests, pipeline tests, custom diagnostic testing, and other scientific services for a purpose as defined by any individual customer. At times we collaborate with large biopharmaceutical companies in an attempt to discover biomarkers that would be helpful in their drug development or marketing. The performance obligations and related revenue for these sales is defined by a written agreement between us and our customer. These services are generally completed upon the delivery of testing results, or other contractually defined milestone(s), to the customer, which is considered the performance obligation. Customers for these services are typically large pharmaceutical companies where collectability is reasonably assured and therefore revenue is accrued upon completion of the performance obligations. Revenue derived from services is often unpredictable and can cause dramatic swings in our overall net revenue line from quarter to quarter.
Operating Expenses
Direct costs and expenses
Cost of diagnostic testing generally consists of cost of materials, direct labor, including bonus, employee benefits, equipment and infrastructure expenses associated with acquiring and processing test samples, including sample accessioning, test performance, quality control analyses, charges to collect and transport samples; curation of test results for physicians; and in some cases, license or royalty fees due to third parties. Costs associated with performing our tests are recorded as the tests are processed regardless of whether revenue was recognized with respect to the tests. Infrastructure expenses include depreciation of laboratory equipment, rent costs, amortization of leasehold improvements and information technology costs. Royalties for licensed technology are calculated as a percentage of revenues generated using the associated technology and recorded as expense at the time the related revenue is recognized. One-time royalty payments related to signing of license agreements or other milestones, such as issuance of new patents, are amortized to expense over the expected useful life of the patents. While we do not believe the technologies underlying these licenses are necessary to permit us to provide our tests, we do believe these technologies are potentially valuable and of possible strategic importance to us or our
90
competitors. Under these license agreements, we are obligated to pay aggregate royalties ranging from 1% to 8% of sales in which the patents or know-how are used in the product or service sold, sometimes subject to minimum annual royalties or fees in certain agreements.
We expect the aggregate cost of diagnostic testing to increase in line with the increase in the number of tests we perform, but the cost per test to decrease modestly over time due to the efficiencies we may gain as test volume increases, and from automation and other cost reductions. Cost of services includes costs incurred for the performance of development services requested by our customers. Costs of development services will vary depending on the nature, timing and scope of customer projects.
Research and development
Research and development expenses consist of costs incurred to develop technology and include salaries and benefits, reagents and supplies used in research and development laboratory work, infrastructure expenses, including allocated facility occupancy and information technology costs, contract services, clinical studies, other outside costs and costs to develop our technology capabilities. Research and development expenses account for a significant portion of our operating expenses and consist primarily of external and internal costs incurred in connection with the discovery and development of our product candidates.
External expenses include: (i) payments to third parties in connection with the clinical development of our product candidates, including contract research organizations and consultants; (ii) the cost of manufacturing products for use in our preclinical studies and clinical trials, including payments to contract manufacturing organizations (CMOs) and consultants; (iii) scientific development services, consulting research fees and for sponsored research arrangements with third parties; (iv) laboratory supplies; and (v) allocated facilities, depreciation and other expenses, which include direct or allocated expenses for IT, rent and maintenance of facilities. External expenses are recognized based on an evaluation of the progress to completion of specific tasks using information provided to us by our service providers or our estimate of the level of service that has been performed at each reporting date. We track external costs by the stage of program, clinical or preclinical.
Internal expenses include employee-related costs, including salaries and related benefits for employees engaged in research and development functions. We do not track internal costs by product candidate because these costs are deployed across multiple programs and, as such, are not separately classified.
Research and development costs are expensed as incurred. Payments made prior to the receipt of goods or services to be used in research and development are deferred and recognized as expense in the period in which the related goods are received or services are rendered. Costs to develop our technology capabilities are recorded as research and development.
We expect our research and development expenses to increase as we continue to innovate and develop additional products and expand our data management resources. As our services revenue grows, an increasing portion of research and development dollars are expected to be allocated to cost of goods for biopharmaceutical service contracts. This expense, though expected to increase in dollars, is expected to decrease as a percentage of revenue in the long term, though it may fluctuate as a percentage of our revenues from period to period due to the timing and extent of these expenses.
Sales, marketing, general and administrative
Our sales and marketing expenses are expensed as incurred and include costs associated with our sales organization, including our direct sales force and sales management, client services, marketing and reimbursement, as well as business development personnel who are focused on our biopharmaceutical customers. These expenses consist primarily of salaries, commissions, bonuses, employee benefits, and travel, as well as marketing and educational activities and allocated overhead expenses. We expect our sales and marketing expenses to increase in dollars as we expand our sales force, increase our presence within the United States, and increase our marketing activities to drive further awareness and adoption of our tests and our future products. These expenses, though expected to increase in dollars, are expected to decrease as a percentage of revenue in the long term, though they may fluctuate as a percentage of our revenues from period to period due to the timing and extent of these expenses.
Our general and administrative expenses include costs for our executive, accounting, finance, legal and human resources functions. These expenses consist principally of salaries, bonuses, employee benefits, and travel, as well as professional services fees such as consulting, audit, tax and legal fees, and general corporate costs and allocated overhead expenses. We expect that our general and administrative expenses will continue to increase in dollars, primarily due to increased headcount and costs associated with operating as a public company, including expenses related to legal, accounting, regulatory, maintaining compliance with exchange listing and requirements of the SEC, director and officer insurance premiums and investor relations. These expenses, though expected to increase in dollars, are expected to decrease as a percentage of revenue in the long term, though they may fluctuate as a percentage from period to period due to the timing and extent of these expenses.
Change in Fair Value of Contingent Consideration
In connection with the purchase transaction of Indi, we recorded contingent consideration pertaining to the amounts potentially payable to Indi shareholder pursuant to the terms of the asset purchase agreement. The fair value of contingent consideration was assessed at each balance sheet date and changes, if any, to the fair value are recognized as operating expenses within the statement of operations.
91
The Company met the gross margin target of $2.0 million for three consecutive months during the three months ended June 30, 2021. Subsequent changes to the contingent consideration following the achievement of the gross margin target are recorded as ‘Interest expense’ in the statements of operations resulting from the passage of time and fixed payment schedule. The significant unobservable inputs used in the measurement of fair value included the probability of successful achievement of the specified product gross margin targets, the period in which the targets were expected to be achieved, and discount rates which ranged from 11% to 13.5%. As a result of the achievement of the gross margin target, the only significant unobservable input used in the measurement of fair value includes the discount rate since all other inputs became fixed and determinable.
Non-Operating Expenses
Interest Expense and Interest Income
Interest expense consists of cash and non-cash interest from our 2021 Term Loan, the 2018 Notes, Paycheck Protection Program loan, convertible debt and changes in the value of our contingent consideration associated with the passage of time subsequent to the achievement of the contingency in the second quarter 2021. Our convertible debt, along with the related accrued interest, was automatically converted to 1,848,280 shares of our common stock upon completion of our IPO in October 2020. Interest income, which is included in ‘Other income, net’ in the statements of operations consists of income earned on our cash and cash equivalents.
Results of Operations
The following table sets forth the significant components of our results of operations for the periods presented (in thousands, except percentages):
|
|
Year Ended December 31, |
|
|
Change |
|
||||||||||
|
|
2021 |
|
|
2020 |
|
|
$ |
|
|
% |
|
||||
Revenues |
|
$ |
54,506 |
|
|
$ |
45,557 |
|
|
$ |
8,949 |
|
|
|
20 |
% |
Operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Direct costs and expenses |
|
|
30,518 |
|
|
|
21,998 |
|
|
|
8,520 |
|
|
|
39 |
% |
Research and development |
|
|
12,789 |
|
|
|
10,818 |
|
|
|
1,971 |
|
|
|
18 |
% |
Sales, marketing, general and administrative |
|
|
50,517 |
|
|
|
34,857 |
|
|
|
15,660 |
|
|
|
45 |
% |
Change in fair value of contingent consideration |
|
|
1,622 |
|
|
|
818 |
|
|
|
804 |
|
|
|
98 |
% |
Total operating expenses |
|
|
95,446 |
|
|
|
68,491 |
|
|
|
26,955 |
|
|
|
39 |
% |
Loss from operations |
|
|
(40,940 |
) |
|
|
(22,934 |
) |
|
|
(18,006 |
) |
|
|
(79 |
)% |
Other (expense) income |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Interest expense |
|
|
(4,508 |
) |
|
|
(7,604 |
) |
|
|
(3,096 |
) |
|
|
(41 |
)% |
Change in fair value of warrant liability |
|
|
— |
|
|
|
(1,252 |
) |
|
|
(1,252 |
) |
|
|
(100 |
)% |
Gain on debt extinguishments, net |
|
|
2,298 |
|
|
|
— |
|
|
|
2,298 |
|
|
|
100 |
% |
Other (expense) income, net |
|
|
(9 |
) |
|
|
440 |
|
|
|
449 |
|
|
|
102 |
% |
Total other expense |
|
|
(2,219 |
) |
|
|
(8,416 |
) |
|
|
(6,197 |
) |
|
|
(74 |
)% |
Net loss |
|
$ |
(43,159 |
) |
|
$ |
(31,350 |
) |
|
$ |
(11,809 |
) |
|
|
(38 |
)% |
Revenues
We generate revenue from our diagnostic tests and services that we provide. Our revenues for the periods indicated were as follows (in thousands, except percentages):
|
|
Year Ended December 31, |
|
|
Change |
|
||||||||||
|
|
2021 |
|
|
2020 |
|
|
$ |
|
|
% |
|
||||
Diagnostic revenue |
|
$ |
48,937 |
|
|
$ |
40,919 |
|
|
$ |
8,018 |
|
|
|
20 |
% |
Services revenue |
|
|
5,569 |
|
|
|
4,638 |
|
|
|
931 |
|
|
|
20 |
% |
Total revenue |
|
$ |
54,506 |
|
|
$ |
45,557 |
|
|
$ |
8,949 |
|
|
|
20 |
% |
Total revenue increased $8.9 million or 20% for the year ended December 31, 2021 compared to the year ended December 31, 2020.
Diagnostic test revenue increased $8.0 million or 20% for the year ended December 31, 2021 compared to the year ended December 31, 2020. The primary driver of the increase in revenue for diagnostic testing was attributable to the success in our targeted strategy of growth in our core lung diagnostic testing. Lung diagnostic revenue grew by $6.1 million or 49% for the year ended December 31, 2021 compared to the same period in 2020 driven primarily from an increase in Nodify XL2 and Nodify CDT tests delivered. We began to see recovery during the fourth quarter 2020 in lung diagnostic testing as our delivered tests exceeded first quarter 2020 delivered tests as healthcare practitioners, including pulmonologists, increasingly returned to pre-pandemic related care. The increase in Diagnostic test revenue was also attributable to the expansion of the sales organization during fiscal year 2021. However, the Company’s sales efforts continued to be impacted by the COVID-19 pandemic and the surge associated with the Delta and Omicron variants which have
92
negatively affected, and we expect may continue to negatively affect, the growth rate of our lung diagnostic testing-related revenue and our clinical studies.
In addition to lung diagnostic testing revenue, our COVID-19 testing revenue increased by $1.9 million, or 7% from $28.3 million in fiscal year 2020 to $30.2 million in 2021 due primarily to the delivery of our COVID-19 tests that were delivered in the first half of 2021. The first quarter 2021 was our peak for COVID-19 testing revenue but we have experienced a substantial decline in COVID-19 testing revenue subsequently as a result of the expiration of our contract with the Big Ten during the second quarter 2021 and as immunizations in the U.S. accelerated. The longevity and extent of COVID-19 revenue is uncertain and may not be indicative of our future revenue and we expect it to vary based on new variants and rates of infection.
Services revenue increased $0.9 million or 20% for the year ended December 31, 2021 compared to the year ended December 31, 2020, due primarily to the recovery in testing volumes from clinical studies and services.
Operating Expenses
Direct costs and expenses
Direct costs and expenses related to revenue increased $8.5 million or 39% for the year ended December 31, 2021 compared to the year ended December 31, 2020. The increase in direct costs and expenses is primarily driven by the increase in lung diagnostic tests delivered during the year ended December 31, 2021 compared to the same period in 2020 and an increase in cost per COVID-19 test.
Research and Development
Research and development expenses increased $2.0 million or 18% for the year ended December 31, 2021 compared to the year ended December 31, 2020. The increase in cost was due primarily to increased employee compensation and benefits costs associated with non-cash stock-based compensation as a result of the transition from a private to a public company and increased headcount of our research and development personnel, partially offset by decreased spending on clinical trials.
