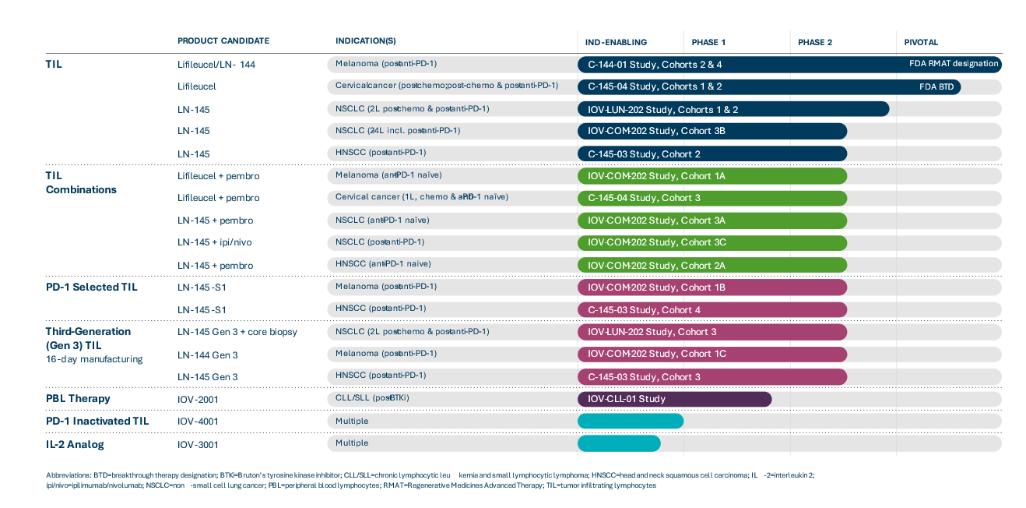
Our current product candidate pipeline is summarized in the figure below:
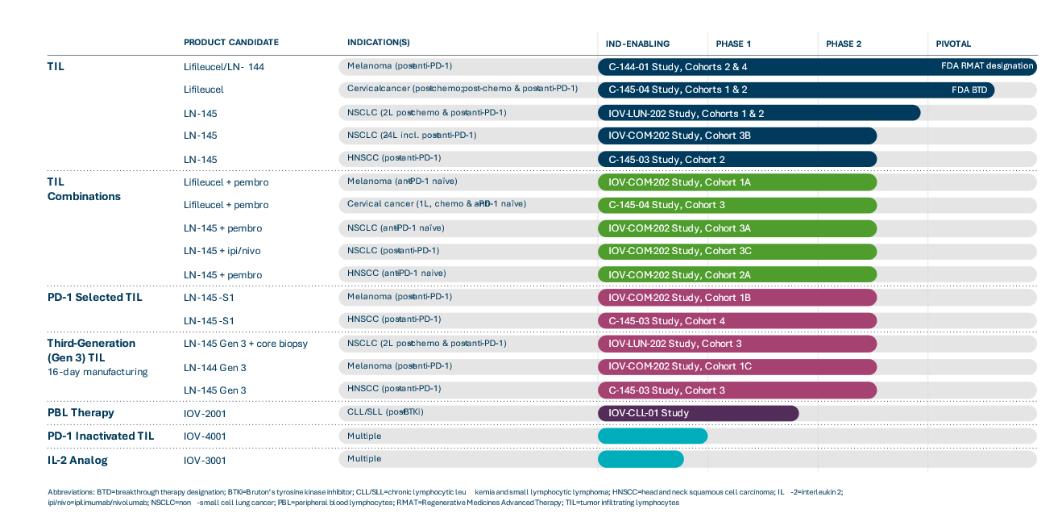
TIL Monotherapy in Metastatic Solid Tumor Cancers
We have investigated TIL monotherapy in metastatic melanoma, cervical cancer, NSCLC and HNSCC. We are conducting a Phase 2 clinical trial, C-144-01, of our lead TIL product candidate, lifileucel, for the treatment of metastatic melanoma. This multicenter pivotal trial enrolled melanoma patients with disease progression following treatment with at least one systemic therapy, including a PD-1 inhibitor and, if BRAF mutated, a BRAF inhibitor, or a combination of BRAF and MEK inhibitors. Cohort 4 of the C-144-01 clinical trial is a single-arm cohort intended to support the submission of a Biologics License Application, or BLA, to the FDA for lifileucel. Cohorts 2 and 4 of the C-144-01 trial use our Gen 2 manufacturing process. We completed and closed enrollment of patients into Cohort 2 of the C-144-01 trial in 2018. Results from Cohort 2 of the C-144-01 clinical trial were initially reported at the American Society of Clinical Oncology annual meeting, or ASCO, in June 2019 and most recently updated at ASCO in June 2021. As of the data extract in April 2021, in 66 patients with metastatic melanoma in Cohort 2, treatment with lifileucel resulted in an objective response rate, or ORR, of 36%, as assessed by investigator, with 3 complete responses and 21 partial responses. The disease control rate, or DCR, was 80.3%. Median duration of response, or DOR, in Cohort 2 had not been reached after 33.1 months of median study follow up. Results from Cohort 2 presented at ASCO in June 2021 also suggest that early intervention with lifileucel at the time of initial progression on anti-PD-1 therapy may maximize benefit. Patients in Cohort 2 were heavily pretreated and had a mean of 3.3 prior therapies. We have previously reported durable responses across a wide age range of metastatic melanoma patients, among those who have received prior anti-CTLA-4 and BRAF targeted treatments, regardless of BRAF mutation status, and in patients with PD-L1 high and low status. The adverse event profile was generally consistent with the underlying advanced disease and the profile of the lymphodepletion and IL-2 regimens. In addition, detailed Cohort 2 data was published in the Journal of Clinical Oncology in May 2021.
Pivotal Cohort 4 of the C-144-01 trial was enrolled to evaluate ORR as read out by an Independent Review Committee, or IRC, as the primary endpoint based on our interpretation of discussions with the FDA as part of an End of Phase 2, or EOP2, meeting held with the FDA in the third quarter of 2018. In October 2018, based on the data provided to the FDA during the EOP2 meeting, we announced that lifileucel had received a Regenerative Medicines Advanced Therapy, or RMAT, designation from the FDA. Enrollment in Cohort 4 of the C-144-01 trial commenced in March 2019 and patient dosing was completed in January 2020. A total of 87 patients were dosed with Gen 2 product released for Cohort 4. In May 2020, we disclosed initial results from Cohort 4 for 68 patients with two radiological assessments, as determined by investigator.
5