The following table summarizes our external and internal costs for the years ended December 31, 2021 and 2020 (in thousands, except percentages):
|
|
Year Ended December 31, |
|
|
Change |
|
|
||||||||||
|
|
2021 |
|
|
2020 |
|
|
$ |
|
|
% |
|
|
||||
External expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Clinical trials and associated costs |
|
$ |
2,061 |
|
|
$ |
2,487 |
|
|
$ |
(426 |
) |
|
|
(17 |
)% |
|
Other external costs |
|
|
3,810 |
|
|
|
2,823 |
|
|
|
987 |
|
|
|
35 |
% |
|
Total external costs |
|
|
5,871 |
|
|
|
5,310 |
|
|
|
561 |
|
|
|
11 |
% |
|
Internal expenses |
|
|
6,918 |
|
|
|
5,508 |
|
|
|
1,410 |
|
|
|
26 |
% |
|
Total research and development expenses |
|
$ |
12,789 |
|
|
$ |
10,818 |
|
|
$ |
1,971 |
|
|
|
18 |
% |
|
Sales, Marketing, General and Administrative
Sales, marketing, general and administrative expenses increased $15.7 million or 45% for the year ended December 31, 2021 compared to the year ended December 31, 2020. This increase was driven primarily by increases in employee compensation and benefits associated with expansion of the Company’s workforce and increased non-cash share-based compensation expense. This increase is also the result of increases in non-employee costs associated with legal and other fees as a result of the transition from a private to a public company.
Change in Fair Value of Contingent Consideration
Change in fair value of contingent consideration increased $0.8 million or 98% for the year ended December 31, 2021 compared to the year ended December 31, 2020. The increase is a result of the change in fair value to reflect the passage of time, changes in discount rate, as well as estimates regarding the period in which targets that trigger the payment of contingent consideration will be achieved and subsequently paid. The gross margin target was met during the three months ended June 30, 2021. The net change to contingent consideration through the date the gross margin target was met is recorded as operating expenses in the statements of operations. Subsequent changes to the contingent consideration following the achievement of the gross margin target are recorded as ‘Interest expense’ in the statements of operations resulting from the passage of time and fixed payment schedule.
93
Non-Operating Expenses
Interest Expense
Interest expense decreased $3.1 million or 41% for the year ended December 31, 2021 compared to the year ended December 31, 2020. This decline was due to a decrease of interest expense related to convertible notes, which were converted to common stock upon the completion of our IPO in October 2020, and a decrease resulting from the refinancing of our term loan, reducing near-term interest costs. Partially offsetting this decline is the increase to contingent consideration recorded as ‘Interest expense’ resulting from the passage of time and fixed payment schedule during the year ended December 31, 2021 of $2.2 million.
Change in Fair Value of Warrant Liability
During the year ended December 31, 2020, we recognized a charge for the change in estimated fair value of our warrant liability related to warrants issued in connection with our Series G preferred stock of $1.3 million. Effective with the closing of our initial public offering in October 2020, the warrants to purchase Series G preferred stock were automatically converted to warrants to acquire common stock and the carrying value of the warrants of $1.6 million were reclassified to additional paid-in capital.
Gain on Debt Extinguishment, net
During the year ended December 31, 2021, we recognized a $2.3 million net gain on debt extinguishment. The Company recorded a gain on debt extinguishment of $3.1 million related to the legal release and forgiveness of the Paycheck Protection Program Loan in full. The gain was partially offset by losses on debt extinguishment of $0.7 million and $0.1 million resulting from the repayment and termination of our 2018 secured promissory note with Innovatus and the write-off of debt issuance costs associated with a $20 million prepayment of our 2021 Term Loan, respectively.
Other (expense) income, net
Other expense, net for the year ended December 31, 2021 is primarily attributable to a loss on disposal of property and equipment. Other income, net for the year ended December 31, 2020 was primarily attributable to sublease income from two subleases that expired in the second and third quarter of 2020 and a gain on disposal of property and equipment.
Liquidity and Capital Resources
We are an emerging growth company and, as such, have yet to generate positive cash flows from operations. We have funded our operating activities primarily through financing activities, which include both debt and equity offerings. In October 2020, we completed an IPO, resulting in net proceeds of approximately $63.8 million after deducting offering costs, underwriting discounts and commissions.
During March 2020, a global pandemic was declared by the World Health Organization related to the rapidly growing outbreak of a novel strain of coronavirus (COVID-19). As a result of the pandemic, the Company diversified its diagnostic testing beyond lung diagnostic testing to include the critical service of COVID-19 diagnostic testing. Beginning in the third quarter 2020, the Company’s COVID-19 testing services began to experience rapid growth with a peak in the first quarter 2021; however, subsequent to this peak, we have experienced a rapid decline in COVID-19 testing revenue primarily as a result of a few significant contracts that have expired as well as the ongoing increase in COVID-19 vaccination rates across the U.S. In addition, the COVID-19 pandemic negatively affected, and we expect may continue to negatively affect, our lung diagnostic testing-related revenue and our clinical studies. We began to see recovery beginning in the fourth quarter 2020 as our core lung diagnostics delivered tests exceeded first quarter 2020 delivered tests and have experienced continued recovery during 2021 as lung diagnostic revenue grew 49% during 2021 as compared to 2020. However, given the continuation of the pandemic through COVID-19 variants, including the associated hospitalization surges and the impact on the medical ecosystem, which include clinical studies and pulmonary practices, the demand for our lung diagnostic testing and biopharmaceutical services has not recovered in line with our expectation from the time of our IPO. While the full outcome of the COVID-19 pandemic is unknown, it continues to negatively impact our ability to grow and scale our business in line with our expectations and disclosures at the time of our IPO. As a result, the items identified above have had an adverse effect on our revenue, results of operations and cash flows.
In March 2021, we completed the closing of our 2021 Term Loan for a principal amount of $30 million and extinguished our prior 2018 term loan for $25.9 million. The 2021 Term Loan contains customary affirmative covenants, including covenants regarding compliance with applicable laws and regulation, payment of taxes, insurance coverage, notice of certain events, and reporting requirements. Further, the 2021 Term Loan contains customary negative covenants limiting the ability to, among other things, incur future debt, transfer assets except for the ordinary course of business, make acquisitions, make certain restricted payments, and sell assets, subject to certain exceptions. The 2021 Term Loan requires the Company to comply with a minimum liquidity ratio covenant (as defined in the 2021 Term Loan) of not less than 0.95 to 1.00, and has a trailing six-month rolling revenue requirement of not less than 70% of the Company’s projected revenue performed at the end each reporting period.
During the second quarter 2021, the Company determined that it would apply for forgiveness under the SBA’s Loan Forgiveness program, a change from its previous intent to repay. Subsequently, in July 2021 the Company applied for loan forgiveness and on August 17, 2021, the Company received legal release and formal notification that the PPP Loan was forgiven in full. As of and for the three
94
months ended September 30, 2021, the Company reduced the ‘Current portion of notes payable’ and recorded a gain on extinguishment in the statements of operations for the $3.1 million forgiven.
On September 30, 2021, we entered into the Consent and First Amendment to Loan and Security Agreement (the 2021 Term Loan Amendment) to, among other things, amend our 2021 Term Loan to eliminate the revenue covenant for the period ended September 30, 2021 and modify the revenue covenant threshold for the three month period ended December 31, 2021. In addition, we agreed to establish a restricted cash collateral account for $15 million for the benefit of our lender if the balance of our cash and cash equivalents declines below $40 million.
On December 30, 2021, the Company raised approximately $16.3 million in gross proceeds from the sale of 3,756,994 common shares at a public offering price of $4.35 per share in an at-the-market offering. The Company received net proceeds of $15.7 million after deducting underwriting discounts and commissions and offering expenses payable by the Company.
On December 31, 2021, we entered into the Consent and Second Amendment to Loan and Security Agreement (Second Amendment) to, among other things, amend our 2021 Term Loan and First Amendment to obtain consent for the $4.6 million January 2022 milestone payment under the Indi APA, repay $20 million in outstanding principal on December 31, 2021, waive the $600,000 prepayment fee on the $20 million Term Loan repayment, and waive the minimum revenue covenant as of December 31, 2021 and modify the minimum revenue requirement to not less 75% for the three months ended March 31, 2022 and not less than 75% on a trailing six month rolling basis for each quarter thereafter of the Company’s projected revenue performed at the end of each reporting period. The Lender agreed to apply the full amount of funds previously established within the restricted cash collateral account to partially prepay the $20 million in outstanding principal, thereby eliminating the restricted cash collateral account. As of December 31, 2021, the Company was in compliance with all restrictive and financial covenants associated with its borrowings.
As of December 31, 2021, we maintained cash and cash equivalents of $32.7 million and we have $10 million in principal balance remaining on our 2021 Term Loan. We have incurred significant losses since inception and, as a result, we have funded our operations to date primarily through the sale of common stock in our IPO in October 2020, the issuance of notes payable, and from our two primary revenue sources: (i) diagnostic testing, which include lung diagnostic testing and COVID-19 testing, and (ii) providing biopharmaceutical companies with development and testing services. In accordance with Accounting Standards Update 2014-15 (ASC Topic 205-40), Presentation of Financial Statements - Going Concern: Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, the Company is required to evaluate whether there is substantial doubt about its ability to continue as a going concern each reporting period, including interim periods. In evaluating the Company’s ability to continue as a going concern, management projected its cash flow sources and needs and evaluated the conditions and events that could raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that these financial statements were issued. Management considered the Company’s current projections of future cash flows, current financial condition, sources of liquidity and debt obligations for at least one year from the date of issuance of this Form 10-K in considering whether it has the ability to meet its obligations.
Our ability to meet our obligations as they come due may be impacted by our ability to remain compliant with financial covenants in our 2021 Term Loan or to obtain waivers or amendments that impact the related covenants. Due to the continued uncertainty caused by the COVID-19 pandemic, significant risks remain with respect to our ability to meet these thresholds and any material adverse effect on our revenues, income and expenses could impact our ability to maintain compliance with these covenants.
Based on our current operating plan, unless we raise additional capital (debt or equity) or obtain waiver from complying with such financial covenants, we expect that we will be unable to maintain our financial covenants under our 2021 Term Loan during the next twelve months, which could result in an Event of Default, as defined, causing an acceleration of the outstanding balance. We have taken steps to improve our liquidity through and the actions noted above and have also undertaken several proactive measures to mitigate the financial and operational impacts of COVID-19 through the reduction of planned capital expenditures and certain operating expenses but we do not expect that these actions alone will be sufficient to maintain our financial covenants. In addition, we have entered into negotiations with certain creditors to modify existing terms of arrangements to delay near term cash requirements and extend the period of payments; however, those negotiations are not final at this time and may not result in final agreement. We plan to raise additional funding through the issuance of equity or debt securities; however, we have not secured such funding at the time of this filing and any such financing activities are subject to market conditions. If we do raise additional capital through public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our existing stockholders’ rights. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. There can be no assurance that additional capital will be available to us or, if available, will be available in sufficient amounts or on terms acceptable to us or on a timely basis nor can there be any assurance that the Company will be a beneficiary of the COVID-19 Action Plan. If adequate capital resources are not available on a timely basis, we intend to consider limiting our operations substantially. This limitation of operations could include a hiring freeze, reductions in our workforce, reduction in cash compensation, deferring capital expenditures, and reducing other operating costs.
The Company’s revenues, results of operations and cash flows have been materially adversely impacted by the items noted above. We expect to continue to incur significant expenses for the foreseeable future and to incur operating losses in the near term while we make
95
investments to support our anticipated growth. Our current operating plan, which is in part determined based on our most recent historical actual results and trends, along with the items noted above, raises substantial doubt about the Company’s ability to continue as a going concern. Our audited financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments that might be necessary should we be unable to continue as a going concern.
Cash Flows
The following summarizes our cash flows for the periods indicated (in thousands):
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Net cash flows (used in) provided by: |
|
|
|
|
|
|
||
Operating activities |
|
$ |
(28,223 |
) |
|
$ |
(21,366 |
) |
Investing activities |
|
|
(2,547 |
) |
|
|
(2,927 |
) |
Financing activities |
|
|
1,262 |
|
|
|
81,131 |
|
Net increase in cash and cash equivalents and restricted cash |
|
$ |
(29,508 |
) |
|
$ |
56,838 |
|
Our cash flows resulted in a net decrease in cash of $29.5 million during the year ended December 31, 2021 compared to a net increase in cash of $56.8 million for the year ended December 31, 2020. For the year ended December 31, 2021, net cash used in operating activities increased by approximately $6.9 million, primarily due to a year-over-year increase in net loss of $11.8 million and decrease in non-cash adjustments to net loss of $4.5 million driven primarily by a decrease in amortization of convertible notes debt discount in 2020 of $4.4 million and an increase in net gain on debt extinguishment in 2021 of $2.3 million. This is partially offset by an increase in stock-based compensation expense of $1.2 million, increase in contingent consideration of $0.8 million, and increase in other non-cash items of $0.2 million. In addition, the Company experienced favorable changes in net working capital of $9.4 million primarily as a result of increased cash collections from customers, partially offset by increased payments to vendors.
Net cash used in investing activities during the year ended December 31, 2021 totaled $2.5 million, a decrease of $0.4 million compared to the same period in 2020. The decrease in net cash used in investing activities was primarily due to payments in 2020 to acquire Oncimmune assets that are not present in 2021. This is partially offset by an increase in purchases of property and equipment during 2021 in support of our operations.
Net cash provided by financing activities during the year ended December 31, 2021 totaled $1.3 million, a decrease of $79.9 million compared to the same period in 2020. The net cash provided by financing activities for the year ended December 31, 2021 primarily resulted from $15.7 million net proceeds from our at-the-money public offering, net proceeds from our 2021 Term Loan of $29.9 million, and proceeds from the issuance of common stock under the ESPP and exercise of stock options of approximately $1.0 million, primarily offset by the repayment of $25.3 million from our 2018 Term Loan and a $20 million prepayment on our 2021 Term Loan. The net cash provided by financing activities for the year ended December 31, 2020 primarily resulted from $72.0 million gross proceeds from our initial public offering, $13.0 million in proceeds from the issuance of convertible notes payable, $1.3 million proceeds from stock option exercises, and $3.1 from proceeds from our paycheck protection program loan, offset by equity financing and other costs associated with our initial public offering of $8.3 million.
Contractual Obligations and Commitments
The following table summarizes our non-cancelable contractual obligations and commitments as of December 31, 2021 (in thousands):
|
|
Payments due by period (1) |
|
|||||||||||||||||
|
|
Total |
|
|
Less than |
|
|
1 to 3 |
|
|
4 to 5 |
|
|
More than |
|
|||||
Operating lease obligations (2) |
|
$ |
937 |
|
|
$ |
775 |
|
|
$ |
158 |
|
|
$ |
4 |
|
|
$ |
— |
|
Borrowings and interest (3) |
|
|
14,386 |
|
|
|
532 |
|
|
|
5,368 |
|
|
|
8,486 |
|
|
|
— |
|
Fair value of contingent consideration (4) |
|
|
37,000 |
|
|
|
18,500 |
|
|
|
18,500 |
|
|
|
— |
|
|
|
— |
|
Total |
|
$ |
52,323 |
|
|
$ |
19,807 |
|
|
$ |
24,026 |
|
|
$ |
8,490 |
|
|
$ |
— |
|
96
Off-Balance Sheet Arrangements
None.
Critical Accounting Policies and Significant Judgments and Estimates
In accordance with accounting principles generally accepted in the United States, we are required to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Certain of these estimates significantly influence the portrayal of our financial condition and results of operations and require us to make difficult, subjective or complex judgments. Our critical accounting policies primarily relate to our fair value estimates, and are described in greater detail in Note 2 to our financial statements in Part 8 of this Annual Report on Form 10-K.
Revenue Recognition
We recognize revenue when our customers obtain control of promised goods or services, in an amount that reflects the consideration which we expect to receive in exchange for our goods or services. To determine revenue recognition for our arrangements with our customers, we perform a five-step process, which includes: (i) identifying the contract(s) with a customer; (ii) identifying the performance obligations in the contract; (iii) determining the transaction price; (iv) allocating the transaction price to the performance obligations in the contract; and (v) recognizing revenue when (or as) we satisfy our performance obligations.
Diagnostic test revenues
Diagnostic test revenues are recognized upon completion of our performance obligation to the deliver test results to our customer, either the prescribing physician or third-party to which we contracted for services to be performed. We consider diagnostic testing to be completed upon the delivery of test results to our customer which is considered the performance obligation. The fees for such services are billed either to a third party such as Medicare, medical facilities, commercial insurance payers, or to the patient. We determine the transaction price related to our contracts by considering the nature of the payer, the historical amount of time until payment by a payer and historical price concessions granted to groups of customers. These estimates require significant judgment by management.
Service revenues
Service revenues are recognized upon completion of our performance obligation to deliver testing results for assay development and testing services. The performance obligations and related revenue for these sales is defined by a written agreement between us and our customer. These services are generally completed upon the delivery of testing results, or other contractually defined milestone(s), to the customer, which is considered the performance obligation. Customers for these services are typically large pharmaceutical companies where collectability is reasonably assured and therefore revenue is accrued upon completion of the performance obligations. Revenue derived from services is often unpredictable and can cause dramatic swings in our overall net revenue line from quarter to quarter.
Share-based Compensation and Grant Date Fair Value
Share-based compensation related to stock options granted to our employees, directors and non-employees is measured at the grant date based on the fair value of the award. For our service-based awards, the fair value of each award is recognized as expense on a straight-line basis over the requisite service period, which is generally the vesting period of the respective awards. Compensation expense for share-based awards with performance conditions is recognized based upon the probability the performance conditions will be met as defined in the grant. Restricted stock units are measured at their grant date fair value using the closing price of our common stock on the date of grant and recognized to expense on a straight-line basis over the vesting period of each award. We estimate forfeitures and adjust these estimates to actual forfeitures as they occur.
We use the Black-Scholes option-pricing model to estimate the fair value of our share-based option awards, which requires assumptions to be made related to expected term of an award, expected volatility, the risk-free rate and expected dividend yield. Following the completion of our initial public offering, our Board of Directors has determined the fair value of our common stock is based on our closing price as reported on the date of grant on the primary stock exchange on which our common stock is traded. Changes in these subjective assumptions can materially affect the estimated value of equity grants and the share-based compensation that we record in our financial statements.
Recently Issued Accounting Pronouncements
In February 2016, the FASB issued ASU No. 2016-02, Leases (ASC Topic 842). The new guidance maintains two classifications of leases: finance leases, which replace capital leases, and operating leases. Lessees will need to recognize a right-of-use asset and a lease liability on the balance sheet for those leases previously classified as operating leases under the old guidance. The liability will be equal to the present value of lease payments. The asset will be based on the liability, subject to adjustment, such as for direct costs. The accounting standard will be effective for the Company beginning January 1, 2022. Based on our current analysis expect the adoption to result in the recognition of approximately $1.5 million of right of use assets and associated lease liabilities, inclusive of both lease and
97
non-lease components, in our balance sheet and do not expect any material impact to our statement of operations or statement of cash flows. We are implementing new processes and internal controls over lease recognition, which will ultimately assist in the application of the new lease standard.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments-Credit Losses: Measurement of Credit Losses on Financial Instruments (ASC Topic 326). This ASU requires measurement and recognition of expected credit losses for financial assets. This guidance will become effective for the Company beginning January 1, 2023 with early adoption permitted. The Company is currently evaluating this guidance and assessing the overall impact on its financial statements.
Implications of Being an Emerging Growth Company and Smaller Reporting Company
We are an “emerging growth company” within the meaning of the Jumpstart Our Business Startups Act (JOBS Act). As an emerging growth company, we may take advantage of certain exemptions from various public company reporting requirements, including the requirement that our internal control over financial reporting be audited by our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), certain requirements related to the disclosure of executive compensation in our periodic reports and proxy statements, the requirement that we hold a nonbinding advisory vote on executive compensation and any golden parachute payments. We may take advantage of these exemptions until we are no longer an emerging growth company. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. In other words, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
We have elected to take advantage of the extended transition period to comply with new or revised accounting standards and to adopt certain of the reduced disclosure requirements available to emerging growth companies. As a result of the accounting standards election, we will not be subject to the same implementation timing for new or revised accounting standards as other public companies that are not emerging growth companies, which may make comparison of our financials to those of other public companies more difficult.
We will remain an emerging growth company until the earliest to occur of (i) the last day of the fiscal year in which we have more than $1.07 billion in annual revenue; (ii) the date we qualify as a “large accelerated filer,” with at least $700 million of equity securities held by non-affiliates; (iii) the date on which we have issued, in any three-year period, more than $1.0 billion in non-convertible debt securities; and (iv) until December 31, 2025 (the year ended December 31st following the fifth anniversary of our initial public offering.
Additionally, we are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of the fiscal year in which: (i) the market value of our common shares held by non-affiliates exceeds $250 million as of the end of that year’s second fiscal quarter, or (ii) our annual revenues exceeded $100 million during such completed fiscal year and the market value of our common shares held by non-affiliates exceeds $700 million as of the end of that year’s second fiscal quarter.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to market risk in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates.
Interest Rate Risk
We are exposed to market risk for changes in interest rates related primarily to our cash and cash equivalents, marketable securities and our indebtedness, including our outstanding 2021 Term Loan. As of December 31, 2021, we had $10 million outstanding on the 2021 Term Loan subject to a floating per annum rate equal to the greater of (i) 2.00% above the prime rate, or (ii) 5.25%. Historically, we have not entered into derivative agreements such as interest rate caps and swaps to manage our floating interest rate exposure.
Periodically throughout the year, we have maintained balances in various operating accounts in excess of federally insured limits. Our cash and cash equivalents are funds held in checking and bank savings accounts, primarily at two U.S. financial institutions. We consider all highly liquid instruments purchased with an original maturity of three months or less to be cash equivalents. We continually monitor our positions with, and the credit quality of, the financial institutions with which we invest.
As of December 31, 2021, a hypothetical 100 basis point increase in interest rates would not have a material impact on our investment portfolio, financial position or results of operations.
Item 8. Financial Statements and Supplementary Data.
The financial statements and supplementary data are as set forth in the index to the financial statements on page F-1.
98
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We maintain "disclosure controls and procedures," as such term is defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, or Exchange Act, that are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in Securities and Exchange Commission rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, management recognized that disclosure controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the disclosure controls and procedures are met. Our disclosure controls and procedures have been designed to meet reasonable assurance standards. Additionally, in designing disclosure controls and procedures, our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible disclosure controls and procedures. The design of any disclosure controls and procedures also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Based on their evaluation as of the end of the period covered by this Annual Report on Form 10-K, our Chief Executive Officer and Chief Financial Officer have concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting for the Company, as such term is defined in Exchange Act Rules 13a-15(f). Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2021 based on the criteria established in "Internal Control – Integrated Framework" issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), using the 2013 framework. Based on our assessment, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2021, the Company's internal control over financial reporting was effective based on the criteria set forth by COSO.
This annual report does not include an attestation report of our registered public accounting firm due to a transition period established by rules of the Securities and Exchange Commission for newly public companies.
Changes in Internal Control over Financial Reporting
During the quarter ended December 31, 2021, there were no changes that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
None.
99
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this item with respect to directors is incorporated by reference from the information under the captions "Board and Corporate Governance Matters—Election of Directors" and "—Director Independence" contained in our Proxy Statement to be filed with the Securities and Exchange Commission within 120 days of the fiscal year ended December 31, 2021 in connection with the solicitation of proxies for our 2022 Annual Meeting of Stockholders (the Proxy Statement). Certain information required by this item concerning executive officers is set forth in the Proxy Statement under the caption "Executive Compensation—Executive Officers" and is incorporated herein by reference.
Item 405 of Regulation S-K calls for disclosure of any known late filing or failure by an insider to file a report required by Section 16(a) of the Exchange Act. This disclosure is contained in the section entitled "Stock Ownership—Delinquent Section 16(a) Reports" in the Proxy Statement and is incorporated herein by reference.
We have adopted a Code of Business Conduct and Ethics that applies to all of our officers and employees, including our President and Chief Executive Officer, our Chief Financial Officer and other employees who perform financial or accounting functions. The Code of Business Conduct and Ethics sets forth the basic principles that guide the business conduct of our employees. Stockholders may request a free copy of our Code of Business Conduct and Ethics by contacting Biodesix, Inc., Attention: Chief Financial Officer, 2970 Wilderness Place, Suite 100, Boulder, Colorado 80301.
To date, there have been no waivers under our Code of Business Conduct and Ethics. We intend to disclose future amendments to certain provisions of our Code of Business Conduct and Ethics or waivers of such Codes granted to executive officers and directors on our website at http://www.biodesix.com within four business days following the date of such amendment or waiver.
Our Board of Directors has appointed an Audit Committee, comprised of Ms. Jean Franchi, as Chairwoman, Mr. Hany Massarany and Dr. Matthew Strobeck. The Board of Directors has determined that Ms. Franchi qualifies as an Audit Committee Financial Expert under the definition outlined by the Securities and Exchange Commission. In addition, each of the members of the Audit Committee qualifies as an "independent director" under the current rules of The NASDAQ Stock Market and Securities and Exchange Commission rules and regulations.
Item 11. Executive Compensation.
The information required by this item is incorporated by reference from the information under "Executive Compensation" contained in the Proxy Statement and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item is incorporated by reference from the information under the captions "Stock Ownership—Security Ownership of Certain Beneficial Owners and Management" and "Executive Compensation—Equity Compensation Plan Information" contained in the Proxy Statement and is incorporated herein by reference.
The information required by this item is incorporated by reference from the information under the caption "Board and Corporate Governance Matters—Certain Relationships and Related Party Transactions" and "—Director Independence" contained in the Proxy Statement and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services.
The information required by this item is incorporated by reference from the information under the caption "Audit Matters—Principal Accountant Fees and Services" contained in the Proxy Statement and is incorporated herein by reference.
100
PART IV
Item 15. Exhibits, Financial Statement Schedules.
Reference is made to the Index to Financial Statements of Biodesix, Inc. included in Item 8 of Part II hereof.
All schedules have been omitted because they are not required, not applicable, or the required information is included in the financial statements or notes thereto.
Item 16. Form 10-K Summary.
Not applicable.
101
Exhibit Index
Exhibit Number |
|
Description |
|
|
|
|
|
3.1** |
|||
3.2** |
|||
4.1** |
|||
4.2** |
|||
4.3** |
|||
4.4** |
|||
4.5** |
|||
10.1+** |
|||
10.2.1+** |
|||
10.2.2+** |
|||
10.2.3+** |
|||
10.3+** |
|||
10.4.1+** |
|||
10.4.2+** |
|||
10.4.3+** |
|||
10.5+** |
|||
10.5.1+** |
|||
10.5.2+** |
|||
102
10.6+** |
|
10.7.1+** |
|
10.7.2+** |
|
10.7.3+** |
|
10.8.1+** |
|
10.8.2+** |
|
10.9+** |
|
10.10.1†+** |
|
10.10.2†+** |
|
10.11.1†+** |
|
10.11.2†+** |
|
10.12+** |
|
10.13.1†** |
|
10.13.2* |
Office Lease between Aero-Tech Investments, LLC and Biodesix, Inc., dated January 24, 2022. |
10.14†** |
|
10.15.1†** |
|
10.15.2†** |
|
10.16** |
103
10.17†** |
|
10.18†** |
|
10.19†** |
|
10.20†** |
|
10.21†** |
|
10.22†** |
|
10.23†** |
|
10.24†** |
|
10.25** |
|
10.26†** |
|
10.27†** |
|
10.28†** |
|
10.29†** |
|
10.30†** |
|
10.31†** |
|
10.32†** |
|
10.33†** |
104
10.34†** |
|
10.35†** |
|
10.36** |
|
10.37** |
|
10.38** |
|
10.39** |
|
10.40** |
|
10.41** |
|
10.42** |
|
23.1* |
|
24.1* |
Power of Attorney (included on the signature page of this Form 10-K). |
31.1* |
|
31.2* |
|
32.1* |
|
32.2* |
|
101.INS* |
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
101.SCH* |
Inline XBRL Taxonomy Extension Schema Document |
101.CAL* |
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
101.DEF* |
Inline XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB* |
Inline XBRL Taxonomy Extension Label Linkbase Document |
101.PRE* |
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
104 |
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
† Portions of this exhibit have been omitted as the Registrant has determined that the omitted information (i) is not material and (ii) would likely cause competitive harm to the Registrant if publicly disclosed.
+ Indicates management contract or compensatory plan.
* Filed herewith.
**Previously filed.
105
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
Biodesix, Inc. |
|
|
|
|
|
Date: March 14, 2022 |
|
By: |
/s/ RYAN H. SIUREK |
|
|
|
Ryan H. Siurek |
|
|
|
Chief Accounting Officer |
POWER OF ATTORNEY
Each person whose signature appears below constitutes and appoints Scott Hutton and Robin Harper Cowie, and each of them, his or her true and lawful agent, proxy and attorney-in-fact, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to file with the Securities and Exchange Commission this Form 10-K and any and all amendments and exhibits thereto, and all documents in connection therewith, granting unto each such attorney-in-fact and agent full power and authority to do and perform each and every act and thing necessary or appropriate to be done, as fully for all intents and purposes as he or she might or could do in person, hereby approving, ratifying and confirming all that such agent, proxy and attorney-in-fact or any of his or her substitutes may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
Name |
|
Title |
|
Date |
|
|
|
|
|
/s/ SCOTT HUTTON |
|
President, Chief Executive Officer (Principal Executive Officer |
|
March 14, 2022 |
Scott Hutton |
|
|
|
|
|
|
|
|
|
/s/ ROBIN HARPER COWIE |
|
Chief Financial Officer, Secretary and Treasurer |
|
March 14, 2022 |
Robin Harper Cowie |
|
|
|
|
|
|
|
|
|
/s/ JOHN PATIENCE |
|
Chairman and Director |
|
March 14, 2022 |
John Patience |
|
|
|
|
|
|
|
|
|
/s/ JEAN FRANCHI |
|
Director |
|
March 14, 2022 |
Jean Franchi |
|
|
|
|
|
|
|
|
|
/s/ JON FAIZ KAYYEM |
|
Director |
|
March 14, 2022 |
Jon Faiz Kayyem, Ph.D. |
|
|
|
|
|
|
|
|
|
/s/ HANY MASSARANY |
|
Director |
|
March 14, 2022 |
Hany Massarany |
|
|
|
|
|
|
|
|
|
/s/ JACK SCHULER |
|
Director |
|
March 14, 2022 |
Jack Schuler |
|
|
|
|
|
|
|
|
|
/s/ MATTHEW STROBECK |
|
Director |
|
March 14, 2022 |
Matthew Strobeck, Ph.D. |
|
|
|
|
|
|
|
|
|
/s/ CHARLES WATTS |
|
Director |
|
March 14, 2022 |
Charles Watts, M.D. |
|
|
|
|
|
|
|
|
|
106
Item 8. Financial Statements and Supplementary Data
BIODESIX, Inc.
INDEX TO FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm ( |
F-2 |
|
|
F-3 |
|
|
|
Statements of Operations for the Years ended December 31, 2021 and 2020 |
F-4 |
|
|
F-5 |
|
|
|
Statements of Cash Flows for the Years ended December 31, 2021 and 2020 |
F-6 |
|
|
F-8 |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Biodesix, Inc.:
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Biodesix, Inc. (the Company) as of December 31, 2021 and 2020, the related statements of operations, convertible preferred stock and stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and needs to raise additional equity or debt capital to fund its operations. These matters raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ KPMG LLP
We have served as the Company’s auditor since 2016
Denver, Colorado
March 14, 2022
F-2
BIODESIX, INC.
Balance Sheets
(in thousands, except share data)
|
|
As of December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Assets |
|
|||||||
Current assets |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Accounts receivable, net of allowance for doubtful accounts of $ |
|
|
|
|
|
|
||
Other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Non‑current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Intangible assets, net |
|
|
|
|
|
|
||
Goodwill |
|
|
|
|
|
|
||
Other long-term assets |
|
|
|
|
|
|
||
Total non‑current assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Liabilities and Stockholders' Equity |
|
|||||||
Current liabilities |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued liabilities |
|
|
|
|
|
|
||
Deferred revenue |
|
|
|
|
|
|
||
Current portion of contingent consideration |
|
|
|
|
|
— |
|
|
Current portion of notes payable |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Non‑current liabilities |
|
|
|
|
|
|
||
Long‑term notes payable, net of current portion |
|
|
|
|
|
|
||
Contingent consideration |
|
|
|
|
|
|
||
Other long-term liabilities |
|
|
|
|
|
|
||
Total non‑current liabilities |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
Stockholders' equity |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
Additional paid‑in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total stockholders' equity |
|
|
|
|
|
|
||
Total liabilities and stockholders' equity |
|
$ |
|
|
$ |
|
||
The accompanying Notes are an integral part of these financial statements.
F-3
BIODESIX, INC.
Statements of Operations
(in thousands, except per share data)
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Revenues |
|
$ |
|
|
$ |
|
||
Operating expenses: |
|
|
|
|
|
|
||
Direct costs and expenses |
|
|
|
|
|
|
||
Research and development |
|
|
|
|
|
|
||
Sales, marketing, general and administrative |
|
|
|
|
|
|
||
Change in fair value of contingent consideration |
|
|
|
|
|
|
||
Total operating expenses |
|
|
|
|
|
|
||
Loss from operations |
|
|
( |
) |
|
|
( |
) |
Other (expense) income: |
|
|
|
|
|
|
||
Interest expense |
|
|
( |
) |
|
|
( |
) |
Change in fair value of warrant liability |
|
|
— |
|
|
|
( |
) |
Gain on debt extinguishments, net |
|
|
|
|
|
— |
|
|
Other (expense) income, net |
|
|
( |
) |
|
|
|
|
Total other expense |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Net loss per share, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
Weighted-average shares outstanding, basic and diluted |
|
|
|
|
|
|
||
The accompanying Notes are an integral part of these financial statements.
F-4
BIODESIX, INC.
Statements of Convertible Preferred Stock and Stockholders' Equity (Deficit)
(in thousands)
|
|
Convertible Preferred Stock |
|
|
Common Stock |
|
|
Additional Paid‑In |
|
|
Accumulated |
|
|
Total Stockholders' Equity |
|
|||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
(Deficit) |
|
|||||||
Balance ‑ January 1, 2020 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||
Issuance of common stock in initial public offering, net of discounts and commission of $ |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Conversion of preferred stock into common stock upon initial public offering |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Conversion of convertible debt into common stock upon initial public offering |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Reclassification of preferred stock warrant liability and put option into additional paid-in capital upon initial public offering |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Exercise of stock options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Stock‑based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance ‑ December 31, 2020 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
Issuance of common stock, net of discounts and commission of $ |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Issuance of common stock under employee stock purchase plan |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Exercise of stock options |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Stock‑based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance - December 31, 2021 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
The accompanying Notes are an integral part of these financial statements.
F-5
BIODESIX, INC.
Statements of Cash Flows
(in thousands)
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Cash flows from operating activities |
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash, cash equivalents, and restricted |
|
|
|
|
|
|
||
Depreciation and amortization |
|
|
|
|
|
|
||
Amortization of convertible notes debt discount |
|
|
— |
|
|
|
|
|
Gain on debt extinguishments, net |
|
|
( |
) |
|
|
— |
|
Stock‑based compensation expense |
|
|
|
|
|
|
||
Change in fair value of warrant liability |
|
|
— |
|
|
|
|
|
Change in contingent consideration |
|
|
|
|
|
|
||
Provision for doubtful accounts |
|
|
|
|
|
|
||
Accrued interest, amortization of debt issuance costs and other |
|
|
|
|
|
|
||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
||
Accounts receivable |
|
|
|
|
|
( |
) |
|
Other current assets |
|
|
|
|
|
( |
) |
|
Other long-term assets and liabilities |
|
|
|
|
|
( |
) |
|
Accounts payable and other accrued liabilities |
|
|
( |
) |
|
|
|
|
Deferred revenue |
|
|
( |
) |
|
|
|
|
Net cash and cash equivalents and restricted cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Cash flows from investing activities |
|
|
|
|
|
|
||
Purchase of property and equipment |
|
|
( |
) |
|
|
( |
) |
Patent costs and intangible asset acquisition, net |
|
|
( |
) |
|
|
( |
) |
Payments to acquire Oncimmune assets |
|
|
— |
|
|
|
( |
) |
Net cash and cash equivalents and restricted cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Cash flows from financing activities |
|
|
|
|
|
|
||
Proceeds from initial public offering |
|
|
— |
|
|
|
|
|
Proceeds from the issuance of common stock |
|
|
|
|
|
— |
|
|
Proceeds from issuance of common stock under employee stock purchase plan |
|
|
|
|
|
— |
|
|
Proceeds from exercise of stock options |
|
|
|
|
|
|
||
Proceeds from issuances of convertible notes payable |
|
|
— |
|
|
|
|
|
Proceeds from term loan and notes payable |
|
|
|
|
|
|
||
Repayment of term loan and notes payable |
|
|
( |
) |
|
|
— |
|
Payment of debt issuance costs |
|
|
( |
) |
|
|
— |
|
Equity financing costs |
|
|
( |
) |
|
|
( |
) |
Other |
|
|
— |
|
|
|
( |
) |
Net cash and cash equivalents and restricted cash provided by financing activities |
|
|
|
|
|
|
||
Net increase (decrease) in cash and cash equivalents and restricted cash |
|
|
( |
) |
|
|
|
|
Cash, cash equivalents, and restricted cash ‑ beginning of period |
|
|
|
|
|
|
||
Cash, cash equivalents, and restricted cash ‑ end of period |
|
$ |
|
|
$ |
|
||
The accompanying Notes are an integral part of these financial statements.
F-6
BIODESIX, INC.
Statements of Cash Flows
(in thousands)
(Continued from the previous page)
Supplemental cash flow information:
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Conversion of preferred stock into common stock |
|
$ |
— |
|
|
$ |
|
|
Conversion of convertible notes and accrued interest into common stock |
|
|
— |
|
|
|
|
|
Reclassification of put option liability to additional paid-in capital |
|
|
— |
|
|
|
|
|
Value of put option recorded at issuance of convertible debt payable |
|
|
— |
|
|
|
|
|
Cash paid for interest |
|
|
|
|
|
|
||
Reclassification of warrant liability to additional paid-in capital |
|
|
— |
|
|
|
|
|
Non-cash debt issuance costs included in Accrued liabilities |
|
|
|
|
|
— |
|
|
The accompanying Notes are an integral part of these financial statements.
F-7
BIODESIX, INC.
Notes to Financial Statements
Note 1 – Organization and Description of Business
Biodesix, Inc. (the “Company”, “Biodesix”, “we” “us” and “our”), formerly Elston Technologies, Inc., was incorporated in Delaware in 2005. The Company’s headquarters are in Colorado, with laboratories in Colorado and Kansas. The Company conducts all of its operations within a single legal entity. Biodesix is a data-driven diagnostic solutions company leveraging state of the art technologies with its proprietary artificial intelligence platform to discover, develop, and commercialize solutions for clinical unmet needs, with a primary focus in lung disease. In addition to diagnostic tests, the Company provides biopharmaceutical companies with services that include diagnostic research, clinical trial testing, and the discovery, development, and commercialization of companion diagnostics.
The Company performs its blood-based diagnostic tests in its laboratory facilities, which are located in Boulder, Colorado and De Soto, Kansas. In May 2020, the Federal Drug Administration (FDA) granted Emergency Use Authorization (EUA) of the Bio-Rad SARS-CoV-2 Droplet Digital™ polymerase chain reaction (ddPCR) test to detect Coronavirus Disease 2019 (COVID-19) infection. In April 2020, the FDA authorized the Platelia SARS-CoV-2 Total Ab test to detect COVID-19 antibodies. Medical products that are granted an EUA are only permitted to commercialize their products under the terms and conditions provided in the authorization. The FDA may revoke an EUA where it is determined that the underlying health emergency no longer exists or warrants such authorization, if the conditions for the issuance of the EUA are no longer met, or if other circumstances make revocation appropriate to protect the public health or safety.
Blood-Based Lung Tests
The Company offers
Diagnosis
Treatment & Monitoring
COVID-19 Tests
We operate and have commercialized the Biodesix WorkSafe™ testing program, under which the Company offers
F-8
These tests under the Biodesix WorkSafe testing program are utilized by healthcare providers, including hospitals and nursing homes, and are also offered to businesses and educational systems to assist in their back-to-work or back-to-school strategies, a crucial element of restarting economic activity.
In developing the Company's products, the Company has built or gained access to unique biorepositories, proprietary technology, and bioinformatics methods that it believes are important to the development of new targeted therapies, determining clinical trial eligibility and guiding treatment selection. The Company’s testing services are made available through its clinical laboratories.
Initial Public Offering
On October 27, 2020, the Company completed its initial public offering (IPO), in which it issued and sold
At-the-Money Offering
On December 30, 2021, the Company raised approximately $
Note 2 – Summary of Significant Accounting Policies
Basis of Presentation and Estimates
The Company’s financial statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP). The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Significant items subject to such estimates include: revenue recognition; the estimation of the fair value of goodwill and other intangible assets; fair value of stock options; income tax uncertainties, including a valuation allowance for deferred tax assets; and contingencies. The Company bases these estimates on historical and anticipated results, trends, and various other assumptions that the Company believes are reasonable under the circumstances, including assumptions as to future events. These estimates form the basis for making judgments about the carrying values of assets and liabilities and recognized revenue and expenses that are not readily apparent from other sources. Actual results could differ from those estimates and assumptions. Certain prior period amounts have been reclassified to conform to the current period presentation.
During March 2020, a global pandemic was declared by the World Health Organization related to the rapidly growing outbreak of a novel strain of coronavirus (COVID-19). The COVID-19 pandemic negatively affected, and we expect will continue to negatively affect, our lung diagnostic testing-related revenue and our clinical studies.
As of December 31, 2021, we maintained cash and cash equivalents of $
Our ability to meet our obligations as they come due may be impacted by our ability to remain compliant with financial covenants in our 2021 Term Loan (see Note 8 – Debt) or to obtain waivers or amendments that impact the related covenants. Due to the continued uncertainty caused by the COVID-19 pandemic, significant risks remain with respect to our ability to meet these thresholds and any material adverse effect on our revenues, income and expenses could impact our ability to maintain compliance with these covenants.
F-9
Based on our current operating plan, unless we raise additional capital (debt or equity) or obtain a waiver from complying with such financial covenants, we expect that we will be unable to maintain our financial covenants under our 2021 Term Loan during the next twelve months, which could result in an Event of Default, as defined, causing an acceleration and repayment of the outstanding balance. We have taken steps to improve our liquidity through raising debt and equity capital during 2021, amendments to our 2021 Term Loan and have also undertaken several proactive measures to mitigate the financial and operational impacts of the COVID-19 pandemic through the reduction of planned capital expenditures and certain operating expenses but we do not expect that these actions alone will be sufficient to maintain our financial covenants. In addition, we have entered into negotiation with certain creditors to modify existing terms of arrangement to delay near term cash requirements and extend the period of payments; however, those negotiations are not final at this time and may not result in final agreement. To maintain an adequate amount of available liquidity and execute our current operating plan, we will need to continue to raise additional funds from external sources, such as through the issuance of equity or debt securities; however, we have not secured such funding at the time of this filing and any such financing activities are subject to market conditions. If we do raise additional capital through public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our existing stockholders’ rights. There can be no assurance that additional capital will be available to us or, if available, will be available in sufficient amounts or on terms acceptable to us or on a timely basis. If adequate capital resources are not available on a timely basis, we intend to consider limiting our operations substantially. This limitation of operations could include a hiring freeze, reductions in our workforce, reduction in cash compensation, deferring capital expenditures, and reducing other operating costs.
The Company’s revenues, results of operations and cash flows have been materially adversely impacted by the items noted above. Our current operating plan, which is in part determined based on our most recent historical actual results and trends, along with the items noted above, raises substantial doubt about the Company’s ability to continue as a going concern. Our audited financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments that might be necessary should we be unable to continue as a going concern.
Segment Reporting
The Company has a operating segment focused on providing diagnostic testing services to customers. All of the Company’s revenue and long-lived assets were derived or located in the United States for the years ended December 31, 2021 and 2020.
Revenue Recognition
The Company generates revenues from (i) diagnostic tests and (ii) assay development and testing services (services revenue).
Diagnostic test revenues consist of blood-based lung tests and COVID-19 tests, which are recognized in the amount expected to be received in exchange for diagnostic tests when the diagnostic tests are delivered. The Company determines the transaction price related to its blood-based lung diagnostic test contracts using a portfolio approach by considering the nature of the payer and historical price concessions granted to groups of customers. The transaction price associated with COVID-19 testing arrangements are determined on the basis of the individual contract with each customer.
Services revenue consists of various types of tests or other scientific services for a purpose as defined by any individual customer, which are often larger biopharmaceutical companies, as defined by a written agreement between the Company and the customer. These services are generally completed upon the delivery of testing results, or achievement of contractual milestone(s) as defined in the customer agreements. Revenue for these services is recognized upon delivery of the completed test results, or upon completion of the contractual milestone(s).
The Company recognizes revenues related to blood-based lung diagnostic billings based on estimates of the amounts ultimately expected to be collected from customers on a portfolio approach as discussed above. In determining the amount to accrue for a delivered test, the Company considers factors such as payment history, payer coverage, whether there is a reimbursement contract between the payer and the Company, payment as a percentage of agreed upon rate (if applicable), amount paid per test and any current developments or changes that could impact reimbursement. Variable consideration, if any, is estimated based on an analysis of historical experience and adjusted as better estimates become available. These estimates require significant judgment by management.
The Company also provides services to patients with whom the Company does not have contracts as defined in Financial Accounting Standards Board (FASB) Accounting Standards Codification 606 (ASC 606). The Company recognizes revenue for these patients when contracts, as defined in ASC 606, are established at the amount of consideration to which it expects to be entitled, or when the Company receives substantially all of the consideration subsequent to satisfaction and delivery of the performance obligations.
Deferred revenue consists of payments received for research, development, and testing services fees received prior to the completion of performance of these tests and services.
See Note 11 — Revenue and Accounts Receivable Credit Concentration for additional information.
F-10
Direct Costs and Expenses
The components of our cost of diagnostic tests and testing services consist of cost of materials, direct labor, including bonus, benefit and stock-based compensation, depreciation of laboratory equipment, rent costs, amortization of leasehold improvements and information technology costs associated with acquiring and processing test samples, including sample accessioning, test performance, quality control analyses, charges to collect and transport samples; curation of test results for physicians; and in some cases, license or royalty fees due to third parties.
Royalties for licensed technology are calculated as a percentage of revenues generated using the associated technology and recorded as expense at the time the related revenue is recognized. One-time royalty payments related to signing of license agreements or other milestones, such as issuance of new patents, are amortized to expense over the expected useful life of the patents. Costs associated with performing tests are expensed as the test is processed regardless of whether and when revenue is recognized with respect to that test.
Research and Development Expenses
Research and development expenses include external and internal costs incurred to develop our technology, collect clinical samples, and conduct clinical studies to develop and support our products. External costs consist primarily of payments to clinical trial sites, sample acquisition costs and laboratory supplies purchased in connection with the Company’s discovery and preclinical activities, process development and clinical development activities. Internal costs consist primarily of salaries and benefits, reagents and supplies used in research and development laboratory work, infrastructure expenses, including allocated facility occupancy and information technology costs.
The Company estimates and accrues its expenses resulting from its obligations under contracts with vendors and consultants in connection with conducting research and development activities. The financial terms of these contracts vary from contract to contract and may result in payments that do not match the periods over which materials or services are provided under such contracts. The Company’s estimates depend on the timeliness and accuracy of the data provided by consultants and vendors regarding the status of each activity. The Company periodically evaluates the estimates to determine if adjustments are necessary or appropriate based on information received. Research and development costs are expensed as incurred.
Sales, Marketing, General and Administrative Expenses
Selling expenses consist primarily of costs associated with our sales organization, including our direct sales force and sales management, client services, marketing, and reimbursement, as well as business development personnel who are focused on our biopharmaceutical customers. These expenses consist primarily of salaries, commissions, bonuses, employee benefits, travel, and stock-based compensation, as well as marketing and educational activities and allocated overhead expenses.
Sales, marketing, general and administrative expenses also include costs for our marketing and sales organizations, and other functions including finance, legal, human resources, and information technology.
These expenses consist principally of salaries, bonuses, employee benefits, travel, stock-based compensation, as well as professional services fees such as consulting, audit, tax and legal fees, and general corporate costs and allocated overhead expenses.
Concentrations of Credit Risk and Other Uncertainties
Substantially all of the Company’s cash and cash equivalents are deposited with two major financial institutions in the United States. The Company continually monitors its positions with, and the credit quality of, the financial institutions with which it holds cash. Periodically throughout the year, the Company has maintained balances in various operating accounts in excess of federally insured limits. The Company has not experienced any losses on its deposits of cash and cash equivalents.
Several of the components of the Company's sample collection kit and test reagents, and certain test systems and related test kits are obtained from single-source suppliers. If these single-source suppliers fail to satisfy the Company's requirements on a timely basis, it could suffer delays in being able to deliver its diagnostic solutions, a possible loss of revenue, or incur higher costs, any of which could adversely affect its operating results.
For a discussion of credit risk concentration of accounts receivable as of December 31, 2021 and 2020, see Note 11 – Revenue and Accounts Receivable Credit Concentration.
Cash and Cash Equivalents
Cash equivalents consist of short-term, highly-liquid instruments with an original maturity of three months or less from the date of purchase.
Restricted Cash
Restricted cash consists of deposits related to the Company’s corporate credit card and a letter of credit related to an operating lease agreement. As of December 31, 2021 and 2020, the Company had $
F-11
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable are stated at the amount management expects to collect from customers based on their outstanding invoices. Management reviews accounts receivable quarterly to determine if any receivable will potentially be uncollectible and to estimate the amount of allowance for doubtful accounts necessary to reduce accounts receivable to its estimated net realizable value based on historical experience, customer creditworthiness, facts, and circumstances specific to outstanding balances, and payment terms.
Inventory
Inventory consists primarily of material supplies, which are consumed in the performance of testing services and charged to 'Direct costs and expenses'. Inventory is stated at cost and reported within ‘Other current assets’ in the balance sheet and were $
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, generally between and
Long-lived assets to be held and used are evaluated for impairment when events or circumstances indicate the carrying value of a long-lived asset or asset group is less than the undiscounted cash flows from the use and eventual disposition over its remaining useful life. The Company assesses recoverability by comparing the sum or projected undiscounted cash flows from the use and eventual disposition of the asset or asset group to its carrying value, and records an impairment loss if the carrying value is greater than the undiscounted future cash flows. There were
Intangible Assets
Intangible assets primarily consist of intangible assets acquired as part of business combinations, external costs associated with patent applications that are probable of future economic benefits, and trademark costs. Finite-lived intangibles are stated at cost, net of accumulated amortization. The Company amortizes finite-lived intangible assets using the straight-line method over their estimated useful lives of
Intangible assets are reviewed for impairment whenever events or changes in circumstances indicate a reduction to fair value below their carrying amounts. There were
Goodwill
Goodwill represents the excess of purchase price over amounts allocated to acquired assets and liabilities assumed in business combinations. The carrying value of goodwill is evaluated for impairment at least annually or more frequently when events or circumstances occur indicate a potential for impairment. The annual impairment test is performed on the last day of our fourth quarter. Prior to performing a quantitative evaluation, an assessment of qualitative factors may be performed to determine whether it is more likely than not that the fair value of the reporting unit exceeds its carrying value. In the event the Company determines that it is more likely than not the carrying value of our single reporting unit is higher than its estimated fair value, quantitative testing is performed comparing recorded values to estimated fair values. If impairment is present, the impairment loss is measured as the excess of the recorded goodwill over its implied fair value. Through December 31, 2021, there were
Fair Value of Financial Instruments
U.S. GAAP for fair value establishes a hierarchy that prioritizes fair value measurements based on the types of inputs used for the various valuation techniques (market approach, income approach, and cost approach). We utilize a combination of market and income approaches to value our financial instruments. Our financial assets and liabilities are measured using inputs from the three levels of the fair value hierarchy. Fair value measurements are categorized within the fair value hierarchy based upon the lowest level of the most significant inputs used to determine fair value. The three levels of the hierarchy and the related inputs are as follows:
Level |
|
Inputs |
1 |
|
Unadjusted quoted prices in active markets for identical assets and liabilities. |
2 |
|
Unadjusted quoted prices in active markets for similar assets and liabilities; |
|
|
Unadjusted quoted prices for identical or similar assets or liabilities in markets that are not active; or |
|
|
Inputs other than quoted prices that are observable for the asset or liability. |
3 |
|
Unobservable inputs for the asset or liability. |
F-12
The carrying amounts of certain financial instruments including cash and cash equivalents, accounts receivable, prepaid expenses and other current assets, accounts payable and accrued liabilities approximate fair value due to their relatively short maturities.
See Note 4 — Fair Value for further discussion related to estimated fair value measurements.
Contingent Consideration
In connection with the purchase transaction with Integrated Diagnostics, Inc. (Indi), the Company recorded contingent consideration for amounts potentially payable to Indi’s shareholder upon attainment of a three-consecutive month gross margin target of $
Warrant Liability
Prior to the completion of the Company’s IPO in October 2020, certain warrants issued to purchase convertible preferred stock were classified as liabilities with changes in the estimated fair value of these liabilities recognized in our results of operations. Upon completion of the Company’s IPO in October 2020, the warrants were automatically converted to warrants to purchase common stock. Accordingly, the warrants were remeasured to an estimate of fair value and recognized in our results of operations and then reclassified to additional paid-in capital.
Put Option Liability
During 2020 and 2019, the Company issued convertible debt with terms that provided for a conversion rate to the convertible debtholders that was more favorable than the price that other investors would pay for common stock in certain situations, including the completion of an IPO. As a result, the convertible debt is a financial instrument that was bifurcated into two instruments, a debt obligation and a put option liability, which represented the estimated fair value of the favorable conversion rate. The estimated fair value of the put option liability was separated from the convertible debt at inception and reported as a liability and debt discount, and amortized to interest expense. As a result of our IPO, the convertible debt was automatically converted to common stock and the put option liability was remeasured to an estimate of fair value and recognized in our results of operations and then reclassified to additional paid-in capital.
Share‑Based Compensation
Stock Options
The Company grants service condition and performance condition stock options. Stock options are granted with exercise prices equal to the fair market value of our common stock on the date of grant. The grant date fair value of each employee stock option is estimated on the date of grant using the Black-Scholes option-pricing model, which requires the use of assumptions, including the expected term of the option, expected volatility of our stock price, expected dividend yield, and the risk-free interest rate, among others. We estimate forfeitures and adjust these estimates to actual forfeitures as they occur. These assumptions involve inherent uncertainties including market conditions and employee behavior that are generally outside of the Company’s control. Service condition stock options are expensed based on the grant date fair value of the awards using the straight-line method over the requisite service period. Performance-condition stock options vest based on achievement of multiple weighted performance goals, certification of performance achievement by the Compensation Committee of the Board of Directors, and continued service. For performance-condition stock options, compensation expense is updated for our expected performance level against performance goals at the end of each reporting period, which involves judgment as to achievement of certain performance metrics.
Restricted Stock Units (RSUs)
The Company grants service-condition RSUs. As a result of our IPO, the grant date fair values of these RSUs are based on the closing market price of our common stock on the grant date. We estimate forfeitures and adjust these estimates to actual forfeitures as they occur. The service-condition RSUs vest based on continued service with compensation expense recognized on a straight-line basis over the requisite service period.
See Note 12 — Share-Based Compensation for additional information related to share-based compensation.
Net Loss per Common Share
Basic net loss per common share is calculated by dividing net loss by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted net loss per common share is computed by dividing net
F-13
loss by the weighted-average number of common share equivalents outstanding for the period, if dilutive, using the treasury stock method. Potentially dilutive securities consisting of options to purchase common stock, warrants to purchase common stock, RSUs and shares subject to purchase under our employee stock purchase plan were excluded from the calculation of diluted net loss per common share because their effect would be anti-dilutive for all periods presented.
Until the Company’s IPO in October 2020, basic and diluted net loss attributable to common stockholders per share was calculated using the two-class method. The net loss was attributable entirely to common stockholders because the participating securities did not have a contractual obligation to share in the Company’s losses. As the Company has reported a net loss for all periods presented, diluted net loss per common share is the same as basic net loss per common share for all periods presented.
Note 3 – Recent Accounting Pronouncements
Recently Issued Accounting Standards Not Yet Adopted
In February 2016, the FASB issued ASU No. 2016-02, Leases (ASC Topic 842). The new guidance maintains two classifications of leases: finance leases, which replace capital leases, and operating leases. Lessees will need to recognize a right-of-use asset and a lease liability on the balance sheet for those leases previously classified as operating leases under the old guidance. The liability will be equal to the present value of lease payments. The asset will be based on the liability, subject to adjustment, such as for direct costs. The accounting standard will be effective for the Company beginning January 1, 2022. Based on our current analysis we expect the adoption to result in the recognition of $
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments-Credit Losses: Measurement of Credit Losses on Financial Instruments. This ASU requires measurement and recognition of expected credit losses for financial assets. This guidance will become effective for the Company beginning January 1, 2023 with early adoption permitted. The Company is currently evaluating this guidance and assessing the overall impact on its financial statements.
Note 4 - Fair Value
Recurring Fair Value Measurements
Our borrowing instruments are recorded at their carrying values in the balance sheets, which may differ from their respective fair values.
|
|
As of December 31, |
|
|||||||||||||
|
2021 |
|
|
2020 |
|
|||||||||||
|
|
Carrying Value |
|
|
Fair Value |
|
|
Carrying Value |
|
|
Fair Value |
|
||||
Borrowings |
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|||||
The financial liabilities that are measured and recorded at estimated fair value on a recurring basis consist of our contingent consideration associated with our acquisition of Indi, and prior to the completion of our IPO in October 2020, the warrant liability, put option liability and contingent value rights granted to certain holders of our convertible preferred stock and debt instruments, which were accounted for as liabilities and remeasured through our statements of operations.
|
|
As of December 31, |
|
|||||
Description |
|
2021 |
|
|
2020 |
|
||
Current portion of contingent consideration |
|
$ |
|
|
$ |
— |
|
|
Contingent consideration |
|
|
|
|
|
|
||
Total contingent consideration |
|
$ |
|
|
$ |
|
||
The following table presents the changes in contingent consideration for the year ended December 31, 2021 (in thousands):
|
|
Year Ended |
|
|
Level 3 Rollforward |
|
December 31, 2021 |
|
|
Beginning balances - January 1, 2021 |
|
$ |
|
|
Changes in fair value |
|
|
|
|
Interest expense |
|
|
|
|
Ending balances - December 31, 2021 |
|
$ |
|
|
F-14
The following table presents the changes in these financial liabilities for the year ended December 31, 2020 (in thousands):
|
|
For the Year Ended December 31, 2020 |
|
|||||||||||||
Level 3 Rollforward |
|
Contingent |
|
|
Put |
|
|
Warrant |
|
|
Contingent |
|
||||
Beginning balances - January 1, 2020 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Additions |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
Changes in fair value |
|
|
|
|
|
— |
|
|
|
|
|
|
( |
) |
||
Reclassified to additional paid-in capital |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
Ending balances - December 31, 2020 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
Contingent Consideration
In connection with the acquisition of Indi in 2018, the Company recorded contingent consideration for amounts contingently payable to Indi's selling shareholders pursuant to the terms of the asset purchase agreement (Indi APA). The contingent consideration arrangement
The Company met the gross margin target of $
The significant unobservable inputs used in the measurement of fair value include the probability of successful achievement of the specified product gross margin targets, the period in which the targets were expected to be achieved, and discount rates which ranged from
Contingent consideration expected to be paid in the next twelve months is recorded in the balance sheets as ‘Current portion of contingent consideration’ while the remaining amount to be paid is recorded as ‘Contingent consideration’ within non-current liabilities. The net change to contingent consideration through the date the gross margin target was met is recorded as operating expenses in the statements of operations. Subsequent changes to the contingent consideration following the achievement of the gross margin target are recorded as ‘Interest expense’ in the statements of operations resulting from the passage of time and fixed payment schedule. The net change to contingent consideration recorded as operating expenses during the years ended December 31, 2021 and 2020 was a loss of $
Put Option Liability
The put option liability was valued based on the calculated returns as a result of the various discounts included in the Company’s convertible notes payable and the related probability assessments of the various settlement scenarios. During 2020, the Company recognized an addition to the put option liability of $
Warrant Liability
In connection with entering into the 2018 Notes, the Company issued to the lender a warrant to purchase
F-15
Contingent Value Rights
In addition to the shares of Series F Preferred Stock that were issued in January 2016, investors who purchased more than their pro‑rata amount in the financing described above received a calculated number of contingent value rights (CVRs). In connection with the Series F financing, the Company issued
Non-Financial Assets and Liabilities
Our non-financial assets, which primarily consist of property and equipment, goodwill, and other intangible assets, are not required to be carried at fair value on a recurring basis and are reported at carrying value. There were no changes to the valuation methods during the periods presented.
Note 5 – Property and Equipment
Property and equipment consist of the following (in thousands):
|
|
As of December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Lab equipment |
|
$ |
|
|
$ |
|
||
Leasehold improvements |
|
|
|
|
|
|
||
Computer equipment |
|
|
|
|
|
|
||
Furniture and fixtures |
|
|
|
|
|
|
||
Software |
|
|
|
|
|
|
||
Vehicles |
|
|
|
|
|
— |
|
|
Construction in process |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Less accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
Total property and equipment, net |
|
$ |
|
|
$ |
|
||
Depreciation expense related to property and equipment was:
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Direct costs and expenses |
|
$ |
|
|
$ |
|
||
Selling, marketing, general and administrative |
|
|
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
||
Note 6 – Goodwill and Intangible Assets
Intangible assets, excluding goodwill, consist of the following (in thousands):
|
|
December 31, 2021 |
|
|
December 31, 2020 |
|
||||||||||||||||||
|
|
Cost |
|
|
Accumulated |
|
|
Net Carrying Value |
|
|
Cost |
|
|
Accumulated |
|
|
Net Carrying Value |
|
||||||
Intangible assets subject to amortization |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Patents |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Purchased technology |
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
Intangible assets not subject to |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Trademarks |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Total |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
F-16
Amortization expense related to definite-lived intangible assets was (in thousands):
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Direct costs and expenses |
|
$ |
|
|
$ |
|
||
Sales, marketing, general and administrative |
|
|
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
||
Future estimated amortization expense of intangible assets is (in thousands):
|
|
As of |
|
|
2022 |
|
$ |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 and thereafter |
|
|
|
|
Total |
|
$ |
|
|
Note 7 – Accrued Liabilities
Accrued liabilities consist of the following (in thousands):
|
|
As of December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Compensation related accruals |
|
$ |
|
|
$ |
|
||
Accrued clinical trial expense |
|
|
|
|
|
|
||
Other expenses |
|
|
|
|
|
|
||
Total accrued liabilities |
|
$ |
|
|
$ |
|
||
Note 8 – Debt
Our long-term debt consists of notes payable associated with our 2021 Term Loan, 2018 Notes and Paycheck Protection Program, each of which is described in further detail below.
|
|
As of December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
2021 Term Loan |
|
$ |
|
|
$ |
— |
|
|
2018 Notes |
|
|
— |
|
|
|
|
|
Paycheck Protection Program |
|
|
— |
|
|
|
|
|
Other |
|
|
|
|
|
— |
|
|
Unamortized debt discount and debt issuance costs |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Less: current maturities |
|
|
|
|
|
|
||
Long-term notes payable |
|
$ |
|
|
$ |
|
||
2021 Term Loan
On March 19, 2021 (Effective Date), the Company entered into a Loan and Security Agreement (the 2021 Term Loan) by and between Silicon Valley Bank (SVB or Lender) and the Company, as borrower, whereby subject to the terms and conditions of the 2021 Term Loan, SVB advanced to the Company an original principal amount of $
F-17
The Company’s final payment, due at maturity on
The Company granted the Lender a security interest in substantially all of the Company’s assets.
The 2021 Term Loan contains certain covenants limiting the ability of the Company to, among other things, incur future debt, transfer assets except for the ordinary course of business, make acquisitions, pay dividends or make other certain restricted payments, or sell assets, subject to certain exceptions, without the prior written consent of the Lender. Failure to comply with the covenants and loan requirements may result in an event of default. As of December 31, 2021,
2018 Notes
In February 2018, the Company issued long-term debt of $
At the time of issuance, the Company paid a facility fee of $
On March 19, 2021, in connection with entering into the 2021 Term Loan agreement with SVB, the Company repaid all outstanding principal, accrued and unpaid interest, and prepayment fees in the amount of $
Paycheck Protection Program Note Payable
In April 2020, the Company entered into a loan pursuant to the Paycheck Protection Program under the CARES Act, as administered by the U.S. Small Business Administration (the SBA). The loan, in the principal amount of $
F-18
All or a portion of the PPP Loan may be forgiven by the SBA upon application by the Company. Under the CARES Act, loan forgiveness is available for the sum of documented payroll costs, covered rent payments, and covered utilities during the eight-week period beginning on the approval date of the PPP Loan. For purposes of the CARES Act, payroll costs exclude compensation of an individual employee earning more than $
Scheduled principal repayments (maturities) of long-term obligations were as follows (in thousands):
|
|
As of |
|
|
2022 |
|
$ |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 and thereafter |
|
|
|
|
Total |
|
$ |
|
|
Note 9 – Warrants to Purchase Convertible Preferred Stock
The Company issued warrants to purchase shares of convertible preferred stock in conjunction with the sale of certain of the convertible preferred shares and issuance of debt. Through the closing of the Company’s IPO, the preferred warrants were classified as liabilities with estimated fair value remeasured at each reporting date reported within in the accompanying statements of operations.
The following table presents the activity for convertible preferred stock warrants (in thousands, except weighted average exercise price):
|
|
Series E |
|
|
Series G (1) |
|
||||||||||
|
|
Warrants |
|
|
Weighted |
|
|
Warrants |
|
|
Weighted |
|
||||
Outstanding ‑ January 1, 2020 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Granted |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Forfeited/canceled |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
Exercised |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Reclassification of warrant liability to additional paid-in capital |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Outstanding ‑ December 31, 2020 |
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
The warrants to purchase Series E convertible preferred stock were not exercised and expired during the three months ended June 2020.
(1) On October 27, 2020, all convertible preferred stock converted to common stock at the completion of our IPO, and as a consequence, the warrants to purchase Series G convertible preferred stock were converted to
Note 10 – Equity
Common Stock
The Company’s Restated Certificate of Incorporation authorizes the Company to issue up to
F-19
Company is currently subject to restrictions on the payment of dividends (see Note 8 - Debt) and
On October 27, 2020, the Company issued and sold
On December 30, 2021, the Company raised approximately $
Preferred Stock
The Company’s Restated Certificate of Incorporation authorizes the Company to issue up to
Note 11 – Revenue and Accounts Receivable Credit Concentration
Diagnostic test revenues consist of blood-based lung tests and COVID-19 tests, which are recognized in the amount expected to be received in exchange for diagnostic tests when the diagnostic tests are delivered. The Company conducts diagnostic tests and delivers the completed test results to the prescribing physician or patient, as applicable. The fees for diagnostic tests are billed either to a third party such as Medicare, medical facilities, commercial insurance payers, or to the patient. The Company determines the transaction price related to its diagnostic test contracts by considering the nature of the payer, historical price concessions granted to groups of customers, and its historical collection experience using a portfolio approach. The Company recognizes revenues for diagnostic tests upon delivery of the tests to the physicians requesting the tests or patient, as applicable.
Services revenue consists of on-market tests, pipeline tests, custom diagnostic testing, and other scientific services for a purpose as defined by any individual customer, which is often with biopharmaceutical companies. The performance obligations and related revenue for these sales is defined by a written agreement between the Company and the customer. These services are generally completed upon the delivery of testing results, or other contractually defined milestone(s), to the customer. Revenue for these services is recognized upon delivery of the completed test results, or upon completion of the contractual milestone(s).
Revenues consisted of the following (in thousands):
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Diagnostic tests |
|
$ |
|
|
$ |
|
||
Services |
|
|
|
|
|
|
||
Total revenue |
|
$ |
|
|
$ |
|
||
Deferred Revenue
Deferred Revenue consists of cash payments from customers received in advance of delivery. As test results are delivered, the Company recognizes the deferred revenue in ‘Revenues’ in the statements of operations. Of the $
The Company’s customers in excess of 10% of total revenue, and their related revenue as a percentage of total revenue were as follows:
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
The Big Ten Conference |
|
|
% |
|
|
% |
||
Centura Healthcare |
|
|
% |
|
|
% |
||
In addition to the above table, we collect reimbursement on behalf of customers covered by Medicare, which accounted for
F-20
risk from its accounts receivable related to services provided to its customers. The Company does not perform evaluations of customers' financial condition and does not require collateral.
The Company’s third-party payors and other customers in excess of 10% of accounts receivable, and their related accounts receivable as a percentage of total accounts receivable were as follows:
|
|
As of December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Medicare |
|
|
% |
|
|
% |
||
Janssen Research and Development, LLC |
|
|
% |
|
|
% |
||
LabCorp DD (formerly Covance) |
|
|
% |
|
|
% |
||
The Big Ten Conference |
|
|
— |
% |
|
|
% |
|
Centura Healthcare |
|
|
— |
% |
|
|
% |
|
Note 12 – Share Based Compensation
Predecessor 2016 and 2006 Equity Incentive Plans
Under the 2006 Equity Incentive Plan (2006 Plan), the Company was authorized to grant incentive stock options, non-statutory stock options, stock appreciation rights, restricted stock awards and RSUs.
In February 2016, the Company adopted the 2016 Equity Incentive Plan (2016 Plan) as a successor to and continuation of the prior 2006 Plan. The 2016 Plan provided for the grant of incentive stock options, non-statutory stock options, stock appreciation rights, restricted stock awards, RSUs, and other stock awards to directors, employees, and consultant. Awards granted under the 2016 Plan or the 2006 Plan that were unallocated, expired or otherwise terminated, or were forfeited, cancelled, or repurchased by the Company, became available for future issuance under the 2016 Plan. In addition, shares subject to an award were withheld to satisfy a participant’s tax withholding obligations, or were reacquired by the Company as consideration for the exercise or purchase price of a stock award also became available for future issuance under the 2016 Incentive Plan.
2020 Equity Incentive Plan
Effective upon the closing of our IPO, the Company’s Board of Directors approved the 2020 Equity Incentive Plan (2020 Plan), which replaced the 2016 Plan. The 2020 Plan provides for the grant of incentive stock options, non-statutory stock options, stock appreciation rights, restricted stock awards, RSUs, performance awards and other stock awards. Officers, directors, employees, consultants, agents, and independent contractors who provide services to the Company may receive awards. The terms of all awards are governed by an agreement between the Company and the recipients, as administered and approved by the Compensation Committee of the Board of Directors. Any awards that expire or are forfeited under the 2016 Plan or 2006 Plan become available for issuance under the 2020 Plan.
The number of shares reserved for issuance under the 2020 Plan is
To the extent an equity award granted under the 2020 Plan (other than any substitute award) or granted under any other equity plan maintained by us under which awards are outstanding as of the effective date of the 2020 Plan (the Prior Plans) expires or otherwise terminates without having been exercised or paid in full, or is settled in cash, the shares subject to such award will become available for future grant under the 2020 Plan. In addition, to the extent shares subject to an award are withheld to satisfy a participant’s tax withholding obligation upon the exercise or settlement of such award (other than any substitute award) or to pay the exercise price of a stock option granted under the 2020 Plan or a Prior Plan, such shares will become available for future grant under the 2020 Plan. The total number of shares available for grant under all Plans was
Employee Stock Purchase Plan
Effective with our IPO in October 2020, the Company’s Board of Directors and its stockholders approved the Company’s Employee Stock Purchase Plan (ESPP). A total of
Subject to any plan limitations, the ESPP allows eligible employees to contribute, normally through payroll deductions, up to
F-21
offering period, whichever is lower.
Description of Awards Granted
The Company has granted incentive stock options, non-statutory stock options, performance-based stock options, and RSUs.
Incentive stock options, which may only be issued to employees, are granted at an exercise price per share equal to the closing market price of the Company’s common stock on the grant date, and vest over time as determined by the Compensation Committee, provided that the term of the options may not exceed
Non-statutory stock options, which may be issued to employees, non-employees and directors, are granted at an exercise price per share equal to the closing market price of the Company’s common stock on the grant date, and vest over time as determined by the Compensation Committee, provided that the term of the options may not exceed ten years from the date of grant. Accelerated vesting may occur in the event of an optionee's death, disability, or other events.
Performance-based stock options are typically granted on an annual basis and consist of a performance-based and service-based component. The performance targets and vesting conditions for performance-condition options are based on achievement of recognized revenue targets.
RSUs and the related terms and conditions are awarded at the discretion of the Compensation Committee. RSU holders have a contractual right to receive a share of common stock when vested. RSUs vest over time as determined by the Compensation Committee. RSU agreements may provide for accelerated vesting in the event of a stock unit holder's death, disability, or retirement or other events.
Our Compensation Committee may grant other stock awards that are based on or related to shares of our common stock, such as awards of shares of common stock granted as bonus and not subject to any vesting conditions, deferred stock units, stock purchase rights, and shares of our common stock issued in lieu of our obligations to pay cash under any compensatory plan or arrangement. To date, the Company has granted service-condition RSUs.
Bonus-To-Options Program
The Company also has a Bonus-to-Options Program (the “Bonus Option Program”), is separate from predecessor Plans and was initially adopted by the Board of Directors in 2008, and subsequently amended and restated in 2010, 2011 and 2015. For fiscal year 2022, the Bonus Option Program is subject to the shares reserved under the 2020 Plan. The Bonus Option Program, which is limited to participation of the Chief Executive Officer, direct reports to the Chief Executive Officer and Vice Presidents of the Company, allows participants who so elect to convert all or a portion of their annual cash bonus into fully vested, non-qualified stock options to purchase shares of common stock (Bonus Options). The exercise price for the options under the Bonus Option Program equals the then current price for the shares of the common stock as of the grant date, as disclosed below under “Fair Value of Common Stock”. Bonus Options issued must be exercised within a
The Company recorded the following activity related to the Bonus Option Program during the year ended December 31, 2021 (in thousands, excepted weighted average exercise price and weighted average contractual life):
|
|
Number of |
|
|
Weighted |
|
|
Weighted |
|
|
Aggregate |
|
||||
Outstanding ‑ January 1, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Granted |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
||
Forfeited/canceled |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
Exercised |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
Outstanding ‑ December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Exercisable ‑ December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
The Company recorded $
F-22
2022 and 2021 by the Compensation Committee of the Board of Directors. In determining the amount of stock compensation to recognize under the Bonus Option Program, the Company estimates the bonus attainment for the year and determines the expected number of options to be delivered to eligible participants. A Black-Scholes option pricing model is used to determine the estimated fair value of the expected number of options to be delivered to eligible participants. The key elements in determining the estimated fair value include assumptions for volatility, the risk-free interest rate, expected dividends and strike price, utilizing the measurement date closing stock price until the grants are authorized.
Share-Based Compensation Expense
Pre-tax share-based compensation expense reported in the Company’s statements of operations was (in thousands):
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Direct costs and expenses |
|
$ |
|
|
$ |
— |
|
|
Research and development |
|
|
|
|
|
|
||
Sales, marketing, general and administrative |
|
|
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
||
The unrecognized remaining share-based compensation expense for options and RSUs was approximately $
Stock Options
Stock option activity during the year ended December 31, 2021, excluding the Bonus Option Program described above, was (in thousands, except weighted average exercise price and weighted average contractual life):
|
|
Number of |
|
|
Weighted |
|
|
Weighted |
|
|
Aggregate |
|
||||
Outstanding - January 1, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Granted |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
||
Forfeited/canceled |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
Exercised |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
Outstanding ‑ December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Exercisable ‑ December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Fair Value of Common Stock
Prior to the Company’s IPO, the fair value of the Company’s common stock underlying the stock options was determined by the Board of Directors with assistance from management and, in part, on input from an independent third-party valuation firm. The Board of Directors determined the fair value of common stock by considering a number of objective and subjective factors, including valuations of comparable companies, sales of convertible preferred stock, operating and financial performance, the lack of liquidity of the Company’s common stock and the general and industry-specific economic outlook. Subsequent to the Company’s IPO, the fair value of the Company’s common stock is determined based on its closing market price on the date of grant.
The estimated grant date fair value of stock options was calculated using the Black-Scholes option-pricing model, based on the following assumptions:
F-23
The fair value of each option grant was estimated on the grant date with the following weighted average assumptions for the years ended December 31, 2021 and 2020:
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Expected term (in years) |
|
|
|
|
|
|
||
Expected volatility |
|
|
% |
|
|
% |
||
Risk‑free rate |
|
|
% |
|
|
% |
||
Expected dividend yield |
|
|
— |
% |
|
|
— |
% |
Restricted Stock Units
During the year ended December 31, 2021, the Company granted
Note 13 – Net Loss per Common Share
Basic earnings per share (EPS) excludes dilution and is computed by dividing net loss attributable to the Company’s stockholders by the weighted-average shares outstanding during the period. Diluted EPS reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised, resulting in the issuance of shares of common stock that would then share in the earnings or losses of the Company.
In connection with the acquisition of Indi in 2018, the Company recorded contingent consideration (See Note 4 – Fair Value) for amounts contingently payable to Indi's selling shareholders pursuant to the terms of the asset purchase agreement. The contingent consideration arrangement requires additional consideration to be paid by the Company to Indi upon attainment of a three-consecutive month gross margin target of $
Basic and diluted loss per share for the years ended December 31, 2021 and 2020 were (in thousands, except per share amounts):
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Numerator |
|
|
|
|
|
|
||
Net loss attributable to common stockholders |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
||
Denominator |
|
|
|
|
|
|
||
Weighted-average shares outstanding used |
|
|
|
|
|
|
||
Net loss per share, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
The potentially dilutive securities as of December 31, 2021 and 2020 primarily represent the shares subject to future issuance under stock options awards, warrants, RSUs, and shares subject to purchase under our employee stock purchase plan and would be subject to the treasury stock method when dilutive the terms of which are described in further detail in Note 12 – Share Based Compensation.
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Options to purchase common stock |
|
|
|
|
|
|
||
Shares committed under ESPP |
|
|
|
|
|
— |
|
|
Warrants |
|
|
|
|
|
|
||
Restricted stock units |
|
|
|
|
|
|
||
Total |
|
|
|
|
|
|
||
F-24
Note 14 - Income Taxes
Since inception, the Company has incurred net taxable losses, and accordingly,
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Federal statutory income tax rate |
|
|
% |
|
|
% |
||
State income taxes, net of federal benefit |
|
|
|
|
|
|
||
Research and developments credits |
|
|
|
|
|
( |
) |
|
Permanent items |
|
|
( |
) |
|
|
( |
) |
Change in valuation allowance |
|
|
( |
) |
|
|
( |
) |
Effective income tax rate |
|
|
— |
% |
|
|
— |
% |
The tax effects of temporary differences that give rise to significant portions of the deferred income tax assets and liabilities are as follows (in thousands):
|
|
As of December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Deferred Tax Assets: |
|
|
|
|
|
|
||
Net operating loss carryforwards |
|
$ |
|
|
$ |
|
||
Research and development tax credits |
|
|
|
|
|
|
||
Interest expense limitation |
|
|
|
|
|
— |
|
|
Property and equipment |
|
|
|
|
|
|
||
Stock-based compensation |
|
|
|
|
|
|
||
Accruals and reserves |
|
|
|
|
|
|
||
Total |
|
|
|
|
|
|
||
Valuation allowance |
|
|
( |
) |
|
|
( |
) |
Total deferred tax assets after valuation allowance |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Deferred Tax Liabilities: |
|
|
|
|
|
|
||
Intangible assets |
|
|
( |
) |
|
|
( |
) |
Total deferred tax liabilities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Net deferred tax assets and liabilities |
|
$ |
— |
|
|
$ |
— |
|
At December 31, 2021, the Company had $
In assessing the realizability of its deferred tax assets, the Company considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. The Company considers the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies in making this assessment. As the Company does not have any historical taxable income, projections of future taxable income over the periods in which the deferred tax assets are deductible, and after consideration of the history of operating losses, the
F-25
During 2021, the Company determined that it now has uncertain tax positions related to its U.S. research and development credits. As of December 31, 2021 and 2020, there was
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Balance at January 1 |
|
$ |
|
|
$ |
— |
|
|
Additions for tax positions related to the current year |
|
|
|
|
|
|
||
Additions for tax positions related to prior years |
|
|
— |
|
|
|
|
|
Reductions for tax positions related to prior years |
|
|
— |
|
|
|
— |
|
Reductions related to settlements |
|
|
— |
|
|
|
— |
|
Reductions related to a lapse of statute |
|
|
— |
|
|
|
— |
|
Balance at December 31 |
|
$ |
|
|
$ |
|
||
The Company monitors proposed and issued tax law, regulations, and cases to determine the potential impact of uncertain income tax positions. At December 31, 2021, the Company had not identified any potential subsequent events that would have a material impact on unrecognized income tax benefits within the next twelve months.
The Company’s federal and state returns for all years remain open to examination by tax authorities.
Note 15 – Commitments and Contingencies
Leases
The Company leases facilities under non‑cancelable operating leases. Rent expense was $
Future minimum lease payments, which do not include amounts for common area maintenance, insurance, or taxes, for operating lease obligations are as follows (in thousands):
|
|
As of |
|
|
2022 |
|
$ |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 and thereafter |
|
|
— |
|
Total |
|
$ |
|
|
Co‑Development Agreement
In April 2014 and amended in October 2016, the Company entered into a worldwide agreement with AVEO to develop and commercialize AVEO's hepatocyte growth factor inhibitory antibody ficlatuzumab with the Company's proprietary companion diagnostic test, BDX004, a version of the Company’s serum protein test that is commercially available to help physicians guide treatment decisions for patients with advanced non‑small cell lung cancer (NSCLC). Under the terms of the agreement, AVEO will conduct a proof‑of‑concept (POC) clinical study of ficlatuzumab for NSCLC in which BDX004 will be used to select clinical trial subjects (the NSCLC POC Trial). Under the agreement, the Company and AVEO would share equally in the costs of the NSCLC POC Trial, and each would be responsible for
In September 2020, the Company exercised its opt-out right with AVEO for the payment of
There were
F-26
License Agreement
In August 2019, we entered into a non-exclusive license agreement with Bio-Rad Laboratories, Inc. (Bio-Rad) (the Bio-Rad License). Under the terms of the Bio-Rad License, the Company received a non-exclusive license, without the right to grant sublicenses, to utilize certain of Bio-Rad’s intellectual property, machinery, materials, reagents, supplies and know-how necessary for the performance of Droplet Digital PCR™ (ddPCR) in cancer detection testing for third parties in the United States. The Company also agreed to purchase all of the necessary supplies and reagents for such testing exclusively from Bio-Rad, pursuant to a separately executed supply agreement (the Supply Agreement) with Bio-Rad. As further consideration for the non-exclusive license, the Company agreed to pay a royalty of
On May 13, 2021 (Effective Date), we reached agreement with CellCarta Biosciences Inc. (formerly Caprion Biosciences, Inc.) (the CellCarta License) on a new royalty bearing license agreement for the Nodify XL2 test. The parties agreed to terminate all prior agreements and replace with this new arrangement, which has a
As part of the acquisition of Oncimmune, the Company entered into several agreements to govern the relationship between the parties. The Company agreed to a license agreement and royalty payment related to an acquired diagnostic test of
Litigation, Claims and Assessments
From time to time, we may become involved in legal proceedings or investigations which could have an adverse impact on our reputation, business and financial condition and divert the attention of our management from the operation of our business. In September 2021, we reached a settlement agreement with the plaintiffs, which received preliminary approval from the Circuit Court of the City of St. Louis, State of Missouri (the Court) on November 10th, regarding a dispute involving the Telephone Consumer Protection Act (TCPA). On January 31, 2022, the Court approved the final settlement payment to third parties of approximately $
Note 16 – Subsequent Events
Common Stock Purchase Agreement
On March 7, 2022 (the Effective Date), the Company entered into a purchase agreement, dated as of March 7, 2022 with Lincoln Park Capital Fund, LLC, an Illinois limited liability company (Lincoln Park), pursuant to which Lincoln Park has committed to purchase up to $
F-27
Lincoln Park has no right to require the Company to sell any shares of common stock to Lincoln Park, but Lincoln Park is obligated to make purchases as the Company directs, subject to certain conditions. In all instances, the Company may not sell shares of its common stock to Lincoln Park under the Purchase Agreement if doing so would result in Lincoln Park beneficially owning more than
Actual sales of shares of common stock to Lincoln Park under the Purchase Agreement will depend on a variety of factors to be determined by the Company from time to time, including, among others, market conditions, the trading price of the common stock and determinations by the Company as to the appropriate sources of funding for the Company and its operations. The net proceeds, if any, under the Purchase Agreement will depend on the frequency and prices at which the Company sells shares of its common stock to Lincoln Park. The Company intends to use any net proceeds from the sale of its common stock to Lincoln Park to advance its growth strategy and for general corporate purposes. On the Effective Date, the Company issued
The Purchase Agreement may be terminated by the Company at any time, at its sole discretion, without any cost or penalty, by giving one business day notice to Lincoln Park to terminate the Purchase Agreement. Lincoln Park has covenanted not to cause or engage in any manner whatsoever, any direct or indirect short selling or hedging of the common stock. Although the Company has agreed to reimburse Lincoln Park for a limited portion of the fees it incurred in connection with the Purchase Agreement, the Company did not pay any additional amounts to reimburse or otherwise compensate Lincoln Park in connection with the transaction, other than the issuance of the Commitment Shares.
Centennial Valley Properties I, LLC Lease Agreement
On March 11, 2022, the Company entered into a Lease Agreement (the Lease) with Centennial Valley Properties I, LLC, a Colorado limited liability company (the Landlord) for office and laboratory space located at 919 West Dillion Road; Louisville, Colorado (the Leased Premises). The purpose of the Lease is to replace the Company’s current leased premises at 2970 Wilderness Place, Suite 100 in Boulder, Colorado and the Company intends to move its corporate headquarters to the Leased Premises by mid-2023.
Under the Lease, the Company will lease approximately
The Lease includes various covenants, indemnities, defaults, termination rights, and other provisions customary for lease transactions of this nature, including maintaining a $
F-28